

Abstract
Background
Patients with hepatic encephalopathy may present with extrapyramidal symptoms and changes in basal ganglia. These changes are similar to those seen in patients with Parkinson's disease. Dopamine agents (such as bromocriptine and levodopa, used for patients with Parkinson's disease) have therefore been assessed as a potential treatment for patients with hepatic encephalopathy.
Objectives
To evaluate the beneficial and harmful effects of dopamine agents versus placebo or no intervention for patients with hepatic encephalopathy.
Search methods
Trials were identified through the Cochrane Hepato‐Biliary Group Controlled Trials Register (January 2014), the Cochrane Central Register of Controlled Trials (CENTRAL) (Issue 12 of 12, 2013), MEDLINE (1946 to January 2014), EMBASE (1974 to January 2014), and Science Citation Index‐Expanded (1900 to January 2014). Manual searches in reference lists, conference proceedings, and online trial registers were also performed.
Selection criteria
Randomised trials were included, irrespective of publication status or language. The primary analyses included data from randomised trials using a parallel‐group design or the first period of cross‐over trials. Paired data from cross‐over trials were included in sensitivity analyses.
Data collection and analysis
Three review authors extracted data independently. Random‐effects meta‐analyses were performed as the result of an expected clinical heterogeneity. Fixed‐effect meta‐analyses, meta‐regression analyses, subgroup analyses, and sensitivity analyses were performed to evaluate sources of heterogeneity and bias (systematic errors). Trial sequential analysis was used to control the risk of play of chance (random errors).
Main results
Five trials that randomly assigned 144 participants with overt hepatic encephalopathy that were published during 1979 to 1982 were included. Three trials assessed levodopa, and two trials assessed bromocriptine. The mean daily dose was 4 grams for levodopa and 15 grams for bromocriptine. The median duration of treatment was 14 days (range seven to 56 days). None of the trials followed participants after the end of treatment. Only one trial reported adequate bias control; the remaining four trials were considered to have high risk of bias. Random‐effects model meta‐analyses showed that dopamine agents had no beneficial or detrimental effect on hepatic encephalopathy in the primary analyses (15/80 (19%) versus 14/80 (18%); odds ratio (OR) 2.99, 95% confidence interval (CI) 0.09 to 100.55; two trials) or when paired data from cross‐over trials were included (OR 1.04, 95% CI 0.75 to 1.43). Clear evidence of intertrial heterogeneity was identified both in the primary analysis (I2 = 65%) and when paired data from cross‐over trials were included (I2 = 40%).
Dopamine agents had no beneficial or harmful effect on mortality (42/144 (29%) versus 38/144 (26%); OR 1.11, 95% CI 0.35 to 3.54; five trials). Trial sequential analyses demonstrated that we lacked information to refute or recommend the interventions for all outcomes. Dopamine agonists did not seem to increase the risk of adverse events.
Authors' conclusions
This review found no evidence to recommend or refute the use of dopamine agents for hepatic encephalopathy. More randomised placebo‐controlled clinical trials without risks of systematic errors and risks of random errors seem necessary to permit firm decisions on dopamine agents for patients with hepatic encephalopathy.
Plain language summary
Dopamine agents for hepatic encephalopathy
Hepatic encephalopathy is a serious complication of severe liver disease. The disease is often fluctuating with a wide spectrum of symptoms ranging from minor, not readily discernible signs to deep coma. Symptoms often develop in connection to stress related to infection, dehydration, obstipation, or gastrointestinal bleeding. The exact underlying mechanisms behind the disease development are not known. Experimental studies suggest that the mental changes seen in hepatic encephalopathy reflect changes in neurotransmitters in the brain.
Dopamine plays a major role in neurotransmission. Several nervous system diseases including Parkinson's disease are caused by a dysfunction in the dopamine system. Some patients with hepatic encephalopathy have symptoms that are similar to those seen in patients with Parkinson's disease (slow cerebration; stiffness of movements; tremor). For patients with Parkinson's disease, the drugs known as dopamine agents (drugs that mimic the effect of the neurotransmitter dopamine) clearly alleviate symptoms. These drugs have also been assessed for patients with hepatic encephalopathy.
We performed the present systematic review to determine the beneficial and harmful effects of dopamine agents for patients with hepatic encephalopathy. Our analyses included five small trials published in 1982 or earlier. All trials but one had high risks of bias (i.e., risks of systematic errors or risks of overestimation of beneficial effects or risks of underestimation of harmful effects). Only 144 patients were included in the five trials, and accordingly risks of random errors (i.e., play of chance) are present. Our analyses showed no significant differences regarding symptoms of hepatic encephalopathy or mortality in patients treated with dopamine agents compared with patients who received an inactive placebo or no intervention. The number of patients with adverse events seemed comparable in the two intervention groups. Based on the available evidence, we conclude that no evidence can be found to recommend or refute the use of dopamine agents for hepatic encephalopathy. More randomised placebo‐controlled clinical trials without risks of systematic errors and risks of random errors seem necessary to obtain firm evidence on dopamine agents for patients with hepatic encephalopathy.
Summary of findings
Summary of findings for the main comparison. Dopamine agonists for hepatic encephalopathy.
| Dopamine agonists versus placebo or no intervention for hepatic encephalopathy | ||||||
| Patient or population: patients with hepatic encephalopathy. Settings: hospitalised patients. Intervention: dopamine agonists versus placebo or no intervention. | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo or no intervention) | Dopamine agonists | |||||
| Mortality Follow‐up: mean one month | Study population | OR 1.11 (0.35 to 3.54) | 144 (five studies) | ⊕⊕⊝⊝ low1,2,3 | ||
| 535 per 1000 | 561 per 1000 (287 to 803) | |||||
| Moderate | ||||||
| 395 per 1000 | 420 per 1000 (186 to 698) | |||||
| Hepatic encephalopathy Follow‐up: mean one month | Study population | OR 2.99 (0.09 to 100.55) | 80 (two studies) | ⊕⊕⊝⊝ low1,2,3 | ||
| 350 per 1000 | 617 per 1000 (46 to 982) | |||||
| Moderate | ||||||
| 184 per 1000 | 403 per 1000 (20 to 958) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1The randomisation methods were classed as adequate in two trials, and three trials were double‐blind. 2The sample size was small, and the statistical power of included trials was weak. 3Because of the small number of trials, tests for publication bias were of limited value.
Background
Description of the condition
Hepatic encephalopathy is a complex neuropsychiatric syndrome seen in severe liver failure (Gitlin 1996; Ferenci 2002). Symptoms range from minor neuropsychiatric changes to deep coma (Conn 1979). Hepatic encephalopathy may be clinically overt or may consist of mild neurocognitive impairments, which have been identified in a substantial percentage of patients with liver disease (Randolph 2009). The course of the disease may be episodic, with recurrent symptoms, or chronic, with more stable symptoms (Bajaj 2011). The exact underlying pathophysiology is not known. Experimental studies suggest that symptoms develop as the result of accumulation of toxic agents that have not been metabolised by the liver (Gitlin 1996). Other potential mechanisms include the generation of false neurotransmitters and an abnormal interaction between astrocytes and other cellular elements with cerebral oedema and alterations in glioneural communication (Haussinger 2000; Cordoba 2001).
Description of the intervention
Many patients with hepatic encephalopathy present with extrapyramidal symptoms and have changes in the basal ganglia, as detected by magnetic resonance imaging and proton spectroscopy (Spahr 2000). These symptoms are comparable with those seen in Parkinson's disease and suggest an impairment of dopamine neurotransmission (Blei 1999; Jover 2003). Patients with Parkinson's disease are less likely to experience dyskinesia and dystonia when treated with levodopa (Stowe 2008). Uncontrolled trials suggest that levodopa or bromocriptine could be beneficial in the treatment of patients with hepatic encephalopathy (Parkes 1970; Jorge 1973). The effects of dopamine agents have also been assessed in randomised clinical trials (Uribe 1979; Michel 1980; Morgan 1980), and previous guidelines suggested that the intervention may be considered in patients with chronic hepatic encephalopathy (Blei 1999; Lizardi‐Cervera 2003).
Why it is important to do this review
We have previously published a systematic review on dopamine agents for hepatic encephalopathy (Als‐Nielsen 2004a). The results of this review were inconclusive. We have been unable to identify any further meta‐analyses or systematic reviews on the topic. To determine the strengths and weaknesses of the current evidence, we have updated our previous review (Als‐Nielsen 2004a).
Objectives
To evaluate the beneficial and harmful effects of dopamine agents versus placebo or no intervention for patients with hepatic encephalopathy.
Methods
Criteria for considering studies for this review
Types of studies
This review included all randomised trials, regardless of publication status, language, or blinding. Unpublished trials were included if the methodology and the data were available in written form. We planned to include observational studies reporting harms, but we identified no observational studies reporting relevant data.
Types of participants
Patients with hepatic encephalopathy were included, irrespective of the aetiology of the underlying liver disease. The diagnostic criteria could include psychometric tests, clinical scoring systems (such as the West‐Haven criteria), electroencephalography (Guerit 2009), or biochemical findings (including ammonia levels). Based on the diagnostic criteria used in the included trials, participants were classified as having overt or minimal hepatic encephalopathy, and the latter was classified further as recurrent or chronic.
Types of interventions
The intervention comparisons assessed were dopamine agents (e.g., levodopa, bromocriptine) versus placebo or no intervention. Studies were included irrespective of the dose or duration of therapy.
Types of outcome measures
Primary outcomes
Mortality (all‐cause).
All cause non‐fatal serious adverse events.
Morbidity. This outcome measure was assessed on the basis of the number of participants who showed no improvement in manifestations of hepatic encephalopathy as defined by the authors of included trials.
Secondary outcomes
All‐cause non‐serious adverse events (number and type) (ICH‐GCP 1997).
Qualitiy of life.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Hepato‐Biliary Group Controlled Trials Register, the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, and Science Citation Index‐Expanded (Royle 2003). Search strategies with time spans of the searches are given in Appendix 1.
Searching other resources
Reference lists in relevant articles and conference proceedings were scanned for additional trials not identified in the electronic searches. We wrote to authors of identified trials and pharmaceutical companies to enquire about additional trials. Ongoing and completed trials were also identified through searches in the World Health Organization Trial Search Portal (www.who.int/trialsearch/).
Data collection and analysis
Selection of studies
All review authors participated in the selection of trials. AEJ listed the potentially eligible trials. Subsequently, trials that fulfilled all inclusion criteria were identified. Excluded trials were listed along with the reasons for exclusion.
Data extraction and management
Three review authors (AEJ, BA‐N, and LLG) extracted data independently. All disagreements were resolved through discussion before analyses.
We extracted data on the design of the trial (country of origin, parallel or cross‐over design, and bias control), participant characteristics (aetiology of underlying liver diseases and type of hepatic encephalopathy, mean age, proportion of men), and the intervention regimen assessed (type, dose, and duration of therapy).
Assessment of risk of bias in included studies
We assessed the risk of bias in the trials independently in accordance with the instructions provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and the Cochrane Hepato‐Biliary Group Module (Gluud 2013). Because of the risk of overestimation of intervention effects in randomised trials with high risk of bias (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Lundh 2012; Savovic 2012, Savovic 2012a), we assessed the influence of risk of bias on trial results using the following domains.
Allocation sequence generation
Low risk of bias: Sequence generation was achieved by using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if performed by an independent person not otherwise involved in the trial.
Uncertain risk of bias: The method of sequence generation was not specified.
High risk of bias: The sequence generation method was not random.
Allocation concealment
Low risk of bias: The participant allocations could not have been foreseen in advance of, or during, enrolment. Allocation was controlled by a central and independent randomisation unit. The allocation sequence was unknown to the investigators (e.g., if the allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
Uncertain risk of bias: The method used to conceal the allocation was not described so that intervention allocations may have been foreseen in advance of, or during, enrolment.
High risk of bias: The allocation sequence was likely to be known to the investigators who assigned the participants.
Blinding of participants, personnel, and outcome assessors
Low risk of bias: Blinding was performed adequately, or the assessment of outcomes was not likely to be influenced by lack of blinding.
Uncertain risk of bias: Information was insufficient to permit assessment of whether blinding was likely to induce bias on the results.
High risk of bias: No blinding or incomplete blinding was performed, and assessment of outcomes was likely to be influenced by lack of blinding.
Incomplete outcome data
Low risk of bias: Missing data were unlikely to make treatment effects depart from plausible values. Sufficient methods, such as multiple imputation, had been employed to handle missing data.
Uncertain risk of bias: Information was insufficient to permit assessment of whether missing data in combination with the method used to handle missing data were likely to induce bias on the results.
High risk of bias: The results were likely to be biased as the result of missing data.
Selective outcome reporting
Low risk of bias: All outcomes were predefined and reported, or all clinically relevant and reasonably expected outcomes were reported. The trial was registered on the www.clinicaltrials.gov web site or on a similar register, or the protocol was published.
Uncertain risk of bias: It was unclear whether all predefined and clinically relevant and reasonably expected outcomes were reported.
High risk of bias: One or more clinically relevant and reasonably expected outcomes were not reported, and data on these outcomes were likely to have been recorded.
Other bias
Low risk of bias: The trial appears to be free of other components that could put it at risk of bias.
Uncertain risk of bias: The trial may or may not be free of other components that could put it at risk of bias.
High risk of bias: Other factors in the trial could put it at risk of bias (e.g., for‐profit involvement, authors conducting trials on the same topic).
Trials with unclear or high risk of bias methodology in one or more of the above domains were considered trials with high risk of bias. The remaining were considered trials with low risk of bias.
Measures of treatment effect
All outcome measures were dichotomised and were expressed using odds ratios (ORs) with 95% confidence intervals (CIs).
Unit of analysis issues
The primary analyses included data from trials using a parallel‐group design and from the first treatment period of cross‐over trials. Additional analyses were performed that included paired data from the cross‐over trials (Becker 1993; Elbourne 2002).
Dealing with missing data
Data on all participants randomly assigned were sought to allow intention‐to‐treat analyses that included participants irrespective of compliance or follow‐up. For participants with missing data, carry‐forward of the last observed response was used. We originally planned to analyse the influence of missing data using imputation (Higgins 2008). We planned to impute missing values as failures, successes, same as control group, same as experimental group, and same as own group (Higgins 2008). We did not perform these analyses because no losses to follow‐up were described.
Assessment of heterogeneity
Intertrial heterogeneity was assessed on the basis of I2 values.
Assessment of reporting biases
We planned to evaluate the risk of reporting bias by comparing trial protocols and published reports. Furthermore, reporting biases were assessed on the basis of the extent to which clinically relevant outcome measures (hepatic encephalopathy, mortality, and adverse events) were reported.
Data synthesis
Analyses were performed in Review Manager 5 (RevMan 2012) and in STATA 12 (STATA 12). Primary meta‐analyses were performed by using random‐effects models because of anticipated variability between trials regarding participants and interventions.
Subgroup analysis and investigation of heterogeneity
Originally, we planned to perform several subgroup analyses to assess sources of intertrial heterogeneity (bias control, participant characteristics, and intervention regimens). However, because of the limited number of trials in the meta‐analyses of the primary outcomes, we were able to perform these subgroup analyses only for the outcome measure of mortality. Likewise, regression analyses (Egger's test) that were planned to estimate the risk of publication bias and other biases (small‐study effects) were performed only for the outcome measure of mortality.
Trial sequential analysis
We performed trial sequential analysis (CTU 2011; Thorlund 2011) to control risks of random errors due to sparse data and repetitive testing of cumulative data (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009; Wetterslev 2009; Thorlund 2010). To minimise the risk of random error, we calculated the required information size, defined as the required sample size necessary to detect or reject intervention effects after adjusting for diversity (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009;Wetterslev 2009; Thorlund 2010). The information size was calculated on the basis of a risk ratio (RR) reduction of 20% or the results of included trials with a low risk of bias (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009;Wetterslev 2009; Thorlund 2010). We presented the results of the analysis in a graph. with individual trials added on the basis of their year of publication. If more than one trial was published in a year, trials were added alphabetically according to the first author's family name. The results of the trials were presented as a cumulative Z‐curve. The trial sequential monitoring boundaries were constructed and the diversity‐adjusted required information size calculated with a type 1 error of 5% and a type 2 error of 20%. The results were displayed as a graph with the cumulative meta‐analysis results entered. The trial sequential analysis shows firm evidence of intervention effects (or no intervention effects) if the cumulative Z‐curve crosses the monitoring boundaries; it also shows that additional trials may be needed if the boundaries are not crossed.
Sensitivity analysis
The robustness of the results was assessed by repeating the meta‐analyses using a fixed‐effect model. No additional sensitivity analyses were performed because of the limited number of trials identified.
Results
Description of studies
Results of the search
In total, 294 references were identified through the literature searches (Appendix 1). After duplicates and clearly irrelevant references (references to papers that did not describe trials of dopaminergic agents for participants with hepatic encephalopathy) were excluded, 17 references were retrieved for further assessment (Figure 1). Of these, eight references referred to five randomised trials that were eligible for inclusion (Uribe 1979; Vij 1979; Michel 1980; Morgan 1980; Koshy 1982). Through correspondence with the authors of two trials (Uribe 1979; Morgan 1980), additional information was obtained on trial results and methods. For the remaining trials, data were gathered from published reports.
1.
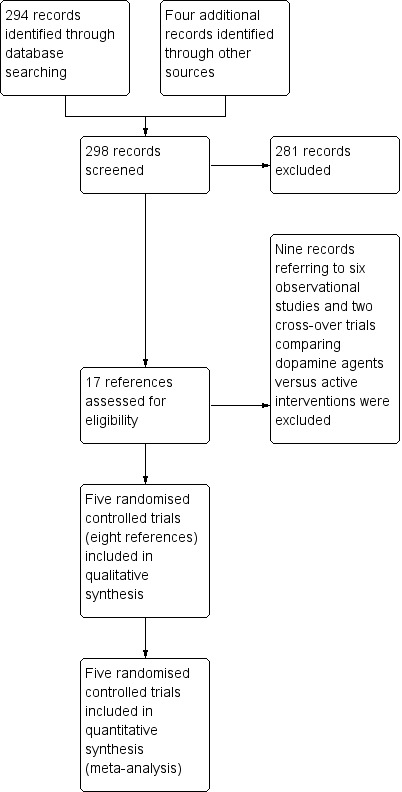
Figure 1. Study flow diagram.
Included studies
All of the included trials were described in at least one full‐paper article published from 1979 to 1982. Three trials used a parallel‐group design (Vij 1979; Michel 1980; Koshy 1982), and two trials used a cross‐over design (Uribe 1979; Morgan 1980).
In total, 144 participants with overt hepatic encephalopathy were included. Three trials (66 participants in the treatment group versus 65 participants in the control group) assessed acute episodes of hepatic encephalopathy (Vij 1979; Michel 1980;Koshy 1982). Two trials (seven participants in the treatment group versus six participants in the control group) assessed chronic hepatic encephalopathy (Uribe 1979; Morgan 1980). Two trials included participants with acute fulminant liver failure due to viral hepatitis (Vij 1979; Koshy 1982). Three trials included participants with cirrhosis (Uribe 1979; Michel 1980; Morgan 1980). The proportion of participants with alcoholic liver disease ranged from 0 to 80%. The proportion of participants with viral hepatitis ranged from 0 to 100%, and mean age ranged from 32 years to 57 years.
Three trials assessed levodopa (Vij 1979; Michel 1980;Koshy 1982), and two trials assessed bromocriptine (Uribe 1979; Morgan 1980). The mean daily dose was 4 grams for levodopa and 15 grams for bromocriptine. The median duration of treatment was 14 days (range seven to 56 days). None of the trials followed participants after the end of treatment. None of the included trials assessed health economics.
Excluded studies
Nine references to eight trials were excluded because they turned out not to be randomised or referred to cross‐over trials that compared dopamine agents versus interventions for hepatic encephalopathy considered potentially active (Characteristics of included studies).
Risk of bias in included studies
All trials had a high risk of bias in the assessment of one or more than one of the bias risk domains.
Allocation
Randomisation methods (allocation sequence generation and allocation concealment) were classed as adequate in two trials (Uribe 1979; Morgan 1980) and unclear in the remaining trials (Figure 2).
2.
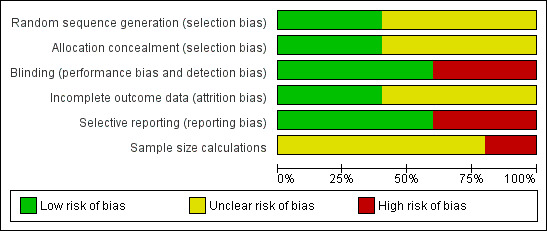
Figure 2. Risk of bias graph: review authors' judgements about all risk of bias items presented as percentages across all included studies.
Blinding
Three trials were blinded using a placebo (Uribe 1979; Michel 1980; Morgan 1980). No blinding was described in the remaining trials.
Incomplete outcome data
Two trials accounted for all participants with missing outcome data (Uribe 1979; Morgan 1980). In the remaining three trials, no dropouts or withdrawals were described, giving the impression that no losses to follow‐up occurred, although this was not specifically stated.
Selective reporting
We were able to extract data on hepatic encephalopathy from only three trials (Uribe 1979; Michel 1980; Morgan 1980).
Other potential sources of bias
No sample size calculations were reported. None of the included trials received industry funding.
Effects of interventions
See: Table 1
Mortality
Random‐effects meta‐analyses found no difference in mortality between participants randomly assigned to dopamine agents versus controls (OR 1.11, 95% CI 0.34 to 3.54; Analysis 1.1). Little intertrial heterogeneity was noted (I2 = 28%). The result was confirmed in a fixed‐effect meta‐analysis (OR 1.24, 95% CI 0.59 to 2.59). No evidence of small‐study effects was identified in regression analysis (Egger's test P value 0.35). In subgroup analyses, no clear differences were seen between trials on participants with acute episodes compared with chronic hepatic encephalopathy (Analysis 1.2) or participants with fulminant liver failure or cirrhosis (Analysis 1.3), trials on levodopa or bromocriptine (Analysis 1.4), trials with a low or unclear risk of bias (Analysis 1.5), or trials using a parallel or cross‐over design (Analysis 1.6).
1.1. Analysis.
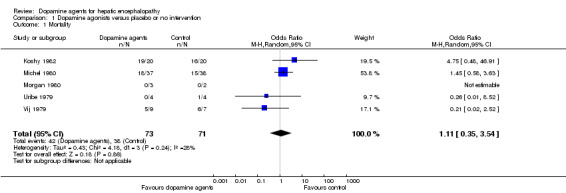
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 1 Mortality.
1.2. Analysis.
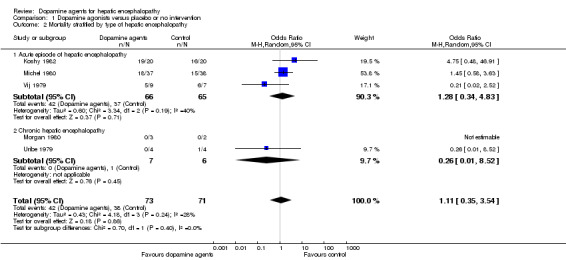
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 2 Mortality stratified by type of hepatic encephalopathy.
1.3. Analysis.
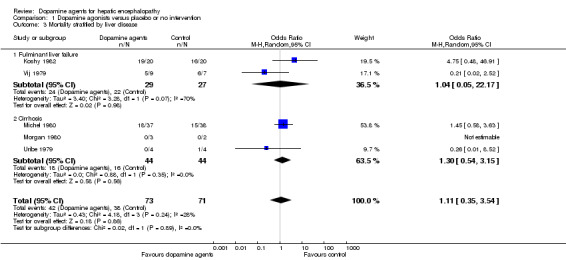
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 3 Mortality stratified by liver disease.
1.4. Analysis.
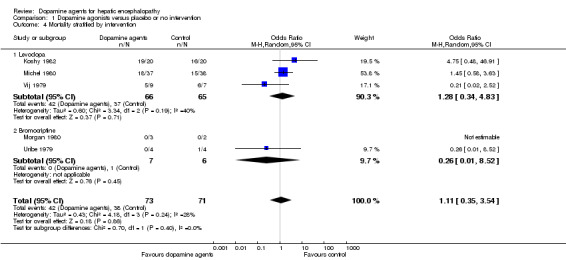
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 4 Mortality stratified by intervention.
1.5. Analysis.
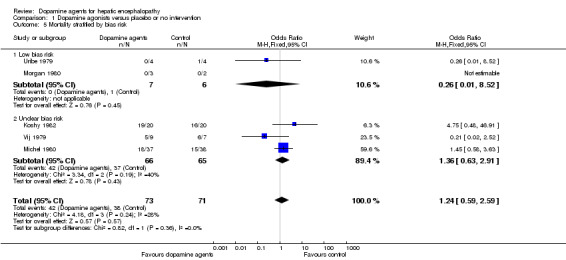
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 5 Mortality stratified by bias risk.
1.6. Analysis.
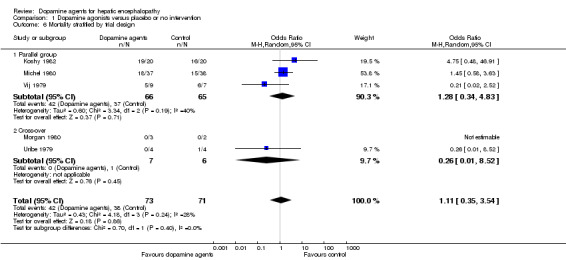
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 6 Mortality stratified by trial design.
The trial sequential analysis graph showed that the cumulative Z‐curve does not cross the monitoring boundary (Figure 3). The analysis showed a diversity‐adjusted required information size of 673 participants (the number of participants needed to reach firm evidence of an intervention effect of 20% risk ratio reduction). The number of participants included corresponds to only 21% of the diversity‐adjusted required information size. Accordingly, we lack evidence to recommend or refute dopamine agents for hepatic encephalopathy.
3.
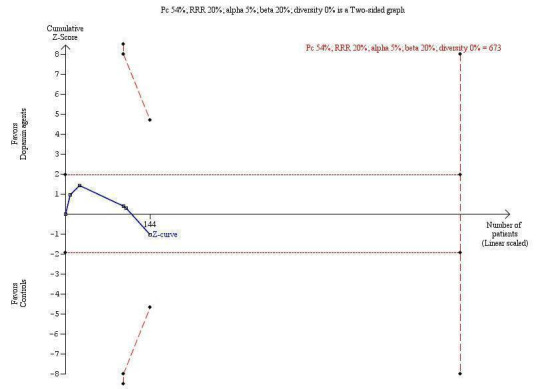
Trial sequential analysis of dopamine agents versus placebo or no intervention in participants with hepatic encephalopathy. The outcome measure is mortality. The analysis was performed with an event rate of 54% (Pc) in the control group, a risk ratio (RR) reduction of 20%, alpha 5%, beta 20%, and diversity 0%. The cumulative Z‐curve does not cross the naive 5% statistical boundaries (dotted horizontal lines) or the trial sequential boundaries for benefits or harms (inward sloping etched lines). The results show that the diversity‐adjusted required information size was 673 participants, corresponding to 21% of the total sample size in the included trials. The programme did not even draw futility boundaries. Accordingly, the meta‐analysis does not recommend or refute an intervention effect; data are simply too few.
Hepatic encephalopathy
The primary random‐effects meta‐analyses showed no significant effects of dopamine agents on hepatic encephalopathy compared with placebo or no intervention when data from parallel‐group trials were analysed (OR 0.33, 95% CI 0.01 to 11.25; Analysis 1.7) or when paired data from the cross‐over trial reporting this outcome measure were included (OR 0.68, 95% CI 0.17 to 2.67; Analysis 1.8). The results were confirmed by fixed‐effect meta‐analyses including data from parallel‐group trials (OR 1.08, 95% CI 0.45 to 2.62), but also when paired data from the two cross‐over trials reporting this outcome measure were included (OR 1.04, 95% CI 0.75 to 1.43).
1.7. Analysis.
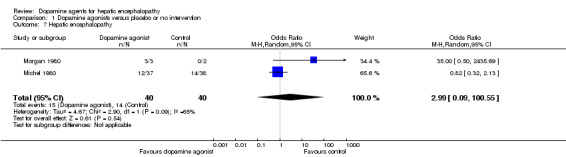
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 7 Hepatic encephalopathy.
1.8. Analysis.
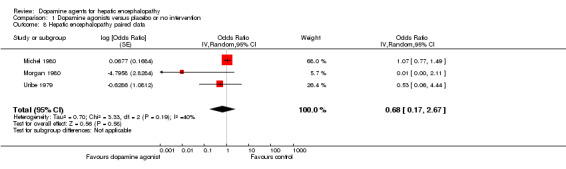
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 8 Hepatic encephalopathy paired data.
Adverse events
We were able to retrieve data on adverse events only from the two cross‐over trials (Uribe 1979; Morgan 1980). In total, seven of 13 participants experienced non‐serious adverse events during treatment with dopamine agents. No adverse events were reported during control periods. No clear difference was observed between intervention and control groups (Analysis 1.9). No serious adverse events were registered. Adverse events included hypomania (n = 1), hallucinations and headache (n = 1), constipation (n = 3), and nausea and vomiting (n = 2).
1.9. Analysis.
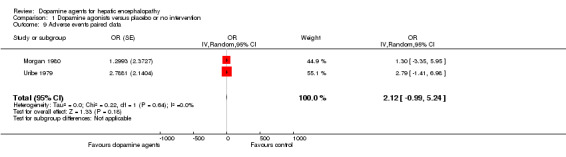
Comparison 1 Dopamine agonists versus placebo or no intervention, Outcome 9 Adverse events paired data.
Quality of life
None of the included trials reported data on quality of life.
Discussion
Summary of main results
Patients with cirrhosis may present with extrapyramidal symptoms similar to those seen in Parkinson's disease (Jover 2003). Further similarities between participants with hepatic encephalopathy and participants with Parkinson's disease include alterations in the basal ganglia (Spahr 2000). In theory, dopamine agents that are effective in Parkinson's disease could alleviate manifestations of hepatic encephalopathy. However, the present systematic review found no evidence to recommend or refute the use of dopamine agents for patients with hepatic encephalopathy. The available evidence includes only a limited number of small trials published before 1983. No clear effects were identified for any of the outcome measures assessed. Additional analyses found no specific subgroups that indicated potential effects when the results of included trials were separated on the basis of the type of hepatic encephalopathy at inclusion, the type of underlying liver disease, or the intervention assessed. The dose and duration of the interventions assessed were similar across trials. Data from participants with Parkinson's disease (Miyasaki 2002) show that the dose of both levodopa and bromocriptine and the duration of the intervention regimens assessed in included trials should be sufficiently high to detect a clinical response. The combined evidence is not promising. However, the statistical power is low, and evidence is insufficient to support or refute beneficial or harmful effects of the interventions assessed.
Overall completeness and applicability of evidence
To ensure completeness of the evidence, we performed extensive literature searches. Our regression analyses showed no clear evidence of publication bias or other small‐study effects. Still, the regression analysis was not sensitive because of the limited number of trials.
The main problem with the included trials is the fact that a number of potentially effective interventions for patients with decompensated liver disease have been identified after the trials were completed. These interventions include treatments for hepatic encephalopathy (Bass 2010), bleeding oesophageal varices (Abraldes 2007), and spontaneous bacterial peritonitis (Wiest 2012). Likewise, the diagnostic assessment and nomenclature for hepatic encephalopathy have been updated (Bajaj 2011). Accordingly, extrapolation of results from the present review to current clinical practice is of limited value.
Quality of the evidence
Adequate internal validity depends on the control of bias and random errors. Because three trials had unclear randomisation (Michel 1980; Morgan 1980; Koshy 1982) and consequently an unclear control of selection bias, the internal validity of their results and of the results of our meta‐analyses can be questioned. The use of a cross‐over design as applied in two of the included trials (Uribe 1979; Morgan 1980) is also debatable. Even chronic hepatic encephalopathy may have a fluctuating course (Basile 1991); therefore, manifestations of hepatic encephalopathy may change during the course of the trial, irrespective of the interventions assessed. The underlying condition and the ability to respond to treatment may not remain stable from the first to the second treatment period. We therefore used only data from the first study period of the cross‐over trials in our primary analyses. Unfortunately, these data were available for only one trial (Morgan 1980). The sensitivity analysis on paired data did not change our overall result.
Potential biases in the review process
Identification and selection of trials are essential to the assessment of bias in the review process. To limit bias in the selection process, we included trials irrespective of language or publication status. We also chose to include trials regardless of the dose or duration of the interventions assessed. This led to a relatively heterogeneous group of trials. We did, however, choose to exclude trials with an active comparison group. This choice was made on the basis of lack of evidence supporting several of the interventions assessed for patients with hepatic encephalopathy. The strategy resulted in the exclusion of two small, low‐quality, cross‐over trials on chronic hepatic encephalopathy (Messner 1982; Uribe 1983). The control groups in these trials received lactulose or neomycin, which could affect the course of hepatic encephalopathy. The total number of participants randomly assigned in these two trials was only 15, and this limits the value of these results.
Agreements and disagreements with other studies or reviews
At present, dopamine agents are not recommended for patients with hepatic encephalopathy. Previous guidelines state that bromocriptine may be considered for patients with chronic hepatic encephalopathy that is unresponsive to other interventions (Blei 1999). In agreement with more recent recommendations (Phongsamran 2010), the present review contradicts these recommendations, suggesting that no evidence is available to support the use of dopamine agents for chronic hepatic encephalopathy.
Authors' conclusions
Implications for practice.
This review does not provide evidence to recommend or refute the use of dopamine agents for patients with hepatic encephalopathy.
Implications for research.
However, we cannot exclude the possibility that dopamine agents may have beneficial effects that were overlooked because of the limited statistical power of the included trials. On the other hand, other interventions for hepatic encephalopathy (such as non‐absorbable disaccharides, branched chain amino acids, and antibiotics) appear potentially more promising than dopamine agents (Als‐Nielsen 2004a; Bass 2010; Les 2011). The value of additional trials on dopamine agents is questionable. Should anyone wish to conduct further trials, we recommend that the dopamine agent used should be tested against placebo in parallel‐group superiority trials conducted according to the SPIRIT guidelines (SPIRIT 2013; SPIRIT 2013a) and reported according to the CONSORT guidelines (www.consort‐statement.org).
What's new
| Date | Event | Description |
|---|---|---|
| 13 January 2014 | New citation required but conclusions have not changed | No new trials fulfilled the inclusion criteria of this review. |
| 13 January 2014 | New search has been performed | Searches were updated January 2014, but no new trials were identified for inclusion in the review. New trials are unlikely to be published in the following four years. The review has been updated based on current methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). |
| 13 January 2012 | Amended | AE Junker is the new, lead author of this first review update. |
Acknowledgements
We thank Marsha Morgan and Misael Uribe for providing additional information on their trials, and Sarah Klingenberg for performing the electronic literature searches.
Peer reviewers: FG Romeiro, Brazil; Diego Sánchez‐Munoz, Spain. Contact editor: Goran Bjelakovic, Serbia.
Appendices
Appendix 1. Search strategies
| Database | Time span | Search strategy |
| Cochrane Hepato‐Biliary Group Controlled Trials Register | January 2014. | (dopa* OR 'dopa decarboxylase' OR levodopa OR bromocriptine) AND ('liver cirrhosis' OR 'hepatic encephalopathy') |
| The Cochrane Central Register of Controlled Trials (CENTRAL) | Issue 12 of 12, 2013. | #1 MeSH descriptor: [Dopamine Agents] explode all trees #2 MeSH descriptor: [Dopa Decarboxylase] explode all trees #3 MeSH descriptor: [Levodopa] explode all trees #4 MeSH descriptor: [Bromocriptine] explode all trees #5 dopa* or dopa decarboxylase or levodopa or bromocriptine #6 #1 or #2 or #3 or #4 or #5 #7 MeSH descriptor: [Liver Cirrhosis] explode all trees #8 MeSH descriptor: [Hepatic Encephalopathy] explode all trees #9 (liver cirrhosis or hepatic encephalopathy) #10 #7 or #8 or #9 #11 #6 and #10 |
| MEDLINE (Ovid SP) | 1946 to January 2014. | 1. exp Dopamine Agents/ 2. exp Dopa Decarboxylase/ 3. exp Levodopa/ 4. exp Bromocriptine/ 5. (dopa* or dopa decarboxylase or levodopa or bromocriptine).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 6. 1 or 2 or 3 or 4 or 5 7. exp Liver Cirrhosis/ 8. exp Hepatic Encephalopathy/ 9. (liver cirrhosis or hepatic encephalopathy).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 10. 7 or 8 or 9 11. 6 and 10 12. (random* or blind* or placebo* or meta‐analysis).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 13. 11 and 12 |
| EMBASE (Ovid SP) | 1974 to January 2014. | 1. exp dopamine receptor stimulating agent/ 2. exp dopamine receptor blocking agent/ 3. exp aromatic levo amino acid decarboxylase/ 4. exp LEVODOPA/ 5. exp BROMOCRIPTINE/ 6. (dopa* or dopa decarboxylase or levodopa or bromocriptine).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 7. 1 or 2 or 3 or 4 or 5 or 6 8. exp liver cirrhosis/ 9. exp hepatic encephalopathy/ 10. (liver cirrhosis or hepatic encephalopathy).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 11. 8 or 9 or 10 12. 7 and 11 13. (random* or blind* or placebo* or meta‐analysis).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 14. 12 and 13 |
| Science Citation Index‐Expanded (http://apps.webofknowledge.com) | 1900 to January 2014. | #5 20 #4 AND #3 #4 1,186,796 TS=(random* or blind* or placebo* or meta‐analysis) #3 205 #2 AND #1 #2 54,464 TS=(liver cirrhosis or hepatic encephalopathy) #1 198,470 TS=(dopa* or dopa decarboxylase or levodopa or bromocriptine) |
Data and analyses
Comparison 1. Dopamine agonists versus placebo or no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 5 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.35, 3.54] |
| 2 Mortality stratified by type of hepatic encephalopathy | 5 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.35, 3.54] |
| 2.1 Acute episode of hepatic encephalopathy | 3 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.34, 4.83] |
| 2.2 Chronic hepatic encephalopathy | 2 | 13 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.01, 8.52] |
| 3 Mortality stratified by liver disease | 5 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.35, 3.54] |
| 3.1 Fulminant liver failure | 2 | 56 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.05, 22.17] |
| 3.2 Cirrhosis | 3 | 88 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.54, 3.15] |
| 4 Mortality stratified by intervention | 5 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.35, 3.54] |
| 4.1 Levodopa | 3 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.34, 4.83] |
| 4.2 Bromocriptine | 2 | 13 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.01, 8.52] |
| 5 Mortality stratified by bias risk | 5 | 144 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.59, 2.59] |
| 5.1 Low bias risk | 2 | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.01, 8.52] |
| 5.2 Unclear bias risk | 3 | 131 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.36 [0.63, 2.91] |
| 6 Mortality stratified by trial design | 5 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.35, 3.54] |
| 6.1 Parallel group | 3 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.34, 4.83] |
| 6.2 Cross‐over | 2 | 13 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.01, 8.52] |
| 7 Hepatic encephalopathy | 2 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 2.99 [0.09, 100.55] |
| 8 Hepatic encephalopathy paired data | 3 | Odds Ratio (Random, 95% CI) | 0.68 [0.17, 2.67] | |
| 9 Adverse events paired data | 2 | OR (Random, 95% CI) | 2.12 [‐0.99, 5.24] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Koshy 1982.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | No blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Losses to follow‐up not described. |
| Selective reporting (reporting bias) | High risk | Hepatic encephalopathy not reported. |
| Sample size calculations | High risk | No. |
Michel 1980.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind placebo‐controlled. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No losses to follow‐up described. |
| Selective reporting (reporting bias) | Low risk | Clinically relevant outcome measures defined and reported. |
| Sample size calculations | Unclear risk | Not reported. |
Morgan 1980.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random number sequence. |
| Allocation concealment (selection bias) | Low risk | Central independent unit. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Administration of identical coded drug container. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted for. |
| Selective reporting (reporting bias) | Low risk | Clinically relevant outcome measures defined and reported. |
| Sample size calculations | Unclear risk | Not reported. |
Uribe 1979.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table. |
| Allocation concealment (selection bias) | Low risk | Central administration of blinded drug containers. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind placebo‐controlled with additional blinded data analyses. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted for. |
| Selective reporting (reporting bias) | Low risk | Clinically relevant outcome measures defined and reported. |
| Sample size calculations | Unclear risk | Not reported. |
Vij 1979.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Losses to follow‐up not described. |
| Selective reporting (reporting bias) | High risk | Hepatic encephalopathy not reported. |
| Sample size calculations | Unclear risk | Not reported. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Catalano 1982 | Observational study. |
| Datta 1976 | Observational study. |
| Jorge 1973 | Observational study. |
| Lunzer 1974 | Observational study. |
| Messner 1982 | Randomised cross‐over trial including 11 participants with chronic hepatic encephalopathy comparing bromocriptine with lactulose. Excluded because the control group received an active intervention. |
| Pascual 1979 | Observational study. |
| Trovato 1982 | Observational study. |
| Ubiria 1980 | Observational study. |
| Uribe 1983 | Randomised cross‐over trial including four participants with chronic hepatic encephalopathy comparing bromocriptine versus neomycin. Excluded because the control group received an active intervention. |
Differences between protocol and review
We have changed the term 'dopaminergic agents' to the MeSH term 'dopamine agents' throughout the review.
Based on reviewer comments, we have omitted the outcome 'Number of participants with hepatic encephalopathy recovery' because the definition of this outcome is highly variable. The outcome of (lack of) improvement in hepatic encephalopathy includes participants with complete as well as partial recovery from hepatic encephalopathy.
In our original protocol, we planned to include health economics as an outcome. This outcome was omitted from our previous and present review on the basis of reviewer comments and evidence concerning the best methods for assessing this outcome. We have gathered data on whether health economics were assessed and have included these data in our table of included trials.
Based on the most recent recommendations regarding the assessment of bias control, we have included bias tables and have assessed the bias control components of allocation (selection bias), blinding (performance bias and detection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), and other potential sources of bias (sample size assessments).
We have included additional analyses on small‐study effects (Egger's test).
Contributions of authors
Anders Ellekær Junker (AEJ) and Lise Lotte Gluud (LLG) drafted the revised version of this updated review with methodology updates based on the most recent recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). AEJ, Bodil Als‐Nielsen (BA‐N), and LLG participated in the literature searches, identified trials eligible for inclusion, extracted data, and performed the statistical analyses. All authors revised the review and have approved the final version.
Sources of support
Internal sources
Copenhagen Trial Unit, Denmark.
External sources
The 1991 Pharmacy Foundation, Denmark.
Danish Center for Evaluation and Health Technology Assessment (DACEHTA), Denmark.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Koshy 1982 {published data only}
- Koshy A, Pratap B, Datta DV. Prospective randomised controlled trial of L‐dopa and hydrocortisone in fulminant hepatitis. Journal of the Association of Physicians of India 1982;30(9):613‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Michel 1980 {published data only}
- Michel H, Cauvet G, Granier PM, Bali JP, Cuilleret A, Cuilleret G. Treatment of cirrhotic hepatic encephalopathy by L‐dopa. A double‐blind study of 58 patients [abstract]. Digestion 1977; Vol. 15:232‐3.
- Michel H, Solere M, Granier P, Cauvet G, Bali JP, Pons F, et al. Treatment of cirrhotic hepatic encephalopathy with L‐dopa. A controlled trial. Gastroenterology 1980;79(2):207‐11. [MEDLINE: ] [PubMed] [Google Scholar]
Morgan 1980 {published data only}
- Morgan MY. Bromocriptine in the treatment of chronic hepatic encephalopathy. In: Capocaccia L, Fischer JE, Rossi‐Fanelli F editor(s). Hepatic Encephalopathy in Chronic Liver Failure. New York: Plenum Press, 1984:255‐65. [Google Scholar]
- Morgan MY, Jakobovits AW, James IM, Lennox R, Sherlock S. Bromocriptine in the treatment of chronic portal systemic encephalopathy [abstract]. Gut 1978; Vol. 19, issue Suppl 2:A453.
- Morgan MY, Jakobovits AW, James IM, Sherlock S. Successful use of bromocriptine in the treatment of chronic hepatic encephalopathy. Gastroenterology 1980;78(4):663‐70. [MEDLINE: ] [PubMed] [Google Scholar]
Uribe 1979 {published data only}
- Uribe M, Farca A, Marquez MA, Garcia Ramos G, Guevara L. Treatment of chronic portal systemic encephalopathy with bromocriptine: a double‐blind controlled trial. Gastroenterology 1979;76(6):1347‐51. [MEDLINE: ] [PubMed] [Google Scholar]
Vij 1979 {published data only}
- Vij JC, Tandon BN. Controlled trial of levodopa in fulminant hepatitis. Indian Journal of Medical Research 1979;69:624‐8. [MEDLINE: ] [PubMed] [Google Scholar]
References to studies excluded from this review
Catalano 1982 {published data only}
- Catalano D, Trovato G, Vancheri F, Mazzone O. Bromocriptine in the treatment of chronic hepatic encephalopathy refractory to conventional treatment: crossover trial with levodopa‐benserazide. Current Therapeutic Research 1982;32:544‐54. [EMBASE 1982249532] [Google Scholar]
Datta 1976 {published data only}
- Datta DV, Maheshwari YK, Aggarwal ML. Levodopa in fulminant hepatic failure: preliminary report. American Journal of the Medical Sciences 1976;272:95‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jorge 1973 {published data only}
- Jorge AD, Sanchez D. Effect of L‐dopa on hepatic coma [Die Wirkung des L‐Dopa im Coma hepaticum]. Schweizerische Rundschau für Medizin Praxis 1973;62(27):868‐72. [MEDLINE: ] [PubMed] [Google Scholar]
Lunzer 1974 {published data only}
- Lunzer M, James IM, Weinman J, Sherlock S. Treatment of chronic hepatic encephalopathy with levodopa. Gut 1974;15(7):555‐61. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Messner 1982 {published data only}
- Messner M, Gall J‐Y, Toulouse P, Javaudin L, Delamaire D, Brissot P, et al. Plasma ratio of branched chain/aromatic amino acids during treatment of chronic hepatic encephalopathy using lactulose/bromocriptine in a double blind procedure [EASL abstract]. Liver 1982;2(3 Pt 2):312. [EMBASE: 1983020208] [Google Scholar]
Pascual 1979 {published data only}
- Pascual Izuel JM, Peces Serrano R, Hernandez Guio C. Role of bromocriptine in the treatment of chronic hepatic encephalopathy [Interes de la bromocriptina en el tratamiento de la encefalopatia hepatica cronica]. Revista Clinica Espanola 1979;155(3):219‐21. [MEDLINE: ] [PubMed] [Google Scholar]
Trovato 1982 {published data only}
- Trovato GM, Catalano D, Vancheri FS, Mazzone O. Successful use of amantadine and levodopa‐benserazide in chronic portal systemic encephalopathy. A crossover trial with conventional therapy. Current Therapeutic Research Clinical and Experimental 1982;31:625‐37. [EMBASE 1982138453] [Google Scholar]
Ubiria 1980 {published data only}
- Ubiria J, Morales M, Vila A, Farre A, Balanzo J. Treatment of chronic portosystemic encephalopathy with bromocriptine [Tratamiento de la encefalopatía crónica portosistémica (E.C.P.S.) con bromocriptina]. Revista Espanola de Las Enfermedades del Aparato Digestivo 1980;58(1):21‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Uribe 1983 {published data only}
- Uribe M, Garcia Ramos G, Ramos M, Valverde C, Marquez MA, Farca A, et al. Standard and higher doses of bromocriptine for severe chronic portal‐systemic encephalopathy. American Journal of Gastroenterology 1983;78(8):517‐22. [MEDLINE: ] [PubMed] [Google Scholar]
- Uribe M, Marquez MA, Guevara L, Garcia Ramos G. Bromocriptine for severe chronic portal systemic encephalopathy [abstract]. Gastroenterology 1982;82:1201. [PubMed] [Google Scholar]
Additional references
Abraldes 2007
- Abraldes JG, Bosch J. The treatment of acute variceal bleeding. Journal of Clinical Gastroenterology 2007;41(Suppl 3):S312‐S317. [PUBMED: 17975482] [DOI] [PubMed] [Google Scholar]
Als‐Nielsen 2004a
- Als‐Nielsen B, Gluud LL, Gluud C. Nonabsorbable disaccharides for hepatic encephalopathy. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD003044.pub2] [DOI] [PubMed] [Google Scholar]
Bajaj 2011
- Bajaj JS, Cordoba J, Mullen KD, Amodio P, Shawcross DL, Butterworth RF, et al. Review article: the design of clinical trials in hepatic encephalopathy ‐ an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Alimentary Pharmacology & Therapeutics 2011;33(7):739‐47. [PUBMED: 21306407] [DOI] [PMC free article] [PubMed] [Google Scholar]
Basile 1991
- Basile AS, Jones EA, Skolnick P. The pathogenesis and treatment of hepatic encephalopathy: evidence for the involvement of benzodiazepine receptor ligands. Pharmacological Reviews 1991;43(1):27‐71. [MEDLINE: ] [PubMed] [Google Scholar]
Bass 2010
- Bass NM, Mullen KD, Sanyal A, Poordad F, Neff G, Leevy CB, et al. Rifaximin treatment in hepatic encephalopathy. New England Journal of Medicine 2010;362(12):1071‐81. [PUBMED: 20335583] [DOI] [PubMed] [Google Scholar]
Becker 1993
- Becker MP, Balagtas CC. Marginal modelling of binary cross‐over data. Biometrics 1993;49:997‐1009. [MEDLINE: ] [PubMed] [Google Scholar]
Blei 1999
- Blei A. Hepatic encephalopathy. In: Bircher J, Benhamou J‐P, McIntyre N, Rizzetto M, Rodés J editor(s). Clinical Hepatology. 2nd Edition. Vol. 1, Oxford: Oxford University Press, 1999:765‐83. [Google Scholar]
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta‐analyses. Journal of Clinical Epidemiology 2008;61:763‐9. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive ‐ Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI] [PubMed] [Google Scholar]
Conn 1979
- Conn H, Lieberthal M. The Hepatic Coma Syndromes and Lactulose. Baltimore: Williams & Wilkins, 1979. [Google Scholar]
Cordoba 2001
- Cordoba J, Alonso J, Rovira A, Jacas C, Sanpedro F, Castells L, et al. The development of low‐grade cerebral edema in cirrhosis is supported by the evolution of (1)H‐magnetic resonance abnormalities after liver transplantation. Journal of Hepatology 2001;35:598‐604. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
CTU 2011
- Copenhagen Trial Unit. TSA ‐ Trial Sequential Analysis. ctu.dk/tsa/ 2011 (accessed 4 September 2012).
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31:140‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ferenci 2002
- Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy ‐ definition, nomenclature, diagnosis, and quantification: final report of the Working Party at the 11th World Congress of Gastroenterology, Vienna, 1998. Hepatology 2002;35:716‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gitlin 1996
- Gitlin N. Hepatic encephalopathy. In: Zakim D, Boyer T editor(s). Hepatology. A Textbook of Liver Disease. 3rd Edition. Vol. 1, Philadelphia: WB Saunders Company, 1996:605‐17. [Google Scholar]
Gluud 2013
- Gluud C, Nikolova D, Klingenberg SL, Alexakis N, Als‐Nielsen B, Colli A, et al. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)). Art. No.: LIVER 2013, issue 12.
Guerit 2009
- Guerit JM, Amantini A, Fischer C, Kaplan PW, Mecarelli O, Schnitzler A, et al. Neurophysiological investigations of hepatic encephalopathy: ISHEN practice guidelines. Liver International 2009;29(6):789‐96. [PUBMED: 19638107] [DOI] [PubMed] [Google Scholar]
Haussinger 2000
- Haussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low‐grade cerebral edema?. Journal of Hepatology 2000;32:1035‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, White IR, Wood AM. Imputation methods for missing outcome data in meta‐analysis of clinical trials. Clinical Trials 2008;5:225‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Whitehead A, Simmonds M. Sequential methods for random‐effects meta‐analysis. Statistics in Medicine 2011;30(9):903‐21. [PUBMED: 21472757] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice CFR and ICH Guidelines. Vol. 1, PA 19063‐2043, USA: Barnett International/PAREXEL, 1997. [Google Scholar]
Jover 2003
- Jover R, Company L, Gutierrez A, Zapater P, Perez‐Serra J, Girona E, et al. Minimal hepatic encephalopathy and extrapyramidal signs in patients with cirrhosis. American Journal of Gastroenterology 2003;98:1599‐604. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Les 2011
- Les I, Doval E, Garcia‐Martinez R, Planas M, Cardenas G, Gomez P, et al. Effects of branched‐chain amino acids supplementation in patients with cirrhosis and a previous episode of hepatic encephalopathy: a randomized study. American Journal of Gastroenterology 2011;106(6):1081‐8. [PUBMED: 21326220] [DOI] [PubMed] [Google Scholar]
Lizardi‐Cervera 2003
- Lizardi‐Cervera J, Almeda P, Guevara L, Uribe M. Hepatic encephalopathy: a review. Annals of Hepatology 2003;2:122‐30. [MEDLINE: ] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Miyasaki 2002
- Miyasaki JM, Martin W, Suchowersky O, Weiner WJ, Lang AE. Practice parameter: initiation of treatment for Parkinson's disease: an evidence‐based review. Report of the Quality Standards Subcommittee of the American Academy of Neurology. American Academy of Neurology 2002;58:11‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
Parkes 1970
- Parkes JD, Sharpstone P, Williams R. Levodopa in hepatic coma. Lancet 1970;2(7687):1341‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Phongsamran 2010
- Phongsamran PV, Kim JW, Cupo Abbott J, Rosenblatt A. Pharmacotherapy for hepatic encephalopathy. Drugs 2010;70(9):1131‐48. [PUBMED: 20518580] [DOI] [PubMed] [Google Scholar]
Randolph 2009
- Randolph C, Hilsabeck R, Kato A, Kharbanda P, Li YY, Mapelli D, et al. Neuropsychological assessment of hepatic encephalopathy: ISHEN practice guidelines. Liver International 2009;29(5):629‐35. [PUBMED: 19302444] [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Savovic 2012
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Savovic 2012a
- Savovic J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Health Technology Assessment 2012;16(35):1‐82. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Spahr 2000
- Spahr L, Vingerhoets F, Lazeyras F, Delavelle J, DuPasquier R, Giostra E, et al. Magnetic resonance imaging and proton spectroscopic alterations correlate with parkinsonian signs in patients with cirrhosis. Gastroenterology 2000;119:774‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
SPIRIT 2013
- Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža‐Jerić K, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials. Annals of Internal Medicine 2013;158:200‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
SPIRIT 2013a
- Chan A‐W, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin J, et al. SPIRIT 2013 Explanation and Elaboration: Guidance for protocols of clinical trials. BMJ (Clinical Research Ed.) 2013;346:e7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
STATA 12 [Computer program]
- STATACorp LP, Texas USA. STATA 12. STATACorp LP, Texas USA, 2012.
Stowe 2008
- Stowe R, Ives N, Clarke CE, van Hilten, Ferreira J, Hawker RJ, et al. Dopamine agonist therapy in early Parkinson's disease. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD006564.pub2; PUBMED: 18425954] [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses. International Journal of Epidemiology 2009;38(1):276‐86. [DOI] [PubMed] [Google Scholar]
Thorlund 2010
- Thorlund K, Anema A, Mills E. Interpreting meta‐analysis according to the adequacy of sample size. An example using isoniazid chemoprophylaxis for tuberculosis in purified protein derivative negative HIV‐infected individuals. Clinical Epidemiology 2010;2:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for Trial Sequential Analysis (TSA). http://ctu.dk/tsa/files/tsa_manual.pdf 2011 (accessed 23 April 2013).
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [PUBMED: 18083463] [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random‐effects model meta‐analyses. BMC Medical Research Methodology 2009;9:86. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiest 2012
- Wiest R, Krag A, Gerbes A. Spontaneous bacterial peritonitis: recent guidelines and beyond. Gut 2012;61(2):297‐310. [PUBMED: 22147550] [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Juni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336(7644):601‐5. [PUBMED: 18316340] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Als‐Nielsen 2004
- Als‐Nielsen B, Gluud LL, Gluud C. Dopaminergic agonists for hepatic encephalopathy. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD003047.pub2] [DOI] [PubMed] [Google Scholar]