

Abstract
All-trans retinoic acid (ATRA) is an anti-cancer differentiation therapy agent effective for acute promyelocytic leukemia (APL) but not acute myeloid leukemia (AML) in general. Using the HL-60 human non-APL AML model where ATRA causes nuclear enrichment of c-Raf that drives differentiation and G1/G0 cell cycle arrest, we now observe that c-Raf in the nucleus showed novel interactions with several prominent regulators of the cell cycle and cell differentiation. One is cyclin-dependent kinase 2 (Cdk2). ATRA treatment caused c-Raf to dissociate from Cdk2. This was associated with enhanced binding of Cdk2 with retinoic acid receptor α (RARα). Consistent with this novel Raf/CDK2/RARα axis contributing to differentiation, CD38 expression per cell, which is transcriptionally regulated by a retinoic acid response element (RARE), is enhanced. The RB tumor suppressor, a fundamental regulator of G1 cell cycle progression or arrest, was also targeted by c-Raf in the nucleus. RB and specifically the S608 phosphorylated form (pS608RB) complexed with c-Raf. ATRA treatment induced S608RB-hypophosphorylation associated with G1/G0 cell cycle arrest and release of c-Raf from RB. We also found that nuclear c-Raf interacted with SMARCD1, a pioneering component of the SWI/SNF chromatin remodeling complex. ATRA treatment diminished the amount of this protein bound to c-Raf. The data suggest that ATRA treatment to HL-60 human cells re-directed c-Raf from its historically pro-proliferation functions in the cytoplasm to pro-differentiation functions in the nucleus.
Keywords: ATRA, APL, Cyclin-dependent kinase 2, RB, SMARCD1
Introduction:
Acute promyelocytic leukemia (APL) is a hematological malignancy that is the M3 subtype of acute myelocytic leukemia (AML) in the French-American-British (FAB) classification system [1, 2]. It accounts for approximately 10% of all cases of AML in adults [3]. APL is cytogenetically characterized by a t(15;17) fusion gene, promyelocytic leukemia-specific retinoic acid receptor alpha (PML-RARα), that blocks transcriptional activation by RARα and arrests granulocyte maturation [4]. It is associated with >98% of APL cases [5]. While APL is susceptible to ATRA based differentiation therapy, most AML are not and remain largely incurable. The human leukemia HL-60 cell line model was established from the peripheral blood leukocytes of a patient diagnosed with acute promyelocytic leukemia (FAB M3) [6] that was retrospectively re-evaluated as a myeloblastic leukemia (FAB M2) and more recently found to represent a hitherto uncharacterized subtype of ATRA responsive AML [7]. HL-60 is ergo a unique model for molecular mechanistic studies of ATRA-induced differentiation of a non-APL AML. It has the potential to reveal how a non-APL AML can be made to differentiate in response to ATRA. It has been an archetype model for studying differentiation induction therapy [8,9].
Although conventional chemotherapy with cytotoxic drugs has been largely unsuccessful for non-APL AML, all-trans retinoic acid (ATRA), a metabolite of vitamin A, could cause a high rate of APL clinical remission by inducing degradation of the PML-RARα fusion protein and the maturation of myeloblasts [10,11]. In this differentiation induction therapy, ATRA acts through binding and activation of two nuclear receptors (NRs), the retinoic acid receptor (RAR) and/or retinoid X receptor (RXR), to transcriptionally regulate target genes with promoters harboring retinoic acid response elements (RARE) [12]. However, transcriptional activation depends on other signaling pathways, notably the mitogen-activated protein kinase (MAPK) pathway that incorporates the Raf/Mek/Erk axis, which are needed to enable RAREs to respond to ATRA [13]. It is thought that MAPK pathway activation provides the signal that enables RARE to respond to ATRA [14]. Targeted promoters are thus acting as signal integrators.
Raf kinase regulates a wide range of physiological responses required for cell proliferation, differentiation, cell death and survival, metabolism and motility [15,16]. In the classical growth factor driven MAPK pathway, stimulation of receptor tyrosine kinases at the cell surface activates a guanine nucleotide exchange factor, stimulating the Ras GTPase, through the exchange of GDP to GTP, which recruits and activates Raf at the plasma membrane. The activated Raf phosphorylates and activates Mek, which phosphorylates and activates Erk, which ultimately activates transcription factors leading to mitogenesis [17,18]. The kinetics of the signal determines the cellular outcome. A fast, transient signal is associated with mitogenesis, whereas a slow, long-lasting signal is seminal to differentiation. In the HL-60 non-APL AML model, ATRA-induced myeloid cell differentiation depends on sustained MAPK pathway activation associated with the up-regulation and unanticipated translocation of c-Raf to the nucleus [19,20,21]. c-Raf in the nucleus targets and activates NFATc3 to promote transcription of genes necessary for differentiation [13]. c-Raf thus has potential targets other than the traditional Mek kinase and its novel nuclear functions may be important to driving differentiation. An important question is whether this is a one off occurrence or reflects a wider basic phenomenon. The targets of nuclear c-Raf and the web of pathways affected are thus of significant interest.
In the nucleus, c-Raf also interacts with the RB tumor suppressor protein [22]. In vitro, it has been reported that RB can be a phosphorylation substrate for Raf-1 [23]. RB governs cell cycle progression by controlling the amount of available E2F, a transcription factor driving G1 to S progression. RB hyperphosphorylation and particularly phosphorylation at serine 608 induces a conformational change that releases E2F sequestered on RB to transcriptionally activate genes that are needed to drive cells beyond the Restriction Point, a G1 cell cycle check point, to S phase [24,25]. ATRA causes RB hypophosphorylation to sequester E2F and block entrance into S phase [26]. Another target of c-Raf in the nucleus is GSK3, a serine threonine kinase, known from pharmacological inhibitor studies to cause diminished RARα transcriptional activity [27]. Nuclear c-Raf increases the phosphorylation at the inhibitory serine-9 and 21 of GSK-3, which dissociates GSK3 from RARα and promotes myeloid cell differentiation in ATRA-treated HL-60 cells [22]. Nuclear c-Raf thus appears to target prominent regulators of proliferation/arrest and differentiation. This motivates interest in identifying nuclear c-Raf targets and gaining insight into emerging anti-proliferation/pro-differentiation nuclear regulatory pathways governed by ATRA-induced nuclear enrichment of c-Raf.
In this study, we aimed to identify novel c-Raf nuclear partners which have a potential role in ATRA-induced HL-60 human myeloblastic leukemia cell differentiation. We ectopically expressed Flag-c-Raf in HEK293T cells and then purified Flag protein was used to troll extracts from HL-60 cells that were either treated or untreated with ATRA.
Mass spectrometry was used to identify the partners of the Flag tagged c-Raf. A number of c-Raf-partner pairs that were ATRA-regulated were identified, and the most prominent and novel candidate parings were tested by immunoprecipitation for confirmation and further study. ATRA induced the enhanced expression of c-Raf in the nucleus and regulated its interaction with several prominent cell cycle/differentiation regulatory proteins, such as Cdk2, RB and SMARCD1. For Cdk2, ATRA treatment diminished the amount of Cdk2 interacting with c-Raf. This was associated with enhanced Cdk2 interaction with RARα. The enhanced Cdk2 interaction with RARα was associated with the enhanced expression of the CD38 cell surface receptor per cell. For RB, ATRA decreased the amount of RB, specifically pS608RB, bound to c-Raf, which was associated with the loss of pS608 phosphorylation and G1/G0 cell cycle arrest. Nuclear c-Raf also interacted with a pioneering component of the SWI/SNF chromatin remodeling complex, SMARCD1; and ATRA treatment diminished the amount of this protein bound to c-Raf. Freed from these arguably pro-proliferation targets, c-Raf could target pro-differentiation targets such as NFATc3 to enable RARE activation as previously reported as a prototype of this paradigm [13]. Our data suggest that ATRA has re-directed the pro-proliferation functions of c-Raf in the cytoplasm to pro-differentiation functions in the nucleus. Our result suggests new targets of susceptibility for therapeutic intervention to enhance the efficiency of all-trans retinoic acid differentiation therapy in AML patients.
Materials and methods:
Cell culture
Human myeloblastic leukemia cells (HL-60) were grown in a humidified atmosphere of 5% CO2 at 37°C and maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (Invitrogen, Carlsbad, CA) and 1% antibiotic antimycotic (Thermo Fisher Scientific, Waltham, MA). The experimental cultures were initiated at a cell density of 0.1×106 cells/ml. Human embryonic kidney cells, i.e. HEK293T and transformed HEK293T-c-Raf-flag, were grown in Dulbecco’s Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and 1% antibiotic-antimycotic (Thermo Fisher Scientific, Waltham, MA) in a 5% CO2 humidified atmosphere at 37°C. Cell growth and viability was monitored with a hemocytometer and 0.2% trypan blue (Invitrogen, Carlsbad, CA) exclusion assay. All-trans retinoic acid (ATRA) (Sigma, St. Louis, MO) was dissolved in 100% ethanol with a stock concentration of 5 mM and used at a final concentration of 1 μM as previously described [28].
Antibodies and reagents
Antibodies for flow cytometric analysis of CD38 and CD11b conjugated with either phycoerythrin (PE) or allophycocyanin (APC) were obtained from BD Biosciences (San Jose, CA). Antibodies for Western blotting including TBP, RB, RARα, GAPDH, CD11b, HRP anti-mouse and anti-rabbit were from Cell Signaling (Danvers, MA). Anti-flag M2 antibody was from Sigma (St. Louis, MO). CD38, c-Raf and BAF60a (SMARCD1) were purchased from BD Biosciences (San Jose, CA). Cdk2, Phospho-Cdk2 (T160), Phospho-Cdk2 (Y15) and Phospho-RB (S608) were from AbCam (Cambridge, MA). Mek1/2 antibody was provided by Santa Cruz Biotechnology (CA, USA). Protein G magnetic beads used for immunoprecipitation were from Millipore (Billerica, MA). M-PER Mammalian protein, NE-PER Nuclear and cytoplasmic extraction reagents were from Pierce Biotechnology (Thermo Scientific, Rockford, IL). Bovine serum albumin (BSA), Nonidet P40 (NP-40), Triton X-100, protease and phosphatase inhibitors were purchased from Sigma (St. Louis, MO).
Construction of the recombinant plasmids
Gateway entry vector (pENTR1A-Flag-His-N2) and destination vector (pLenti-CMV-Puro-Dest, Addgene #17452) were used for construction of plasmid. c-Raf primers were designed by adding a CAGGTACC overhang (KPN1) at the 5’ end and GTGGATCCC overhang (BamHI) at the 3’ end of the forward and reverse primers, respectively. For generation of pENTR1A-c-Raf-Flag-His, the coding sequence (CDS) of c-Raf gene (Accession No: NM_001354689.1) was amplified from HL-60 cells by conventional PCR with a pair of primers mentioned in Tab.1. Amplified PCR products were cloned into entry vector and then recombined into destination vector that carried the desired back bone and drug selection marker. To produce the lentiviral particles, packaging plasmid i.e. psPAX2 (Addgene # 12260) and envelope plasmid i.e. pMD2.G (Addgene # 12259) were used.
Tab. 1.
Oligonucleotide primers for amplification of c-Raf cDNA.
| Gene | Target Primer Sequence | TM (°C) | CDS (bp) |
|---|---|---|---|
| c-Raf (F) | CAGGTACCATGGAGCACATACAGGGAGCT | 61 | 1947 |
| c-Raf (R) | GTGGATCCCGAAGACAGGCAGCCTCGGGGA |
Virus preparation and transduction
Lentiviral particles were produced using 2.5 μg of pMD2.g, 7.5 μg of psPAX2, and 10 μg of CMV-c-Raf-Flag-His plasmid. HEK293T cells were co-transfected with these plasmids at roughly 50-60% confluence in 10 cm cell culture dishes with DMEM and 10% FBS using polyethylenimine (PEI) (Sigma, St. Louis, MO) according to the manufacturer’s protocol. After 48 h, media containing viral particles was collected and 5 mL of additional media was added to the dish for 24 h until final collection. Total lentiviral media was concentrated using Amicon Ultra (Millipore, Billerica, MA) centrifugal filters. Concentrated viral media was stored at −80°C until use. Lentivirus particles were transduced into HEK293T cells in a six-well plate. 100 μL concentrated viral particles was added to 5 × 104 cells in 1 mL DMEM with 10% heat inactivated FBS. After 72 h, transduced cells were trypsinized and cultured in DMEM with 10% heat inactivated FBS and selected for 3 weeks in 3 μg/mL puromycin.
Recombinant proteins and trolling
To extract the c-Raf-Flag protein, transfected cells were lysed in lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 1mM EDTA, pH 8.0, 1% NP-40) supplemented with 1/100 volume of protease and phosphatase inhibitors (Sigma, St. Louis, MO) and clarified through centrifugation at 14,800 rpm (10 min at 4°C). Trolling procedure was adapted from Shen et al. [29] with some modifications. Briefly, 2 mg of extracted c-Raf Flag protein was immunoprecipitated with anti-flag M2 Affinity Gel (Sigma, St. Louis, MO). The pulldown samples were extensively washed in lysis buffer, 1M NaCl and 0.1% NP-40, to remove proteins that interacted with recombinant c-Raf Flag protein. Samples were either used immediately or frozen at −80°C until required.
HL-60 cells were either untreated or ATRA-treated for 72 h, and total cell lysates were prepared as described by Martin et al. [30]. Briefly, 8 × 106 cells were washed twice with phosphate-buffered saline (PBS), pH 7.2, followed by a single wash with 10 ml of cell extraction buffer (CEB). Cells were pelleted, the supernatant aspirated, and the cells were then transferred to a 2 ml glass Dounce homogenizer in the remaining droplet of buffer. Cells were then allowed to swell by addition of an equal volume of CEB to the volume occupied by the packed cell pellet followed by incubation on ice for 20 minutes. Cells were gently lysed with 20 strokes of a B-type pestle (Dounce homogenizer) and lysis was monitored by staining a small aliquot of the lysate with 0.2% trypan blue (Invitrogen, Carlsbad, CA) which was examined using a light microscope. The lysate was then transferred to a 1 ml Eppendorf tube and centrifuged at 4°C for 20 min at 14,800 rpm. The clear supernatant was carefully removed and then diluted to 7.5-15 mg/ml with extraction dilution buffer (EDB; 10 mM HEPES, 50 mM NaCl, 2 mM MgCl2, 5 mM EGTA, 1 mM DTT). Extracts were either used immediately or frozen at −80°C for later use.
Total cell lysates from HL-60 cells, either treated with ATRA or untreated, were incubated overnight with Flag protein extracts from HEK293T-c-Raf-Flag cells. As a control, HEK293T cells were also trolled.
LC-MS/MS analysis
After 24 h of incubation, samples were washed with 50 mM ammonium bicarbonate, pH 8.0, subsequently resuspended in 500 ng trypsin and incubated at 37°C for 4-6 h. Samples were centrifuged, and supernatants were collected. 250 ng trypsin was further added to supernatant and incubated overnight at 37°C. On the next day, samples were acidified with 50% formic acid and vacuum dried. Lyophilized sample was resuspended in 20 μL buffer A (0.1% formic acid and 3% acetonitrile in water) and stored at −80 °C until required. All samples (either ATRA treated or untreated) were analyzed using a Q Exactive HF mass spectrometer (Thermo Scientific, Rockford, IL) to identify putative c-Raf partners. LC-MS/MS analysis was adapted from Wang et al. [31]. Briefly, upon desalting by C18 stage-tips, the obtained peptides were reconstituted in 20 μl of 0.1% formic acid and separated by an Easy-nLC 1200 system coupled to a Q Exactive HF mass spectrometer (Thermo Scientific, Rockford, IL). 1 μl peptides were injected and analyzed on a reverse phase C18 column (75 μm i.d. × 15 cm, 3 μm particle size) at a flow rate of 250 nL/min. Mobile phase A (0.1% formic acid in ultrapure water) with an eluting buffer as mobile phase B (0.1% formic acid in 80% acetonitrile) was used to establish a linear 50-min gradient of 7–25% buffer B. Peptides were then ionized by electrospray at 1.5 kV. The mass spectrometer was operated in positive ion mode acquiring at a resolution of 120,000, with a full MS spectrum (m/z = 350-1800) using an automatic gain control (AGC) target of 3 × 106. The 12 most intense ions were selected for higher-energy collisional dissociation (HCD) fragmentation (normalized collision energy 27) and MS/MS spectra were generated with an AGC target of 1 × 105 at a resolution of 30,000. The dynamic exclusion time was set to 30 s.
Database analysis
The raw data were created by XCalibur 4.0.27 software (Thermo Scientific, Rockford, IL) and processed with Proteome Discoverer (PD) software suite 2.1 (Thermo Scientific, Rockford, IL), against Uniprot human protein database in Sequest HT node. The precursor and fragment mass tolerances were set to 10 ppm and 0.02 Da, respectively.
Reversed database searches were used to evaluate false discovery rate (FDR) of peptide and protein identifications. The maximum of two missed cleavage sites of trypsin was allowed. Carbamidomethylation (C) was set as a static modification, and oxidation (M) and acetyl (protein N-terminal) were set as variable modifications. False discovery rate (FDR) of peptide spectrum matches (PSMs) and peptide identification were determined using the percolator algorithm at 1% based on q-value.
Western blotting and immunoprecipitation
Immunoprecipitation followed by Western blot experiments were performed to validate the mass spectrometry data. For this, HL-60 cells were either untreated or treated with ATRA for 72 h and then washed and pelleted. Nuclear and cytoplasmic protein fractions were made with NE-PER extraction kit (Thermo Scientific, Rockford, IL) following the manufacturer’s instruction with the addition of protease and phosphatase inhibitors. The purity of the nuclear and cytoplasmic fractionations was assessed using clathrin as a cytoplasmic marker and TATA binding protein (TBP) as a nuclear marker. The nuclear fractions used were verified as TBP positive and clathrin negative by Western blotting. Protein concentration was determined using the Pierce BCA protein assay (Thermo Scientific, Rockford, IL) according to the manufacturer’s protocol. For immunoprecipitation experiments, equal amounts of protein were pre-cleared with PureProteome protein G magnetic beads (Millipore, Billerica, MA) and then incubated overnight with beads and appropriate antibodies. Bead/antibody/protein slurries were then washed and subjected to standard SDS-PAGE analysis. For Western blotting, 25 μg of protein was resolved by SDS-PAGE using 12% polyacrylamide gel. The electrotransfer was done onto a PVDF membrane (Millipore, Billerica, MA) at 400 mA.
The membranes were blocked in dry nonfat milk before being incubated with the indicated primary antibody overnight at 4°C. Images were captured on a Bio-Rad ChemiDoc XRS Molecular Imager. Band density was determined using Image J and normalized to the loading control of the lane. For scaling in the bar graphs, the lowest normalized value is arbitrarily set to one and the values for other bands normalized to that and shown relative to the lowest value, which was typically the untreated control unless there was no detectable signal then the lowest detectable signal was used. When normalized values diverge with statistical significance, namely p less than or equal to 0.05, in the blots, then they are shown. However, all trends shown in the representative blot and the bar graphs synopsizing repeats, are reproducible; nevertheless western blotting is not strictly quantitative and the variation in amplitude for blot to blot repeats of expression level of the same molecule can have variability so that the number of repeats did not achieve a p less than or equal to 0.05. The values from at least three biological repeats were tabulated and statistically evaluated using GraphPad Prism 6.01.
Flow cytometric phenotypic analysis
Expression of cell surface differentiation markers was quantified by flow cytometry (Becton Dickinson LSR II flow cytometer) (San Jose, CA). 0.5 × 106 cells were harvested by centrifugation at 700 rpm for 5 minutes. Pelleted cells were resuspended in 200 μL PBS containing 2.5 μL of phycoerythrin (PE)-conjugated anti-CD38 and allophycocyanin (APC)-conjugated anti-CD11b antibodies. Following incubation for 1h at 37°C, cells were analyzed by flow cytometry. Gates to determine the percentage of positive cells were set to exclude 95% of control cells. Cell cycle analysis was performed as previously described (32).
Statistical analysis
Experiments were biological replicates in triplicate, and the results are shown as mean with standard deviation (SD). A multiple t-test using the Holm-Sidak method was used assess differences between groups. A p value less than 0.05 was considered significant.
Results:
c-Raf cloning and expression in HEK293T cells
A vector expressing the Flag-tagged c-Raf trolling protein was generated. A Flag-tagged c-Raf cDNA expression vector was constructed and ectopically expressed in HEK293T cells. Conventional PCR amplification of coding sequence (CDS) of c-Raf from HL-60 cells was performed with primers given in Tab.1. The product was 1947 bp consistent with the expected molecular mass of c-Raf (Fig. 1A). A KPNI and BAMHI restriction digest yielded appropriate 2420 bp and 1947 bp fragments (Fig. 1B). Colony PCR and DNA Sanger sequencing further confirmed the correct plasmid construction (data not shown). We then constructed a pLenti-CMV-c-Raf-flag plasmid using gateway recombination cloning kit (Invitrogen, Carlsbad, CA), and expressed it in HEK293T cells.
Fig. 1.
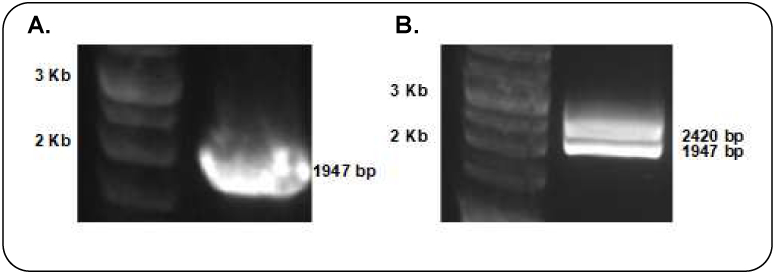
Gel electrophoresis results of c-Raf cloning. (A) PCR amplification of c-Raf, (B) restriction enzyme digestion of c-Raf cloned plasmid, (C) Flag expression in lysate of HEK293T-c-Raf-Flag cells by Western blot.
HEK293T-c-Raf-Flag cells were collected for Western blot analysis following 48 h of virus transduction and 72 h of puromycin selection. As shown in Fig. 1C, recombinant cells showed significantly increased levels of flag expression, while untransfected parental cells showed no flag expression.
c-Raf partners identified by Flag-c-Raf trolling and mass spectrometry
Lysates from HL-60 cells that were either untreated or treated with ATRA were incubated with the recombinant Flag-tagged c-Raf. To test the trolling, the anticipated binding of c-Raf and Mek was confirmed. Mek1/2 immunoprecipitated with c-Raf (Fig. 2). This verified that the trolling detected c-Raf partners as c-Raf kinase is known to complex with Mek1/2 [33]. After validation, mass spectrometry was used to detect c-Raf partners complexed to flag tagged c-Raf after trolling cell extracts from HL-60 cells that were either untreated or ATRA-treated. After screening using a Venn diagram tool for partners expressed in ATRA-treated cells, we found 19 potential c-Raf partners that were regulated during ATRA treatment. Tab. 2 shows the results. Three partners with substantively higher scores than the rest were selected for further study. These candidates were cycle-dependent kinase 2 (Cdk2), retinoblastoma-associated protein (RB1), and SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member 1 (SMARCD1). Each of these has historically been implicated with regulation of cell proliferation and differentiation. Interestingly, the fourth highest scoring candidate was histone H3, and H3K9ac expression has recently been implicated as promoting proliferation and inhibiting differentiation in non-APL AML (including HL-60) cells [34]. While we did not further pursue histone H3 here, the report is consistent with the notion proposed here that nuclear Raf targets are of importance.
Fig. 2.
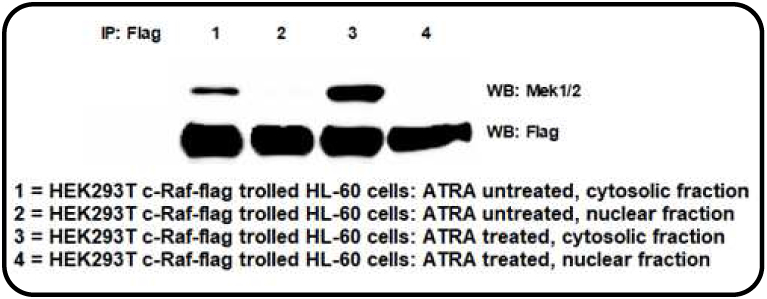
Co-immunoprecipitation followed by Western blot validation of the trolling. c-Raf-Flag was pulled down from HEK293T-c-Raf-flag recombinant cells lysates and used to troll either ATRA treated or untreated HL-60 cells. The pull down partners were resolved by Western blotting and probed for MEK1/2.
Tab.2:
Mass spectrometry data showed the putative partners of c-Raf that were ATRA regulated. Lysate from untreated and ATRA treated cells were trolled with Flag tagged c-Raf and analyzed by LC-MS/MS.
| Sr. No. |
Protein | AN Uniprot |
Gene |
‡ Average Score Sequest HT: Sequest HT |
Intracellular localization, Uniprot database annotation |
|---|---|---|---|---|---|
| 1. | Cyclin-dependent kinase 2 |
P24941 | CDK2 | 12.35 | Cytoplasm, Cytoskeleton, Endosome, Nucleus |
| 2. | Retinoblastoma- associated protein |
P06400 | RB1 | 7.665 | Nucleus |
| 3. | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member 1 |
Q96GM5 | SMARCD1 | 7.655 | Nucleus |
| 4. | Histone H3.1 | P68431 | HIST1H3A | 7.22 | Chromosome, Nucleosome core, Nucleus |
| 5. | Serine/threonine- protein phosphatase 4 regulatory subunit 1 |
Q8TF05 | PPP4R1 | 5.27 | Cytoplasm, Nucleus |
| 6. | RNA helicase aquarius |
O60306 | AQR | 4.95 | Nucleus, Spliceosome |
| 7. | Methylthioribose-1- phosphate isomeras |
Q9BV20 | MRI1 | 4.325 | Cell projection, Cytoplasm, Nucleus |
| 8. | Proliferating cell nuclear antigen |
P12004 | PCNA | 4.07 | Nucleus |
| 9. | Sorting nexin-3 | O60493 | SNX3 | 3.6 | Cytoplasmic vesicle, Endosome |
| 10. | Rho GTPase- activating protein 15 |
Q53QZ3 | ARHGAP15 | 3.35 | Cytoplasm, Membrane |
| 11. | Integrator complex subunit 8 |
Q75QN2 | INTS8 | 3.05 | Nucleus |
| 12. | Vacuolar protein sorting-associated protein 4B |
O75351 | VPS4B | 2.85 | Endosome, Membrane |
| 13. | WD40 repeat- containing protein SMU1 |
Q2TAY7 | SMU1 | 2.635 | Cytoplasm, Nucleus |
| 14. | Schlafen family member 11 |
Q7Z7L1 | SLFN11 | 2.345 | Chromosome, Nucleus |
| 15. | Tyrosine--tRNA ligase, cytoplasmic |
P54577 | YARS | 1.83 | Cytoplasm |
| 16. | Mitochondrial import inner membrane translocase subunit Tim13 |
Q9Y5L4 | TIMM13 | 1.415 | Membrane, Mitochondrion, Mitochondrion inner membrane |
| 17. | AT-rich interactive domain-containing protein 2 |
Q68CP9 | ARID2 | 1.215 | Nucleus |
| 18. | Nuclear cap- binding protein subunit 1 |
Q09161 | NCBP1 | 1.08 | Cytoplasm, Nucleus |
| 19. | Neuropathy target esterase |
Q8IY17 | PNPLA6 | 0.99 | Endoplasmic reticulum, Extracellular, Cytoskeleton |
Score Sequest HT- It is the sum of the scores of the individual peptides from the Sequest HT search
Novel c-Raf-Cdk2 interaction in nucleus and its role in ATRA induced myeloid cell differentiation
Pursuing the c-Raf/Cdk2 and Cdk2/RARα interactions, we first detected the expression of these molecules in ATRA treated or untreated cells using Western blotting. After 72 h of ATRA treatment, we collected nuclear cell lysates and analyzed the expression of c-Raf, total Cdk2, phospho-Y15Cdk2 and phospho-T160Cdk2 by Western blot. ATRA-treated cells showed enhancement of c-Raf expression (Fig. 3A). We also found that ATRA treatment led to ~50% decrease in the amount of nuclear cyclin E1 and Cdk2 with concomitant decreases in the Cdk2 kinase phosphorylated at both activating T160 and inhibitory Y15 sites (Fig. 3B-E). The p27 Kip1 cyclin dependent kinase inhibitor (CDKI) was also likewise analyzed. Western blot analysis showed an increase in p27 Kip1 protein at 72 h of ATRA treatment. (Fig. 3F). These results indicate that ATRA treatment affects regulatory molecules that control G1/G0 cell cycle arrest (Fig. 4H shows flow cytometric DNA histograms confirming that these changes are associated with the anticipated G1/0 arrest at 72 h. The observed ATRA driven reduction in cyclin E1 and Cdk2 levels may contribute to increased p27Kip1 stability since cyclin E-CDK2 complexes can target p27Kip1 for degradation through phosphorylation of Thr187 (47,48). We also found that ATRA enhances the expression of RARα protein, a known driver of ATRA-induced differentiation (Fig. 3G, Fig. 3H shows the TBP control).
Fig. 3.
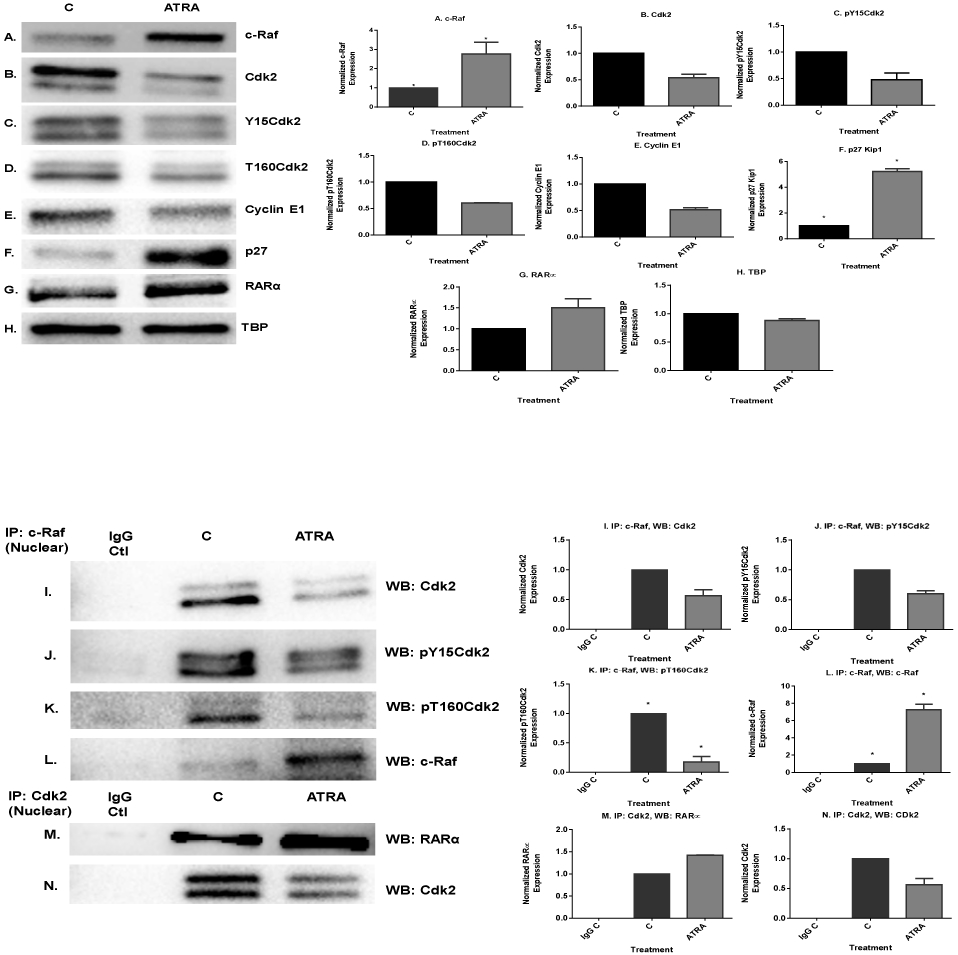
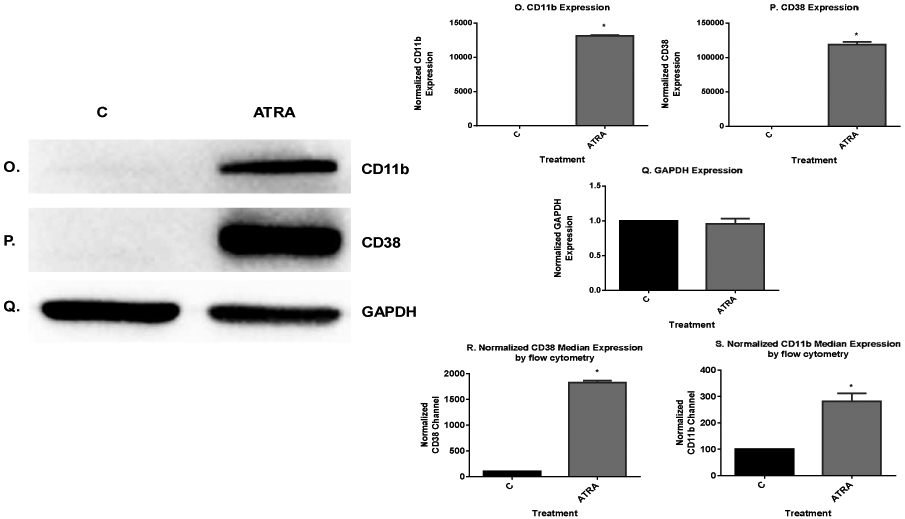
ATRA induced modulation of nuclear molecules in wild-type HL-60 cells. A. Western blot of c-Raf in HL-60 cells cultured with ATRA for 72 h showed that ATRA upregulated nuclear c-Raf. B-E. Western blots of nuclear lysate from control and ATRA-treated cells show that ATRA treatment led to decrease in the amount of cyclin E1 and Cdk2 with its phospho residues, pY15CDK2 and pT160CDK2. F. ATRA upregulates the CDKI p27Kip1 to promote G1/G0 arrest. Western blots of nuclear lysate show that ATRA upregulates the p27Kip1. G. Western blot of nuclear lysate showing that ATRA enhanced the amount of the nuclear RARα protein. H. TATA binding protein (TBP) was the loading control. I-L. Co-immunoprecipitation results show the ATRA-induced decrease in the amount of nuclear c-Raf-Cdk2, c-Raf-Y15Cdk2 and c-Raf-T160Cdk2 complexes. M-N. Co-immunoprecipitation show that ATRA increased the amount of Cdk2 bound to RARα. CDK2 was immunoprecipitation bait and blot of immunoprecipitate was probed for RARα and CDK2, control to confirm CDK2 immunoprecipitated. O & P. ATRA-treated cells showed enrichment of CD38 and CD11b expression by Western blot. Q. GAPDH loading control. R & S. ATRA treatment enhanced median CD38 and CD11b expression per cell. Median expression of CD38 and CD11b per cell was assessed by flow cytometry at 72 h in untreated control and ATRA-treated HL-60 cells. Bar graph of median CD38 and CD11b expression per cell showing that ATRA treated cells significantly increases expression per cell of CD38 and CD11b (*p< 0.05). All experiments shown are representative of three replicates.
Fig. 4.

ATRA treatment enhances the cell cycle arrest at G1/G0 by modulating the nuclear molecules. A. Western blot of c-Raf in HL-60 cells cultured with ATRA for 72 h showed that ATRA upregulated c-Raf compared to control. B & C. ATRA downregulates total RB and pS608 phosphorylated RB. D. TATA binding protein (TBP) is the loading control. E-G. Immunoprecipitation results showed that ATRA induced downregulation of the amount of nuclear c-Raf complexed with RB and specifically with its serine 608 phosphorylated form (pS608 RB). Using untreated and ATRA-treated cells, an equal amount of pre-cleared nuclear lysates collected 72 h post treatment were incubated overnight with 2.5 μg of the precipitating antibody with magnetic beads and resolved on 12 % polyacrylamide gels. Blots were probed for RB, pS608 RB and c-Raf, control to confirm that c-Raf immunoprecipitated. H. Cell cycle distribution at 72 h showing the percentage of cells in G1/G0, S and G2/M was analyzed using flow cytometry with propidium iodide staining. ATRA induced G1/G0 cell cycle arrest betrayed by relative increase in G1/0 cells in treated (right) compared to untreated control (left) cells. All experiments shown are representative of three replicates.
Using immunoprecipitation, we found that ATRA regulated a novel interaction of c-Raf with Cdk2 in the nucleus. Immunoprecipitation using c-Raf as bait was done with nuclear lysate from ATRA-treated and untreated cells and probed for Cdk2. This revealed a c-Raf-Cdk2 interaction. ATRA reduced the amount of c-Raf/Cdk2 complex. The c-Raf complexed with both pY15Cdk2 and pT160Cdk2, the former being canonically associated with Cdk2 inhibition and the latter activation. ATRA-induced decrease in the amount of c-Raf/Cdk2, c-Raf/pY15Cdk2 and c-Raf/pT160Cdk2 complexes. This occurred with the reduction in nuclear Cdk2. (Fig. 3I-K, Fig. 3L verifies anticipated presence of the Raf bait control). We also found that the ATRA-induced decrease in the amount of Cdk2 bound to c-Raf was associated with an increase in amount of Cdk2 bound to retinoic acid receptor alpha (RARα). We performed co-immunoprecipitation using Cdk2-specific antibody in both ATRA-treated and untreated cells and probed for RARα. ATRA enhanced the amount of Cdk2 bound to RARα. Thus, ATRA treatment caused c-Raf in the nucleus to dissociate from Cdk2, and this was associated with enhanced binding of Cdk2 with RARα (Fig. 3M & N).
We sought evidence of increased expression of RARα target genes associated with the increase in bound Cdk2. CD38 is one of the earliest characterized ATRA-induced differentiation antigens, and its expression propels differentiation [35,36,37]. Expression of CD38 is dependent on RARα acting through a RARE in the promoter of the gene [38]. We thus use it here as an endogenous promoter-reporter for RARE activation. We measured the expression of the CD38 membrane protein in untreated and ATRA-treated cells. ATRA increased the amount of CD38 expressed (Fig. 3O). The expression of CD38 significantly differed (p=0.0002) between the control and ATRA-treated group by flow cytometry (Fig. 3R). To explore another potential endogenous promoter–reporter gene [39], we measured the expression of CD11b, a later differentiation marker of progressive ATRA-induced differentiation, and we found that ATRA also increased the amount of CD11b expression by Western blot and flow cytometry (p=0.013) (Fig. 3P and S. Fig. 3Q is the GAPDH loading control). This result is consistent with the predicted increased transcription driven by enhanced RARα activity. The data support the role of the Raf/Cdk2/RARα axis for promoting ATRA-induced differentiation.
Role of the c-Raf-RB interaction in ATRA-induced differentiation and G1/G0 arrest
We sought to confirm and expand the observation of the interaction of c-Raf and RB detected by mass spectrometry using immunoprecipitation. We first established the expression of c-Raf, RB, and pS608 RB, the hinge region phosphorylation that betrays E2F dissociation, in the nucleus of ATRA treated or untreated cells using Western blotting. Nuclear lysates were isolated from cells that were untreated or ATRA-treated for 72 h. We reported that ATRA-induced differentiation is driven by a sustained activation of MAPK signal associated with the unanticipated enrichment of c-Raf in the nucleus detected by imaging cytometry [19,20,21]. After 72 h of ATRA treatment, Western blotting results showed that there was more c-Raf in the nucleus (Fig. 4A). At the same time ATRA caused downregulation of total RB and diminished the amount of pS608-RB (Fig. 4B & C. Fig. 4D is the nuclear loading control.). Immunoprecipitation of c-Raf followed by immunoblotting showed that nuclear c-Raf complexed with RB and specifically with pS608 RB. ATRA treatment reduced the amount of RB and pS608 RB bound with c-Raf (Fig. 4E-G). This ATRA-induced reduction in c-Raf bound to RB and RB hypo-phosphorylation [28] is associated with cell cycle arrest as shown by flow cytometric DNA histograms (Fig. 4H). Hence it appears that the ATRA-induced liberation of Raf sequestered on RB associated with cell cycle arrest frees more c-Raf for the pro-differentiation targets indicated above.
Nuclear c-Raf interacts with SWI/SNF complex protein SMARCD1
We sought confirmation of the novel ATRA-regulated c-Raf interaction with SMARCD1 in the nucleus detected by mass spectrometry using co-immunoprecipitation. SMARCD1 is the pioneering subunit of the SWI/SNF chromatin remodeling machine. SMARCD1 regulates the transcriptional activation or repression of cellular genes by chromatin remodeling [40]. Cells were untreated or ATRA-treated for 72 h and nuclear lysates were generated. Western blotting showed a slight, but not significant, downregulation of SMARCD1 in response to ATRA. (Fig. 5A. Fig. 5B is the TBP nuclear protein loading control). Immunoprecipitation revealed that nuclear c-Raf complexed with SMARCD1. ATRA diminished the interaction of SMARCD1 and c-Raf in the nucleus (Fig. 5c & D). Hence c-Raf interacted with another prominent cell cycle regulator and that interaction was ATRA-regulated. The SWI/SNF machine is known to bind RB [41], too, suggesting that c-Raf is interacting with prominent components of chromatin that govern cell cycle and differentiation.
Fig. 5.
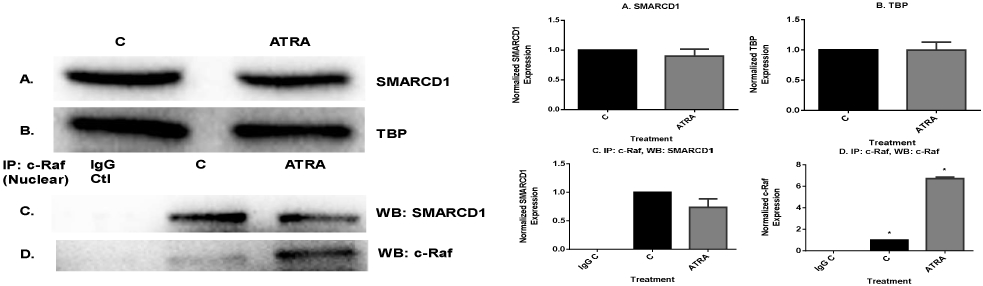
Nuclear c-Raf interacts with SWI/SNF complex protein, SMARCD1. A. Western blots of nuclear lysate showed that ATRA diminished SMARCD1 level slightly. B. TATA binding protein (TBP) was the loading control. C & D. Co-immunoprecipitation results show that ATRA diminished nuclear c-Raf interaction with SMARCD1 protein. An equal amount of pre-cleared nuclear lysate collected after 72 h of ATRA treatment were incubated overnight with 2.5 μg of the precipitating antibody with magnetic beads and resolved on 12% polyacrylamide gels. Blots were probed for SMARCD1 and c-Raf, control to confirm c-Raf was immunoprecipitated.
Discussion:
All-trans retinoic acid therapy has been used successfully in the treatment of the APL subtype of AML but has been largely unsuccessful for AML in general. Combination therapy has been considered an option to make differentiation therapy a reality for these other AML and the cases of resistance in APL. Combination therapy, however, requires mechanistic insight into targets that could be therapeutically exploited with ATRA. Around 90% of newly diagnosed APL patients achieve complete remission with differentiation therapy using ATRA [42,43]. However, 10% of the patients develop resistance to treatment and have been unresponsive to ATRA. Here, we attempted to increase our understanding of signaling related to the novel finding of c-Raf in the nucleus. In ATRA treated HL-60 AML cells, nuclear c-Raf was enriched and interacted with several novel partners. We have reported that nuclear c-Raf targets potentially significant regulatory proteins, namely retinoblastoma (RB), NFATc3 and GSK-3, which regulate the transcription of various genes and play a pivotal role in driving differentiation [13,22,44]. Knowing there were such targets made it important to illuminate pathways that were implicated, and we undertook a search for the rest of the partners to identify and characterize these new nuclear regulatory pathways. We expressed a c-Raf-flag protein in HEK293T stable transfectants, extracted the recombinant protein and trolled lysates from HL-60 cells that were either treated or untreated with ATRA. Mass spectrometry was then used to identify the c-Raf partners. After screening using Venn analysis software for partners expressed in ATRA-treated cells that were also ATRA regulated, we found 19 potential c-Raf partners that were regulated during ATRA treatment. The highest ranked of these nuclear c-Raf partners, cyclin-dependent kinase 2 (Cdk2), Retinoblastoma protein 1 (RB1), and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 1 (SMARCD1), were selected for further studies to see if these have roles in ATRA-induced myeloid cell differentiation.
These nuclear c-Raf targets are functionally related. Cdk2 is activated by Cyclin E in the Cyclin E-Cdk2 complex to drive the G1/S transition of the cell cycle by controlling RB phosphorylation in a classical paradigm of cell cycle regulation. G1/G0 cell cycle arrest involves the coordinate regulation of signals that negatively control cell cycle machinery and inhibit the G1/S transition. Growth arrest associated with cell differentiation could ergo be driven by several mechanisms including the downregulation of cyclins or upregulation of cyclin-dependent kinase inhibitors (CKIs) or both [45]. ATRA causes G1/G0 cell cycle arrest of HL-60 AML cells [28,46] and the present study shows that this reflects reduced levels of cyclin E1, down-regulation of CDK2, pY15CDK2 and pT160CDK2 and dephosphorylation of RB tumor suppressor protein. Expression of p27 Kip1 also increased in cells treated with ATRA. The reduction in cyclin E1 levels likely contributes to increased p27 Kip1 stability after ATRA treatment because cyclin E-CDK2 complexes can target p27 Kip1 for degradation through phosphorylation on Thr187 [47,48]. An increase in hypophosphorylated RB protein seen in cells treated with ATRA, was seen too for RARα plus RXR selective non-metabolizable retinoids, pointing to the significance of RARα as a driver of ATRA-induced differentiation [28]. We observe that this hypophosphorylation is associated with loss of pS608-RB. During cell cycle progression, cyclin E-CDK2 complexes phosphorylate RB protein late in the G1 phase, thereby preventing E2F binding [49,50]. Hypophosphorylated RB protein could potentially prevent entry into S phase and establish an irreversible cell cycle exit in the terminally differentiated cells [45]. This result relates to the earlier data above to suggest that c-Raf mediates a coupling between growth arrest and differentiation. c-Raf bound the S608 phosphorylated RB, which does not bind the G1-S cell cycle promoting transcription factor E2F. ATRA-induced upregulation of p27 Kip1 CDKI with downregulation of Cdk2 culminating in loss of pS608 RB causes RB to sequester E2F and divorce sequestered c-Raf. This frees the c-Raf in the nucleus from sequestration by RB and changes stoichiometry to favor other targets such as those that drive differentiation, for example NFATc3 [13]. Our data now suggest that ATRA also uses c-Raf to regulate differentiation via Cdk2. We found a novel c-Raf-Cdk2 (pY15Cdk2 and pT160Cdk2) complex in the nucleus which diminished after ATRA treatment. Phosphorylation at Y15 is associated with Cdk2 inhibition while phosphorylation at T160 is associated with activation, hence it is enigmatic that ATRA caused downregulation of both forms. Immunoprecipitation showed that ATRA treatment causes c-Raf bound to Cdk2 to divorce. More Cdk2 becomes available for binding to RARα, and more targets of RARα regulation increase expression. At the same time, c-Raf freed from the arguably pro-proliferation target (Cdk2) and target pro-differentiation targets such as NFATc3 to enable RARE activation [13]. Hence ATRA results in c-Raf shifting from pro-proliferation to pro-differentiation partners. An emerging prominent nuclear function of c-Raf appears to be to enable RARα to work better to drive differentiation.
SMARCD1 (also known as BAF60a), a member of SWI/SNF (switch/sucrose non-fermentable) chromatin remodeling complex family, has been known to interact with transcription factors, including RB, to govern changes in local chromatin structure and transcriptional activation [51]. Our finding revealed c-Raf in the nucleus interacts with SWI/SNF complex protein SMARCD1. ATRA treatment dissociated the interaction between c-Raf and SMARCD1. SMARCD1 recruits various transcription factors (TFs) to the SWI/SNF chromatin remodeling complex [52,53,54]. c-Raf in the nucleus thus interacts with a group of associated regulatory molecules that control cell proliferation and differentiation.
In conclusion, the current study showed that ATRA induces the enrichment of c-Raf in the nucleus where it regulates the interaction with several targets that have prominent roles in controlling cell cycle and differentiation (Fig. 6). We found evidence suggesting new nuclear signaling pathways that use traditionally cytoplasmic signaling molecules imported to the nucleus by ATRA. One is a novel nuclear Raf/Cdk2/RARα axis. These novel nuclear pathways illuminate novel targets for therapeutic intervention in cancer differentiation therapy.
Fig. 6.

Schematic diagram of ATRA-induced nuclear translocation of c-Raf and its interaction with prominent regulators of cell proliferation/arrest and differentiation.
Acknowledgments
This work was supported in part by NIH (RO1 CA152870) (AY) and by Guangdong Provincial Science and Technology Project (2017B020226001), Basic Research Projects of Shenzhen Knowledge Innovation Program (JCYJ20170818095453642 and JCYJ20180307123759162) and General Research Fund of Hong Kong Research Grant Council (CityU_11303815) (M.Y). Authors are also grateful to Dr. John Babish and Paracelsian for generous support.
Footnotes
Conflict of interest
Authors declare that they have no conflicts of interest with the contents of this article.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Riester M, Stephan-Otto Attolini C, Downey RJ, Singer S & Michor F. (2010). A differentiation-based phylogeny of cancer subtypes. PLoS Comput. Biol 6 (5). e1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thuler LCS & Pombo-de-Oliveira MS. (2016). Acute promyelocytic leukaemia is highly frequent among acute myeloid leukaemias in Brazil: a hospital-based cancer registry study from 2001 to 2012. Ann. Hematol 96 (3). 1–8. [DOI] [PubMed] [Google Scholar]
- 3.Iriyama N, Yoshino Y, Yuan B, Horikoshi A, Hirabayashi Y, Hatta Y, Toyoda H & Takeuchi J. (2012). Speciation of arsenic trioxide metabolites in peripheral blood and bone marrow from an acute promyelocytic leukemia patient. J Hematol Oncol. 5:1. doi: 10.1186/1756-8722-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo-Coco F & Hasan SK. (2014). Understanding the molecular pathogenesis of acute promyelocytic leukemia. Best Pract Res Clin Haematol. 27(1):3–9. [DOI] [PubMed] [Google Scholar]
- 5.de Thé H, Le Bras M & Lallemand-Breitenbach V. (2012). The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J Cell Biol. 198(1): 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins SJ, Gallo RC & Gallagher RE. (1977). Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 270(5635):347–349. [DOI] [PubMed] [Google Scholar]
- 7.Bunaciu RP, MacDonald RJ, Gao F, Johnson LM, Varner JD, Wang X, Nataraj S, Guzman ML & Yen A. (2018). Potential for subsets of wt-NPM1 primary AML blasts to respond to retinoic acid treatment. Oncotarget. 9(3):4134–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breitman TR, Selonick SE & Collins SJ. (1980). Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A. 77(5):2936–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yen A (1990). HL-60 cells as a model of growth control and differentiation: The significance of variant cells. Hematol Rev. 4(1); 5–46. [Google Scholar]
- 10.Vitaliano-Prunier A, Halftermeyer J, Ablain J, de Reynies A, Peres L, Le Bras M, Metzger D, & de Thé H. (2014). Clearance of PML/RARA-bound promoters suffice to initiate APL differentiation. Blood. 124 (25). 3772–3780. [DOI] [PubMed] [Google Scholar]
- 11.Gianni M, Fratelli M, Bolis M, Kurosaki M, Zanetti A, Paroni G, Rambaldi A, Borleri G, Rochette-Egly C, Terao M & Garattini E. (2017). RARalpha2 and PML-RAR similarities in the control of basal and retinoic acid induced myeloid maturation of acute myeloid leukemia cells. Oncotarget. 8(23):37041–37060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rochette-Egly C & Germain P. (2009). Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl Recept Signal. 7: e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geil WM & Yen A. (2014). Nuclear Raf-1 kinase regulates the CXCR5 promoter by associating with NFATc3 to drive retinoic acid-induced leukemic cell differentiation. FEBS J. 281(4):1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J & Yen A. (2004). A novel retinoic acid-responsive element regulates retinoic acid-induced BLR1 expression. Mol Cell Biol. 24(6):2423–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, & Kolch W. (2011). Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2(3): 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desideri E, Cavallo AL & Baccarini M. (2015). Alike but different: RAF paralogs and their signaling outputs. Cell. 161(5): 967–970. [DOI] [PubMed] [Google Scholar]
- 17.McKay MM & Morrison DK. (2007). Integrating signals from RTKs to ERK/MAPK. Oncogene. 26(22): 3113–3121. [DOI] [PubMed] [Google Scholar]
- 18.Schneider T, Martinez-Martinez A, Cubillos-Rojas M, Bartrons R, Ventura F & Rosa JL. (2018). The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget. 9(59):31531–31548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong HY, Varvayanis S & Yen A. (2001). Retinoic acid causes MEK-dependent RAF phosphorylation through RAR alpha plus RXR activation in HL-60 cells. Differentiation. 68(1): 55–66. [DOI] [PubMed] [Google Scholar]
- 20.Wang J & Yen A. (2008). A MAPK-positive feedback mechanism for BLR1 signaling propels retinoic acid-triggered differentiation and cell cycle arrest. J Biol Chem. 283(7):4375–4386. [DOI] [PubMed] [Google Scholar]
- 21.Smith J, Bunaciu RP, Reiterer G, Coder D, George T, Asaly M & Yen A. (2009). Retinoic acid induces nuclear accumulation of Raf1 during differentiation of HL-60 cells. Exp Cell Res. 315(13):2241–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace AS, Supnick HT, Bunaciu RP & Yen A. (2016). RRD-251 enhances all-trans retinoic acid (RA)-induced differentiation of HL-60 myeloblastic leukemia cells. Oncotarget. 7(29):46401–46418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dasgupta P, Sun J, Wang S, Fusaro G, Betts V, Padmanabhan J, Sebti SM & Chellappan SP. (2004). Disruption of the Rb-Raf-1 interaction inhibits tumor growth and angiogenesis. Mol Cell Biol. 24(21):9527–9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen A & Pardee AB. (1978). Exponential 3T3 cells escape in mid-G1 from their high serum requirement. Exp Cell Res. 116(1):103–13. [DOI] [PubMed] [Google Scholar]
- 25.Burke JR, Deshong AJ, Pelton JG & Rubin SM. (2010). Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem. 285(21):16286–16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikeda MA, Jakoi L & Nevins JR. (1996). A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A. 93(8):3215–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta K, Stefan T, Ignatz-Hoover J, Moreton S, Parizher G, Saunthararajah Y & Wald DN. (2016). GSK-3 Inhibition Sensitizes Acute Myeloid Leukemia Cells to 1,25D-Mediated Differentiation. Cancer Res. 76(9):2743–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks SC 3rd, Kazmer S, Levin AA & Yen A. (1996). Myeloid differentiation and retinoblastoma phosphorylation changes in HL-60 cells induced by retinoic acid receptor- and retinoid X receptor-selective retinoic acid analogs. Blood. 87(1):227–237. [PubMed] [Google Scholar]
- 29.Shen Y, Liu P, Jiang T, Hu Y, Au FKC & Qi RZ. (2017). The catalytic subunit of DNA polymerase δ inhibits γTuRC activity and regulates Golgi-derived microtubules. Nat Commun. 8(1):554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin SJ, Newmeyer DD, Mathias S, Farschon DM, Wang HG, Reed JC, Kolesnick RN & Green DR. (1995). Cell-free reconstitution of Fas-, UV radiation- and ceramide-induced apoptosis. EMBO J. 14(21):5191–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang R, Huang M, Li L, Kaneko T, Voss C, Zhang L, Xia J & Li SSC. (2018). Affinity Purification of Methyllysine Proteome by Site-Specific Covalent Conjugation. Anal Chem. 90(23):13876–13881. [DOI] [PubMed] [Google Scholar]
- 32.Shen M & Yen A. (2008). c-Cbl interacts with CD38 and promotes retinoic acid-induced differentiation and G0 arrest of human myeloblastic leukemia cells. Cancer Res. 68(21):8761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leicht DT, Balan V, Zhu J, Kaplun A, Bronisz A, Rana A & Tzivion G. (2013). MEK-1 activates C-Raf through a Ras-independent mechanism. Biochim Biophys Acta. 1833(5):976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kahl M, Brioli A, Bens M, Perner F, Kresinsky A, Schnetzke U, Hinze A, Sbirkov Y, Stengel S, Simonetti G, Martinelli G, Petrie K, Zelent A, Böhmer FD, Groth M, Ernst T, Heidel FH, Scholl S, Hochhaus A & Schenk T. (2019). The acetyltransferase GCN5 maintains ATRA-resistance in non-APL AML. Leukemia. 33(11):2628–2639. [DOI] [PubMed] [Google Scholar]
- 35.Lamkin TJ, Chin V, Varvayanis S, Smith JL, Sramkoski RM, Jacobberger JW & Yen A. (2006). Retinoic acid-induced CD38 expression in HL-60 myeloblastic leukemia cells regulates cell differentiation or viability depending on expression levels. J Cell Biochem. 97(6):1328–38. [DOI] [PubMed] [Google Scholar]
- 36.Shen M & Yen A. (2009). c-Cbl tyrosine kinase-binding domain mutant G306E abolishes the interaction of c-Cbl with CD38 and fails to promote retinoic acid-induced cell differentiation and G0 arrest. J Biol Chem. 284(38):25664–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Congleton J, Jiang H, Malavasi F, Lin H & Yen A. (2011). ATRA-induced HL-60 myeloid leukemia cell differentiation depends on the CD38 cytosolic tail needed for membrane localization, but CD38 enzymatic activity is unnecessary. Exp Cell Res. 317(7):910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kishimoto H, Hoshino S, Ohori M, Kontani K, Nishina H, Suzawa M, Kato S & Katada T. (1998). Molecular mechanism of human CD38 gene expression by retinoic acid. Identification of retinoic acid response element in the first intron. J Biol Chem. 1998 273(25):15429–15434. [DOI] [PubMed] [Google Scholar]
- 39.Hickstein DD, Baker DM, Gollahon KA & Back AL. (1992). Identification of the promoter of the myelomonocytic leukocyte integrin CD11b. Proc Natl Acad Sci U S A. 89(6):2105–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito T, Yamauchi M, Nishina M, Yamamichi N, Mizutani T, Ui M, Murakami M & Iba H. (2001). Identification of SWI.SNF complex subunit BAF60a as a determinant of the transactivation potential of Fos/Jun dimers. J Biol Chem. 276(4):2852–2857. [DOI] [PubMed] [Google Scholar]
- 41.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, Wan C, Vlasblom J, Dar VU, Bezginov A, Clark GW, Wu GC, Wodak SJ, Tillier ER, Paccanaro A, Marcotte EM & Emili A. (2012). A census of human soluble protein complexes. Cell. 150(5):1068–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, Specchia G, Sica S, Divona M, Levis A, Fiedler W, Cerqui E, Breccia M, Fioritoni G, Salih HR, Cazzola M, Melillo L, Carella AM, Brandts CH, Morra E, von Lilienfeld-Toal M, Hertenstein B, Wattad M, Lübbert M, Hänel M, Schmitz N, Link H, Kropp MG, Rambaldi A, La Nasa G, Luppi M, Ciceri F, Finizio O, Venditti A, Fabbiano F, Döhner K, Sauer M, Ganser A, Amadori S, Mandelli F, Döhner H, Ehninger G, Schlenk RF & Platzbecker U. (2013). Gruppo Italiano Malattie Ematologiche dell'Adulto; German-Austrian Acute Myeloid Leukemia Study Group; Study Alliance Leukemia. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 369(2):111–121. [DOI] [PubMed] [Google Scholar]
- 43.Jalili M, Salehzadeh-Yazdi A, Mohammadi S, Yaghmaie M, Ghavamzadeh A & Alimoghaddam K. (2017). Meta-Analysis of Gene Expression Profiles in Acute Promyelocytic Leukemia Reveals Involved Pathways. Int J Hematol Oncol Stem Cell Res. 11(1):1–12. [PMC free article] [PubMed] [Google Scholar]
- 44.Supnick HT, Bunaciu RP & Yen A. (2018). The c-Raf modulator RRD-251 enhances nuclear c-Raf/GSK-3/VDR axis signaling and augments 1,25-dihydroxyvitamin D3-induced differentiation of HL-60 myeloblastic leukemia cells. Oncotarget. 9(11):9808–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dimberg A, Bahram F, Karlberg I, Larsson LG, Nilsson K & Oberg F. (2002). Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1). Blood. 99(6):2199–2206. [DOI] [PubMed] [Google Scholar]
- 46.Congleton J, MacDonald R & Yen A. (2012). Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 26(6):1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheaff RJ, Groudine M, Gordon M, Roberts JM & Clurman BE. (1997). Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11(11):1464–1478. [DOI] [PubMed] [Google Scholar]
- 48.Vlach J, Hennecke S & Amati B. (1997). Phosphorylation dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 16(17): 5334–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dyson N (1998). The regulation of E2F by pRB-family proteins. Genes Dev. 12(15): 2245–2262. [DOI] [PubMed] [Google Scholar]
- 50.Lundberg AS & Weinberg RA. (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 18(2):753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang P, Li L, Bao Z & Huang F. (2016). Role of BAF60a/BAF60c in chromatin remodeling and hepatic lipid metabolism. Nutr Metab (Lond). 13:30. doi: 10.1186/s12986-016-0090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L, Fulcoli FG, Ferrentino R, Martucciello S, Illingworth EA & Baldini A. (2012). Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 8(3): e1002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh J, Sohn DH, Ko M, Chung H, Jeon SH & Seong RH. (2008). BAF60a interacts with p53 to recruit the SWI/SNF complex. J Biol Chem. 283(18):11924–11934. [DOI] [PubMed] [Google Scholar]
- 54.Hsiao PW, Fryer CJ, Trotter KW, Wang W & Archer TK. (2003). BAF60a mediates critical interactions between nuclear receptors and the BRG1 chromatin-remodeling complex for transactivation. Mol Cell Biol. 23(17):6210–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]