

Key Points
-
•
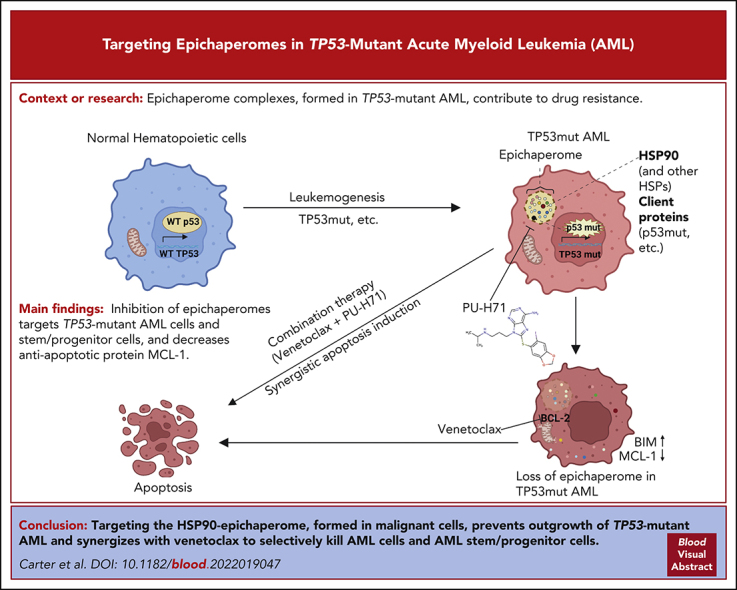
The HSP90 epichaperome is detected in TP53-mutant AML and AML stem/progenitor cells but not in healthy BM.
-
•
Targeting the epichaperome prevents outgrowth of TP53-mutant AML and kills AML bulk and stem/progenitor but not healthy BM cells.
Visual Abstract
Abstract
TP53-mutant acute myeloid leukemia (AML) remains the ultimate therapeutic challenge. Epichaperomes, formed in malignant cells, consist of heat shock protein 90 (HSP90) and associated proteins that support the maturation, activity, and stability of oncogenic kinases and transcription factors including mutant p53. High-throughput drug screening identified HSP90 inhibitors as top hits in isogenic TP53–wild-type (WT) and -mutant AML cells. We detected epichaperomes in AML cells and stem/progenitor cells with TP53 mutations but not in healthy bone marrow (BM) cells. Hence, we investigated the therapeutic potential of specifically targeting epichaperomes with PU-H71 in TP53-mutant AML based on its preferred binding to HSP90 within epichaperomes. PU-H71 effectively suppressed cell intrinsic stress responses and killed AML cells, primarily by inducing apoptosis; targeted TP53-mutant stem/progenitor cells; and prolonged survival of TP53-mutant AML xenograft and patient-derived xenograft models, but it had minimal effects on healthy human BM CD34+ cells or on murine hematopoiesis. PU-H71 decreased MCL-1 and multiple signal proteins, increased proapoptotic Bcl-2-like protein 11 levels, and synergized with BCL-2 inhibitor venetoclax in TP53-mutant AML. Notably, PU-H71 effectively killed TP53-WT and -mutant cells in isogenic TP53-WT/TP53-R248W Molm13 cell mixtures, whereas MDM2 or BCL-2 inhibition only reduced TP53-WT but favored the outgrowth of TP53-mutant cells. Venetoclax enhanced the killing of both TP53-WT and -mutant cells by PU-H71 in a xenograft model. Our data suggest that epichaperome function is essential for TP53-mutant AML growth and survival and that its inhibition targets mutant AML and stem/progenitor cells, enhances venetoclax activity, and prevents the outgrowth of venetoclax-resistant TP53-mutant AML clones. These concepts warrant clinical evaluation.
Epichaperome complexes, comprising heat shock protein 90 (HSP90) and associated proteins, act to maintain malignant phenotypes and are enriched in acute myeloid leukemia (AML) stem cells. After identifying HSP90 inhibitors as top hits in a high-throughput drug screen, Carter et al found that targeting the epichaperome with the HSP90 inhibitor PU-H71 boosts the effectiveness of venetoclax in treating AML, inhibiting the outgrowth of venetoclax-resistant TP53-mutant AML in vivo in murine models. These data support clinical investigation of PU-H71 with venetoclax in patients with TP53-mutant AML, where prognosis has remained dismal despite the introduction of targeted therapies.
Introduction
TP53-mutant acute myeloid leukemia (AML) has an extremely poor prognosis and, essentially, does not respond to available therapies,1, 2, 3, 4 including BCL2 inhibition by venetoclax combined with hypomethylating agents or chemotherapy.5,6 After exposure to therapeutic agents, TP53 mutations become dominant, conferring resistance to multiple therapies.7,8 Molecular analyses of venetoclax/hypomethylating agent–treated samples from patients with AML helped identify mutations in TP53, FLT3, PTPN11, and RAS as key determinants of resistance and early relapse.5 Thus, therapies that are effective regardless of the mutation status of TP53 and kinases in the cells constitute an urgent unmet need.
Heat shock protein 90 (HSP90) chaperone is a key regulator of proteostasis in healthy cells and assists in the folding and maturation of “client” proteins, including kinases and transcription factors.9,10 Epichaperome complexes, formed in malignant cells,11, 12, 13 consist of HSP90, other HSPs, cochaperones, and associated proteins that support disease phenotypes by stabilizing oncogenic kinases, including FLT3, SHP2, and RAF,14, 15, 16, 17 and transcription factors such as mutant p53.9,18,19 Thus, epichaperomes maintain aberrant signaling networks essential for the growth, survival, and therapy resistance of malignant cells. In AML, many oncogenic kinases20, 21, 22, 23 are associated with and supported by epichaperomes. Moreover, stem cells from TP53-mutant AML have activated JAK/STAT signaling,24 and epichaperomes can potentiate oncogenic mutations in TP53-mutants,18,25 thereby contributing to leukemogenesis and drug resistance. We previously reported that HSP90 regulator heat shock factor (HSF)-1 can be targeted in AML and that HSP90 inhibition effectively targets FLT3-mutant AML, even those with acquired venetoclax resistance.26,27
Although HSP90 has been recognized as a promising anticancer target for decades, previous efforts to target HSP90 clinically had limited success, partially, because of toxicities related to the lack of specificity for cancer cells.28 Unlike other adenosine triphosphate–competitive HSP90 inhibitors, PU-H71 (zelavespib) selectively binds to the altered adenosine triphosphate–binding site of HSP90 when incorporated into epichaperomes, with greatly reduced binding affinity for free HSP90. PU-H71 potently kills cancer cells by targeting cancer-specific signaling pathways and has a broader therapeutic window in animal models than nonselective HSP90 inhibitors.13,29, 30, 31, 32, 33, 34, 35 Importantly, PU-H71 has limited activity against healthy CD34+ cells and peripheral blood (PB) leukocytes,13,35 and it was well tolerated in clinical trials, in which changes in cancer-associated protein levels demonstrated its target engagement.36,37 A phase 1b clinical trial of PU-H71 in combination with ruxolitinib in patients with myelofibrosis was conducted at MD Anderson Cancer Center.38 Additionally, a patient with relapsed/refractory AML with a PML-SYK fusion gene who had progressed from myeloproliferative neoplasm achieved long-lasting complete remission after receiving compassionate PU-H71 treatment.39 These data support therapeutically targeting epichaperomes with PU-H71 in TP53-mutant AML, an activity that has not been investigated.
In this study, we identified epichaperomes in TP53-mutant AML and AML stem/progenitor cells and demonstrated that selective targeting of epichaperomes with PU-H71 has antileukemia activity, synergizes with venetoclax in AML and AML stem/progenitor cells with TP53-mutations, and has minimal effects on normal hematopoiesis in vitro and in vivo. Importantly, we mimicked the clinical situation of initially infrequent TP53-mutant cells that outgrow under therapeutic selection pressure and demonstrated that PU-H71, but not BCL-2 or MDM2 inhibition, was able to suppress both TP53–wild-type (WT) and -mutant cells.
Methods
Cells, cell culture, and treatment
Molm13 cells (FLT3-mutant) were purchased from the German Collection of Microorganisms and Cell Cultures. TP53-R248W Molm13 cells,40 MCL-1 overexpression/knockdown cells,41 and TP53-knockout (KO) and TP53-mutant (R175H, Y220C, M237I, R248Q, R273H, and R282W) Molm13 and K562 cells42 were generated as previously described. Samples from patients with TP53-mutant AML (supplemental Table 1; available on the Blood website) and healthy bone marrow (BM) donors were obtained after written informed consent following protocols approved by MD Anderson’s institutional review board and in accordance with the Declaration of Helsinki. Healthy BM-derived mesenchymal stromal cells (MSCs) were obtained as described previously.43 Cell lines and mononuclear cells isolated from primary samples via density-gradient centrifugation were cultured as previously described.44 Cell lines and primary cells cocultured with MSCs (AML:MSC ratio, 4:1) were treated with PU-H71, venetoclax, venetoclax/PU-H71, or nutlin3a. PU-H71, fluorescein isothiocyanate (FITC)-PU-H71, and FITC-control were provided by Samus Therapeutics Inc. Clonogenic assay was conducted as described.45
High-throughput drug screening
We performed high-throughput drug screening to identify compounds that kill cells independent of TP53 mutation status. TP53-WT and -mutant (R175H and R248Q, respectively) Molm13 cells42 were treated with 2398 compounds/drugs (supplemental Table 2) as detailed in the supplemental Methods.
Epichaperome detection
Epichaperomes were identified as previously described,46 using FITC-PU-H71 as probe. Results were expressed as the mean fluorescence intensity difference between FITC-PU-H71 and FITC-control.
Cell viability was determined as previously described.44,47 For AML cells cocultured with MSCs, annexin V/7-aminoactinomycin D positivity was determined in leukemia cells (CD45+) and stem/progenitor cells (CD34+CD38–/CD34+CD38+) and expressed as specific apoptosis as previously described.48
Immunoblot analysis
Protein levels were determined via immunoblot analysis, as previously described.44 Sources of antibodies are listed in supplemental Table 3.
High-parametric cell death assay
We used a high-parametric cell death assay,49 modified from the one previously described,50 to assess cell fates. Briefly, cells treated with PU-H71 at specified concentrations were stained with a collection of antibodies for detecting various cell death modes and cell stress responses and subjected to multiparametric flow cytometry analysis. Details are shown in the supplemental Methods.
In vivo experiments
Mouse experiments were conducted following MD Anderson’s Institutional Animal Care and Use Committee–approved protocols. NSG mice (females, aged 9-10 weeks) injected with luciferase-/green fluorescent protein (GFP)-labeled TP53-R248W Molm13 cells (0.45 × 106 cells) via tail veins were treated with vehicle (n = 4), 30 mg/kg PU-H71 (n = 3), or 50 mg/kg PU-H71 (n = 3) (intraperitoneal) every other day (3 times per week) after engraftment confirmed via noninvasive luciferase imaging, using the IVIS-200 system (Xenogen). Disease progression and treatment responses were monitored via the imaging. Mouse survival was tracked. Leukemia-free NSG mice (6- or 8-week-old females) were treated with vehicle or 50 mg/kg PU-H71 (n = 3/group), 3 times per week. Red and white blood cell counts and hemoglobin levels in PB were determined using an ABX Pentra 60 C+ hematology analyzer (Horiba Medical). NSG mice (6- or 8-week-old females) injected with luciferase-/GFP-labeled TP53-WT Molm13 (0.40 × 106 cells) plus blue fluorescent protein (BFP)-labeled TP53-R248W Molm13 cells (0.04 × 106 cells) were treated with vehicle, 50 mg/kg venetoclax (5 days with and 2 days without it, oral gavage), 50 mg/kg PU-H71 (3 times per week), or the combination (n = 5 per group). Disease progression and treatment responses were monitored with luciferase imaging. Additionally, mice (n = 3 per group) were treated as described earlier and euthanized at the end of the 3-week treatment. GFP+ and BFP+ cells in the PB and BM were determined via flow cytometry. NSGS mice (6- or 8-week-old males) bearing TP53-mutant patient-derived xenograft cells (2 × 106 cells per mouse) were treated with vehicle, venetoclax, PU-H71, or the combination (n = 10 per group) after engraftment.48 Disease progression and treatment responses were monitored via flow cytometry of human CD45+ cells. All mice were obtained from The Jackson Laboratory.
Statistics
Cell line experiments were performed in triplicates, unless otherwise specified; results were expressed as mean ± standard errors of the mean. We used CalcuSyn software to determine the mean combination index (CI) values51 for the median effective dose (ED50), ED75, and ED90. CI < 1 indicated a synergistic; CI = 1, an additive; and CI > 1, an antagonistic effect. Drug screening was performed in duplicates. Results were expressed as area over the curve lethal dose, a value that describes the toxic effect of the drug, with 0 as nontoxic and 1 as highly toxic, calculated by integrating the values over the curve and dividing them by the lethality area. Student t test was used to assess differences between groups, and P values ≤ .05 were considered statistically significant. Mouse survival was estimated using the Kaplan–Meier method. Survival data were analyzed using the log-rank test.
Results
HSP90 is a therapeutic target in AML cells independent of TP53 status
High-throughput drug screening revealed ∼200 agents out of 2400 with pan-toxicity in TP53-WT, TP53-R175H, and TP53-R248Q Molm13 cells (ranked from the most to the least effective; supplemental Table 2). HSP90 inhibitors AT13387, PU-H71, and 17-AAG were ranked as 2.1%, 3.4%, and 3.6% of the top-most active drugs based on the area over the lethal (ie, negative) region of the growth rate index vs concentration response curves (area over the curve lethal dose; Figure 1A). This metric specifically emphasizes drugs that can suppress viable cell counts from baseline and not those that are cytostatic.
Figure 1.

HSP90 is a therapeutic target independent of TP53 gene mutation status, and the epichaperome is present in TP53-mutant AML cells and AML stem/progenitor cells. (A) High-throughput screening of ∼2400 approved drugs identified ∼200 compounds with activity in TP53-WT, TP53-R248Q, and TP53-R175H Molm13 cells independent of TP53 mutation status (left, red indicates HSP90 inhibitors). The error bars are the SEM of AOC_LD of the 3 cell lines. Three agents targeting HSP90 are top hits (right). Viable cell counts were normalized to the median cell count at the time of drug addition. A value of 1 represents negative control-like growth, values between 0 and 1 represent cyto-suppression, and negative values denote loss of cells from baseline. (B-D) The epichaperome was identified in Molm13 cells with variable TP53 status: (B) (left) a representative experiment and (right) quantification of triplicate experiments in primary AML cells and stem progenitor cells from patients; (C) a representative result from a patient with TP53-mutation (left) and results from TP53-mutant (right), TP53-/RAS-mutant, or kinase-mutant samples and samples without TP53/kinase mutations; and in normal BM controls (D). The epichaperome was identified via flow cytometry using FITC-labeled PU-H71 as probe. AOC_LD, area over the curve lethal dose; MFI, mean fluorescence intensity; SEM, standard error of the mean.
The epichaperome is present in TP53-mutant AML cells and stem/progenitor cells
Using FITC-PU-H71 as a probe, we identified epichaperomes in TP53-WT Molm13, consistent with FLT3-internal tandem duplication (ITD)–driving high signaling activity in Molm13 cells. Epichaperome levels were even higher in TP53-KO or -mutant Molm13 cells (Figure 1B). Immunoblot analysis revealed high levels of HSP90 in Molm13 and even higher levels of HSP90 and several signaling proteins in TP53-KO and -mutant Molm13 cells (supplemental Figure 1). Epichaperomes were detected in bulk, CD34+, and CD34+CD38– AML stem/progenitor cells from patients (supplemental Table 1) with TP53-mutant AML (TP53-mutant, n = 6; TP53/RAS-mutant, n = 4) and in samples with oncogenic kinase mutations (n = 7), again consistent with epichaperome-dependency of AML with hyperactive signalosomes.35 Epichaperome levels were significantly higher in TP53-/kinase-mutant AML than in AML without TP53/kinase mutations (Figure 1C). Importantly, healthy BM or stem/progenitor cells lack epichaperomes (Figure 1D).
Targeting epichaperomes effectively kills TP53-mutant AML and AML stem/progenitor cells and prevents their outgrowth
PU-H71, at subclinical doses, effectively induced cell death in both TP53-WT and TP53-R248W cells, whereas apoptosis induced by BCL-2 (venetoclax) or MCL-1 (AZD5991) inhibitors, at doses effective in TP53-WT cells, lost activity in TP53-R248W cells (Figure 2A). PU-H71 also effectively induced cell death and decreased viable cell numbers in TP53-WT, TP53-KO, and various hotspot TP53-mutant Molm13 and K562 cells as well as leukemia cells with or without TP53 mutations, largely independent of TP53 mutation status (Figure 2B; supplemental Figure 2).
Figure 2.

PU-H71 effectively induces cell death in TP53-mutant AML cells and stem/progenitor cells and prevents the outgrowth of TP53-mutant AML cells. (A) TP53-WT and TP53-R248W Molm13 cells were treated with PU-H71 or BH3 mimics targeting either BCL-2 (venetoclax [VEN]) or MCL-1 (AZD5991). (B-D) TP53-WT, -KO, and -mutant Molm13 cells (B), TP53-mutant primary AML cells and stem/progenitor cells (C), and healthy BM and BM stem/progenitor cells (D) were treated with PU-H71. After 48 hours of treatment, cell death and cell counts were determined via flow cytometry. Primary cells were cocultured with MSCs. (E-F) A mixture of GFP-labeled TP53-WT and BFP-labeled TP53-R248W Molm13 cells (1000:1 ratio) was treated with nutlin3a or PU-H71 (E) and flow cytometry was used to assess cell death and surviving TP53-WT (GFP) and TP53-R248W (BFP) cells or treated with VEN or PU-H71 (F), and cell viability and cell numbers were assessed via flow cytometry in the presence of counting beads. Fresh drugs and cell culture medium were added 2 or 3 times per week. 7AAD, 7-aminoactinomycin D; AnnV, annexin V; VEN, venetoclax.
Next, primary TP53-mutant AML PB cells (n = 3) (supplemental Table 1) and healthy BM cells (n = 3) cocultured with MSCs were treated with PU-H71. PU-H71 induced cell death and decreased viable cells in AML and AML stem/progenitor cells (Figure 2C). However, it had minimal toxicity in normal BM and stem/progenitor cells (Figure 2D), in agreement with the presence of epichaperomes in TP53-mutant AML cells and stem/progenitor cells and the lack thereof in healthy BM cells and stem/progenitor cells.
To mimic the clinical situation that a minor TP53-mutant clone within a majority of TP53-WT AML cells becomes dominant after exposure to therapies and determine whether PU-H71 effectively kills and prevents the outgrowth of TP53-mutant cells, we treated isogenic GFP-labeled TP53-WT and BFP-labeled TP53-R248W Molm13 cell mixtures (1000:1 ratio) with either PU-H71 or the MDM2 inhibitor nutlin3a. Nutlin3a effectively induced apoptosis and decreased TP53-WT cells initially. However, its activity diminished over time. On day 11, and more so on day 28, fewer cells were apoptotic, and TP53-R248W cells were dominant. In stark contrast, PU-H71 potently induced cell death and eliminated both TP53-WT and TP53-R248W cells by day 28 (Figure 2E). We then treated the cell mixture with PU-H71 or venetoclax. Although initially decreasing because of venetoclax, the cell numbers subsequently rose even with increased venetoclax concentrations. The starting numbers of TP53-WT cells and TP53-R248W cells were 10 × 106 and 10 × 103, respectively, but after 24 days of venetoclax treatment they increased to 16 × 106 and 8 × 106, respectively. In contrast, after 24 days of PU-H71 treatment, only 335 TP53-WT and 26 TP53-R248W cells remained (Figure 2F). Hence, PU-H71 was able to almost eradicate both isogenic TP53-WT and -mutant cells, whereas nutlin3a and venetoclax became completely inefficient.
Baseline cellular stress and proliferation state, not TP53 status, determine sensitivity to PU-H71–induced cell death
To assess PU-H71–driven stress responses and cell death modes, PU-H71–treated TP53-WT, TP53-KO, and TP53-mutant (R175H and Y220C) Molm13 cells were subjected to high-parametric flow cytometry analysis. Singlets from 16 experimental conditions (4 cell types, untreated or treated with 40, 100, or 250 nM PU-H71) were pooled and subjected to uniform manifold approximation and projection (UMAP) dimension reduction to project and visualize cells into 2 dimensions, followed by clustering using FlowSOM to identify cell communities with similar proteomic profiles (Figure 3A; supplemental Figure 3). FlowSOM identified 10 different cellular populations with 2 distinct clusters, CL1 and CL2, corresponding to live cells (Figure 3A-C). We observed that expression patterns of features of interest differed across and within live and dead cell compartments (Figure 3C). ATF4, RIP3, Ki-67, LC3B, and γ-H2AX expressions were mainly localized in the live-cell compartment. Live untreated cells exhibited DNA damage response (γ-H2AX), particularly in proliferating cells with Ki-67 expression, endoplasmic reticulum (ER) stress (ATF4), and autophagy (LC3B), illustrating the baseline cellular stress response present in the cells. Live-cell clusters CL1 and CL2 were detected at varying frequencies before and after treatment. No other live-cell clusters emerged or significantly increased in samples treated with PU-H71 (Figure 3B), suggesting that PU-H71 did not induce a distinct, divergent stress response based on cellular features assessed in the multiparametric assay.
Figure 3.

PU-H71 targets baseline cellular stress responses to induce cell death. (A) Isogenic TP53-WT, -KO, and -mutant (R175H and Y220C) Molm13 cells were untreated or treated with 40, 100, or 250 nM PU-H71; stained with an array of antibodies; and subjected to flow cytometry analysis. Cells from all experiments were subjected to the FlowSOM algorithm to identify clusters. Cells were then subjected to UMAP dimensional reduction and projected on 2-dimensional plots with live (blue) and dead (gray) cells. (B) Bubble plot of the FlowSOM cluster frequencies from panel A across TP53-WT, -KO, and -mutant (R175H and Y220C) Molm13 cells. (C) UMAP plots for the indicated markers. (D) The FlowSOM cluster frequencies shown in panel B were used for UMAP dimension reduction to map similarities and dissimilarities in the response of isogenic TP53-WT, -KO, and -mutant (R175H and Y220C) leukemia cells to PU-H71 treatment. Shapes indicate the cell types and colors indicate the treatment conditions. (E) Single-cell protein expression heatmap showing expression of the indicated markers (rows) across 16 different experimental conditions (TP53-WT, -KO, -R175H, and -Y220C leukemia cells that were untreated or treated with 40, 100, and 250 nM PU-H71) and the 10 FlowSOM clusters identified in panel A. (F) Volcano plot showing differentially expressed markers between CL1 and CL2 live cells. Dashed vertical lines indicate the fold change cutoff ratio of 0.5. (G) Volcano plot showing differentially expressed markers between untreated CL1 cells and CL1 cells treated with 40 or 100 nM PU-H71. Dashed vertical lines indicate the fold change cutoff ratio of 0.5. (H) Violin plots summarize expression of the indicated markers in untreated CL1 cells (bluish gray) and CL1 cells treated with 40 nM (yellow) or 100 nM (blue) PU-H71. (I) Log2-transformed CL2/CL1 ratios in TP53-WT, -KO, and -mutant leukemia cells plotted against 4 different treatment conditions. The FlowSOM CL1 and CL2 frequencies, shown in panel B, of untreated and PU-H71-treated TP53-WT, -KO and -mutant leukemia cells were used to calculate CL2/CL1 ratios. (J) Single-cell protein expression heatmap showing expression of the indicated markers (rows) across control CL2 cells and residual CL2 cells detected after treatment of TP53-KO and -mutant (R175H and Y220C) leukemia cells with 250 nM PU-71. The scale bar indicates scaled marker expression levels. (K) Violin plots summarize expression of the indicated markers in CL2 clusters from untreated TP53-KO, and -R175H and -Y220C mutant leukemia cells (bluish gray) vs residual CL2 in TP53-KO, and -R175H and -Y220C mutant leukemia cells treated with 250 nM PU-H71.
We quantified the fractions of clusters identified via FlowSOM clustering (Figure 3A-B), and clustering analysis revealed that the PU-H71 concentration was the main driver for grouping of leukemia cells; that is, cells treated with the same PU-H71 concentration clustered together irrespective of their TP53 status (Figure 3D). Untreated isogenic TP53-WT, -KO, and -mutant leukemia cells and cells treated with 40 nM PU-H71 clustered together, indicating that therapy pressure at this dose had no significant effect on cluster composition. Treatment with 100 and 250 nM PU-H71 resulted in the clustering of leukemia cells distinct from untreated cells (Figure 3D). Particularly, 250 nM PU-H71 significantly depleted live cells (CL1 and CL2) across all leukemia cells. These findings indicate that PU-H71 elicited similar proteomic alterations in leukemia cells irrespective of TP53 status, which provides the rationale for using PU-H71 in the treatment of TP53-deficient AML, regardless of gene loss or specific mutations.
To assess the proteomic alterations induced by different PU-H71 concentrations, we extracted data from 653 392 single cells (both live and dead) from 16 different experimental conditions (Figure 3B) and generated a heatmap of single-cell proteomic expression levels to delineate the proteomic architecture, map therapy–driven proteomic alterations, and identify PU-H71 associated stress responses and cell death modes (Figure 3E). TP53-WT leukemia cells treated with ONC-201, cytarabine, or both were used in parallel as controls because these agents are known to induce ER stress, autophagy, DNA damage, and cell cycle arrest (supplemental Figure 4).52 We observed that dead cell compartments contained hierarchically organized, distinct subpopulations displaying differential expression of cleaved caspase-3, cleaved poly(ADP-ribose)polymerases (PARP), γ-H2AX, RIP3, and live/dead aqua dye. The induction of active caspase-3 and cleaved PARP indicated that the mode of cell death induced by PU-H71 is mainly apoptosis. Dampened DNA damage response, measured based on γ-H2AX levels, and reduced RIP3 expression in treated compared with that in untreated live cells ruled out parthanatos and necroptosis,50 respectively, as the mechanisms of PU-H71–induced cell death (Figure 3E). PU-H71–induced cell death was significantly suppressed by caspase but was largely unaffected by necroptosis or ferroptosis inhibition in TP53-WT, -KO, or -mutant (R175H, Y220C) Molm13 cells (supplemental Figure 5), further supporting that apoptosis is the primary mechanism of cell death, independent of TP53 status. We previously reported that γ-H2AX upregulation marks the earliest event in cell death through trajectory analysis.52 As expected, we identified γ-H2AXhi cells in hierarchically organized dead cell compartment.
The live-cell proteomic landscape was heterogeneous (Figure 3C), and the expression of features (ATF4, RIP3, Ki-67, LC3B, p21, cleaved PARP, and γ-H2AX) varied across live cells and treatment conditions. CL1, the major live-cell cluster, differed from CL2, a less dominant live-cell cluster, and expressed higher levels of Ki-67 and ER stress, assessed using ATF4 (Figure 3F). We, then, mapped how PU-H71 altered proteomic profiles of CL1 and CL2 cells. To this end, we filtered live cells, CL1 and CL2, and performed differential expression analysis to compare the proteomic profiles of untreated TP53-WT, -KO, and -mutant cells vs those of PU-H71–treated cells (40 and 100 nM). Because treatment with 250 nM PU-H71 induced considerable cell death and significantly depleted CL1 and CL2 clusters, we excluded them from differential expression analysis. We found that 40 and 100 nM PU-H71 induced neither cell death nor reduced live-cell counts (Figure 3C), which permitted an assessment of PU-H71–driven proteomic alterations. We observed at either 40 or 100 nM PU-H71 more pronounced changes in the expression of ATF4, RIP3, p21, and LC3B (Figure 3G) in CL1 cells (treatment vs control) than in CL2 cells, resulting in the downregulation and compromise of autophagy and ER stress/unfolded protein response (Figure 3H; supplemental Figure 6). CL1 cells were proliferating with high Ki-67 expression (Figures 3C and 4E-F) and higher levels of baseline ER stress. Therefore, we concluded that proliferating cells with higher ER stress are most susceptible to PU-H71 than those with low ER stress. Collectively, PU-H71 suppresses baseline cell stress responses, leading to apoptosis induction.
Figure 4.

PU-H71 has antileukemia activity in NSG mice bearing TP53-mutant AML xenografts with minimal toxicity in normal hematopoiesis. NSG mice bearing TP53-R248W Molm13 cells received 30 or 50 mg/kg PU-H71, and leukemia-free NSG mice received 50 mg/kg PU-H71. (A) Disease progression and treatment responses in NSG mice bearing TP53-R248W Molm13 cells were assessed via in vivo luciferase imaging; (left) imaging of individual mice and (right) quantification of luciferase imaging of all mice per group. (B) Survival of NSG mice bearing TP53-R248W Molm13 cells were either vehicle- or PU-H71–treated. Arrows indicate treatment times. (C) RBC counts, WBC counts, and hemoglobin levels in leukemia-free NSG mice treated with 50 mg/kg PU-H71. Shaded areas indicate the treatment period. (D) Measurement of the epichaperome in healthy BM cells and Lin–Sca1+c-Kit+ BM cells collected from leukemia-free NSG mice. RBC, red blood cell; WBC, white blood cell.
A fraction of Ki-67–low CL2 cells survived, whereas CL1 cells were more sensitive to PU-H71 treatment. The finding highlights that PU-H71, at 40 and 100 nM, elicited only marginal proteomic alterations in CL2 cells. Higher fractions of TP53-KO (supplemental Figure 6) and -mutant CL2 cells survived with 250 nM PU-H71 treatment (Figures 3B and 4I) than TP53-WT cells. Of note, CL1 cells in all groups were almost completely eliminated independent of TP53 status, and we were unable to detect TP53-WT CL2 cells after 250 nM PU-H71 (Figure 3B,I). Next, we delineated the proteomic profiles of CL2 cells that survived 250 nM PU-H71 (resistant cells) by pooling the surviving TP53-KO and -mutant CL2 cells and generating a single-cell protein expression heatmap (Figure 3J). Untreated CL2 cells were used as the control. Remarkably, surviving/PU-H71 resistant TP53-KO or -mutant cells had significantly lower levels of ER stress, autophagy, and DNA damage responses (Figure 3J-K). This may suggest that apoptosis induction in cells with compromised baseline cell stress response is partially p53-dependent, and loss of p53 function could confer a marginal survival advantage under PU-H71 therapy pressure.
PU-H71 has antileukemia activity and extends the survival of mice bearing TP53-mutant AML xenografts, with minimal toxicity for normal hematopoiesis
PU-H71 decreased leukemia burden in NSG mice bearing TP53-R248W Molm13 (Figure 4A). The median survival of mice treated with 30 or 50 mg/kg PU-H71 were 32 and 35 days, respectively; the latter was significantly longer than that of controls (27 days) (P = .012; Figure 4B). Assessments of red and white blood cell counts and hemoglobin levels in leukemia-free NSG mice revealed that treatment with PU-H71 (50 mg/kg; 4 weeks) had only minimal effects on normal hematopoiesis (Figure 4C). We did not detect epichaperomes in either bulk or Lin–Sca1+c-Kit+ stem/progenitor BM cells from leukemia-free NSG mice (Figure 4D).
PU-H71 plus venetoclax synergistically induces cell death in AML cells and stem/progenitor cells independent of TP53 mutation status
Immunoblot analysis of BCL-2 proteins showed that PU-H71 decreased MCL-1 and increased Bcl-2-like protein 11 (BIM) in TP53-WT and -mutant Molm13 cells (Figure 5A), suggesting that PU-H71 can enhance venetoclax activity. Indeed, compared with either agent alone, the combination induced significantly more cell death in TP53-R175H Molm13 cells (Figure 5B). To validate the role of MCL-1, we treated MCL-1 overexpressing Molm13 cells and found that MCL-1 overexpression reduced not only venetoclax but also PU-H71 and venetoclax/PU-H71 activity (Figure 5C); conversely, MCL-1 knockdown in both TP53-WT and -R175H Molm13 cells sensitized to venetoclax, PU-H71, and venetoclax/PU-H71 (Figure 5D), supporting the notion of MCL-1 as a resistance factor and that PU-H71–mediated MCL-1 reduction contributes to the observed synergy. Additionally, PU-H71 treatment decreased the levels of multiple signaling proteins in both TP53-WT and -mutant cells, including p-STAT3, p-protein kinase B (AKT), phosphorylated extracellular signal-regulated kinase, c-Myc, heat shock factor (HSF)-1, and hypoxia inducing factor (HIF)-1α (supplemental Figure 7).
Figure 5.

PU alters the levels of BCL-2 proteins and synergizes with VEN to induce cell death in AML independent of TP53 mutation status. (A) BCL-2 family protein levels were determined in TP53-WT and -mutant Molm13 cells treated with PU-H71. Results of a representative immunoblotting (left) and quantitative analysis of 3 independent measurements (right). (B-F) TP53-R175H Molm13 cells (B), control and MCL-1 overexpressing Molm13 cells (C), and MCL-1 knockdown (KD) by small interfering RNA TP53-WT or TP53-R175H Molm13 cells (D), (F) were treated with PU, VEN, or both for 48 hours. Cell death was determined via flow cytometry. CON, control; PU, PU-H71.
In TP53-mutant primary AML samples cocultured with MSCs (n = 9; many from patients with comutations and/or venetoclax resistance/relapse; supplemental Table 1), treatments with PU-H71 or venetoclax significantly induced cell death. The combination was significantly more effective (Figure 6A) and highly synergistic in both bulk and AML stem/progenitor cells (CI < 1; supplemental Table 4). This synergism was also observed in a sample (#13) from a patient who relapsed and was resistant to venetoclax-based therapy and had failed compassionate PU-H71 monotherapy (Figure 6A; supplemental Tables 1 and 4). Similarly, PU-H71 suppressed blast colony formation, which was significantly enhanced when combined with venetoclax (Figure 6B; n = 3; supplemental Table 1). Additionally, although more effective in Ki-67–high primary cells, PU-H71 also killed Ki-67–low cells. The combination with venetoclax was more effective in both Ki-67–low and –high AML cells and stem/progenitor cells than each single agent (Figure 6C). Each agent and their combination had minimal toxicity against healthy BM CD45+ and CD34+ stem/progenitor cells and did not reduce colony numbers (Figure 6D-E).
Figure 6.

PU and VEN combination synergistically targets AML cells and stem/progenitor cells with TP53 mutations but with limited activities against healthy BM and BM stem/progenitor cells. PB cells from patients with primary AML with TP53 mutations and healthy BM cells were treated with PU, VEN, or both. (A-B) Cell death of AML cells and stem/progenitor cells at 48 hours treatments (A) and clonogenic assay (B) in primary samples from patients. (C) Viable cells in Ki-67–low (solid markers) and –high (open markers) AML cells and stem/progenitor cells from patients with TP53 mutations. (Left) Ki-67 staining of 1 of the samples. (Middle and right) Viable CD45+ cells or CD34+CD38– cells in various treatment groups compared with the untreated control. Patient samples used for various treatments and patient characteristics are shown in supplemental Table 1. (D-E) Cell death at 48-hour treatments (D) and clonogenic assays (E) in healthy BM samples. Primary cells were cocultured with MSCs during treatments. For colony assays, error bars represent mean ± SEM of 3 plating × 2 counting of each sample. ∗P ≤ .05; ∗∗P ≤ .01; ∗∗∗P ≤ .001; ∗∗∗∗P ≤ .0001. BFU-E, burst forming unit-erythroid.
Targeting epichaperomes has antileukemia activity and enhances venetoclax efficacy in vivo in TP53-mutant AML
We examined the in vivo effects of PU-H71, venetoclax, and their combination in TP53-WT cells spiked with isogenic TP53-mutant cells. After 3-week treatment, in vivo imaging demonstrated (Figure 7A) significantly reduced leukemia burden in PU-H71–treated mice compared with that in controls (P = .025), which was further reduced in mice treated with PU-H71/venetoclax (P = .0077). Leukemia burden of mice treated with venetoclax was not significantly different (P = .33). After 4 weeks, 4 PU-H71–treated and all combination-treated mice with significantly lower leukemia burden (P = .030) were still alive. After 5 weeks, all mice had died, except 2 combination-treated mice. In a separate experiment, flow cytometry analysis demonstrated that PU-H71 and PU-H71/venetoclax significantly suppressed circulating and BM-resident TP53-WT and TP53-R248W AML cells after 3-week treatment (n = 3 per group). Moreover, PU-H71/venetoclax was significantly more effective than venetoclax alone against TP53-WT and TP53-R248W cells in PB and BM (Figure 7B). These findings suggest that inhibition of epichaperomes enhances venetoclax efficacy independent of TP53 mutation status. In a patient-derived xenograft model generated from a chemorefractory/multitherapy resistant patient who was also resistant to venetoclax with TP53/NRAS/KRAS mutations and complex cytogenetics, PU-H71 prolonged survival (80 vs control 67 days; P = .0001) and was significantly more effective than venetoclax in decreasing circulating leukemia cells (P = .0005) and spleen weight (P = .007) while increasing survival (P = .004; Figure 7C). Although effectively reducing circulating leukemia cells and spleen weight, PU-H71/venetoclax did not extend survival of NSGS mice, likely because of toxicity, because weight loss was observed (supplemental Figure 8).
Figure 7.

PU plus VEN has enhanced antileukemia activity against both TP53-WT and -mutant AML in vivo. NSG mice injected with a mixture of luciferase-/GFP-labeled TP53-WT Molm13 cells and BFP-labeled TP53-R248W Molm13 cells (10:1 ratio) or NSGS mice injected with a PDX TP53-mutant cells (2 × 106 cells per mouse) were treated with PU (50 mg/kg), VEN (50 mg/kg), or both. (A) Luciferase imaging of leukemia burden in NSG mice: (left) images of individual mice and (right) quantification of luciferase imaging of all mice per treatment group). (B) Flow cytometry was used to identify TP53-WT and -R248W Molm13 cells in PB and BM after 3-week treatment. (C) Circulating blasts and spleen weight after 4-week treatment (left) and mouse survival in NSGS mice (right). PDX, patient-derived xenograft.
Discussion
We demonstrate that epichaperomes are essential for TP53-mutant AML growth and survival and can be targeted by epichaperome-specific inhibitor PU-H71. PU-H71 kills TP53-mutant AML and AML stem/progenitor cells, synergizes with venetoclax, and prevents the outgrowth of venetoclax-resistant TP53-mutant AML clones, with limited toxicity to healthy cells. Our data support the clinical development of PU-H71 plus venetoclax as a treatment in TP53-mutant AML.
A previous study demonstrated that mutant TP53 promotes cell proliferation, invasion, and motility by stimulating signaling pathways, which, in turn, induce HSP90 expression that further stabilizes signaling proteins and mutant TP53.53 We have shown that STAT3 and mutant TP53 engage in a positive feedback loop involving HSP90.54 Mutant TP53 prevents SHP2-mediated STAT3 deactivation.25 Consistent with previous studies, we demonstrate that TP53-mutant and -KO cells have higher levels of epichaperomes and signaling proteins such as p-STAT3 than TP53-WT cells, which can be inhibited by PU-H71.
A high-parametric cell death assay and single-cell analysis reveal that PU-H71 suppresses baseline cellular stress responses and induces apoptosis as prevalent mode of cell death and proliferating cells are more sensitive to PU-H71, suggesting that agents that induce cell proliferation or augment cell stress could enhance PU-H71 efficacy and target quiescent leukemia cells. Indeed, we demonstrate that cotargeting epichaperomes and BCL-2 is synergistic in inducing death of both Ki-high and -low AML cells and stem/progenitor cells and further decreases colony-forming capacities.
Epichaperome inhibition by PU-H71 was reported to alter BCL-2 family protein levels and overcome BCL-2–mediated resistance in cancer cells.55 We observed that PU-H71 decreases antiapoptotic MCL-1 while increasing proapoptotic BIM, thereby contributing to the synergistic effect of PU-H71/venetoclax. We validated MCL-1 reduction as a mechanism of PU-H71/venetoclax synergism by genetically altering its expression. PU-H71 alters BCL-2 protein levels likely by decreasing epichaperome-regulated signaling proteins. Indeed, PU-H71 decreased p-AKT and AKT levels, in agreement with a recent report showing that other HSP90 inhibitors increase BIM and decrease p-AKT levels.56 Furthermore, PU-H71 decreases many signaling proteins in both TP53-WT and -mutant cells including HIF1α, which is known to be regulated by HSP90.57 HIF1α reduction may also contribute to PU-71–incuded cell death, supported by previous studies showing that HIF1α inhibition targets TP53-mutant AML.58
Clinically, an agent that targets cells independent of specific mutations, such as PU-H71, has a distinct advantage over agents that selectively target only cells with certain mutations, and, thus, it has potentially broader applications in AML and cancer therapy in general, because it prevents the outgrowth of subclones driven by additional mutations. This is particularly relevant in the context of BCL-2 and FLT3 inhibition, in which subclones evolve with lethal driver mutations. Epichaperome targeting may also suppress cells with mutations other than TP53 and FLT3, and these mechanisms are presently under investigation.
Conflict-of-interest disclosure: B.W. and R.A.M. are employees of Samus Therapeutics Inc. B.L.E. has received research funding from Celgene, Deerfield, Novartis, and Calico and consulting fees from GRAIL; he is a member of the scientific advisory board and shareholder for Neomorph Inc, TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Joe Munch, MD Anderson’s Research Medical Library, for editing the manuscript and Deanna Alexander for help in submitting the manuscript.
This work was supported, in part, by the National Institutes of Health, National Cancer Institute through MD Anderson's Cancer Center Support grant P30 CA016672, the University of Texas MD Anderson Cancer Center MDS/AML Moon Shot, and the Paul and Mary Haas Chair in Genetics (M.A.) and grants from the Cancer Prevention and Research Institute of Texas (RP190581, supporting the Gulf Coast Consortia High-Throughput Flow Cytometry Program, and RP200668, supporting the Combinatorial Drug Discovery Program, both located at the Texas A&M Health Science Center Institute of Biosciences and Technology).
Authorship
Contribution: B.Z.C. conceptualized the study, analyzed data, and wrote the manuscript; P.Y.M., M.M., W.T., B.K., J.P., A.D.B., L.B.O., Y.N., S.I., M.S., N.N., R.T.P., M.M.-M., C.S., M.B., N.P., S.B., B.L.E., E.J.S., B.W., and R.A.M. performed experiments, analyzed data, provided materials and/or data, and edited the manuscript; G.I.K. helped design the study and edited the manuscript; U.M.M. helped conceptualize the study, reviewed the experimental data, and edited the manuscript; and M.A. conceptualized the study, planned and analyzed experiments, and edited the manuscript.
Footnotes
Materials described in the manuscript, including relevant raw data, are available to researchers wishing to use them for noncommercial purposes, without breaching participant confidentiality, on request from the corresponding authors, Bing Z. Carter (bicarter@mdanderson.org) and Michael Andreeff (mandreef@mdanderson.org).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Bing Z. Carter, Email: bicarter@mdanderson.org.
Michael Andreeff, Email: mandreef@mdanderson.org.
Supplementary Material
References
- 1.Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5(7):e331. doi: 10.1038/bcj.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. doi: 10.1038/leu.2016.263. [DOI] [PubMed] [Google Scholar]
- 3.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–3491. doi: 10.1002/cncr.30203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689. doi: 10.1182/bloodadvances.2020003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. doi: 10.1182/blood.2019003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K, Maiti A, Loghavi S, et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer. 2021;127(20):3772–3781. doi: 10.1002/cncr.33689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan B, Chen Q, Shimada K, et al. Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia. Leukemia. 2019;33(4):931–944. doi: 10.1038/s41375-018-0279-6. [DOI] [PubMed] [Google Scholar]
- 9.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 10.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 11.Rodina A, Wang T, Yan P, et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature. 2016;538(7625):397–401. doi: 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillarsetty N, Jhaveri K, Taldone T, et al. Paradigms for precision medicine in epichaperome cancer therapy. Cancer Cell. 2019;36(5):559–573.e7. doi: 10.1016/j.ccell.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moulick K, Ahn JH, Zong H, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003;9(12):4483–4493. [PubMed] [Google Scholar]
- 15.Chen J, Yu WM, Daino H, Broxmeyer HE, Druker BJ, Qu CK. SHP-2 phosphatase is required for hematopoietic cell transformation by Bcr-Abl. Blood. 2007;109(2):778–785. doi: 10.1182/blood-2006-04-019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou F, Yuan LD, Zhu JJ. Heat shock protein 90 indirectly regulates ERK activity by affecting Raf protein metabolism. Acta Biochim Biophys Sin (Shanghai) 2005;37(7):501–505. doi: 10.1111/j.1745-7270.2005.00069.x. [DOI] [PubMed] [Google Scholar]
- 17.Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrova EM, Yallowitz AR, Li D, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523(7560):352–356. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagn F, Lagleder S, Retzlaff M, et al. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat Struct Mol Biol. 2011;18(10):1086–1093. doi: 10.1038/nsmb.2114. [DOI] [PubMed] [Google Scholar]
- 20.Brady A, Gibson S, Rybicki L, et al. Expression of phosphorylated signal transducer and activator of transcription 5 is associated with an increased risk of death in acute myeloid leukemia. Eur J Haematol. 2012;89(4):288–293. doi: 10.1111/j.1600-0609.2012.01825.x. [DOI] [PubMed] [Google Scholar]
- 21.Gilliland DG, Griffin JD. Role of FLT3 in leukemia. Curr Opin Hematol. 2002;9(4):274–281. doi: 10.1097/00062752-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Kornblau SM, Womble M, Qiu YH, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108(7):2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tabe Y, Jin L, Tsutsumi-Ishii Y, et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res. 2007;67(2):684–694. doi: 10.1158/0008-5472.CAN-06-3166. [DOI] [PubMed] [Google Scholar]
- 24.Antony ML, Noble-Orcutt K, Lee Y, et al. JAK/STAT inhibition targets TP53 altered primary human acute myeloid leukemia stem cells [abstract] Blood. 2020;136(suppl 1):27–28. [Google Scholar]
- 25.Schulz-Heddergott R, Stark N, Edmunds SJ, et al. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell. 2018;34(2):298–314.e7. doi: 10.1016/j.ccell.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beeharry N, Landrette S, Gayle S, et al. LAM-003, a new drug for treatment of tyrosine kinase inhibitor-resistant FLT3-ITD-positive AML. Blood Adv. 2019;3(22):3661–3673. doi: 10.1182/bloodadvances.2019001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishida Y, Zhao R, Heese LE, et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia. 2021;35(9):2469–2481. doi: 10.1038/s41375-021-01308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, Picard D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones. 2018;23(4):467–482. doi: 10.1007/s12192-018-0877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culjkovic-Kraljacic B, Fernando TM, Marullo R, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood. 2016;127(7):858–868. doi: 10.1182/blood-2015-05-645069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darby JF, Workman P. Chemical biology: many faces of a cancer-supporting protein. Nature. 2011 Oct 19;478(7369):334–335. doi: 10.1038/478334b. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein RL, Yang SN, Taldone T, et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J Clin Invest. 2015;125(12):4559–4571. doi: 10.1172/JCI80714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kucine N, Marubayashi S, Bhagwat N, et al. Tumor-specific HSP90 inhibition as a therapeutic approach in JAK-mutant acute lymphoblastic leukemias. Blood. 2015;126(22):2479–2483. doi: 10.1182/blood-2015-03-635821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nayar U, Lu P, Goldstein RL, et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood. 2013;122(16):2837–2847. doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taldone T, Ochiana SO, Patel PD, Chiosis G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci. 2014;35(11):592–603. doi: 10.1016/j.tips.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zong H, Gozman A, Caldas-Lopes E, et al. A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Rep. 2015;13(10):2159–2173. doi: 10.1016/j.celrep.2015.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunphy MPS, Pressl C, Pillarsetty N, et al. First-in-human trial of epichaperome-targeted PET in patients with cancer. Clin Cancer Res. 2020;26(19):5178–5187. doi: 10.1158/1078-0432.CCR-19-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speranza G, Anderson L, Chen AP, et al. First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile. Invest New Drugs. 2018;36(2):230–239. doi: 10.1007/s10637-017-0495-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pemmaraju N, Gundabolu K, Pettit K, et al. Phase 1b study of the epichaperome inhibitor PU-H71 administered orally with ruxolitinib continuation for the treatment of patients with myelofibrosis [abstract] Blood. 2019;134(suppl 1):4178. [Google Scholar]
- 39.Sugita M, Wilkes DC, Bareja R, et al. Targeting the epichaperome as an effective precision medicine approach in a novel PML-SYK fusion acute myeloid leukemia. NPJ Precis Oncol. 2021;5(1):44. doi: 10.1038/s41698-021-00183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishida Y, Montoya RH, Morita K, et al. Clonal expansion of mutant p53 clones by MDM2 inhibition in acute myeloid leukemias [abstract] Blood. 2020;136(suppl 1):27–28. [Google Scholar]
- 41.Pan R, Ruvolo V, Mu H, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell. 2017;32(6):748–760.e6. doi: 10.1016/j.ccell.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365(6453):599–604. doi: 10.1126/science.aax3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. [PubMed] [Google Scholar]
- 44.Carter BZ, Mak PY, Tao W, et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica. 2020;107(1):58–76. doi: 10.3324/haematol.2020.260331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng Z, Samudio IJ, Zhang W, et al. Simultaneous Inhibition of PDK1/AKT and Fms-Like Tyrosine Kinase 3 Signaling by a Small-Molecule KP372-1 Induces Mitochondrial Dysfunction and Apoptosis in Acute Myelogenous Leukemia. Cancer Res. 2006;66(7):3737–3746. doi: 10.1158/0008-5472.CAN-05-1278. [DOI] [PubMed] [Google Scholar]
- 46.Merugu S, Sharma S, Kaner J, et al. Chemical probes and methods for single-cell detection and quantification of epichaperomes in hematologic malignancies. Methods Enzymol. 2020;639:289–311. doi: 10.1016/bs.mie.2020.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter BZ, Mak PY, Tao W, et al. Maximal activation of apoptosis signaling by cotargeting antiapoptotic proteins in BH3 mimetic-resistant AML and AML stem cells. Mol Cancer Ther. 2022;21(6):879–889. doi: 10.1158/1535-7163.MCT-21-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Mak PY, Mu H, et al. Combinatorial inhibition of focal adhesion kinase and BCL-2 enhances antileukemia activity of venetoclax in acute myeloid leukemia. Mol Cancer Ther. 2020;19(8):1636–1648. doi: 10.1158/1535-7163.MCT-19-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muftuoglu M, Mak PY, Ruvolo V, et al. High dimensional interrogation of stress response patterns and cell death modes in AML [abstract] Blood. 2020;136(suppl 1):15. [Google Scholar]
- 50.Bergamaschi D, Vossenkamper A, Lee WYJ, Wang P, Bochukova E, Warnes G. Simultaneous polychromatic flow cytometric detection of multiple forms of regulated cell death. Apoptosis. 2019;24(5-6):453–464. doi: 10.1007/s10495-019-01528-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 52.Muftuoglu M, Ruvolo V, Nishida Y, et al. Single-cell mapping of stress response and cell death pathways in acute myeloid leukemia reveals stressor-specific alterations and distinct response patterns [abstract] Blood. 2019;134(suppl 1):882. [Google Scholar]
- 53.Alexandrova EM, Marchenko ND. Mutant p53 - heat shock response oncogenic cooperation: a new mechanism of cancer cell survival. Front Endocrinol (Lausanne) 2015;6:53. doi: 10.3389/fendo.2015.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romeo MA, Gilardini Montani MS, Benedetti R, Santarelli R, D'Orazi G, Cirone M. STAT3 and mutp53 engage a positive feedback loop involving HSP90 and the mevalonate pathway. Front Oncol. 2020;10:1102. doi: 10.3389/fonc.2020.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta. 2013;1833(6):1356–1366. doi: 10.1016/j.bbamcr.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 56.Kim SH, Cho YK, Huh JH, et al. Heat shock protein 90 inhibitors AUY922, BIIB021 and SNX5422 induce bim-mediated death of thyroid carcinoma cells. Anticancer Res. 2020;40(11):6137–6150. doi: 10.21873/anticanres.14634. [DOI] [PubMed] [Google Scholar]
- 57.Minet E, Mottet D, Michel G, et al. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Wang Y, Li J, Che G. Therapeutic targeting of TP53-mutated acute myeloid leukemia by inhibiting HIF-1α with echinomycin. Oncogene. 2020;11(10):3015–3019. doi: 10.1038/s41388-020-1201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.