

Abstract
The delivery of functional proteins remains a major challenge in advancing biological and pharmaceutical sciences. Herein, we describe a powerful, simple, and highly effective strategy for the intracellular delivery of functional cargoes. Previously, we demonstrated that cell-penetrating peptide (CPP) additives equipped with electrophilic thiol-reactive moieties temporarily attach to the cellular membrane, thereby facilitating the cellular uptake of protein- and antibody-CPP cargoes through direct membrane transduction at low concentrations. Now, we hypothesize that CPP-additives with an increased retention on the cellular membrane will further enhance intracellular uptake. We discovered that adding a small hydrophobic peptide sequence to an arginine-rich electrophilic CPP-additive further improved the uptake of protein-CPP conjugates, whereas larger hydrophobic anchors showed increased cytotoxicity. Cell viability and membrane integrity measurements, structure–activity relationship studies, and quantitative evaluation of protein-CPP uptake revealed important design principles for cell-surface-retained CPP-additives. These investigations allowed us to identify a nontoxic, thiol-reactive CPP-additive containing the hydrophobic ILFF sequence, which can deliver fluorescent model proteins at low micromolar concentrations. This hydrophobic CPP-additive allowed the addition of protein cargoes for intracellular delivery after initial additive incubation. Time-lapse fluorescence microscopy and membrane tension analysis of cells treated with fluorescent ILFF-CPP-additives supported the claim of increased cell surface retention and suggested that the protein-CPP cargoes enter the cell through a mechanism involving lowered cell membrane tension. Finally, we demonstrated that our newly engineered hydrophobic CPP-additive enabled the uptake of a functional macrocyclic peptidic MDM2-inhibitor and a recombinant genome editing protein. This indicates that the developed hydrophobic CPP-additive holds promise as a tool to enhance the intracellular delivery of peptide and protein cargoes.
Introduction
Recent developments have positioned biopharmaceuticals as highly effective treatment options for a wide range of diseases.1−3 In particular, peptide and protein-based diagnostics and drugs are gaining increasing significance in modern medicine due to their vast structural and functional diversity.4,5 Despite considerable success for extracellular targets, most peptide/protein-based drugs, which are often hydrophilic and thus cell impermeable, have difficulty in accessing intracellular targets due to inefficient delivery into cells.6 Consequently, the cell impermeability of proteins remains a major obstacle for protein-drug manufacturers and the molecular life sciences community at large. As a result, researchers from across disciplines have proposed several ingenious intracellular delivery strategies to address this limitation.
Various reported methods for intracellular delivery rely on either physically disrupting the cell membrane (e.g., particle bombardment,7 fluid shear forces8) or utilizing biological carrier (e.g., viral vectors9)- or chemical carrier-based systems (e.g., dendrimers, cationic lipids). While physical disruption has the potential to import extracellular cargoes directly to the cytosol of individual cells in vitro, its applications in vivo are limited due to the high cytotoxicity caused by irreparable damage to the plasma membrane and the risk of nonspecific delivery. In contrast, carrier-based systems are considered to be more cost-effective, easier to perform, and biologically friendly alternatives. These systems have greatly improved the delivery of nucleic acid therapeutics.2 However, the same success has not been achieved in delivering peptide/protein-based therapeutics. Despite the development of promising approaches such as cell-permeable miniproteins,10,11 cell surface thiol-mediated uptake,12,13 and endosome release,14−16 the number of effective and efficient strategies for direct intracellular delivery into the cytosol of a wide range of biofunctional macromolecules remains limited.17
Cell-penetrating peptides (CPPs) are particularly prominent carrier systems that have been utilized for delivering a variety of cargoes.18 The truncated human immunodeficiency virus (HIV)-1 transcription-transactivating protein (TAT) was the first highly cationic CPP to be identified.19 Cationic CPPs, 8–10-mer nontoxic peptides consisting of mostly arginine or lysine residues, are among the most recognizable and widely used types of CPPs.20 Due to their ability to enter cells, CPPs have been exploited for delivering functional proteins into cells.21,22 The entry mechanism of proteins conjugated with CPPs (protein-CPPs) roughly depends on their size and concentration.23 At high molecular weights or low concentrations, protein-CPPs predominantly enter via endocytosis and remain trapped in endosomes, leading to a decrease in the effective concentration of the cargo within the cell.24 Several strategies have been reported to release the trapped cargoes via endosome release; however, these methods do not ensure complete release of the cargo into the cytosol.16,25,26 On the other hand, direct transduction allows delivered proteins to be readily available within the cytosol, where they can exert their physiological functions.27 Initially, it was believed that bidentate hydrogen-bonding interactions between the guanidinium group of arginines and the membrane phosphate groups, along with membrane potential, were key to direct transduction.28 Subsequent studies revealed that ionic gradients,29 proteoglycan interactions,30 and cell membrane lipid packing31,32 also contribute to this process. However, only small protein-CPPs (<20 kDa) at high concentrations (>20 μM) have been shown to enter cells through direct transduction. Developing systems that enable the direct transduction of larger protein-CPP cargoes at lower concentrations remains an important challenge to tackle.
Different research groups, including ours, have proposed strategies to accomplish direct transduction for larger protein-CPPs.33−36 An effective approach involves the cyclization of CPPs, leading to improved and rapid uptake of protein-CPPs (Figure 1A).37 This enhancement can be attributed to the optimal presentation of positive charges on the cyclic peptide. Cyclic CPPs, such as cyclic TAT and cyclic R10, have been successfully employed for delivering diverse protein cargoes, including peptide macrocycles,38 fluorescent proteins,39 and nanobodies.40 Despite the broad applications, the minimum concentration required for effective transduction still falls within the range of 10–50 μM. To further improve transduction efficiencies at lower concentrations, several strategies have been explored. For instance, the addition of a penetration acceleration sequence (Pas) or a 4-((4-(dimethylamino)phenyl)azo)benzoic acid (DABCYL) moiety has shown increased uptake efficiency at treatment concentrations below 10 μM of cargo peptide or small proteins, respectively.41−43 Other studies have proposed the use of counterions44,45 and membrane-remodeling amphiphilic peptides,31,32 which create membrane lipid packing defects to enhance the direct transduction of CPPs and protein-CPP conjugates.
Figure 1.

Schematic diagram illustrating the state-of-the-art and aims of this article. (A) Current method of delivering proteins by conjugating cyclic CPPs. (B) Developed “CPP-additive technology” employs an excess amount of free CPP with an electrophilic thiol-reactive moiety, which facilitates the cellular uptake of protein cargo. (C) Proposed improvement of CPP-additives through the addition of a hydrophobic “anchor” moiety, aiming to enhance intracellular uptake and delivery efficiency of protein-CPP cargoes.
Recently, we introduced the “CPP-additive technology”, in which the protein-CPP cargo is added to cells in the presence of 2 to 5-fold excess amounts of cell-surface anchoring CPPs (Figure 1B).46 This technology relies on the critical functions of these CPP-additives, including (1) temporal cell surface anchoring enabled by functional groups, such as maleimides or electrophilic disulfides, that covalently attach to thiol-containing proteins or other macromolecules on the cell membrane, (2) formation of nucleation zones (areas on the cell membrane considered as nonendocytic entry sites for CPPs47,48), and (3) reducing the membrane tension–priming the cell membrane for the subsequent permeation of the protein-CPP cargo. Through this strategy, we achieved efficient cytosolic delivery of various protein-CPP cargoes, including nanobodies for endogenous targeting, even at low micromolar concentrations (∼5 μM).49 Notably, during our studies, we observed rapid entry of both protein-CPP and the CPP-additives into cells, indicating a limited time of engagement on the cell membrane.46
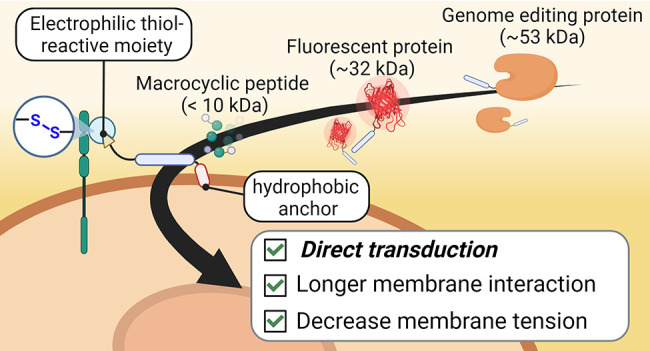
In this study, we aimed to design CPP-additives capable of longer retention on the cell surface to augment their effect on protein delivery (Figure 1C). Taking inspiration from previous research, in which the incorporation of hydrophobic amino acids or moieties on CPPs led to improved uptake properties, particularly by enhancing endosomal release,41,50 we proposed equipping the original CPP-additives with an additional hydrophobic “anchor” motif. Our investigations confirmed that the hydrophobic motif increased engagement with the cell membrane without compromising cell viability. Most importantly, the hydrophobic CPP-additive outperformed the original CPP-additive, exhibiting increased uptake of a model fluorescent protein and allowing the subsequent addition of protein cargoes after initial CPP-additive incubation. Consequently, this enhancement facilitated the efficient functional delivery of both a peptide macrocycle and a recombinant genome editing protein.
Result and Discussion
Designing Enhanced CPP-Additives through Increased Hydrophobicity
Amphipathic CPPs containing hydrophobic amino acids or motifs usually demonstrate better efficiency in terms of cellular uptake, resulting from increased cell surface/lipid interactions.30,50 Based on this, we hypothesized that incorporating hydrophobic elements could enhance the retention of cell-anchoring CPP-additives carrying electrophilic disulfides at the membrane. In this study, we designed CPP(R10)-additives containing 5-thio-2-nitrobenzoic acid-disulfide (TNB) with additional hydrophobic moieties. These motifs include small peptides hydrophobic amino acid sequences, namely ILFF (transposed version of the Pas peptide)41 in TNB-R10-ILFF 2 and FWF50 motifs in TNB-R10-FWF 3, which were previously shown to enhance the uptake of conjugated CPPs and proteins, respectively, primarily through enhanced endosome release. To compare the difference between aromatic and aliphatic hydrophobic tags, we prepared CPP-additives with hydrophobic tags having all aliphatic amino acids, namely, TNB-R10-ILIL 4 and TNB-R10-ILLL 5. We also prepared CPP-additives with unnatural amino acids containing polycyclic aromatic hydrocarbon (PAH) side chains, substituting either one or two of the l-phenylalanine residues with l-naphthylalanine residues in TNB-R10-Nal 6 or TNB-R10-Nal2 7. l-Naphthylalanines were previously reported to improve membrane interaction of cyclic CPPs within endosomes.14,51 Moreover, large hydrophobic domains, such as pyrenes on previously reported counterions that enhanced poly arginine CPP uptake, showed favorable membrane interaction.44 Hence, we coupled it to the side chain of a lysine with a pyrene moiety (TNB-R10-Py 8). Last, we synthesized hydrophobic CPP-additives with lipids conjugated to a C-terminal lysine residue in TNB-R10-C18 9 and TNB-R10-Chol 10, containing lipid molecules that are components of the cell membrane (Figure 2A).52,53 These CPP-additives were synthesized with a C-terminal amide by standard Fmoc-based solid phase peptide synthesis, and the TNB conjugation was performed on resin before cleavage and purification using reverse phase-high performance liquid chromatography.
Figure 2.

Design and screening of CPP-additives with a hydrophobic motif. (A) Synthesized hydrophobic CPP-additives 1–10 with different hydrophobic motifs. (B) Cell viability as determined by WST-1 mitochondrial reduction assay after treating HeLa cells with 2.5, 5, or 10 μM hydrophobic CPP-additives 1–10 for 1 h in serum-free FluoroBrite DMEM. Red dotted line indicates 100% viability. (C) Analysis of membrane integrity following CPP-additive treatment as determined by measuring the release of LDH into the cell medium. Data presented as mean ± standard error (SE) of three biological replicates. (D) Rationale of the protein-CPP delivery assay. Without CPP-additives, NLS-mCherry-R10 I enters cells via endocytosis, while in the presence of CPP-additives, direct cytosolic uptake should occur and the NLS-mCherry-R10 I accumulates on the nucleolus. (E) Confocal microscopy images of cells treated with NLS-mCherry-R10 I in the presence or absence of indicated CPP-additives (5 μM) for 1 h in serum-free FluoroBrite DMEM. (F) Individual nuclear fluorescence intensities of 300 cells across three biological replicates following cotreatment with NLS-mCherry-R10 and CPP-additive. Red bar corresponds to the median fluorescence intensity of each treatment. P-values were determined by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis. (G) Nucleolar staining in different cell lines treated with I (5 μM) and 2 (5 μM) for 1 h in serum-free FluoroBrite DMEM. Scale bars: 20 μm.
We expected that cell surface-anchored CPP-additives with large hydrophobic entities could disrupt membranes, causing the release of intracellular components or cell death. To assess this, we analyzed the cytotoxicity and membrane integrity of HeLa cells treated with an increasing concentration of the synthesized CPP-additives. Cytotoxicity was assessed by measuring the ability of cellular mitochondrial dehydrogenase in live cells to reduce tetrazolium salts (WST1) into formazan, while membrane integrity was evaluated by monitoring the release of lactate dehydrogenase (LDH) into the cell culture media. The results, shown in Figure 2B,C, indicate that TNB-R10-ILFF 2 displayed minimal cytotoxicity across these concentrations. Moreover, at 10 μM, 2 led only to a marginal release of LDH. On the other hand, TNB-R10-FWF 3 showed no cytotoxicity at 5 μM; however, LDH was significantly released at this concentration, suggesting that this additive may not be suitable for further applications. We found that the CPP-additives 4 and 5 with aliphatic hydrophobic tags showed no cytotoxic effect and no LDH release from treated cells, while a notable increase in cytotoxicity and a high degree of LDH release were observed for all other CPP-additives 6–10 at 5 μM. At 2.5 μM, peptides 6–10 showed minimal cytotoxicity in the WST1 assay; however, significant LDH release was still detected, raising concerns about their suitability for subsequent cellular applications even at this concentration.
We probed whether the new CPP-additives facilitate the cytosolic uptake of a fluorescent model protein into HeLa cells. To achieve this, we employed a mCherry (32 kDa, −3 net charge) fused with a nuclear localization sequence (NLS), and linked it with a linear R10-peptide through maleimide labeling as protein cargo I. These modifications ensured that successful delivery into the cytosol led to distinct nucleolar localization, consistent with previous findings (Figure 2D).40,46,54 Confocal microscopy images revealed that in the absence of any CPP-additives, the NLS-mCherry-R10-conjugate I at a concentration of 5 μM was taken up through endocytosis and entrapped in the endocytic vesicle, indicated by a bright punctuate signal along the perinuclear region. However, coadministration of I (5 μM) with CPP-additives 1 or 2 (5 μM) in HeLa cells for 1 h resulted in nucleolar signals, as evident in microscopy images. Notably, cells cotreated with I and 2 exhibited higher nucleolar fluorescence (yellow asterisks, Figure 2E). In addition, cotreatment of HeLa cells with I (5 μM) and CPP-additives 4 and 5 (5 μM) also resulted in nucleolar signals, albeit to a lesser extent than 2 (Figure S5). This result fits well with the Wimley–White hydrophobicity scale, in which aromatic amino acids (Phe and Trp) were found to more likely interact with the membrane interface than aliphatic residues (Ile and Leu).55 The result also fits well with our hypothesis of improving CPP-additives with hydrophobic tags. We coincubated HeLa cells with I and 2 for 1 h, changed the supernatant to fresh culture media, and conducted cytotoxicity assessments of the treated cells after 24 h (Figure S6). We observed no cytotoxic effects under these conditions. To confirm the necessity of R10 on the cargo protein when utilizing the CPP-additive strategy, we coincubated HeLa cells with unconjugated NLS-mCherry (5 μM) with 2 (5 μM) and I with 2. The treatment with NLS-mCherry and 2 showed no uptake, neither endosomal nor cytosolic (Figure S7). These data ascertain the importance of conjugating or fusing the protein of interest with a CPP to benefit from the CPP-additive approach. We also observed that nucleolar localization persisted after 24 h following treatment with I and 2, which was not observed in the condition without 2 (Figure S8).
Since we measured low cytotoxicity for all peptides at 2.5 μM, we also probed the ability of CPP-additives 6–8 at this concentration. Only 2, 6, and 7 demonstrated the successful delivery of I (5 μM) into the cytosol (Figure S9). However, we observed extracellular protein aggregation for additives 6 and 7, manifested as intense fluorescent signals. Interestingly, pyrene-conjugated additive 8 exhibited only minimal enhancement in the cytosolic uptake of I.
Next, we examined the effects of 2 on the delivery of mCherry-derivatives containing either linear (R10, II) or cyclic (cR10, III) linked via disulfide bridges. The CPPs were expected to be cleaved intracellularly. Due to the presence of the NLS sequence on the mCherry, successful cytosolic delivery of these conjugates should lead to their accumulation in the nuclear space but not on the nucleolus.54 Cotreatment of II or III (5 μM) with 2 (5 μM) resulted in cells having distinct nuclear fluorescence (Figure S10).
We quantified and compared the efficiencies of CPP-additives 1 and 2 in facilitating the uptake of I. To do this, we measured the resulting fluorescence intensities within cell nuclei, where more intense nuclear/nucleolar staining indicates improved uptake efficiency. The individual nuclear fluorescence intensity in treated cells was quantified using the Fiji script provided in the Supporting Information. Consistent with the microscopy images, the nuclei of cells cotreated with I and 2 exhibited significantly higher fluorescence intensity compared to conditions without any CPP-additives and to cotreatment with 1 (Figure 2F).
Last, we investigated whether 2 facilitates the cellular uptake of NLS-mCherry-R10 I into different cell lines. Cotreatment of HeLa (cervical adenocarcinoma), HEK293T (human embryonic kidney), SKBR3 (HER2-positive breast cancer), SJSA1 (human osteosarcoma), MCF7 and MDAMB468 (HER2-negative breast adenocarcinoma), and SW480 (colorectal adenocarcinoma) with I (5 μM) in the presence of 2 (5 μM) for 1 h resulted in a high degree of nucleolar staining in all tested cell lines (Figure 2G), whereas none of the treated cells showed nucleolar signals when treated with NLS-mCherry-R10 I alone (Figure S11). These data suggest the applicability of 2 for the delivery of protein-CPPs in diverse cell types.
Structure–Activity Relationship of Hydrophobic CPP-Additives
We proceeded to interrogate key structural components of hydrophobic CPP-additive 2. A crucial element of CPP-additives is their cell surface anchoring moiety.46 To explore this, we examined a maleimide-containing analogue 11(56) and a chloroacetamide alkylated analogue 12 as a negative control in addition to the TNB-conjugated 2 (Figure 3A). When HeLa cells were incubated with I (5 μM) along with either 2 or 11 (5 μM), we observed nucleolar labeling. However, cells treated with alkylated peptide 12 did not exhibit nucleolar labeling [Figure 3A(i)]. This result suggests that the hydrophobic ILFF motif cannot replace the thiol-reactive cell surface anchoring moiety, which remains crucial for the activity of CPP-additives. Moreover, we observed that the disulfide-forming TNB additive 2 led to a greater enhancement of I uptake compared to maleimide compound 11 [Figure 3A(ii)].
Figure 3.

Structure–activity relationship of TNB-R10-ILFF. Confocal images of HeLa cells treated with I (5 μM) and with analogues of 2 having [A(i)] different thiol-reactive moieties and [A(ii)] its corresponding quantification. Graph shows individual nuclear fluorescence intensities of 600 cells across six biological replicates. (B) Different CPP sequences and (C) different distances between the R10 and hydrophobic ILFF motif in serum-free FluoroBrite DMEM for 1 h. Scale bars, 20 μm. Graphs show individual nuclear fluorescence intensities of 300 cells across three biological replicates following cotreatment with I and CPP-additive. Red bars on the graphs correspond to the median fluorescence intensity of each treatment. P-values were determined by one-way analysis (ANOVA) followed by Tukey’s post hoc analysis. All uptake experiments were performed at 37 °C.
We probed the significance of the polyarginine (R10) CPP sequence on hydrophobic CPP-additives. For this, we synthesized and tested analogues with different cationic CPPs, replacing the R10 sequence, namely, the arginine-rich cationic Tat (TNB-Tat-ILFF 13) and polylysine (TNB-K10-ILFF 14). In addition, we prepared an analogue lacking the R10 sequence (TNB-ILFF 15). When cells were cotreated with I (5 μM) and 13, 14, or 15 (5 μM), we observed the absence of nucleolar staining (Figure 3B). These data confirm the importance of the polyarginine stretch in the design of CPP-additives. Previous works report a strong interaction of polyarginines with the cell membrane57,58 and the self-association propensity of polyarginines.59 Considering these findings, we assume that the polyarginine stretch in the hydrophobic CPP-additives served two critical functions (1) aids membrane interaction and (2) enables improved association with protein-CPP. We further investigated this by synthesizing and utilizing the scrambled analogue 16 (TNB-RRRIRFRRRLRRRF). Despite having the same amino acid composition as 2, HeLa cells cotreated with I and 16 did not show any nucleolar staining (Figure 3B). This finding reinforces the significance of the polyarginine stretch in the design of an effective CPP-additive.
To analyze the importance of linkers between the R10 and hydrophobic ILFF motifs, we prepared analogues of 2 with varying lengths of a polyethylene glycol (PEG)-based linker [n = 1 (17), n = 2 (18), n = 3 (19), n = 4(20)], thereby increasing the distance between the R10 and ILFF sequences. We found that cotreating HeLa cells with I (5 μM) and these analogues (5 μM) resulted in a similar degree of nuclear fluorescence when compared to cotreatment with 2 (Figure 3C). This finding indicates that the varying distances between the R10 and ILLF motifs do not significantly impact the activity of the hydrophobic CPP-additive. Further, we explored a transposed version of 2, in which the hydrophobic motif is presented on the N-terminal (FFLI-R10-TNB 21). CPP-additive 21 showed a similar ability to enhance the uptake of I into cells, adding further clarity to the design of the new hydrophobic CPP-additive (Figure S12). The fact that the transposed version maintains its effectiveness in facilitating cellular uptake underscores the robustness and modularity of the hydrophobic motif, regardless of its specific location within the CPP-additive sequence.
To scrutinize the minimum required concentration of protein-CPP, we cotreated HeLa cells with decreasing concentrations of I in the presence of 2 (5 μM). Our observations revealed a linear concentration-dependent correlation and that 2 retains its ability to facilitate the uptake of I until 1.25 μM of treatment concentration (Figure S13A). Additionally, we assessed the total fluorescence within cell nuclei after treatment with various concentrations of I in the presence of 1 or 2 and at various incubation times. Consistent with our findings above, these data showed that cotreating cells with I and 2 led to higher nuclear fluorescence compared to cotreatment with 1 (Figure S13B). We also found an increase of nuclear fluorescence in cells at longer incubation times (Figure S13C). Saturation points of delivery might occur at higher concentrations. However, investigation toward the saturation point also results in high toxicity, affecting the overall experimental result.
Cellular delivery efficiencies of CPPs and protein-CPPs are often diminished in the presence of serum due to interactions with serum proteins.60 To assess whether the hydrophobic CPP-additive 2 retains its capacity to enhance the delivery in serum-containing media, we cotreated cells with I (5 μM) and 1 or 2 (5 and 10 μM) for 1 h in FluoroBrite DMEM with varying serum concentrations. Confocal images show higher fluorescence intensity, particularly in the nucleolar region, in cells cotreated with I and 2 (Figure S14). Notably, even at 5% serum, 2 (5 μM) enhanced the delivery of I into cells. Nucleolar fluorescence remains visible even at 10% serum, albeit to a lesser degree. The decrease in nucleolar fluorescence at higher serum concentrations could be attributed to the interaction of either the CPP-additive or the protein-CPP cargo with serum proteins. Importantly, prolonged incubation (24 h) with I and 2 in the presence of serum did not result in cytotoxicity (Figure S14C). However, we did not observe nucleolar signals in HeLa cells treated with I and 1 or 2 (10 μM) in medium with 20% serum (Figure S14D). These data suggest the potential practical application of 2 for delivering protein-CPP cargo under conditions with up to 10% serum. Further improvements on the CPP-additives are needed to enable efficient delivery in high serum conditions.
Finally, we probed the performance of the ILFF tag as part of the protein cargo conjugate. For this, we conjugated NLS-mCherry with maleimide-R10-ILFF peptide to generate NLS-mCherry-R10-ILFF IV. We found that IV alone can enter the cytosol of the cells only at concentrations above 10 μM (Figure S15A). Cotreatment of IV (5 or 2.5 μM) with the hydrophobic CPP-additive 2 (5 μM) showed significant delivery and a high degree of nucleolar fluorescence. A direct comparison of the uptakes of I and IV (5 μM) with the same amount of 2 showed an increased uptake of IV in the presence of 2 as compared to I with 2 (Figure S15B,C). Previously, it was observed that octaarginines with a hydrophobic Pas motif (PasR8) were efficient in delivering small peptide-CPP conjugates via direct transduction but faced limitations in delivering larger conjugates.61 Hence, the enhanced uptake observed when the hydrophobic ILFF motif is present in both the CPP-additive and the protein-CPP conjugate might originate from cooperative binding between the protein cargo and the additive.46 Further analyses and investigations are warranted to understand the underlying mechanism in detail. To isolate the contribution of the hydrophobic ILFF-tag to the activity of the CPP-additive, we continued using NLS-mCherry-R10 I as the model protein.
Cell Surface Retention of Hydrophobic CPP-Additives and Time-Course Analysis of Protein-CPP Uptake
To elucidate the performance of the hydrophobic CPP-additives over time, we synthesized Cy5-labeled analogues of 1 and 2, TNB-Cy5-R10 22 and TNB-Cy5-R10-ILFF 23 (Figure 4). Time-lapse microscopy revealed that TNB-Cy5-R10 22 initially localizes at the membrane but quickly enters the cytosol after peptide addition (Figure S16A, upper panel). In contrast, TNB-Cy5-R10-ILFF 23 remained on the cell membrane even after 600 s (Figure S16A, lower panel). The line-scan profile showed that the fluorescence intensity of 22 was mainly observed within the interior of cells after 10 min, while 23 remained concentrated around the cell periphery (Figure S16B). Extending the time-lapse imaging, we found that cell-surface-associated CPP-additives 23 were internalized into endosomes (dot-like signals) only after 30 min, possibly due to membrane recycling (Figure S17). Our data align with a previous study that found hydrophobicity-tagged polyarginines [e.g., Alexa488 and Hemagglutinin (HA) peptide] exhibiting enhanced interactions with cell membranes compared to unmodified polyarginines.48 In our case, we observed a similar trend of increased cell membrane interaction of cell-surface-anchored hydrophobic CPP-additives.
Figure 4.

Cell membrane interaction and decreased membrane tension induced by hydrophobic CPP-additive. [A(i)] Chemical structure of TNB-Cy5-R10 22. [A(ii)] Cell surface accumulation on cells treated with 1 μM of the peptide after 5 and 10 min. [A(iii)] Line-scan profile showing the distribution of Cy5 fluorescence intensity. [B(i)] Chemical structure of TNB-Cy5-R10-ILFF 23. [B(ii)] Cell surface accumulation on cells treated with 23 (1 μM) after 5 and 10 min. [B(iii)] Line-scan profile showing Cy5 fluorescence remaining within the cell periphery after prolonged incubation. (C) Time lapse images of HeLa cells treated with I (5 μM) and 23 (1 μM) in serum-free FluoroBrite DMEM for 15 min. I and 23 were added to the cells after 60 s of initial incubation. Images were taken at 60 s intervals. Yellow asterisks indicate the emergence of nucleolar signals. Scale bar: 10 μm. (D) Images showing the change in fluorescence lifetime (τ) in ns of Flipper-TR probe in HeLa cells treated with 23 (1 μM). Time-lapse images were captured every 15 s. Enlarged regions show the change of τ (white arrows) within areas with a high accumulation of 23. Scale bar, 5 μm. (E) Graphs showing individual data points indicating the change in τ (Δτ) after (i) 15, (ii) 30, (iii) 45, and (iv) 60 s of 30 selected regions-of-interest (ROIs) across three biological replicates. Δτ is calculated as the difference between the τ of a ROI after n seconds of peptide addition (τns) minus the initial τ before peptide addition (τ0s).
Next, we investigated the behavior of 22 and 23 (1 μM) in the presence of I (5 μM). After 5 min of cotreatment, 22 along with I accumulated on the cell membrane, as observed through confocal time-lapse microscopy [Figure 4A(ii); upper panel] and line–scan profile analyses [Figure 4A(iii)]. Similarly, 23 also exhibited cell membrane localization after 5 min [Figure 4B(ii); upper panel]. However, unlike 22, the cell membrane accumulation of 23 persisted even after 10 min, accompanied by a distinct nucleolar signal from I [Figure 4B(ii); lower panel, yellow asterisk]. Montages of time-lapse images (60 s intervals) of cells cotreated with I and 22 or 23 are found in Figures S18 and 4C, respectively. It is noteworthy that nucleolar mCherry signals were observed after 15 min in cells cotreated with I (5 μM) and 22 (1 μM) (Figure S18, yellow asterisk). In contrast, cells cotreated with I and 23 (1 μM) already displayed visible nucleolar staining after 3 min (Figure 4C, yellow asterisk), supporting that the hydrophobic CPP-additive 23 achieved the uptake of I more rapidly compared to 22). This rapid intracellular delivery, in combination with the absence of bright fluorescent dot-like signals (that would indicate endosomal-entrapped additives and cargoes), points toward a nonendosomal delivery. An alkylated analogue was incapable of both interacting with the cell membrane and facilitating the uptake of protein-CPPs (Figure S19). Taken together, these findings demonstrate that 23 remains longer on the cell membrane compared to 22, likely due to the combination of the hydrophobic ILFF tag and cell-surface cross-linking TNB motifs. While both are capable of mediating the uptake of I, hydrophobic CPP-additive 23 requires less time to achieve observable signals in the nucleolus.
Encouraged by these results, we further investigated the role of hydrophobicity in the interaction of CPP-additives with the cell membranes. Previous studies have revealed that polyarginine CPPs enter the cell by direct transduction through areas of membrane lipid packing defects.32 This effect can be further enhanced by treatment with membrane-remodeling small molecules [e.g., pyrenebutyrate (PyB)] and amphipathic peptides (e.g., EpN18).32 We previously showed that regions where CPP-additives accumulate experience reduced membrane tension.46 To examine the effect of the hydrophobic CPP-additive, we used the mechanosensitive Flipper-TR probe62 and conducted fluorescence lifetime image microscopy (FLIM) on cells treated with 22 or 23 (1 μM). Time-lapse FLIM images of a region of 23 accumulation show a decrease in fluorescence lifetime (τ) over time, represented by a red-to-green change in pixel colors (Figure 4D, white arrows). Plotting the obtained τ values of CPP-additive accumulation regions over time showed a reduction of τ for cells treated with 22 compared to 23, while control cells (treated with cell culture media) showed no significant change (Figure S20).
We quantified the change in membrane tension (Δτ) by recording the τ of CPP-additive accumulation regions before (τ0s) and after n seconds (τns) of treatment. A negative Δτ value indicates decreased membrane tension, while a positive value indicates increased tension.63Figure 4E presents the Δτ of 30 individual sites of 22 or 23 accumulation after (i) 15, (ii) 30, (iii) 45, and (iv) 60 s. Regions of 23 accumulation already showed significantly lower Δτ compared to control cells from 15 s after peptide treatment [Figure 4E(i)], indicating an early decrease in membrane tension. This decrease in tension was sustained throughout the measurement. In contrast, accumulation regions of 22 exhibited a significant reduction of Δτ after 30 s postpeptide addition.
To further understand the performance of protein-CPPs in the presence of hydrophobic CPP-additives, we additionally evaluated uptake experiments with longer incubation times of the CPP-conjugates. During our studies, we detected endosome-trapped NLS-mCherry, observed as bright dot-like signals, when HeLa cells were cotreated with I and 2 for 1 h at 37 °C (Figures 2E and S21). To halt endocytosis, we conducted the delivery assay at 4 °C. Confocal images of HeLa cells treated with I (5 μM) and 2 (5 μM) still showed nucleolar staining after incubation at 4 °C, indicating that nonendocytic direct transduction entry plays a role in the observed protein uptake in the presence of 2 (Figure S22). Furthermore, under these conditions, cotreatment with 2 led to more pronounced nucleolar staining as compared to using CPP-additive 1. Nevertheless, the overall nuclear fluorescence intensity after 4 °C incubation was lower than that after 37 °C incubation.
The observed discrepancy between the efficiencies at 4 and 37 °C can be rationalized by different scenarios. First, since disulfide formation is reduced at lower temperatures,64 the TNB-containing additive 2 might be less effective. Second, lipid membranes have increased rigidity and are less dynamic at lower temperatures.65 Last but not the least, the uptake mechanism facilitated by 2 combines direct transduction and endocytosis, followed by endosome release at extended incubation times of the CPP-conjugates. Along those lines, it was reported previously that incorporating a hydrophobic tag into a protein-CPP fusion protein could enhance endosomal release, leading to improved intracellular delivery.50 Similarly, another work utilized two units of the penetration acceleration peptide (FFLI) conjugated to the N-terminus of a dodecaarginine (R12) to facilitate the delivery of unmodified proteins through endocytosis and potentially subsequent endosome release.66
To investigate if the hydrophobic CPP-additives 2 could be responsible for releasing entrapped protein cargoes, we preloaded endosomes with NLS-mCherry-R10 I (5 μM) for 1 h to serve as fluorescent markers (1st treatment) following a previously reported study.67 Subsequently, we washed the cells and treated them with hydrophobic CPP-additive 2 (5 μM) for another 1 h (2nd treatment). We hypothesized that if 2 facilitates endosome release, I would be released into the cytosol and then translocate into the nucleus; however, upon analysis, the preloaded cells treated with 2 showed no nucleolar staining compared to untreated cells (Figure S23). These data, together with the 4 °C experiment above, suggest that while some level of endosome release may have occurred, the proteins-CPP cargoes are delivered primarily through direct transduction.
We tested whether hydrophobic CPP-additive preassociated on the cell membrane remains capable of enabling the direct transduction of I. After incubating cells with 1 or 2 (5 μM) for 5 min, we washed off excess CPP-additives before adding I (5 μM) (Figure S24A). Confocal microscopy revealed nucleolar staining exclusively in cells pretreated with compound 2 (Figure S24B). Considering that Figure S17 demonstrated the internalization of membrane-associated CPP-additives after 30 min, direct transduction could only have occurred within this time frame. This uptake is soon followed by endocytosis, resulting in the observation of NLS-mCherry-containing endosomes. Importantly, our data reveal that protein-CPP cargoes could be applied following pretreatment of hydrophobic CPP-additive 2, suggesting an alternative application scheme.
To further elucidate the performance of cell-surface-retained hydrophobic CPP-additive, we compared 2 with the established small-molecule counterion pyrenebutyrate (PyB). Previous work reports PyB as a counterion with strong membrane interactions that facilitates the uptake of R8-EGFP into HeLa cells.45 We pretreated cells with PyB (50, 10, and 5 μM) or 2 (5 μM) for 5 min, followed by the addition of cargo-protein I to obtain a final concentration of 10 or 5 μM and further incubated for 1 h. Cells pretreated with PyB (50 μM) or the hydrophobic CPP-additive 2 (5 μM) followed by I treatment (10 μM) showed a similar degree of nucleolar signal (Figure S25). Upon decreasing the concentration of PyB to 10 μM and lower, we observed only endosomal uptake. Additionally, lowering the concentration of I led to the loss of enhanced uptake of I by PyB even at 50 μM of the counterion. While PyB was previously reported to associate with the cell surface and enhance protein-CPP uptake,32,45 the hydrophobic CPP-additive 2 demonstrated a stronger enhancement of NLS-mCherry-R10 I uptake compared to PyB.
Based on these results, the combination of (1) better cell membrane accumulation, (2) early decrease in membrane tension, and (3) sustained membrane interaction highlights the significant improvements brought about by incorporating the hydrophobic “ILFF” motif onto the CPP-additive. It would be in the interest of the community to perform more detailed biophysical evaluations to give further explanation of the detailed mechanism of CPP-additives-mediated direct transduction through the cell membrane.
TNB-R10-ILFF Improves the Cellular Delivery of Functional Macrocyclic Peptides and a Recombinant Genome Editing Protein
Finally, we applied hydrophobic CPP-additive 2 to the functional delivery of peptide and protein cargoes into cells. CMR19 (4 kDa, 0 net charge) is a recently discovered macrocyclic peptide, identified by mRNA display, that binds strongly to the Leu- and Trp- pockets of the MDM2 protein to block p53 interactions. As this peptide is cell impermeable, we used cyclic CPP (cR10)-conjugation to CMR19 via an intracellularly cleavable disulfide bridge to enable cell uptake and reduce cell proliferation of MDM2-dependent SJSA1 cells.38 We hypothesized that CPP-additives, particularly the hydrophobic additive 2, would further enhance the uptake of CMR19-cR10. We monitored this effect by treating SJSA-1 cells with CMR19-cR10 with or without CPP-additives 1 and 2 (Figure 5A).
Figure 5.

Delivery of CMR19-cR10. (A) CMR19 is cell impermeable. Conjugation with cyclic R10 (CMR19-cR10) enables cell entry and a physiological effect. Cotreatment in the presence of 2 should improve the uptake resulting in improved cell death by apoptosis. (B) Cell viability after incubating SJSA1 cells with culture media (Control), p53/MDM2 protein–protein interaction inhibitor CMR19, or CMR19-cR10 (5 μM) in the presence of CPP-additive (5 μM) for 24 h in RPMI1640 with 2.5% FCS. (C) Apoptosis induction following peptide treatment. [C(i)] Flow cytometry showing mean percentage of live, apoptotic, and necrotic cells across biological replicates ± SEM and [C(ii)] luminescence-based caspase 3/7 activation assay. The data are presented as mean ± SEM of three biological replicates (N = 3). The indicated P-values are calculated using one-way ANOVA followed by Tukey’s post hoc analysis.
First, we conducted a cell viability assay to determine the impact of cotreatment of CMR19-cR10 and 2 on cell proliferation. As previously reported,38 CMR19-cR10 (5 μM) alone did not decrease the viability of the treated SJSA1 cells after 24 h (Figure 5B). Cotreating SJSA1 cells with CMR19-cR10 and 1 (5 μM) did not show a significant decrease in cell proliferation compared to the control. However, cotreatment with 2 (5 μM) resulted in a significant decrease in cell viability. Control setups, in which we tested the CPP-additives 1 and 2 alone, remained at ∼95% viability, demonstrating that the decreased cell viability is not caused by the CPP-additives (Figure S26A).
Second, we confirmed that the observed decrease in cell viability is due to apoptosis resulting from the recovery of p53 function. We performed annexin V–propidium iodide (PI) staining and caspase 3/7 activity assays. Annexin V binds to phosphatidylserines (PS) exposed on apoptotic cells, while cell-impermeable PIs intercalate with the DNA of cells with impaired cell membrane integrity.68 By flow cytometry, we differentiated necrotic cells [Q1: annexin V (−), PI (+)], late and early apoptotic cells [Q2: annexin V (+), PI (+); Q3: annexin V (+), PI (−)], and live cells [Q3: annexin V (−), PI (−)] (Figure S26B). We found that treatment with CMR19-cR10 alone resulted in ∼75% living cells and ∼15% apoptotic cells [Figure 5C(i)]. Cotreatment with CPP-additive 1 showed a result, while cotreatment with 2 led to a higher number of apoptotic cells (∼55%). We also assessed the activation of executioner caspases 3/7, which occurs during the later stages of apoptosis, through a luminescence-based assay. Only cells treated with CMR19-cR10 showed increased luminescence, where cotreatment with 2 led to statistically higher caspase 3/7 activation [Figure 5C(ii)]. Taken together, we provide evidence of improved CMR19-cR10 uptake into SJSA1 cells in the presence of 2 in a serum-containing medium.
Last, we used the hydrophobic peptide additive 2 to enhance the delivery of the functional genome-editing enzyme Cre recombinase (43 kDa, +25 net charge). To assess the delivery efficiency, we established a HeLa cell line stably transfected with the CreSTOPLight plasmid (HeLa-CreSTOP) to act as reporter cells.46,69 The reporter sequence in HeLa-CreSTOP cells contains a ZsGreen coding sequence flanked by loxP sites, positioned upstream of an mScarlet coding region. Reporter cells initially express ZsGreen. Upon successful cytosolic delivery of Cre, the loxP sites along with the ZsGreen coding region are recombined, allowing the expression of mScarlet, leading to a green-to-red fluorescence emission change in reporter cells (Figure 6A). We cotreated HeLa-CreSTOP cells with NLS-CreR8 (5 μM) only or in the presence of 1 or 2 (5 μM) in DMEM with 5% FCS. After 24 h of incubation, we changed the media to fresh DMEM with 10% FCS and continued incubation for another 24 h. Confocal tile-scan images of HeLa-CreSTOP treated with NLS-CreR8 showed only minimal mScarlet-expressing cells, whereas there were noticeably more red fluorescent cells in the presence of CPP-additives 1 and 2 (Figure 6B). It is worth mentioning that cotreatment of reporter cells with NLS-CreR8 (5 μM) and 2 (5 μM) resulted in more mScarlet-expressing cells with a decreased number of ZsGreen-expressing cells.
Figure 6.

Delivery of genome editing NLS-CreR8. (A) Schematic diagram of CreSTOPLight assay to determine successful cytosolic delivery of Cre recombinase. (B) Tile-scan confocal microscopy images of HeLa-CreSTOP cells cotreated with NLS-CreR8 (2.5 μM) with no additives or indicated CPP-additives (5 μM) after 24 h incubation in DMEM with 2.5% FCS. Inset shows magnified images (scale bar, 20 μm). [C(i)] Representative scatter plot showing EGFP (y-axis) and mScarlet (x-axis) of HeLa-CreSTOP cells cotreated with NLS-CreR8 only (top), NLS-CreR8 with 1 (middle) or 2 (bottom) analyzed by flow cytometry. [C(ii)] Graph showing the percentage of mScarlet-expressing cells among 20,000 gated events. Data presented as mean ± SEM of three biological replicates (N = 3). Indicated P-values are calculated by one-way ANOVA followed by Tukey’s post hoc analysis.
We validated the microscopy images with quantitative flow cytometry data. Scatter plot graphs were used to represent the expression levels of each fluorescent protein (x-axis, mScarlet expression; y-axis, ZsGreen expression) in individual cells within the population [Figure 6C(i)]. The population of reporter cells treated with NLS-CreR8 alone had a high ZsGreen signal but a low mScarlet signal [Figure 6C(i); top panel]. In contrast, the population of cells treated with both NLS-CreR8 and 1 or 2 showed a higher mScarlet signal [Figure 6C(i); middle and bottom]. The percentages of mScarlet-expressing cells after cotreatment of NLS-CreR8 and 1 or 2 are at 80 and 95%, respectively, with the latter being significantly higher than the former [Figure 6C(ii)].
Conclusions
We report that the addition of a small hydrophobic tag to the cell-surface-anchoring CPP-additives greatly enhances the delivery of functional peptide and protein payloads. Through careful screening, we identified the hydrophobic CPP-additive 2 as having enhanced performance in facilitating the uptake protein-CPP into cells compared to other hydrophobic anchors, which showed high cytotoxicity and loss of membrane integrity. Here, we demonstrate that this hydrophobic CPP-additive efficiently delivered larger CPP-protein cargoes, such as the nonpermeable NLS-mCherry model protein (∼32 kDa) at low μM concentrations compared to the nontagged version.
Structure–activity relationship studies of 2 established the following key features: (1) a cell-surface anchoring moiety in the form of an electrophilic disulfide, (2) a polyarginine sequence, and (3) a local hydrophobic stretch. Interestingly, the distance between the polyarginine sequence and the hydrophobic ILFF-peptide, as well as transposing the sequence to present the hydrophobic motif on the N-terminus, did not affect the protein uptake efficiency. Additionally, cells cotreated with 2 and an NLS-mCherry conjugated with the ILFF-containing R10 IV showed a higher uptake as compared to the original NLS-mCherry-R10 I, suggesting a potential cooperative effect of hydrophobic CPP-conjugated protein with hydrophobic CPP-additives. Further investigations are ongoing to better understand the contribution of hydrophobic motifs to both protein-CPP and CPP-additives in enhancing cellular delivery.
Our time-lapse microscopy experiments demonstrated that a Cy5-labeled hydrophobic CPP-additive remains strongly associated with the cell membrane, while an alkylated control and the nontagged analogue showed reduced membrane interaction. Furthermore, cells pretreated with the hydrophobic CPP-additive 2 for 5 min remained capable of allowing the direct transduction of NLS-mCherry-R10 I, in contrast to cells pretreated with CPP-additive 1. This implies that cell surface retention of CPP-additives is crucial in facilitating the uptake of protein-CPP cargoes.
Hydrophobic CPP-additives enhanced the uptake of protein-CPP cargoes in living cells at 37 °C. The rapid appearance of the nucleolar signal after cotreatment with NLS-mCherry-R10 I and hydrophobic CPP-additive 23 and the absence of entrapped fluorescent cargoes or additives below 10 min strongly suggest, that direct transduction is primarily involved in the uptake of protein-CPPs at this point (Figure 4C). Additionally, delayed addition of hydrophobic CPP-additives 2 did not result in the release of entrapped proteins (Figure S23). Performing the uptake experiments at 4 °C also pointed toward nucleolar localization of protein cargo I via direct transduction, although with reduced efficiency compared to 37 °C (Figure S22). While this lower efficiency could be explained by a mixed mechanism of direct transduction and endosomal escape (for example, if the protein cargo I and the hydrophobic CPP-additive 2 undergo simultaneous endosomal uptake), diminished reactivity toward disulfide-bond formation as well as reduced membrane dynamics could also account for this observation.
Examining the local effect of retained CPP-additives revealed that hydrophobic CPP-additive decreased the cell membrane tension. By comparing the change in membrane tension at CPP-additive accumulation sites on the cell membrane, we observed a significant decrease in membrane tension at accumulation sites of 23 after 15 s of incubation. This indicates that the hydrophobic tagged-CPP-additives allow faster cell membrane association and interaction, which together with sustained membrane interaction rationalizes the improved NLS-mCherry-R10 I uptake. This work pioneers the use of a hydrophobic moiety installed on CPP-additives to facilitate direct protein transduction, which involves prolonged retention on the cell membrane and better priming of protein-CPP entry sites. Additional biophysical characterizations will shed light on the precise effects and modes of action of hydrophobic CPP-additives.
Finally, we applied improved hydrophobic CPP-additive 2 to deliver functional peptide and protein payloads. Cotreatment of the macrocyclic peptide inhibitor of MDM2 (CMR19-cR10) with 2 resulted in higher cell death by apoptosis in SJSA1 cells, showing enhanced delivery compared to 1. Hydrophobic CPP-additive 2 also enhanced the uptake of the recombinant genome editing protein, NLS-CreR8, into reporter cells, leading to more cells with the expected genetic recombination. These findings demonstrate that codelivery of cargo-CPP conjugate and 2 leads to significantly higher functional readout than using 1, further exemplifying the improvement provided by the hydrophobic motif.
In conclusion, our work presents the next-generation of CPP-additive technology achieved through the simple addition of a hydrophobic ILFF tag. This enhancement results in longer cell membrane retention, faster effects on membrane tension, and improved direct transduction of protein-CPP. We are currently working toward adding cell specificity to CPP-additives and understanding the cooperative effects between CPP-additives and protein-CPPs, which could lead to the application of hydrophobic CPP-additive technology in a more complex biological system. This research holds promise for advancing the field of cellular delivery and has potential for various biotechnological and therapeutic applications.
Acknowledgments
We thank Dr. Martin Lehmann and Dr. Agata Witkowska (FMP-Berlin) for their valuable input for the microscopy experiments and fluorescence lifetime imaging, respectively. We also thank Dr. Christopher Wolff (FMP-Berlin) for his assistance in creating the quantification script used throughout the paper. We also thank Dr. Andreas L. Marzinzik (Novartis) and Dr. Anselm Schneider for their aid in acquiring the CMR19 and CMR19-cR10 peptides. For general peptide synthesis, we thank Ines Kretzschmar. J.V.V.A. is funded by the Alexander von Humboldt Fellowship for Postdocs. C.P.R.H., J.F., and L.F. acknowledge the support from the DFG (RTG2473 “Bioactive Peptides”, Project-number 392923329 and CRC1349, Project-number 387284271). J.G.-G. is funded by Margarita Salas Fellowship from the Spanish Ministry of Universities. C.P.R.H. and J.F. thank the Fonds der Chemischen Industrie (FCI) for the Chemiefonds fellowship. Figures 1, 2D, 5A, and 6A, and TOC were created using https://www.biorender.com/.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c05365.
Materials and methods, peptide structures and UPLC chromatograms (ZIP)
The authors declare the following competing financial interest(s): The technology described in this article for the delivery of cargoes into cells using cell-surface-reactive peptides is part of a patent application (WO2022180242A2) by J.V.V.A. and C.P.R.H. published in 2022.09.01.
Supplementary Material
References
- Zinn S.; Vazquez-Lombardi R.; Zimmermann C.; Sapra P.; Jermutus L.; Christ D. Advances in Antibody-Based Therapy in Oncology. Nat. Cancer 2023, 4 (2), 165–180. 10.1038/s43018-023-00516-z. [DOI] [PubMed] [Google Scholar]
- Paunovska K.; Loughrey D.; Dahlman J. E. Drug Delivery Systems for RNA Therapeutics. Nat. Rev. Genet. 2022, 23 (5), 265–280. 10.1038/s41576-021-00439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesik-Brodacka M. Progress in Biopharmaceutical Development. Biotechnol. Appl. Biochem. 2018, 65 (3), 306–322. 10.1002/bab.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillard S.; Minter R. R.; Jackson R. H. Engineering Therapeutic Proteins for Cell Entry: The Natural Approach. Trends Biotechnol. 2015, 33 (3), 163–171. 10.1016/j.tibtech.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Esfandiari A.; Cassidy S.; Webster R. M. Bispecific Antibodies in Oncology. Nat. Rev. Drug Discovery 2022, 21 (6), 411–412. 10.1038/d41573-022-00040-2. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Qin X.; Kong F.; Chen P.; Pan G. Improving Cellular Uptake of Therapeutic Entities through Interaction with Components of Cell Membrane. Drug Delivery 2019, 26 (1), 328–342. 10.1080/10717544.2019.1582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N. S.; Burkholder J.; Roberts B.; Martinell B.; McCabe D. In Vivo and in Vitro Gene Transfer to Mammalian Somatic Cells by Particle Bombardment. Proc. Natl. Acad. Sci. U.S.A. 1990, 87 (24), 9568–9572. 10.1073/pnas.87.24.9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenac A.; Schilberth F. L.; Wagner E.; Wixforth A.; Lächelt U.; Westerhausen C. Transient Permeabilization of Living Cells: Combining Shear Flow and Acoustofluidic Trapping for the Facilitated Uptake of Molecules. Processes 2021, 9 (6), 913. 10.3390/pr9060913. [DOI] [Google Scholar]
- Morshedi Rad D.; Alsadat Rad M.; Razavi Bazaz S.; Kashaninejad N.; Jin D.; Ebrahimi Warkiani M. A Comprehensive Review on Intracellular Delivery. Adv. Mater. 2021, 33 (13), 2005363. 10.1002/adma.202005363. [DOI] [PubMed] [Google Scholar]
- Smith B. A.; Daniels D. S.; Coplin A. E.; Jordan G. E.; McGregor L. M.; Schepartz A. Minimally Cationic Cell-Permeable Miniature Proteins via α-Helical Arginine Display. J. Am. Chem. Soc. 2008, 130 (10), 2948–2949. 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- Daniels D. S.; Schepartz A. Intrinsically Cell-Permeable Miniature Proteins Based on a Minimal Cationic PPII Motif. J. Am. Chem. Soc. 2007, 129 (47), 14578–14579. 10.1021/ja0772445. [DOI] [PubMed] [Google Scholar]
- Gasparini G.; Sargsyan G.; Bang E.-K.; Sakai N.; Matile S. Ring Tension Applied to Thiol-Mediated Cellular Uptake. Angew. Chem., Int. Ed. 2015, 54 (25), 7328–7331. 10.1002/anie.201502358. [DOI] [PubMed] [Google Scholar]
- Laurent Q.; Martinent R.; Lim B.; Pham A.-T.; Kato T.; López-Andarias J.; Sakai N.; Matile S. Thiol-Mediated Uptake. JACS Au 2021, 1 (6), 710–728. 10.1021/jacsau.1c00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; LaRochelle J. R.; Jiang B.; Lian W.; Hard R. L.; Selner N. G.; Luechapanichkul R.; Barrios A. M.; Pei D. Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery. Biochemistry 2014, 53 (24), 4034–4046. 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo-Oliveras A.; Najjar K.; Dayani L.; Wang T.-Y.; Johnson G. A.; Pellois J.-P. Protein Delivery into Live Cells by Incubation with an Endosomolytic Agent. Nat. Methods 2014, 11 (8), 861–867. 10.1038/nmeth.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akishiba M.; Takeuchi T.; Kawaguchi Y.; Sakamoto K.; Yu H.-H.; Nakase I.; Takatani-Nakase T.; Madani F.; Gräslund A.; Futaki S. Cytosolic Antibody Delivery by Lipid-Sensitive Endosomolytic Peptide. Nat. Chem. 2017, 9 (8), 751–761. 10.1038/nchem.2779. [DOI] [PubMed] [Google Scholar]
- Du S.; Liew S. S.; Li L.; Yao S. Q. Bypassing Endocytosis: Direct Cytosolic Delivery of Proteins. J. Am. Chem. Soc. 2018, 140 (47), 15986–15996. 10.1021/jacs.8b06584. [DOI] [PubMed] [Google Scholar]
- Bechara C.; Sagan S. Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand?. FEBS Lett. 2013, 587 (12), 1693–1702. 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- Vivès E.; Brodin P.; Lebleu B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272 (25), 16010–16017. 10.1074/JBC.272.25.16010. [DOI] [PubMed] [Google Scholar]
- Futaki S.; Nakase I. Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization. Acc. Chem. Res. 2017, 50 (10), 2449–2456. 10.1021/acs.accounts.7b00221. [DOI] [PubMed] [Google Scholar]
- Fawell S.; Seery J.; Daikh Y.; Moore C.; Chen L. L.; Pepinsky B.; Barsoum J. Tat-Mediated Delivery of Heterologous Proteins into Cells. Proc. Natl. Acad. Sci. U.S.A. 1994, 91 (2), 664–668. 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futaki S.; Suzuki T.; Ohashi W.; Yagami T.; Tanaka S.; Ueda K.; Sugiura Y. Arginine-Rich Peptides. J. Biol. Chem. 2001, 276 (8), 5836–5840. 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- Tünnemann G.; Martin R. M.; Haupt S.; Patsch C.; Edenhofer F.; Cardoso M. C. Cargo-dependent Mode of Uptake and Bioavailability of TAT-containing Proteins and Peptides in Living Cells. FASEB J. 2006, 20 (11), 1775–1784. 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- Futaki S.; Arafiles J. V. V.; Hirose H. Peptide-Assisted Intracellular Delivery of Biomacromolecules. Chem. Lett. 2020, 49 (9), 1088–1094. 10.1246/cl.200392. [DOI] [Google Scholar]
- Pei D.; Buyanova M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30 (2), 273–283. 10.1021/acs.bioconjchem.8b00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar K.; Erazo-Oliveras A.; Brock D. J.; Wang T.-Y.; Pellois J.-P. An L- to d-Amino Acid Conversion in an Endosomolytic Analog of the Cell-Penetrating Peptide TAT Influences Proteolytic Stability, Endocytic Uptake, and Endosomal Escape. J. Biol. Chem. 2017, 292 (3), 847–861. 10.1074/jbc.M116.759837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani F.; Lindberg S.; Langel Ü.; Futaki S.; Gräslund A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 1–10. 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard J. B.; Jessop T. C.; Lewis R. S.; Murray B. A.; Wender P. A. Role of Membrane Potential and Hydrogen Bonding in the Mechanism of Translocation of Guanidinium-Rich Peptides into Cells. J. Am. Chem. Soc. 2004, 126 (31), 9506–9507. 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- Moghal M. M. R.; Islam M. Z.; Hossain F.; Saha S. K.; Yamazaki M. Role of Membrane Potential on Entry of Cell-Penetrating Peptide Transportan 10 into Single Vesicles. Biophys. J. 2020, 118 (1), 57–69. 10.1016/j.bpj.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobin M.-L. L.; Blanchet M.; Henry S.; Chaignepain S.; Manigand C.; Castano S.; Lecomte S.; Burlina F.; Sagan S.; Alves I. D. The Role of Tryptophans on the Cellular Uptake and Membrane Interaction of Arginine-Rich Cell Penetrating Peptides. Biochim. Biophys. Acta, Biomembr. 2015, 1848 (2), 593–602. 10.1016/j.bbamem.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Pujals S.; Miyamae H.; Afonin S.; Murayama T.; Hirose H.; Nakase I.; Taniuchi K.; Umeda M.; Sakamoto K.; Ulrich A. S.; Futaki S. Curvature Engineering: Positive Membrane Curvature Induced by Epsin N-Terminal Peptide Boosts Internalization of Octaarginine. ACS Chem. Biol. 2013, 8 (9), 1894–1899. 10.1021/cb4002987. [DOI] [PubMed] [Google Scholar]
- Murayama T.; Masuda T.; Afonin S.; Kawano K.; Takatani-Nakase T.; Ida H.; Takahashi Y.; Fukuma T.; Ulrich A. S.; Futaki S. Loosening of Lipid Packing Promotes Oligoarginine Entry into Cells. Angew. Chem., Int. Ed. 2017, 56 (26), 7644–7647. 10.1002/anie.201703578. [DOI] [PubMed] [Google Scholar]
- Tietz O.; Cortezon-Tamarit F.; Chalk R.; Able S.; Vallis K. A. Tricyclic Cell-Penetrating Peptides for Efficient Delivery of Functional Antibodies into Cancer Cells. Nat. Chem. 2022, 14 (3), 284–293. 10.1038/s41557-021-00866-0. [DOI] [PubMed] [Google Scholar]
- Du S.; Liew S. S.; Zhang C.; Du W.; Lang W.; Yao C. C. Y.; Li L.; Ge J.; Yao S. Q. Cell-Permeant Bioadaptors for Cytosolic Delivery of Native Antibodies: A “Mix-and-Go” Approach. ACS Cent. Sci. 2020, 6 (12), 2362–2376. 10.1021/acscentsci.0c01379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Lau S. Y.; Lim Z. W.; Chang S. C.; Ghadessy F.; Partridge A.; Miserez A. Phase-Separating Peptides for Direct Cytosolic Delivery and Redox-Activated Release of Macromolecular Therapeutics. Nat. Chem. 2022, 14 (3), 274–283. 10.1038/s41557-021-00854-4. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wan T.; Li B.; Pan Q.; Xin H.; Qiu Y.; Ping Y. Rational Design of Poly(Disulfide)s as a Universal Platform for Delivery of CRISPR-Cas9Machineries toward Therapeutic Genome Editing. ACS Cent. Sci. 2021, 7 (6), 990–1000. 10.1021/acscentsci.0c01648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichart F.; Horn M.; Neundorf I. Cyclization of a Cell-Penetrating Peptide via Click-Chemistry Increases Proteolytic Resistance and Improves Drug Delivery. J. Pept. Sci. 2016, 22 (6), 421–426. 10.1002/psc.2885. [DOI] [PubMed] [Google Scholar]
- Schneider A. F. L.; Kallen J.; Ottl J.; Reid P. C.; Ripoche S.; Ruetz S.; Stachyra T.-M.; Hintermann S.; Dumelin C. E.; Hackenberger C. P. R.; Marzinzik A. L. Discovery, X-Ray Structure and CPP-Conjugation Enabled Uptake of P53/MDM2Macrocyclic Peptide Inhibitors. RSC Chem. Biol. 2021, 2 (6), 1661–1668. 10.1039/D1CB00056J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischan N.; Herce H. D.; Natale F.; Bohlke N.; Budisa N.; Cardoso M. C.; Hackenberger C. P. R. Covalent Attachment of Cyclic TAT Peptides to GFP Results in Protein Delivery into Live Cells with Immediate Bioavailability. Angew. Chem., Int. Ed. 2015, 54 (6), 1950–1953. 10.1002/anie.201410006. [DOI] [PubMed] [Google Scholar]
- Herce H. D.; Schumacher D.; Schneider A. F. L.; Ludwig A. K.; Mann F. A.; Fillies M.; Kasper M.-A.; Reinke S.; Krause E.; Leonhardt H.; Cardoso M. C.; Hackenberger C. P. R. Cell-Permeable Nanobodies for Targeted Immunolabelling and Antigen Manipulation in Living Cells. Nat. Chem. 2017, 9 (8), 762–771. 10.1038/nchem.2811. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Nakase I.; Michiue H.; Takeuchi T.; Tomizawa K.; Matsui H.; Futaki S. Enhanced Intracellular Delivery Using Arginine-Rich Peptides by the Addition of Penetration Accelerating Sequences (Pas). J. Controlled Release 2009, 138 (2), 128–133. 10.1016/j.jconrel.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Mann G.; Satish G.; Brik A. Enhanced Live-Cell Delivery of Synthetic Proteins Assisted by Cell-Penetrating Peptides Fused to DABCYL. Angew. Chem., Int. Ed. 2021, 60 (13), 7333–7343. 10.1002/anie.202016208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A.; Mandal S.; Arafiles J. V. V.; Gómez-González J.; Hackenberger C. P. R.; Brik A. Structure-Uptake Relationship Study of DABCYL Derivatives Linked to Cyclic Cell-Penetrating Peptides for Live-Cell Delivery of Synthetic Proteins. Angew. Chem., Int. Ed. 2022, 61 (47), e202207551 10.1002/anie.202207551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara M.; Perret F.; Takeuchi T.; Futaki S.; Lazar A. N.; Coleman A. W.; Sakai N.; Matile S. Arginine Magic with New Counterions up the Sleeve. Org. Biomol. Chem. 2005, 3 (9), 1659. 10.1039/b501472g. [DOI] [PubMed] [Google Scholar]
- Takeuchi T.; Kosuge M.; Tadokoro A.; Sugiura Y.; Nishi M.; Kawata M.; Sakai N.; Matile S.; Futaki S. Direct and Rapid Cytosolic Delivery Using Cell-Penetrating Peptides Mediated by Pyrenebutyrate. ACS Chem. Biol. 2006, 1 (5), 299–303. 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- Schneider A. F. L.; Kithil M.; Cardoso M. C.; Lehmann M.; Hackenberger C. P. R. Cellular Uptake of Large Biomolecules Enabled by Cell-Surface-Reactive Cell-Penetrating Peptide Additives. Nat. Chem. 2021, 13 (6), 530–539. 10.1038/s41557-021-00661-x. [DOI] [PubMed] [Google Scholar]
- Duchardt F.; Fotin-Mleczek M.; Schwarz H.; Fischer R.; Brock R. A Comprehensive Model for the Cellular Uptake of Cationic Cell-Penetrating Peptides. Traffic 2007, 8 (7), 848–866. 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Hirose H.; Takeuchi T.; Osakada H.; Pujals S.; Katayama S.; Nakase I.; Kobayashi S.; Haraguchi T.; Futaki S. Transient Focal Membrane Deformation Induced by Arginine-Rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20 (5), 984–993. 10.1038/mt.2011.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A. F. L.; Benz L. S.; Lehmann M.; Hackenberger C. P. R. Cell-Permeable Nanobodies Allow Dual-Color Super-Resolution Microscopy in Untransfected Living Cells. Angew. Chem., Int. Ed. 2021, 60 (40), 22075–22080. 10.1002/anie.202103068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönn P.; Kacsinta A. D.; Cui X.-S.; Hamil A. S.; Kaulich M.; Gogoi K.; Dowdy S. F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6 (1), 32301. 10.1038/srep32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Liu T.; Liu Y.-Y.; Briesewitz R.; Barrios A. M.; Jhiang S. M.; Pei D. Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS Chem. Biol. 2013, 8 (2), 423–431. 10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K.; Michibata J.; Hirai Y.; Ide A.; Ikitoh A.; Takatani-Nakase T.; Futaki S. Potentiating the Membrane Interaction of an Attenuated Cationic Amphiphilic Lytic Peptide for Intracellular Protein Delivery by Anchoring with Pyrene Moiety. Bioconjugate Chem. 2021, 32 (5), 950–957. 10.1021/acs.bioconjchem.1c00101. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y.; Arafiles J. V. V.; Kawaguchi Y.; Nakase I.; Hirose H.; Futaki S. Stearylated Macropinocytosis-Inducing Peptides Facilitating the Cellular Uptake of Small Extracellular Vesicles. Bioconjugate Chem. 2022, 33 (5), 869–880. 10.1021/acs.bioconjchem.2c00113. [DOI] [PubMed] [Google Scholar]
- Schneider A. F. L.; Wallabregue A. L. D.; Franz L.; Hackenberger C. P. R. Targeted Subcellular Protein Delivery Using Cleavable Cyclic Cell-Penetrating Peptides. Bioconjugate Chem. 2019, 30 (2), 400–404. 10.1021/acs.bioconjchem.8b00855. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. Experimentally Determined Hydrophobicity Scale for Proteins at Membrane Interfaces. Nat. Struct. Mol. Biol. 1996, 3 (10), 842–848. 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Hansen S.; Arafiles J. V. V.; Ochtrop P.; Hackenberger C. P. R. Modular Solid-Phase Synthesis of Electrophilic Cysteine-Selective Ethynyl-Phosphonamidate Peptides. Chem. Commun. 2022, 58 (60), 8388–8391. 10.1039/D2CC02379B. [DOI] [PubMed] [Google Scholar]
- Takechi Y.; Tanaka H.; Kitayama H.; Yoshii H.; Tanaka M.; Saito H. Comparative Study on the Interaction of Cell-Penetrating Polycationic Polymers with Lipid Membranes. Chem. Phys. Lipids 2012, 165 (1), 51–58. 10.1016/j.chemphyslip.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Robison A. D.; Sun S.; Poyton M. F.; Johnson G. A.; Pellois J.-P.; Jungwirth P.; Vazdar M.; Cremer P. S. Polyarginine Interacts More Strongly and Cooperatively than Polylysine with Phospholipid Bilayers. J. Phys. Chem. B 2016, 120 (35), 9287–9296. 10.1021/acs.jpcb.6b05604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesei G.; Vazdar M.; Jensen M. R.; Cragnell C.; Mason P. E.; Heyda J.; Skepö M.; Jungwirth P.; Lund M. Self-Association of a Highly Charged Arginine-Rich Cell-Penetrating Peptide. Proc. Natl. Acad. Sci. U.S.A. 2017, 114 (43), 11428–11433. 10.1073/pnas.1712078114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuge M.; Takeuchi T.; Nakase I.; Jones A. T.; Futaki S. Cellular Internalization and Distribution of Arginine-Rich Peptides as a Function of Extracellular Peptide Concentration, Serum, and Plasma Membrane Associated Proteoglycans. Bioconjugate Chem. 2008, 19 (3), 656–664. 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Hirose H.; Tanaka G.; Pujals S.; Katayama S.; Nakase I.; Futaki S. Effect of the Attachment of a Penetration Accelerating Sequence and the Influence of Hydrophobicity on Octaarginine-Mediated Intracellular Delivery. Mol. Pharmaceutics 2012, 9 (5), 1222–1230. 10.1021/mp200518n. [DOI] [PubMed] [Google Scholar]
- Colom A.; Derivery E.; Soleimanpour S.; Tomba C.; Molin M. D.; Sakai N.; González-Gaitán M.; Matile S.; Roux A. A Fluorescent Membrane Tension Probe. Nat. Chem. 2018, 10 (11), 1118–1125. 10.1038/s41557-018-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Calvo J.; López-Andarias J.; Maillard J.; Mercier V.; Roffay C.; Roux A.; Fürstenberg A.; Sakai N.; Matile S. HydroFlipper Membrane Tension Probes: Imaging Membrane Hydration and Mechanical Compression Simultaneously in Living Cells. Chem. Sci. 2022, 13 (7), 2086–2093. 10.1039/D1SC05208J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M.; Ignasiak M. T.; Chan B.; Croft A. K.; Radom L.; Schiesser C. H.; Pattison D. I.; Davies M. J. Reactivity of Disulfide Bonds Is Markedly Affected by Structure and Environment: Implications for Protein Modification and Stability. Sci. Rep. 2016, 6 (1), 38572. 10.1038/srep38572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los D. A.; Murata N. Membrane Fluidity and Its Roles in the Perception of Environmental Signals. Biochim. Biophys. Acta, Biomembr. 2004, 1666 (1–2), 142–157. 10.1016/j.bbamem.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Okuda A.; Tahara S.; Hirose H.; Takeuchi T.; Nakase I.; Ono A.; Takehashi M.; Tanaka S.; Futaki S. Oligoarginine-Bearing Tandem Repeat Penetration-Accelerating Sequence Delivers Protein to Cytosol via Caveolae-Mediated Endocytosis. Biomacromolecules 2019, 20 (5), 1849–1859. 10.1021/acs.biomac.8b01299. [DOI] [PubMed] [Google Scholar]
- Erazo-Oliveras A.; Najjar K.; Truong D.; Wang T.-Y.; Brock D. J.; Prater A. R.; Pellois J.-P. The Late Endosome and Its Lipid BMP Act as Gateways for Efficient Cytosolic Access of the Delivery Agent DfTAT and Its Macromolecular Cargos. Cell Chem. Biol. 2016, 23 (5), 598–607. 10.1016/j.chembiol.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S. E.; Elgendy M.; Martin S. J. Expression, Purification and Use of Recombinant Annexin V for the Detection of Apoptotic Cells. Nat. Protoc. 2009, 4 (9), 1383–1395. 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- Yang Y. S.; Hughes T. E. Cre Stoplight: A Red/Green Fluorescent Reporter of Cre Recombinase Expression in Living Cells. Biotechniques 2001, 31 (5), 1036–1041. 10.2144/01315st03. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.