

Abstract
Lenvatinib, a multi-kinase inhibitor, serves a crucial role in the treatment of unresectable hepatocellular carcinoma (HCC). However, >50% of patients receiving lenvatinib therapy experience tumor growth or metastasis within 1 year, highlighting the need to address acquired resistance as a critical clinical challenge. To elucidate the factors associated with acquired resistance to lenvatinib, a lenvatinib-resistant HCC cell line (JHH-7_LR) was established by exposing a lenvatinib-sensitive HCC cell line, JHH-7, to lenvatinib. The changes in protein expression associated with the development of resistance were analyzed using a proteomic approach, detecting 1,321 proteins and significant changes in the expression of 267 proteins. Using Ingenuity Pathway Analysis bioinformatics software, it was revealed that the activity of multiple signaling pathways varied alongside the changes in expression of these proteins, and c-SRC was identified as a protein involved in a number of these signaling pathways, with its activity varying markedly upon the acquisition of resistance. When co-administering dasatinib, a c-SRC inhibitor, the partial restoration of lenvatinib sensitivity in the JHH-7_LR cell line was observed. The present study demonstrated that increased c-SRC expression was partially associated with HCC resistance to lenvatinib, suggesting that c-SRC inhibition could reduce the resistance of HCC to lenvatinib.
Keywords: lenvatinib, hepatocellular carcinoma, c-SRC, dasatinib, proteome
Introduction
Liver cancer is the third leading cause of cancer-related death worldwide and has a poor prognosis, and thus the development of more effective treatments is strongly desired (1). It is estimated that over 1 million people will be affected by liver cancer annually by 2025 (2), and hepatocellular carcinoma (HCC), as the most common form of liver cancer, accounts for approximately 90% of cases (3). In most cases, due to the lack of tumor-specific symptoms, HCC is diagnosed at an advanced stage (Barcelona Clinic Liver Cancer classification C), and the mainstay of treatment is chemotherapy (4). In addition, the recurrence rate in the remnant liver, even after R0 resection, has been reported to be up to 80% (5), and the main treatment is shifted to chemotherapy within 2 years from diagnosis in over 50% of cases diagnosed as early-stage HCC and treated radically with surgery or ablation (6). For unresectable advanced HCC, molecular-targeted therapy using lenvatinib (7), sorafenib (8,9), atezolizumab, and bevacizumab (10) is reported to be effective. In particular, oral molecular targeted drugs such as lenvatinib and sorafenib are used as crucial drugs in the first-line chemotherapy of HCC.
Lenvatinib is an oral multi-kinase inhibitor that inhibits mainly vascular endothelial growth factor receptor 1–3, fibroblast growth factor receptor (FGFR) 1–4, platelet-derived growth factor receptor (PDGFR) α, c-KIT, and RET (11). In terms of the disease control rate (complete response + partial response + stable disease ratio), response rate (complete response + partial response ratio), median progression-free survival, and median time to progression, lenvatinib shows significantly better results compared with sorafenib, another oral multi-kinase inhibitor (75.5 vs. 60.5%, 24.1 vs. 9.2%, 7.4 vs. 3.7 months, 8.9 vs. 3.7 months, respectively) (7). In contrast, tumor growth or metastasis occurs in more than 50% of patients who receive lenvatinib therapy within 1 year (7), and the development of acquired resistance is a crucial clinical problem to be resolved (12).
The factors underlying primary resistance to lenvatinib have been investigated widely and identified, and include low protein levels of fibroblast growth factor 19 (FGF19), which is a ligand of FGFR4, the main reaction pathway of lenvatinib, and Kelch-like ECH-associated protein 1 (13–15); activation of the hepatocyte growth factor/c-MET pathway (16); and high protein levels of stomatin-like protein 2 and epidermal growth factor receptor (17,18).
Few studies have investigated acquired resistance to lenvatinib. Possible mechanisms for developing acquired resistance to anti-cancer drugs include mutations of genes encoding proteins that are the targets of drugs or are present in downstream pathways of proteins inhibited by drugs as well as the activation of collateral pathways (19,20). Genetic variations associated with developing resistance to many anti-cancer drugs have also been investigated. However, the genetic variations responsible for developing acquired resistance to lenvatinib have yet to be elucidated. In 2021, Myojin et al (14) established a cell line that acquired resistance to lenvatinib from a lenvatinib-sensitive cell line, Hep3B, by continuous exposure to lenvatinib, and revealed that FGF19 overexpression restored lenvatinib susceptibility to this cell line. However, in their study, no significant changes in FGF19 protein levels were observed with the acquisition of resistance, and we cannot conclude that the reduced expression of FGF19 is the cause of acquired resistance to lenvatinib.
In order to investigate the drug resistance mechanism with the activation of alternative pathways in detail, a comprehensive analysis of proteins derived from cancer cells and identification of signaling pathways whose activity significantly changes before and after the development of resistance to anti-cancer drugs should be performed. Recently, the usefulness of a comprehensive protein analytic approach has been revealed in many studies such as the analysis of predictive markers for the efficacy of erlotinib in non-small cell lung cancer (21–25). In the present study, we established a lenvatinib-resistant HCC cell line, JHH-7_LR, from a lenvatinib-sensitive HCC cell line by continuous exposure to lenvatinib. Then, to clarify the factors related to the acquisition of lenvatinib resistance, we identified the proteins with significant changes in their expression before and after the development of resistance. In addition, we analyzed the signaling pathways composed by the proteins with significant changes in expression. Furthermore, we assessed the impact of inhibiting those signaling pathways on the efficacy of lenvatinib in the JHH-7_LR cell line.
Materials and methods
Reagents
A lenvatinib-sensitive human hepatocellular carcinoma cell line, JHH-7, was purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). William's E Medium and GlutaMAX™ Supplement were purchased from Thermo Fisher Scientific K.K. Lenvatinib and dasatinib were purchased from LC Laboratories and Cayman Chemical, respectively. A Cell Proliferation Kit I was purchased from Merck. The other cell culture and sample preparation reagents were purchased from Fujifilm Wako Pure Chemical Corporation. All other reagents were obtained from commercial sources, and those used for proteomic analysis were graded for high-performance liquid chromatography or liquid chromatography-mass spectrometry (LC/MS).
Establishment of a lenvatinib-acquired resistance cell line
JHH-7 cells were cultured in a humidified incubator at 37°C with 5% CO2. A lenvatinib-acquired resistance cell line (JHH-7_LR) was generated upon continuous exposure of JHH-7 cells to lenvatinib; the exposure concentration started at 0.5 µmol/l and was increased to 40 µmol/l for over 1 year.
The acquisition of lenvatinib resistance was verified by a cytotoxicity assay using a Cell Proliferation Kit I. The cells were plated in a 96-well plate at 1.0×103 cells/well in medium supplemented with 10% fetal bovine serum. After overnight incubation, the medium was changed to 100 µl fresh medium containing 0 or 0.005–50 µmol/l of lenvatinib and cultured for another 5 days. Then, 10 µl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution (5 mg/ml) was added to each well. After incubation for 4 h, 10% SDS solution was added to stop the reaction, and absorbance at 550 and 690 nm of each well was measured immediately. The growth inhibition ratio (Ir) was calculated by equation 1:
Ir = (AbsLB - AbsS)/(AbsLB - AbsM) (1)
where AbsLB, AbsS, and AbsM represent the values of Abs550-Abs690 nm of the sample incubated without lenvatinib, sample incubated with lenvatinib, and medium, respectively.
The calculated Ir and lenvatinib concentrations were applied to equation 2, and the nonlinear least-squares MULTI program was used to estimate the inhibition of 50% cancer cell growth (IC50):
Ir = 100 x Imax/(1 + Exp (-S x (C - IC50) (2)
where S, C, and Imax represent the sigmoid variable, lenvatinib concentration, and maximum inhibition rate, respectively.
Proteome analysis
Cytoplasmic proteins of each cell line were extracted using a Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies), and the concentrations of the extracted proteins were measured using a DC™ Protein Assay Kit (Bio-Rad Laboratories). The extracts were diluted to 0.25 mg/ml with phosphate-buffered saline, and 100 µl of the diluted samples was incubated at 37°C for 60 min with 100 mg urea and 10 µl of 650 mmol/l dithiothreitol in 8 mol/l urea/0.5 mol/l Tris HCL (pH 8.5). After spiking with 10 µl of 1 mol/l iodoacetamide in 8 mol/l urea/0.5 mol/l Tris HCL (pH 8.5), the samples were incubated at 37°C for 30 min. Subsequently, to digest the proteins, 12 µl of 1 mg/ml trypsin in 20 mmol/l acetic acid was added and incubated at 37°C for 3 h. Finally, the trypsinized samples were desalted and concentrated using an ISOLUTE C18 (EC) column (Biotage Japan Ltd., Tokyo, Japan) and applied to LC/MS.
LC/MS analysis was performed with an EksigentNanoLC 425 coupled to a Triple TOF 6600 (AB Sciex). First, 10 µl of the sample was loaded on a trap column (Acclaim PepMap 100 C18, 5 µm, 0.2 mm I.D. ×10 mm; Thermo Fisher Scientific K.K.) and then separated using an analytical column (Acclaim PepMap 100 C18, 3 µm, 0.075 mm I.D. ×250 mm; Thermo Fisher Scientific K.K.) with a gradient from 2 to 32% solvent B at a flow rate of 300 nl/min for 120 min (solvent A: 0.1% formic acid in water; solvent B: 0.1% formic acid in acetonitrile). Ion source parameters were set as follows: ion source voltage, 2,350 V; ion source gas (GS1 and GS2), 5 and 0; interface heater temperature, 150°C; declustering potential, 80 V.
Sequential window acquisition of all theoretical fragment ion spectra (SWATH) was acquired using the 100 SWATH variable window method (AB Sciex Pte. Ltd.) from m/z 100–1,800 with each 25 ms accumulation time. Library samples were prepared by mixing all samples to be analyzed equally and measured three times by data-dependent acquisition, selecting the top 25 highest peaks. UniProt (uniprot_sprot.fasta) was used for library data preparation. The ion chromatograms were analyzed for five transitions per peptide and five peptides per protein and then processed with a peptide confidence threshold of 99% and a false discovery rate of <1%. ProteinPilot ver. 5.0.1, SWATH Acquisition Micro App ver. 2.0 (AB Sciex), and Peak View ver. 2.2 (AB Sciex) were used for analysis.
Pathway analysis
The differences in protein expression levels between the lenvatinib-sensitive and -resistant cell lines were analyzed by a t-test, and the P-value was adjusted by the Benjamini-Hochberg method (q-value). Using the UniProt code, expression level ratio, P-value, and q-value of proteins with q<0.05 and those with a change of expression in the acquired resistance cell line of more than 2- or 0.5-fold compared to the original cell line, core analysis was performed with bioinformatics software (Ingenuity Pathway Analysis ver. Winter 2021; Qiagen, Venlo, Netherlands).
Effect of dasatinib on lenvatinib sensitivity
The JHH-7_LR and JHH-7 cell lines were plated in 96-well plates at 1.0×103 cells/well in medium supplemented with 10% fetal bovine serum. After overnight incubation, the medium was changed to 100 µl fresh medium containing lenvatinib (0 or 0.005–50 µmol/l) with or without dasatinib (2.5 or 5 µmol/l). After incubation for another 5 days, IC50 was estimated as described earlier.
Statistical analysis
Statistical analysis was performed on the difference between two groups with unpaired t-test. For multiple comparisons, one-way ANOVA was first performed, followed by Dunnett's test. Multiple comparisons were controlled for the false discovery rate based on the Benjamini-Hochberg method. SPSS ver. 28 (IBM Japan Co., Ltd.) was used for statistical analysis.
Results
Establishment of a lenvatinib-resistant cell line
We established a lenvatinib-resistant cell line, JHH-7_LR, after exposing the parental HCC cell line JHH-7 to step-wise increasing concentrations of lenvatinib up to 40 µmol/l. Lenvatinib inhibited the proliferation of both the JHH-7 and JHH-7_LR HCC cells in a concentration-dependent manner. The IC50 of JHH-7_LR cells to lenvatinib was approximately 70 times higher than that of JHH-7 cells (41.3 and 0.56 µmol/l, respectively) (Fig. 1).
Figure 1.
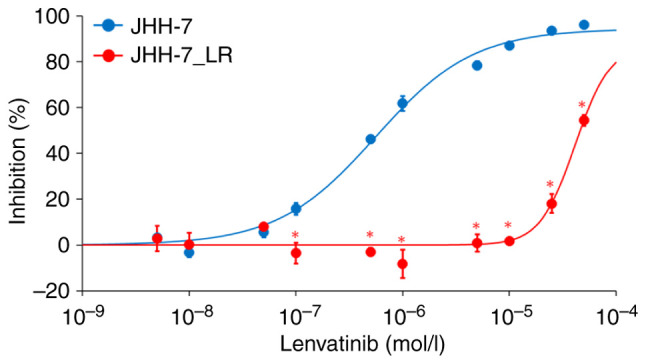
Inhibition of JHH-7 and JHH-7_LR cell proliferation by lenvatinib. Each symbol and vertical bar represent the mean and standard deviation, respectively (n=3). Solid lines are the fitted lines using the least squares method. *P<0.01 vs. JHH-7 cells.
Proteome analysis
We performed SWATH analysis to investigate the changes in protein expression levels associated with lenvatinib resistance. SWATH analysis detected 4,013 peptides and identified 1,323 proteins. Among them, 1,321 proteins for which quantitative values were obtained in all samples were analyzed. Along with the development of resistance, the expression levels of 115 proteins were more than doubled and significantly increased (q<0.05). However, the expression levels of 152 proteins were reduced by half and significantly decreased (q<0.05) (Fig. 2).
Figure 2.
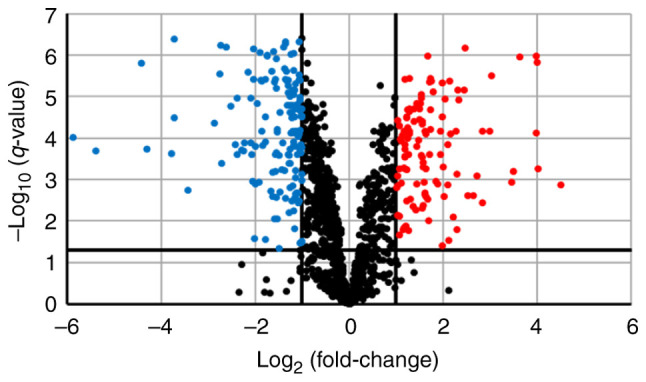
Fluctuations in protein expression levels associated with the development of resistance. Volcano plot showing the relationship between the magnitude of protein expression change [log2 (fold-change); x-axis] and statistical significance of this change [-log10 (q-value); y-axis] in a comparison of JHH-7 and JHH-7_LR cells. Red dots (n=115) indicate proteins that were significantly increased by a factor of ≥2 (q<0.05). Blue dots (n=152) indicate proteins that were significantly decreased by a factor of ≥2 (q<0.05).
Pathway analysis
Pathway analysis was performed using 267 proteins with substantial changes in expression with the acquisition of resistance to lenvatinib as well as 124 signaling pathways were detected in which activity was affected by changes in the expression of those proteins (Fig. 3). For the proteins involved in the top 10 signaling pathways with the highest -log(P-value), calculated based on the number and proportion of proteins contained in the signaling pathway, the top 10 proteins with the most significant change of expression with the acquisition of resistance to lenvatinib were extracted as lenvatinib resistance-related protein candidates (Tables I and II). Among those proteins, c-SRC, Ras-related C3 botulinum toxin substrate 1, nucleoside diphosphate kinase A, serine hydroxymethyltransferase (cytosolic), and serine hydroxymethyltransferase (mitochondrial) were found to have enzymatic activity and are involved in multiple pathways. The changes in their expression with the acquisition of resistance were 5.44-, 0.45-, 0.34-, 0.42-, and 2.0-fold, respectively. The changes in cadherin-2, NAD(P)H dehydrogenase [quinone] 1, and Filamin-A expressions were more significant than those in c-SRC expression upon acquisition of resistance. However, those proteins were contained in only one signaling pathway extracted by pathway analysis.
Figure 3.

Top significantly enriched canonical pathways determined using Ingenuity Pathway Analysis bioinformatics software. Columns with diagonal lines indicate the canonical pathways that include c-SRC. EIF2, eukaryotic initiation factor 2; ILK, integrin linked kinase; NRF2, nuclear factor erythroid 2-related factor 2.
Table I.
Top 10 proteins exhibiting increased expression following the development of lenvatinib resistance.
| Expression levelsa | Ratio of expression levelsb | ||||
|---|---|---|---|---|---|
|
|
|
||||
| UniProt code | Name | JHH-7 | JHH-7_LR | JHH-7_LR/JHH-7 | P-valuec |
| P19022 | Cadherin-2 | 13,995 | 222,657 | 15.91 | 2.4×10−8 |
| P12931 | Proto-oncogene tyrosine-protein kinase Src | 103,614 | 563,152 | 5.44 | 3.2×10−7 |
| P04179 | Superoxide dismutase [Mn], mitochondrial | 45,934 | 226,645 | 4.93 | 3.1×10−7 |
| P07305 | Histone H1.0 | 21,414 | 105,396 | 4.92 | 8.5×10−3 |
| P78330 | Phosphoserine phosphatase | 3,982,655 | 13,190,082 | 3.31 | 10.0×10−8 |
| O43707 | α-actinin-4 | 1,885,027 | 5,750,919 | 3.05 | 7.7×10−6 |
| P35221 | Catenin α-1 | 51,001 | 150,172 | 2.94 | 2.1×10−3 |
| P35222 | Catenin β-1 | 26,094 | 76,507 | 2.93 | 4.8×10−6 |
| Q14651 | Plastin-1 | 97,393 | 274,947 | 2.82 | 3.5×10−6 |
| P08263 | Glutathione S-transferase A1 | 1,866,497 | 5,041,783 | 2.70 | 2.9×10−5 |
Average total area sums from the peak signals derived from all quantitated peptides in a protein across three replicates.
Ratio of the average total area sums from JHH-7 and JHH-7_LR samples.
P-value was calculated using an unpaired t-test.
Table II.
Top 10 proteins exhibiting decreased expression following the development of lenvatinib resistance.
| Expression levelsa | Ratio of expression levelsb | ||||
|---|---|---|---|---|---|
|
|
|
||||
| UniProt code | Name | JHH-7 | JHH-7_LR | JHH-7_LR/JHH-7 | P-valuec |
| P15559 | NAD(P)H dehydrogenase [quinone] 1 | 1,145,340 | 27,193 | 0.02 | 4.1×10−5 |
| P21333 | Filamin-A | 1,611,699 | 221,586 | 0.14 | 4.4×10−6 |
| Q99615 | DnaJ homolog subfamily C member 7 | 323,245 | 93,505 | 0.29 | 2.8×10−6 |
| P11586 | C-1-tetrahydrofolate synthase, cytoplasmic | 2,096,222 | 627,168 | 0.30 | 1.6×10−8 |
| Q9Y3U8 | 60S ribosomal protein L36 | 405,738 | 121,794 | 0.30 | 1.2×10−8 |
| P35579 | Myosin-9 | 773,819 | 251,904 | 0.33 | 9.7×10−4 |
| P18206 | Vinculin | 1,547,743 | 505,010 | 0.33 | 7.4×10−9 |
| Q13885 | Tubulin beta-2A chain | 491,799 | 165,287 | 0.34 | 1.7×10−4 |
| Q06830 | Peroxiredoxin-1 | 3,870,952 | 1,313,386 | 0.34 | 5.5×10−5 |
| P15531 | Nucleoside diphosphate kinase A | 276,722 | 94,293 | 0.34 | 2.7×10−7 |
Average total area sums from the peak signals derived from all quantitated peptides in a protein across three replicates.
Ratio of the average total area sums from JHH-7 and JHH-7_LR samples.
P-value was calculated using an unpaired t-test.
Effect of dasatinib on lenvatinib sensitivity
We examined the involvement of c-SRC in lenvatinib sensitivity. The concomitant use of dasatinib, a c-SRC inhibitor, increased the sensitivity of JHH-7_LR cells to lenvatinib in a dose-dependent manner. However, dasatinib had no effect on the lenvatinib sensitivity of JHH-7 cells (Fig. 4).
Figure 4.
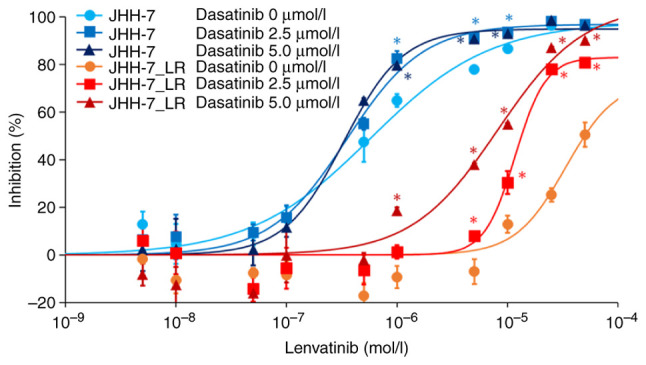
Inhibition of c-SRC activity increases the sensitivity of JHH-7_LR cells to lenvatinib. Symbols and vertical bars represent the mean and standard deviation, respectively (n=3). Solid lines are the fitted lines using the least squares method. *P<0.01 vs. 0 µmol/l dasatinib for each cell line.
Discussion
In the present study, we found that multiple signaling pathways were associated with the development of resistance to lenvatinib. Among them, the expression levels of many proteins that constitute the signaling pathways linked with c-SRC changed significantly with the acquisition of resistance to lenvatinib, and the expression level of c-SRC increased by approximately 5-fold with the acquisition of resistance.
c-SRC is a non-receptor tyrosine kinase encoded by SRC, which is homologous to the Rous sarcoma virus proto-oncogene γ-Src (26–28). c-SRC is located downstream of receptor tyrosine kinases such as epidermal growth factor receptor (29–31), FGFR (32), PDGFR (33), HER2/neu (29), and c-MET (34), and it is involved in cell proliferation and metastasis in various carcinomas (35–37) by activation of the RAS/RAF/MEK/ERK pathway (38), PI3K/Akt/mTOR pathway (39), integrin/FAK pathway (40,41), and JAK/STAT pathway (39,40). In addition, c-SRC is reported to be involved in the acquisition of resistance to other tyrosine kinase inhibitors. Yoshida et al (42) found that multiple phosphorylation reactions change with the acquisition of resistance, and c-SRC was identified as a factor related to these phosphorylation reactions in vitro using a gefitinib-resistant cell line, established from a gefitinib-sensitive non-small cell lung cancer cell line, PC-9. In our data, changes in cadherin-2 expression were more significant than those in c-SRC expression upon acquisition of resistance. However, it has been reported that the expression of N-cadherin encoded by CDH2 is affected by c-SRC (43), and we speculated that the variation in its expression level was affected by the variation in c-SRC activity.
Given that our data suggested the possibility that acquired resistance to lenvatinib is induced by enhancement of c-SRC-related pathways, we assessed the effect of a c-SRC inhibitor on the sensitivity of JHH-7_LR cells to lenvatinib. Of the drugs used in the clinical setting, dasatinib, which is widely used for treating chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia, is known to inhibit c-SRC activity (44,45). Dasatinib inhibits SRC family kinases, BCR-ABL, PDGFRβ, and c-Kit among tyrosine kinases and is reported as a potent inhibitor of c-SRC (IC50: 0.5 nmol/l) (46). In our study, dasatinib increased the sensitivity of JHH-7_LR cells to lenvatinib in a dose-dependent manner. Similarly, Murakami et al (47) reported that dasatinib partially restored the afatinib sensitivity of afatinib-resistant non-small cell lung cancer cell lines established by continuous exposure to afatinib. However, although the concentration of dasatinib in the medium was set at sufficiently high levels (2.5 and 5.0 µmol/l), which were more than 10-fold higher than the IC50 value of dasatinib against c-SRC (0.5 nmol/l) and the maximum blood concentration of dasatinib (approximately 0.2 µmol/l) after continuous oral administration, dasatinib did not completely restore the sensitivity of acquired drug resistance cell lines to lenvatinib and afatinib. There are two possible reasons why dasatinib is unable to fully restore lenvatinib sensitivity. First, because the concentration of dasatinib in the medium required for inhibiting c-SRC phosphorylation varies depending on the cell line (48), it may not have been able to inhibit c-SRC in JHH-7_LR cells sufficiently. For this point, evaluating the impact of a more potent c-SRC inhibitor on the lenvatinib sensitivity of JHH-7_LR cells is required. Next, it is possible that pathways other than the c-SRC-related pathways simultaneously influence the development of resistance to lenvatinib. For this point, the involvement of other pathways needs to be studied in more detail.
This study has some limitations. First, in the proteome analysis, we were unable to detect changes in FGF19 expression, which has been reported as one of the factors affecting the development of resistance in previous studies, nor were we able to assess its effects on the lenvatinib sensitivity of the JHH-7_LR cell line. On the other hand, the expression level of N-cadherin, which has been reported to be decreased by knocking down FGF19 in hepatocellular carcinoma-derived cell lines (49), was confirmed to increase with the development of resistance in our study. In addition, Myojin et al (14) also reported that protein levels of FGF19 did not change with the development of resistance, unlike the change in its mRNA level. Thus, although we could not detect FGF19 in our study, we considered that the decrease in FGF19 expression did not cause the development of resistance to lenvatinib. Next, we used only one type of HCC cell line; therefore, conducting similar studies using multiple cell lines and clinical samples is necessary. In the future, we aim to construct a lenvatinib-resistant cell line derived from other multi-cell lines. In addition, we should assess the contribution of the change in c-SRC expression to developing resistance to lenvatinib in the clinical setting in detail. Thirdly, we could not analyze the change of expression of proteins, which could not be detected by comprehensive protein expression analysis using LC-MS/MS. For this point, additional analysis, such as comprehensive mRNA analysis, could provide helpful information. In the subsequent study, we will analyze this point in detail and elucidate the factors that cause resistance to lenvatinib in more detail.
In conclusion, the activation of c-SRC may be an essential factor in the development of lenvatinib resistance induced by long-term exposure to lenvatinib. We also found that exposure to dasatinib, a c-SRC inhibitor, could partially eliminate the resistance of the JHH-7_LR cell line to lenvatinib. We believe that detailed examinations of the mechanism of c-SRC activation associated with lenvatinib resistance will lead to the development of a more effective method for overcoming lenvatinib resistance.
Acknowledgements
Not applicable.
Funding Statement
This work was supported by JSPS KAKENHI (grant no. JP18K06743).
Availability of data and materials
The raw mass spectrometry data and peptide/protein identification results generated and/or analyzed during the current study are available in the jPOST repository, https://repository.jpostdb.org/entry/JPST002307. The other datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
KY, TA, DN, HY and AN conceived and designed the experiments. MT performed the experiments. MT and TA analyzed the data and wrote the paper. MT and TA confirm the authenticity of all the raw data. KY, TA, DN, HY and AN revised the paper. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C, Al-Raddadi R, Alvis-Guzman N, Amoako Y, Artaman A, et al. The burden of primary liver cancer and underlying etiologies From 1990 to 2015 at the global, regional, and national level: Results from the global burden of disease study 2015. JAMA Oncol. 2017;3:1683–1691. doi: 10.1001/jamaoncol.2017.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park JW, Chen M, Colombo M, Roberts LR, Schwartz M, Chen PJ, Kudo M, Johnson P, Wagner S, Orsini LS, Sherman M. Global patterns of hepatocellular carcinoma management from diagnosis to death. Liver Int. 2015;35:2155–2166. doi: 10.1111/liv.12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kokudo N, Takemura N, Hasegawa K, Takayama T, Kubo S, Shimada M, Nagano H, Hatano E, Izumi N, Kaneko S, et al. Clinical practice guidelines for hepatocellular carcinoma: The Japan society of hepatology 2017 (4th JSH-HCC guidelines) 2019 update. Hepatol Res. 2019;49:1109–1113. doi: 10.1111/hepr.13411. [DOI] [PubMed] [Google Scholar]
- 6.Tabrizian P, Jibara G, Shrager B, Schwartz M, Roayaie S. Recurrence of hepatocellular cancer after resection: Patterns, treatments, and prognosis. Ann Surg. 2015;261:947–955. doi: 10.1097/SLA.0000000000000710. [DOI] [PubMed] [Google Scholar]
- 7.Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 8.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 9.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, et al. Efficacy and safety of sorafenib in patients in the Asia-pacific region with. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 10.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 11.Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, Uenaka T, Asada M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–671. doi: 10.1002/ijc.23131. [DOI] [PubMed] [Google Scholar]
- 12.Ao J, Chiba T, Shibata S, Kurosugi A, Qiang N, Ma Y, Kan M, Iwanaga T, Sakuma T, Kanzaki H, et al. Acquisition of mesenchymal-like phenotypes and overproduction of angiogenic factors in lenvatinib-resistant hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2021;549:171–178. doi: 10.1016/j.bbrc.2021.02.097. [DOI] [PubMed] [Google Scholar]
- 13.Matsuki M, Hoshi T, Yamamoto Y, Ikemori-Kawada M, Minoshima Y, Funahashi Y, Matsui J. Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med. 2018;7:2641–2653. doi: 10.1002/cam4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myojin Y, Kodama T, Maesaka K, Motooka D, Sato Y, Tanaka S, Abe Y, Ohkawa K, Mita E, Hayashi Y, et al. ST6GAL1 is a novel serum biomarker for lenvatinib-susceptible FGF19-driven hepatocellular carcinoma. Clin Cancer Res. 2021;27:1150–1161. doi: 10.1158/1078-0432.CCR-20-3382. [DOI] [PubMed] [Google Scholar]
- 15.Zheng A, Chevalier N, Calderoni M, Dubuis G, Dormond O, Ziros PG, Sykiotis GP, Widmann C. CRISPR/Cas9 genome-wide screening identifies KEAP1 as a sorafenib, lenvatinib, and regorafenib sensitivity gene in hepatocellular carcinoma. Oncotarget. 2019;10:7058–7070. doi: 10.18632/oncotarget.27361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol. 2020;37:24. doi: 10.1007/s12032-020-01350-4. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Y, Huang C, Lu L, Yu K, Zhao J, Chen M, Liu L, Sun Q, Lin Z, Zheng J, et al. STOML2 potentiates metastasis of hepatocellular carcinoma by promoting PINK1-mediated mitophagy and regulates sensitivity to lenvatinib. J Hematol Oncol. 2021;14:16. doi: 10.1186/s13045-020-01029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin H, Shi Y, Lv Y, Yuan S, Ramirez CFA, Lieftink C, Wang L, Wang S, Wang C, Dias MH, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595:730–734. doi: 10.1038/s41586-021-03741-7. [DOI] [PubMed] [Google Scholar]
- 19.Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee N, Bivona TG. Polytherapy and targeted cancer drug resistance. Trends Cancer. 2019;5:170–182. doi: 10.1016/j.trecan.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung CH, Seeley EH, Roder H, Grigorieva J, Tsypin M, Roder J, Burtness BA, Argiris A, Forastiere AA, Gilbert J, et al. Detection of tumor epidermal growth factor receptor pathway dependence by serum mass spectrometry in cancer patients. Cancer Epidemiol Biomarkers Prev. 2010;19:358–365. doi: 10.1158/1055-9965.EPI-09-0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pitteri SJ, Amon LM, Buson TS, Zhang Y, Johnson MM, Chin A, Kennedy J, Wong CH, Zhang Q, Wang H, et al. Detection of elevated plasma levels of epidermal growth factor receptor before breast cancer diagnosis among hormone therapy users. Cancer Res. 2010;70:8598–8606. doi: 10.1158/0008-5472.CAN-10-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrisi VM, Bongarzone I, Mangia A, Cremona M, De Bortoli M, Vaghi E, Galetta D, Pastorino U, Quaranta M, Abbate I, Paradiso A. Characterization of a serum protein pattern from NSCLC patients treated with Gefitinib. Clin Biochem. 2011;44:936–940. doi: 10.1016/j.clinbiochem.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Lazzari C, Spreafico A, Bachi A, Roder H, Floriani I, Garavaglia D, Cattaneo A, Grigorieva J, Viganò MG, Sorlini C, et al. Changes in plasma mass-spectral profile in course of treatment of non-small cell lung cancer patients with epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Oncol. 2012;7:40–48. doi: 10.1097/JTO.0b013e3182307f17. [DOI] [PubMed] [Google Scholar]
- 25.Gregorc V, Novello S, Lazzari C, Barni S, Aieta M, Mencoboni M, Grossi F, De Pas T, de Marinis F, Bearz A, et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): A biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014;15:713–721. doi: 10.1016/S1470-2045(14)70162-7. [DOI] [PubMed] [Google Scholar]
- 26.Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. 1911;13:397–411. doi: 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 28.Czernilofsky AP, Levinson AD, Varmus HE, Bishop JM, Tischer E, Goodman HM. Nucleotide sequence of an avian sarcoma virus oncogene (src) and proposed amino acid sequence for gene product. Nature. 1980;287:198–203. doi: 10.1038/287198a0. [DOI] [PubMed] [Google Scholar]
- 29.Luttrell DK, Lee A, Lansing TJ, Crosby RM, Jung KD, Willard D, Luther M, Rodriguez M, Berman J, Gilmer TM. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc Natl Acad Sci USA. 1994;91:83–87. doi: 10.1073/pnas.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao W, Irby R, Coppola D, Fu L, Wloch M, Turner J, Yu H, Garcia R, Jove R, Yeatman TJ. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene. 1997;15:3083–3090. doi: 10.1038/sj.onc.1201496. [DOI] [PubMed] [Google Scholar]
- 32.LaVallee TM, Prudovsky IA, McMahon GA, Hu X, Maciag T. Activation of the MAP kinase pathway by FGF-1 correlates with cell proliferation induction while activation of the Src pathway correlates with migration. J Cell Biol. 1998;141:1647–1658. doi: 10.1083/jcb.141.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Courtneidge SA, Fumagalli S, Koegl M, Superti-Furga G, Twamley-Stein GM. The Src family of protein tyrosine kinases: Regulation and functions. Dev Suppl. 1993:57–64. [PubMed] [Google Scholar]
- 34.Rahimi N, Hung W, Tremblay E, Saulnier R, Elliott B. c-Src kinase activity is required for hepatocyte growth factor-induced motility and anchorage-independent growth of mammary carcinoma cells. J Biol Chem. 1998;273:33714–33721. doi: 10.1074/jbc.273.50.33714. [DOI] [PubMed] [Google Scholar]
- 35.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–5642. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 36.Zhao R, Wu Y, Wang T, Zhang Y, Kong D, Zhang L, Li X, Wang G, Jin Y, Jin X, Zhang F. Elevated Src expression associated with hepatocellular carcinoma metastasis in northern Chinese patients. Oncol Lett. 2015;10:3026–3034. doi: 10.3892/ol.2015.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito Y, Kawakatsu H, Takeda T, Sakon M, Nagano H, Sakai T, Miyoshi E, Noda K, Tsujimoto M, Wakasa K, et al. Activation of c-Src gene product in hepatocellular carcinoma is highly correlated with the indices of early stage phenotype. J Hepatol. 2001;35:68–73. doi: 10.1016/S0168-8278(01)00077-0. [DOI] [PubMed] [Google Scholar]
- 38.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem. 2000;275:27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 39.Yu H, Jove R. The STATs of cancer-new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 40.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 41.Lau GM, Lau GM, Yu GL, Gelman IH, Gutowski A, Hangauer D, Fang JW. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci. 2009;54:1465–1475. doi: 10.1007/s10620-008-0519-0. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, Fang B, Koomen J, Rawal B, Fisher KJ, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res. 2014;20:4059–4074. doi: 10.1158/1078-0432.CCR-13-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Debiais F, Lemonnier J, Hay E, Delannoy P, Caverzasio J, Marie PJ. Fibroblast growth factor-2 (FGF-2) increases N-cadherin expression through protein kinase C and Src-kinase pathways in human calvaria osteoblasts. J Cell Biochem. 2001;81:68–81. doi: 10.1002/1097-4644(20010401)81:1<68::AID-JCB1024>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 44.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 45.Ottmann O, Dombret H, Martinelli G, Simonsson B, Guilhot F, Larson RA, Rege-Cambrin G, Radich J, Hochhaus A, Apanovitch AM, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: Interim results of a phase 2 study. Blood. 2007;110:2309–2315. doi: 10.1182/blood-2007-02-073528. [DOI] [PubMed] [Google Scholar]
- 46.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino) thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 47.Murakami Y, Sonoda K, Abe H, Watari K, Kusakabe D, Azuma K, Kawahara A, Akiba J, Oneyama C, Pachter JA, et al. The activation of SRC family kinases and focal adhesion kinase with the loss of the amplified, mutated EGFR gene contributes to the resistance to afatinib, erlotinib and osimertinib in human lung cancer cells. Oncotarget. 2017;8:70736–70751. doi: 10.18632/oncotarget.19982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang AY, Wang M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer. 2013;13:267. doi: 10.1186/1471-2407-13-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao H, Lv F, Liang G, Huang X, Wu G, Zhang W, Yu L, Shi L, Teng Y. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3β/β-catenin signaling cascade via FGFR4 activation. Oncotarget. 2016;7:13575–13586. doi: 10.18632/oncotarget.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw mass spectrometry data and peptide/protein identification results generated and/or analyzed during the current study are available in the jPOST repository, https://repository.jpostdb.org/entry/JPST002307. The other datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.