

Abstract
Currently, there are no effective therapies for intestinal and hepatic fibrosis representing a considerable unmet need. Breakthroughs in pathogenesis have accelerated the development of anti‐fibrotic therapeutics in recent years. Particularly, with the development of nanotechnology, the harsh environment of the gastrointestinal tract and inaccessible microenvironment of fibrotic lesions seem to be no longer considered a great barrier to the use of anti‐fibrotic drugs. In this review, we comprehensively summarize recent preclinical and clinical studies on intestinal and hepatic fibrosis. It is found that the targets for preclinical studies on intestinal fibrosis is varied, which could be divided into molecular, cellular, and tissues level, although little clinical trials are ongoing. Liver fibrosis clinical trials have focused on improving metabolic disorders, preventing the activation and proliferation of hepatic stellate cells, promoting the degradation of collagen, and reducing inflammation and cell death. At the preclinical stage, the therapeutic strategies have focused on drug targets and delivery systems. At last, promising remedies to the current challenges are based on multi‐modal synergistic and targeted delivery therapies through mesenchymal stem cells, nanotechnology, and gut‐liver axis providing useful insights into anti‐fibrotic strategies for clinical use.
Keywords: anti‐fibrotic strategies, challenges and prospects, hepatic fibrosis, intestinal fibrosis, nanotechnology
Translational Impact Statement.
The review describes the pathological mechanisms underlying intestinal and hepatic fibrosis, systematically summarizing and discussing anti‐fibrotic targets and the corresponding anti‐fibrotic strategies from the view of pathological mechanisms based on molecular, cellular, and tissue levels. The challenges and opportunities for developing anti‐fibrotic therapeutics are highlighted and discussed in detail. According to this, the potential opportunities and breakthroughs of anti‐fibrotic therapeutic methods are discussed, providing an incentive for clinical translation.
1. INTRODUCTION
Fibrosis is characterized by the excessive activation, proliferation, and differentiation of fibroblasts, the deposition of large amounts of extracellular matrix (ECM), and the destruction of tissue structure which can impair the function of almost any organ. 1 Fibrosis has long been considered an irreversible disease, however, because of the progress in pathology studies and preventative therapeutic strategies, the reversal of fibrosis has come into focus in recent years. 2 , 3 The intestine and liver, are physiologically interconnected via the gut‐liver axis. The quality of life and even survival would declined for individuals following a diagnosis of intestinal or liver fibrosis. Although there are currently no effective therapies for intestinal or hepatic fibrosis, some therapeutic agents are currently being used in clinical trials for hepatic fibrosis at ClinicalTrials.gov. (Table 1). As the mechanisms underlying primary fibrosis are similar in different organs and include chronic inflammation and immune imbalance leading to the overactivation and proliferation of ECM‐producing cells, inspiration can be obtained from the latest anti‐fibrosis research. 4 For example, Rurik et al. developed messenger RNA (mRNA)‐loaded lipid nanoparticles targeted to T cells in vivo to generate transient anti‐fibrotic chimeric antigen receptors, which were found to reduce fibrosis and restore cardiac function after injury. 5 Long et al. developed a nanodecoy that mimicked fibroblasts using poly lactic‐co‐glycolic acid (PLGA) nanoparticles modified by an autologous skin fibroblast membrane. The nanodecoy carries a variety of cytokine receptors on its surface, which bind the corresponding fibrogenic cytokines. 6 Another study used the peptide library carried by the phage to adhere to and isolate myofibroblasts in vivo. After amplification and screening in vitro, the polypeptide sequences targeting myofibroblasts were screened from cells. The targeted peptides enabled the precise delivery of sorafenib for anti‐renal fibrosis. 7 Besides, several studies found a similarity between inflammatory bowel diseases (IBDs) and non‐alcoholic fatty liver disease (NAFLD), major causes of intestinal fibrosis and liver fibrosis respectively, in epidemiology across geographic areas over time, suggesting some close association between intestinal fibrosis and liver fibrosis. 8 And this kind of association also indicated a therapeutic strategy on intestinal and hepatic fibrosis, which is discussed in Section 5.3. Here, this review will describe the pathological mechanisms underlying intestinal and hepatic fibrosis, systematically summarizing and discussing anti‐fibrotic targets and the corresponding anti‐fibrotic strategies from the view of pathological mechanisms based on molecular, cellular, and tissue levels. The challenges and opportunities for developing anti‐fibrotic therapeutics are highlighted and discussed. Lastly, the potential opportunities and breakthroughs of anti‐fibrotic therapeutic methods are discussed, providing an incentive for clinical translation.
TABLE 1.
Examples of compounds in clinical phases II or III of development aimed at reducing hepatic fibrosis.
| Target | Therapeutics | Modality | Phase | Primary outcome | NCT identifier |
|---|---|---|---|---|---|
| Dual agonist of GLP‐1 and GCGR | Cotadutide | Biomacromolecules | IIb/III | No results posted. | NCT05364931 |
| Dual agonist of PPARα and PPARδ | Elafibranor | Small‐molecule | III | The primary endpoint of NASH remission for patients with considerable liver fibrosis was not achieved when treated with Elafibranor at 120 mg/d for 72 weeks. | NCT02704403 |
| FGF21 analogues |
Pegbelfermin BMS‐986036 |
Biomacromolecules | II | Pegbelfermin was generally well tolerated in the advanced NASH population. However, it did not meet the primary endpoint (≥1 stage). Improvement in the NASH‐CRN fibrosis score was assessed via biopsy. | NCT03486912 |
| SCD1 inhibitor | Aramchol | Small‐molecule | III | A histological improvement in fibrosis (≥1 stage) was demonstrated for 39% of the patients according to NASH‐CRN. | NCT02279524 |
| THR‐ β agonist | Resmetirom MGL‐3196 | Small‐molecule | III | Resmetirom achieved both primary and critical secondary endpoints in phase III clinical trials (MAESTRO‐NASH) with good tolerance. The NAFLD activity score decreased by more than two points and was markedly higher than that in the placebo group. | NCT03900429 |
| PNPLA3 gene inhibitor | AZD2693 | Oligonucleotides | II | No results posted. | NCT05809934 |
| CBP/β‐catenin inhibitor | PRI‐724 | Small‐molecule | I/II | One out of every three patients who received the recommended PRI‐724 suffered a serious adverse reaction possibly related to PRI‐724. In addition, hepatic fibrosis was not decreased with any statistical significance after 12 weeks. | NCT03620474 |
| HSC proliferation inhibitor | Hydronidone | Small‐molecule | II | There was a markedly significant improvement in liver fibrosis scores in the hydronidone group with good tolerance compared to the placebo group at week 52. In addition, the best improvement in the Ishak score was with the 90 mg/tid (270 mg/day) dosage. | NCT02499562 |
| HSP47 mRNA | BMS‐986263 | siRNA‐loaded LNP | II | BMS‐986263 was well tolerated after 12 weeks and showed improvement in METAVIR and Ishak scores in patients with chronic hepatitis C‐related fibrosis progression. | NCT03420768 |
| LOXL2 inhibitor | Simtuzumab | Antibody | II | There was no therapeutic effect in decreasing hepatic collagen content or hepatic venous pressure gradient in phase IIb trials for patients with bridging fibrosis or compensated cirrhosis associated with nonalcoholic steatohepatitis. | |
| Cyclophilin B inhibitor | Rencofilstat | Small‐molecule | II | Rencofilstat achieved the primary endpoint in ALTITUDE‐NASH markedly improving liver function in patients with advanced NASH. In addition, Rencofilstat achieved all secondary endpoints. | NCT05402371 |
| CCR2 and CCR5 inhibitor | Cenicriviroc | Small‐molecule | II | Cenicriviroc was well tolerated, and significantly improved fibrosis without worsening NASH after 1 year of treatment compared with the placebo. | NCT02217475 |
| Pan‐caspase inhibitor | Emricasan IDN‐6556 | Small‐molecule | II | There was no improvement in liver histology in patients with NASH fibrosis. Although treatment with Emricasan lowered serum ALT in the short‐term, it may have led to alternative mechanisms of cell death. | NCT02138253 |
Abbreviations: CCR2, C‐C chemokine receptor 2; CCR5, C‐C chemokine receptor 5; FGF21, fibroblast growth factor 21; GCGR, glucagon receptor; GLP1, glucagon‐like peptide‐1; HSC, hepatic stellate cell; HSP47, heat shock protein 47; LNP, lipid nanoparticle; LOXL2, lysyl oxidase‐like 2; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PNPLA3, patatin‐like phospholipase domain containing 3; SCD1, stearoyl‐coA desaturase; THR, thyroid hormone receptors.
2. INTESTINAL FIBROSIS
Intestinal fibrosis is a complication following chronic intestinal inflammation leading to the thickening of the gut wall and lumen strictures and which ultimately results in functional loss. 9 , 10 IBD, especially Crohn's disease (CD), involves chronic inflammation of the gastrointestinal (GI) tract and is the main cause of intestinal fibrosis. 11
2.1. Pathogenesis of intestinal fibrogenesis
Repeated intestinal injury activates innate and adaptive immune responses because of chronic inflammation. 12 The excessive immune response generates pro‐inflammatory and pro‐fibrotic molecules triggering the recruitment, proliferation, differentiation, and activation of ECM‐producing cells, which are central to the pathogenesis of fibrosis 10 (Figure 1a). ECM‐producing cells include myofibroblasts, smooth muscle cells, and pericytes, but myofibroblasts are the primary ECM‐producing cells. In addition to fibroblasts, epithelial and endothelial cells are important sources of myofibroblasts. Epithelial‐to‐mesenchymal transition (EMT) is a pathological process of cellular transdifferentiation in which epithelial cells acquire mesenchymal features, a fibroblast‐like profile, and show downregulated epithelial and upregulated mesenchymal markers. 13 Endothelial‐to‐mesenchymal transition (EndMT) is also a pathological process of cellular transdifferentiation, however, in this case, the endothelium acquires mesenchymal features, a fibroblast‐like profile, and shows downregulated endothelial markers and upregulated mesenchymal markers. 10 Both EMT and EndMT promote the development of intestinal fibrosis. 14
FIGURE 1.
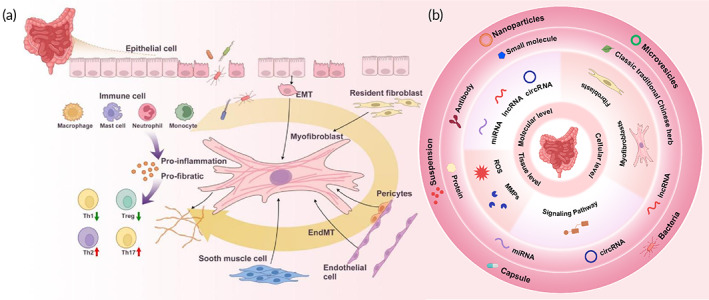
(a) Schematic illustration of the pathogenesis of intestinal fibrogenesis. As a result of chronic inflammation, the excessive immune response generates pro‐inflammatory and pro‐fibrotic molecules triggering the recruitment, proliferation, differentiation, and activation of ECM‐producing cells, which are central to the pathogenesis of fibrosis. The pro‐inflammatory and pro‐fibrotic factors released from the activated fibroblasts and immune cells produce a feedback loop, leading to an inflammation‐facilitated fibrosis process. (b) Anti‐fibrotic targets at different levels are summarized. Based on the understanding of the mechanisms underlying intestinal fibrogenesis, anti‐fibrotic targets at molecular, cellular, and tissue levels are summarized, and the corresponding therapeutic strategies are discussed.
2.2. Conventional therapeutic approaches
Since intestinal fibrosis is often associated with inflammation, traditional anti‐fibrotic therapeutic approaches depend on anti‐inflammatory agents. However, anti‐inflammatory therapies have demonstrated limited therapeutic effects on fibrosis. Corticosteroids may exert an indirect anti‐fibrotic effect beneficial for some fibrotic conditions by decreasing collagen synthesis and slowing the wound healing process. 15 However, patients with fibrotic strictures often require surgery, and corticosteroids are associated with an increased risk of postoperative complications. 16 Small‐molecule immunosuppressants may improve pulmonary function and reduce pulmonary fibrosis in patients with systemic sclerosis indicating that this class of drugs may prevent, or at least delay, intestinal fibrosis progression. 17 However, there is no evidence to support the use of small‐molecule immunosuppressants, often used in the treatment of IBD, as an anti‐fibrotic drug in intestinal fibrosis. 18 Studies investigating anti‐tumor necrosis factor α (TNF‐α) antibody effects on fibrosis have produced inconsistent results. 19 , 20 It is generally accepted that anti‐TNF‐α antibodies have no obvious therapeutic effect on intestinal fibrosis.
2.3. Targets and new therapeutic strategies
Although no drugs have been approved for the clinical treatment of intestinal fibrosis, an increasing number of studies are investigating therapeutic strategies. With the new drug delivery systems, the harsh GI tract environment is no longer a barrier for anti‐fibrotic drugs, particularly those developed from nucleic acid and biological macromolecules. 21 Based on the current understanding of intestinal fibrogenesis, anti‐fibrotic targets, and the corresponding therapeutic strategies can be summarized at the molecular, cellular, and tissue levels (Figure 1b).
2.3.1. Molecular level
Biomolecules, such as nucleic acids and proteins, are the basic functional units in cells that carry out important biological functions. Some aberrant events occur during intestinal fibrosis at the molecular level. 22 In the following sub‐sections, these molecular level changes are summarized along with their corresponding therapeutics (Table 2).
TABLE 2.
Targets and new therapeutic strategies for intestinal fibrosis.
| Targets | MoA | Therapeutics | Modality | Delivery system | References | |
|---|---|---|---|---|---|---|
| Molecular level | Noncoding RNAs | Reduction of abnormal expression of colonic fibronectin collagen I protein | MiR‐200b | RNA | Microvesicles | 29 |
| Signaling pathway | Inhibition of TGF‐β1 pathway | Vivomixx® | Multi‐strain probiotic | Capsule | 36 | |
| Inhibition of TGF‐β1 pathway | ACE‐I | Small‐molecule | PEG‐suspension | 37 | ||
| Inhibition of Wnt/β‐catenin signaling | ICG‐001 | Small‐molecule | N | 39 | ||
| Inhibition of HIF pathway | BAH | Small‐molecule | N | 41 | ||
| Inhibition of AXL pathway | BGB324 | Small‐molecule | N | 42 | ||
| Protein level | Competitive binding with IL‐36 receptors | Antibodies against IL36R | Antibody | N | 44 | |
| Induction of high tittered antibodies to IL‐12 and IL‐23 | p40 peptide based vaccine | Peptide | N | 45 | ||
| Reduction of IL‐13 and TGF‐β levels | pXYCYT:Hsp65 | Bacteria | N | 46 | ||
| Cellular level | Myofibroblasts | Suppression on activation of myofibroblasts | Triptolide | Small‐molecule | N | 48 |
| Disturbance of myofibroblast function | GED‐0507‐34 Levo | Small‐molecule | N | 49 | ||
| Inhibition of myofibroblast accumulation and autophagy potentiation | AMA0825 | Small‐molecule | N | 50 | ||
| Inhibition of human colonic myofibroblast proliferation was reduced | Ang (1–7); Captopril | Small‐molecule | N | 51 | ||
| Fibroblasts | Inhibition of fibroblasts proliferation | Pirfenidone | Small‐molecule | N | 53 | |
| Inhibition of accelerated fibroblast migration | Vitamin D | Small‐molecule | N | 54 | ||
| Inhibition of fibroblast proliferation and activation |
Wu‐Mei‐Wan |
A traditional Chinese herb | N | 55 | ||
| Tissue level | Neutralization of excessive MMP‐9 | Anti‐MMP‐9 antibody | Antibody | N | 59 | |
| Regulation of TIMP/MMP balance | Thalidomide | Small‐molecule | N | 60 | ||
| ECM homeostasis reconstitution | Anti‐FAP antibody | Antibody | N | 61 | ||
| Reduction in ROS levels | D‐CeO2 nanozyme | Small‐molecule | Dextran‐coated nanoparticle | 63 |
Abbreviations: ACE‐I, angiotensin converting enzyme inhibitors; FAP, fibroblast activating protein; MMP, matrix metalloproteinase; MoA, mechanism of action; TIMP, tissue inhibitors of metalloproteinases.
Noncoding RNAs as anti‐fibrotic targets
Several studies have shown that noncoding RNAs (ncRNAs) present various modulatory functions in fibrogenesis. ncRNAs, such as microRNAs (miRNAs), long‐noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), modulate fibrosis, and influence protein expression. 23 The application of delivery systems has increased the feasibility of ncRNA manipulation in vivo making ncRNAs feasible therapeutic targets for various diseases. 24 , 25 miRNAs can be knocked down or complemented through the administration of antisense oligonucleotides or the delivery of miRNA mimics. 26 , 27 In IBD, various miRNAs are upregulated or downregulated at the intestinal fibrotic site compared to their expression in healthy tissues supporting the use of miRNA‐based therapeutics for intestinal fibrosis. The most widely known examples of miRNAs are the miRNA‐29 and miRNA‐200 families which are downregulated in the stricture mucosa of patients with CD. 28 Increasing miRNA levels in the mucosa of lesions could be beneficial in intestinal fibrosis. Jia et al. developed miR‐200b‐loaded microvesicles (miR‐200b‐MVs) shown to protect miRNAs from degradation, transfer miRNAs to target cells, and attenuate colitis and intestinal fibrosis. Compared to a PBS (phosphate buffered saline)‐treated control, miR‐200b‐MVs reduced the expression of colonic fibronectin and collagen I by 28.08% and 51.65%, respectively, in rats with intestinal fibrosis. 29 lncRNAs are long‐chain (>200 nucleotides) ncRNAs that play important roles in gene transcription, mRNA processing, and translation. 30 The lncRNA HOX transcript antisense RNA (HOTAIR) has been shown to play a significant role in intestinal fibrosis through transforming growth factor (TGF‐β) signaling and EndMT modulation. 31 CircRNA, a single‐stranded covalently closed circular RNA, is another subclass of ncRNAs. The functions of circRNAs are similar to those of lncRNAs and include modulating gene transcription and mRNA translation. 32 It has been reported that Hsa_circRNA_102610 promotes TGF‐β1‐induced EMT by sponging miR‐130a‐3p in fibrosis. 33 lncRNA and circRNA may also be prospective targets for intestinal anti‐fibrotic therapy, although this scenario has yet to be investigated. However, it is necessary to distinguish whether changes in ncRNA levels are causative or consequential. The same ncRNAs often modulate the expression of multiple gene targets creating the potential for off‐target effects. 34 , 35 When considering ncRNAs as a target for intestinal anti‐fibrotic treatment, the therapeutic agent, which is often also an ncRNA, must be able to enter the cell, and sometimes the nucleus, at sufficient concentrations which makes drug delivery a significant challenge.
Signaling pathways as anti‐fibrotic targets
Various signaling pathways participate in intestinal fibrosis development and several anti‐fibrotic targets have been identified. Vivomixx®, a mixed probiotic formulation, was found to prevent a fibrotic phenotype based on cellular and molecular parameters through inhibition of the TGF‐β1/Smad pathways (Figure 2a). 36 Zhang et al. developed a chitosan‐modified PLGA nanoparticle for co‐delivering patchouli alcohol and simvastatin to alleviate colonic fibrosis by preventing the TGF‐β/Smad2/3 pathway. This nanoparticle effectively alleviated colonic fibrosis in a DSS‐induced intestinal fibrosis model. 37 TGF‐β activates various pro‐fibrotic pathways in the intestine, however, it also possesses anti‐inflammatory activity and can regulate immunity through inducing Treg/Th17 cell differentiation, inhibiting dendritic cell proliferation, and promoting the anti‐inflammatory transformation of macrophages. Therefore, the complete elimination of TGF‐β is not required. Wnt/β‐catenin signaling was also demonstrated to be involved in the fibrotic process. 38 Lewis et al. found that ICG‐001, an inhibitor of Wnt, could restrain fibrogenesis in CCD‐18Co cells and emphasized that cross‐talk between the Wnt and TGF‐β signaling pathways was involved in the pathogenesis of fibrogenesis 39 making ICG‐001 a potential anti‐fibrotic therapeutic agent of multiple targets. It has been reported that hypoxic conditions are characteristic of IBD‐related fibrosis. 40 Accordingly, betulinic acid hydroxamate (BAH), an inhibitor of hypoxia‐inducing factor (HIF) prolyl‐hydroxylases, was developed as a novel anti‐fibrotic therapy for IBD. 41 BAH dose‐dependently activated the HIF pathway in both the 2,4,6‐trinitrobenzene sulfonic acid (TNBS)‐ and DSS‐induced fibrosis models. Continuous administration of BAH for 17 days significantly downregulated the expression of fibrotic markers, including collagen I, tissue inhibitors of metalloproteinases (TIMP‐1), and α‐smooth muscle actin (α‐SMA) (Figures 2b and 3c). Additionally, the AXL pathway was suggested to promote myofibroblast activation. Bemcentinib (BGB324), a small‐molecule inhibitor of the AXL pathway, was found to block the fibrotic process and prevent matrix‐stiffness in human intestinal organoids promoting myofibroblast apoptosis 42 suggesting inhibition of the AXL pathway as a candidate for anti‐intestinal fibrosis.
FIGURE 2.
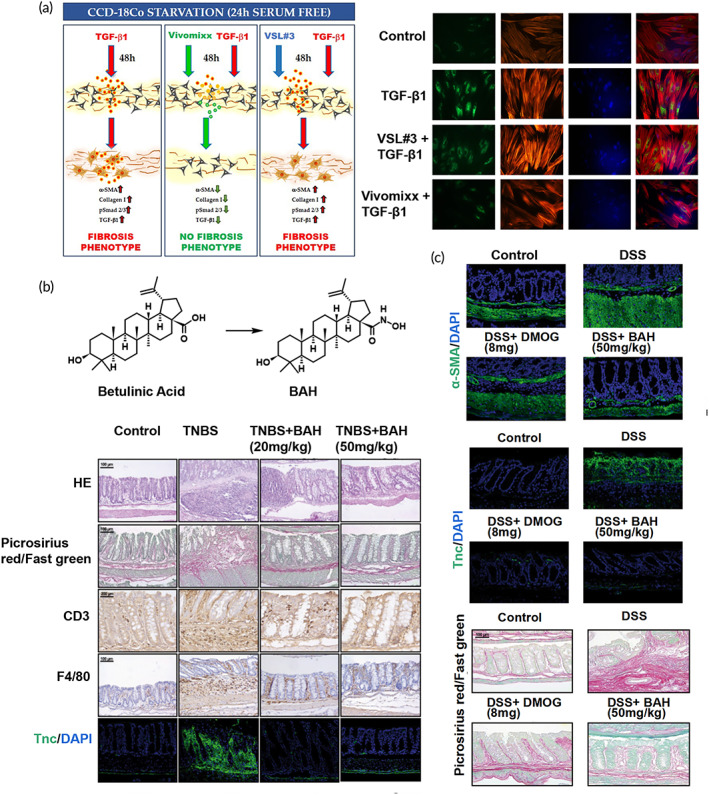
Strategies for anti‐intestinal fibrogenesis. (a) The lysates of multi‐strain probiotic formulations improve intestinal fibrosis in CCD‐18Co cells by inhibiting the TGF‐β1 pathway. Reproduced with permission from Reference 36, copyright 2021, Multidisciplinary Digital Publishing Institute. (b) and (c) Chemical structures of betulinic acid and betulinic acid hydroxamate (BAH); BAH attenuates fibrosis in the TNBS‐ and DSS‐induced fibrosis model. Reproduced with permission from Reference 41, copyright 2021, Springer Nature.
FIGURE 3.
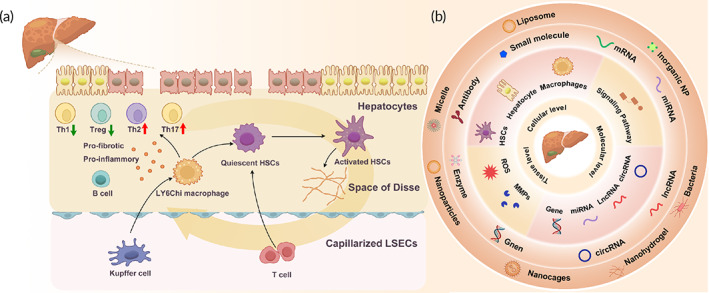
(a) Schematic illustration of the pathogenesis of hepatic fibrogenesis. In chronic liver disease, damaged hepatocytes, Kupffer cells, and macrophages release chemokines and cytokines to activate hepatic stellate cells (HSCs), which produce excessive extracellular matrix. The activated HSCs aggravate hepatocyte injuries and induce capillarization and accumulation of liver sinusoidal endothelial cells. The proinflammatory and pro‐fibrotic factors released from the HSCs and immune cells create a positive feedback loop. (b) Summary of anti‐fibrotic targets at different biological scales. Anti‐fibrotic targets at molecular, cellular, and tissue levels and their corresponding therapeutic strategies are summarized based on the understanding of the mechanisms underlying intestinal fibrogenesis.
Ligands and receptors as anti‐fibrotic targets
Some ligands produce pro‐fibrotic effects through cell membrane receptors that could be considered anti‐fibrotic targets. Concerning the important roles of interleukins (ILs) in activating and regulating immunity, some studies have found that ILs participate in intestinal fibrosis and many researchers are committed to developing anti‐fibrotic strategies against these targets. As a target for anti‐intestinal fibrosis, IL‐36 is the most widely studied. 43 Scheibe et al. developed anti‐IL‐36 antibodies that bind competitively to IL‐36R and inhibit its downstream biological functions. Furthermore, they found that neutralizing antibodies against IL‐36R could reduce fibrosis and inflammation in the DSS‐ or TNBS‐induced fibrosis models suggesting that the inhibition of IL‐36 could be a strategy for anti‐intestinal fibrosis. 44 Guan et al. showed that administration of a p40 peptide vaccine generated neutralizing antibodies against IL‐12 and IL‐23 producing an immunomodulatory effect by modifying the Treg/Th1 and Treg/Th17 ratios in the lamina propria to reduce TNBS‐induced intestinal fibrosis in mice. 45 Furthermore, the level of the pro‐fibrotic cytokine IL‐13 could be regulated by the gut microbiota. Cunha et al. showed that oral administration of Lactococcus lactis NCDO2118 FnBPA+ (pXYCYT:Hsp65) reduced the levels of IL‐13 and attenuated the degree of fibrosis in mice with TNBS‐induced colitis. 46
2.3.2. Cellular level
Fibroblasts have long been recognized as an important source of myofibroblasts which are considered the chief effector cell in fibrogenesis. 47 Considering the important roles of these cells in the occurrence and development of intestinal fibrosis, myofibroblast‐ and fibroblast‐based anti‐fibrotic strategies have been widely investigated (Table 2).
Myofibroblasts as anti‐fibrotic targets
Since activated myofibroblasts are the main ECM‐producing cells and represent the central mediators of intestinal fibrosis, strategies that prevent myofibroblast activation, block their function, or induce their death may decrease the production of ECM and improve fibrosis. Triptolide exerts anti‐inflammatory and immunomodulatory effects, however, its anti‐fibrotic effects remain unclear. In one study, a rat model of colonic fibrosis, induced by the administration of TNBS for 6 weeks, was treated with triptolide every day. PBS solution was used as the control. Triptolide inhibited the activation and suppressed the differentiation of myofibroblasts in rats with colonic fibrosis resulting in decreased ECM deposition in the colon. 48 Speca et al. explored the anti‐fibrogenic efficiency of PPAR‐γ modulator agonists in the intestine and found that GED‐0507‐34 Levo disturbed myofibroblast function and suppressed fibrosis‐associated protein synthesis, suggesting that PPAR‐γ agonists may be a potential therapeutic drug candidate for intestinal fibrosis. 49 Rho kinases (ROCKs) are highly activated in myofibroblasts. A study showed that prophylactic local administration of a ROCK inhibitor (AMA0825) prevented fibrogenesis in DSS‐ and adoptive T‐cell transfer‐induced fibrosis models. Therapeutic administration of AMA0825 reversed intestinal DSS‐induced fibrosis, likely because ROCK inhibition inhibited myofibroblast accumulation and potentiated autophagy. 50 An increasing number of studies demonstrate that the renin‐angiotensin system (RAS) participates in regulating inflammation, fibrosis, and cell proliferation. In a study to assess the effects of RAS on intestinal fibrosis, the level of human colonic myofibroblast proliferation and collagen secretion was determined after treatment with angiotensin (Ang) II or Ang (1–7) solution. 51 The results revealed that collagen secretion and the proliferation of human colonic myofibroblasts were reduced by Ang (1–7) and captopril demonstrating that therapeutic agents targeting RAS are very likely to improve fibrosis.
Fibroblasts as anti‐fibrotic targets
Strategies preventing fibroblast activation, accumulation, and transition into myofibroblasts would be beneficial for fibrosis treatment. 52 In a study, pirfenidone (PFD) solution was added to fibroblasts isolated from biopsies taken from patients with CD. PFD inhibited the proliferation of the fibroblasts in the inflamed colonic mucosa suggesting that PFD may be applicable in intestinal fibrosis treatment. 53 Gisbert‐Ferrándiz et al. found that vitamin D (VD) receptor expression decreased while fibroblast migration increased in intestinal stenosis tissue from patients with CD compared to healthy tissue. Following treatment with a VD solution, VD receptor levels increased preventing the accumulation of fibroblasts and the subsequent development of intestinal fibrosis induced by heterotopic transplant in mice, suggesting beneficial VD effects on intestinal fibrosis. 54 In addition, Wu et al. found that the administration of Wu–Mei–Wan (WMW), a classic traditional Chinese herb medicine, could inhibit fibroblast proliferation and activation by decreasing ECM deposition and inhibiting TNBS‐induced intestinal fibrosis mice model compared to a placebo. 55
2.3.3. Tissue level
The tissue microenvironment has attracted increasing attention because it has unique features that are different from normal physiological conditions. 56 , 57 The microenvironment of intestinal fibrosis exhibits special characteristics which could be used to develop anti‐fibrotic therapies (Table 2). Matrix metalloproteinases (MMPs) are a group of proteases that degrade the ECM. Tissue inhibitors of metalloproteinase (TIMPs) are the primary endogenous MMP inhibitors. In normal intestinal tissue, the expression of MMPs and TIMPs is balanced, but in fibrosis this balance is disturbed contributing to collagen deposition. MMP‐9 is an MMP that can degrade ECM including gelatin and collagen. 58 Goffin et al. found that the levels of the MMP‐9 degradation products were high in the serum of patients with CD and evaluated the effects of anti‐MMP‐9 antibodies in an intestinal fibrosis model. Anti‐MMP‐9 antibodies were found to neutralize excessive MMP‐9 reducing collagen deposition, suggesting that anti‐MMP‐9 antibodies may be an effective method for dampening fibrosis. 59 Thalidomide, an immunomodulator, also has been demonstrated to inhibit intestinal fibrosis by regulating the TIMP/MMP protein balance and degradation of ECM. 60 Truffi et al. further explored the differences between fibrotic and nonfibrotic mucosa in patients with CD and found that fibroblast activating protein (FAP) was upregulated in myofibroblasts from fibrotic mucosa compared to that from nonstenotic mucosa. Isolated bowel tissue cultures were treated with an anti‐FAP antibody revealing that the ECM in CD strictures was remodeled by TIMP‐1 expression inhibition and that reduced collagen production improved intestinal fibrosis. 61 FAP expression is restricted to fibrotic areas and is not expressed in non‐strictured tissue making it suitable for identifying targets unique to pathological myofibroblasts. By targeting FAP on myofibroblasts, toxic drugs could be delivered to myofibroblasts, inducing their death and decreasing ECM production, without affecting normal intestinal tissues presenting new strategies for intestinal fibrosis treatment.
Metabolites such as the reactive oxygen species (ROS) have been demonstrated to play an important role in aggravating fibrosis. Evidence indicates that ROS activates and promotes TGF‐β signaling, in turn, TGF‐β1 boosts ROS production and decreases its elimination, which promotes fibrogenesis. 62 Cao et al. developed a dextran‐coated cerium oxide (D‐CeO2) nanozyme by chemical precipitation which has potent ROS scavenging abilities to alleviate intestinal fibrosis. The D‐CeO2 nanozyme efficiently scavenged ROS and regulated the fibrosis‐related microenvironment in TNBS‐ and DSS‐induced intestinal fibrosis mice models, providing a practical use for nanozymes with ROS scavenging capabilities in anti‐fibrotic strategies. 63
3. HEPATIC FIBROSIS
Hepatic fibrosis is characterized by excessive accumulation of the ECM and hepatocellular necrosis, followed by cirrhosis and hepatic failure which leads to approximately one million deaths annually. 1 The pathogenesis of hepatic fibrosis includes viral infections, drug‐induced toxicity, metabolic disorders, cholestasis, and autoimmune disorders. 64 Although there is currently no cure for hepatic fibrosis, some drugs have been effective at reducing symptoms, particularly those combined with nanoparticle delivery systems.
3.1. Pathogenesis of hepatic fibrogenesis
The effect of hepatic fibrosis during chronic liver disease involves inflammatory cytokines released by inflammatory cells which promotes the activation and proliferation of hepatic stellate cells (HSCs). HSCs are the primary abnormal ECM‐producing cells in the liver. Other ECM‐producing cells include fibroblasts and mesenchymal cells. 65 HSCs, which account for 5%–8% of liver non‐parenchymal cells, are located in the subendothelial space between hepatocytes and sinusoidal endothelial cells, and are responsible for the storage of vitamin A and lipid droplets under healthy conditions. 66 Upon liver injury, the HSCs are transformed from a quiescent condition to a myofibroblast‐like phenotype in two stages. First, chronic liver damage induces oxidative stress, resulting in the loss of lipid droplets from HSCs and their activation. Second, HSCs continuously release fibrogenic cytokines, and along with inflammatory cells, establish a pro‐fibrogenic environment at the tissue level (Figure 3a). 67
3.2. Conventional therapeutic approaches
Therapeutic approaches for hepatic fibrosis include etiological and anti‐fibrotic treatments. Conventional treatment is aimed at treating the primary disease, such as antiviral drugs in viral hepatitis, anti‐inflammatory drugs in autoimmune liver disease, and antiparasitic drugs in schistosomiasis. Although the etiological treatment of liver fibrosis is fundamental, it does not prevent its progression and presents a limited therapeutic effect. Some studies have demonstrated that interferon‐γ (IFN‐γ) and IL‐10 possess anti‐hepatic fibrotic effects in vitro. However, the effects in vivo are limited. 68 Chinese herbal medicines, such as Fuzheng huayu capsules, have also been used to treat anti‐hepatic fibrosis. 69 , 70 It is necessary to develop new therapeutic strategies with good efficacy and few side effects to treat hepatic fibrosis.
3.3. Targets and new therapeutic strategies
An increasing number of studies are investigating therapies for hepatic fibrosis, particularly combined with nano‐delivery technology. The application of the nanoparticle system is a unique advantage for liver disease since approximately 30%–99% of unmodified nanoscale particles preferentially accumulate in the liver following intravenous (IV) administration. 71 Moreover, some receptors are overexpressed during the process of hepatic fibrosis. In the following sections, anti‐fibrotic targets are summarized at different levels and the corresponding therapeutic strategies are discussed (Figure 3b).
3.3.1. Molecular level
Aberrant molecular events occur in genome transcription, translation, and protein metabolism in the occurrence and development of hepatic fibrosis. 72 The extensive knowledge of these differential pathogenic molecular events has led to the identification of several promising therapeutic targets (Table 3).
TABLE 3.
Targets and new therapeutic strategies for hepatic fibrosis.
| Targets | MoA | Therapeutics | Modality | Delivery systems | References | |
|---|---|---|---|---|---|---|
| Molecular level | Gene | Silenced the HMGB1 gene | HMGB1‐siRNA | siRNA | NAL nanoparticles | 73 |
| Regulation of pro‐fibrotic genes in HSCs | miRNA‐29b, miRNA‐122 | miRNA | Micelle | 74 | ||
| Noncoding RNAs | Knockdown of miR‐221‐3p | TuD‐miR‐221‐3p | miRNA | AAV8 | 76 | |
| Dysregulated miR‐34a | Pterostilbene | Small‐molecule | N | 78 | ||
| Increased circRNA SCAR expression located in the mitochondria | CircRNA SCAR expression vectors | pcD‐ciR | Mitochondria‐targeting NPs | 84 | ||
| Signaling pathway | Inhibited TGF‐β1/Smad3 signaling | Umbelliferone | Small‐molecule | N | 85 | |
| Inhibited hedgehog signaling | Vismodegib | Small‐molecule | cRGDyK‐liposome | 87 | ||
| Inhibited TGF‐β signaling | TGF‐βR blocker | Protein | Lactococcus lactis | 88 | ||
| Cellular level | Hepatocyte | Hepatocyte protection | Betulinic acid | Small‐molecule | Chitosan NPs | 93 |
| Improved fibrotic primary hepatocyte functions | HNF4A mRNA | mRNA | Lipid NPs | 94 | ||
| HSCs | Inhibited HSC activation | Imatinib | Small‐molecule | Liposomes | 96 | |
| HSC cytotoxicity activated | GMO, miR‐29b | Small‐molecule, miRNA | PEG‐PLGA NPs | 97 | ||
| Inhibited HSC proliferation and activation | Quercetin | Small‐molecule | Nanocages | 98 | ||
| Transformed aHSCs to quiescent phenotype | Relaxin‐plasmid, miR‐30a‐5p mimic | Gene, miRNA | LPH NPs | 99 | ||
| Destroyed the Golgi structure and downregulated collagen I production | Retinoic acid, DOX | Small‐molecule | Micelles | 100 | ||
| Broke down the dense collagen stroma and inhibited aHSC | Collagenase, silibinin | Enzyme, small‐molecule | NPs | 101 | ||
| Thermal ablation | Gold | Inorganic material | Nanorods | 102 | ||
| Hepatic immune cells | Macrophage repolarized toward an anti‐fibrotic M1 phenotype | CSF‐1R siRNA | siRNA | Nanohydrogel | 104 | |
| Modulated M2 macrophages | TSG‐6 | Cytokine | CaP@BSA NPs | 105 | ||
| Tissue level | Reduced ROS levels | PD‐MC | Material | Micelle | 107 | |
| Reduced MMP‐9 mRNA expression | MMP‐9 siRNA | siRNA | Vitamin A‐liposome | 108 |
Abbreviations: GMO, Germacrone; HMGB1, human high mobility group box‐1; HNF4A, hepatocyte nuclear factor 4 alpha; MMP, matrix metalloproteinase; MoA, mechanism of action.
Genes as anti‐fibrotic targets
Genes are the most basic structures supporting life, storing all information about the process of life. Gene therapies have great potential in hepatic anti‐fibrotic treatment. The human high mobility group box‐1 (HMGB1) protein is a fibroblast chemokine that promotes hepatic inflammation and fibrosis. Zhang et al. developed HMGB1‐siRNA‐loaded lipid nanoparticles for the targeted silencing of the HMGB1 gene in HSCs. The accumulation of lipid nanoparticles was markedly higher in liver tissue after modification with a pPB peptide. The activation and proliferation of HSCs was inhibited by the nanoparticles in vitro. Furthermore, this approach decreased collagen secretion in the liver and greatly increased the survival time in the TAA‐ and carbon tetrachloride (CCl4)‐induced hepatic fibrosis mouse model (Figure 4a,b). 73 In another study, a cationic micelle assembled from a vitamin A (VA)‐conjugated copolymer was developed for the targeted delivery of miRNA‐29b and miRNA‐122 to HSCs. The results showed that the expression of fibrosis‐related genes was significantly downregulated by miRNA‐29b and miRNA‐122, improved hepatic function, and decreased the degree of fibrosis (Figure 4c). 74
FIGURE 4.
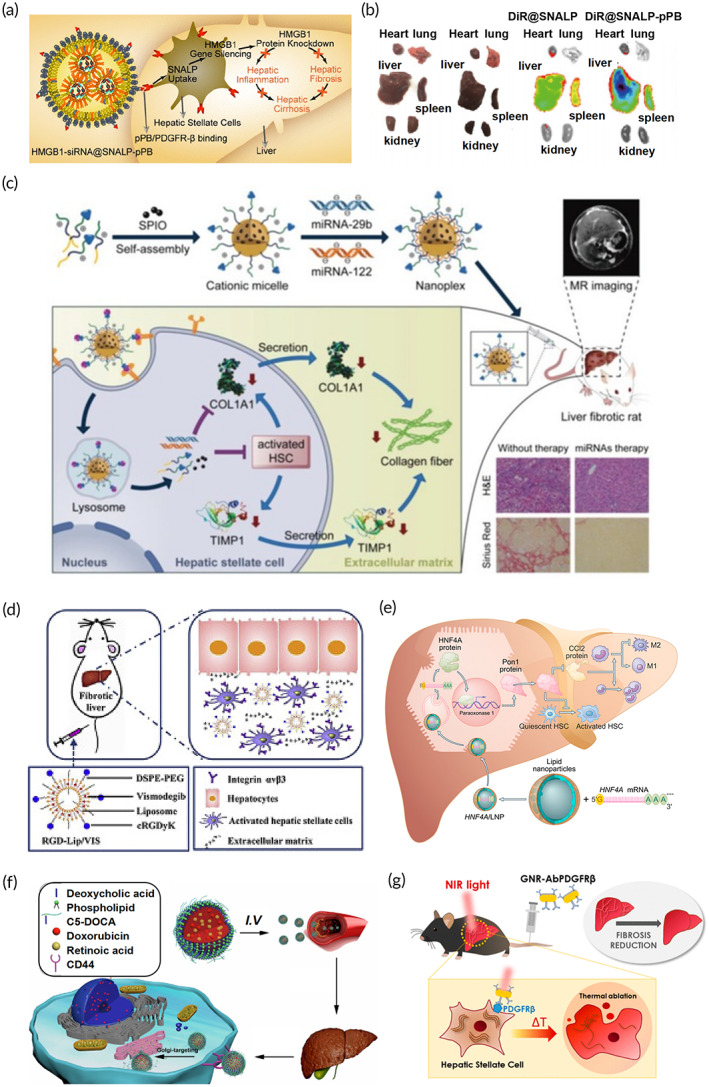
Strategies for anti‐hepatic fibrogenesis. (a) Human high mobility group box‐1‐siRNA@SNALP‐pPB effectively alleviate liver fibrosis. (b) The modification of pPB increases the accumulation of SNALP in the liver. Reproduced with permission from Reference 73, copyright 2020, American Chemical Society. (c) Schematic illustration of the miRNA loaded vitamin A‐modification of superparamagnetic iron oxide alleviating liver fibrosis. Reproduced with permission from Reference 74, copyright 2019, Wiley. (d) The Vismodegib loaded RGDLip/VIS reduced hepatic fibrosis by inhibiting hedgehog signaling in bile duct ligation and thioacetamide mice models. Reproduced with permission from Reference 87, copyright 2019, Elsevier. (e) Nanoparticles designed to deliver mRNA for recovery of hepatocytes. Reproduced with permission from Reference 94, copyright 2021, Elsevier. (f) The assembly of doxorubicin‐retinoic acid‐chondroitin sulfate micelles targeting the Golgi apparatus by N‐acetylgalactosaminyltransferase decoration. Reproduced with permission from Reference 100, copyright 2019, American Chemical Society. (g) The schematic illustration of gold nanorods‐PDGFRβ‐mediated photothermal therapy decreases carbon tetrachloride induced hepatic fibrosis in a mice model. Reproduced with permission from Reference 102, copyright 2021, American Chemical Society.
Noncoding RNAs as anti‐fibrotic targets
It has been reported that miRNA‐101 family members weaken TGF‐β signaling and miRNA‐223 inhibited fibrotic progression by targeting Cxcl10 and Taz in hepatocytes and macrophages. 75 Researchers have found that miRNA‐221‐3p promoted liver fibrosis in the hepatocytes of mouse models, subsequently developing into AAV TuD‐miR‐221‐3p to downregulate its expression. The expression level of miR‐221‐3p decreased significantly in the AAV TUD‐miR‐221‐3p‐treated group compared to that in the control group. Additionally, fewer fibrotic areas and injuries in the liver were observed in mouse models of fibrosis. 76 Dysregulation of miR‐34a has also been shown to be highly correlated with liver fibrosis. 77 Pterostilbene has been demonstrated to downregulate miR‐34a expression, attenuating hepatocyte EMT, which may improve liver fibrosis. 78 These results support the regulatory role of miRNAs in hepatic fibrosis. Moreover, some studies have shown that lncRNAs, such as H19, growth arrest‐specific transcript 5, Gm5091, and HIF 1 alpha‐antisense RNA 1 (HIF1A‐AS1) may inhibit the progression of liver fibrosis. 79 , 80 Thus, external supplementation of these lncRNAs provides a potential method to treat liver fibrosis. On the contrary, some lncRNAs have been demonstrated to promote liver fibrosis, including nuclear paraspeckle assembly transcript 1 (NEAT1), HOTAIR, and liver‐enriched fibrosis‐associated lncRNA1 (lnc‐LFAR1). 81 , 82 , 83 The downregulation of overexpressed lncRNAs in patients could improve fibrosis. circRNA was also investigated in the treatment of liver fibrosis. Zhao et al. found that mitochondrial ROS production was inhibited by the mitochondrial steatohepatitis‐associated circRNA ATP5B regulator (SCAR), which prevented fibroblast activation. When circRNA SCAR expression vectors were constructed and encapsulated into mitochondria‐targeting nanoparticles, the nanoparticles alleviated the cirrhosis associated with a high‐fat diet in vivo, suggesting circRNAs as a novel target in the process of fibrosis in nonalcoholic steatohepatitis (NASH), and supporting the potential for circRNA as an anti‐fibrotic target. 84 The use of ncRNAs as targets in liver fibrosis is challenging, but also promising.
Signaling pathways as anti‐fibrotic targets
Various fibrotic signaling pathways have been highlighted as important anti‐fibrotic targets in hepatic fibrosis. Umbelliferone (UMB), also known as 7‐hydroxycoumarin, demonstrated efficacy in the prevention of hepatic fibrogenesis in CCl4‐induced liver fibrosis by inhibiting TGF‐β1/Smad3 signaling and downregulating α‐SMA and collagen I expression. 85 Letrozole has been demonstrated to ameliorate liver fibrosis by inhibiting the downstream pathway of CTGF. 86 Vismodegib was loaded onto cRGDyK‐modified liposomes to inhibit the activation of HSCs by the inhibition of the hedgehog pathway. The cRGDyK‐modified liposomes showed affinity to αvβ3 on aHSC in vitro and in vivo, increased accumulation on aHSCs rather than on quiescent HSCs, and inhibited the progression of fibrosis in bile duct ligatonin thioacetamide mice models (Figure 4d). 87 Bacteria have also been used as a “delivery tool” to produce therapeutic proteins. Yuan et al. constructed a recombinant L. lactis expressing the TGF‐βR extracellular domain bound to the TGF‐βR, which reduced hepatic fibrosis in CCl4‐treated mice. Recombinant bacteria with intrinsic biocompatibility represent a potential therapeutic strategy, particularly for the oral administration of biomacromolecules, reducing the degree of liver fibrosis. 88 Because bacteria can produce and deliver target proteins, their use can markedly reduce the costs associated with drug production. 89
3.3.2. Cellular level
Hepatocytes, HSCs, and hepatic macrophages are key participants in the progression of whole liver fibrosis, and could be selected as therapeutic targets according to the following roles: (1) protecting hepatocytes from damage, reducing their apoptosis, and maintaining their function; (2) reversing the activated state of HSCs, inhibiting their proliferation, and promoting their apoptosis; and (3) promoting the transformation of hepatic macrophages to an anti‐fibrotic phenotype, and inhibiting the production of pro‐fibrotic factors (Table 3). 90
Hepatocytes as targets
As hepatocyte necrosis is a primary mechanism underlying fibrosis, preventing hepatocyte damage via delivery of liver‐protecting drugs has potential in liver fibrosis treatment. Some receptors are highly expressed on hepatocytes during fibrogenesis and may be targeted for drug delivery. 91 For example, the asialoglycoprotein receptor (ASGP‐R) has become a popular ligand for hepatocyte‐targeting, as it is highly expressed on the surface of hepatocytes in the process of fibrosis. 92 Galactose, which specifically targets ASGP‐R, was modified on betulinic acid (BA)‐loaded chitosan nanoparticles to reduce the degree of liver injury in a liver fibrosis experimental model. BA is a pentacyclic triterpenoid acid that protects hepatocytes from damage. Galactosylated nanoparticles demonstrated increased drug accumulation in hepatocytes via galactose‐ASGP‐R interaction. 93 ASGP‐R can also be viewed as an index to reflect the level of liver fibrosis giving galactose‐modified delivery systems potential in anti‐fibrotic therapies for diagnosis of the degree of liver fibrosis based on the accumulation of galactose in hepatocytes. In one study, Yang et al. reported the recovery of hepatocytes following hepatocyte nuclear factor 4 alpha (HNF4A) mRNA transfection in vitro. These authors developed a method of lipid nanoparticle‐mediated HNF4A mRNA delivery in hepatocytes which greatly inhibited fibrogenesis in models of liver fibrosis induced by hepatotoxin or cholestasis 94 (Figure 4e).
HSCs as targets
Activated HSCs are the primary ECM‐producing cells in hepatic fibrosis and are regarded as central mediators of hepatic fibrosis. HSCs are also considered the primary target cells for many anti‐hepatic fibrosis strategies. 95 Specific receptors and ligands expressed on the surface of HSCs during fibrogenesis could be targeted to increase delivery efficiency. Although the activation of HSCs was inhibited by imatinib in vitro, free imatinib therapy is hampered by low concentrations at the HSCs. Imatinib‐loaded liposomes modified with VA (ILC) were prepared, and when compared to free imatinib, the ILC presented strong anti‐fibrotic effects with reduced cytotoxicity. 96 Germacrone (GMO) and miR‐29b were found to suppress the proliferation of activated HSCs and promote their death. Ji et al. developed PEG‐PLGA nanoparticles modified with cyclic RGD peptides (cRGD), which are specifically bound to integrin αvβ3, co‐loading GMO and miR‐29b for anti‐fibrotic liver treatment. The accumulation of cRGD‐modified PEG‐PLGA nanoparticles in the activated HSCs significantly reduced the production of collagen I causing considerable anti‐fibrotic effects in the mice model of CCl4‐induced liver fibrosis. 97 Zhang et al. also developed RGD‐modified hepatitis B core protein nanocages to target quercetin delivery to HSCs. The nanocages accumulated in HSCs via the affinity of the RGD domain to integrin αvβ3 and transformed HSCs into a quiescent phenotype, reducing ECM secretion in vitro and in vivo. 98 Relaxin (RLX) is a small‐molecule polypeptide with anti‐fibrotic activity. It inhibits the activation and proliferation of fibroblasts and enhances the degradation of collagen fibers. Hu et al. developed lipid‐protamine‐hyaluronic acid (LPH) nanoparticles co‐encapsulating the relaxin gene and a miR‐30a‐5p mimic targeted to the sigma‐1 receptor of aHSCs. The LPH nanoparticles delivered a combination therapy involving the crosstalk between macrophages and aHSCs to achieve a synergistic therapeutic effect in mice with fibrosis. 99 In another study, doxorubicin (DOX) and retinoic acid were encapsulated together into chondroitin sulfate nanomicelles (CS micelles) to target aHSCs in an anti‐fibrotic liver. This in vitro study showed that CS micelles were selectively endocytosed by activated HSCs through CD44 receptors and displayed synergistic therapeutic effects in a CCl4‐induced fibrosis model (Figure 4f). 100 Luo et al. developed collagenase‐ and silibinin encapsulated nanoparticles coated with chondroitin sulfate to protect the stability of collagenase and target CD44 on aHSCs in vivo. Compared with normal hepatocytes, the nanoparticles accumulated in aHSCs via the chondroitin sulfate targeting function. Furthermore, collagenase broke down the dense collagen stroma and facilitated the penetration of silibinin into aHSCs in vivo, inhibiting fibrosis in mice. 101 Ribera et al. developed anti‐PDGFRβ‐modified gold nanorods targeted at aHSCs. The nanorods produced thermal ablation under near‐infrared light inducing fibrosis regression in mice with CCl4‐induced fibrosis (Figure 4g). 102
Macrophages as targets
Kupffer cells (KCs) are resident macrophages in the liver that participate in the fibrotic process by altering the cell phenotype. During liver fibrosis, KCs transform from an anti‐fibrotic M1 phenotype into a pro‐fibrotic M2 phenotype. 103 Specific receptors highly expressed in activated KCs provide a rationale for the delivery of drugs to KCs via ligand‐modified delivery systems that precipitate macrophage repolarization. Kaps et al. developed a nanohydrogel equipped with mannose residues (ManNP), which targeted the delivery of siRNA to M2 polarized macrophages to knockdown the CSF‐1 receptor (CSF‐1R) mRNA in vivo. The siRNA‐loaded ManNPs exhibited good biocompatibility and accumulation in M2‐type macrophages. 104 In another study, tumor necrosis factor‐stimulated gene 6 (TSG‐6) was found to ameliorate liver fibrosis. Furthermore, Wang et al. developed calcium phosphate nanoparticles (CaP@BSA NPs) for loading TSG‐6 with high loading efficacy. They found that the CaP@BSA NPs improved liver fibrosis by modulating M2 macrophages demonstrating promising anti‐fibrotic effects. 105
3.3.3. Tissue level
The liver microenvironment exhibits special characteristics during the process of liver fibrogenesis which could be considered as targets against liver fibrosis. 106 These environmental characteristics could also be considered as sensors for targeted drug delivery and responsive release in the fibrotic microenvironment (Table 3). In one study, a ROS and pH dual‐sensitivity polydatin‐encapsulated micelle (PDPA) was prepared. The micelles fractured in acidic lysosomes, releasing polydatin, which is a promising anti‐fibrotic drug. Moreover, the micelle presented synergistic ROS scavenging activity resulting in the inhibition of liver fibrosis, by preventing hepatocyte apoptosis and the activation of pro‐fibrotic cells in vitro and in vivo. 107 MMPs degrade ECM proteins and participate in the progression of liver fibrosis, exerting different roles in different stages. MMP‐9 has been found to have an important role in the early stage of liver fibrosis. In another study, Graphene nanostars linked to the PAMAM‐G5 dendrimer were developed to deliver a plasmid encoding for MMP‐9 to cause MM‐9 overexpression and lead to collagen breakdown. 108 The overexpression of MMP‐targeted therapy reduced the production of collagen in fibrotic tracts markedly reducing hepatic injury.
4. CHALLENGES
Although different anti‐fibrotic strategies at the molecular, cellular, and tissue levels have been discussed in this review, there are still challenges to developing safe, effective, and low‐toxicity anti‐fibrotic therapies (Figure 5).
FIGURE 5.
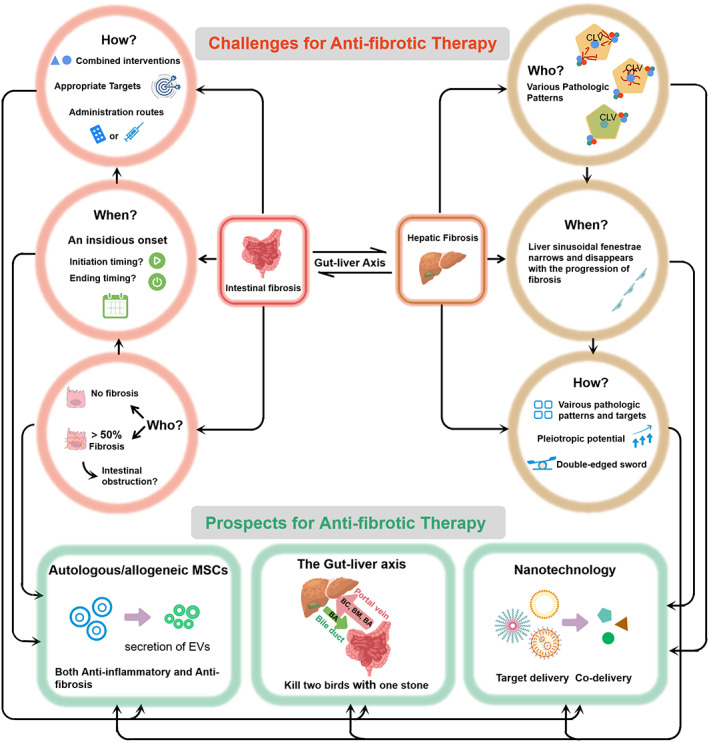
Challenges and opportunities for developing anti‐fibrotic therapeutics for intestinal and hepatic fibrosis. For intestinal fibrosis, the relationship between inflammation and fibrosis should be clarified for patient screening and a personalized therapeutic schedule should be provided. In addition, more than onethird of patients with inflammatory bowel disease experienced nonalcoholic fatty liver disease suggesting a close association between the two diseases, and which is likely influenced by the intestinal microbiome and gut barrier destruction. For hepatic fibrosis, various pathologic patterns pose great difficulty in developing anti‐hepatic fibrosis therapies. The narrowing of the liver sinusoidal fenestrae with fibrosis progression is a challenge currently complicating effective drug delivery. Effective therapies should have multiple targets at different stages. (BC, bacterial composition; BM, bacterial metabolites; BA, bile acid).
4.1. Intestinal fibrosis
4.1.1. Patient screening for anti‐fibrosis therapies
To date, the relationship between inflammation and fibrosis is still unclear. Local inflammation is known to trigger intestinal fibrosis, however, with the use of biological agents, progress has been achieved regarding anti‐inflammatory treatments. Regardless, there has been no marked decrease in the incidence of intestinal fibrosis. In addition, some inflammatory gut diseases, such as celiac disease and lymphocytic colitis, are not complicated by fibrotic lesions, suggesting that non‐inflammatory factors, such as microorganisms and bacterial products, are also drivers of fibrogenesis. 109 Inflammation is necessary for fibrosis, but it most likely does not affect fibrotic progression. It is unclear as to which type of patient should receive anti‐fibrotic treatment because not all patients with inflammation develop intestinal fibrosis and obstruction. A method for early identification of patients at high risk for progression to fibrosis is still lacking and is one of the difficulties in anti‐fibrotic therapy. 44 It is also unclear as to whether patients with complete bowel obstruction should receive anti‐fibrotic therapy. Once fibrous stenosis occurs, the patient must undergo balloon dilatation or strictureplasty to remove the block and it remains unknown whether these interventions impact anti‐fibrotic treatment. Intestinal fibrosis has an insidious onset and its progression is difficult to detect and evaluate until the formation of intestinal stenosis and obstruction. Thus, the exact time of initiation of therapy is unclear. It is crucial to understand the pathophysiology of CD‐associated strictures and predictors of stricturing with accuracy and sensitivity to allow early anti‐fibrotic treatment to slow down the fibrotic process, and possibly even reverse intestinal fibrosis. Additionally, it is difficult to determine when therapy should end due to a lack of accepted clinical trial endpoints and detected responses while undergoing anti‐fibrotic therapy.
4.1.2. The appropriate strategy
As mentioned, previously, it is probable that anti‐inflammatory or anti‐fibrotic treatments alone would not prevent or reverse fibrosis, and should be used in combination with anti‐inflammatory treatments. However, it is unclear as to how “combined interventions” should be developed, including the order and ratio of anti‐inflammatory or anti‐fibrotic drugs. Some studies have shown that the cause of fibrous stenosis is not only the proliferation and deposition of fibrous tissue, but also the proliferation and hypertrophy of intestinal smooth muscle cells (SMCs) within the submucosa and muscularis propria of intestinal stenosis, which also promotes the EMT process in intestinal fibrosis leading to the production of collagen. 110 The proliferation of SMCs seems to be a unique feature of intestinal stenosis and the underlying cause of increased thickness of the bowel wall stricture, as changes in the SMCs in fibrosis in other organs are not obvious. It is not enough to design and develop a treatment strategy focusing on the process of intestinal fibrosis while ignoring smooth muscle hyperplasia. Treatments targeting SMCs in intestinal fibrosis may be particularly attractive as the strategy would benefit anti‐fibrotic processes by inhibiting EMT, while also directly decreasing the thickness of the bowel wall stricture by preventing smooth muscle proliferation. 22
The excessive accumulation of ECM components and the proliferation of SMCs increase the density and stiffness of fibrotic lesions, which develops an inaccessible microenvironment and hampers the distribution and accumulation of therapeutic agents. 111 The effective delivery of drugs into a hypovascularized fibrotic tissue is a substantial challenge. During IV administration, the thickening of the smooth muscle and the increase in fat around the intestinal cavity impair the ability of drugs to reach the mucosal layer where fibroblasts and myofibroblasts are distributed. However, when administering oral preparations, the intestinal mucosal side of the fibrotic site usually exhibits barrier damage, enabling drugs that are targeted to fibroblasts or myofibroblasts to enter the mucosal layer. Consequently, oral or local administration is more suitable for targeting fibroblasts, myofibroblasts, or epithelial interstitial cells and IV administration may be more practical for drug delivery to SMCs which are mainly distributed in the submucosa.
4.2. Hepatic fibrosis
4.2.1. Patient screening for anti‐fibrosis therapies
Chronic liver diseases associated with hepatic fibrosis mainly progress through one of three patterns: (1) Chronic viral hepatitis leading to a “post necrotic” pattern, which is the main feature of porto‐central progression, accompanied by sinusoidal arterialization and neoangiogenesis; (2) Chronic alcoholic hepatitis and NASH leading to a “pericentral fibrosis” pattern; or (3) Cholestasis caused by primary biliary cholangitis and primary sclerosing cholangitis, leading to biliary fibrosis. 112 , 113 These various patterns most likely differ in their progression to hepatic fibrosis, which is necessary for making individualized clinical decisions on anti‐fibrotic therapy. The key issues are the identification of which patients qualify to be treated more urgently, as well as the recognition of low‐, intermediate‐, or high‐risk advanced fibrosis. Biopsies have been the gold standard for fibrosis assessment, but its limitations are well‐known, including invasiveness, potential complications, sampling errors, and high cost making it non‐attractive to clinicians and patients. Therefore, a precise, non‐invasive, and inexpensive method of screening patients for individualized fibrosis assessment and treatment is still lacking.
Both the free‐drug and the nanoparticle‐based therapies for liver sinusoidal fenestrae easily accumulate in the normal liver following IV administration. Particles of this size can also reach hepatocytes or HSCs, while larger particles are easily phagocytized by KCs. 114 However, this window of time narrows and disappears with the progression of fibrosis. The size of the delivery system should be designed considering the manner in which it passes through the liver endothelial fenestrae to reach the target cells and achieve maximum concentration. Fibrotic treatment should be administered before the liver sinusoidal fenestrae becomes narrow and disappears. Once the liver sinusoidal fenestrae closes, it is difficult for drugs to reach the hepatocytes or HSCs. Recently, Li et al. utilized a pretreatment with Riociguat which reversed the liver sinusoid capillarization, followed by a targeted delivery of peptide‐nanoparticles encapsulated with the anti‐fibrosis agent JQ1. This strategy highlights the importance of the liver sinusoidal endothelium in anti‐liver fibrosis. 115
4.2.2. The appropriate strategy
The increased hardness and density of the ECM in fibrosis is the primary barrier in fibrotic tissue to drug delivery and should be considered in the development of delivery systems targeting drugs to fibrotic organs, regardless of the target cell. In addition to the ECM, there are also alterations in the biochemical features of the liver, such as the liver sinusoidal fenestrae, which present challenges to delivery. Furthermore, when targeting liver immune cells for liver fibrosis, the facilitation of nanoparticle endocytosis by immune cells becomes a double‐edged sword. Although macrophages are easily targeted, there is also a probability of them being cleared before they reach liver macrophages. If the targets are missed, then endocytosis by macrophages is reduced. In liver fibrosis, HSCs are located in the space of Disse near the endothelial cells. Such a location makes it difficult to target HSCs, as the delivery system might also be internalized by KCs and liver sinusoidal endothelial cells before reaching HSCs. 116 Emerging evidence in hepatic fibrosis suggests that different pathologies are involved in the different subtypes of fibrogenic cells. These pathologies may have diverse anti‐fibrotic targets, respond differently to clinical management, and need specific therapeutic drugs. The various liver zones and etiologies may also indicate differences in the rate of liver fibrosis progression, the dynamics of the necroinflammatory infiltration, and the onset and progression of portal hypertension, suggesting barriers to drug delivery systems that remain unclear. Finally, even if the target is specific to one cell type, the pleiotropic potential should not be neglected. The target cell exists in different stages, including activation, proliferation, pro‐fibrogenic factors, and collagen release, but to date, research has targeted only a single mode of action. 117 Liver fibrogenesis is a complicated process with a complex underlying network. Accordingly, more research is needed to identify the appropriate treatment strategies.
5. PROSPECTS
Hepatic fibrosis therapeutics currently under exploration have a broad range of targets based on the mechanism of action (MoA) (Table 1) and include (i) improving metabolic disorders, (ii) preventing the activation and proliferation of HSCs, (iii) promoting the degradation of collagen, and (iv) reducing inflammation and cell death. The complex pathophysiology underlying hepatic fibrosis and its interplay with metabolic syndrome processes is unmistakable, although the link is incompletely understood. It is generally believed that hepatic insulin resistance contributes to the development of hepatic fibrosis, as hepatocytes lose their ability to suppress glucose production in response to insulin while preserving their capacity to generate lipids. In addition, metabolic dysregulation further expands the inflammation cascade, contributing to hepatic fibrosis development. 118 Targeting the regulation of glycometabolism and lipid metabolism to improve hepatic fibrosis, with treatments including Cotadutide, Elafibranor, Pegbelfermin, Aramchol, Resmetirom, and AZD2693 is a hot research topic. However, Elafibranor and Pegbelfermin have shown no improvement in hepatic fibrosis in phase II and III trials. Hydronidone targeted to HSC proliferation has been shown to reach the primary endpoint with good tolerance in phase II clinical trials. It has also been used during an initial phase III study on hepatitis B with liver fibrosis and is expected to provide the medical demand for hepatitis B liver fibrosis treatment. It is also an important target for the inhibition of excessive collagen deposition in the treatment of hepatic fibrosis. Simtuzumab (SIM) a monoclonal antibody against the enzyme lysyl LOXL‐2, was tested in phase IIb trials. However, this clinical trial has been halted due to its ineffectiveness. 119 Finally, a chemokine receptor‐2/5 (CCR2/5) antagonist (Cenicriviroc) developed to primarily target inflammation showed good anti‐fibrotic effects in a phase IIb trials (CENTAUR study). In Table 1, the therapeutic in phase clinical trials were based on the specific pathway involved during the hepatic fibrosis process compared to conventional therapeutic approaches. In addition, nanodrugs and gene drugs are gradually being developed and promise to have fewer side effects while requiring lower dosages. However, only a small portion of the phase II trial therapeutics have progressed to phase III which generates doubts on single‐agent therapies. In addition, even when focusing on the same MoAs, trial outcomes for different compounds could be different. We speculate that this is likely due to insufficient concentrations of the therapeutics at the action in vivo requiring multi‐modal synergistic therapy and targeted delivery technology for clinical translation. Besides, several promising therapeutic strategies were summarized as follows (Figure 5).
5.1. Mesenchymal stem cells
Mesenchymal stem cells (MSCs) are early undifferentiated cells with the ability of self‐renewal and multi‐directional differentiation. MSC‐based therapies are becoming a hot topic, particularly in autoimmune diseases. Autologous and allogeneic BM‐MSCs have been shown to reduce IBD inflammation in clinical trials. 120 In addition, clinical studies have demonstrated that autologous or allogeneic adipose‐derived MSCs exert promising therapeutic effects on anal fistulae, closely related to fibrosis. 121 , 122 In a recent phase I–II clinical study, local injections of MSCs were found to reverse intestinal fibrosis and resolve intestinal stenosis. 123 This effect was most likely dependent on their immunoregulatory effects and inhibition of TGF‐β signaling 124 demonstrating an exciting prospect of simultaneous anti‐inflammatory and anti‐fibrosis effects from MSC therapies. EVs derived from MSCs may also regulate intestinal fibrosis through cargo loading. 125
MSCs have shown therapeutic effects on liver inflammation and fibrosis through self‐differentiation and EV secretion. 126 Another study reported that allogeneic BM‐MSCs were able to migrate to the location of liver injury, increase the M2/M1 macrophage ratio, and promote HSC apoptosis revealing that BM‐MSCs are able to relieve liver fibrosis in a CCl4 model. 127 The cytokines and EVs secreted by MSCs can also alleviate liver fibrosis. Rong et al. showed that BM‐MSC derived EVs were effective in a CCl4 rat model and were able to protect hepatocytes. Other studies have suggested that the anti‐fibrotic effect of EVs derived from MSCs is even greater than that of the MSCs themselves. 128 The most attractive aspect of MSCs‐based therapy is that there is no need to distinguish which patient type would develop fibrosis because EV‐ and MSC‐based therapies are both anti‐inflammatory and anti‐fibrotic.
5.2. Application of nanotechnology
Since its first application in the clinic, there has been great progress in the application of nanotechnology as a drug vehicle. One advantage of nanotechnology is the ability to target specific parts and single cells within an organ for drug delivery, maximizing drug activity while minimizing side effects. The free‐drug possesses promising anti‐fibrotic properties, but its non‐targeted accumulation induces mucosal damage at healthy sites. 129 There is real opportunity to exploit nanotechnology for targeted drug delivery to intestinal lesions by manipulating the specific pathological differences between normal and fibrotic intestinal tissues. The markers expressed at high levels on fibrotic intestinal tissues, such as α‐SMA, FAP, PDGFR, and FGFR, could be considered as specific targets recognized by ligands modified on the nano‐delivery system. Fibrosis is a complex disorder and there is an urgent need to focus on combination therapies that have different mechanisms of action. Owing to their nanoscale size and structure, nanoparticle delivery systems are especially suitable for the co‐delivery of drugs. Combination therapies using nanoparticles could target a variety of mechanisms, or multiple disease states simultaneously, to provide synergistic therapeutic effects for effective future anti‐fibrotic therapies.
5.3. The gut‐liver axis
Several studies found a close association between NAFLD, a major cause of liver fibrosis, and IBD, as up to one‐third of patients with IBD experienced NAFLD. 130 , 131 Although the mechanism for the association is unclear, the gut‐liver axis is a link between the intestine and liver, involving a crucial component of the bidirectional relationship. The interaction is created through the portal vein, which transports gut microbial components and metabolites directly to the liver, which then transports bile and metabolites to the intestine. 132 The interaction between the intestinal flora and the bile acids have been demonstrated to either directly or indirectly affect intestinal and liver diseases. 133 Bile acids transform into secondary bile acids following metabolization by intestinal flora and are reabsorbed by the liver through intestinal liver circulation. The insufficient flow of bile acids, resulting in deficient luminal levels, can affect the composition of intestinal flora, subsequently inducing abnormalities in the intestinal microbiota. Increased levels of secondary bile acids in the gut lumen can induce alterations in the microbiota, resulting in reduced intestinal FXR signaling, the loss of tight junctions between epithelial cells, thinning of the mucous layer, and a decreased production of anti‐microbial peptides, which contribute to the interaction of pathobionts with mucosal immune systems and lead to intestinal inflammation and fibrosis. When the gut barrier is destroyed, the gut microbial components and metabolites, and even the structure‐altered gut microbiota itself, can easily transfer to the liver promoting liver inflammation and fibrosis. Moreover, the overproduction of secondary bile acids reabsorbed by the liver induces abnormal liver immunity and activates HSCs provoking liver damage and promoting liver fibrosis. 134 , 135 The gut‐liver axis should be investigated in intestinal and hepatic diseases. Therapeutics targeting the gut‐liver axis may improve intestinal and hepatic fibrosis through regulation of the intestinal flora and bile acids producing dual benefits.
6. CONCLUSIONS
This review comprehensively discussed the pathogenesis, targets, and therapies for intestinal and hepatic fibrosis to highlight the important advances, challenges, and opportunities for anti‐fibrotic therapies. A deeper understanding of the mechanisms of fibrosis, along with more advanced drug delivery technologies, creates an increased need for clinicians, pharmacists, and researchers to further develop integrated strategies encompassing new agents utilizing different delivery systems for effective individualized clinical prospects that treat patients with fibrosis and enhance their quality of life.
AUTHOR CONTRIBUTIONS
Xin Li: Conceptualization (lead); resources (lead); writing – original draft (lead). Mengli Yu: Writing – original draft (supporting). Qingwei Zhao: Writing – review and editing (supporting). Yang Yu: Writing – review and editing (lead).
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that may influence the work reported in this review.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (82270692, 81803449), the Natural Science Foundation of Zhejiang Province (LYY20H300003), the Hospital Pharmacy Project of Zhejiang Pharmaceutical Association (2020ZYY01), and the Medical Science and Technology Project of Zhejiang Province (2022488040).
Li X, Yu M, Zhao Q, Yu Y. Prospective therapeutics for intestinal and hepatic fibrosis. Bioeng Transl Med. 2023;8(6):e10579. doi: 10.1002/btm2.10579
Contributor Information
Xin Li, Email: 11219017@zju.edu.cn.
Qingwei Zhao, Email: qwzhao@zju.edu.cn.
Yang Yu, Email: yuyang201507@swu.edu.cn.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75(2):473‐488. [DOI] [PubMed] [Google Scholar]
- 2. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587(7835):555‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sofias AM, De Lorenzi F, Peña Q, et al. Therapeutic and diagnostic targeting of fibrosis in metabolic, proliferative and viral disorders. Adv Drug Deliv Rev. 2021;175:113831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sisto M, Ribatti D, Lisi S. Organ fibrosis and autoimmunity: the role of inflammation in TGFβ‐dependent EMT. Biomolecules. 2021;11(2):310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rurik JG, Tombácz I, Yadegari A, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Long Q, Liu ZH, Shao QW, et al. Autologous skin fibroblast‐based PLGA nanoparticles for treating multiorgan fibrosis. Adv Sci. 2022;9(21):e2200856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng HT, Huang HC, Lee TY, et al. Delivery of sorafenib by myofibroblast‐targeted nanoparticles for the treatment of renal fibrosis. J Control Release. 2022;346:169‐179. [DOI] [PubMed] [Google Scholar]
- 8. Sakamoto N, Ogawa K, Suda G, et al. Clinical phase 1b study results for safety, pharmacokinetics and efficacy of ND‐L02‐s0201, a novel targeted lipid nanoparticle delivering HSP47 SIRNA for the treatment of Japanese patients with advanced liver fibrosis. J Hepatol. 2018;68:S242. [Google Scholar]
- 9. D'Alessio S, Ungaro F, Noviello D, Lovisa S, Peyrin‐Biroulet L, Danese S. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat Rev Gastroenterol Hepatol. 2022;19(3):169‐184. [DOI] [PubMed] [Google Scholar]
- 10. Wang J, Lin SN, Brown JM, van Wagoner DV, Fiocchi C, Rieder F. Novel mechanisms and clinical trial endpoints in intestinal fibrosis. Immunol Rev. 2021;302(1):211‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'haens G, Rieder F, Feagan BG, et al. Challenges in the pathophysiology, diagnosis, and management of intestinal fibrosis in inflammatory bowel disease. Gastroenterology. 2022;162(1):26‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu XM, Lin XX, Tan JY, et al. Cellular and molecular mechanisms of intestinal fibrosis. Gut Liver. 2023;17(3):360‐374. doi: 10.5009/gnl220045 Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lovisa S, Genovese G, Danese S. Role of epithelial‐to‐mesenchymal transition in inflammatory bowel disease. J Crohns Colitis. 2019;13(5):659‐668. [DOI] [PubMed] [Google Scholar]
- 14. Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. 2019;50(4):992‐1006. [DOI] [PubMed] [Google Scholar]
- 15. Singal AK, Mathurin P. Diagnosis and treatment of alcohol‐associated liver disease: a review. Jama. 2021;326(2):165‐176. [DOI] [PubMed] [Google Scholar]
- 16. Cushing K, Higgins PDR. Management of Crohn disease: a review. Jama. 2021;325(1):69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Broen JCACA, van Laar JM. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol. 2020;16(3):167‐178. [DOI] [PubMed] [Google Scholar]
- 18. Rieder F, Reinisch W. Thiopurines and the natural course of Crohn's disease: did we finally find the right therapeutic target? Am J Gastroenterol. 2014;109(7):1037‐1040. [DOI] [PubMed] [Google Scholar]
- 19. Vande Casteele NV, Jeyarajah J, Jairath V, Feagan BG, Sandborn WJ. Infliximab exposure‐response relationship and thresholds associated with endoscopic healing in patients with ulcerative colitis. Clin Gastroenterol Hepatol. 2019;17(9):1814‐1821.e1. [DOI] [PubMed] [Google Scholar]
- 20. Chen BX, Han ZM, Zhou Q, Liu HB, Xu PC, Zhi FC. Efficacy of infliximab in treatment‐naive patients with stricturing small bowel Crohn's disease. Scand J Gastroenterol. 2021;56(7):812‐819. [DOI] [PubMed] [Google Scholar]
- 21. Li XX, Yang YY, Wang ZB, et al. Multistage‐responsive nanocomplexes attenuate ulcerative colitis by improving the accumulation and distribution of oral nucleic acid drugs in the colon. ACS Appl Mater Interfaces. 2022;14(1):2058‐2070. [DOI] [PubMed] [Google Scholar]
- 22. Lin SN, Mao R, Qian CC, et al. Development of antifibrotic therapy for stricturing Crohn's disease: lessons from randomized trials in other fibrotic diseases. Physiol Rev. 2022;102(2):605‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghafouri‐Fard S, Abak A, Talebi SF, et al. Role of miRNA and lncRNAs in organ fibrosis and aging. Biomed Pharmacother. 2021;143:112132. [DOI] [PubMed] [Google Scholar]
- 24. Wang MN, Zhao JZ, Jiang H, Wang XM. Tumor‐targeted nano‐delivery system of therapeutic RNA. Mater Horiz. 2022;9(4):1111‐1140. [DOI] [PubMed] [Google Scholar]
- 25. Li MY, Li Y, Li SQ, et al. The nano delivery systems and applications of mRNA. Eur J Med Chem. 2022;227:113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kara G, Calin GA, Ozpolat B. RNAi‐based therapeutics and tumor targeted delivery in cancer. Adv Drug Deliv Rev. 2022;182:114113. [DOI] [PubMed] [Google Scholar]
- 27. Huang PS, Liao CJ, Huang YH, et al. Functional and clinical significance of dysregulated microRNAs in liver cancer. Cancers. 2021;13(21):5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou LY, Lin SN, Rieder F, Chen MH, Zhang SH, Mao R. Noncoding RNAs as promising diagnostic biomarkers and therapeutic targets in intestinal fibrosis of Crohn's disease: the path from bench to bedside. Inflamm Bowel Dis. 2021;27(7):971‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang J, Zhou CZ, Zhu R, et al. miR‐200b‐containing microvesicles attenuate experimental colitis associated intestinal fibrosis by inhibiting epithelial‐mesenchymal transition. J Gastroenterol Hepatol. 2017;32(12):1966‐1974. [DOI] [PubMed] [Google Scholar]
- 30. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wasson CW, Abignano G, Hermes H, et al. Long non‐coding RNA HOTAIR drives EZH2‐dependent myofibroblast activation in systemic sclerosis through miRNA 34a‐dependent activation of NOTCH. Ann Rheum Dis. 2020;79(4):507‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675‐691. [DOI] [PubMed] [Google Scholar]
- 33. Yin J, Ye YL, Hu T, et al. Hsa_circRNA_102610 upregulation in Crohn's disease promotes transforming growth factor‐β1‐induced epithelial‐mesenchymal transition via sponging of hsa‐miR‐130a‐3p. World J Gastroenterol. 2020;26(22):3034‐3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winkle M, El‐Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics—challenges and potential solutions. Nat Rev Drug Discov. 2021;20(8):629‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gupta A, Andresen JL, Manan RS, Langer R. Nucleic acid delivery for therapeutic applications. Adv Drug Deliv Rev. 2021;178:113834. [DOI] [PubMed] [Google Scholar]
- 36. Lombardi F, Augello FR, Palumbo P, et al. Soluble fraction from lysate of a high concentration multi‐strain probiotic formulation inhibits TGF‐β1‐induced intestinal fibrosis on CCD‐18Co cells. Nutrients. 2021;13(3):882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang JX, Ou A, Tang XP, et al. “two‐birds‐one‐stone” colon‐targeted nanomedicine treats ulcerative colitis via remodeling immune microenvironment and anti‐fibrosis. J Nanobiotechnology. 2022;20(1):389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deshmukh AP, Vasaikar SV, Tomczak K, et al. Identification of EMT signaling cross‐talk and gene regulatory networks by single‐cell RNA sequencing. Proc Natl Acad Sci USA. 2021;118(19):e2102050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lewis A, Sánchez S, Berti G, et al. Small‐molecule Wnt inhibitors are a potential novel therapy for intestinal fibrosis in Crohns disease. Clin Sci. 2022;136(19):1405‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strowitzki MJ, Ritter AS, Kimmer G, Schneider M. Hypoxia‐adaptive pathways: a pharmacological target in fibrotic disease? Pharmacol Res. 2019;147:104364. [DOI] [PubMed] [Google Scholar]
- 41. Prados ME, García‐Martín A, Unciti‐Broceta JD, et al. Betulinic acid hydroxamate prevents colonic inflammation and fibrosis in murine models of inflammatory bowel disease. Acta Pharmacol Sin. 2021;42(7):1124‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steiner CA, Rodansky ES, Johnson LA, et al. AXL is a potential target for the treatment of intestinal fibrosis. Inflamm Bowel Dis. 2021;27(3):303‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elias M, Zhao S, Le HT, et al. IL‐36 in chronic inflammation and fibrosis—bridging the gap? J Clin Invest. 2021;131(2):e144336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scheibe K, Kersten C, Schmied A, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. 2019;156(4):1082‐1097.e11. [DOI] [PubMed] [Google Scholar]
- 45. Guan QD, Weiss CR, Wang SH, et al. Reversing ongoing chronic intestinal inflammation and fibrosis by sustained block of IL‐12 and IL‐23 using a vaccine in mice. Inflamm Bowel Dis. 2018;24(9):1941‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. da Cunha VPD, Preisser TM, Santana MP, Machado DCC, Pereira VB, Miyoshi A. Mycobacterial Hsp65 antigen delivered by invasive Lactococcus lactis reduces intestinal inflammation and fibrosis in TNBS‐induced chronic colitis model. Sci Rep. 2020;10(1):20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao XJ, Yang WJ, Yu TM, et al. Th17 cell‐derived amphiregulin promotes colitis‐associated intestinal fibrosis through activation of mTOR and MEK in intestinal myofibroblasts. Gastroenterology. 2023;164(1):89‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tao QS, Wang BC, Zheng Y, Li GW, Ren JN. Triptolide ameliorates colonic fibrosis in an experimental rat model. Mol Med Rep. 2015;12(2):1891‐1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Speca S, Rousseaux C, Dubuquoy C, et al. Novel PPARγ modulator GED‐0507‐34 levo ameliorates inflammation‐driven intestinal fibrosis. Inflamm Bowel Dis. 2016;22(2):279‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Holvoet T, Devriese S, Castermans K, et al. Treatment of intestinal fibrosis in experimental inflammatory bowel disease by the pleiotropic actions of a local rho kinase inhibitor. Gastroenterology. 2017;153(4):1054‐1067. [DOI] [PubMed] [Google Scholar]
- 51. Garg M, Royce SG, Tikellis C, et al. Imbalance of the renin‐angiotensin system may contribute to inflammation and fibrosis in IBD: a novel therapeutic target? Gut. 2020;69(5):841‐851. [DOI] [PubMed] [Google Scholar]
- 52. Lenti MV, Di Sabatino AD. Intestinal fibrosis. Mol Aspects Med. 2019;65:100‐109. [DOI] [PubMed] [Google Scholar]
- 53. Kurahara LH, Hiraishi K, Hu Y, et al. Activation of myofibroblast TRPA1 by steroids and pirfenidone ameliorates fibrosis in experimental Crohn's disease. Cell Mol Gastroenterol Hepatol. 2018;5(3):299‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gisbert‐Ferrándiz L, Cosín‐Roger J, Hernández C, et al. Diminished vitamin D receptor protein levels in Crohn's disease fibroblasts: effects of vitamin D. Nutrients. 2020;12(4):973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu F, Shao QQ, Hu ML, et al. Wu‐Mei‐Wan ameliorates chronic colitis‐associated intestinal fibrosis through inhibiting fibroblast activation. J Ethnopharmacol. 2020;252:112580. [DOI] [PubMed] [Google Scholar]
- 56. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151‐172. [DOI] [PubMed] [Google Scholar]
- 57. Li MZ, Yin H, Yan Z, et al. The immune microenvironment in cartilage injury and repair. Acta Biomater. 2022;140:23‐42. [DOI] [PubMed] [Google Scholar]
- 58. Bormann T, Maus R, Stolper J, et al. Role of matrix metalloprotease‐2 and MMP‐9 in experimental lung fibrosis in mice. Respir Res. 2022;23(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goffin L, Fagagnini S, Vicari A, et al. Anti‐MMP‐9 antibody: a promising therapeutic strategy for treatment of inflammatory bowel disease complications with fibrosis. Inflamm Bowel Dis. 2016;22(9):2041‐2057. [DOI] [PubMed] [Google Scholar]
- 60. Chen HJ, Xu HX, Luo LJ, et al. Thalidomide prevented and ameliorated pathogenesis of Crohn's disease in mice via regulation of inflammatory response and fibrosis. Front Pharmacol. 2019;10:1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Truffi M, Sorrentino L, Monieri M, et al. Inhibition of fibroblast activation protein restores a balanced extracellular matrix and reduces fibrosis in Crohn's disease strictures ex vivo. Inflamm Bowel Dis. 2018;24(2):332‐345. [DOI] [PubMed] [Google Scholar]
- 62. Ferro D, Baratta F, Pastori D, et al. New insights into the pathogenesis of non‐alcoholic fatty liver disease: gut‐derived lipopolysaccharides and oxidative stress. Nutrients. 2020;12(9):2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cao YM, Cheng K, Yang M, et al. Orally administration of cerium oxide nanozyme for computed tomography imaging and anti‐inflammatory/anti‐fibrotic therapy of inflammatory bowel disease. J Nanobiotechnology. 2023;21(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151‐166. [DOI] [PubMed] [Google Scholar]
- 65. Zhao MY, Wang LQ, Wang MZ, et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct Target Ther. 2022;7(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fondevila MF, Fernandez U, Heras V, et al. Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J Hepatol. 2022;77(1):15‐28. [DOI] [PubMed] [Google Scholar]
- 67. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease—novel insights into cellular communication circuits. J Hepatol. 2022;77(4):1136‐1160. [DOI] [PubMed] [Google Scholar]
- 68. Damiris K, Tafesh ZH, Pyrsopoulos N. Efficacy and safety of anti‐hepatic fibrosis drugs. World J Gastroenterol. 2020;26(41):6304‐6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu MB, Rong PQ, Jin TY, Zhang PP, Liang HY, Zheng GQ. Chinese herbal medicine for Wilson's disease: a systematic review and meta‐analysis. Front Pharmacol. 2019;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li H. Advances in anti hepatic fibrotic therapy with traditional Chinese medicine herbal formula. J Ethnopharmacol. 2020;251:112442. [DOI] [PubMed] [Google Scholar]
- 71. Haroon HB, Hunter AC, Farhangrazi ZS, Moghimi SM. A brief history of long circulating nanoparticles. Adv Drug Deliv Rev. 2022;188:114396. [DOI] [PubMed] [Google Scholar]
- 72. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537‐2564. [DOI] [PubMed] [Google Scholar]
- 73. Zhang JF, Shen HW, Xu JJ, et al. Liver‐targeted siRNA lipid nanoparticles treat hepatic cirrhosis by dual antifibrotic and anti‐inflammatory activities. ACS Nano. 2020;14(5):6305‐6322. [DOI] [PubMed] [Google Scholar]
- 74. Wu J, Huang JS, Kuang SC, et al. Synergistic microRNA therapy in liver fibrotic rat using MRI‐visible nanocarrier targeting hepatic stellate cells. Adv Sci. 2019;6(5):1801809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. He Y, Hwang S, Cai Y, et al. MicroRNA‐223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. 2019;70(4):1150‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsay HC, Yuan QG, Balakrishnan A, et al. Hepatocyte‐specific suppression of microRNA‐221‐3p mitigates liver fibrosis. J Hepatol. 2019;70(4):722‐734. [DOI] [PubMed] [Google Scholar]
- 77. Pan Y, Wang J, He L, Zhang FC. MicroRNA‐34a promotes EMT and liver fibrosis in primary biliary cholangitis by regulating TGF‐β1/smad pathway. J Immunol Res. 2021;2021:6890423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Song L, Chen TY, Zhao XJ, et al. Pterostilbene prevents hepatocyte epithelial‐mesenchymal transition in fructose‐induced liver fibrosis through suppressing miR‐34a/Sirt1/p53 and TGF‐β1/Smads signalling. Br J Pharmacol. 2019;176(11):1619‐1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu RP, Li XJY, Zhu WW, et al. Cholangiocyte‐derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology. 2019;70(4):1317‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang ZF, Yang XK, Gui SY, et al. The roles and mechanisms of lncRNAs in liver fibrosis. Front Pharmacol. 2021;12:779606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ye JF, Lin YQ, Yu Y, Sun D. LncRNA NEAT1/microRNA‐129‐5p/SOCS2 axis regulates liver fibrosis in alcoholic steatohepatitis. J Transl Med. 2020;18(1):445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. El‐Kharashi OA, Mohamed DI, Khairy E, Ezzat SF, Zaki WS. Exenatide promotes cardiac lncRNAs HOX transcript antisense RNA (HOTAIR) in Wistar rats with liver cirrhosis; a novel role of GLP‐1 receptor agonists in cirrhotic cardiomyopathy. Eur J Pharmacol. 2019;855:294‐304. [DOI] [PubMed] [Google Scholar]
- 83. Zhang K, Shi ZM, Zhang MX, et al. Silencing lncRNA Lfar1 alleviates the classical activation and pyoptosis of macrophage in hepatic fibrosis. Cell Death Dis. 2020;11(2):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhao QY, Liu JY, Deng H, et al. Targeting mitochondria‐located circRNA SCAR alleviates NASH via reducing mROS output. Cell. 2020;183(1):76‐93.e22. [DOI] [PubMed] [Google Scholar]
- 85. Mahmoud AM, Hozayen WG, Hasan IH, Shaban E, Bin‐Jumah M. Umbelliferone ameliorates CCl4‐induced liver fibrosis in rats by upregulating PPARγ and attenuating oxidative stress, inflammation, and TGF‐β1/Smad3 signaling. Inflammation. 2019;42(3):1103‐1116. [DOI] [PubMed] [Google Scholar]
- 86. Sakai N, Kamimura K, Miyamoto H, et al. Letrozole ameliorates liver fibrosis through the inhibition of the CTGF pathway and 17β‐hydroxysteroid dehydrogenase 13 expression. J Gastroenterol. 2023;58(1):53‐68. [DOI] [PubMed] [Google Scholar]
- 87. Li Y, Pu S, Liu Q, et al. An integrin‐based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis. J Control Release. 2019;303:77‐90. [DOI] [PubMed] [Google Scholar]
- 88. Yuan SL, Dong M, Zhang HL, et al. Oral delivery of a Lactococcus lactis expressing extracellular TGFβR2 alleviates hepatic fibrosis. Appl Microbiol Biotechnol. 2021;105(14–15):6007‐6018. [DOI] [PubMed] [Google Scholar]
- 89. Li ZT, Wang YX, Liu J, et al. Chemically and biologically engineered bacteria‐based delivery systems for emerging diagnosis and advanced therapy. Adv Mater. 2021;33(38):e2102580. [DOI] [PubMed] [Google Scholar]
- 90. Cai JJ, Zhang XJ, Li HL. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. 2019;70(3):1026‐1037. [DOI] [PubMed] [Google Scholar]
- 91. Kumar S, Duan QH, Wu RX, Harris EN, Su QZ. Pathophysiological communication between hepatocytes and non‐parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Deliv Rev. 2021;176:113869. [DOI] [PubMed] [Google Scholar]
- 92. Wang JQ, Li LL, Hu A, et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature. 2022;608(7922):413‐420. [DOI] [PubMed] [Google Scholar]
- 93. Wu ZC, Liu XY, Liu JY, Piao JS, Piao MG. Preparation of betulinic acid galactosylated chitosan nanoparticles and their effect on liver fibrosis. Int J Nanomedicine. 2022;17:4195‐4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yang TH, Poenisch M, Khanal R, et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J Hepatol. 2021;75(6):1420‐1433. [DOI] [PubMed] [Google Scholar]
- 95. Yang W, He H, Wang TT, et al. Single‐cell transcriptomic analysis reveals a hepatic stellate cell‐activation roadmap and myofibroblast origin during liver fibrosis in mice. Hepatology. 2021;74(5):2774‐2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. El‐Mezayen NS, El‐Hadidy WF, El‐Refaie WM, Shalaby TI, Khattab MM, El‐Khatib AS. Hepatic stellate cell‐targeted imatinib nanomedicine versus conventional imatinib: a novel strategy with potent efficacy in experimental liver fibrosis. J Control Release. 2017;266:226‐237. [DOI] [PubMed] [Google Scholar]
- 97. Ji D, Wang QH, Zhao Q, et al. Co‐delivery of miR‐29b and germacrone based on cyclic RGD‐modified nanoparticles for liver fibrosis therapy. J Nanobiotechnology. 2020;18(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang Q, Xu D, Guo QY, et al. Theranostic quercetin nanoparticle for treatment of hepatic fibrosis. Bioconjug Chem. 2019;30(11):2939‐2946. [DOI] [PubMed] [Google Scholar]
- 99. Hu MY, Wang Y, Liu ZS, et al. Hepatic macrophages act as a central hub for relaxin‐mediated alleviation of liver fibrosis. Nat Nanotechnol. 2021;16(4):466‐477. [DOI] [PubMed] [Google Scholar]
- 100. Luo JW, Zhang P, Zhao T, et al. Golgi apparatus‐targeted chondroitin‐modified nanomicelles suppress hepatic stellate cell activation for the management of liver fibrosis. ACS Nano. 2019;13(4):3910‐3923. [DOI] [PubMed] [Google Scholar]
- 101. Luo JW, Zhang ZW, Zeng YC, Dong Y, Ma LX. Co‐encapsulation of collagenase type I and silibinin in chondroitin sulfate coated multilayered nanoparticles for targeted treatment of liver fibrosis. Carbohydr Polym. 2021;263:117964. [DOI] [PubMed] [Google Scholar]
- 102. Ribera J, Vilches C, Sanz V, et al. Treatment of hepatic fibrosis in mice based on targeted plasmonic hyperthermia. ACS Nano. 2021;15(4):7547‐7562. [DOI] [PubMed] [Google Scholar]
- 103. Wen YK, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases‐diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18(1):45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kaps L, Leber N, Klefenz A, et al. In vivo siRNA delivery to immunosuppressive liver macrophages by α‐mannosyl‐functionalized cationic nanohydrogel particles. Cell. 2020;9(8):1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang M, Zhang M, Fu LH, et al. Liver‐targeted delivery of TSG‐6 by calcium phosphate nanoparticles for the management of liver fibrosis. Theranostics. 2020;10(1):36‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411‐428. [DOI] [PubMed] [Google Scholar]
- 107. Lin LT, Gong HY, Li R, et al. Nanodrug with ROS and pH dual‐sensitivity ameliorates liver fibrosis via multicellular regulation. Adv Sci. 2020;7(7):1903138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Melgar‐Lesmes P, Luquero A, Parra‐Robert M, et al. Graphene‐dendrimer nanostars for targeted macrophage overexpression of metalloproteinase 9 and hepatic fibrosis precision therapy. Nano Lett. 2018;18(9):5839‐5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Franzè E, Dinallo V, Laudisi F, et al. Interleukin‐34 stimulates gut fibroblasts to produce collagen synthesis. J Crohns Colitis. 2020;14(10):1436‐1445. [DOI] [PubMed] [Google Scholar]
- 110. Mao R, Doyon G, Gordon IO, et al. Activated intestinal muscle cells promote preadipocyte migration: a novel mechanism for creeping fat formation in Crohn's disease. Gut. 2022;71(1):55‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xing L, Chang X, Shen LJ, et al. Progress in drug delivery system for fibrosis therapy. Asian J Pharm Sci. 2021;16(1):47‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rosenthal SB, Liu X, Ganguly S, et al. Heterogeneity of HSCs in a mouse model of NASH. Hepatology. 2021;74(2):667‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Carter J, Wang S, Friedman SL. Ten thousand points of light: heterogeneity among the stars of NASH fibrosis. Hepatology. 2021;74(2):543‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang AX, Meng KY, Liu YD, et al. Absorption, distribution, metabolism, and excretion of nanocarriers in vivo and their influences. Adv Colloid Interface Sci. 2020;284:102261. [DOI] [PubMed] [Google Scholar]
- 115. Li FF, Zhao Y, Cheng ZX, et al. Restoration of sinusoid fenestrae followed by targeted nanoassembly delivery of an anti‐fibrotic agent improves treatment efficacy in liver fibrosis. Adv Mater. 2023;35(17):e2212206. doi: 10.1002/adma.202212206 [DOI] [PubMed] [Google Scholar]
- 116. Tranah TH, Edwards LA, Schnabl B, Shawcross DL. Targeting the gut‐liver‐immune axis to treat cirrhosis. Gut. 2021;70(5):982‐994. [DOI] [PubMed] [Google Scholar]
- 117. Mahdinloo S, Kiaie SH, Amiri A, Hemmati S, Valizadeh H, Zakeri‐Milani P. Efficient drug and gene delivery to liver fibrosis: rationale, recent advances, and perspectives. Acta Pharm Sin B. 2020;10(7):1279‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet. 2021;398(10308):1359‐1376. [DOI] [PubMed] [Google Scholar]
- 119. Moon HJ, Finney J, Ronnebaum T, Mure M. Human lysyl oxidaselike 2. Bioorg Chem. 2014;57:231‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Forbes GM, Sturm MJ, Leong RW, et al. A phase 2 study of allogeneic mesenchymal stromal cells for luminal Crohn's disease refractory to biologic therapy. Clin Gastroenterol Hepatol. 2014;12(1):64‐71. [DOI] [PubMed] [Google Scholar]
- 121. Serrero M, Grimaud F, Philandrianos C, Visée C, Sabatier F, Grimaud JC. Long‐term safety and efficacy of local microinjection combining autologous microfat and adipose‐derived stromal vascular fraction for the treatment of refractory perianal fistula in Crohn's disease. Gastroenterology. 2019;156(8):2335‐2337.e2. [DOI] [PubMed] [Google Scholar]
- 122. Barnhoorn MC, Wasser MNJM, Roelofs H, et al. Long‐term evaluation of allogeneic bone marrow‐derived mesenchymal stromal cell therapy for Crohn's disease perianal fistulas. J Crohns Colitis. 2020;14(1):64‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vieujean S, Loly JP, Boutaffala L, et al. Mesenchymal stem cell injection in Crohn's disease strictures: a phase I‐II clinical study. J Crohns Colitis. 2022;16(3):506‐510. [DOI] [PubMed] [Google Scholar]
- 124. Li SN, Wu JF. TGF‐β/SMAD signaling regulation of mesenchymal stem cells in adipocyte commitment. Stem Cell Res Ther. 2020;11(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gómez‐Ferrer M, Amaro‐Prellezo E, Dorronsoro A, et al. HIF‐overexpression and pro‐inflammatory priming in human mesenchymal stromal cells improves the healing properties of extracellular vesicles in experimental Crohn's disease. Int J Mol Sci. 2021;22(20):11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang YF, Huang B, Jin T, Ocansey DKW, Jiang J, Mao F. Intestinal fibrosis in inflammatory bowel disease and the prospects of mesenchymal stem cell therapy. Front Immunol. 2022;13:835005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu YW, Dong YT, Wu XJ, Xu XT, Niu JQ. The assessment of mesenchymal stem cells therapy in acute on chronic liver failure and chronic liver disease: a systematic review and meta‐analysis of randomized controlled clinical trials. Stem Cell Res Ther. 2022;13(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rong XL, Liu JZ, Yao X, Jiang TC, Wang YM, Xie F. Human bone marrow mesenchymal stem cells‐derived exosomes alleviate liver fibrosis through the Wnt/beta‐catenin pathway. Stem Cell Res Ther. 2019;10(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Passaro F, Tocchetti CG, Spinetti G, et al. Targeting fibrosis in the failing heart with nanoparticles. Adv Drug Deliv Rev. 2021;174:461‐481. [DOI] [PubMed] [Google Scholar]
- 130. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. 2020;158(7):1913‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zou ZY, Shen B, Fan JG. Systematic review with meta‐analysis: epidemiology of nonalcoholic fatty liver disease in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2019;25(11):1764‐1772. [DOI] [PubMed] [Google Scholar]
- 132. Chopyk DM, Grakoui A. Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology. 2020;159(3):849‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Smirnova E, Muthiah MD, Narayan N, et al. Metabolic reprogramming of the intestinal microbiome with functional bile acid changes underlie the development of NAFLD. Hepatology. 2022;76(6):1811‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20(1):40‐54. [DOI] [PubMed] [Google Scholar]
- 135. Jones RM, Neish AS. Gut microbiota in intestinal and liver disease. Annu Rev Pathol. 2021;16:251‐275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.