

Abstract

Modern therapeutic development often involves several stages that are interconnected, and multiple iterations are usually required to bring a new drug to the market. Computational approaches have increasingly become an indispensable part of helping reduce the time and cost of the research and development of new drugs. In this Perspective, we summarize our recent efforts on integrating molecular modeling and machine learning to develop computational tools for modulator design, including a pocket-guided rational design approach based on AlphaSpace to target protein–protein interactions, delta machine learning scoring functions for protein–ligand docking as well as virtual screening, and state-of-the-art deep learning models to predict calculated and experimental molecular properties based on molecular mechanics optimized geometries. Meanwhile, we discuss remaining challenges and promising directions for further development and use a retrospective example of FDA approved kinase inhibitor Erlotinib to demonstrate the use of these newly developed computational tools.
Introduction
The cost of research and development of new drugs has risen over the past decade.1−3 There is great interest in advancing computational methods, including molecular modeling and machine learning (ML), to greatly accelerate the process of drug discovery and development.4−10 The goal of drug discovery is to find small molecules that inhibit the disease-related target and, therefore, modulate the disease. Meanwhile, the pharmacokinetic properties including absorption, distribution, metabolism, excretion, and toxicity (ADME/T) of small molecules also need to be considered in the drug development process.11 The success of drug development requires both good efficacy and the desired pharmacokinetic properties of the drug molecules. Computer-aided drug design (CADD) offers valuable tools in finding candidate compounds in all stages of drug discovery and development.
It should be noted that a modern therapeutic development process often involves multiple nonlinearly interconnected stages.12Figure 1A shows a simplified “pipeline” of therapeutic development where CADD tools are playing important roles. Hit identification is the first step which involves finding the compounds with desired bioactivity.13 Ligand based methods use prior knowledge to predict new drug compounds with similar bioactivities (Figure 1B).14 Similarity search,15 pharmacophore modeling,16,17 and quantitative structure–activity relationships18,19 are common tools for ligand based drug screening. Structure based methods such as molecular docking20−26 and molecular dynamics simulations27−32 are common computational tools used to predict the binding affinity between the protein and ligand (Figure 1C). Binding site prediction tools33−39 offer information on the protein surface where a ligand molecule binds to produce the desired output (activation, inhibition, or modulation).5 If the structure of the protein is not solved experimentally, homology modeling40−43 and ab initio protein structure modeling44−47 approaches can be used to predict the protein structure.
Figure 1.

Roles of computational models at the stages of drug discovery and development.
After discovering the hit compounds, subsequent steps involve producing more potent and selective compounds with desirable physicochemical properties.13 It is estimated that over 40% of drug candidates are withdrawn in preclinical tests due to ADME/T concerns.48 To address the problem, a number of computational models have been developed to predict the ADME/T properties of small molecules.49−60 The accurate prediction of molecular properties requires models to learn more robust molecular representations for 1D SMILES,61 2D graph,62 and 3D geometry.63
As computational tools at all stages of drug discovery and development have been extensively reviewed recently,64,65 in this perspective, we focus on the recent progress related to our group. Specifically, we first summarize protein pocket identification and analysis methods based on fragment-centric topographical mapping. Subsequently, we discuss recent advances and promising directions in developing ML and deep learning (DL)-based molecular docking models. Finally, we describe our recent efforts to improve small molecule representation learning and explore chemical space66 more efficiently.
1. Protein Pocket Identification and Representation
1.1. Protein Pocket Identification: Fragment-Centric Topographical Mapping
In contemporary approaches to structure-based drug design, focus is placed on the analysis of concave regions found on biomolecular surfaces. These regions can vary from well-defined small-molecule binding sites to extensive protein–protein interaction (PPI) interfaces.67 Gaining a deeper understanding of concave biomolecular surfaces holds immense potential for various applications in modulator design, such as lead optimization, enhanced ligand screening, and even the identification of previously undiscovered druggable sites.67−69 Furthermore, comparisons of these surfaces investigate ligand selectivity, off-target effects, and polypharmacological activity.70,71
One of the main tasks in analyzing the protein surfaces is identifying binding sites. There are a number of algorithms that do this, and these can be broadly classified as geometry-35,37,72−76 or energy-based methods.68,77−81 Geometry-based methods map out the concavities on the surface using grids or alpha-spheres, while energy-based methods calculate the interaction energy or surface accessibility between the protein and diverse small-molecule probes. The next step usually requires ML methods to characterize the binding site. For example, some methods use clustering to agglomerate data points into binding pockets and others use regression to score the concavities on the protein surface for their ligandability.82 Most methods can correctly predict and rank the voluminous and well-defined ligand binding pockets in holo structures.72,83,84
The alpha-sphere geometry-based representation of the concavities is an established strategy that relies on Voronoi diagrams.34,35,73,74,85−87 Voronoi diagrams form polyhedra defined by the planes that bisect adjacent atoms, resulting in a tessellation of the concave region. Alpha-spheres are placed at the vertices of the Voronoi diagram. These alpha-spheres are then clustered to represent binding pockets. Rooklin et al. developed a python program named AlphaSpace, an alpha-sphere-based method developing on the popular method fpocket,35 that leverages concepts of fragment-based drug discovery and caters to the analysis of PPI (Figure 2).34 To this aim, AlphaSpace uses a tuned clustering method to demarcate pockets that reflect on average one fragment per pocket and defines alpha-atoms, a theoretical ligand atom whose properties reflect a docked fragment. It represents entire PPI surfaces by retaining shallower fragment-centric pockets during the evaluation stage and suggests targetable pockets near the binding partner for ligand optimization. Furthermore, Katigbak et al. developed AlphaSpace 2.0, a python package that clusters the alpha-atoms to a β-cluster, a pseudomolecular representation of the concave site comprised of β-atoms (Figure 2).33 Each β-atom is replaced with a probe atom that iterates through different Vina atom types. The AutoDock Vina score is calculated for each atom type of each probe atom. Then the best Vina score for each probe atom is summed to generate the overall β-score. This β-score reflects the maximum theoretical docking score of the β-cluster.
Figure 2.

Workflow of AlphaSpace to optimize a ligand to bind to a protein begins with a selected PDB structure and follows the purple arrows until the optimization is validated experimentally. The additions introduced by AlphaSpace2.0 follow the orange arrows.
Both AlphaSpace and AlphaSpace2 offer alignment-free and alignment-based pocket matching protocols, respectively, allowing for comparisons of binding sites between structures within the alpha-sphere framework.33,34 Both methods assess the change in the inhibitor binding site of PPI interfaces using the curated 2P2I database, which provides structures of a protein in the apo, PPI complex, or inhibitor bound PPI complex states. In both cases they can observe the binding pockets on the apo structure that corresponds to the inhibitor binding site, and AlphaSpace2 reports similar ligand-ability estimates of that binding site across structures. However, on the CryptoSite database consisting of structures where a pocket forms in the holo state but not the apo state, AlphaSpace2 reports differences in ligand-ability estimates.
DL methods are also gaining prominence due to their ability to learn complex relationships from raw protein data and predict binding surfaces.82,88 DL architectures can learn the representation and functional roles of the binding pockets without first preprocessing the data to explicitly detect the pockets. Mylonas et al. developed DeepSurf, a 3D-convolutional neural network (CNN) trained on voxels placed on the protein surface.89 It had superior performance compared to other methods tested in the SHREC 2022 contest at identifying druggable sites on diverse data sets of apo and holo structures.83,89 Gainza et al. proposed MaSIF, a surface representation framework that uses overlapping radial patches characterized by a chemical and geometric fingerprint.90,91 These patches can be input into geometric neural network architectures and trained to perform particular tasks, including predicting PPI and ligand sites. Most recently, Smith et al. developed Graph Attention Site Prediction (GrASP) which uses a graph attention network (GAT) architecture to encode the binding site from a protein structure graph. This model is trained on a modified version of sc-PDB to predict the likelihood of an atom to be part of the binding site. These atoms are then clustered and aggregated into binding sites with ligand-ability scores.92 This results in superior precision and recall compared to another graph neural network (GNN)-based method, P2Rank, on the COACH420 and HOLO4K benchmarks.92
1.2. Experimentally Validating Predictions of Underutilized but Targetable Regions
AlphaSpace has guided the optimization of minimal protein mimetics that target PPIs by expanding mimetics to target secondary binding sites and incorporating noncanonical amino acids to reach untargeted binding pockets
(93−95) Guided by the binding pocket analysis, researchers introduced a terminal tryptophan of a p53 mimetic to interact with a previously untargeted secondary site which resulted in an ∼10-fold improvement of binding affinity to MdmX and Mdm2.95 This interaction was verified to bind to the secondary site through NMR. In another case, optimization steps were able to reverse the loss of binding affinity resulting from truncating the full length protein to a minimal mimetic. The truncated helical MML-mimetic bound poorly to the KIX domain of the p300/CBP coactivator (Kd: >100 μm), but a MML mimetic mutated with noncanonical amino acids was optimized to a similar binding affinity (Kd: 3.3 ± 0.5 μm) as the full-length wildtype MML (Kd: 1.0 ± 0.3 μm).93 Moreover, suggested mutations to the cross-linked helix dimers of the NEMO coiled coil reversed the binding affinity loss due to the aforementioned. These mimetics were also able to inhibit the native PPI in vivo.94 In these cases, the role of AlphaSpace was to suggest initial fragments for improvement, but expertise and further experimental optimization were necessary to generate stable mimetics and maximize protein–ligand binding affinity.93−96
AlphaSpace has also guided the analysis of protein structures for targetable binding sites to design small molecule inhibitors and understand their binding mechanisms. Some computational experiments qualitatively studied the interaction mechanisms and binding modes by docking inhibitors to the most targetable pockets of the transmembrane protein ABCG2 and by running molecular dynamics (MD) simulations of the complexes.97,98 Other binding pocket analyses led to successful prospective docking studies. Hou et al. detected a cryptic pocket in a metastable state of the apo striatal-enriched protein tyrosine phosphatase (STEP) from MD simulations. Ensemble docking to this state at the cryptic pocket resulted in 11 novel inhibitors (IC50 = 9.7–47.7 μm).99 These experimental validations highlight the utility and role that AlphaSpace can play in identifying pockets to develop ligands with improved binding.
2. Protein–Ligand Docking in the Machine-Learning Era
Employing computational methods to discover bioactive compounds is becoming an indispensable technique to accelerate the process of drug discovery,100−107 leading to the successful discovery of several approved drugs.104,108−110 The drug discovery process encompasses two major categories: ligand-based drug discovery (LBDD) and structure-based drug discovery (SBDD). LBDD is an indirect approach that employs quantitative structure–activity relationship methods, molecular similarity, and pharmacophore modeling to predict various properties of compounds.14 On the other hand, SBDD involves using the three-dimensional structures of the protein and small molecule to identify or optimize drug compounds.101,111 SBDD focuses on recognizing binding sites and essential interactions crucial to the biological functions of the target. By doing so, it suggests therapeutic compounds that can compete with these crucial interactions, thereby disrupting biological pathways effectively.
Virtual screening (VS) is a method within SBDD where extensive libraries of commercially available, druglike chemical compounds are computationally screened against target proteins with known or predicted 3D structures in a high-throughput manner. The primary goal of VS is to identify potential compounds that exhibit a high predicted binding affinity for the target protein. These selected compounds are then subjected to experimental testing to validate their actual binding capabilities.112,113
Molecular docking is a widely used computational procedure in VS protocols.114−117 It operates based on the lock-and-key118 model of drug action and predicts the noncovalent interactions between the target and small molecules. In structure-based VS, molecular docking methods are commonly used to assess large libraries of molecules.26
2.1. Protein–Ligand Scoring Functions
Molecular docking programs rely on a scoring function to evaluate the binding affinity and optimize the binding orientation between protein and ligand.21−23,25,119−121 The scoring function plays a pivotal role in accurately predicting how well the ligand binds to the target protein. Consequently, developing a robust, efficient, and accurate scoring function is a critical component of molecular docking.107
A rigorous theoretical prediction of protein–ligand binding affinity requires the calculation of the free energy difference between the bound and unbound state. However, existing methods for free energy calculation, such as free energy perturbation122,123 and thermodynamic integration,124,125 are computationally expensive, making them impractical for virtual screening over a large set of compounds. To overcome this limitation, scoring functions serve as fast and approximate computational methods to evaluate the strength of noncovalent interactions between proteins and ligands, based on their 3D binding poses.126−128 They are widely used in SBDD to determine the binding mode of ligands (docking power), predict the binding affinity between proteins and ligands (scoring power), identify hits from a large compound library for given protein targets (screening power), and perform structure–activity relationship analyses for hit-to-lead and lead optimization.105,129,130 It is important to note that scoring functions provide estimations of ligand binding affinity, and these predicted scores may not align closely with binding affinities determined through rigorous methods, such as free energy perturbation. For an in-depth understanding of terms such as “scoring power”, “docking power”, and “screening power”, we direct readers to the comprehensive definitions provided in the paper “Comparative Assessment of Scoring Functions: The CASF-2016 Update”.131
Classical scoring functions use a linear combination of force-field or interaction descriptors to evaluate the binding affinity between proteins and ligands. They can be categorized into different types based on their underlying principles. Physics-based scoring functions use the fundamental molecular physics terms derived from experimental data and/or QM calculations;21,119,121 knowledge-based scoring functions use the statistical potentials derived from experimentally determined protein–ligand structures;132−135 and empirical scoring functions make predictions by fitting experimentally measured binding affinity data using linear regression on a set of physical descriptors.21−23,120,136−139
In recent years, ML-based scoring functions have garnered increasing attention due to the rapid growth of experimental data and advancements in computational infrastructure.140,141 Unlike classical scoring functions that adopt linear forms, ML-based scoring functions have the capability to learn more complex functional forms using ML algorithms such as support vector machine (SVM),142 random forest (RF),143 and extreme gradient boosting (XGB).144 In 2010, Ballester et al. introduced RF-score which is the first ML-based scoring function that outperforms classical scoring functions in terms of scoring power.145 In 2013, Zillian et al. developed SFCscoreRF which achieved significantly higher scoring power on common benchmarks.146 However, both models performed much worse when performing docking and screening tasks compared to classical scoring functions.147,148 The reader is directed to this review by Chao et al.107 for more in-depth discussion on scoring functions.
The Δ-ML strategy improves the scoring-ranking-docking-screening power of ML-based scoring functions
To address this problem, Wang et al. applied a Δ-ML strategy, which involves using ML to predict the correction between experimental binding affinity and computationally predicted binding affinity. The final predicted score is obtained by adding this ML correction to classical scoring functions. The first model to employ this strategy is ΔVinaRF20,149 a random forest model that predicts the corrections to the Vina score. It uses pharmacophore-based solvent-accessible surface area descriptors and AutoDock Vina features as input. ΔVinaRF20 demonstrated superior performance in all power tests on both CASF-2013 and CASF-2007 benchmarks compared to classical scoring functions.150,151 Subsequently, Lu et al. have proposed the ΔVinaXGB model which uses XGB trees.152,153 This model incorporates features from explicit water molecules, metal ions, and ligand conformational stability in addition to the ΔVinaRF20 features.152 The ΔVinaXGB model outperformed other scoring functions on the CASF-2016 benchmark data set.131 Recently, Yang et al. have developed the ΔLin_F9XGB model154 based on a classical scoring function named Lin_F9.139 Lin_F9 is a linear empirical scoring function that builds on AutoDock Vina, overcoming some of Vina’s limitations by introducing new empirical terms for better scoring accuracy. By applying the Δ-ML strategy to the Lin_F9 scoring function, and improving feature selection and the training set, ΔLin_F9XGB achieves superior scoring, ranking, and screening performance on the CASF-2016 benchmark.154
In addition to the CASF-2016 benchmark data set, previous models have undergone extensive testing on the LIT-PCBA data set, specifically designed for virtual screening and ML purposes.155 Preliminary results have shown that the LIT-PCBA data set is challenging due to the high imbalance of active/inactive compounds, common molecular properties shared between active and inactive compounds, and weak potencies of the active compounds.154,156 The average top 1% enrichment factor achieved by ΔLin_F9XGB is 5.55, which outperforms other scoring methods such as AutoDock Vina, ΔVinaRF20, and Lin_F9.
Learning distance likelihood potential boosts the docking and screening performance of DL-based scoring functions
Aside from traditional ML-based scoring functions, the emergence of DL methods has introduced new strategies for developing scoring functions in drug discovery. Neural network-based models use hand-crafted features as input, including protein–ligand interactions and ligand-dependent descriptors.147,157,158 Convolutional neural network (CNN)-based scoring functions adopt a different approach by using vectorized 3D grids that cover the protein–ligand binding region as input. These grids are processed through CNN layers, followed by readout layers to make predictions about the binding affinity.159−164 Graph neural networks (GNNs) represent molecules as graphs and can learn on the protein–ligand binding system through graph convolutions.165−169 However, despite the increasing popularity of DL-based scoring functions, it is worth noting that these models have not significantly outperformed traditional ML-based scoring functions on well-established benchmark data sets like CASF.107
Recently, Méndez-Lucio et al. proposed a geometric DL-based model named DeepDock170 which learns the probability density distribution of the distance between ligand atom and the protein surface. This approach showed competitive performance in docking and screening tasks when compared to those with well-established scoring functions. Building on the concept of distance likelihood, Shen et al. developed RTMScore.171 Unlike DeepDock, RTMScore overcomes the need for protein surface triangulation by using the probability density distribution of distances between ligand atoms and protein amino acids at the residue level. To achieve this, RTMScore represents proteins as undirect graphs at the residue level and leverages graph transformers to learn the representation of both proteins and ligands. As a result, RTMScore achieves top docking and screening power on the CASF-2016 data set, outperforming state-of-the-art ML- and DL-based scoring functions.
However, RTMScore utilizes biased protein pockets generated from known binder structures as the input protein structure. For example, when evaluating the binding score of the ligand 1A30 on the protein 1E66 (Figure 3A), only the protein structure within 10 Å of the binder ligand 1E66 is used along with the docked ligand 1A30 structure as model input (Figure 3B). This selection method may lead to two potential problems. First, this method requires the binder structure to cut the protein, while in real world virtual screening scenarios, the protein may not have a known binder, or the binder structure may be unknown. Second, the selected pocket is biased toward the binder ligand, potentially neglecting essential interactions between the decoy ligands and the protein when the decoy structures are far from the binder ligand (Figure 3B and C).
Figure 3.

Biased protein pockets when evaluating the docking power and screening power on the CASF-2016 data set. (A) Protein and ligand structures. (B) The biased protein pocket resulting from using the binder ligand structure to cut the protein. (C) The unbiased protein pocket without using the binding structure.
The influence of the biased pocket selection was investigated by using docked decoy structures to cut the proteins (Figure 3C) and retesting RTMScore on the CASF-2016 data set. The results, as presented in Table 1, revealed a performance drop when the unbiased pocket was used as the model input for RTMScore models. Despite this drop in performance, the unbiased model still outperformed most of the state-of-the-art models in terms of docking and screening power. This finding highlights the importance of addressing the issue of biased pocket selection in DL-based scoring functions. Although the performance of RTMScore was affected by the unbiased pocket, the fact that it still outperformed many state-of-the-art models indicates that learning distance likelihood potential is a promising direction to improve the docking and screening performance of DL scoring functions.
Table 1. Docking Power and Screening Power of RTMScore on CASF-2016 Data Set When Using the Unbiased Pocket Selection Method, Compared with Other State-of-the-Art Models.
| Docking
Power (Success Rate) |
Forward
Screening Power |
||||
|---|---|---|---|---|---|
| Model Name | Without native poses | With native poses | EF1% | Top1 success rate | Reference |
| RTMScore1-unBiased | 0.877 | 0.909 | 24.84 | 0.509 | This work |
| RTMScore1-Biased | 0.937 | 0.986 | 28.78 | 0.737 | (171) |
| DeepDock | 0.870 | – | 16.41 | 0.439 | (170) |
| PIGNet | 0.870 | – | 19.6 | 0.554 | (172) |
| DeepBSP | 0.872 | 0.885 | – | – | (173) |
| OnionNet-SFCT | – | 0.937 | 15.50 | 0.421 | (174) |
| ΔLin_F9XGB | – | 0.867 | 12.61 | 0.404 | (154) |
| ΔVinaXGB | – | 0.916 | 13.14 | 0.368 | (152) |
| ΔVinaRF20 | 0.849 | 0.891 | 11.73 | 0.421 | (149) |
| KORP-PL | 0.856 | 0.891 | 22.23 | 0.421 | (175) |
| GlideScore-SP | 0.846 | 0.877 | 11.44 | 0.368 | (23) |
| ChemPLP@GOLD | 0.832 | 0.860 | 11.91 | 0.351 | (176) |
| AutoDock Vina | 0.846 | 0.902 | 7.70 | 0.298 | (22) |
The very recent work by Shen et al. extends the RTMScore model to the GenScore framework.177 By introducing an adjustable binding affinity term during the training of the probability density distribution, GenScore successfully addresses the issue of low scoring and ranking power experienced by RTMScore, achieving balanced scoring, ranking, docking, and screening power on the CASF-2016 data set. Furthermore, the evaluation of GenScore on the LIT-PCBA data set for real-world screening power is particularly noteworthy. Their best-performing GenScore model exhibits an impressive average top 1% enhancement factor of 6.80 among all targets, demonstrating an excellent screening power.
However, it is important to acknowledge that the biased pocket problem, as mentioned earlier, is not explicitly addressed by GenScore. The reliance on pocket selection using binder ligand structures limits its applicability in real-world virtual screening protocols, especially for novel targets for which binder ligands might not be known or available. Addressing the biased pocket problem would undoubtedly enhance the utility and versatility of RTMScore, GenScore, and other DL-based scoring functions in real-world virtual screening scenarios, making them more effective in identifying potential drug candidates for novel targets with unknown binder structures. Future research in this direction may help overcome this limitation and further establish DL-based scoring functions as valuable tools in drug discovery efforts.
2.2. Molecular Docking in the Deep Learning Era
Traditional molecular docking methods such as AutoDock Vina22 and Lin_F9139 rely on the scoring functions and optimization algorithms that search for the global maximum of the scoring functions. The optimization algorithm rotates the rotatable bonds in the molecule, translates the molecule, and rotates the whole molecule until it finds the best conformation with the highest score predicted by the scoring function. In principle, the optimization algorithms can also be applied to DL-based scoring functions. However, in practice, the optimization algorithms often fail to converge when using DL-based scoring functions, especially for those compounds with a high number of rotatable bonds.170 Recently, researchers have developed DL-based docking models to predict binding poses in one shot by treating molecular docking as a regression task.178,179 Although these models are faster than traditional docking methods, they do not significantly outperform traditional docking methods in terms of accuracy.
In late 2022, Corso et al. introduced DiffDock,180 a diffusion generative model which learns the ligand pose distribution from ligand and protein structures. The model employs a diffusion process over the ligand center position, orientation, and torsional angles of each rotatable bond. By iteratively refining ligand poses through updates of translations, rotations, and torsion angles, DiffDock can sample binding poses from random initial ligand conformations.
On the PDBBind blind docking task, DiffDock achieves a 38% top-1 accuracy, significantly outperforming state-of-the-art molecular docking models.180 This success demonstrates the potential of DL methods in improving the ligand binding pose prediction. By combination of DiffDock with modern DL-based scoring functions, a more robust virtual screening protocol can be developed for real-world drug discovery applications.
3. Molecular Representation Learning with Deep Learning Methods
3.1. Exploring Chemical Space Efficiently with Force-Field Optimized 3D Geometry and 2D Molecular Graphs
In theoretical chemistry, the Schrödinger equation (SE) serves
as the “standard model” because it represents the physics
of the charged particles that make up almost all of the molecules
and materials. Unfortunately, the analytical solution to SE is only
available for systems with a few degrees of freedom. Numerical solutions
such as high-accurate quantum mechanics (QM) calculations impose prohibitive
computational cost.181−184 To overcome this challenge, ML approaches to predict molecular energies
have been crafted to circumvent the task of solving SE of the system 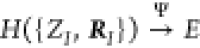 by learning the mapping from the geometry
to the energy
by learning the mapping from the geometry
to the energy  through supervised learning.185−191 With advances in computer infrastructure and the availability of
organized chemistry data sets, DL192 methods
are gaining popularity. DL methods utilize the atomic coordinates
{RI} and
atom types {ZI} to predict
molecular energetics directly, avoiding the need for hand-crafted
molecular descriptors used in ML methods.193−203 In the past few years, many DL models204−213 with distinct model architectures have been developed for the prediction
of molecular energetics with better performance on the QM9 benchmark
data set.214−216 This data set contains minimized structures
and molecular properties calculated using the density functional theory217 (DFT) method for 133,885 molecules composed
of H, C, N, O, and F atoms with up to nine heavy atoms. However, despite
the progress, many DL models developed on the QM9 data set still require
DFT-optimized geometries as input for prediction, which leads to the
original problem of computational expense not being fully addressed.
through supervised learning.185−191 With advances in computer infrastructure and the availability of
organized chemistry data sets, DL192 methods
are gaining popularity. DL methods utilize the atomic coordinates
{RI} and
atom types {ZI} to predict
molecular energetics directly, avoiding the need for hand-crafted
molecular descriptors used in ML methods.193−203 In the past few years, many DL models204−213 with distinct model architectures have been developed for the prediction
of molecular energetics with better performance on the QM9 benchmark
data set.214−216 This data set contains minimized structures
and molecular properties calculated using the density functional theory217 (DFT) method for 133,885 molecules composed
of H, C, N, O, and F atoms with up to nine heavy atoms. However, despite
the progress, many DL models developed on the QM9 data set still require
DFT-optimized geometries as input for prediction, which leads to the
original problem of computational expense not being fully addressed.
Force-field optimized geometries can be used to predict DFT-level molecular energetics
In 2019, Lu et al. have demonstrated the feasibility of using Merck Molecular Force Field (MMFF)183 optimized geometry for gas phase DFT-level electronic energy prediction.209 They first train a model called DTNN_7ib on the QM9 data set using DFT-optimized geometries as input and then retrain the readout layer of the model on their prepared MMFF-optimized geometries. The trained DTNN_7ib achieves a mean-absolute-error (MAE) of 0.79 kcal/mol on DFT-level electronic energy by using MMFF-optimized geometry.
In 2021, Lu et al. extended the effectiveness of MMFF-optimized geometry beyond the QM9 benchmark data set by constructing the Frag20 data set.63 These data consisted of DFT-calculated properties for over half a million molecules, with both MMFF and DFT-optimized geometries, and consisted of elements H, B, C, O, N, F, P, S, Cl, and Br, with no more than 20 heavy atoms. They train a model named sPhysNet on the Frag20 data set and achieve better than or close to chemical accuracy (1 kcal/mol) on multiple test sets, including two external test sets based on experimental crystal structures. The work indicated the potential of using MMFF-optimized geometries as a more computationally efficient alternative to explore chemical space66 compared to solely relying on DFT-optimized geometries.
MMFF geometry also proves useful for predicting experimental properties
In 2022, Xia et al.51 developed a DL model named sPhysNet-MT-ens5 for predicting experimental hydration free energy and octanol/water partition coefficient (logP) using MMFF optimized geometry. Figure 4 shows the overall workflow using the model for prediction. For each compound, up to 300 conformations are generated using EDKTG218 implemented in RDKit219 and then optimized with MMFF. The conformation with the lowest MMFF energy is then selected as the input geometry for the prediction of experimental hydration free energy and logP, as well as other properties of the compound. An ensemble of five independently trained sPhysNet-MT models is used for prediction, and the average of the five predictions is used as the ensemble prediction. The model achieves a root-mean-square error (RMSE) of 0.620 kcal/mol on the FreeSolv220 data set for experimental hydration free energy prediction, as well as an RMSE of 0.393 on the PHYSPROP18,221−223 data set for experimental logP prediction.
Figure 4.

Overall workflow using sPhysNet-MT-ens5 for energetics prediction. (A) MMFF geometry generation protocol. (B) The prediction of electronic energies and transfer free energies with sPhysNet-MT-ens5.
Spatial information computed from MMFF geometry enhances 2D molecular representations
Unlike models that use 3D molecular geometry as input, many DL models use 2D molecular graphs to predict various chemical properties.224−231 While they have shown success in these tasks, their limitations in representing molecules with 2D graphs have been widely discussed.232−236 This is because models based solely on 2D connection graphs may not distinguish between molecules with 3D spatial structures. For example, message passing GNNs cannot distinguish decaprismane237 C20H20 from dodecahedrane238 C20H20 purely based on their 2D graphs.235
To address this limitation, models that incorporate geometry information can achieve better performance.52,239 In 2022, Zhang et al. developed A3D-PNAConv52 which uses both 2D and 3D information on molecules. They demonstrate that incorporating 3D atomic features obtained from MMFF-optimized geometries enhances the learning power of GNN models in predicting calculated solvation free energies. The 3D atomic features are calculated by atom-centered symmetry functions (ACSFs),200 which are sets of many-body interaction functions encoding atomic environments in 3D structures. The GNNs use the 3D atomic features as the initial atomic embedding and then perform several iterations of message passing on the 2D molecular graphs. In comparison, they have also trained GNNs without 3D atomic features. The GNNs with 3D atomic features as initialization outperform their 2D counterpart for predicting calculated water solvation free energy on their Frag20-Aqsol-100k data set. This indicates that using 3D atomic features obtained from MMFF geometries can enhance the learning capability of GNN-based models.
3.2. Enhancing Downstream Tasks through Transfer Learning with Pretrained Molecular Modeling Data
In the early stages of the drug discovery process, data scarcity is the most common issue for DL models in the prediction of experimental physicochemical properties. DL models heavily rely on the size and quality of the training data, and they tend to overfit on small data sets, leading to poor generalizability and performance.240 In contrast, molecular modeling data can be obtained in larger quantities within a reasonable time frame, thanks to advancements in high-performance computing. As shown in Table 2, publicly available data sets generated using molecular modeling methods are significantly larger than experimental data sets in terms of size. To address the scarcity of experimental values, calculated data sets should be fully utilized when training DL models on experimental data.
Table 2. Overview of Common Experimental and Calculated Data Sets for Energetic-Related Properties.
| Type | Name | Data set size | #Heavy Atoms | Properties | Reference |
|---|---|---|---|---|---|
| Calculated | QM9 | 133,885 | 1–9 | Electronic energy, HOMO, LUMO, etc. | (215) |
| ANI-1 | 24,687,809 | 1–8 | Electronic energy | (241) | |
| Frag20 | 566,296 | 1–20 | Electronic energy | (63) | |
| Frag20-solv-678k | 678,916 | 1–20 | Electronic energies (gas water and octanol) | (51) | |
| Experimental | FreeSolv | 643 | 1–24 | Hydration Free Energy | (220) |
| PHYSPROP-logP | 14,174 | 3–85 | Water/octanol partition coefficient | (221,223) | |
| MNSOL | 3,037 | 0–28 | Solvation free energies and transfer free energies in 92 solvents | (242) |
Pretraining on DFT-calculated energetics aids in improving the prediction of downstream tasks on related experimental properties
The fine-tuning strategy is commonly employed in training DL models. First, a model is pretrained on a source task, and then, its trained weights are transferred as the initialization for a related target task. This fine-tuning approach often yields better performance compared to random initialization.243 For instance, Zhang et al. developed a data set named Frag20-Aqsol-100k which contains 100k diverse molecules sampled from Frag20 and CSD20 data sets, along with their calculated hydration free energy using their SMD_B3LYP protocol.52 They pretrained DL models on the Frag20-Aqsol-100k data set and fine-tuned the models on the FreeSolv data set, which contains 640 compounds with experimentally measured hydration free energy. Their fine-tuned model achieved state-of-the-art performance and can even achieve chemical accuracy with a very small training data size of just 100 molecules.52
Xia et al.51 extended the work and develop Frag20-solv-678k which contains 678,916 conformations and their calculated energetics in gas, water, and octanol phases. They pretrained a DL model on all three electronic energetics and the transfer free energies between the three phases, and fine-tuned the model on a combined data set composed of molecules from FreeSolv with experimental hydration free energy and PHYSPROP with experimental logP. The fine-tuned model predicts experimental hydration free energy with an RMSE of 0.620 kcal/mol on the FreeSolv data set, as well as experimental logP with an RMSE of 0.393 on the PHYSPROP data set, achieving state-of-the-art performance on both tasks simultaneously (Figure 5).
Figure 5.

Model performance comparison between sPhysNet-MT and other state-of-the-art models on the experimental hydration free energy and logP prediction. Results are adapted from the sPhysNet-MT manuscript.51
3.3. Improving Molecular Representations through Mutual Learning with Multitask Training
There are a wide range of ML and DL models that have been developed for a variety of chemical tasks. Although some chemical tasks are related, they are treated by different models with distinct model architectures. For example, the models developed for the prediction of high-level QM calculated molecular energetics204−212 and the models for predicting of experimental transfer free energies52,225,228,229,239,244,245 are often developed separately. While separate models may be more effective for accurate predictions for weakly related tasks,210,211 multitask models can be advantageous when tasks are well-related.246,247
Multitask training on related tasks leads to more robust molecular representation learning
Motivated by the fact that many molecular modeling methods use the direct relationship between absolute energies and transfer energies to predict experimental properties with satisfactory results,248−251 Xia et al. developed a multitask DL model named sPhysNet-MT.51 This model simultaneously predicts electronic energies of molecules in gas, water, and octanol as well as the transfer free energies between these phases. On their calculated data set, which includes molecules with energetics computed at the DFT-level, the sPhysNet-MT model achieves chemical accuracy on all tasks. Additionally, on the experimental data set containing molecules with experimental hydration free energy and logP, the model achieves state-of-the-art performance on both tasks simultaneously (Figure 5).
Similarly, Lu et al. developed the T5Chem model for multitask reaction prediction, which encompasses forward reaction prediction, reactant prediction, reagent prediction, reaction yield prediction, and reaction type classification.252 On the test set of their reaction prediction data set called USPTO_500_MT, T5Chem achieves top-1 accuracy of 97.5%, 72.9%, and 24.9% for forward prediction, retrosynthesis, and reagents prediction, respectively. The model also achieves an accuracy of 99.4% in reaction type classification, R2 of 0.22, and MAE of 17.8 in reaction yield prediction, showing competitive performance compared to models trained solely on individual tasks.
4. Case Study: 1M17
To illustrate the computational tools described in this paper, we show a case study using the crystal structure of the FDA approved inhibitor Erlotinib bound to the kinase domain of the epidermal growth factor receptor (EGFR) from PDB 1M17 (Figure 6).
Figure 6.

Case study of PDB 1M17 with models mentioned in the paper. (A) AlphaSpace and AlphaSpace 2.0 binding pocket analysis. (B) Binding affinity prediction was done using Lin_F9 and ΔLinF9XGB. (C) Prediction of physicochemical properties of the ligand molecule using sPhysNet-MT.
We first use AlphaSpace to analyze the binding of Erlotinib to EGFR kinase. The crystal receptor and crystal ligand are the inputs for AlphaSpace and AlphaSpace2.0 analysis. To calculate the AlphaSpace 2.0 β-score, hydrogens and partial charges are added to the ligand and protein input files. The AlphaSpace output characterizes the pockets that are in contact with the ligand, provides a binding score, and suggests undertargeted pockets (Figure 6A). The alpha-atoms and β-atoms are colored by the pockets which they belong to.
We then apply ΔLin_F9XGB for the prediction of the binding affinity. Similarly to the preparation of the AlphaSpace 2.0 input files, we add hydrogens and generate the partial charges to the crystal ligand and crystal protein input files. We apply Lin_F9 local optimization on the ligand binding orientation and get the Lin_F9 binding score. The Vina, solvent accessible surface area, water, metal, and ligand descriptors are calculated for the optimized geometry. These are input to an ensemble of 10 XGB models, the average output of which is used as correction of the Lin_F9 binding affinity. The sum of the Lin_F9 and XGB correction is the final ΔLin_F9XGB score or the predicted binding affinity. The predicted binding affinity of Erlotnib to EGFR kinase from the structure 1M17 (pKpredd = 8.46, Kpredd = 3.5 nM, Figure 6B) closely matches the experimental binding affinity found in BindingDB (with an average of pKexpd and pKexpi = 8.9 ± 0.9).253 It is worth noting that BindingDB contains 45 entries for Ki (ranging from 0.1 to 136 nM) and Kd (ranging from 0.35 to 190 nM), indicating a wide range of experimentally measured binding affinities. Our predicted Kd value of 3.5 nM falls comfortably within this spectrum of binding affinities.
Finally, we use sPhysNet-MT to predict the physicochemical properties of the ligand molecule Erlotinib. Starting from the SMILES of Erlotinib, we first generate up to 300 conformations using EDKTG and optimize them with MMFF. 291 conformations are successfully generated, and the one with the lowest MMFF energy is selected as the input geometry for sPhysNet-MT. The average output of an ensemble of 5 independently trained sPhysNet-MT models is used as the final prediction. The model predicts the logP of the molecule to be 2.76 which is close to the experimental measured value queried from the DrugBank database, 2.7 (Figure 6C).254 The detailed scripts to carry out these calculations and analysis can be found at: https://github.com/SongXia-NYU/drug_discovery_perspective.
5. Concluding Remarks and Perspective
The success of developing computational tools for drug design requires the integration of both molecular modeling and ML techniques. Models for small molecules, protein surfaces, and protein–ligand interactions are all required during different stages of the drug design process, and there is still room for development and research in these methods.
The AlphaSpace methods can be more robust and applicable. Further improvements can account for the role of protein conformational stability in pocket ligand-ability. This can make pocket comparisons more robust, especially when large conformational changes or comparisons between apo and holo structures are involved. When apo structures result in lower ligand-ability estimates, it limits confidence in using apo structures for downstream structure-based design. Quantified comparisons between different proteins and between orthosteric and allosteric pockets can also provide insights into ligand selectivity and off-target effects. Future versions can address these limitations by leveraging deep learning approaches to learn the representation of these relationships and improve ligand-ability estimates. Smith et al. suggest that there are opportunities to apply advanced deep learning image vision techniques to this research question.92 AlphaSpace2 and future versions can be tested on recently developed benchmarks for protein–ligand binding site prediction such as SHREC,83 HOLO4K, COACH420,78 and for binding site comparisons such as ProSPECCTs.70,255 Additionally, a Web server can be developed to give medicinal chemists a more user-friendly experience.
Despite the improved performance of ML- and DL-based molecular docking and scoring methods on the CASF-2016 data set, these methods tend to have a high false positive rate and lack generalizability on real-world VS data sets. More consideration should be given to the curation of the training data sets and to introducing regularization techniques to current VS protocol. Computationally generated decoy poses and training data sets should be generated carefully to prevent the model from having noncausal bias, an issue where the model learns specific data patterns instead of meaningful ligand–protein interactions.256,257 Training data sets tend to have many positive labels that lack diversity and have few or biased negative labels.83,88,258,259 Non- and poor-binders are under-reported, and computationally selected nonbinders should be verified experimentally.260,261 There is a lack of quality and standardized data for some targets. Methods such as data set debiasing262 and introducing specific models to classify and rank actives from inactives171,263−265 are promising regularization techniques to tackle this problem. Furthermore, it is necessary to test models in fair benchmarking data sets akin to real-world conditions.155,266
The size and quality of the training data are also important considerations for the prediction of molecular properties with DL models. More diverse data sets are required to further explore the chemical space. The Frag20 data preparation protocol63 can be used to carefully select diverse molecules, and modern high performance computing platforms can with the help generate labels for DFT calculations for calculated data sets. Collaboration with experimental groups is needed to enlarge and standardize experimental training sets. More careful pretraining protocols and model architecture designs should be developed to fully take advantage of the existing experimental data. Another disadvantage of sPhysNet-MT as well as many other DL-based models is that they can run into problems when considering charged molecules, because they do not explicitly express molecular charge in the model. For example, when predicting pKa, the molecule may be ionized and the model may have problems learning the molecular representation without knowing the total charge of the system.
Additionally, structure-based drug design methods are limited by the exclusion of protein dynamics in most current protein–ligand representations. Protein flexibility can range from slight perturbation of the ligand binding pockets to reconstitution of the binding site to large-scale conformational changes. In the case of binding site prediction, the introduction of the protein dynamics to the model can lead to the detection of cryptic pockets that are not seen in the static crystal structures and to better predictors of binding site ligandability.82 Cryptic pockets can be teased out using standard MD simulations or specialized MD simulations where the solvent molecules are altered to act as probes to tease out these pockets.80 Researchers are also using ML models that have learned the aspects of protein flexibility that lead to cryptic pockets from MD simulations to predict cryptic sites.267,268
Researchers have addressed this limitation of protein flexibility within the context of docking and binding affinity prediction as well.269 Some methods introduce side chain flexibility to the receptor binding pocket,270 others include MD simulations to probe the stability of the predicted binding pose,271 while some DL methods learn the features that drive protein–ligand interactions in MD simulations to predict binding affinity.272 The most common method is ensemble docking, which performs docking on a diverse selection of static receptor structures.273,274 Although rigorous in concept, ensemble docking can actually lead to a decrease in performance when using too many structures and thus researchers propose additional ML methods to select the best subset of structures.275−277 The use of technology, like cryo-EM that can determine diverse receptor structures, enhanced sampling MD simulations that speed conformational sampling, and DL protein structure prediction methods that can predict previously undetermined protein structures and conformations will drive the research in understanding the role of protein conformation in ligand binding. The decision to introduce protein flexibility to structure-based drug design comes with additional computational cost, a factor that must be aligned with the scope and goals of every screening campaign and currently does not guarantee improved performance.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (R35-GM127040). S.X. and E.C. were also partially supported by a graduate fellowship from the Simons Center for Computational Physical Chemistry (SCCPC) at NYU. We thank NYU-ITS for providing computational resources.
Author Contributions
∥ S.X. and E.C. contributed equally.
The authors declare no competing financial interest.
References
- Brown D. G.; Wobst H. J. A decade of FDA-approved drugs (2010–2019): trends and future directions. Journal of medicinal chemistry 2021, 64 (5), 2312–2338. 10.1021/acs.jmedchem.0c01516. [DOI] [PubMed] [Google Scholar]
- Mullard A. 2020 FDA drug approvals. Nat. Rev. Drug Discovery 2021, 20 (2), 85–91. 10.1038/d41573-021-00002-0. [DOI] [PubMed] [Google Scholar]
- Wouters O. J.; McKee M.; Luyten J. Research and development costs of new drugs—reply. JAMA 2020, 324 (5), 518–518. 10.1001/jama.2020.8651. [DOI] [PubMed] [Google Scholar]
- Shaker B.; Ahmad S.; Lee J.; Jung C.; Na D. In silico methods and tools for drug discovery. Computers in Biology and Medicine 2021, 137, 104851. 10.1016/j.compbiomed.2021.104851. [DOI] [PubMed] [Google Scholar]
- Katsila T.; Spyroulias G. A.; Patrinos G. P.; Matsoukas M.-T. Computational approaches in target identification and drug discovery. Computational and Structural Biotechnology Journal 2016, 14, 177–184. 10.1016/j.csbj.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Puhl A. C.; Zorn K. M.; Lane T. R.; Russo D. P.; Klein J. J.; Hickey A. J.; Clark A. M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18 (5), 435–441. 10.1038/s41563-019-0338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A. Artificial Intelligence for Drug Discovery, Biomarker Development, and Generation of Novel Chemistry. Mol. Pharmaceutics 2018, 15 (10), 4311–4313. 10.1021/acs.molpharmaceut.8b00930. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Srivastava D.; Sahu M.; Tiwari S.; Ambasta R. K.; Kumar P. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Molecular Diversity 2021, 25 (3), 1315–1360. 10.1007/s11030-021-10217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan R. S. K.; Kihlberg J.; Cross J. B.; Poongavanam V. Enhancing preclinical drug discovery with artificial intelligence. Drug Discovery Today 2022, 27 (4), 967–984. 10.1016/j.drudis.2021.11.023. [DOI] [PubMed] [Google Scholar]
- Patel L.; Shukla T.; Huang X.; Ussery D. W.; Wang S. Machine learning methods in drug discovery. Molecules 2020, 25 (22), 5277. 10.3390/molecules25225277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran B.; Abed S. N.; Al-Attraqchi O.; Kuche K.; Tekade R. K.. Chapter 21 - Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In Dosage Form Design Parameters; Tekade R. K., Ed.; Academic Press: 2018; pp 731–755. [Google Scholar]
- Wagner J. A.; Dahlem A. M.; Hudson L. D.; Terry S. F.; Altman R. B.; Gilliland C. T.; DeFeo C.; Austin C. P. Application of a dynamic map for learning, communicating, navigating, and improving therapeutic development. Clinical and Translational Science 2018, 11 (2), 166–174. 10.1111/cts.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. P.; Rees S.; Kalindjian S. B.; Philpott K. L. Principles of early drug discovery. British journal of pharmacology 2011, 162 (6), 1239–1249. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya C.; Coop A.; Polli J. E.; MacKerell A. D. Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Current computer-aided drug design 2011, 7 (1), 10–22. 10.2174/157340911793743547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A.; Jenkins J. L.; Scheiber J.; Sukuru S. C. K.; Glick M.; Davies J. W. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49 (1), 108–119. 10.1021/ci800249s. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45 (1), 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lai L. Pocket v. 2: further developments on receptor-based pharmacophore modeling. J. Chem. Inf. Model. 2006, 46 (6), 2684–2691. 10.1021/ci600246s. [DOI] [PubMed] [Google Scholar]
- Leo A.; Hoekman D.. Exploring QSAR; American Chemical Society: 1995. [Google Scholar]
- Verma J.; Khedkar V. M.; Coutinho E. C. 3D-QSAR in drug design-a review. Current topics in medicinal chemistry 2010, 10 (1), 95–115. 10.2174/156802610790232260. [DOI] [PubMed] [Google Scholar]
- Yuriev E.; Agostino M.; Ramsland P. A. Challenges and advances in computational docking: 2009 in review. Journal of Molecular Recognition 2011, 24 (2), 149–164. 10.1002/jmr.1077. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins: Struct., Funct., Bioinf. 2003, 52 (4), 609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. Journal of medicinal chemistry 2003, 46 (4), 499–511. 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. Journal of Computer-Aided Molecular Design 2007, 21 (5), 281–306. 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- Miteva M. A.; Lee W. H.; Montes M. O.; Villoutreix B. O. Fast structure-based virtual ligand screening combining FRED, DOCK, and Surflex. Journal of medicinal chemistry 2005, 48 (19), 6012–6022. 10.1021/jm050262h. [DOI] [PubMed] [Google Scholar]
- Hansson T.; Oostenbrink C.; van Gunsteren W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12 (2), 190–196. 10.1016/S0959-440X(02)00308-1. [DOI] [PubMed] [Google Scholar]
- Kaya S.; Tüzün B.; Kaya C.; Obot I. B. Determination of corrosion inhibition effects of amino acids: quantum chemical and molecular dynamic simulation study. Journal of the Taiwan Institute of Chemical Engineers 2016, 58, 528–535. 10.1016/j.jtice.2015.06.009. [DOI] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; Van Der Spoel D. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29 (7), 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. T.; Humphrey W.; Gursoy A.; Dalke A.; Kalé L. V.; Skeel R. D.; Schulten K. NAMD: a parallel, object-oriented molecular dynamics program. International Journal of Supercomputer Applications and High Performance Computing 1996, 10 (4), 251–268. 10.1177/109434209601000401. [DOI] [Google Scholar]
- Pearlman D. A.; Case D. A.; Caldwell J. W.; Ross W. S.; Cheatham T. E. III; DeBolt S.; Ferguson D.; Seibel G.; Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91 (1–3), 1–41. 10.1016/0010-4655(95)00041-D. [DOI] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. Journal of computational chemistry 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Katigbak J.; Li H.; Rooklin D.; Zhang Y. AlphaSpace 2.0: Representing Concave Biomolecular Surfaces Using β-Clusters. J. Chem. Inf. Model. 2020, 60 (3), 1494–1508. 10.1021/acs.jcim.9b00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooklin D.; Wang C.; Katigbak J.; Arora P. S.; Zhang Y. AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein-Protein Interaction Interfaces. J. Chem. Inf. Model. 2015, 55 (8), 1585–1599. 10.1021/acs.jcim.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10 (1), 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.; Chen C.; Lei X.; Zhao J.; Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic acids research 2018, 46 (W1), W363–W367. 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf Model 2009, 49 (2), 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Wass M. N.; Kelley L. A.; Sternberg M. J. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic acids research 2010, 38 (suppl_2), W469–W473. 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalidas Y.; Chandra N. PocketDepth: a new depth based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol. 2008, 161 (1), 31–42. 10.1016/j.jsb.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Eswar N.; Eramian D.; Webb B.; Shen M.-Y.; Sali A. Protein structure modeling with MODELLER. Structural proteomics: high-throughput methods 2008, 426, 145–159. 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; de Beer T. A. P.; Rempfer C.; Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research 2018, 46 (W1), W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U.; Eswar N.; Davis F. P.; Braberg H.; Madhusudhan M. S.; Rossi A.; Marti-Renom M.; Karchin R.; Webb B. M.; Eramian D. MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic acids research 2006, 34 (90001), D291–D295. 10.1093/nar/gkj059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A.; Mezulis S.; Yates C. M.; Wass M. N.; Sternberg M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols 2015, 10 (6), 845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin C.; Pogorelov T. V.; Luthey-Schulten Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 2002, 12 (2), 176–181. 10.1016/S0959-440X(02)00306-8. [DOI] [PubMed] [Google Scholar]
- Yang J.; Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic acids research 2015, 43 (W1), W174–W181. 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. E.; Chivian D.; Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic acids research 2004, 32 (suppl_2), W526–W531. 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Zhang J.; Roy A.; Zhang Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins: Struct., Funct., Bioinf. 2011, 79 (S10), 147–160. 10.1002/prot.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy T. Managing the drug discovery/development interface. Drug discovery today 1997, 2 (10), 436–444. 10.1016/S1359-6446(97)01099-4. [DOI] [Google Scholar]
- Tetko I. V.; Tanchuk V. Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. Journal of chemical information and computer sciences 2002, 42 (5), 1136–1145. 10.1021/ci025515j. [DOI] [PubMed] [Google Scholar]
- Nunes A. M. V.; de Andrade F. d. C. P.; Filgueiras L. A.; de Carvalho Maia O. A.; Cunha R. L.; Rodezno S. V.; Maia Filho A. L. M.; de Amorim Carvalho F. A.; Braz D. C.; Mendes A. N. preADMET analysis and clinical aspects of dogs treated with the Organotellurium compound RF07: A possible control for canine visceral leishmaniasis?. Environmental Toxicology and Pharmacology 2020, 80, 103470. 10.1016/j.etap.2020.103470. [DOI] [PubMed] [Google Scholar]
- Xia S.; Zhang D.; Zhang Y. Multitask Deep Ensemble Prediction of Molecular Energetics in Solution: From Quantum Mechanics to Experimental Properties. J. Chem. Theory Comput. 2023, 19 (2), 659–668. 10.1021/acs.jctc.2c01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Xia S.; Zhang Y. Accurate Prediction of Aqueous Free Solvation Energies Using 3D Atomic Feature-Based Graph Neural Network with Transfer Learning. J. Chem. Inf. Model. 2022, 62 (8), 1840–1848. 10.1021/acs.jcim.2c00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 2017, 7 (1), 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S. K.; Singla D.; Mondal A. K.; Raghava G. P. DrugMint: a webserver for predicting and designing of drug-like molecules. Biology Direct 2013, 8 (1), 1–12. 10.1186/1745-6150-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker B.; Yu M.-S.; Song J. S.; Ahn S.; Ryu J. Y.; Oh K.-S.; Na D. LightBBB: computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37 (8), 1135–1139. 10.1093/bioinformatics/btaa918. [DOI] [PubMed] [Google Scholar]
- Lee H.-M.; Yu M.-S.; Kazmi S. R.; Oh S. Y.; Rhee K.-H.; Bae M.-A.; Lee B. H.; Shin D.-S.; Oh K.-S.; Ceong H. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC bioinformatics 2019, 20, 67–73. 10.1186/s12859-019-2814-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Kapoor P.; Chaudhary K.; Gautam A.; Kumar R.; Consortium O. S. D. D.; Raghava G. P. In silico approach for predicting toxicity of peptides and proteins. PloS one 2013, 8 (9), e73957 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Eckert A. O.; Schrey A. K.; Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic acids research 2018, 46 (W1), W257–W263. 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N. K.; Singla D.; Agarwal S.; Raghava G. P. ToxiPred: a server for prediction of aqueous toxicity of small chemical molecules in T. Pyriformis. Journal of Translational Toxicology 2014, 1 (1), 21–27. 10.1166/jtt.2014.1005. [DOI] [Google Scholar]
- Dong J.; Wang N.-N.; Yao Z.-J.; Zhang L.; Cheng Y.; Ouyang D.; Lu A.-P.; Cao D.-S. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. Journal of cheminformatics 2018, 10, 1–11. 10.1186/s13321-018-0283-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M. A.; Stacey R. G.; Wishart D. S.; Foster L. J. Chemical language models enable navigation in sparsely populated chemical space. Nature Machine Intelligence 2021, 3 (9), 759–770. 10.1038/s42256-021-00368-1. [DOI] [Google Scholar]
- Mahmood O.; Mansimov E.; Bonneau R.; Cho K. Masked graph modeling for molecule generation. Nat. Commun. 2021, 12 (1), 3156. 10.1038/s41467-021-23415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Xia S.; Lu J.; Zhang Y. Dataset Construction to Explore Chemical Space with 3D Geometry and Deep Learning. J. Chem. Inf. Model. 2021, 61 (3), 1095–1104. 10.1021/acs.jcim.1c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou B.; Zhu Z.; Merkurjev E.; Ke L.; Chen L.; Jiang J.; Zhu Y.; Liu J.; Zhang B.; Wei G.-W. Machine Learning Methods for Small Data Challenges in Molecular Science. Chem. Rev. 2023, 123 (13), 8736–8780. 10.1021/acs.chemrev.3c00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadybekov A. V.; Katritch V. Computational approaches streamlining drug discovery. Nature 2023, 616 (7958), 673–685. 10.1038/s41586-023-05905-z. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48 (3), 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Nisius B.; Sha F.; Gohlke H. Structure-based computational analysis of protein binding sites for function and druggability prediction. J. Biotechnol. 2012, 159 (3), 123–134. 10.1016/j.jbiotec.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Macari G.; Toti D.; Polticelli F. Computational methods and tools for binding site recognition between proteins and small molecules: from classical geometrical approaches to modern machine learning strategies. Journal of Computer-Aided Molecular Design 2019, 33 (10), 887–903. 10.1007/s10822-019-00235-7. [DOI] [PubMed] [Google Scholar]
- Rehman A. U.; Lu S.; Khan A. A.; Khurshid B.; Rasheed S.; Wadood A.; Zhang J. Hidden allosteric sites and De-Novo drug design. Expert Opinion on Drug Discovery 2022, 17 (3), 283–295. 10.1080/17460441.2022.2017876. [DOI] [PubMed] [Google Scholar]
- Ehrt C.; Brinkjost T.; Koch O. A benchmark driven guide to binding site comparison: An exhaustive evaluation using tailor-made data sets (ProSPECCTs). PLOS Computational Biology 2018, 14 (11), e1006483 10.1371/journal.pcbi.1006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt C.; Brinkjost T.; Koch O. Impact of Binding Site Comparisons on Medicinal Chemistry and Rational Molecular Design. J. Med. Chem. 2016, 59 (9), 4121–4151. 10.1021/acs.jmedchem.6b00078. [DOI] [PubMed] [Google Scholar]
- Clark J. J.; Orban Z. J.; Carlson H. A. Predicting binding sites from unbound versus bound protein structures. Sci. Rep 2020, 10 (1), 15856.(acccessed 2023/06/11/20:18:15). From www.nature.com 10.1038/s41598-020-72906-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski T. A.; Naghibzadeh S.; Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31 (13), 3352–3355. (acccessed 7/9/2023) 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Woodward C.; Edelsbrunner H. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998, 7 (9), 1884–1897. (acccessed 2023/07/09) 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M.; Rippmann F.; Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. Journal of Molecular Graphics and Modelling 1997, 15 (6), 359–363. 10.1016/S1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- Laurie A. T. R.; Jackson R. M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21 (9), 1908–1916. (acccessed 7/9/2023) 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- Hernandez M.; Ghersi D.; Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic acids research 2009, 37 (suppl_2), W413–W416. 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivák R.; Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics 2018, 10 (1), 39. 10.1186/s13321-018-0285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngan C.-H.; Hall D. R.; Zerbe B.; Grove L. E.; Kozakov D.; Vajda S. FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2012, 28 (2), 286–287. 10.1093/bioinformatics/btr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J.; Wang Q.; Wu F.; Huang Z. In Silico Methods for Identification of Potential Active Sites of Therapeutic Targets. Molecules 2022, 27, 7103. 10.3390/molecules27207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche D. B.; Brackenridge D. A.; McGuffin L. J. Proteins and Their Interacting Partners: An Introduction to Protein-Ligand Binding Site Prediction Methods. International Journal of Molecular Sciences 2015, 16, 29829–29842. 10.3390/ijms161226202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma F.; Abate C.; Decherchi S.; Cavalli A. Ligandability and druggability assessment via machine learning. WIREs Computational Molecular Science 2023, 13, e1676(acccessed 2023/07/10) 10.1002/wcms.1676. [DOI] [Google Scholar]
- Gagliardi L.; Raffo A.; Fugacci U.; Biasotti S.; Rocchia W.; Huang H.; Amor B. B.; Fang Y.; Zhang Y.; Wang X.; et al. SHREC 2022: Protein-ligand binding site recognition. Computers & Graphics 2022, 107, 20–31. 10.1016/j.cag.2022.07.005. [DOI] [Google Scholar]
- Schmidtke P.; Souaille C.; Estienne F.; Baurin N.; Kroemer R. T. Large-Scale Comparison of Four Binding Site Detection Algorithms. J. Chem. Inf. Model. 2010, 50 (12), 2191–2200. 10.1021/ci1000289. [DOI] [PubMed] [Google Scholar]
- Richards F. M. The interpretation of protein structures: total volume, group volume distributions and packing density. Journal of molecular biology 1974, 82 (1), 1–14. 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- Ccgi M.Molecular operating environment (MOE), 2013.08. Chemical Computing Group Inc., Montreal 2016, 354. [Google Scholar]
- Aggarwal R.; Gupta A.; Chelur V.; Jawahar C. V.; Priyakumar U. D. DeepPocket: Ligand Binding Site Detection and Segmentation using 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2022, 62 (21), 5069–5079. 10.1021/acs.jcim.1c00799. [DOI] [PubMed] [Google Scholar]
- Özçelik R.; van Tilborg D.; Jiménez-Luna J.; Grisoni F. Structure-Based Drug Discovery with Deep Learning**. ChemBioChem. 2023, 24 (13), e202200776(acccessed 2023/07/17) 10.1002/cbic.202200776. [DOI] [PubMed] [Google Scholar]
- Mylonas S. K.; Axenopoulos A.; Daras P. DeepSurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 2021, 37 (12), 1681–1690. (acccessed 7/10/2023) 10.1093/bioinformatics/btab009. [DOI] [PubMed] [Google Scholar]
- Gainza P.; Sverrisson F.; Monti F.; Rodolà E.; Boscaini D.; Bronstein M. M.; Correia B. E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17 (2), 184–192. 10.1038/s41592-019-0666-6. [DOI] [PubMed] [Google Scholar]
- Sverrisson F.; Feydy J.; Correia B. E.; Bronstein M. M.. Fast end-to-end learning on protein surfaces. bioRxiv: 2020. [Google Scholar]
- Smith Z.; Strobel M.; Vani B. P.; Tiwary P. Graph Attention Site Prediction (GrASP): Identifying Druggable Binding Sites Using Graph Neural Networks with Attention. bioRxiv 2023, 10.1101/2023.07.25.550565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooklin D.; Modell A. E.; Li H.; Berdan V.; Arora P. S.; Zhang Y. Targeting Unoccupied Surfaces on Protein-Protein Interfaces. J. Am. Chem. Soc. 2017, 139 (44), 15560–15563. (acccessed 2023/06/11/21:32:54). From ACS Publications 10.1021/jacs.7b05960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadek J.; Wuo M. G.; Rooklin D.; Hauenstein A.; Hong S. H.; Gautam A.; Wu H.; Zhang Y.; Cesarman E.; Arora P. S. Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics. Nat. Commun. 2020, 11 (1), 1786.(acccessed 2023/06/11/21:28:12). From www.nature.com 10.1038/s41467-020-15576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torner J. M.; Yang Y.; Rooklin D.; Zhang Y.; Arora P. S. Identification of Secondary Binding Sites on Protein Surfaces for Rational Elaboration of Synthetic Protein Mimics. ACS Chem. Biol. 2021, 16 (7), 1179–1183. (acccessed 2023/06/11/20:40:38). From ACS Publications 10.1021/acschembio.1c00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell A. E.; Marrone F. III; Panigrahi N. R.; Zhang Y.; Arora P. S. Peptide Tethering: Pocket-Directed Fragment Screening for Peptidomimetic Inhibitor Discovery. J. Am. Chem. Soc. 2022, 144 (3), 1198–1204. (acccessed 2023/06/11/21:20:39). From ACS Publications 10.1021/jacs.1c09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Gao X.-Y.; Chen X.-H.; Zhang S.-L.; Wang W.-J.; Sheng X.-H.; Chen D.-Z. Fragment-centric topographic mapping method guides the understanding of ABCG2-inhibitor interactions. RSC Adv. 2019, 9 (14), 7757–7766. (acccessed 2023/06/11/21:43:13). From pubs.rsc.org 10.1039/C8RA09789E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.-J.; Chen X.-H.; Zeng J.-C.; Duan L.-L.; Liu Z.-H.; Sheng X.-H. Theoretical insight into the multiple interactions of quinazoline inhibitors with breast cancer resistance protein (BCRP/ABCG2). J. Biomol. Struct. Dyn. 2020, 38 (14), 4336–4343. 10.1080/07391102.2019.1677503. [DOI] [PubMed] [Google Scholar]
- Hou X.; Sun J.-p.; Ge L.; Liang X.; Li K.; Zhang Y.; Fang H. Inhibition of striatal-enriched protein tyrosine phosphatase by targeting computationally revealed cryptic pockets. Eur. J. Med. Chem. 2020, 190, 112131.(acccessed 2023/06/11/21:37:08). From ScienceDirect 10.1016/j.ejmech.2020.112131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K.; Nagata N.; Takahashi Y. De Novo Design of Drug-Like Molecules by a Fragment-Based Molecular Evolutionary Approach. J. Chem. Inf. Model. 2014, 54 (1), 49–56. 10.1021/ci400418c. [DOI] [PubMed] [Google Scholar]
- Lionta E.; Spyrou G.; Vassilatis D.; Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Current topics in medicinal chemistry 2014, 14 (16), 1923–1938. 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgulla C.; Boeszoermenyi A.; Wang Z.-F.; Fischer P. D.; Coote P. W.; Padmanabha Das K. M.; Malets Y. S.; Radchenko D. S.; Moroz Y. S.; Scott D. A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580 (7805), 663–668. 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpfe D.; Bajorath J. Current Trends, Overlooked Issues, and Unmet Challenges in Virtual Screening. J. Chem. Inf. Model. 2020, 60 (9), 4112–4115. 10.1021/acs.jcim.9b01101. [DOI] [PubMed] [Google Scholar]
- Talele T. T.; Khedkar S. A.; Rigby A. C. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Current topics in medicinal chemistry 2010, 10 (1), 127–141. 10.2174/156802610790232251. [DOI] [PubMed] [Google Scholar]
- Stein R. M.; Kang H. J.; McCorvy J. D.; Glatfelter G. C.; Jones A. J.; Che T.; Slocum S.; Huang X.-P.; Savych O.; Moroz Y. S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579 (7800), 609–614. 10.1038/s41586-020-2027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Drie J. H. Computer-aided drug design: the next 20 years. Journal of Computer-Aided Molecular Design 2007, 21 (10), 591–601. 10.1007/s10822-007-9142-y. [DOI] [PubMed] [Google Scholar]
- Yang C.; Chen E. A.; Zhang Y. Protein-Ligand Docking in the Machine-Learning Era. Molecules 2022, 27 (14), 4568. 10.3390/molecules27144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman G. D.; Egbertson M. S.; Halczenko W.; Laswell W. L.; Duggan M. E.; Smith R. L.; Naylor A. M.; Manno P. D.; Lynch R. J. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35 (24), 4640–4642. 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- Greer J.; Erickson J. W.; Baldwin J. J.; Varney M. D. Application of the Three-Dimensional Structures of Protein Target Molecules in Structure-Based Drug Design. J. Med. Chem. 1994, 37 (8), 1035–1054. 10.1021/jm00034a001. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Vondrasek J. INHIBITORS OF HIV-1 PROTEASE: A Major Success of Structure-Assisted Drug Design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27 (1), 249–284. (acccessed 2023/06/22) 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- Jhoti H.; Leach A. R.. Structure-based drug discovery; Springer: 2007. [Google Scholar]
- Lavecchia A.; Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 2013, 20 (23), 2839–2860. 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- Reddy A. S.; Pati S. P.; Kumar P. P.; Pradeep H.; Sastry G. N. Virtual screening in drug discovery-a computational perspective. Current Protein and Peptide Science 2007, 8 (4), 329–351. 10.2174/138920307781369427. [DOI] [PubMed] [Google Scholar]
- Ferreira L. G.; Dos Santos R. N.; Oliva G.; Andricopulo A. D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20 (7), 13384–13421. 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ruyck J.; Brysbaert G.; Blossey R.; Lensink M. F. Molecular docking as a popular tool in drug design, an in silico travel. Advances and Applications in Bioinformatics and Chemistry 2016, Volume 9, 1–11. 10.2147/AABC.S105289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres P. H. M.; Sodero A. C. R.; Jofily P.; Silva-Jr F. P. Key Topics in Molecular Docking for Drug Design. International Journal of Molecular Sciences 2019, 20 (18), 4574. 10.3390/ijms20184574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Fu A.; Zhang L. Progress in molecular docking. Quantitative Biology 2019, 7, 83–89. 10.1007/s40484-019-0172-y. [DOI] [Google Scholar]
- Koshland Jr D. E. The key-lock theory and the induced fit theory. Angewandte Chemie International Edition in English 1995, 33 (23–24), 2375–2378. 10.1002/anie.199423751. [DOI] [Google Scholar]
- Allen W. J.; Balius T. E.; Mukherjee S.; Brozell S. R.; Moustakas D. T.; Lang P. T.; Case D. A.; Kuntz I. D.; Rizzo R. C. DOCK 6: Impact of new features and current docking performance. Journal of computational chemistry 2015, 36 (15), 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47 (7), 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: applications of AutoDock. Journal of molecular recognition 1996, 9 (1), 1–5. . [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Thomas L. L. Perspective on Free-Energy Perturbation Calculations for Chemical Equilibria. J. Chem. Theory Comput. 2008, 4 (6), 869–876. 10.1021/ct800011m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorian F.; Perez-Benito L.; Lenselink E. B.; Congreve M.; van Vlijmen H. W. T.; Mason J. S.; Graaf C. d.; Tresadern G. Accurate Prediction of GPCR Ligand Binding Affinity with Free Energy Perturbation. J. Chem. Inf. Model. 2020, 60 (11), 5563–5579. 10.1021/acs.jcim.0c00449. [DOI] [PubMed] [Google Scholar]
- Bhati A. P.; Wan S.; Wright D. W.; Coveney P. V. Rapid, Accurate, Precise, and Reliable Relative Free Energy Prediction Using Ensemble Based Thermodynamic Integration. J. Chem. Theory Comput. 2017, 13 (1), 210–222. 10.1021/acs.jctc.6b00979. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Nilsson I.; Ryde U. Binding Affinities of Factor Xa Inhibitors Estimated by Thermodynamic Integration and MM/GBSA. J. Chem. Inf. Model. 2011, 51 (4), 947–958. 10.1021/ci100458f. [DOI] [PubMed] [Google Scholar]
- Li J.; Fu A.; Zhang L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdisciplinary Sciences: Computational Life Sciences 2019, 11 (2), 320–328. 10.1007/s12539-019-00327-w. [DOI] [PubMed] [Google Scholar]
- Böhm H. J.; Stahl M. The use of scoring functions in drug discovery applications. Reviews in computational chemistry 2002, 18, 41–87. 10.1002/0471433519.ch2. [DOI] [Google Scholar]
- Huang S.-Y.; Grinter S. Z.; Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12 (40), 12899–12908. 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J.; Wang S.; Balius T. E.; Singh I.; Levit A.; Moroz Y. S.; O’Meara M. J.; Che T.; Algaa E.; Tolmachova K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566 (7743), 224–229. 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele T. T.; Arora P.; Kulkarni S. S.; Patel M. R.; Singh S.; Chudayeu M.; Kaushik-Basu N. Structure-based virtual screening, synthesis and SAR of novel inhibitors of hepatitis C virus NS5B polymerase. Bioorg. Med. Chem. 2010, 18 (13), 4630–4638. 10.1016/j.bmc.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M.; Yang Q.; Du Y.; Feng G.; Liu Z.; Li Y.; Wang R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59 (2), 895–913. 10.1021/acs.jcim.8b00545. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Hendlich M.; Klebe G. Knowledge-based scoring function to predict protein-ligand interactions11Edited by R. Huber. J. Mol. Biol. 2000, 295 (2), 337–356. 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- Huang S. Y.; Zou X. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. Journal of computational chemistry 2006, 27 (15), 1866–1875. 10.1002/jcc.20504. [DOI] [PubMed] [Google Scholar]
- Huang S. Y.; Zou X. An iterative knowledge-based scoring function to predict protein-ligand interactions: II. Validation of the scoring function. Journal of computational chemistry 2006, 27 (15), 1876–1882. 10.1002/jcc.20505. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Martin Y. C. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. J. Med. Chem. 1999, 42 (5), 791–804. 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- Böhm H. J. A novel computational tool for automated structure-based drug design. Journal of Molecular Recognition 1993, 6 (3), 131–137. 10.1002/jmr.300060305. [DOI] [PubMed] [Google Scholar]
- Böhm H.-J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. Journal of Computer-Aided Molecular Design 1994, 8 (3), 243–256. 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- Wang R.; Lai L.; Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. Journal of Computer-Aided Molecular Design 2002, 16 (1), 11–26. 10.1023/A:1016357811882. [DOI] [PubMed] [Google Scholar]
- Yang C.; Zhang Y. Lin_F9: A Linear Empirical Scoring Function for Protein-Ligand Docking. J. Chem. Inf. Model. 2021, 61 (9), 4630–4644. 10.1021/acs.jcim.1c00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ain Q. U.; Aleksandrova A.; Roessler F. D.; Ballester P. J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdisciplinary Reviews: Computational Molecular Science 2015, 5 (6), 405–424. 10.1002/wcms.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Sze K. H.; Lu G.; Ballester P. J. Machine-learning scoring functions for structure-based virtual screening. Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11 (1), e1478 10.1002/wcms.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C.; Vapnik V. Support-vector networks. Machine Learning 1995, 20 (3), 273–297. 10.1007/BF00994018. [DOI] [Google Scholar]
- Liaw A.; Wiener M. Classification and regression by randomForest. R news 2002, 2 (3), 18–22. [Google Scholar]
- Chen T.; Guestrin C.. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 2016; pp 785–794.
- Ballester P. J.; Mitchell J. B. O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26 (9), 1169–1175. (acccessed 6/24/2023) 10.1093/bioinformatics/btq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilian D.; Sotriffer C. A. SFCscoreRF: A Random Forest-Based Scoring Function for Improved Affinity Prediction of Protein-Ligand Complexes. J. Chem. Inf. Model. 2013, 53 (8), 1923–1933. 10.1021/ci400120b. [DOI] [PubMed] [Google Scholar]
- Ashtawy H. M.; Mahapatra N. R. Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model. 2018, 58 (1), 119–133. 10.1021/acs.jcim.7b00309. [DOI] [PubMed] [Google Scholar]
- Wójcikowski M.; Ballester P. J.; Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep 2017, 7 (1), 46710. 10.1038/srep46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Zhang Y. Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. Journal of computational chemistry 2017, 38 (3), 169–177. 10.1002/jcc.24667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Han L.; Liu Z.; Wang R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54 (6), 1717–1736. 10.1021/ci500081m. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Li X.; Li Y.; Liu Z.; Wang R. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model. 2009, 49 (4), 1079–1093. 10.1021/ci9000053. [DOI] [PubMed] [Google Scholar]
- Lu J.; Hou X.; Wang C.; Zhang Y. Incorporating Explicit Water Molecules and Ligand Conformation Stability in Machine-Learning Scoring Functions. J. Chem. Inf. Model. 2019, 59 (11), 4540–4549. 10.1021/acs.jcim.9b00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Guestrin C.. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA; 2016.
- Yang C.; Zhang Y. Delta Machine Learning to Improve Scoring-Ranking-Screening Performances of Protein-Ligand Scoring Functions. J. Chem. Inf. Model. 2022, 62 (11), 2696–2712. 10.1021/acs.jcim.2c00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran-Nguyen V.-K.; Jacquemard C.; Rognan D. LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model. 2020, 60 (9), 4263–4273. 10.1021/acs.jcim.0c00155. [DOI] [PubMed] [Google Scholar]
- Tran-Nguyen V.-K.; Bret G.; Rognan D. True accuracy of fast scoring functions to predict High-Throughput screening data from docking poses: The simpler the better. J. Chem. Inf. Model. 2021, 61 (6), 2788–2797. 10.1021/acs.jcim.1c00292. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. NNScore: A Neural-Network-Based Scoring Function for the Characterization of Protein-Ligand Complexes. J. Chem. Inf. Model. 2010, 50 (10), 1865–1871. 10.1021/ci100244v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf. Model. 2011, 51 (11), 2897–2903. 10.1021/ci2003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach I.; Dzamba M.; Heifets A.. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv preprint arXiv:1510.02855 2015. [Google Scholar]
- Ragoza M.; Hochuli J.; Idrobo E.; Sunseri J.; Koes D. R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57 (4), 942–957. 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniewska-Dziubinska M. M.; Zielenkiewicz P.; Siedlecki P. Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 2018, 34 (21), 3666–3674. (acccessed 6/29/2023) 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez J.; Škalič M.; Martínez-Rosell G.; De Fabritiis G. KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58 (2), 287–296. 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- Zheng L.; Fan J.; Mu Y. OnionNet: a Multiple-Layer Intermolecular-Contact-Based Convolutional Neural Network for Protein-Ligand Binding Affinity Prediction. ACS Omega 2019, 4 (14), 15956–15965. 10.1021/acsomega.9b01997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Zheng L.; Liu Y.; Qu Y.; Li Y.-Q.; Zhao M.; Mu Y.; Li W. OnionNet-2: a convolutional neural network model for predicting protein-ligand binding affinity based on residue-atom contacting shells. Frontiers in Chemistry 2021, 9, 913. 10.3389/fchem.2021.753002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg E. N.; Sur D.; Wu Z.; Husic B. E.; Mai H.; Li Y.; Sun S.; Yang J.; Ramsundar B.; Pande V. S. PotentialNet for Molecular Property Prediction. ACS Central Science 2018, 4 (11), 1520–1530. 10.1021/acscentsci.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.; Ryu S.; Park K.; Choe Y. J.; Ham J.; Kim W. Y. Predicting Drug-Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59 (9), 3981–3988. 10.1021/acs.jcim.9b00387. [DOI] [PubMed] [Google Scholar]
- Karlov D. S.; Sosnin S.; Fedorov M. V.; Popov P. graphDelta: MPNN Scoring Function for the Affinity Prediction of Protein-Ligand Complexes. ACS Omega 2020, 5 (10), 5150–5159. 10.1021/acsomega.9b04162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Zhou J.; Xu T.; Huang L.; Wang F.; Xiong H.; Huang W.; Dou D.; Xiong H.. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, 2021; pp 975–985.
- Son J.; Kim D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PloS one 2021, 16 (4), e0249404 10.1371/journal.pone.0249404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Lucio O.; Ahmad M.; del Rio-Chanona E. A.; Wegner J. K. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nature Machine Intelligence 2021, 3 (12), 1033–1039. 10.1038/s42256-021-00409-9. [DOI] [Google Scholar]
- Shen C.; Zhang X.; Deng Y.; Gao J.; Wang D.; Xu L.; Pan P.; Hou T.; Kang Y. Boosting Protein-Ligand Binding Pose Prediction and Virtual Screening Based on Residue-Atom Distance Likelihood Potential and Graph Transformer. J. Med. Chem. 2022, 65 (15), 10691–10706. 10.1021/acs.jmedchem.2c00991. [DOI] [PubMed] [Google Scholar]
- Moon S.; Zhung W.; Yang S.; Lim J.; Kim W. Y. PIGNet: a physics-informed deep learning model toward generalized drug-target interaction predictions. Chemical Science 2022, 13 (13), 3661–3673. 10.1039/D1SC06946B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J.; He X.; Zhang J. Z. H. DeepBSP—a Machine Learning Method for Accurate Prediction of Protein-Ligand Docking Structures. J. Chem. Inf. Model. 2021, 61 (5), 2231–2240. 10.1021/acs.jcim.1c00334. [DOI] [PubMed] [Google Scholar]
- Zheng L.; Meng J.; Jiang K.; Lan H.; Wang Z.; Lin M.; Li W.; Guo H.; Wei Y.; Mu Y.. Improving protein-ligand docking and screening accuracies by incorporating a scoring function correction term. Briefings in Bioinformatics 2022, 23 ( (3), ). 10.1093/bib/bbac051 (acccessed 7/16/2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadukova M.; Machado K. d. S.; Chacón P.; Grudinin S. KORP-PL: a coarse-grained knowledge-based scoring function for protein-ligand interactions. Bioinformatics 2021, 37 (7), 943–950. 10.1093/bioinformatics/btaa748. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49 (1), 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Hou T.; Shen C.; Zhang X.; Hsieh C.-Y.; Deng Y.; Wang D.; Xu L.; Wu J.; Li D.; Kang Y. A generalized protein-ligand scoring framework with balanced scoring, docking, ranking and screening powers. Chemical Science 2023, 14, 8129. 10.1039/D3SC02044D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stärk H.; Ganea O.; Pattanaik L.; Barzilay D. R.; Jaakkola T.. EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction. In Proceedings of the 39th International Conference on Machine Learning, Proceedings of Machine Learning Research; 2022.
- Lu W.; Wu Q.; Zhang J.; Rao J.; Li C.; Zheng S. TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction. bioRxiv 2022, 2022.2006.2006.495043. 10.1101/2022.06.06.495043. [DOI] [Google Scholar]
- Corso G.; Stärk H.; Jing B.; Barzilay R.; Jaakkola T.. Diffdock: Diffusion steps, twists, and turns for molecular docking. arXiv preprint arXiv:2210.01776 2022. [Google Scholar]
- Rossi M.; Chutia S.; Scheffler M.; Blum V. Validation Challenge of Density-Functional Theory for Peptides—Example of Ac-Phe-Ala5-LysH+. J. Phys. Chem. A 2014, 118 (35), 7349–7359. 10.1021/jp412055r. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D. Conformation Generation: The State of the Art. J. Chem. Inf. Model. 2017, 57 (8), 1747–1756. 10.1021/acs.jcim.7b00221. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17 (5–6), 520–552. . [DOI] [Google Scholar]
- Bannwarth C.; Ehlert S.; Grimme S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15 (3), 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- Keith J. A.; Vassilev-Galindo V.; Cheng B.; Chmiela S.; Gastegger M.; Müller K.-R.; Tkatchenko A. Combining Machine Learning and Computational Chemistry for Predictive Insights Into Chemical Systems. Chem. Rev. 2021, 121 (16), 9816–9872. 10.1021/acs.chemrev.1c00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lilienfeld O. A.; Muller K.-R.; Tkatchenko A. Exploring chemical compound space with quantum-based machine learning. Nature Reviews Chemistry 2020, 4 (7), 347–358. 10.1038/s41570-020-0189-9. [DOI] [PubMed] [Google Scholar]
- Musil F.; Grisafi A.; Bartók A. P.; Ortner C.; Csányi G. b.; Ceriotti M. Physics-Inspired Structural Representations for Molecules and Materials. Chem. Rev. 2021, 121 (16), 9759–9815. 10.1021/acs.chemrev.1c00021. [DOI] [PubMed] [Google Scholar]
- Langer M. F.; Goeßmann A.; Rupp M. Representations of molecules and materials for interpolation of quantum-mechanical simulations via machine learning. npj Computational Materials 2022, 8 (1), 41. 10.1038/s41524-022-00721-x. [DOI] [Google Scholar]
- Haghighatlari M.; Li J.; Heidar-Zadeh F.; Liu Y.; Guan X.; Head-Gordon T. Learning to Make Chemical Predictions: The Interplay of Feature Representation, Data, and Machine Learning Methods. Chem. 2020, 6 (7), 1527–1542. 10.1016/j.chempr.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; von Lilienfeld O. A. Ab Initio Machine Learning in Chemical Compound Space. Chem. Rev. 2021, 121 (16), 10001–10036. 10.1021/acs.chemrev.0c01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dral P. O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11 (6), 2336–2347. 10.1021/acs.jpclett.9b03664. [DOI] [PubMed] [Google Scholar]
- LeCun Y.; Bengio Y.; Hinton G. Deep learning. Nature 2015, 521 (7553), 436–444. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Rupp M.; Tkatchenko A.; Muller K.-R.; von Lilienfeld O. A. v. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108 (5), 058301. 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Montavon G.; Rupp M.; Gobre V.; Vazquez-Mayagoitia A.; Hansen K.; Tkatchenko A.; Müller K.-R.; Lilienfeld O. A. v. Machine learning of molecular electronic properties in chemical compound space. New J. Phys. 2013, 15 (9), 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- Hansen K.; Montavon G.; Biegler F.; Fazli S.; Rupp M.; Scheffler M.; Lilienfeld O. A. v.; Tkatchenko A.; Müller K.-R. Assessment and Validation of Machine Learning Methods for Predicting Molecular Atomization Energies. J. Chem. Theory Comput. 2013, 9 (8), 3404–3419. 10.1021/ct400195d. [DOI] [PubMed] [Google Scholar]
- Hirn M.; Poilvert N.; Mallat S.. Quantum Energy Regression using Scattering Transforms. arXiv:1502.02077 2015. [Google Scholar]
- Hansen K.; Biegler F.; Ramakrishnan R.; Pronobis W.; von Lilienfeld O. A.; Müller K.-R.; Tkatchenko A. Machine Learning Predictions of Molecular Properties: Accurate Many-Body Potentials and Nonlocality in Chemical Space. J. Phys. Chem. Lett. 2015, 6 (12), 2326–2331. 10.1021/acs.jpclett.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 2010, 104 (13), 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Bartók A. P.; Kondor R.; Csányi G. On representing chemical environments. Phys. Rev. B 2013, 87 (18), 184115. 10.1103/PhysRevB.87.184115. [DOI] [Google Scholar]
- Behler J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134 (7), 074106. 10.1063/1.3553717. [DOI] [PubMed] [Google Scholar]
- Behler J. Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations. Phys. Chem. Chem. Phys. 2011, 13 (40), 17930–17955. 10.1039/c1cp21668f. [DOI] [PubMed] [Google Scholar]
- Behler J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Quantum Chem. 2015, 115 (16), 1032–1050. 10.1002/qua.24890. [DOI] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chemical Science 2017, 8 (4), 3192–3203. 10.1039/C6SC05720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K. T.; Arbabzadah F.; Chmiela S.; Müller K. R.; Tkatchenko A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8 (1), 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer J.; Schoenholz S. S.; Riley P. F.; Vinyals O.; Dahl G. E.. Neural Message Passing for Quantum Chemistry. In Proceedings of the 34th International Conference on Machine Learning, Proceedings of Machine Learning Research; 2017.
- Schütt K. T.; Sauceda H. E.; Kindermans P. J.; Tkatchenko A.; Müller K. R. SchNet - A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148 (24), 241722. 10.1063/1.5019779. [DOI] [PubMed] [Google Scholar]
- Lubbers N.; Smith J. S.; Barros K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 2018, 148 (24), 241715. 10.1063/1.5011181. [DOI] [PubMed] [Google Scholar]
- Lu C.; Liu Q.; Wang C.; Huang Z.; Lin P.; He L. Molecular Property Prediction: A Multilevel Quantum Interactions Modeling Perspective. Proceedings of the AAAI Conference on Artificial Intelligence 2019, 33, 1052–1060. 10.1609/aaai.v33i01.33011052. [DOI] [Google Scholar]
- Lu J.; Wang C.; Zhang Y. Predicting Molecular Energy Using Force-Field Optimized Geometries and Atomic Vector Representations Learned from an Improved Deep Tensor Neural Network. J. Chem. Theory Comput. 2019, 15 (7), 4113–4121. 10.1021/acs.jctc.9b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unke O. T.; Meuwly M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J. Chem. Theory Comput. 2019, 15 (6), 3678–3693. 10.1021/acs.jctc.9b00181. [DOI] [PubMed] [Google Scholar]
- Gasteiger J.; Groß J.; Günnemann S.. Directional Message Passing for Molecular Graphs. arXiv:2003.03123 2020. [Google Scholar]
- Klicpera J.; Giri S.; Margraf J. T.; Günnemann S.. Fast and Uncertainty-Aware Directional Message Passing for Non-Equilibrium Molecules. arXiv:2011.14115 2020. [Google Scholar]
- Fang Y.; Zhang Q.; Zhang N.; Chen Z.; Zhuang X.; Shao X.; Fan X.; Chen H. Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nature Machine Intelligence 2023, 5 (5), 542–553. 10.1038/s42256-023-00654-0. [DOI] [Google Scholar]
- Ruddigkeit L.; Deursen R. v.; Blum L. C.; Reymond J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52 (11), 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; Lilienfeld O. A. v. Quantum chemistry structures and properties of 134 kilo molecules. Scientific Data 2014, 1 (1), 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavatskikh M.; Leguy J.; Hunault G.; Cauchy T.; Mota B. D. Dataset’s chemical diversity limits the generalizability of machine learning predictions. Journal of Cheminformatics 2019, 11 (1), 69. 10.1186/s13321-019-0391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136 (3B), B864–B871. 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- Riniker S.; Landrum G. A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model. 2015, 55 (12), 2562–2574. 10.1021/acs.jcim.5b00654. [DOI] [PubMed] [Google Scholar]
- RDKit: Open-Source Cheminformatics Software; 2016. http://www.rdkit.org (accessed 2022-06-15).
- Mobley D. L.; Guthrie J. P. FreeSolv: a database of experimental and calculated hydration free energies, with input files. Journal of Computer-Aided Molecular Design 2014, 28 (7), 711–720. 10.1007/s10822-014-9747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauman J.; Howard P.. Physprop database. Syracuse Research, Syracuse, NY, USA, 1995. [Google Scholar]
- Fourches D.; Muratov E.; Tropsha A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50 (7), 1189–1204. 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunova M.; Ginsburg B.; Tropsha A.; Isayev O. OpenChem: A Deep Learning Toolkit for Computational Chemistry and Drug Design. J. Chem. Inf. Model. 2021, 61 (1), 7–13. 10.1021/acs.jcim.0c00971. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Wu Z.; Hsieh C.-Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. Journal of Cheminformatics 2021, 13 (1), 12. 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: a benchmark for molecular machine learning. Chemical Science 2018, 9 (2), 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvenaud D. K.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R. P.. Convolutional Networks on Graphs for Learning Molecular Fingerprints; 2015. [Google Scholar]
- Coley C. W.; Barzilay R.; Green W. H.; Jaakkola T. S.; Jensen K. F. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction. J. Chem. Inf. Model. 2017, 57 (8), 1757–1772. 10.1021/acs.jcim.6b00601. [DOI] [PubMed] [Google Scholar]
- Xiong Z.; Wang D.; Liu X.; Zhong F.; Wan X.; Li X.; Li Z.; Luo X.; Chen K.; Jiang H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63 (16), 8749–8760. 10.1021/acs.jmedchem.9b00959. [DOI] [PubMed] [Google Scholar]
- Yang K.; Swanson K.; Jin W.; Coley C.; Eiden P.; Gao H.; Guzman-Perez A.; Hopper T.; Kelley B.; Mathea M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59 (8), 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak Y.; Laghuvarapu S.; Mehta S.; Priyakumar U. D. Chemically Interpretable Graph Interaction Network for Prediction of Pharmacokinetic Properties of Drug-Like Molecules. Proceedings of the AAAI Conference on Artificial Intelligence 2020, 34 (01), 873–880. (acccessed 2023/06/21) 10.1609/aaai.v34i01.5433. [DOI] [Google Scholar]
- Vermeire F. H.; Green W. H. Transfer learning for solvation free energies: From quantum chemistry to experiments. Chemical Engineering Journal 2021, 418, 129307. 10.1016/j.cej.2021.129307. [DOI] [Google Scholar]
- Xu K., ; Hu W., ; Leskovec J., ; Jegelka S., . How powerful are graph neural networks? arXiv preprint arXiv:1810.00826. 2018 [Google Scholar]
- Dehmamy N.; Barabási A.-L.; Yu R.. Understanding the representation power of graph neural networks in learning graph topology. Advances in Neural Information Processing Systems 2019, 32. [Google Scholar]
- Garg V.; Jegelka S.; Jaakkola T.. Generalization and representational limits of graph neural networks. In International Conference on Machine Learning, 2020; PMLR: pp 3419–3430. [Google Scholar]
- Sato R.A survey on the expressive power of graph neural networks. arXiv preprint arXiv:2003.04078 2020. [Google Scholar]
- Corso G.; Cavalleri L.; Beaini D.; Liò P.; Veličković P. Principal neighbourhood aggregation for graph nets. Advances in Neural Information Processing Systems 2020, 33, 13260–13271. [Google Scholar]
- Schultz H. P. Topological organic chemistry. polyhedranes and prismanes. Journal of Organic Chemistry 1965, 30 (5), 1361–1364. 10.1021/jo01016a005. [DOI] [Google Scholar]
- Paquette L. A.; Ternansky R. J.; Balogh D. W.; Kentgen G. Total synthesis of dodecahedrane. J. Am. Chem. Soc. 1983, 105 (16), 5446–5450. 10.1021/ja00354a043. [DOI] [Google Scholar]
- Cho H.; Choi I. S. Enhanced Deep-Learning Prediction of Molecular Properties via Augmentation of Bond Topology. ChemMedChem. 2019, 14 (17), 1604–1609. 10.1002/cmdc.201900458. [DOI] [PubMed] [Google Scholar]
- Mater A. C.; Coote M. L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59 (6), 2545–2559. 10.1021/acs.jcim.9b00266. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Scientific Data 2017, 4 (1), 170193. 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenich A. V.; Kelly C. P.; Thompson J. D.; Hawkins G. D.; Chambers C. C.; Giesen D. J.; Winget P.; Cramer C. J.; Truhlar D. G.. Minnesota solvation database (MNSOL) version 2012. 2020.
- Cai C.; Wang S.; Xu Y.; Zhang W.; Tang K.; Ouyang Q.; Lai L.; Pei J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63 (16), 8683–8694. 10.1021/acs.jmedchem.9b02147. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Wu Z.; Hsieh C. Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform 2021, 13 (1), 12. 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; From NLM PubMed-not-MEDLINE.
- Chen D.; Gao K.; Nguyen D. D.; Chen X.; Jiang Y.; Wei G.-W.; Pan F. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat. Commun. 2021, 12 (1), 3521. 10.1038/s41467-021-23720-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.; Zhao Z.; Wang R.; Wei G. W. TopP-S: Persistent homology-based multi-task deep neural networks for simultaneous predictions of partition coefficient and aqueous solubility. Journal of computational chemistry 2018, 39 (20), 1444–1454. 10.1002/jcc.25213. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Wang R.; Wang M.; Gao K.; Nguyen D. D.; Wei G.-W. Boosting Tree-Assisted Multitask Deep Learning for Small Scientific Datasets. J. Chem. Inf. Model. 2020, 60 (3), 1235–1244. 10.1021/acs.jcim.9b01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt A.; Eckert F.; Arlt W. COSMO-RS: An Alternative to Simulation for Calculating Thermodynamic Properties of Liquid Mixtures. Annu. Rev. Chem. Biomol. Eng. 2010, 1 (1), 101–122. 10.1146/annurev-chembioeng-073009-100903. [DOI] [PubMed] [Google Scholar]
- Warnau J.; Wichmann K.; Reinisch J. COSMO-RS predictions of logP in the SAMPL7 blind challenge. J. Comput. Aid Mol. Des 2021, 35 (7), 813–818. 10.1007/s10822-021-00395-5. [DOI] [PubMed] [Google Scholar]
- Kundi V.; Ho J. Predicting Octanol-Water Partition Coefficients: Are Quantum Mechanical Implicit Solvent Models Better than Empirical Fragment-Based Methods?. J. Phys. Chem. B 2019, 123 (31), 6810–6822. 10.1021/acs.jpcb.9b04061. [DOI] [PubMed] [Google Scholar]
- Bannan C. C.; Calabró G.; Kyu D. Y.; Mobley D. L. Calculating Partition Coefficients of Small Molecules in Octanol/Water and Cyclohexane/Water. J. Chem. Theory Comput. 2016, 12 (8), 4015–4024. 10.1021/acs.jctc.6b00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Zhang Y. Unified Deep Learning Model for Multitask Reaction Predictions with Explanation. J. Chem. Inf. Model. 2022, 62 (6), 1376–1387. 10.1021/acs.jcim.1c01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic acids research 2016, 44 (D1), D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Guo A. C.; Lo E. J.; Marcu A.; Grant J. R.; Sajed T.; Johnson D.; Li C.; Sayeeda Z.; et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46 (D1), D1074–d1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]; From NLM.
- Eguida M.; Rognan D. Estimating the Similarity between Protein Pockets. International Journal of Molecular Sciences 2022, 23 (20), 12462. 10.3390/ijms232012462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scantlebury J.; Brown N.; Von Delft F.; Deane C. M. Data Set Augmentation Allows Deep Learning-Based Virtual Screening to Better Generalize to Unseen Target Classes and Highlight Important Binding Interactions. J. Chem. Inf. Model. 2020, 60 (8), 3722–3730. 10.1021/acs.jcim.0c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Shen C.; Liao B.; Jiang D.; Wang J.; Wu Z.; Du H.; Wang T.; Huo W.; Xu L.; et al. TocoDecoy: A New Approach to Design Unbiased Datasets for Training and Benchmarking Machine-Learning Scoring Functions. J. Med. Chem. 2022, 65 (11), 7918–7932. 10.1021/acs.jmedchem.2c00460. [DOI] [PubMed] [Google Scholar]
- van Tilborg D.; Alenicheva A.; Grisoni F. Exposing the Limitations of Molecular Machine Learning with Activity Cliffs. J. Chem. Inf. Model. 2022, 62 (23), 5938–5951. 10.1021/acs.jcim.2c01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.; Tilahun E. L.; Reid T.-E.; Zhang L.; Wang X. S. Benchmarking methods and data sets for ligand enrichment assessment in virtual screening. Methods 2015, 71, 146–157. 10.1016/j.ymeth.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réau M.; Langenfeld F.; Zagury J.-F.; Lagarde N.; Montes M. Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Frontiers in Pharmacology 2018, 9, Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-López E.; Fernández-de Gortari E.; Medina-Franco J. L. Yes SIR! On the structure-inactivity relationships in drug discovery. Drug Discovery Today 2022, 27 (8), 2353–2362. 10.1016/j.drudis.2022.05.005. [DOI] [PubMed] [Google Scholar]
- Sundar V.; Colwell L. The Effect of Debiasing Protein-Ligand Binding Data on Generalization. J. Chem. Inf. Model. 2020, 60 (1), 56–62. 10.1021/acs.jcim.9b00415. [DOI] [PubMed] [Google Scholar]
- Wang L.; Shi S.-H.; Li H.; Zeng X.-X.; Liu S.-Y.; Liu Z.-Q.; Deng Y.-F.; Lu A.-P.; Hou T.-J.; Cao D.-S. Reducing false positive rate of docking-based virtual screening by active learning. Briefings in Bioinformatics 2023, 24 (1), bbac626.(acccessed 7/22/2023) 10.1093/bib/bbac626. [DOI] [PubMed] [Google Scholar]
- Adeshina Y. O.; Deeds E. J.; Karanicolas J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (31), 18477–18488. (acccessed 2023/07/22) 10.1073/pnas.2000585117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H.; Kim H.; Allen J. E.; Wulff H. Pose Classification Using Three-Dimensional Atomic Structure-Based Neural Networks Applied to Ion Channel-Ligand Docking. J. Chem. Inf. Model. 2022, 62 (10), 2301–2315. 10.1021/acs.jcim.1c01510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg J.; Flachsenberg F.; Rarey M. In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2019, 59 (3), 947–961. 10.1021/acs.jcim.8b00712. [DOI] [PubMed] [Google Scholar]
- Cimermancic P.; Weinkam P.; Rettenmaier T. J.; Bichmann L.; Keedy D. A.; Woldeyes R. A.; Schneidman-Duhovny D.; Demerdash O. N.; Mitchell J. C.; Wells J. A.; et al. CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites. J. Mol. Biol. 2016, 428 (4), 709–719. 10.1016/j.jmb.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller A.; Ward M.; Borowsky J.; Kshirsagar M.; Lotthammer J. M.; Oviedo F.; Ferres J. L.; Bowman G. R. Predicting locations of cryptic pockets from single protein structures using the PocketMiner graph neural network. Nat. Commun. 2023, 14 (1), 1177. 10.1038/s41467-023-36699-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feixas F.; Lindert S.; Sinko W.; McCammon J. A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. 10.1016/j.bpc.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath P. A.; Forli S.; Goodsell D. S.; Olson A. J.; Sanner M. F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLOS Computational Biology 2015, 11 (12), e1004586 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guterres H.; Im W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60 (4), 2189–2198. 10.1021/acs.jcim.0c00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Y.; Wei Y.; Wang P.; Wang X.; Li H.; Wu N.; Bauer S.; Zheng S.; Shi Y.; Wang Y.; et al. From Static to Dynamic Structures: Improving Binding Affinity Prediction with a Graph-Based Deep Learning Model. arXiv e-prints 2022, 10.48550/arXiv.2208.10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E.; Baudry J.; Chodera J.; Demir Ö.; McCammon J. A.; Miao Y.; Smith J. C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114 (10), 2271–2278. 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. F. Flexible receptor docking for drug discovery. Expert Opinion on Drug Discovery 2015, 10 (11), 1189–1200. 10.1517/17460441.2015.1078308. [DOI] [PubMed] [Google Scholar]
- Chandak T.; Mayginnes J. P.; Mayes H.; Wong C. F. Using machine learning to improve ensemble docking for drug discovery. Proteins: Struct., Funct., Bioinf. 2020, 88 (10), 1263–1270. (acccessed 2023/07/24) 10.1002/prot.25899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Lopez J.; Aguila S. A.; Gilson M. K.; Brizuela C. A. Improving Structure-Based Virtual Screening with Ensemble Docking and Machine Learning. J. Chem. Inf. Model. 2021, 61 (11), 5362–5376. 10.1021/acs.jcim.1c00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi S.; Narimani Z.; Ashouri M.; Firouzi R.; Karimi-Jafari M. H. Ensemble learning from ensemble docking: revisiting the optimum ensemble size problem. Sci. Rep 2022, 12 (1), 410. 10.1038/s41598-021-04448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]