

Abstract
Spastic paraplegia type 11 (SPG11) is a common autosomal recessive form of hereditary spastic paraplegia (HSP) characterized by the degeneration of cortical motor neuron axons, leading to muscle spasticity and weakness. Impaired lipid trafficking is an emerging pathology in neurodegenerative diseases including SPG11, though its role in axonal degeneration of human SPG11 neurons remains unknown. Here, we established a pluripotent stem cell-based SPG11 model by knocking down the SPG11 gene in human embryonic stem cells (hESCs). These stem cells were then differentiated into cortical projection neurons (PNs), the cell types affected in HSP patients, to examine axonal defects and cholesterol distributions. Our data revealed that SPG11 deficiency led to reduced axonal outgrowth, impaired axonal transport, and accumulated swellings, recapitulating disease-specific phenotypes. In SPG11-knockdown neurons, cholesterol was accumulated in lysosome and reduced in plasma membrane, revealing impairments in cholesterol trafficking. Strikingly, the liver-X-receptor (LXR) agonists restored cholesterol homeostasis, leading to the rescue of subsequent axonal defects in SPG11-deficient cortical PNs. To further determine the implication of impaired cholesterol homeostasis in SPG11, we examined the cholesterol distribution in cortical PNs generated from SPG11 disease-mutation knock-in hESCs, and observed a similar cholesterol trafficking impairment. Moreover, LXR agonists rescued the aberrant cholesterol distribution and mitigated the degeneration of SPG11 disease-mutated neurons. Taken together, our data demonstrate impaired cholesterol trafficking underlying axonal degeneration of SPG11 human neurons, and highlight the therapeutic potential of LXR agonists for SPG11 through restoring cholesterol homeostasis.
Keywords: Hereditary spastic paraplegias, Axonal degeneration, Cortical projection neuron, Human pluripotent stem cells, Cholesterol intracellular trafficking, LXR agonist
1. Introduction
Axonal degeneration of human motor neurons underlies debilitating motor neuron diseases including amyotrophic lateral sclerosis and hereditary spastic paraplegias (HSPs). HSPs are a heterogeneous group of neurogenetic diseases characterized by progressive lower limb spasticity due to the length-dependent degeneration of cortical motor neuron axons (Blackstone et al., 2011; Fink, 2006; Harding, 1983). Since the identification of the first HSP gene, SPAST, scientists have identified over 80 genetic loci associated with HSP (Blackstone, 2018; Hedera, 1993; Martinuzzi et al., 2021). Based on genetic inherence mode, HSPs are further divided into autosomal dominant, autosomal recessive and X-linked inheritance (Finsterer et al., 2012). SPG11 is one of the most frequent autosomal recessive forms of HSP caused by mutations in the SPG11 gene that encodes spatacsin protein (Denora et al., 2013; Fink, 2013; Stevanin et al., 2008). In addition to typical symptoms of HSP such as lower limb spasticity and weakness, SPG11 patients often exhibit cognitive impairment, mental retardation, early-onset parkinsonism, and thin corpus callosum (Franca Jr. et al., 2012; Kara et al., 2016; Stevanin et al., 2008). The SPG11 protein is a binding partner of the SPG15 protein, another common recessive form of HSP (Hirst et al., 2013). Though both SPG11 and SPG15 proteins have been shown to regulate autophagy lysosome reformation (Renvoise et al., 2014), how impaired lysosomal function leads to axonal degeneration remains elusive.
Cholesterol is highly enriched in the central nerve system (CNS), critical for maintaining axonal and synaptic functions (Korinek et al., 2020; Pfrieger, 2003). A recent study showed that the spatacsin protein can regulate cholesterol trafficking and prevent abnormal cholesterol accumulation in lysosomes (Branchu et al., 2017). Spatacsin protein activates clathrin downstream and recruits dynamin to exocytose gangliosides and lipid from lysosomes (Boutry et al., 2018). By knocking down SPG11 in fibroblast cells, a study reported that the loss of spatacsin inhibits the formation of tubules and causes cholesterol accumulation on the lysosome membrane (Boutry et al., 2019). Cholesterol, a major component of membranes, plays an important role in maintaining neuronal function (Korinek et al., 2020). Though these recent findings suggest that the impaired lipid trafficking caused by lysosomal dysfunction is induced in SPG11 deficiency fibroblast cells, whether the impaired cholesterol trafficking underlies axonal defects and whether axonal degeneration can be rescued by targeting the cholesterol trafficking defects are unknown.
Currently, there is no effective cure for SPG11, partially due to the lack of human neuronal models to study this disease and to test drugs. After the successful establishment of human embryonic stem cell (hESC) lines in 1998, these pluripotent stem cells that can differentiate into any cell types including neurons, provide unique resources to generate various neuronal subtypes to study neurological diseases (Ming et al., 2011; Tao and Zhang, 2016; Thomson et al., 1998; Zhao and Bhattacharyya, 2018). In this study, we established hESC-based SPG11 model by generating SPG11-Knockdown hESC lines and establishing disease-mutation knock-in hESC lines. These stem cells were differentiated into cortical projection neurons (PNs) (Boisvert et al., 2013; Denton et al., 2016a; Li et al., 2009), the cell type affected in patients. We then examined the disease-relevant axonal defects and tested the rescue of these axonal defects by targeting cholesterol trafficking using LXR agonists. LXR agonists significantly improve cholesterol homeostasis and mitigate disease-specific axonal defects and apoptosis, demonstrating a new strategy to rescue axonal defects in SPG11 through the restoration of cholesterol homeostasis using LXR agonists.
2. Methods
2.1. Lentivirus production and transfection of hESCs
To knockdown spatacsin, shRNA sequences that target SPG11 and luciferase (as controls) were cloned into pLKO.1. shRNAs that target two different areas of the SPG11 gene (shRNA-A and B) were utilized to minimize off-target effects. SPG11 shRNA-A targets SPG11 sequences GAGGAACCCATAGAGCTTAAATG, and SPG11 shRNA-B targets SPG11 sequences GCTCATTGACGATCAAGATATCA (Supplemental Fig. 1). Lentiviruses were packaged using calcium phosphate transfection method as described previously (Denton et al., 2014). Briefly, pLKO.1 shRNA plasmid, psPAX2 packaging plasmid and pMD2.G envelope plasmid were mixed and then incubated in calcium phosphate solution with 2× Hepes buffered saline for 1 min. The mixture was transferred into the dishes for co-culturing the cocktail with HEK293T cells. After 2 to 3 days, the viral particles from the cell culture medium were collected through 20,000 rpm ultracentrifugation (SW28 rotor, Beckman) at 16 °C for 2 h and the pellet was resuspended in hESC medium. For stable transfection of H9 ESCs (WA09, WiCell Research Institute), cells were dissociated to small clusters and then incubated with viruses at 37 °C for 30 min. With the Blasticidin selection, the hESCs with stable transfection would survive while other cells including feeder cells would gradually die. The Blasticidin resistant cells were then dissociated to select clonal SPG11 knockdown hESC cell lines. With knockdown efficacy confirmed by qPCR, both SPG11 knockdown and Luciferase knockdown lines (as controls) were generated for differentiation.
2.2. Neural differentiation of pluripotent stem cells
Human ESCs were maintained in ESC medium and differentiated into cortical PNs as described previously (Boisvert et al., 2013; Li et al., 2009). Stem cells were cultured on a feeder layer in the hESC medium and 10 μg/mL bFGF. After being dissociated and detached from the feeder, the stem cell aggregates/clusters were cultured in suspension with hESC medium to initiate the differentiation (day 0 of the neural differentiation process). These hESC aggregates were then cultured in Neural Induction Medium (NIM) and attached to culture dishes as we previously described (Boisvert et al., 2013). On around the 15th day of differentiation, the neuroepithelial clusters were isolated and cultured in suspension culture. The neurospheres were cultured in suspension for 3–4 weeks and were then plated onto polyornithine-coated coverslips for terminal differentiation. At about 2 days after plating (total 6 weeks), the cortical PNs were fixed to examine early axonal outgrowth and extension. With further culture, the long-term cultured cortical PNs (over 10 weeks) were used to analyze axonal degeneration. Cells from three independent coverslips in each group were measured and compared (all data points were plotted in the figures).
2.3. Immunostaining and image acquisition
Immunostaining analyses were performed as we previously described (Denton et al., 2014). Coverslips were washed with PBS and then fixed in 4% paraformaldehyde (PFA). Primary antibodies used in this study included anti-Ctip2 (Abcam, Rat IgG), anti-Tau (Sigma, rabbit IgG), anti-LAMP1 (Abcam, Rabbit IgG). After incubation with primary antibody at 4 °C overnight, cells were subsequently washed and incubated with corresponding fluorescence-conjugated secondary antibodies. With cells stained with Hoechst (Sigma) as indicator, coverslips were mounted on glass slides.
2.4. Western blot
To collect samples for western analysis, cells were lysed in RIPA buffer supplemented with PMSF and proteinase inhibitor cocktails. Then 20 μg samples were run on 8% SDS-PAGE gels and subsequently incubated with primary antibodies in 3% BSA overnight at 4 °C. This was followed by the incubation with secondary antibodies. After adding enhanced chemiluminescence substrate, immunoreactivity was revealed using the Bio-Rad ChemiDocTMMP Imaging System. The primary antibodies used in this study were anti-SPG11 (Sigma-Aldrich, rabbit IgG) and anti-β-actin (Sigma-Aldrich, mouse IgG A5316). For the secondary antibodies, we utilized HRP-conjugated IgG from Jackson ImmunoResearch.
2.5. Colocalization analysis of cholesterol and lysosome staining
Similar to immunostaining, coverslips were fixed in 4% PFA for 20 min at room temperature first. They were treated with 0.05 mg/mL Filipin III (Cayman Chemical) in PBS with 10% FBS for 2 h at room temperature. Cells were incubated in primary antibody, anti-LAMP-1, with 5% BSA in PBS overnight at 4°C. Then cells were processed for secondary antibody incubation in 5% BSA for 45 min at room temperature and mounted on microscope slides. Triplicate coverslips were used to acquire images by using 60× objective of the fluorescence microscope. The colocalization images were analyzed using ImageJ software (Boutry et al., 2019). Specifically, the LAMP1-positive lysosomal area (first) and the soma of the cell (second area) were selected and added to ROI manager. We quantified the total fluorescent intensity of the first and second selection area under the blue channel (cholesterol stained). These areas represent the relative cholesterol level in the lysosome and soma, respectively. The ratio between two values is the proportion of cholesterol accumulation in the lysosome (Boutry et al., 2019). The proportion of cholesterol accumulated in lysosome was calculated by % = (filipin intensity in selected area of LAMP1+) / (filipin intensity in whole cell) × 100%.
2.6. Plasma membrane labeling and analysis of PM cholesterol levels
Plasma was stained with Deep Red CellMask™ plasma membrane stain (Invitrogen, 1:1000) for 10 min at 37 °C. After cells were washed, fixed and stained with Filipin III using cell-based cholesterol detection kit following the manufacture’s protocol (Cayman Chemical). Images were then taken using Keyence microscope. To analyze membrane cholesterol levels, plasma membrane was selected in ImageJ and then Filipin level was measured by overlapping with plasma membrane area.
2.7. Neurite outgrowth analysis
After dissociating and plating the neurospheres on coverslips for 2 days, cells were fixed and subjected for neurite outgrowth analysis (Denton et al., 2016b; Zhu et al., 2014). The longest process, which also had the greatest Tau intensity, was measured by using “Simple Neurite Tracer” plugin in ImageJ software. In particular, we stained the neurons with both anti-Tau and anti-Ctip2 antibodies, the latter serves as a cortical neuron marker.
2.8. Axonal swelling analysis
To analyze axonal swellings, cortical neuron cultures at around 3 months were stained with the axonal marker, Tau. Axonal swellings are defined as a diameter >2 times of the contiguous axons (Cheng et al., 2013; Lauria et al., 2003). The swelling density was calculated by swelling number over axonal length in the selected area by using ImageJ software as we described previously (Mou et al., 2020a). At least 3 randomly selected areas per coverslips from three independent coverslips in each group were analyzed.
2.9. Gene expression analysis
Total RNA was isolated and 1 μg of RNA was taken for reverse transcription using iScriptcDNA Synthesis Kit (BioRad). For quantitative PCR (qPCR), reactions were performed in a 20 μL mixture containing cDNA, primers, and SYBR Green Master Mix in the QuantStudio 6 Flex Real-Time PCR System per the manufacturer’s instructions (Applied Biosystems). Gene expression levels were compared with the housekeeping gene GAPDH, and calculated using the comparative CT method. Primers used for qPCR include SPG11, 5′- CTCCTAGTGTCCTGCCATCTGA, 5′- GGGCTAGAGAAATGTGGGAGATG; GAPDH, 5′- ATGACATCAAGAAGGTGGTG, 5′- CATACCAGGAAATGAGCTTG.
2.10. Caspase 3/7 activity assay
The caspase 3 and 7 activities are highly related to apoptosis levels in cells and were examined using Caspase-Glo 3/7 Assay kit (Promega) following the manufacturer’s instructions. Briefly, neurons were dissociated with Accutase, a mild enzyme that shows increased survival for neural cells (Wachs et al., 2003), into single cell suspension. Cells were then seeded onto 96-well plates with the density of 5000 cells/well in 50 μl, lysed and incubated with illuminance substrate by adding 50 μl of caspase-3/7 reagents following the manufacture’s protocol (Promega). After incubation for 45 min at room temperature, luminescence from each well was measured using a Gen5 microplate reader (BioTek).
2.11. Live cell imaging with MitoTracker dye
To examine mitochondrial transport, neurons (about 10-week) were stained with 50 nM MitoTracker Red for 4 min and then the coverslips were washed with image medium. The coverslips were picked up and placed upside down in 35 mm glass bottom dishes with pre-added 200 μl image medium. Live-cell imaging was performed by the Olympus IX83 microscope equipped with an incubation chamber. The cells were kept in 37 °C with 5% CO2 while imaging. Images were taken every 5 s for 5 min. The mitochondrial motile percentage was analyzed by ImageJ software with MultiKymograph plugin and Bio-formats Package, as described previously (Mou et al., 2020b).
2.12. Generation of SPG11 mutation knock-in hESC lines using CRISPR-cas9 mediated gene editing
To further examine the role of SPG11 disease mutation, isogenic H9 hESC lines with the disease specific SPG11 mutation were established (Chen et al., 2022). Briefly, using CRISPR-cas9 mediated gene editing, a c.118C > T (p. Gln40X) homozygous mutation of the SPG11 gene (Supplemental Fig. 1) was knocked-into H9 hESC lines by the Cell and Genome Engineering Core at the University of Connecticut Health Center. H9 hESCs serve as isogenic controls. Both H9 control and H9-Gln40X were then differentiated into cortical PNs as we described in the above section for the examination of the effects caused by this disease-specific mutation.
2.13. Statistical analysis
The Shapiro-Wilk test, skewness and kurtosis were used to assess if the data were normally or approximately normally distributed (Kim, 2013). A two-tailed Student t-test was used to compare mean values between two groups, after confirming that the data were normally or approximately normally distributed. When comparing mean values among three or more groups, a one-way ANOVA was conducted to assess groupwise differences for normally or approximately normally distributed data; alternatively, a Kruskal-Wallis H test (also known as one-way ANOVA on ranks) was used for data that were not normally or approximately normally distributed. Post hoc Dunnett tests were performed to further compare the differences between the treatment groups to the control group. The statistical significance level was set at p < 0.05 and SAS 9.4 (SAS Institute Inc., Cary, NC) was used for statistical analysis.
2.14. Study approval
All experiments involving hESCs were approved by the Embryonic Stem Cell Research Oversight Committee (ESCRO) and Institutional Biosafety Committee (IBC) at the University of Illinois.
3. Results
3.1. Generation and differentiation of SPG11 knockdown hESC lines
To examine the effects of loss of SPG11 function on human neurons, we established SPG11 knockdown hESC lines by combining lentiviruses and RNA interference (RNAi). pLKO.1 vectors were adopted to express small hairpin RNA (shRNA) sequences that target SPG11 (SPG11 shRNA) and luciferase (Luc shRNA, as controls) (Fig. 1a). Two different SPG11 RNAi sequences, SPG11 RNAi-A (SPG11-A) and SPG11 RNAi-B (SPG11-B) were utilized to minimize off-target effects. The constructed vectors were first transfected into HEK293T cells (transient transfection) to check the knockdown efficacy (Fig. 1b). After confirming the knockdown efficiency of SPG11 expression by both shRNAs, the lentiviruses containing SPG11 and Luciferase shRNA were produced and collected from HEK293T cells, which infected hESCs to generate RNAi hESC lines. After drug selection and passages, clonal hESC lines with uniform GFP expression that indicates the shRNA expression were generated (Fig. 1c). The protein expression of the SPG11 protein, Spatacsin, was dramatically reduced in SPG11 knockdown hESCs (Supplemental Fig. 2). The hESCs were expanded by those GFP positive clusters and differentiated into neurons. We confirmed that hESCs were differentiated into cortical projection neurons by checking the expression of Ctip2 (Fig. 1d), a marker for subcerebral cortical PNs (Arlotta et al., 2005; Li et al., 2009). During the differentiation process (Fig. 1e), knockdown efficacy was monitored by quantifying the mRNA expression levels (Fig. 1f). Those data showed that both SPG11-shRNA knockdown groups maintained a significant reduction in SPG11 mRNA transcripts through the differentiation process, revealing the successful establishment of a hESC-based model of SPG11.
Fig. 1.
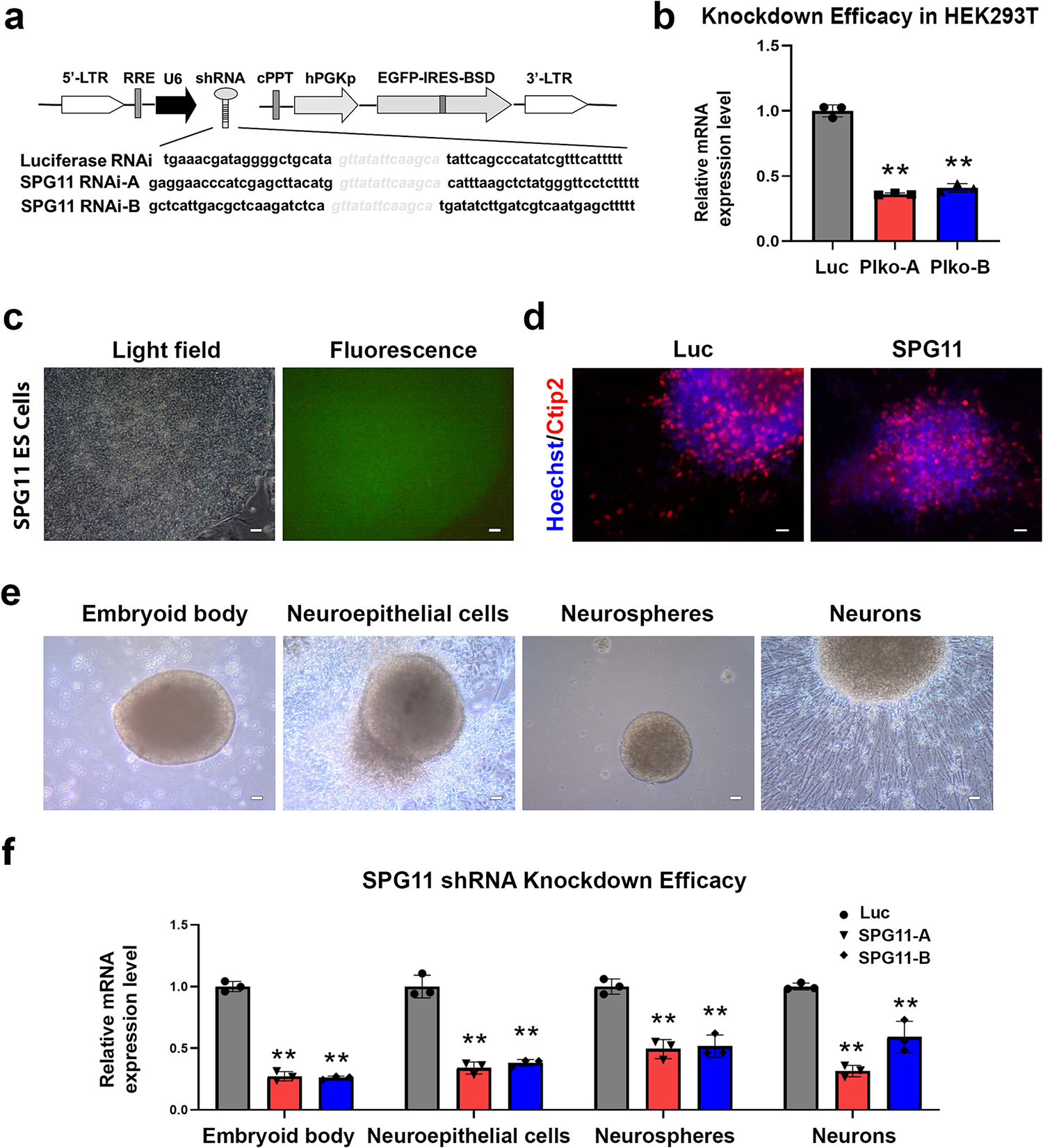
SPG11 hESC-based model construction and knockdown efficacy test. (a) Schematic map of the pLKO.1 vector containing the SPG11 RNAi sequences. (b) To examine the knockdown efficiency, HEK293 cells were transfected with lentiviral vectors, followed by separation of RNA and RT-qPCR. Quantification of the mRNA expression of SPG11 confirmed the significant knockdown effects by both shRNAs. (c) Fluorescence images of GFP expression in SPG11 RNAi-expressing ES cells showed uniform expression of GFP, indicating the expressing of shRNA in the stem cell colonies. Scale bar = 40 μm. (d) Immunostaining showing the generation of Ctip2+ cortical PNs (7-week) from luciferase and SPG11-knockdown hESCs. Scale bar = 20 μm. (e) Light field images showing typical stages during differentiation of cortical PNs from hESCs. Scale bar = 40 μm. (f) qPCR showing the expression of SPG11 genes at 4 stages during the cortical neuron differentiation from Luciferase and SPG11 knockdown (SPG11-A, SPG11-B) hESCs. Data were presented as Mean ± SD, **p < 0.01 versus luciferase RNAi group by Dunnett’s test after ANOVA.
3.2. Characterization of SPG11 axonal defects in hESC-derived neurons
To observe the effects of SPG11 on axonal development, hESCs were differentiated into cortical projection neurons by using the established protocols (Boisvert et al., 2013; Li et al., 2009). The axons are generally derived by the longest neurite, which involves in the formation of the complex neuronal architecture during development (Tosney and Landmesser, 1985). We then examined axonal development and measured axonal length of Ctip2+ cortical PNs (Fig. 2a, Supplemental Fig. 3). Our data showed that the average length of Tau+ axons was significantly decreased in SPG11 knockdown neurons compared to that in Ctip2+ cortical PNs derived from Luc control hESCs (Fig. 2b). Similar reductions of axonal length were observed in knockdown neurons in both shRNA-A and shRNA-B groups, suggesting that neurite outgrowth in neurons is sensitive to spatacsin levels.
Fig. 2.
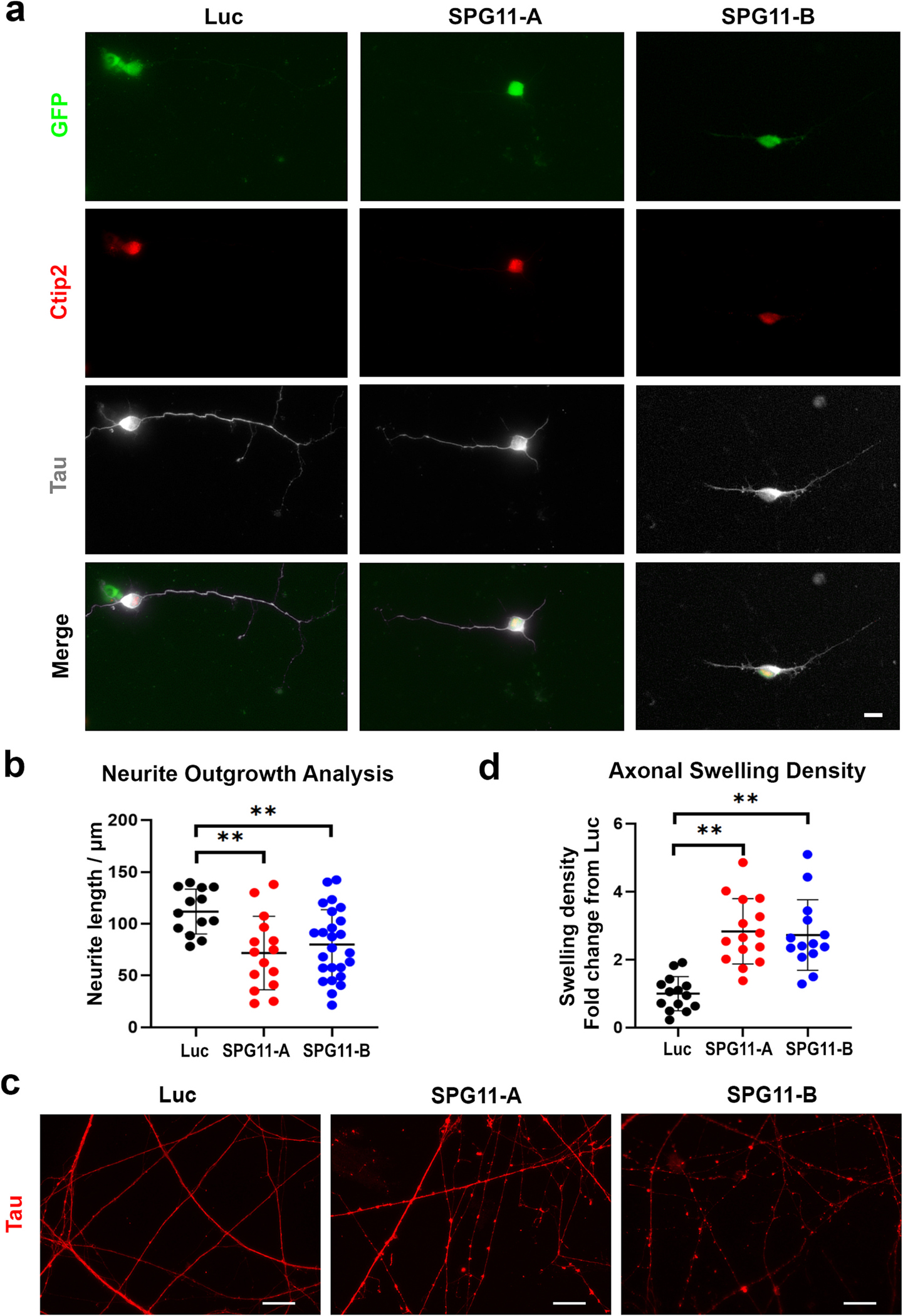
SPG11 neurons display axonal defects in two independent knockdown groups. (a) Immunostaining showing the expression of Tau and Ctip2 in 6-week cortical PNs derived from Luciferase shRNA, SPG11 shRNA-A, and SPG11 shRNA-B hESCs. Scale bar = 10 μm. (b) Neurite outgrowth quantification of 6-week neurons revealed a significant reduction of axonal length in SPG11 knockdown neurons compared to control neurons. (c) Immunostaining of Tau showing the formation of axonal swellings along axons in SPG11 knockdown cultures. Scale bar = 20 μm. (d) Quantification revealed a significant increase in the density of axonal swellings in week 11 SPG11-knockdown (both shRNA-A and shRNA-B groups) neurons compared to control neurons. Data were presented as Mean ± SD. The differences between knockdown groups and Luciferase RNAi control group were analyzed by Dunnett’s test after ANOVA. **p < 0.01 versus luciferase group.
Next, we compared the density of axonal swellings, a marker for degenerated axons. Accumulation of axonal swellings is a characteristic pathological change observed in HSP, which is formed by the accumulation of transported cargos caused by impaired transport (Carpenter, 1968). To determine if the knockdown of SPG11 in neurons leads to axonal phenotypes, Tau immunostaining was performed to visualize axonal swellings (Fig. 2c). The number of axonal swellings was quantified and divided by axonal length as we described before (Denton et al., 2014; Mou et al., 2020a). Under control conditions, neurons mostly exhibited long smooth neurites. In SPG11-knockdown neural cultures, accumulated axonal swellings were observed. The quantification results revealed a significant increase in the number of axonal swellings in neurons from both SPG11-A and SPG11-B groups, as compared to control neurons (Fig. 2d). These data suggest that loss of spatacsin function leads to impaired axonal transport and accumulated axonal swellings in human neurons.
3.3. Loss of spatacsin results in abnormal cholesterol distribution in neurons
Given that spatacsin is important for maintaining normal lysosomal tubule structure and cholesterol trafficking (Boutry et al., 2019), we hypothesized that loss of spatacsin in human neurons results in cholesterol accumulation in lysosomes, disrupting cholesterol distribution to plasma membrane. To test this hypothesis, we first checked the cholesterol distribution in neurons by using Filipin III (Vienken et al., 2017), a dye stained for free cholesterol in Luc- and SPG11-shRNA hESC-derived neurons (Fig. 3a). We observed clear membrane Filipin staining in control neurons, which was disrupted in SPG11-deficient neurons. To confirm the aberrant cholesterol in plasma membrane of SPG11-knocodwn neurons, we used Deep Red CellMask™ to label plasma membrane, and found a significant reduction of cholesterol in plasma membrane of SPG11 shRNA-A and shRNA-B neurons (Supplemental Fig. 4). These data suggest that the loss of spatacsin leads to an alteration of cholesterol distribution in SPG11 neurons.
Fig. 3.
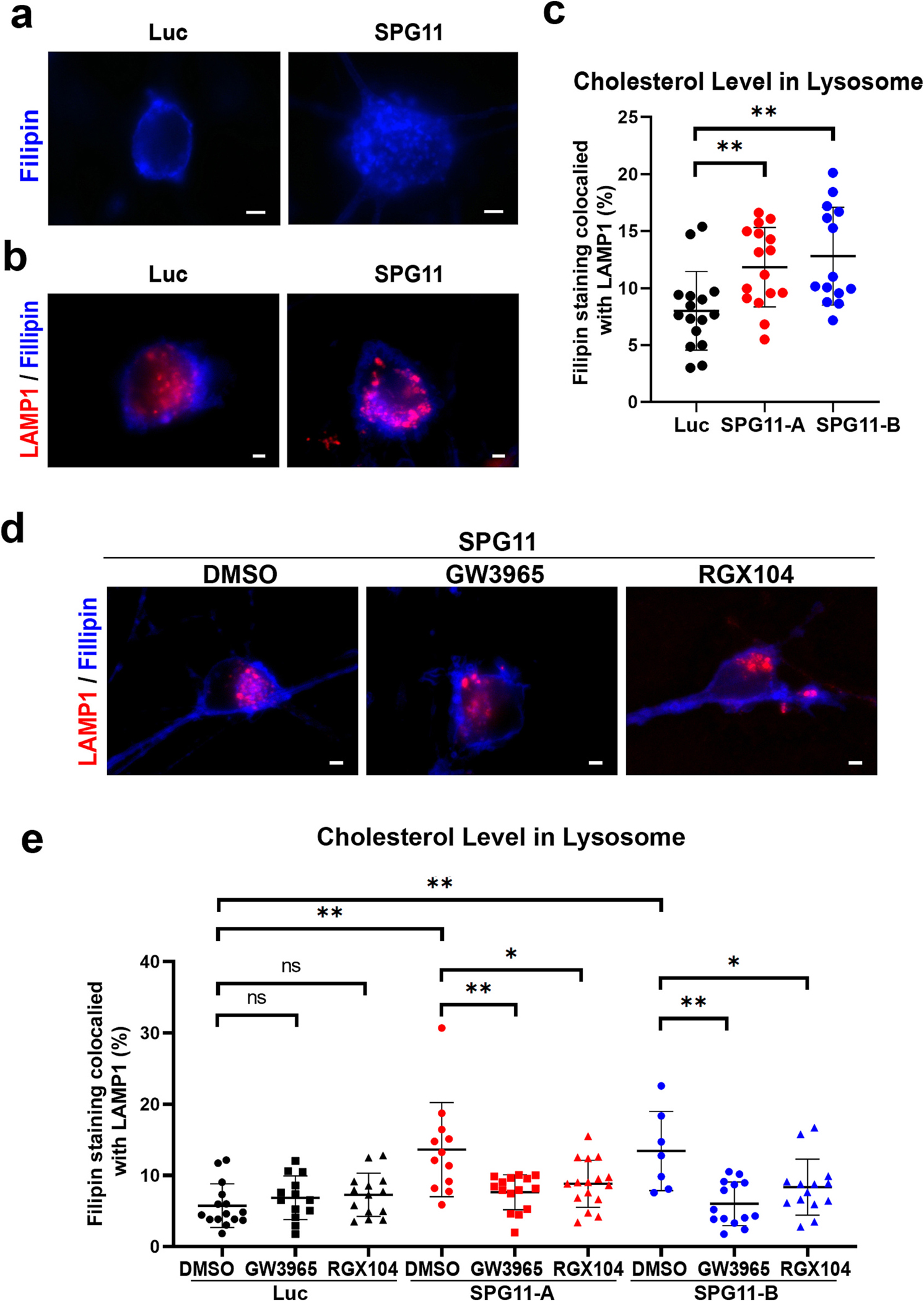
Impaired distribution of cholesterol in SPG11 neurons. (a) Staining of cholesterol using Filipin cholesterol kit in week 8 SPG11 knockdown and control neurons. The cholesterol distributions among the soma were showed. Scale bar = 2 μm. (b) Double staining of cholesterol marker filipin and lysosomal marker LAMP1 showed the accumulated cholesterol in lysosome of SPG11 neurons. The lysosomal cholesterol was shown as overlapping parts. Scale bar = 2 μm. (c) Quantification of the amount of filipin staining colocalized with LAMP1 in two independent SPG11 knockdown groups. (d) Staining of cholesterol marker filipin and lysosomal marker LAMP1 in SPG11 neurons treated with vehicle (DMSO) or drugs (GW3965 and RGX104). The lysosomal cholesterol was shown as the overlapping parts. Scale bar = 2 μm. (e) Quantification of the amount of filipin staining colocalized with LAMP1 in SPG11 knockdown groups treated with GW3965, RGX104, or DMSO (vehicle control). The proportion of cholesterol in lysosome was significantly increased in SPG11 neurons, which was mitigated by the treatment of either LXR agonists. Data were presented as Mean ± SD. *p < 0.05, **p < 0.01 versus DMSO group by Dunnett’s test after ANOVA. n.s. = not significant.
To further examine the localization of cholesterol, we labelled cholesterol with the main SPG11-targeted organelle, lysosomes, for cholesterol accumulation quantification (Varga et al., 2015). After the double staining of filipin and LAMP1 (Fig. 3b), the colocalization analysis was performed to quantify the cholesterol in lysosome. The proportion of cholesterol accumulated in lysosome was compared between SPG11 knockdown and control neurons (Fig. 3c). Our data showed that the proportion of cholesterol in lysosome is significantly increased in SPG11 neurons compared to control neurons (Fig. 3c). Together with the filipin analysis, these data suggest that spatacsin deficiency impairs cholesterol homeostasis and leads to the accumulation of cholesterol in lysosomes and reduction of cholesterol in plasma membranes.
3.4. LXR agonists reduce lysosomal cholesterol accumulation in SPG11 cortical neurons
Given that the liver X receptors (LXRs) can regulate cholesterol homeostasis, which has been shown to enhance the efflux of free form cholesterol from the late endosomes/lysosomes (Rigamonti et al., 2005), it is possible that LXR agonists may rescue axonal defects through regulating cholesterol trafficking. To test this hypothesis, we examined the effects of LXR agonists on cholesterol trafficking in SPG11 neurons. Given that SPG11 is critical for lysosomal function and cholesterol can accumulate in lysosome in SPG11 knockdown neurons, we examined the effect of LXR agonists on cholesterol distribution in lysosome after applying 1 μM GW3965 or RGX104, every 2 days for 1 week (Fig. 3d). Interestingly, the increased proportion of cholesterol in lysosome was significantly mitigated by the application of LXR agonists in SPG11-kncokdown neurons. Both GW3965 and RGX104 significantly reduced the accumulated cholesterol in neurons derived from both SPG11-shRNA-A and SPG11-shRNA-B groups (Fig. 3e). Furthermore, in control luciferase-knockdown neurons, treatment of GW 3965 and RGX104 did not significantly alter the lysosomal cholesterol contents in these normal neurons. Together, these data confirm that LXR activators can restore cholesterol homeostasis.
3.5. Synthetic LXR agonists rescue axonal outgrowth and transport defects in SPG11 knockdown neurons
Given that LXR agonist can restore normal cholesterol trafficking and distribution, we examined whether LXR agonists have any protective effects against axonal defects of SPG11 knockdown neurons. Previously, we showed that the absence of spatacsin impaired the neurite outgrowth (Fig. 2b). To evaluate the effects of synthetic LXRs agonists in SPG11 neurons, the SPG11 neurons were treated with cholesterol-targeting drugs (GW3965 or RGX104 at 1 μM final concentration) (Collins et al., 2002; Tavazoie et al., 2018) and vehicle control (DMSO), and were cultured for 2 days (Fig. 4a). By measuring the average length of Tau+ axons, both GW3965 and RGX104 significantly increase the neurite length of SPG11 neurons to a level similarly as control neurons (Fig. 4b). In addition, there was no significant difference in neurite length between normal neurons treated with and without those drugs (Fig. 4b), suggesting that those drugs did not display distinct effect to healthy neurite outgrowth during neuronal development. Taken together, these data revealed that LXR agonists can ameliorate the neurite outgrowth abnormalities observed in hESC-derived SPG11 neurons.
Fig. 4.
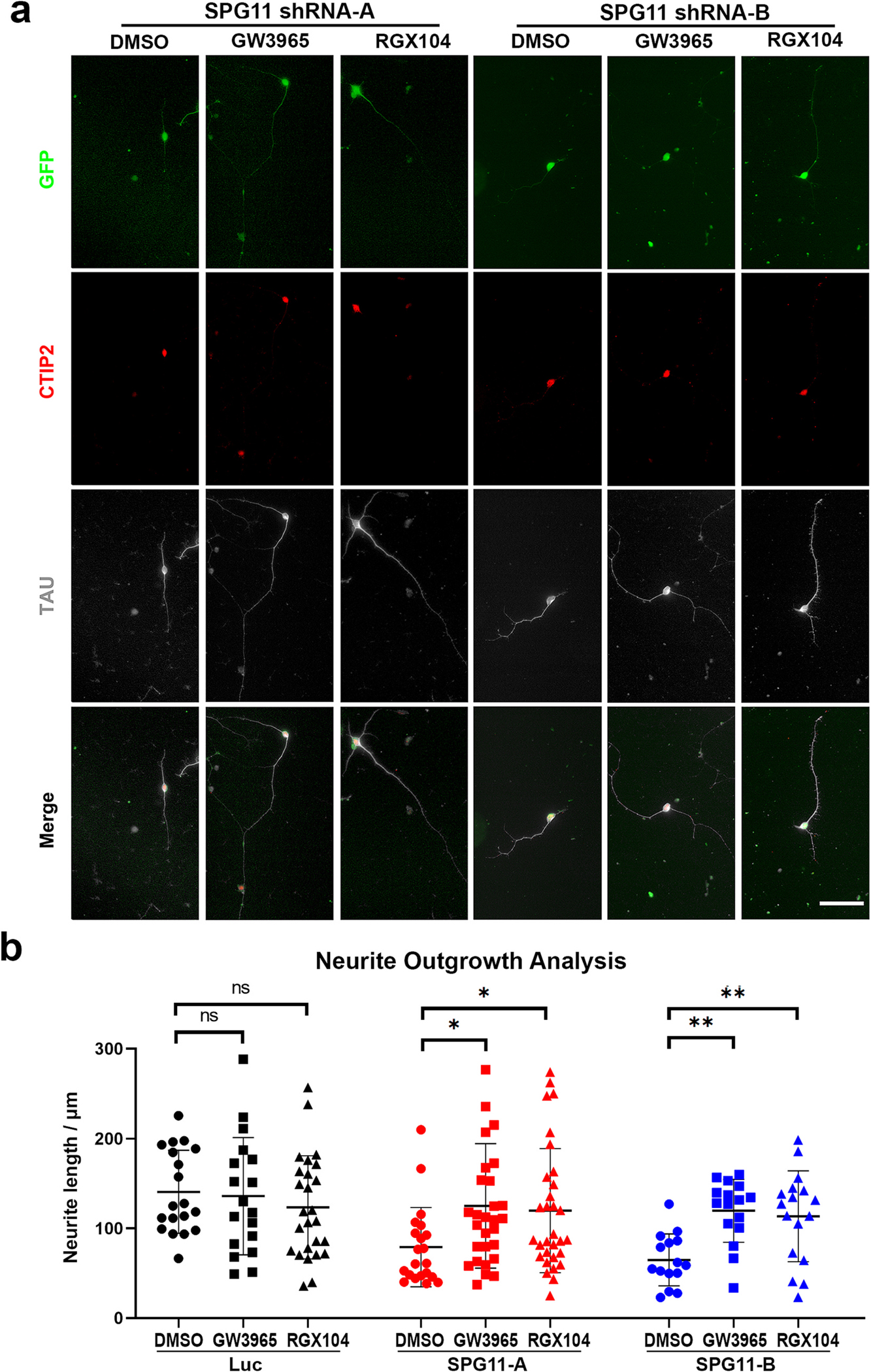
Rescue of SPG11 neurite outgrowth abnormalities by LXR agonists. (a) Immunostaining of Tau protein showing the neurite outgrowth in cortical PNs (CTIP2+) derived from SPG11 shRNA-A and SPG11 shRNA-B hESCs. Cortical PNs were treated with LXR agonists (GW3865 or RGX104, 1 μM), or vehicle control (DMSO) for 48 h followed by the immunostaining to examine the neurite outgrowth. Scale bar = 20 μm. (b) Neurite outgrowth quantification revealed a significant elongation of Tau+ axons in SPG11 knockdown neurons after the treatment of GW3965, RGX104, or DMSO (Vehicle control). Data were presented as Mean ± SD. Specific p values, *p < 0.05, **p < 0.01 versus DMSO group by Dunnett’s test after ANOVA. n.s. = not significant.
Considering that aberrant transport along axons is a characteristic change in SPG11, we then investigated the effects of LXR agonists on axonal transport. Increased axonal swellings in SPG11 knockdown neurons suggests that loss of spatacsin impairs axonal transport. To confirm this, we first examined the axonal transport of mitochondria to examine the fast axonal transport in live neuron cultures (Hollenbeck and Saxton, 2005; Mandal and Drerup, 2019). Cortical PNs derived from Luc-, SPG11-shRNA-A and SPG11-shRNA-B hESCs were stained with MitoTracker dye followed by washing and live cell imaging. The kymographs of mitochondria were analyzed, which can track the trajectories of each mitochondrion within the selected axons segment (Fig. 5a). The result showed that there was a significant reduction in the percentage of motile mitochondria in SPG11 neurons (Fig. 5b); over 30% of the mitochondria were motile in control (luciferase knockdown) neurons, whereas in SPG11 neurons, only around 20% of the mitochondria were motile. Thus, these data suggested that loss of spatacsin reduced axonal transport of mitochondria and decreased the proportion of motile mitochondria. After treating with 1 μM GW3965 or RGX104 for 1 week, the mitochondrial motility of SPG11 neurons was measured (Fig. 5c). There were significant increases in the percentage of motile mitochondria in both GW3965- and RGX104-treated groups compared with vehicle-treated SPG11-shRNA-A groups (Fig. 5d). Treatment of Vehicle (DMSO) alone did not affect the mitochondrial motility (Supplemental Fig. 5). Moreover, a similar increase of motile mitochondria by GW3965 and RGX104 was observed in SPG11-shRNA-B groups (Fig. 5d). Taken together, both GW3965 and RGX104 can significantly raise the percentage of motile mitochondria, revealing that the disruption of axonal transport can be rescued by LXR agonists.
Fig. 5.
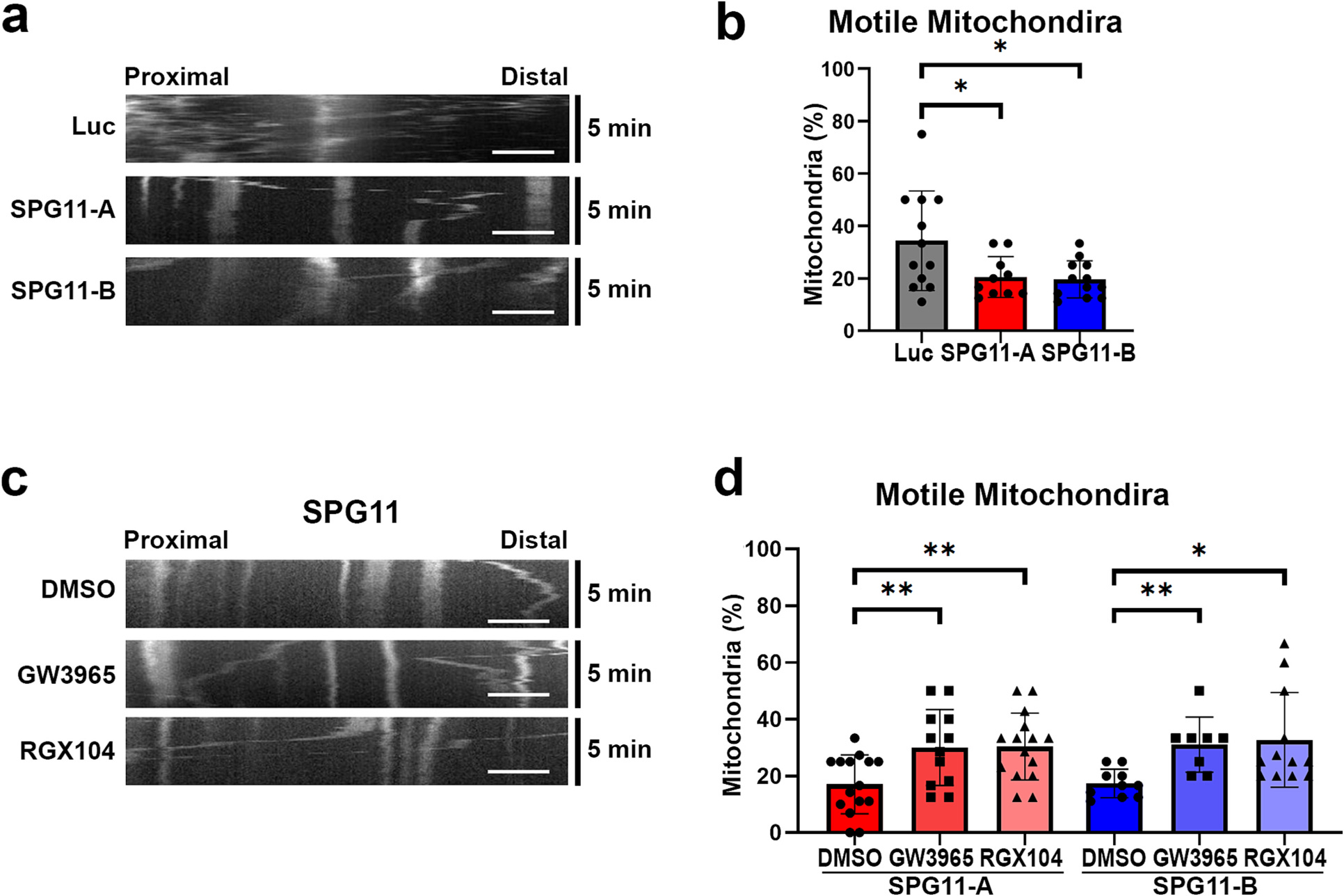
LXR agonists restore the reduced motile mitochondria in SPG11 neurons. (a) Representative kymographs showing the mitochondrial movement in SPG11 neurons comparing to luciferase group. X-axis represents distance, and Y-axis refers to time (a 5-min recording). Scale bar = 5 μm. (b) Quantification of motile mitochondria percentage revealed a significant reduction of motile mitochondria in week 10 SPG11 knockdown neurons. Data were presented as Mean ± SD, *p < 0.05 versus luciferase shRNA (control) group. (c) Mitochondrial movement in SPG11 neurons after the treatment of GW3965, RGX104, and DMSO (vehicle control). Representative kymographs showing the mitochondrial movement in these groups. (d) Quantification of motile mitochondria percentage in SPG11 shRNA-A and shRNA-B groups after the treatments with GW3965, RGX104, or DMSO control. Scale bar = 5 μm. Data were presented as Mean ± SD, *p < 0.05 and **p < 0.01 versus DMSO group by Dunnett’s test after ANOVA.
3.6. LXR agonists mitigate axonal swellings and apoptosis in SPG11 long-term cultures
In long-term cultures, we further examined the protective effects of LXR agonists on axonal swellings levels as we previously described (Denton et al., 2014). SPG11 knockdown and control neurons were treated with 1 μM RGX104 and GW3965 for 1 week. Interestingly, both RGX104 and GW3965 treatment significantly reduced the number of swellings in SPG11 knockdown neurons (Fig. 6a and b). The increased axonal swellings in both shRNA-A and shRNA-B groups can be effectively inhibited by the treatment of RGX104 or GW3965. These data demonstrate that relieving the disruption of axonal transport by the treatment of LXR agonists can mitigate accumulated axonal swellings in SPG11 neurons.
Fig. 6.
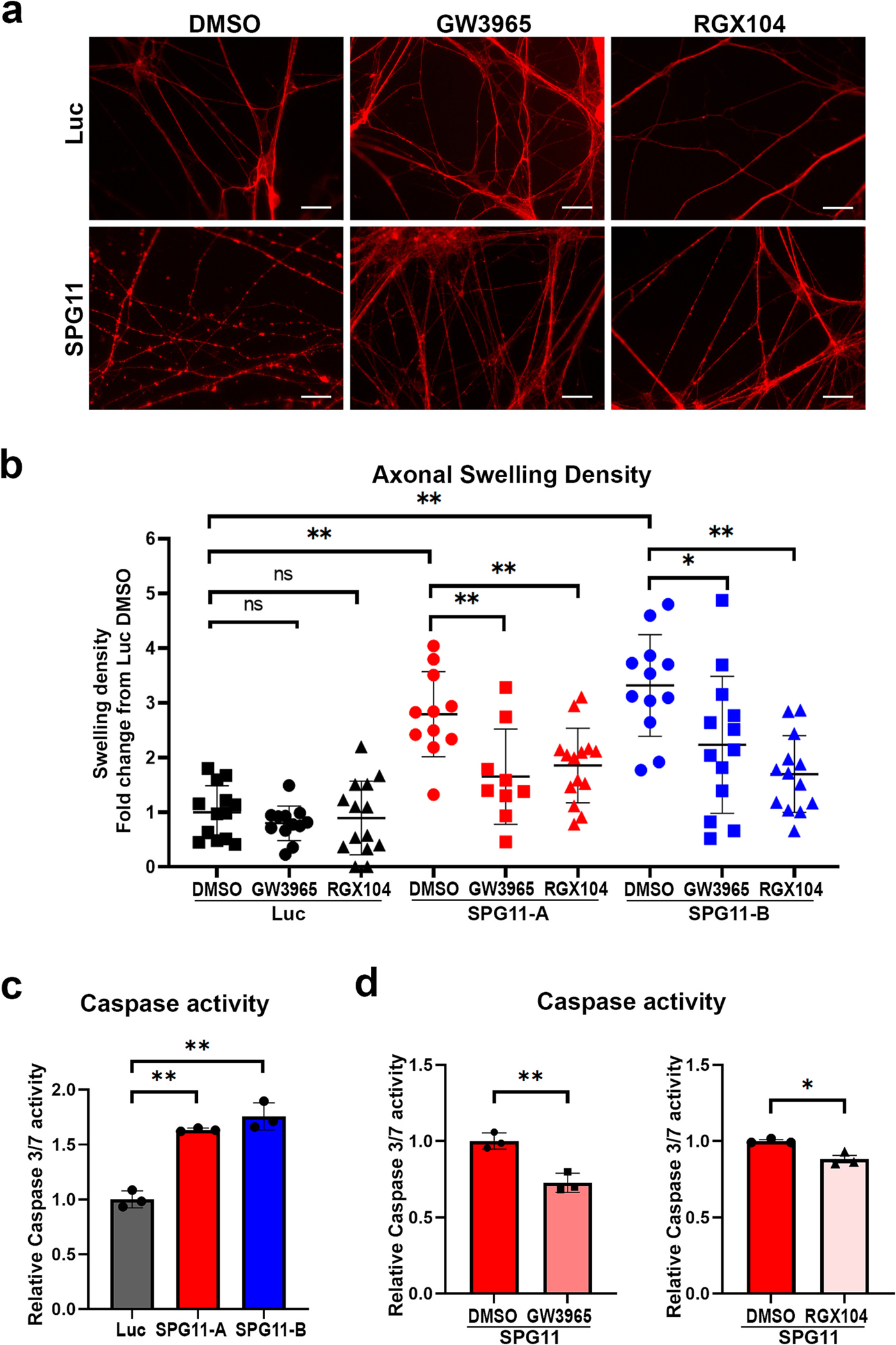
LXR agonists reduce the accumulated axonal swelling and apoptosis in SPG11 neurons. (a) Immunostaining of Tau protein showing SPG11 and control neuronal axons in long-term cultures with or without drug treatments. Scale bar = 20 μm. (b) Quantification of axonal swelling density in long-term cultures revealed a significant reduction of increased axonal swellings in SPG11 knockdown groups (SPG11-A and SPG11-B) by the treatment of GW3965 and RGX104. Both drugs did not show significant effects on axonal swellings density in control Luc neurons. Data were presented as Mean ± SD, *p < 0.05, **p < 0.01 versus SPG11 DMSO group by Dunnett’s test after ANOVA. (c) Relative Caspase 3/7 activity was significantly increased in two independent SPG11 knockdown groups (SPG11-A and SPG11-B) compared to Luc control neurons. Data were presented as Mean ± SD, ***p < 0.01 versus luciferase group by Dunnett’s test. (d) Relative Caspase 3/7 activity in both SPG11 knockdown treated with different drugs for 1 week. After treatment with GW3965 or RGX104, the caspase activity was significantly decreased. Data were presented as Mean ± SD, *p < 0.05, **p < 0.01, versus DMSO group by two-sided t-test.
In addition, to study the protection effect and toxic potency of those drugs in SPG11 neurons, one direct way is to determine apoptosis levels. Given that both caspases 3 and 7 are known as executioner caspases and are activated universally during apoptosis process (Walsh et al., 2008), the caspase 3/7 activities were compared between control and SPG11 neurons. Our results showed that the caspase 3/7 activity in SPG11 neurons was significantly higher than control neurons (Fig. 6c). To evaluate the effects of GW3965 and RGX104, the apoptosis level of neurons was determined to check the mortality rate with treatments. The apoptosis levels in SPG11 neurons were significantly reduced after GW3965 treatment (Fig. 6d). Similarly, the Caspase 3/7 levels were much lower in RGX104-treated group compared to that in Vehicle (DMSO)-treated group, confirming the ability of attenuating caspase 3/7 activity by LXR agonists in SPG11 neurons. Taken together, these data revealed that both LXR agonists, GW3965 and RGX104, are able to mitigate axonal defects and partially rescue the mortality rate of SPG11 cortical neurons.
3.7. SPG11 disease-causing mutation results in aberrant cholesterol accumulation and degeneration in human cortical neurons
SPG11 is a common dominant recessive form of HSP, and can be caused by compound or homozygous mutations of the SPG11 gene (Southgate et al., 2010; Stevanin et al., 2008). To confirm that SPG11 disease-specific mutation can directly impair cholesterol trafficking in SPG11 patients, we knocked in the disease-causing mutation (Franca Jr. et al., 2012) (c. 118C > T, p.Gln40×) in H9 hESCs using CRSPR-cas9 mediated gene editing. After drug selection and gene sequencing confirmation as we reported before, we successfully generated H9 SPG11 Gln40× homozygous lines (Chen et al., 2022). These H9 SPG11 Gln40× lines, as well as normal H9 (isogenic control), were efficiently differentiated into CTIP2+ cortical PNs (Fig. 7a). The expressions of SPG11 mRNA were significantly reduced along differentiation at both neural progenitors and neuron stages, confirming that loss of Spatacsin function is implicated in SPG11 (Fig. 7b). Notably, in SPG11 Gln40× neurons, the localization of cholesterol in lysosomes, as indicated by the co-localization of Filipin (stained for total Cholesterol) and LAMP1 (marker for lysosomes), was significantly increased (Fig. 7c and d). This data confirms the involvement of aberrant cholesterol accumulation in lysosome in SPG11, and suggests that impaired cholesterol homeostasis underlies the degeneration of these SPG11 neurons.
Fig. 7.
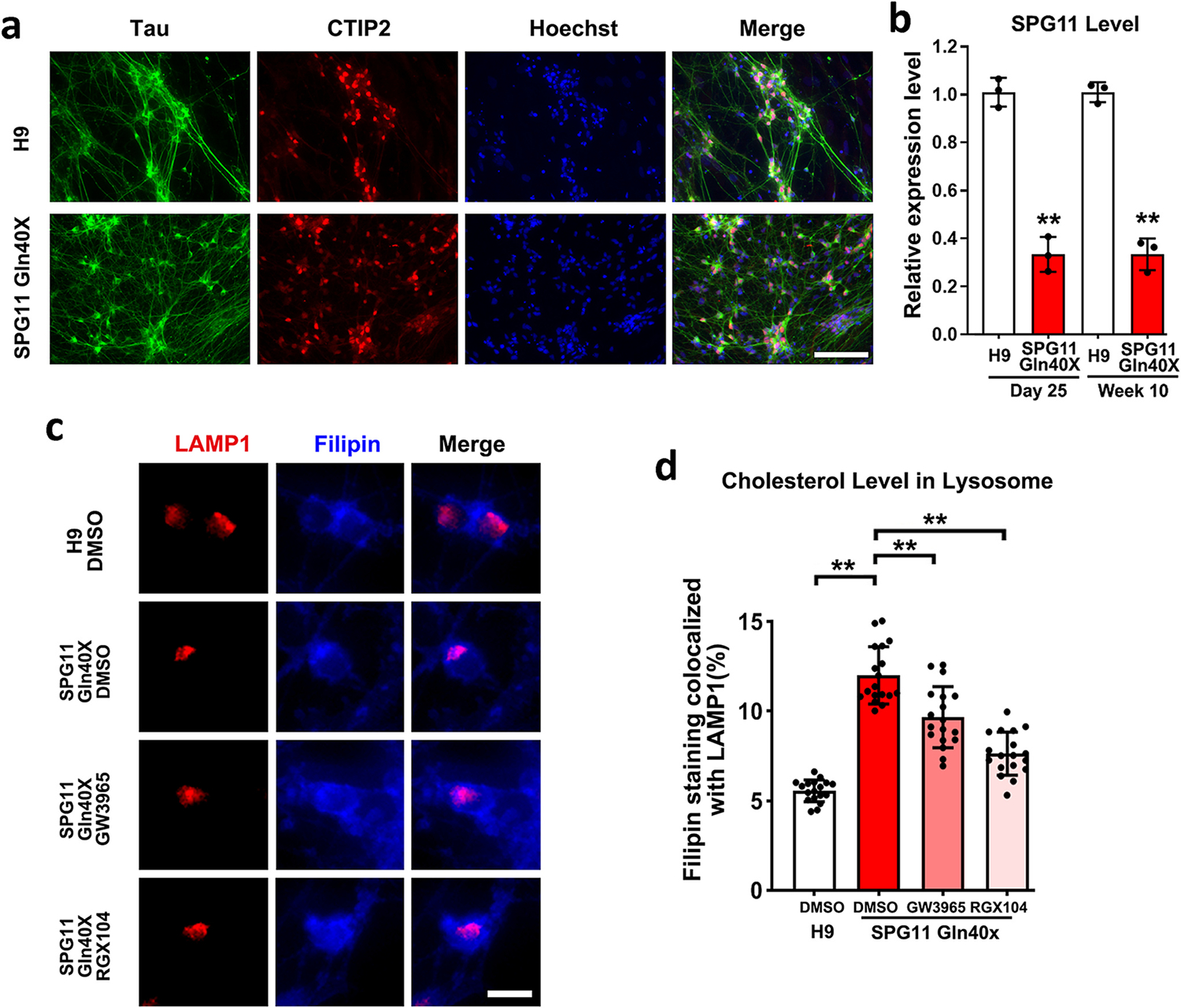
SPG11 disease-causing mutation impairs cholesterol distribution in lysosomes. (a) H9 p.Gln40× hESCs, as well as H9 control hESCs (isogenic control), were differentiated into cortical PNs that were positive for CTIP2 and Tau. (b) qPCR showing the expression of SPG11 (Spatacsin) mRNA at two different stages (day 25, progenitors; and 10-week, mature neurons) during neural differentiation. (c) Immunostaining showing cholesterol marker filipin and lysosomal marker LAMP1 in SPG11 neurons treated with vehicle (DMSO) or drugs (GW3965 and RGX104). Scale bar = 10 μm. (d) Quantification of the percentage of filipin staining colocalized with LAMP1 in H9 control and SPG11 mutated neurons treated with GW3965, RGX104, or DMSO (vehicle control). Data were presented as Mean ± SD. **p < 0.01 versus H9 control by two-sided t-test (b); **p < 0.01 versus SPG11 DMSO group (d) by Dunnett’s test after ANOVA.
To further test this hypothesis, we applied LXR agonists to SPG11 Gln40X neurons, and examined whether their degeneration can be rescued through restoring cholesterol homeostasis. SPG11 Gln40X neurons were treated with GW3965 (1 μM), RGX104 (1 μM), or DMSO (Vehicle control). At 1-week after treatment, the localization of cholesterol in lysosome was compared between different groups. Our data revealed that the percentage of Filipin staining co-localized with LAMP1 was significantly reduced after GW3965 or RGX treatment (Fig. 7c and d). To further determine the effects of LXR agonists on cholesterol distribution, we examined the cholesterol levels in plasma membrane in H9 and SPG11 Gln40X neurons by performing CellMask™ plasma membrane Deep Red and Filipin staining. Filipin intensity overlapped with CellMask™ plasma membrane Deep Red stain was then determined using Image J and compared between groups. Interestingly, the PM cholesterol levels were significantly reduced in SPG11 Gln40X neurons (Fig. 8a and b), and this reduction was mitigated in SPG11 neurons after GW3965 and RGX104 treatment (Fig. 8a and b). These data suggest that GW3965 and RGX104 treatment mitigate the aberrant cholesterol accumulation in lysosome and restore cholesterol homeostatic in SPG11-mutated neurons.
Fig. 8.
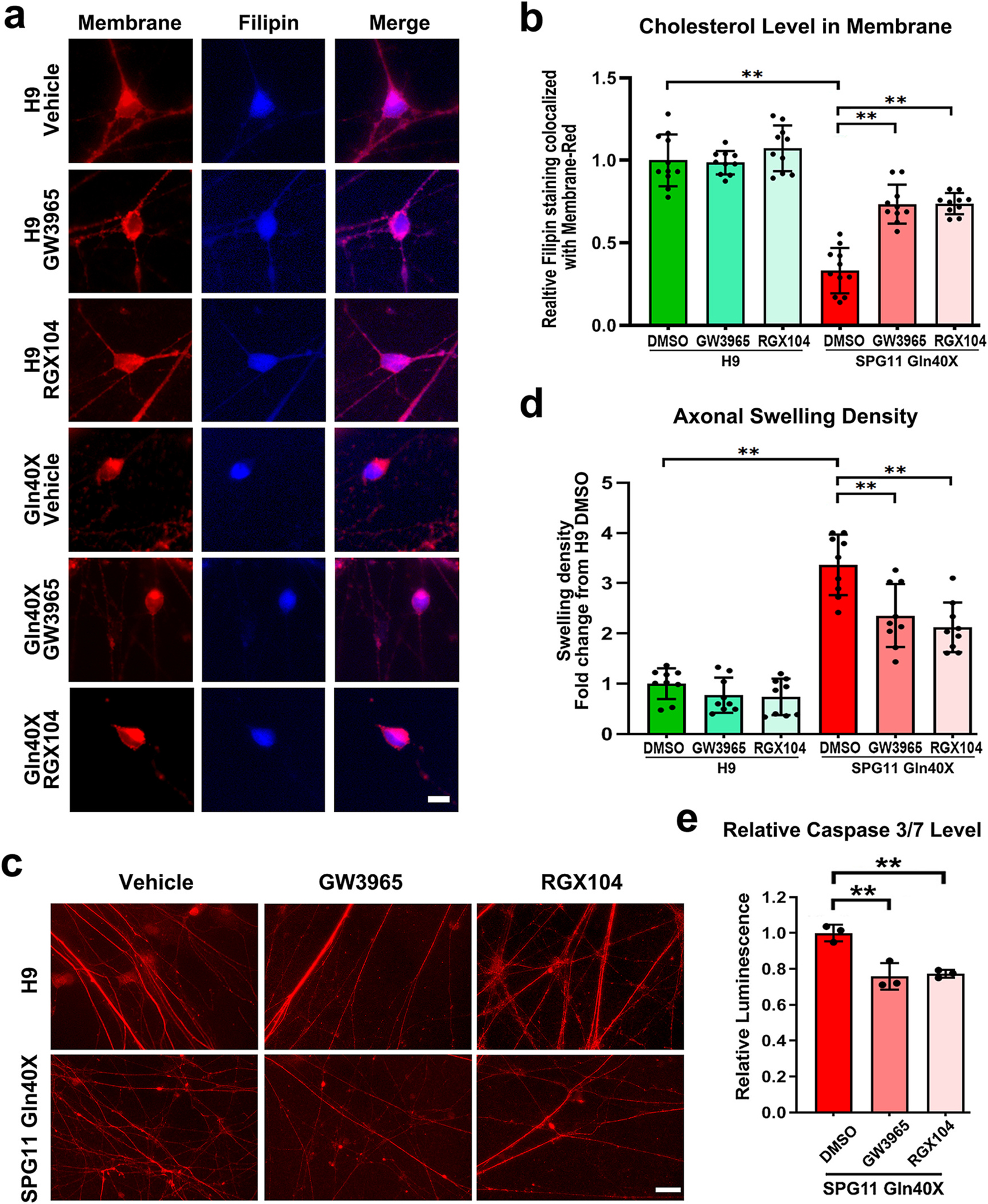
Effects of LXR agonists on axonal swellings and apoptosis in SPG11-mutated neurons. (a) Immunostaining of cholesterol using Filipin cholesterol kit (blue) and plasma membrane using CellMask™ plasma membrane Deep Red stain (red) in cortical neurons derived from H9 and SPG11 40× iPSCs. Scale bar = 10 μm. (b) Quantification of the relative intensity of filipin staining in plasma membrane (PM) of cortical neurons after treatment of GW3965, RGX104, and DMSO (vehicle). (c) Immunostaining of Tau protein showing SPG11 and control neuronal axons in long-term cultures with or without drug treatments. Scale bar = 20 μm. (d) Quantification of axonal swelling density in long-term cultures revealed a significant reduction of increased axonal swellings in SPG11-mutated neurons after the treatment of GW3965 and RGX104. Both drugs did not show significant effects on axonal swellings density in control Luc neurons. (e) After treatment with LXR agonists and vehicle, the apoptosis levels in SPG11 p.Gln40X neurons were examined by analyzing the Caspase 3/7 activities. Data were presented as Mean ± SD. **p < 0.01 versus SPG11 DMSO group by Dunnett’s test after ANOVA.
Finally, to identify the role of LXR agonists on axonal degeneration, we compared the accumulation of axonal swelling in H9 and SPG11 40X cortical neurons after treatment with GW3965, RGX104, and DMSO (Vehicle control). There was a significant increase in the axonal swelling density in cortical neurons derived from SPG11 40X iPSCs compared to normal H9 cortical neurons (Fig. 8c and d). After the treatment of GW3965 and RGX104, the accumulated axonal swellings were significantly reduced (Fig. 8c and d), confirming that LXR agonist can ameliorate axonal degeneration in long-term neurons. Furthermore, the apoptosis of these neurons, as indicated by the Caspase 3/7 levels, was also significantly inhibited by GW3965 or RGX treatment (Fig. 8e). Taken together, our data reveal that aberrant cholesterol accumulation underlies the degeneration of SPG11 neurons; and LXR agonists can rescue the degeneration of SPG11 neurons through regulating cholesterol homeostasis.
4. Discussion
Impaired axonal development and degeneration are implicated in debilitating neurodegenerative diseases including HSPs, though the detailed mechanisms underlying axonal defects of human cortical neurons in HSP are largely unclear. Using cortical PNs derived from SPG11 knockdown hESCs, we observed reduced axonal length, insufficient axonal transport, accumulated axonal swellings in these neurons. These data recapitulate disease-specific axonal defects of HSP and reveal the role of spatacsin in maintaining axonal length and transport. Movement of mitochondria in axons is not only a general representative for cargo and organelle transport, but also an indication of how fast the energy provider can arrive for high energy demands in axons (Mandal and Drerup, 2019). The significant reduction in the percentage of motile mitochondria implicates that axonal transport is impaired and becomes less efficient in SPG11 neurons. While the cause-and-effect relationship between impaired axonal transportation and mitochondrial dynamics is unclear, both contribute to axonal dysfunction in SPG11 neurons (Chevalier-Larsen and Holzbaur, 2006; Millecamps and Julien, 2013). Increased axonal swellings caused by the accumulation of transport cargos were observed in SPG11-deficiency neurons, further supporting the impairment of axonal transport in these neurons. Although most neurodegeneration is not directly correlated to defects of motor proteins or cytoskeleton production in axonal transport, the slight difference of homeostasis or interrupted intracellular signaling pathway impairs the axonal transport mechanisms, which causes a slow-down of transported cargos in axons (Carpenter, 1968; Che et al., 2016).
Lipid dysfunction has been observed in common neurodegenerative diseases including Parkinson’s disease and amyotrophic lateral sclerosis (Tracey et al., 2018; Valadas et al., 2018). As one of the critical lipids distributed in plasma and axonal membranes of neuronal cells, cholesterol is important for axonal and synaptic functions. A recent study showed that loss of spatacsin can alter cholesterol distribution in fibroblast cells (Boutry et al., 2019). Using SPG11-knockdown hESCs, as well as SPG11-disease-mutant hESCs (p.Gln40X), our study revealed abnormal cholesterol distribution in SPG11-deficiency human cortical neurons. The cholesterol level is significantly increased in cytoplasm, while in plasma membrane, conversely, it is decreased. Specifically, the colocalization analysis revealed that there is a high proportion of cholesterol accumulation in lysosomes, revealing the impaired intracellular transport of cholesterol in SPG11 neurons.
Notably, our findings indicate a novel strategy of rescuing SPG11 by adopting synthetic LXR agonists which restore cellular morphologies and cholesterol homeostasis in hESC-derived SPG11-knockdown neurons. The impaired outgrowth and extension of axons could also lead to thin corpus callosum, a developmental defect complicated with SPG11. Treating with synthetic LXR agonists, we found that the decreased axonal outgrowth activity, reduced axonal transport and increased axonal swellings can be partially restored in SPG11 neurons. We have tested two LXR agonists, GW3965 (Collins et al., 2002) and RGX104 (Tavazoie et al., 2018), and both are effective in reducing the axonal defects and restoring cholesterol trafficking. LXR agonists has been shown to regulate cholesterol trafficking in human macrophage (Rigamonti et al., 2005) and our data revealed the similar effects of LXR agonist in human neurons. Though the detailed mechanism of regulating cholesterol trafficking by LXR agonists and the efficacy of these LXR agonists in vivo await further investigation, our data demonstrate that impaired cholesterol trafficking underlies axonal defects in SPG11, and serves as a therapeutic target for regulating axonal degeneration in SPG11.
In conclusion, this is the first study of using synthetic LXR agonists to restore impaired cholesterol trafficking in spatacsin-deficiency human neurons. Our data demonstrate that loss of spatacsin results in cholesterol accumulation in lysosome, leading to axonal degeneration in cortical neurons. After the treatment with LXR agonists, SPG11 pathologies including axonal defects and apoptosis are partially relieved, providing a potential therapeutic approach through targeting the lipid trafficking defects for the rescue of neurodegenerative pathologies in SPG11 patients.
Supplementary Material
Evidence before this study.
SPG11 is a common form of hereditary spastic paraplegias, a group of neurodegenerative diseases caused by axonal degeneration of cortical motor neurons. SPG11 protein spatacsin can bind with SPG15 protein to form a complex, which plays an important role in autophagy lysosome reformation (Chang et al., J Clin Invest, 2014, 124:5249–62. doi: https://doi.org/10.1172/JCI77598). Further studies on SPG11 revealed that spatacsin can regulate cholesterol trafficking in fibroblast cells (Boutry et al., Commun Biol. 2019, 2:380. doi: https://doi.org/10.1038/s42003-019-0615-z), and loss of spatacsin can lead to lysosomal cholesterol accumulation in mouse motor neurons (Branchu et al., Neurobiol Dis. 2017, 102:21–37. doi: https://doi.org/10.1016/j.nbd.2017.02.007). However, it remains unknown whether impaired cholesterol trafficking underlies axonal degeneration of SPG11 human neurons, and more importantly, whether targeting cholesterol trafficking by small molecules (e.g., LXR agonists) recues axonal degeneration of SPG11 neurons.
Added value of this study.
This is the first study of using synthetic LXR agonists to restore aberrant cholesterol trafficking in SPG11 neurons. Our results linked impaired cholesterol homeostasis to axonal degeneration in human SPG11 neurons, and demonstrated the protective effects of LXR agonists against SPG11 pathologies through targeting cholesterol trafficking.
Implications of all the available evidence.
Our study demonstrated that impaired cholesterol trafficking underlies axonal degeneration of human SPG11 neurons. Moreover, LXR agonists are effective in rescuing aberrant cholesterol trafficking and subsequent axonal degeneration in SPG11 neurons, offering a potential therapeutic approach for SPG11 patients.
Acknowledgments
This work has been partially supported by the National Institutes of Health grants (R21NS089042 and RO1 NS118066) and the Blazer Foundation.
Footnotes
Declaration of Competing Interest
The authors have declared that no conflict of interest exists.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106293.
Data availability
The data that support the findings of this study are available in the paper or from the corresponding author upon reasonable request.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD, 2005. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45, 207–221. [DOI] [PubMed] [Google Scholar]
- Blackstone C, 2018. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol. 51, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C, O’Kane CJ, Reid E, 2011. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat. Rev. Neurosci. 12, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert EM, Denton K, Lei L, Li XJ, 2013. The specification of telencephalic glutamatergic neurons from human pluripotent stem cells. J. Vis. Exp. 5, 16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutry M, Branchu J, Lustremant C, Pujol C, Pernelle J, Matusiak R, Seyer A, Poirel M, Chu-Van E, Pierga A, et al. , 2018. Inhibition of lysosome membrane recycling causes accumulation of gangliosides that contribute to neurodegeneration. Cell Rep. 23, 3813–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutry M, Pierga A, Matusiak R, Branchu J, Houllegatte M, Ibrahim Y, Balse E, El Hachimi KH, Brice A, Stevanin G, et al. , 2019. Loss of spatacsin impairs cholesterol trafficking and calcium homeostasis. Commun. Biol. 2, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchu J, Boutry M, Sourd L, Depp M, Leone C, Corriger A, Vallucci M, Esteves T, Matusiak R, Dumont M, et al. , 2017. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 102, 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, 1968. Proximal axonal enlargement in motor neuron disease. Neurology 18, 841–851. [DOI] [PubMed] [Google Scholar]
- Che DL, Chowdary PD, Cui B, 2016. A close look at axonal transport: cargos slow down when crossing stationary organelles. Neurosci. Lett. 610, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chai E, Mou Y, Roda RH, Blackstone C, Li XJ, 2022. Inhibiting mitochondrial fission rescues degeneration in hereditary spastic paraplegia neurons. Brain. 145, 4016–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Dauch JR, Porzio MT, Yanik BM, Hsieh W, Smith AG, Singleton JR, Feldman EL, 2013. Increased axonal regeneration and swellings in intraepidermal nerve fibers characterize painful phenotypes of diabetic neuropathy. J. Pain 14, 941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL, 2006. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, et al. , 2002. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J. Med. Chem. 45, 1963–1966. [DOI] [PubMed] [Google Scholar]
- Denora PS, Santorelli FM, Bertini E, 2013. Hereditary spastic paraplegias: one disease for many genes, and still counting. Handb. Clin. Neurol. 113, 1899–1912. [DOI] [PubMed] [Google Scholar]
- Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ, 2014. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32, 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton KR, Xu C, Shah H, Li XJ, 2016a. Modeling axonal defects in hereditary spastic paraplegia with human pluripotent stem cells. Front. Biol. (Beijing) 11, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton KR, Xu CC, Li XJ, 2016b. Modeling axonal phenotypes with human pluripotent stem cells. Methods Mol. Biol. 1353, 309–321. [DOI] [PubMed] [Google Scholar]
- Fink JK, 2006. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 6, 65–76. [DOI] [PubMed] [Google Scholar]
- Fink JK, 2013. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G, 2012. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 318, 1–18. [DOI] [PubMed] [Google Scholar]
- Franca MC Jr., Yasuda CL, Pereira FR, D’Abreu A, Lopes-Ramos CM, Rosa MV, Cendes F, Lopes-Cendes I, 2012. White and grey matter abnormalities in patients with SPG11 mutations. J. Neurol. Neurosurg. Psychiatry 83, 828–833. [DOI] [PubMed] [Google Scholar]
- Harding AE, 1983. Classification of the hereditary ataxias and paraplegias. Lancet 1, 1151–1155. [DOI] [PubMed] [Google Scholar]
- Hedera P (1993). Hereditary spastic paraplegia overview. In GeneReviews((R)), Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, and Amemiya A, eds. (Seattle (WA)). [Google Scholar]
- Hirst J, Borner GH, Edgar J, Hein MY, Mann M, Buchholz F, Antrobus R, Robinson MS, 2013. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell 24, 2558–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM, 2005. The axonal transport of mitochondria. J. Cell Sci. 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, Chelban V, Manole A, Hamed SA, Haridy NA, et al. , 2016. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 139, 1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, 2013. Statistical notes for clinical researchers: assessing normal distribution (2) using skewness and kurtosis. Restor. Dent. Endod. 38, 52–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek M, Gonzalez-Gonzalez IM, Smejkalova T, Hajdukovic D, Skrenkova K, Krusek J, Horak M, Vyklicky L, 2020. Cholesterol modulates presynaptic and postsynaptic properties of excitatory synaptic transmission. Sci. Rep. 10, 12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G, Morbin M, Lombardi R, Borgna M, Mazzoleni G, Sghirlanzoni A, Pareyson D, 2003. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology 61, 631–636. [DOI] [PubMed] [Google Scholar]
- Li XJ, Zhang X, Johnson MA, Wang ZB, Lavaute T, Zhang SC, 2009. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development 136, 4055–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Drerup CM, 2019. Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 13, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinuzzi A, Blackstone C, O’Kane CJ, Stevanin G, 2021. Editorial: hereditary spastic paraplegias: at the crossroads of molecular pathways and clinical options. Front. Neurosci. 15, 708642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP, 2013. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. [DOI] [PubMed] [Google Scholar]
- Ming GL, Brustle O, Muotri A, Studer L, Wernig M, Christian KM, 2011. Cellular reprogramming: recent advances in modeling neurological diseases. J. Neurosci. 31, 16070–16075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou Y, Dong Y, Chen Z, Denton KR, Duff MO, Blackstone C, Zhang SC, Li XJ, 2020a. Impaired lipid metabolism in astrocytes underlies degeneration of cortical projection neurons in hereditary spastic paraplegia. Acta Neuropathol. Commun. 8, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou Y, Mukte S, Chai E, Dein J, Li XJ, 2020b. Analyzing mitochondrial transport and morphology in human induced pluripotent stem cell-derived neurons in hereditary spastic paraplegia. J. Vis. Exp. 156, e60548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, 2003. Cholesterol homeostasis and function in neurons of the central nervous system. Cell. Mol. Life Sci. 60, 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, Mankodi A, Vanderver A, Schindler A, Toro C, Gahl WA, et al. , 2014. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 1, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti E, Helin L, Lestavel S, Mutka AL, Lepore M, Fontaine C, Bouhlel MA, Bultel S, Fruchart JC, Ikonen E, et al. , 2005. Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ. Res. 97, 682–689. [DOI] [PubMed] [Google Scholar]
- Southgate L, Dafou D, Hoyle J, Li N, Kinning E, Critchley P, Nemeth AH, Talbot K, Bindu PS, Sinha S, et al. , 2010. Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 11, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, Rosa AL, Lerer I, Hamri A, Alegria P, et al. , 2008. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131, 772–784. [DOI] [PubMed] [Google Scholar]
- Tao Y, Zhang SC, 2016. Neural subtype specification from human pluripotent stem cells. Cell Stem Cell 19, 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, et al. , 2018. LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172 (825–840), e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM, 1998. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Tosney KW, Landmesser LT, 1985. Development of the major pathways for neurite outgrowth in the chick hindlimb. Dev. Biol. 109, 193–214. [DOI] [PubMed] [Google Scholar]
- Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST, 2018. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadas JS, Esposito G, Vandekerkhove D, Miskiewicz K, Deaulmerie L, Raitano S, Seibler P, Klein C, Verstreken P, 2018. ER lipid defects in Neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron 98 (1155–1169), e1156. [DOI] [PubMed] [Google Scholar]
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, Koch N, Hennings JC, Franzka P, Huebner AK, et al. , 2015. In vivo evidence for lysosome depletion and impaired Autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 11, e1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vienken H, Mabrouki N, Grabau K, Claas RF, Rudowski A, Schomel N, Pfeilschifter J, Lutjohann D, van Echten-Deckert G, Meyer Zu Heringdorf D, 2017. Characterization of cholesterol homeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts reveals a Niemann-pick disease type C-like phenotype with enhanced lysosomal ca(2+) storage. Sci. Rep. 7, 43575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachs FP, Couillard-Despres S, Engelhardt M, Wilhelm D, Ploetz S, Vroemen M, Kaesbauer J, Uyanik G, Klucken J, Karl C, et al. , 2003. High efficacy of clonal growth and expansion of adult neural stem cells. Lab. Investig. 83, 949–962. [DOI] [PubMed] [Google Scholar]
- Walsh JG, Cullen SP, Sheridan C, Luthi AU, Gerner C, Martin SJ, 2008. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U. S. A. 105, 12815–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bhattacharyya A, 2018. Human models are needed for studying human neurodevelopmental disorders. Am. J. Hum. Genet. 103, 829–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PP, Denton KR, Pierson TM, Li XJ, Blackstone C, 2014. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet. 23, 5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the paper or from the corresponding author upon reasonable request.