

Abstract
Bromination of high-pressure, high-temperature (HPHT) nanodiamond (ND) surfaces has not been explored and can open new avenues for increased chemical reactivity and diamond lattice covalent bond formation. The large bond dissociation energy of the diamond lattice–oxygen bond is a challenge that prevents new bonds from forming, and most researchers simply use oxygen-terminated NDs (alcohols and acids) as reactive species. In this work, we transformed a tertiary-alcohol-rich ND surface to an amine surface with ~50% surface coverage and was limited by the initial rate of bromination. We observed that alkyl bromide moieties are highly labile on HPHT NDs and are metastable as previously found using density functional theory. The strong leaving group properties of the alkyl bromide intermediate were found to form diamond–nitrogen bonds at room temperature and without catalysts. This robust pathway to activate a chemically inert ND surface broadens the modalities for surface termination, and the unique surface properties of brominated and aminated NDs are impactful to researchers for chemically tuning diamond for quantum sensing or biolabeling applications.
Graphical Abstract
The addition of bromine to small molecules, macro-molecules, and carbon nanomaterials typically includes a bromine source such as Br2, HBr, or N-bromosuccinimide and can lead to bromine bonds with sp3- or sp2-hybridized carbon atoms. Bromine bound to carbon serves as a good leaving group and enables a catalogue of reactions to be performed.1 Powerful demonstrations include Ullman coupling and Sonogashira reactions that use the enhanced leaving group properties of halogens in general and bromine in particular to form new chemical transformations.2,3 The synthesis of highly ordered nanoarchitectures with halogenated porphyrin rings takes advantage of the temperature-dependent dissociation of iodine and bromine to direct assembly on metal surfaces with a high level of fidelity.4,5 Bromine-functionalized graphene and carbon nanotubes have allowed robust chemical routes to be used for band engineering and sensing applications.6–9 In contrast, bromination on the surface of bulk diamond and detonation nanodiamonds (NDs) has not been widely explored—for good reason.10,11 Bromine has an atomic radius of 112 pm,12 which is large compared to the densely packed facet of the 111 diamond surface with 18 atoms/nm2.13 For that reason, bromination of Si and Ge is more common due to the increase in crystal lattice spacing and decreased steric hindrance.14–16 One previous bromination protocol with N-bromosuccinimide or NBS in CCl4 was used with detonation NDs, and a single X-ray photoelectron spectroscopy (XPS) spectrum is recorded with water being used during purification cycles.17 The XPS in Martin et al. shows broad Br 3d features from 67.5 to 77.5 eV and has low signal-to-noise, maybe due to the near-complete rehydroxylation of their sample during purification. In this work, we show the following: alkyl bromide formation on high-pressure, high-temperature (HPHT) NDs is possible using thionyl bromide (SOBr2); it displays enhanced chemical reactivity compared to that of brominated small molecule analogues, and the reaction products must be carefully handled under inert conditions to retain alkyl bromide moieties. This work addresses the difficulty in chemically activating HPHT ND surfaces, provides a new platform for atomic and molecular control, and will be of interest to researchers using diamond and other ultrahard materials for quantum sensing applications.18–20
Understanding this work is based on the knowledge that open-air aerobic oxidation of HPHT ND powders results in alcohol (hydroxyl) groups terminating the surface in a fashion similar to that for bulk diamond on the {111} facet and not carboxylic acid termination as found on detonation NDs.21,22 Alcohol termination of bulk diamond is widely accepted within the diamond research community, but this fact has not been widely accepted by researchers studying HPHT NDs. Kono, Wang, and Loh discuss and reference evidence for alcohol termination on bulk diamond based on surface energy, crystallographic structure, and sterics.23–25 This conclusion is supported by spectroscopic data and is rationalized by the understanding that the {111} facet is predominantly exposed in HPHT NDs due to ball milling. Similar to bulk diamond, {111} facets are single-dangling-bond surfaces and may form one sp3-hybridized bond per surface carbon atom.26 HPHT of 30–50 and 5 nm detonation NDs does not have the same surface termination after aerobic oxidation and must be addressed separately. The differences between HPHT and detonation NDs are a result of the production method, their resultant surface-to-volume ratio, and exposed crystallographic facets. HPHT NDs of 30–100 nm are produced in a top-down approach through ball milling of bulk single-crystal diamonds by manufacturers such as Microdiamant (Switzerland) and are commercially sold for polishing applications.27 Ball milling predominantly cleaves the {111} crystallographic planes based on facet strength and in turn exposes the single-dangling-bond surface.28 The dangling sp3 bond can then support alcohol bond formation during aerobic oxidation, as seen in bulk 111-terminated diamond.26 In handling bulk diamond, a jeweler cleaves a rough diamond stone along the (111) direction to produce a well-cleaved diamond, and this phenomenon has been empirically known for centuries.29 Density functional theory (DFT) calculations by Telling clearly showed that diamond cleavage proceeds as a function of strain from {111} → {110} → {100} with maximum strengths of 90, 130, and 225 GPa, respectively, and is caused by strength anisotropy.30 Electron microscopy of 30–100 nm HPHT NDs has verified the cleavage process, with irregularly shaped particles that resemble shards of broken crystallites.31 In contrast, detonation NDs are produced through a bottom-up approach wherein hexogen or trinitrotoluene (TNT) is ignited in stainless steel vessels, resulting in faceted spherical particles with diameters of ~5 nm.32 Detonation NDs have a larger surface area (270–315 m2/g)33 than HPHT NDs (57–140 m2/mg), a diverse range of carbon–oxygen moieties from ethers to acid anhydrides,34 and a high concentration of sp2 groups formed by dangling bonds,35 as confirmed via Raman spectroscopy.36 Only very recent comparative studies of sub-5 nm HPHT and detonation NDs at the same oxidation conditions (450 °C for 30 min) showed similar surface structures, which were a mixture of C–O and C=O bonding environments.37,38
In most settings where surface control is required, HPHT NDs (ranging from 30–100 nm) are first aerobically oxidized to remove amorphous carbon from the surface and produce an alcohol-rich surface (ND–OH).22 Chemical modification of the tertiary alcohol surface is a critical step for the construction of functional sensors using nitrogen vacancy centers (NVCs) or as a catalytic substrate, but modifying or removing the C–O–H bonds is challenging. The difficulty in modifying oxidized diamond surfaces is due to the large carbon–oxygen bond dissociation energies (1442 kJ/mol),39 high atomic surface density (18.2 atoms/nm2 on the 111 facet), and steric hindrance. In contrast, chemical modifications of detonation NDs typically target surface carboxylate groups via amidic coupling or reactive sp2 structures, which can be created by thermal annealing of detonation NDs. Much work has been accomplished with the more reactive 5 nm detonation materials and has been reviewed thoroughly.32,34,40 HPHT NDs can be converted from tertiary alcohols to carboxylic acids with acid–base–acid chemistry and provide a pathway for surface linking chemistry;41 however, the surface density of carboxylic acids can reach only a few percent of the surface carbon atoms and is inhomogeneous.42 Growth of silica (SiO2) shells on ND cores is a demonstrated route for chemical modification of HPHT NDs, and background-free cellular imaging, real-time magnetic sensing, and coating with tailored lipidic bilayers have been demonstrated.43–46 With silica shell growth, the carbon–oxygen groups are not removed from the diamond surface but instead used to form a priming layer to generate silica growth in a modified Stöber method.
For long-term advancements with HPHT NDs and ultradense materials such as silicon carbide, we frame this work as important, because a reactive surface intermediate is prepared at room temperature and could be used for any nucleophilic species to generate new diamond lattice covalent bonds. Direct amine bond formation of HPHT NDs has not been demonstrated and is a clear example of a useful surface termination approach for biolabeling applications due to the enhanced reactivity of amines. Previously, direct bulk diamond lattice–nitrogen bond formation is limited; for example, Stacey and co-workers used nitrogen-rich plasma treatment to terminate (001) diamond surfaces and produced a N–N-rich surface.47 Although these surfaces are expected to induce a positive electron affinity, they left behind minimal surface states within the bandgap and allowed the near-surface carbon atoms to exhibit bulk-like electronic states. In a related study, Sotowa et al. used gas-phase hydrogenation, chlorination, and amination chemistry of diamond powders in a tube furnace but yielded low carbon–nitrogen bonding rates that were dominated by the formation of imines and not amines.48 Zhu and Hamers converted hydrogen-terminated bulk diamond to an amine-terminated form using ammonia plasma and further established a negative electron affinity surface similar to hydrogen-terminated diamond.49 Simple wet chemistry of oxidized HPHT NDs with bromine and amine termination can open unique pathways for low-cost chemical transformations, surface dipole engineering, and NVC sensing and as a source of solvated electrons for chemistry.50–52
Here, we demonstrate that room temperature and catalysis-free diamond-lattice-to-nitrogen bond formation of aerobically oxidized HPHT NDs is possible through a highly labile alkyl bromide (ND–Br) intermediate. Surface analysis of the NDs revealed that ~50% of the surface carbon atoms were aminated, which is due to the upper limit of bromine termination previously described by DFT.53 We describe a reaction pathway that chemically activates the ND–OH, converting the alcohol groups to alkyl bromides; moreover, we found that the ND–Br surfaces have enhanced reactivity and that debromination occurs within seconds under open-air conditions and at 90 °C under inert conditions. Debromination under open-air conditions did not immediately yield new alcohol groups, suggesting that a long-lived “radical carbon” or other intermediate is present at the ND surface. Generating amine bonds on the diamond lattice is motivated by the enhanced reactivity of amines, the vast library of chemical modifications based on amines, and their negative electron affinity surface. Conjugation of ND–NH3 with folic acid to form amide bonds was performed, which established that reaction of these amine groups with small molecules was successful. By using mild nucleophiles (amines), we show that reactions with ND–Br under conditions at 25 °C and without catalysts are possible. Additionally, a reaction of ND–Br and propargylamine yielded a polyimine-coated ND core via a Sonogashira-type reaction and an 11-fold increase in the nitrogen content. With these reactive ND–Br constructs, researchers in chemistry, bioengineering, and materials science could use a catalogue of nucleophiles to generate new diamond–heteroatom bonds. Robust chemical and electronic structure analysis of the NDs using overlapping spectroscopic techniques under inert, temperature-dependent, and open-air conditions provided definitive characterization. This new surface pathway will likely be impactful for researchers using nanoscale diamonds with nitrogen vacancy centers (NVCs) for quantum sensing and biolabeling.
Hydrophilic ND–OH surface and water desorption via TPD-DRIFTS.
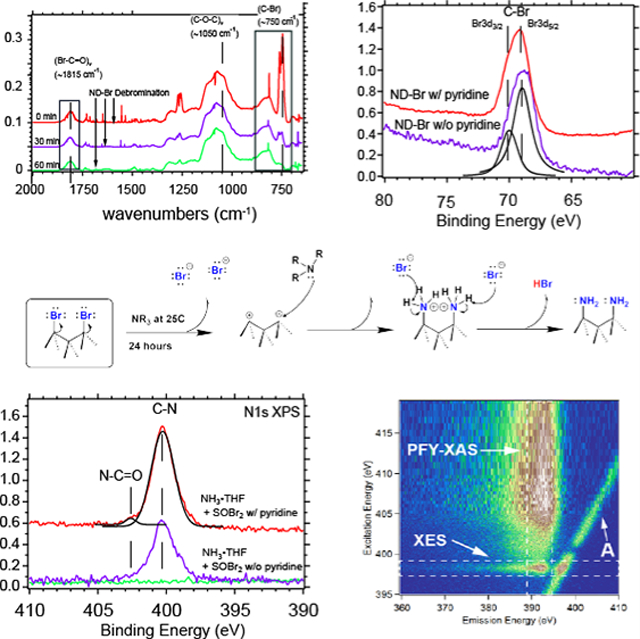
Key spectroscopic features of aerobically oxidized ND–OH can yield important information about hydrophilicity/hydrophobicity and the preparation of ND constructs prior to bromination chemistry. Aerobic oxidation of HPHT NDs rendered the 30–50 nm ND–OH particles hydrophilic and tannish in color after removal of dark amorphous carbon. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) provided surface vibrational modes of the ND–OH samples under open-air, inert atmosphere and temperature-dependent conditions. ND–OH samples have a pronounced peak at 1105 cm−1 assigned to the (C–O)ν band of tertiary alcohols, the bending mode of adsorbed water (O–H)δ at ~1630 cm−1, a carboxylic acid peak at 1780 cm−1, and a broad O–H stretching band (O–H)ν at 3000–3500 cm−1 due to adsorbed water and alcohol groups (Figure 1A). Using Kubelka–Munk transforms, we estimate that there are ~15:1 alcohol groups for each carboxylic acid observed after oxidation at 525 °C for 5 h, and this ratio can be estimated via integration and the available extinction coefficients of tertiary alcohols and carboxylic acids from NIST.22,54 Previous work on HPHT NDs mistakenly assigned the 1630 cm−1 peak to C=O-containing groups such as ketones, and we wished to clarify that this feature is not due to oxygen-terminated diamond.55 We confirmed the decrease of the adsorbed water spectral component at 1630 cm−1 via temperature-programmed desorption (TPD)-DRIFTS at 25, 100, 200, and 300 °C under open-air conditions. The observed water stretching band and bending mode simultaneously decreased as expected when water left the surface, and the stretching modes of (C–O)ν and (C=O)ν on the ND–OH surface remained (Figure 1A). The quantitative removal of adsorbed water was found to be 72, 97, and 100% at 100, 200, and 300 °C, respectively. Here, the 1630 cm−1 peak was used as a quantifiable signature for the dryness of our samples prior to bromination chemistry with SOBr2. During DRIFTS data collection, the observation of the adsorbed water band is a key metric in determining the hydrophilicity, hydrophobicity, and adsorbed water content during bromination and amination reactions. The materials and procedures of all samples are detailed in the Supporting Information (SI) and show ND–OH, ND–Br, and ND–NH3 sample preparation.
Figure 1.
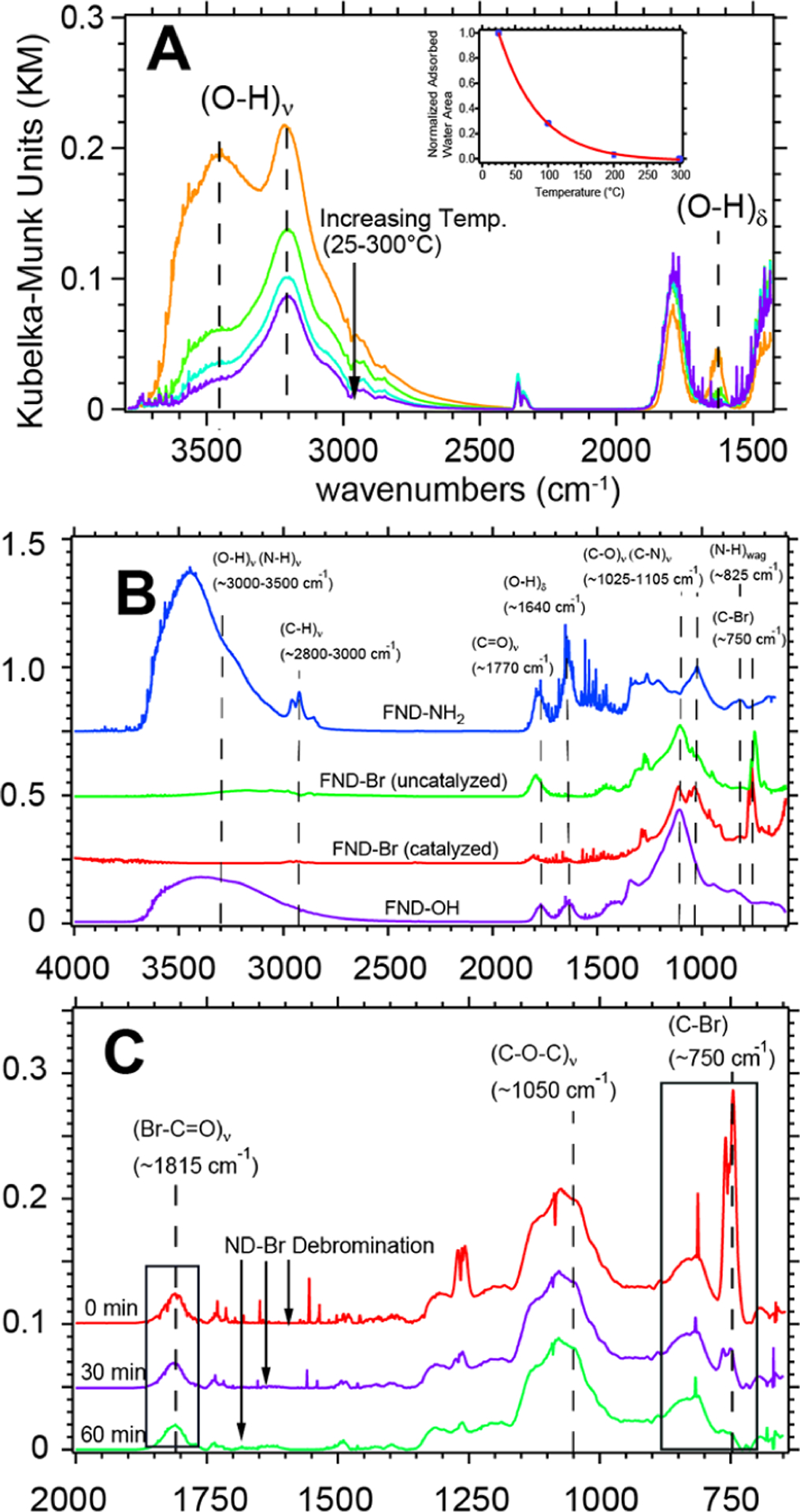
DRIFTS data showing the transition from ND–OH → ND–Br → ND–NH2 under open-air and inert atmosphere conditions. TPD-DRIFTS data from 25–300 °C confirm the contribution of (O–H)ν stretching modes due to both adsorbed water and the hydroxyl-terminated diamond (panel A). The (O–H)ν band from 3000–3500 cm−1 decreases proportionally to the elimination of (O–H)δ at 1630 cm−1. The inset shows the integrated (O–H)δ signal in Kubelka–Munk units as a function of temperature. DRIFTS spectra reveal a strong (C–Br)ν signal at 750 cm−1 that confirms alkyl bromide formation on the ND surface after addition of SOBr2 for 24 h with and without the presence of pyridine (panel B). After amination chemistry, ND–NH2 becomes highly hydrophilic; the (C–N)ν mode becomes prominent at 1025 cm−1, a small (N–H)wag signal at 825 cm−1 is observed, and the (C–Br)ν peak is absent (Panel B). In situ reactivity of the alkyl bromides under open-air conditions was tracked by opening two small valves on an inert atmosphere DRIFTS chamber and monitoring the decrease in the (C–Br)ν intensity at 750 cm−1 with pseudo-first-order kinetics (panel C). Note: During the debromination process, the (C–O–C)ν signal at 1075 cm−1 remained unchanged and did not convert readily back to the representative alcohol peak position of 1100 cm−1.
Synthesis and inert DRIFTS characterization of ND–Br.
The conversion of ND–OH (tertiary alcohols) to ND–Br (alkyl-bromide-terminated) constructs was accomplished with SOBr2 at room temperature, wherein the diamond lattice bromine bond is likely formed via an SN1 (substitution type 1) mechanism, and the addition of pyridine increases the ND–Br rate (see Figure S2 for a mechanism, and the SI for materials and procedures). ND–Br formation was confirmed with a strong doublet peak arising at 750 cm−1 due to the (C–Br)ν stretching mode (Figure 1B), which was in a region that was vibrationally silent in ND–OH. The minor (Br–C=O)ν of an acid bromide was also observed with a shift from 1780 → 1815 cm−1 and is understood by the few acids on the HPHT ND surface. We predict that the bromination of acid-rich HPHT NDs after acid–base–acid chemistry would yield a few percent of acid bromides consistent with ref 40, yet would be less reactive than the metastable alkyl bromide bonds based on our findings. Mechanistically (see Figure S2 in Supporting Information), ND–Br reactions with SOBr2 can proceed via a bromosulfite ether intermediate, a release of bromide ions via pyridine nucleophilic attack, and finally carbocation inter-mediate formation, resulting in nucleophilic attack by Br− on the diamond surface. Bromination of diamond with an SN2 (substitution type 2) mechanism would not be possible due to the saturation of all carbon–carbon bonds in the diamond subsurface and therefore should follow an SN1 or E1 mechanism during bromination with a carbocation intermediate. The mechanism depicted in Figure S2 is hypothetical and depicts the most likely route for bromination. ND–Br bond formation proceeded without pyridine within 24 h, with easier purification and a lower ND–Br yield (Figures 1B,C and S3 in Supporting Information). The use of pyridine produced a viscous side product of pyridinium perbromide (NC5H5·HBr·Br2), which was removed with a dimethyl sulfoxide (DMSO) washing step. An increased rate of ND–Br formation with pyridine was observed when comparing 2 and 24 h reactions and tracking the emergence of the (C–Br)ν stretching mode at 750 cm−1 (Figure S3 in Supporting Information). The champion ND–Br sample (orange trace in Figure 1C) was produced after only a single washing step and remained stable for 24 h, and the (C–Br)ν signal at 750 cm−1 dominated the spectra. The champion ND–Br sample lacks reproducibility due to instability associated with an increased level of bromination and is under investigation.
There is evidence of a unique intracrystallite Williamson-ether-like reaction occurring as a byproduct of the reaction of alkyl bromides and alcohols adjacent to one another on the diamond surface. Significant surface features are modified after the bromination reaction, including complete removal of the (O–H)ν band at 3000–3500 cm−1 from adsorbed water and removal of alcohol groups. While desorption of water was predicted due to desiccating the sample prior to bromination, the elimination of alcohol (C–O–H) groups was unexpected and suggested that ether functionalities (C–O–C) were formed during the SOBr2 treatment through a substitution reaction. The formation of ethers is traditionally understood to proceed by an intramolecular Williamson-type ether rearrangement whereby an alkoxide reacts with an alkyl halide to yield an ether.56,57 Based on that understanding, ether bridges would be formed by adjacent alcohols reacting with nearest neighbor alkyl bromides along with the formation of HBr (see Figure S2 for mechanistic details). The evidence includes the emergence of a peak at ~1025 cm−1 after bromination, which we assign to the (C–O–C)ν stretching mode and lack of (O–H)ν signal from alcohols at ~3200 cm−1; however, the details of the chemical kinetics and quantification are beyond the scope of this Letter.
Confirmation of alkyl bromides and surface coverage.
X-ray photoelectron spectroscopy (XPS), a surface-sensitive and element-specific technique confirmed the transition from an alcohol-rich surface to an alkyl-bromide-terminated surface, in agreement with our DRIFTS results. An inert transfer module was used for all XPS data to ensure the retention of alkyl bromide termination and eliminate H2O and O2 exposure. The C 1s XPS data of ND–OH showed peaks at 284.5, 286.5, and 288.5 eV, which were assigned to bulk diamond (C–C), alcohols (C–O), and carboxylic acids (COOH), respectively (Figure 2A). Previous quantitative analysis has shown the alcohol-to-carboxylate ratio to be ~15:1, suggesting that SOBr2 reacts primarily with hydroxyl moieties on the diamond surface, in agreement with the DRIFTS results.22 ND–OH treated with SOBr2 produced an ND–Br construct that exhibited convolved features attributable to the C–C, C–Br, and C–O bonding environments at 284.5, 286.0, and 286.5 eV, respectively. We interpret the results as being consistent with a partially brominated surface that retains either alcohol or ether moieties due to the intracrystallite Williamson-ether-like reaction. Br 3d XPS spectra confirmed alkyl bromide formation with convolution of the Br 3d3/2 and Br 3d5/2 spin states at 70.0 and 69.0 eV, respectively, with an energy difference of ~1.0 eV and a ratio of 0.6 eV (Figure 2B).58 SOBr2 with pyridine from 2 → 24 h reaction times showed an increased Br 3d signal, and peak positions remained consistent at 69.0 and 70.0 eV, indicating no change in bonding environment (Figures 1B, 2B, and S3). The addition of pyridine plays two active roles: harvesting protons generated during the nucleophilic attack of the alcohol moiety on the sulfur center and activating the release of bromide ions from the intermediate bromosulfite ester complex bound to the diamond surface. The XPS and DRIFTS data indicate that the rate of alkyl bromide formation increased by ~60% over the course of the reaction with pyridine.
Figure 2.
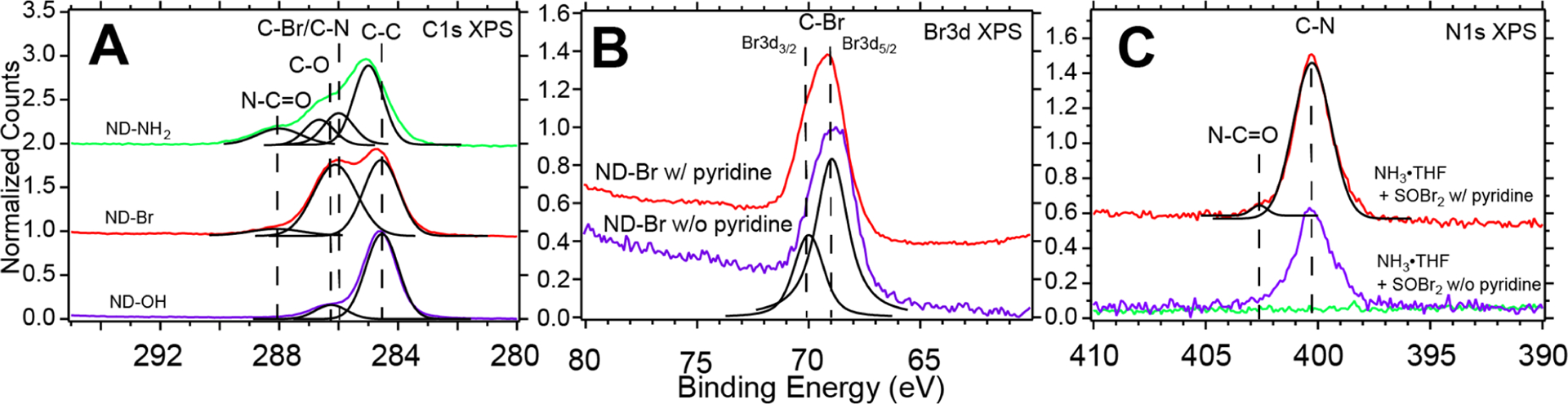
Br 3d, N 1s, and C 1s XPS spectra confirming the conversion of alcohol-rich diamond surfaces (ND–OH) to alkyl-bromide-terminated ND–Br and amine-terminated ND–NH2. ND–Br constructs show strong Br 3d XPS peaks at ~69 and 70 eV, which are consistent with C–Br bond formation, and Br 3d3/2 and Br 3d5/2 spin–orbit contributions are Gaussian-fitted with ΔE = 1.04 eV. The rising edge in the Br 3d XPS spectrum is due to the Au substrate, on which the ND–Br is deposited (A). The 400.3 eV peak in the N 1s spectrum is assigned to formed C–N bonds, while a minor peak at 402.7 eV is assigned to an amide bond (B). The C 1s spectrum shows the evolution of ND–OH → ND–Br → ND–NH2 with contributions from C–O, C–Br, and C–N bonds at each stage, respectively.
The quantification of surface groups is based on a model in which all C 1s XPS signals are from diamond with an inelastic mean free path of approximately 1.87 nm and where the N 1s, O 1s, and Br 3d signals originate from only the surface moieties. Based on analysis of the XPS survey scans as well as inelastic mean free electron escape depths of C, N, O, and Br photoelectrons and their respective ionization cross sections, we estimate that 36–52% of all surface sites were brominated in examining three different samples (Section 4 in Supporting Information). Br 3d XPS data of ND–Br samples were originally underestimated due to spontaneous debromination after synthesis and debromination under ultrahigh-vacuum conditions. A typical survey scan with a Tougaard background of ND–Br showed C 1s, O 1s, and Br 3d atomic percentages of 73, 23.5, and 2.5%, respectively. When originally modeling the surface termination with a 111 facet (18.2 atoms/nm2), we calculated that ~3 C–Br bonds/nm2 were formed, and without the addition of pyridine, the bromination reached ~0.5% or 0.6 C–Br bonds/nm2. The bromination rates were corrected when considering the yield of C–N bond formation that proceeds after successful bromination. Based on the nitrogen atomic percentage of 5.4–7.8% after amination chemistry and a 1:1 alkyl bromide → amine mechanism, we conclude that Br levels of 36–52% were achieved (Tables 2 and 3 in Supporting Information). The bromination of 36–52% of the surface sites reached the maximum of 50% predicted by Tiwari, yet these DFT calculations did not account for the intracrystallite surface chemistry of the Williamson-ether-like reaction.53 Previous DFT calculations showed that the addition of bromine atoms to a clean C-terminated (001)-(2 × 1) surface at 50% coverage had an adsorption energy of 1.82 eV/atom, which was lower than that for the addition of hydrogen, fluorine, and chlorine across a (111)-(1 × 1) surface, with 2.17, 4.72, and 2.22 eV/atom, respectively.53,59 Lower bromination rates are predicted in comparison to those with hydrogen (31 pm), fluorine (64 pm), and chlorine (97 pm) due to the large atomic radii of bromine (112 pm). For comparison, the C–H, C–F, C–Cl, and C–Br bond distances on diamond were calculated to be 110, 136, 171, and 192 pm, respectively.53,59 It should be noted that the bromine termination study by Tiwari was carried out on (001) surfaces, yet the same authors also conducted the earlier hydrogen, fluorine, and chlorine termination study with (111) and showed they were consistent when calculating surface coverage, adsorption energies, and bond distance. Our experimental evidence of the upper limits of ND–Br coverage are well supported theoretically with DFT.
Diamond–bromine bond dissociation studies in open air.
The ND–Br samples exposed to air resulted in fast alkyl bromide dissociation, representing potential evidence of a “radical carbon” on the diamond 111 surface.60 The ND–Br surface is extremely labile in comparison to that of many brominated substrates, including 1-bromoadamantane. Instantaneous debromination of ND–Br under open-air conditions at 25 °C was tracked with DRIFTS and showed a t1/2 value of ~12 min when air was allowed into an inert atmosphere DRIFTS chamber (Figure S4 in Section 2 of Supporting Information). Alkyl bromide dissociation was found to have pseudo-first-order kinetics of k′ = 7.67 × 10−7 1/s based on the local relative humidity at the time of the experiments. During the 80 min experiment, there was no evidence that the diamond surface formed new alcohols, as no increase in the 1105 cm−1 peak was observed. Notably, the (Br–C=O)ν mode at 1815 cm−1, which shifted 35 cm−1 from its nominal (C=O)ν position, did not change during the air exposure study. We hypothesize that sterically unhindered acid bromides at edges and defect sites are more stable than alkyl bromides. Alkyl bromides are sterically hindered and energetically destabilizing on the diamond facets as calculated by DFT; therefore, acid bromides did not readily undergo debromination during these air exposure experiments. Debromination was also observed instantly at 90 °C under inert conditions; it is detailed in Figure S6 and further supports our findings of a metastable ND–Br surface.
The prediction of a carbocation-like state with brominated diamond was described by Larsson and Lunell in 1997.60 Their DFT calculations showed a surface bond angle of 112.9° that was interpreted as having sp2 character and described as a “radical carbon.” We interpret our findings as a possible conformation of this unique bonding environment found on brominated diamond. For small molecules, an analogous system would be that of 8,9-dehydro-2-adamantyl with a stable carbocation at −120 °C, as confirmed via 1H and 13C nuclear magnetic resonance (NMR) spectroscopy.61 However, tracking carbocations with solution or solid-state NMR techniques is not possible due to the lack of sensitivity of NMR spectroscopy to trace surface groups on HPHT ND surfaces. The lability of the C–Br bond, the unique properties of diamond, and the low concentration of water molecules under atmospheric conditions have aided this finding. Based on these observations and analysis, the conclusion is that rehydroxylation is slow compared to the debromination kinetics at 60% relative humidity and 25 °C. The debromination side products are likely HBr and Br2 but were not analyzed, as the ND surface structure was our primary focus. In a control experiment to test the rehydroxylation kinetics of ND–Br, we reacted the ND–Br samples with 18 MΩ water [55.5 M] for 1 min, purified by centrifugation and probed with DRIFTS. We found the reemergence of an alcohol-rich surface within the 1 min reaction time (Figure S5 in Supporting Information). The pseudo-first-order rate was calculated to be k′ = 0.055 1/s with neat water, an increase of 7.2 × 104 in reaction rate, and we estimate that the rehydroxylation reaction is complete in approximately 66 ms under these conditions.
Direct catalyst-free amine formation at room temperature.
Demonstration of amine insertion via nucleophilic attack of ND–Br was accomplished at room temperature using ammonia in tetrahydrofuran (NH3·THF) and was found to convert all brominated surface species. The use of NH3·THF is rationalized to ensure that all XPS and X-ray absorption spectroscopy (XAS) spectroscopic signatures for carbon–nitrogen bond formation are a result of new amine moieties. ND–NH3 inner-shell chemistry (covalent bonds directly with the diamond lattice) was verified via laboratory and synchrotron-based surface-sensitive spectroscopies, as we show with the following evidence. DRIFTS spectra showed that the (C–Br)ν stretching mode at 750 cm−1 was completely removed, and new peaks related to amine functionalization appeared at 1025 and 820 cm−1, corresponding to (C–N)ν and (N–H)wag modes, respectively (Figure 1B). An unambiguous conversion from a hydrophobic ND–Br surface to a hydrophilic ND–NH3 surface after amination chemistry was evident through the large increase in both the (O–H)ν and (O–H)δ modes of adsorbed water observed at 3000–3500 and 1630 cm−1, respectively. When comparing ND–OH versus ND–NH3, the adsorbed water signal increases, and we conclude that the hydrophilicity qualitatively increased when amine termination is present at 36–52% surface coverage (see SI Section 4). The colloidal solutions of ND–NH3 were stored in 18 MΩ water prior to further analysis, and conjugation to folic acid via N-hydroxysulfosuccinimide/1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (sulfo-NHS/EDC) coupling is described in the Materials and Procedures section. Conformation of amide bond formation with ND–NH3 constructs was a demonstration that surface amines were chemically accessible for biolabeling protocols and surface engineering when fluorescent NDs are used.
Ambiguity in assigning chemical moieties with DRIFTS is common, and XPS allowed for a definitive confirmation of C–N bond formation after NH3·THF treatments. We found that successful amine bond formation was a function of C–Br density (catalyzed and uncatalyzed ND–Br) on the ND surface, as observed in Figure 3B, whereby the N 1s photoemission spectra show enhanced C–N resonance at 400.3 eV and a minor C=N feature at 402.6 eV, consistent with amination of diamond films with NH3 plasma and CN thin films.49,62 The C 1s photoemission peaks after amination chemistry also show a deconvolved C–N resonance at 286.0 eV, C–C resonance at 284.8 eV, and contributions from C–O and C=O at 286.5 and 288.0 eV, respectively. Based on the XPS atomic % results, we conclude that the conversion of ND–Br → ND–NH2 is unity, with a typical survey scan showing C 1s, O 1s, and N 1s percentages of 69.4, 22.8, and 7.8%, respectively. No Br 3d was detected at the atomic concentration sensitivity levels of the XPS of ~0.1%. Among the probed ND–NH3 samples, we estimate that 36–52% of surface carbon atoms were aminated, which translates to 6.5–9.5 amines/nm2 (see SI Section 4). The disparity in the maximum Br 3d/N 1s (2.5%:7.8%) signal is due to instantaneous debromination as a function of time after synthesis and under ultrahigh-vacuum conditions. No Br 3d photoelectrons were detected in either survey scans or high-resolution scans after the amination and purification steps. The rate of C–N bond formation was proportional to the concentration of C–Br bonds on the surface, and we conclude that catalyst-free amine bond formation is a consequence of the dissociation of alkyl bromides and the reactive intermediate that was observed using DRIFTS (Section 2 in Supporting Information).
Figure 3.
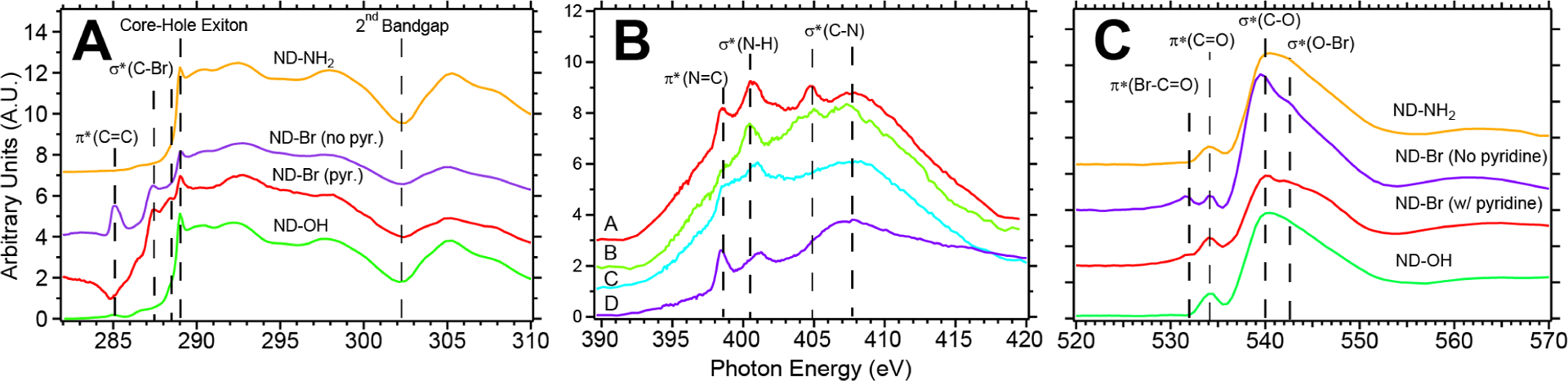
XAS spectra of ND–OH, ND–Br and ND–NH2 probing the excited- and ground-state electronic structure of the nanodiamond constructs. C 1s and O 1s XAS data show the emergence of σ*(C–Br) and π*(Br–C=O) transitions after bromination chemistry and its elimination after amination chemistry (A and B). N 1s XAS spectra of ND–NH2 constructs produced by reaction with NH3•THF (plots A/B), hydrazine (C) and amide bond formation to folic acid via sulfo-NHS/EDC. π*(C=N), σ*(N–H) and σ*(C–N) transitions are observed at 398.5 eV, 400.5 eV and 405/407.8 eV, respectively.
Soft X-ray spectroscopy of ND–OH, ND–Br, and ND–NH3.
XAS measurements of NDs are essential in this work, because they provide simultaneous conformation of the presence of diamond (core–hole exciton and second absolute bandgap) while examining the surface termination of nitrogen or oxygen moieties independently (Figure 3). XAS is an element-specific spectroscopy technique that provides information on the electronic structure of molecules and solids, including the chemical state, bond order, bond length, and orientation of adsorbates.63 In the soft X-ray regime, when collecting in total electron yield (TEY) mode, TEY provides surface and bulk information with a mean probing depth of approximately 5–10 nm. Figure 3A shows the C 1s XAS spectra for ND–OH and the characteristic features of bulk diamond; we observe little sp2-like carbon near 285 eV, a strong sp3 core–hole exciton peak at 289.0 eV, and a second absolute bandgap at 302 eV.64,65 The C 1s spectra of ND–OH have weak signals associated with π*(C=C) (approximately 285.3 eV) due to amorphous carbon and carbonyl (π*(C=O) transitions, covering 286.5–288 eV) prior to the large core–hole exciton peak at 289.0 eV. After bromination, ND–Br shows new features in the 286–288.5 eV region with strong resonances at 287.2 and 288.4 eV and is assigned to the σ*(C–Br) electronic states due to unoccupied Br p-orbitals bound to the diamond surface.53 The amplitude of the carbon–bromine resonances is increased for a surface species in TEY mode and is due to the larger photon absorption cross section of the 1s → p transitions of brominated diamond. O 1s data for ND–OH are dominated by the σ*(C–O) at 540 eV due to alcohols and small π*(C=O) transitions due to carbonyls at 534 eV. After bromination, minor O 1s features arise at 532 and 542–550 eV due to π*(Br=O) and σ*(O–Br) and resonances, respectively. Due to the atomically dense diamond surface, we consider that both alkyl bromides and different carbon–oxygen–bromine bonding environments may exist simultaneously.
N 1s XAS presents key signatures of C=N, C–N, and N–H bond formation after catalyst-free amination chemistry at room temperature. Challenges exist in performing N 1s XAS on diamond due to the large C 1s background at high energy (380–420 eV) that the N 1s near-edge features are convolved with. First, we assign the weak low-energy resonance at 398.5 eV to the π*(N–C=O) of a carboxyamide and reinforce this assignment by probing a control sample. An amide was prepared with a silica-coated ND functionalized with amines (3-aminopropyl-triemethoxysilane, i.e., APTES), conjugated to folic acid via sulfo-NHS/EDC coupling, and produced a strong π*(N–C=O) peak at 398.2 eV (Figure 4B and SI Section 5). The feature at 398.5 eV is assigned to a carboxyamide on the ND surface and has enhanced π-character due to the nitrogen lone-pair delocalization with the carbonyl.66 Low-energy N 1s peaks (398–400 eV) are typically assigned to 1s → π* transitions, and the presence of C=N features has been observed in six-ring heterocycles such as cytosine and pyrazinecarboxamide.67 Our findings are consistent with Graf et al., who studied APTES on silica and assigned N 1s signals at ~388 eV to π*(C=N) due to radiation damage-induced deprotonation, and we observed similar features (Section 5 in Supporting Information). The feature at 400.5 eV is assigned to the σ*(N–H) of a primary or secondary amine and is consistent across amination with NH3·THF-, hydrazine-, and folic-acid-conjugated samples.66,68 The higher-energy features at 404.8 and 407.6 eV are assigned to the σ*(C–N) bonding environment. ND–NH3 1s → σ* transitions are due to a primary amine C–N and possibly a C–N–C bridging configuration similar to a secondary amine. When ND–NH3 samples are conjugated to folic acid and amide bond formation occurs, several key features change as the nitrogen lone-pair electrons contribute to the delocalized bonding environment. The π*(C=N) resonance at 398.5 eV increases in intensity, σ*(N–H) remains at 400.5 eV, the σ*(C–N) peak becomes a broadened peak at 407 eV, and double peaks are no longer present. These assignments are reinforced by the control experiments with amine-functionalized silica bound to folic acid, and the conversion is evident (Section 5 in Supporting Information). Recent N 1s XAS studies of gas phase and aqueous NH3 and NH4+ show similar resonances to our study and report that distinct lower-energy features at 401–402 eV are assigned to NH3, while NH4+ features are not observed until 405 eV and above.69,70 We highlight these new reports, because the N 1s spectra reported in Figure 3B may contain contributions of ND–NH2 and ND–NH3 due to a tightly bound water layer and hydrogen bonding that may exist on the ND surface. ND–NH3 samples were not desiccated at high temperature in vacuo prior to deposition in the same fashion that ND–OH samples were prepared.
Figure 4.
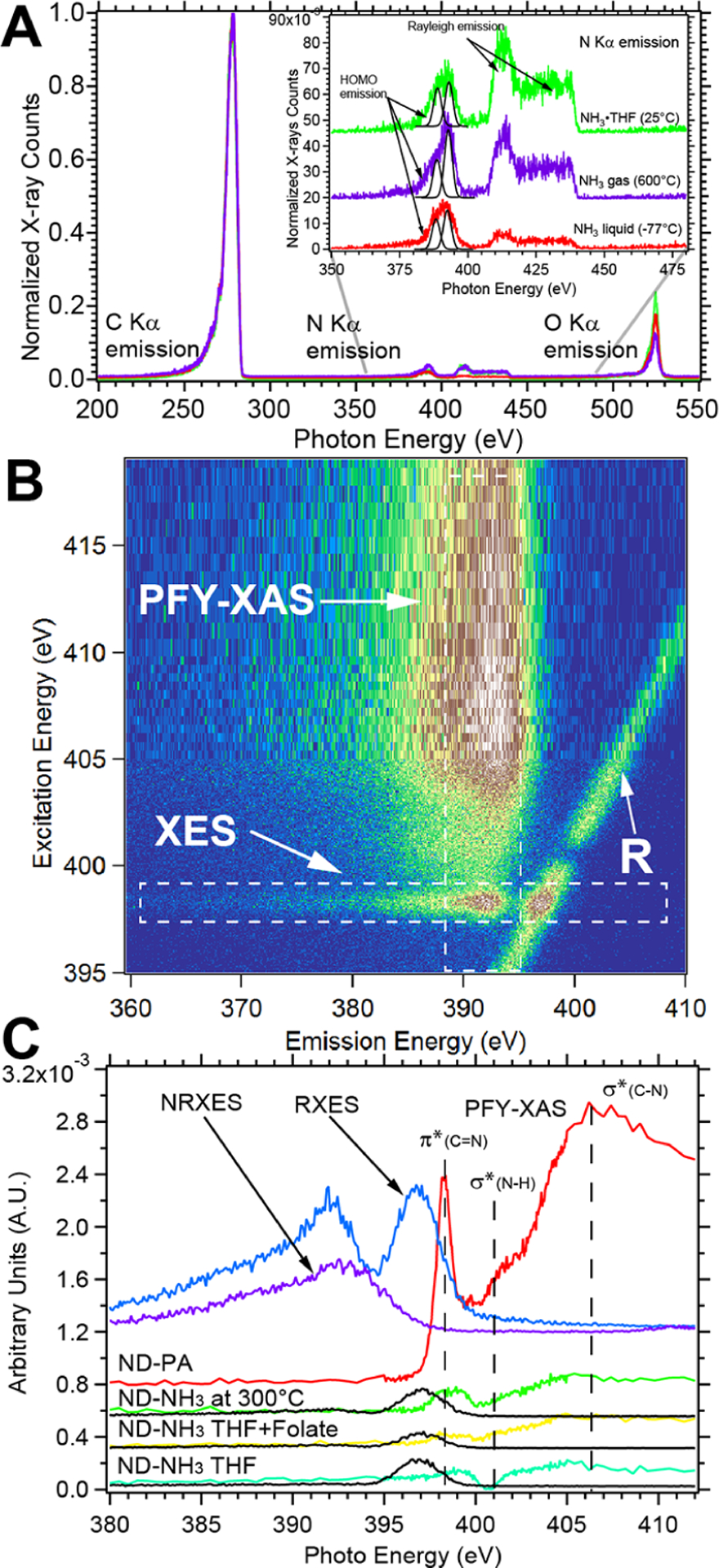
XES, RIXS, and PFY-XAS spectra of ND–OH, ND–Br, and ND–NH2 probing the excited- and ground-state electronic structures of the nanodiamond constructs, collected with the TES detector. The N 1s RIXS map of propargylamine-treated ND–Br samples shows an 11×-fold increase in the amine signal (B). PFY-XAS and XES plots were extracted with integration, as illustrated by the white dashed boxes. The PFY-XAS spectrum shows a π*(C=N) transition at 398.1 eV and a broad σ*(C–N) transition at 406.5 eV, while the XES spectrum shows peak emission at 392.5 eV and decreasing spectator emission to 380 eV (B). Other aminated samples with NH3·THF, NH3 gas at 300 °C, and after folic acid conjugation are shown for comparison. Off-resonance XES signals (integration from 410–440 eV excitation energy) show distinct above- and below-bandgap emission for amination at −77, 25, and 600 °C (A).
Transition edge sensor, RIXS, PFY-XAS, and XES.
The transition edge sensor (TES) detector housed at beamline 10–1 at the Stanford Synchrotron Radiation Lightsource (SSRL) provides background-free X-ray emission data and electronic structure information on the valence (occupied) and conduction (unoccupied) density of states (DOS) of both the ND and surface moieties.71,72 Resonant inelastic X-ray scattering or RIXS maps provide the complete DOS with extraction of X-ray emission spectroscopy (XES) data and partial fluorescence yield X-ray absorption spectroscopy (PFY-XAS). XES and PFY-XAS spectra are produced with an integration window with a static emission or excitation energy as highlighted with the dashed white box in Figure 4B. Figure 4A shows C, N, and O Kα emission data after amination chemistry performed at −77, 25, and 600 °C with condensed NH3, NH3·THF, and gaseous NH3, respectively. C Kα X-ray emission was consistent with past studies of diamond, with bandgap emission beginning at ~284 eV and extending to 250 eV.73 The C Kα emission features above the bandgap (289–312 eV) are different for each ND–NH2 construct and are potentially due to vibronic coupling to the diamond lattice, surface moieties, or Rayleigh scattering.73 Importantly, the N Kα emission data are unique in intensity for each of the chemistries used, extend from 365–440 eV, and include both X-ray emission features and Rayleigh scattering. Because XAS/XES data of aminated diamond are scarce, we compare our spectra with model systems of ammonia and N-doped graphene.74,75 Our standard NH3·THF chemistry of ND–Br constructs yielded three distinct regions of emission from 380–402 eV (region A), 408–420 eV (region B), and 420–440 eV (region C). The last two regions show step-like emission and are due to Rayleigh scattering and participator emission, respectively.76 Chemical treatment of ND–Br with NH3·THF at 25 °C yields amination rates similar to those of NH3 gas at 600 °C, yet the mechanism is expected to be different due to temperature-dependent debromination and surface reconstruction. Nucleophilic addition of NH3 to the tertiary carbocation or reactive intermediate on the diamond surface at 25 °C should proceed as depicted in Figure S8 with production of an HBr side product. NH3 gas chemistry at 600 °C should proceed through thermal debromination at temperatures above 90 °C, desorption of alcohol groups above 500 °C, and Pandey reconstruction of the diamond surface.77
We model our XES spectra focusing on the dominant C–N and N–H occupied density of states and compare the spectra to ammonia and glycine. NH3·THF treatment yielded a dominant spectator N Kα peak at 398.3 eV, which is assigned to the lone-pair electrons on nitrogen or C–N bonds and is supported by similar XES features in glycine and DFT calculations of the highest occupied molecular orbital (HOMO) isodensity surfaces.76 The 402.5 eV peak is assigned to the N–H HOMO orbitals and is very close to a resonant excitation of the σ*(N–H) transition. The lower-energy tail from 380–393 eV is assigned to a mixture of delocalized HOMO states described as HOMO–8/9/10 in ref 74. Weinhardt et al. used the StoBe-DeMon package to calculate the transition probabilities based on the ground-state Kohn–Sham eigenstates, which accurately calculated the XES spectra of glycine, diglycine, and triglycine. Our gas-phase amination chemistry using NH3/N2 mixtures at 600 °C produced similar features at 398.3 eV (lone-pair electrons amines/C–N) and 402.5 eV (N–H) with reduced intensity in comparison to that for the NH3·THF treatments. A new feature arose at 393.7 eV, which we assign to HOMOs of imides (C=N), consistent with increased π*(C=N) XAS transitions at 399.5 eV (data not shown). At temperatures above 90 and 500 °C, alkyl bromides and alcohols will desorb and produce sp2-like Pandey chains due to uncoordinated surface sites, and nitrogen insertion is likely to occur.77 Gas-phase NH3 chemistry at high temperatures is more complex with competing kinetics of thermal desorption, surface reconstruction, and new amine/imide bond formation. Amination chemistry at −77 °C with liquid ammonia only showed N Kα features representative of spectator emission, with an asymmetric peak from 380–407 eV. The lower kinetic rate of amination and shorter reaction time of 30 min resulted in less C–N bond formation but did highlight that catalyst-free bond formation still occurred at −77 °C (Section 7 in Supporting Information for RIXS maps).
Propargyl amine reacted with ND–Br.
In a demonstration of an uncatalyzed Sonogashira-type coupling reaction,78 ND–Br was reacted at 25 °C with propargyl amine (PA), an alkyne, and produced an 11-fold increase in the nitrogen signal in comparison to that of other ND–NH3 constructs (Figure 4B,C). The original intent of the PA reaction was to form a new C–N surface bond with an outward facing alkyne for click chemistry; yet, the reaction did not proceed in the fashion envisioned. A RIXS map details both the occupied and unoccupied electronic structures of the PA-reacted ND samples (Figure 4B). The RIXS map was generated by scanning the X-ray monochromator (excitation energy on the y-axis) and recording the emitted X-rays using the TES detector. The linear feature marked “R” is the Rayleigh scattering of the incident X-rays and is a common feature in RIXS. The RIXS map details a high reactivity level between ND–Br and PA without a metal-based catalyst and should be compared to RIXS maps in Section 7 of the Supporting Information. Our findings are similar to the uncatalyzed reaction of a brominated precursor and an alkyne by Liu and Li using UV irradiation, which proceeded through a carbocation intermediate.79 PFY-XAS reveals this increase in nitrogen content of the ND–NH3 samples compared to other amination routes (Figure 4C). The PFY-XAS data are produced by integrating the emission intensity as a function of excitation photon energy, are complementary to traditional XAS, and eliminate the high-energy background signal of carbon. PFY-XAS peaks at 398.3 eV, a shoulder at 401.0 eV, and a broad resonance at 406.2 eV are assigned to the π*(C=N), σ*(N–H), and σ*(C–N) resonances, respectively. The sharp and intense π*(C=N) feature is evidence that PA polymerized on the ND surface, as explained below.
N Kα emissions from PA-treated samples have much higher count rates than single carbon–nitrogen bonds on the ND surface with NH3·THF or NH3(condensed or gas) chemistry and are evidence of polymerization (Section 6 in Supporting Information). Comparison of the N Kα resonant X-ray emission (RXES-blue trace) and nonresonant X-ray emission (NRXES-purple trace) of ND-PA differ in the intense emission at 396.8 eV for the RXES overlapping with the Rayleigh scattering line (marked R) and the lower-energy peak positions from 391–392 eV. RXES data were produced by integrating emission counts from 397–400 eV excitation energies (white box in Figure 4B), while NRXES data were obtained by integrating from 410–440 eV. The RXES and NRXES peaks are red-shifted 1.5 and 6.3 eV from the C=N resonance at 398.3 eV, respectively. The RXES peak that overlaps with the Rayleigh line is due to either vibronic coupling or a large overlap between the electron wave function and core hole produced by the exciton, leading to higher emission rates, as observed previously with NH3 and ND3.74,80 The vibrational fine structure of the propargyl amine sample was not observed in this study as was observed with NH3. The integrated XES signals (black traces) of NH3·THF, NH3 gas at 300 °C, and folic-acid-conjugated NDs are included for comparison. These results are consistent with reactions and polymerizations of aminoalkynes with transition metal catalysts and yield imines and enamines.81 Because there is a minor π*(C=C) resonance observed in the PA reaction, we conclude that a polyimine, not an enamine, is the dominant reaction product.
Importantly, the C 1s XAS of PA-treated samples does not show the core–hole exciton and second bandgap of diamond and is due to a shell of polyimine formed on the ND core via polymerization (Section 6 in Supporting Information). The suppression of the core–hole exciton in conjunction with the intense nitrogen signal, clear π*(C=N) PYF-XAS signatures caused by imines, and σ*(C–N) N 1s PFY-XAS resonance reinforces that multiple layers of reacted PA reside on the ND surface.82 We propose two possible mechanisms in which the alkyne or amine reacts with the carbocation after debromination and continues to polymerize in the presence of HBr. A control reaction of ND–Br with N-Boc PA yielded low amination rates, and the presence of the diamond electronic structure was observed (data not shown), showing that suppression of the polymerization reaction had occurred. This Sonogashira-like reaction has not been reported on any ND surface and highlights the level of reactivity of ND–Br under mild conditions and in the absence of a metal cocatalyst. Future studies can control the growth of the PA shell by stopping the reactions at various time intervals and characterizing the ND constructs.
In summary, we realized the first bromination and subsequent amination of HPHT NDs, provided detailed observations of the diamond–bromine bond, and realized subsequent carbon–nitrogen bond formation at 25 °C without the use of catalysts. The formation of alkyl bromides was found to be very labile and sensitive to water, and we observed evidence of an intracrystallite Williamson-ether-like surface side product. The bromination rates were found to be 36–52% of the available surface carbon atoms based on XPS analysis and were near the predicted value of 50% surface coverage based on DFT. The bromination yields were found indirectly by analyzing the atomic % concentrations of the final ND–NH3 constructs and was necessary due to the decomposition of ND–Br as a function of time. The lability of the carbon–bromine bond on diamond has been confirmed experimentally here, and this bond is highly unstable compared to that in brominated small molecule analogues. Uncatalyzed carbon–nitrogen bond formation is understood by the good leaving group properties of ND–Br, readily forming a reactive intermediate that subsequently leads to covalent amine termination. As a proof of concept relevant for biolabeling with NV diamond, we conjugated folic acid to ND–NH3 constructs and found evidence of amide bond formation. The use of synchrotron spectroscopy and the TES detector allowed detailed examination of the occupied and unoccupied electronic states of the ND surface termination, and we found that propargylamine reacted with ND–Br to yield polyimines in an uncatalyzed Sonogashira-type reaction. This work is impactful for researchers who wish to explore heteroatom chemistry on diamond and other ultrahard, ultradense materials (e.g., silicon carbide or boron carbide). Applications would include quantum sensing with HPHT NDs or as a source of free electrons for chemical transformations.
Supplementary Material
ACKNOWLEDGMENTS
A.W. would like to acknowledge financial support through the National Institutes of Health NIGMS office (1SC3GM125574-01) and Army Research Office (W911NF1810453 and W911NF17S000205) through the Department of Defense. P.C. would like to acknowledge financial support through the Czech Science Foundation Project No. 18-17071 S (to P.C.), MSM Project No. 8C18004 (NanoSpin) (to P.C., H.R.), European Regional Development Fund; OP RDE; Projects: Chem-BioDrug (No. CZ.02.1.01/0.0/0.0/16_019/0000729) (to P.C., H.R.), and CARAT (No. CZ.02.1.01/0.0/0.0/16_026/0008382) (to P.C., H.R.). Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The TES spectrometer was developed with funding from the Department of Energy, Laboratory Directed Research and Development program under Contract No. DE-AC02-76SF00515. This work was supported by the U.S. Department of Energy Office of Basic Energy Sciences Proposal No. 100487. The use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. S.S. acknowledges funding from the Walter Ahlström Foundation. S.S. also received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No 841621. P.J.S., J. Valenzuela, and G.J. would like to acknowledge support through the MARC and RISE program at SJSU (5T34GM008253-33 and 5R25GM071381-13).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jpclett.1c04090
The authors declare no competing financial interest.
Contributor Information
Cynthia Melendrez, Department of Chemistry, San José State University, San José, California 95192, United States.
Jorge A. Lopez-Rosas, Department of Chemistry, San José State University, San José, California 95192, United States.
Camron X. Stokes, Department of Chemistry, San José State University, San José, California 95192, United States.
Tsz Ching Cheung, Department of Chemistry, San José State University, San José, California 95192, United States.
Sang-Jun Lee, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States.
Charles James Titus, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States; Department of Physics, Stanford University, Palo Alto, California 94025, United States.
Jocelyn Valenzuela, Department of Chemistry, San José State University, San José, California 95192, United States.
Grace Jeanpierre, Department of Chemistry, San José State University, San José, California 95192, United States.
Halim Muhammad, Department of Chemistry, San José State University, San José, California 95192, United States.
Polo Tran, Department of Chemistry, San José State University, San José, California 95192, United States.
Perla Jasmine Sandoval, Department of Chemistry, San José State University, San José, California 95192, United States.
Tyanna Supreme, Department of Chemistry, San José State University, San José, California 95192, United States.
Virginia Altoe, The Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Jan Vavra, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 166 10 Prague 6, Czech Republic.
Helena Raabova, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 166 10 Prague 6, Czech Republic.
Vaclav Vanek, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 166 10 Prague 6, Czech Republic.
Sami Sainio, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States; Microelectronics Research Unit, Faculty of Information Technology and Electrical Engineering, University of Oulu, Oulu, Finland 90014.
William B. Doriese, Quantum Electromagnetics Division, National Institute of Standards and Technology, Boulder, Colorado 80305, United States
Galen C. O’Neil, Quantum Electromagnetics Division, National Institute of Standards and Technology, Boulder, Colorado 80305, United States
Daniel S. Swetz, Quantum Electromagnetics Division, National Institute of Standards and Technology, Boulder, Colorado 80305, United States
Joel N. Ullom, Quantum Electromagnetics Division, National Institute of Standards and Technology, Boulder, Colorado 80305, United States
Kent Irwin, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States; Department of Physics, Stanford University, Palo Alto, California 94025, United States.
Dennis Nordlund, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States.
Petr Cigler, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 166 10 Prague 6, Czech Republic.
Abraham Wolcott, Department of Chemistry, San José State University, San José, California 95192, United States.
REFERENCES
- (1).Saikia I; Borah AJ; Phukan P Use of Bromine and Bromo-Organic Compounds in Organic Synthesis. Chem. Rev 2016, 116, 6837–7042. [DOI] [PubMed] [Google Scholar]
- (2).Ullmann F; Bielecki J Ueber Synthesen in Der Biphenylreihe. Berichte der deutschen chemischen Gesellschaft 1901, 34, 2174–2185. [Google Scholar]
- (3).Chinchilla R; Nájera C Recent Advances in Sonogashira Reactions. Chem. Soc. Rev 2011, 40, 5084–5121. [DOI] [PubMed] [Google Scholar]
- (4).Lafferentz L; Eberhardt V; Dri C; Africh C; Comelli G; Esch F; Hecht S; Grill L Controlling on-Surface Polymerization by Hierarchical and Substrate-Directed Growth. Nat. Chem 2012, 4, 215–220. [DOI] [PubMed] [Google Scholar]
- (5).Grill L; Dyer M; Lafferentz L; Persson M; Peters MV; Hecht S Nano-Architectures by Covalent Assembly of Molecular Building Blocks. Nat. Nanotechnol 2007, 2, 687–691. [DOI] [PubMed] [Google Scholar]
- (6).Dresselhaus MS; Jorio A; Hofmann M; Dresselhaus G; Saito R Perspectives on Carbon Nanotubes and Graphene Raman Spectroscopy. Nano Lett. 2010, 10, 751–758. [DOI] [PubMed] [Google Scholar]
- (7).Jankovsky O; Simek P; Klimova K; Sedmidubsky D; Matejkova S; Pumera M; Sofer Z Towards Graphene Bromide: Bromination of Graphite Oxide. Nanoscale 2014, 6, 6065–6074. [DOI] [PubMed] [Google Scholar]
- (8).Bulusheva LG; Okotrub AV; Flahaut E; Asanov IP; Gevko PN; Koroteev VO; Fedoseeva YV; Yaya A; Ewels CP Bromination of Double-Walled Carbon Nanotubes. Chem. Mater 2012, 24, 2708–2715. [Google Scholar]
- (9).Hines D; Rümmeli MH; Adebimpe D; Akins DL High-Yield Photolytic Generation of Brominated Single-Walled Carbon Nanotubes and Their Application for Gas Sensing. Chem. Commun 2014, 50, 11568–11571. [DOI] [PubMed] [Google Scholar]
- (10).Ikeda Y; Saito T; Kusakabe K; Morooka S; Maeda H; Taniguchi Y; Fujiwara Y Halogenation and Butylation of Diamond Surfaces by Reactions in Organic Solvents. Diamond Relat. Mater 1998, 7, 830–834. [Google Scholar]
- (11).Raymakers J; Haenen K; Maes W Diamond Surface Functionalization: From Gemstone to Photoelectrochemical Applications. Journal of Materials Chemistry C 2019, 7, 10134–10165. [Google Scholar]
- (12).Zhang B; Yan J; Shang Y; Wang Z Synthesis of Fluorescent Micro- and Mesoporous Polyaminals for Detection of Toxic Pesticides. Macromolecules 2018, 51, 1769–1776. [Google Scholar]
- (13).Field J The Properties of Diamond; Academic Press, 1979. [Google Scholar]
- (14).Lee JY; Kang MH First-Principles Study of the Cl and Br Adsorbed Si(100) Surfaces. Phys. Rev. B 2004, 69, 113307–1. [Google Scholar]
- (15).Bedzyk M; Materlik G X-Ray Standing Wave Analysis for Bromine Chemisorbed on Germanium. Surf. Sci 1985, 152, 10–16. [Google Scholar]
- (16).D’Evelyn MP; Yang YL; Cohen SM Adsorption, Desorption, and Decomposition of Hcl and Hbr on Ge(100) - Competitive Pairing and near-First-Order Desorption-Kinetics. J. Chem. Phys 1994, 101, 2463–2475. [Google Scholar]
- (17).Martin R; Heydorn PC; Alvaro M; Garcia H General Strategy for High-Density Covalent Functionalization of Diamond Nanoparticles Using Fenton Chemistry. Chem. Mater 2009, 21, 4505–4514. [Google Scholar]
- (18).Mamin HJ; Kim M; Sherwood MH; Rettner CT; Ohno K; Awschalom DD; Rugar D Nanoscale Nuclear Magnetic Resonance with a Nitrogen-Vacancy Spin Sensor. Science 2013, 339, 557–560. [DOI] [PubMed] [Google Scholar]
- (19).Staudacher T; Raatz N; Pezzagna S; Meijer J; Reinhard F; Meriles CA; Wrachtrup J Probing Molecular Dynamics at the Nanoscale Via an Individual Paramagnetic Centre. Nat. Commun 2015, 6, 8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Karaveli S; Gaathon O; Wolcott A; Sakakibara R; Shemesh OA; Peterka DS; Boyden ES; Owen JS; Yuste R; Englund D Modulation of Nitrogen Vacancy Charge State and Fluorescence in Nanodiamonds Using Electrochemical Potential. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 3938–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).de Theije FK; Reedijk MF; Arsic J; van Enckevort WJP; Vlieg E Atomic Structure of Diamond {111} Surfaces Etched in Oxygen Water Vapor. Phys. Rev. B 2001, 64, No. 085403. [Google Scholar]
- (22).Wolcott A; Schiros T; Trusheim ME; Chen EH; Nordlund D; Diaz RE; Gaathon O; Englund D; Owen JS Surface Structure of Aerobically Oxidized Diamond Nanocrystals. J. Phys. Chem. C 2014, 118, 26695–26702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kono S; et al. Carbon 1s X-Ray Photoelectron Spectra of Realistic Samples of Hydrogen-Terminated and Oxygen-Terminated Cvd Diamond (111) and (001). Diamond Relat. Mater 2019, 93, 105–130. [Google Scholar]
- (24).Wang XF; Ruslinda AR; Ishiyama Y; Ishii Y; Kawarada H Higher Coverage of Carboxylic Acid Groups on Oxidized Single Crystal Diamond (001). Diamond Relat. Mater 2011, 20, 1319–1324. [Google Scholar]
- (25).Loh KP; Xie XN; Yang SW; Zheng JC Oxygen Adsorption on (111)-Oriented Diamond: A Study with Ultraviolet Photoelectron Spectroscopy, Temperature-Programmed Desorption, and Periodic Density Functional Theory. J. Phys. Chem. B 2002, 106, 5230–5240. [Google Scholar]
- (26).de Theije FK; van Veenendaal E; van Enckevort WJP; Vlieg E Oxidative Etching of Cleaved Synthetic Diamond {111} Surfaces. Surf. Sci 2001, 492, 91–105. [Google Scholar]
- (27).Chang YR; et al. Mass Production and Dynamic Imaging of Fluorescent Nanodiamonds. Nat. Nanotechnol 2008, 3, 284–288. [DOI] [PubMed] [Google Scholar]
- (28).Scholze A; Schmidt WG; Bechstedt F Structure of the Diamond (111) Surface: Single-Dangling-Bond Versus Triple-Dangling-Bond Face. Phys. Rev. B 1996, 53, 13725–13733. [DOI] [PubMed] [Google Scholar]
- (29).Ramaseshan S The Cleavage Properties of Diamond. Proc. Indian Acad. Sci. A 1946, 24, 114. [Google Scholar]
- (30).Telling RH; Pickard CJ; Payne MC; Field JE Theoretical Strength and Cleavage of Diamond. Phys. Rev. Lett 2000, 84, 5160–5163. [DOI] [PubMed] [Google Scholar]
- (31).Rehor I; Cigler P Precise Estimation of Hpht Nanodiamond Size Distribution Based on Transmission Electron Microscopy Image Analysis. Diamond Relat. Mater 2014, 46, 21–24. [Google Scholar]
- (32).Mochalin VN; Shenderova O; Ho D; Gogotsi Y The Properties and Applications of Nanodiamonds. Nat. Nanotechnol 2012, 7, 11–23. [DOI] [PubMed] [Google Scholar]
- (33).Costa GCC; Shenderova O; Mochalin V; Gogotsi Y; Navrotsky A Thermochemistry of Nanodiamond Terminated by Oxygen Containing Functional Groups. Carbon 2014, 80, 544–550. [Google Scholar]
- (34).Krueger A; Lang D Functionality Is Key: Recent Progress in the Surface Modification of Nanodiamond. Adv. Funct. Mater 2012, 22, 890–906. [Google Scholar]
- (35).Osswald S; Yushin G; Mochalin V; Kucheyev SO; Gogotsi Y Control of Sp(2)/Sp(3) Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air. J. Am. Chem. Soc 2006, 128, 11635–11642. [DOI] [PubMed] [Google Scholar]
- (36).Osswald S; Mochalin VN; Havel M; Yushin G; Gogotsi Y Phonon Confinement Effects in the Raman Spectrum of Nanodiamond. Phys. Rev. B 2009, 80, No. 075419. [Google Scholar]
- (37).Stehlik S; et al. Size and Purity Control of Hpht Nanodiamonds Down to 1 Nm. J. Phys. Chem. C Nanomater Interfaces 2015, 119, 27708–27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Stehlik S; et al. Size Effects on Surface Chemistry and Raman Spectra of Sub-5 Nm Oxidized High-Pressure High-Temperature and Detonation Nanodiamonds. J. Phys. Chem. C 2021, 125, 5647–5669. [Google Scholar]
- (39).Borodich FM; Korach CS; Keer LM Modeling the Tribochemical Aspects of Friction and Gradual Wear of Diamond-Like Carbon Films. Journal of Applied Mechanics 2007, 74, 23–30. [Google Scholar]
- (40).Kruger A; Liang YJ; Jarre G; Stegk J Surface Functionalisation of Detonation Diamond Suitable for Biological Applications. J. Mater. Chem 2006, 16, 2322–2328. [Google Scholar]
- (41).Chang BM; Lin HH; Su LJ; Lin WD; Lin RJ; Tzeng YK; Lee RT; Lee YC; Yu AL; Chang HC Highly Fluorescent Nanodiamonds Protein-Functionalized for Cell Labeling and Targeting. Adv. Funct. Mater 2013, 23, 5737–5745. [Google Scholar]
- (42).Nguyen TTB; Chang HC; Wu VWK Adsorption and Hydrolytic Activity of Lysozyme on Diamond Nanocrystallites. Diamond Relat. Mater 2007, 16, 872–876. [Google Scholar]
- (43).Bumb A; Sarkar SK; Billington N; Brechbiel MW; Neuman KC Silica Encapsulation of Fluorescent Nanodiamonds for Colloidal Stability and Facile Surface Functionalization. J. Am. Chem. Soc 2013, 135, 7815–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Rehor I; et al. Fluorescent Nanodiamonds Embedded in Biocompatible Translucent Shells. Small 2014, 10, 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rehor I; et al. Plasmonic Nanodiamonds: Targeted Core-Shell Type Nanoparticles for Cancer Cell Thermoablation. Adv. Healthcare Mater 2015, 4, 460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Vavra J; Rehor I; Rendler T; Jani M; Bednar J; Baksh MM; Zappe A; Wrachtrup J; Cigler P Supported Lipid Bilayers on Fluorescent Nanodiamonds: A Structurally Defined and Versatile Coating for Bioapplications. Adv. Funct. Mater 2018, 28, 1803406. [Google Scholar]
- (47).Stacey A; et al. Nitrogen Terminated Diamond. Advanced Materials Interfaces 2015, 2, 1500079. [Google Scholar]
- (48).Sotowa K-I; Amamoto T; Sobana A; Kusakabe K; Imato T Effect of Treatment Temperature on the Amination of Chlorinated Diamond. Diamond Relat. Mater 2004, 13, 145–150. [Google Scholar]
- (49).Zhu D; Bandy JA; Li S; Hamers RJ Amino-Terminated Diamond Surfaces: Photoelectron Emission and Photocatalytic Properties. Surf. Sci 2016, 650, 295–301. [Google Scholar]
- (50).Jirasek V; Cech J; Kozak H; Artemenko A; Cernak M; Kromka A Plasma Treatment of Detonation and Hpht Nano-diamonds in Diffuse Coplanar Surface Barrier Discharge in H-2/N-2 Flow. Physica Status Solidi a-Applications and Materials Science 2016, 213, 2680–2686. [Google Scholar]
- (51).Simon N; Charrier G; Goncalves AM; Aureau D; Gautier P; Ndjeri M; Etcheberry A Direct Amination of Diamond Surfaces by Electroless Treatment in Liquid Ammonia Solution. Electrochem. Commun 2014, 42, 17–20. [Google Scholar]
- (52).Zhu D; Zhang LH; Ruther RE; Hamers RJ Photo-Illuminated Diamond as a Solid-State Source of Solvated Electrons in Water for Nitrogen Reduction. Nat. Mater 2013, 12, 836–841. [DOI] [PubMed] [Google Scholar]
- (53).Tiwari AK; Goss JP; Briddon PR; Wright NG; Horsfall AB; Rayson MJ Bromine Functionalisation of Diamond: An Ab Initio Study. Physica Status Solidi a-Applications and Materials Science 2012, 209, 1703–1708. [Google Scholar]
- (54).Chu PM; Guenther FR; Rhoderick GC; Lafferty WJ The Nist Quantitative Infrared Database. J. Res. Natl. Inst. Stand. Technol 1999, 104, 59–81. [Google Scholar]
- (55).Ryan RG; Stacey A; O’Donnell KM; Ohshima T; Johnson BC; Hollenberg LCL; Mulvaney P; Simpson DA Impact of Surface Functionalization on the Quantum Coherence of Nitrogen-Vacancy Centers in Nanodiamonds. ACS Appl. Mater. Interfaces 2018, 10, 13143–13149. [DOI] [PubMed] [Google Scholar]
- (56).Williamson A Xlv. Theory of Ætherification. London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 1850, 37, 350–356. [Google Scholar]
- (57).Johnstone RAW; Rose ME Rapid, Simple, and Mild Procedure for Alkylation of Phenols, Alcohols, Amides and Acids. Tetrahedron 1979, 35, 2169–2173. [Google Scholar]
- (58).Smykalla L; Shukrynau P; Korb M; Lang H; Hietschold M Surface-Confined 2d Polymerization of a Brominated Copper-Tetraphenylporphyrin on Au(111). Nanoscale 2015, 7, 4234–4241. [DOI] [PubMed] [Google Scholar]
- (59).Tiwari AK; Goss JP; Briddon PR; Wright NG; Horsfall AB; Jones R; Pinto H; Rayson MJ Calculated Electron Affinity and Stability of Halogen-Terminated Diamond. Phys. Rev. B 2011, 84, 245305. [Google Scholar]
- (60).Larsson K; Lunell S Stability of Halogen-Terminated Diamond (111) Surfaces. J. Phys. Chem. A 1997, 101, 76–82. [Google Scholar]
- (61).Olah GA; Liang G; Babiak KA; Ford TM; Goff DL; Morgan TK; Murray RK Structure of Cyclopropylcarbinyl and Cyclobutyl Cations - 8,9-Dehydro-2-Adamantyl and 2,5-Dehydro-4-Protoadamantyl Cations. J. Am. Chem. Soc 1978, 100, 1494–1500. [Google Scholar]
- (62).Marton D; Boyd KJ; Albayati AH; Todorov SS; Rabalais JW Carbon Nitride Deposited Using Energetic Species - a 2-Phase System. Phys. Rev. Lett 1994, 73, 118–121. [DOI] [PubMed] [Google Scholar]
- (63).Stöhr J NEXAFS Spectroscopy; Springer-Verlag: Berlin, 1992; p 404. [Google Scholar]
- (64).Morar JF; Himpsel FJ; Hollinger G; Hughes G; Jordan JL Observation of a C-1s Core Exciton in Diamond. Phys. Rev. Lett 1985, 54, 1960–1963. [DOI] [PubMed] [Google Scholar]
- (65).Himpsel FJ; van der Veen JF; Eastman DE Experimental Bulk Energy Bands for Diamond Using $H\Ensuremath{\Nu}$-Dependent Photoemission. Phys. Rev. B 1980, 22, 1967–1971. [Google Scholar]
- (66).Zubavichus Y; Shaporenko A; Korolkov V; Grunze M; Zharnikov M X-Ray Absorption Spectroscopy of the Nucleotide Bases at the Carbon, Nitrogen, and Oxygen K-Edges. J. Phys. Chem. B 2008, 112, 13711–13716. [DOI] [PubMed] [Google Scholar]
- (67).Leinweber P; Kruse J; Walley FL; Gillespie A; Eckhardt K-U; Blyth RIR; Regier T Nitrogen K-Edge Xanes - an Overview of Reference Compounds Used to Identify ‘Unknown’ Organic Nitrogen in Environmental Samples. Journal of Synchrotron Radiation 2007, 14, 500–511. [DOI] [PubMed] [Google Scholar]
- (68).Bottcher S; Vita H; Weser M; Bisti F; Dedkov YS; Horn K Adsorption of Water and Ammonia on Graphene: Evidence for Chemisorption from X-Ray Absorption Spectra. J. Phys. Chem. Lett 2017, 8, 3668–3672. [DOI] [PubMed] [Google Scholar]
- (69).Reinholdt P; Vidal ML; Kongsted J; Iannuzzi M; Coriani S; Odelius M Nitrogen K-Edge X-Ray Absorption Spectra of Ammonium and Ammonia in Water Solution: Assessing the Performance of Polarizable Embedding Coupled Cluster Methods. J. Phys. Chem. Lett 2021, 12, 8865–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ekimova M; Quevedo W; Szyc Ł; Iannuzzi M; Wernet P; Odelius M; Nibbering ETJ Aqueous Solvation of Ammonia and Ammonium: Probing Hydrogen Bond Motifs with Ft-Ir and Soft X-Ray Spectroscopy. J. Am. Chem. Soc 2017, 139, 12773–12783. [DOI] [PubMed] [Google Scholar]
- (71).Titus CJ; et al. L-Edge Spectroscopy of Dilute, Radiation-Sensitive Systems Using a Transition-Edge-Sensor Array. J. Chem. Phys 2017, 147, 214201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Lee SJ; et al. Soft X-Ray Spectroscopy with Transition-Edge Sensors at Stanford Synchrotron Radiation Lightsource Beamline 10–1. Rev. Sci. Instrum 2019, 90, 113101. [DOI] [PubMed] [Google Scholar]
- (73).Ma Y; Skytt P; Wassdahl N; Glans P; Guo J; Nordgren J Core Excitons and Vibronic Coupling in Diamond and Graphite. Phys. Rev. Lett 1993, 71, 3725–3728. [DOI] [PubMed] [Google Scholar]
- (74).Weinhardt L; Weigand M; Fuchs O; Bar M; Blum M; Denlinger JD; Yang W; Umbach E; Heske C Nuclear Dynamics in the Core-Excited State of Aqueous Ammonia Probed by Resonant Inelastic Soft X-Ray Scattering. Phys. Rev. B 2011, 84, 104202. [Google Scholar]
- (75).Wang X; Hou Z; Ikeda T; Oshima M; Kakimoto M -a.; Terakura, K. Theoretical Characterization of X-Ray Absorption, Emission, and Photoelectron Spectra of Nitrogen Doped Along Graphene Edges. J. Phys. Chem. A 2013, 117, 579–589. [DOI] [PubMed] [Google Scholar]
- (76).Weinhardt L; et al. Local Electronic Structure of the Peptide Bond Probed by Resonant Inelastic Soft X-Ray Scattering. Phys. Chem. Chem. Phys 2019, 21, 13207–13214. [DOI] [PubMed] [Google Scholar]
- (77).Pandey KC New Dimerized-Chain Model for the Reconstruction of the Diamond (111)-(2 × 1) Surface. Phys. Rev. B 1982, 25, 4338–4341. [Google Scholar]
- (78).Sonogashira K; Tohda Y; Hagihara N Convenient Synthesis of Acetylenes - Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]
- (79).Liu WB; Li L; Li CJ Empowering a Transition-Metal-Free Coupling between Alkyne and Alkyl Iodide with Light in Water. Nat. Commun 2015, 6, 6526. [DOI] [PubMed] [Google Scholar]
- (80).Weinhardt L; et al. Electron-Hole Correlation Effects in Core-Level Spectroscopy Probed by the Resonant Inelastic Soft X-Ray Scattering Map of C60. J. Chem. Phys 2011, 135, 104705. [DOI] [PubMed] [Google Scholar]
- (81).Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- (82).Shearer J; Callan PE; Masitas CA; Grapperhaus CA Influence of Sequential Thiolate Oxidation on a Nitrile Hydratase Mimic Probed by Multiedge X-Ray Absorption Spectroscopy. Inorg. Chem 2012, 51, 6032–6045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.