

Abstract
In mammals there are two 3-oxo-4-ene steroid reductases that generate either A/B-trans or A/B cis-ring junctions in the steroid nucleus known as steroid 5α- and 5β- reductases, respectively. There is only one steroid 5β- reductase in each species and these are members of the aldo-keto-reductase (AKR) protein superfamily. The corresponding human enzyme is AKR1D1, and it plays an essential role in bile-acid biosynthesis. Germline mutations in AKR1D1 give rise to bile-acid deficiency. Because of its central role in steroid metabolism and need for detailed structure-function studies there is a need to purify the enzyme to homogeneity and in high yield. We report the purification of milligram amounts of crystallographic quality homogeneous recombinant protein for structure-function studies and its characterization.
1. Introduction
5α- and 5β-Dihydrosteroids are produced by the NADPH-dependent double bond reduction of Δ4-3-ketosteroids and they are major metabolites of the androstane, pregnane, cholane and cholestane series of steroids on route to the corresponding tetrahydrosteroids, as shown in Fig. 1. The 5α-dihydrosteroids are produced by steroid 5α-reductases (SRD5A1-2) (Russell & Wilson, 1994) whereas the 5β-dihydrosteroids are produced by steroid 5β-reductase (Russell & Stechell, 1992; Russell, 2003). Steroid 5β-reductase is a member of the aldo-keto reductase (AKR) protein superfamily and the human and murine enzymes correspond to AKR1D1 and AKR1D4, respectively (Hyndman et al., 2003; Jez, Bennett, et al., 1997; Jez, Flynn, et al., 1997). These enzymes are unique in steroid biochemistry since 5β-reduction results in steroids that are non-planar in which the A-ring is now perpendicular to the remainder of the steroid nucleus due to an A/B cis-ring fusion (Fig. 1). The enzymes are unique to the AKR superfamily since they catalyze double bond reduction and not carbonyl reduction. The bent shape in the steroid product is an important feature of all bile acids since this results in an amphipathic steroid essential for the emulsification of fats. Steroid 5β-reductase deficiency is an inborn error of steroid metabolism that results in bile acid deficiency in which the neonate can no longer absorb fats and fat-soluble vitamins and is neonatal fatal (Kimura et al., 1998; Lemonde et al., 2003; Setchell et al., 1988; Shneider et al., 1994; Ueki et al., 2009). Expression, purification and characterization of AKR1D1, aids crystallographic studies, mechanistic studies, site-directed mutagenesis to study the effect of germline mutations (Di Costanzo et al., 2008; Di Costanzo, 2009; Drury et al., 2010), and enables the use of the enzyme as a synthon to produce 5β-dhydrostetroids which are otherwise difficult to generate chemically. This chapter will describe the purification of recombinant AKR1D1 and its biochemical characterization.
Fig. 1.
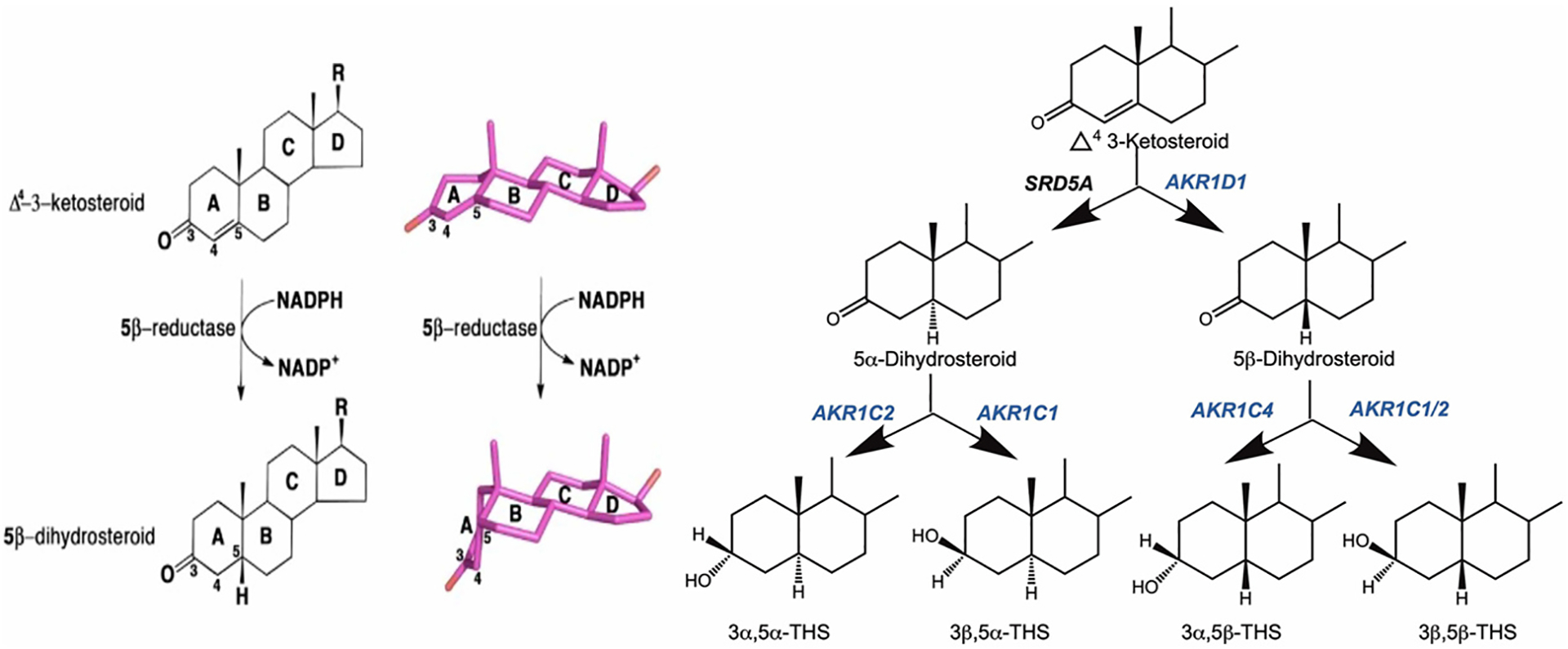
Metabolism of Δ4-3-Ketosteroids to Tetrahydrosteroids. Left panel shows introduction of A/B cis ring fusion by 5β-reductase; right panel shows formation of tetrahydrosteroids.
2. General overview of methods
Homogeneous AKR1D1 is purified as a His-tagged protein expressed in E. coli using a single Ni-Sepharose column chromatography step in high yield by open column chromatography. The method is superior to using sequential purification on DEAE-Sepharose and Blue-Sepharose routinely used for other AKR1C enzymes because this approach is low yielding. The purified protein can be characterized for NADPH binding, steady state kinetic constants for substrate utilization and inhibition studies (Chen et al., 2011) as well as crystallographic studies (Di Costanzo et al., 2008) and transient state kinetics (Chen et al., 2015). The enzyme can also be purified by FPLC using HisTrap column chromatography and both methods are described.
3. Equipment
Incubator cabinet at 37 °C
Incubator with shaking at 37 °C
GE Pharmacia Fraction Collector Fra-100 with 3-way valve -Open column chromatography
Pharmacia Single Path Monitor UV-1-Open column chromatography
Gilson Miniplus 2 Peristaltic Pump 2-Open column chromatography
AKTA purifier with UV-900 detector- FPLC
GE AKTA Frac-920 fraction collector-FPLC
Beckman DU 640 UV/Vis spectrophotometer
Fluorescence spectrophotometer F-4500 (Hitachi America, Ltd., New York, NY)
Bioscan Imaging Scanner System 200-IBM with AutoChanger 3000; Bioscan, Washington, DC
Sorvall RC6 Plus centrifuge
Thermo Fiberlite F12-6 × 500 LEX Fixed Angle Rotor (096–62375)
Thermo Fiberlite F13–14 × 50cy Fixed Angle Rotor (096–145007)
QSonica Q500 sonicator with 1/2″ probe (4406)
4. Materials
4.1. Bacterial growth
OverExpress C41(DE3) Chemically Competent cells #CMC0017
AKR1D1 cDNA subcloned in Novagen pET-16b vector (6xHisTag N-terminus and 10 × His Tag N-terminus)
BD Difco Dehydrated Culture Media LB #DF00446-17-3
BD Difco Dehydrated Culture Media LB Agar #DF0445-17-4
Teknova 2x 2YT broth powder #Y0210
Sigma Aldrich Ampicillin Sodium Salt #A9518
Roche Isopropyl β-D-1-thiogalactopyranoside #11411446001
Two 4-L Erlenmeyer flasks
4.2. Protein purification
Ni Sepharose 6 Fast flow was purchased from Amersham-Open column chromatography
HisTrap FF (5 mL pre-packed) FPLC
HiTrap FF (5 mL pre-packed) FPLC
Biosciences Fisherbrand Round Bottom Borosilicate Glass Tubes (For chromatographic fractions) #14-961-29
Fisher brand Round Bottom Borosilicate Glass Tubes (For Hitrap fractions) #14-961-27
Spectra Por Dialysis Membranes MWCO 10 kD #132572
Thermo Scientific Pierce BCA Protein Assay Kit #23227
Fisher brand Sterile Syringe Filter 0.45 μm PES #09720514
Nalgene Rapid Flow Filter Unit 0.2 μm 1000 mL #567-0020
MP Biomedicals TRIS Ultra-Pure #819638
Imidazole
Invitrogen UltraPure 0.5 M EDTA pH 8.0 #15575020
Fisher BioReagents 2-Mercaptoethanol (β-mercaptoethanol) #BP176100
Fisher BioReagents Sodium Chloride #BP358-1
Fisher Chemical Potassium Chloride #P330–500
Fisher BioReagents Potassium Phosphate Dibasic #BP363
Sigma Aldrich Potassium Phosphate Monobasic #P-5655
Sigma Aldrich Sodium Hydroxide #S5881
Amicon Ultra-15 Centrifugal Filter Unit MWCO 10 kDa #UFC901008
USA Scientific Articice cryogenic tubes #1418–7310
Thermo Scientific Coomassie Brilliant Blue R-250 Dye #20278
Sigma Aldrich Glacial Acetic Acid #A6283
4.3. Specific activity measurements
Roche Diagnostics NADPH Grade
[4-14C]-Testosterone (50 mCi/mmol) PerkinElmer Life and Analytical Sciences
Microcapillary tubes Drummond Microcaps 1–5 μL capacity #1-000-0010/CA, #P1-000-0020/CA, #P1-000-0030, #P1-000-0040, #P1-000-0050/CA
5. Stock solutions
5.1. Bacterial growth
5.1.1. Reagent storage
Ampicillin and IPTG can be stored at −80 °C for up to one year. LB plates can be stored for up to one month at 4 °C. LB and 2YT broth can be stored for up to one week, add Ampicillin directly before use.
- 100 mg/mL Ampicillin
- Dissolve 2 g Ampicillin in 20 mL milliQ water, filter by 0.45 μm syringe filter.
- 1 M Isopropyl β-D-1-thiogalactopyranoside (IPTG)
- Dissolve 1.2 g IPTG in 5 mL milliQ water, filter by 0.45 μm syringe filter.
- 2 × 1.5 L 2YT broth (2 L total) + 100 μg/mL Ampicillin
- Dissolve 45.5 g 2YT broth powder in 1.5 L milliQ water in 4-L flasks.
- Autoclave and let cool to room temperature before adding 1.5 mL of 100 mg/mL Ampicillin.
- LB agar plates +100 μg/mL Ampicillin
- Dissolve 4 g LB agar powder in 100 mL milliQ water.
- Autoclave and let cool to 50 °C before adding 100 μL of 100 mg/mL Ampicillin, pour into dishes and let cool completely.
- LB broth +100 μg/mL Ampicillin
- Dissolve 2.5 g LB broth powder in 100 mL milliQ water.
- Autoclave and let cool to room temperature before adding 100 μL of 100 mg/mL Ampicillin.
5.2. Protein purification
5.2.1. Reagent storage
Ni-Sepharose and imidazole stock buffers, potassium phosphate monobasic and dibasic stocks can be stored for up to one month at 4 °C. Ni-Sepharose column buffer should be made fresh the day of use, add β-mercaptoethanol directly before use. Coomassie gel stain and destain can be stored at room temperature for up to three months.
- Ni-Sepharose buffer: 1 M Tris-HCl pH 7.9 @ 25 °C, pH 8.6 @ 4 °C
- Dissolve 121.4 g Tris-HCl in 800 mL milliQ water. Fill volume to 1 L with milliQ water and adjust pH.
- Filter through 0.2μm vacuum filter.
Sonication buffer: 20 mM Tris-HCl pH 7.9 plus 5 mM imidazole in milliQ water
- 1 M Potassium phosphate dibasic stock
- Dissolve 136.1 g potassium phosphate dibasic in 1 L milliQ water.
- Filter through 0.2 μm vacuum filter.
- 1 M Potassium phosphate monobasic stock
- Dissolve 174.2 g potassium phosphate monobasic in 1 L milliQ water.
- Filter through 0.2 μm vacuum filter
- Column Buffers:
- 20 mM Tris-HCl pH 7.9 plus 20 mM imidazole plus 0.5 M NaCl
- 20 mM Tris-HCl pH 7.9 plus 400 mM imidazole plus 0.5 M NaCl
Protein storage buffer: 20 mM potassium phosphate pH 7.0 @ 25 °C, 1 mM β-mercaptoethanol, 1 mM EDTA and 30% glycerol
- Column Clean Buffers
- 0.1 M NaOH
- 70% EtOH
- 20 mM Tris-HCl pH 7.9 plus 20 mM imidazole plus 0.5 M NaCl
- 20% EtOH
Ni-Sepharose column storage buffer: 20% EtOH in milliQ water.
- Coomassie gel stain
- Dissolve 1 g Coomassie R250 in 300 mL MeOH, 650 mL milliQ water, 50 mL acetic acid.
- Filter through 0.2 μm vacuum filter.
- Coomassie destain
- Mix 100 mL Acetic acid, 500 mL MeOH, and 400 mL milliQ water.
- Filter through 0.2 μm vacuum filter.
5.3. Substrates
5.3.1. Reagent storage
NADPH and testosterone substrates should be made fresh. Potassium phosphate pH 7 buffer can be stored for up to 1 month at 4 °C.
5.3.2. 2.3 mM NADPH
Dissolve 8.6 mg NADPH in 1 mL milliQ water to obtain a 10 mM stock solution. Dilute a small amount of the original solution 10-fold and titrate the concentration using the molar extinction coefficient 6220 M−1cm−1 at absorbance at 340 nm. Adjust the total volume of the stock or amount added to reaction accordingly.
5.3.3. Testosterone
Testosterone (Steraloids, Cat:A6950-000 Lot:B1313) FW: 288.42. Dissolve 34.6 mg in 12 mL acetonitrile to a final concentration of 10 mM. Check concentration using the molar extinction coefficient of 17,200 M−1 cm−1 at 242 nm. Dilute into assay as required. For 5 assays need 10 nmoles/mL plus 200,000 dpm [4-14C]-Testosterone.
6. Protein purification procedure for His-tagged proteins
Prepare reagents for bacterial growth, protein purification, and specific activity assays according to recipes detailed above.
6.1. Preparation of plasmids
Prepare the plasmids ahead of time. The GeneAmp RNA PCR Core Kit was used to reverse transcribe polyA (+) RNA from human liver. cDNA was synthesized using the RNA mixed with MuLV reverse transcriptase, Oligo d(T)16 and dNTP, and incubated at 42 °C for 30 min. The cDNA was then amplified for 25 cycles of PCR to produce a fragment containing AKR1D1 flanked by two unique restriction sites. Each cycle had a denaturation step (30 s at 95 °C), an annealing step (120 s at 60 °C), and an elongation step (30 s at 72 °C). The forward primer contained an NcoI restriction site upstream from the initiation codon, 5′-GGG CCA TGG GAT CTC AGT GCT GCA AG-3′, and the reverse primer contained a BamHI site downstream from the termination codon, 5′-CCC GGA TCC AGT ATT CAT GAA ATG g-3′, in which the underlined nucleotides indicate restriction sites. The PCR fragment was then ligated into the PCR cloning vector pCR4-TOPO vector. The vector was digested with NcoI and BamHI, and the resultant fragment was subcloned into the expression vector pET16b. The AKR1D1 sequence was verified by dideoxy sequencing.
Since attempts to purify AKR1D1 using sequential ion-exchange chromatography and Blue-Sepharose did not produce a high yield a tagged approach was taken (Table 1). AKR1D1 was PCR amplified from the pET16b-AKR1D1 construct using the same conditions as before (Section 2.2.1) except the forward primer was replaced with one containing an NdeI site, 5′-GGG CAT ATG GAT CTC AGT GCT GCA AG-3′. After digestion with NdeI and BamHI, the resultant fragment was subcloned into the expression vector pET16b. The AKR1D1 sequence was verified by dideoxy sequencing. The difference between this pET16b-AKR1D1 construct and the previous version is the inclusion of an N-terminal 10x His-Tag. A similar strategy can be used to generate a pET16b-AKR1D1 construct with a N-terminal 6x His-Tag.
Table 1.
Purification of recombinant pET16b AKR1D1 (minus His-Tag).a
| Sonicate | 27 | 110.8 | 953 | 8.6 | 1 | 100 |
| Dialysate | 15 | 112.2 | 954 | 8.5 | 1 | 100 |
| Pooled DEAE cellulose | 26 | 9.8 | 588 | 60 | 7.0 | 62 |
| Dialyzed DEAE cellulose | 25 | 9.7 | 495 | 51 | 5.9 | 52 |
| Dialyzed Blue Sepharose | 0.6 | 1.2 | 96 | 80 | 9.2 | 10 |
All activity measurements were performed under radiometric assay conditions.
Open column chromatography.
1 L culture.
6.2. Expression and purification of 10x-His-tagged-AKR1D1 from E. coli by open column chromatography
Day 1. Streak plates.
Timing: 15 min
Streak C41 (DE3) cells containing pET16b AKR1D1 vector onto pre-warmed LB agar plates and place in 37 °C incubator overnight.
Day 2: Inoculate cultures.
Timing: 10 h, induce and grow overnight.
- In the morning make three inoculation cultures by transferring a single colony from the streaked plate into 15 mL LB broth + 100 μg/mL Ampicillin. Shake at 37 °C at 220 rpm.
- Two of the cultures will be used to inoculate 1.5 L cultures, the third can be used to make a fresh glycerol stock.
At midday, place 4-L flasks with 1.5 L 2YT broth + ampicillin in 37 °C shaker to prewarm.
Check for turbidity in 15 mL inoculation cultures eight hours after beginning growth.
- When inoculation cultures are turbid, transfer one 15 mL culture into each flask of 1.5 L 2YT broth, shake at 37 °C.
- Note: Remove 1 mL from one 4-L flask to use as a blank when measuring culture optical density (OD).
Check the OD of each flask periodically until 0.6–0.7 OD is reached, approximately 3 h.
When target OD is reached, add 1.5 mL of 1 M IPTG (final concentration 1 mM IPTG) and shake overnight for 14 h at 37 °C.
Day 3: Wash and sonicate bacterial pellet.
Timing: 2–3 h.
Note: Keep bacterial pellet and lysate on ice throughout processing steps.
Spin down bacterial pellet in 500-mL centrifuge bottles at 6500 rpm for 15 min at 4 °C. Discard supernatant and repeat until all culture has been pelleted.
To wash the pellet, resuspend each pellet in 50 mL of sonication buffer, then pool slurries into two bottles and centrifuge at 6500 rpm for 15 min at 4 °C.
-
Resuspend both pellets again in 50 mL of sonication buffer, then pool and spin down slurries in one centrifuge bottle.
Pause Point: Washed pellet can be stored at − 80 °C until ready for sonication and column purification. When proceeding to column chromatography steps from a frozen pellet, thaw the pellet on ice before resuspending in sonication buffer.
Resuspend the pellet in 60 mL of sonication buffer, transfer to a glass beaker for sonication.
Sonicate the resuspended pellet using the following parameters: 30 s cycle of 2 s on followed by 1 s off. Run the cycle 6 times with 90 s in between cycles to prevent overheating. Keep the lysate on ice between cycles.
Transfer the bacterial lysate into 50 mL conical tubes and centrifuge at 12,000 rpm for 30 min at 4 °C.
- Combine supernatant in a glass beaker and filter through 0.45 μm syringe filter.
- Note: Save 10–20 mg of remaining lysed pellet. Record the exact weight and store at −80 °C.
Dialyze supernatant overnight into 20 mM Tris-HCl pH 7.9 plus 5 mM imidazole and 0.5 M NaCl.
Precondition Ni-Sepharose 3–4 h, equilibrate overnight in column buffer
Resin with a flow rate of 0.5 mL/min.
Day 4. Column Chromatography.
The dialyzed fraction was loaded onto a Ni-Sepharose column equilibrated with the dialysis buffer and the column was washed with the same buffer. Bound protein was eluted with a linear imidazole gradient from 20 mM to 400 mM. Active fractions containing AKR1D1 were identified by monitoring the conversion of [4-14C]-testosterone to 5β-dihydrotestosterone (5β-DHT) by discontinuous assay using standard assay conditions and by visualization of the protein content of each fraction by SDS-PAGE.
- Open column chromatography, see Table 2 for purification summary. Collect fractions throughout the entire run.
- Load lysate volume onto packed Ni-Sepharose column at a flow rate of 0.8 mL/min.
- Wash the column with 3 CV of loading buffer 0.8 mL/min to wash unbound protein from the column or until A280 absorbance is less than 0.1 OD units.
- Elute bound protein from the column over a 5 CV gradient starting at 20 mM Imidazole and ending in 400 mM Imidazole.
- Peak fractions were pooled and dialyzed overnight in 20 mM potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, and 30% glycerol.
Table 2.
Purification of recombinant pET16b AKR1D1 (plus 10-His-Tag).
| Sonicate | 30.2 | 99.7 | 818 | 8.2 | 1.0 | 100 |
| Dialysate | 28.0 | 100.8 | 796 | 7.9 | 1.0 | 97 |
| Ni Sepharose | 4.0 | 5.7 | 462 | 81 | 9.9 | 56 |
Day 5.
The purified protein was obtained in 56% yield and had a final specific activity of 80 nmoles of testosterone reduced/min/mg protein under standard assay conditions (Table 2).
All activity measurements were performed under radiometric assay conditions. Purification is from 1- to 1.5-L culture.
6.3. Purification of pET16b-AKR1D1 (6-His-Tag) from E. coli by FPLC
Similar success can be observed using 6-His-Tagged constructs with a variety of HisTrap columns using FPLC. As follows:
6.3.1. Equivalent to day 3 preceding method
The cell paste from a 3 L culture is thawed at room temperature for 10 min and then transferred on ice. The cell paste is resuspended in 60 mL of lysis buffer (20 mM Tris, pH 7.9, 20 mM imidazole, 500 mM NaCl) supplemented with 1 mg/mL lysozyme and 1 mg/mL DNase. The suspension is incubated on ice for 30 min.
The cell suspension is transferred to glass beaker and sonicated using 1 s pulse, 0.1 s rest for 30 s followed by 60 s of rest for 7 cycles. A 300 μL sample of the lysate was reserved after sonication for further analysis. The rest of the cell lysate was clarified by centrifugation at 4 °C 8000 rpm for 20 min. The supernatant was centrifuged again at 4 °C 15,000 × g for 30 min to remove any remaining cell debris.
The lysate supernatant was injected in three 20 mL portions on three different metal charged HisTrap (5.0 mL) columns: Ni, Co, and Zn.
For each column, the same elution protocol was used. The sample was loaded on the HisTrap FF 5 mL column 2.5 mL/min. The unbound protein was washed with 6CV of 20 mM Tris, pH 7.9, 20 mM imidazole, 500 mM NaCl @ 2.5 mL/min followed by 3 CV of 20 mM Tris, pH 7.9, 60 mM imidazole, 500 mM NaCl @ 5 mL/min. The column was eluted with a 60–400 mM imidazole gradient in 10 CV. The column was then washed with 400 mM imidazole solution for 5 CV and re-equilibrated with starting buffer.
The purity of the fractions was assessed by SDS-PAGE (10 μL) as shown in Fig. 2.
The combined fractions of the HisTrap column were dialyzed against 10 mM KPO4 pH 7 for 1 h then overnight and dialyzed in 10 mM KPO4 pH 7 with 1 mM EDTA for 2 h. Precipitate was observed after overnight dialysis. The combined fractions were stored in − 80 °C for future purification, see Fig. 2.
After storage, the protein was thawed in the 4 °C fridge. Assayed for activity and purified on 1 mL HiTrap Blue Sepharose affinity column. The protein was loaded in 1 mL/min 10 mM KPO4 pH 7, 1 mM 2-mercaptoethanol, 1 mM EDTA. The unbound protein was washed with 3 CV of the same buffer and eluted with a 5CV gradient from 0 M KCl to 1 M KCl followed by 30 CV of 1 M KCl.
The fractions after HiTrap column were analyzed by SDS PAGE, combined, and dialyzed in 20 mM KPO4 pH 7.4,1 mM EDTA, 20% glycerol 2 L twice (1 h and overnight) and then in 20 mM KPO4 pH 7.4, 1 mM EDTA, 30% glycerol plus 1 mM β-mercaptoethanol 2 L for 1 h, as shown in Fig. 2.
Fig. 2.
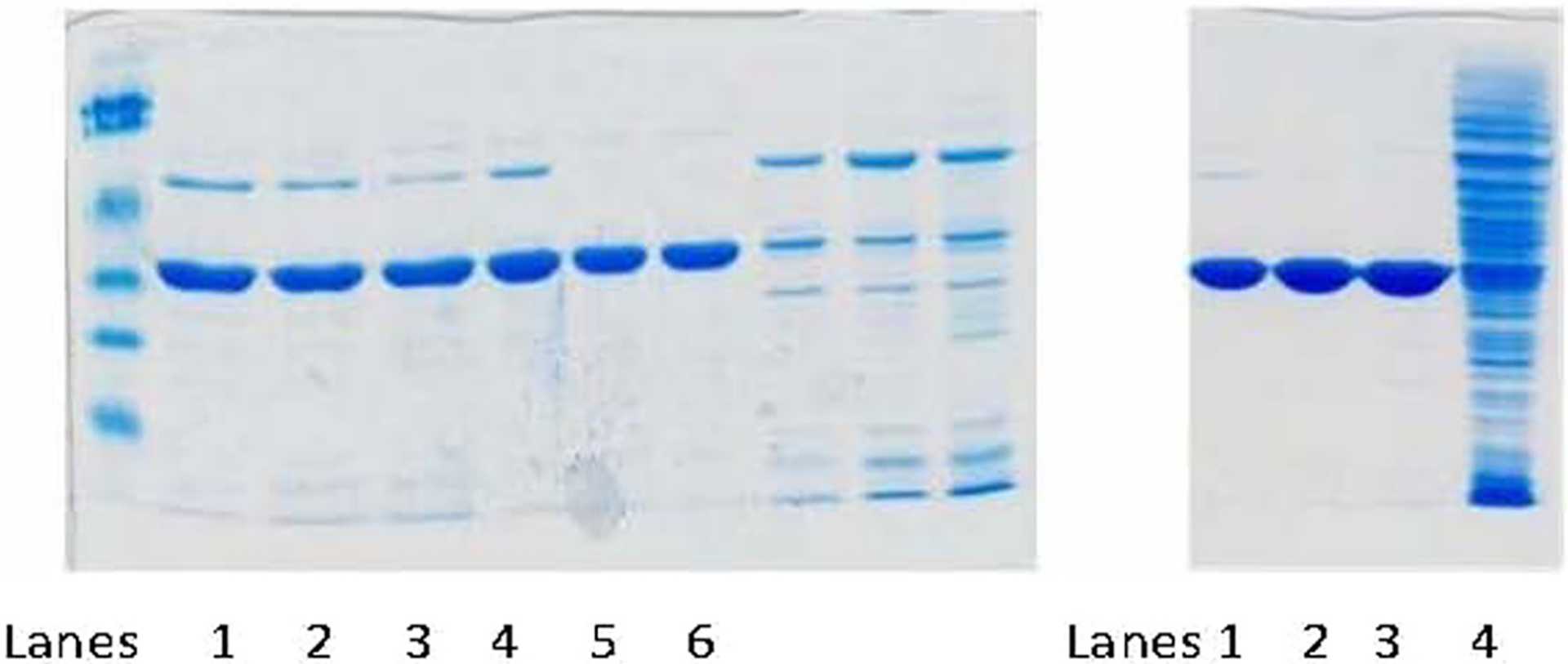
Purity of 6-His-Tagged-AKR1D1 generated by HisTrap Column Chromatography. (Left) After HisTrap. Lanes 1–3, Ni column combined fractions, Co column combined fractions, Zn-column combined fractions, respectively; Lanes 4–6, Ni column combined fractions after dialysis, Co-column combined fractions after dialysis, Zn- column combined fractions after dialysis, respectively. (Right) Pooled fractions after HiTrap-Blue Sepharose column chromatography, Lanes 1–3 analysis of HiTrap pooled fractions after Ni, Co, and Zn His-Trap chromatography, respectively.
6.4. Measuring specific activity for AKR1D1
There are two assays for measuring enzyme activity a discontinuous radiometric assay and a continuous fluorometric assay. Both can be used during purification to monitor enzyme activity.
6.4.1. Radiometric assay
The reduction of [4-14C]-testosterone was used to monitor 5β-reductase activity collected during purification and for product profiling. Reactions contained 2 μM [4-14C]-testosterone (40,000 dpm), 8 μM cold testosterone, 5% acetonitrile, 200 μM NADPH and 100 mM potassium phosphate buffer, pH 6.0, in a final volume of 200 μL. Reactions were initiated by the addition of enzyme and performed at 37 °C. Reaction mixture cocktails can be made to reduce inaccuracies by multiple pipetting. Reactions were quenched using 1 mL of ice-cold water-saturated ethyl acetate at different time intervals. The aqueous phase was extracted with ethyl acetate, vacuum-dried and redissolved in 50 μL ethyl acetate. Resuspensions were applied to LK6D Silica TLC (thin layer chromatography) plates (Whatman International Ltd) and developed twice in toluene/acetone (80:20 v/v). Radiochromatograms were scanned with an automatic TLC-linear analyzer (Bioscan Imaging Scanner System 200-IBM with AutoChanger 3000; Bioscan, Washington, DC) and the relative percentage of peaks were compared and quantitated as nmoles of product formed assuming that each steroid was recovered with the same efficiency. The 5β-dihydrotestosterone (5β-DHT) product was identified by co-chromatography with the authentic standard which was visualized by spraying with acetic acid/sulfuric acid/anisaldehyde (100:2:1 v/v) and heat.
Note: When measuring activity during purification steps, perform the reaction in duplicate with 10 μL of the lysate or fraction being tested to see sufficient activity, see Tables 1–3.
Table 3.
Purification of 6-His-Tagged AKR1D1 on different HisTrap columns.
| Sonicate/Histrap | 21 | 7.42 | 155.82 | 44.4 | 6.0 | 100 | 1 | 931,980 | 100 |
| Ni Before dialysis | 19 | 0.58 | 11.02 | 40.3 | 70.0 | 7.07 | 11.70 | 766,401 | 82.23 |
| Co Before dialysis | 23 | 0.53 | 12.19 | 32.8 | 61.9 | 7.82 | 10.36 | 754,566 | 80.96 |
| Zn Before dialysis | 28 | 0.51 | 14.28 | 33.0 | 64.3 | 9.16 | 10.75 | 922,621 | 99.00 |
| Ni after dialysis | 19 | 0.51 | 9.69 | 28.7 | 55.9 | 6.22 | 9.34 | 545,681 | 58.55 |
| Co after dialysis | 24 | 0.39 | 9.36 | 24.8 | 63.7 | 6.01 | 10.66 | 594,510 | 63.79 |
| Zn after dialysis | 29 | 0.41 | 11.89 | 24.6 | 60.2 | 7.63 | 10.07 | 714,430 | 76.66 |
| Ni-His Trap plus HiTrap Blue Sepharose | 5 | 0.95 | 4.75 | 70.7 | 74.0 | 3.05 | 12.38 | 353,475 | 37.93 |
| Co-His Trap plus HiTrap Blue Sepharose | 5 | 1.22 | 6.10 | 94.5 | 77.5 | 3.91 | 12.96 | 472,558 | 50.70 |
| Zn-His Trap plus HiTrap Blue Sepharose | 6 | 1.37 | 8.22 | 107.1 | 79.1 | 5.28 | 13.23 | 642,553 | 68.94 |
| 10xHis Tag Protein | 1.02 | 80.46 |
Assays measured by fluorimetry.
Note: When measuring specific activity of the final purified protein, first dilute the stock to 0.5 mg/mL using 100 mM Potassium phosphate pH 7.0 buffer. Add the enzyme using microcaps and perform the assay in duplicate using a range of 0–5 μL of enzyme (final concentration 0–2.5 μg/mL), see Tables 2 and 3.
6.4.2. Fluorometric assay
The principle of the assay is to measure the AKR1D1 dependent loss of NADPH fluorescence in the presence of testosterone. A NADPH calibration curve is constructed from a stock solution of 1 mM NADPH to cover the final concentration range of 0–10 μM with Excitation at 340 nM and Emission at 460 nM using a slit width of 10 nm. Curves of FU versus NADPH amount are constructed. Assay systems of 1.0 mL contain 100 mM potassium phosphate pH 6.0, 8.5 μM NADPH, 10 μM testosterone, 4% acetonitrile and 0.05 μM enzyme. After adding NADPH and steroid to the buffer, the decay of NADPH is measured for 5 min (non-enzymatic rate) in FU/min. The reaction is initiated by the addition of enzyme and reactions run for 5 min at 37 °C and FU/min are measured. Subtraction of the non-enzymatic rate gives the enzymatic rate which can then be converted to pmoles NADPH consumed per min using the calibration curve. This assay gives the same specific activity for purified enzyme as the radiometric assay.
7. Other considerations
7.1. Advantages and limitations
The FPLC protocol is expected to provide 6 mg of protein from a 1 L prep. Monitoring the % yield after each step by activity measurement can identify where loss is occurring.
7.2. Optimization and troubleshooting
- Problem: protein is not eluted in a single peak or comes out in the flow through/waste.
- Potential solution: ensure column is fully equilibrated and check the pH of the column buffers. Incorrect pH can affect protein charge and subsequent column binding/retention.
- Problem: bacterial lysate has little to no specific activity.
- Potential solution: Cells may not be completely lysed, or protein may be aggregating in the pellet. Run the saved portion of the lysed pellet on an SDS-PAGE gel to determine if a large fraction of the protein remains in the pellet after lysis. If the protein is present in large quantities in the pellet, the sonication procedure may be insufficient to lyse the cells. Try sonicating the pellet for additional cycles, resuspending in a larger volume of sonication buffer, or use an alternative lysis technique such as French press or microfluidics lysis. Alternatively, the protein may be aggregating and partitioning to the insoluble fraction. Try inducing growth for less time or at a slightly lower temperature to prevent aggregation.
- Problem: high pressure limit reached.
- Potential solutions: Inline FPLC filters may be clogged. Try replacing and/or cleaning solvent inlet and inline filters. Make sure that bacterial lysate is filtered through 0.45 μm syringe filter before placing on column. Filter all column buffers through 0.2 μm vacuum filters before flowing through FPLC.
8. Characterization of recombinant protein
8.1. Reagents
NADPH-Roche
All steroids-Steraloids
Triple buffer system comprising of equimolar sodium phosphate, sodium pyrophosphate, and 3-[(1,1-dimethyl-2-hydroxyethyl)amino]-2-hydroxypropanesulfonic acid
Indomethacin, Ursodeoxycholate and mefenamic acid-ICN Biomedicals, Inc
4-Bnzolybenzoic acid Sigma-Aldrich, Inc
8.2. pH optima and stability
pH optima and stability studies were performed on AKR1D1 using the fluorometric assay. The studies used a triple buffer system to cover the pH range from 5 to 9 while maintaining constant ionic strength. pH optima studies were performed under normal assay conditions (10 μM testosterone) except 50 mM triple buffer was used in place of potassium phosphate buffer. The optimal pH for reduction of testosterone was found to be 6.0 (as shown in Fig. 3, ▲). To test the stability of the enzyme over the time course of the assay (5 min), pH stability studies were performed by preincubating the enzyme in the triple buffer system at varying pH. The amount of activity remaining was estimated by diluting an aliquot into the standard assay. The enzyme was stable in the pH range 5 – 9 (see Fig. 3 ■). The depressed kcat in the pH optima studies is therefore not due to a decrease in enzyme stability and instead reflects the effect of pH on one or more rate-determining steps. Independent measurements performed at pH 9.0 showed that there was no effect on Km for testosterone thus the effect reported was solely based on kcat. These studies were repeated using cortisone and gave similar results.
Fig. 3.
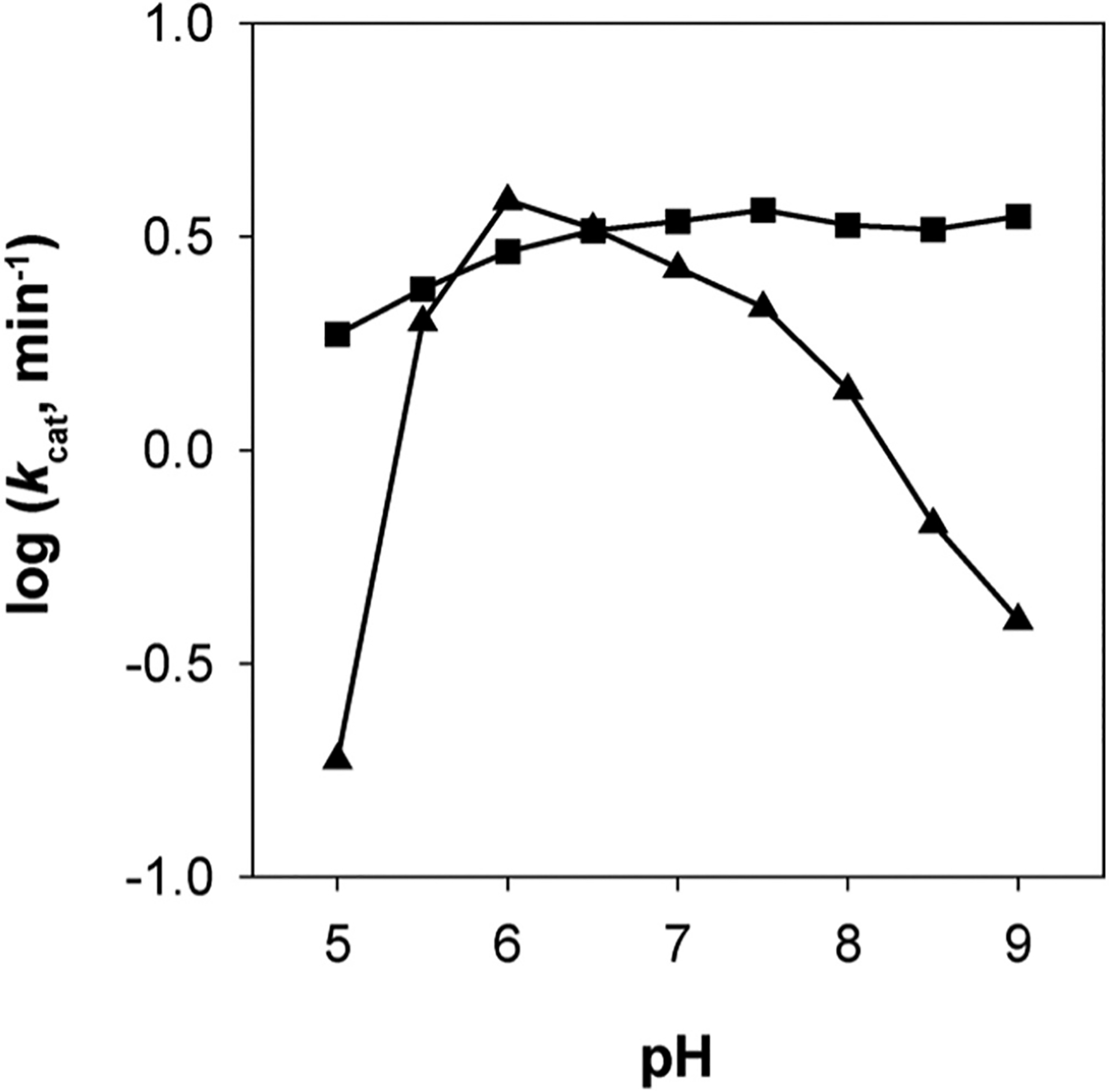
pH effects on AKR1D1 kcat. pH stability of the enzyme was tested from pH 5–9 in a triple buffer system using the standard assay (■). pH optima of the enzyme was tested from pH 5–9 in a modified standard assay using the triple buffer system (▲). Results are replotted as kcat versus pH, where assays were performed with saturating testosterone. This research was originally published in Steroids. Chen et al., (2011).
8.3. NADPH binding and Kd determination
AKR1D1 contains 5 tryptophan residues and has intrinsic fluorescence when excited at 295 nm. Incremental addition of NADPH quenched the fluorescence emission signal at 340 nm and generated an energy transfer band at 460 nm (Fig. 4A). An isosbestic point was observed at 405 nm indicating a product precursor relationship between the emission signal and the energy transfer band. This energy transfer band likely results from the interaction of the nicotinamide ring with Trp89 based on identical experiments with AKR1C9 and homology considerations (Jin & Penning, 2006). Plots of ΔF/ΔFmax versus [NADPH] were fitted to the Morrison equation to obtain the dissociation constant (Kd) of NADPH for wild-type AKR1D1 (Kd = 43 ± 16 nM) (Fig. 4B). This tight binding of NADPH is a characteristic property of other steroid transforming AKRs (Cooper et al., 2007; Jin & Penning, 2006) but is less than that seen with aldose reductase AKR1B1 (Kd = 9.0 nM).
Fig. 4.
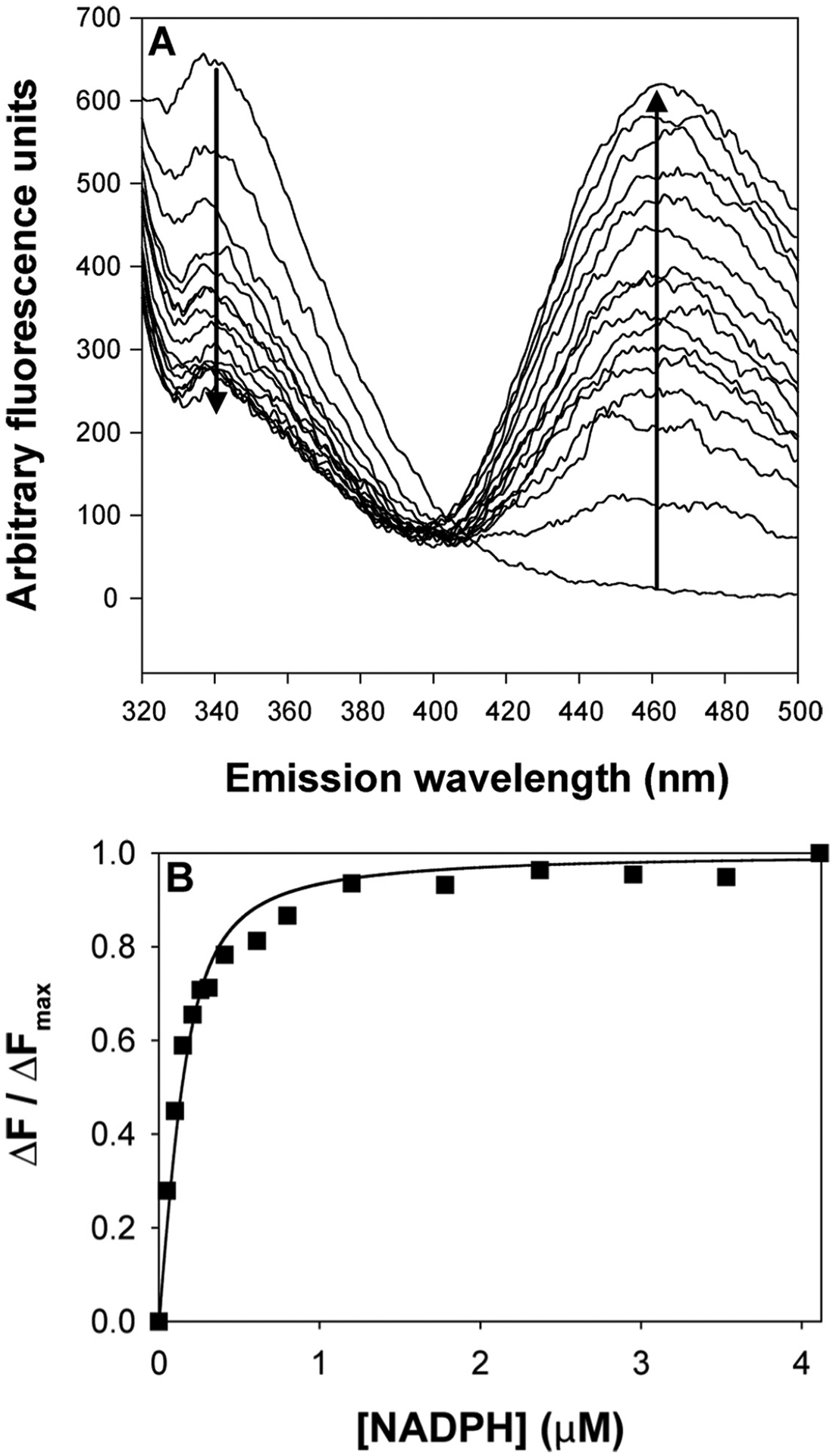
Titration of the fluorescence emission spectra of AKR1D1 with increasing [NADPH]. (Panel A) Emission spectrum of recombinant AKR1D1 excited at 295 nm following the addition of increasing concentrations of NADPH from 0 to 4 μM. A decrease in emission is seen at 340 nm and an increase in emission is seen at 460 nm with an isosbestic point at 405 nm. (Panel B) Plot of ΔF/ΔFmax versus [NADPH], wild type (■). This research was originally published in the Journal of Biological Chemistry. Drury et al., (2010).
8.4. Substrate specificity
The ability of AKR1D1 to catalyze the 5β-reduction of a range of Δ4-3-ketosteroids was determined fluorometrically by using the standard spectrofluorimetric assay in which steroid substrate concentration was varied (0.75 μM – 40 μM). Kinetic analyses of initial velocities obtained were performed using the Henri-Michaelis-Menten equation v = (Vmax × [S])/(Km + [S]) and fit using the program GraphFit. The iterative fits provided estimates of Vmax and Km and associated standard errors. Vmax was converted to kcat values using the predicted molecular weight of the enzyme.
5β-reduction was observed with C18-C27 steroids (Table 4). kcat values ranged from 0.5 min−1 to 9.9 min−1 indicating that there were differences in the rate-determining steps. Similarly large differences in Km were observed and these were most apparent with the glucocorticoids cortisol and cortisone. However, this difference was not seen in corticosterone suggesting that the introduction of a C17 side-chain that resembles glycerol has a large effect on Km. Catalytic efficiencies also varied by 30-fold. The bile acid precursor Δ4-cholesten-7α-ol-3-one gave a catalytic efficiency of 2.13 min−1 μM−1 and was similar to that seen with other steroid hormones such as testosterone (2.63 min−1 μM−1) and corticosterone (0.73 min−1 μM−1). The catalytic efficiencies for 4-estren-17β-ol-3-one (0.77 min−1 μM−1) and 1,4-androstadien-17β-ol-3-one (0.56 min−1 μM−1) showed that while the majority of substrates tested had an angular methyl group at C10 and a single double bond in the A-ring, these were not prerequisites for 5β-reduction to proceed. Substrate inhibition was observed for C18 and C19 substrates. A representative trace showing substrate inhibition by testosterone as the concentration is increased is shown in Fig. 5. A modified equation was used to analyze the initial velocities of those substrates exhibiting inhibition, v = (Vmax ⁎ [S])/(Km + [S] + [S]2/Ki). Many synthetic steroids also contain a Δ−1,4-diene in the A-ring. Using 1,4-androstadien-17β-ol-3-one as substrate, co-chromatography with authentic standards was used to identify the product of the reaction. The reaction has three possible outcomes: testosterone from reduction of the Δ1–2-ene; 5β-androst-1-en-17β-ol-3-one from reduction of the Δ4–5-ene; or 5β-DHT from reduction of the Δ−1,4-diene. The product did not co-migrate with either testosterone or 5β-DHT, suggesting that the enzyme was unable to reduce the Δ1–2-ene and preferred the Δ4–5-ene (Table 4).
Table 4.
Substrate specificity of AKR1D1.
| 4-Estren-l7β-ol-3-one | C18 | 2.3 ± 0.2 | 3.0 ± 0.5 | 18.6 ± 3.4 | 0.77 |
| Δ4-Androstene-3,17-dione | C19 | 5.1 ± 0.7 | 0.9 ± 0.2 | 4.3 ± 1.3 | 5.67 |
| Testosterone | C19 | 7.1 ± 1.7 | 2.7 ± 1.2 | 14.5 ± 5.8 | 2.63 |
| Epitestosterone | C19 | 5.1 ± 0.6 | 2.9 ± 0.6 | 10.0 ± 2.2 | 1.76 |
| 1,4-Androstadien-17β-ol-3-one | C19 | 1.8 ± 0.1 | 3.2 ± 0.2 | 0.56 | |
| Corticosterone | C21 | 1.6 ± 0.1 | 2.2 ± 0.5 | N.D. | 0.73 |
| Cortisol | C21 | 2.3 ± 0.1 | 13.1 ± 1.8 | N.D. | 0.18 |
| Cortisone | C21 | 9.9 ± 0.1 | 15.1 ± 0.3 | N.D. | 0.66 |
| 4-Cholesten-7α-ol-3-one | C27 | 1.7 ± 0.1 | 0.8 ± 0.2 | N.D. | 2.13 |
| Cholestenone | C27 | 0.5 ± 0.03 | 0.3 ± 0.1 | N.D. | 1.67 |
N.D., Not determined.
All activity measurements were determined fluorometrically.
This Table was originally published in Steroids Chen, M., Drury, J. E., Penning, T. M. (2011). Substrate specificity and inhibitor analysis of human steroid 5β-reductase. Steroids, 76, 484–490. © the Author(s).
Fig. 5.
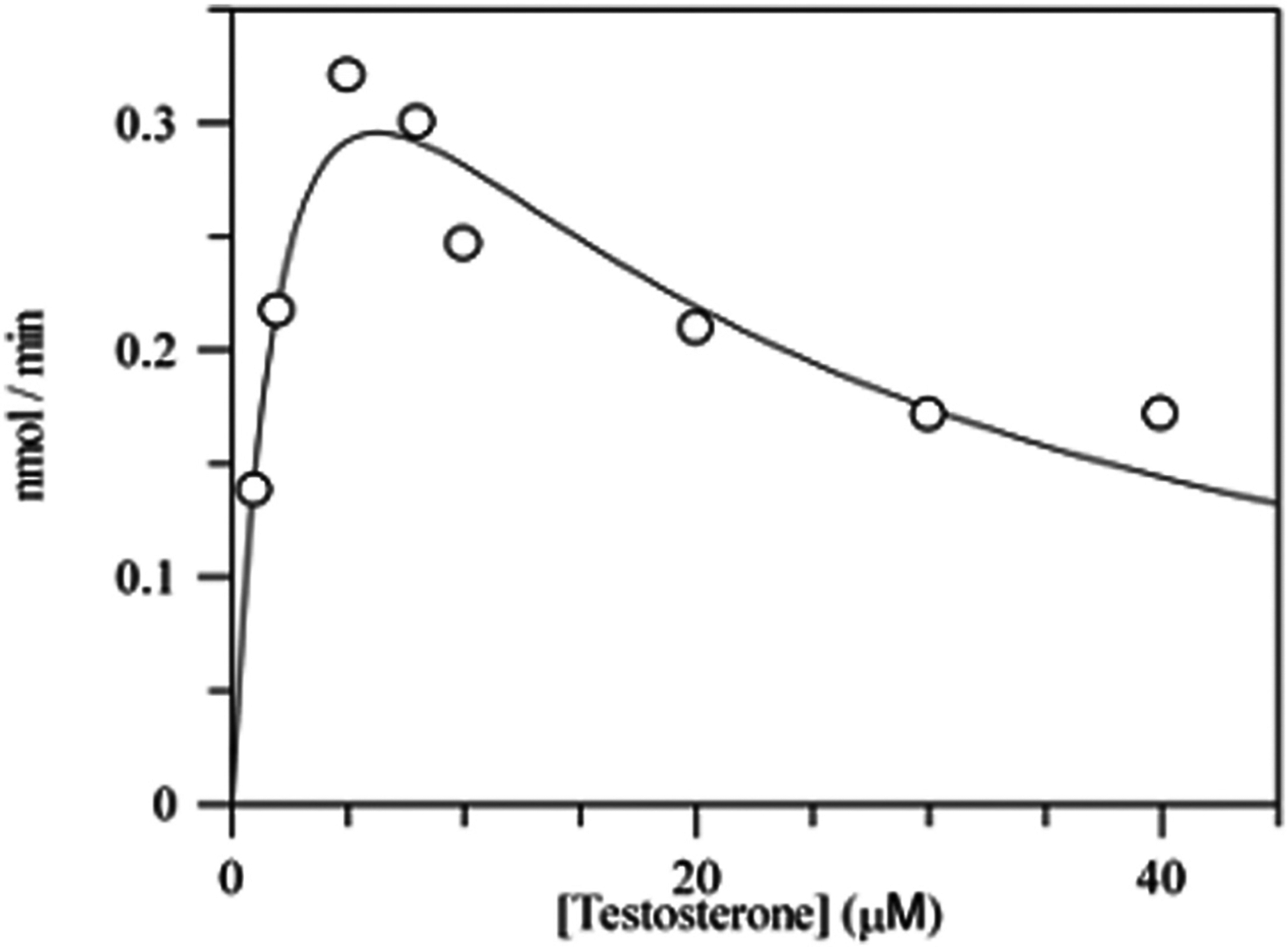
Substrate inhibition of AKR1D1 by testosterone. Velocity versus substrate plot for AKR1D1 showing substrate inhibition using testosterone. Assays contained 0.078 μM enzyme, 1–40 μM testosterone, 12 μM NADPH, 4% acetonitrile in 100 mM potassium phosphate buffer, pH 6.0, in a final volume of 1 mL. Reactions were monitored fluorometrically by using an excitation wavelength of 340 nm (slit-width 5 nm) and an emission wavelength set at 450 nm (slit-width 10 nm) at 37 °C. Kinetic analysis of initial velocities obtained was performed using the Henri-Michaelis-Menten equation for uncompetitive substrate inhibition v = (Vmax × [S])/(Km + [S] + [S]2/Ki) and fit using the program GraphFit. The iterative fits provided estimates of Vmax, Km and Ki and associated standard errors. This research was originally published in the Journal of Biological Chemistry. Di Costanzo et al., (2008).
8.5. Inhibition studies
Non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to inhibit the human AKR1Cs (Bauman et al., 2005; Byrns et al., 2008). The ability of these compounds to inhibit the AKR1D1 dependent reduction of [4-14C]-testosterone was measured (Table 5). Indomethacin, an AKR1C3 selective inhibitor, and mefenamic acid and 4-benzoylbenzoic acid, which are nonspecific AKR1C isoform inhibitors, were ineffective. Bile acids, e.g., chenodeoxycholic acid and its 7β-isomer ursodeoxycholic acid, are potent (nanomolar affinity) selective inhibitors of AKR1C2. These compounds were weak non-competitive inhibitors of AKR1D1 yielding Ki values of 3.2 μM and 9.8 μM respectively. Finasteride, which is a mechanism-based inhibitor of 5α-reductase with sub-nanomolar affinity, was also tested (Bull et al., 1996). Finasteride acted as a competitive inhibitor with a Ki value of 2.1 μM under conditions of reversible enzyme inhibition. Upon preincubation of 5β-reductase with finasteride, no time-dependent inhibition was observed either in the presence or absence of NADPH. By contrast inhibition constants for finasteride for 5α-reductase type 2 (SRD5A2) are 26 nM and less than 1 nM under reversible and time dependent-inhibition studies respectively (Bull et al., 1996; Russell & Wilson, 1994).
Table 5.
Inhibition of AKR1D1 by isoform selective inhibitors for AKR1C and 5α-reductase enzymes.
| Indomethacin | AKR1C3 | 8.2 (C)b | >100 n.i. |
| Mefenamic acid | AKR1C1, 1C2, 1C3 | 0.8, 0.22, 0.30 (C)c | >100 n.i. |
| 4-Benzoylbenzoic acid | AKR1C1, 1C2, 1C3 | 12.5, 13.3, 1.9 (C)c | >100 n.i. |
| Chenodeoxycholic acid | AKR1C1, 1C2, 1C3 | 8.3, 0.017, 10 (C)c | 3.2 (NC) |
| Ursodeoxycholic acid | AKR1C1, 1C2, 1C3 | 4.9, 0.011, 1.3 (C)c | 9.8 (NC) |
| Finasteride | 5α-reductase | 0.026 (C), « 0.001 (MBI)d | 2.1 (C) |
C, Competitive inhibition; NC, non-competitive inhibition; MBI, mechanism-based inhibition; n.i., no inhibition detected at the concentration of inhibitor used.
The Ki values were determined by measuring the inhibition of testosterone reduction.
This Table was originally published in Steroids Chen, M., Drury, J. E., Penning, T. M. (62011). Substrate specificity and inhibitor analysis of human steroid 5β-reductase. Steroids, 76, 484–490. c the Author(s).
References
- Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, & Penning T (2005). Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Molecular Pharmacology, 67(1), 60–68. 10.1124/mol.104.006569 [DOI] [PubMed] [Google Scholar]
- Bull H, Garcia-Calvo M, Andersson S, Baginsky WF, Chan H, Ellsworth D, … Harris G (1996). Mechanism-based inhibition of human steroid 5α-reductase by finasteride: Enzyme-catalyzed formation of NADP−dihydrofinasteride, a potent bisubstrate analog inhibitor. Journal of the American Chemical Society, 118, 2359–2365. [Google Scholar]
- Byrns M, Steckelbroeck S, & Penning T (2008). An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochemical Pharmacology, 75(2), 484–493. 10.1016/j.bcp.2007.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Drury J, & Penning T (2011). Substrate specificity and inhibitor analyses of human steroid 5β-reductase (AKR1D1). Steroids, 76, 484–490. 10.1016/j.steroids.2011.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Jin Y, & Penning T (2015). In-depth dissection of the P133R mutation in steroid 5β-reductase (AKR1D1): A molecular basis of bile acid deficiency. Biochemistry, 54, 6343–6351. 10.1021/acs.biochem.5b00816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper W, Jin Y, & Penning T (2007). Elucidation of a complete kinetic mechanism for a mammalian hydroxysteroid dehydrogenase (HSD) and identification of all enzyme forms on the reaction coordinate: The example of rat liver 3alpha-HSD (AKR1C9). The Journal of Biological Chemistry, 282(46), 33484–33493. 10.1074/jbc.M703414200 [DOI] [PubMed] [Google Scholar]
- Di Costanzo L, Drury J, Penning T, & Christianson D (2008). Crystal structure of human liver D4–3-ketosteroid 5b-reductase (AKR1D1) and implications for substrate binding and catalysis. The Journal of Biological Chemistry, 283, 16830–16839. 10.1074/jbc.M801778200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Costanzo L, Drury JE, Christianson DW, & Penning TM (2009). Structure and catalytic mechanism of steroid 5b-reductase (AKR1D1). Molecular and Cellular Endocrinology, 301, 191–198. 10.1016/j.mce.2008.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury JE, Mindnich R, & Penning T (2010). Characterization of disease-related 5b-reductase (AKR1D1) mutations reveals their potential to cause bile acid deficiency. The Journal of Biological Chemistry, 285(32), 24529–24537. 〈http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20522910〉. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyndman D, Bauman D, Heredia V, & Penning T (2003). The aldo-keto reductase superfamily homepage. Chemico-Biological Interactions, 143–144, 621–631. 10.1016/s0009-2797(02)00193-x [DOI] [PubMed] [Google Scholar]
- Jez J, Bennett M, Schlegel B, Lewis M, & Penning T (1997). Comparative anatomy of the aldo-keto reductase superfamily. Biochemical Journal, 326(Pt 3), 625–636. 10.1042/bj3260625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jez J, Flynn T, & Penning T (1997). A new nomenclature for the aldo-keto reductase superfamily. Biochemical Pharmacology, 54, 639–647. 10.1016/s0006-2952(97)84253-0 [DOI] [PubMed] [Google Scholar]
- Jin Y, & Penning T (2006). Multiple steps determine the overall rate of the reduction of 5alpha-dihydrotestosterone catalyzed by human type 3 3alpha-hydroxysteroid dehydrogenase: Implications for the elimination of androgens. Biochemistry, 45(43), 13054–13063. 10.1021/bi060591r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Kondo K-H, Okuda K, Higashi S, Suzuki M, Kurosawa T, … Setoguchi T (1998). Diagnosis of the first Japanese patient with 3-oxo-delta-4-steroid 5b-reductase deficiency by use of immunoblot analysis. European Journal of Pediatrics, 157, 386–390. 10.1007/s004310050835 [DOI] [PubMed] [Google Scholar]
- Lemonde H, Custard E, Bouquet J, Duran M, Overmars H, Scambler P, & Clayton P (2003). Mutations in SRD5B1 (AKR1D1), the gene encoding delta-4–3-oxosteroid 5b-reductase, in hepatitis and liver failure in infancy. Gut, 1494–1499. 10.1136/gut.52.10.1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, & Stechell KD (1992). Bile acid biosynthesis. Biochemistry, 31, 4737–4749. 10.1021/bi00135a001 [DOI] [PubMed] [Google Scholar]
- Russell DW, & Wilson J (1994). Steroid 5 alpha-reductase: Two genes/two enzymes. Annual Review of Biochemistry, 63, 25–61. 10.1146/annurev.bi.63.070194.000325 [DOI] [PubMed] [Google Scholar]
- Russell DW (2003). The enzymes, regulation and genetics of bile acid synthesis. Annual Review of Biochemistry, 72, 137–174. 10.1146/annurev.biochem.72.121801.161712 [DOI] [PubMed] [Google Scholar]
- Setchell KD, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubl J, & Balistreri W (1988). Delta-4–3-oxosteroid 5b-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. The Journal of Clinical Investigation, 2148–2157. 10.1172/JCI113837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider B, Setchell K, Whitington P, Neilson K, & Suchy F (1994). D4-3-oxosteroid 5b-reductase deficiency causing neonatal liver failure and hemochromatosis. The Journal of Pediatrics, 124, 234–238. 10.1016/s0022-3476(94)70310-8 [DOI] [PubMed] [Google Scholar]
- Ueki I, Kimura A, Chen HL, Yorifuji T, Mori J, Itoh S, … T M (2009). SRD5B1 gene analysis needed for the accurate diagnosis of primary 3-oxo-Delta4-steroid 5b-reductase deficiency. Journal of Gastroenterology and Hepatology, 24(5), 776–785, https://doi.org/JGH5669 [pii]10.1111/j.1440-1746.2008.05669.x. [DOI] [PubMed] [Google Scholar]