

INTRODUCTION
Aquaporin-4-IgGpositive neuromyelitis optica spectrum disorder (AQP4+NMOSD) and myelin-oligodendrocytes glycoprotein antibody-associated disease (MOGAD) are recently identified antibody-mediated autoimmune disorders of the central nervous system (CNS).1,2
Biomarkers of these diseases are antibodies targeting the aquaporin-4 (AQP4) water channel on the astrocyte end-feet in AQP4+NMOSD,3 and myelin-oligodendrocyte glycoprotein (MOG) on the outermost myelin sheath layer in MOGAD.4
For a long time, the clinical and radiological overlap have hampered the recognition of these two diseases as separate entities. Initially, these disorders were considered variants of multiple sclerosis (MS), given their similar predilection for the optic nerve and spinal cord. Later, after the discovery of the AQP4-IgG, it was recognized that some patients with an NMOSD phenotype were negative for AQP4-IgG. After the discovery of MOG-IgG it became apparent that some of these AQP4-IgG seronegative NMOSD cases were positive for MOG-IgG.
However, the phenotype of MOGAD is much broader than that of NMOSD and only a minority of MOGAD patients fulfilled these criteria.5 Moreover, recent investigations have highlighted substantial prognostic differences in terms of relapse-risk and disability accrual during the disease course supporting a separate pathophysiology for each. There are also important differences in demographics, clinical, radiologic and pathologic features that resulted in the need for separate criteria for MOGAD from NMOSD. This ultimately led to the publication of separate diagnostic criteria for MOGAD in 2023 to capture these patients and no longer label them as seronegative NMOSD.1
Epidemiology of AQP4+NMOSD and MOGAD
AQP4+NMOSD and MOGAD are rare disorders. The estimated annual incidence of AQP4+NMOSD is 0.4–7.3/million people;6,7 it is largely unknown in MOGAD, although a few European studies estimated it at 1.6–3.4/million people.8,9 AQP4+NMOSD mainly affects middle-aged females (40–60 years, 9:1 female to male ratio)6,7 with a predilection for Afro-Caribbean or Asian individuals.6,7 MOGAD incidence has a biphasic behavior, with a peak of incidence in children (reported up to three times higher)9 and later in young adults (20–30 years).10–12 No clear sex preference or high-risk ethnicities have been identified in MOGAD thus far.
Pathophysiology of AQP4+NMOSD and MOGAD (Figure 1)
Figure 1. Schematic representation of AQP4+NMOSD and MOGAD pathogenesis.
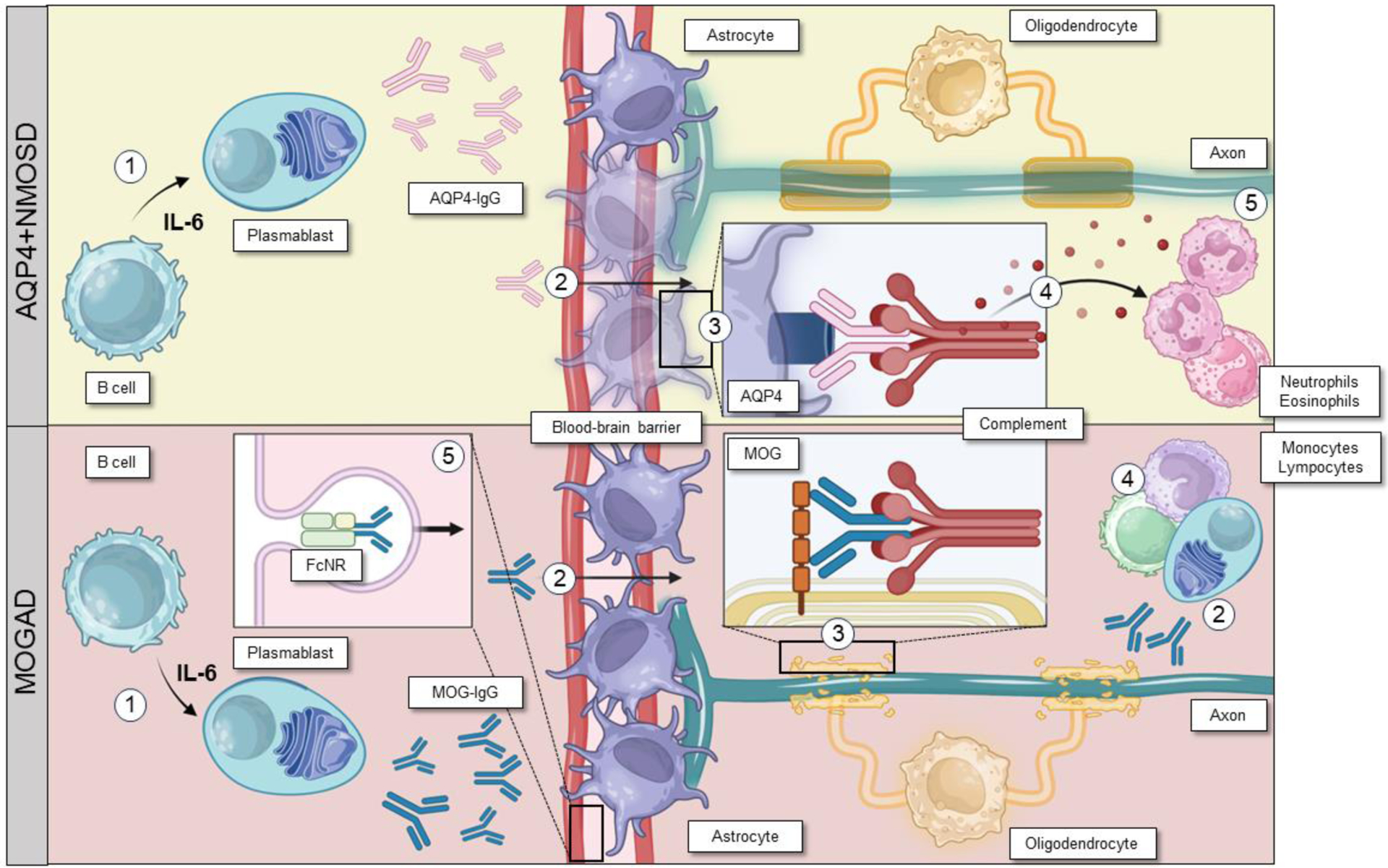
AQP4+NMOSD: 1. Interleukin-6 promotes differentiation of B cells into AQP4-IgG secreting plasmablasts; 2. AQP4-IgGs reach the blood stream and cross the blood-brain barrier; 3. AQP4-IgGs bind to AQP4 on astrocytes and activate the complement cascade through the classical pathway leading to astrocyte damage; 4. The release of anaphylatoxins after complement activation recruit granulocytes, which will ultimately damage neurons and eventually, although not primarily, oligodendrocytes (5).
MOGAD: 1. Interleukin-6 promotes differentiation of B cells into MOG-IgG secreting plasmablasts; 2. MOG-IgGs reach the blood stream and cross the blood-brain barrier, but recent evidence suggest they might also be produced intrathecally; 3. MOG-IgGs bind to MOG on oligodendrocytes and activate the complement cascade through the classical pathway leading to olygodendrocyte damage; 4. local inflammation recruits T cells and monocytes/macrophages; 5. MOG-IgGs recycling in the blood stream seems to contribute to the persistence of the mechanism of damage. Figure created with Biorender.com.
Abbreviations: AQP4=aquaporin-4; AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; IL-6=interleukin-6; MOG=myelin oligodendrocyte; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease.
Like most autoimmune disorders, the first step of the pathophysiological cascade is represented by an unknown mechanism of loss of self-tolerance, which occurs in the periphery. B cells differentiate into antibody-producing plasmablasts, that secrete the pathological autoantibody which eventually enter the CNS.13,14 Antibody production and entry into the CNS may be facilitated by high levels of a pro-inflammatory cytokine called interleukin-6 (IL-6), which increases blood-brain barrier permeability and promotes differentiation of B cells into plasmablasts to enhance antibody-production.15 Alternatively, CNS regions free of the blood-brain barrier such as the area postrema, may offer an alternative route of entry, especially in AQP4+NMOSD. However, intrathecal MOG-IgG production is reported in MOGAD but not AQP4+NMOSD.16–20
Other major differences in pathophysiology emerge once the respective antibodies reach the CNS. AQP4-IgGs bind to the water channel on astrocytes at the blood-brain barrier.3 The binding between the antibody and its target activates the classical pathway of the complement cascade, with primary damage to the astrocytes through the formation of the membrane attack complex and antibody-dependent cellular cytotoxicity.13 Meanwhile, secondary products of complement activation, such as the C5a anaphylatoxin, act as a chemo-attractant for granulocytes, which are locally recruited and cause secondary axonal loss and demyelination in bystander tissue.13
This pathogenesis is supported by pathology findings in AQP4+NMOSD showing: (1) antibody and complement deposition; (2) astrocyte damage or loss (even outside of lesioned tissue) with reduced AQP4 expression; (3) granulocyte infiltration; (4) secondary demyelination and axonal loss in the white matter and grey matter.21
The mechanism of CNS damage has yet to be fully elucidated in MOGAD. One reason is that human MOG-IgG do not usually cross-react with rodent MOG, making studies of animal models more challenging. The selective loss of MOG is also inconsistent in human pathology samples,22,23 raising doubts on the pathogenicity of MOG-IgG. However, MOG-IgG pathogenicity was supported by using the small proportion of MOG-IgG that does cross-react to MOG rodent epitopes, and showing intrathecal MOG-IgG induces a similar disease to humans in murine models.24
According to the most recent hypothesis, in the CNS the binding between MOG-IgG and myelin may lead to increased local production of IL-6 and B-cell activating factor (BAFF), with recruitment of CD4+ T cells and macrophages that will ultimately damage neurons and oligodendrocytes.14 Complement may also contribute to MOGAD pathophysiology, as supported by pre-clinical models,25 evidence of complement deposition with antibody-dependent cellular phagocytosis on pathology samples,22,23,26 and higher activation of both the classic and alternative complement pathways in patients than healthy individuals27 However, complement activation seems less effective with MOG-IgG than AQP4-IgG, possibly because most patients have bivalent binding MOG-IgG, which are known to be less effective in complement activation.28 In addition, MOG-IgGs were able to induce demyelination also by activating the neonatal Fc-receptor pathway, which enhanced the activation and tissue infiltration by T cells in animal models.29 The involvement of CD4+ T cells represents one of the main differences with MS, where CD8+ T cells are usually predominant on pathology samples.22
Cytokine profiling is similar in AQP4+NMOSD and MOGAD but different to MS, showing upregulation of T helper 17-related and some T helper 1-related molecules.30
Differences and similarities in AQP4+NMOSD and MOGAD pathophysiology are summarized in Table 1 and graphically shown in Figure 1.
Table 1.
Differences and similarities in AQP4+NMOSD and MOGAD pathophysiology.
| AQP4+NMOSD | MOGAD | |
|---|---|---|
| Targets | ||
| Antigen | AQP4 | MOG |
| Cell | Astrocyte | Oligodendrocyte |
| Site of antibody production | ||
| Periphery | Yes | Yes |
| CNS | No | Yes |
| Cytokines | ||
| IL-6 | Yes | Yes |
| IL-10 | Yes | Yes |
| IL-17a | Yes | Yes |
| G-CSF | Yes | Yes |
| TNF-alfa | Yes | Yes |
| BAFF/APRIL | Yes | Yes |
| Effectors of damage | ||
| Complement | Yes | Yes, but less prominent |
| Cell infiltrates | Granulocytes | CD4+T cells, macrophages/microglia |
| Outcomes | ||
| Neuronal loss | Yes | Yes, but less severe |
| Astrocytic damage | Yes | No |
| Oligodendrocyte damage | Not prominent | Yes |
| Demyelination | Yes | Yes |
| Damage Biomarkers | ||
| Neurofilament light chain | High (during attacks) | High (during attack) |
| GFAP | High | Normal |
| Myelin basic protein | Normal | High |
Abbreviations: AQP4=aquaporin-4; APRIL= a proliferation-inducing ligand; BAFF=B-cell activating factor; CD4=cluster of differentiation 4; G-CSF=granulocytes colony-stimulating factor; GFAP= Glial fibrillary acidic protein; IL=interleukin; MOG=myelin oligodendrocyte glycoprotein; MOGAD= myelin oligodendrocyte glycoprotein antibody-associated disease; AQP4+NMOSD=neuromyelitis optica spectrum disorder; TNF-alfa=tumor necrosis factor-alfa.
AQP4+NMOSD AND MOGAD DIAGNOSIS
Summary of core clinical manifestations
AQP4+NMOSD and MOGAD share several core clinical features, namely the presence of optic neuritis and myelitis, and are discussed below:
Optic neuritis is associated with variable degrees of visual loss, eye pain worsened by eye movements, and dyschromatopsia. At disease onset, it is the most common presentation in adult MOGAD (50–65%)14 and relatively common in AQP4+NMOSD (35%).31 It can occur in isolation, in association with myelitis, or in the context of acute disseminated encephalomyelitis (ADEM).1,2 In contrast to MS, bilateral simultaneous involvement of the optic nerves is common in both AQP4+NMOSD (17–82%)32,33 and MOGAD (50–84%).33,34 Visual loss at nadir is usually severe with a median visual acuity of hand movement in AQP4+NMOSD and between hand movements and count fingers in MOGAD.35,36 Clues suggesting a diagnosis of MOGAD may be the presence of eye pain before the onset of visual loss (often mistaken for headache, especially in children)37 and evidence of optic disc edema at fundoscopy (86–90%),11,36 that is often moderate to severe and sometimes accompanied by peripapillary hemorrhages.36 At follow-up, recovery is usually complete or almost complete in MOGAD (visual acuity of 20/30 or 20/25),35,36 while recovery can be partial or absent in AQP4+NMOSD (median visual acuity of count fingers).35 Residual permanent blindness in at least one eye (i.e., visual acuity of ≤20/200) is rare in MOGAD (6–12%)35,36 and relatively common in AQP4+NMOSD (60–69%).32,35 However, a non-negligible proportion of patients with MOGAD (16%)36 may develop a steroid-dependent chronic form of optic neuropathy, which relapses at steroid-withdrawal or tapering (chronic relapsing inflammatory optic neuropathy, CRION).
Myelitis: these episodes are characterized by acute/subacute onset of motor, sensory, and autonomic symptoms indicating an involvement of the spinal cord, including but not limited to para/tetraparesis or plegia, sensory level across the trunk, Lhermitte’s phenomenon, sphincteric urgency or retention, and sexual dysfunction. Myelitis is the most common presentation in AQP4+NMOSD patients (50%),31 but also occurs in 20–40% of adult and 15–20% of pediatric MOGAD.14 Similar to optic neuritis, attacks are usually moderate or severe at nadir, with a median EDSS of 7.0 in AQP4+NMOSD and 5.5 in MOGAD.38 Over 30% of patients are wheelchair dependent at nadir in both diseases39 and there is a potential need for admission to the intensive care unit (ICU) for mechanic ventilation due to respiratory failure, especially in AQP4+NMOSD (2–7%).40,41 In the long term, only 6–7% of MOGAD compared to 37–44% of AQP4+NMOSD will need a gait aid.38,39 In MOGAD, residual sphincteric dysfunction may persist over time in more than 50% of patients with history of myelitis with many requiring ongoing intermittent urinary catheterization.38 Accompanying itch42 and the development of painful paroxysmal tonic spasms43 should prompt AQP4-IgG testing, since they are more typical of AQP4+NMOSD. In contrast, the presence of acute flaccid weakness with areflexia may suggest MOGAD and could reflect the involvement of the anterior grey matter, mimicking the acute flaccid myelitis that has been reported to follow enterovirus infection.39
Besides the clinical involvement of the optic nerve and the spinal cord, AQP4+NMOSD and MOGAD patients can also manifest with symptoms related to infratentorial or cerebral involvement:
(Acute) brainstem/cerebellar syndromes: signs or symptoms referable to infratentorial involvement can be observed in both AQP4+NMOSD and MOGAD. In AQP4+NMOSD the area postrema syndrome, characterized by intractable vomiting or hiccups for days to several weeks, is the most frequent manifestation of brainstem involvement (16–60% of patients).1,44 It is usually associated with evidence of a lesion in the area postrema, sometimes representing the extension of a cervical spinal cord lesion.45 In MOGAD, brainstem or cerebellar symptoms usually occur in the context of polyfocal cerebral involvement or ADEM, and are mainly represented by ataxia (45%) or diplopia (26%).44 Attacks of isolated facial numbness and diplopia and trigeminal neuralgia are all much more common in MS than AQP4+NMOSD or MOGAD.
Cerebral manifestations: The frequency and manifestations of cerebral involvement are very different between AQP4+NMOSD and MOGAD.
Approximately 3% of AQP4+NMOSD patients may present with symptoms of diencephalic involvement (e.g., narcolepsy, inappropriate antidiuretic hormone secretion syndrome, hyperphagia, thermic homeostasis dysregulation, and dysfunction of the hypothalamus-hypophysis axis).46,47 Other cerebral manifestations, including encephalopathy, ADEM, posterior-reversible encephalopathy (PRES), and seizures have been reported as well, but are rare.48,49
In MOGAD, ADEM represents the most common presenting manifestation in pediatric patients (20–60%), especially in those less than 12 years old.2,14 It is defined by the concomitant presence of polyfocal CNS symptoms, unexplained encephalopathy, and large poorly demarcated lesions in the grey and white matter at magnetic resonance imaging (MRI).50 Severe encephalopathy or status epilepticus can lead to inability to protect the airway and the need for mechanical ventilation.41 Despite the potential severity of the acute phase, recovery is usually good although deficits in cognition have been reported.51–53
Finally, MOGAD patients may present with cerebral cortical encephalitis, a recently described phenotype characterized by clinical manifestations (i.e., headache [79%], seizures [68%], encephalopathy [63%], and fever [42%])54 and typical T2-FLAIR cortical hyperintensity with corresponding leptomeningeal or cortical gadolinium enhancement.54,55 It is observed in almost 7% of all patients but is more common in children (13.5%) than adults (3.6%).54 Cerebral cortical encephalitis often precedes other short-term MOGAD attacks. Radiological signs resolve in over 90% of patients,54 and can occasionally improve without acute immunotherapy.56
Major MRI features
Magnetic resonance imaging (MRI) is useful when it comes to differentiating between AQP4+NMOSD and MOGAD. Details are provided below:
Optic nerve imaging: It is essential to order orbital MRI with fat saturated images to have sufficient sensitivity to confirm optic neuritis but also to be able to adequately identify discriminators as conventional MRI brain is inadequate for the evaluation of optic neuritis. Optic neuritis is frequently bilateral and severe in both AQP4+NMOSD and MOGAD. In both cases long-segments of inflammation (i.e., T2-hyperintensity or gadolinium enhancement involving over half the distance from the orbit to the chiasm) and optic nerve swelling is common and non-specific.1,2,57 However, lesions usually involve the anterior portion of the optic nerves in MOGAD (sometimes with optic nerve head swelling visible on MRI),2 and are commonly posteriorly located involving the chiasm and the optic tracts in AQP4+NMOSD.1,33 Isolated optic chiasm involvement is more characteristic of AQP4+NMOSD, but MOGAD optic nerve enhancement may extend to involve the chiasm relatively frequently with MOGAD optic neuritis.58 Enhancement of the optic nerve sheath (perioptic enhancement/optic peri-neuritis) and extension to the orbital fat can also be observed in 50% of MOGAD-related optic neuritis36 and may help discriminate from MS.59 In both disorders asymptomatic enhancement may be observed at the site of prior optic neuritis in approximately 20% of patients, possibly representing subclinical blood-brain barrier leakage or residual inflammation.60,61 Chronic atrophy of the optic nerve or optic disc occurs in 12–83% of AQP4+NMOSD,57,62 and can be clinically observed in MOGAD. Examples of acute and chronic MRI findings are shown in Figure 2, with the corresponding schematic representation in Figure 3.
Spinal cord imaging: during acute myelitis AQP4+NMOSD and MOGAD involve similar regions of the cord although conus involvement favors MOGAD.39,63 Approximately 85% of AQP4+NMOSD and 70% of MOGAD patients with acute myelitis demonstrate longitudinally extensive spinal cord T2-lesions,39 which by definition extend over at least three vertebral segments on sagittal T2-weighted images.2,46 By contrast, longitudinally-extensive lesions in MS myelitis occurs in <1%, although occasionally coalescence of multiple short lesions can artifactually appear longitudinally-extensive in MS while in chronic MS sometimes more hazy longitudinally extensive T2-hyperintensity can be encountered.64 T2-lesions are more likely to be solitary in AQP4+NMOSD and multiple in MOGAD.39 Acute gadolinium enhancement (elongated ring-like, patchy) is almost invariably present in AQP4+NMOSD, but less frequent and more faint in MOGAD;39,65,66 leptomeningeal enhancement can be observed in both diseases.65,67 To note, around 10% of acute myelitis in MOGAD initially have a normal MRI, which will usually disclose spinal cord abnormalities after a median delay of six days.68 T2-lesions on axial images are usually central and involve the grey and the white matter,2,69 although T2-hyperintensity restricted to the grey matter in an H-shaped fashion (“H-sign”) is more frequent in MOGAD than AQP4+NMOSD.39 Marked central canal T2-hyperintensity may occur with AQP4+NMOSD and MOGAD but is rare in MS and this signal change usually resolves in follow up.70 It may reflect a potential space from incomplete closure of the central canal that becomes apparent in the setting of central spinal cord inflammation and swelling.70 Another transient radiological sign helpful in differentiating AQP4+NMOSD from MS and potentially MOGAD, is the evidence of spinal cord lesions with areas of T2-hyperintensity at least equal to the cerebrospinal fluid (“brighter spotty lesions”), which tend to be more extensive than just an enlarged central canal and are more common in AQP4+NMOSD.71–73
Figure 2. MRI examples of optic neuritis in patients with MOGAD, AQP4+NMOSD, and MS.

Top row shows MRI findings during the acute phase (post-contrast T1-weighted images with fat saturation), while follow-up up imaging is displayed in the bottom row (pre-contrast T1-weighted images). Unless otherwise specified, images are all shown in axial view.
MOGAD: Bilateral anterior optic neuritis (A, arrows) extending over 50% of optic nerve length on the right side (i.e., long optic neuritis), and short on the left side, with no or minimal residual optic nerve atrophy (B). AQP4+NMOSD: Bilateral optic neuritis (C, arrows) involving the chiasm (zoom-in picture, coronal detail) with mild residual atrophy (D). MS: Unilateral short left optic neuritis (E, arrow) with mild residual focal atrophy (F).
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; Gd= post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease; MS=multiple sclerosis; T1=pre-contrast T1-weighted images.
Figure 3. Schematic representation of optic neuritis in patients with MOGAD, AQP4+NMOSD, and MS.
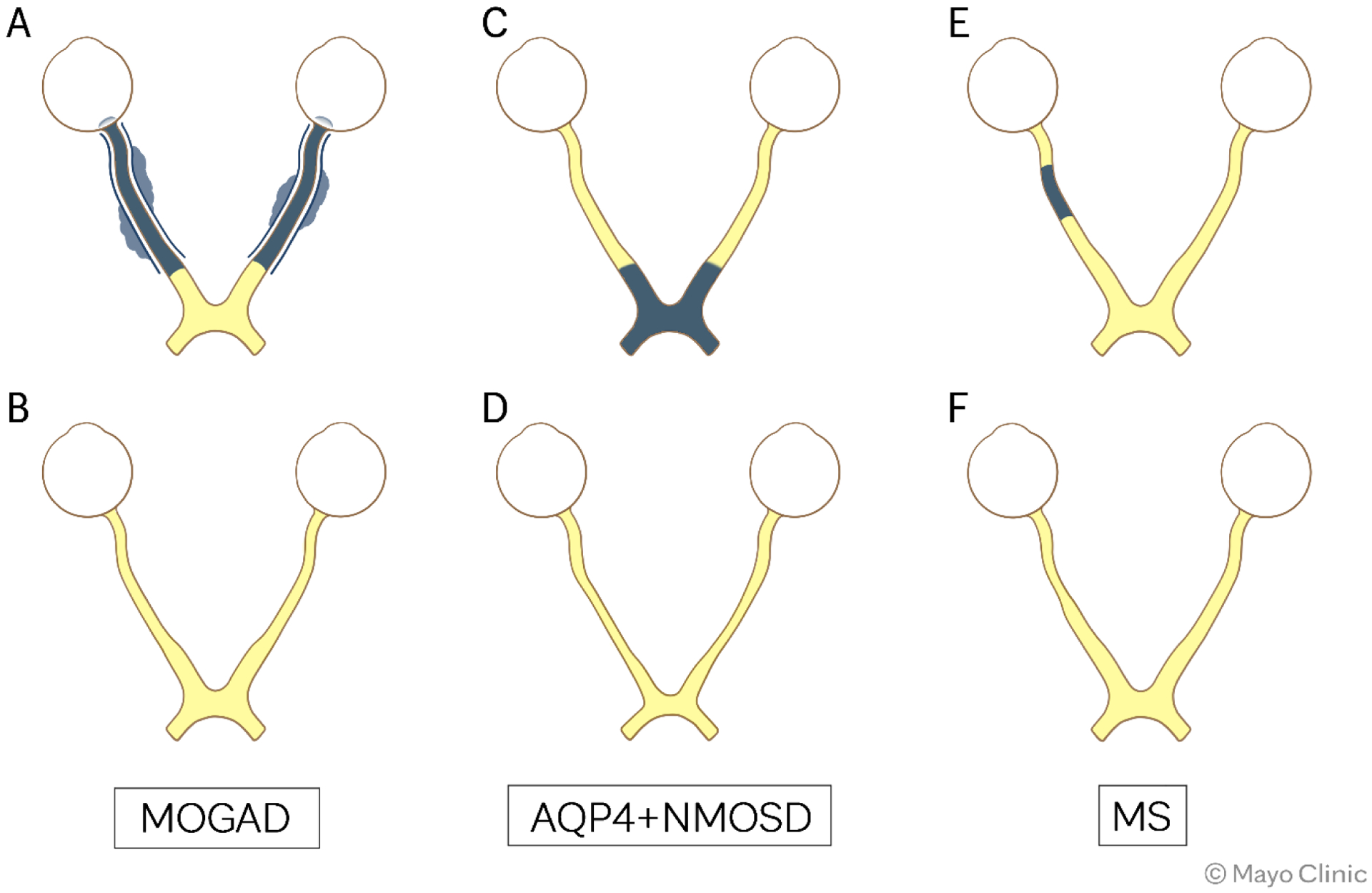
Top row shows schematic representation of the optic nerve during the acute phase, while follow-up up is displayed in the bottom row. All images are shown in axial view.
MOGAD: Bilateral anterior optic neuritis with accompanying optic disc edema extending over 50% of optic nerve length bilaterally with optic nerve sheaths and perioptic fat involvement (A) and minimal residual optic nerve atrophy (B). AQP4+NMOSD: Bilateral optic neuritis involving the chiasm (C) with residual atrophy (D). MS: Unilateral short right optic neuritis (E) with residual focal atrophy (F).
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; Gd= post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease; MS=multiple sclerosis.
The severity of chronic atrophy is proportional to the number of myelitis in AQP4+NMOSD and MOGAD,74 is mainly lesional rather than diffuse, and long segments of atrophy can be a clue to AQP4+NMOSD diagnosis.38,69
Examples of acute and chronic MRI findings are shown in Figure 4, with the corresponding schematic representation in Figure 5.
Figure 4. MRI examples of myelitis in patients with MOGAD, AQP4+NMOSD, and MS.
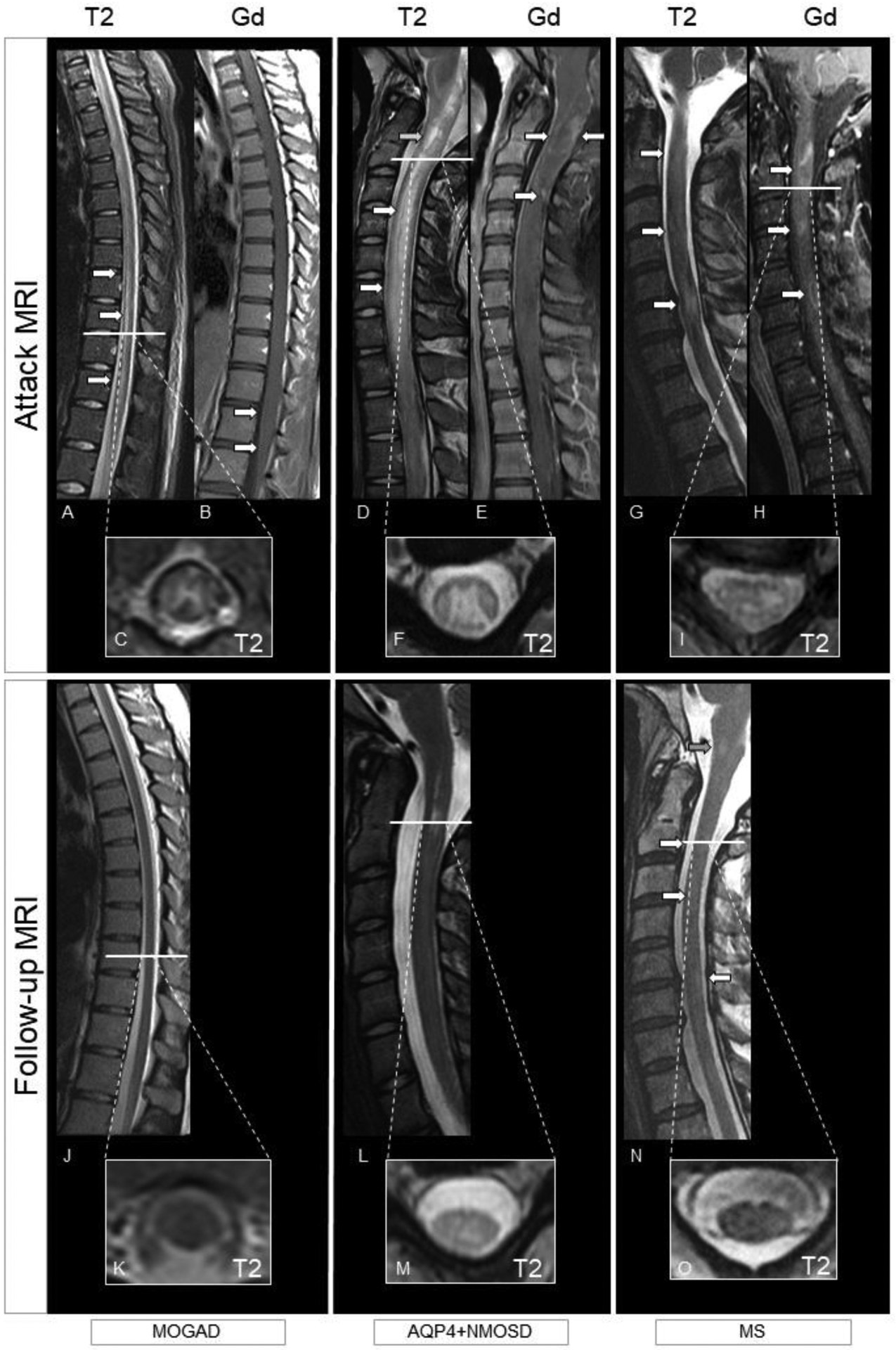
Top row shows MRI findings during the acute phase (T2-weighted images and post-contrast T1-weighted images), while follow-up up imaging is displayed in the bottom row (T2-weighted images). MOGAD: Longitudinally extensive myelitis (i.e., T2-lesion extending over at least three continuous vertebral segments) with a linear appearance involving the thoracic cord down to the conus (A, arrows, sagittal view). There is associated H-sign (i.e., exclusive involvement of the grey matter; C, axial view). Enhancement is absent, except for a mild leptomeningeal enhancement of the conus (B, arrows, sagittal view). The T2-lesion completely resolved on T2-weighted images at follow-up (J, sagittal view and K, axial view), with no evident atrophy. AQP4+NMOSD: Longitudinally extensive myelitis with a T2-lesion starting from the area postrema and involving the cervical cord (D, arrows, sagittal view) with associated swelling. Intralesional increased focal T2-hyperintensity similar to the CSF (i.e., brighter spotty lesion) is also present (D, green arrow). The T2-lesion is centrally located in both the grey and the white matter (F). Enhancement is inhomogeneous (E, arrows, sagittal view). At follow-up, the T2-lesion reduced in size on T2-weighted images (L, sagittal and M axial view) although still present. Residual atrophy of the cord is particularly evident on axial view (M). MS: Multiple focal short spinal cord T2-lesions (G, arrows, sagittal view) located in the peripheral white matter (I, axial view). All lesions enhance, with the bottom lesion showing a ring-pattern of enhancement (H, arrows, sagittal view). T2-lesions reduce in size and prominence of T2-hyperintensity persist on follow-up T2-weighted images (N, arrows, sagittal view and O, axial view). The patient also developed an interval T2-lesion (N, green arrow).
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; Gd=post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease; MS=multiple sclerosis; T2=T2-weighted images.
Figure 5. Schematic representation of myelitis in patients with MOGAD, AQP4+NMOSD, and MS.

Top row shows spinal cord findings during the acute phase (T2-weighted images and post-contrast T1-weighted images), while follow-up up imaging is displayed in the bottom row (T2-weighted images). MOGAD: Longitudinally extensive myelitis with a linear T2-lesion appearance involving the lower cervical and upper to middle thoracic cord and another lesion in the conus (A, sagittal view). There is associated H-sign with the T2-lesion restricted to grey matter (C, axial view). Minimum linear enhancement and leptomeningeal enhancement of the conus (B, sagittal view). The T2-lesion completely resolved on T2-weighted images at follow-up (J, sagittal view and L, axial view), with no evident atrophy. Gadolinium enhancement resolved (K). AQP4+NMOSD: Longitudinally extensive myelitis with a T2-lesion involving the cervical and thoracic cord (D, sagittal view) with elongated ring enhancement (E). The T2-lesion is centrally located in both the grey and the white matter (F, axial view). At follow-up, the lesion is smaller on T2-weighted images (M, sagittal and O axial view) although still present. Gadolinium enhancement resolved (N). MS: Multiple focal short spinal cord T2-lesions (G, sagittal view) located in the peripheral white matter (I, axial view). One lesion shows homogeneous nodular enhancement (H, sagittal view). T2-lesions reduce in size and persist on follow-up T2-weighted images (P, sagittal view and R, axial view) with development of focal left sided spinal cord atrophy particularly evident on axial images (R). A new interval T2-lesion is also present (P). Gadolinium enhancement resolved (Q).
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; Gd=post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease; MS=multiple sclerosis; T2=T2-weighted images.
Brain imaging: Brain lesions are observed in up to 80% of AQP4+NMOSD patients.75 MRI findings have been extensively analyzed and classified in 2015, with the definition of typical and non-specific lesions.46 Typical lesions are usually observed at periependymal level,46 following regions of high AQP4 expression.76 Among them, periependymal lesions along the lateral ventricles are the most common (12–40%),46 especially in the course of cerebral attacks.77 Corresponding pencil-thin linear ependymal enhancement is typical of AQP4+NMOSD and not found in MOGAD or MS.78
During the acute phase, lesions may demonstrate typical patterns of heterogeneous appearance (“marbled pattern”) or homogeneous involvement of the splenium (“arch bridge pattern”), which may help diagnosis.46 Of note, callosal lesions can also be observed in patients with MOGAD at a similar frequency, but their size rarely exceeds 2.5 cm (11%) and the extracallosal brain involvement is common (55%).77 Callosal lesions can resolve in the chronic phase, although with a higher rate in MOGAD than AQP4+NMOSD (56% vs. 15%).77 The shape of callosal lesions may also help differentiate MS, where lesions are usually focal ovoid, with sharp margins, and with the major axis perpendicular to the lateral ventricles.79
Other periependymal lesions in AQP4+NMOSD may surround the third ventricle resulting in diencephalic involvement (i.e., thalamus, hypothalamus, and anterior border of the midbrain) that may be asymptomatic. Diencephalic lesions favor AQP4+NMOSD over MS, although rarely encountered (6% of AQP4+NMOSD patients).75 Finally, periependymal lesions around the cerebral aqueduct and the fourth ventricle are also relatively frequent (7–46% of AQP4+NMOSD patients), and those in the dorsal medulla can involve the area postrema causing the hallmark clinical syndrome with intractable nausea, vomiting and hiccups.46 Other brain lesions considered typical of AQP4+NMOSD are those in the cerebral peduncles and corticospinal tracts and large hemispheric lesions in the white matter (i.e., with maximum transverse diameter of >3 cm, often spindle-like or with a radial shape). Similar lesions have also been reported in patients with MOGAD.80 Tumefactive lesions (≥2 cm) are more frequent in MOGAD than AQP4+NMOSD (22% vs 5%).81
Small non-specific lesions (i.e., <3mm) in the subcortical and deep white matter, similar to those encountered in aging, small vessel disease or migraine, are the most common type of brain lesions in AQP4+NMOSD patients (35–84%).46
Other than the ependymal enhancement, also cloud-like, nodular, and leptomeningeal enhancement were considered typical of AQP4+NMOSD. However, more recent investigations suggest that cloud-like and nodular enhancement may be encountered with a similar frequency also in MOGAD and MS,78,81 while the leptomeningeal enhancement is much more common in MOGAD (46% of cerebral attacks) and can actually help discriminate from AQP4+NMOSD (7%) and MS (4%).78 Persistent enhancement over three months is rare in all these disorders.78 Similar to the variety of cerebral manifestations, the radiological features of brain MRI in MOGAD are heterogeneous. Brain lesions can be found in 42–53% of patients with MOGAD,82 and include lesions in the deep grey matter, cortical lesions, subcortical or juxtacortical lesions, brainstem and cerebellar lesions, large hemispheric lesions, and, rarely, leukodystrophy-like patterns.63,82,83 Among all these locations, lesions in the deep grey matter63,82 and large lesions in the middle cerebellar peduncles,44 are the most characteristic and more common in MOGAD than AQP4+NMOSD. Diffuse involvement of the pons and/or adjacent to the fourth ventricle (anterior location) may also favor MOGAD over AQP4+NMOSD, although not confirmed in all studies.44,82
Brain lesions in this disease are usually poorly demarcated (“fluffy”),84 in line with what is observed in patients with ADEM,50 of which conversely 50% test positive for MOG-IgG.85 Transient faint T1-hypointensity can occur in the acute phase of MOGAD but chronic T1 hypointensities are rare and are much more suggestive of MS.81 Cortical lesions in MOGAD usually occur during episodes of cerebral cortical encephalitis, are visible on FLAIR images and involve large cortical areas.54,55 This contrasts with MS, where the assessment of cortical lesions may be less visible on conventional sequences and are better identified using advanced sequences such as double inversion recovery (DIR), phase sensitive inversion recovery (PSIR), or magnetization-prepared rapid gradient-echo (MPRAGE).86,87 Furthermore, evidence of cortical lesions also favors MOGAD over AQP4+NMOSD, as in the latter cortical involvement was absent75,88,89 or rarely90,91 found.
Examples of acute and chronic MRI findings with the corresponding schematic representation are shown in Figures 6–7 (MOGAD), 8–9 (AQP4+NMOSD), and 10–11 (MS).
Figure 6. MRI examples of brain lesions in patients with MOGAD.

Top row shows MRI findings during the acute phase, while follow-up up imaging is displayed in the bottom row. Images are in axial view.
Poorly defined (i.e., fluffy) T2-lesions in the entire medulla and cerebellum (A, arrows), completely resolving at follow-up imaging (E). Bilateral fluffy T2-lesions in the middle cerebellar peduncles (B, arrows) with reduction in size but persistence at follow-up (F, arrows) with accompanying fourth ventricle ex vacuo enlargement. Bilateral fluffy T2-lesions of the thalami (C, arrows) in a patient with prominent leptomeningeal enhancement (zoom-in picture, post-contrast T1-weighted sequence) undergoing complete resolution at follow-up (G). Patient with cerebral cortical encephalitis showing an extensive cortical T2-lesion (D, arrow) with focal enhancement (zoom-in picture, post-contrast T1-weighted sequence), completely resolved at follow-up (H).
Abbreviations: FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease.
Figure 7. Schematic representation of brain lesions in patients with MOGAD.

Top row shows brain findings during the acute phase, while follow-up up is displayed in the bottom row. Images are all shown on axial view.
T2-lesion involving the entire medulla (A), completely resolving at follow-up (F). Bilateral fluffy T2-lesions in the middle cerebellar peduncles (B) resolved at follow-up (G). Bilateral fluffy T2-lesions of the thalami and additional lesions in the white matter (C) undergoing complete resolution at follow-up (H). Cerebral cortical encephalitis with an extensive cortical T2-lesion (D) accompanied by leptomeningeal enhancement (E). Both cortical lesion and enhancement completely resolved at follow-up (I, J).
Abbreviations: FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images; MOGAD=myelin oligodendrocyte glycoprotein antibody-associated disease.
Figure 8. MRI examples of brain lesions in patients with AQP4+NMOSD.

Top row shows MRI findings during the acute phase, while follow-up up imaging is displayed in the bottom row. Images are shown in axial view.
T2-lesion in the area postrema (A, arrow) almost invisible but still present at follow-up (F, zoomed-in picture, arrow). T2-lesion involving the dorsal pons abutting to the fourth ventricle (B, arrow) with complete resolution at follow-up (G). Periependymal T2-lesion (C, arrow) with corresponding linear ependymal enhancement (zoom-in picture, post-contrast T1-weighted sequence), persisting at follow-up (H, arrow). T2-lesion involving the splenium of the corpus callosum in another patient (D, arrow), significantly reduced in size but still visible at follow-up (I, arrow). Multiple small nonspecific T2-lesions in the subcortical white matter (E, arrows), persisting unchanged at follow-up (J, arrows). Additional interval T2-lesions are observed as well (J, green arrows).
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images.
Figure 9. Schematic representation of brain lesions in patients with AQP4+NMOSD.

Top row shows brain findings during the acute phase, while follow-up up is displayed in the bottom row. Images are all shown on axial view.
T2-lesion in the area postrema (A) smaller but still present at follow-up (F). Posterior T2-lesion abutting to the fourth ventricle (B) smaller but still present at follow-up (G). T2-lesion in the corticospinal tract and splenium of the corpus callosum (C) smaller but still present at follow-up (H). Multiple small nonspecific T2-lesions in the subcortical white matter (D), persisting unchanged at follow-up (I). Additional interval T2-lesions are observed as well (I). The presence of linear ependymal enhancement (E), resolving at follow-up (J) is typical of AQP4+NMOSD.
Abbreviations: AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder; FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images.
Figure 10. MRI examples of brain lesions in patients with MS.
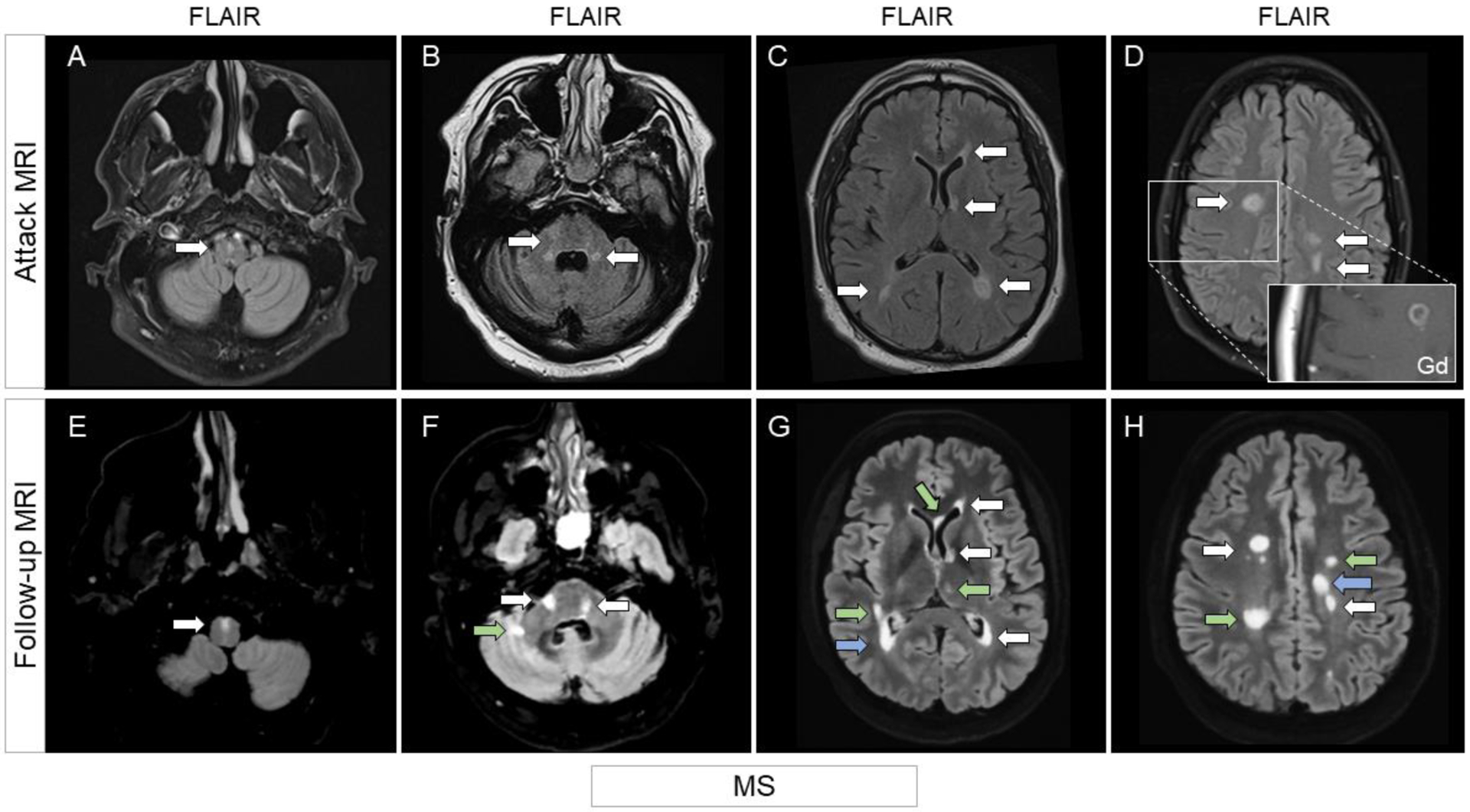
Top row shows MRI findings during the acute phase, while follow-up up imaging is displayed in the bottom row. Unless otherwise specified, images are all shown on FLAIR sequences, axial view.
Small T2-lesion in the anterior medulla (A, arrow), unchanged at follow-up (E, arrow). Multiple T2-lesions in the peripheral pons and abutting on the fourth ventricles (B, arrows), still visible at follow-up (F, arrows) with additional interval T2-lesions (F, green arrow). Multiple ovoid periventricular T2-lesions abutting on the lateral ventricles (C, arrows). T2-lesions persisted at follow-up (G, arrows), at times increasing in size (G, blue arrow). Additional interval T2-lesions (G, green arrows) are shown as well. White matter T2-lesions (D, arrows), one showing ring enhancement (zoom-in picture, post-contrast T1-weighted sequence). T2-lesions persisted at follow-up (H, arrows), at times increasing in size (H, blue arrow). Additional interval T2-lesions (H, green arrows) are shown as well.
Abbreviations: FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images; MS=multiple sclerosis.
Figure 11. Schematic representation of brain lesions in patients with MS.

Top row shows brain findings during the acute phase, while follow-up up imaging is displayed in the bottom row. Images are all shown on axial view. Small peripheral T2-lesion in the anterior medulla (A), substantially unchanged at follow-up with additional interval T2-lesion (F).
Multiple T2-lesions in the peripheral pons, trigeminal nerve, and abutting on the fourth ventricles (B), substantially unchanged at follow-up with additional interval lesion (G). Multiple ovoid periventricular, juxtacortical and deep white matter T2-lesions (C). T2-lesions persisted at follow-up (H), at times increasing in size. Additional juxtacortical interval T2-lesions are also visible (H). White matter T2-lesions and one cortical T2-lesion (D) persisting at follow-up with additional interval lesions development (I). Two of the lesions shown in C demonstrate open or closed ring enhancement, which is typically observed in MS (E) and resolves at follow-up (J).
Abbreviations: FLAIR=fluid-attenuated inversion recovery; Gd=post-contrast T1-weighted images; MS=multiple sclerosis.
Remission MRI
Finally, although not specifically covered by the diagnostic criteria, remission MRI may be relevant for the differentiation of AQP4+NMOSD, MOGAD, and MS given their different behavior in terms of T2-lesion resolution and accumulation of asymptomatic lesions.
After the acute event brain T2-lesion resolution is very common in MOGAD (60–79%),2,81,92–94 can occasionally be observed in AQP4+NMOSD (14–27%),44,81,93,95 and is very rare in MS (0–17%).44,81,92,93 Similar findings are observed in the spinal cord, where 67–79% of lesions will ultimately resolve in MOGAD.92–94 T2-lesion resolution in MOGAD occurs at approximately 3 months from appearance, but can be faster for small lesions.94 Steroid administration favors resolution of large lesions (i.e., ≥ 1 cm), but is not a necessary condition since spontaneous resolution was observed in over half T2-lesions not undergoing any acute treatment.94 In contrast, concomitant T1-hypointensity during the acute phase reduced the likelihood of lesions resolving over time.94 Altogether these observations (i.e., spontaneous resolution, steroid response, effect of T1-hypointensity, and timing of resolution) suggest that this phenomenon is part of MOGAD natural history and relies on at least three factors: edema reabsorption, mild tissue damage, and post-acute healing processes like, for instance, remyelination.94
Large reductions and progressive fragmentation is typical in AQP4+NMOSD although complete resolution is rare,92,93,96 and persistence of T2-lesions in the rule in MS.92,93
Surveillance MRI outside of attacks is standard of care in MS as new asymptomatic lesions or enlarging T2-lesions are well recognized to occur particularly with low or moderate efficacy medication and their presence may lead to treatment escalation. However, new asymptomatic T2-lesions or enlarging T2-lesions are far less common in MS when high efficacy treatments are used. The presence of new or enlarging T2-lesions has been commonly used in MS clinical trials as a surrogate end-point.97. In AQP4+NMOSD and MOGAD the frequency of new or enlarging asymptomatic T2-lesions is rare and estimated between 3–13%98–101 and 3–14%100–102, respectively. This has implications for clinical practice as surveillance MRIs are generally not recommended in AQP4+NMOSD or MOGAD. Also, it has implications for upcoming clinical trials in these disorders as this will be a less useful clinical trial endpoint.
Table 2 summarizes the main radiological features of AQP4+NMOSD, MOGAD, and MS.
Table 2.
Imaging findings in AQP4+NMOSD and MOGAD. MS findings are also shown for comparison.
| AQP4+NMOSD | MOGAD | MS | |
|---|---|---|---|
| Optic nerve | |||
| Bilateral involvement | ++ | ++ | − |
| Longitudinally extensive lesions (>50% length of optic nerve) | ++ | ++ | − |
| Location | Posterior with chiasm | Anterior | Anterior/middle |
| Optic nerve enhancement | +++ | +++ | +++ |
| Optic nerve sheath enhancement | − | ++ | − |
| Perioptic fat enhancement | − | ++ | − |
| Spinal cord | |||
| Multiple lesions | − | ++ | +++ |
| Longitudinally extensive lesions | +++ | +++ | − |
| Location (axial) | Central | Central | Peripheral |
| Grey matter involved | +++ | +++ | + |
| White matter involved | +++ | + | +++ |
| Location (sagittal) | Cervico-thoracic | Cervico-thoracic | Cervico-thoracic |
| Conus involved | + | ++ | + |
| Parenchymal enhancement | Lens-shape, heterogeneous | Faint, ill-defined | Ring, nodular |
| Leptomeningeal enhancement | + | ++ | − |
| Brain | |||
| Shape | Along white matter tracts | Poorly demarcated | Ovoid |
| Cortical lesions | − | + | ++ |
| Juxtacortical lesions | − | ++ | +++ |
| Subcortical lesions | ++ | + | − |
| Periventricular lesions | Peri-3rd/4th ventricle and peri-ependymal lateral ventricles | − | Dawson’s fingers |
| Corpus callosum lesions | ++ | ++ | +++ |
| Deep grey matter lesions | + | ++ | − |
| Diffuse pons/middle cerebellar peduncle lesions | + | +++ | |
| T1-hypointense lesions | + | + | +++ |
| Ring enhancement | − | − | ++ |
| Ependymal enhancement | ++ | − | − |
| Leptomeningeal enhancement | − | ++ | −1 |
| T2-lesion resolution | + | +++ | − |
| Inter-attack asymptomatic accumulation of new T2-lesions | − | − | ++2 |
Note that: “−“ indicates rare findings (<5%), “+” infrequent findings (5–30%), “++” common findings (30–69%), “+++” very common findings (>70%).
May be seen with more sophisticated MRI techniques (e.g., post-contrast Fluid-attenuated inversion recovery or at 7.0 T field strength)
Depdendent on treatment and is common with low or moderate efficacy MS disease modifying treatments but rare with high efficacy treatments
Abbreviations: MOGAD=myelin-oligodendrocyte glycoprotein antibody-associated disease; MS=multiple sclerosis; AQP4+NMOSD=aquaporin-4-IgG positive neuromyelitis optica spectrum disorder.
AQP4-IgG and MOG-IgG testing
Some basic knowledge of the antibody testing methodology, the optimal specimen and technique, and potential pitfalls is crucial given their importance in diagnosis of AQP4+NMOSD and MOGAD. The cell-based assay (CBA) technique and analysis in serum is generally recommended for AQP4-IgG and MOG-IgG, with live CBA conferring some advantages over the fixed technique.2,103
AQP4-IgG testing:
Live or fixed CBA with immunofluorescence or flow cytometry (FACS)-based detection or quantification are recommended for AQP4-IgG testing, since they demonstrated high sensitivity (69.7–100.0%) and the highest specificity (85.8–100.0%) in independent cohorts.95,104,105 Older generation techniques using mouse tissue based immunofluorescence have lower sensitivity although reasonably high specificity.106 The enzyme-linked immunosorbent assay has lower sensitivity and a false positive rate fivefold higher than CBA, particularly with low positive results.104,105 False positives are usually low titers and confirmatory testing with one or more different assays is recommended.1 However, evidence low positive results in the context of live CBA assays can be considered reliable, as different live CBA assays demonstrated a strong agreement irrespective of antibody titer (100% concordance in high positive and 79% in low positive patients).107
False negatives may be also encountered (especially in patients receiving immunosuppressive treatments or when tested just after plasma exchange)1 and retesting in these scenario’s should be considered,1 although less than 1% of individuals initially testing negative for AQP4-IgG will subsequently seroconvert to AQP4-IgG-positive.108
Finally, although AQP4-IgG can also be found in the CSF,16,109 antibody testing with modern CBA or FACS on serum is more sensitive.16 The presence of CSF positivity appears related to high AQP4-IgG titers in serum (higher likelihood of a CSF positive test in patients with AQP4-IgG titer in serum >1:100) and is more common during clinical attacks.16 Therefore, serum testing is generally sufficient for AQP4-IgG testing, although in highly suspicious cases CSF AQP4-IgG can be considered, but isolated CSF AQP4-IgG positives are exceedingly rare.16
MOG-IgG testing:
In contrast with AQP4-IgG, MOG-IgG testing is more complex, and has historically represented a challenge. In fact, initial reports using Western Blot or ELISA targeting denaturated MOG proteins, yielded the presence of MOG-IgG in serum of many patients with MS and healthy controls,110–112 leading to the misconception that these antibodies may represent an epiphenomenon of demyelination. The association between MOG-IgG and specific demyelinating phenotypes such as ADEM and optic neuritis (but not MS), was first highlighted by using a laboratory assay expressing MOG in its native tridimensional conformation4 and was subsequently confirmed by CBAs expressing full-length human MOG.113,114 ELISA testing demonstrated a poor diagnostic performance and reproducibility, and is therefore not recommended or suitable for diagnosing MOGAD.2,107
However, in contrast with AQP4-IgG testing,104,107 an excellent agreement between different CBA assays was only reached with clear positive (82%) and clear negative results (97.5%), with a slight improvement when only live CBA were considered.107 The agreement on borderline positives was poor (33%).107 Caution is needed with low positive CBA results given MOG-IgG can be found at low titer in 1–2% of disease controls.107,115,116 The positive predictive value of MOG-IgG increases when ordered in high probability situations and with higher antibody titers.115 The MOG-IgG is still a very useful test with high specificity (≈98–99%) and this is exemplified by a recent study that showed no MOG-IgG positive among 703 pediatric healthy controls.117
These observations highlight two fundamental principles which should be considered when testing MOG-IgG: (1) CBA methodologies providing quantitative or semiquantitative data can be useful, and (2) testing should be reserved to patients with a high a priori probability of having MOGAD. Universal testing of all MS patients is not recommend given the potential for 1–2% to have low positive results that may lead to confusion about the diagnosis. It is preferred to select those with features that are suggestive of MOGAD and avoid testing in patients with classic features of MS.
Finally, recent investigations have highlighted a role for CSF testing in MOGAD. In fact, although serum is overall more sensitive to MOG-IgG detection, concomitant detection of MOG-IgG in serum and CSF occurs in 41–87% of patients.17–20,118–120 CSF positivity can be observed in isolation in 3–29%17–20,118–120 and in suspicious cases negative for MOG-IgG in serum, CSF MOG-IgG testing should be undertaken. Patients with evidence of intrathecal synthesis of MOG-IgG or CSF MOG-IgG positivity seem to have a worse clinical prognosis.19,20 As false positive MOG-IgG in CSF can be rarely encountered in MS and other diseases,19,20 the result should always be put into clinical context.
Additional cerebrospinal fluid analysis and other laboratory features in MOGAD and AQP4+NMOSD:
Other than disease-specific antibody testing, additional laboratory analysis can be helpful in the differential diagnosis process, although not included in the diagnostic criteria. CSF usually reveals pleocytosis in over 50% of patients with MOGAD (median 31–40 cells/ul)121,122 and AQP4+NMOSD (median 19 cells/ul)123 but rarely in MS. Cells are usually predominantly lymphocytes121–123 although also monocytes (MOGAD),121,122 neutrophils (both AQP4+NMOSD and MOGAD)121–123 or eosinophils (AQP4+NMOSD)123 can be found. In MOGAD, CSF abnormalities may vary by phenotype and are more common in patients with brain and/or spinal cord lesions.124 CSF oligoclonal bands are rarely encountered in patients with MOGAD and AQP4+NMOSD (approximately 10–20%)121–124 compared to 88% of patients with MS.125
Approximately 2–3% of AQP4+NMOSD patients can have coexistent myasthenia gravis.126,127 Although AQP4+NMOSD diagnosis usually follows that of myasthenia,128 anti-acetylcholine receptor antibody in serum should be checked in case of compatible clinical manifestations. Rarely MOG-IgG were also found coexisting with AQP4-IgG and in most cases is likely related to its background rate being found in 1–2% of disease controls. Most patients with dual AQP4-IgG and MOG-IgG positivity had high-titer AQP4-IgG and low titer MOG-IgG and a phenotype more suggestive of AQP4+NMOSD.129
The laboratory features and antibody testing of AQP4+NMOSD and MOGAD are summarized in Table 3.
Table 3.
Recommendations and laboratory features of AQP4+NMOSD and MOGAD.
| AQP4+NMOSD | MOGAD | |
|---|---|---|
| Antibody | AQP4-IgG1 | MOG-IgG1 |
| Sample | ||
| Serum | Yes (preferred) | Yes (preferred) |
| CSF | No (isolated CSF AQP4-IgG extremely rare) | Yes (≈10% isolated CSF MOG-IgG) |
| Test assay | ||
| Live CBA | Yes (gold standard) | Yes (gold standard) |
| Fixed CBA | Yes | Yes |
| Murine tissue-based assays | Intermediate sensitivity but very good specificity | May have white matter staining when CSF tested but very insensitive24 |
| ELISA | Good performance but reduced sensitivity and risk of false positives at low titer vs cell-based assays | Not recommended due to inconsistent results |
| Quantitative results important | No | Yes (risk of false positives at low titer) |
| Seroconversion important | No | Yes (relapse-risk) |
| CSF findings | ||
| Pleocytosis | ++ | ++ |
| High protein | ++ | ++ |
| Oligoclonal bands | + (<20%) | + (<20%) |
Note that: “−“ indicates rare findings (<5%), “+” infrequent findings (5–30%), “++” common findings (30–69%), “+++” very common findings (>70%).
Abbreviations: AQP4=aquaporin-4; CBA=cell-based assay; CSF=cerebrospinal fluid; ELISA= enzyme linked immunosorbent assay; MOG=myelin oligodendrocyte glycoprotein; MOGAD=myelin-oligodendrocyte glycoprotein antibody-associated disease; AQP4+NMOSD= neuromyelitis optica spectrum disorder.
Diagnostic criteria
The latest diagnostic criteria for AQP4+NMOSD and MOGAD are dated 2015 and 2023, respectively, and are summarized in Table 4.
Table 4.
Diagnostic criteria of AQP4+NMOSD and MOGAD.
| AQP4+NMOSD | MOGAD | ||
|---|---|---|---|
| Antibody test (CBA) positive (AQP4+NMOSD) or clear positive (MOGAD) | Core clinical features | Optic neuritis | Optic neuritis |
| Myelitis | Myelitis | ||
| Area postrema syndrome Acute brainstem syndrome | Brainstem or cerebellar deficits | ||
| Symptomatic cerebral syndrome with AQP4+NMOSD-typical brain MRI lesions | ADEM | ||
| Symptomatic narcolepsy or acute diencephalic clinical syndrome with AQP4+NMOSD-typical diencephalic MRI lesions | Cerebral monofocal or polyfocal deficits | ||
| Cerebral cortical encephalitis often with seizures | |||
| Antibody test (CBA) negative/unknown (AQP4+NMOSD) or low positive/positive without titre in serum or negative in serum but CSF positive (MOGAD) | Supporting features | ||
| Optic neuritis | Normal findings or only nonspecific white matter lesions in the brain | Bilateral simultaneous clinical involvement | |
| Longitudinal optic nerve involvement (> 50% length of the optic nerve on T2 or postgadolinium T1) | Longitudinal optic nerve involvement (> 50% length of the optic nerve) | ||
| Optic chiasm | Perineural optic sheath enhancement | ||
| Optic disc oedema | |||
| Myelitis | Longitudinally extensive myelitis | Longitudinally extensive myelitis | |
| Longitudinally extensive focal spinal cord atrophy in patients with history compatible with acute myelitis | Central cord lesion or H-sign | ||
| Conus lesion | |||
| Area postrema syndrome | Dorsal medulla/area postrema lesions | - | |
| Brain, brainstem, or cerebral syndromes | Periependymal brainstem lesions | Multiple ill-defined T2 hyperintense lesions in supratentorial and often infratentorial white matter | |
| Deep grey matter involvement | |||
| Ill-defined T2-hyperintensity involving pons, middle cerebellar peduncle, or medulla | |||
| Cortical lesion with or without lesional and overlying meningeal enhancement | |||
| Exclusion of better diagnoses including multiple sclerosis | Yes | Yes |
Abbreviations: CBA=cell-based assay; CSF=cerebrospinal fluid; MOGAD= myelin oligodendrocyte glycoprotein antibody-associated disease; AQP4+NMOSD= aquaporin-4-IgG positive neuromyelitis optica spectrum disorder.
In both cases, the diagnostic algorithm starts from the evidence of core clinical features, and then dichotomizes based on antibody-serostatus. Clear evidence of the pathogenetic antibody in a patient with typical clinical manifestations allows the achievement of the diagnosis; alternatively, additional clinical or MRI requirements are needed. The comparison of AQP4+NMOSD and MOGAD diagnostic criteria highlights several differences. First, for AQP4+NMOSD diagnosis but not MOGAD there is a seronegative or unknown antibody status category.1 As MOG-IgG is found in up to 30% of seronegative NMOSD patients,130,131 future iterations of AQP4-IgG seronegative NMOSD will likely require a negative MOG-IgG test. The reason for this is that MOG-IgG positive patients with a compatible syndrome should be diagnosed with MOGAD rather than AQP4-IgG seronegative NMOSD. MOGAD diagnostic criteria, but not AQP4+NMOSD, have additional requirements for low positive or CSF MOG-IgG positive tests given the challenges with low positive results and limited data on CSF MOG-IgG, respectively.
AQP4+NMOSD AND MOGAD TREATMENT
Treatment in autoimmune disorders has two main goals: (1) promote recovery after an acute attack, and (2) prevent subsequent relapses. In this section we will describe the main treatment strategies in AQP4+NMOSD and MOGAD.
Acute treatment of attacks
Acute treatment in both AQP4+NMOSD and MOGAD is similar to MS. It mainly includes intravenous steroids and plasma exchange although occasionally, intravenous immunoglobulin (IVIg) is also used. The details are summarized in Table 5. There is evidence that early treatment (i.e., <7 day-delay from symptoms onset) reduces the likelihood of residual deficits in both AQP4+NMOSD and MOGAD.132 Also, the use of both steroids and plasma exchange may be more common in these conditions, given the greater severity of symptoms at nadir and the high efficacy of early plasma exchange or apheresis by immunoadsorption in patients with AQP4+NMOSD.133–135 In AQP4+NMOSD transitional corticosteroids for a few weeks are often used while awaiting attack-prevention treatments to work and the duration varies depending on the type of immunosuppressant used.103,136 In MOGAD longer steroid tapers for many months have sometimes been used to prevent early relapses but the majority of patients will not have an early relapse and prolonged steroids has a large side effect burden, particularly in growing children.10,137,138 Therefore, using such an approach in all patients is problematic and further studies are needed to determine the role of a more prolonged corticosteroid taper.
Table 5.
Main treatment protocols in AQP4+NMOSD and MOGAD.
| Protocol | AQP4+NMOSD | MOGAD | |
|---|---|---|---|
| Acute treatment of attacks | |||
| Importance | − | Residual disability | Residual disability |
| Steroids | Intravenous methylprednisolone 1,000 mg/day for 5 days1 | +++ | +++ |
| Plasma exchange2 | Every other day for 5–7 cycles | +++ | ++ |
| Steroid tapering | Oral steroids 20–40 mg followed by a taper | Weeks3 | Weeks-months |
| Chronic attack-preventive treatment 4 | |||
| Importance | − | Affects long-term prognosis | Unknown |
| Start at first clinical attack | − | +++ | + |
| Complement inhibitors | |||
| Eculizumab | 900 mg intravenous every week for the first 4 weeks, then 1,200 mg every two weeks | +++ | Not tried in trials |
| Ravulizumab | Body-weight-based intravenous loading dose (2,400–3,000mg) plus a body-weight-based maintenance dose (3,000–3,600mg) on day 15, then once every 8 weeks | +++ | Not tried in trials |
| B cells depletants | |||
| Rituximab | 375 mg/m2 intravenous every week for the first 4 weeks, or 1,000 mg x 2 doses 2 weeks apart and then 1,000 mg 2 weeks apart every 6 months | +++ | Trial ongoing |
| Inebilizumab | 300 mg intravenous every 15 days x 2 doses, and then every 6 months | +++ | Limited data |
| IL-6 receptors inhibitors | |||
| Satralizumab | 120 mg subcutaneously every 4 weeks | +++ | Trial ongoing |
Note that: “−“ indicates rare (<5%), “+” infrequent (5–30%), “++” common (30–69%), “+++” very common/very high efficacy (>70%).
Alternatively, oral steroids bioequivalent (i.e., prednisone 1,250 mg) may be considered given its similar efficacy in patients with optic neuritis.163
Intravenous immunoglobulins may be sometimes administered instead of PLEX.
Duration may vary depending on steroid-sparing treatment making effect.
We focused here on level 1 evidence of efficacy, although biosimilars of rituximab, tocilizumab, mycophenolate, and azathioprine may be used and a trial on rozanolixizumab is ongoing in MOGAD.
Chronic attack-preventive treatment
Since AQP4-IgG positive AQP4+NMOSD patients at first clinical attack are at high risk of relapses in the first year (70%) and disability worsening is strongly associated with acute attacks, all newly diagnosed patients should undergo a chronic treatment aimed to prevent attacks.103 In contrast with AQP4+NMOSD, approximately 50% of MOGAD patients will ultimately have a relapsing disease course.12 Therefore, treatment is usually recommended only after the second clinical event in MOGAD patients, although exceptions may be made in those with a severe first attack with residual disability.
AQP4+NMOSD represents one of the few neurological disorders with tailored proven treatments available, where drugs target key elements of disease pathophysiology, namely (1) IL-6 (satralizumab), (2) B cells and their subsets (inebilizumab and rituximab), and (3) complement (eculizumab, ravulizumab). The very high efficacy of these biologic drugs was demonstrated in phase 3 clinical trials (level 1 evidence of efficacy), although only in AQP4+NMOSD. Biosimilars of rituximab are potential alternatives to reduce costs. In resource limited settings, azathioprine and mycophenolate mofetil have been used but their efficacy appears to be less,139 and they have drawbacks including need for at least 6 months of concomitant corticosteroids with its high side effect burden and risk of secondary lymphoproliferative disorders with long-term use.140,141 Tocilizumab is another off-label IL-6 blocking medications with some data supporting its use.142 In this review we will focus on treatments that have class 1 evidence. A summary of treatment efficacy and the main side effects during the trials and in the open label phase (if available) are provided below and in Table 5.
Complement inhibitors
Eculizumab:
This humanized monoclonal antibody inhibits C5 preventing the membrane attack complex formation. The phase-3 trial included only AQP4+NMOSD adult patients and the treatment showed a 94% relapse-risk reduction compared to the placebo arm,143 and similar results during the open-label extension.144 Although eculizumab was administered as add-on therapy in most patients, efficacy was confirmed in the subgroup of patients receiving eculizumab monotherapy145 and was not influenced by the main demographic/clinical variables, including age, sex, race, disease duration, annualized relapse rate, EDSS, prior treatment with rituximab, and additional autoimmune conditions.146
All patients received N. meningitidis vaccination prior to eculizumab administration to reduce the risk of capsulated bacteria infection, with no evidence of meningitis during the trial and its extension so far,144 although one case occurred in the initial phase 2 open label trial of this medication.147 Adverse effects were generally mild or moderate. Headache, upper respiratory tract infections, nasopharyngitis, and urinary tract infections were the most common.143,144 One single death from pulmonary empyema was registered.143,144
Ravulizumab:
Ravulizumab is a humanized monoclonal antibody similar to eculizumab that also targets C5. The main difference with eculizumab is that the complementary binding region and the neonatal fragment crystallizable region were modified in ravulizumab to lengthen its half-life. It was administered to AQP4-IgG seropositive AQP4+NMOSD patients in a phase-3 open label trial using the placebo arm of the PREVENT trial as the control group.148 No patients relapsed during the trial, leading to a 98.6% reduction in relapse-risk. In addition, investigators found a significant improvement of the ambulation index at study end.148 Similar to eculizumab, adverse events were generally mild to moderate and included COVID-19 infection, headache, back pain, arthralgia, and urinary tract infections. 148However, despite prior vaccination, two patients developed meningococcal meningitis, but recovered with treatment.148
B cell depletion
Rituximab:
Rituximab is a chimeric monoclonal antibody targeting the surface CD20 biomarker expressed on B cells. Its efficacy was proven in AQP4+NMOSD patients in a randomized placebo-controlled clinical trial (RIN-1). In this study, no treated patients relapsed although the total number of patients (19 in each study arm) was smaller than some of the other clinical trials. In addition, an improvement in the quantification of nerve and spinal cord impairment (QOSI) scale was observed. No deaths occurred. Infusion reactions, headache, nasopharyngitis, and upper respiratory tract infections were the most common adverse effects, confirming rituximab’s good safety profile.149
Inebilizumab:
This humanized monoclonal antibody targets surface CD19, which is a B-cell lineage biomarker with a wider expression than CD20, since it is also present on plasmablasts and, to a lesser extent, plasma cells. It was administered in monotherapy to both AQP4+NMOSD and AQP4-IgG seronegative NMOSD patients (18 patients), showing an overall relapse-risk reduction of 77% when in AQP4+NMOSD patients.150 Secondary endpoints such as the risk of EDSS worsening, radiological activity and hospitalization were also met.150,151 No significant benefit of treatment were observed among AQP4-IgG seronegative patients,150 although a post-hoc analysis suggested that treated patients had a lower annualized-relapse rate at study end than pre-enrollment.152
Post-hoc analyses also provided interesting insights into treatment response showing that patients with early and persisting B cell depletion (≤4 cells/ul at 6 months) were clinically stable for over 2 years.153
Adverse events mainly included urinary tract infections, arthralgias, and infusion-related reactions. In the open-label phase a decrease of immunoglobulins over time was observed, and further long-term analysis will be needed to determine its frequency and impact on risk of infections.154 Two deaths due to respiratory failure within a relapse, and an indeterminate neurological condition occurred.150
IL-6 receptor inhibitors
Satralizumab:
Satralizumab is a humanized monoclonal antibody inhibiting the IL-6 receptor. It was administered monotherapy or add-on in AQP4+NMOSD and AQP4-IgG seronegative NMOSD patients, demonstrating an overall relapse-risk reduction of 74–79% in AQP4+NMOSD patients.155,156 In contrast to the other medications, satralizumab administration is a subcutaneous injection, so can be given at home. The adverse effects included upper respiratory or urinary infections and injection-related reactions. Efficacy and safety results were confirmed in the open label extension of the trial, and no deaths were reported.157 Seronegative patients did not show a benefit, but larger cohort studies are needed.155,156
So far, no level 1 evidence of treatment efficacy are available in patients with MOGAD and empiric treatment decisions are based on retrospective analyses and expert opinion.158,159 Treatment currently administered in clinical practice include old immunosuppressants (azathioprine, mycophenolate mofetil), B cell depleting therapies (rituximab), IL-6 receptor inhibitors (tocilizumab), and IVIg.160 Among them, multicenter retrospective studies demonstrated that rituximab and intravenous immunoglobulins administration161 are effective maintenance treatments. However, rituximab seems less effective in MOGAD than AQP4-IgG positive AQP4+NMOSD, despite B cell depletion.162 Thus, IVIg or tocilizumab are often favored in clinical practice. Clinical trials evaluating the efficacy of rituximab (NCT05545384), satralizumab (NCT05271409) and rozanolixizumab (an inhibitor of the neonatal Fc receptor, NCT05063162) versus placebo are currently ongoing.
SUMMARY
The recent identification of AQP4+NMOSD and MOGAD as separate disorders has prompted the development of separate diagnostic criteria for MOGAD diagnosis. The stratification of patients based on the presence/absence of AQP4-IgG in NMOSD and stratification by antibody-titer in MOGAD represents the main differences between the two criteria. Using CBA in serum, AQP4-IgG and MOG-IgG are sensitive and highly specific but caution is needed with low positive MOG-IgG which is found in 1–2% of disease controls. In addition, specific MRI features of the diseases are described and included as supportive criteria in uncertain cases. NMOSD negative for AQP4-IgG and MOG-IgG likely represents a heterogenous group of disorders that should be a focus of research to potentially discover new antibodies that associate with demyelination. A number of highly effective attack-prevention treatments are now available for AQP4+NMOSD and should be started promptly at diagnosis. Empiric attack prevention treatments in MOGAD are generally reserved for those with two or more attacks. However, level 1 evidence is lacking in MOGAD. The approach to developing treatments in AQP4+NMOSD will be a useful template for MOGAD and randomized clinical trials are now underway with the hope for a proven attack-prevention treatment in the near future.
Key points:
The recognition of AQP4+NMOSD and MOGAD as distinct disorders led to separate diagnostic criteria for each.
AQP4-IgG is highly specific for NMOSD diagnosis at any titer. In contrast, caution is needed with low-titer MOG-IgG which can be encountered with other diseases.
Recognition of the MRI features of AQP4+NMOSD and MOGAD is helpful as there are important discriminators between each other and MS.
Maintenance treatment should be started after the 1st attack in AQP4+NMOSD but is generally not started until the 2nd attack in MOGAD given the latter can have a monophasic course in over half of cases.
Studies elucidating the pathophysiology of AQP4+NMOSD led to the development of proven targeted treatments in AQP4+NMOSD and similar analyses in MOGAD are underway in an attempt to develop attack-prevention treatments in that disease.
SYNOPSIS.
Aquaporin-4-IgG positive neuromyelitis optica spectrum disorder (AQP4+NMOSD) and myelin-oligodendrocyte glycoprotein antibody-associated disease (MOGAD) are antibody-associated diseases targeting astrocytes and oligodendrocytes, respectively. Their recognition as distinct entities has led to each having its own diagnostic criteria which require a combination of clinical, serologic, and MRI features. The therapeutic approach to acute attacks in AQP4+NMOSD and MOGAD is similar. There is now class 1 evidence to support attack-prevention medications for AQP4+NMOSD. MOGAD lacks proven treatments although clinical trials are now underway. In this review, we will outline similarities and differences between AQP4+NMOSD and MOGAD in terms of diagnosis and treatment.
CLINICS CARE POINTS.
AQP4+NMOSD and MOGAD are separate antibody-mediated CNS disorders with different antigenic targets.
Bilateral optic neuritis and extensive myelitis are common features of both AQP4+NMOSD and MOGAD; cerebral involvement can be encountered with both diseases, although ADEM favors MOGAD.
It is important to recognize the MRI features of MOGAD and AQP4+NMOSD as it helps to select those that should be tested for MOG-IgG and AQP4-IgG.
Caution is needed with low positive MOG-IgG as it can occur in 1–2% of other diseases.
Long-term attack-prevention treatment is required after the 1st attack in AQP4+NMOSD, but not MOGAD where it is typically reserved for those with 2 or more attacks.
In AQP4+NMOSD, level 1 evidence of efficacy is available for complement inhibitors (eculizumab, ravulizumab), B cell depleting agents (rituximab, inebilizumab), and IL-6 receptor inhibitors (satralizumab).
High level evidence of treatment efficacy is not available for MOGAD yet, but clinical trials are now underway.
Disclosure statement:
Laura Cacciaguerra received speaker and consultant honoraria from ACCMED, Roche, BMS Celgene, Sanofi and travel support for conferences by Merck Serono.
Eoin P. Flanagan was a site primary investigator in a randomized clinical trial on Inebilizumab in neuromyelitis optica spectrum disorder run by Medimmune/Viela-Bio/Horizon Therapeutics, has received funding from the NIH (R01NS113828), and is a member of the medical advisory board of the MOG project. Dr. Flanagan is an editorial board member of the Journal of the Neurological Sciences and Neuroimmunology Reports, and a patent has been submitted on DACH1-IgG as a biomarker of paraneoplastic autoimmunity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banwell B, Bennett JL, Marignier R, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023;22(3):268–282. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Connor KC, McLaughlin KA, De Jager PL, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13(2):211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunchok A, Chen JJ, Saadeh RS, et al. Application of 2015 Seronegative Neuromyelitis Optica Spectrum Disorder Diagnostic Criteria for Patients With Myelin Oligodendrocyte Glycoprotein IgG-Associated Disorders. JAMA Neurol. 2020;77(12):1572–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanagan EP, Cabre P, Weinshenker BG, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79(5):775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papp V, Magyari M, Aktas O, et al. Worldwide Incidence and Prevalence of Neuromyelitis Optica: A Systematic Review. Neurology. 2021;96(2):59–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Connell K, Hamilton-Shield A, Woodhall M, et al. Prevalence and incidence of neuromyelitis optica spectrum disorder, aquaporin-4 antibody-positive NMOSD and MOG antibody-positive disease in Oxfordshire, UK. J Neurol Neurosurg Psychiatry. 2020;91(10):1126–1128. [DOI] [PubMed] [Google Scholar]
- 9.de Mol CL, Wong Y, van Pelt ED, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler. 2020;26(7):806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128–3138. [DOI] [PubMed] [Google Scholar]
- 11.Ramanathan S, Mohammad S, Tantsis E, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobo-Calvo A, Ruiz A, Rollot F, et al. Clinical Features and Risk of Relapse in Children and Adults with Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. Ann Neurol. 2021;89(1):30–41. [DOI] [PubMed] [Google Scholar]
- 13.Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11(6):535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marignier R, Hacohen Y, Cobo-Calvo A, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20(9):762–772. [DOI] [PubMed] [Google Scholar]
- 15.Fujihara K, Bennett JL, de Seze J, et al. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2020;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol Neuroimmunol Neuroinflamm. 2016;3(3):e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mariotto S, Gajofatto A, Batzu L, et al. Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology. 2019;93(20):e1867–e1872. [DOI] [PubMed] [Google Scholar]
- 18.Pace S, Orrell M, Woodhall M, et al. Frequency of MOG-IgG in cerebrospinal fluid versus serum. J Neurol Neurosurg Psychiatry. 2022;93(3):334–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon YN, Kim B, Kim JS, et al. Myelin Oligodendrocyte Glycoprotein-Immunoglobulin G in the CSF: Clinical Implication of Testing and Association With Disability. Neurol Neuroimmunol Neuroinflamm. 2022;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carta S, Cobo Calvo A, Armangue T, et al. Significance of Myelin Oligodendrocyte Glycoprotein Antibodies in CSF: A Retrospective Multicenter Study. Neurology. 2023;100(11):e1095–e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125(Pt 7):1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139(5):875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020;143(5):1431–1446. [DOI] [PubMed] [Google Scholar]
- 24.Spadaro M, Winklmeier S, Beltran E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. 2018;84(2):315–328. [DOI] [PubMed] [Google Scholar]
- 25.Kohyama K, Nishida H, Kaneko K, Misu T, Nakashima I, Sakuma H. Complement-dependent cytotoxicity of human autoantibodies against myelin oligodendrocyte glycoprotein. Front Neurosci. 2023;17:1014071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yandamuri SS, Filipek B, Obaid AH, et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keller CW, Lopez JA, Wendel EM, et al. Complement Activation Is a Prominent Feature of MOGAD. Ann Neurol. 2021;90(6):976–982. [DOI] [PubMed] [Google Scholar]
- 28.Macrini C, Gerhards R, Winklmeier S, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. 2021;144(8):2375–2389. [DOI] [PubMed] [Google Scholar]
- 29.Mader S, Ho S, Wong HK, et al. Dissection of complement and Fc-receptor-mediated pathomechanisms of autoantibodies to myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci U S A. 2023;120(13):e2300648120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneko K, Sato DK, Nakashima I, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry. 2018;89(9):927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(9):1176–1180. [DOI] [PubMed] [Google Scholar]
- 32.Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107–1114. [DOI] [PubMed] [Google Scholar]
- 33.Ramanathan S, Prelog K, Barnes EH, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. 2016;22(4):470–482. [DOI] [PubMed] [Google Scholar]
- 34.Chen JJ, Bhatti MT. Clinical phenotype, radiological features, and treatment of myelin oligodendrocyte glycoprotein-immunoglobulin G (MOG-IgG) optic neuritis. Curr Opin Neurol. 2020;33(1):47–54. [DOI] [PubMed] [Google Scholar]
- 35.Jitprapaikulsan J, Chen JJ, Flanagan EP, et al. Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein Autoantibody Status Predict Outcome of Recurrent Optic Neuritis. Ophthalmology. 2018;125(10):1628–1637. [DOI] [PubMed] [Google Scholar]
- 36.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin Oligodendrocyte Glycoprotein Antibody-Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol. 2018;195:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology. 2006;67(2):258–262. [DOI] [PubMed] [Google Scholar]
- 38.Mariano R, Messina S, Kumar K, Kuker W, Leite MI, Palace J. Comparison of Clinical Outcomes of Transverse Myelitis Among Adults With Myelin Oligodendrocyte Glycoprotein Antibody vs Aquaporin-4 Antibody Disease. JAMA Netw Open. 2019;2(10):e1912732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubey D, Pittock SJ, Krecke KN, et al. Clinical, Radiologic, and Prognostic Features of Myelitis Associated With Myelin Oligodendrocyte Glycoprotein Autoantibody. JAMA Neurol. 2019;76(3):301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elsone L, Goh YY, Trafford R, Mutch K, Jacob A. HOW OFTEN DOES RESPIRATORY FAILURE OCCUR IN NEUROMYELITIS OPTICA? Journal of Neurology, Neurosurgery & Psychiatry. 2013;84(11):e2–e2.25346970 [Google Scholar]
- 41.Zhao-Fleming HH, Valencia Sanchez C, Sechi E, et al. CNS Demyelinating Attacks Requiring Ventilatory Support With Myelin Oligodendrocyte Glycoprotein or Aquaporin-4 Antibodies. Neurology. 2021;97(13):e1351–e1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elsone L, Townsend T, Mutch K, et al. Neuropathic pruritus (itch) in neuromyelitis optica. Mult Scler. 2013;19(4):475–479. [DOI] [PubMed] [Google Scholar]
- 43.Usmani N, Bedi G, Lam BL, Sheremata WA. Association between paroxysmal tonic spasms and neuromyelitis optica. Arch Neurol. 2012;69(1):121–124. [DOI] [PubMed] [Google Scholar]
- 44.Banks SA, Morris PP, Chen JJ, et al. Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubey D, Pittock SJ, Krecke KN, Flanagan EP. Association of Extension of Cervical Cord Lesion and Area Postrema Syndrome With Neuromyelitis Optica Spectrum Disorder. JAMA Neurol. 2017;74(3):359–361. [DOI] [PubMed] [Google Scholar]
- 46.Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84(11):1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Etemadifar M, Nouri H, Khorvash R, Salari M, Ghafari K, Aghababaee A. Frequency of diencephalic syndrome in NMOSD. Acta Neurol Belg. 2022;122(4):961–967. [DOI] [PubMed] [Google Scholar]
- 48.Magana SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology. 2009;72(8):712–717. [DOI] [PubMed] [Google Scholar]
- 49.McKeon A, Lennon VA, Lotze T, et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008;71(2):93–100. [DOI] [PubMed] [Google Scholar]
- 50.Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler. 2013;19(10):1261–1267. [DOI] [PubMed] [Google Scholar]
- 51.Bartels F, Baumgartner B, Aigner A, et al. Impaired Brain Growth in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Acute Disseminated Encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. 2023;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahn CD, Miles BS, MacGregor DL, Blaser SI, Banwell BL, Hetherington CR. Neurocognitive outcome after acute disseminated encephalomyelitis. Pediatr Neurol. 2003;29(2):117–123. [DOI] [PubMed] [Google Scholar]
- 53.Kuni BJ, Banwell BL, Till C. Cognitive and behavioral outcomes in individuals with a history of acute disseminated encephalomyelitis (ADEM). Dev Neuropsychol. 2012;37(8):682–696. [DOI] [PubMed] [Google Scholar]
- 54.Valencia-Sanchez C, Guo Y, Krecke KN, et al. Cerebral Cortical Encephalitis in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. Ann Neurol. 2023;93(2):297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogawa R, Nakashima I, Takahashi T, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. 2017;4(2):e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Budhram A, Sechi E, Nguyen A, Lopez-Chiriboga AS, Flanagan EP. FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis With Seizures (FLAMES): Is immunotherapy always needed to put out the fire? Mult Scler Relat Disord. 2020;44:102283. [DOI] [PubMed] [Google Scholar]
- 57.Tatekawa H, Sakamoto S, Hori M, et al. Imaging Differences between Neuromyelitis Optica Spectrum Disorders and Multiple Sclerosis: A Multi-Institutional Study in Japan. AJNR Am J Neuroradiol. 2018;39(7):1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tajfirouz D, Padungkiatsagul T, Beres S, et al. Optic chiasm involvement in AQP-4 antibody-positive NMO and MOG antibody-associated disorder. Mult Scler. 2022;28(1):149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. 2018;15(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shah SS, Morris P, Buciuc M, et al. Frequency of Asymptomatic Optic Nerve Enhancement in a Large Retrospective Cohort of Patients With Aquaporin-4+ NMOSD. Neurology. 2022;99(8):e851–e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pacheco JM, Tajfirouz D, Madhavan A, et al. Asymptomatic Optic Nerve Enhancement in Myelin Oligodendrocyte Glycoprotein Associated Disease. Investigative Ophthalmology & Visual Science. 2022;63(7):1215 – A0215–1215 – A0215. [Google Scholar]
- 62.Srikajon J, Siritho S, Ngamsombat C, Prayoonwiwat N, Chirapapaisan N, Siriraj Neuroimmunology Research G. Differences in clinical features between optic neuritis in neuromyelitis optica spectrum disorders and in multiple sclerosis. Mult Scler J Exp Transl Clin. 2018;4(3):2055217318791196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. 2014;71(3):276–283. [DOI] [PubMed] [Google Scholar]
- 64.Asnafi S, Morris PP, Sechi E, et al. The frequency of longitudinally extensive transverse myelitis in MS: A population-based study. Mult Scler Relat Disord. 2020;37:101487. [DOI] [PubMed] [Google Scholar]
- 65.Fadda G, Alves CA, O’Mahony J, et al. Comparison of Spinal Cord Magnetic Resonance Imaging Features Among Children With Acquired Demyelinating Syndromes. JAMA Netw Open. 2021;4(10):e2128871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cacciaguerra L, Sechi E, Rocca MA, Filippi M, Pittock SJ, Flanagan EP. Neuroimaging features in inflammatory myelopathies: A review. Front Neurol. 2022;13:993645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asgari N, Flanagan EP, Fujihara K, et al. Disruption of the leptomeningeal blood barrier in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2017;4(4):e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sechi E, Krecke KN, Pittock SJ, et al. Frequency and characteristics of MRI-negative myelitis associated with MOG autoantibodies. Mult Scler. 2021;27(2):303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cacciaguerra L, Valsasina P, Mesaros S, et al. Spinal Cord Atrophy in Neuromyelitis Optica Spectrum Disorders Is Spatially Related to Cord Lesions and Disability. Radiology. 2020;297(1):154–163. [DOI] [PubMed] [Google Scholar]
- 70.Webb LM, Cacciaguerra L, Krecke KN, et al. Marked central canal T2-hyperintensity in MOGAD myelitis and comparison to NMOSD and MS. J Neurol Sci. 2023;450:120687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hyun JW, Lee HL, Park J, et al. Brighter spotty lesions on spinal MRI help differentiate AQP4 antibody-positive NMOSD from MOGAD. Mult Scler. 2022;28(6):989–992. [DOI] [PubMed] [Google Scholar]
- 72.Hyun JW, Kim SH, Jeong IH, Lee SH, Kim HJ. Bright spotty lesions on the spinal cord: an additional MRI indicator of neuromyelitis optica spectrum disorder? J Neurol Neurosurg Psychiatry. 2015;86(11):1280–1282. [DOI] [PubMed] [Google Scholar]
- 73.Yonezu T, Ito S, Mori M, et al. “Bright spotty lesions” on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult Scler. 2014;20(3):331–337. [DOI] [PubMed] [Google Scholar]
- 74.Chien C, Scheel M, Schmitz-Hubsch T, et al. Spinal cord lesions and atrophy in NMOSD with AQP4-IgG and MOG-IgG associated autoimmunity. Mult Scler. 2019;25(14):1926–1936. [DOI] [PubMed] [Google Scholar]
- 75.Cacciaguerra L, Meani A, Mesaros S, et al. Brain and cord imaging features in neuromyelitis optica spectrum disorders. Ann Neurol. 2019;85(3):371–384. [DOI] [PubMed] [Google Scholar]
- 76.Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63(7):964–968. [DOI] [PubMed] [Google Scholar]
- 77.Chia NH, Redenbaugh V, Chen JJ, Pittock SJ, Flanagan EP. Corpus callosum involvement in MOG antibody-associated disease in comparison to AQP4-IgG-seropositive neuromyelitis optica spectrum disorder and multiple sclerosis. Mult Scler. 2023;29(6):748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elsbernd P, Cacciaguerra L, Krecke KN, et al. Cerebral enhancement in MOG antibody-associated disease. J Neurol Neurosurg Psychiatry. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai MT, Zhang YX, Zheng Y, Fang W, Ding MP. Callosal lesions on magnetic resonance imaging with multiple sclerosis, neuromyelitis optica spectrum disorder and acute disseminated encephalomyelitis. Mult Scler Relat Disord. 2019;32:41–45. [DOI] [PubMed] [Google Scholar]
- 80.Mastrangelo V, Asioli GM, Foschi M, et al. Bilateral extensive corticospinal tract lesions in MOG antibody-associated disease. Neurology. 2020;95(14):648–649. [DOI] [PubMed] [Google Scholar]
- 81.Cacciaguerra L, Morris P, Tobin WO, et al. Tumefactive Demyelination in MOG Ab-Associated Disease, Multiple Sclerosis, and AQP-4-IgG-Positive Neuromyelitis Optica Spectrum Disorder. Neurology. 2023;100(13):e1418–e1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cobo-Calvo A, Ruiz A, Maillart E, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology. 2018;90(21):e1858–e1869. [DOI] [PubMed] [Google Scholar]
- 83.Chen C, Liu C, Fang L, et al. Different magnetic resonance imaging features between MOG antibody- and AQP4 antibody-mediated disease: A Chinese cohort study. J Neurol Sci. 2019;405:116430. [DOI] [PubMed] [Google Scholar]
- 84.Jurynczyk M, Geraldes R, Probert F, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. 2017;140(3):617–627. [DOI] [PubMed] [Google Scholar]
- 85.Lopez-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG Serostatus With Relapse After Acute Disseminated Encephalomyelitis and Proposed Diagnostic Criteria for MOG-IgG-Associated Disorders. JAMA Neurol. 2018;75(11):1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wattjes MP, Ciccarelli O, Reich DS, et al. 2021 MAGNIMS-CMSC-NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. 2021;20(8):653–670. [DOI] [PubMed] [Google Scholar]
- 87.Nelson F, Poonawalla A, Hou P, Wolinsky JS, Narayana PA. 3D MPRAGE improves classification of cortical lesions in multiple sclerosis. Mult Scler. 2008;14(9):1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Calabrese M, Oh MS, Favaretto A, et al. No MRI evidence of cortical lesions in neuromyelitis optica. Neurology. 2012;79(16):1671–1676. [DOI] [PubMed] [Google Scholar]
- 89.Sinnecker T, Dorr J, Pfueller CF, et al. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology. 2012;79(7):708–714. [DOI] [PubMed] [Google Scholar]
- 90.Tahara M, Ito R, Tanaka K, Tanaka M. Cortical and leptomeningeal involvement in three cases of neuromyelitis optica. Eur J Neurol. 2012;19(5):e47–48. [DOI] [PubMed] [Google Scholar]
- 91.Kim W, Lee JE, Kim SH, et al. Cerebral Cortex Involvement in Neuromyelitis Optica Spectrum Disorder. J Clin Neurol. 2016;12(2):188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Redenbaugh V, Chia NH, Cacciaguerra L, et al. Comparison of MRI T2-lesion evolution in pediatric MOGAD, NMOSD, and MS. Mult Scler. 2023:13524585231166834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sechi E, Krecke KN, Messina SA, et al. Comparison of MRI Lesion Evolution in Different Central Nervous System Demyelinating Disorders. Neurology. 2021;97(11):e1097–e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cacciaguerra L, Redenbaugh V, Chen JJ, et al. Timing and Predictors of T2-Lesion Resolution in Patients With Myelin-Oligodendrocyte-Glycoprotein-Antibody-Associated Disease. Neurology. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Redenbaugh V, Montalvo M, Sechi E, et al. Diagnostic value of aquaporin-4-IgG live cell based assay in neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. 2021;7(4):20552173211052656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tackley G, Kuker W, Palace J. Magnetic resonance imaging in neuromyelitis optica. Mult Scler. 2014;20(9):1153–1164. [DOI] [PubMed] [Google Scholar]
- 97.Sormani MP, Bruzzi P. MRI lesions as a surrogate for relapses in multiple sclerosis: a meta-analysis of randomised trials. Lancet Neurol. 2013;12(7):669–676. [DOI] [PubMed] [Google Scholar]
- 98.Paolilo RB, Rimkus CM, da Paz JA, Apostolos-Pereira SL, Callegaro D, Sato DK. Asymptomatic MRI lesions in pediatric-onset AQP4-IgG positive NMOSD. Mult Scler Relat Disord. 2022;68:104215. [DOI] [PubMed] [Google Scholar]
- 99.Lee MY, Yong KP, Hyun JW, Kim SH, Lee SH, Kim HJ. Incidence of interattack asymptomatic brain lesions in NMO spectrum disorder. Neurology. 2020;95(23):e3124–e3128. [DOI] [PubMed] [Google Scholar]
- 100.Camera V, Holm-Mercer L, Ali AAH, et al. Frequency of New Silent MRI Lesions in Myelin Oligodendrocyte Glycoprotein Antibody Disease and Aquaporin-4 Antibody Neuromyelitis Optica Spectrum Disorder. JAMA Netw Open. 2021;4(12):e2137833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.S BS-M, Chen JJ, Morris P, et al. Frequency of New or Enlarging Lesions on MRI Outside of Clinical Attacks in Patients With MOG-Antibody-Associated Disease. Neurology. 2022;99(18):795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fadda G, Banwell B, Waters P, et al. Silent New Brain MRI Lesions in Children with MOG-Antibody Associated Disease. Ann Neurol. 2021;89(2):408–413. [DOI] [PubMed] [Google Scholar]
- 103.Wingerchuk DM, Lucchinetti CF. Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2022;387(7):631–639. [DOI] [PubMed] [Google Scholar]
- 104.Waters P, Reindl M, Saiz A, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2016;87(9):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prain K, Woodhall M, Vincent A, et al. AQP4 Antibody Assay Sensitivity Comparison in the Era of the 2015 Diagnostic Criteria for NMOSD. Front Neurol. 2019;10:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78(9):665–671; discussion 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reindl M, Schanda K, Woodhall M, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. 2020;7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Majed M, Sanchez CV, Bennett JL, et al. Alterations in aquaporin-4-IgG serostatus in 986 patients: a laboratory-based longitudinal analysis. Ann Neurol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berger T, Rubner P, Schautzer F, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349(2):139–145. [DOI] [PubMed] [Google Scholar]
- 111.Kuhle J, Pohl C, Mehling M, et al. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N Engl J Med. 2007;356(4):371–378. [DOI] [PubMed] [Google Scholar]
- 112.Lampasona V, Franciotta D, Furlan R, et al. Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology. 2004;62(11):2092–2094. [DOI] [PubMed] [Google Scholar]
- 113.Chan A, Decard BF, Franke C, et al. Serum antibodies to conformational and linear epitopes of myelin oligodendrocyte glycoprotein are not elevated in the preclinical phase of multiple sclerosis. Mult Scler. 2010;16(10):1189–1192. [DOI] [PubMed] [Google Scholar]
- 114.Ketelslegers IA, Van Pelt DE, Bryde S, et al. Anti-MOG antibodies plead against MS diagnosis in an Acquired Demyelinating Syndromes cohort. Mult Scler. 2015;21(12):1513–1520. [DOI] [PubMed] [Google Scholar]
- 115.Sechi E, Buciuc M, Pittock SJ, et al. Positive Predictive Value of Myelin Oligodendrocyte Glycoprotein Autoantibody Testing. JAMA Neurol. 2021;78(6):741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Held F, Kalluri SR, Berthele A, Klein AK, Reindl M, Hemmer B. Frequency of myelin oligodendrocyte glycoprotein antibodies in a large cohort of neurological patients. Mult Scler J Exp Transl Clin. 2021;7(2):20552173211022767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gaudioso CM, Mar S, Casper TC, et al. MOG and AQP4 Antibodies among Children with Multiple Sclerosis and Controls. Ann Neurol. 2023;93(2):271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akaishi T, Takahashi T, Misu T, et al. Difference in the Source of Anti-AQP4-IgG and Anti-MOG-IgG Antibodies in CSF in Patients With Neuromyelitis Optica Spectrum Disorder. Neurology. 2021;97(1):e1–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Matsumoto Y, Kaneko K, Takahashi T, et al. Diagnostic implications of MOG-IgG detection in sera and cerebrospinal fluids. Brain. 2023. [DOI] [PubMed] [Google Scholar]
- 120.Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. 2016;13(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jarius S, Lechner C, Wendel EM, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 2: Results from 108 lumbar punctures in 80 pediatric patients. J Neuroinflammation. 2020;17(1):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jarius S, Pellkofer H, Siebert N, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation. 2020;17(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jarius S, Paul F, Franciotta D, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. 2011;306(1–2):82–90. [DOI] [PubMed] [Google Scholar]
- 124.Sechi E, Buciuc M, Flanagan EP, et al. Variability of cerebrospinal fluid findings by attack phenotype in myelin oligodendrocyte glycoprotein-IgG-associated disorder. Mult Scler Relat Disord. 2021;47:102638. [DOI] [PubMed] [Google Scholar]
- 125.Dobson R, Ramagopalan S, Davis A, Giovannoni G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: a meta-analysis of prevalence, prognosis and effect of latitude. J Neurol Neurosurg Psychiatry. 2013;84(8):909–914. [DOI] [PubMed] [Google Scholar]
- 126.McKeon A, Lennon VA, Jacob A, et al. Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve. 2009;39(1):87–90. [DOI] [PubMed] [Google Scholar]
- 127.Kunchok A, Flanagan EP, Snyder M, et al. Coexisting systemic and organ-specific autoimmunity in MOG-IgG1-associated disorders versus AQP4-IgG+ NMOSD. Mult Scler. 2021;27(4):630–635. [DOI] [PubMed] [Google Scholar]
- 128.Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology. 2012;78(20):1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibodies in Adult and Pediatric Patients. JAMA Neurol. 2020;77(2):257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82(6):474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carnero Contentti E, Lopez PA, Pettinicchi JP, et al. What percentage of AQP4-ab-negative NMOSD patients are MOG-ab positive? A study from the Argentinean multiple sclerosis registry (RelevarEM). Mult Scler Relat Disord. 2021;49:102742. [DOI] [PubMed] [Google Scholar]
- 132.Stiebel-Kalish H, Hellmann MA, Mimouni M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm. 2019;6(4):e572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206–216. [DOI] [PubMed] [Google Scholar]
- 134.Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346–351. [DOI] [PubMed] [Google Scholar]
- 135.Kleiter I, Gahlen A, Borisow N, et al. Apheresis therapies for NMOSD attacks: A retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm. 2018;5(6):e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Costello F Neuromyelitis Optica Spectrum Disorders. Continuum (Minneap Minn). 2022;28(4):1131–1170. [DOI] [PubMed] [Google Scholar]
- 137.Nosadini M, Eyre M, Giacomini T, et al. Early Immunotherapy and Longer Corticosteroid Treatment Are Associated With Lower Risk of Relapsing Disease Course in Pediatric MOGAD. Neurol Neuroimmunol Neuroinflamm. 2023;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huda S, Whittam D, Jackson R, et al. Predictors of relapse in MOG antibody associated disease: a cohort study. BMJ Open. 2021;11(11):e055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang W, Wang L, Xia J, et al. Efficacy and safety of azathioprine, mycophenolate mofetil, and reduced dose of rituximab in neuromyelitis optica spectrum disorder. Eur J Neurol. 2022;29(8):2343–2354. [DOI] [PubMed] [Google Scholar]
- 140.O’Neill BP, Vernino S, Dogan A, Giannini C. EBV-associated lymphoproliferative disorder of CNS associated with the use of mycophenolate mofetil. Neuro Oncol. 2007;9(3):364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kandiel A, Fraser AG, Korelitz BI, Brensinger C, Lewis JD. Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut. 2005;54(8):1121–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang C, Zhang M, Qiu W, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020;19(5):391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2019;381(7):614–625. [DOI] [PubMed] [Google Scholar]
- 144.Wingerchuk DM, Fujihara K, Palace J, et al. Long-Term Safety and Efficacy of Eculizumab in Aquaporin-4 IgG-Positive NMOSD. Ann Neurol. 2021;89(6):1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pittock SJ, Fujihara K, Palace J, et al. Eculizumab monotherapy for NMOSD: Data from PREVENT and its open-label extension. Mult Scler. 2022;28(3):480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Palace J, Wingerchuk DM, Fujihara K, et al. Benefits of eculizumab in AQP4+ neuromyelitis optica spectrum disorder: Subgroup analyses of the randomized controlled phase 3 PREVENT trial. Mult Scler Relat Disord. 2021;47:102641. [DOI] [PubMed] [Google Scholar]
- 147.Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12(6):554–562. [DOI] [PubMed] [Google Scholar]
- 148.Pittock SJ, Barnett M, Bennett JL, et al. Ravulizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. Ann Neurol. 2023. [DOI] [PubMed] [Google Scholar]
- 149.Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19(4):298–306. [DOI] [PubMed] [Google Scholar]
- 150.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352–1363. [DOI] [PubMed] [Google Scholar]
- 151.Marignier R, Bennett JL, Kim HJ, et al. Disability Outcomes in the N-MOmentum Trial of Inebilizumab in Neuromyelitis Optica Spectrum Disorder. Neurol Neuroimmunol Neuroinflamm. 2021;8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marignier R, Pittock SJ, Paul F, et al. AQP4-IgG-seronegative patient outcomes in the N-MOmentum trial of inebilizumab in neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. 2022;57:103356. [DOI] [PubMed] [Google Scholar]
- 153.Bennett JL, Aktas O, Rees WA, et al. Association between B-cell depletion and attack risk in neuromyelitis optica spectrum disorder: An exploratory analysis from N-MOmentum, a double-blind, randomised, placebo-controlled, multicentre phase 2/3 trial. EBioMedicine. 2022;86:104321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rensel M, Zabeti A, Mealy MA, et al. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: Analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for ⩾4 years in the N-MOmentum trial. Mult Scler. 2022;28(6):925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yamamura T, Kleiter I, Fujihara K, et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2019;381(22):2114–2124. [DOI] [PubMed] [Google Scholar]
- 156.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yamamura T, Weinshenker B, Yeaman MR, et al. Long-term safety of satralizumab in neuromyelitis optica spectrum disorder (NMOSD) from SAkuraSky and SAkuraStar. Mult Scler Relat Disord. 2022;66:104025. [DOI] [PubMed] [Google Scholar]
- 158.Thakolwiboon S, Zhao-Fleming H, Karukote A, Mao-Draayer Y, Flanagan EP, Avila M. Meta-analysis of effectiveness of steroid-sparing attack prevention in MOG-IgG-associated disorder. Mult Scler Relat Disord. 2021;56:103310. [DOI] [PubMed] [Google Scholar]
- 159.Spagni G, Sun B, Monte G, et al. Efficacy and safety of rituximab in myelin oligodendrocyte glycoprotein antibody-associated disorders compared with neuromyelitis optica spectrum disorder: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2023;94(1):62–69. [DOI] [PubMed] [Google Scholar]
- 160.Sechi E, Cacciaguerra L, Chen JJ, et al. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front Neurol. 2022;13:885218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen JJ, Huda S, Hacohen Y, et al. Association of Maintenance Intravenous Immunoglobulin With Prevention of Relapse in Adult Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol. 2022;79(5):518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Durozard P, Rico A, Boutiere C, et al. Comparison of the Response to Rituximab between Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibody Diseases. Ann Neurol. 2020;87(2):256–266. [DOI] [PubMed] [Google Scholar]
- 163.Morrow SA, Fraser JA, Day C, et al. Effect of Treating Acute Optic Neuritis With Bioequivalent Oral vs Intravenous Corticosteroids: A Randomized Clinical Trial. JAMA Neurol. 2018;75(6):690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]