

Abstract
Background:
Lymphatic valves play a critical role in ensuring unidirectional lymph transport. Loss of lymphatic valves or dysfunctional valves are associated with several diseases including lymphedema, lymphatic malformations, obesity, and ileitis. Lymphatic valves first develop during embryogenesis in response to mechanotransduction signaling pathways triggered by oscillatory lymph flow. In blood vessels, eNOS (gene name: Nos3) is a well characterized shear stress signaling effector, but its role in lymphatic valve development remains unexplored.
Methods:
We used global Nos3−/− mice and cultured hdLECs to investigate the role of eNOS in lymphatic valve development, which requires oscillatory shear stress (OSS) signaling.
Results:
Our data reveal a 45% reduction in lymphatic valve specification cell clusters and that loss of eNOS protein inhibited activation of β-catenin and its nuclear translocation. Genetic knockout or knockdown of eNOS led to downregulation of β-catenin target proteins in vivo and in vitro. However, pharmacological inhibition of NO production did not reproduce these effects. Coimmunoprecipitation and proximity ligation assays reveal that eNOS directly binds to β-catenin and their binding is enhanced by OSS. Finally, genetic ablation of the Foxo1 gene enhanced FOXC2 expression and partially rescued the loss of valve specification in the eNOS knockouts.
Conclusion:
In conclusion, we demonstrate a novel, nitric oxide-independent role for eNOS in regulating lymphatic valve specification and propose a mechanism by which eNOS directly binds β-catenin to regulate its nuclear translocation and thereby transcriptional activity.
Keywords: VE-cadherin, FOXO1, mechanotransduction, shear stress, lymph flow
GRAPHICAL ABSTRACT
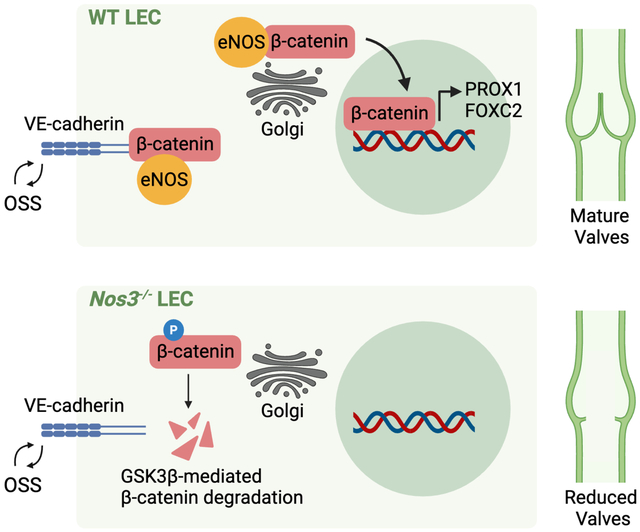
INTRODUCTION
Lymphatic vessels are required to maintain tissue fluid homeostasis by absorbing interstitial fluid and delivering it to the blood vessels1. Lymph transport against an increasing pressure gradient is facilitated by the growth of intraluminal, bicuspid valves during embryonic development that prevent lymph backflow2. Several diseases are associated with the loss of lymphatic valves or dysfunctional valves, including congenital lymphedema, cancer-associated lymphedema, obesity, ileitis, and lymphatic malformations3–8.
Lymphatic valve development in the mesentery occurs in response to the onset of lymph flow at embryonic day E15.59. Lymph flow reversal at the branchpoints of the mesenteric lymphatic network generates an oscillatory shear stress (OSS) on the surface of the lymphatic endothelial cells (LECs)9,10. In response to OSS, the transcription factor β-catenin translocates to the nucleus and upregulates the genes, PROX1 and FOXC2, in subsets of LECs, marking the specification of LEC clusters that later form valves11.
Mechanotransduction signaling in the lymphatic vasculature was recently shown to lead to AKT phosphorylation in a VE-cadherin-dependent manner and the AKT signaling pathway was partially responsible for lymphatic valve development12. A major target of AKT in the lymphatic valve leaflets is FOXO1, which is a repressor of lymphatic valve formation13. However, in blood vessels, it is well established that mechanotransduction signaling leads to phosphorylation of AKT and the S1177 residue of endothelial nitric oxide synthase (eNOS)14. While a previous study showed that eNOS expression is induced shortly after the onset of lymph flow and before the appearance of PROX1high valve specification clusters in the mouse mesentery9, it remains unclear whether and how eNOS signaling regulates lymphatic valve development. Here, we show that eNOS plays an early role in lymphatic valve specification by regulating the transcription factor β-catenin and elucidate a mechanism by which eNOS directly binds β-catenin to regulate its transcriptional activity.
METHODS
In accordance with the Transparency and Openness Promotion (TOP) Guidelines, all supporting data and materials are available from the authors upon reasonable request.
Mice:
Mice were housed under a 12:12 hour light/dark cycle and fed a standard laboratory diet (Teklad Global 18% Protein Rodent Diet, #2918). The Nos3−/− and Foxo1flox strains were obtained from the Jackson Laboratory. The Prox1-GFP, Ctnnb1flox, Prox1CreERT2 (Tmak), and Ctnnb1ex3(loxP) strains were described previously15–18. All mice were maintained on a pure C57BL/6J background and both sexes were used. Tamoxifen (TM) was dissolved in sunflower oil and ethanol for a final stock of 20mg/mL with 5% ethanol. Embryonic deletion of Foxo1 and exon 3 of β-catenin was induced by intraperitoneal injection of 5mg TM into pregnant dams on E14.5. Postnatal deletion of β-catenin was induced by subcutaneous injection of 100ug of TM at P1 and P3. Postnatal recombination of exon 3 of β-catenin was induced by subcutaneous injection of 100μg of TM once on P0. Since all experiments were done in embryonic or postnatal mice, the sex was not determined and data from both sexes were combined. All animal experiments were performed in accordance with the guidelines approved by the University of South Florida (USF) Institution of Animal Care and Use Committee (IACUC).
Generation of Prox1CreERT2 (Jscal) Strain
Difficulty breeding a previous Prox1CreERT2 with Ctnnb1flox to homozygosity indicated that these two alleles resided on the same chromosome. Therefore, we generated a new Prox1CreERT2 strain by inserting a CreERT2 followed by a SV40 polyA signal right after the start codon in the RP23–360I16 BAC clone. Random transgenesis generated 4 founders and the highest efficiency founder was selected for propagation. With repeated breeding, homozygous Prox1CreERT2;Ctnnb1flox/flox mice were eventually obtained with both strains, presumably due to crossover events.
Quantification of valve specification clusters, lymphatic valves and branchpoints:
Mesenteries from embryos or postnatal pups were harvested along with the intestine and pinned onto a Sylgard 170 cushion as previously described19. After immunostaining with PROX1 and FOXC2, the mesenteries were imaged on an inverted fluorescence microscope (Axio Observer Z1, Zeiss). Alternatively, tissues expressing the Prox1GFP reporter were imaged under a Zeiss Axio Zoom V16 fluorescence stereoscope. PROX1hi cell clusters (cell clusters having 10 or more cells expressing high levels of PROX1) or PROX1hi valves were quantified with Fiji (NIH ImageJ) software, which was also used to measure length of the large collecting vessels and the thinner pre-collecting vessels located near the intestinal wall. Between 4–5 arcades, defined as a large collecting vessel with its associated pre-collecting vessels, were imaged in each mesentery for later valve analysis.
Quantification of PROX1 and FOXC2 expression in vivo:
Mesenteries from P3 pups were collected and immunostained with PROX1, FOXC2 and VE-cadherin. High magnification confocal images of lymphangion and valve areas were obtained on a Leica TCS SP8 laser scanning confocal microscope. Using Fiji software, a ROI was drawn around each lymphangion or valve and added to ‘ROI manager’. The area of each ROI was the same for all lymphangion and valve regions. The mean pixel intensity was measured in each ROI in each image. Between 4–6 lymphangion or valve areas were imaged and analyzed from 5–6 control and knockout littermates.
Whole-mount immunostaining procedure
All steps were carried at out 4°C on an orbital shaker (Belly Dancer, IBI Scientific) unless otherwise mentioned. The mesentery along with the intestine was harvested and pinned onto a Sylgard 170 cushion and subsequently fixed overnight in 1% paraformaldehyde. The tissues were washed the next day with PBS three times 10 minutes each after which the whole-mount immunostaining procedure was carried out in the following steps: (1) Permeabilize the tissues with PBS + 0.3% Triton X-100 (PBST) for one hour; (2) Block the tissues with 3% donkey serum in PBST for two hours; (3) Incubate the tissues with primary antibodies dissolved in PBST overnight; (4) Wash the tissues with PBST five times 15 minutes each; (5) Incubate the tissues with secondary antibodies dissolved in PBST for 2 hours at room temperature; (6) Wash the tissues with PBST five times 15 minutes each; (7) Mount tissues on glass slides (Superfrost Plus Microscope slides, Fisherbrand) using ProLong Diamond Antifade Mountant containing DAPI (Invitrogen) and store slides at 4°C overnight. Tissues were imaged on a fluorescence microscope (Zeiss Axio Zoom V16 or Zeiss Axio Observer Z1) or confocal microscope (Leica TCS SP8). The images were acquired with Zen 2 Pro software and analyzed with Fiji software. Figures were created with Adobe Photoshop and Fiji software.
Cell culture, shRNA and OSS
Primary hdLECs were cultured on fibronectin-coated six-well plates in EBM-2 MV media and used at passage 6–7 for all experiments. To knock down eNOS, cells were infected with lentiviral particles expressing an shRNA targeting NOS3 or a control scramble construct expressing GFP for 48 hours. The lentiviral shNOS3 (VectorBuilder Inc.) target sequence is CCGGAACAGCACAAGAGTTAT.
The cells were exposed to OSS when they reached 95% confluency. Cells cultured in 6-well plates were exposed to OSS using a test tube rocker (Thermolyne Speci-Mix aliquot mixer model M71015, Barnstead International) with a preset frequency of 18 rpm, 0.3Hz11. Cells cultured in 10cm plates were exposed to OSS using a horizontal bidirectional shaker (MS-NOR-30, Major Science) that switched between clockwise and counterclockwise rotation at a frequency of 100rpm, 0.5Hz20. All OSS experiments were performed inside a humidified cell culture incubator with 5% CO2.
For experiments detecting phospho-eNOS, hdLECs were cultured to reach over 95% confluency after which the medium was switched from EBM-2 MV containing 5% serum to 0.5% serum medium for starvation. After 6 hours of starvation, cells were exposed to OSS for the indicated time points after which lysates were collected. For the SC-79 treatment experiment, cells were starved for 6 hours and then treated with 8μg/mL SC-79 for 30 minutes which was previously reported to lead to increased phospho-AKT levels in hdLECs13. DMSO was used as a control.
For L-NAME (100μM and 1mM), L-NMMA (100μM), and KT5823 (5μM and 10μM) inhibitors, hdLECs were cultured to reach over 95% confluency and then the indicated drugs were added to the media. Cells were exposed to static or OSS conditions for 48 hours after which, protein lysates were collected for western blot. For L-NAME and L-NMMA, water was used as a vehicle control. For KT5823, DMSO was used as a vehicle control.
Western blot
hdLEC lysates were collected using RIPA buffer (Pierce™, Thermo Scientific™) and the total protein concentration was measured using a BCA Protein Assay Kit (Pierce™, Thermo Scientific™). Protein gel electrophoresis was performed with a 4%−12% Blot™ Bis Tris Plus polyacrylamide gel and the Invitrogen™ Mini Gel Tank using 20–25μg of lysate per lane. Dry transfer onto a PVDF membrane was performed using the Invitrogen™ iBlot™ 2 Gel Transfer Device. For blots containing phospho-eNOS, wet transfer onto a PVDF membrane was performed using the Invitrogen™ Mini Blot Module and Invitrogen™ Bolt™ Transfer Buffer. The iBind™ Western Device was used to incubate the membrane with primary and secondary antibodies. The SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific™) was used to develop the membrane for protein visualization using the Bio-rad ChemiDoc imaging system.
Co-Immunoprecipitation (Co-IP)
The Co-IP procedure was carried out using the Pierce™ Co-IP kit (Thermo Scientific™). hdLECs lysates were collected using IP/Lysis Wash Buffer (Pierce™, Thermo Scientific™) and protein concentration was measured using a BCA Protein Assay Kit (Pierce™, Thermo Scientific™). For each Co-IP reaction, an antibody-coupled resin was prepared in a Pierce™ Spin Column by mixing 50ug of antibody with the AminoLink™ Plus Coupling Resin and incubating on a rotator (Labnet™ Revolver™ Tube Mixer) at room temperature for 2 hours. Lysate (1mg) was pre-cleared by incubating with the Pierce™ Control Agarose Resin at 4°C for 30 minutes with constant rotation on a rotator. The supernatant was then incubated with the antibody-coupled resin at 4°C overnight with constant rotation on a rotator. The next day, the samples were washed 4 times with IP/Lysis Wash Buffer (Pierce™, Thermo Scientific™). The protein complex was then eluted using the elution buffer provided in the kit and western blot with dry protein transfer was performed as described above.
Proximity Ligation Assay (PLA)
The PLA procedure was carried out using the Duolink PLA Fluorescence Protocol (Millipore Sigma). hdLECs were cultured on glass coverslips and upon reaching 95% confluency, the cells were exposed to static or OSS conditions for 48 hours. The cells were then fixed with 4% paraformaldehyde for 12 minutes at room temperature. Next, the cells were washed with PBS three times 5 minutes each and subsequently, the PLA procedure was carried out according to the following steps: (1) Permeabilize the cells with PBS + 0.1% Triton X-100 (PBST) for 20 minutes at room temperature; (2) Block the cells with Duolink Blocking Solution for 1 hour in a humidified cell culture incubator at 37°C; (3) Incubate the cells with antibodies diluted in Duolink Antibody Diluent overnight at 4°C; (4) Wash the cells with prepared 1X Wash Buffer A 2 times 5 minutes each on an orbital shaker at room temperature; (5) Incubate the cells with anti-rabbit MINUS and anti-mouse PLUS Duolink PLA probes diluted in Duolink Antibody Diluent for 1 hour in a humidified cell culture incubator at 37°C; (6) Wash the cells with 1X Wash Buffer A 2 times 5 minutes each on an orbital shaker at room temperature; (7) Incubate the cells with ligation solution prepared according to manufacturer’s instructions for 30 minutes in a humidified cell culture incubator at 37°C; (8) Wash the cells with 1X Wash Buffer A 2 times 5 minutes each on an orbital shaker at room temperature; (9) Incubate the cells with amplification solution prepared according to manufacturer’s instructions for 100 minutes in a humidified cell culture incubator at 37°C; (10) Wash the cells with prepared 1X Wash Buffer B 2 times 10 minutes each followed by 0.01X Wash Buffer B once for 1 minute on an orbital shaker at room temperature; (11) Mount the cells on glass slides (Superfrost Plus Microscope slides, Fisherbrand) using Duolink In Situ Mounting Medium with DAPI. Cells were subsequently imaged on a confocal microscope (Leica TCS SP8).
Statistics
All data are represented as mean ± SEM. An unpaired 2-sided Student’s t-test was performed to determine significant differences between 2 groups. One-way ANOVA followed by Tukey’s multiple-comparison test was performed to determine significant differences between 3 groups. Two-way ANOVA followed by Tukey’s multiple-comparison test was used to determine signficant differences in data sets with 2 independent variables. p<0.05 was considered significant. Graphpad Prism software (version 6) was used for all statistical analyses and to plot quantitative data.
Resources
Please see the Major Resources Table in the Supplemental Materials.
RESULTS
eNOS is upregulated and activated in lymphatic valve LECs
To investigate eNOS expression in developing and mature valves, we harvested mesenteries from WT embryos and performed whole-mount immunostaining for PROX1 and eNOS. At E16.5, many PROX1high valve specification clusters were observed in the mesentery as previously reported9 and eNOS was highly expressed in these PROX1high valve LECs compared to the surrounding PROX1low lymphangion LECs (Supplemental Figure 1A). At later developmental stages, eNOS was highly expressed in the LECs of mature valves at E18.5 (Figure 1A), P3 (Supplemental Figure 1B) and P7 (Supplemental Figure 1C). Whole-mount immunostaining for eNOS was performed in E16.5 Nos3−/− mice as a negative control revealing an absence of the protein (data not shown). We failed to detect PROX1 in adjacent arteries which served as a negative control.
Figure 1: eNOS is upregulated and activated in response to OSS.
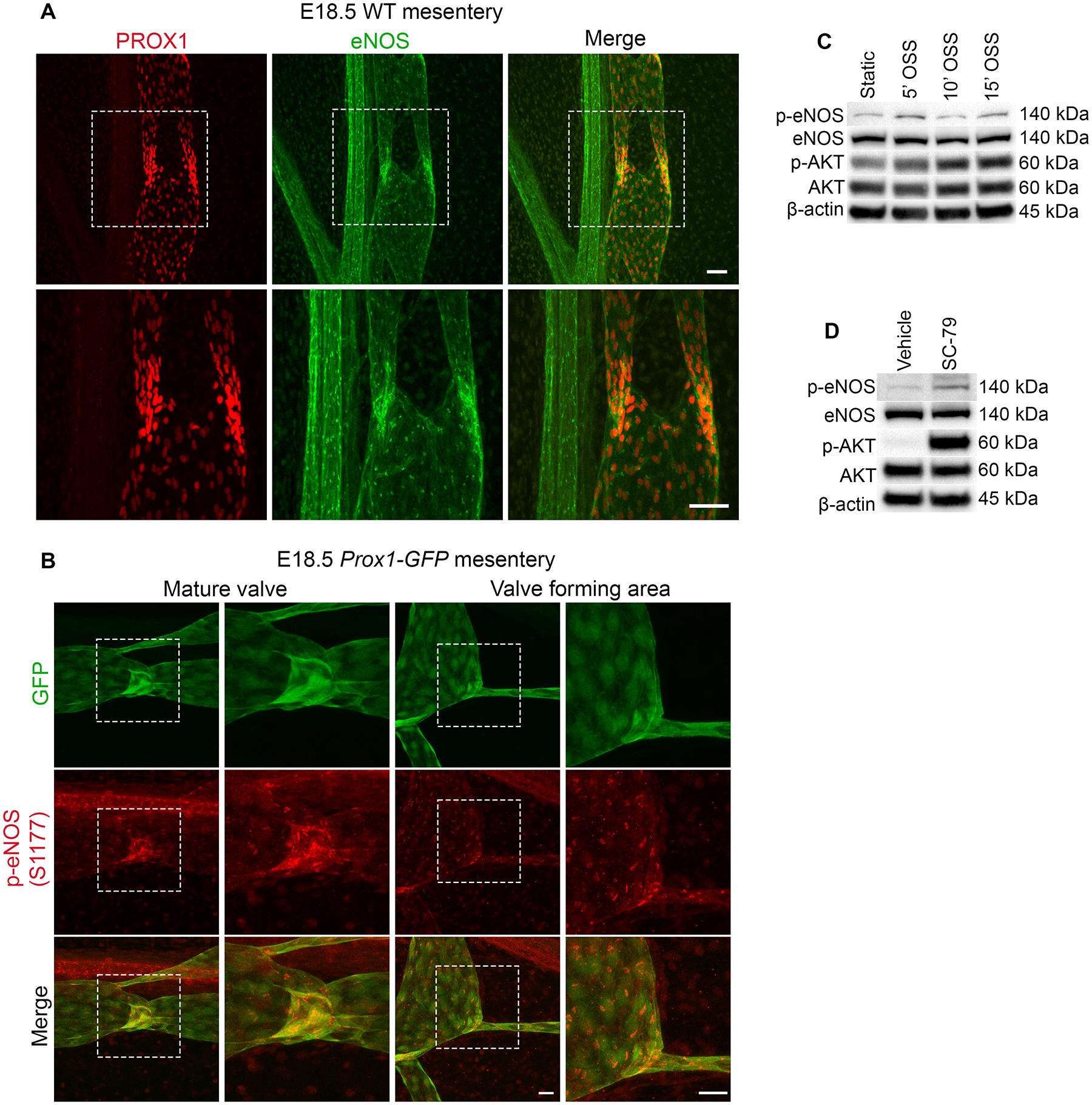
(A) Whole-mount immunostaining of PROX1 (red) and eNOS (green) in WT mesenteries at E18.5. The bottom images are high-magnification images of the white boxed areas. (B) Whole-mount immunostaining of GFP (green) and phospho-Ser1177 eNOS (red) in WT mesenteries at E18.5 showing a mature valve and valve forming area. The images on the right are high magnification images of the white boxed areas. (C) Western blot for indicated proteins using lysates from hdLECS exposed to static conditions or OSS conditions for 5, 10 and 15 mins. (D) Western blot for indicated proteins using lysates from hdLECS treated with vehicle or SC-79. Scale bars are 50μm in A and 25μm in B.
Although eNOS is highly expressed in developing and mature valve LECs, it is unknown whether eNOS is phosphorylated and activated in response to OSS during embryonic valve specification. To address this, we analyzed mesenteries at E18.5, a timepoint when fully mature valves can be found, from Prox1-GFP reporter mice that allow convenient visualization of lymphatic valve morphology15. Whole-mount immunostaining for phospho-Ser1177 of eNOS revealed strong phospho-eNOS staining in the GFPhigh LECs of mature valves and valve forming areas (Figure 1B). We failed to detect phospho-Ser1177 of eNOS in E18.5 Nos3−/− mice which served as a negative control (data not shown). To investigate whether OSS induces phosphorylation of eNOS in LECs, we exposed cultured human dermal LEC (hdLECs) to OSS for 5, 10 and 15 minutes or static conditions as a control and performed western blot. We found that eNOS phosphorylation peaked at 5 and 15 minutes of OSS, whereas AKT phosphorylation steadily increased from 5 to 15 minutes of OSS (Figure 1C). To confirm that eNOS is a downstream target of AKT, we treated hdLECs with the AKT activator SC-79 or vehicle and performed western blot. We found that SC-79 treatment led to an increase in phosphorylated AKT and phosphorylated eNOS (Figure 1D). These results suggest that eNOS is activated by AKT in response to OSS.
eNOS regulates lymphatic valve development during embryogenesis
To investigate whether eNOS regulates valve specification, we performed whole-mount immunostaining for PROX1 and FOXC2 in WT and Nos3−/− mesenteries at E16.5. We observed several cell clusters expressing high levels of PROX1 and FOXC2 in WT mesenteries and these were reduced in Nos3−/− mesenteries (Figure 2A). We failed to detect FOXC2 in Foxc2 knockout animals which served as a negative control (data not shown). Since FOXC2 expression is high in the adjacent arterial smooth muscles21, we used PROX1 staining to quantify specification clusters in both groups and normalized the number to vessel length. We found 45% fewer PROX1high clusters per millimeter in Nos3−/− mesenteries compared to WT controls (Figure 2B) while there was no significant difference in the total length of lymphatic vessels between the two groups (Figure 2C).
Figure 2: Nos3−/− embryos have a defect in lymphatic valve development.
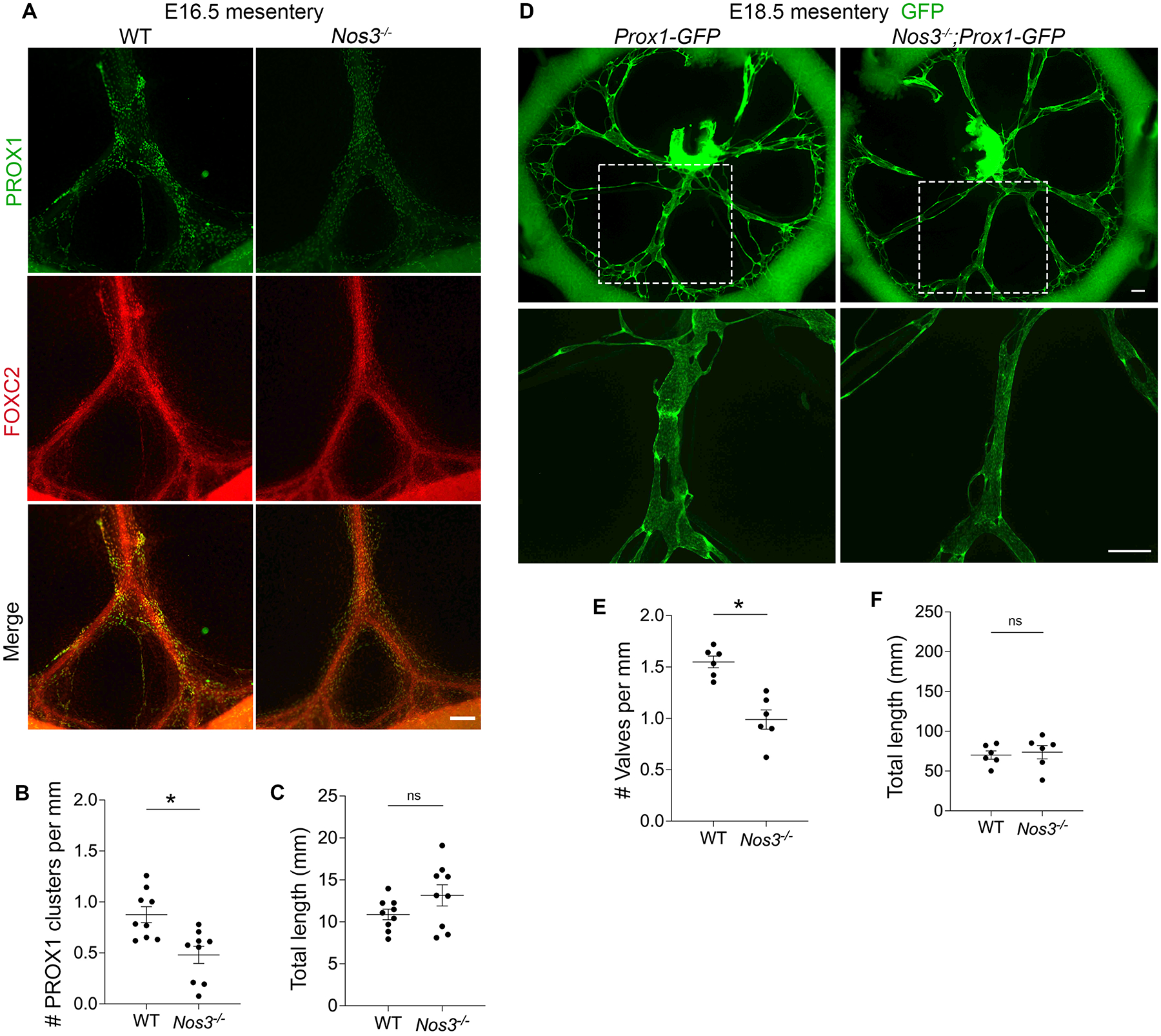
(A) Whole-mount immunostaining of PROX1 (green) and FOXC2 (red) in WT and Nos3−/− E16.5 mesenteries. (B) PROX1 clusters per millimeter from each E16.5 mesentery. (C) Total length of lymphatic vessels from each E16.5 mesentery. (B,C) All values are means ± SEM of n=9 littermates per genotype. (D) Fluorescence imaging of Prox1-GFP (green) mesenteries at E18.5. The bottom images are high-magnification images of the white boxed areas. (E) Valves per millimeter from each E18.5 mesentery. (F) Total length of lymphatic vessels from each E18.5 mesentery. (E,F) All values are means ± SEM of n=6 littermates per genotype. *P<0.05, unpaired Student’s t-test. Scale bars are 150μm in A and 300μm in D. Sex was not determined and data from both sexes were combined.
Next, we wanted to determine whether the valve specification defects led to fewer mature lymphatic valves at E18.5, a timepoint when fully formed valves first appear in the mesentery. We harvested mesenteries from Nos3−/−;Prox1-GFP embryos and Prox1-GFP controls at E18.5 and counted the GFPhigh lymphatic valves in both groups (Figure 2D). Quantification of the GFPhigh lymphatic valves revealed that Nos3−/−;Prox1-GFP mice had 36% fewer valves per millimeter compared to controls (Figure 2E) and there was no significant difference in the total length of lymphatic vessels between the two groups (Figure 2F).
Nos3−/− mice have fewer lymphatic valves at P3
To investigate whether the valve specification defects lead to fewer valves postnatally, we quantified all of the GFPhigh valves in Nos3−/−;Prox1-GFP and Prox1-GFP control mesenteries at postnatal day P3. As expected, we found that Nos3−/− mice had 60% fewer total valves compared to controls (Figure 3, A and C) while there was no significant difference in the total length of lymphatic vessels between the two groups (Figure 3D).
Figure 3: Nos3−/− animals have fewer lymphatic valves postnatally.
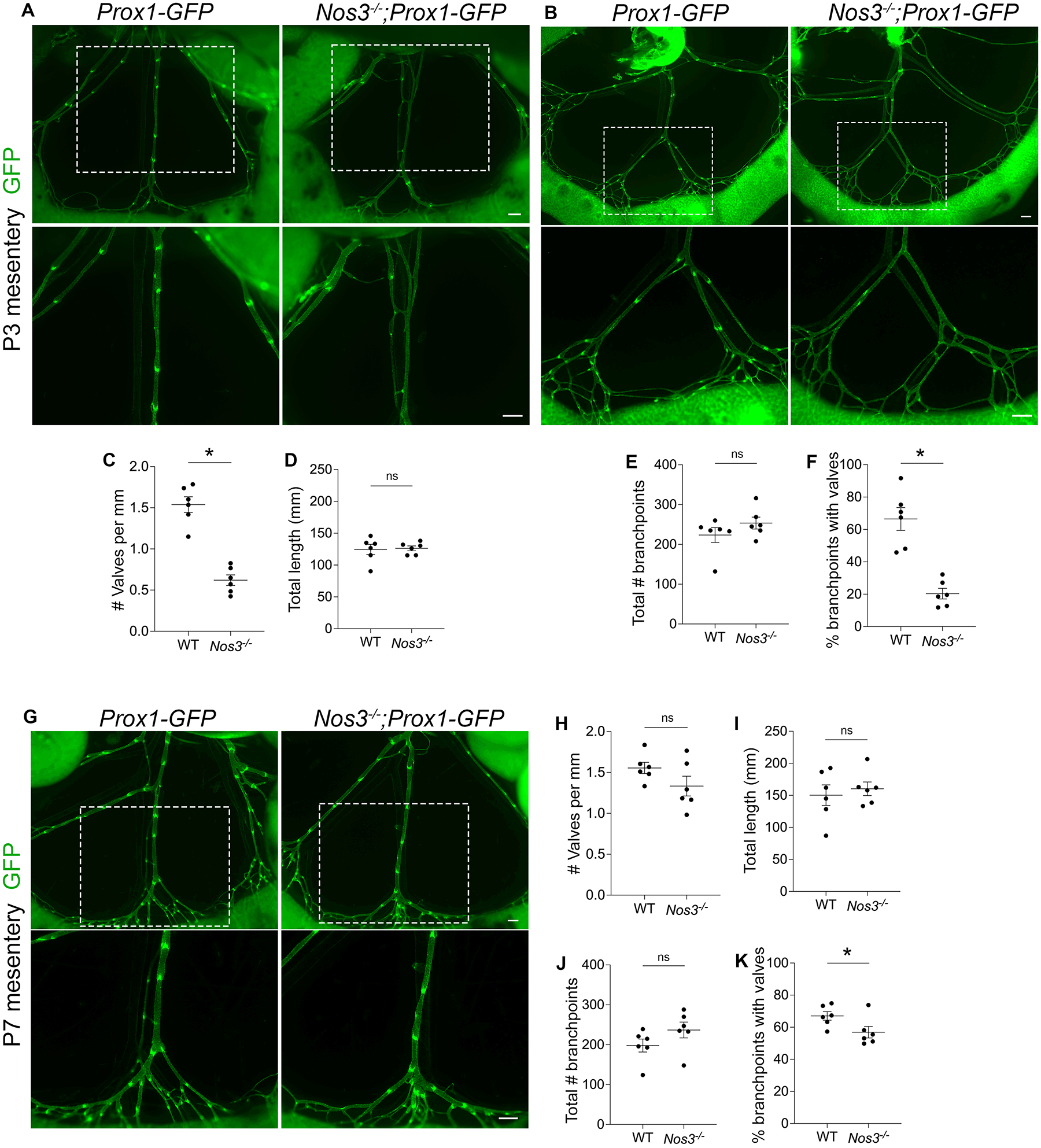
(A,B) Fluorescence imaging of Prox1-GFP (green) mesenteries at P3. The bottom images are high-magnification images of the white boxed areas. (C) Valves per millimeter from each mesentery at P3. (D) Total length of lymphatic vessels from each mesentery at P3. (E) Total number of branchpoints from each mesentery at P3. (F) Percentage of total branchpoints containing valves from each mesentery at P3. (G) Fluorescence imaging of Prox1-GFP (green) mesenteries at P7. The bottom images are high-magnification images of the white boxed areas. (H) Valves per millimeter from each mesentery at P7. (I) Total length of lymphatic vessels from each mesentery at P7. (J) Total number of branchpoints from each mesentery at P7. (K) Percentage of total branchpoints containing valves from each mesentery at P7. All values are means ± SEM of n=6 littermates per genotype. *P<0.05, unpaired Student’s t-test. Scale bars are 300μm in A, B and G. Sex was not determined and data from both sexes were combined.
Since lymphatic valves preferentially grow in areas of lymph flow reversal such as branchpoints9, we quantified the total branchpoints and the branchpoints containing valves in Nos3−/−;Prox1-GFP mice and Prox1-GFP controls. We found that there was no significant difference in the total number of branchpoints between the two groups (Figure 3, B and E), indicating that Nos3−/− mice did not have a defect in vessel branching. In contrast, only 20% of the branchpoints in Nos3−/−;Prox1-GFP mice had valves, which was significantly less than the 66% of branchpoints that had valves in controls (Figure 3F).
To determine whether the valve defects persisted at even later timepoints, we harvested mesenteries from Nos3−/−;Prox1-GFP mice and Prox1-GFP controls at P7 and quantified valves. Unexpectedly, we found that there was no significant difference in valve number and total length of lymphatic vessels between the two groups (Figure 3, G–I). At P7, 57% of branchpoints in Nos3−/−;Prox1-GFP mice had valves and this was significantly lower than controls, in which 67% of branchpoints had valves (Figure 3K), and there was no significant difference in the total number of branchpoints between the two groups (Figure 3J). Interestingly, the percentage of branchpoints with valves in Nos3−/−;Prox1-GFP mice increased from 20% at P3 to 57% at P7, suggesting that Nos3−/− mice experience a delay in valve development.
eNOS regulates expression of the valve genes Prox1 and Foxc2
Valve LECs are specified when OSS activates signaling pathways that lead to upregulation of PROX1 and FOXC29,11. Since Nos3−/− mice experience a defect in valve specification, we wanted to investigate whether eNOS regulated the expression of PROX1 and FOXC2. We harvested mesenteries from Nos3−/− mice and WT littermate controls at P3, performed whole-mount immunostaining for PROX1, FOXC2 and VE-cadherin and took high magnification confocal images of the lymphangion and valve areas. We observed that Nos3−/− mice had reduced PROX1 and FOXC2 expression compared to WT controls in the lymphangion LECs (Figure 4A). Interestingly, we observed no change in expression of PROX1 and FOXC2 in the valve LECs between the two groups (Figure 4A). When the pixel intensity was quantified, it revealed a significant decrease in PROX1 and FOXC2 expression in the lymphangion LECs in Nos3−/− mice compared to WT controls (Figure 4B). Conversely, there was no significant difference in PROX1 and FOXC2 expression in valve LECs between the two groups (Figure 4B). We failed to detect VE-cadherin in Cdh5 knockout animals which served as negative controls (data not shown).
Figure 4: eNOS regulates PROX1 and FOXC2 expression.
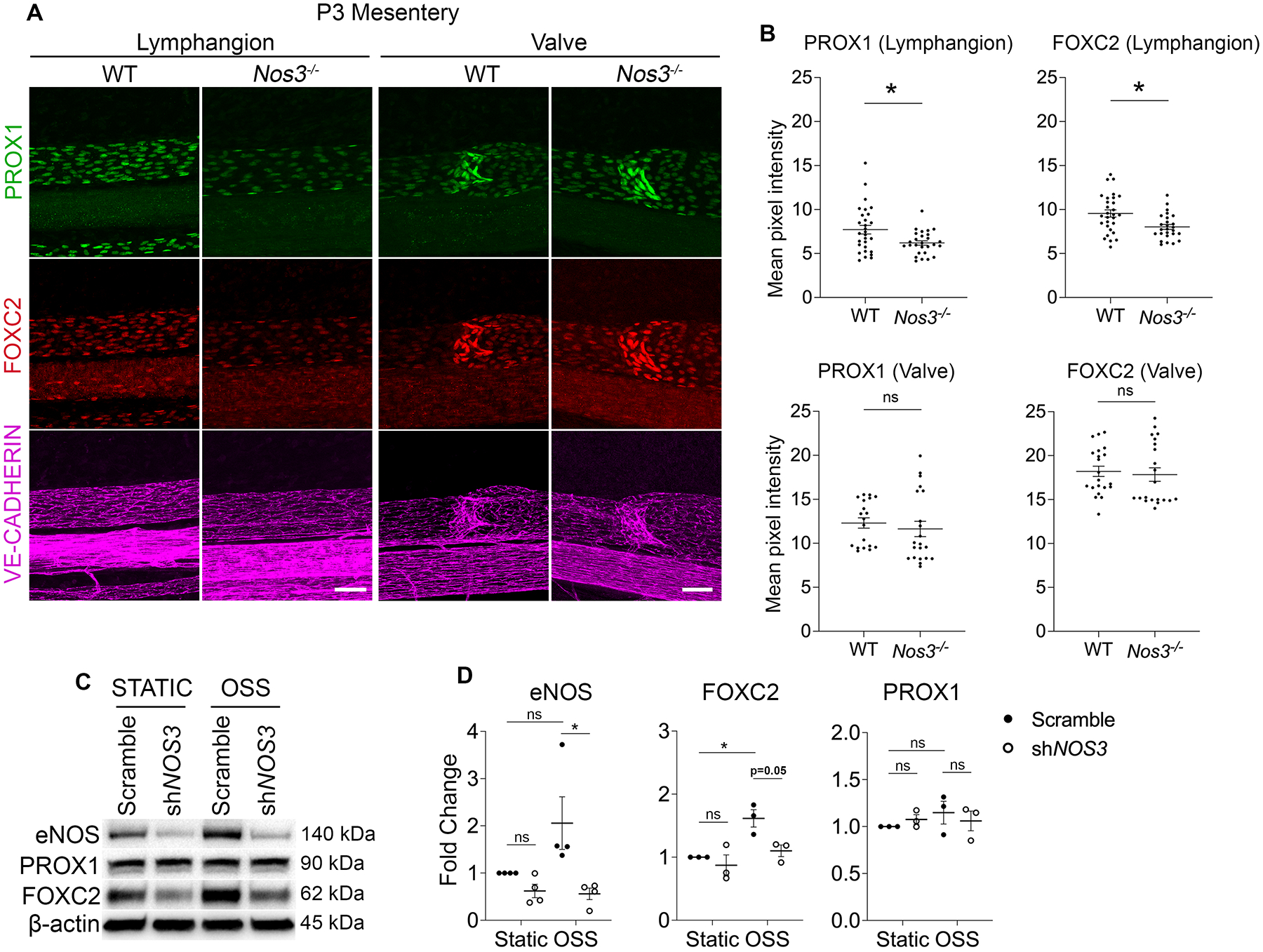
(A) Whole-mount immunostaining of PROX1 (green), FOXC2 (red) and VE-Cadherin (magenta) in WT and Nos3−/− P3 mesenteries. (B) Mean pixel intensity measurements of PROX1 and FOXC2 in lymphangion or valve LECs from each mesentery. (C) Western blot for indicated proteins using lysates from hdLECS cultured under static or OSS conditions for 48 hours and transfected with a control scramble or shRNA against NOS3. (D) Quantification of the indicated proteins probed by western blot. All values are means ± SEM of (B) n=4–6 lymphangion or valve areas from N=5–6 littermates per genotype and (D) n=3–4 independent experiments. *P<0.05, unpaired Student’s t-test in (B) and Two-way ANOVA followed by Tukey’s multiple-comparison test in (D). Scale bars are 50μm in A. Sex was not determined and data from both sexes were combined.
Next, we infected cultured hdLECs with a lentivirus expressing an shRNA against NOS3 or a scramble control shRNA and exposed them to static or OSS conditions. Western blot analysis showed significantly reduced eNOS protein in the shNOS3 LECs compared to scramble LECs under OSS conditions, confirming efficient knockdown by the shRNA (Figure 4, C and D). Consistent with previous reports, eNOS and FOXC2 expression was increased in scramble LECs in response to OSS (Figure 4, C and D)9. In support of our in vivo data, we observed reduced FOXC2 expression in shNOS3 LECs compared to scramble-infected LECs in both static and OSS conditions (Figure 4, C and D). PROX1 expression did not change significantly in response to OSS in scramble-infected LECs as previously published11 and was not affected by NOS3 knockdown (Figure 4, C and D).
eNOS regulates β-catenin signaling
Valve specification is regulated by the transcription factor β-catenin, which directly binds the promoter regions of Prox1 and Foxc2 and upregulates their expression in response to OSS11. Since our data showed that eNOS regulates PROX1 and FOXC2 expression, we investigated whether eNOS regulates β-catenin signaling. In endothelial cells, β-catenin signaling is regulated by a multiprotein complex which phosphorylates cytoplasmic β-catenin and consequently marks it for proteasomal degradation, thereby limiting the fraction of β-catenin that can translocate to the nucleus and transcribe target genes22. We harvested mesenteries from Nos3−/− and WT embryos at E18.5 and performed whole-mount immunostaining using antibodies against PROX1 and non-phosphorylated β-catenin, which represents the pool of β-catenin that is spared by the destruction complex and is free to transcribe target genes. We found that in WT embryos, non-phosphorylated β-catenin localized at the cell membrane and colocalized with PROX1 in the nucleus (Figure 5A). Conversely, in Nos3−/− embryos, non-phosphorylated β-catenin was mainly localized at the cell membrane with no visible nuclear staining (Figure 5A). Immunostaining of hdLECs showed that under static conditions there was no detectable β-catenin in shNOS3 hdLECs compared to scramble hdLECs (Figure 5B). Scramble hdLECs exposed to OSS led to increased accumulation of nuclear β-catenin compared to static conditions as previously reported (Figure 5B)11. In support of our in vivo data, shNOS3 hdLECs exposed to OSS showed no visible nuclear β-catenin staining (Figure 5B), indicating that eNOS is responsible for the nuclear translocation of β-catenin.
Figure 5: eNOS regulates β-catenin signaling.
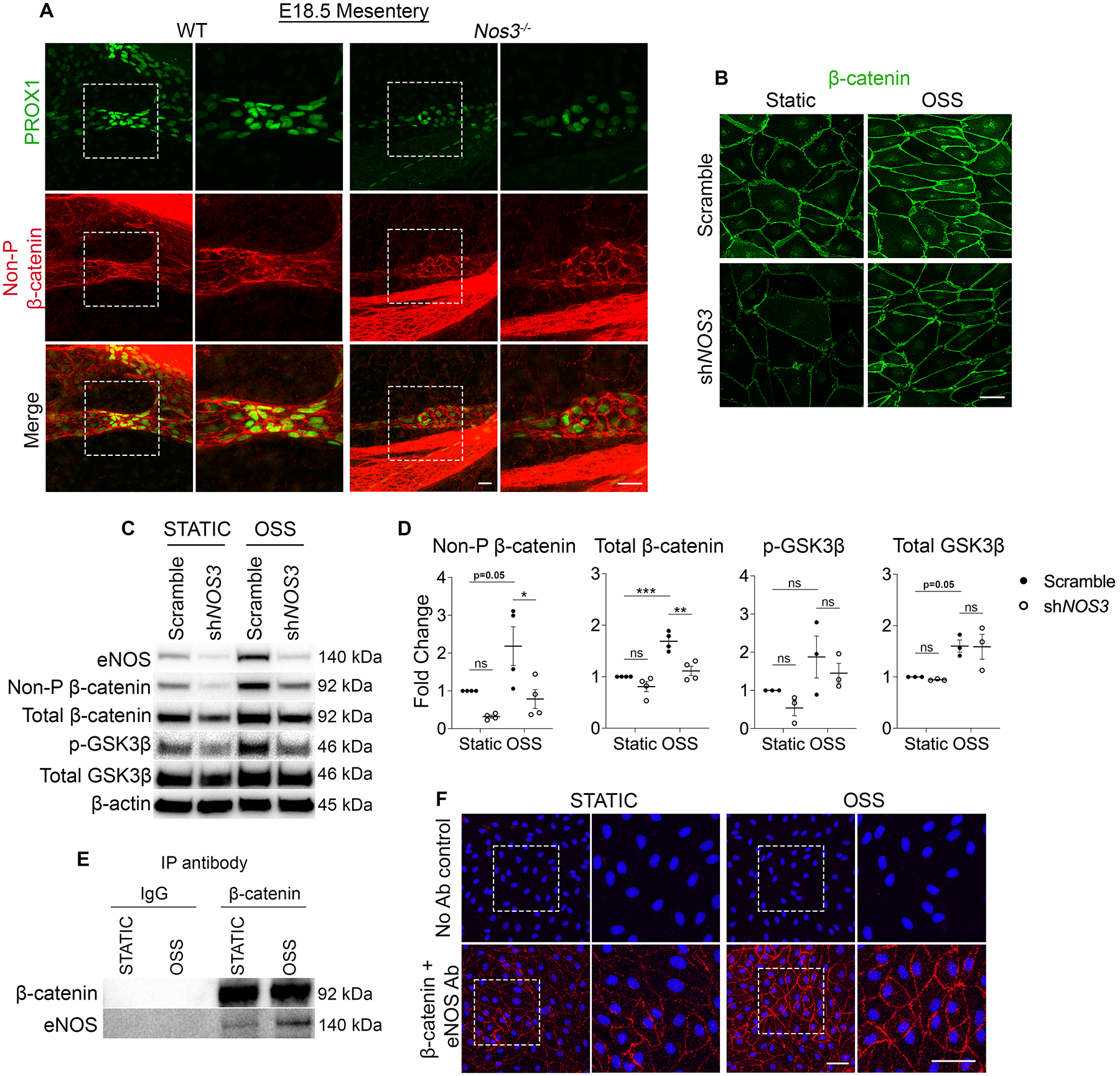
(A) Whole-mount immunostaining of PROX1 (green) and Non-phosphorylated β-catenin (referred to as Non-P β-catenin) (red) in WT and Nos3−/− E18.5 mesenteries. The images on the right are high-magnification images of the white boxed areas. (B) Immunostaining of β-catenin (green) in hdLECs cultured under static or OSS conditions for 48 hours and transfected with a control scramble or shRNA against NOS3. (C) Western blot for indicated proteins using lysates from scramble or shNOS3 hdLECS cultured under static or OSS conditions for 48 hours. (D) Quantification of the indicated proteins probed by western blot. All values are means ± SEM of n=3–4 independent experiments. *P<0.05, **P<0.01, ***P<0.001, Two-way ANOVA followed by Tukey’s multiple-comparison test. (E) Lysates from hdLECS cultured under static or OSS conditions were immunoprecipitated (IP) using anti-β-catenin antibody or mouse IgG control antibody followed by western blot for indicated proteins. (F) PLA was carried out in hdLECs exposed to static or OSS conditions using antibodies against β-catenin and eNOS or using no antibodies as a negative control. The images on the right are high-magnification images of the white boxed areas. Scale bars are 25μm in A and, 50μm in B and F.
Because phosphorylation and subsequent degradation of β-catenin inhibits its transcriptional activity22, we performed western blot for non-phosphorylated β-catenin and total β-catenin on lysates collected from scramble and shNOS3 hdLECs exposed to static or OSS. In response to OSS, scramble LECs showed increased non-phosphorylated β-catenin and total β-catenin levels as previously published (Figure 5, C and D)11. In support of our in vivo data, we found that NOS3-knockdown LECs showed significantly reduced non-phosphorylated β-catenin and total β-catenin levels compared to scramble LECs under OSS conditions (Figure 5, C and D). Put together, our data indicate that in the absence of eNOS, there is more degradation of β-catenin that blunts the response to flow.
eNOS regulates GSK3β activity
Next, we investigated the mechanism by which eNOS regulates β-catenin signaling. A study in HUVECs showed that eNOS activation and subsequent nitric oxide (NO) production can enhance nuclear translocation of β-catenin23. GSK3β is part of the β-catenin destruction complex, and the study showed that NO-mediated activation of protein kinase G (PKG) resulted in phosphorylation of GSK3β, which inhibits GSK3β activity22,23. To test whether eNOS regulates GSK3β activity in hdLECs, we performed western blot for phospho-GSK3β. In response to OSS, we observed a two-fold increase in phospho-GSK3β in scramble hdLECs, suggesting that GSK3β activity may be inhibited by OSS (Figure 5, C and D). Furthermore, we found that NOS3-knockdown hdLECs showed reduced phospho-GSK3β levels compared to scramble hdLECs in OSS conditions (Figure 5, C and D). This suggests that GSK3β could be degrading β-catenin in the absence of eNOS.
eNOS regulates β-catenin signaling independent of NO production
Since we observed phospho-eNOS in valve LECs, we tested whether eNOS regulated β-catenin signaling via NO production. We exposed hdLECs to static or OSS conditions in the presence of L-NG-Nitro arginine methyl ester (L-NAME), a pan-NOS inhibitor, or vehicle control. Western blot analysis showed no change in levels of non-phosphorylated β-catenin, total β-catenin, phospho-GSK3β and total GSK3β in hdLECs treated with 100μM and 1mM L-NAME compared to vehicle-treated hdLECs, in both static and OSS conditions (Supplemental Figure 2, A and B). We also treated hdLECs with an irreversible pan-NOS inhibitor, L-NG-Monomethyl Arginine (L-NMMA) and consistently found no change in non-phosphorylated β-catenin, total β-catenin, phospho-GSK3β and total GSK3β between vehicle and 100μM L-NMMA-treated hdLECs, in both static and OSS conditions (Supplemental Figure 2C).
To test whether eNOS regulates β-catenin signaling through NO-PKG signaling, we treated hdLECs with a PKG inhibitor, KT5823 or vehicle and exposed them to static or OSS conditions. Western blot analysis showed that in static conditions there was no change in total β-catenin, non-phosphorylated β-catenin, phospho-GSK3β and total GSK3β levels between 5μM or 10μM KT5823-treated hdLECs and vehicle-treated hdLECs (Supplemental Figure 2D). In OSS conditions, there was no change in total β-catenin, phospho-GSK3β and total GSK3β levels between 5μM or 10μM KT5823-treated hdLECs and vehicle-treated hdLECs (Supplemental Figure 2D). Non-phosphorylated β-catenin levels did not change in 5μM KT5823-treated hdLECs but slightly increased in 10μM KT5823-treated hdLECs compared to vehicle-treated hdLECs (Supplemental Figure 2D). This is likely an off-target effect of the drug since the 10μM dosage is approximately 200 times higher than the IC50.
eNOS forms a complex with β-catenin in hdLECs
Since loss of eNOS protein but not NO inhibition was found to affect β-catenin, we decided to test whether eNOS forms a complex with β-catenin to regulate its signaling. Evidence for eNOS binding β-catenin has been reported in HUVECs but has not been investigated in LECs23. We immunoprecipitated β-catenin from hdLEC lysates that were exposed to static or OSS conditions (Figure 5E). A Co-IP western blot revealed the presence of β-catenin under static and OSS conditions as a positive control. While eNOS protein was present in the static condition, its presence was enhanced by OSS, suggesting more binding of these two proteins in flow. As a negative control, the Co-IP experiment was repeated with an IgG antibody, which revealed an absence of β-catenin and eNOS (Figure 5E). These data suggest that eNOS forms a complex with β-catenin, either directly or indirectly to regulate its activity.
To determine whether eNOS directly bound β-catenin, we performed an in situ proximity ligation assay (PLA) in fixed cells that were exposed to static or OSS conditions. Amplification products were detected in both static- and OSS-exposed hdLECs, indicating a close proximity (<40nm) and direct binding of the two proteins (Figure 5F). In comparison to static hdLECs, the amplification products were more abundant in OSS hdLECs and were highly localized at the cell membrane with sparse distribution in the cytoplasm and nucleus (Figure 5F). As expected, no amplification products were detected in control hdLECs lacking primary antibodies (Figure 5F). To further probe the cellular compartment where eNOS and β-catenin colocalize, we performed immunostaining for the two proteins in hdLECs exposed to static or OSS conditions (Supplemental Figure 3). Consistent with the PLA staining, we found that eNOS and β-catenin colocalize at the cell membrane and this was more abundant in the OSS conditions compared to static conditions (Supplemental Figure 3). Interestingly, we also observed a pool of eNOS and β-catenin that colocalized in the perinuclear golgi region specifically under OSS conditions (Supplemental Figure 3). Taken together, our findings suggest that eNOS binds β-catenin to regulate its nuclear translocation and transcriptional activity, and this binding is enhanced under OSS conditions.
Postnatal deletion of β-catenin does not lead to a complete loss of lymphatic valves
Embryonic lymphatic-specific deletion of β-catenin has been previously shown to result in a complete loss of valve specification clusters at E16.5 and mature valves at E18.511. However, the role of β-catenin in postnatal lymphatic valve development has not been investigated. To delete β-catenin in a lymphatic-specific manner postnatally, we generated a transgenic mouse line expressing tamoxifen-inducible Cre recombinase (CreERT2) that is under the control of the Prox1 gene promoter. This strain was interbred with Ctnnb1flox/flox and Prox1-GFP mice to generate Prox1CreERT2;Ctnnb1flox/flox;Prox1-GFP mice (hereafter: Ctnnb1LEC-KO (Jscal)) and control littermates lacking Cre recombinase (Ctnnb1fl/fl)16. We administered tamoxifen (TM) at P1 and P3 and analyzed the mesentery at P8 (Figure 6A). Whole-mount immunostaining for β-catenin revealed ~100% efficient deletion of the protein from lymphatic vessels (Figure 6B). We further verified efficiency and specificity of the Prox1CreERT2 (Jscal) line by crossing it with the R26-mTmG Cre-reporter and analyzing reporter gene expression at P7, P14 and P21 following TM administration at P1 and P3 (Supplemental Figure 4A). We observed that Cre-mediated GFP expression labeled nearly all lymphatic endothelial cells in the mesentery, diaphragm, and ear skin (Supplemental Figure 4, B–G). As expected, the lymphatic vessels in littermate controls that lacked Prox1CreERT2 expressed tdTomato instead of GFP, further confirming the functionality of our Cre line (Supplemental Figure 4, B–D). We found that Ctnnb1LEC-KO (Jscal) mice had 58% fewer lymphatic valves at P8 compared to controls (Figure 6, C and E) while there was no significant difference in the total length of lymphatic vessels between the two groups (Figure 6H). To further validate our Prox1CreERT2 strain, we crossed the Ctnnb1flox/flox and Prox1-GFP mice to a previously published Prox1CreERT2 strain17 to again generate Prox1CreERT2;Ctnnb1flox/flox;Prox1-GFP mice (hereafter: Ctnnb1LEC-KO (Tmak)) and control littermates lacking Cre recombinase (Ctnnb1fl/fl). Following TM administration at P1 and P3, we analyzed the mesentery at P8 and found that Ctnnb1LEC-KO (Tmak) mice had 55% fewer lymphatic valves compared to controls (Figure 6, D and F) while there was no significant difference in the total length of lymphatic vessels between the two groups (Figure 6I). There was no significant difference in the valve number and length of lymphatic vessels between Ctnnb1LEC-KO (Jscal) and Ctnnb1LEC-KO (Tmak) mice (Figure 6, G and J). In contrast to the requirement for β-catenin in embryonic specification, our data show that β-catenin maintains approximately 50% of postnatal valves.
Figure 6: Postnatal β-catenin deletion does not lead to a complete loss of valves.
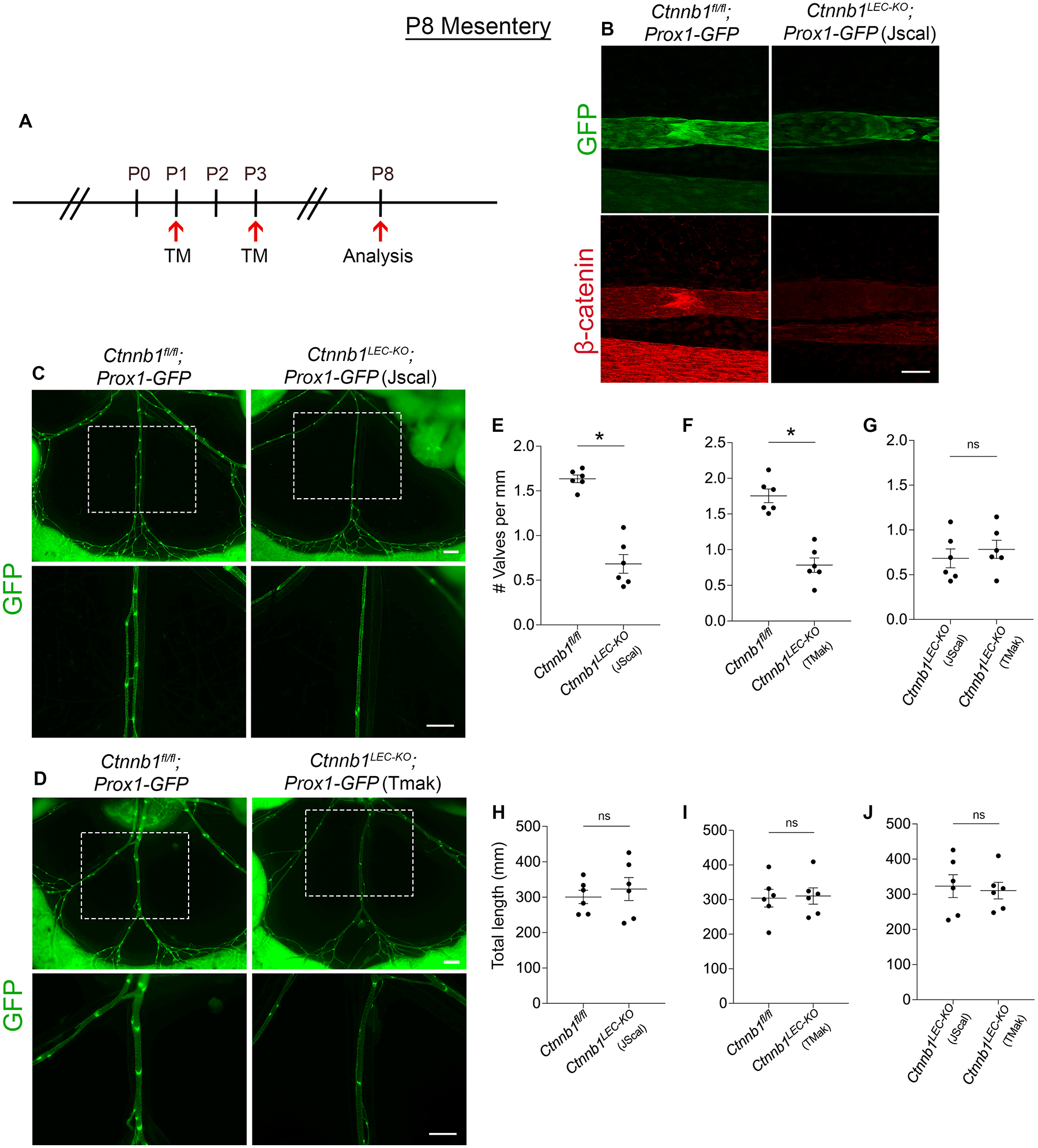
(A) Tamoxifen injection schedule for postnatal deletion of Ctnnb1. (B) Whole-mount immunostaining of GFP (green) and β-catenin (red) in Ctnnb1fl/fl;Prox1-GFP and Ctnnb1LEC-KO;Prox1-GFP (Jscal) P8 mesenteries. (C-D) Fluorescence imaging of Prox1-GFP (green) mesenteries at P8. The bottom images are high-magnification images of the white boxed areas. (E-G) Valves per millimeter from each mesentery at P8. (H-J) Total length of lymphatic vessels from each mesentery at P8. All values are means ± SEM of n=6 littermates per genotype. *P<0.05, unpaired Student’s t-test. Scale bars are 50μm in B and, 500μm in C and D. Sex was not determined and data from both sexes were combined.
β-catenin gain-of-function allele does not rescue valve loss in Nos3−/− mice
Since our data suggests that eNOS regulates β-catenin signaling to regulate lymphatic valve development, we decided to test whether enhancing β-catenin signaling could rescue the valve defects in Nos3−/− mice. To genetically enhance β-catenin signaling, we utilized a previously published mouse model in which exon 3 of the β-catenin gene is floxed (Ctnnb1lox(ex3))18. Deletion of exon 3 prevents β-catenin phosphorylation and targeting for proteasomal degradation, thereby leading to its accumulation and gain-of-function (GOF)18. To induce the β-catenin-GOF allele in Nos3−/− mice in a lymphatic specific manner, we generated Prox1CreERT2;Ctnnb1+/lox(ex3);Nos3−/−;Prox1-GFP mice (hereafter referred to as Nos3−/−;Ctnnb1LEC-GOF). Littermates lacking Cre recombinase (hereafter referred to as Nos3−/−;Ctnnb1+/lox(ex3)) served as controls expected to phenocopy Nos3−/− mice. First, we induced embryonic expression of the β-catenin-GOF allele at E14.5 and analyzed both groups at E18.5 (Supplemental Figure 5A). Unexpectedly, we found that β-catenin GOF did not lead to an increase in the number of GFPhigh valve areas in Nos3−/− embryos (Supplemental Figure 5B). Quantification revealed no significant difference in valve number between the two groups (Supplemental Figure 5C) and a 40% increase in length of lymphatic vessels in Nos3−/−;Ctnnb1LEC-GOF compared to Nos3−/−;Ctnnb1+/lox(ex3) mice (Supplemental Figure 5D). Next, we induced postnatal expression of the β-catenin-GOF allele at P0 and analyzed mesenteries from both groups at P3 (Supplemental Figure 5E). In contrast to our embryonic data, we found that postnatal β-catenin GOF led to a significant loss of valves in Nos3−/− mice (Supplemental Figure 5F). Quantification revealed a 60% decrease in valve number in Nos3−/−;Ctnnb1LEC-GOF mice compared to Nos3−/−;Ctnnb1+/lox(ex3) mice (Supplemental Figure 5G) with no difference in total length of lymphatic vessels between the two groups (Supplemental Figure 5H).
Foxo1 deletion partially rescues valve loss in Nos3−/− embryos
Since our data showed that eNOS regulates the expression of PROX1 and FOXC2, we decided to test whether directly inducing FOXC2 expression could rescue the valve defects in Nos3−/− mice. FOXO1 is a transcription factor that was shown to repress the expression of valve genes and directly bound the Foxc2 promoter as a repressor13. The same study showed that lymphatic-specific deletion of Foxo1 stimulated lymphatic valve formation and rescued the loss of valves in Foxc2 heterozygous mice13. Additionally, a recent study showed that inhibition of FOXO1 using the drug AS1842865 increased the nuclear translocation of β-catenin in hdLECs through an unidentified mechanism24. Thus, we generated Prox1CreERT2;Foxo1fl/fl; Nos3−/−;Prox1-GFP mice (hereafter: Nos3−/−;Foxo1LEC-KO). Littermates lacking Cre recombinase (hereafter: Nos3−/−;Foxo1fl/fl) were expected to phenocopy Nos3−/− mice. Foxo1+/fl or Foxo1fl/fl mice lacking Cre recombinase served as controls. We induced embryonic deletion of Foxo1 in a lymphatic-specific manner by injecting tamoxifen in pregnant dams at E14.5 and analyzing mesenteries at E18.5 (Figure 7A). Similar to our previous data, we observed a 33% decrease in valve number in Nos3−/−;Foxo1fl/fl mice compared to controls (Figure 7, B and C). Furthermore, we found a 24% increase in valve number in Nos3−/−;Foxo1LEC-KO mice compared to Nos3−/−;Foxo1fl/fl mice, indicating that embryonic deletion of Foxo1 partially rescues the valve loss phenotype in Nos3−/− embryos (Figure 7, B–C). There was no difference in the total length of lymphatic vessels between all three groups (Figure 7D).
Figure 7: Foxo1 deletion partially rescues valve loss in Nos3−/− embryos.
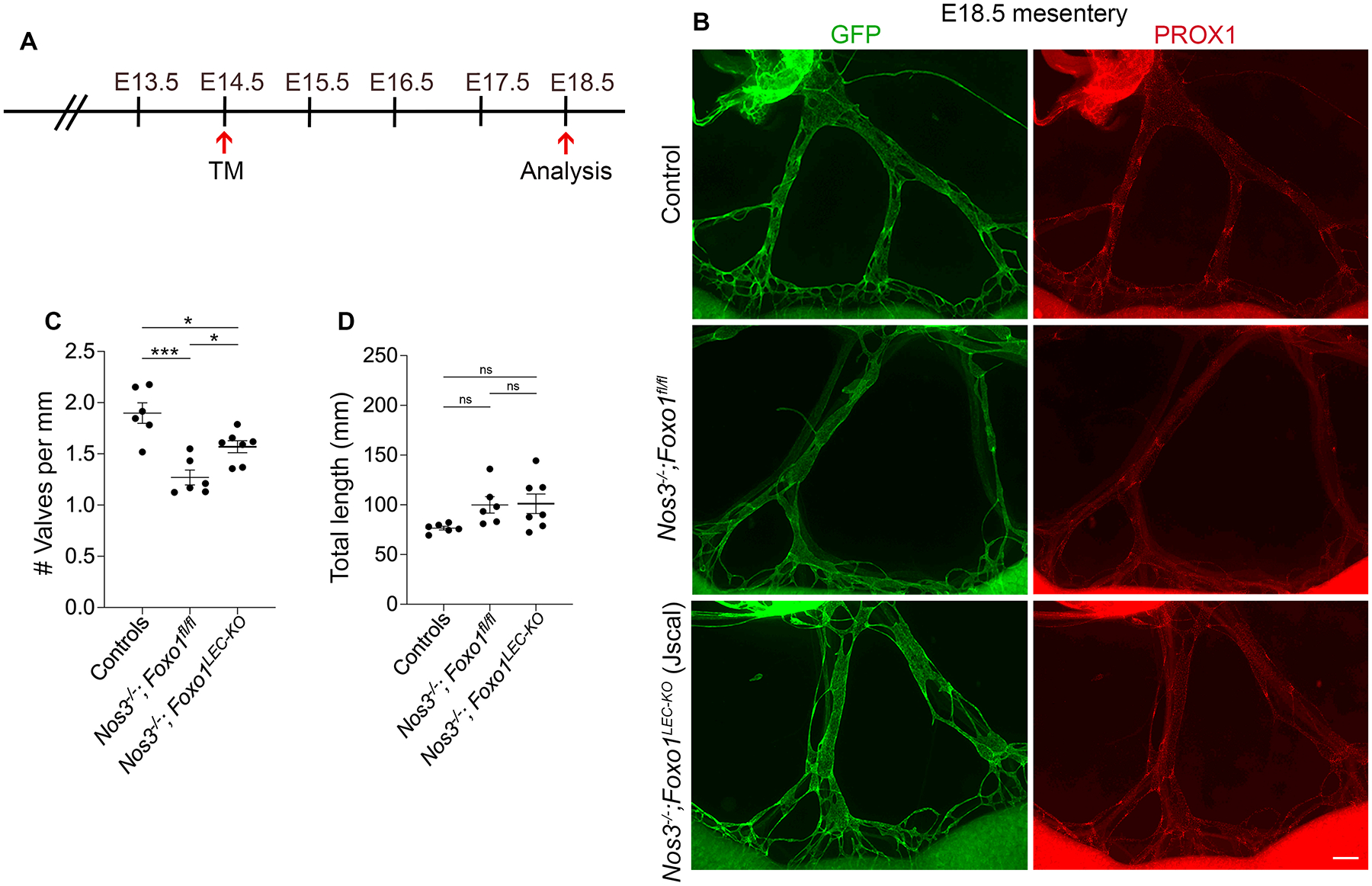
(A) Tamoxifen injection schedule for embryonic deletion of Foxo1. (B) Whole-mount immunostaining of E18.5 Prox1-GFP mesenteries from controls (Foxo1fl/fl or Foxo1+/fl), Nos3−/−;Foxo1fl/fl and Nos3−/−;Foxo1LEC-KO (Jscal). (C) Valves per millimeter from each mesentery at E18.5. (D) Total length of lymphatic vessels from each mesentery at E18.5. All values are means ± SEM of n=6–7 embryos per genotype. *P<0.05, **P<0.01, ***P<0.001, One-way ANOVA followed by Tukey’s multiple-comparison test. Scale bar is 300μm in B. Sex was not determined and data from both sexes were combined.
DISCUSSION
OSS generated by lymph flow activates mechanotransduction signaling pathways to regulate lymphatic valve development9–13. However, the exact signaling pathways that are activated by OSS have not been fully identified. As eNOS is a well-studied shear stress signaling molecule in BECs, we investigated its role in lymphatic valve development. Previous work showed that eNOS expression is induced in LECs shortly after the onset of lymph flow9. Consistent with this study, we show that eNOS is upregulated in the LECs of valve specification clusters and continues to be highly expressed in developing and mature valves. Furthermore, we show that eNOS regulates lymphatic valve specification by forming a complex with the transcription factor β-catenin to regulate its nuclear translocation and binding to the Prox1 and Foxc2 promoters.
eNOS regulates expression of the valve genes PROX1 and FOXC2
LEC clusters that are specified to form lymphatic valves express high levels of PROX1 and FOXC2 compared to the surrounding LECs and are first observed at E16.5 in the mouse mesentery9. We found that Nos3−/− mice experience a defect in valve specification that led to fewer valves at E18.5 and P3. Since lymphatic valves preferentially grow at branchpoints, we confirmed that Nos3−/− mice have a valve development defect and not a branching defect by showing that Nos3−/− mice had normal branching but failed to grow valves at the branchpoints.
Consistent with a defect in valve specification, we found that Nos3−/− mice have significantly reduced levels of PROX1 and FOXC2 in the lymphangion LECs compared to WT controls. This was recapitulated in vitro where NOS3 knockdown led to reduced FOXC2 expression at the protein level. NOS3 knockdown did not affect PROX1 levels in vitro, however this was not surprising because previous reports have established that the in vitro OSS model does not modulate PROX1 expression11–13. Altogether, our data indicate that Nos3−/− LECs have a blunted ability to upregulate PROX1 and FOXC2 in response to OSS to specify valve LECs. Interestingly, we saw that there was no difference in PROX1 and FOXC2 expression levels in the LECs of fully formed valves in WT and Nos3−/− mice. We reason that LECs of mature valves must have adequate levels of PROX1 and FOXC2 if they were able to form a valve. Other signaling pathways that can modulate PROX1 and FOXC2 expression independently of eNOS that may give rise to the limited number of valves that we observed in the Nos3−/− mice13.
eNOS regulates lymphatic valve development independent of NO production
In BECs, AKT has been shown to phosphorylate the Serine1177 residue of eNOS in response to fluid shear stress, which leads to its activation and subsequent NO production14. Moreover, we previously published that OSS induced AKT activation in cultured hdLECs and injecting an AKT activator in WT mice led to ectopic lymphatic valve growth in the mice12. Based on this we initially developed the hypothesis that eNOS-NO signaling acts downstream of AKT activation in regulating lymphatic valve development. We detected phospho-Ser1177 eNOS in the LECs of developing and mature valves in vivo and found that in cultured hdLECs, OSS led to the phosphorylation of eNOS coinciding with the phosphorylation of AKT. In contrast to blood vessel shear signaling, our data show that pharmacological inhibition of NO production does not have an effect on β-catenin activity. Similarly, we show that in hdLECS, NOS3 knockdown led to reduced phospho-GSK3β, however, the pharmacological inhibition of NO production or PKG activation did not affect GSK3β phosphorylation. Thus, we show that eNOS regulates β-catenin activity independent of NO production. As an alternative explanation, we show that eNOS forms a complex with β-catenin and their binding is enhanced by exposure to OSS. Additionally, our PLA and immunostaining imaging suggest that eNOS interacts with β-catenin in the cell membrane and golgi. Future studies are needed to explain the localization of this interaction and address whether disrupting binding can interfere with mechanotransduction signaling.
eNOS regulates β-catenin nuclear translocation
In LECs, the adherens junction protein VE-cadherin binds β-catenin at the cell membrane12. During valve development, VE-cadherin releases β-catenin in response to OSS, thus allowing it to translocate to the nucleus and upregulate valve genes12. Additionally, the fraction of cellular β-catenin that can translocate to the nucleus is regulated by a multiprotein destruction complex involving GSK3β22. The complex sequentially phosphorylates β-catenin, targeting it for polyubiquitination and proteasomal degradation22. We found that in Nos3−/− embryos non-phosphorylated β-catenin was restricted to the membrane and absent in the nucleus. Consistent with this, we failed to detect nuclear β-catenin in NOS3-knockdown hdLECs exposed to either static or OSS flow conditions. We further showed that NOS3-knockdown hdLECs had increased GSK3β activity which led to lower levels of total β-catenin. Altogether, our data suggests the eNOS regulates β-catenin activity by preventing β-catenin degradation and promoting its nuclear translocation.
Since our data showed that eNOS regulates β-catenin activity, we attempted to rescue the valve defects in Nos3−/− mice by deleting exon 3 of the β-catenin gene which would prevent β-catenin from getting phosphorylated and degraded. Unexpectedly, we found that genetic overexpression of β-catenin in such a manner failed to increase the valve number in Nos3−/− mice at E18.5. It is possible that eNOS forms a complex with β-catenin to chaperone its nuclear translocation and would explain why preventing β-catenin degradation alone in the absence of eNOS would be insufficient to rescue valve formation in Nos3−/− embryos.
Distinct signaling pathways regulate valve development during embryonic and postnatal stages
Unexpectedly, we found that at P7 there is no difference in lymphatic valves between WT and Nos3−/− mice, indicating that Nos3−/− mice experience a delay in valve development due to defective valve specification. Our data show that the percentage of branchpoints with valves increases in Nos3−/− mice from P3 to P7, indicating that Nos3−/− mice are actively growing new valves to make up for the initial delay in valve development. Since this rapid valve growth is independent of eNOS, we propose that other signaling pathways become active after birth to enable normal valve growth. Additionally, we show that postnatal loss of β-catenin only leads to a 55% loss of lymphatic valves whereas embryonic loss of β-catenin was reported to lead to a complete loss of valves11. This argues that a pathway independent of β-catenin is regulating postnatal valve development. Indeed, we recently published that FOXO1 is a target of OSS-induced AKT, so it may represent one alternative pathway regulating valve development postnatally13. FOXO1 acts as a repressor of valve genes such as FOXC2, GATA2, KLF4 and KLF2 and genetic deletion of Foxo1 was found to promote new valve growth embryonically as well as postnatally13. Consistent with this, we were able to partially rescue the valve number in Nos3−/− embryos by genetically deleting Foxo1. The differences in signaling pathways that regulate lymphatic valve development in embryonic and postnatal mice could be governed by differences in lymph shear stress values between the two stages. The average lymph flow shear stress was measured to be 0.64 dynes/cm2 with peaks of 4–12 dynes/cm2 in adult rat mesenteric lymphatic vessels25. During embryonic development, a lower blood pressure would lead to a lower interstitial fluid load and thus a lower shear stress than that experienced postnatally after a rapid rise in blood pressure26–28. Our findings could imply that more FOXO1 is phosphorylated and excluded from the nucleus after birth to augment lymphatic valve development and explains the distinct phenotypes seen between embryonic and postnatal stages upon genetic deletion of β-catenin and explains how Nos3−/− mice are able grow new valves after birth. In conclusion, we provide a novel role for eNOS in regulating lymphatic valve specification and describe a mechanism by which eNOS directly binds β-catenin to regulate its transcriptional activity.
Limitations
A potential limitation of our study is the use of global Nos3−/− knockout mice which are a well characterized model of hypertension. The earliest timepoint of recorded blood pressure in Nos3−/− mice is at 2 months of age, at which point they exhibit significantly elevated systolic blood pressure compared to their age-matched controls29–32. The valve defects that we show are present during embryonic development and are unlikely to be the result of increased blood pressure. Further, we primarily mated Nos3+/− x Nos3+/− mice, so pregnant females were heterozygous and presumed normotensive throughout embryonic development. Furthermore, we rule out a role for NO and instead show that eNOS regulates valve specification by directly binding β-catenin in LECs. The ability to restore valve specification by knocking out Foxo1 further supports that valve defects are not due to NO signaling.
The in vitro OSS model used in our study is another potential limitation as it does not reproduce the same magnitude of shear stress being exerted in vivo. However, this model of OSS provides enough material for western blot and qRT-PCR, and yields clear immunofluorescence images. In several papers, this model of OSS has allowed us to elucidate signaling mechanisms, many of which were confirmed in vivo11–13,20.
Supplementary Material
HIGHLIGHTS.
Nos3−/− mice experience a defect in lymphatic valve specification and fail to upregulate the valve genes, PROX1 and FOXC2, in response to oscillatory shear stress.
eNOS regulates lymphatic valve specification by directly binding β-catenin and promoting its nuclear translocation and transcriptional activity.
Genetic deletion of Foxo1 partially rescues the valve defects in Nos3−/− mice.
ACKNOWLEDGMENTS
The authors thank Drs. Taija Makinen, Young-Kwon Hong and Makoto Taketo for kindly providing the Prox1CreERT2, Prox1-GFP and Ctnnb1lox(ex3) strains respectively. Additionally, we thank Dr. Brant Isakson for helpful discussion regarding the interaction between eNOS and β-catenin. The graphical abstract was created with Biorender.com.
SOURCES OF FUNDING
This work was supported by NIH NHLBI grants R01 HL142905 and R01 HL164825 (to J.P.S), R01 HL145397 (to Y.Y.), and AHA Predoctoral Fellowship 827540 (to D.I).
Non-standard Abbreviations and Acronyms
- OSS
Oscillatory shear stress
- LEC
Lymphatic endothelial cell
- eNOS
Endothelial Nitric Oxide Synthase
- TM
Tamoxifen
- hdLEC
Human dermal lymphatic endothelial cell
- NO
Nitric oxide
- Co-IP
Co-immunoprecipitation
- PLA
Proximity ligation assay
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1.Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol. 2011;193:607–618. doi: 10.1083/jcb.201012094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis MJ, Rahbar E, Gashev AA, Zawieja DC, Moore JE, Jr. Determinants of valve gating in collecting lymphatic vessels from rat mesentery. Am J Physiol Heart Circ Physiol. 2011;301:H48–60. doi: 10.1152/ajpheart.00133.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer D, Jannaway M, Yang Y, J PS. Lymphatic Valves and Lymph Flow in Cancer-Related Lymphedema. Cancers (Basel). 2020;12. doi: 10.3390/cancers12082297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czepielewski RS, Erlich EC, Onufer EJ, Young S, Saunders BT, Han YH, Wohltmann M, Wang PL, Kim KW, Kumar S, et al. Ileitis-associated tertiary lymphoid organs arise at lymphatic valves and impede mesenteric lymph flow in response to tumor necrosis factor. Immunity. 2021;54:2795–2811.e2799. doi: 10.1016/j.immuni.2021.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geng X, Cha B, Mahamud MR, Srinivasan RS. Intraluminal valves: development, function and disease. Dis Model Mech. 2017;10:1273–1287. doi: 10.1242/dmm.030825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapinski PE, Lubeck BA, Chen D, Doosti A, Zawieja SD, Davis MJ, King PD. RASA1 regulates the function of lymphatic vessel valves in mice. J Clin Invest. 2017;127:2569–2585. doi: 10.1172/JCI89607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castorena-Gonzalez JA. Lymphatic Valve Dysfunction in Western Diet-Fed Mice: New Insights Into Obesity-Induced Lymphedema. Front Pharmacol. 2022;13:823266. doi: 10.3389/fphar.2022.823266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Homayun-Sepehr N, McCarter AL, Helaers R, Galant C, Boon LM, Brouillard P, Vikkula M, Dellinger MT. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight. 2021;6. doi: 10.1172/jci.insight.149831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, et al. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell. 2012;22:430–445. doi: 10.1016/j.devcel.2011.12.020 [DOI] [PubMed] [Google Scholar]
- 10.Sweet DT, Jimenez JM, Chang J, Hess PR, Mericko-Ishizuka P, Fu J, Xia L, Davies PF, Kahn ML. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J Clin Invest. 2015;125:2995–3007. doi: 10.1172/JCI79386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha B, Geng X, Mahamud MR, Fu J, Mukherjee A, Kim Y, Jho EH, Kim TH, Kahn ML, Xia L, et al. Mechanotransduction activates canonical Wnt/beta-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 2016;30:1454–1469. doi: 10.1101/gad.282400.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Cha B, Motawe ZY, Srinivasan RS, Scallan JP. VE-Cadherin Is Required for Lymphatic Valve Formation and Maintenance. Cell Rep. 2019;28:2397–2412 e2394. doi: 10.1016/j.celrep.2019.07.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scallan JP, Knauer LA, Hou H, Castorena-Gonzalez JA, Davis MJ, Yang Y. Foxo1 deletion promotes the growth of new lymphatic valves. The Journal of Clinical Investigation. 2021;131. doi: 10.1172/JCI142341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisslthaler B, Dimmeler S, Hermann C, Busse R, Fleming I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand. 2000;168:81–88. doi: 10.1046/j.1365-201x.2000.00627.x [DOI] [PubMed] [Google Scholar]
- 15.Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B, et al. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood. 2011;117:362–365. doi: 10.1182/blood-2010-07-298562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253 [DOI] [PubMed] [Google Scholar]
- 17.Bazigou E, Lyons OT, Smith A, Venn GE, Cope C, Brown NA, Makinen T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J Clin Invest. 2011;121:2984–2992. doi: 10.1172/JCI58050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo j. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee R, Knauer LA, Yang Y. Protocol for in vivo and in vitro study of lymphatic valve formation driven by shear stress signaling pathway. STAR Protoc. 2023;4:102141. doi: 10.1016/j.xpro.2023.102141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi D, Park E, Jung E, Cha B, Lee S, Yu J, Kim PM, Lee S, Hong YJ, Koh CJ, et al. Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance. JCI Insight. 2019;4. doi: 10.1172/jci.insight.125068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kume T, Jiang H, Topczewska JM, Hogan BL. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001;15:2470–2482. doi: 10.1101/gad.907301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimelman D, Xu W. β-Catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055 [DOI] [PubMed] [Google Scholar]
- 23.Warboys CM, Chen N, Zhang Q, Shaifta Y, Vanderslott G, Passacquale G, Hu Y, Xu Q, Ward JP, Ferro A. Bidirectional cross-regulation between the endothelial nitric oxide synthase and beta-catenin signalling pathways. Cardiovasc Res. 2014;104:116–126. doi: 10.1093/cvr/cvu173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogunsina O, Banerjee R, Knauer LA, Yang Y. Pharmacological inhibition of FOXO1 promotes lymphatic valve growth in a congenital lymphedema mouse model. Front Cell Dev Biol. 2022;10:1024628. doi: 10.3389/fcell.2022.1024628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon JB, Greiner ST, Gashev AA, Cote GL, Moore JE, Zawieja DC. Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation. 2006;13:597–610. doi: 10.1080/10739680600893909 [DOI] [PubMed] [Google Scholar]
- 26.Ishiwata T, Nakazawa M, Pu WT, Tevosian SG, Izumo S. Developmental changes in ventricular diastolic function correlate with changes in ventricular myoarchitecture in normal mouse embryos. Circ Res. 2003;93:857–865. doi: 10.1161/01.Res.0000100389.57520.1a [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Guo X, Kassab GS. Axial nonuniformity of geometric and mechanical properties of mouse aorta is increased during postnatal growth. Am J Physiol Heart Circ Physiol. 2006;290:H657–664. doi: 10.1152/ajpheart.00803.2005 [DOI] [PubMed] [Google Scholar]
- 28.Le VP, Kovacs A, Wagenseil JE. Measuring left ventricular pressure in late embryonic and neonatal mice. J Vis Exp. 2012. doi: 10.3791/3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Vliet BN, Chafe LL, Montani JP. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J Physiol. 2003;549:313–325. doi: 10.1113/jphysiol.2003.041897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang XP, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice: cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24 [DOI] [PubMed] [Google Scholar]
- 31.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0 [DOI] [PubMed] [Google Scholar]
- 32.De Moudt S, Hendrickx JO, De Meyer GRY, Martinet W, Fransen P. Basal Vascular Smooth Muscle Cell Tone in eNOS Knockout Mice Can Be Reversed by Cyclic Stretch and Is Independent of Age. Front Physiol. 2022;13:882527. doi: 10.3389/fphys.2022.882527 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.