

Abstract
Background:
Oxidative stress contributes to thrombosis in atherosclerosis, inflammation, infection, aging, and malignancy. Oxidant-induced cysteine modifications, including sulfenylation, can act as a redox-sensitive switch that controls protein function. Protein disulfide isomerase (PDI) is a prothrombotic enzyme with exquisitely redox-sensitive active-site cysteines.
Objectives:
We hypothesized that PDI is sulfenylated during oxidative stress, contributing to the prothrombotic potential of PDI.
Methods:
Biochemical and enzymatic assays using purified proteins, platelet and endothelial cell assays, and in vivo murine thrombosis studies were used to evaluate the role of oxidative stress in PDI sulfenylation and prothrombotic activity.
Results:
PDI exposure to oxidants resulted in the loss of PDI reductase activity and simultaneously promoted sulfenylated PDI generation. Following exposure to oxidants, sulfenylated PDI spontaneously converted to disulfided PDI. PDI oxidized in this manner was able to transfer disulfides to protein substrates. Inhibition of sulfenylation impaired disulfide formation by oxidants, indicating that sulfenylation is an intermediate during PDI oxidation. Agonist-induced activation of platelets and endothelium resulted in the release of sulfenylated PDI. PDI was also sulfenylated by oxidized low-density lipoprotein (oxLDL). In an in vivo model of thrombus formation, oxLDL markedly promoted platelet accumulation following an arteriolar injury. PDI oxidoreductase inhibition blocked oxLDL-mediated augmentation of thrombosis.
Conclusion:
PDI sulfenylation is a critical posttranslational modification that is an intermediate during disulfide PDI formation in the setting of oxidative stress. Oxidants generated by vascular cells during activation promote PDI sulfenylation, and interference with PDI during oxidative stress impairs thrombus formation.
Keywords: cysteine, disulfide, oxidation-reduction, protein disulfide isomerase, sulfenylation, thrombosis
1 |. INTRODUCTION
Thrombus formation often occurs during oxidative stress, as with atherosclerosis, infection, inflammatory states, aging, sickle cell disease, and cancer. It is well-established that oxidant production contributes to thrombus formation [1–3]. Yet, how oxidative stress contributes to thrombus formation remains an area of active investigation. Direct modification by oxidants of proteins involved in thrombus formation is a potential prothrombotic mechanism and has been described for von Willebrand factor, thrombomodulin, and protein C [4]. Prothrombotic protein disulfide isomerase (PDI) family proteins may also undergo oxidant-dependent modifications [5]. However, how oxidants that are commonly generated in vascular pathology modify PDI during thrombus formation is unknown.
PDI is the archetypal member of a thiol isomerase family that promotes oxidative protein folding in the endoplasmic reticulum (ER). PDI supports protein folding by 4 thioredoxin-like domains, termed a, b, b′, and a′, with a flexible x-linker flanked by the b′ and a′ domains. The PDI active sites contain the redox-sensitive cysteine-glycine-histidine-cysteine (CGHC) motif and are located within the a and a′ domains. The b and b′ domains include hydrophobic regions and participate in substrate recognition. The b′ domain contains 2 additional cysteines at positions 312 and 343. Many factors influence the redox-sensitive nature of PDI, including the redox potential of the active-site cysteines [6], electrostatic potentials governed by the environment and pKa of the active-site cysteines [7,8], and the flux of oxidants relative to the buffering capacity of antioxidants [9]. PDI oxidation resulting in disulfide bond formation in the active site has marked effects on both the PDI structure [10,11] and function, converting a reductase into an oxidase and isomerase.
A role for PDI in thrombus formation has been demonstrated in several different models of thrombus formation using PDI-directed antibodies, PDI small molecule antagonists, and peptides that inhibit PDI function [4,12]. Studies on thrombus formation in mice lacking platelet PDI or expressing PDI mutants have confirmed a role of PDI in thrombus formation [13,14]. PDI oxidoreductase inhibition as an antithrombotic strategy has even advanced to clinical trials and appears to be effective in reducing cancer-associated thrombosis [15]. Yet, despite this evidence that PDI participates in thrombus formation, little is known about cysteine modifications in prothrombotic PDI.
Our recent studies demonstrated that cysteine sulfenylation contributes to thrombus formation in hyperlipidemia [16]. Cysteine sulfenylation is a labile and transient posttranslational sulfur oxoform generated by the oxidation of cysteines with 2-electron reactive oxygen species. Sulfenylation is at the crossroad of many oxidative cysteine modifications [17,18]. In thrombotic disorders, cysteine sulfenylation of platelet proteins is an important regulator of platelet reactivity under oxidative stress conditions [16,19]. The sulfenic oxoform can be selectively detected using beta dicarbonyl–based carbon nucleophilic probes [20], and proteome-based screens designed to detect sulfenylated proteins have putatively identified PDI family proteins [21,22].
In this study, we report that PDI undergoes cysteine sulfenylation by 2-electron oxidants. Sulfenylation of the active-site cysteines is an intermediary to disulfide formation and promotes PDI oxidase activity. Secreted PDI from endothelial cells and platelets is sulfenylated, suggesting their potential to be oxidized and to contribute to thrombus formation. We further show that incubation with oxidized low-density lipoprotein (oxLDL) results in PDI sulfenylation and that oxLDL augmentation of thrombus formation is prevented by PDI oxidoreductase inhibition in an in vivo model of thrombus formation.
2 |. METHODS
2.1 |. PDI expression and purification
PDI was expressed and purified as previously described [11,23]. In brief, the cDNA for human PDI was inserted between the FLAG and streptavidin binding peptide in a pT7-FLAG-SBP-1 vector (ThermoFisher). PDI was expressed in BL21 (DE3) Escherichia coli (New England Biolabs) and grown in LB-ampicillin broth at 37 °C until induced overnight at 25 °C with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The cells were lysed by sonication, and the clarified lysates were poured into a high-capacity streptavidin-coated agarose column. The column was washed with 150 mL of a wash buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), and 5 mM DTT. The protein was eluted with 200 mM biotin before dialysis against phosphate-buffered saline (PBS) and concentrated and stored at −80 °C until use. In a sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) electrophoresis, reduced PDI resolves at 60 to 70 kDa due to the N-terminal FLAG tag and the C-terminal streptavidin binding peptide. PDI fragments were expressed and purified as previously described [24]. A detailed description of the circular dichroism analysis of PDI wild-type (PDI-CCCC) and catalytically inactive mutant PDI-AAAA is provided in the Supplementary material.
2.2 |. PDI reductase activity assay
For the PDI reductase activity assay, 0.2 μM PDI was treated with increasing concentration of the indicated oxidants in a phosphate buffer (39 mM KH2PO4, 61 mM K2HPO4, and 2 mM EDTA; pH 7.0) for 1 hour at 37 °C. Following this incubation, residual reductase activity of PDI was monitored using a plate reader by following PDI-mediated cleavage of 0.3 μM di-eosin-GSSG (Supplementary Figure S1) at 545 nm for up to 30 minutes at room temperature. As no reducing agents were added, the PDI-catalyzed reaction proceeded as a single turn-over event, and the fluorescence in the latter 15 minutes was averaged. The data were plotted as the percentage of reductase activity in the absence of oxidant vs oxidant concentration, and fit to the following equation using GraphPad Prism v9:
2.3 |. Oxidative cysteine modification and disulfide detection
Reduced recombinant human PDI was diluted to 5 μM in PBS, followed by treatment with hydrogen peroxide for up to 1 hour at 37 °C. At the indicated time points, 0.2 mM maleimide-PEG2-biotin was added to alkylate the free cysteines and thus quench the reaction. For disulfide detection, PDI was treated with 100 μM H2O2 for 30 minutes at 37 °C in the presence or absence of 1 mM arsenite, followed by quenching the reaction and blocking all free thiols with 0.2 mM N-ethylmaleimide for 15 minutes at room temperature. Unreacted N-ethylmaleimide was then removed by desalting through a MicroBio Spin P6 column. 1× Laemmli sample buffer containing 10% (v/v) β-mercaptoethanol and 0.2 mM maleimide-PEG2-biotin was added before boiling the samples for SDS-PAGE, Western blotting, and detection of biotin using streptavidin-HRP. A detailed description of the disulfide kinetic analysis by mass spectrometry is previously described [25] and provided in the Supplementary material.
2.4 |. Cysteine sulfenylation detection with purified proteins
Recombinant human PDI was diluted to 5 μM with a Tris buffer (50 mM Tris and 150 mM NaCl; pH 7.4). The protein was prereduced with 200 μM tris(2-carboxyethyl)phosphine (TCEP) for 15 minutes at room temperature before desalting into Tris buffer using Micro Bio-Spin-6 columns (Bio-Rad). Five micromolar PDI was then oxidized with 100 μM H2O2 in the presence of 0.1 mM benzo[c][1,2]thiazine–based probe containing an alkyne arm (BTD) [20] for up to 1 hour at 37 °C. The probe-labeled protein was subjected to click chemistry using 100 μM biotin-PEG3-azide or 100 μM Alexa Fluor488-azide, 1 mM TCEP, 0.1 mM BTTP, and 1 mM Cu(II)SO4 to covalently link the azide to the alkyne. The reactions were rocked for 60 minutes at 37 °C. Excess click reagents were removed by acetone precipitation overnight, and the protein was pelleted by centrifugation at 14 000 RCF at 4 °C for 10 minutes. The protein pellet was resolubilized in prewarmed PBS containing 1% (w/v) sodium dodecyl sulfate (SDS) and sonicated briefly using a Branson 2800 sonicator. The solubilized samples were treated with 1× Laemmli sample buffer with 10% (v/v) β-mercaptoethanol and boiled for 10 minutes at 100 °C before resolving on a 4% to 15% TGX SDS-polyacrylamide gel (Bio-Rad). The in-gel fluorescence was detected with a Bio-Rad ChemiDoc followed by InstantBlue Coomassie staining for 1 hour. Before imaging, the Coomassie-stained gel was destained 3 times for 10 minutes each with water.
2.5 |. PDI oxidase/isomerase activity assay
Bovine RNAse was reduced and denatured in a 100 mM Tris buffer (pH 8) containing 140 mM dithiothreitol and 6 M guanidinium hydrochloride overnight at 4 °C. Denatured scrambled RNAse was desalted using a PD MiniTrap G-25 column preequilibrated with Tris buffer. The concentration of RNAse was determined by absorbance at 280 nm with a molar extinction coefficient of 9.3 mM−1cm−1 [26]. Before the oxidative renaturing of ribonuclease activity, 13 μM PDI was reduced with 0.1 mM TCEP for 15 minutes at room temperature, followed by desalting using a PD MiniTrap G-25 or PD-10 column preequilibrated with Tris buffer. In some experiments, the protein was treated with up to 5 mM sodium arsenite before oxidation. PDI was oxidized or reduced with 0.1 mM H2O2 or TCEP, respectively, for 1 hour at 37 °C. For PDI oxidase/isomerase activity, 4 μM reduced or oxidized PDI was added to 4.5 mM 2’,3’-cyclic cytidine monophosphate (cCMP) before initiating the reaction with 1.33 μM scRNAse. The oxidative renaturation of ribonuclease activity was measured spectrophotometrically by the hydrolysis of cCMP to CMP at 295 nm, and proceeded without a redox buffer.
2.6 |. Laser-injury thrombosis model
Thrombus formation in response to laser injury was measured as described previously [27–30]. Briefly, cremaster muscle arterioles were injured using a MicroPoint Laser System (Photonics Instruments). Platelet accumulation was measured by infusion of Dylight 649-labeled antiplatelet antibodies (CD42b; 0.1 mg/kg body weight; Emfret Analytics) through a jugular vein catheter. Data were acquired before and after laser injury using the 640/670 nm channel. Images were captured for 250 seconds at 0.5 frames/s using a CCD camera (Hamamatsu). Data were analyzed using Slidebook 6.0 (Intelligent Imaging Innovations). Data from 16 to 30 thrombi were used to determine the median value of the integrated fluorescence intensity to account for variability in thrombus formation under any given experimental condition. AUC was calculated for individual thrombi and normalized to injury lengths to evaluate statistical significance. Injury lengths were determined as previously described [29].
3 |. RESULTS
3.1 |. Oxidative cysteine modifications control PDI reductase activity
Thrombosis typically occurs in an oxidative environment. Since PDI is highly redox-sensitive and critical for thrombus formation, we evaluated the sensitivity of PDI enzymatic activity to a variety of oxidants. An oxidant panel was tested for influence on PDI reductase activity using di-eosin–oxidized glutathione (di-eosin-GSSG) as a PDI substrate. This nonfluorescent probe becomes fluorescent upon reduction to reduced eosin-glutathione. The 2-electron oxidant, hydrogen peroxide, inhibited PDI reductase activity (half-maximal inhibitory concentration [IC50] = 3.1 ± 0.5 μM; Figure 1A). Other oxidants showed varying degrees of inhibition with IC50 values ranging from 0.70 μM for the peroxynitrite donor SIN-1, and those from 5.4 to 42 μM for hydrogen sulfide donors DATS, GYY4137, ADT-OH, and the nitrosating agents SNAP and DEA-NONOate (Supplementary Figure S2).
FIGURE 1.
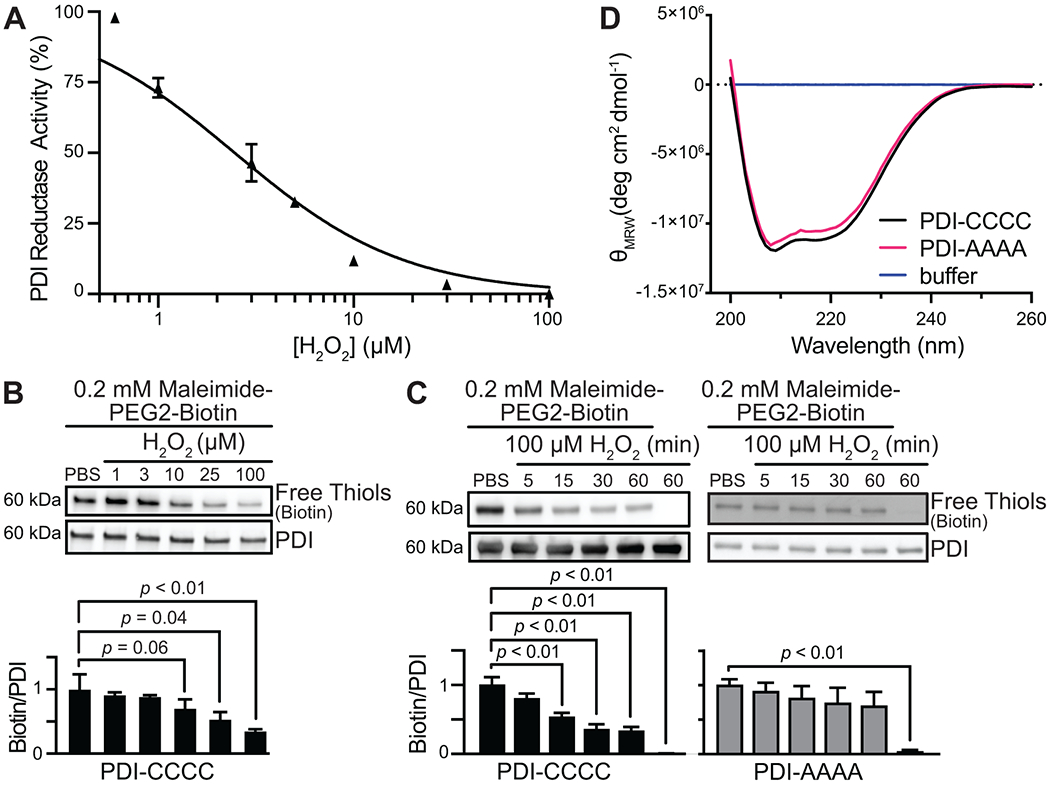
PDI reductase activity is controlled by oxidative cysteine modification. (A) Eosin-glutathione fluorescence from PDI-mediated di-eosin oxidized glutathione reduction is concentration-dependently inhibited by H2O2. The percentage of maximum PDI reductase activity was plotted, and the IC50 value was calculated by plotting H2O2 vs. normalized response and fitting by nonlinear regression. (B) H2O2 concentration-dependently promoted oxidative cysteine modification on PDI as detected by a loss of maleimide-PEG2-biotin alkylation to the free thiol. (C) H2O2 promoted a loss of free thiols over time when all active-site cysteines (53CGHC56 and 397CGHC400, respectively) are present (PDI-CCCC), but not when the cysteines are mutated to alanine (PDI-AAAA). (D) PDI-AAAA is properly folded and structurally similar to PDI wild-type despite being catalytically inactive. The intensity and shape of the far-UV CD spectra of PDI wild-type (PDI-CCCC) (black) and PDI-AAAA (magenta) are consistent with those of folded proteins containing ordered secondary structure. The spectra are also similar to those reported in previous studies for properly folded and catalytically active PDI protein [11]. p-values were determined by one-way ANOVA with Dunnett’s post hoc analysis. Data represented as mean ± SEM.
Mechanistically, PDI oxidoreductase activity is controlled by active-site cysteines that are susceptible to oxidation [31]. To test whether free thiols of PDI are oxidized by hydrogen peroxide, we used a biotin-conjugated electrophilic probe maleimide, which targets the free sulfhydryl group of reduced cysteine residues. In the presence of H2O2, PDI exhibited a concentration-dependent loss of free thiol with maximal loss observed at 100 μM H2O2 (Figure 1B). H2O2 promoted sustained loss of free thiol within 15 minutes (Figure 1C). Significant changes in maleimide binding were not observed when all the active-site cysteines of the CGHC motif were mutated to alanine (PDI-AAAA). PDI-AAAA is properly folded and structurally similar to wild-type PDI (PDI-CCCC), as detected by circular dichroism (Figure 1D). These studies suggest that the catalytic cysteines within the CGHC motif of PDI are targets of H2O2 and that H2O2-mediated modification of these active-site cysteines inhibits PDI reductase activity.
3.2 |. Active-site cysteines within PDI are sulfenylated
The cysteine thiolate anion is susceptible to oxidation to sulfenic acid in an appropriate environment and in the presence of suitable electrophiles [17]. Two electron oxidants, including H2O2, peroxynitrite, and lipid hydroperoxide, could mediate the transition of the thiolate to a sulfenic oxoform based on their electrophilic nature [17]. Given the reactivity of free thiols within the active-site motif of PDI, we tested the hypothesis that sulfenic acids are generated by oxidation of the active-site cysteines. As the sulfenic acid state is electrophilic, selective detection of this form was achieved using an alkyne-containing benzothiazine-based carbon nucleophilic probe (BTD) followed by coupling to AlexaFluor 488-azide using copper(I)–mediated azide-alkyne cycloaddition reactions (“click chemistry”). In inverse correlation to the maleimide labeling shown in Figure 1, sulfenic acid formation increased on PDI over time, with the most BTD labeling at 60 minutes relative to PBS treatment (Figure 2A). A control condition without the probe showed no labeling. Using the PDI-AAAA mutant, we found no significant increase in BTD labeling over time despite Cys312 and Cys343 being present. These observations are consistent with the hypothesis that only the active-site cysteines on PDI are sensitive to oxidants.
FIGURE 2.
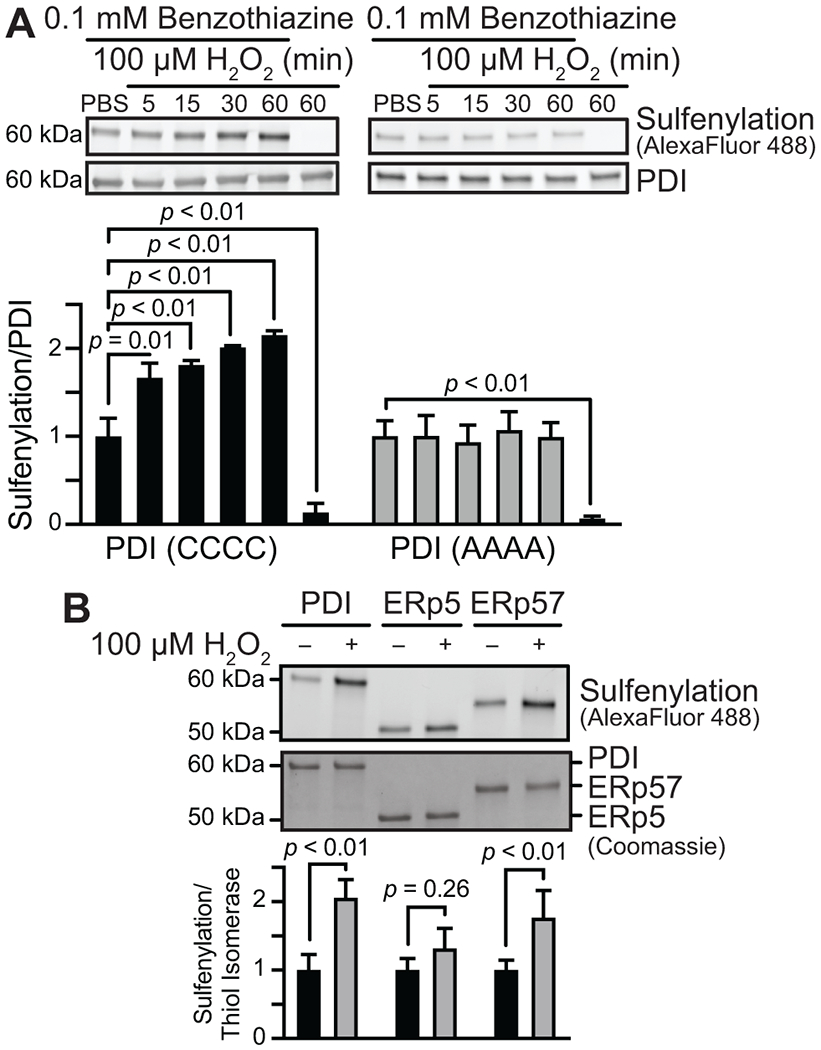
PDI and ERp57, but not ERp5, are sulfenylated by H2O2. (A) The active-site cysteines of PDI (PDI-CCCC) are sulfenylated over time by H2O2. No increase in sulfenylation was observed when these cysteines were mutated to alanine (PDI-AAAA). (B) Evaluation of H2O2-induced sulfenylation using BTD labeling shows that PDI and ERp57 are sulfenylated, while ERp5 is not. p-values were determined by unpaired Student’s t-test. Data represented as mean ± SEM.
As the CGHC motif is also present on the active site of different thiol isomerase family members, we next determined whether sulfenylation occurs after oxidant exposure in other thiol isomerases. ERp57 also has a domain organization of a-b-b′-a′ and is structurally similar to PDI. In contrast, ERp5 has a domain structure of a-a0-b and shows less structural homology, but nonetheless, it contains thioredoxin folds containing a CGHC motif. Similar to PDI, ERp57 showed a significant increase in BTD labeling in the presence of H2O2, whereas ERp5 reacted less efficiently with BTD (Figure 2B). These results indicate that sulfenylation shows a preference for certain thiol isomerase family members and that cysteines within the CGHC motif are only one of the requirements in order for this reaction to occur.
PDI is a multidomain protein. To test whether the domain structure affects sulfenylation, we evaluated the ability of isolated PDI fragments to undergo sulfenylation. Both the a and a′ domains of PDI contain active-site cysteines within a CGHC motif. Yet, the a domain is connected to the b domain by a 4-amino acid linker, while the a′ domain is connected to the b′ domain by a 19-amino acid segment termed x-linker. The x-linker affords the a′ domain considerably more flexibility than the a domain, resulting in dynamic conformational changes between the b′ and a′ domains [11,32]. Previous studies have demonstrated the differences between the a and a′ domains in their ability to undergo oxidation [33]. In addition, the b′ domain contains free thiols beyond the CGHC motif that could be sulfenylated in isolation. We first determined whether isolated PDI domains are sulfenylated upon incubation with H2O2. Of the isolated domains, only the a domain was significantly sulfenylated (Figure 3A); however, the extent of sulfenylation upon H2O2 exposure (1.3 ± 0.10-fold) was modest compared to wild-type PDI (2.0 ± 0.25-fold). Addition of the b domain to a (i.e., the abb′ fragment) enhanced H2O2-induced sulfenylation (1.8 ± 0.10-fold), and addition of bb′ to a (i.e., the abb′ fragment) increased H2O2-induced sulfenylation to levels comparable to that observed in full-length PDI (2.1 ± 0.31-fold; Figure 3B). In contrast, adding bb′ to the a′ domain failed to significantly enhance H2O2-induced sulfenylation (Figure 3C). These studies show that sulfenylation occurs preferentially on the a domain and that interactions of a with the b and b′ domains, but especially the b domain, enhance its sulfenylation.
FIGURE 3.
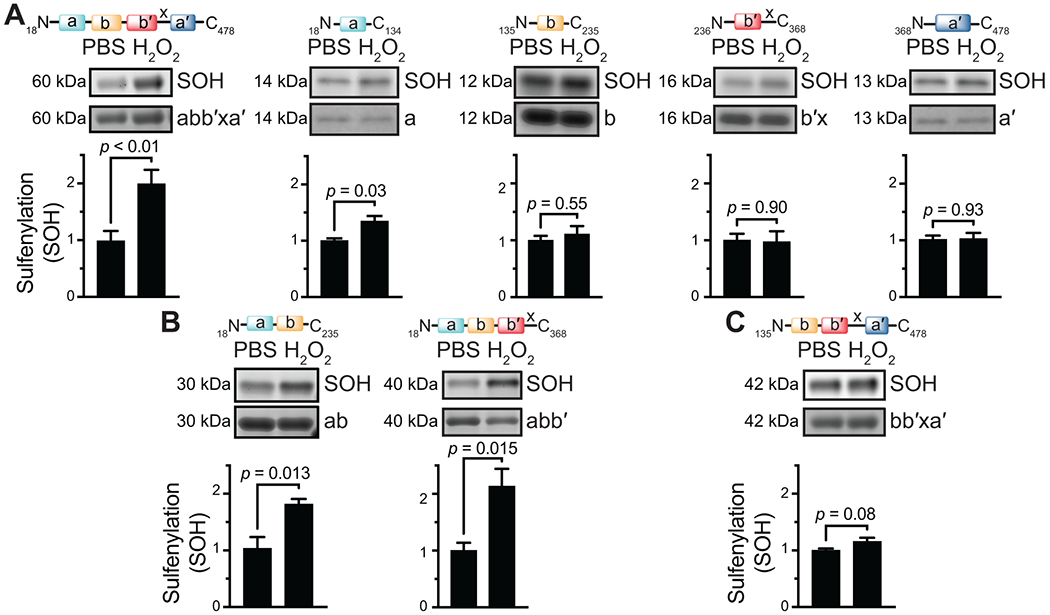
PDI sulfenylation occurs only within the a domain and requires substrate binding domains for maximal sulfenic acid formation. The indicated domains were exposed to H2O2 and evaluated for sulfenylation using BTD labeling. (A) Evaluation of WT PDI and isolated domains showed that the a domain is the only isolated domain to demonstrate significant sulfenylation. (B) Sulfenylation within the a domain is augmented by addition of the b domain or the bb′ fragment. (C) No significant sulfenylation is observed in the absence of the a domain. p-values were determined by unpaired Student’s t-test. Data represented as mean ± SEM.
Since fragments might behave differently in the context of full-length proteins, we repeated these experiments using PDI-CCAA and PDI-AACC variants, in which the catalytic cysteines were removed from the a and a′ domains, respectively (Supplementary Figure S3A). Consistently with the fragment studies, BTD labeling of PDI-CCAA was similar to that of wild-type PDI, whereas labeling of the PDI-AACC variant was delayed.
3.3 |. Sulfenylation by H2O2 promotes PDI disulfide formation
As sulfenic acid is a precursor to many other sulfur oxoforms, including disulfides, we determined whether disulfides are present following peroxide oxidation and, if so, whether disulfide formation in this setting is sulfenic acid–dependent. To measure the effect of peroxide oxidation on disulfide formation, we used a differential maleimide labeling method comprising 3 sequential steps. In the first step, the sample is labeled with Cy3-maleimide, which blocks free thiols. In the second step, the Cy3-treated sample was then reduced using tris(2-carboxyethyl)phosphine (TCEP) to reduce disulfides and other reversible oxidative modifications, including sulfenylation. In the third and final step, the newly formed free thiols were labeled with Cy5-maleimide (Figure 4A). Using this method, we found that the disulfide content in PDI significantly increased after H2O2 exposure compared to untreated conditions (Figure 4B). We used arsenite to determine whether sulfenic acids are a necessary intermediate for PDI disulfide formation. Arsenite reduces sulfenic acids to free thiols and does not reduce disulfides when oxidized through a sulfenylation-independent mechanism (Supplementary Figure S3B) [34–36]. Following incubation with arsenite, the increase in PDI disulfide formation induced by H2O2 exposure was prevented (Figure 4C). These results suggest that the sulfenic acid state is an important intermediary sulfur oxoform for disulfides that forms in PDI upon H2O2 exposure.
FIGURE 4.
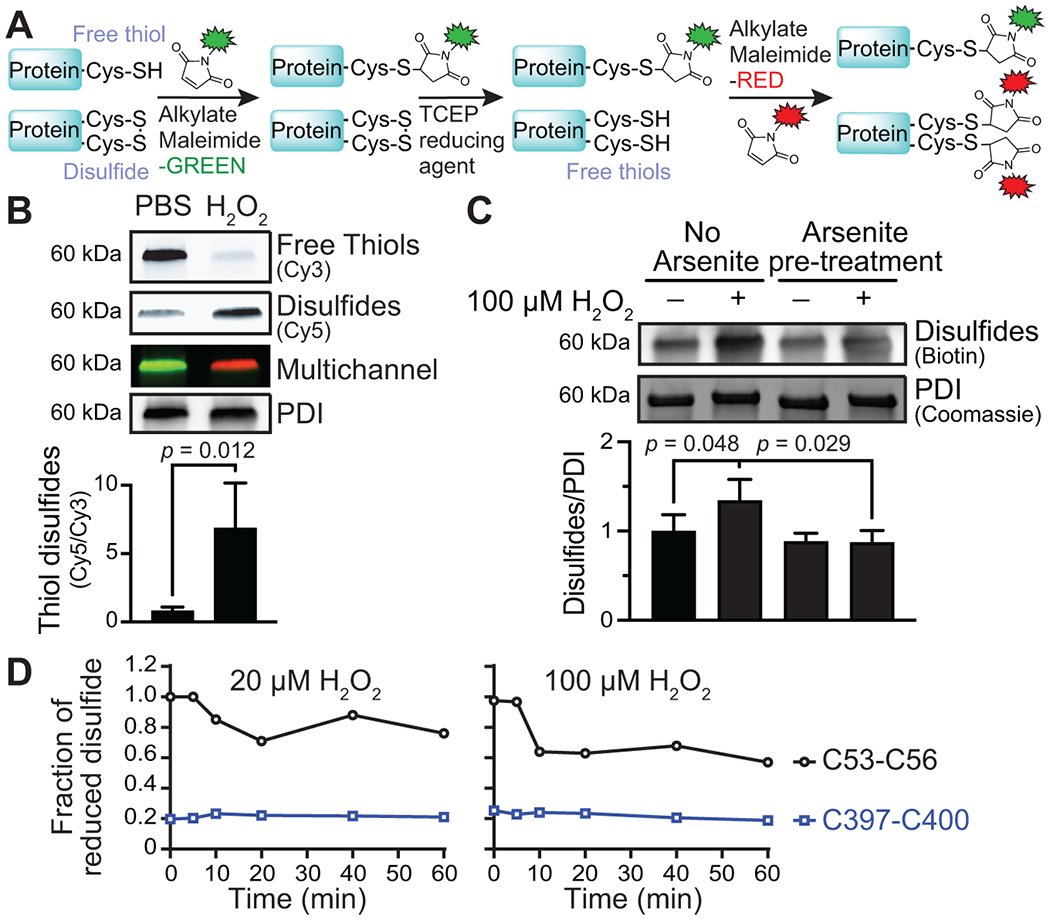
Sulfenylation by H2O2 promotes PDI disulfide formation. (A) Differential alkylation to detect PDI disulfide formation. The free cysteine thiols were labeled and blocked with maleimide-Cy3. Cysteine disulfides and other reversible oxidative modifications, including sulfenylation, were reduced with TCEP, and the newly available free thiols were labeled with maleimide-Cy5. (B) H2O2 promoted disulfide formation on PDI compared to buffer-treated conditions. (C) H2O2-mediated disulfide formation on PDI was prevented by arsenite. (D) Redox state of the PDI a (C53-C56) and a′ (C397-C400) active-site cysteines determined by differential cysteine alkylation and mass spectrometry. p-values were determined by unpaired Student’s t-test in (B) and one-way ANOVA with Tukey’s post hoc analysis in (C). Data represented as mean ± SEM.
To further characterize disulfide formation in the a or a′ domain by H2O2 exposure, we performed differential cysteine alkylation followed by mass spectrometry. Oxidation of the a domain catalytic cysteines was detected after a 10-minute incubation with 20 or 100 μM of H2O2. The C53-C56 motif was 25% and 40% oxidized after a 60-minute incubation with 20 or 100 μM of H2O2, respectively (Figure 4D). H2O2 oxidation of the a′ domain was represented as the fraction of reduced C397-C400 relative to the untreated sample (zero-time point). The basal redox state of the a′ domain catalytic cysteines was 80% oxidized despite incubation with excess reduced glutathione. Aberrant disulfide bonds may have formed with neighboring b′ domain unpaired cysteines Cys312 and/or Cys343 that were refractive to reduction. Cys312 and Cys343 were 74% and 33% oxidized, respectively. These data suggest that the PDI a domain C53-C56 motif was more readily oxidized by H2O2.
3.4 |. PDI sulfenylation promotes PDI oxidase activity
PDI oxidoreductase activity is controlled by its redox status. Its oxidized state allows PDI to function as an oxidase and isomerase, whereas its reduced form allows PDI to reduce disulfide bonds [4]. To determine how sulfenylation-mediated disulfide formation affects PDI isomerase activity, we used a protein refolding assay with reduced and denatured RNAse (thiol-scrambled RNAse; scRNase). In this assay, PDI oxidizes and refolds scRNAse to generate a functional RNAse. RNase activity was then detected spectrophotometrically by measuring the hydrolysis of cCMP to CMP. Native RNAse showed fast kinetics, whereas scRNAse did not cleave cCMP (Figure 5A). In the presence of TCEP-reduced PDI, scRNAse was not refolded; therefore, no activity was present. However, H2O2-oxidized PDI promoted scRNAse refolding and restoration of RNAse activity (Figure 5A).
FIGURE 5.
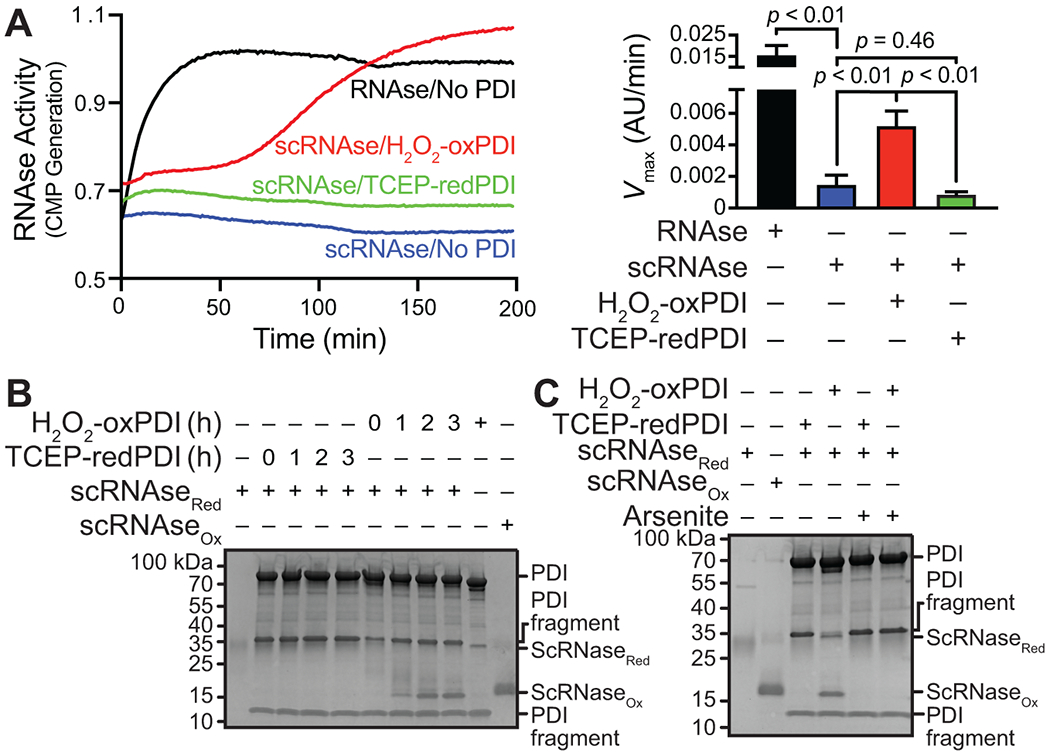
PDI sulfenylation promotes PDI oxidase activity. (A) Oxidized PDI promotes oxidative renaturing of ribonuclease activity. (B) Oxidized PDI, but not reduced PDI, increases scRNase oxidation as detected by the loss of acetamido maleimidylstilbene disulfonic acid (AMS). Fully reduced RNAse resolves at 30 kDa, whereas fully oxidized RNAse resolves at 15 kDa. Oxidized intermediates may be observed between 15 and 30 kDa. (C) Arsenite prevents H2O2-mediated oxidation of PDI and thus prevents PDI-mediated oxidation of scrambled and reduced RNAse. p-values were determined by one-way ANOVA with Tukey’s post hoc analysis. Data represented as mean ± SEM.
To determine whether sulfenic acids are important for disulfided PDI to function as an oxidase, we added arsenite to PDI before oxidant treatment and observed concentration-dependent inhibition of RNAse activity (Supplementary Figure S3C). These results were validated with a discontinuous gel-based assay wherein the reduced state of RNAse could be detected using an acetamido maleimidylstilbene disulfonic acid (AMS) probe, which labels free thiols and shifts the protein to a higher apparent molecular weight. As a control, scRNAse (scRNAseRed) appeared at 30 kDa, whereas fully functional and oxidized RNAse (scRNAseox) appeared at 15 kDa (Figure 5B). TCEP-reduced PDI did not increase RNAse oxidation over time (no bands at 15 kDa). Consistent with the enzymatic assay for scRNAse refolding, H2O2-oxidized PDI promoted scRNAse oxidation to the same molecular weight as the scRNAseox control with maximal oxidation observed between 2 and 3 hours (Figure 5B). As arsenite prevented the H2O2-oxidized PDI refolding of RNAse, we also confirmed using the discontinuous gel-based assay that arsenite treatment before H2O2-mediated PDI oxidation abrogated scRNAse oxidation, demonstrating that scRNAse remained reduced upon arsenite exposure (Figure 5C). These data suggest the intermediary sulfenic acid formed upon oxidant exposure promotes PDI oxidase and isomerase activities.
3.5 |. Platelets and endothelial cells secrete PDI that is sulfenylated
PDI is secreted from activated endothelial cells and platelets [37–39]. However, the redox state of secreted PDI has not been previously evaluated. We hypothesized that agonist activation resulted in the secretion of sulfenylated PDI. Using human umbilical vein endothelial cells (HUVECs) as a model, we stimulated the cells with thrombin in the presence of BTD. Thrombin is a potent activator of the protease-activated G-protein coupled receptor 1 (PAR1) on HUVECs, inducing reactive oxygen species generation and promoting exocytosis of many proteins, including PDI [40–44]. Following exposure of HUVECs to thrombin in the presence of BTD, PDI was immunoprecipitated from the conditioned media. Click chemistry with biotin-azide was performed on the BTD-labeled PDI to detect protein labeling with the alkyne-containing BTD probe. These studies showed that upon stimulation with thrombin, HUVECs released sulfenylated PDI, whereas PDI was not sulfenylated, and consequently, little sulfenylated PDI was released under untreated conditions (Figure 6A). This finding suggests that the redox state of PDI secreted from activated endothelial cells includes sulfenylated species.
FIGURE 6.
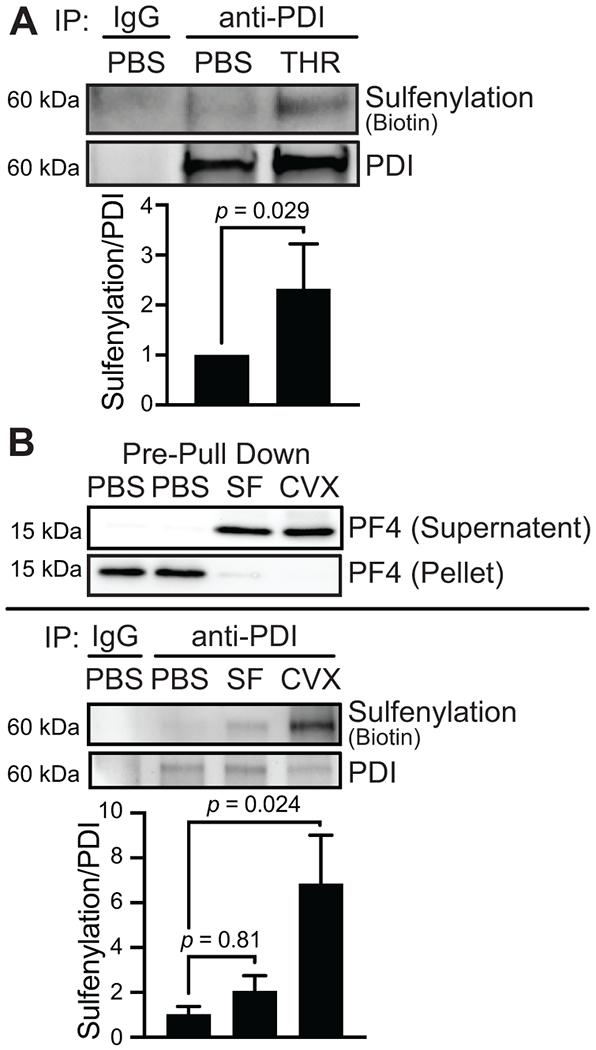
Endothelial cells and platelets secrete sulfenylated PDI. (A) PDI secreted from activated endothelial cells is sulfenylated. HUVECs were stimulated with 1 U/mL human a thrombin (THR) in the presence of 0.1 mM BTD for 1 h before inactivation with 5 U/mL hirudin and media collection for immunoprecipitation. The media was concentrated before immunoprecipitating PDI with an anti-PDI or control IgG antibody. Click chemistry was performed to attach biotin-PEG3-azide onto the alkyne arm of BTD, and biotin was detected by western blotting and chemiluminescence. (B) PDI secreted from activated platelets is sulfenylated. Washed human platelets (1 × 109/mL) were stimulated with 50 μM SFLLRN (SF) or 500 ng/mL convulxin (CVX) for 15 min in the presence of 0.1 mM BTD. The supernatant containing the releasate was collected after centrifuging the platelets, and a biotin-PEG3-azide was conjugated to the alkyne of BTD by click chemistry. The releasate was desalted, and PDI was immunoprecipitated with an anti-PDI or control IgG antibody. Biotin was detected by western blotting and chemiluminescence. A control blot to assess platelet factor 4 (PF4) as a marker of platelet secretion was performed in the releasate and the cell pellet before immunoprecipitation. p-values were determined by unpaired Student’s t-test (A) and one-way ANOVA with Dunnett’s post hoc analysis (B). Data represented as mean ± SEM.
We also determined whether PDI exocytosed from activated human platelets is sulfenylated. To achieve secretion from platelets, we stimulated platelets with SFLLRN, an activating peptide for PAR, and convulxin, a snake venom that targets the glycoprotein VI receptor signaling pathway. SFLLRN and convulxin both promoted platelet factor 4 secretion to the supernatant, as detected by Western blot analysis (Figure 6B). Immunoprecipitation of secreted PDI showed that convulxin stimulation promoted significantly more PDI sulfenylation than SFLLRN. In contrast, immunoprecipitation of PDI in platelets was not significantly cysteine-sulfenylated relative to control unstimulated platelets, as detected by BTD labeling (Supplementary Figure S4). These data support the hypothesis that sulfenylated PDI is secreted by platelets and raise the possibility that the degree of PDI sulfenylation varies by agonists.
3.6 |. oxLDL elicits PDI sulfenylation and promotes PDI-dependent thrombosis
Circulating oxLDL is elevated in patients with coronary artery disease [45,46]. In addition to the contributions of oxLDL to atherosclerosis, oxLDL can stimulate signaling downstream of scavenger receptors such as CD36 in platelets, promoting thrombus formation [47,48]. We found that incubation of PDI with oxLDL resulted in the loss of free thiols, indicating oxidation (Figure 7A). PDI exposure to oxLDL also resulted in sulfenylation comparable with that observed for H2O2 or SIN-1 (Figure 7B). PDI infused into oxLDL-treated mice showed increased BTD labeling relative to saline treatment (Figure 7C), which indicates oxidative cysteine modification in vivo. These observations suggest that oxLDL promotes PDI oxidation via a sulfenic acid intermediate.
FIGURE 7.
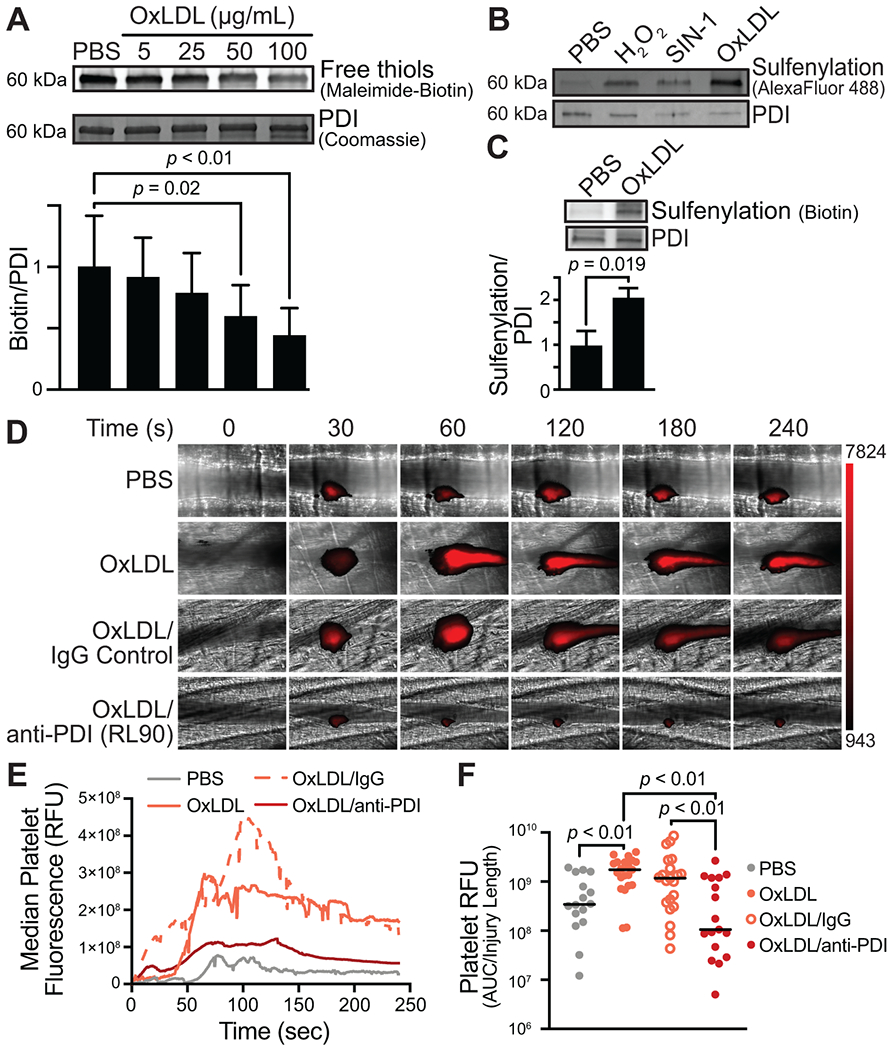
Oxidized LDL sulfenylates PDI and PDI oxidoreductase activity inhibition prevents oxLDL-induced platelet accumulation. (A) Incubation of recombinant PDI with increasing concentrations of oxLDL results in loss of free PDI thiols. (B) Incubation with oxidants, including H2O2, SIN-1, and oxLDL, results in PDI sulfenylation (top row). Total PDI is indicated by Western blot analysis (bottom row). (C) Intravenous injection of PDI postoxLDL infusion is cysteine sulfenylated in vivo. (D, E) Intravital microscopy images of the cremasteric artery from intravenously injected PBS or 2.5 mg/kg oxLDL-injected mice exposed to 1 mg/kg nonimmune IgG or anti-PDI antibody (RL90). (E) Median integrated fluorescence intensity over time of platelets is shown. (F) Quantification of the normalized platelet accumulation as area under the curve (AUC) with the median shown. PBS, N = 16 injuries; oxLDL, N = 21 injuries; oxLDL + IgG, N = 21 injuries; oxLDL + RL90, N = 17 injuries. p-values were determined by nonparametric one-way ANOVA with Dunnett’s post hoc analysis.
We tested the effect of oxLDL in a thrombosis model to determine whether infusion of this oxidant augmented thrombus formation and, if so, whether augmentation of thrombus formation by oxLDL depended on PDI. For these studies, the cremaster vasculature of mice was dissected, and cremaster arterioles were exposed. Laser injury–initiated thrombus formation and platelet accumulation were monitored to assess the kinetics of thrombus formation over time. To model systemic oxidative stress, we infused oxLDL in the mice before initiating thrombus formation. oxLDL infusion increased platelet accumulation by 5.1-fold compared with saline infusion in mice (AUC PBS vs AUC oxLDL, P < .01; Figure 7D–F, Supplementary Videos S1 and S2). We used an anti-PDI antibody to determine whether extracellular PDI contributed to the augmentation of platelet accumulation observed with systemic oxLDL exposure. Infusion of the anti-PDI antibody RL90 blocked oxLDL-augmented platelet accumulation by 90.9% (AUC RL90, then oxLDL vs oxLDL, P < .01). In contrast, oxLDL-mediated platelet accumulation was unaffected by infusion of nonimmune IgG (AUC RL90, then oxLDL vs oxLDL, P = .21; Supplementary Videos S3 and S4). Median injury sizes in all 4 groups were similar, demonstrating that the differences observed with oxLDL and anti-PDI antibodies did not result from the differences in vascular injury extent (Supplementary Figure S5). These studies indicate that oxLDL-mediated augmentation of platelet accumulation is at least partially dependent on PDI function.
4 |. DISCUSSION
The active-site cysteines within the CGHC motif of PDI are among the most reactive cysteines in the entire proteome [49,50]. Cysteines within this motif are subject to various redox modifications, including reduction, oxidation, nitrosation, glutathionylation, and others [4,51], each with different implications for thrombus formation. In this study, we show that PDI undergoes sulfenylation and that this modification promotes the formation of a disulfided state. Reduced PDI cleaves several prothrombotic substrates [52–55], and oxidized PDI promotes platelet aggregation [56,57]. The nitrosation of PDI (SNO-PDI) inhibits PDI oxidoreductase activity and blocks thrombus formation when infused into mice [51]. Glutathionylation and succination are reported to inhibit PDI oxidoreductase function [58–60]. We now show that in oxidative stress, PDI is sulfenylated and this posttranslational modification promotes PDI oxidase activity.
Our studies indicate that PDI sulfenylation by peroxides occurs selectively, affecting cysteines within the active-site CGHC motif but not other free cysteines and preferentially sulfenylating the a domain. Studies showing no significant change in sulfenylation following H2O2 incubation with constructs in which both active-site motifs are mutated (PDI-AAAA) suggest that the other 2 free cysteines within processed PDI (Cys312, Cys343) are not sulfenylated in response to H2O2 (Figure 2). Given the background signals in PDI-AAAA in the absence of peroxides, however, Cys312 and Cys343 may be oxidized in the basal state. Indeed, Oximouse datasets indicate Cys343 as a hit for PDI thiol oxidation [61], which is further supported by our mass spectrometry experiments demonstrating that both Cys312 and Cys343 are refractory to reduction. Alternatively, this background signal could be from Michael acceptor-based probes such as maleimide reacting with free amines [62]. PDI nitrosation can occur at a free cysteine outside the CGHC motif, Cys343, within the b′ domain [63]. Previously, we found that PDI persulfidation occurs at sites other than the CGHC motifs since this cysteine modification can be detected in the PDI-AAAA mutant [64]. However, persulfidation is further enhanced in wild-type PDI [64]. Although ERp57 is also sulfenylated following peroxide exposure, ERp5 showed minimal sulfenylation. PDI and ERp57 contain domains that are similarly positioned (a-b-b′-x-a′-c). In contrast, ERp5 has a distinct architecture (a-a0-b). ERp5 may be sulfenylated depending on the oxidant and the local environment in which its active-site cysteines are deprotonated; however, further studies are required to understand the basis of selectivity among PDI family members. Nonetheless, our evaluation of PDI fragments and PDI variants PDI-CCAA and PDI-AACC supports the premise that the domain structure is an important determinant of peroxide-induced sulfenylation. These studies show that H2O2-mediated sulfenylation in PDI primarily targets the a domain (Figure 3). Augmentation of a domain sulfenylation within the abb′ fragment indicates a role for communication between the a and bb′ domains. Neither the isolated a′ domain nor the bb′a′ fragment is significantly sulfenylated, yet PDI-AACC displayed some level of sulfenylation. Removal of the a domain may influence the accessibility of the a′ domain to sulfenylation by modification of the PDI conformation [11]. In this regard, H2O2-mediated PDI sulfenylation demonstrates unexpected specificity as a cysteine modification.
Protein sulfenylation is increasingly recognized as a critical cysteine modification in controlling vascular function. Sulfenylation of several vascular proteins is a switch to control their activity and modify cell function [21,65,66]. Inhibition of CD36-dependent sulfenylation in platelets blocks aggregation and procoagulant phosphatidylserine exposure [16]. Src family kinase activity is regulated by sulfenylation [67], and we recently showed src kinase sulfenylation during platelet activation downstream of CD36 [16]. Just as sulfenylation acts as a molecular switch in several other vascular proteins, PDI sulfenylation by oxidants converts PDI from a reductase into an oxidase.
The primary role of PDI is oxidative folding. PDI is present in the ER at very high levels, and the ability of PDI to perform oxidative folding of nascent proteins is essential to life. Whereas deletion of active-site cysteines in the a′ domain interfered with thrombus formation, mutations of active-site cysteines in the a domain were incompatible with survival [13]. In the oxidative environment of ER, Ero1α-mediated enzymatic PDI oxidation is an important mechanism for PDI disulfide formation. Ero1α preferentially oxidizes the PDI a′ domain [33,68] and produces H2O2 from O2 during PDI oxidation [69]. The basal redox state of the a′ domain is oxidized (Figure 4). H2O2 created from Ero1α could oxidize active-site cysteines in the PDI a domain to disulfides via a sulfenic acid intermediate. This putative physiologic role of protein sulfenylation may be maladapted under pathological conditions where PDI escapes into the extracellular environment. Extracellular PDI is associated with various pathologies [70–72]. This extracellular PDI could be oxidized by Ero1α, which has been demonstrated on the platelet surface [73]. Alternatively, oxidant levels that promote sulfenic acid formation on PDI active-site cysteines could be achieved outside cells [9]. Sulfenic acids thus formed would be short-lived and undergo further oxidation to sulfinic acid or react with adjacent thiols to form disulfide bonds [74,75]. This disulfide could be transferred to substrate proteins, as evidenced by the fact that PDI exposure to H2O2 enables PDI to fold RNAse in a manner that requires a sulfenylated intermediate (Figure 5). Oxidized PDI released into the circulation could modify substrate proteins on vascular cells.
Oxidant-induced PDI sulfenylation with subsequent disulfide bond formation has implications for thrombosis. Our previous studies showed that alkylating sulfenylation impairs thrombus formation in the presence, but not the absence, of oxidative stress [16]. However, which vascular proteins are sulfenylated during oxidative stress and whether they contribute to thrombosis in this context were not evaluated. The sulfenylation of PDI released from platelets and endothelial cells suggests a conversion to oxidized PDI associated with activation (Figure 6). PDI may be sulfenylated intracellularly, but detection of intracellular PDI sulfenylation may be negligible if binding of the probe is out-competed by the kinetics of loss of the sulfenic acid moiety to alterantive oxoforms or by the elimination of the oxidant generated through cellular antioxidant defense mechanisms. Recent studies indicate that oxidized PDI, but not reduced PDI, promotes platelet aggregation [56,57]. Oxidant-induced disulfide formation in PDI through a sulfenic acid intermediate could thus contribute to platelet accumulation during thrombus formation. Anti-PDI antibodies inhibit thrombus formation in several models [4], including oxLDL-augmented thrombosis (Figure 7), underscoring the importance of PDI release into the extracellular environment for PDI function in thrombosis. These studies provide a model for the role of PDI in thrombus formation during oxidative stress. The premise is that oxidant-induced PDI sulfenylation is an important mechanism for generating prothrombotic oxidized PDI during oxidative stress. Levels of H2O2 and other oxidants capable of eliciting PDI sulfenylation are achieved at sites of vascular inflammation prone to thrombosis [9]. These oxidants could lead to the formation of oxidized, prothrombotic PDI. Such oxidants could enhance PDI oxidation in both intracellular and extracellular compartments. Activation of vascular cells results in the release of sulfenylated PDI, which may be converted to oxidized PDI and may contribute to platelet activation and thrombus formation.
Supplementary Material
Essentials.
Thrombus formation typically occurs during oxidative stress.
Oxidant-induced protein disulfide isomerase (PDI) sulfenylation is an intermediate during PDI disulfide formation.
PDI oxidoreductase inhibition blocks oxidant-induced augmentation of thrombus formation in vivo.
PDI contributes to the conversion of oxidants into a thrombotic response.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant numbers R35HL135775 [to R.F.], U01HL143365 [to R.F.], T32HL134643 [to M.Y.], T32HL007917 [to M.Y. and R.F.], K99HL164888 [to M.Y.], R01HL150146 [to N.Pozzi], R35GM128840 [to B.C.S.], R01GM102187 [to K.S.C.]), the American Society of Hematology Scholar Award (to M.Y.), the Medical College of Wisconsin Cardiovascular Center’s A. O. Smith Fellowship Scholars Program (to M.Y.), a Senior Research Grant (to P.J.H.) from the NSW Cardiovascular Research Capacity Program, and the Sarnoff Cardiovascular Research Foundation (to A.P.).
Funding information
National Institutes of Health; NIH, Grant Numbers: R35HL135775, U01HL143365, T32HL134643, T32HL007917, K99HL164888, R01HL150146, R35GM128840, R01GM102187. American Society of Hematology Scholar Award. Medical College of Wisconsin Cardiovascular Center A. O. Smith Fellowship Scholars Program. NSW Cardiovascular Research Capacity Program. Sarnoff Cardiovascular Research Foundation.
Footnotes
DECLARATION OF COMPETING INTERESTS
R.F. is a founder and consultant for Platelet Diagnostics. The remaining authors have no competing interests to disclose.
SUPPLEMENTARY MATERIAL
The online version contains supplementary material available at https://doi.org/10.1016/j.jtha.2023.03.034
REFERENCES
- [1].Day SM, Duquaine D, Mundada LV, Menon RG, Khan BV, Rajagopalan S, Fay WP. Chronic iron administration increases vascular oxidative stress and accelerates arterial thrombosis. Circulation. 2003;107:2601–6. [DOI] [PubMed] [Google Scholar]
- [2].Kenet G, Freedman J, Shenkman B, Regina E, Brok-Simoni F, Holzman F, Vavva F, Brand N, Michelson A, Trolliet M, Loscalzo J, Inbal A. Plasma glutathione peroxidase deficiency and platelet insensitivity to nitric oxide in children with familial stroke. Arterioscler Thromb Vasc Biol. 1999;19:2017–23. [DOI] [PubMed] [Google Scholar]
- [3].Fuentes E, Gibbins JM, Holbrook LM, Palomo I. NADPH oxidase 2 (NOX2): a key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc Med. 2018;28:429–34. [DOI] [PubMed] [Google Scholar]
- [4].Yang M, Flaumenhaft R. Oxidative cysteine modification of thiol isomerases in thrombotic disease: a hypothesis. Antioxid Redox Signal. 2021;35:1134–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nagarkoti S, Kim YM, Ash D, Das A, Vitriol E, Read TA, Youn SW, Sudhahar V, McMenamin M, Hou Y, Boatwright H, Caldwell R, Essex DW, Cho J, Fukai T, Ushio-Fukai M. Protein disulfide isomerase A1 as a novel redox sensor in VEGFR2 signaling and angiogenesis. Angiogenesis. 2023;26:77–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Quan S, Schneider I, Pan J, Von Hacht A, Bardwell JCA. The CXXC motif is more than a redox rheostat. J Biol Chem. 2007;282:28823–33. [DOI] [PubMed] [Google Scholar]
- [7].Karala AR, Lappi AK, Ruddock LW. Modulation of an active-site cysteine pKa allows PDI to act as a catalyst of both disulfide bond formation and isomerization. J Mol Biol. 2010;396:883–92. [DOI] [PubMed] [Google Scholar]
- [8].Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–83. [DOI] [PubMed] [Google Scholar]
- [10].Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, Wang CC. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal. 2013;19:36–45. [DOI] [PubMed] [Google Scholar]
- [11].Chinnaraj M, Flaumenhaft R, Pozzi N. Reduction of protein disulfide isomerase results in open conformations and stimulates dynamic exchange between structural ensembles. J Biol Chem. 2022;298:102217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gaspar RS, Gibbins JM. Thiol isomerases orchestrate thrombosis and hemostasis. Antioxid Redox Signal. 2021;35:1116–33. [DOI] [PubMed] [Google Scholar]
- [13].Zhou J, Wu Y, Wang L, Rauova L, Hayes VM, Poncz M, Essex DW. The C-terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest. 2015;125:4391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim K, Hahm E, Li J, Holbrook LM, Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM, Cho J. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood. 2013;122:1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zwicker JI, Schlechter BL, Stopa JD, Liebman HA, Aggarwal A, Puligandla M, Caughey T, Bauer KA, Kuemmerle N, Wong E, Wun T, McLaughlin M, Hidalgo M, Neuberg D, Furie B, Flaumenhaft R, CATIQ Investigators. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight. 2019;4:e125851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang M, Li W, Harberg C, Chen W, Yue H, Ferreira RB, Wynia-Smith SL, Carroll KS, Zielonka J, Flaumenhaft R, Silverstein RL, Smith BC. Cysteine sulfenylation by CD36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv. 2020;4:4494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta. 2014;1840:847–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49:7748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sonego G, Le TM, Crettaz D, Abonnenc M, Tissot JD, Prudent M. Sulfenylome analysis of pathogen-inactivated platelets reveals the presence of cysteine oxidation in integrin signaling pathway and cytoskeleton regulation. J Thromb Haemost. 2021;19:233–47. [DOI] [PubMed] [Google Scholar]
- [20].Gupta V, Yang J, Liebler DC, Carroll KS. Diverse redoxome reactivity profiles of carbon nucleophiles. J Am Chem Soc. 2017;139:5588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang Y, Li Z, Zhang L, Tang H, Zhang H, Wang C, Chen SY, Bu D, Zhang Z, Zhu Z, Yuan P, Li K, Yu X, Kong W, Tang C, Jung Y, Ferreira RB, Carroll KS, Du J, Yang J, et al. Endogenous SO2-dependent Smad3 redox modification controls vascular remodeling. Redox Biol. 2021;41:101898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Akter S, Fu L, Jung Y, Conte ML, Lawson JR, Lowther WT, Sun R, Liu K, Yang J, Carroll KS. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat Chem Biol. 2018;14:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bekendam RH, Flaumenhaft R. Assays of thiol isomerase enzymatic activity. Methods Mol Biol. 2019;1967:133–48. [DOI] [PubMed] [Google Scholar]
- [24].Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, Xue G, Yuan C, Furie BC, Flaumenhaft R, Huang M, Furie B. Quercetin-3-rutinoside inhibits protein disulfide isomerase by binding to its b’x domain. J Biol Chem. 2015;290:23543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chiu J. Measurement of redox states of the β3 integrin disulfide bonds by mass spectrometry. Bio Protoc. 2019;9:e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lyles MM, Gilbert HF. Mutations in the thioredoxin sites of protein disulfide isomerase reveal functional nonequivalence of the N- and C-terminal domains. J Biol Chem. 1994;269:30946–52. [PubMed] [Google Scholar]
- [27].De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, Chaudhry SA, Dockendorff C, Parikh SM, Ingber DE, Flaumenhaft R. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A. 2018;115:E982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koseoglu S, Dilks JR, Peters CG, Fitch-Tewfik JL, Fadel NA, Jasuja R, Italiano JE Jr, Haynes CL, Flaumenhaft R. Dynamin-related protein-1 controls fusion pore dynamics during platelet granule exocytosis. Arterioscler Thromb Vasc Biol. 2013;33:481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grover SP, Bendapudi PK, Yang M, Merrill-Skoloff G, Govindarajan V, Mitrophanov AY, Flaumenhaft R. Injury measurements improve interpretation of thrombus formation data in the cremaster arteriole laser-induced injury model of thrombosis. J Thromb Haemost. 2020;18:3078–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Revollo L, Merrill-Skoloff G, De Ceunynck K, Dilks JR, Guo S, Bordoli MR, Peters CG, Noetzli L, Ionescu A, Rosen V, Italiano JE, Whitman M, Flaumenhaft R. The secreted tyrosine kinase VLK is essential for normal platelet activation and thrombus formation. Blood. 2022;139:104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Peixoto ÁS, Geyer RR, Iqbal A, Truzzi DR, Soares Moretti AI, Laurindo FRM, Augusto O. Peroxynitrite preferentially oxidizes the dithiol redox motifs of protein-disulfide isomerase. J Biol Chem. 2018;293:1450–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chinnaraj M, Barrios DA, Frieden C, Heyduk T, Flaumenhaft R, Pozzi N. Bioorthogonal chemistry enables single-molecule FRET measurements of catalytically active protein disulfide isomerase. ChemBioChem. 2021;22:134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang L, Niu Y, Zhu L, Fang J, Wang X, Wang L, Wang CC. Different interaction modes for protein-disulfide isomerase (PDI) as an efficient regulator and a specific substrate of endoplasmic reticulum oxidoreductin-1alpha (Ero1alpha). J Biol Chem. 2014;289:31188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–50. [PubMed] [Google Scholar]
- [35].Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG 3rd, Lo DC, Stockwell BR. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci U S A. 2015;112:E2245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reinhardt C, von Brühl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic-Terpo A, Shen B, Ushio-Fukai M, Cho J, Du X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sonkar VK, Kumar R, Jensen M, Wagner BA, Sharathkumar AA, Miller FJ Jr, Fasano M, Lentz SR, Buettner GR, Dayal S. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019;3:1272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Birch KA, Ewenstein BM, Golan DE, Pober JS. Prolonged peak elevations in cytoplasmic free calcium ions, derived from intracellular stores, correlate with the extent of thrombin-stimulated exocytosis in single human umbilical vein endothelial cells. J Cell Physiol. 1994;160:545–54. [DOI] [PubMed] [Google Scholar]
- [43].Birch KA, Pober JS, Zavoico GB, Means AR, Ewenstein BM. Calcium/calmodulin transduces thrombin-stimulated secretion: studies in intact and minimally permeabilized human umbilical vein endothelial cells. J Cell Biol. 1992;118:1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hanss M, Collen D. Secretion of tissue-type plasminogen activator and plasminogen activator inhibitor by cultured human endothelial cells: modulation by thrombin, endotoxin, and histamine. J Lab Clin Med. 1987;109:97–104. [PubMed] [Google Scholar]
- [45].Toshima S, Hasegawa A, Kurabayashi M, Itabe H, Takano T, Sugano J, Shimamura K, Kimura J, Michishita I, Suzuki T, Nagai R. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler Thromb Vasc Biol. 2000;20:2243–7. [DOI] [PubMed] [Google Scholar]
- [46].Gao S, Zhao D, Wang M, Zhao F, Han X, Qi Y, Liu J. Association between circulating oxidized LDL and atherosclerotic cardiovascular disease: a meta-analysis of observational studies. Can J Cardiol. 2017;33:1624–32. [DOI] [PubMed] [Google Scholar]
- [47].Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Berger M, Naseem KM. Oxidised low-density lipoprotein-induced platelet hyperactivity-receptors and signalling mechanisms. Int J Mol Sci. 2022;23:9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, Yokoyama M, Konrad DB, Lum KM, Simon GM, Kemper EK, Lazear MR, Yin S, Blewett MM, Dix MM, Nguyen N, et al. An activity-guided map of electrophile-cysteine interactions in primary human T cells. Cell. 2020;182:1009–26.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bekendam RH, Iyu D, Passam F, Stopa JD, De Ceunynck K, Muse O, Bendapudi PK, Garnier CL, Gopal S, Crescence L, Chiu J, Furie B, Panicot-Dubois L, Hogg PJ, Dubois C, Flaumenhaft R. Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J Thromb Haemost. 2018;16:2322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hotchkiss KA, Matthias LJ, Hogg PJ. Exposure of the cryptic Arg–Gly–Asp sequence in thrombospondin-1 by protein disulfide isomerase. Biochim Biophys Acta. 1998;1388:478–88. [DOI] [PubMed] [Google Scholar]
- [53].Bowley SR, Fang C, Merrill-Skoloff G, Furie BC, Furie B. Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat Commun. 2017;8:14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Essex DW, Li M. Redox modification of platelet glycoproteins. Curr Drug Targets. 2006;7:1233–41. [DOI] [PubMed] [Google Scholar]
- [55].Li J, Kim K, Jeong SY, Chiu J, Xiong B, Petukhov PA, Dai X, Li X, Andrews RK, Du X, Hogg PJ, Cho J. Platelet protein disulfide isomerase promotes glycoprotein ibalpha-mediated platelet-neutrophil interactions under thromboinflammatory conditions. Circulation. 2019;139:1300–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang L, Wang X, Lv X, Jin Q, Shang H, Wang CC, Wang L. The extracellular Ero1alpha/PDI electron transport system regulates platelet function by increasing glutathione reduction potential. Redox Biol. 2022;50:102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Przyborowski K, Kurpinska A, Wojkowska D, Kaczara P, Suraj-Prazmowska J, Karolczak K, Malinowska A, Pelesz A, Kij A, Kalvins I, Watala C, Chlopicki S. Protein disulfide isomerase-A1 regulates intraplatelet reactive oxygen species-thromboxane A2-dependent pathway in human platelets. J Thromb Haemost. 2022;20:157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Manuel AM, Walla MD, Faccenda A, Martin SL, Tanis RM, Piroli GG, Adam J, Kantor B, Mutus B, Townsend DM, Frizzell N. Succination of protein disulfide isomerase links mitochondrial stress and endoplasmic reticulum stress in the adipocyte during diabetes. Antioxid Redox Signal. 2017;27:1281–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Halloran M, Parakh S, Atkin JD. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int J Cell Biol. 2013;2013:797914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR Jr, Hutchens S, Tew KD. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69:7626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, Heppner DE, Li J, Long J, Mills EL, Szpyt J, He Z, Du G, Garrity R, Reddy A, Vaites LP, Paulo JA, Zhang T, Gray NS, Gygi SP, Chouchani ET. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell. 2020;180:968–83.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hill BG, Reily C, Oh JY, Johnson MS, Landar A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic Biol Med. 2009;47:675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ogura J, Ruddock LW, Mano N. Cysteine 343 in the substrate binding domain is the primary S-Nitrosylated site in protein disulfide isomerase. Free Radic Biol Med. 2020;160:103–10. [DOI] [PubMed] [Google Scholar]
- [64].Shieh M, Ni X, Xu S, Lindahl SP, Yang M, Matsunaga T, Flaumenhaft R, Akaike T, Xian M. Shining a light on SSP4: a comprehensive analysis and biological applications for the detection of sulfane sulfurs. Redox Biol. 2022;56:102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sánchez-Gómez FJ, Calvo E, Bretón-Romero R, Fierro-Fernández M, Anilkumar N, Shah AM, Schröder K, Brandes RP, Vázquez J, Lamas S. NOX4-dependent hydrogen peroxide promotes shear stress-induced SHP2 sulfenylation and eNOS activation. Free Radic Biol Med. 2015;89:419–30. [DOI] [PubMed] [Google Scholar]
- [66].Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T, Ushio-Fukai M. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 2018;23:3565–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Heppner DE, Dustin CM, Liao C, Hristova M, Veith C, Little AC, Ahlers BA, White SL, Deng B, Lam YW, Li J, van der Vliet A. Direct cysteine sulfenylation drives activation of the Src kinase. Nat Commun. 2018;9:4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB, Wang CC. Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a′ domains of protein-disulfide isomerase. J Biol Chem. 2009;284:199–206. [DOI] [PubMed] [Google Scholar]
- [69].Matsusaki M, Okuda A, Matsuo K, Gekko K, Masuda T, Naruo Y, Hirose A, Kono K, Tsuchi Y, Urade R. Regulation of plant ER oxidoreductin 1 (ERO1) activity for efficient oxidative protein folding. J Biol Chem. 2019;294:18820–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–7. [DOI] [PubMed] [Google Scholar]
- [71].Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, Stockwell BR. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Benham AM. The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal. 2012;16:781–9. [DOI] [PubMed] [Google Scholar]
- [73].Swiatkowska M, Padula G, Michalec L, Stasiak M, Skurzynski S, Cierniewski CS. Ero1alpha is expressed on blood platelets in association with protein-disulfide isomerase and contributes to redox-controlled remodeling of alphaIIbbeta3. J Biol Chem. 2010;285:29874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013;288:26480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Beedle AE, Lynham S, Garcia-Manyes S. Protein S-sulfenylation is a fleeting molecular switch that regulates non-enzymatic oxidative folding. Nat Commun. 2016;7:12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.