

Abstract
We conducted the present study to investigate protein expression and functioning of A2A and A2B adenosine receptors (ARs) in neutrophils of patients affected by systemic sclerosis (SSc). The presence of A2A and A2B ARs was assessed by immunoblotting using specific antibodies. Equilibrium A2A and A2B ARs binding parameters were evaluated by radioligand binding assay. Functional studies were conducted to investigate coupling of the A2B AR to the adenylyl cyclase pathway. This is the first report of the use of Western blot analysis to confirm the presence of A2A and A2B ARs in human neutrophils. No significant changes in A2A AR binding parameters or expression levels were detected between SSc patients and healthy control individuals. A significant decrease (65%) in the maximum density of A2B AR binding sites occurred in SSc neutrophils, whereas no changes in the affinity constant values were found. Moreover, a decrease in A2B AR mediated adenylyl cyclase activity was observed in patients with SSc. Our findings demonstrate the occurrence of selective alterations in A2B AR density and signalling in SSc.
Keywords: adenosine, A2 adenosine receptors, neutrophils, receptor binding, systemic sclerosis
Introduction
Systemic sclerosis (SSc), also known as scleroderma, is a connective tissue disease of unknown aetiology. Possibly an autoimmune disorder, it is accompanied in the vast majority of cases by the presence of antinuclear antibodies [1]. SSc may affect virtually any organ of the body, including skin, gastrointestinal tract, lungs, heart, kidneys, and musculoskeletal system. Altered connective tissue metabolism can cause either localized or diffuse thickening of the skin, while inflammation is associated with endothelial damage. Clinically, microvascular disturbance, teleangiectasia, Raynaud's phenomenon, polyarthralgia and polyarthritis, as well as oesophageal hypomobility, visceral muscolaris mucosa damage and pulmonary fibrosis, have been described [2].
The mechanisms leading to endothelial damage, inflammation and fibrosis are unclear. Reactive oxygen species in neutrophils may increase the extent of inflammation and fibrosis during the respiratory burst and could be involved in endothelial damage [3]. The endothelial cells of microvessels are deficient in the synthesis of catalase, which provides natural defence against superoxide damage, and appear to be particularly susceptible to superoxide injury during reperfusion [4].
Adenosine is an important endogenous regulator of neutrophil functioning. It is released intracellularly and modulates neutrophil activity by interacting with specific surface receptors [5]. Distinct adenosine receptor (AR) subtypes A1, A2A, A2B and A3 have been identified and their functions characterized in neutrophils. Specifically, activation of A1 ARs enhances chemotaxis, phagocytosis and adherence [6,7]; A2A ARs inhibit reactive oxygen species generation, phagocytosis and adherence [8-10]; and A2A and A3 ARs inhibit neutrophil degranulation [11-14]. Adenosine has been shown to prevent the release of vascular endothelial growth factor from neutrophils via A2B AR activation [15]. Because activation of ARs reduces both immune and inflammatory responses, adenosine release has been hypothesized to be a possible mechanism of cell self-protection from activated neutrophils [5]. An increase in adenosine deaminase activity has been described in patients with SSc, suggesting an alteration in adenosine control mechanisms in this disease [16,17].
In the present study we analyzed A2A and A2B AR subtypes in neutrophils from patients affected by SSc by means of expression analysis, radioligand binding assays and functional studies.
Methods
Chemicals and reagents
Bacitracine, benzamidine, trypsin inhibitor, sodium orthovanadate, Nonidet P-40, SDS, phenylsulfonylfluoride, aprotinin and adenosine deaminase (ADA) were purchased from Sigma (St. Louis, MO, USA). Unlabelled AR agonists/antagonists and the anti-β-actin antibody were supplied by RBI/Sigma (St. Louis, MO, USA). [3H]CGS21680 (CGS21680 = [2-p-(2-carbowyethyl)phenylethylamino]-5'-N-ethylcarboxamidoadenosine), [3H]NECA (NECA = 5'-N-ethylcarboxamidoadenosine), and [32P]α-ATP were supplied by NEN Life Sciences (Köln, Germany). Electrophoresis reagents were purchased from BioRad (Munchen, Germany). A2AAR and A2BAR antibodies were supplied by Alpha Diagnostic (San Antonio, TX, USA). All other chemicals were from standard commercial sources.
Patients
Twenty-six patients affected by SSc were included in the study (22 women and 4 men; mean age ± standard deviation 53.0 ± 11.3 years). They all fulfilled standard criteria of the American College of Rheumatology for SSc. Sixteen patients were anticentromere antibody positive and four were SCL-70 positive. Limited symptoms of disease, involving skin thickness alterations to the face, hands and feet, were present in 18 patients (mean disease duration <5 years, skin score range [according to the modified Rodnan total skin thickness score] 10–21). Diffuse symptoms with more extensive skin involvement were present in eight patients (mean disease duration <5 years, total skin thickness score range 27–30). The activity score [18] varied between 0.5 and 3.5 and the severity score [19] between 2 and 6. The erythrocyte sedimentation rate was 24 ± 23 mm/hour (mean ± standard deviation).
Control samples were obtained from 26 healthy volunteers, who were similar to the patients included in the study in terms of sex distribution and age (20 women and 6 men; mean age ± standard deviation 49.0 ± 9.2 years). Informed consent to participate in the study was obtained from all individuals.
Sample collection and neutrophil preparation
Venous blood (20 ml) was drawn between 08:00 and 09:00 a.m. from fasting individuals by antecubital venipuncture, collected in heparinized (10 IU/L) plastic tubes and processed immediately. Neutrophils were isolated following the Boyum method [20] with some modifications.
Western blot analysis
Neutrophils were lysed in RIPA buffer (150 mmol/l NaCl, 50 mmol/l Tris-HCl, pH 8, 0.5% sodium deoxhycolate, 1% Nonidet P-40, 1 mmol/l phenylsulfonylfluoride, 10 μg/ml aprotinin, 100 μmol/l sodium orthovanadate) for 1 hour at 4°C. After centrifugation at 15,000 g for 30 min, soluble fractions were assayed for protein content using BioRad protein assay. Equivalent amounts of proteins (50 μg/sample) were analyzed by SDS-PAGE, using 10% (weight/vol) polyacrylamide resolving gels. Protein bands were transferred to nitrocellulose and probed with 0.1 μg/ml rabbit anti-human A2A AR or A2B AR antibodies.
A2A AR antibody is an affinity-purified rabbit polyclonal antibody raised against a peptide mapping to the carboxyl-terminus of A2A AR. It specifically reacts with human, bovine, rat and pig A2A receptors and does not cross-react with A1, A2B, or A3 AR subtypes. A2B AR antibody is an affinity-purified rabbit polyclonal antibody raised against a region that corresponds to the second extracellular domain of A2B AR of human origin.
After washing, membranes were incubated with anti-rabbit secondary antibody conjugated to horseradish peroxidase for 2 hours at room temperature, and bands were visualized by chemiluminescence, in accordance with the manufacturer's instructions (Sigma-Aldrich). Membranes were re-probed with an anti-β-actin antibody for normalization.
Binding assay
For membrane preparation, cells were washed twice with 10 mmol/l Tris-HCl buffer, pH 7.4, containing 10 mmol/l MgCl2, in the presence of protease inhibitors (200 μg/ml bacitracine, 160 μg/ml benzamidine, 20 μg/ml trypsin inhibitor [T1]) and centrifuged at 48,000 g for 15 min at 4°C. Pellets were diluted in 20 volumes of T1 buffer, treated with ADA (2 IU/ml) for 60 min at 37°C to remove endogenous adenosine, and washed twice with 50 mmol/l Tris-HCl buffer, pH 7.4, containing 10 mmol/l MgCl2 (T2).
A2A AR binding assay was performed by using a specific radiolabelled A2A AR agonist, namely [3H]CGS21680. Aliquots of neutrophil membranes (0.2–0.3 mg protein) were incubated with different [3H]CGS21680 concentrations (5–30 nmol/l) in a final volume of 250 μl of T2 buffer. Nonspecific binding was determined in the presence of 100 μmol/l NECA. After 90 min incubation at 25°C, the binding reaction was terminated by vacuum filtration through Whatman GF/C glass fibre filters (Whatman, Maidstone, UK), accompanied by three washes with ice-cold T2 buffer (4 ml). A2A AR specificity was evaluated through competition experiments, using different AR ligands.
A2B AR binding assay was performed using 20 nmol/l [3H]NECA in the presence of 50 nmol/l cyclopentyladenosine (CPA) and 100 nmol/l SCH58261 (SCH58261 = 5-amino-7-[phenylethyl]-2-[2-furyl]-pyrazolo [4,3-e]-1,2,4-triazolo [1,5-c]pyrimidine) to prevent [3H]NECA binding to A1 and A2A ARs, respectively [21]. Scatchard analysis was performed on competition experiments carried out in the presence of unlabelled NECA at concentrations ranging from 50 nmol/l to 2 mmol/l. Aliquots of neutrophil membranes (0.2–0.4 mg proteins) were incubated in a final volume of 250 μl T2 buffer. Nonspecific binding was evaluated in the presence of 100 μmol/l NECA. After 90 min incubation at 0°C, the reaction was terminated either by vacuum filtration through Whatman GF/C glass fibre filters, accompanied by three washes with ice-cold T2 buffer (4 ml), or by centrifugation at 2900 g for 15 min at 4°C. A2B AR specificity was evaluated through competition experiments, using different AR ligands.
Adenylyl cyclase assay
Neutrophils were homogenized in buffer solution containing 10 mmol/l Hepes, 1 mmol/l EGTA and 10 mmol/l NaCl2, and then centrifuged at 46,500 g for 20 min at 4°C. Pellets were resuspended in 10 volumes of 10 mmol/l Hepes, containing protease inhibitors (200 μg/ml bacitracine and 160 μg/ml benzamidine), incubated for 30 min at 30°C with 2 U/ml ADA, and centrifuged. Adenylyl cyclase (AC) activity was measured as described by Salomon [22] and Johnson and Salomon [23], with some modifications. NECA-mediated stimulation of AC activity was assessed by incubating aliquots of membranes with increasing NECA concentrations from 0.01 nmol/l to 10 μmol/l. The reaction was started by adding membrane aliquots (10–50 μg proteins/tube), conducted for 15 min at 24°C, and then stopped by transferring samples on ice and adding 500 μl ice-cold stop solution (120 mmol/l zinc acetate, 144 mmol/l Na2CO3). The stop solution contained [3H]cAMP (10,000–15,000 cpm/sample) to monitor column recovery. Newly formed ZnCO3 allowed precipitation of residual ATP, discarded through centrifugation at 2700 g for 8 min. Supernatants containing both [32P]α-cAMP and [3H]cAMP were further purified by double-step Dowex-Alumina chromatography and counted by means of a β-counter (Packard Tricarb 1600; Perkin Elmer, Wellesley, MA, USA).
To evaluate A2B AR mediated cAMP accumulation, the reaction was carried out in the presence of selective A2A antagonist SCH58261 at a concentration (100 nmol/l) able to block A2A receptors completely [21].
Data and statistical analysis
Affinity constant values (Kd) and maximum number of binding sites (Bmax) were calculated using the nonlinear multipurpose curve-fitting computer program Graph-Pad Prism The 50% inhibitory concentration values were calculated using the same program and converted to Ki values through the Cheng and Prusoff equation.
A GS-670-BIO-RAD imaging densitometer was used for semiquantitative analysis of immunoblots. Partial F test (P < 0.01) was used to determine binding data with the best fit to a one-site or two-site model. Differences in binding parameters between SSc patients and control individuals were evaluated by one-way analysis of variance.
Results
In both control and SSc neutrophils, Western blot analysis identified two specific immunoreactive bands of 45 kDa and 50 kDa, corresponding to A2A and A2B ARs, respectively (Fig. 1). This confirmed the presence of both AR subtypes in human neutrophils.
Figure 1.
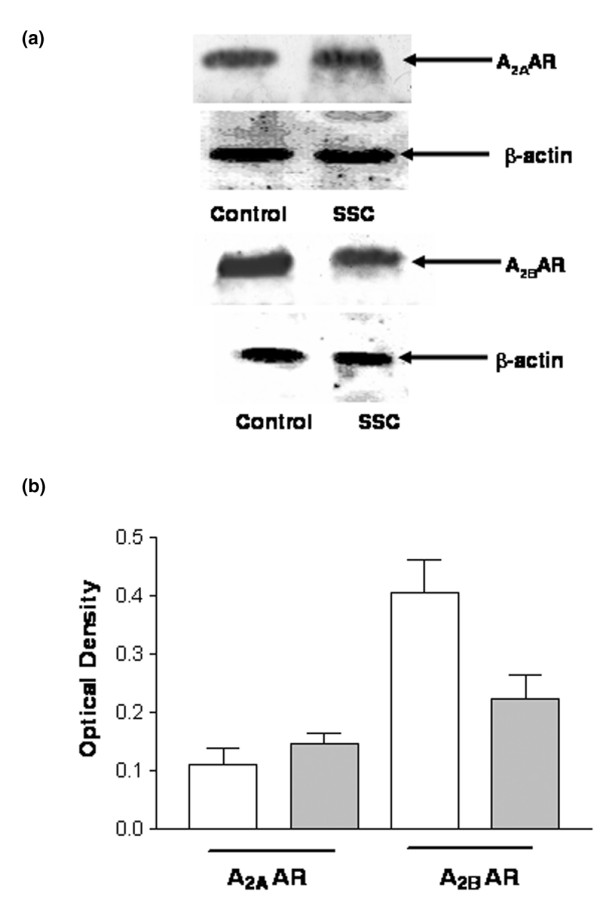
Immunoblotting analysis of A2A and A2B adenosine receptors (ARs) from systemic sclerosis (SSc) neutrophils and controls. Cells obtained from 26 healthy volunteers and 26 SSc patients were lysed as described in the Methods section. Equal amounts of protein (50 μg) were separated on polyacrylamide gel, blotted and probed with 0.1 μg/ml rabbit anti-human A2A AR or A2B AR antibodies. Immunoreactive bands were visualized according to electrogenerated chemiluminescence protocol. A2A and A2B AR antibodies recognized immunoreactive bands of 45 kDa and 50 kDa, respectively. (a) Representative experiment performed on neutrophils from one healthy volunteer and one SSc patient. (b) Densitometric analysis of A2A and A2B AR immunoreactive bands from 26 healthy volunteers and 26 SSc patients. Graph bars: mean ± standard error of band density, normalized to β-actin. White bars are controls; grey bars are SSc patients.
To characterize ARs, binding assays were conducted in neutrophil membrane fractions. SSc patients were randomly divided into two subgroups in order to obtain large amounts of protein, as required by the experiments.
The selective A2A AR agonist [3H]CGS21680 identified a homogenous population of binding sites in control individuals. Kd and Bmax values were 25 ± 1.3 nmol/l and 35 ± 2.4 fmol/mg protein, respectively (Fig. 2). Competition experiments using [3H]CGS21680 in combination with a variety of A2A ligands revealed a pharmacological profile typical for A2A ARs (R-PIA [R-N6-phenylisopropyladenosine] > teofilline > NECA > SCH58261; data not shown). Scatchard analysis for SSc neutrophils revealed no significant differences in Kd and Bmax between patients (mean values: Kd = 23 ± 1.8 nmol/l, Bmax = 40 ± 3.2 fmol/mg protein) and healthy control individuals (P > 0.05; Fig. 2), suggesting that no alteration in A2A binding sites occurs in SSc. In agreement with this, densitometric analysis of immunoblots showed no significant changes in A2A AR immunoreactive bands in SSc neutrophils relative to controls (optical density: 0.11 ± 0.03 for patients versus 0.15 ± 0.02 for controls).
Figure 2.
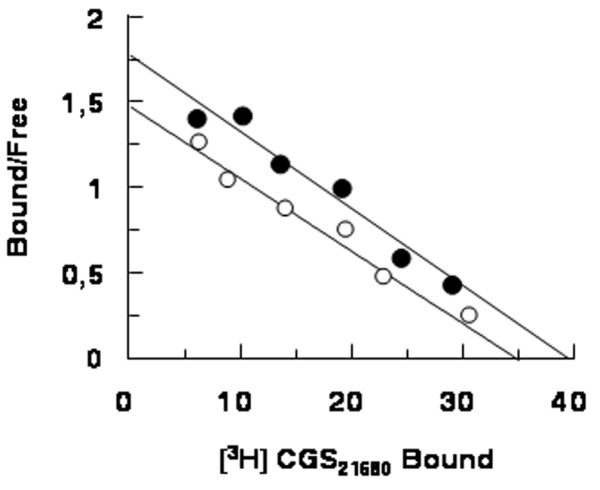
Representative Scatchard plot of [3H]CGS21680 saturation binding data. Empty circles indicate neutrophil membranes from healthy volunteers (affinity constant [Kd] = 25 ± 1.3 nmol/l; maximum number of binding sites [Bmax] = 35 ± 2.4 fmol/mg); filled circles indicate neutrophil membranes from systemic sclerosis (SSc) patients overall (Kd = 23 ± 1.8 nmol/l; Bmax = 40 ± 3.2 fmol/mg). Assays were performed in triplicate.
A2B AR binding sites were identified using [3H]NECA as radioligand in the presence of 50 nmol/l CPA and 100 nmol/l SCH58261, to prevent nonspecific binding to A1 and A2A AR subtypes. We performed competition experiments using a wide range (50 nmol/l to 2 mmol/l) of [3H]NECA concentrations to allow the identification of A2B AR low-affinity binding sites. Data analysis revealed that the one-site model produced a significantly better fit than the two-site model (P < 0.05), both in control and SSc neutrophils. In our experimental conditions, control neutrophils exhibited the presence of low-affinity binding sites with Kd and Bmax values of 476 ± 34 nmol/l and 3696 ± 210 fmol/mg, respectively (Fig. 3). Competition experiments using [3H]NECA in combination with a variety of AR ligands revealed a pharmacological profile typical for A2B ARs (R-PIA > teofilline > SCH58261 = MRS1220 > DPCPX > 2Cl-adenosine > NECA > MRS1706; Table 1). Scatchard analysis for SSc neutrophils showed no significant differences in Kd and Bmax between the two subgroups of patients. However, a significant alteration in Bmax was found relative to controls, whereas Kd values remained unaltered. Overall, mean values for Kd and Bmax in SSc were 469 ± 35 nmol/l and 1292 ± 98 fmol/mg protein, respectively (P < 0.05; Fig. 3). Moreover, experiments conducted in individual patients using a concentration of NECA of 500 nmol/l showed similar specific binding values (expressed as fmol/mg protein), confirming the homogeneity of A2B AR sites between SSc subgroups (Fig. 4). The alteration in A2B AR levels in SSc patients was confirmed by immunoblotting assay. Densitometric analysis of immunoreactive bands showed a reduction in A2B expression in SSc patients (optical density 0.22 ± 0.04) as compared with controls (optical density 0.40 ± 0.06; P < 0.05; Fig. 1).
Figure 3.
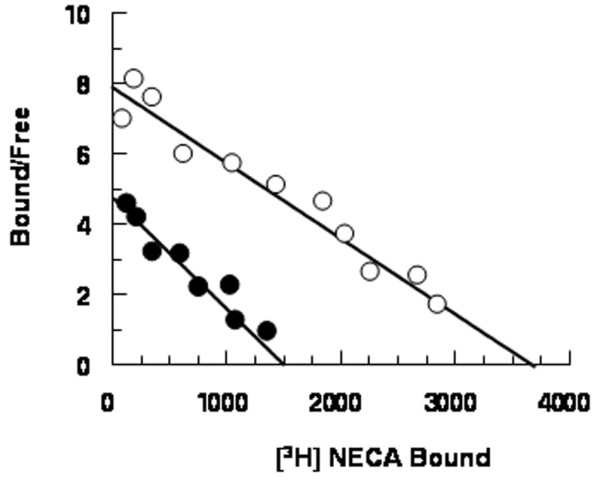
Representative Scatchard plot of [3H]NECA saturation binding data. Competition binding experiments were performed, incubating aliquots of neutrophil membranes with 20 nmol/l [3H]NECA and different NECA concentrations (50 nmol/l to 2 mmol/l), in the presence of 50 nmol/l CPA and 100 nmol/l SCH58261. Empty circles indicate neutrophil membranes from healthy volunteers (affinity constant [Kd] = 476 ± 34 nmol/l, maximum number of binding sites [Bmax] = 3696 ± 210 fmol/mg); filled circles indicate neutrophil membranes from systemic sclerosis (SSc) patients overall (Kd = 469 ± 35 nmol/l, Bmax = 1292 ± 98 fmol/mg). Assays were performed in triplicate.
Table 1.
Specificity of [3H]NECA binding to A2B adenosine receptors in control neutrophil membranes
| [3H]NECA Ki (μmol/l) | |
| NECA | 0.315 ± 0.028 |
| 2 Cl-adenosine | 0.954 ± 0.600 |
| R-PIA | 1000 ± 86 |
| SCH58261 | >10 |
| Teofilline | 47 ± 3.5 |
| MRS1706 | 0.005 ± 0.0003 |
| DPCPX | 2 ± 0.12 |
| MRS1220 | >10 |
Competition experiments were performed, incubating aliquots of neutrophil membranes with 20 nmol/l [3H]NECA (plus 50 nmol/l CPA and 100 nmol/l SCH58261) in the presence of increasing ligand concentrations. Ki values are expressed as mean ± SEM of three separate experiments. Ki values were calculated from IC50 values (concentration of drug causing 50% inhibition of specific binding) using the Cheng and Prusoff equation.
Figure 4.
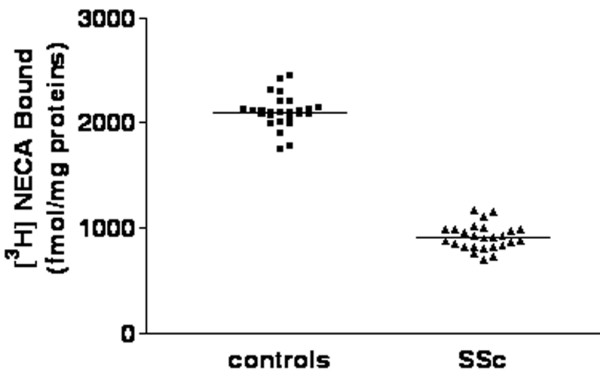
A2B adenosine receptor binding experiments performed in individual patients using NECA at 500 nmol/l concentration. Neutrophils were obtained from healthy volunteers (n = 26) and systemic sclerosis (SSc) patients (n = 26). Horizontal lines indicate the mean values.
Functional coupling of A2B ARs to stimulatory G proteins in neutrophil membranes was assessed by evaluating the effects of the agonist NECA (in the presence of 100 nmol/l SCH58261) on AC activity. NECA stimulated AC activity in a concentration dependent manner. Dose-response curves revealed significant differences between SSc patients (EC50 = 373 ± 26 nmol/l; Emax = 35 ± 2.9%) and controls (EC50 = 165 ± 9.3 nmol/l; Emax = 43 ± 3.2%), suggesting an alteration in A2B AR responsiveness in SSc (Fig. 5).
Figure 5.

A2B adenosine receptor (AR)-mediated stimulation of adenylyl cyclase activity in control (empty circles) and systemic sclerosis (SSc; filled circles) neutrophil membranes. Membranes were incubated with different NECA concentrations (ranging from 10 nmol/l to 100 μmol/l) and the activity of adenylyl cyclase, expressed as pmol/min per mg protein, was evaluated. Values are expressed as mean ± standard error of three indipendent experiments. EC50 values were 165 ± 9.3 for control versus 373 ± 26 nmol/l for SSc.
Discussion
In the present study we analyzed A2A and A2B AR subtypes in neutrophils of patients affected by SSc, by means of Western blot, radioligand binding techniques and functional studies. This is the first report of use of Western blot analysis to confirm the presence of A2A and A2B ARs in human neutrophils.
A2A and A2B AR equilibrium binding parameters were measured using radioligand binding assays. Scatchard analysis of [3H]CGS21680 saturation binding to A2A AR showed no significant difference in Bmax or Kd between SSc neutrophils and controls, suggesting that the A2A AR subtype remained unaltered in SSc. Conversely, when A2B AR was analyzed a reduction in Bmax (65%) was observed, with no significant change in Kd values.
A2B ARs are known to be low-affinity adenosine binding sites. Competition experiments using a variety of A2B AR agonists and antagonists revealed a pharmacological profile typical of A2B ARs, which is consistent with studies conducted in transfected cell models. Our findings represent the first characterization of A2B ARs in neutrophils with binding experiments.
In order to analyze a population of nonhomogenous patients and to evaluate the impact of the disease on A2 ARs, SSc patients were randomly divided into two subgroups. No difference was found when the two groups were compared, suggesting that different degrees of disease severity and activity had no impact on the assays, but that the disease per se is required to modulate levels and functioning of A2B receptors.
Functional studies were performed to investigate whether the decrease in level of A2B ARs was accompanied by alterations in receptor responsiveness. An evaluation of the ability of NECA to increase AC activity revealed functional coupling of A2B receptors to G proteins. In SSc patients a significant reduction (by more than 50%) in NECA potency was observed, without any effect on agonist efficacy.
Our findings suggest that a selective reduction in A2B AR levels and responsiveness occurred in SSc. Alterations in the expression and functionality of A2B ARs (low-affinity ARs) in patients with SSc may be responsible for the increase in free oxygen radicals, and consequent oxidative damage, that characterizes SSc. This would account for impaired control of hypoxic and inflammatory processes.
In neutrophils it has long been known that adenosine and its analogues inhibit O2 - generation, phagocytosis and cell adherence by occupying specific A2 ARs. Because hypoxia, ischaemia and inflammation can stimulate adenosine production, A2 AR regulation has been postulated to be a self-protective mechanism for cells from activated neutrophils [24]. Eltzschig and coworkers [25] reported that A2B ARs are selectively upregulated in endothelial cells by hypoxia (more than fivefold increase in mRNA), which is associated with ATP hydrolysis and release of adenosine. Taken together, these findings show some coordination between AR transcription and nucleoside signalling at the vascular interface during hypoxia. We might speculate that chronic inflammatory conditions in SSc patients impaired regulatory mechanisms mediated by the anti-inflammatory effects of adenosine via A2B AR activation. In addition, it was reported by Visser and coworkers [26] that increases in cAMP in activated neutrophils play an anti-inflammatory role. The reduced activation of cAMP we observed in SSc patients might be correlated with the inability of these patients to control the inflammatory process.
It was no surprise to find an alteration in adenosinergic system responsiveness in SSc. In fact, adenosine produces a constellation of responses, including anti-inflammatory actions and vasodilatation, mediated through interactions with high-affinity receptor subtype A2A and low-affinity receptor subtype A2B. Moreover, in SSc and related disorders, alterations in adenosine metabolism have been suggested. Indeed, purine analogue 2-chlorodeoxyadenosine, which is utilized for the treatment of such chronic disorders [27,28], appears to reduce the number of abnormal fibroblasts.
A2B ARs were initially thought to be of lesser physiological relevance because of their relatively low affinity for adenosine, and it was only recently that important functions attributable to A2B ARs were discovered. A pivotal role for them was postulated in inflammatory pathological conditions, when adenosine is released at high levels (up to the micromolar range). In light of our findings, a closer examination of A2B AR functions may be valuable because of the potential therapeutic importance of these receptors as targets for treatment with selective agents.
Conclusion
Our findings demonstrated a reduction in A2 low-affinity (A2B) AR density and functioning in neutrophils of patients affected by SSc, suggesting an alteration in adenosinergic system responsiveness. This reduction could relate to the increased production of free oxygen radicals and consequent oxidative damage that characterize SSc, highlighting an impairment in the ability of neutrophils to control hypoxia and inflammation.
No differences between two randomly selected subgroups of SSc patients were found, thus suggesting that different degrees of disease severity and activity had no impact on the degree of A2B AR reduction. Consequently, the functional status of A2B ARs may be considered a marker of the disease, making it worthwhile to characterize a larger cohort of patients, including their closest relatives and patients with early SSc.
Abbreviations
AC = adenylyl cyclase; ADA = adenosine deaminase; AR = adenosine receptor; Bmax = maximum number of binding sites; CGS21680 = (2-p-[2-carbowyethyl]pheylethylamino)-5'N-ethylcarboxamidoadenosine; CPA = cyclopentyladenosine; Kd = affinity constant; NECA = 5'-N-ethylcarboxamidoadenosine; R-PIA = R-N6-phenylisopropyladenosine; SCH58261 = 5-amino-7-(phenylethyl)-2-(2-furyl)-pyrazolo(4,3-e)-1,2,4-triazolo(1,5-c)pyrimidine; SSc = systemic sclerosis.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
LB organized the study design and recruited the patients. LT carried out the binding experiments and statistical analysis. AR participated in the immunoblotting experiments and helped to draft the manuscript. FdF participated in the collection of human samples. AL participated in the coordination of the study and helped with problem solving. SB participated in the coordination of the study and in planning the manuscript. CM participated in the coordination of the study and designed the AC assay. All authors read and approved the final manuscript.
Contributor Information
Laura Bazzichi, Email: l.bazzichi@int.med.unipi.it.
Letizia Trincavelli, Email: ltrincavelli@farm.unipi.it.
Alessandra Rossi, Email: rossi@farm.unipi.it.
Francesca De Feo, Email: fdefeo@farm.unipi.it.
Antonio Lucacchini, Email: lucas@farm.unipi.it.
Stefano Bombardieri, Email: s.bombardieri@int.med.unipi.it.
Claudia Martini, Email: cmartini@farm.unipi.it.
References
- Seibold JR. Scleroderma. In: Ruddy S, Harris ED, Sledge CB, editor. Kelley's Textbook of Rheumatology. 6. Vol. 2. Philadelphia: WB Saunders; 2000. pp. 1211–1239. [Google Scholar]
- Medsger TA., Jr . Systemic sclerosis (scleroderma): clinical aspects. In: Koopman W, editor. Arthritis and Allied Conditions: Textbook of Rheumatology. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 1590–1624. [Google Scholar]
- Murrel D. A radical proposal for the pathogenesis of scleroderma. J Am Acad Dermatol. 1993;28:78–85. doi: 10.1016/0190-9622(93)70014-k. [DOI] [PubMed] [Google Scholar]
- Shingo M, Yoshioka K, Nobunaga M. Human vascular smooth muscle cells and endothelial cells lack catalase activity and are susceptible to hydrogen peroxide. Inflammation. 1985;9:309–320. doi: 10.1007/BF00916279. [DOI] [PubMed] [Google Scholar]
- Schrier DJ, Imre KM. The effects of adenosine agonists on human neutrophil function. J Immunol. 1986;137:3284–3289. [PubMed] [Google Scholar]
- Rose FR, Hirschhorn R, Weissmann G, Cronstein BN. Adenosine promotes neutrophil chemotaxis. J Exp Med. 1988;167:1186–1194. doi: 10.1084/jem.167.3.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN, Levin RI, Philips MR, Hirschhorn R, Abramson SB, Weissman G. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J Immunol. 1992;148:2201–2206. [PubMed] [Google Scholar]
- Cronstein BN, Kubersky SM, Weissman G, Hirschhorn R. Engagement of adenosine receptors inhibits hydrogen peroxide (H2O2) release by activated human neutrophils. Clin Immunol Immunopathol. 1987;42:76–85. doi: 10.1016/0090-1229(87)90174-7. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, Levin RI, Belanoff J, Weissman G, Hirschhorn R. Adenosine: an endogenous inhibitor of neutrophil-mediated injury to endothelial cells. J Clin Invest. 1986;78:760–770. doi: 10.1172/JCI112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN, Haines KA. Stimulus-response uncoupling in the neutrophil. Adenosine A2-receptor occupancy inhibits the sustained, but not the early, events of stimulus transduction in human neutrophils by a mechanism independent of actin-filament formation. Biochem J. 1992;281:631–635. doi: 10.1042/bj2810631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, Bai TR. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- Bouma MG, Jeunhomme TM, Boyle DL, Dentener MA, Voitenok NN, Van den Wildenberg FA, Buurman WA. Adenosine inhibits neutrophil degranulation in activated human whole blood: involvement of adenosine A2 and A3. J Immunol. 1997;158:5400–5408. [PubMed] [Google Scholar]
- Ezeamuzie CI. Involvement of A3 receptors in the potentiation by adenosine of the inhibitory effect of theophylline on human eosinophil degranulation: possible novel mechanism of the anti-inflammatory action of theophylline. Biochem Pharmacol. 2001;61:1551–1559. doi: 10.1016/S0006-2952(01)00613-X. [DOI] [PubMed] [Google Scholar]
- Ezeamuzie CI, Philips E. Positive coupling of atypical adenosine A3 receptors on human eosinophils to adenylyl cyclase. Biochem Biophys Res Commun. 2003;300:712–718. doi: 10.1016/S0006-291X(02)02910-8. [DOI] [PubMed] [Google Scholar]
- Wakai A, Wang JH, Winter DC, Street JT, O'Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock. 2001;15:297–301. doi: 10.1097/00024382-200115040-00008. [DOI] [PubMed] [Google Scholar]
- Meunier P, Filipe P, Emerit I, Freitas J, Guerra Rodrigo F, Manso C. Adenosine deaminase in progressive systemic sclerosis. Acta Derm Venereal. 1995;75:297–299. doi: 10.2340/0001555575297299. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Nakajima H. Serum adenosine deaminase activity in systemic sclerosis (scleroderma) and related disorders. J Am Acad Dermatol. 1992;27:411–414. doi: 10.1016/0190-9622(92)70209-x. [DOI] [PubMed] [Google Scholar]
- Valentini G, Della Rossa A, Bombardieri S, Bencivelli W, Silman AJ, D'Angelo S, Cerinic MM, Belch JF, Black CM, Bruhlmann P, et al. European multicentre study to define disease activity criteria for systemic sclerosis. II. Identification of disease activity variables and development of preliminary activity indexes. Ann Rheum Dis. 2001;60:592–598. doi: 10.1136/ard.60.6.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medsger TA, Jr, Silman AJ, Steen VD, Black CM, Akesson A, Bacon PA, Harris CA, Jablonska S, Jayson MI, Jimenez SA, et al. A disease severity scale for systemic sclerosis: development and testing. J Rheumatol. 1999;26:2159–2167. [PubMed] [Google Scholar]
- Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of mononuclear cells by centrifugation and sedimentation at 1 × g. Scand J Clin Lab Invest. 1968;21:77–89. [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. Pharmacological characterization of adenosine A2B receptors. Studies in human mast cells co-expressing A2A and A2B adenosine receptors subtypes. Biochem Pharmacol. 1998;55:627–633. doi: 10.1016/S0006-2952(97)00512-1. [DOI] [PubMed] [Google Scholar]
- Salomon Y. Adenylate cyclase assay. Adv Cyclic Nucleotide Res. 1979;10:35–55. [PubMed] [Google Scholar]
- Johnson RA, Salomon Y. Determination of adenylyl cyclase catalytitc activity using a single and double column procedure. In: Johnson RA, Corbin JD, editor. Methods in Enzymology. Vol. 195. New York: Academic Press; 1994. pp. 1–21. [DOI] [PubMed] [Google Scholar]
- Martini C, Trincavelli L, Fiorini M, Nardi M, Bazzichi L, Lucacchini A. Effect of FMLP stimulation on [3H] NECA binding to adenosine receptors in neutrophil membranes. Adv Exp Med Biol. 1998;431:89–94. doi: 10.1007/978-1-4615-5381-6_17. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenosine nucletoide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser S, Theron AJ, Ramafi G, Ker JA, Anderson R. Apparent involvement of the A2A subtype adenosine receptor in the anti-inflammatory interactions of CGS 21680, cyclopentyladenosine, and IB-MECA with human neutrophils. Biochem Pharmacol. 2000;60:993–999. doi: 10.1016/S0006-2952(00)00414-7. [DOI] [PubMed] [Google Scholar]
- Davis LS, Sanal S, Sangueza OP. Treatment of scleromyxedema with 2-chlorodeoxyadenosine. J Am Academy Dermatol. 1996;35:288–290. doi: 10.1016/S0190-9622(96)90650-7. [DOI] [PubMed] [Google Scholar]
- Majewski S, Skopinska M, Blaszcyk M, Ryba M, Grieb P, Chorzelski T, Jablonska S. Systemic administration of 2-chloro-2'-deoxyadenosine (2-CdA) in patients with systemic scleroderma. Arch Immunol Ther Exp (Warsz) 1994;42:33–34. [PubMed] [Google Scholar]