

Abstract
Signaling by the kinase cascade composed of Raf, MEK, and ERK is critical for animal development and is often inappropriately activated in human malignancies. We sought to identify factors that control signaling mediated by the Caenorhabditis elegans Raf ortholog LIN-45. A genetic screen showed that the degradation of LIN-45 required the E3/E4 ubiquitin ligase UFD-2. Both UFD-2 and its partner, the ATP-dependent segregase CDC-48, were required for the developmental regulation of LIN-45 protein abundance. We showed that UFD-2 acted in the same pathway as the E3 ubiquitin ligase SCFSEL−10 to decrease LIN-45 abundance in cells in which Raf-MEK-ERK signaling was most highly active. UFD-2 also reduced the protein abundance of activated LIN-45 carrying a mutation equivalent to the cancer-associated BRAF(V600E) variant. Our structure-function studies showed that the disruption of LIN-45 domains that mediate protein-protein interactions, including the conserved cysteine-rich domain and 14–3-3–binding motifs, were required for UFD-2–independent degradation of LIN-45. We propose a model in which UFD-2 and CDC-48 act downstream of SCFSEL−10 to remove LIN-45 from its protein interaction partners and facilitate proteasomal targeting and degradation. These findings imply that UFD-2 and CDC-48 may be important for Raf degradation during normal and oncogenic Ras and MAPK signaling in mammalian cells.
One-Sentence Summary:
A pathway that degrades wild-type and mutant forms of the C. elegans Raf ortholog LIN-45 is identified.
INTRODUCTION
The Raf family of protein kinases are effectors of Ras signaling that are stimulated by GTP-bound Ras (1). Raf directly phosphorylates the kinase MEK, which in turn phosphorylates the MAP kinase ERK. In humans, BRAF, RAF1, and ARAF are Raf family members, while in the nematode Caenorhabditis elegans, LIN-45 is the sole ortholog (2). Raf proteins share an N-terminal Ras-binding domain (RBD), a cysteine-rich domain (CRD), an intermediate “hinge” region, and a C-terminal kinase domain. Inactive Raf is a monomer and found in the cytosol in an autoinhibited conformation imposed by interactions between the CRD and 14–3-3 proteins bound to the hinge and C-terminal regions (3). Raf activation is stimulated by GTP-Ras binding to the RBD, which relieves it from its autoinhibited conformation and recruits it to the plasma membrane. In this state, Raf proteins dimerize through their kinase domains, which enables their autophosphorylation and activation (3, 4). A major mechanism of Raf inactivation involves negative feedback through direct phosphorylation by ERK, resulting in hyperphosphorylated, monomeric, and signaling-incompetent Raf (5). The ERK phosphorylation site T401 in BRAF (T432 in C. elegans) is required for negative feedback (6, 7). This site is necessary for the function of a Cdc4-phosphodegron (CPD) motif, which is recognized by the Skp-Cullin-F-box (SCF) E3 ubiquitin ligase SCFFBXW7 (termed SCFSEL−10 in C. elegans and this work) (8, 9).
Dysregulated Raf activation is considered a driver event in melanomas, in which the activating mutation BRAF(V600E) is commonly found in both malignant melanomas and benign, melanocytic nevi (10). Despite constitutive kinase activation of BRAF(V600E) protein (11), increased ERK activity is rarely found in nevi carrying the BRAF(V600E) mutation, while it is more common in BRAF(V600E) melanomas (12–14). These results suggest that malignant transformation may involve losses to negative regulation of BRAF(V600E) signaling.
In C. elegans, Raf signaling impacts multiple developmental and physiological processes (15); one example is the patterning of cell fates in the hermaphrodite vulva, which involves the specification of six vulval precursor cells (VPCs) of epithelial origin, termed P3.p-P8.p (16). During the L2 larval stage, production of an EGF-like inductive signal by the Anchor Cell (AC) stimulates EGF receptor (EGFR), Ras, and Raf-MEK-ERK signaling in the nearest VPC, termed P6.p (17). In the L3 stage, activation of the C. elegans ERK ortholog MPK-1 in P6.p promotes a primary (1º) vulval cell fate. In neighboring cells P5.p and P7.p, activation of Notch and Ral promote an alternative secondary (2º) cell fate. Signaling by MPK-1 and Notch mutually inhibit one another (18), which results in a reproducible pattern of 1º and 2º cell fates and ensures the formation of one vulva. Mutations that hyperactivate Ras or Raf signaling result in formation of more than one vulva, or the Multivulva (Muv) phenotype (19).
Using C. elegans, we exploited the easily screened Muv phenotype to discover negative regulators of an activated LIN-45 modeled on BRAF(V600E) and identified the highly conserved E3/E4 ubiquitin ligase UFD-2. We showed that mutants lacking ufd-2 enhance the effects of the activated mutant of LIN-45, manifested as elevated MPK-1 activity and developmental fate transformation in VPCs. We showed that loss of UFD-2 resulted in increased LIN-45 protein abundance in VPCs where MPK-1 is active, and that this regulation is post-transcriptional. Our findings implicate UFD-2 as a component of the previously described MPK-1 and SCFSEL−10-dependent negative feedback reducing LIN-45 protein abundance. Using RNAi knockdown, we showed that LIN-45 protein abundance was also regulated by CDC-48, the C. elegans ortholog of human VCP, a conserved ATPase that unfolds and extracts protein substrates from large structures such as protein complexes and membranes. We hypothesized that UFD-2 acts on LIN-45 present in multiprotein complexes. We screened for mutants of LIN-45 that abolished the requirement for UFD-2 in LIN-45 protein degradation. We identified mutations in the conserved CRD, and in the phosphorylation-regulated 14–3-3 binding sites, that bypass the requirement for UFD-2. Because these mutations disrupt known 14–3-3 interactions, we propose a model that UFD-2 facilitates proteasome targeting and degradation by removing LIN-45 from multiprotein complexes with 14–3-3 proteins.
RESULTS
Isolation of ufd-2 mutant as an enhancer of lin-45(V627E)
To understand the cellular and molecular mechanisms that control signaling by BRAF(V600E), we generated C. elegans strains carrying integrated, single-copy transgenes expressing the equivalent mutation, lin-45(V627E), in the conserved Raf activation segment (Fig. S1A). A small proportion of adult hermaphrodites carrying lin-45(V627E) transgenes displayed the Muv phenotype; however, the majority were wild-type in appearance (Fig. 1A). In a forward genetic screen, we isolated a recessive mutant that displayed a highly penetrant Muv phenotype in combination with lin-45(V627E) (Fig. S1B). We mapped and sequenced this mutation and determined that this strain carried a nonsense mutation in ufd-2 (Fig. S1C,D). UFD-2 is the sole C. elegans ortholog of human UBE4E, a U-box protein with E3/E4 ubiquitin ligase activities (20, 21).
Fig. 1: UFD-2 is a negative regulator of LIN-45.
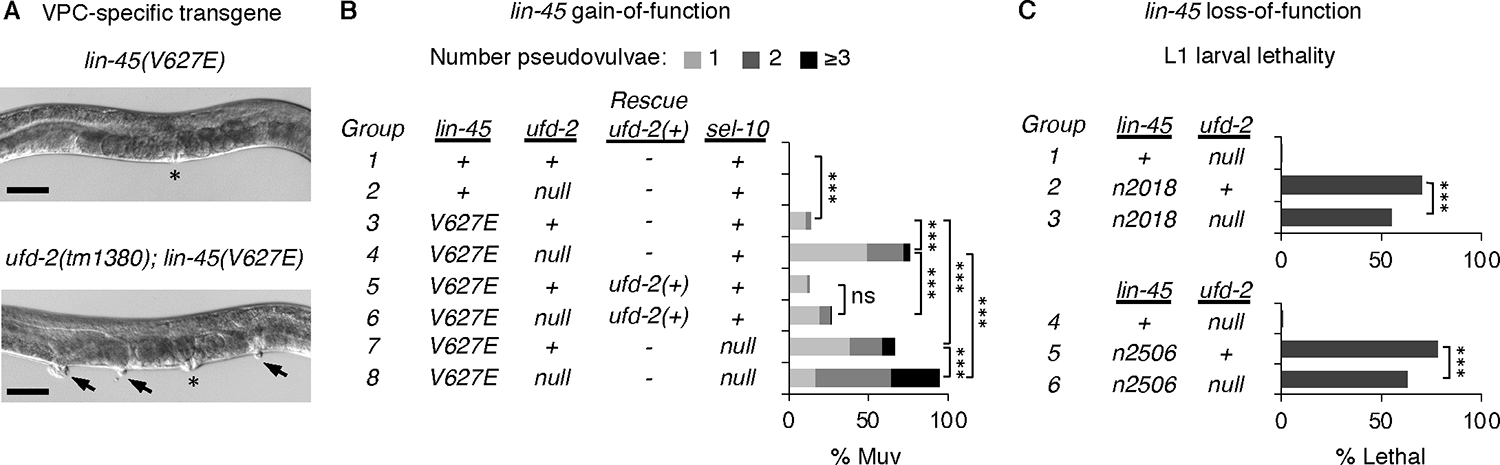
A) Top: adult hermaphrodite that expresses lin-45(V627E). Bottom: Multivulva (Muv) phenotype of ufd-2(tm1380); lin-45(V627E) adult. Asterisk indicates location of vulva; arrowheads indicate pseudovulvae. Scale bar, 50 μm. B) Muv phenotype, shown as percentage of total adults. Genotypes of groups labeled 1–8 are indicated in the table. Genotypes carrying a VPC-specific transgene expressing wild-type ufd-2(+) are labeled Rescue ufd-2(+). The numbers of adults scored (n) listed by group are: 1 (n=88), 2 (n=114), 3 (n=184), 4 (n=199), 5 (n=120), 6 (n=111), 7 (n=139), 8 (n=142). C) L1 larval lethality, shown as percentage of total larvae. Genotypes of groups labeled 1–6 are indicated in the table. The numbers of larvae scored (n) listed by group are: 1 (n=459), 2 (n=1159), 3 (n=880), 4 (n=397), 5 (n=531), 6 (n=434). For all panels, comparisons were made using the Fisher’s exact test. ns not significant, ***P<0.0001.
To confirm the role of ufd-2, we examined mutants carrying the null allele ufd-2(tm1380) (22), which we refer to as ufd-2(null) throughout this work. For our analysis, we used a lin-45(V627E) transgene that caused the Muv phenotype in 14% of adults (Fig. 1B). ufd-2(null) mutants were not Muv in the presence of lin-45(+); however, ufd-2(null) mutants carrying the lin-45(V627E) transgene displayed a highly penetrant Muv phenotype affecting 76% of adults (Fig. 1A–B, Table S1). In ufd-2(null) mutants, the penetrance was significantly reduced by the introduction of a single-copy wild-type ufd-2(+) transgene expressed specifically in VPCs (Fig. 1B), confirming that ufd-2 status determines the Muv phenotype seen in animals expressing lin-45(V627E). As previously reported (9), the Muv phenotype of lin-45(V627E) animals was also enhanced by the null sel-10(ok1632) mutation, referred to here as sel-10(null) (Fig. 1B). In animals lacking both ufd-2 and sel-10, penetrance of the Muv phenotype in lin-45(V627E) animals was significantly increased compared to either single mutant (Fig. 1B), suggesting that UFD-2 and SCFSEL−10 function in parallel for some roles.
Our results using lin-45(V627E) suggest that UFD-2 functions to inhibit LIN-45 activity. As a negative regulator, we expect that ufd-2 loss would also suppress phenotypes resulting from reduced LIN-45 activity. We examined two alleles of lin-45, termed n2018 and n2506, that alter the LIN-45 RBD and result in partial loss of function and lethality at the L1 larval stage caused by failure of duct cell specification (23, 24). Compared to either lin-45 loss-of-function mutant alone, the ufd-2(null); lin-45 double mutants displayed significantly reduced lethality (Fig. 1C, Table S2). These results support a role for UFD-2 in negatively regulating LIN-45 and exclude the possibility that its regulation is limited to vulval induction.
UFD-2 negatively regulates Raf-MEK-ERK signaling in VPCs
In VPCs, patterned fate specification is initiated by an EGF-like/LIN-3 inductive signal from the AC (19). Consequently, activation of the Raf-MEK-ERK orthologs LIN-45, MEK-2, and MPK-1 occurs in P6.p (Fig. 2A). To test whether ufd-2 impacts MPK-1 activity, we used ERK-KTR, a fluorescent Kinase Translocation Reporter (KTR) (25). In the presence of active MPK-1, ERK-KTR is localized to the cytoplasm; in the absence of MPK-1 activity, ERK-KTR accumulates in the nucleus. This subcellular distribution can be expressed as a ratio of Cytoplasm/Nucleus (C/N) intensity. As we previously reported (17), by the early L3 stage, ERK-KTR was more cytoplasmic in P6.p compared to P4.p, P5.p, P7.p, and P8.p (Fig. 2B). A comparison of the ERK-KTR C/N ratio in each VPC of ufd-2(null) mutants and ufd-2(+) controls showed no significant difference, indicating that loss of UFD-2 alone did not affect MPK-1 activation (Fig. 2C, Table S3). However, in animals expressing lin-45(V627E), the C/N ratio observed in ufd-2(null) mutants was significantly increased compared to ufd-2(+) controls in P4.p, P5.p, P7.p, and P8.p (Fig. 2D, Table S3). These results show that loss of ufd-2 function results in greater MPK-1 activation in the presence of LIN-45(V627E). Because MPK-1 activation results from LIN-45 and MEK-2 signaling, this result is consistent with a role for UFD-2 in regulating MPK-1, either directly or indirectly through LIN-45 or MEK-2.
Fig. 2: UFD-2 negatively regulates LIN-45 signaling.
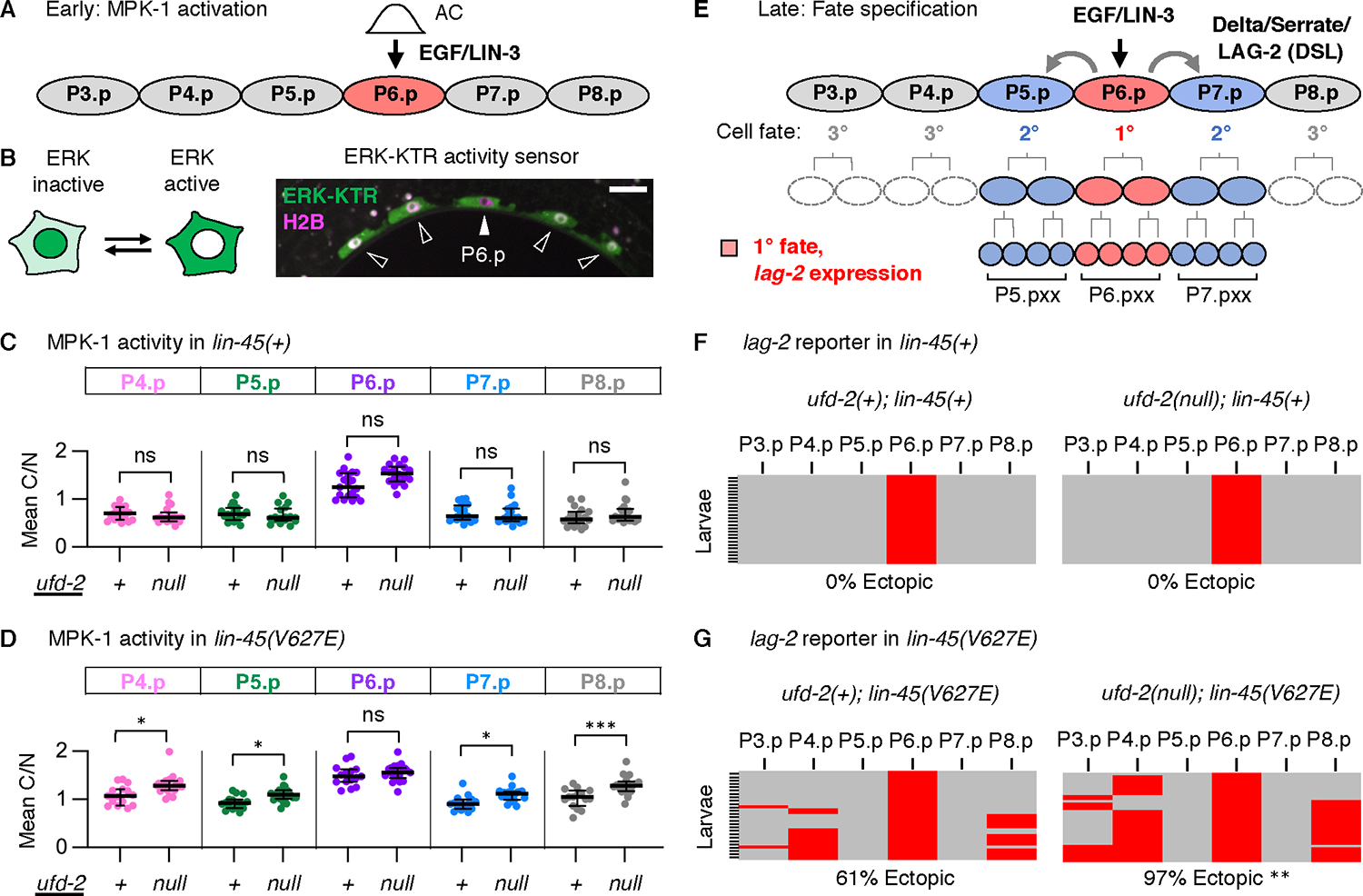
A) A schematic of early VPC fate patterning, initiated at the L2 larval stage by activation of MPK-1 in P6.p. B) The ERK-KTR accumulates in the nucleus when MPK-1 is inactive and the cytoplasm when MPK-1 is active. ERK-KTR (green) localization in VPCs (arrows) of a representative wild-type L2 stage larva. The histone H2B (magenta) was used as a nuclear marker. Scale bar, 10 μm. C-D) All values represent a ratio of ERK-KTR intensity in Cytoplasm/Nucleus (C/N) for individual cells. C) MPK-1 activity in lin-45(+). The numbers of larvae scored (n) by genotype were: ufd-2(+); lin-45(+) (n=19) and ufd-2(null); lin-45(+) (n=18). D) MPK-1 activity in lin-45(V627E). The numbers of larvae scored (n) by genotype were: ufd-2(+); lin-45(V627E) (n=15) and ufd-2(null); lin-45(V627E) (n=19). Groups were compared using a one-way ANOVA followed by Šídák’s multiple comparisons test. ns not significant, *P <0.01, ***P <0.0001. E) At the L3 larval stage, MPK-1 signaling in P6.p results in 1° fate (red). Expression of lag-2 in P6.p promotes 2° fate in P5.p and P7.p (blue). F-G) All panels show expression of a lag-2 reporter in L3 stage VPCs, where expression is positive (red) or negative (gray) for descendants of VPCs (columns) for multiple larvae (rows). The percentage of larvae displaying expression in a VPC other than P6.p is shown (% Ectopic). The numbers of larvae scored (n) by genotype were: ufd-2(+); lin-45(+) (n=37), ufd-2(null); lin-45(+) (n=36), ufd-2(+); lin-45(V627E) (n=31), or ufd-2(null); lin-45(V627E) (n=36). The ufd-2(null); lin-45(V627E) and ufd-2(+); lin-45(V627E) genotypes were compared using a Fisher’s exact test. **P <0.001.
In VPCs, activated MPK-1 phosphorylates the Elk1 ortholog LIN-1 (26), resulting in 1º cell fate through the derepression of LIN-1 target genes, including the Delta-like gene lag-2 (27) (Fig. 2E). In developing larvae, expression of lag-2 can be monitored using arIs222, a reporter that contains a lag-2 promoter fragment and drives expression of 2xNLS-tagRFP exclusively in P6.p, as well as its descendants after fate commitment, cells we term “P6.pxx” (Fig. 2E) (28). Additionally, this reporter is expressed ectopically in VPCs other than P6.p in genotypes with increased Raf-MEK-ERK signaling (29). In the presence of lin-45(+), we did not observe a change in the P6.pxx expression pattern in ufd-2(null) mutants (Fig. 2F). However, in animals expressing lin-45(V627E), the frequency of ectopic lag-2 reporter expression in ufd-2(null) mutants was significantly increased compared to ufd-2(+) controls (Fig. 2G, Table S4).
UFD-2 negatively regulates LIN-45 protein abundance
We previously showed that LIN-45 protein is downregulated in VPCs by a mechanism involving SCFSEL−10 (9). Because it has E3/E4 ubiquitin ligase activities, we reasoned that UFD-2 may be required in this process. To test whether ufd-2 loss impacts LIN-45 protein abundance, we generated N- and C-terminal GFP-3xFLAG knock-in alleles of lin-45 (Fig. S2A–D) and examined the expression of tagged protein in P5.pxx, P6.pxx, and P7.pxx cells. When tagged at either the N-terminus or C-terminus, GFP-LIN-45 protein is evident in P5.pxx and P7.pxx and reduced in P6.pxx (Fig. 3A, Fig. S2D). Increased accumulation of GFP-LIN-45 in P6.pxx cells was seen in 68% of ufd-2(null) mutants, a significantly greater portion than in wild-type animals (Fig. 3A,B, Table S5). Loss of sel-10 resulted in accumulation of GFP-LIN-45 in P6.pxx in 100% of sel-10(null) mutants (Fig. 3B, Fig. S2C). Using Western blot analysis of protein from whole animals, we confirmed that the GFP-lin-45 allele produced a full-length FLAG-tagged protein and ufd-2(null) mutants showed a reproducible increase in GFP-3xFLAG-LIN-45 abundance compared to ufd-2(+) controls (Fig. S2E,F).
Fig. 3. UFD-2 is required for LIN-45 protein degradation.
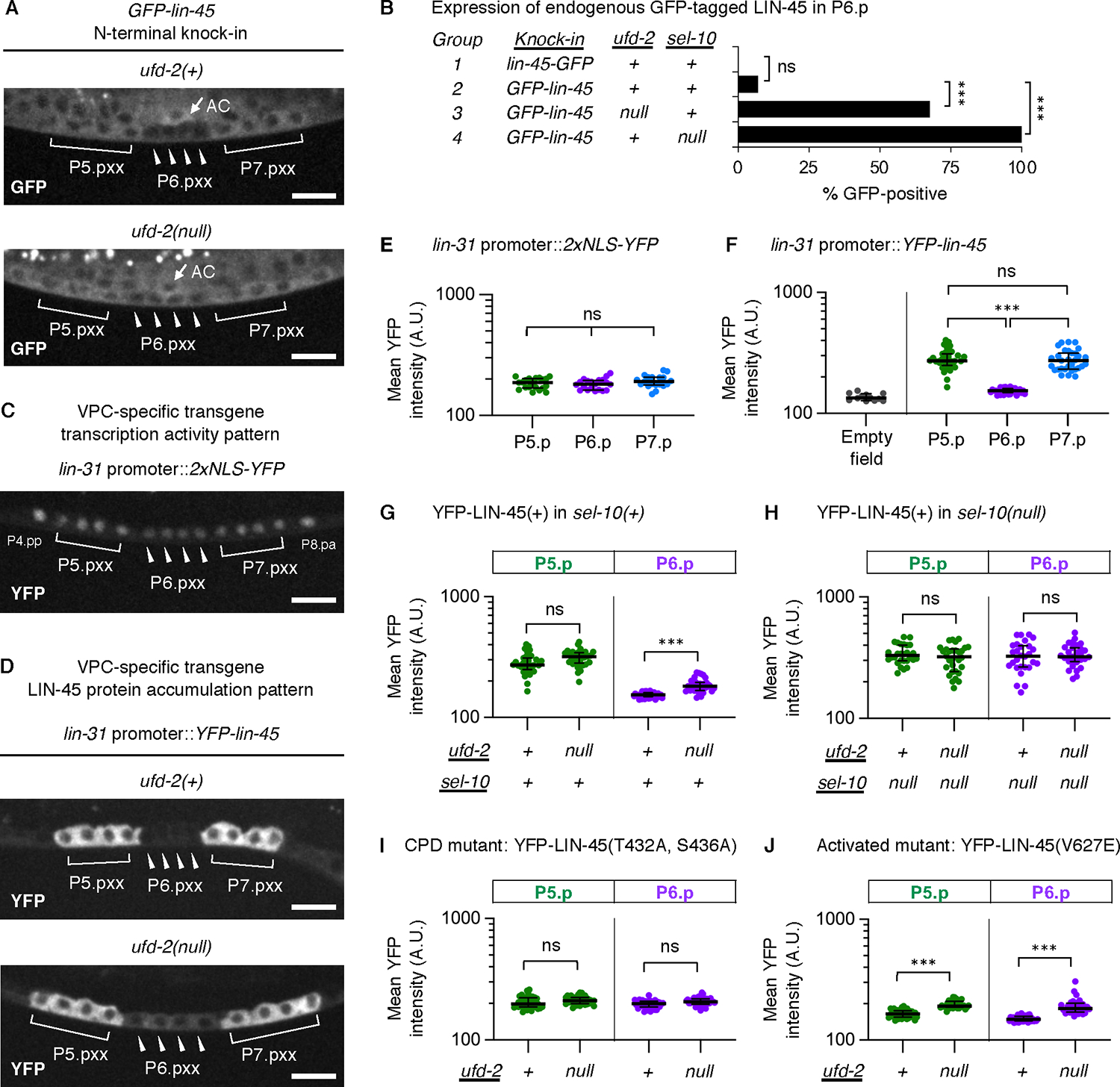
All micrographs show L3 stage larvae. Indicated are: cells descended from VPCs (labeled P5.pxx, P6.pxx, and P7.pxx) and the anchor cell (AC). Scale bar, 10 μm. A) Expression of the GFP-lin-45 knock-in allele in ufd-2(+) and ufd-2 null genotypes. B) Expression of GFP-lin-45 knock-in alleles in P6.pxx cells, shown as percentage of larvae. The total numbers of larvae (n) scored are: lin-45-GFP (n=30), GFP-lin-45 (n=28), ufd-2(null); GFP-lin-45 (n=34), GFP-lin-45; sel-10(null) (n=30). Comparisons were made using the Fisher’s exact test. ns not significant; ***P<0.0001. C) Expression of 2xNLS-YFP driven by lin-31 promoter elements. D) Expression of YFP-LIN-45 driven by the same lin-31 promoter as in C), in ufd-2(+) and ufd-2 null genotypes. E-J) For all measurements of YFP intensity, the cells analyzed are P5.pxx (green), P6.pxx (purple), P7.pxx (blue). Values represent intensities in arbitrary units (A.U.) from cells; the number scored (n) represents different larvae analyzed. E) 2xNLS-YFP (n=20). F) Empty field (n=12) and YFP-LIN-45 (n=32). G) YFP-LIN-45 in ufd-2(+); sel-10(+) and ufd-2(null); sel-10(+) (n=33) genotypes. H) YFP-LIN-45 in ufd-2(+); sel-10(null) (n=28) and ufd-2(null); sel-10(null) (n=30). I) Intensities for the mutant YFP-LIN-45(T432A, S436A) in ufd-2(+) (n=35) and ufd-2(null) (n=37) genotypes. J) Intensities for the mutant YFP-LIN-45(V627E) in ufd-2(+) (n=36) and ufd-2(null) (n=32) genotypes. In all panels, groups were compared using a one-way ANOVA followed by Šídák’s multiple comparisons test. ns not significant, ***P <0.0001.
To quantify LIN-45 protein abundance specifically in P6.pxx during the brief developmental window it is regulated by SCFSEL−10 (30), we used integrated, single-copy transgenes specifically expressed in VPCs. Expression in VPCs and their descendants is accomplished using a promoter fragment derived from the lin-31 gene (referred to here as lin-31p) capable of driving uniform expression in all VPCs from larval stages L1 through late L3 (31). When used to drive the expression of YFP alone, protein was observed in all six VPCs and accumulated to the same amount in P5.pxx, P6.pxx, and P7.pxx (Fig. 3C,E, Table S6). As shown previously (9), when the same lin-31p was used to express YFP-LIN-45, protein abundance in P6.pxx was significantly reduced compared to either P5.pxx or P7.pxx (Fig. 3D,F, Table S6). Because uniform accumulation was observed when only YFP was expressed, this result suggested that YFP-LIN-45 is downregulated in P6.pxx by a post-transcriptional mechanism. We note that YFP-LIN-45 protein amounts in P5.pxx and P7.pxx did not differ significantly (Fig. 3F), and we report the amounts in P5.pxx in all further experiments. The lin-31p::yfp-lin-45 transgene fully rescued vulval development of a lin-45 null mutant (Fig. S3A) and did not cause phenotypes when expressed in wild-type animals (Fig. S3B) indicating that YFP-tagged LIN-45 is both functional and not hyperactive.
In ufd-2(null) mutants, YFP-LIN-45 abundance in P6.pxx of animals carrying the lin-31p::yfp-lin-45 transgene was significantly increased compared to ufd-2(+) controls, while amounts in P5.pxx and P7.pxx were unaffected (Fig. 3D,G, Fig. S2G, Table S7). We tested whether contributions made by UFD-2 and SCFSEL−10 were additive, a result we would expect if each ubiquitin ligase regulated LIN-45 by different pathways. However, YFP-LIN-45 abundance was not significantly different in the sel-10(null) single mutants and ufd-2(null); sel-10(null) double mutants (Fig. 3H, Table S7), suggesting that UFD-2 and SCFSEL−10 act in the same pathway. Our previous work showed that LIN-45 degradation in P6.pxx is dependent on the CPD residues T432 and S436 (9). In ufd-2(null) mutants, we found no significant difference in the abundance of a mutant, YFP-LIN-45(T432A, S436A), in which CPD function is abrogated (Fig. 3I, Table S8) (9). Because SCFSEL−10 targets its substrates through this degron (32), this result is also consistent with UFD-2 and SCFSEL−10 acting in the same pathway.
Because the post-transcriptional downregulation occurred in P6.pxx, where MPK-1 is active, we tested the effect of ufd-2 on LIN-45(V627E), a mutant where MPK-1 activity is elevated ectopically in other VPCs that would not normally activate it (30). In ufd-2(null) mutants, amounts of the YFP-LIN-45(V627E) in P5.pxx, P6.pxx, and P7.pxx were significantly increased compared to ufd-2(+) controls (Fig. 3J, Fig. S2H, Table S8).
CDC-48 negatively regulates LIN-45 protein abundance
In C. elegans, UFD-2 can cooperate with chaperones and promote degradation of a substrate targeted by the E3 ubiquitin ligase CHN-1, ortholog of human STUB1 (20, 22). Additionally, UFD-2 interacts with the highly conserved ATP-dependent CDC-48 segregase, ortholog of yeast Cdc48 and human VCP proteins (33). To understand the mechanistic details of UFD-2-mediated degradation of LIN-45, we assessed the effect of chn-1 mutation and cdc-48 knock-down.
The viable allele chn-1(by155) is a deletion that eliminates the CHN-1 U-box, a domain required for E3 ligase activity and binding to UFD-2 (22). We assessed YFP-LIN-45 protein abundance in P6.pxx of chn-1 mutants and found no significant difference from controls (Fig. S4A).
To test the role of CDC-48, we used RNAi to simultaneously deplete cdc-48.1 and cdc-48.2, two highly similar genes encoding the C. elegans orthologs (34). Compared to animals treated with lacZ RNAi as a negative control, amounts of YFP-LIN-45 were significantly increased in both P5.pxx and P6.pxx depleted for cdc-48.1 and cdc-48.2 (Fig. 4A, Table S9). To control for off-target effects, we tested RNAi clones targeting non-overlapping 5’ regions (set #1) and 3’ regions (set #2) of cdc-48.1 and cdc-48.2 genes. Both RNAi conditions had similar effects, validating that cdc-48 knockdown increased YFP-LIN-45 accumulation in VPCs (Fig. 4A, Table S9). In all experiments, double cdc-48.1/2 knockdown animals failed to develop to adulthood. However, VPC fate specification at the L3 stage appeared to be normal, as P5.p, P6.p, and P7.p all completed two cell divisions.
Fig. 4. LIN-45 downregulation requires the CDC-48 segregase.
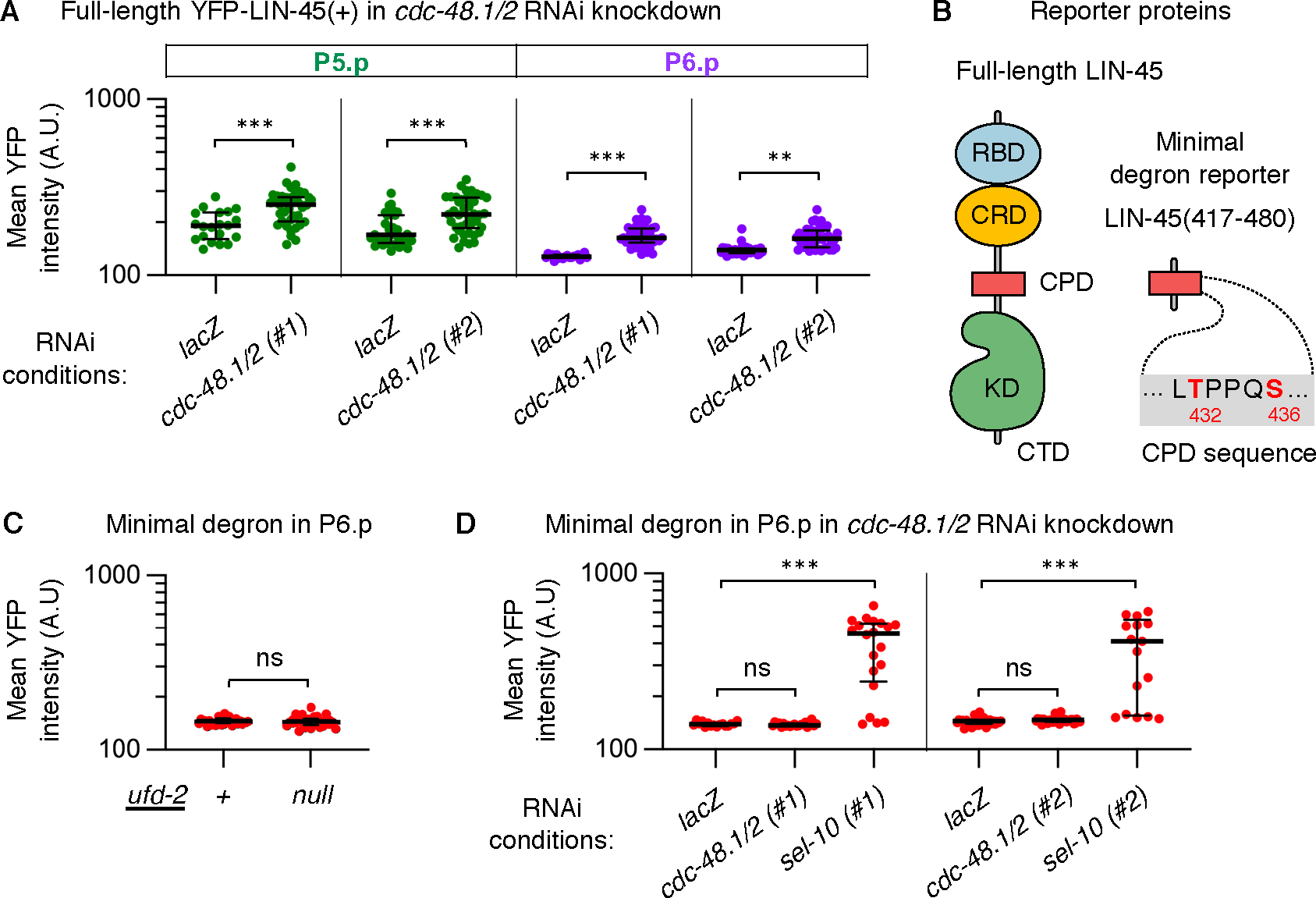
For all panels, values represent YFP intensities in arbitrary units (A.U.) from individual cells; the number scored (n) represents different larvae analyzed. A) YFP-LIN-45 full-length protein in P5.pxx (green) and P6.pxx (purple) is shown for RNAi knockdown of either the negative control lacZ or simultaneous knockdown of the cdc-48.1 and cdc-48.2 genes, labeled cdc-48.1/2. The RNAi conditions and number of larvae scored were: lacZ (n=20), 5’ regions of cdc-48.1/2 (#1) (n=42), lacZ (n=33), and 3’ regions of cdc-48.1/2 (#2) (n=42). B) Left: Diagram of LIN-45 domains: the RBD (blue), CRD (yellow), CPD (red), kinase domain (KD, green), and C-terminal tail (CTD). Right: Diagram of the minimal degron of LIN-45, which contains residues 417–480 and a canonical CPD motif. C) The minimal degron YFP-LIN-45(417–480) in P6.pxx (red) in ufd-2(+) (n=31) and ufd-2(null) (n=31) genotypes. D) The minimal degron YFP-LIN-45(417–480) in P6.pxx (red) is shown for RNAi knockdown. The RNAi conditions and number of larvae scored were: lacZ (n=16), the 5’ regions of cdc-48.1/2 (#1) (n=21), the 5’ region of sel-10 (#1) (n=20), lacZ (n=31), 3’ regions of cdc-48.1/2 (#2) (n=36), and sel-10 (#2) (n=17). In all panels, groups were compared using a one-way ANOVA followed by Šídák’s multiple comparisons test. ns not significant, **P <0.001, ***P <0.0001.
Studies of yeast and human CDC-48 suggest that its substrates are often well-folded, or part of multiprotein complexes, or localized to membranes. By unfolding such proteins, CDC-48 is thought to prepare them for degradation by the proteasome (35). We reasoned that if normal downregulation of LIN-45 protein requires its unfolding, then this step may be bypassed by a protein derived from LIN-45 but lacking its well-folded domains. To test this prediction, we used a reporter expressing a YFP-tagged 64-amino acid region of LIN-45 encompassing the CPD motif termed the “minimal degron,” or YFP-LIN-45(417–480) (Fig. 4B). As previously shown (30), the minimal degron was sufficient to produce the normal pattern of LIN-45, meaning it accumulated visibly in P5.pxx and P7.pxx and was absent in P6.pxx (Fig. S4B) (30). In contrast to full-length protein, protein abundance of YFP-LIN-45(417–480) in P6.pxx was not affected in ufd-2(null) mutants (Fig. 4C, Fig. S4B, Table S10), or in animals depleted of cdc-48.1 and cdc-48.2 (Fig. 4D, Table S11). However, accumulation of this reporter was significantly increased by the depletion of sel-10 (Fig. 4D), confirming that the reduction in protein accumulation of the minimal degron still relied on SCFSEL−10 activity. The results using this shorter, minimal degron suggest that native, full-length LIN-45 contains sequence features or domains that impede degradation in the absence of UFD-2 or CDC-48.
LIN-45 protein interaction domains are required for UFD-2 dependence
In yeast and C. elegans, CDC-48 is known to target substrates within protein complexes (33, 36, 37). We hypothesized that UFD-2 and CDC-48 act in C. elegans to target LIN-45 in multiprotein complexes, for example LIN-45 in an autoinhibited monomer and/or the Ras-bound activated dimer. If a protein interaction in wild-type LIN-45 causes its relative abundance to rely on UFD-2, then we expect that removal of that interaction may render LIN-45 downregulation UFD-2-independent. We tested this possibility by systematically perturbing LIN-45 domains. LIN-45 contains several domains present in all three human Raf proteins (Fig. 5A). Among these are the RBD, the CRD, a 14–3-3 binding site located within the “hinge” conserved region 2 (CR2), the C-terminal kinase domain, and a second 14–3-3 binding site located near the C-terminus (1).
Figure 5. LIN-45 mutant forms bypass the requirement for UFD-2.
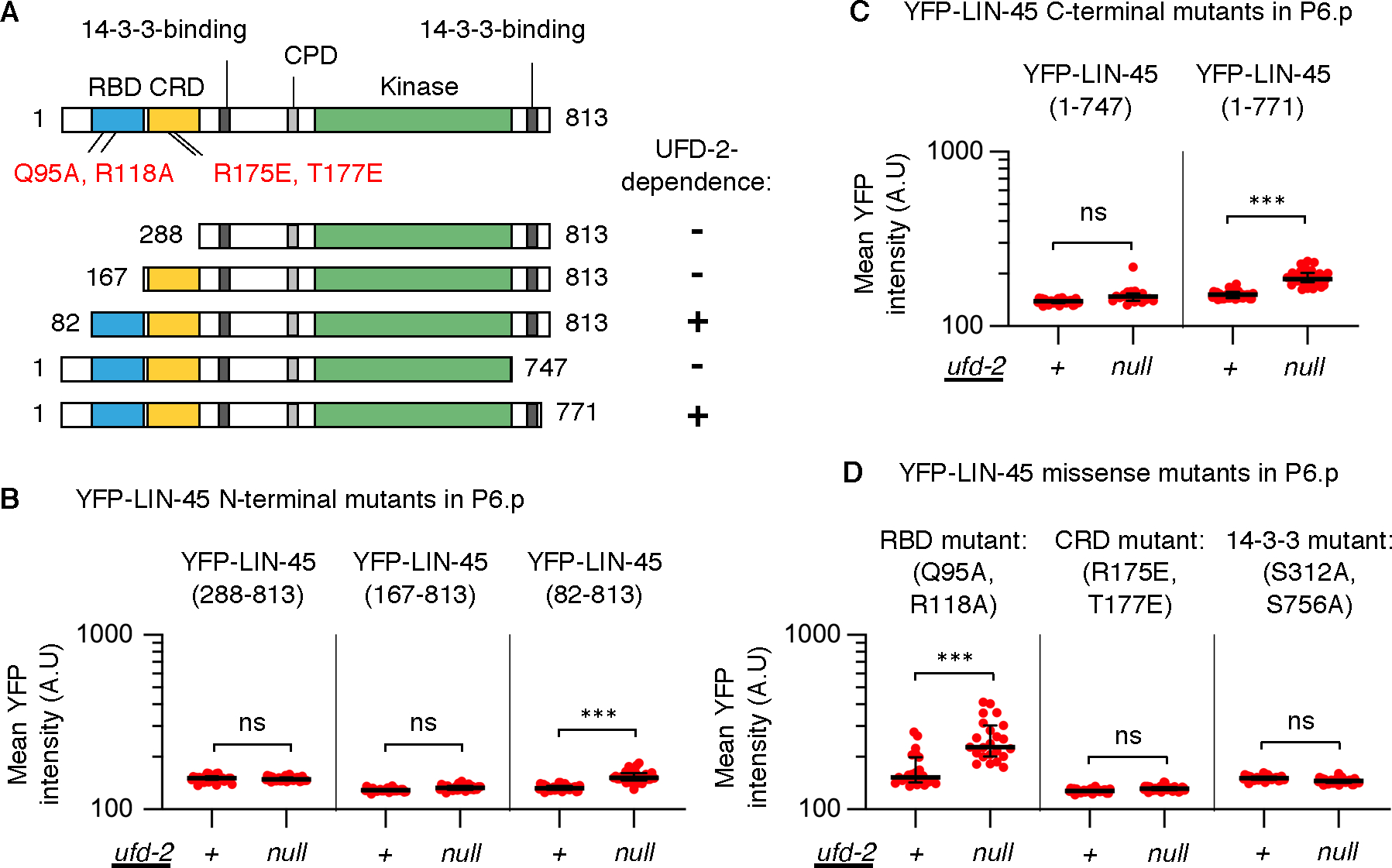
Mutant forms of YFP-LIN-45 were examined in ufd-2(+) and ufd-2(null) genotypes. A) Map of LIN-45 protein and domains, including RBD (blue), CRD (yellow), kinase domain (KD, green), and 14–3-3 binding motifs (dark gray), and locations of mutations and truncations tested. For mutants tested, protein abundance in P6.pxx cells was UFD-2-dependent (+) or independent (–), as indicated at right. B-D) For all panels, values represent YFP intensities in arbitrary units (A.U.) from P6.pxx cells; the number scored (n) represents different larvae analyzed. B) N-terminal truncations were: YFP-LIN-45(288–813), in ufd-2(+) (n=36) and ufd-2(null) (n=32); YFP-LIN-45(167–813), in ufd-2(+) (n=28) and ufd-2(null) (n=29); YFP-LIN-45(82–813), in ufd-2(+) (n=31) and ufd-2(null) (n=36). C) C-terminal truncations were: YFP-LIN-45(1–747), in ufd-2(+) (n=31) and ufd-2(null) (n=19); YFP-LIN-45(1–771), in ufd-2(+) (n=35) and ufd-2(null) (n=36). D) Missense mutations scored were YFP-LIN-45(Q95A, R118A), in ufd-2(+) (n=23) and ufd-2(null) (n=23), LIN-45(R175E, T177E) in ufd-2(+) (n=29) and ufd-2(null) (n=40), and LIN-45(S312A, S756A) in ufd-2(+) (n=33) and ufd-2(null) (n=33). In all panels, groups were compared using a one-way ANOVA followed by Šídák’s multiple comparisons test. ns not significant, ***P <0.0001.
To test whether LIN-45 N-terminal domains impact its UFD-2 dependence, we analyzed three truncations within the N-terminus (Fig. 5A). The longest of the three, YFP-LIN-45(82–813), retained both the RBD and CRD. Like full-length YFP-LIN-45, abundance of this form in P6.pxx was increased in ufd-2(null) mutants (Fig. 5B, Table S12), indicating that it was UFD-2-dependent. A second truncation, YFP-LIN-45(288–813), removed the RBD, while the third, YFP-LIN-45(167–813), removed both the RBD and CRD. For these mutants, protein amounts in P6.pxx were not significantly increased in ufd-2(null) mutants (Fig. 5B), indicating that they bypassed the requirement for UFD-2.
Next, we tested whether the LIN-45 C-terminus impacts UFD-2 dependence (Fig. 5A). The truncated form YFP-LIN-45(1–771) retained the C-terminal 14–3-3 binding site but lacks the remaining C-terminus. Like full-length YFP-LIN-45, abundance of this form in P6.pxx was increased in ufd-2(null) mutants (Fig. 5C, Table S12), indicating that it was UFD-2-dependent. In contrast, the truncation YFP-LIN-45(1–747) lacked all sequences C-terminal to the kinase domain. Protein abundance of this form in P6.pxx was not significantly increased in ufd-2(null) mutants (Fig. 5C), indicating that this mutant bypassed UFD-2.
To define with higher resolution the residues of LIN-45 required for UFD-2 dependence, we generated missense mutations in full-length YFP-LIN-45 that disrupt well-characterized protein interactions. Recruitment of Raf to the plasma membrane requires interaction with Ras through the Raf RBD. We ablated this interaction by mutating LIN-45 residues Gln95 and Arg118 (Fig. S5A), residues where mutations in human RAF1 prevent Ras binding (38, 39). Protein accumulation of the mutant YFP-LIN-45(Q95A, R118A) in P6.pxx was significantly increased in ufd-2(null) mutants (Fig. 5D, Table S12), indicating that the Ras-RBD interaction is not required for UFD-2-dependence. To confirm that mutations at Gln95 and Arg118 indeed abrogate function, we examined lin-45 null animals expressing YFP-LIN-45(Q95A, R118A) and found that this transgene was incapable of rescuing vulval development (Fig. S3A).
Raf conformation and activity is controlled by 14–3-3 protein interactions with the Raf CRD, and two phosphorylation-regulated interactions with the CR2 and C-terminal domains (3, 40, 41). We disrupted the CRD by generating mutations at LIN-45 Arg175 and Thr177 (Fig. S5A), conserved residues where mutation of human RAF1 prevents CRD-14–3-3 binding (42). We also tested mutations at LIN-45 Ser312 and Ser756, residues within highly conserved CR2 and C-terminal 14–3-3 binding sites (Fig. S5B,C). The CR2 and C-terminal Serine residues are phosphorylated in human Raf proteins and their mutation in RAF1 or BRAF abolishes 14–3-3 binding (43, 44). Protein abundance of YFP-LIN-45(R175E, T177E) in P6.pxx was not significantly different between ufd-2(+) and ufd-2(null) mutants (Fig. 5D, Table S12). Similarly, abundance of YFP-LIN-45(S312A, S756A) in P6.pxx was not significantly different between ufd-2(+) and ufd-2(null) mutants (Fig. 5D). These results indicate that mutants known to abolish 14–3-3 protein interactions cause LIN-45 to bypass the requirement for UFD-2. We tested the functional consequence of each mutant by assessing rescue of the lin-45 null mutant. The YFP-LIN-45(R175E, T177E) transgene partially rescued normal vulval induction (50% of animals) while 25% had a Muv phenotype (Fig. S3A), indicating that this LIN-45 form is functional in some animals, and rendered ligand-independent in others. The YFP-LIN-45(S312A, S756A) mutant transgene rescued vulval induction, indicating that this mutant is functional (Fig. S3A).
In summary, our genetic data using truncations and missense mutations of YFP-LIN-45 demonstrate that conserved protein-protein interaction sites within LIN-45 confer dependence on UFD-2 in reducing LIN-45 protein abundance (Fig. 6). Specifically, we identified the conserved CRD, CR2, and C-terminal 14–3-3-binding sites as domains required in LIN-45 for UFD-2-dependent degradation.
Fig. 6. Model of UFD-2 requirement in LIN-45 abundance.
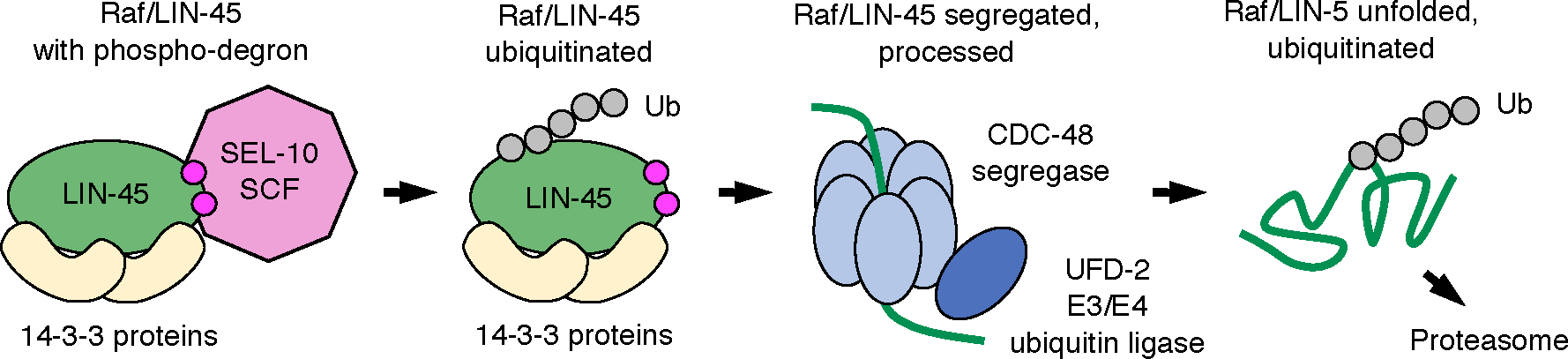
We propose a model that UFD-2 and CDC-48 facilitate degradation to reduce LIN-45 protein abundance. Raf proteins, shown here as LIN-45 (green), exist in multiprotein complexes containing 14–3-3 proteins (yellow). Our previous work showed that the LIN-45 phospho-degron and SCFSEL−10 E3 ubiquitin ligase (pink) are required for downregulation of LIN-45. SCFSEL−10 is known to produce K48-linked polyubiquitin chains (Ub), a signal for both the proteasome and the CDC-48 complex. We propose that efficient transfer of ubiquitinated LIN-45 to the proteasome requires its unfolding by CDC-48 and ubiquitination by UFD-2.
DISCUSSION
We identified UFD-2 and CDC-48 as negative regulators of LIN-45 protein abundance in C. elegans. During larval development, LIN-45 protein amounts were downregulated by a post-transcriptional mechanism in P6.pxx, a VPC where MPK-1 is highly active. Loss of either ufd-2 or cdc-48 resulted in increased LIN-45 accumulation in these cells. However, the requirement for ufd-2 was bypassed by LIN-45 mutations that abrogated domains conserved across Raf proteins: the CRD and 14–3-3 binding sites. In this discussion, we address how these results relate to the previously described degradation of LIN-45 by MPK-1 and SCFSEL−10 and propose a model for the roles of UFD-2 and CDC-48 based on their known molecular activities.
In human cells, ERK directly phosphorylates BRAF and RAF1 (5, 7) and in some contexts, stimulates BRAF ubiquitination and degradation (8). In developing C. elegans, the spatial and temporal pattern of LIN-45 protein in VPCs is controlled by the ERK ortholog MPK-1, as well as the kinases GSK-3 and CDK-2 (30). Our previous work established that LIN-45 contains a phosphorylation-regulated CPD, and that its downregulation in P6.pxx requires C. elegans SCFSEL-10. In this work, we found evidence that UFD-2 acts in the same pathway as SCFSEL−10 to decrease LIN-45 protein abundance. Two epistasis results support this model. First, LIN-45 protein amounts in a ufd-2; sel-10 double mutant were unchanged from those in a sel-10 mutant. Secondly, loss of ufd-2 did not affect protein abundance for LIN-45 containing mutations within its CPD motif. We note that these ubiquitin ligases employ different substrate targeting mechanisms, with SCFSEL−10 directly bound to substrates at a phosphorylated CPD and UFD-2 indirectly recruited to substrates through its association with chaperones or CDC-48.
In VPCs, one consequence of MPK-1’s role in stimulating LIN-45 downregulation is that LIN-45 protein amounts are lowest in P6.pxx where MPK-1 activity is highest (30). A second consequence is that disruption of LIN-45 regulation, such as in a sel-10 mutant, only affects protein abundance in P6.pxx. In this work, we showed that loss of ufd-2 behaved in the same manner, causing increased LIN-45 abundance only in P6.pxx cells where MPK-1 activity is highest. If this mechanism acts as negative feedback, then we predict that hyperactive Raf mutants found in cancers would stimulate ERK activation and cause Raf protein downregulation. In C. elegans, animals expressing the activated LIN-45(V627E) have elevated MPK-1 activity in VPCs other than P6.pxx (30). Here, we showed that loss of ufd-2 led to increased LIN-45(V627E) protein abundance in P5.pxx, P6.pxx, and P7.pxx. This result, and our initial isolation of ufd-2 as a negative regulator of the activated allele lin-45(V627E), lead us to believe that ufd-2 is an important point of regulation in cells and tissues where Raf kinase activity is elevated.
Studies of yeast and human CDC-48 and UFD-2 proteins show they act in an ATP-dependent segregase complex that unfolds protein substrates, disassembles protein complexes, and facilitates substrate targeting to the proteasome (45). Cryo-electron microscopy reveals that protein substrates are unfolded by CDC-48 as they translocate through a central pore formed within a hexameric ring of ATPase domains (35, 46). Substrate recruitment to the complex is accomplished by adaptor proteins termed Ufd1 and Npl4 in yeast (46). Finally, a substrate’s ubiquitin chains can be extended or trimmed through the activities of UFD-2 ubiquitin ligase or a deubiquitylase, respectively (47, 48). The physical interaction between CDC-48 and UFD-2 is highly conserved in yeast, human, and C. elegans (33, 47).
Natural, cellular substrates of Cdc48 share some characteristics: they are modified by K48-linked polyubiquitin chains, and in many cases, they exist in high molecular weight complexes or are localized to a membrane. Examples of substrates in large complexes include C. elegans UNC-45, a co-chaperone that forms a muscle filament associated with HSP90 and myosin (33, 49), and yeast Rpb1, a subunit of the RNA Polymerase II holoenzyme (36). In many cases, ubiquitin modification of substrates is supplied by another ubiquitin ligase, yet their efficient degradation still requires Ufd2 activity. For example, UNC-45 is modified by the CHN-1 E3 ubiquitin ligase, but its degradation requires UFD-2 (22, 33). In yeast, the cell-cycle kinase Mps1 is ubiquitinated by the anaphase-promoting complex, yet its degradation requires Ufd2 (50). One model to account for the need for UFD-2 in addition other ubiquitin ligases is that UFD-2 is responsible for extending ubiquitin chains of newly unfolded substrates as they exit the CDC-48 complex, a step ensuring efficient transfer to the proteasome (45).
Given the well-defined biochemical activities of CDC-48 and UFD-2, we propose that these proteins are responsible for unfolding and ubiquitinating LIN-45 (Fig. 6). Our previous work established that LIN-45 is regulated in a post-transcriptional manner that requires SCFSEL−10 and a LIN-45 CPD motif. Because the function of SCFSEL−10 in producing a proteasome polyubiquitin signal is well-understood, we think it is likely that post-transcriptional decreases of LIN-45 abundance result from degradation. LIN-45 ubiquitination by SCFSEL−10 may also serve as a signal allowing it to be targeted by CDC-48, which would be expected to extract substrates from their protein complexes and unfold their polypeptides. Finally, UFD-2 may further modify unfolded LIN-45 to promote efficient proteasome targeting (Fig. 6). In light of this model, our experiments identifying mutations in LIN-45 that bypass UFD-2 are informative. Specifically, mutation of the conserved CRD or 14–3-3 binding sites each eliminated the need for UFD-2 in reducing LIN-45 protein abundance. We propose that efficient degradation of LIN-45 protein is prevented by interactions through its 14–3-3-binding domains. While the exact composition of UFD-2-dependent LIN-45 complexes is not yet known, it is possible that they represent 14–3-3-bound LIN-45 in its autoinhibited or activated states, or possibly high molecular weight complexes in which Raf proteins are found in response to activation (51), or in complex with chaperones (52). These findings add to the repertoire of molecular mechanisms that regulate Raf protein activity in diverse cellular contexts.
MATERIALS AND METHODS
C. elegans genetics
The complete genotypes of C. elegans strains used in this work are listed in Table S13. The following alleles were obtained from the Caenorhabditis Genetics Center. LGII: ufd-2(tm1380). LGIV: lin-45(n2018), lin-45(n2506). LGV: sel-10(ok1632). The RNAi-sensitized mutant nre-1(hd20) lin-15B(hd126) displays enhanced RNAi effectiveness in VPCs (53). The transgenes arIs222 [lag-2p::2xNLS-tagRFP::unc-54 3′UTR] (28) and arTi4 [lin-31p::YFP-lin-45(417–480)] (30) were previously described.
Plasmids and transgenes used for expression in C. elegans
Plasmids used for transgenes are described in Table S14. lin-45 transgenes contained lin-45 cDNA, Wormbase sequence Y73B6A.5a. Mutations were generated using PCR and cloned by Gibson assembly. For YFP reporters, the YFP and lin-45 coding sequences were fused in frame to produce a sequence encoding N-terminally tagged YFP-LIN-45 protein. The ufd-2 transgene contained ufd-2 cDNA, sequence T05H10.5b.1. For expression in VPCs, coding sequences were cloned into pCC395, a vector derived from pCFJ910 (Addgene #44481) (54), containing the visible pharynx marker myo-2p::tagRFP, a Neomycin resistance gene, regulatory elements of the lin-31 gene (31) and a 3′ UTR derived from the unc-54 gene. All YFP-LIN-45 reporters were made using the same lin-31 promoter strategy. Some transgenes were cloned into pCC249 (de la Cova, et al. 2020), which has identical sequences but lacks myo-2p::tag-RFP.
Single-copy miniMos insertion transgenes were generated by germline injection of N2 strain hermaphrodites with 10 ng/μL transgene plasmid and a co-injection mixture of pCFJ601, pCFJ90, pGH8, and pMA122, and strains carrying integrated reporters were isolated as described previously (54). Briefly, transformant progeny of injected hermaphrodites were selected using G418 disulfate (2.5 μg/mL). Surviving progeny without visible co-injection markers were screened for transgene integration based on Neomycin resistance and YFP expression.
C. elegans alleles made by gene editing
Allele cov37 inserted GFP-3xFLAG at the start codon of lin-45 isoform Y73B6A.5a, resulting in an N-terminal fusion encoding GFP-3xFLAG-LIN-45. Allele cov40 inserted GFP-3xFLAG at the stop codon shared by all lin-45 isoforms, resulting in a C-terminal fusion encoding LIN-45–3xFLAG-GFP. All guide RNAs used in this work were designed using the software SapTrap Builder (55) and are listed in Table S15. The repair templates plasmids contained GFP-3xFLAG and a self-excising cassette and are derived from pDD282 (Addgene #66823) (56). The plasmid pKS4 was used for cov37 and contained 700 bp genomic fragments flanking the lin-45 start codon. The plasmid pAD2 was used for cov40 and contained 700 bp fragments flanking the lin-45 stop codon.
Gene-edited alleles were generated by germline injection of N2 strain hermaphrodites with 10 ng/μL of repair template plasmid and a ribonucleoprotein mixture as described previously (57), specifically: 250 ng/μL Cas9 protein (Macrolabs, University of California Berkeley), 100 ng/μL Alt-R-Crispr-Cas9 tracrRNA (Integrated DNA Technologies catalog 1072532), and 55 ng/μL custom gene-specific Alt-R crRNA synthesized by Integrated DNA Technologies, 2.5 ng/μL pCFJ90, and 20 ng/μL pGH8. Isolation of new gene-edited strains was performed as described previously (56). Briefly, transformant progeny of injected hermaphrodites were selected using Hygromycin B (250 μg/mL). Surviving progeny that displayed a Roller phenotype without visible co-injection markers were screened for genome editing based on GFP expression. All alleles were validated by PCR and Sanger DNA sequencing of the GFP coding sequence and 300 bp upstream and downstream of knock-in sites. All primers used for genotype validation are listed in Table S16.
Assessment of Multivulva and lethality phenotypes
All animals scored for Multivulva and lethality phenotypes were grown at 20°C. To assess the Multivulva phenotype, L4 hermaphrodites from uncrowded cultures were picked to fresh plates; adults were examined ~24 h later using a dissecting microscope and scored for the presence of a normal vulva and the number of any pseudovulvae. To assess L1 lethality of lin-45 hypomorphic mutants, L4 hermaphrodites were picked individually and transferred to fresh plates daily. At ~48 h after egg laying, all progeny were examined using a dissecting microscope and scored as live larvae, dead larvae, or unhatched eggs. All genotypes for the lin-45(n2018) experiment carried the mutation dpy-20(e1282); all genotypes for the lin-45(n2506) experiment carried the mutation unc-24(e138).
Imaging of VPCs
For imaging of VPCs during larval development, larvae were mounted on an agarose pad in M9 buffer containing 10 mM levamisole. Unless otherwise specified, adults were allowed to lay eggs for 12–16 hours and larvae were grown at 20°C for approximately 2 days, a time point when larvae of early, mid and late L3 stage were easily found. To further refine the developmental stage, only L3 stage larvae in which it was evident that VPCs had undergone either one or two cell divisions were scored and reported here.
Qualitative scoring of lag-2 reporter expression was performed using a Zeiss Axio Imager microscope equipped with widefield LED fluorescent illumination (Excelitas Technologies) and CMOS camera (Zeiss). The locations of VPC descendants after division were determined using DIC optics. Images of RFP expression from arIs222 [lag-2p::2xNLS-tagRFP] were acquired using a 40x objective, 500 ms exposure, and 100% LED power. Expression was scored on a positive/negative basis. Descendants of P3.p, P4.p, and P8.p which had divided and fused with the hypodermis were scored as negative.
Images used for quantitation were acquired using a Nikon Ti inverted microscope equipped with a spinning disk confocal system (Crest Optics) and dual sCMOS cameras (Teledyne Photometrics). To quantify YFP-tagged LIN-45, all Z-stacks of YFP were acquired using a 40x objective; for all images, an exposure time of 700 ms and laser power of 25% was used. To assess ERK-KTR localization in VPCs before cell division in the early L3 stage, animals were synchronized using 2-hour egg collections, grown at 25°C, and imaged at ~28 hours after egg collection. For all ERK-KTR experiments, Z-stacks of mClover and mCherry were acquired using dual cameras and a 60x objective; for all images, exposure time and laser power used for mClover and mCherry were equal: 500 ms and 25% laser power. Blank images were acquired using the same parameters.
RNAi treatment
Plasmids used in RNAi knock-down experiments are derived from the RNAi feeding vector L4440 (Addgene #1654). The lacZ and sel-10 RNAi plasmids were a gift from Iva Greenwald. Non-overlapping genomic sequences to target the 5’ and 3’ regions of cdc-48.1 and cdc-48.2 genes were amplified using primers listed in Table S17. Genomic fragments were cloned in the L4440 vector by Gibson assembly.
For each RNAi condition, an HT115(DE3) E. coli strain carrying the relevant RNAi plasmid was grown in liquid culture and spread on NGM plates containing IPTG, as previously described (58). Simultaneous knock-down of cdc-48.1 and cdc-48.2 was achieved by plating a 1:1 mixture of two strains, carrying cdc-48.1 and cdc-48.2 plasmids. All C. elegans genotypes treated with RNAi carried the mutations nre-1(hd20) lin-15B(hd126), and were synchronized by a standard bleach/sodium hydroxide protocol to prepare eggs (59). Approximately 200 eggs were placed on each plate and grown at 20°C. L3 stage larvae were examined ~44 hours after egg preparation.
Image quantitation and analysis
The subcellular localization of the ERK-KTR biosensor was quantified using methods described in de la Cova, et al. (2017) and the Nikon NIS-Elements software. Briefly, illumination correction and background subtraction was performed using the blank images obtained during each experiment. Image segmentation and manual curation was performed in NIS-Elements to create regions of interest (ROIs) for the nucleus (excluding the nucleolus) and cytoplasm of VPCs P4.p, P5.p, P6.p, P7.p, and P8.p. (P3.p fuses with the hyp7 syncytium in approximately half of animals and was not analyzed.) Mean fluorescence intensity for mClover within the nucleus and cytoplasm ROIs was determined for up to five of the most equatorial Z slices per cell. Data presented is a ratio of the mean cytoplasmic mClover intensity/mean nuclear mClover intensity, referred to as “C/N” ratio, per individual cell.
Expression of YFP-tagged LIN-45 protein reporters was quantified using NIS-Elements software. ROIs representing the cytoplasm of P5.pxx, P6.pxx, and P7.pxx cells were created manually. While all other YFP-LIN-45 reporters were exclusively cytoplasmic, the truncated YFP-LIN-45(417–480) form was both cytoplasmic and nuclear. For this reason, the nucleus was included in the quantification of this mutant. Data presented is the mean YFP fluorescence intensity for the most equatorial Z slice, per set of VPC descendants P5.pxx, P6.pxx, and P7.pxx.
Data processing and statistical analysis
All statistical analyses were performed using GraphPad Prism software. Descriptions of tests used, groups compared, and P-values are in Tables S1–S12.
For qualitative scoring of the Muv, lethal, lag-2 reporter, and GFP-LIN-45 protein accumulation phenotypes, we made planned, pairwise comparisons. The Fisher’s exact test was used to calculate a two-tailed P-value and adjusted using Bonferroni correction.
For quantitative measurements of ERK-KTR C/N ratio and YFP intensities, we transformed data values to Log10(Y). Groups were compared using one-way ANOVA tests, followed by planned, pairwise comparisons to compare the mean values observed for different genotypes in the same VPC. Adjusted P-values were calculated using Šídák’s multiple comparisons test.
Supplementary Material
Singling out Raf.
The Raf family of kinases are integral components of Ras-MEK-ERK signaling cascades and have important roles in cancer progression. Townley et al. used a genetic screen to identify regulators of the Raf ortholog LIN-45 in Caenorhabditis elegans. The authors found that the E3/E4 ubiquitin ligase UFD-2 reduced the abundance of both wild-type LIN-45 and a form with an equivalent mutation to the cancer associated variant BRAF(V600E). Degradation of LIN-45 required the ATPase CDC-48, which removed LIN-45 from a complex with 14–3-3 proteins to make it accessible to UFD-2. These findings have implications for our understanding of Raf degradation in mammalian cells and strategies for the blockade of mutant Raf signaling. -Amy E. Baek
Acknowledgments:
The authors thank Hannes Bülow, Jennifer Gutzman, and Christopher Quinn for their comments and discussions of this manuscript. We thank Jagadeesh Uppala for assistance in quantifying protein abundance, Jordan Gonnering and Teresa Buchanan for facilities support, and Wormbase for C. elegans genome data. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Funding:
University of Wisconsin Discovery and Innovation grant 101X404 (CCD)
National Institutes of Health grant R03CA248684 (CCD)
University of Wisconsin-Milwaukee scholarship (CSRT)
Footnotes
Competing interests:
Authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Materials created in this work are available upon request to the corresponding author.
References and Notes:
- 1.Terrell EM, Morrison DK, Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb Perspect Med 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han M, Golden A, Han Y, Sternberg PW, elegans lin-C. 45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 363, 133–140 (1993). [DOI] [PubMed] [Google Scholar]
- 3.Park E et al. , Architecture of autoinhibited and active BRAF-MEK1–14-3–3 complexes. Nature 575, 545–550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo Y et al. , Cryo-EM structure of a dimeric B-Raf:14–3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dougherty MK et al. , Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell 17, 215–224 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Eisenhardt AE et al. , Phospho-proteomic analyses of B-Raf protein complexes reveal new regulatory principles. Oncotarget 7, 26628–26652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritt DA, Monson DM, Specht SI, Morrison DK, Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 30, 806–819 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saei A et al. , Loss of USP28-mediated BRAF degradation drives resistance to RAF cancer therapies. J Exp Med 215, 1913–1928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de la Cova C, Greenwald I, SEL-10/Fbw7-dependent negative feedback regulation of LIN-45/Braf signaling in C. elegans via a conserved phosphodegron. Genes Dev 26, 2524–2535 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damsky WE, Bosenberg M, Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene 36, 5771–5792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H et al. , Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Uribe P, Andrade L, Gonzalez S, Lack of association between BRAF mutation and MAPK ERK activation in melanocytic nevi. J Invest Dermatol 126, 161–166 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Houben R et al. , Phospho-ERK staining is a poor indicator of the mutational status of BRAF and NRAS in human melanoma. J Invest Dermatol 128, 2003–2012 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Venesio T et al. , In melanocytic lesions the fraction of BRAF V600E alleles is associated with sun exposure but unrelated to ERK phosphorylation. Mod Pathol 21, 716–726 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Sundaram MV, Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook, 1–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin H, Reiner DJ, The Signaling Network Controlling C. elegans Vulval Cell Fate Patterning. J Dev Biol 6, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Cova C, Townley R, Regot S, Greenwald I, A Real-Time Biosensor for ERK Activity Reveals Signaling Dynamics during C. elegans Cell Fate Specification. Dev Cell 42, 542–553 e544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo AS, Bais C, Greenwald I, Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science 303, 663–666 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Sternberg PW, Vulval development. WormBook, 1–28 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellerschmied D et al. , UFD-2 is an adaptor-assisted E3 ligase targeting unfolded proteins. Nat Commun 9, 484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koegl M et al. , A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Hoppe T et al. , Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell 118, 337–349 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Abdus-Saboor I et al. , Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development 138, 3545–3555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu V, Zobel CL, Lambie EJ, Schedl T, Kornfeld K, Caenorhabditis elegans lin-45 raf is essential for larval viability, fertility and the induction of vulval cell fates. Genetics 160, 481–492 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW, High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs D, Beitel GJ, Clark SG, Horvitz HR, Kornfeld K, Gain-of-function mutations in the Caenorhabditis elegans lin-1 ETS gene identify a C-terminal regulatory domain phosphorylated by ERK MAP kinase. Genetics 149, 1809–1822 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Greenwald I, Spatial regulation of lag-2 transcription during vulval precursor cell fate patterning in Caenorhabditis elegans. Genetics 188, 847–858 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sallee MD, Greenwald I, Dimerization-driven degradation of C. elegans and human E proteins. Genes Dev 29, 1356–1361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Underwood RS, Deng Y, Greenwald I, Integration of EGFR and LIN-12/Notch Signaling by LIN-1/Elk1, the Cdk8 Kinase Module, and SUR-2/Med23 in Vulval Precursor Cell Fate Patterning in Caenorhabditis elegans. Genetics 207, 1473–1488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de la Cova CC, Townley R, Greenwald I, Negative feedback by conserved kinases patterns the degradation of Caenorhabditis elegans Raf in vulval fate patterning. Development 147, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan PB, Lackner MR, Kim SK, MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell 93, 569–580 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Yumimoto K, Nakayama KI, Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol 67, 1–15 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Janiesch PC et al. , The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol 9, 379–390 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Mouysset J, Kahler C, Hoppe T, A conserved role of Caenorhabditis elegans CDC-48 in ER-associated protein degradation. J Struct Biol 156, 41–49 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Twomey EC et al. , Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verma R, Oania R, Fang R, Smith GT, Deshaies RJ, Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell 41, 82–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maric M, Maculins T, De Piccoli G, Labib K, Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346, 1253596 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Block C, Janknecht R, Herrmann C, Nassar N, Wittinghofer A, Quantitative structure-activity analysis correlating Ras/Raf interaction in vitro to Raf activation in vivo. Nat Struct Biol 3, 244–251 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Harding A, Hsu V, Kornfeld K, Hancock JF, Identification of residues and domains of Raf important for function in vivo and in vitro. J Biol Chem 278, 45519–45527 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Cookis T, Mattos C, Crystal Structure Reveals the Full Ras-Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biomolecules 11, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez Fiesco JA, Durrant DE, Morrison DK, Zhang P, Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat Commun 13, 486 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark GJ et al. , 14–3-3 zeta negatively regulates raf-1 activity by interactions with the Raf-1 cysteine-rich domain. J Biol Chem 272, 20990–20993 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Brummer T et al. , Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene 25, 6262–6276 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Pandit B et al. , Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 39, 1007–1012 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Bodnar N, Rapoport T, Toward an understanding of the Cdc48/p97 ATPase. F1000Res 6, 1318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bodnar NO et al. , Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat Struct Mol Biol 25, 616–622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richly H et al. , A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Ernst R, Mueller B, Ploegh HL, Schlieker C, The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell 36, 28–38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gazda L et al. , The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell 152, 183–195 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu C et al. , Ubiquitin ligase Ufd2 is required for efficient degradation of Mps1 kinase. J Biol Chem 286, 43660–43667 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mysore VP et al. , A structural model of a Ras-Raf signalosome. Nat Struct Mol Biol 28, 847–857 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diedrich B et al. , Discrete cytosolic macromolecular BRAF complexes exhibit distinct activities and composition. EMBO J 36, 646–663 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng Y, Luo KL, Shaye DD, Greenwald I, A Screen of the Conserved Kinome for Negative Regulators of LIN-12 Negative Regulatory Region (“NRR”)-Missense Activity in Caenorhabditis elegans. G3 (Bethesda) 9, 3567–3574 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frokjaer-Jensen C et al. , Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods 11, 529–534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwartz M, Jorgensen E, SapTrap Builder: a desktop utility for CRISPR edit design. MicroPubl Biol 2018, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickinson DJ, Pani AM, Heppert JK, Higgins CD, Goldstein B, Streamlined Genome Engineering with a Self-Excising Drug Selection Cassette. Genetics 200, 1035–1049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dokshin GA, Ghanta KS, Piscopo KM, Mello CC, Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics 210, 781–787 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamath RS, Ahringer J, Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Stiernagle T, Maintenance of C. elegans. WormBook, 1–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O, C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O, CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.