

Abstract
Thrombin is a key factor in the stimulation of fibrin deposition, angiogenesis, proinflammatory processes, and proliferation of fibroblast-like cells. Abnormalities in these processes are primary features of rheumatoid arthritis (RA) in synovial tissues. Tissue destruction in joints causes the accumulation of large quantities of free hyaluronic acid (HA) in RA synovial fluid. The present study was conducted to investigate the effects of HA and several other glycosaminoglycans on antithrombin, a plasma inhibitor of thrombin. Various glycosaminoglycans, including HA, chondroitin sulfate, keratan sulfate, heparin, and heparan, were incubated with human antithrombin III in vitro. The residual activity of antithrombin was determined using a thrombin-specific chromogenic assay. HA concentrations ranging from 250 to 1000 μg/ml significantly blocked the ability of antithrombin to inhibit thrombin in the presence of Ca2+ or Fe3+, and chondroitin A, B and C also reduced this ability under the same conditions but to a lesser extent. Our study suggests that the high concentration of free HA in RA synovium may block antithrombin locally, thereby deregulating thrombin activity to drive the pathogenic process of RA under physiological conditions. The study also helps to explain why RA occurs and develops in joint tissue, because the inflamed RA synovium is uniquely rich in free HA along with extracellular matrix degeneration. Our findings are consistent with those of others regarding increased coagulation activity in RA synovium.
Keywords: antithrombin, glycosaminoglycan, hyaluronic acid, rheumatoid arthritis, thrombin
Introduction
Thrombin is a multifunctional protease that can activate hemostasis and coagulation through the cleavage of fibrinogen to form fibrin clots. Increasing fibrin deposition is a predominant feature of rheumatoid arthritis (RA) in synovial tissue, which contributes to chronic inflammation and progressive tissue abnormalities [1]. Thrombin also acts as a mitogen to stimulate the abnormal proliferation of synovial cells during RA pathogenesis. In this regard, thrombin can elevate the expression of nuclear factor-κB, interleukin-6, and granulocyte colony-stimulating factor in fibroblast-like cells of the RA synovium [2,3]. By a similar mechanism, thrombin can upregulate the transcription of vascular endothelial growth factor receptor and thereby induce the permeability, proliferation, and migration of capillary endothelial cells or their progenitors during angiogenesis [4-6]. Thrombin also plays an important role in the proinflammatory process by stimulating neutrophil adhesion to vessel walls and releasing prostacyclin [7]. Thus, thrombin is essential for enhancing synovial thickness and inflammation during the pathogenesis of RA.
The principal plasma inhibitor of thrombin is antithrombin, a single-chain 51 kDa glycoprotein that is synthesized in liver. The inhibitory activity of antithrombin on thrombin is significantly enhanced by heparin, a type of glycosaminoglycan (GAG) [8]. The GAG family comprises large anionic polysaccharides with similar disaccharide repeats of uronic acid and hexosamine. Physiologically important GAGs include hyaluronic acid (HA), chondroitin sulfates, keratan sulfate (KS), heparin, and heparan, which are the major components of joint cartilage, synovial fluid, and other soft connective tissues [9,10]. Along with the destruction of RA joint tissue, a remarkable quantity of various GAG molecules, especially HA, are released from the extracellular matrix of the synovium [9,10], which is a key feature of RA progression. Because GAGs and heparin share a similar molecular structure, we investigated how HA and other GAGs affect antithrombin activity.
Methods
Highly purified HA, chondroitin sulfate A (CSA), chondroitin sulfate B (CSB), chondroitin sulfate C (CSC), KS, heparin, or heparan (Seikagaku, Tokyo, Japan) were incubated for 24 hours with human antithrombin III at 150 μg/ml (Sigma, St. Louis, MO, USA) at 37°C in working buffer (100 mmol/l Tris-HCl, pH 7.5) containing 5 mmol/l CaCl2 or FeCl3. The concentration of antithrombin was determined according to its physiologic level in synovial fluid [11,12]. The reaction was stopped with EDTA. Residual activity of antithrombin was analyzed using the chromogenic Actichrome AT III (American Diagnostica, Greenwich, CT, USA) kit, which quantifies antithrombin III activity as follows. After exposure to GAGs, antithrombin was incubated with the thrombin reagent provided with the kit and residual thrombin activity was determined by incubation with the thrombin-specific chromogenic substrate in the kit. Absorbance was measured at a wavelength of 405 nm. Hence, the inhibitory ability of antithrombin on thrombin was inversely proportional to the residual thrombin activity. This assay method is usually used in the clinical setting. We prepared a series of control tests in which HA, CSA, CSB, CSC, and KS were digested in 0.1 mol/l phosphate buffer (prepare 100 ml of the buffer with 94 ml of 0.1 M KH2PO4 and 6 ml of 0.1 M K2HPO4, pH 6.2) at 37°C for 2 hours with 0.1 units/ml hyaluronidase (Seikagaku, Japan) before incubation with antithrombin. Hyaluronidase preferentially digests HA rather than other GAGs.
To determine whether HA can prevent heparin from stimulating antithrombin, we simultaneously incubated heparin (10 μg/ml) and various concentrations of HA with antithrombin (150 μg/ml) at 37°C for 24 hours in the presence of 5 mmol/l CaCl2. To investigate the effect of HA on antithrombin in the presence of other metal ions, we incubated HA (1 mg/ml) and human antithrombin III (150 μg/ml) at 37°C for 24 hours in the presence of CaCl2, FeCl3, KCl, MgCl2, and NaCl at various concentrations. Residual antithrombin activity was measured as described above.
Results
In the absence of heparin, antithrombin partly inhibited thrombin activity. Low concentrations of HA did not significantly affect antithrombin activity, regardless of the presence or absence of Ca2+ or Fe3+. However, HA concentrations above 250 μg/ml considerably suppressed the inhibitory ability of antithrombin against thrombin in the presence of Ca2+ or Fe3+, and 1 mg/ml HA completely blocked antithrombin activity under the same conditions. Consequently, thrombin activity was gradually elevated by increasing HA concentrations between 250 and 1000 μg/ml. However, HA at concentrations above 1000 μg/ml progressively lost the ability to prevent inhibition of thrombin activity by antithrombin. Furthermore, HA after digestion with hyaluronidase inhibited antithrombin activity at relatively low concentrations (100 μg/ml) in the presence of Ca2+. This observation indicated that the inhibitory effect of HA on antithrombin was not caused by impurities in the reagent. The control without antithrombin indicated that HA does not directly affect thrombin (Fig. 1).
Figure 1.
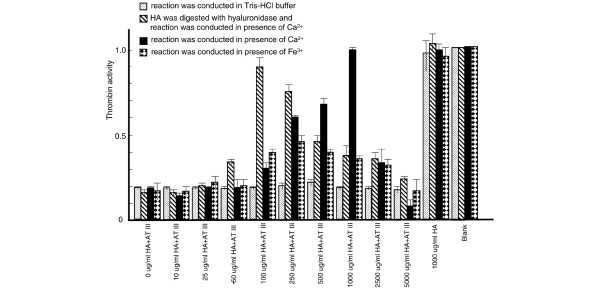
Effect of hyaluronic acid (HA) on antithrombin (AT). Various concentrations of HA, digested or not with hyaluronidase, were incubated with antithrombin in the presence of 5 mmol/l CaCl2 or FeCl3. Thrombin activity in the absence of both HA and antithrombin (blank) was considered as 1 and the activities of the other tests were normalized based on comparisons with blank. Values are expressed as mean ± standard deviation of data from triplicate experiments.
CSA, CSB, and CSC also inhibited the antithrombin effect in the presence of Ca2+ but to a lesser extent than did HA (Fig. 2). KS did not significantly affect antithrombin activity. Exposing CSs and KS to hyaluronidase did not clearly change this effect, indicating that CSs themselves inhibit antithrombin (data not shown). In contrast to HA, heparin and heparan clearly stimulated thrombin inhibition by antithrombin (Fig. 2). However, the stimulatory effect of heparin was considerably decreased in the presence of HA and Ca2+. Moreover, the ability of HA to prevent heparin activity was progressively strengthened with increased concentrations of HA within the range 250–1000 μg/ml (Fig. 3). Other metal ions, including K+, Mg2+, and Na+, did alter the effect of HA on antithrombin (Fig. 4).
Figure 2.
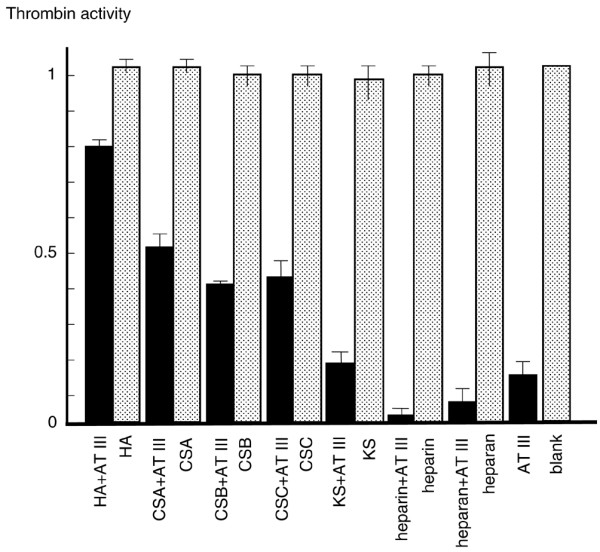
Effects of various glycosaminoglycans (GAGs) on antithrombin (AT). Hyaluronic acid (HA), chondroitin sulfate A (CSA), chondroitin sulfate B (CSB), chondroitin sulfate C (CSC), keratan sulfate (KS), heparin, or heparan (500 μg/ml) was incubated with 150 μg/ml antithrombin and 5 mmol/l CaCl2. Controls consisted of only GAG or AT and blank (working buffer only). Thrombin activity of blank was considered as 1 and the activities of other tests were normalized based on comparisons with blank. Values are expressed as mean ± standard deviation of data from triplicate experiments.
Figure 3.
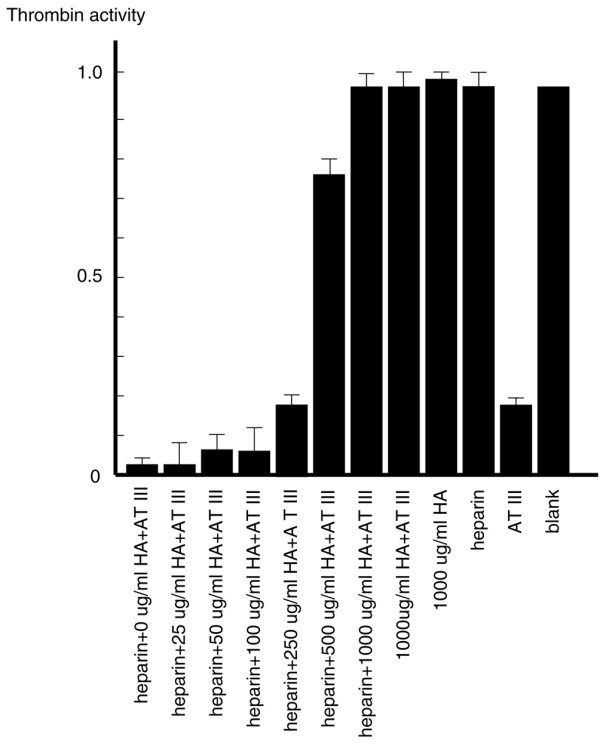
Heparin stimulates antithrombin (AT) activity in the presence of hyaluronic acid (HA). Heparin (10 μg/ml) and various concentrations of HA were incubated with 150 μg/ml antithrombin in presence of 5 mmol/l CaCl2. Thrombin activity of blank (reaction buffer only) was considered as 1 and the activities of other tests were normalized based on comparisons with blank. Values are expressed as mean ± standard deviation of data from triplicate experiments.
Figure 4.
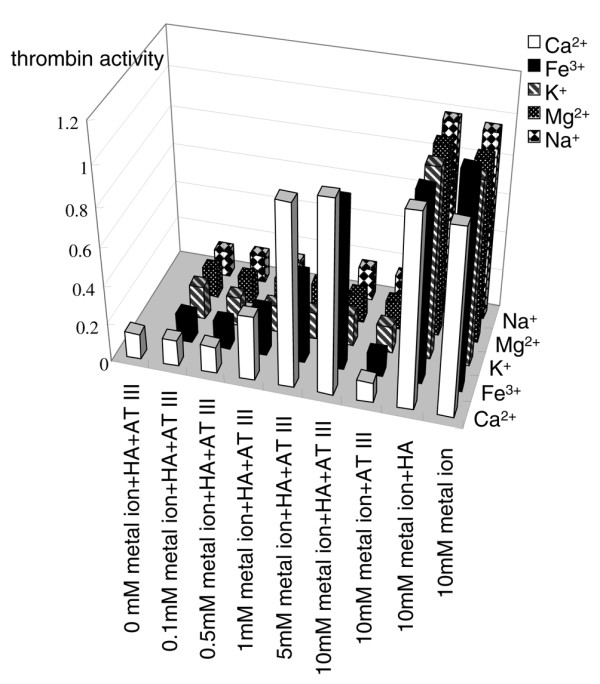
Effects of various metal ions on ability of hyaluronic acid (HA) to inhibit the activity of antithrombin (AT). HA (1000 μg/ml) and antithrombin (150 μg/ml) were incubated with various concentrations of CaCl2, FeCl3, KCl, MgCl2, or NaCl. Thrombin activity of blank (reaction buffer only) was considered as 1 and the activities of other tests were normalized based on comparisons with blank. Values are expressed as mean ± standard deviation of data from triplicate experiments.
Discussion
The destruction of joint tissue is a primary feature of RA. In the inflamed RA synovium, proliferating macrophages and colonizing lymphocytes, together with persistent angiogenesis, produce large amounts of matrix metalloproteinases that destroy the surrounding cartilage and extracellular matrix of connective tissue [13]. Because GAGs are the basic structural components of joint cartilage, synovial fluid, and soft tissues [9,10], the RA synovium produces an abundance of free GAGs during tissue destruction. Among these, HA is a predominant component of the articular surface and synovial fluid, in which the HA concentration is between 1500 and 2500 μg/ml [14,15]. Pitsillides and coworkers [14] found that the ratio of free HA to bound HA was significantly increased in the RA (4.53 ± 0.40) as compared with the healthy (1.87 ± 0.42) synovium, although the total concentration of hyaluronan was not increased in the rheumatoid synovium. Their histochemical staining also showed that hyaluronan was concentrated in the lining layer of noninflamed synovial membrane but was more uniformly distributed throughout rheumatoid samples. On the other hand, the HA level is very low among various other tissues. For example, the concentration of serum HA from healthy individuals averages 16 ng/ml, which is 1 × 105 fold lower than that in synovial fluid [16,17].
The present study found that HA at concentrations between 250 and 1000 μg/ml significantly blocked the ability of antithrombin to inhibit thrombin. This finding helps to explain why RA occurs and develops in joint tissue, because the inflamed RA synovium is uniquely rich in free HA and other GAGs, along with extracellular matrix degeneration. Although the HA levels are higher in RA than in healthy sera [18], we demonstrated that the relatively low levels of HA do not prevent antithrombin activity and thus cannot cause blood clots in the circulation. Hence, only the conditions in the RA synovium can drive the pathogenesis of thrombin-related RA, which includes abnormal angiogenesis, extreme proliferation of fibroblast-like cells, excessive fibrin deposition, and proinflammatory processes. Thus, thrombin-related RA worsens because of the snowball effect of HA release in inflamed joints.
Our notion is supported by many other studies. Jones and coworkers [11] found that antithrombin activity is selectively depressed in RA synovial fluid as compared with that in osteoarthritis, although the concentration of the antithrombin–thrombin complex was significantly increased. Ohba and coworkers [12] also found high levels of thrombin activity in RA synovial fluid. These findings support the notion that inhibiting antithrombin activity plays an essential role in RA pathogenesis. Wang and coworkers [10] recently constructed a model of arthritis by injecting various GAGs into mice. We postulate that the injected GAGs significantly disrupted the inhibition of thrombin by antithrombin, which therefore caused connective tissue disease through abnormally activated angiogenesis, proinflammatory processes, and fibrin deposition. On the other hand, heparan, which has an almost identical structure to that of heparin but contains fewer sulfates, stimulated antithrombin activity in a similar manner to heparin. These observations indicate that the diverse effects of GAGs on antithrombin are due to differences in their molecular configurations. Heparin pentasaccharide can form complexes with antithrombin and expose a reactive proteinase binding loop on the protein surface [19,20]. Because the molecular structure of HA is analogous to that of heparin, HA might exert its effect by binding to the heparin-binding region of antithrombin. However, such binding did not stimulate the activity of antithrombin as did heparin and heparan; in fact, it blocked the ability of antithrombin to inhibit thrombin. In the present study, the stimulatory effect of heparin on antithrombin was considerably decreased in the presence of HA, supporting the notion that HA could compete with heparin for the heparin-binding region of antithrombin.
Remarkably, HA affected the inhibition by antithrombin only within the range 250–1000 μg/ml. At concentrations above 2000 μg/ml, HA either lost its inhibitory effect or elevated the ability of antithrombin to inhibit thrombin. The physiologic level of free HA in the RA synovium is just within the range 500–1000 μg/ml [14]. Some clinical studies have shown that injecting HA into articular rheumatoid joints can ameliorate inflammation [21,22]. Although further investigation is required to elucidate the exact mechanism by which HA inhibits antithrombin, the results of the present study do not refute the notion that optimal proteoglycan uptake can improve overall articular function in patients with arthritis.
Why HA inhibited antithrombin more after than before hyaluronidase digestion remains obscure. Perhaps the small HA molecule can easily bind and thus exert a more inhibitory role on antithrombin. Nagaya and coworkers [23] found high hyaluronidase activity in the synovial fluid and serum of RA patients, implying an abundance of small HA molecules in the RA synovium. Maneirio and coworkers [24] reported that HA at various molecular weights had different effects on the interleukin-1 induced synthesis of both nitric oxide and prostaglandin E2 in chondrocytes. How Ca2+ and Fe3+ are involved in inhibiting antithrombin by HA is also poorly understood. Some investigators found that Ca2+ dramatically promotes the ability of heparin to drive antithrombin activity [8,25,26]. Thus, both Ca2+ and Fe3+ ions might play similar roles in HA-induced changes in the configuration of antithrombin.
Synovial fluid from RA patients contains a far greater abundance of free iron than that from patients with osteoarthritis [27,28]. It was reported that Fe3+ stored in the RA synovium perpetuates inflammation by supporting the production of oxygen radicals and by promoting hyaluronic acid degradation, as well as the release of lysosomal enzymes [29]. Telfer and coworkers [30] recently found that proinflammatory cytokines produced in the RA synovium increased the accumulation of iron in synovial fluid. On other hand, Davies and coworkers [31] reported that neutrophils from synovial fluid and the circulation of RA patients could increase the release of free Ca2+ at inflammatory sites. Caruthers and coworkers [32] also showed that calcium signaling is altered in T lymphocytes from RA patients.
Genome-wide single nucleotide polymorphism analysis has shown that peptidylarginine deiminase (PADI4), an enzyme that post-translationally catalyzes peptidyl arginine to citrulline, is closely associated with RA [33]. We recently found that recombinant human PADI4 protein inactivated human antithrombin III via citrullination in vitro. We also detected an increased level of citrullinated antithrombin in the plasma of RA patients [34]. PADI4 is extensively expressed in RA synovial tissue [35,36]. Thus, we suggested that the citrullination of antithrombin is one potential pathway through which PADI4 contributes to the pathogenesis of RA [34]. This notion does not contradict the current findings. We postulate that the genetic, single nucleotide polymorphism-associated disorder of PADI4 and its excessive citrullination of antithrombin play important roles in initiating the RA pathogenic process, whereas inhibition of antithrombin by HA contributes to the development of RA rather than its initiation, because free HA in the synovium achieves high concentrations along with RA progression. Because of abundant Fe3+ and altered Ca2+ metabolism together with significant hyaluronidase activity in the RA synovium, thrombin-related RA specifically worsens in joint tissue as a result of antithrombin inactivation by local PADI4 and free HA (Fig. 5).
Figure 5.
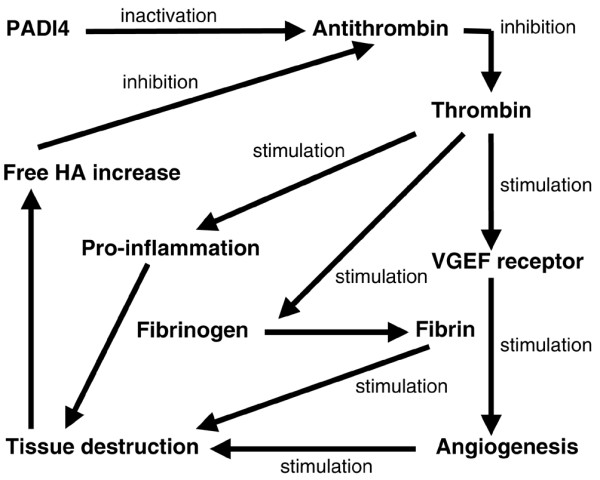
Proposed mechanism of involvement of hyaluronic acid (HA) and peptidylarginine deiminase (PADI4) in the pathogenesis of rheumatoid arthritis. VEGF, vascular endothelial growth factor.
HA is an important component of the extracellular matrix. Thrombin and antithrombin play key roles in hemostasis and are involved in the pathogenic processes of many diseases [6,37,38]. The findings presented here should also be useful in investigating the nature of other diseases.
Conclusion
At concentrations of 250–1000 μg/ml in vitro, HA blocked the thrombin-inhibitory ability of antithrombin in the presence of Ca2+ and Fe3+. This finding suggested that the high concentration of free HA in diseased RA synovium locally blocks antithrombin under physiologic conditions and thereby deregulates the activity of thrombin. These processes in turn drive the thrombin-related pathogenesis of RA, which includes extensive fibrin deposition, extreme angiogenesis, and abnormal fibroblast-like cell proliferation. Our findings are consistent with those of previous reports regarding increased coagulation activity in the RA synovium.
Abbreviations
CS = chondroitin sulfate; GAG = glycosaminoglycan; HA = hyaluronic acid; KS = keratan sulfate; PADI = peptidylarginine deiminase; RA = rheumatoid arthritis.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
XC designed and executed the study and prepared the manuscript. RY and KY supervised the project, evaluated data, and assisted in preparing the manuscript.
Acknowledgments
Acknowledgements
We thank every member of the Rheumatology Diseases Laboratory of Riken for their general contribution to making this study possible.
References
- Carmassi F, de Negri F, Morale M, Song KY, Chung SI. Fibrin degradation in the synovial fluid of rheumatoid arthritis patients: a model for extravascular fibrinolysis. Semin Thromb Hemost. 1996;22:489–496. doi: 10.1055/s-2007-999049. [DOI] [PubMed] [Google Scholar]
- Shin H, Kitajima I, Nakajima T, Shao Q, Tokioka T, Takasaki I, Hanyu N, Kubo T, Maruyama I. Thrombin receptor mediated signals induce expressions of interleukin 6 and granulocyte colony stimulating factor via NF-kappa B activation in synovial fibroblasts. Ann Rheum Dis. 1999;58:55–60. doi: 10.1136/ard.58.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Nakajima T, Kitajima I, Shigeta K, Abeyama K, Imamura T, Okano T, Kawahara K, Nakamura T, Maruyama I. Thrombin receptor-mediated synovial proliferation in patients with rheumatoid arthritis. Clin Immunol Immunopathol. 1995;76:225–233. doi: 10.1006/clin.1995.1120. [DOI] [PubMed] [Google Scholar]
- Tsopanoglou NE, Maragoudakis MEJ. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J Biol Chem. 1999;274:23969–23976. doi: 10.1074/jbc.274.34.23969. [DOI] [PubMed] [Google Scholar]
- Maragoudakis ME, Tsopanoglou NE, Andriopoulou P. Mechanism of Thrombin-induced angiogenesis. Biochem Soc Trans. 2002;30:173–177. doi: 10.1042/0300-5127:0300173. [DOI] [PubMed] [Google Scholar]
- Narayanan S. Multifunctional roles of thrombin. Ann Clin Lab Sci. 1999;29:275–280. [PubMed] [Google Scholar]
- Morris R, Winyard PG, Brass LF, Blake DR, Morris CJ. Thrombin in inflammation and healing' relevance to rheumatoid arthritis. Ann Rheum Dis. 1994;53:72–79. doi: 10.1136/ard.53.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe EM, Stafford AR, Fredenburgh JC, Weitz JI. Mechanism of catalysis of inhibition of factor IXa by antithrombin in the presence of heparin or pentasaccharide. J Biol Chem. 2003;278:35767–35774. doi: 10.1074/jbc.M304803200. [DOI] [PubMed] [Google Scholar]
- Lozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- Wang JY, Roehrl MH. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc Natl Acad Sci USA. 2002;99:14362–14367. doi: 10.1073/pnas.222536599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HW, Bailey R, Zhang Z, Dunne KA, Blake DR, Cox NL, Morris CJ, Winyard PG. Inactivation of antithrombin III in synovial fluid from patients with rheumatoid arthritis. Ann Rheum Dis. 1998;57:162–165. doi: 10.1136/ard.57.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba T, Takase Y, Ohhara M, Kasukawa R. Thrombin in synovial fluid of paients with rheumatoid arthritis mediates proliferation of synovial fibroblast-like cells by induction of plate derived growth factor. J Rheumatol. 1996;23:1505–1511. [PubMed] [Google Scholar]
- Jain A, Nanchahal J, Troeberg L, Green P, Brennan F. Production of cytokines, vascular endothelial growth factor, matrix metalloproteinases, and tissue inhibitor of metalloproteinases 1 by tenosynovium demonstrates its potential for tendon destruction in rheumatoid arthritis. Arthritis Rheum. 2001;44:1754–1760. doi: 10.1002/1529-0131(200108)44:8<1754::AID-ART310>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Pitsillides AA, Worrall JG, Wilkinson LS, Bayliss MT, Edwards JC. Hyaluronan concentration in non-inflamed and rheumatoid synovium. Br J Rheumatol. 1994;33:5–10. doi: 10.1093/rheumatology/33.1.5. [DOI] [PubMed] [Google Scholar]
- Nakayama Y, Shirai Y, Yoshihara K, Uesaka S. Evaluation of glycosaminoglycans levels in normal joint fluid of the knee. J Nippon Med Sch. 2000;67:92–95. doi: 10.1272/jnms.67.92. [DOI] [PubMed] [Google Scholar]
- Takei YG, Honma T, Ito A. Quantitation of hyaluronic acid in serum with automated microparticle photometric agglutination assay. J Immunoassay Immunochem. 2002;23:85–94. doi: 10.1081/IAS-120002276. [DOI] [PubMed] [Google Scholar]
- Wyatt HA, Dhawan A, Cheeseman P, Mieli-Vergani G, Price JF. Serum hyaluronic acid concentrations are increased in cystic fibrosis patients with liver disease. Arch Dis Child. 2002;86:190–193. doi: 10.1136/adc.86.3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partsch G, Leeb B, Stancikova M, Raffayova H, Eberl G, Hitzelhammer H, Smolen JS. Low serum hyaluronan in psoriatic arthritis patients in comparison to rheumatoid arthritis patients. Clin Exp Rheumatol. 1996;14:381–386. [PubMed] [Google Scholar]
- Skinner R, Abrahams JP, Whisstock JC, Lesk AM, Carrell RW, Wardell MR. The 2.6 A structure of antithrombin indicates a conformational change at the heparin binding site. J Mol Biol. 1997;266:601–609. doi: 10.1006/jmbi.1996.0798. [DOI] [PubMed] [Google Scholar]
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Matsuzaka S, Yoshida Y, Miyauchi S, Wada Y, Moriya H. The effects of intraarticularly injected sodium hyaluronate on levels of intact aggrecan and nitric oxide in the joint fluid of patients with knee osteoarthritis. Osteoarthritis Cartilage. 2004;12:536–542. doi: 10.1016/j.joca.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Moreland LW. Intra-articular hyaluronan (hyaluronic acid) and hylans for the treatment of osteoarthritis: mechanisms of action. Arthritis Res Ther. 2003;5:54–67. doi: 10.1186/ar623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaya H, Yamagata T, Yamagata S, Iyoda K, Ito H, Hasegawa Y, Iwata H. Examination of synovial fluid and serum hyaluronidase activity as a joint marker in rheumatoid arthritis and osteoarthritis patients (by zymography) Ann Rheum Dis. 1999;58:186–188. doi: 10.1136/ard.58.3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneiro E, de Andres MC, Fernandez-Sueiro JL, Galdo F, Blanco FJ. The biological action of hyaluronan on human osteoartritic articular chondrocytes: the importance of molecular weight. Clin Exp Rheumatol. 2004;22:307–312. [PubMed] [Google Scholar]
- Rezaie AR. Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism. Evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J Biol Chem. 1998;273:16824–16827. doi: 10.1074/jbc.273.27.16824. [DOI] [PubMed] [Google Scholar]
- Bedsted T, Swanson R, Chuang YJ, Bock PE, Bjork I, Olson ST. Heparin and calcium ions dramatically enhance antithrombin reactivity with factor IXa by generating new interaction exosites. Biochemistry. 2003;42:8143–8152. doi: 10.1021/bi034363y. [DOI] [PubMed] [Google Scholar]
- Blake DR, Gallagher PJ, Potter AR, Bell MJ, Bacon PA. The effect of synovial iron on the progression of rheumatoid disease. A histologic assessment of patients with early rheumatoid synovitis. Arthritis Rheum. 1984;27:495–501. doi: 10.1002/art.1780270503. [DOI] [PubMed] [Google Scholar]
- Ahmadzadeh N, Shingu M, Nobunaga M. Iron-binding proteins and free iron in synovial fluids of rheumatoid arthritis patients. Clin Rheumatol. 1989;8:345–351. doi: 10.1007/BF02030347. [DOI] [PubMed] [Google Scholar]
- Morris CJ, Blake DR, Wainwright AC, Steven MM. Relationship between iron deposits and tissue damage in the synovium: an ultrastructural study. Ann Rheum Dis. 1986;45:21–26. doi: 10.1136/ard.45.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telfer JF, Brock JH. Proinflammatory cytokines increase iron uptake into human monocytes and synovial fibroblasts from patients with rheumatoid arthritis. Med Sci Monit. 2004;10:BR91–BR95. [PubMed] [Google Scholar]
- Davies EV, Williams BD, Whiston RJ, Cooper AM, Campbell AK, Hallett Altered Ca2+ signalling in human neutrophils from inflammatory sites. Ann Rheum Dis. 1994;53:446–449. doi: 10.1136/ard.53.7.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers DM, Arrol HP, Bacon PA, Young SP. Dysregulated intracellular Ca2+ stores and Ca2+ signaling in synovial fluid T lymphocytes from patients with chronic inflammatory arthritis. Arthritis Rheum. 2000;43:1257–1265. doi: 10.1002/1529-0131(200006)43:6<1257::AID-ANR8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- Chang X, Yamada R, Sawada T, Suzuki A, Yamamoto K. The inhibition of antithrombin by peptidylarginine deiminase 4 may contribute to pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2004. [DOI] [PubMed]
- Vossenaar ER, Radstake TR, Van Der Heijden A, Van Mansum MA, Dieteren C, De Rooij DJ, Barrera P, Zendman AJ, Van Venrooij WJ. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis. 2004;63:373–381. doi: 10.1136/ard.2003.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Yamada R, Suzuki A, Sawada T, Yoshino S, Tokuhiro S, Yamamoto K. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology (Oxford) 2004. [DOI] [PubMed]
- van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997;34:188–204. [PubMed] [Google Scholar]
- Ishiguro K, Kojima T, Kadomatsu K, Nakayama Y, Takagi A, Suzuki M, Takeda N, Ito M, Yamamoto K, Matsushita T, et al. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. 2000;106:873–878. doi: 10.1172/JCI10489. [DOI] [PMC free article] [PubMed] [Google Scholar]