

Abstract
建立了基于超高效液相色谱-四极杆/静电场轨道阱质谱快速测定保健食品中13种β-受体阻滞剂的分析方法,并对其质谱裂解规律进行了分析研究。实验对质谱条件、色谱条件、提取溶剂及基质效应等进行了详细探究,使用甲醇对样品进行稀释提取,高速离心、超声处理,用Acquity UPLC BEH C18柱(100 mm×2.1 mm, 1.7 μm)分离,以乙腈和0.1%(v/v)甲酸水溶液作为流动相进行梯度洗脱,采用正离子模式检测,数据采集使用一级母离子全扫描和数据依赖的二级离子扫描(Full MS/dd-MS2)模式,在10 min内实现了保健食品中13种β-受体阻滞剂的分离和高精度一级、二级扫描,得到准确的质量数和准确碎片离子信息。通过方法学验证,13种β-受体阻滞剂在0.5~100 μg/L内线性关系良好,相关系数(r)均≥0.9912,检出限为1~10 μg/kg。空白加标样品中13种β-受体阻滞剂的平均回收率为75.3%~108.4%,相对标准偏差为0.9%~10.0%(n=6)。用本方法对市售的30批次保健食品进行筛查,均未检出13种β-受体阻滞剂。该方法检测速度快,准确性强,灵敏度高,可用于保健食品中β-受体阻滞剂的快速测定。
Keywords: 超高效液相色谱-四极杆/静电场轨道阱质谱法, β-受体阻滞剂, 保健食品
Abstract
A method based on ultra performance liquid chromatography-quadrupole/electrostatic field orbitrap mass spectrometry was developed for the rapid determination of 13 β-blockers in health foods. The MS fragmentation pathways of the analytes were subsequently investigated. The optimal MS conditions, extraction solvents, mobile phases, and matrix effects were evaluated in detail. The samples were extracted with methanol, filtered by high-speed centrifugation and ultrasonic treatment, and then separated on an Acquity UPLC BEH C18 column (100 mm×2.1 mm, 1.7 μm) with gradient elution using acetonitrile and 0.1% (v/v) formic acid aqueous solution as mobile phases. MS analysis was conducted in positive-ion mode, and the data were collected using full mass and data-dependent MS2 scans (Full MS/dd-MS2). The efficient separation and high-precision primary and secondary scanning of the 13 β-blockers in health foods were realized within 10 min, and accurate mass numbers and fragment-ion information were obtained. The methodological validation showed good linear relationships in the range of 0.5-100 μg/L, with correlation coefficients (r)≥0.9912. The limits of detection ranged from 1 to 10 μg/kg. When the standard substances were added to the blank sample in the amount of 10-200 μg/kg, the recoveries were in the range of 75.3%-108.4%, and the relative standard deviations ranged from 0.9% to 10.0% (n=6). The method was used to screen 30 batches of commercially available health foods, and none of the 13 β-blockers was detected. The proposed method is fast, accurate, and sensitive, and can be used for the rapid determination of β-blockers in health foods.
Keywords: ultra performance liquid chromatography-quadrupole/electrostatic field orbitrap mass spectrometry, β-blockers, health foods
当前,随着健康意识的增强,人们更倾向通过饮食来达到降血压的目的,许多声称具有降血压功能的保健食品也不断涌向市场[1,⇓-3]。然而,为了追求利益,部分不法生产者会在一些保健食品中非法添加β-受体阻滞剂类药物以吸引消费者而获利。不当食用含有β-受体阻滞剂类药物的食品将会导致心肌耗氧量和血管阻力增加,氧自由基和心肌细胞凋亡[4,5],对人体带来严重的健康风险。因此,开发一种保健食品中β-受体阻滞剂类药物的检测方法,并对市售的相关产品展开风险监测,以确保市场流通保健食品的质量安全成为当务之急。
当前,对于β-受体阻滞剂的检测,报道的方法主要有气相色谱-质谱法(GC-MS)[6,⇓-8]、毛细管电泳法[9,10]、荧光光谱法[11]、酶联免疫法[12]、液相色谱-串联质谱法(LC-MS/MS)[13,⇓,⇓,⇓,⇓-18]、高效液相色谱法(HPLC)[19,20]、解吸附电晕束离子源耦合离子阱质谱法(DCBI-MS)[21]等,这些方法覆盖的β-受体阻滞剂范围较少,且在应用于食品样品检测时都面临着基质干扰无法有效消除的难题。近年来,高分辨质谱作为一种食品中非法添加物筛查的有效手段,已在食品检测中广泛使用,但目前还少有报道使用高分辨质谱进行保健食品中β-受体阻滞剂检测的方法,难以满足实际检测的需要。
本研究通过优化色谱及质谱条件,建立了一种超高效液相色谱-四极杆/静电轨道阱高分辨质谱快速测定食品中13种β-受体阻滞剂的分析方法,并成功应用于实际样品的测定,方法快速便捷,准确率高,为监管部门对保健食品中非法添加的分析检测提供了重要支撑。
1 实验部分
1.1 仪器、试剂与材料
Q Exactive四极杆/静电场轨道阱质谱系统及Dionex UltiMate 3000超高效液相色谱系统(美国Thermo Fisher公司);分析天平XP 105 (瑞士Mettler公司); Milli-Q Advantage A10超纯水机(美国Millipore公司)。
13种β-受体阻滞剂标准物质:阿替洛尔(atenolol, CAS号29122-68-7)、醋丁洛尔(acebutolol, CAS号37517-30-9)、普萘洛尔(propranolol, CAS号318-98-9)、纳多洛尔(nadolol, CAS号42200-33-9)、拉贝洛尔(labetalol, CAS号36894-69-6)、比索洛尔(bisoprolol, CAS号66722-44-9)、塞利洛尔(celiprolol, CAS号56980-93-9)、卡维地洛(celiprolol, CAS号72956-09-3)、阿罗洛尔(arotinolol, CAS号68377-92-4)、艾司洛尔(esmolol, CAS号81147-92-4)、美托洛尔(metoprolol, CAS号37350-58-6)、奈必洛尔(nebivolol, CAS号99200-09-6)、贝凡洛尔(bevantolol, CAS号59170-23-9),纯度均大于99%,均购自天津阿尔塔科技有限公司;甲醇、乙腈(LC-MS级,德国Merck公司);甲酸(LC-MS级,赛默飞世尔科技有限公司)。
30批食品样品:2022年7月分别从电商及实体店铺购买,其中10批次为固态片剂食品,20批次为液态口服液类样品。
1.2 实验条件
1.2.1 溶液配制
分别准确称取/移取13种β-受体阻滞剂标准物质适量,用甲醇溶解并定容至10 mL,配制成质量浓度为100 μg/mL的标准储备液,-18 ℃避光保存。
分别准确吸取13种标准储备液适量,用甲醇定容至10 mL,配制成质量浓度为1.0 μg/mL的混合标准溶液,-18 ℃避光保存。
标准工作液:根据需要吸取适量混合标准溶液,用甲醇稀释成所需浓度,现配现用。
1.2.2 样品制备
对于固态片剂样品,取适量混匀研磨后,精密称取1 g(精确至0.001 g),置于50 mL具塞离心管中,加入甲醇10 mL,涡旋1 min使其混合均匀,在25 ℃下通过超声波(40 kHz, 250 W)提取30 min,放冷至室温。提取后,混合物以8000 r/min离心5 min,收集上清液至20 mL容量瓶中,用上述方法重复提取一次,合并2次提取液,定容至刻度,摇匀。过0.22 μm有机滤膜后上机测定。
对于液态样品,经充分混匀后准确吸取1.0 mL置于50 mL具塞离心管中,加甲醇40 mL,在25 ℃下超声(40 kHz, 250 W)提取15 min,放冷至室温。用甲醇定容,过0.22 μm有机滤膜后上机测定。
1.2.3 色谱条件
色谱柱:Waters Acquity UPLC BEH C18(100 mm×2.1 mm, 1.7 μm);流动相:0.1%(v/v)甲酸水溶液(A)-乙腈(B);流速0.2 mL/min,进样体积3 μL,柱温35 ℃。梯度洗脱程序:0~3.0 min, 98%B; 3.0~10.0 min, 98%B~5%B; 10.0~15.0 min, 5%B; 15.0~20.0 min, 5%B~98%B。
1.2.4 质谱条件
离子源:加热电喷雾离子源(HESI),正离子模式;喷雾电压:3.0 kV;毛细管温度:320 ℃;加热器温度:50 ℃;鞘气:氮气,流速为40 arb;辅助气:氮气,流速为5 arb;扫描模式:一级母离子全扫描和数据依赖的二级子离子扫描(Full MS/dd-MS2)模式。一级母离子全扫描分辨率:7×104半峰宽(FWHM);质谱扫描范围:m/z 100~1200;自动增益控制(AGG)及最大注入时间(IT)分别为1×106、100 ms。数据依赖二级子离子扫描分辨率:1.75×104 FWHM;触发阈值:1×105;归一化碰撞能量设定为15%、30%、45%。13种β-受体阻滞剂的质谱分析参数见表1。
表 1.
13种β-受体阻滞剂的质谱参数
| Compound | Molecular formula |
tR/ min |
Structural formula | Theoretical [M+H]+ |
Measured [M+H]+ |
Fragment ions (m/z) |
|---|---|---|---|---|---|---|
| Atenolol | C14H22N2O3 | 6.23 |

|
267.1703 | 267.1694 | 225.1227, 208.0975, 116.1077, 98.0973 |
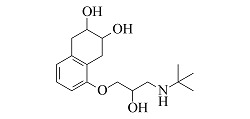
| Nadolol | C17H27NO4 | 6.77 |
|
310.2013 | 310.2017 | 205.0868, 149.0240, 119.7833 |
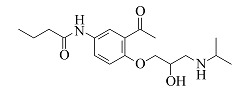
| Acebutolol | C18H28N2O4 | 7.25 |
|
337.2122 | 337.2108 | 319.2004, 260.1272, 116.1070, 98.0967 |
| Arotinolol | C15H21N3O2S3 | 7.26 |

|
372.0869 | 372.0856 | 316.0232, 298.9968, 74.0607 |
| Metoprolol | C15H25NO3 | 7.39 |

|
268.1907 | 268.1898 | 191.1060, 159.0799, 116.1069, 98.0967 |
| Celiprolol | C20H33N3O4 | 7.55 |

|
380.2544 | 380.2527 | 307.1648, 251.1021, 119.7495 |
| Esmolol | C16H25NO4 | 7.57 |

|
296.1856 | 296.1846 | 219.1008, 145.0644, 116.1070, 98.0967 |
| Labetalol | C19H24N2O3 | 7.77 |

|
329.1860 | 329.1848 | 311.1743, 294.1479, 162.0544, 91.0546 |
| Bisoprolol | C18H31NO4 | 7.86 |

|
326.2326 | 326.2314 | 116.1070, 98.0967, 74.0607 |
| Propranolol | C16H21NO2 | 8.01 |

|
260.1645 | 260.1637 | 183.0799, 157.0644, 116.1070, 98.0967 |
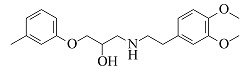
| Bevantolol | C20H27NO4 | 8.13 |
|
346.2013 | 346.2000 | 165.0906, 150.0671 |
| Carvedilol | C24H26N2O4 | 8.35 |

|
407.1965 | 407.1953 | 283.1430, 224.1273, 100.0759 |
| Nebivolol | C22H25F2NO4 | 8.55 |

|
406.1824 | 406.1814 | 388.1701, 151.0550, 123.0603 |
2 结果与讨论
2.1 质谱条件的优化
本研究采用Q Exactive高分辨质谱的Full MS/dd-MS2模式进行13种β-受体阻滞剂的分析,这些化合物的一级离子及二级碎片离子信息见表1。通过使用目标化合物的精确分子离子质量数,结合二级碎片离子谱的分析,能够实现在没有标准品的情况下,对样品中有无上述13种β-受体阻滞剂类化合物进行快速筛查。使用超高效液相色谱-四极杆/静电场轨道阱质谱对13种β-受体阻滞剂的分子离子提取色谱图见图1。
图1. 13种β-受体阻滞剂的提取离子色谱图.

2.2 流动相及提取溶剂的选择
实验考察了0.1%(v/v)甲酸水溶液-甲醇、0.1%(v/v)甲酸水溶液-乙腈、0.02 mol/L乙酸铵溶液-乙腈、0.02 mol/L乙酸铵溶液(含0.1%(v/v)甲酸)-乙腈等流动相体系在最优的分离条件下各化合物的峰形及分离情况,发现0.1%(v/v)甲酸水溶液-乙腈作为流动相,梯度洗脱后13种化合物在10 min内能够得到较好的分离。
参考已有相关文献[22,⇓-24]报道,实验考察了以甲醇、乙腈、80%(v/v)甲醇水溶液、1%(v/v)乙酸甲醇、1%(v/v)乙酸乙腈为提取溶剂时空白加标样品中13种β-受体阻滞剂类的加标回收率,如图2所示。结果表明,甲醇作为提取溶剂时样品中13种β-受体阻滞剂的回收率在80.4%~116.4%范围内,提取效果整体较优,故本实验选择甲醇为提取溶剂。
图2. 不同提取溶剂对13种β-受体阻滞剂回收率的影响.

2.3 基质效应考察
选择典型空白样品基质,制备质量浓度为20 μg/L的空白基质加标溶液,上机测定,制备同样浓度的标准品溶液,通过二者响应强度的百分比来确定基质效应[24]。结果表明,13种β-受体阻滞剂的基质效应在87.4%~107.3%范围内,空白基质对目标物定量影响较小。
2.4 灵敏度、准确度和精密度
以质量浓度(μg/L)为横坐标,峰面积为纵坐标制作标准曲线,13种β-受体阻滞剂在0.5~100 μg/L范围内呈良好的线性关系,相关系数(r)均≥0.9912。向空白样品中添加逐级稀释的标准品溶液,经前处理后,上机测定,确定有信号检出的最低含量为该目标物的检出限,为1~10 μg/kg,详细结果见表2。
表 2.
13种β-受体阻滞剂的回归方程、相关系数、检出限和定量限
| Compound | Regression equation | r | LOD/ (μg/kg) |
LOQ/ (μg/kg) |
|---|---|---|---|---|
| Atenolol | y=2.31×105x-4.81×105 | 0.9970 | 4 | 10 |
| Nadolol | y=4.91×105x+5.50×105 | 0.9912 | 10 | 20 |
| Acebutolol | y=6.08×105x+1.10×106 | 0.9937 | 2 | 5 |
| Arotinolol | y=4.89×105x-7.25×105 | 0.9957 | 1 | 3 |
| Metoprolol | y=6.97×105x-6.26×105 | 0.9930 | 1 | 3 |
| Celiprolol | y=7.24×105x-7.98×105 | 0.9962 | 1 | 3 |
| Esmolol | y=9.86×105x-6.33×105 | 0.9958 | 4 | 10 |
| Labetalol | y=4.58×105x-8.50×105 | 0.9942 | 1 | 3 |
| Bisoprolol | y=6.96×105x-6.59×105 | 0.9957 | 1 | 3 |
| Propranolol | y=1.01×106x-6.70×105 | 0.9951 | 1 | 3 |
| Bevantolol | y=7.50×105x-7.39×105 | 0.9960 | 1 | 3 |
| Carvedilol | y=6.72×105x-5.65×105 | 0.9972 | 1 | 3 |
| Nebivolol | y=5.24×105x-7.82×105 | 0.9956 | 1 | 3 |
y: peak area; x: mass concentration, μg/L.
分别称取空白样品各1 g,在低、中、高3个水平下分别添加13种β-受体阻滞剂标准物质进行加标回收试验,每个水平进行6次平行分析,回收率结果见表3。13种β-受体阻滞剂的平均回收率为75.3%~108.4%,相对标准偏差(RSD)为0.9%~10.0%(n=6),其准确度和精密度能满足保健食品检测的要求,表明该分析方法准确、可靠,适合此类物质的定量分析。
表 3.
13种β-受体阻滞剂的加标回收率及相对标准偏差(n=6)
| Compound | Added/(μg/kg) | Recovery/% | RSD/% |
|---|---|---|---|
| Atenolol | 10 | 105.2 | 0.9 |
| 20 | 105.8 | 4.9 | |
| 200 | 93.8 | 6.5 | |
| Nadolol | 20 | 78.4 | 3.6 |
| 50 | 77.8 | 4.1 | |
| 200 | 78.0 | 3.1 | |
| Acebutolol | 10 | 79.4 | 4.4 |
| 20 | 82.8 | 5.7 | |
| 200 | 81.0 | 7.1 | |
| Arotinolol | 10 | 80.6 | 3.6 |
| 20 | 77.2 | 2.9 | |
| 200 | 76.0 | 5.1 | |
| Metoprolol | 10 | 91.9 | 4.2 |
| 20 | 89.6 | 2.5 | |
| 200 | 102.2 | 5.6 | |
| Celiprolol | 10 | 86.5 | 4.4 |
| 20 | 75.3 | 5.7 | |
| 200 | 81.7 | 4.4 | |
| Esmolol | 10 | 80.9 | 10.0 |
| 20 | 95.9 | 8.6 | |
| 200 | 88.9 | 2.7 | |
| Labetalol | 10 | 87.5 | 1.5 |
| 20 | 104.8 | 5.2 | |
| 200 | 79.6 | 6.1 | |
| Bisoprolol | 10 | 91.8 | 6.5 |
| 20 | 108.4 | 5.9 | |
| 200 | 89.0 | 2.8 | |
| Propranolol | 10 | 86.6 | 8.2 |
| 20 | 78.3 | 1.8 | |
| 200 | 82.0 | 5.1 | |
| Bevantolol | 10 | 89.3 | 3.4 |
| 20 | 84.0 | 3.6 | |
| 200 | 84.8 | 6.3 | |
| Carvedilol | 10 | 78.9 | 9.4 |
| 20 | 77.3 | 4.1 | |
| 200 | 77.6 | 4.8 | |
| Nebivolol | 10 | 89.0 | 2.5 |
| 20 | 81.3 | 1.9 | |
| 200 | 90.3 | 1.2 |
2.5 β-受体阻滞剂类化合物的质谱裂解规律
β-受体阻滞剂是一类以苯乙醇胺结构为母核、苯环连接碱性β-轻胺(仲胺)的苯乙胺类药物。本实验选用的β-受体阻滞剂为芳氧丙醇胺类化合物,含有以N为中心的碱性基团。
由表1中各化合物的二级碎片离子可见,阿替洛尔、醋丁洛尔、普萘洛尔、比索洛尔、艾司洛尔和美托洛尔均产生了m/z 116.1070和m/z 98.0965的碎片离子,推断其裂解规律为这6种化合物在碳氧键处断裂失去其芳香结构,形成碎片离子C6H14NO+(m/z 116.1070),再进一步脱水得到碎片离子C6H12N+(m/z 98.0965),以醋丁洛尔为例,其二级质谱裂解机理如图3所示。卡维地洛和贝凡洛尔因含有电负极性较强的间苯二醚结构极易从中间断裂,以卡维地洛为例,其二级质谱裂解机理如图4所示。塞利洛尔、纳多洛尔、拉贝洛尔、阿罗洛尔及萘必洛尔分子结构式中的特征官能团与其他目标物虽然各不相同,但其裂解规律相似。
图3. 醋丁洛尔二级质谱裂解机理.

图4. 卡维地洛的二级质谱裂解机理.

通过研究β-受体阻滞剂的质谱裂解规律,找出其中的特征碎片离子,可以对未知的此类化合物实现非靶标筛查,这也有助于监管部门及时发现该类药物的结构类似物在保健食品中的非法添加。
2.6 实际样品测定
利用本方法对随机抽取的30批次市售片剂、口服液类保健食品进行快速筛查检测,均未检出上述13种β-受体阻滞剂。
3 结论
采用超高效液相色谱-四极杆/静电场轨道阱质谱技术,建立了食品中13种β-受体阻滞剂的快速精准检测方法,与目前所报道的方法相比,本方法不仅灵敏度满足要求,同时抗干扰能力强,速度快,成本低,可应用于实际样品中非法添加β-受体阻滞剂类药物的快速筛查,为监管部门提供了有效的技术支撑。
参考文献:
- [1]. Chen D Y, Zhang H, Feng J L, et al. Chinese Journal of Chromatography, 2020, 38(8): 880 [DOI] [PubMed] [Google Scholar]; 陈东洋, 张昊, 冯家力, et al. 色谱, 2020, 38(8): 880 [DOI] [PubMed] [Google Scholar]
- [2]. Zhou Y L, Xi Z, Kang J, et al. Journal of Food Safety and Quality, 2022, 13(9): 2908 [Google Scholar]; 周亚兰, 席彰, 康靖, et al. 食品安全质量检测学报, 2022, 13(9): 2908 [Google Scholar]
- [3]. Sung I K, Park S J, Karig K, et al. Korean J Food Sci Anim Resour, 2015, 135(1): 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Regal P, Monica D B, Rocio B, et al. Food Addit Contam, 2017, 34(4): 10 [Google Scholar]
- [5]. Cao Y C, Feng Y X. Journal of Huaihai Medicine, 2019, 39(6): 659 [Google Scholar]; 曹永春, 冯燕娴. 淮海医药, 2019, 39(6): 659 [Google Scholar]
- [6]. Fan W S, Zhang L, Zhu J K, et al. Occupation and Health, 2023, 39(7): 904 [Google Scholar]; 范雯谡, 张蕾, 朱吉凯, et al. 职业与健康, 2023, 39(7): 904 [Google Scholar]
- [7]. Yue H X, Lei W, Du X N, et al. Journal of Chinese Mass Spectrometry Society, 2018, 39(1): 61 [Google Scholar]; 岳韩笑, 雷雯, 杜晓宁, et al. 质谱学报, 2018, 39(1): 61 [Google Scholar]
- [8]. Liu X J, Zhang Y Y, Liu T T, et al. Chemical Analysis and Meterage, 2023, 32(4): 63 [Google Scholar]; 刘晓晶, 张媛媛, 刘彤彤, et al. 化学分析计量, 2023, 32(4): 63 [Google Scholar]
- [9]. Peng J W, Xu L, Li X C, et al. Chemical Industry Times, 2016, 30(8): 1 [Google Scholar]; 彭佳伟, 徐蕾, 李小晨, et al. 化工时刊, 2016, 30(8): 1 [Google Scholar]
- [10]. Wang J J, Zheng G J, Wang J L, et al. Chinese Journal of Analytical Chemistry, 2001, 29(6): 671 [Google Scholar]; 王建军, 郑国钧, 王佳亮, et al. 分析化学, 2001, 29(6): 671 [Google Scholar]
- [11]. Tan X P. [MS Dissertation]. Chongqing: Southwest University, 2016. [Google Scholar]; 谭选平. [硕士学位论文]. 重庆: 西南大学, 2016. [Google Scholar]
- [12]. Cai W J, Ying Y F, Lu C B, et al. Chinese Journal of Veterinary Drug, 2017, 51(8): 26 [Google Scholar]; 蔡文金, 应永飞, 陆春波, et al. 中国兽药杂志, 2017, 51(8): 26 [Google Scholar]
- [13]. Hu S J, Li Y, Zhou Y, et al. Chinese Journal of Chromatography, 2019, 37(7): 701 [DOI] [PubMed] [Google Scholar]; 胡胜杰, 李优, 周莹, et al. 色谱, 2019, 37(7), 701 [DOI] [PubMed] [Google Scholar]
- [14]. Zhao K X, Chai M J, Song X, et al. Journal of Food Safety and Quality, 2019, 10(11): 3481 [Google Scholar]; 赵孔祥, 柴铭骏, 宋旭, et al. 食品安全质量检测学报, 2019, 10(11): 3481 [Google Scholar]
- [15]. Liu X H, Shi Y H, Ding W H, et al. Quality and Safety of Agro-products, 2020(5): 68 [Google Scholar]; 刘新辉, 史永晖, 丁文慧, et al. 农产品质量与安全, 2020(5): 68 [Google Scholar]
- [16]. Yuan L J, Zong W. Journal of Food Safety and Quality, 2022, 13(20): 6518 [Google Scholar]; 袁利杰, 纵伟. 食品安全质量检测学报, 2022, 13(20): 6518 36316354 [Google Scholar]
- [17]. Zeng X, Zhao T T, Liu C S, et al. Journal of Food Science and Technology, 2023, 23(2): 288 [Google Scholar]; 曾曦, 赵甜甜, 刘春生, et al. 中国食品学报, 2023, 23(2): 288 [Google Scholar]
- [18]. Nie X M, Dong X Y, Xu X L, et al. Chinese Journal of Chromatography, 2019, 37(9): 1011 [DOI] [PubMed] [Google Scholar]; 聂雪梅, 董旭阳, 许秀丽, et al. 色谱, 2019, 37(9): 1011 [DOI] [PubMed] [Google Scholar]
- [19]. Ni Z, Zhang C S, Xie X C. Chinese Journal of Pharmaceutical Analysis, 2019, 39(5): 895 [Google Scholar]; 倪赞, 张崇生, 谢循策. 药物分析杂志, 2019, 39(5): 895 31613952 [Google Scholar]
- [20]. Zhang Q, Zhao P Z, Qu L, et al. Food Science, 2014, 35(20): 202 [Google Scholar]; 张旖, 赵普贞, 曲栗, et al. 食品科学, 2014, 35(20): 202 [Google Scholar]
- [21]. Wang H, Zhao Y, Liao P, et al. Chemical Journal of Chinese Universities, 2013, 34(3): 556 [Google Scholar]; 王华, 赵勇, 廖鹏, et al. 高等学校化学学报, 2013, 34(3): 556 [Google Scholar]
- [22]. Liu B, Bao Y, Lang L, et al. Journal of Food Safety and Quality, 2019, 10(6): 1511 [Google Scholar]; 刘斌, 包懿, 郎乐, et al. 食品安全质量检测学报, 2019, 10(6): 1511 [Google Scholar]
- [23]. Xu H B, Zhang S P, Du R Y, et al. Chinese Journal of Chromatography, 2022, 40(6): 531 [DOI] [PMC free article] [PubMed] [Google Scholar]; 徐红斌, 张申平, 杜茹云, et al. 色谱, 2022, 40(6): 531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Tan H J, Guo C C, Xing S, et al. Chinese Journal of Chromatography, 2019, 37(9): 969 [DOI] [PubMed] [Google Scholar]; 谭会洁, 郭常川, 邢晟, et al. 色谱, 2019, 37(9): 969 [DOI] [PubMed] [Google Scholar]