

Abstract
Polyadenylation is an essential process for the stabilization and export of mRNAs to the cytoplasm and the polyadenylation signal hexamer (herein referred to as hexamer) plays a key role in this process. Yet, only 14 Mendelian disorders have been associated with hexamer variants. This is likely an under-ascertainment as hexamers are not well defined and not routinely examined in molecular analysis. To facilitate the interrogation of putatively pathogenic hexamer variants, we set out to define functionally important hexamers genome-wide as a resource for research and clinical testing interrogation. We identified predominant polyA sites (herein referred to as pPAS) and putative predominant hexamers across protein coding genes (PAS usage >50% per gene). As a measure of the validity of these sites, the population constraint of 4532 predominant hexamers were measured. The predominant hexamers had fewer observed variants compared to non-predominant hexamers and trimer controls, and CADD scores for variants in these hexamers were significantly higher than controls. Exome data for 1477 individuals were interrogated for hexamer variants and transcriptome data were generated for 76 individuals with 65 variants in predominant hexamers. 3′ RNA-seq data showed these variants resulted in alternate polyadenylation events (38%) and in elongated mRNA transcripts (12%). Our list of pPAS and predominant hexamers are available in the UCSC genome browser and on GitHub. We suggest this list of predominant hexamers can be used to interrogate exome and genome data. Variants in these predominant hexamers should be considered candidates for pathogenic variation in human disease, and to that end we suggest pathogenicity criteria for classifying hexamer variants.
Keywords: polyadenylation, bioinformatics, variant classification, RNA seq
Introduction
Most genes use polyadenylation (polyA) to maintain the stability of nascent mRNA and to export mRNA to the cytoplasm. This process is directed by the polyadenylation signal, which comprises a set of sequences in the 3′ untranslated region. These sequences initiate cleavage of the 3′ end of the mRNA at the PAS followed by addition of a polyA tail [1]. The most well-defined component of these sequences is the polyadenylation signal hexamer (herein referred to as hexamer) motif, typically 10–30 bases upstream of the PAS [2]. The canonical hexamer AATAAA (and variants thereof) is critical in facilitating this process [3].
Few hexamer variants have been associated with Mendelian disease. In the 2023.1 version of the HGMD [4] database, there were only 34 hexamer variants in 14 genes associated with Mendelian disorders (Table 1). For comparison, in HGMD overall, there were 1058 disease-associated variants in start methionine (AUG) codons and 323 661 disease-associated variants of any type. We hypothesized that Mendelian disease-associated hexamer variants are under-ascertained because of the manifold challenges of identifying such variants.
Table 1.
Pathogenicity classifications of 33 polyadenylation signal hexamer disease-associated variants in 14 genes.
| Gene | WT Hexamers | Hexamers cDNA Position | GRCh37 Position & Variation | Descriptor | Variant hexamers | CADD score | HGMD ID | Classification | Pathogenicity Criteria |
|---|---|---|---|---|---|---|---|---|---|
| BMP1 | AGTAAA | c.*239_*244 | Chr8:22058957 T > C | NM_001199.4:c.*241 T > C | AGCAAA | 17.97 | CR150372 | Likely Path | PS3_Mod, PS4_Mod, PM3_Sup, PP3, PP4 |
| F9 | AATAAA | c.*1365_*1370 | ChrX:138645597 T > G | NM_000133.4: c.*1367 T > G | AAGAAA | 5.32 | CR2219631 | VUS | PS4_Sup, PM1_Sup, PP4 |
| ChrX:138645598A > G | NM_000133.4: c.*1368A > G | AATGAA | 2.70 | CR005437 | VUS | PS4_Sup, PM1_Sup, PP4, BP4 | |||
| ChrX:138645598A > C | NM_000133.4: c.*1368A > C | AATCAA | 2.37 | CR2219630 | VUS | PM1_Sup, BP4 | |||
| FOXP3 | AATAAA | c.*873_*878 | ChrX:49106919 T > C | NM_014009.4:c.*876A > G | AATGAA | 13.02 | CR014834 | Likely Path | PS4_Mod, PM1_Sup, PP1_Strong, PP3, PP4 |
| ChrX:49106917 T > C | NM_014009.4:c.*878A > G | AATAAG | 12.77 | CR097218 | VUS | PS3_Sup, PS4_Sup, PM1_Sup, PP3, PP4 | |||
| HBA2 | AATAAA | c.*89_*94 | Chr16:223690-223691del | NM_000517.4:c.*91_*92del | AAAAGT | 12.61 | CD2026691 | VUS | PS4_Sup, PM1_Sup, PP3, PP4 |
| Chr16:223691A > G | NM_000517.4:c.*92A > G | AATGAA | 11.73 | CR920785 | Likely Path | PS4_Mod, PM1_Sup, PM3_Sup, PM5_Sup, PP1_Mod, PP3, PP4 | |||
| Chr16:223692-223693del | NM_000517.6:.*93_*94del | AATAGT | 11.84 | CD941949 | Likely Path | PS4_Mod, PM1_Sup, PM3, PM5_Sup, PP3, PP4 | |||
| Chr16:223693A > G | NM_000517.6:c.*94A > G | AATAAG | 12.77 | CR830007 | Pathogenic | PS3_Sup, PS4, PM1_Sup, PM3, PP3, PP4 | |||
| Chr16:223693A > C | NM_000517.6:c.*94A > C | AATAAC | 12.24 | CR106042 | Likely Path | PM1_Sup, PM3, PM5_Sup, PP3, PP4 | |||
| HBB | AATAAA | c.*108_*113 | Chr11:5246720 T > G | NM_000518.5:c.*108A > C | CATAAA | 15.14 | CR016252 | VUS | PS4_Sup, PM1_Sup, PP3 |
| Chr11:5246720 T > C | NM_000518.5: c.*108A > G | GATAAA | 15.27 | CR127145 | VUS | PS4_Mod, PM1_Sup, PP3, PP4 | |||
| Chr11:5246718A > T | NM_000518.5:c.*110 T > A | AAAAAA | 14.07 | CR045224 | VUS | PS4_Sup, PM1_Sup, PM5_Sup, PP3, PP4 | |||
| Chr11:5246718A > G | NM_000518.5:c.*110 T > C | AACAAA | 14.17 | CR850010 | Pathogenic | PS3_Sup, PS4, PM1_Sup, PM3, PM5_Sup, PP1, PP3, PP4 | |||
| Chr11:5246718A > C | NM_000518.5:c.*110 T > G | AAGAAA | 14.03 | CR014260 | VUS | PS4_Sup, PM1_Sup, PM5_Sup, PP3, PP4 | |||
| Chr11:5246717-5256718del | NM_000518.5:c.*110_*111del | AAAAAA | 13.15 | CD951735 | VUS | PS4_Sup, PM1_Sup, PM5_Sup, PP3, PP4 | |||
| Chr11:5246714-5246718del | NM_000518.5:c.*110_*114del | AAAACA | 14.11 | CD920867 | Likely Path | PS3_Sup, PS4_Mod, PM1_Sup, PM3_Sup, PM5_Sup, PP3, PP4 | |||
| Chr11:5246717 T > C | NM_000518.5:c.*111A > G | AATGAA | 14.04 | CR900265 | Pathogenic | PS4, PM1_Sup, PM3, PM5_Sup, PP3, PP4 | |||
| Chr11:5246716 T > C | NM_000518.5:c.*112A > G | AATAGA | 14.73 | CR900266 | Likely Path | PM1_Sup, PM3, PM5_Sup, PP3, PP4 | |||
| Chr11:5246716 T > A | NM_000518.5:c.*112A > T | AATATA | 14.64 | CR057232 | VUS | PS4_Sup, PM1_Sup, PP3, PP4 | |||
| Chr11:5246715 T > C | NM_000518.5:c.*113A > G | AATAAG | 13.23 | CR880076 | Pathogenic | PS3_Sup, PS4, PM1_Sup, PM3, PP3, PP4 | |||
| HBD | AATAAA | c.*106_*111 | Chr11:5254085 T > A | NM_000519.4: c.*109A > T | AATTAA | 13.00 | CR109506 | VUS | PS4_Sup, PM1_Sup, PP3, PP4 |
| IL2RG | AATAAA | c.*303-*308 | ChrX:70327278 T > C | NM_000206.3: c.*308A > G | AATAAG | 15.05 | CR0910465 | VUS | PS3_Sup, PS4_Sup, PM1_Sup, PP3, PP4 |
| INS | AATAAA | Chr11:2181023 T > C | NM_000207.2:c.*59A > G | AATAAG | 12.87 | CR101141 | VUS | PS3_Sup, PS4_Sup, PM1_Sup, PP3 | |
| ITGA2B | AATAAA | c.*163_*168 | Chr17:42449567A > G | NM_000419.5c.*165 T > C | AACAAA | 12.82 | CR153724 | VUS | PS4_Sup, PM1_Sup, PP3 |
| NAA10 | AATAAA | c.*38_*43 | ChrX:153195400 T > C | NM_003491.4:c.*39A > G | AGTAAA | 12.95 | CR1913378 | Likely Path | PS3_Sup, PS4_Sup, PM1_Sup, PP1_Mod, PP3 |
| ChrX:153195401 T > C | NM_003491.4:c.*40A > G | GATAAA | 14.35 | CR1913377 | VUS | PS4_Sup, PM1_Sup, PP3 | |||
| ChrX:153195397 T > C | NM_003491.4:c.*43A > G | AATAGA | 14.18 | CR1913376 | Likely Path | PS3_Sup, PS4_Sup, PM1_Sup, PP1_Strong, PP3 | |||
| RNASEH2C | AATAAA | c.*78_*83 | Chr11:65487176 T > C | NM_032193: c.*78A > G | GATAAA | 16.68 | CR2027419 | VUS | PS4_Sup, PM1_Sup, PP3 |
| STUB1 | AATAAA | c.*238_*243 | Chr16:732729 T > C | NM_005861.3c.*240 T > C | AACAAA | 14.22 | CR1815154 | VUS | PS3_Sup, PS4_Sup, PM1_Sup, PP1, PP3 |
| UROD | AATAAA | c.*57_*62 | Chr1:45481230-45481231del | NM_000374.4:c.*62_*63del | AATAAG | 12.67 | CD122215 | VUS | PS4_Sup, PM1_Sup, PP3 |
| GLA | ATTAAA | c.1272_1277 | ChrX:100652809-810del | NM_000169.2:c.1277_1278del | ATTAGA | 23.7 | CD031841 | VUS | PS3_Sup, PS4_Sup, PM1_Sup, PP3, PP4 |
While there are numerous PAS and hexamer databases, these databases provide lists of all potential sites without consideration as to importance or functionality [5–8]. For example, the PolyASite 2.0 database specifies >560 000 PAS and >860 000 hexamers based on 3′ end sequencing of mRNA molecules for 32 494 genes. A challenge of hexamer analysis is that genes can have numerous candidate PAS that may not be biologically relevant. Additionally, each PAS can have multiple hexamer motifs that represent candidate hexamers. While there have been several efforts to globally assess hexamers and the consequences of variation in those sites, those efforts were focused on common variants associated with susceptibility traits, rather than Mendelian, single gene disorders [9, 10]. Similarly, previous efforts to identify hexamers used inclusion and exclusion criteria to select for hexamers without the consideration for identifying candidate hexamers for Mendelian disorders ([11], preprint). Here, we used the most comprehensive list of PAS and hexamers available to date, PolyA Site 2.0, and aimed to identify hexamers that are candidates for Mendelian disorder associations by defining pPAS and identifying corresponding hexamers. Importantly, our work focuses on hexamers in genes associated with Mendelian disease, which should be useful to clinical laboratories.
To identify hexamers that are candidates for association with Mendelian disease variation, we hypothesized that such hexamers would 1) be the hexamers associated with the pPAS within a given gene, 2) exhibit a strong population constraint signal, 3) demonstrate aberrant scores from in silico evaluations of variants, and 4) a sample set of such variants would be associated with perturbations in mRNA processing. We set out to define a set of PAS and corresponding hexamers that met these criteria that could be used as a candidate list for Mendelian gene-associated pathogenic variation. We also propose variant classification criteria for clinical genomic testing laboratories.
Results
Selection of pPAS and predominant hexamers
The workflow for identifying pPAS and their associated predominant hexamers is outlined in Fig. 1. To identify pPAS, we used all PAS in PolyASite 2.0 as the starting point. This database provides a comprehensive list of PAS identified by performing a meta-analysis of 29 3′ end sequencing studies. The total number of unique PAS after filtering and re-annotating was 280 148. The average number of PAS per protein-coding gene was 14.2, with 85% of protein coding genes having more than one.
Figure 1.
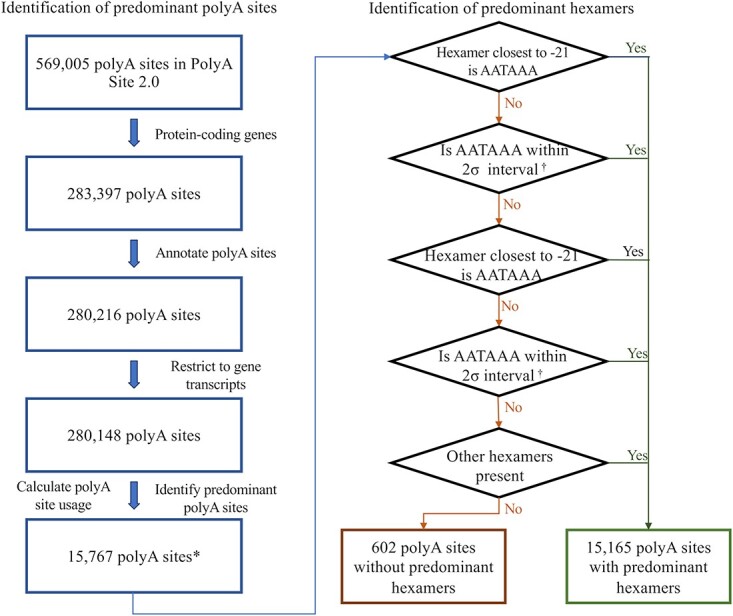
The overview of the workflow to identify predominant polyA sites and predominant hexamers. * denotes the number of unique polyA sites, inclusive of polyA sites that may be shared by overlapping genes. The total number of genes is 16 004. † refers to 2 standard deviations from the mean of the distribution of corresponding lone hexamers downstream of polyA sites.
We used relative TPM values to assess the usage of each PAS for all sites identified in a gene model. For genes that had >50% usage of one site, that PAS was defined as the pPAS. Most PAS (>77%) had <1% usage (Fig. S1). Across 18 580 protein coding genes, 15 767 pPAS were identified in 16 004 genes and 197 of these were associated with more than one gene (Fig. 1). For 2576 protein coding genes, no pPAS was identified (Fig. S1, Additional File 1).
Next, we aimed to identify a predominant hexamer(s) for each pPAS. For 15 767 pPAS, 45% had more than one candidate hexamer, with an average of 2.5 candidate hexamers for each pPAS (Fig. 2A). To identify the predominant hexamers, we considered both the strength of the hexamers and the proximity of the hexamers to the pPAS [12, 13].
Figure 2.

The summary of predominant hexamers selected upstream of 15 767 predominant polyA sites. A: The landscape of the predominant hexamers. B: The sequence of all predominant hexamers and their respective counts for 15 767 predominant polyA sites.
In total, 15 212 predominant hexamers were identified for 15 165 pPAS and no hexamers were identified for 602 pPAS. In most, we identified the canonical AATAAA hexamer motif (61.6%) with the next most common ATTAAA in 15%. The remaining 23.4% of the predominant hexamers comprised one of 16 other hexamer motifs (Fig. 2B).
Of 5376 clinically important genes, we identified pPAS for 4532 genes (84%) (Fig. 3A and B). For the remaining 844 genes, 744 (88%) had an average of >30 PAS per gene with an average highest PAS usage of 37.6%. The remaining 100 genes did not have any identified PAS.
Figure 3.
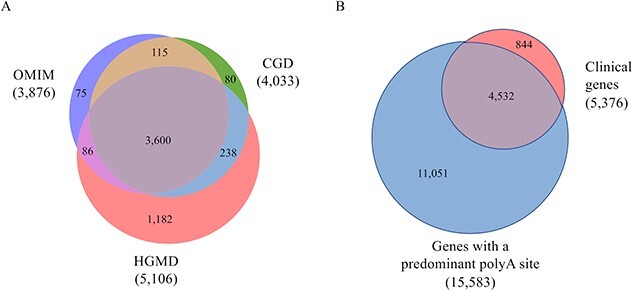
Identification of clinical genes and corresponding predominant polyA sites. In parentheses are the number of genes. A: The overlap of clinical genes from HGMD, OMIM, and CGD databases. B: The overlap of clinical genes found in HGMD, OMIM, and CGD and genes with a predominant polyA site.
Predominant hexamers are less tolerant to variation than control sequences
We hypothesized that predominant hexamers would be critical for gene function and thus less tolerant to population variation. To test this, we compared the occurrence of variants in gnomAD in predominant hexamers versus non-predominant hexamers and versus upstream control sequences (Fig. 4A, see Supplementary Methods). There were 7150 predominant hexamers, 141 561 non-predominant hexamers and 5144 sets of trimers included in this analysis. Q-Q plots showed significant deviation from the expected distributions (Fig. 4B and C). The distribution of allele frequencies in the predominant hexamers were significantly lower than non-predominant hexamers (Mann–Whitney U test p-value < 2.22 × 10−16, Kolmogorov–Smirnov test p-value = 1.08 × 10−09) and the trimer controls (Mann–Whitney U test p-value = 1.93 × 10−09, Kolmogorov–Smirnov test p-value = 1.36 × 10−07). We also observed greater evolutionary conservation in predominant hexamers. Predominant hexamers have significantly higher PhyloP [14] scores compared to secondary hexamers, other hexamers, and trimer controls (Mann–Whitney U test p-value < 2.2 × 10−16, Kolmogorov–Smirnov test p-value < 2.2 × 10−16) (Fig. S2). This indicated that predominant hexamers are likely to be functionally important.
Figure 4.

Constraint analysis and in silico prediction of deleterious variants in predominant hexamers and controls. A: Diagram of predominant hexamers and control groups. B: Q-Q plot of the distribution of minor allele frequency (MAF) of variants observed in predominant hexamers compared to non-predominant hexamer regions (Secondary hexamers and Other hexamers). C: Q-Q plot of the distribution of MAF of variants observed in predominant hexamer regions compared to trimer controls. D, E: Comparison of CADD and FATHMM-MKL scores, respectively, between predominant hexamers, secondary hexamers, other hexamers, other strong hexamers, and trimer controls.
Additionally, we have observed that the number of variants leading to preserved hexamers was significantly higher (Fischer’s Exact test p-value < 0.00001) in predominant hexamers than in non-predominant hexamers, consistent with findings from Findlay et al. ([11], preprint). Of the 2591 variants occurring in predominant hexamers, 1651 (64%) were preserving variants and 940 (36%) were disrupting variants. Of the 49 911 variants occurring in non-predominant hexamers, 18 467 variants (37%) led to preserved hexamers whereas 31 444 variants (63%) led to disrupted hexamers. This indicated that disrupting variants are more constrained in the population in predominant hexamers compared to non-predominant hexamers.
Predominant hexamer variants show higher deleteriousness scores
We hypothesized that a non-coding in silico metapredictor would yield significantly higher deleteriousness scores for variants in predominant hexamers compared to variants in non-predominant hexamers and trimer controls. To test this, we compared CADD and non-coding FATHMM-MKL scores of all possible combinations of SNVs occurring in predominant hexamers, secondary hexamers, other hexamers, other strong hexamers, and trimer controls as defined in the methods (Fig. 4A). Other ‘strong’ hexamers were a subset of other hexamers that only included AATAAA and ATTAAA hexamers. This was to ensure that the deleteriousness scores for the strong hexamers were not diluted due to the inclusion of other weak hexamers. After quality control, 7148 predominant hexamers, 3401 secondary hexamers, 134 104 other hexamers, 50 710 other strong hexamers, and 5142 trimer controls sets were analyzed.
Both CADD and FATHMM-MKL scores were higher for variants in predominant hexamers when compared to each of the control groups (p-values for comparisons had Mann–Whitney U test and Kolmogorov–Smirnov test p-value < 2.22 × 10−16) (Fig. 4D and E). The median CADD score for predominant hexamers was 14.2, whereas the median scores for secondary hexamers, other hexamers, other strong hexamers, and trimer controls were 12.7, 6.4, 6.4, and 7.4, respectively. This pattern was also observed for FATHMM-MKL predictions, with scores >0.5 suggesting deleteriousness and scores <0.5 suggesting neutral effect [15]. The median score for variants in predominant hexamers was 0.93, suggesting deleteriousness of variants. The median scores for secondary hexamers, other hexamers, other strong hexamers, and the trimer controls were 0.89, 0.20, 0.20, and 0.23, respectively. Low CADD and FATHMM-MLK scores for other hexamers, other strong hexamers, and the trimer controls suggest neutral effects of the variants in these regions. Interestingly, both predictions showed that the secondary hexamers upstream of pPAS may be functionally important.
Predominant hexamer variants are associated with perturbations in mRNA processing
To understand the effects of variants occurring in predominant hexamers, 76 individuals from the ClinSeq® cohort were selected for RNA-sequencing based on having at least one hexamer variant (see Materials and Methods). Only variants in predominant hexamers where the wild type hexamer sequence was either AATAAA or ATTAAA were considered. Two types of RNA-sequencing (3′ end sequencing and standard RNA sequencing) were performed. RNA sequencing data that passed QC were available for 65 variants in 64 genes (Table S1, Additional File 2). PAS usage was determined by 3′ end sequencing and analysis of extended RNA products (non-polyadenylated) and overall gene expression was determined by RNA-sequencing. For controls, we randomly selected 60 non-predominant hexamer 3′ UTR variants from the same dataset.
The UCSC browser [16] was used to view aligned 3′ end sequencing data (from control individuals without hexamer variants) and the presence of background APA was manually assessed for the 64 genes with identified predominant hexamer variants and the 60 genes with identified non-predominant hexamer 3′ UTR variants in the ClinSeq® cohort. Overall, >70% (89/125) of the genes used a single PAS for polyadenylation and APA was observed in <30% of the genes (Fig. 5A), this did not differ in the two sets of genes. This would be consistent with the hypothesis that most genes use a single PAS. The changes in PAS usage was determined for the 65 predominant hexamer variants versus 60 control 3′ UTR variants (Fig. S9, Additional File 1). Twenty-five genes (25/64, 39%) with predominant hexamer variants showed changes in APA (Fig. 5B, Table S1, Additional File 2). The changes in polyadenylation were observed irrespective of whether a gene typically used a single PAS or APA was common for the gene. In contrast, no changes in polyadenylation were observed in the 60 controls (Fig. 5B). We conclude that changes in polyadenylation site usage are associated with predominant hexamer variants.
Figure 5.

Comparing the effect of variants in 3′ UTR regions. A: The proportion of genes with single polyA site usage vs. multiple polyA site usage for 64 case genes with hexamer variants and 60 control genes with control variants. B: The effect of predominant hexamer variants and control variants on alternative polyadenylation (APA). Multiple site genes refers to genes with multiple polyA sites, and single site genes refers to genes with one polyA site. C: The effect of predominant hexamer and control variants on mRNA. D: Gene expression analyses in 65 predominant hexamer variants. The lines from top to bottom denote Bonferroni corrected p-value and p-value = 0.05, respectively.
The effect of hexamer variation on RNA transcript extension and gene expression was also determined for these genes and variants. Extended RNA transcripts were observed for eight predominant hexamer variants (8/65, 12%) but none of the genes with control 3′ UTR variants (0/60) (Fig. 5C). Interestingly, these extended transcripts were not supported by additional PAS in 3′ end sequencing data. This suggested that the predominant hexamer variants resulted in loss of polyadenylation, consequentially resulting in longer non-polyadenylated transcript that could be subject to nonsense mediated decay or degradation by miRNA.
No change of expression level was noted for any of the predominant hexamer variants in the dataset (Fig. 5D), however, the predominant hexamer variants identified in MS4A6A, TP53, TRAPPC3, SHISA5, ATP5F1E, ERAP1, DHRS7, IK, SPTLC1, and TMEM176A were previously identified eQTL variants and/or variants showing allele-specific expression [17]. This suggests our cohort size of n = 76 may have been underpowered to detect small expression changes from eQTL variants.
Of the 65 variants identified in the 76 samples with RNA sequence data only three variants were in genes associated with a disorder inherited in an autosomal dominant pattern (PMP22, SPTLC1, and TP53) and all three variants were too common in gnomAD (>1% popmax) to be classified as pathogenic.
In silico predictions in classification of hexamer variants for pathogenicity
Next, we sought to assess if FATHMM-MKL and CADD can be used to discriminate pathogenic variants vs common variants in predominant hexamers. We collected 34 variants in the HGMD database that were considered hexamer variants, one variant in IGF1 was not in the vicinity of the 3′ end of the mRNA and was removed from all analyses. Evidence of pathogenicity was sought for each variant (see methods). Ten variants had sufficient evidence to be classified as likely pathogenic or pathogenic without considering PP3 (bioinformatic prediction of pathogenicity). For control variants, gnomAD variants in predominant hexamer regions with MAF >1% were included.
While the number of available, recognized pathogenic variants is small, we observed higher CADD and FATHMM-MKL scores for pathogenic variants compared to the control variants (Fig. 6A and B). Using a CADD score of ≥10, the likelihood ratio was 2.5 with 100% sensitivity and 60% specificity. For FATHMM-MKL, the likelihood ratio at the score ≥0.7 was 2.86 with 100% sensitivity, and 65% specificity. This suggested that in silico prediction can aid in identifying pathogenic variants at these score thresholds.
Figure 6.
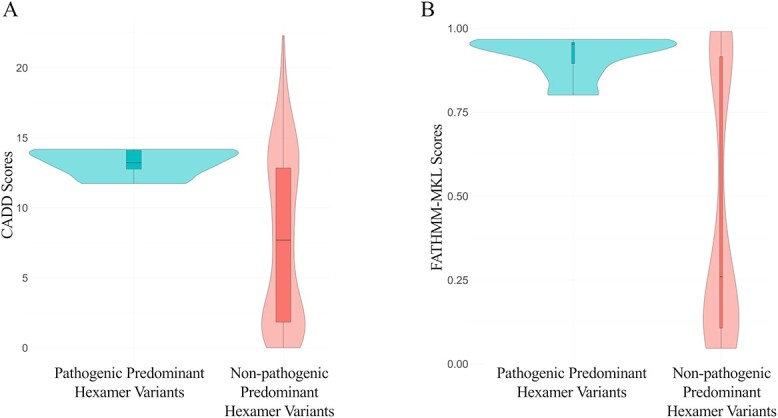
A, B: Comparison of CADD and FATHMM-MKL scores of pathogenic and non-pathogenic predominant hexamer variants, respectively.
Classification of reported hexamer variants according to the adapted ACMG/AMP criteria
Thirty-three hexamer variants noted to be “disease mutations (DM)” in HGMD were annotated for evidence that supported pathogenicity according to ACMG/AMP pathogenicity criteria [18] specified for polyadenylation variants as noted in the methods (IGF1 variant not considered due to insufficient evidence of a hexamer). Nine of the variants had sufficient evidence to support a likely pathogenic classification and four had sufficient evidence to support a pathogenic classification, eight of these variants were reported in multiple unrelated cases. Of the 20 variants that had insufficient evidence, the majority were single case reports with limited case data. Eleven variants would move from VUS to likely pathogenic with the addition of moderate functional data.
Phenotypes in individuals with predominant hexamer variants
Thirty-five individuals in the ClinSeq® cohort had predominant hexamer variants considered to be variants of interest based on frequency in gnomAD, inheritance pattern of associated disease and disease mechanism. Analysis of personal and family history taken at the time of enrollment did not identify associated phenotypes for 32 of the participants. For three participants, potentially related conditions were evident in the personal or family history. An individual with a variant in the ALK gene, associated with susceptibility to neuroblastoma, had a benign brain tumor. An individual with a variant in the NBN gene, associated with uterine smooth muscle tumors and ovarian cancer (among other phenotypes), had a family history of uterine fibroids and uterine cancer. Finally, an individual with a variant in the ABCA1 gene, associated with abnormal cholesterol, had high cholesterol.
Discussion
The underrepresentation of hexamer variants associated with Mendelian disease is likely due to several interrelated factors. First, functionally relevant hexamers are not well-defined for clinical or research laboratories to interrogate. Second, many exomes do not include 3′ UTR regions. Finally, it may be the case that few genes are biologically sensitive to the changes caused by hexamer variants. While this may be a truly uncommon class of pathogenic genetic variation, we strongly suspect that there are more than 14 genes susceptible to this variation. This under-ascertainment could be mitigated as future advances in sequencing technology shifts from exome to genome sequencing, where the 3′ UTR regions would be readily available for interrogation. However, the lack of well-defined, functionally relevant hexamers hinders discovery of pathogenic hexamer variants and advancing the understanding of the role of these variants in disease. Here we have defined a set of predominant hexamers as candidates for Mendelian disease-associated variation, reasoning that predominant hexamers are more likely to be functionally relevant. We have shown predominant hexamers to be constrained, have higher deleteriousness scores by in silico prediction tools, and demonstrated that variants in these predominant hexamers in the ClinSeq® cohort contribute to APA and mRNA extension. The key resource we have provided is a set of predominant hexamers that can be interrogated in clinical and research sequencing, and we have also suggested adaptations of the ACMG criteria to support pathogenicity classification of hexamer variants.
Several databases provide information on PAS and hexamers without consideration of importance or functionality [5–8]. While identification of all potential PAS and hexamers is useful for understanding the mechanism of RNA processing and regulation, for the purpose of interrogation of exome and genome sequence data for association with Mendelian diseases, focusing on highly relevant hexamers will facilitate assessment of variant pathogenicity. To define a list of hexamers for clinical interrogation we used the PolyASite 2.0 database for identification of all PAS and corresponding hexamers and then used a 50% threshold to define a set of pPAS and hexamers. The large tissue diversity in the PolyASite 2.0 database (221 samples across multiple tissue types) allowed the identification of pPAS and related hexamers across tissue types for the majority of protein-coding genes. We suggest that this set of highly used PAS, and the corresponding hexamers, are likely to be functionally relevant and therefore useful for clinical interrogation. Indeed, of the 33 variants identified in HGMD associated with Mendelian diseases, 32/33 are in predominant hexamers. This list of predominant hexamers should be considered tissue-agnostic with the understanding that hexamers that are tissue-specific may be underrepresented. However, work by Shulman et al. showed 69% of polyadenylation QTLs affected more than one tissue with consistent effects across tissues, which may mitigate this limitation [9]. It is likely, however, that some biologically relevant PAS and hexamers were missed in our analyses because they did not meet our threshold of >50% use (2576 or 12.7% of protein coding genes) or genes were not expressed in the tissues in the PolyASite 2.0 database (1664 or 8.2% of protein coding genes). The former could occur if the gene has high APA. As our understanding of gene regulation increases, these PAS can be further investigated.
The predominant hexamers we identified were significantly constrained with higher deleteriousness scores compared to control sequences supporting the identification of functionally important PAS using TPM across datasets. Unexpectedly, we also observed higher constraint and deleteriousness scores for secondary hexamers compared to other control sequences. This suggested that sequences beyond the predominant hexamers near pPAS are conserved and functionally important. It is possible that these secondary hexamers overlap motifs aiding polyadenylation [19–21]. While our study focused on identifying predominant hexamers, the effect of secondary hexamers or APA on the deleteriousness of predominant hexamer variants remains to be investigated.
To understand the implication of variants in predominant hexamers, we analyzed exome and RNA sequence data for the effect of such variants on RNA expression and processing. The variants we identified in predominant hexamers did not alter gene expression, however, 45% of variants resulted in either APA or extended RNA suggesting these predominant hexamers are important in RNA cleavage and polyadenylation. While it is surprising that variation in APA and/or RNA cleavage were not accompanied by changes in RNA expression, RNA expression may not be necessary for clinically relevant perturbation of gene function. A hexamer variant in STUB1 has been shown to affect protein levels without affecting gene expression and polyadenylation [22]. It is also possible that changes in gene expression were present but not large enough to be recognized in this study based on the small sample size as our study was underpowered to detect eQTLs. The remaining 55% of predominant hexamer variants in the sample set did not have a noticeable effect on RNA processing. This may be due to the creation of an alternative functional hexamer sequence as seen with SHISA5 gene (Fig. S3, Additional File 1). Of the 40 variants that resulted in no apparent changes, 31 variants created an alternate hexamer sequence. Finally, the use of peripheral blood for RNA analysis may have masked RNA processing or expression changes that are tissue specific. While the list of predominant hexamers is tissue agnostic and thus generally applicable, it may not capture tissue specific predominant hexamers that may still be of clinical interest.
The functional relevance of predominant hexamers is supported by our findings that all but one of the 33 ‘DM’ hexamer variants reported in HGMD are in a predominant hexamer (the variant in BMP1 is not in the predominant hexamers). A previously proposed set of hexamers for mining clinical variants, captured all 33 ‘DM’ hexamer variants reported in HGMD [10], but included 229 014 hexamers. While such a brute force search of hexamer variants may increase yield, manually interpreting a large number of variants is costly and may not be feasible in a clinical setting. While such an inclusive list can be useful for advancements of understanding polyadenylation mechanism, a more focused list of functionally important and thus clinically relevant hexamers is needed. Our approach identified 15 767 pPAS from >560 000 PAS in the PolyASite 2.0 database, removing ~97% of PAS that are less likely to be clinically important. Our list of predominant hexamers is efficient for researchers focused on diseases with unexplained etiology to interrogate for clinically relevant variants in their disease cohorts.
To identify predominant hexamer variants with clinical effect we analyzed exome data from 1477 individuals. Thirty-five predominant hexamer variants were identified in genes where loss of function variants might contribute to disease. Personal and family history collected at the time of enrollment did not support a contribution to the participant’s phenotype for 32 individuals. However, three individuals had phenotypes that could be related to the gene in question. As polyA variants might be expected to be hypomorphic, rather than complete loss of function, it is possible that resultant phenotypes may be less penetrant or less severe. Looking at the hexamer variants in HGMD, over half of the variants are in globin genes [17], where the levels of gene products are precisely regulated. Of the remaining 14 variants, eight are on the X-chromosome and may be more sensitive to partial loss of function. We suggest this list of predominant hexamers may be especially useful in interrogating patient cohorts where phenotypes suggest specific genetic etiologies and the causative variants have yet to be identified, including individuals with disorders inherited in an autosomal recessive pattern, where the affected individual has only a single identified pathogenic variant.
To support pathogenicity classification of hexamer variants we have suggested specifications to the ACMG/AMP pathogenicity criteria. The ACMG/AMP pathogenicity criteria were meant to be generally applicable to all genes, however, some criteria are not applicable to non-coding variants (PVS1, PS1, PM4, PM5, PP2). As pathogenicity classification is dependent on the number of criteria that can be applied to a variant, dropping criteria from consideration reduces the ability to classify a variant as pathogenic or likely pathogenic. It is useful in the case of non-coding variants to consider if certain criteria can be amended rather than dropped. Ellingford et al. have provided general recommendations for non-coding variants [23] and here we specified them further for polyadenylation signal hexamers. We have suggested a specification for PM5 (same amino acid, specified as same hexamer) that recognizes prior evidence of a different pathogenic variant in the same hexamer. Identification of a pathogenic variant in a hexamer supports the importance of the hexamer in gene function. PM1 and PP3 were also specified. While limited pathogenic variants were available to set a cutoff for a CADD score, comparison of CADD scores of common hexamer variants to pathogenic hexamer variants suggested a cutoff of ≥10 which was implemented. However, as PP3 for non-coding variants considers conservation, which is related to presence in a functional domain (PM1), it was determined that PP3 and PM1 should not both be applied together at full strength until further evidence was available to correctly weight these two criteria for hexamers. We have suggested using PM1 at supporting strength for variants in predominant hexamers. PP3 can be applied for variants with a CADD score ≥10.
Using the ACMG criteria to classify hexamer variants identified in HGMD, 13 were classified as pathogenic or likely pathogenic. All four variants classified as pathogenic were awarded PS4 at full strength for multiple unrelated affected individuals with the variant. Twenty variants were supported by a single case. Case data including segregation (six variants), phenotype specific for gene (23 variants) and presence with a second pathogenic variant in recessively inherited disease (ten variants) were important in classifying variants as likely pathogenic or pathogenic. Ten variants had functional studies of patient cells that contributed toward their classification. While additional functional data could move 11 variants from VUS to likely pathogenic, the opportunity for ex vivo functional studies is limited with many variants being supported by a single case. Thirty-two variants were present in a predominant hexamer and received PM1_Supporting for presence in a functional domain. Finally, 30 variants received PP3 for a CADD score ≥10. We suggest that classification of hexamer variants as pathogenic or likely pathogenic is limited by patient data and could be aided by in vitro functional studies not typically pursued for hexamer variants.
Using the hexamer variants that were classified as pathogenic or likely pathogenic based on our adapted ACMG criteria, we tested CADD and FATHMM-MKL to determine whether in silico predictions can predict pathogenic hexamer variants. We suggest that deleteriousness scores with the specified thresholds (CADD score = 10, FATHMM-MKL score = 0.70) support pathogenicity. While a CADD score of 10 seems low in comparison to the threshold typically used for coding variants, unlike FATHMM-MKL non-coding scores that only considers non-coding variant properties, CADD considers both coding and non-coding variant properties (e.g. amino acid change), and scores are measured in respect to deleteriousness of all variants [24, 25]. Thus, we expect that the CADD scores for pathogenic hexamer variants will be lower than CADD scores for pathogenic coding variants and therefore require different thresholds as compared to coding variants to be considered as evidence for pathogenicity. When classifying variants using the adapted ACMG criteria we suggest using a CADD score  10 in support of pathogenicity. As more pathogenic hexamer variants are identified, the use of CADD versus other bioinformatic predictors, and the exact threshold to be used, can be reassessed.
10 in support of pathogenicity. As more pathogenic hexamer variants are identified, the use of CADD versus other bioinformatic predictors, and the exact threshold to be used, can be reassessed.
In summary, we have identified a set of 15 212 hexamers that are candidates for variation that may be associated with Mendelian genetic disorders. These sites are supported by high polyadenylation usage and are conserved and population constrained. We encourage researchers and clinicians who have access to genome sequencing data to evaluate these sites for disease association using the resources we have provided.
Materials and methods
Identification of pPAS and predominant hexamers for protein-coding genes
To identify pPAS, we used the PolyASite 2.0 database (https://polyasite.unibas.ch/atlas/) [5]. Only the PAS with gene annotations as protein-coding genes defined by Ensembl [26] release 96 were examined. Quality control included re-annotation and removal of intergenic PAS (see Supplementary Methods).
To measure the relative usage of PAS within a gene, we used the quantification of polyA site abundance from PolyA Site 2.0, which was quantified in Transcripts Per Million (TPM). For each polyA site, TPM values were averaged across 221 samples. Usage for each polyA site was calculated as 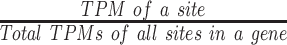 . The pPAS were defined as >50% PAS usage reasoning that PAS likely to cause Mendelian disease would be used in a large percentage of transcripts. Additionally, of the 33 known hexamer variants in HGMD, 32 hexamer variants were associated with PAS with usage >50%. If no PAS reached >50% usage, the gene was not further considered.
. The pPAS were defined as >50% PAS usage reasoning that PAS likely to cause Mendelian disease would be used in a large percentage of transcripts. Additionally, of the 33 known hexamer variants in HGMD, 32 hexamer variants were associated with PAS with usage >50%. If no PAS reached >50% usage, the gene was not further considered.
To identify the candidate hexamers for each pPAS, hexamers occurring within the −60 to +10 region around each pPAS representative position (the position that had the highest number of supporting reads in PolyASite 2.0) were examined. The predominant hexamers were selected based on the strength of the hexamer sequence in polyadenylation [12], relative distance (−21 bp) from the representative position [5, 13, 27], and the distribution of the distances at which the hexamer sequence occurred as the lone hexamer (Fig. 1, see Supplementary Methods).
Constraint analysis of hexamer variants
To assess the population constraint within predominant hexamers, we compared the distribution of minor allele frequencies (MAF) in predominant hexamer regions vs. control regions. For this analysis, we used gnomAD v3.0 and only the single nucleotide variants (SNV) were included. The control regions were defined as Secondary hexamers (hexamers upstream of pPAS that are not the predominant hexamer), Other hexamers (hexamers upstream of non-pPAS), and trimer controls as described in the methods (Fig. 4A). For each predominant hexamer, two trimers from the 3′ UTR upstream of the predominant hexamer, with the same nucleotide composition as the predominant hexamer were selected as controls (see Supplementary Methods, Fig. 4). The distribution of variant frequencies in all positions (inclusive of positions with 0% VAF) within predominant hexamers, non-predominant hexamers (secondary hexamers and other hexamers), and the trimer controls were compared using the Mann–Whitney U test and the Kolmogorov–Smirnov test.
To determine the difference in the number of preserving and disrupting variant locations in predominant hexamers and non-predominant hexamers, we defined a preserving variant as a variant leading one of the 18 recognized hexamers found in PolyA Site 2.0 (See Table S1). Disrupting variants were defined as variants leading to any other hexamer. Multiallelic variants and hexamers with more than one variant in gnomAD were removed. Fischer’s exact test was performed to compare the number of variant locations observed when comparing the two groups.
To assess evolutionary conservation of our predominant hexamers, pre-computed PhyloP [14] scores were downloaded from UCSC (https://hgdownload.cse.ucsc.edu/goldenPath/hg38/phyloP100way/hg38.phyloP100way.bw) and converted to bed format. The phyloP bed file was then intersected with the high quality predominant hexamer, secondary hexamer, other hexamer, and trimer control regions described previously using bedtools.
Key Points
Predominant polyadenylation site (pPAS): Polyadenylation site with >50% usage in a given gene.
Non-predominant polyadenylation site (Non-pPAS): Polyadenylation site with ≤50% usage in a given gene.
Hexamer: Six nucleotide polyadenylation signal sequence motif within 60 nucleotides upstream of a PAS.
Predominant hexamer: Hexamer that is associated with the pPAS.
Other hexamers: Hexamers that are associated with Non-pPAS of a gene.
Other strong hexamers: Subgroup of other hexamers that are either AATAAA or ATTAAA sequence motif.
Secondary hexamers: Hexamers upstream of pPAS that are not predominant hexamers.
Non-predominant hexamers: All hexamers that are not predominant hexamers including secondary hexamers and other hexamers.
Sequencing and data processing
Exome data for 1477 ClinSeq® participants were queried for variants in hexamers. Based on the presence of variants in hexamers 76 individuals were selected for RNA sequencing. Two types of RNA sequencing were performed on the 76 samples to study the effects of predominant hexamer variants. RNA-sequencing (TruSeq Stranded mRNA kit, Illumina) was performed to examine gene expression and extension of mRNA. 3′-end sequencing (Quantseq_REV, Lexogen, Greenland, NH) was performed to identify and measure the PAS usage (see Supplementary Methods). Total RNA was isolated from whole blood using the PAXgene Blood RNA system (Qiagen, Gaithersburg, MD), prepared as described in the Supplementary Methods, and sequenced at the NIH Intramural Sequencing Center (NISC) on a NovaSeq 6000 with v1.0 reagents (Illumina).
FASTQ files were aligned to hg19 using STAR v.2.7.3a [28]. For RNA sequencing, BAM files were aligned to the transcriptome. Duplicate reads were marked with Picard v.2.22.2 [29], and gene expression was quantified using RSEM v.1.3.2. For 3′ end sequencing, the PolyASite 2.0 pipeline was used to identify PAS and hexamers (see Supplementary Methods).
Assessing the effect of hexamer SNVs
The effect of predominant hexamer SNVs was compared to selected control SNVs as described in the Supplementary Methods. For each SNV, the following were examined: 1) the usage of alternative polyadenylation (APA), 2) mRNA extension, 3) the effect of the SNV on gene expression. The usage of APA referred to alternative polyadenylation which resulted in either shorter or longer transcripts. The mRNA extension referred to the elongated extension of mRNA beyond the defined transcript. The usage of APA and the extension of mRNA due to the SNV was examined on the UCSC Genome Browser. The gene expression analyses of samples with and without the SNVs were compared by TPM values of RNA sequencing data using ANOVA. Usage of APA was compared to baseline APA for the gene. A gene was considered to have baseline APA when a second peak was observed at a non-pPAS, in ≥10% of control samples, with the peak height ≥10% of the height of the primary peak in the UCSC Genome Browser. The threshold of 10% of the height of the primary peak was set to consider any noise or artifactual peaks that may arise. In cases where peaks overlapped two identified PAS, that peak was counted as a single peak.
Known allele-specific expression variants and eQTLs were determined using GTEx data. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal (https://gtexportal.org/home/datasets) on 04/30/2018 and dbGaP accession number phs000424.v8.p2 on 08/20/2019. eQTLs were retrieved from the GTEx Portal v.8 https://storage.googleapis.com/gtex_analysis_v8/single_tissue_qtl_data/GTEx_Analysis_v8_eQTL.tar) and were intersected with hexamers using bedtools. WASP-corrected ASE expression matrices were downloaded from dbGAP.
Assessing deleteriousness of predominant hexamer variants using in silico tools
To assess the potential deleteriousness of variants in our predominant hexamers compared to variants in control regions, we compared the score distributions from FATHMM-MKL [15] and CADD [24, 25]; two widely used in silico tools used to predict the deleteriousness of non-coding variants. Both these tools take conservation into account when predicting the deleteriousness of variants. The control regions were defined as secondary hexamers, strong (AATAAA and ATTAAA) hexamers upstream of non-pPAS, other hexamers upstream of non-pPAS, and the trimer controls as described in the methods (Fig. 4A). Each group was filtered and lifted over to hg19 as described in methods. Control hexamers that overlapped pPAS and predominant hexamers were removed. Phred-scaled CADD v1.6 and non-coding FATHM-MKL scores were retrieved using Ensembl VEP v103 [30]. Every possible combination of SNVs at each unique position was included.
To assess the ability of FATHMM-MKL and CADD tools to predict the pathogenicity of predominant hexamer variants, we defined a set of pathogenic/likely pathogenic predominant hexamer variants and non-pathogenic predominant hexamer variants and compared the scores by calculating the likelihood ratio. Predominant hexamer variants listed as disease mutations in HGMD [4] v.2021.3 were assessed for pathogenicity classification as described below (Table 1). Ten variants that reached a classification of pathogenic/likely pathogenic without the use of bioinformatic evidence (PP3) were used in this analysis. Non-pathogenic predominant hexamer variants were defined as variants in predominant hexamers in gnomAD v 2.1.1 with MAF >1%, excluding variants with MAF >50%. As FATHMM-MKL predicts deleteriousness scores for SNVs only, eight SNVs (excluding two indel variants) were included for non-coding FATHMM-MKL, and all ten SNVs and indels were included for CADD prediction scores.
Determination of clinically significant genes
Genes from HGMD release 2023.1 (https://my.qiagendigitalinsights.com/bbp/ last accessed April 6, 2020), OMIM [31] (https://omim.org/downloads), and CGD [32] (https://research.nhgri.nih.gov/CGD/download/CGD.txt) were downloaded and converted to HGNC IDs. The list of clinically significant genes included OMIM genes with an inheritance pattern and phenotype, HGMD genes with a ‘DM’ variant annotation, and all CGD genes.
Classification of variant pathogenicity using ACMG/AMP pathogenicity criteria
Hexamer variants designated as “DM, Disease Mutations” in HGMD were classified using the American College of Medical Genetics and Genomics and Association for Molecular Pathology rules for pathogenicity classifications, Table 1. ClinVar was queried for hexamer variants (October 13, 2021), however, no additional variants assessed as likely pathogenic or pathogenic were identified. Specifications to the ACMG/AMP criteria [23] used to assess variants can be found in Table 2. Table 2 details all criteria considered for pathogenicity assessment of hexamers including the original ACMG/AMP criteria, recommendations from the ClinGen Sequence Variant Interpretation Working Group (SVI web page https://clinicalgenome.org/working-groups/sequence-variant-interpretation/) and ClinGen Variant Curation Expert Panels (Cardiology [33], Malignant Hyperthermia [34] and specifications specific to polyA signal hexamers. Of note, Ellingford et al. [23] has published general specifications for non-coding variants, many of which overlap with our recommendations; however, our recommendations are further specified for polyA hexamer variants. Several criteria do not apply to hexamer variants including PVS1 for loss of function variants, PS1 for same amino acid change, PM4 for protein length change, PP2 for missense variant in gene with low rate of missense variation, BP1 for missense variant in gene where loss of function is the known mechanism of disease, BP3 for in-frame insertions or deletions, and BP7 for synonymous variants. Several other criteria were specified for variants in hexamers. For PS3/BS3, functional studies that show reduced RNA or protein levels in patient cells can be used as evidence to support pathogenicity. For genes where haploinsufficiency is a known mechanism of disease, PS3_moderate can be awarded when three or more heterozygous cell lines from unrelated individuals show a >25% reduction in mRNA and/or protein levels as compared to wildtype; PS3_Supporting can be awarded when one or two heterozygous cell lines from unrelated individuals show a >25% reduction in mRNA and/or protein levels as compared to wildtype. If protein and/or mRNA levels in cell lines from two unrelated individuals are shown to be comparable to wildtype, BS3_Moderate can be awarded. If protein and/or mRNA levels in a cell line from a single individual is shown to be comparable to wildtype, BS3_Supporting can be awarded. For PS3 and BS3 it may be important to make sure the tissue available for testing is the relevant tissue for disease [23]. For PM5, typically used for missense variants in the same codon, it was determined that a variant in a hexamer where a previous variant had been classified as pathogenic can be awarded PM5 at a supporting level assuming the new hexamer sequence is predicted to have equivalent or less polyadenylation activity as compared to the previously classified pathogenic variant (see Table 3). Insertion and deletion variants that recreate a variant hexamer should be interpreted the same as single nucleotide variants, insertion and deletion variants that disrupt the hexamer can be awarded PM5_Supporting. PM1, presence in a well-established functional domain, is awarded at supporting for variants in predominant hexamers. PP3 can be awarded for variants with a CADD score ≥10. A CADD score <5 can be used in support of benign status, BP4. Recent guidance by ClinGen has focused on setting criteria strength levels using likelihood ratios based on representative benign and pathogenic variants. An adequate truth set of variants are not available for hexamer variants so these suggested criteria should be revisited over time as more pathogenic hexamer variants are identified.
Table 2.
ACMG/AMP criteria used for polyadenylation signal hexamer variant pathogenicity classification (see Supplementary Information for full explanations).
| Criteria | Criteria Description | Modification |
|---|---|---|
| Pathogenic Criteria | ||
| PVS1 | Loss of function allele in a gene where loss of function is a known mechanism of disease. | Not Applicable |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | Not Applicable |
| PS2/PM6 |
De novo occurrence in an individual with disease and no family history. Each proven de novo case, count for 2 points, each assumed de novo case, count for 1 point. Very Strong: ≥8 points Strong: 4–7 points Moderate: 2–3 points Supporting: 1 point |
Nonea |
| PS3 | Well-established functional studies supportive of a damaging effect on gene or gene product. Moderate: For genes where loss of function is a known mechanism of disease, decreased RNA or protein levels (<75% of WT) in three or more cell lines from unrelated individuals who harbor that variant Supporting: For genes where loss of function is a known mechanism of disease, decreased RNA or protein levels (<75% of WT) in cells from affected individual who harbors the variant |
Variant type Specific |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls. If specifications have been provided by a ClinGen expert panel, case counting should consider their recommendation for strength. Popmax MAF in gnomAD should be <0.0006 to use these guidelines. For odds ratio calculations gnomAD can be used as a control set. Strong: ≥7 unrelated cases with associated condition. For variants with ≥7 unrelated cases an odds ratio can be calculated to determine strength level, an odds ratio ≥18.3 allows for PS4 to be used at strong. Moderate: 2–6 unrelated cases with associated condition. For variants with 2–6 unrelated cases an odds ratio can be calculated to determine strength level, an odds ratio ≥4.8 allows for PS4 to be used at moderate. Supporting: One case with associated condition. |
Strengthb |
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain. Downgraded to avoid overcounting with PP3. Supporting: Use for variants in predominant hexamers. |
Variant type Specific |
| PM2 | Absent from controls (gnomAD). Incorporated into PS4, do not consider separately unless ClinGen expert panel specifications for PS4 are used and PM2 is incorporated into those specifications. |
Not Applicable |
| PM3 | Variants in trans with pathogenic variant for recessive disorders. An individual cannot be counted for both PM3 and PS4. Example weighting below, see ClinGen Sequence Variant Interpretation Recommendation for in trans Criterion for complete explanation. Moderate: Identified with pathogenic variant in trans, phase known OR identified in homozygous state in two unrelated affected individuals. Supporting: Identified homozygous state in an affected individual. |
Nonea |
| PM4 | Protein length change. | Not Applicable |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. Consider novel changes in hexamer sequence. Supporting: Single nucleotide variant in a hexamer where a different single nucleotide variant was previously determined to be likely pathogenic. New hexamers must be in lower functional group as predicted by Sheets et alc Previously established likely pathogenic variant must reach a classification of pathogenicity without PM5. |
Variant type Specific |
| PP1 | Co-segregation with disease in multiple affected family members. Strong: Co-segregation with disease in ≥7 reported meioses Moderate: Co-segregation with disease in 5–6 reported meioses Supporting: Co-segregation with disease in 3–4 reported meioses |
Noned |
| PP2 | Missense variant in gene with low rate of benign missense variants | Not Applicable |
| PP3 | Computational evidence suggests impact on gene or gene product. Supporting: CADD score of ≥10. |
Variant type Specific |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology. | Nonee |
| Benign Criteria | ||
| BA1 | Allele frequency is >0.05 in any general continental population dataset of at least 2000 observed alleles and found in a gene without a gene- or variant-specific BA1 modification. If specifications have been provided by an expert panel BA1 should be determined as set by the expert panel for the gene. |
Nonee |
| BS1 | Popmax allele frequency greater than expected for the disorder. If specifications have been provided by an expert panel BS1 should be determined as set by the expert panel for the gene. |
Nonee |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder with full penetrance expected at an early age. | Nonee |
| BS3 | Well-established functional studies show no damaging effect on protein function Moderate: No reduction in RNA or protein level in three or more cell lines from unrelated individuals who harbor the variant. Supporting: No reduction in RNA or protein level in cells from an individual who harbors the variant. |
Variant type Specific |
| BS4 | Lack of segregation in family members. | Nonee |
| BP1 | Missense variant in a gene for which loss of function is known mechanism of disease. | Not Applicable |
| BP2 | Observed in cis with a pathogenic variant in any inheritance pattern. | Nonee |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function. | Not Applicable |
| BP4 | Computational evidence suggests no impact on gene or gene product. Supporting: CADD score of <5.0. |
Variant type Specific |
| BP5 | Variant found in a case with an alternate molecular basis for disease. | Nonee |
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved | Not Applicable |
Criteria that have been modified for polyA variants are noted in the modification column. For criteria included in the original ACMG/AMP framework but not used in these specified criteria, the row is shown in gray.
bJohnston et al. [34].
cSee Sheets et al. Table 3.
dKelly et al. [33].
eRichards et al. [18].
Table 3.
Polyadenylation activity for 17 variant hexamers as compared to AAUAAA, hexamers have been grouped according to polyadenylation activity.
| Sequence | Polyadenylation (% AAUAAA) | Classification Group |
|---|---|---|
| AAUAAA | 100 | Group 6 |
| AUUAAA | 77 ± 4.7 | Group 5 |
| AGUAAA | 29 ± 8.1 | Group 4 |
| CAUAAA | 18 ± 6.4 | Group 3 |
| UAUAAA | 17 ± 3.0 | Group 3 |
| ACUAAA | 11 ± 6.0 | Group 2 |
| GAUAAA | 11 ± 1.0 | Group 2 |
| AAUACA | 11 ± 2.3 | Group 2 |
| AAUAUA | 10 ± 2.3 | Group 2 |
| AAGAAA | 6.0 ± 1.0 | Group 1 |
| AACAAA | 4.0 ± 2.0 | Group 1 |
| AAAAAA | 4.6 ± 3.7 | Group 1 |
| AAUGAA | 4.3 ± 0.6 | Group 1 |
| AAUCAA | 4.0 ± 1.7 | Group 1 |
| AAUAAC | 3.7 ± 1.5 | Group 1 |
| AAUAGA | 3.3 ± 1.5 | Group 1 |
| AAUUAA | 2.3 ± 0.6 | Group 1 |
| AAUAAG | 1.7 ± 0.6 | Group 1 |
PS1/PM5 can be awarded at a moderate level when a variant creates a hexamer that falls into the same (or lower) group as the previously classified variant. Sheets et al.
Statistical analyses and liftover
Statistical analyses (Mann–Whitney U test, Kolmogorov–Smirnov test, ANOVA) and graph generation was performed using R v.3.6.2 [35]. Box plots, violin plots, and Q-Q plots were generated using R library ggplot2 v.3.3.1 [36]. Likelihood ratios were calculated as described [37]. Liftover was performed using Crossmap v.0.5.4 [38].
Examining for associated phenotype in individuals with predominant hexamer variants
Exome data was available on 1477 ClinSeq® participants and was assessed for rare variants in predominant hexamers (<0.2% in gnomAD popmax, 194 variants). Variants were further filtered to remove variants in genes primarily associated with disorders with autosomal recessive inheritance (69 variants), variants in genes associated with childhood disease unlikely to be present in an adult cohort (32 variants), variants common in the cohort (<0.2% in cohort, four variants) and variants in genes with limited loss of function variants reported as “disease mutations (DM)” in HGMD (54 variants). Thirty-five variants were determined to be variants of interest. Personal and family history of participants with a variant of interest were assessed for associated disease.
Supplementary Material
Acknowledgements
The authors thank the members of the ClinGen Sequence Variant Interpretation Working group for productive discussions on the variant pathogenicity criteria. The authors thank Peter Stenson for his work in performing the search of existing disease variants in hexamers.
Contributor Information
Henoke K Shiferaw, Center for Precision Health Research, National Human Genome Research Institute, National Institutes of Health, 50 South Drive, Bethesda, MD 20892, United States.
Celine S Hong, Center for Precision Health Research, National Human Genome Research Institute, National Institutes of Health, 50 South Drive, Bethesda, MD 20892, United States.
David N Cooper, Institute of Medical Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, United Kingdom.
Jennifer J Johnston, Center for Precision Health Research, National Human Genome Research Institute, National Institutes of Health, 50 South Drive, Bethesda, MD 20892, United States.
NISC, NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, National Institutes of Health, Bethesda, MD 20892, United States.
Leslie G Biesecker, Center for Precision Health Research, National Human Genome Research Institute, National Institutes of Health, 50 South Drive, Bethesda, MD 20892, United States.
Conflict of Interest statement: L.G.B. is a member of the Illumina Medical Ethics Committee, receives research support from Merck, Inc., and is a compensated editor for Cold Spring Harbor Laboratory Press and Wolters-Kluwer.
Funding
This work was supported by the National Human Genome Research Institute [HG200359-12 to HS, CH, JJJ and LGB].
Data availability
The refined list of pPAS and corresponding predominant hexamers are included in Supplementary Material with this publication, publicly available for download in BED file format on our github page (https://github.com/BieseckerLab/PolyAProject), and for view through UCSC public track hubs at https://genome.ucsc.edu/cgi-bin/hgHubConnect under the name “Predominant PAS and predominant hexamers“. ClinSeq® sequencing data are available on dbGaP (phs000971.v3.p1).
References
- 1. Colgan DF, Manley JL. Mechanism and regulation of mRNA polyadenylation. Genes Dev 1997;11:2755–66. [DOI] [PubMed] [Google Scholar]
- 2. Proudfoot N. Poly(A) signals. Cell 1991;64:671–74. [DOI] [PubMed] [Google Scholar]
- 3. Keller W, Bienroth S, Lang KM. et al. Cleavage and polyadenylation factor CPF specifically interacts with the pre-mRNA 3′ processing signal AAUAAA. EMBO J 1991;10:4241–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stenson PD, Mort M, Ball EV. et al. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herrmann CJ, Schmidt R, Kanitz A. et al. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res 2020;48:D174–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. You L, Wu J, Feng Y. et al. APASdb: a database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res 2015;43:D59–D67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang R, Nambiar R, Zheng D. et al. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res 2018;46:D315–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muller S, Rycak L, Afonso-Grunz F. et al. APADB: a database for alternative polyadenylation and microRNA regulation events. Database (Oxford) 2014;2014. 10.1093/database/bau076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shulman ED, Elkon R. Systematic identification of functional SNPs interrupting 3′ UTR polyadenylation signals. PLoS Genet 2020;16:e1008977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen M, Wei R, Wei G. et al. Systematic evaluation of the effect of polyadenylation signal variants on the expression of disease-associated genes. Genome Res 2021;31:890–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Findlay SD, Romo L, Burge CB. Quantifying negative selection in human 3′ UTRs uncovers constrained targets of RNA-binding proteins. bioRxiv. in press 2022; 2022.2011.2030.518628. [DOI] [PMC free article] [PubMed]
- 12. Sheets MD, Ogg SC and Wickens MP. Point mutations in AAUAAA and the poly (a) addition site: effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res 1990;18:5799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gruber AJ, Schmidt R, Gruber AR. et al. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res 2016;26:1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pollard KS, Hubisz MJ, Rosenbloom KR. et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shihab HA, Rogers MF, Gough J. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015;31:1536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kent WJ, Sugnet CW, Furey TS. et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Consortium, G.T . The genotype-tissue expression (GTEx) project. Nat Genet 2013;45:580–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darmon SK and Lutz CS. Novel upstream and downstream sequence elements contribute to polyadenylation efficiency. RNA Biol 2012;9:1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nunes NM, Li W, Tian B. et al. A functional human Poly(A) site requires only a potent DSE and an A-rich upstream sequence. EMBO J 2010;29:1523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hall-Pogar T, Zhang H, Tian B. et al. Alternative polyadenylation of cyclooxygenase-2. Nucleic Acids Res 2005;33:2565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turkgenc B, Sanlidag B, Eker A. et al. STUB1polyadenylation signal variant AACAAA does not affect polyadenylation but decreasesSTUB1translation causing SCAR16. Hum Mutat 2018;39:1344–48. [DOI] [PubMed] [Google Scholar]
- 23. Ellingford JM, Ahn JW, Bagnall RD. et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med 2022;14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kircher M, Witten DM, Jain P. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rentzsch P, Schubach M, Shendure J. et al. CADD-splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 2021;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cunningham F, Allen JE, Allen J. et al. Ensembl 2022. Nucleic Acids Res 2022;50:D988–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beaudoing E, Freier S, Wyatt JR. et al. Patterns of variant polyadenylation signal usage in human genes. Genome Res 2000;10:1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobin A, Davis CA, Schlesinger F. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broad Institute . Picard Tools. Broad Institute, GitHub Repository. 2019. http://broadinstitute.github.io/picard/.
- 30. McLaren W, Gil L, Hunt SE. et al. The Ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amberger JS, Bocchini CA, Schiettecatte F. et al. Omim.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res 2015;43:D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solomon BD, Nguyen AD, Bear KA. et al. Clinical genomic database. Proc Natl Acad Sci U S A 2013;110:9851–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kelly MA, Caleshu C, Morales A. et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen's Inherited Cardiomyopathy Expert Panel. Genet Med 2018;20:351–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnston JJ, Dirksen RT, Girard T. et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet Med 2021;23:1288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Team, R.C . R Foundation for Statistical Computing. Vienna: Austria, 2017, in press. [Google Scholar]
- 36. Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag, 2016. [Google Scholar]
- 37. Pejaver V, Byrne AB, Feng BJ. et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet 2022;109:2163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao H, Sun Z, Wang J. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014;30:1006–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The refined list of pPAS and corresponding predominant hexamers are included in Supplementary Material with this publication, publicly available for download in BED file format on our github page (https://github.com/BieseckerLab/PolyAProject), and for view through UCSC public track hubs at https://genome.ucsc.edu/cgi-bin/hgHubConnect under the name “Predominant PAS and predominant hexamers“. ClinSeq® sequencing data are available on dbGaP (phs000971.v3.p1).