

Abstract
The tumor suppressor p53-like protein p63 is required for self-renewal of epidermal tissues. Loss of p63 or exposure to ultraviolet (UV) irradiation triggers terminal differentiation in keratinocytes. However, it remains unclear how p63 diverts epidermal cells from proliferation to terminal differentiation, thereby contributing to successful tissue self-renewal. Here, we used bottom-up proteomics to identify the proteome at the chromatin in normal human epidermal keratinocytes (NHEK) following UV irradiation and p63 depletion. We found that loss of p63 increased DNA damage, and that UV irradiation recruited the cyclin-dependent kinase CDK12 and the serine/threonine protein kinase SMG1 to chromatin only in the presence of p63. A post-translational modification analysis of ΔNp63α with mass spectrometry revealed that phosphorylation of T357/S358 and S368 was dependent on SMG1, whereas CDK12 increased phosphorylation of ΔNp63α at S66/S68 and S301. Indirect phosphorylation of ΔNp63α in the presence of SMG1 enabled ΔNp63α to bind to the tumor suppressor p53-specific DNA recognition sequence, whereas CDK12 rendered ΔNp63α less responsive to UV irradiation and was not required for specific DNA binding. CDK12 and SMG1 are known to regulate the transcription and splicing of RNAs and the decay of nonsense RNAs, respectively, and a subset of p63 specific protein-protein interactions at the chromatin also linked p63 to RNA transcription and decay. We observed that in the absence of p63, UV irradiation resulted in more ORF1p. ORF1p is the first protein product of the intron-less non-LTR retrotransposon LINE-1, indicating a derailed surveillance of RNA processing and/or translation. Our results suggest that p63 phosphorylation and transcriptional activation might correspond to altered RNA processing and/or translation to protect proliferating keratinocytes from increased genotoxic stress.
Keywords: DNA damage, MAPK1, ERK2, PRKM1, PRKM2, ATX, KIAA0421, LIP, CRK7, CRKRS, cyclin-dependent kinase 12
Graphical Abstract
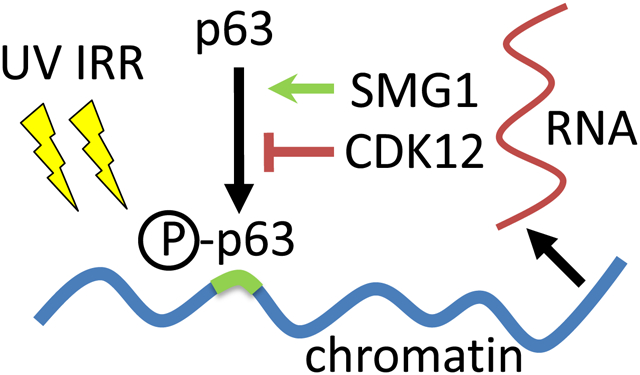
Introduction
Incomplete DNA repair in somatic cells stabilizes the tumor suppressor p53. Subsequent binding of the transcription factor p53 to its specific DNA recognition sequence triggers programmed cell death, which ultimately prevents the accumulation of somatic cells that are at higher risk for malignant transformation1. The pathways used by p53 to minimize tumor formation in adult multicellular organisms2 are well defined, but the precise molecular function of the evolutionarily older p53 family members p63 and p73 remain less well characterized. The transcription factors p53, p63 and p73 share high sequence identity within the central DNA binding domain, suggesting that p63 and p73 may also function in tumor suppression3,4. In vitro transactivation assays confirm that p63 and p73 bind to the canonical p53 DNA response element of p21CIP/WAF, a DNA consensus sequence necessary for the transcriptional activation of p21CIP/WAF during p53-mediated apoptosis. Both p63 and p73 can drive expression of p53 target genes, but the expression efficiency depends on the specific protein variants of p63 and p73. Genetic evidence has shown that loss of p63 and p73 can result in a failure of cells to undergo apoptosis in response to DNA damage5. However, mice that are null for p53, p63 or p73 differ dramatically in their developmental phenotypes. P53−/− mice develop normally and show high tumor incidence6, but mice null for either p63 or p73 die shortly after birth due to severe developmental defects. P73 is required for normal brain and immune system development7 whereas mice null for p63 fail to maintain stem cells in stratified epithelia8–11.
Here, we analyzed ΔNp63, the most abundant p63 proteoform in keratinocytes (Figure 1A). TAp63, the other major p63 proteoform, arises from the use of alternative promoters within the p63 gene12 and protects the female germ line from DNA damage13. TAp63 proteoforms include a N-terminal transactivation domain that is conserved in p53 but absent in ΔNp63 proteoforms. All p63 proteoforms include a core DNA binding domain and an oligomerization domain that allows p63 to tetramerize, bind to DNA, and activate gene transcription. Differential splicing events of the p63 RNA downstream of the tetramerization domain give rise to several alternative C-termini. The longest and most conserved C-terminal variant (p63α) includes a short sequence of six glutamines, a sterile alpha motif domain (SAM) and an outermost C-terminal transactivation inhibition domain (TID) which blocks N-terminal TAp63-mediated transcriptional activation of genes14. A specific phosphorylation event in response to DNA damage releases the intramolecular inhibition and enables specific DNA binding of TAp63α13, whereas the shorter C-terminal proteoforms p63β and p63γ constitutively activate gene transcription because they are devoid of the outmost TID domain15,16.
Figure 1: Phosphorylation of ΔNp63α is increased in UV irradiated human epidermal keratinocytes (NHEK).

(A) Schematic representation of the major p63 proteoform, ΔNp63α. Alternate proteoforms are indicated by a vertical bar and labeled TA, β, and γ. Boxes in grey highlight protein domains with specific function. TA stands for transactivation and SAM for sterile alpha motif. (B) NHEK were infected with lentiviral p63si1 or scrambled (scr., control) shRNA, grown for additional three days, UV irradiated, and after 1h of recovery, the whole cell lysate was analyzed by Western blotting. The Western blot shows knock down of ΔNp63α in NHEK and phase contrast images visualize changes in NHEK morphology. (C) Proteins γH2A.X, p63, and βActin were detected in low (<3) or high (>4) passaged NHEK. Three differently phosphorylated ΔNp63α proteoforms ΔNp63α-[a], [b], and [c] (arrows) can be distinguished by denaturing 8 % polyacrylamide gel electrophoresis. The upper bar graph depicts ΔNp63α and γH2A.X protein levels following normalization to βActin and control (no UV, scr.) protein levels. Mass spectrometry was used to determine the amount of ΔNp63α in the chromatin associated proteome and shows enhanced chromatin association of phosphorylated p63 (lower bar graph). (D) The schematic summarizes the experimental design that was used to quantify the chromatin associated proteome in NHEK in response to UV irradiation. HaCaT cells were SILAC labeled with 13C15N-lysine and 13C15N-arginine to serve as internal standard for comparison between the four different experimental NHEK conditions (+/− p63si1 shRNA, +/− UV irradiation). (E) The networks summarize potential interactions between proteins that are enriched at the chromatin following UV irradiation in the presence of p63 but not in its absence. The protein-protein networks highlight potential interactions (left), whereas the protein levels are indicated in the graphs (right). Colors of the protein nodes match the colors in the graphs.
TAp63 protein variants with the shorter C-termini p63β and p63γ induce strong target gene transcription in vitro; ΔNp63 with C-termini p63β and p63γ show significantly weaker transactivation12,17. Notably, specific gene transcription by the ΔNp63 variant is further reduced by the p63α C-terminus when overexpressed in a heterologous expression system, suggesting that a dynamic regulation of ΔNp63α specific DNA binding or transcriptional activation of target genes is possible. ΔNp63 mRNA transcript and protein levels vastly outnumber TAp63 in most somatic tissues12,18. TAp63 mRNA transcripts are barely detectable on the mRNA level in skin keratinocytes19 and TAp63 protein is virtually absent in normal human epidermal keratinocytes (NHEK) in primary cell culture when detected by Western blotting with a TAp63-specific monoclonal antibody20. Rather, TAp63 is predominantly expressed in germ cells during spermatogenesis and oogenesis, which argues for a tissue-specific use of the two alternative promoters in vertebrates. Nevertheless, conditional knock out of TAp63 in the epidermis induces genomic instability and accelerated skin aging10 and TAp63 suppresses tumorigenesis in a p53−/− background in vivo21. In contrast, high levels of ΔNp63α maintain the proliferative potential of basal keratinocytes and contribute to the expansion of transient amplifying suprabasal keratinocytes, as concluded from the study of p63 knock out mice11,22–24. ΔNp63α is essential for terminal differentiation of keratinocytes in vivo9,25,26, and ΔNp63 specific knock out in the epidermis leads to defects in terminal differentiation27. However, this observation is not recapitulated upon loss of the C-terminal p63α-specific protein sequence alone which increases gene transactivation by ΔNp63 to ΔNp63β/γ-like levels28. Additional in vitro experiments suggest that ΔNp63α drives expression of keratinocyte differentiation genes in transactivation assays in vitro29,30. However, ΔNp63α expression also diminishes within days during terminal differentiation31, suggesting that p63-driven expression of genes is limited to a short time window during terminal differentiation. Thus, the p63α C-terminus may directly contribute to functional regulation of ΔNp63α specific DNA binding and subsequent gene transactivation.
Previous reports concluded that the phosphorylation status of ΔNp63α marks different epidermal keratinocyte pools. Clonal expansion assays of keratinocytes in primary cell culture showed phosphorylation of ΔNp63α S66/S68 during transition of epidermal stem cells to progenitor cells32 and upon TGFβR1 activation33. Phosphorylation of p63 at multiple sites by the kinase c-Abl slows p63 protein turn over and increases cell viability in response to genotoxic stress34. Moreover, ΔNp63 is phosphorylated by the serine/threonine kinase ATM in response to cisplatin treatment of squamous cell carcinoma cells35. While numerous genetic studies indicated p63’s requirement for self-renewal of epidermal stem cells and suggested a possible involvement in terminal differentiation of keratinocytes, the molecular mechanisms by which ΔNp63α dynamically responds to genotoxic stress in keratinocytes and thus secures faithful self-renewal of the epidermis remain less well defined.
Here, we show that the kinases CDK12 and SMG1 are recruited to the chromatin upon UV irradiation only in the presence of p63 and independently influence p63 phosphorylation. CDK12 is associated with activation of gene transcription36 and RNA splicing37. SMG1 regulates genome stability38 and mRNA decay39 and is part of the SURF complex consisting of SMG1-UPF1-eRF1-eRF, which can associate with the exon junction complex (EJC) through UPF2. SMG1 triggers UPF1 phosphorylation and induces non-sense mRNA decay40. Furthermore, we find three differentially phosphorylated proteoforms of p63 in response to UV irradiation, and we provide evidence that the intron-less protein product ORF1p of the element LINE-1 is upregulated upon UV irradiation in the absence of p63.
Materials and Methods
Purification of the chromatin associated proteome
Nuclei were extracted from cells according to Berkowitz et al41 (with modifications). Briefly, cells were lysed in lysis buffer (320 mM Sucrose, 2 mM MgCl2, 1 mM Potassium phosphate at pH 7.0). Following centrifugation, the protein pellet was incubated in hypotonic buffer for 15 min on ice (10 mM NaCl, 1 mM Potassium phosphate, pH 7.0, 800 g, 10 min), and resuspended in modified lysis buffer (320 mM Sucrose, 1 mM MgCl2, 2 mM Potassium phosphate at pH 7.0, 0.3 % IGEPAL CA 630, 1 x Complete Proteinase Inhibitor Cocktail, EDTA-free (Roche), 1 x Phosstop Phosphatase Inhibitor Cocktail (Roche). Cells were carefully disrupted twice in a dounce tissue grinder with ten strokes each of a loose and a tight pestle. Finally, nuclei were collected by centrifugation at 800 g, 10 min.
For extraction of chromatin associated proteins, the nuclei pellet was carefully resuspended in No-Salt-Extraction buffer (10 mM Potassium Phosphate, pH 7.0, 10 mM EDTA, 1 mM EGTA, 1 x Proteinase and Phosphatase inhibitors) and incubated at 4 °C while mixing. The nuclei fragments were collected by centrifugation (4,000 rpm, 10 min in a tabletop centrifuge at 4 °C), resuspended in High-Salt-Extraction buffer (No-Salt-Extraction buffer with NaCl added to 2 M final concentration, 0.5 % Triton X-100), passed twenty times through a needle (gauge 20) to shear genomic DNA, and incubated for an additional 3h at 4 °C while mixing. The lysate was cleared by ultracentrifugation (100,000 g, 30 min at 4 °C) and the supernatant containing chromatin-bound factors was stored at −80 °C.
Preparation of the chromatin associated proteome in human primary keratinocytes with SILAC labeled standard
HaCaT (immortalized human keratinocytes42) were SILAC labeled (13C15N-lysine and 13C15N-arginine, Cambridge Isotope Laboratories) in DMEM (Pierce) for six cell passages and nuclei were extracted as described above. Immortalized HaCaT cells were labeled with SILAC because NHEKs quickly differentiated within the six cell passages that were used to achieve complete stable isotope labeling. Labeling efficiency of >98 % was verified by mass spectrometry and aliquots of HaCaT nuclei were snap frozen in liquid nitrogen and stored at −80 °C. Following isolation of NHEK nuclei, the amount of protein was determined with a Bradford assay. SILAC-labeled HaCaT nuclei were mixed with the non-labeled nuclei 1:1 (w:w), and chromatin-associated proteins were extracted as described above. Biological triplicate experiments were performed throughout all conditions.
Protein digestion
The combined light and heavy labeled, chromatin-bound proteome was methanol:chloroform precipitated (methanol:chloroform, 4:1 (v:v), 16,000 rpm at 4 °C, 10 min), washed in methanol (16,000 rpm at 4 °C, 5 min), and the non-dried protein pellet was carefully resuspended in 2 % RapiGest (Waters). The sample was diluted in 100 mM Tris-HCl, pH 8.5 to 0.4 % RapiGest, redissolved by sonication for 1 h, and subsequently diluted to 0.2 % RapiGest in 100 mM Tris-HCl, pH 8.5. Disulfide bonds were reduced by incubation with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Pierce, 37 °C for 20 min, protected from light) and free sulfhydryl residues were alkylated with 10 mM Iodoacetamide (37 °C, 30 min). Proteins were digested with recombinant sequencing grade porcine trypsin (Promega, 1:30 (w:w), 37 °C, 18 h, mixing) or chymotrypsin or AspN. RapiGest was subsequently inactivated by incubation in formic acid (9 % formic acid, 37 °C, 3 h, mixing). The sample volume was reduced to near complete dryness under vacuum and the sample was subsequently re-suspended in the hydrophilic liquid chromatography buffer A (94.9 % H2O, 5 % acetonitrile, 0.1 % formic acid).
Peptide separation and mass spectrometry
Mass spectrometric identification and quantification of proteins was performed with MudPIT43. The sample was pressure-loaded (400 psi) onto a reversed phase resin (Aqua 5, C18, 5 μm, 125 Å pore size, Phenomenex, length 2.4 cm in a fused silica capillary with an inner diameter (id) of 250 μm, Polymicro Technologies). Peptides were subsequently trapped on a strong cation exchange resin (Partisphere SCX, Whatman, 2 cm column length, id 250 μm) and then sequentially eluted from the cation exchange column in 12 steps with increasing percentages of buffer C (500 mM ammonium acetate, 5 % acetonitrile, 0.1 % formic acid) ranging from 0 % to 90 %. Peptides eluted in each step were separated by hydrophobicity on a second reversed phase column (11.5 cm column length of Aqua 3 resin, C18, 3 μm, 125 Å pore size, Phenomenex) using a 120 min gradient of buffer B (80 % acetonitrile, 0.1 % formic acid). Specifically, peptides were separated with high pressure liquid chromatography (HPLC) with an Agilent 1100 or 1200 Quaternary HPLC (Agilent) over a 92 min or 95 min gradient ranging from 0 % to 40 % or 45 % buffer B, respectively. The nominal flow rate before the split-flow was 150 μl/min (~800 psi) which resulted in a final flow rate at 1 to 2 μl/min at the at ESI tip. Alternatively, peptides were separated with an Eskigent nanoLC HPLC (Eskigent) which delivered a 104 min linear gradient of 0% to 45% buffer B at a flow rate of 0.5 μl/min.
Peptides were nano-electrospray ionized at 2.5 kV in front of a 200 °C or 250 °C or 275 °C heated ion inlet as they eluted from the open tip (id < 1 μm) of the analytical column. Positively charged peptides in the electrospray current were detected in an LTQ-Orbitrap, LTQ-Orbitrap XL, LTQ-Orbitrap Velos, LTQ-Orbitrap Velos pro, or LTQ-Orbitrap Elite mass spectrometer (Thermo Finnigan, Bremen, Germany) in data-dependent acquisition mode. Survey mass spectra were taken at a resolution of R = 60,000 or 120,000 and covered a m/z range of m/z 400 to m/z 1800 or m/z 2000. The 6, 7 or 13 most abundant precursor ions with a charge of z ≥ +2 were selected for collision induced dissociation (CID). A minimal signal of 500 or 1000 counts was required for CID. Precursor ions were isolated with a ± 1 Da mass width and fragmented with a normalized collision energy of 35.0 %, an activation time of 30 ms, and a Q value of 0.25. On the Orbitrap elite mass spectrometer, fragment ion spectra of phosphorylated peptides were monitored for a neutral loss of phosphate (m/z 32.70, m/z 49.00, and m/z 98.00), with a product ion peak within the 3 most intense ions. Peaks were then selected for a second, subsequent CID based fragmentation (MS3).
Precursor ions were selected once or on the LTQ-Orbitrap Elite 3 times with an expiration S/N > 3.0. Selected precursor ions were subsequently excluded for a 20 s or 30 s. The dynamic exclusion list held either 300 or 500 entries and the maximal duration of exclusion was set to 60 s. The exclusion mass window was set to either m/z ±1.5 or m/z −0.5 to m/z +1.5. The maximal ion injection time was limited to 500 ms for survey scans in the Orbitrap Fourier transform mass spectrometer (FTMS) and to 100 ms for CID fragment ion scans in the linear trap quadrupole (LTQ) mass spectrometer.
Data analysis of SILAC labeled proteome
Tandem mass spectra were searched with the human IPI or Uniprot databases (IPI: v 2010 and uniprot: v 2010_06) with prolucid44 and filtered with DTASelect45. In order to identify novel open reading frames in the human genome with proteomics, the human genome (GRCh37) was 6-frame translated, and open reading frames (ORFs) with amino acid sequences of ≥ 30 amino acid length were considered for endoproteolytic cleavage in silico with the endoprotease trypsin and resulting peptide sequences of ≥6 amino acids length were stored in a peptide sequence database. The search results were scored for the presence or absence in either experimental condition (p63 knock down or UV irradiated) and expressed ORFs in the genome, that were not located within exons, were further analyzed with blastp46 to reveal sequence homology to known transposable elements, retroelements or retroviruses in the genome.
Subsequent quantitative data analysis was performed using standard procedures as outlined in the IP2 analysis software (Integrated Proteomics Applications) with the following modifications: individual protein ratios were normalized to the average of all measured ratios to account for differences in protein abundance between HaCaT and NHEK. Subsequent data analysis (two-experiment comparisons as well as k-means clustering) was performed using the biostatistics package of MATLAB (Mathworks). Proteins regulated across all four conditions were K-means clustered with distance measured by correlation, with 100 maximal iterations and 10 replications using new initial cluster centroid positions. K-means clustered protein groups were analyzed with GeneMANIA or BioGrid plug-in tools in Cytoscape (ww.cytoscape.org) or searched for enrichment in GO categories with GOMiner47.
Cell culture of normal human epidermal keratinocytes and shRNA-mediated knock down of proteins in human primary keratinocytes
Normal human epidermal keratinocytes (NHEK) isolated from human neonatal skin were received from Lonza and expanded in keratinocyte growth medium (KGM-2) to passage 3 or 5 prior to infection with Mission shRNA (Sigma) lentiviral particles generated in HEK293T cells and incubated for 16 h. 48 h post infection keratinocytes were UV irradiated (120 J/m2 to 1200 J/m2) in the absence of cell culture media and lysed after 1 h recovery in the original cell culture media. Human keratinocytes in primary cell culture represent a heterogeneous cell population that displays limited clonal expansion capacity because keratinocyte subpopulations transition from stem cell-like keratinocytes to transient amplifying keratinocytes to terminally differentiated. Early (<3 passages) and late (>4 passages) primary keratinocyte cell cultures are specifically denoted here to loosely reflect the overall status of the primary keratinocyte cell culture even before terminal differentiation.
Western blot analysis
NHEK were lysed in 2x SDS gel loading buffer (Sigma), heated for 5 min to 95 °C and spun at 16,000 rpm at room temperature for 15 min. Protein lysates were subjected to SDS-gel electrophoresis (Life Technologies) and SDS-gels were immunoblotted. p63 protein variants were detected with the monoclonal p63 antibody 4A4 (Ventana) following separation of proteins in non-gradient 8 % Tris-Glycine SDS-gels. Monoclonal antibodies were used for the detection of γH2A.X (Cell Signaling) and βActin (Sigma), and polyclonal antisera were used for detection of S*66/68 (corresponding to S160/162 in TAp63, Cell Signaling) and for LINE-1 ORF1p. Horseradish peroxidase signals were recorded with X-ray films (Kodak) and quantified with ImageJ (National Institutes of Health).
ΔNp63α-myc/his phosphorylation analysis
The cDNA sequence of human ΔNp63α was cloned into a pcDNA3.1A/Myc-His A vector backbone (Invitrogen) as described previously12, and ΔNp63α-myc/his tagged protein was overexpressed in HEK293 cells following plasmid transfection with Fugene 6 or HD (Promega). SMG1 or CDK12 expression plasmids (Origene) or pcDNA3.1-GFP expression plasmids were co-transfected in 1:1 molar stoichiometry to the pcDNA3.1A-ΔNp63αmyc/his plasmid. Cells were lysed 24 h later in lysis buffer (6 M guanidinium chloride, 500 mM NaCl, 20 mM sodium phosphate, pH 7.8) and ΔNp63α-myc/his protein was purified with NiNTA resin according to manufacturer’s instructions (Thermofisher). Proteins were precipitated with methanol:chloroform (water:methanol:chloroform, 1:1:1, v:v:v), resuspended in digestion buffer, and reduced and alkylated as described above. Each sample was split into three equal aliquots and digested separately with trypsin, chymotrypsin, and AspN. Following reversed phase purification (C18 ZipTip, Sigma) according to manufacturer recommendations, eluted peptides from each aliquot were combined in 100 mM Triethylammonium bicarbonate (TEAB) buffer, pH 8.5, and labeled light, medium, and heavy for each experimental condition (GFP, SMG1, or CDK12) with isotope defined dimethyl groups on free amines (lysine and N-terminus) according to48. Labeled peptides of all three experimental conditions were combined and subjected to reverse phase chromatographic separation and mass spectrometry on an LTQ Orbitrap Elite (Thermo). Data analysis was performed with IP2 (Integrated Proteomics) and chromatographic peaks for phosphorylated peptides were extracted and quantified with Census49. The average difference of CRK7 versus control (GFP) or SMG1 versus control (GFP) was calculated based on the intensity of the isotope specific chromatographic peaks. Relative changes of phosphorylated peptides that cover the same phosphorylation site were averaged.
DNA binding assay
Whole cell lysate was incubated on ice with a biotinylated, p21 responsive element50 containing double stranded oligonucleotides for 1 h, and p63 bound to the p21 responsive element was detected with the monoclonal antibody 4A4 using the protocol described in the TransFactor chemiluminiscent kit (Clontech).
Results
P63 phosphorylation upon UV irradiation of keratinocytes
First, we analyzed the proteoform pattern of p63 in NHEK upon UV irradiation and increased number of cell passages. As a control, p63 proteoforms were depleted by more than 90 % with the splice variant insensitive lentiviral shRNA p63si1, which slightly altered cell morphology (Figure 1B). We monitored p63 protein levels on Western blot with the splice-variant independent monoclonal antibody 4A4 and with mass spectrometry and, as previously described, we detected only ΔNp63α12,19,31. In control NHEK ΔNp63α migrated faster during SDS-gel electrophoresis than upon UV irradiation with 1200 J/m2 which slowed its migration (Figure 1C). A λ protein phosphatase treatment and subsequent Western blot analysis confirmed hyperphosphorylation of ΔNp63α upon genotoxic stress (Figure S1), as observed previously51.
Based on the electrophoretic migration pattern of ΔNp63α upon UV irradiation, we differentiated 3 proteoforms of ΔNp63α (Figure 1C). We refer to ΔNp63α in control NHEK as “ΔNp63α-[c]” to distinguish it from the hyperphosphorylated ΔNp63α-[a] and from ΔNp63α-[b]. ΔNp63α-[b] displayed a higher apparent molecular weight than ΔNp63α-[c] but lower than ΔNp63α-[a]. ΔNp63α-[b] was the most predominant p63 proteoform in late passage NHEK that had not yet terminally differentiated, and ΔNp63α-[b] was present as residual p63 in NHEK upon knock down of p63 with p63si1. UV irradiation of early passage NHEK hyperphosphorylated ΔNp63α-[c] to ΔNp63α-[a] but failed to phosphorylate ΔNp63α-[b] to ΔNp63α-[a] in late passage NHEK (Figure 1C, right panel). Thus, ΔNp63α-[b] may reflect a p63 proteoform that is not regulated by or not sensitive to UV irradiation. We evaluated the genotoxic stress in NHEK based on phosphorylated histone H2A.X (γH2A.X) which marks the sites of DNA damage. As expected, γH2A.X was increased in normal NHEK upon UV irradiation (Figure 1C). A knock down of p63 further elevated γH2A.X levels in control and UV irradiated NHEK. γH2A.X was already overall increased in late passage NHEK, and a further elevation upon UV irradiation was independent of the p63 expression status.
Recruitment of proteins to chromatin in response to UV irradiation
Next, we identified proteins that were recruited to chromatin in response to UV irradiation to prevent increased DNA damage. NHEK were UV irradiated and the chromatin associated proteome was isolated according to Berkowitz et al41 and quantitatively compared to controls. The relative abundance of proteins at the chromatin was determined with quantitative mass spectrometry52 (Figure 1D and Figure S2). Immortalized epidermal keratinocytes (HaCaT) were stable isotope labeled by heavy lysine and arginine in cell culture (SILAC)53 which allowed for a direct comparison between experimental conditions (e.g. NHEK +/− UV irradiation and/or +/− p63si1 shRNA). Relative quantification of ΔNp63α levels with mass spectrometry revealed that hyperphosphorylated ΔNp63α-[a] strongly associated with chromatin following UV irradiation (>20 fold). ΔNp63α-[b] in late passage NHEK was already present at the chromatin and remained almost unaltered upon UV irradiation (Figure 1C).
Chromatin-associated proteins clustered into 9 individual groups depending on their relative presence in the four conditions (control, p63si1, UV, and UV-p63si1, Figure S3). We distinguished 9 groups to allow for differential chromatin binding of proteins depending on the experimental conditions UV irradiation and p63 depletion. While proteins bound to chromatin upon UV irradiation and independent of p63 status for example, we focused our analysis on group 8 which included p63 and subsumed proteins that were recruited to the chromatin upon UV irradiation and only in presence of p63 but not upon p63-depletion (Table S1). Group 8 was enriched for the GO term “chromatin binding” and an in-depth analysis revealed that three protein kinases (CDK12, SMG1 and MAPK1 (ERK2, PRKM1, PRKM2)) relocated to chromatin in the presence of p63 only. The kinases increased at the chromatin in the presence of p63 and UV irradiation. Protein network analysis showed that the proteins in Group 8 may form several small protein-protein interaction networks according to known protein-protein interactions curated in Ingenuity Pathway Analysis54,55 (QUIAGEN IPA, Figure 1E and Figure S4). Protein interaction networks included either CDK12 or SMG1 but not MAPK1, and CDK12 may interact indirectly with p63. CDK12 is a potential binding partner of the pre-mRNA splicing factor PRPF40A (Yeast two hybrid screens and AP-MS56,57). PRPF40A can associate with E3 SUMO ligase PIAS156 which in turn interacts with p63 to co-regulate transcriptional transactivation58. P63 can also bind to IGFBP259, which associates with proteins involved in RNA metabolism. Group 8 included the splicing factors SRSF1/SF2, which may initiate splicing of the nascent RNA. CDK12 can phosphorylate SRSF1/SF2 and the C-terminal hepta-peptide repeat domain (CTD) of the RNA polymerase IIb subunit RBP, which activates transcription elongation. RBP is part of the RNA-polymerase IIb protein complex present in Group 8. Thus, potentially direct protein-protein interactions may associate p63 with CDK12 and RNA splicing as well as transcriptional regulation.
The phosphatidylinositol 3 kinase-related serine/threonine-protein kinase SMG1 was recruited to the chromatin in the presence of p63. The protein-protein interaction network in Group 8 suggested that SMG1 might associate with RBMA8, which marks mRNAs that were successfully spliced by the Exon Junction Complex (EJC). RBMA8 can also interact with RNA-polymerase IIb proteins. Additionally, the network included RNA-polymerase IIb protein complexes that may associate with other proteins, including XAB2 (which is involved in transcription-coupled DNA-repair60), RNGTT (which caps the 5’-terminus of the nascent mRNA), and SUB1 (which binds to single stranded DNA in order to stabilize the multi-protein transcription complex). Mitogen activated protein kinase 1 MAPK1 was also recruited to the chromatin and can phosphorylate p63 according to61. MAPK1 transduces extracellular stimuli through the MAPK1 signaling cascade to the nucleus; however, MAPK1 has not been described as interacting with any of the proteins in Group 8.
SMG1 and CDK12 modulate p63 phosphorylation
By evaluating ΔNp63α phosphorylation upon UV irradiation in a time-course experiment, we tested whether MAPK1, SMG1 or CDK12 might dynamically phosphorylate p63 at specific sites (Figure 2). UV irradiation (360 J/m2) hyperphosphorylated ΔNp63α-[c] to ΔNp63α-[a] within 40 min (Figure 2A) and altered ΔNp63α turnover. p63 protein abundance increased and peaked within 1 h, subsequently diminishing to almost complete absence at 12 h post UV irradiation. As previously observed, residual ΔNp63α-[b] in p63si1-treated NHEK was not hyperphosphorylated to ΔNp63α-[a] following UV irradiation. ΔNp63α-[b] levels were barely increased 1 h following UV irradiation and completely diminished 4 h post UV irradiation. We determined the phosphorylation sites and levels in p63 with a phosphor-S66/S68 specific monoclonal antibody and Western blot to monitor ΔNp63α phosphorylation at serine S66/S68 1 h after low dose UV irradiation (120 J/m2) as previously reported62 (Figure 2B and Figure S5B). We found that S66/S68 specific phosphorylation was not altered by depletion of MAPK1 or SMG1. Knock down of CDK12 prevented S66/S68 phosphorylation, indicating that CDK12 either directly or indirectly induces S66/S68 phosphorylation upon low dose UV irradiation.
Figure 2: Cyclin-dependent kinase 12 (CDK12) and serine/threonine protein kinase SMG1 regulate phosphorylation of p63 in human keratinocytes following UV irradiation.
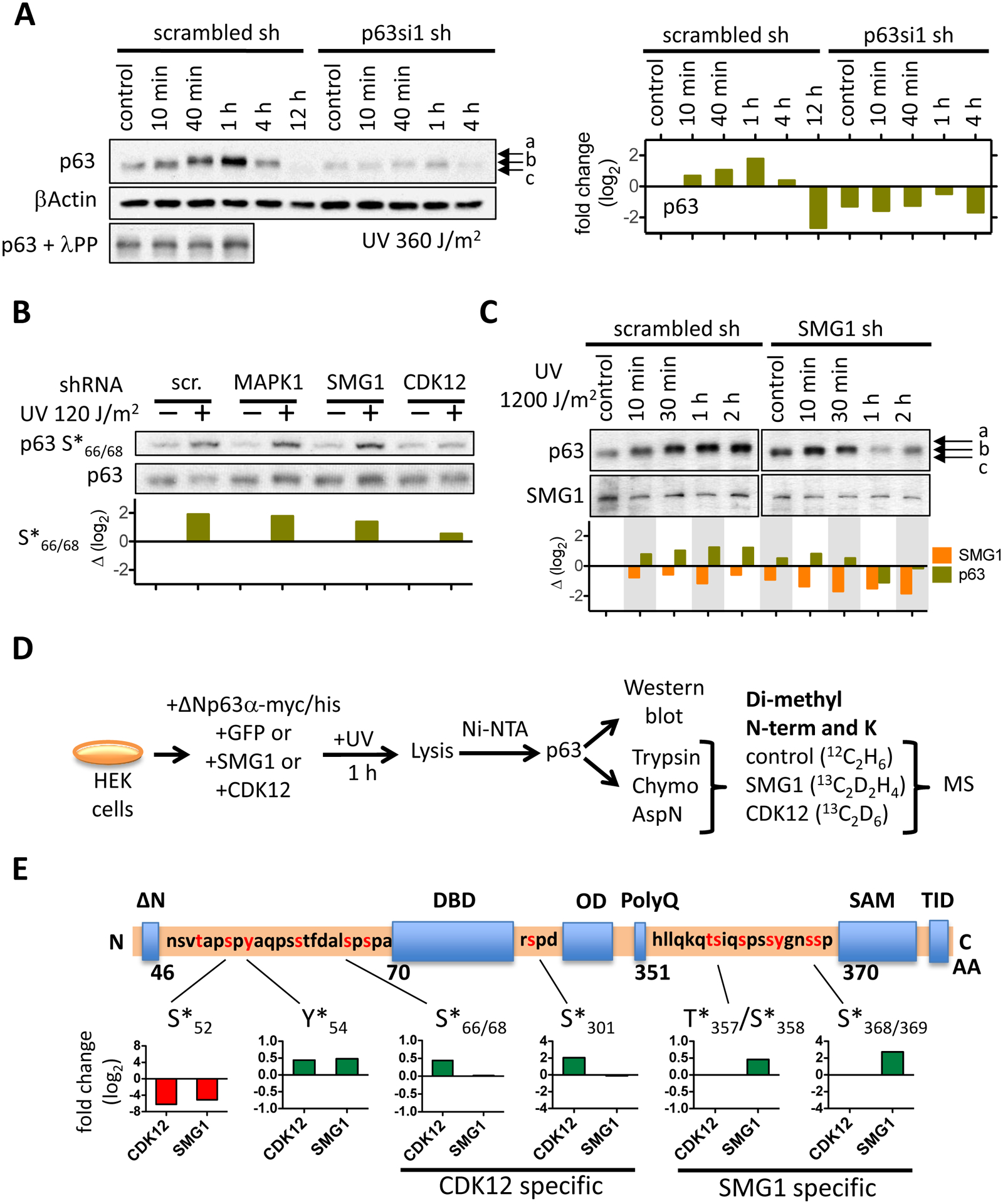
(A) The Western blot shows ΔNp63α phosphorylation following UV irradiation with 360 J/m2 in a time-course experiment. An aliquot of control and of each of the first three time points were digested with λ protein phosphatase to show that the electrophoretic shift of p63 is dependent on phosphorylation. Relative ΔNp63α protein levels are depicted in the bar graph (right). (B) MAPK1 or SMG1 or CDK12 was depleted in NHEK with specific shRNAs, NHEK UV irradiated (120 J/m2), ΔNp63α phosphorylation at S66/68 detected by Western blot. The bar graph depicts the fold change of ΔNp63α phosphorylation in the kinase knock-down cells relative to control. (C) ΔNp63α protein levels and its SDS-gel electrophoretic migration pattern was determined in NHEK following SMG1 knock-down and UV irradiation. Relative protein levels are indicated in the bar graph below the Western blot. (D) The schematic depicts the detection of ΔNp63α phosphorylation with mass spectrometry. ΔNp63α was transiently overexpressed in human embryonic kidney cells (HEK293) and phosphorylation in presence of either CDK12 or SMG1 quantified following digest with different proteolytic enzymes, isotope labeling of peptides by reductive methylation and quantitative mass spectrometry. (E) Phosphorylation of ΔNp63α by SMG1 and CDK12 is localized within the ΔNp63α protein. The relative change in phosphorylation at specific sites is normalized to the phosphorylation level of ΔNp63α in control HEK293 cells that were transfected with GFP. The bar graphs depict the relative change in phosphorylation at the amino acid sites that are indicated.
We knocked down SMG1 and monitored p63 phosphorylation following high dose UV irradiation (1200 J/m2) to assess whether SMG1 influenced hyperphosphorylation of p63 (Figure 2C and Figure S1C). In control, ΔNp63α-[c] shifted to ΔNp63α-[a] within 1 h, which corresponded with a peak in protein levels (Figure 2C, left panel). Partial knock down of SMG1 with an SMG1-specific shRNA prevented phosphorylation of ΔNp63α-[c] to ΔNp63α-[a]. Instead, ΔNp63α-[b] was observed with a peak in protein levels within 10 min instead of 1 h after UV irradiation (Figure 2C, right panel). The results showed that the presence of SMG1 shifted ΔNp63α-[c] to ΔNp63α-[a] and stabilized elevated levels of phosphorylated ΔNp63α in NHEK in response to UV irradiation. We utilized quantitative mass spectrometry that was specifically tailored to analyze p63 phosphorylation to determine the sites that were phosphorylated in the presence of SMG1 (Methods, Figure 2D). To this end, a myc/his-tagged version of ΔNp63α was overexpressed in HEK239 cells with either SMG1 or CDK12 or GFP (control). ΔNp63α-myc/his was purified with Ni-NTA resin, aliquots were digested separately with trypsin, chymotrypsin, and AspN, and the resulting peptides of each condition were isotope-labeled with reductive methylation for relative quantification48. Over 90 % of ΔNp63α-myc/his amino acid sequence was detected by mass spectrometry, revealing 25 distinct phosphorylation sites including previously identified sites like Y*5463–65 and S*66/68 (Figure S5A). SMG1 and CDK12 altered relative phosphorylation at several distinct sites (Figure 2E). Both kinases reduced phosphorylation of the N-terminal S52 in ΔNp63α and increased phosphorylation of Y54, which is specifically phosphorylated by the tyrosine kinase c-Abl63–65. CDK12-mediated phosphorylation of S66/S68 was confirmed by mass spectrometry, and CDK12 overexpression promoted phosphorylation of p63 at amino acid S301, which is located between the DNA binding and oligomerization domain of p63. The phosphorylation sites S66/S68 and S301 flank the central DNA binding domain of p63. Overexpression of SMG1 induced phosphorylation of T357/S358 and S368 in a section of p63α that is devoid of any functional annotation. Several additional sites between amino acids 351 and 370 were also phosphorylated, indicating that this segment of ΔNp63α is extensively post-translationally modified. SMG1 belongs to the family of protein kinases that preferentially phosphorylate S/TQ motifs. However, T357/S358 and S368 do not match the S/TQ motif. Neither mass spectrometric measurements detected phosphorylation at three out of the four possible S/TQ sites present in ΔNp63α (Figure S5A) nor did Western blot reveal increased S/TQ phosphorylation with a S*/T*Q specific antibody following UV irradiation (Figure S5B), suggesting that either SMG-1 has an indirect effect on ΔNp63α phosphorylation or that SMG1 recognizes a sequence motif other than S/TQ.
SMG1 but not CDK12 induces p63 DNA binding
Given that ΔNp63α was part of the chromatin associated proteome following UV irradiation in NHEK, and because phosphorylation-induced binding of p63 to the p53 specific DNA recognition sequence correlates with apoptosis13, we tested whether UV irradiation in the absence of CDK12 or SMG1 altered the ability of ΔNp63α to bind to the conserved p53-specific DNA recognition element. We knocked down SMG1 or CDK12 in NHEK, lysed cells 1 h post UV irradiation, and quantified the amount of ΔNp63α that bound to the p53 DNA recognition sequence in an in vitro transcription factor binding assay (Figure 3A). Depletion of p63 confirmed that the signal was specific to p63, and p63 binding was about two-fold above background signal, which was determined using a mutated p53 DNA binding sequence (mut p53-BS). Low dose UV irradiation (120 J/m2) of NHEK did not alter DNA binding of ΔNp63α in control (scrambled shRNA) or in CDK12-depleted NHEK (Figure 3B, left panel), indicating that knock-down of CDK12 did not limit p63 binding to the specific p53 DNA recognition sequence. In contrast, knock-down of SMG1 reduced DNA binding of ΔNp63α, suggesting that p63’s ability to bind to the specific DNA sequence in response to UV irradiation is at least in part dependent on SMG1. High dose UV irradiation (1200 J/m2) reduced ΔNp63α DNA binding in control or in CDK12 knock-down NHEK (Figure 3B, right panel). UV irradiation induced DNA damage activates p53 which competes with p63 for the p53 DNA recognition sequence. Because whole cell lysates were used to test for DNA specific binding, activated p53 (which is also hyperphosphorylated) might out-compete ΔNp63α at the p53-specific DNA binding site, thereby explaining a reduced DNA binding of ΔNp63α in control and CDK12-depleted NHEK. Loss of SMG1 did not alter the binding of ΔNp63α to the specific DNA binding sequence upon high dose UV irradiation, most likely because loss of SMG1 also prevents p53 activation (SMG1 is known to phosphorylate and activate p53 in response to genotoxic stress66). To reduce potential competition of activated p53 for the p53 specific DNA binding sequence in the in vitro transcription factor binding assay, we sequestered p53 protein from UV irradiated NHEK lysates with a bead-conjugated anti-p53 antibody prior to testing the DNA binding capability of p63 (Figure 3C). Following p53 depletion, ΔNp63α specific DNA binding increased upon UV irradiation in control or in CDK12-depleted NHEK. Knock-down of SMG1 now prevented an increase in DNA binding of ΔNp63α. Thus, we conclude that SMG1, but not CDK12, is necessary to induce specific DNA binding of ΔNp63α upon UV irradiation.
Figure 3: Presence of SMG1 regulates ΔNp63α DNA binding following UV irradiation.

(A) The schematic shows the experimental setup to detect ΔNp63α binding to the specific recognition sequence of p53 (p53-BS) in the p21 promoter. (B) The specific DNA binding of ΔNp63α in UV irradiated NHEK was quantified following CDK12 or SMG1 knock- down. Values are normalized to control (scrambled shRNA). (C) NHEK were UV irradiated and p53 was depleted prior to measuring the relative DNA binding of ΔNp63α to the specific p53 DNA sequence. CDK12 (CDK12 si1), SMG1 (SMG-1 si1), p63 (p63 si1), or control (scr.) shRNA was applied in NHEK and DNA binding of ΔNp63α subsequently measured. NHEK treated with scrambled shRNA were incubated with a mutated sequence of the p53 DNA binding site (mut p53-BS) to show binding specificity of the assay. (Student’s t-test; *, p < 0.1; **, p < 0.05; ***, p < 0.001).
Altered RNA surveillance and increased DNA damage upon loss of p63
If p63 balances p53 based activation of DNA damage response pathways and apoptosis at low dose UV irradiation with the help of SMG1 kinase, we asked whether additional proteins at the chromatin might regulate p63 function. Relative quantification of proteins in the chromatin-associated proteome indicated that ZFR, EXSC3, and RXRA levels were reduced, albeit statistically insignificant, at the chromatin following ΔNp63α-depletion (Figure 4A) The Zinc Finger RNA-binding protein ZFR binds to double-stranded RNA and is required for normal epidermal development in mice67. The RNA Exosome Component EXOSC3 is part of the RNA surveillance pathway68,69 and the Retinoid X Receptor α RXRA regulates keratinocyte proliferation70. Thus, diminished ΔNp63α levels were accompanied by a loss of proteins involved in RNA surveillance (ZFR and EXOSC3) and keratinocyte proliferation (RXRA). ΔNp63α depletion led to increased chromatin binding of DTX3L, CDC5L, P26s4, TIFβ, FANCI, Lsh, and UBTF, all of which either silence RNA transcription or support DNA damage repair. The Fanconi Anemia protein 1 (FANCI) is involved in the p53-mediated DNA damage response71. The nucleolar transcription factor 1 UBTF and the TATA box-binding protein-associated factor RNA-polymerase I subunit β TIFβ control gene transcription and CDC5L is associated with the spliceosome. The Lymphoid-specific helicase (Lsh) was identified as a direct target of ΔNp63 transcription factor activity72 and facilitates DNA-methylation of repetitive and microsatellite DNA sequences to silence transcription73.
Figure 4: ΔNp63α depletion in NHEK alters the presence of transcription factors and DNA repair proteins at the chromatin and induces expression of retrotransposon protein LINE-1 ORF1p.

(A) Proteins differentially associate with the chromatin following knock-down of p63 in NHEK. The volcano plot shows all proteins that were quantified with bottom-up proteomics at the chromatin in control and p63 knock-down NHEK. Proteins that are either increased (red quadrant) or decreased (green quadrant) are highlighted. Eleven significantly changed proteins are labeled with the respective Uniprot gene name. (B) The bar graph shows spectral counts (SpC) for ORF1p of the non-LTR retrotransposon LINE-1 in NHEK or HaCaT cells (IEF fraction 10) upon p63 knock down and UV irradiation. (C) Monomeric and trimeric ORF1p protein levels were determined in control and p63 shRNA treated NHEK after UV irradiation. The left panel shows ORF1p levels following different doses of UV irradiation (20 to 1200 J/cm2). The right panel displays ORF1p protein in a time-course experiment (10 min to 12 h) following UV irradiation of NHEK with 360 J/m2. Bar graphs below indicate the relative change of monomeric and trimeric ORF1p as well as ΔNp63α after normalization to βActin.
Because we observed enhanced γH2A.X and recruitment of the DNA damage response protein FANCI to the chromatin, we searched our proteomic data for protein or protein fragments (Methods) that would indicate a loss of transcriptional control and increased DNA damage in the absence of p63 in NHEK. UV irradiation is known to activate viruses and retroviruses that integrate into the host DNA and to activate transposable elements (or remnants thereof) that are widely distributed in intergenic regions of the genome. In our dataset we found that UV irradiation of NHEK specifically induced ORF1p of the non-LTR retrotransposon LINE-1 in the absence of p63. Furthermore, peptides derived from the protein ORF1p were identified in a ΔNp63α enriched, native protein fraction isolated from the immortalized human keratinocyte cell line HaCaT (Figure 4B). We used Western blotting to detect and differentiate monomeric from trimeric ORF1p that can bind to intron-less LINE-1 RNA74 (Figure 4C). The Western blot analysis showed that ORF1p levels were higher in UV irradiated NHEK in the absence of p63 than in controls, as initially observed in the proteomic dataset. The amount of ORF1p remained almost unaltered at different doses of UV irradiation (20 to 1200 J/m2) in control NHEK (scrambled shRNA), whereas knock-down of ΔNp63α resulted in a strong upregulation of monomeric and trimeric ORF1p with increasing UV dose (Figure 4C, left panel). In control NHEK, trimeric ORF1p protein levels peaked 10 min after 360 J/m2 dose of UV-irradiation (dose used in Figure 2A) and returned to baseline levels 12 h later (Figure 4C, right panel). In the absence of ΔNp63α, trimeric ORF1p accumulated more gradually with a peak 1 h after UV irradiation. Because ΔNp63α suppressed the accumulation of the LINE1 RNA binding, trimeric version of ORF1p in control and UV irradiated NHEK, we used RNAseq to determine LINE-1 RNA levels in p63 depleted NHEK and found a small but insignificant increase in LINE1 and ERV3-1 RNAs in the absence of p63.
Discussion
We used Western blot and mass spectrometry to discern three different phosphorylated ΔNp63α proteoforms in NHEK that may be present simultaneously in skin. ΔNp63α-[c] represents a basally phosphorylated ΔNp63α proteoform. ΔNp63α-[b] is low in abundance but remains present in either higher passage NHEK or as remnant p63 proteoform upon knock down of p63 in NHEK. We found that ΔNp63α-[a] is hyperphosphorylated following UV irradiation in a time- and UV dose-dependent manner and localized p63 to chromatin. Mass spectrometric analysis of the chromatin associated proteome determined that CDK12 and SMG1 were recruited to chromatin in the presence of p63 and impacted the phosphorylation status of p63. CDK12 induced p63 phosphorylation at S66/S68 and S301, which frame the DNA binding domain and potentially reduce specific DNA binding of p63. CDK12-mediated phosphorylation most likely shifted ΔNp63α-[c] to ΔNp63α-[b], and ΔNp63α-[b] protein levels diminished rapidly upon UV irradiation. Residual ΔNp63α-[b] in p63 knock-down cells failed to shift to ΔNp63α-[a] at intermediate doses of UV irradiation, therefore we propose that at least a subset of the ΔNp63α-[b] variants is less responsive to UV irradiation. Phosphorylation of S66/S68 was previously shown to correlate inversely with the proliferative capacity of keratinocyte stem cells in vitro75. The presence of SMG1 resulted in phosphorylation of the p63α specific C-terminus at S357/T358 and S368/369 and shifted ΔNp63α-[c] to ΔNp63α-[a]. Hyperphosphorylated ΔNp63α-[a] was recruited to chromatin and bound to the specific DNA recognition sequence. It remains unknown whether CDK12 and SMG1 phosphorylate p63 directly or indirectly through activation of other kinases that were not identified here. SMG1 might phosphorylate p63 directly, as other kinases of the phosphatidyl inositol protein kinase family recognize sequence motifs beyond S/TQ (such as MTOR). For example, the SMG1-related kinase ATM phosphorylates p63 in squamous cell carcinoma cells upon cisplatin treatment, albeit in a different phosphorylation pattern76.
We further show that high levels of UV irradiation activated ΔNp63α’s specific DNA binding. UV irradiation also activated the p63 homologue and tumor suppressor protein p53, which partially overrode binding of ΔNp63α to the p53 specific DNA sequence. Indeed, NHEK respond to high levels of UV irradiation with the appearance of pyknotic, sunburned cells which enter a rapid apoptosis instead of terminal differentiation. Reduced p63 protein levels recruited the DNA damage responsive protein FANCI and the chromatin remodeling protein Lsh to chromatin, and increased levels of γH2A.X indicated ongoing DNA damage and repair. Higher trimeric ORF1p protein levels in p63-depleted NHEK may be a consequence of reduced turnover of intron-less retrotransposon-derived RNAs, however LINE-1 RNA levels did not increase in response to p63-depletion alone.
The recruitment of CDK12 and SMG1 to chromatin and (indirect) phosphorylation of p63 might allow ΔNp63α to surveil the transcriptome. While RNA surveillance regulates cell differentiation (for example, in the immune and nervous system)77, less is known about RNA surveillance in terminal differentiation of keratinocytes. Perturbances of the transcriptome might divert CDK12 from regulating RNA splicing to activating ΔNp63α-[c] to ΔNp63α-[b], thereby restricting NHEK proliferative capabilities32,78. Excessive DNA damage may redirect SMG1 from non-sense RNA decay to converting ΔNp63α-[c] to hyperphosphorylated ΔNp63α-[a] which specifically binds to the p53-DNA consensus sequence to potentially suppress apoptosis and trigger terminal differentiation79,80. In line with this interpretation, we observed increased trimeric, RNA-binding ORF1p protein in the absence of p63 and following UV irradiation. Thus, p63 may sense alterations in the cellular RNA pool which increase the risk of genome instability to protect proliferating keratinocytes form genotoxic stress and secure seamless self-renewal of the epidermis.
Supplementary Material
Proteins co-regulated with ΔNp63α at the chromatin (Table_S1.xlsx).
Figure S1 Western blot analysis of ΔNp63α following UV irradiation.
Figure S2 Enrichment of the chromatin associated proteome.
Figure S3 Clustering of proteins upon p63 los and/or UV irradiation.
Figure S4 Additional protein-protein interaction networks in Group 8.
Figure S5 Phosphorylated Amino acid sites in ΔNp63α.
Material S1 Representative spectra of phosphorylated peptides in p63(Material_S1_Phospho_peptide_spectra.pdf).
Material S2 Images of Western blots (Material_S2_WesternBlots_Manuscript_p63.pdf).
Acknowledgements
The authors are grateful to Tao Xu who wrote lockmasscorrector and Sun Kyung Park for access to IP2. The authors thank Dr. Gerhard Schumann for the polyclonal anti-ORF1p rabbit serum and Dr. William Dalton for the HA-SMG1 plasmid. We are indebted to Claire Delahaunty for critically reading the manuscript.
Funding
The work was funded by NIH 5P41RR011823 and NIH 5P01AG031097.
Abbreviations
- NHEK
normal human epidermal keratinocytes
- UV
ultraviolet
- LINE
long interspersed element-1
- MS
mass spectrometry
- WB
Western blot
- Chymo
Chymotrypsin
- ΔN
Conserved N-terminal sequence specific to ΔNp63
- DBD
DNA binding domain
- OD
oligomerization domain
- PolyQ
Poly-glutamine sequence
- SAM
sterile α motif domain
- TID
transactivation inhibitory domain
- N-term
N-terminus of peptide
- K
lysine residue
- BS
DNA binding site
- RE
responsive element
- HRP
horseradish peroxidase
- SILAC
stable isotope labeled by amino acids in cell culture
- Ni-NTA
Nickel-Nitrilotriacetic acid
- λPP
λ Protein Phosphatase
- GFP
Green Fluorescent Protein
Data availability
All proteomic experiments are available in Massive/ProteomeXchange with the following accession number MSV000088064/PDX028222.
References
- 1.Vazquez A, Bond EE, Levine AJ & Bond GL The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 7, 979–987, (2008). [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ & Oren M The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9, 749–758, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leslie M Brothers in arms against cancer. Science 331, 1551–1552, (2011). [DOI] [PubMed] [Google Scholar]
- 4.Bell HS et al. A p53-derived apoptotic peptide derepresses p73 to cause tumor regression in vivo. J Clin Invest 117, 1008–1018, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores ER et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416, 560–564, (2002). [DOI] [PubMed] [Google Scholar]
- 6.Donehower LA et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221, (1992). [DOI] [PubMed] [Google Scholar]
- 7.Yang A et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404, 99–103, (2000). [DOI] [PubMed] [Google Scholar]
- 8.Yang A et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718, (1999). [DOI] [PubMed] [Google Scholar]
- 9.Mills AA et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713, (1999). [DOI] [PubMed] [Google Scholar]
- 10.Su X et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 5, 64–75, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senoo M, Pinto F, Crum CP & McKeon F p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 129, 523–536, (2007). [DOI] [PubMed] [Google Scholar]
- 12.Bamberger C & Schmale H Identification and tissue distribution of novel KET/p63 splice variants. FEBS letters 501, 121–126, (2001). [DOI] [PubMed] [Google Scholar]
- 13.Suh EK et al. p63 protects the female germ line during meiotic arrest. Nature 444, 624–628, (2006). [DOI] [PubMed] [Google Scholar]
- 14.Straub WE et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis 1, e5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deutsch GB, Zielonka EM, Coutandin D & Dotsch V Quality control in oocytes: domain-domain interactions regulate the activity of p63. Cell Cycle 10, 1884–1885, (2011). [DOI] [PubMed] [Google Scholar]
- 16.Straub WE et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis 1, e5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang A et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2, 305–316, (1998). [DOI] [PubMed] [Google Scholar]
- 18.Beyer U, Moll-Rocek J, Moll UM & Dobbelstein M Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc Natl Acad Sci U S A 108, 3624–3629, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bamberger C, Hafner A, Schmale H & Werner S Expression of different p63 variants in healing skin wounds suggests a role of p63 in reepithelialization and muscle repair. Wound Repair Regen 13, 41–50, (2005). [DOI] [PubMed] [Google Scholar]
- 20.Yang A et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell 24, 593–602, (2006). [DOI] [PubMed] [Google Scholar]
- 21.Guo X et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol 11, 1451–1457, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liefer KM et al. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res 60, 4016–4020, (2000). [PubMed] [Google Scholar]
- 23.Parsa R, Yang A, McKeon F & Green H Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol 113, 1099–1105, (1999). [DOI] [PubMed] [Google Scholar]
- 24.Romano RA et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 139, 772–782, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koster MI et al. p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci U S A 104, 3255–3260, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koster MI & Roop DR The role of p63 in development and differentiation of the epidermis. J Dermatol Sci 34, 3–9, (2004). [DOI] [PubMed] [Google Scholar]
- 27.Chakravarti D et al. Induced multipotency in adult keratinocytes through down-regulation of DeltaNp63 or DGCR8. Proc Natl Acad Sci U S A 111, E572–581, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki D, Sahu R, Leu NA & Senoo M The carboxy-terminus of p63 links cell cycle control and the proliferative potential of epidermal progenitor cells. Development 142, 282–290, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borrelli S et al. The p63 target HBP1 is required for skin differentiation and stratification. Cell Death Differ 17, 1896–1907, (2010). [DOI] [PubMed] [Google Scholar]
- 30.Dimri GP, Itahana K, Acosta M & Campisi J Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol 20, 273–285, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bamberger C, Pollet D & Schmale H Retinoic acid inhibits downregulation of DeltaNp63alpha expression during terminal differentiation of human primary keratinocytes. J Invest Dermatol 118, 133–138, (2002). [DOI] [PubMed] [Google Scholar]
- 32.Suzuki D & Senoo M Increased p63 phosphorylation marks early transition of epidermal stem cells to progenitors. J Invest Dermatol 132, 2461–2464, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherukuri P et al. Phosphorylation of DeltaNp63alpha via a novel TGFbeta/ALK5 signaling mechanism mediates the anti-clonogenic effects of TGFbeta. PLoS One 7, e50066, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan M, Luong P, Hudson C, Gudmundsdottir K & Basu S c-Abl phosphorylation of DeltaNp63alpha is critical for cell viability. Cell Death Dis 1, e16, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratovitski EA Phospho-DeltaNp63alpha-dependent microRNAs modulate chemoresistance of squamous cell carcinoma cells to cisplatin: at the crossroads of cell life and death. FEBS Lett 587, 2536–2541, (2013). [DOI] [PubMed] [Google Scholar]
- 36.Chen HH, Wang YC & Fann MJ Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol 26, 2736–2745, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko TK, Kelly E & Pines J CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J Cell Sci 114, 2591–2603, (2001). [DOI] [PubMed] [Google Scholar]
- 38.Brumbaugh KM et al. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell 14, 585–598, (2004). [DOI] [PubMed] [Google Scholar]
- 39.Yamashita A, Ohnishi T, Kashima I, Taya Y & Ohno S Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev 15, 2215–2228, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kashima I et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20, 355–367, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berkowitz DM, Kakefuda T & Sporn M A simple and rapid method for the isolation of enzymatically active HeLa cell nuclei. J Cell Biol 42, 851–854, (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boukamp P et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106, 761–771, (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb KJ, Xu T, Park SK & Yates JR 3rd. Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. Journal of proteome research 12, 2177–2184, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cociorva D, D LT & Yates JR Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics Chapter 13, Unit 13 14, (2007). [DOI] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J Mol Biol 215, 403–410, (1990). [DOI] [PubMed] [Google Scholar]
- 47.Zeeberg BR et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biology 4, R28, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S & Heck AJ Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nature protocols 4, 484–494, (2009). [DOI] [PubMed] [Google Scholar]
- 49.Park SK & Yates JR 3rd. Census for proteome quantification. Curr Protoc Bioinformatics Chapter 13, Unit 13 12 11–11, (2010). [DOI] [PubMed] [Google Scholar]
- 50.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW & Vogelstein B Definition of a consensus binding site for p53. Nat Genet 1, 45–49, (1992). [DOI] [PubMed] [Google Scholar]
- 51.Westfall MD, Joyner AS, Barbieri CE, Livingstone M & Pietenpol JA Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of DeltaNp63alpha. Cell Cycle 4, 710–716, (2005). [DOI] [PubMed] [Google Scholar]
- 52.Washburn MP, Wolters D & Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature biotechnology 19, 242–247, (2001). [DOI] [PubMed] [Google Scholar]
- 53.Geiger T, Cox J, Ostasiewicz P, Wisniewski JR & Mann M Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods 7, 383–385, (2010). [DOI] [PubMed] [Google Scholar]
- 54.Montojo J et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warde-Farley D et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–220, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colland F et al. Functional proteomics mapping of a human signaling pathway. Genome Res 14, 1324–1332, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingham RJ et al. WW domains provide a platform for the assembly of multiprotein networks. Mol Cell Biol 25, 7092–7106, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Megidish T, Xu JH & Xu CW Activation of p53 by protein inhibitor of activated Stat1 (PIAS1). J Biol Chem 277, 8255–8259, (2002). [DOI] [PubMed] [Google Scholar]
- 59.Lunardi A et al. A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network. Proc Natl Acad Sci U S A 107, 6322–6327, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakatsu Y et al. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J Biol Chem 275, 34931–34937, (2000). [DOI] [PubMed] [Google Scholar]
- 61.Huang Y et al. Global tumor protein p53/p63 interactome: making a case for cisplatin chemoresistance. Cell Cycle 11, 2367–2379, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westfall MD, Mays DJ, Sniezek JC & Pietenpol JA The Delta Np63 alpha phosphoprotein binds the p21 and 14–3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 23, 2264–2276, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonfloni S et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 15, 1179–1185, (2009). [DOI] [PubMed] [Google Scholar]
- 64.Wei J et al. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 134, 1412–1423, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leong CO, Vidnovic N, DeYoung MP, Sgroi D & Ellisen LW The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 117, 1370–1380, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gewandter JS, Bambara RA & O’Reilly MA The RNA surveillance protein SMG1 activates p53 in response to DNA double-strand breaks but not exogenously oxidized mRNA. Cell Cycle 10, 2561–2567, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meagher MJ & Braun RE Requirement for the murine zinc finger protein ZFR in perigastrulation growth and survival. Mol Cell Biol 21, 2880–2890, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Q, Greimann JC & Lima CD Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127, 1223–1237, (2006). [DOI] [PubMed] [Google Scholar]
- 69.Preker P et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854, (2008). [DOI] [PubMed] [Google Scholar]
- 70.Li M et al. Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature 407, 633–636, (2000). [DOI] [PubMed] [Google Scholar]
- 71.Smogorzewska A et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 39, 36–47, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keyes WM et al. DeltaNp63alpha Is an Oncogene that Targets Chromatin Remodeler Lsh to Drive Skin Stem Cell Proliferation and Tumorigenesis. Cell Stem Cell 8, 164–176, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun LQ et al. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev 18, 1035–1046, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Callahan KE, Hickman AB, Jones CE, Ghirlando R & Furano AV Polymerization and nucleic acid-binding properties of human L1 ORF1 protein. Nucleic Acids Res 40, 813–827, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suzuki D & Senoo M Increased p63 Phosphorylation Marks Early Transition of Epidermal Stem Cells to Progenitors. J Invest Dermatol, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang Y et al. ATM kinase is a master switch for the Delta Np63 alpha phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 7, 2846–2855, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laffleur B & Basu U Biology of RNA Surveillance in Development and Disease. Trends Cell Biol 29, 428–445, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki D & Senoo M Expansion of epidermal progenitors with high p63 phosphorylation during wound healing of mouse epidermis. Exp Dermatol 22, 374–376, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Truong AB & Khavari PA Control of keratinocyte proliferation and differentiation by p63. Cell Cycle 6, 295–299, (2007). [DOI] [PubMed] [Google Scholar]
- 80.De Laurenzi V et al. p63 and p73 transactivate differentiation gene promoters in human keratinocytes. Biochem Biophys Res Commun 273, 342–346, (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proteins co-regulated with ΔNp63α at the chromatin (Table_S1.xlsx).
Figure S1 Western blot analysis of ΔNp63α following UV irradiation.
Figure S2 Enrichment of the chromatin associated proteome.
Figure S3 Clustering of proteins upon p63 los and/or UV irradiation.
Figure S4 Additional protein-protein interaction networks in Group 8.
Figure S5 Phosphorylated Amino acid sites in ΔNp63α.
Material S1 Representative spectra of phosphorylated peptides in p63(Material_S1_Phospho_peptide_spectra.pdf).
Material S2 Images of Western blots (Material_S2_WesternBlots_Manuscript_p63.pdf).
Data Availability Statement
All proteomic experiments are available in Massive/ProteomeXchange with the following accession number MSV000088064/PDX028222.