

Gerstmann‐Sträussler‐Scheinker disease (GSS) is a rare genetic prionopathy caused by pathogenic variations in the prion‐related protein gene (PRNP). Its clinical manifestations typically include progressive cerebellar ataxia, pyramidal signs, and dementia. However, GSS displays significant phenotypic variability. 1 , 2 In this article, we describe three patients with early‐onset GSS, characterized by juvenile variation mimicking an anticipation phenomenon.
We conducted whole exome sequencing (WES) in a large series of undetermined ataxia cases. Among them, 32 patients with GSS were clinically and genetically defined. We identified a missense variant in Exon 2 at codon 102 of the PRNP gene (c.305C > T(p.Pro102Leu)). This variant is absent in population databases (gnomAD), but has been observed in individuals with GSS syndrome, being one of the three most frequent variants—which account for ~85% of cases. 3
The mean age of onset for GSS associated with the P102L variant is 53.7 ± 10.6. 3 However, within our cohort of 32 GSS patients, we encountered three cases of exceptionally early‐onset GSS at ages 15, 17, and 18 (Video 1 and Figs. S1 and S2). Such cases demonstrate an anticipation‐like effect reminiscent of diseases caused by expansion‐repeat mutations. In parallel with other neurodegenerative conditions, such as juvenile‐onset Parkinson's disease, where genetic susceptibility factors can influence an early age of onset (before 20 years), we postulate that similar factors could contribute to the earlier presentation in these patients. Hence, we suggest the term juvenile‐onset GSS for such instances.
Video 1.
Case 1: At the age of 21 (onset at 17), the patient exhibits appendicular ataxia, extensor plantar reflex, and an ataxic‐spastic gait. Case 3: At the age of 19 (onset at 18), the patient presents with mild gait impairment and ataxia that is more pronounced in the lower limbs than in the upper limbs.
Gait impairment was the first symptom noted in all patients, followed by lower limb paresthesia and pyramidal signs (Table 1). We did not find any predictor in our data for the early presentation. It was not influenced by parents' age of onset, maternal or paternal inheritance, first symptom, or disease progression.
TABLE 1.
Clinical data of juvenile‐onset Gerstmann‐Sträussler‐Scheinker cases
| Case 1 | Case 2 b | Case 3 | |
|---|---|---|---|
| Gender | F | M | F |
| Age at onset | 17 | 15 | 18 |
| Age at death | N/A | 20 | N/A |
| Inheritance | Paternal | Paternal | Maternal |
| Parent's age at onset | 31 | 36 | 26 |
| Parent's age at death | 33 | 43 | 31 |
| Mutation | c.305C > T (p.Pro102Leu) | c.305C > T (p.Pro102Leu) | c.305C > T (p.Pro102Leu) |
| Codon 129 | M/M | Unknown | Unknown |
| Presenting symptom | Gait impairment | Gait impairment | Gait impairment |
| Hearing loss | − | + | + |
| Lower limb paresthesia | + | + | + |
| Pain | + | + | − |
| Depression | + | + | − |
| Anxiety | + | − | + |
| Hallucinations | − | + | − |
| Dementia (later) | − | + | − |
| Weakness | + | + | + |
| Spasticity | + | Unknown | + |
| Appendicular ataxia | + | Unknown | − |
| Axial ataxia | + | Unknown | + |
| Slurred speech | + | + | + |
| Age at cognitive assessment (years since onset) | 23 (6) | N/A | 22 (4) |
| MMSE /30 | 24 | N/A | 26 |
| ACE‐R /100 | 67 a | N/A | 81 |
| Attention and orientation /18 | 15 | N/A | 14 |
| Memory /26 | 14 | N/A | 19 |
| Fluency /14 | 8 | N/A | 7 |
| Language /26 | 22 | N/A | 25 |
| Visuospatial /16 | 8 a | N/A | 16 |
Note: Clinical features of three cases of juvenile‐onset Gerstmann‐Sträussler‐Scheinker.
Abbreviations: F, female; M, male; N/A, not available; M/M, homozygosity for methionine; MMSE, Mini‐Mental State Examination; ACE‐R, Addenbrooke's Cognitive Examination‐Revised.
Due to ataxia, some cognitive tasks might not be properly evaluated, underestimating patient cognition in tasks that require fine motor skills such as writing and drawing.
Data collected retrospectively from medical charts. No information on neurological examination was available, and genetic variants were inferred post‐mortem from the analysis of DNA from relatives.
Predicting the age of onset for genetic prion diseases remains a challenge. In GSS, the increased lifetime risk is not associated with a specific age of onset, but rather with a moderate risk accumulating over an individual's lifespan. Previous large‐scale statistical analyses failed to identify significant determinants of the age of onset for P102L GSS. 2 , 3 Similarly, the established associations between codon 129 polymorphisms and phenotypic presentation and disease duration in Creutzfeldt‐Jakob disease do not seem to correlate with disease onset or duration in P102L GSS. This lack of correlation extends to factors such as sex, parental age at onset, and parental age at the child's birth. 3
The factors that potentially influence the age of onset for genetic prion diseases, as well as early biomarkers, have gained increased attention, particularly because of preclinical studies demonstrating improved survival rates in prion protein‐infected mice with early treatment compared to delayed treatment. 4 , 5 Determining the optimal timing for intervention in presymptomatic individuals poses a major challenge in conducting therapeutic clinical trials for prion diseases. 3
These cases contribute to our understanding of the phenotypic variability in GSS. Further investigations are necessary to elucidate the underlying mechanisms influencing juvenile‐onset GSS. Advancing our understanding of juvenile‐onset GSS will enhance diagnostic accuracy, facilitate early approach, and ultimately improve patient outcomes in GSS and related prion diseases.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique; (4) Table: A. Conception B. Review and Critique; (5) Video: A. Recording B. Editing C. Review and Critique; (6) Figure: A. Conception B. Review and Critique.
T.Y.T.S.: 1A, 1B, 1C, 2A, 2B, 3A, 5A, 6A
M.V.O.M.: 1A, 1B, 1C, 2A, 2B, 3A, 4A, 5A, 5B, 6A
E.Z.: 1A, 1B, 1C, 2A, 2B, 2C, 3B, 4B, 5C, 6B
J.L.P.: 1A, 1B, 1C, 2A, 2B, 2C, 3B, 4B, 5C, 6B
O.G.P.B.: 1A, 1B, 1C, 2A, 2B, 2C, 3B, 4B, 5C, 6B
Disclosures
Ethical Compliance Statement: This study was approved by the ethics committee number 68—Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, HCFM/USP. Informed consent was obtained. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of interest: The authors declare that there are no conflicts of interest relevant to this work. Financial disclosure and funding support: No specific funding was received for this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Supporting information
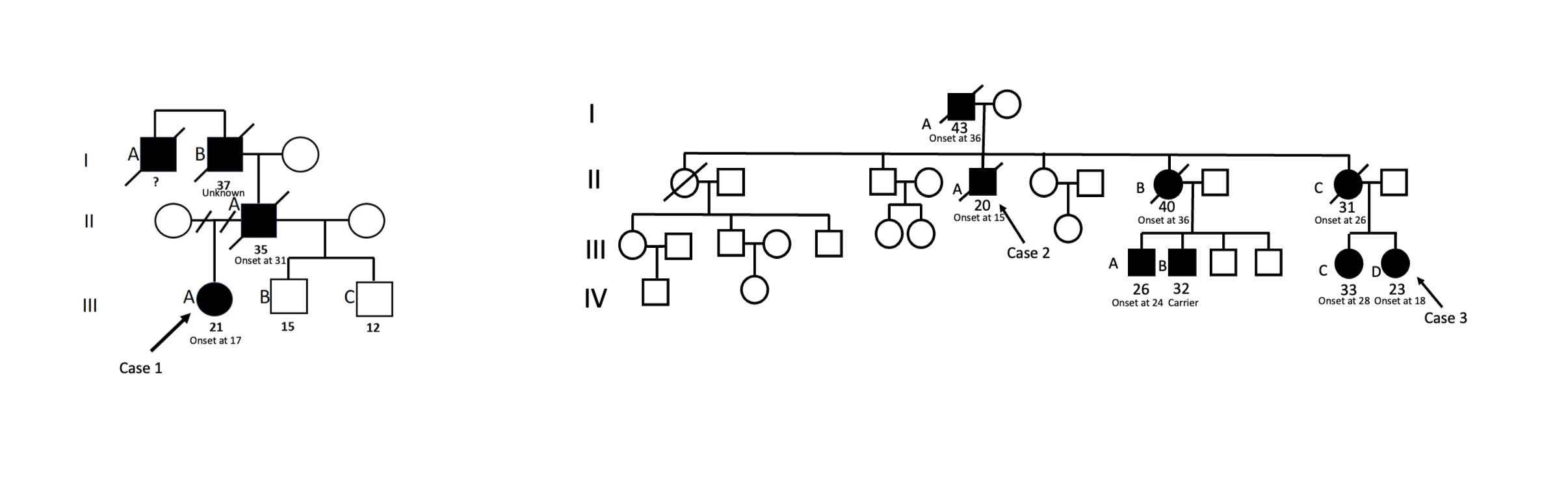
FIG. S1. Genealogical tree of the patients reported with age of onset: Case 1. The patient (III‐A), her father (II‐A), her grandfather(I‐B), and her grand‐uncle(I‐A) presented complex neurological features, including cerebellar ataxia, parkinsonism, pyramidal signs, and cognitive decline.The patient from case 2 (II‐A), his father (I‐A), and his two sisters (II‐B and II‐C) presented complex neurological features, including cerebellar ataxia, parkinsonism, pyramidal signs, and cognitive decline. Her niece, the patient of case 3 (III‐D), presented her first symptoms at 18 years old. Three other nephews and nieces (III‐A, III‐B, III‐C) were affected. We can see a mimic of an anticipation phenomenon within these kindred.
{kind=link}
FIG. S2. Case 1: Brain MRI at the age of 17. The image is typically normal early in the course of the disease, with cortical and/or cerebellar atrophy often appearing with disease progression.
{kind=link}
Relevant disclosures and conflict of interest are listed at the end of this article.
References
- 1. Takada LT, Kim MO, Cleveland RW, et al. Genetic prion disease: experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet 2017;174:39–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tesar A, Matej R, Kukal J, et al. Clinical variability in P102L Gerstmann‐Sträussler‐Scheinker syndrome. Ann Neurol 2019;86:643–652. [DOI] [PubMed] [Google Scholar]
- 3. Minikel EV, Vallabh SM, Orseth MC, et al. Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology 2019;93:e125–e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vallabh SM, Minikel EV, Schreiber SL, Lander ES. Towards a treatment for genetic prion disease: trials and biomarkers. Lancet Neurol 2020;19(4):361–368. [DOI] [PubMed] [Google Scholar]
- 5. Giles K, Berry DB, Condello C, et al. Different 2‐aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J Pharmacol Exp Ther 2015;355:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIG. S1. Genealogical tree of the patients reported with age of onset: Case 1. The patient (III‐A), her father (II‐A), her grandfather(I‐B), and her grand‐uncle(I‐A) presented complex neurological features, including cerebellar ataxia, parkinsonism, pyramidal signs, and cognitive decline.The patient from case 2 (II‐A), his father (I‐A), and his two sisters (II‐B and II‐C) presented complex neurological features, including cerebellar ataxia, parkinsonism, pyramidal signs, and cognitive decline. Her niece, the patient of case 3 (III‐D), presented her first symptoms at 18 years old. Three other nephews and nieces (III‐A, III‐B, III‐C) were affected. We can see a mimic of an anticipation phenomenon within these kindred.
FIG. S2. Case 1: Brain MRI at the age of 17. The image is typically normal early in the course of the disease, with cortical and/or cerebellar atrophy often appearing with disease progression.