

Abstract
Bone marrow niches (endosteal and perivascular) play important roles in both normal bone marrow function and pathological processes such as cancer cell dormancy. Unraveling the mechanisms underlying these events in humans has been severely limited by models that cannot dissect dynamic events at the niche level. Utilizing microfluidic and stem cell technologies, we present a 3D in vitro model of human bone marrow that contains both the perivascular and endosteal niches, complete with dynamic, perfusable vascular networks. We demonstrate that our model can replicate in vivo bone marrow function, including maintenance and differentiation of CD34+ hematopoietic stem/progenitor cells, egress of neutrophils (CD66b+), and niche-specific responses to doxorubicin and granulocyte-colony stimulating factor. Our platform provides opportunities to accelerate current understanding of human bone marrow function and drug response with high spatial and temporal resolution.
Keywords: Tissue engineering, Organ-on-a-chip, Hematopoiesis, Breast cancer
1. Introduction
The bone marrow is comprised of many cell types and has a unique sinusoidal blood supply; together, these features generate distinct microenvironments or niches [1,2] that are small (on the order of 100 μm) and in close proximity. The perivascular and endosteal niches are of particular interest because they support hematopoiesis [3,4], a primary function of bone marrow, and impact disease processes, including tumor metastasis and dormancy [1,5,6]. Current understanding of these niche-specific phenomena has largely been gleaned from small animal models, which do not necessarily recapitulate human bone marrow function and have limited spatiotemporal resolution.
The endosteal niche is thought to be comprised of osteoblast cells at the surface of bone, where hematopoietic stem/progenitor cells (HSPC) reside, and adjacent blood vessels [7,8]. Similarly, the space immediately adjacent to the abluminal surface of a blood vessel comprises the perivascular niche [4,9]. The sinusoidal blood vessels of the bone marrow, found in both niches, are surrounded by many different cell types including mesenchymal stem cells (MSC), specialized CXCL12 abundant reticular cells, HSPC, leukocytes at different stages of differentiation, and adipocytes [10]. Bone marrow stromal cells (BMSC) are known to secrete chemotactic signals such as stem cell factor (SCF) [4] and CXCL-12/SDF-1 [11]. Osteoblasts express osteopontin—implicated in maintaining HSPC quiescence—in addition to VCAM and CXCL-12 [7,12]. Endothelial cells (EC) in the bone marrow upregulate specialized ligands on the luminal surface such as E-selectin, ICAM, and VCAM [3,13] compared to other tissues [14,15]. These chemokines and adhesion molecules work cooperatively to facilitate homing of circulating HSPC to bone marrow [3,13,16]. Moreover, the higher level of E-selectin expression by EC in the bone marrow may play an important role in regulating HSPC cell cycling [17] and has been implicated as a regulator of breast cancer metastasis to bone [13,18].
Advances in tissue engineering and microfluidics have led to “organ-on-a-chip” technologies, which generate 3D microphysiological tissues through co-culture of human cells in hydrogels; this technology provides specific advantages compared to traditional 2D culture and mouse models. For example, microfluidic technology can capture the spatial scale and cell-specific features of the adjacent endosteal and perivascular bone marrow microenvironments. Although several labs have utilized similar tissue engineering and microfluidic approaches to recreate aspects of the perivascular and endosteal niches [1,5,19,20], these models lack vital aspects of in vivo tissue, such as cell egress from the bone marrow into the peripheral circulation. Here, we present a unified model of the perivascular and endosteal human bone marrow niches, which includes the perfusable vascular network that is essential for niche maintenance and function [4,21]. We demonstrate hematopoiesis (maintenance of CD34+ HSPC cells, proliferation and differentiation of these cells into myeloid and erythroid lineages) and egress of neutrophils (CD66b+) through the microvascular network. Our model also mimics the in vivo drug response to both doxorubicin and G-CSF, which respectively decrease and increase neutrophil concentrations in the peripheral circulation. Our model platform is uniquely situated to simulate the bone marrow response to a range of stimuli and be validated with observable endpoints (e.g., peripheral blood) in humans.
2. Materials and methods
2.1. Device design and modeling
The microfluidic device consists of two hexagonal chambers connected by three symmetric two-way ports (Fig. 1A). The port geometry was designed based on the capillary burst valve concept, and thus enables sequential loading of fibrin hydrogels without leaking into the adjacent chamber [22,23]. The ports also allow diffusion of soluble signaling molecules and migration of cells between the two hexagonal chambers. Each hexagonal chamber is 865 μm wide and the ports are 80 μm long; this scale is appropriate for simulating the bone marrow’s adjacent niches. A third bottom chamber, adjacent to the hexagonal chambers, enables loading of a third cell type at a later time point (Fig. 1A and B). The bottom chamber connects to the hexagonal chambers through asymmetric one-way valves designed to prevent fibrin gel from the hexagonal chambers from entering the bottom chamber during initial loading.
Fig. 1. Characterization of transport phenomena in a three chambered-microfluidic device.

(A) A CAD drawing depicts the overall device design with connecting fluidic lines. (B) Actual size of the device. (C) COMSOL simulation of interstitial flow through the fluidic device predicts flow of ~10 μm/s (D) FRAP experiments in the top (+) and middle (•) of the device (panel C) showed an interstitial flow velocity of 22.58 μm/s and 4.48 μm/s, respectively. Average interstitial flow was 17.7 ± 4.7 μm/s, n = 3. (E) COMSOL modelling of 70 kDa dextran through the right side of the device predicts limited diffusion of dextran through the ports. (F) 70 kDa TRITC-dextran and FITC-dextran perfused through a 10 mg/mL fibrin gel in the device showed limited diffusion between chambers and an even distribution of dextran in the bottom central chamber. (G) Mean fluorescent intensity of an arbitrary line (horizontal white dotted line on the image insets) through the middle ports of the simulated device. Experimentally measured values (blue circles) mapped closely to the theoretical concentration profile (yellow line).
To model molecular transport in the device, the device design was imported into COMSOL Multiphysics 5.2. The steady state interstitial fluid flow was modelled using the transport through porous media with a no-slip boundary condition at the walls and tissue chambers filled with acellular fibrin gel. Differential fluid levels in the device’s fluidic lines created a hydrostatic pressure difference responsible for driving fluid through the tissue (Fig. S1). To mimic soluble factor transport in the device, we analyzed transport of 70 kDa dextran by coupling the transport of diluted species and the transport through porous media modules of COMSOL. The pressure heads were matched with experimental conditions. The following physical constants were used: diffusion coefficient of dextran, 7 × 10−11 m2/s; porosity of fibrin gel, 0.99; and hydraulic permeability of fibrin gel, 1.5 × 10−13 m2 [23,24].
2.2. Device preparation
A master mold of the device, comprised of SU-8 on a silicon wafer, was prepared using standard soft photolithography techniques as previously described [25,26]. Briefly, a 100 μm layer of SU-8 3050 was spun onto a Si-wafer. Then, the microfluidic design was patterned onto the wafer via a single mask photolithography step. After silanizing the mold, polydimethylsiloxane (PDMS) was mixed with curing agent (Dow Corning) in a 10:1 ratio, degassed, and poured onto the mold. The PDMS-filled molds were baked at 60 °C overnight; then, the cured PDMS was removed from the mold, punched with fluidic lines and loading ports, and plasma bonded to a glass cover slip (Fisher Scientific) or a glass slide (Fisher Scientific) [27]. Devices were exposed to ultraviolet (UV) light for at least 30 min to ensure sterility before loading.
2.3. Dextran diffusion and fluorescence recovery after photobleaching (FRAP)
A fibrin gel (10 mg/mL) was injected into all three chambers of the device and polymerized for 30 min at 37 °C. Fluidic lines were coated with 1% gelatin and then filled with EGM-2 (Lonza) for up to 24 h. Prior to imaging, EGM-2 in the top fluidic lines was replaced with either 70 kDa TRITC- or FITC-dextran. Dextran diffusion through the fibrin gel was observed every 5 min for 60 min (13 sequential images). For FRAP experiments (performed on an Olympus FV3000), 70 kDa FITC-dextran was loaded into the top fluidic line (Fig. S1). A circular region was photobleached for 2 s and 20 images were acquired at a rate of 0.56 or 0.76 s/frame. The images were thresholded and the center of the bleached region was identified using the circle tool in FIJI. The coordinates of the centroid were used to measure displacement of the centroid over time and calculate interstitial velocity through the fibrin gel. This process was repeated at multiple locations in the device.
2.4. Cell culture
Endothelial colony-forming cells (EC) were isolated from cord blood as previously described by our group and others [25,28,29]. All cord blood samples were collected in accordance with the University of California, Davis and Washington University in St. Louis School of Medicine Institutional Review Board regulations. Briefly, EC were cultured on 1% gelatin in EGM-2 (Lonza). All cells were maintained at 37.5 °C in a 5% CO2 incubator and used between passages 5 and 7.
HSPC (CD34+) were isolated from cord blood using the EasySep™ Human Cord Blood CD34 Positive Selection Kit II (StemCell Technologies) according to the manufacturer’s guidelines. Sorting efficiency was assessed via flow cytometry on a small aliquot of isolated cells. Isolated cells were cryopreserved in CryoStor (StemCell Technologies) for future use. Cells were thawed the day of use and kept in a 1:1 blend of fully supplemented EGM-2 and SFEM StemSpan II (STEMCELL Technologies) + 1x CD34 expansion +1 μM SR1 before loading into the microfluidic device. To quantify the number of CD34+ cells loaded into each chamber, cells in some samples were loaded with 1 μM of CellTracker Green CMFDA (Invitrogen) per the manufacturer’s instructions and imaged using an IX83 inverted microscope (Olympus).
The human fetal osteoblast (hFOB) 1.19 cell line (ATCC, CRL-11372) was expanded at 33.5 °C in DMEM/F12 media (Gibco) containing 10% FBS (Invitrogen) and 0.3 mg/mL of G418 (Invitrogen) prior to cryopreservation. After thawing, osteoblasts were cultured for up to 7 days at 37.5 °C and 5% CO2 and passaged a maximum of one time before use. Bone marrow stromal cells (BMSC, Lonza) were cultured in Myelocult 5100 (StemCell Technologies) supplemented with 1 μM hydrocortisone (StemCell Technologies), 2 mM L-glutamine (ThermoFisher), and 50 U/mL Penicillin-Streptomycin (ThermoFisher). Cells were maintained at 37.5 °C in a 5% CO2 incubator and used between passages 3 and 5. MDA-MB-231 breast cancer cells expressing red fluorescent protein (RFP) (Cell Biolabs) were cultured in DMEM (Gibco) with 10% FBS (Gibco), 2 mM L-Glutamine (Gibco), and 1% Penicillin-Streptomycin (Gibco).
2.5. Bone marrow-on-a-chip (BMoaC) model
Our group has significant experience utilizing fibrin hydrogels to create vascularized microphysiological systems [25,30–32]. In addition, fibrinogen is found around blood vessels in the bone marrow, making it a suitable matrix for this study [33]. Fibrinogen (Sigma) was prepared in Dulbecco’s Phosphate-Buffered Saline (DPBS, Invitrogen) and used at a final concentration of 10 mg/mL. To generate the perivascular niche, EC (1 × 107 cells/mL) and BMSC (1 × 107 cells/mL) were mixed in a 1:1 ratio in fibrinogen (10 mg/mL final); this suspension was mixed with thrombin for a final concentration of 3 U/mL and loaded into the device. Similarly, to generate the endosteal niche, EC (1 × 107 cells/mL) were mixed with OB (1 × 107 cells/mL) at a 1:1 ratio in fibrinogen. Loaded gels were allowed to sit for 3–5 min prior to loading the other side of the device. Devices were polymerized for 20–30 min in the incubator, after which time the fluidic lines were coated with 1% gelatin. To investigate hematopoiesis in the BMoaC, CD34+ (1 × 106 cells/mL) were mixed at a ratio of 1:10:10 with the EC and respective stromal cell (BMSC or hFOB) in fibrogen, mixed with thrombin, and injected into the device. SN controls were formed by loading both sides of the device with either hFOB or BMSC with EC and CD34+ cells.
Devices were cultured over a period of 14 days. To encourage uniform vascular development within chambers and anastomosis with the ports, the pressure heads were flipped every day for the first 4 days; then, pressures were held constant [22,34]. Devices received a 1:1 blend of fully supplemented EGM-2 and SFEM StemSpan II + 1x CD34 expansion +1 μM SR1 unless otherwise noted. Vessel permeability was characterized with 70 kDa dextran as previously described [23,34]. Briefly, 70 kDa TRITC-dextran (5 μg/mL) was introduced through the top fluidic lines and allowed to perfuse through the device for 15 min before acquiring a series of time lapse images. The permeability (P) was calculated by quantifying the average background fluorescence intensity (Ib), initial fluorescence intensity (Ii), and final fluorescence intensity (If) during a time period (Δt) in a blood vessel of a particular diameter (D):
Prior to experiments, devices were observed, paired by morphology, and then assigned to the control or experimental group; this protocol ensured consistent sample quality in each group. Unless otherwise indicated, all replicates were technical.
On days 7, 10, and 14, devices were assigned to either tissue analysis via immunofluorescence microscopy or individual cell analysis via flow cytometry and Methocult CFU assays. Devices designated for microscopy were fixed in 10% formalin for no more than 2 h. Devices designated for flow cytometry were digested with nattokinase (20 U/mL, Japan Bio Science Laboratory), pooled, and placed in a gel-coated well for 1 h to deplete stromal cells from the sample prior to analysis [35]. Egressed cells were collected from each niche by removing media from the fluidic lines (2 fluidic lines per niche, 4 total) and analyzed by flow cytometry.
To study cancer cell migration, tissues in the hexagonal chambers were cultured for 4 days; then, MDA-MB-231 (1 × 106 cells/mL) in a fibrin gel (10 mg/mL) were loaded into the bottom chamber and devices were cultured for an additional 8 days. Control devices were filled with fibrin gel and cultured with media for four days prior to loading cancer cells. Devices were imaged the day cancer cells were loaded (Day 0) and every 48 h until the end of the experiment (Day 8). To quantify observations, images were thresholded and assigned regions of interest (ROIs) in the hexagonal and bottom chambers (areas near ports were avoided to minimize artefacts); then, fluorescent area in the ROIs was measured. In order to calculate whether the niches bias cancer cell growth, the bottom chamber was divided into two equal parts, the fluorescent area in the perivascular side (PBC) was subtracted from the fluorescent area on the endosteal side (EBC) [26], and this value was divided by total fluorescent area in the bottom chamber (EBC + PBC) [27]:
2.6. Monolayer controls
Monolayer control studies were adapted from previously published reports [36,37]. Briefly, hFOB or BMSC were seeded with EC in a 1:1 ratio at a density of 800 cells/cm2 in a 6-well plate; 103 CD34+ cells were seeded into the same plates three days later. As an additional control, BMSC monolayers were seeded at a density of 800 cells/cm2 in a 6-well plate. hFOB + EC were cultured in EGM-2; BMSC + EC and BMSC were cultured in a 1:1 blend of fully supplemented EGM-2 and BMSC media. Monolayers were cultured for three days; then, media was aspirated, wells were rinsed with Dulbecco’s PBS (Gibco), and CD34+ cells were added. Subsequently, monolayers were cultured in a 1:1 blend of fully supplemented EGM-2 and SFEM StemSpan II + 1x CD34 expansion +1 μM SR1. At Days 7 and 10, half the media and the non-adherent cells were removed from the well and the media was refreshed. All non-adherent cells were collected at Day 14. Cells were analyzed via Methocult assays and flow cytometry.
2.7. Phosphorescence lifetime imaging microscopy (PhLIM)
Phosphorescence lifetime image microscopy (PhLIM) was used to measure O2 tension within the device, as detailed in previous publications [38,39]. Briefly, our FV1200 confocal microscope (Olympus), upgraded with a phosphorescent lifetime instrument (ISS, Urbana-Champaign, IL), was modulated at 10 kHz and 5% duty cycle to excite Oxyphor G4 dye (O2 Enterprises, Philadelphia, PA) via a 635 nm laser. Beam emission was collected by a miniTDU with two Hamamatsu 7422p-50 detectors attached directly to the confocal head. Our confocal microscope has a stage-top incubator (Okolab, San Bruno, CA) for O2 and temperature control.
BMoaC were loaded with 20 μM Oxyphor G4 dye on Days 6, 9 and 13, and O2 concentrations were measured on Days 7, 10 and 14 at 37 °C room air. A calibration curve correlating O2 concentration with Oxyphor G4 lifetime was generated for each device by measuring room air oxygen and lifetime measurements taken upstream of feeding lines. O2 concentration is expected to be close to the environmental O2. Five measurements were averaged to generate the phosphorescent lifetime of each pixel; this value was rank ordered to eliminate outliers (e.g., upper outlier is > 1.5*(IQ)+Q3 where IQ is the interquartile range and Q3 is the value of 3rd quartile). The remaining data was converted to percent O2 using the calibration curve. Finally, a mean O2 percentage was calculated for each tissue chamber.
2.8. Methocult assay
A Methocult assay (MethoCult™ H4034 Optimum, StemCellM Technologies) was performed per modified manufacturer’s instructions. Briefly, cells isolated from the device after nattokinase digestion were resuspended in IMDM (Gibco) with 2% FBS (Gibco) at a concentration of 1.3 × 103 cells/mL, mixed with MethoCult media, and plated in 60 mm2 Gridded Scoring Dishes (Corning) at a density of 360 cells/plate. Cells were cultured for 10 days and resulting colonies were imaged on an IX83 inverted microscope (Olympus). Colony types were identified manually by blinded researchers through comparison to gold-standard images provided by the vendor. To account for variation in seeding density and proliferation rates, we normalized our cell counts for each trial and condition by comparing Day 10 and Day 14 to the respective Day 7 CD45+ population. We refer to this correction as the dilution factor (DF).
2.9. Immunofluorescence
Devices were blocked overnight with 0.5 M Glycine (Sigma Aldrich) and 2% bovine serum albumin (BSA, Sigma Aldrich) in DPBS. Antibodies in 0.1% BSA were delivered through the fluidic lines and devices were stained overnight. Antibodies were chosen based on expected expression of targets in the native bone marrow (Table S1) and were used at 1:100 unless otherwise noted. Devices stained for osteopontin and leptin were permeabilized with 0.1% Tween-20. Devices were rinsed with DPBS between primary and secondary antibodies. Nuclei were counterstained with DAPI at a final concentration of 5 μg/mL or SytoxOrange (Invitrogen) at 5 μM. Before imaging, devices were rinsed and incubated with ProLong™ Antifade (ThermoFisher) per the manufacturer’s instructions to reduce photobleaching. Fluorescent and time-lapse images were acquired with an IX83 inverted microscope (Olympus) and multi-area confocal images were collected with an FV3000 Confocal (Olympus). For the confocal, devices were imaged with Multi-Area-Time-Lapse (MATL) acquisition so that both niches could be imaged simultaneously. Image post-processing was done either with FV310 or in FIJI. Device images were stitched using the FIJI pairwise stitching plug-in Ref. [40] or by hand in PowerPoint. CD34+ cell quantification was done in FIJI by cropping the image to the hexagonal chamber, thresholding, utilizing the watershed tool, and analyzing particles that were greater than 1002 pixels with a circularity of 0.1–1.
For the stromal cell migration experiments, hFOB and BMSC were labeled with Cell Tracker™ Violet BMQC and Deep Red (Thermo Fisher Scientific), respectively, according to the manufacturer’s instructions. Labeled stromal cells were mixed with EC and loaded into the BMoaC; images of the devices were collected on Day 4 using the confocal microscope. Devices loaded with unstained stromal cells were used as a control. Cell fluorescent area was calculated by thresholding the image, inverting, and analyzing particles. Obvious pieces of auto-fluorescent debris in the fluidic line were removed when appropriate.
2.10. Ki-67 staining and quantification for BMoaC cancer cell migration
Devices were permeabilized with 0.3% Triton X-100. Nuclei were counterstained with DAPI before blocking with 0.5 M Glycine (Sigma Aldrich) and 2% bovine serum albumin (BSA, Sigma Aldrich) in DPBS. Devices were counterstained with a monoclonal rabbit anti-human CD34 (Abcam) at 1:100 followed by a secondary AlexaFluor 488 goat anti-rabbit (Invitrogen). BMoaC and fibrin only controls were counterstained with conjugated AlexaFluor 647 rabbit anti-human Ki-67 (Cell Signaling Technologies) at 1:50. All staining steps were performed overnight at 4 °C. Rinses were performed between each step at room temperature. Z-stack images were acquired every 10 μm in the Z-plane to ensure all the cells were captured with little to no overlap, and MATL acquisition enabled the entire device to be imaged.
Custom ImageJ macros were written to separately quantify Ki-67 in the endosteal, perivascular, and cancer chambers. First, a mask was created from the RFP signal and used to separate cancer cells from other cell types in the tissue. Next, DAPI signal within the cancer cells was identified through particle analysis. Then, Ki-67 signal was thresholded and compared to DAPI signal from cancer cells; colocalization of Ki-67 and DAPI signals identified Ki-67+ cancer cells. The number of Ki-67+ and Ki-67- cancer cells was recorded in each Z-stack and chamber.
2.11. Flow Cytometry
Cells were blocked in 100 μL of a 0.1% BSA solution for 15 min at 4 °C and then stained with a panel of conjugated antibodies for specific target antigens (provided in Table S2) for 1 h at 4 °C. Cells were washed with PBS and then exposed to a fixable live/dead marker for 30 min. Cells were washed again in PBS and analyzed using an Attune NxT Acoustic Focusing Cytometer (ThermoFisher #A24858). Compensation controls were generated using single-stained populations of positive beads and negative beads (Spherotech #CMIgP-30). Data analysis and compensation correction was performed using FlowJo software. Target cell populations were gated using fluorescent minus one (FMO) controls in cryopreserved peripheral blood mononuclear cells (PBMCs).
2.12. Statistical analysis
All statistical analysis was performed using GraphPad Prism 8. T-tests were performed where indicated. Analysis of variance (ANOVA) followed by a Tukey’s or Sidak’s test for post-hoc comparisons were performed as indicated in the paper. All data is represented as the mean with the standard deviation included.
3. Results
3.1. Microfluidic device recreates scale and physical environment of the bone marrow
The microfluidic device was designed to recreate the scale of the bone marrow’s endosteal and perivascular microenvironments while maintaining the tissue’s fundamental biological function - hematopoiesis. To this end, the device consists of two adjacent hexagonal chambers connected by symmetrical two-way ports, which enable niche-niche communication and cell trafficking (Fig. S1), but also facilitate sequential loading of the cellularized fibrinogen gel and limit leakage into the adjacent microfluidic lines [41,42]. A third bottom chamber is connected to both hexagonal chambers by one-way ports; the design (Fig. 1A and B) allows the bottom chamber to be kept dry while the top two chambers have active tissue cultures. At a later timepoint, the bottom chamber can be loaded with a tissue and the ports allow transport of media and cells.
Our lab has previously demonstrated that interstitial flow in microfluidic devices facilitates formation of dynamic, perfusable microvascular networks by vasculogenesis [25,41]. As a first step toward creating microvascular networks in the bone marrow device, the interstitial flow was modelled using COMSOL to identify conditions favorable for vasculogenesis (Fig. 1C) [23,41]. In this simulation, pressures ranging from 4 to 20 mm H2O in the fluidic lines feeding each hexagonal chamber (Fig. S1) produced interstitial flow ranging from 6 to 20 μm/s. The interstitial flow varied with position in the chamber, with slower flow in the middle of the chamber and higher flow near the ports (Figs. 1C and S1). These flow rates support vasculogenesis and capillary formation in microfluidic devices [25,41]. To verify these theoretical interstitial flows, we observed transport of a bleached circular region (Fig, 1D), which was created using fluorescence recovery after photobleaching (FRAP) on an Olympus FV3000 confocal microscope (Fig. 1D). We measured experimental interstitial velocities of 17.7 μm/s (±4.71 μm/s, n = 3) and 4.48 μm/s (n = 1) adjacent to the high-pressure ports and in the middle of the chamber, respectively. These values align with the theoretical values measured in the same regions.
Additionally, the transport of molecules similar in size to secreted factors was modelled in COMSOL (Fig. 1E) and measured experimentally using 70 kDa FITC-dextran and TRITC-dextran introduced into the left and right hexagonal chambers, respectively (Fig. 1F). Due to the symmetric design of the chambers and the operating conditions, convective flow between the chambers is near zero (Fig. 1E and F) and the mode of molecular transport is primarily diffusion. This convective isolation also occurs in the adjacent bottom chamber, with a sharp spatial transition (concentration gradient) in the middle of the chamber (Fig. 1E–G). The mean fluorescence intensity of the FITC-dextran in the bottom chamber (horizontal dashed line in Fig. 1E and F) mapped closely to the theoretical concentration profile (Fig. 1G).
3.2. Bone marrow-on-a-chip (BMoaC) recapitulates in vivo markers of bone marrow
To model the human bone marrow niches, we focused on the tissue’s major features: a vascular network, each niche’s characteristic stromal cell population, and HSPC. The stromal cell is a key regulator of the niches; it secretes soluble molecules and maintains the extracellular matrix. We and others have also demonstrated that stromal cells are a necessary component to create and maintain a 3D vascular network [28, 43]. Leveraging this expertise, we generated perfusable vascular networks by combining human cord blood derived-EC with a stromal cell (1:1 ratio) in a fibrin matrix, where the perivascular and endosteal niches were distinguished by the identity of the stromal cell population. To model the osteoblast – the major stromal cell in the endosteal niche – we chose the human fetal osteoblast cell line hFOB 1.19 (ATCC, CRL-11372) based on simplicity of culture conditions, ease of dissemination, and ability to differentiate into mature osteoblasts. For the perivascular niche, we chose primary bone marrow stromal cells (BMSC, Lonza), which contain a variety of stromal cells (e.g., fibroblast, mesenchymal stem cells) but no osteoblasts. To complete our bone marrow model, we simply added HSPC (CD34+) sourced from human cord blood to the device.
First, we characterized the phenotype of the stromal cell populations as monolayers and as a co-culture with EC in the bone marrow-on-a-chip (BMoaC). hFOB and two BMSC donors were characterized as individual cell populations by flow cytometry prior to injection into the device. Both populations of stromal cells had low levels of CD31 (<0.1%), CD34 (<5%), CD45 (<2.5%), CD36 (~1.4%), and CXCL-12 (~5%); and expressed similar levels of CD44 (~96%), CD105 (~98%), and CD90 (~56%) (Fig. 2A). The hFOB and two BMSC donors also expressed CD73, NG2, CD10, and ALP (Fig. 2A), with some differences between the donors.
Fig. 2. Stromal cells cultured in BMoaC express proteins found in native marrow.
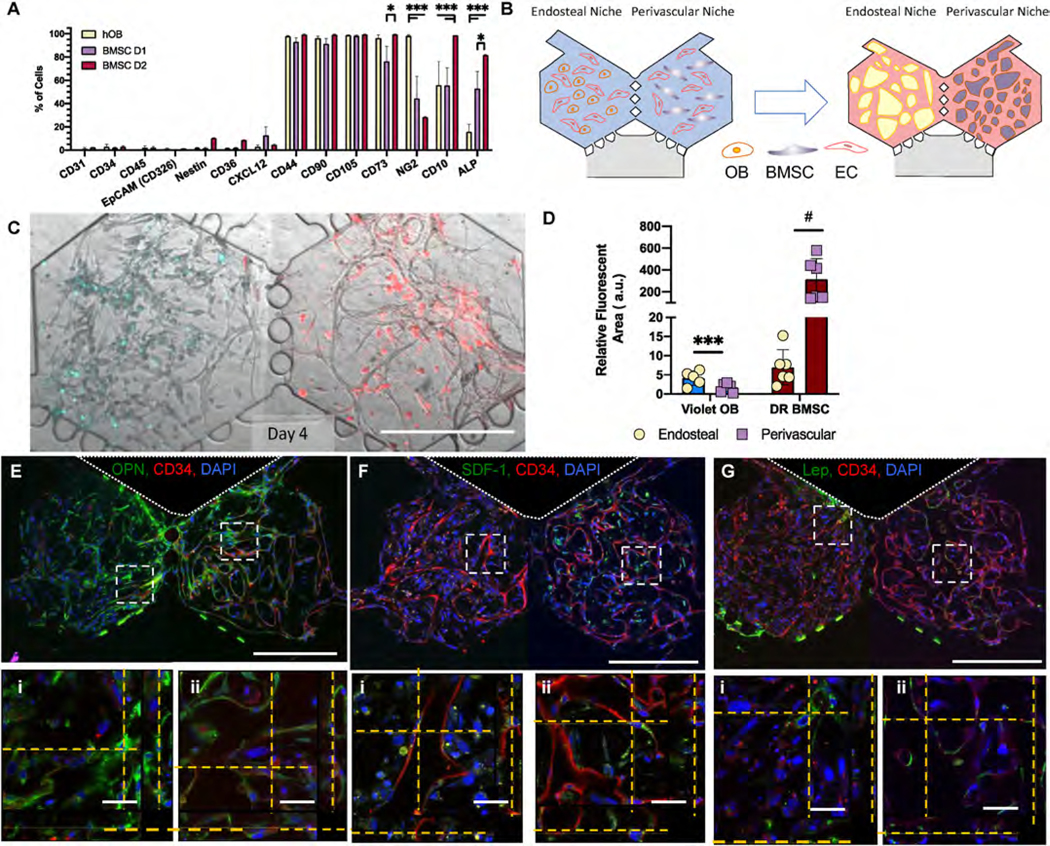
(A) Marker expression of independently cultured monolayers of hFOB and two BMSC donors as determined by flow cytometry, n = 4. (B) Cartoon schematic of experimental design. Cord blood-derived endothelial cells (EC) seeded with either hFOB (OB, Endosteal Niche) or BMSC (Perivascular Niche) form microvascular networks in a period of 4–7 days. (C) BMoaC cultured with CellTracker™ Violet hFOB and Deep Red BMSC demonstrated stromal separation by niche. (D) Relative fluorescent area (RFA) of Violet hFOB in the endosteal niche compared to unstained BMSC within the perivascular niche and RFA of unstained hFOB in the endosteal niche to Deep Red BMSC in the perivascular niche. Fluorescent area was determined by normalizing to an unstained control. *p < 0.05, ***p < 0.001 by two-way ANOVA. (E) Osteopontin (green) was expressed in the (i) endosteal and (ii) perivascular niches. EC (red) counterstained with CD34. (F) SDF-1 was detected adjacent to EC (red) in the (i) endosteal niche with enhanced expression of SDF-1 in the (ii) perivascular niche. (G) EC (red) were adjacent to stromal cells that express leptin as seen in the (i) endosteal and (ii) perivascular niche. Nuclei counterstained with DAPI (blue). Scale bars: 500 μm and 50 μm for low and high magnification images, respectively.
To generate the vascular networks in each niche, EC were mixed with either hFOB or BMSC in a fibrin hydrogel and injected into either the left or right hexagonal chamber, respectively, to generate a 3D tissue (Fig. 2B). Formation of vascular networks was observed between Day 4 and Day 7 (Fig. S2A). Networks formed in the endosteal niche had significantly shorter total and average vessel length (Student’s t-test p < 0.05; Figs. S2B) and a higher number of vessel endpoints (p < 0.001) compared to the networks within the perivascular niche. To determine whether stromal cells migrate between chambers, we labeled hFOB and BMSC with the membrane dyes CellTracker™ Violet and Deep Red, respectively, prior to injection into the chambers. Following formation of vascular networks, we imaged the niches using fluorescence microscopy and observed that the Violet hFOB and Deep Red BMSC remain primarily within their respective niches (Fig. 2C and D).
Next, we characterized the phenotype of stromal cells in each niche. After tissue development in the device, both niches stained positive for osteopontin (OPN, Fig. 2E); OPN+ cells were closely associated with EC in the perivascular but not the endosteal niche. Both niches also stained positively for SDF-1/CXCL-12 (Fig. 2F). Very few stromal cells were leptin+ in either niche (Fig. 2G). As an important control, we also created single niche (SN) devices in which both hexagonal chambers contained either the endosteal niche or the perivascular niche. The SN control devices retained the protein expression observed in the BMoaC (Fig. S3), with the exception of SDF-1/CXCL-12, which appeared lower in the SN perivascular niche control than when both niches were present in the BMoaC.
Then, we assessed whether EC cultured in BMoaC expressed biochemical factors and proteins that are associated with bone marrow endothelium in vivo. After 7 days of culture, BMoaC were fixed and stained with antibodies against the basement membrane marker laminin (Fig. 3A); the transmembrane proteins Stem Cell Factor (SCF) (Fig. 3B), ICAM-1 (CD54) (Fig. 3C), VCAM-1 (CD106) (Fig. 3D), and E-selectin (CD62E) (Fig. 3E); and either CD34 or CD31, which are EC markers (Fig. 3A–F). EC expressed SCF on the abluminal side of the vessel wall in both niches (Fig. 3B). hFOB and some BMSC also stained for SCF throughout the niche. EC in both niches also expressed the cellular adhesion markers ICAM-1, VCAM-1, and E-Selectin (Fig. 3 C-E) without stimulation by inflammatory factors like TNFα; this phenotype is typical of EC found in the bone marrow [14,15]. The basement membrane protein laminin was clearly seen on the abluminal side of the vessel wall in both niches (Fig. 3B). Co-expression of these markers with an EC-specific marker (CD34 or CD31) was easily detected at high magnification (Fig. 3A–F). hFOB also expressed VCAM-1. Vascular networks were also positive for nestin (Fig. 3F), which identifies arterioles in the bone marrow [3], and were perfusable, as measured using 70 kDa TRITC-dextran (Fig. 3G., Sup. Vid. 1). Vessel networks in the endosteal niche were more permeable than those in the perivascular niche (17.0 × 10−7 ± 8.90 × 10−7 cm/s and 5.2 × 10−7 ± 3.82 × 10−7 cm/s, respectively, p < 0.05)).
Fig. 3. Endothelial cells in BMoaC expressed proteins found in native bone marrow.
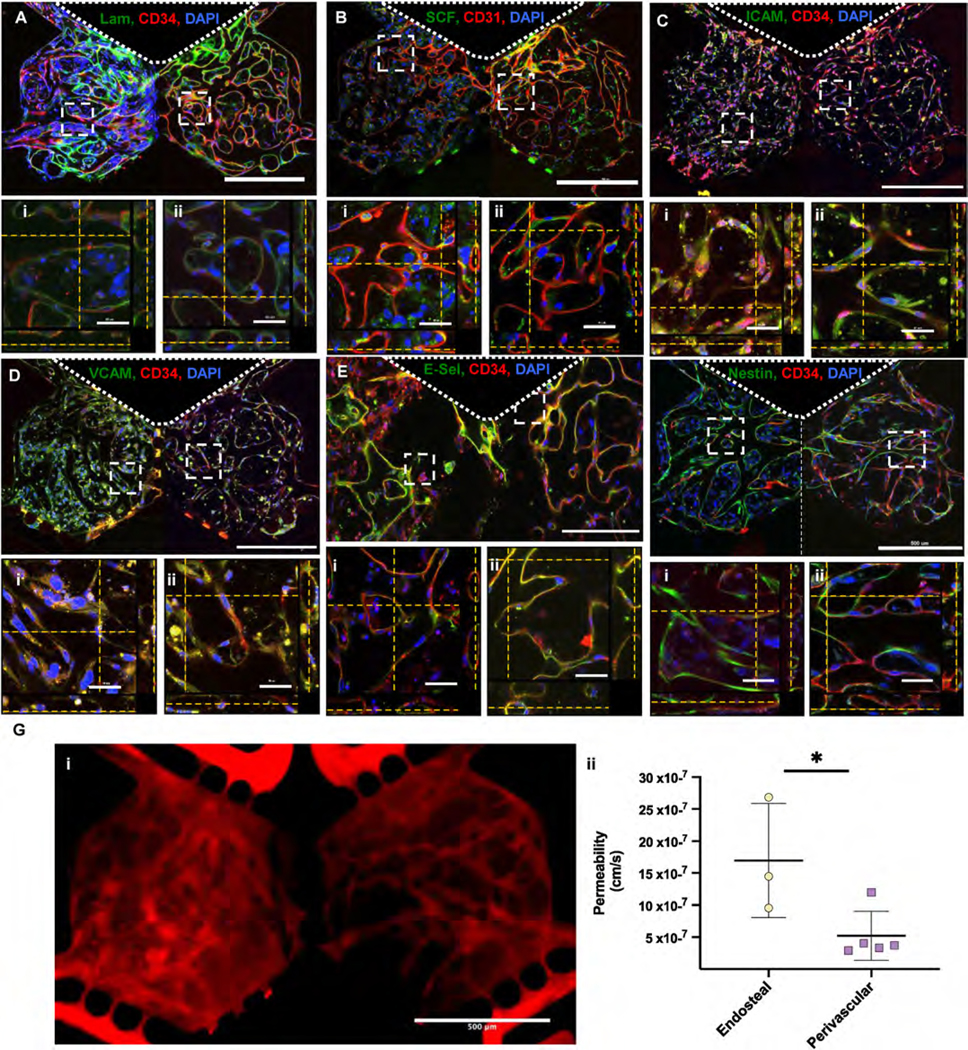
(A)–(F) Endothelial cell (red, CD31 or CD34) (A) laminin in the basement membrane, (B) SCF, (C) I-CAM, (D) V-CAM, (E) E-Selectin/CD62E and (F) Nestin. ROIs show z-stacks in either the (i) endosteal or (ii) perivascular niche. Nuclei were counterstained with DAPI (Blue). Scale bars: 500 μm and 50 μm for low and high magnification views, respectively. (G) BMoaC microvascular networks were i) perfused with 70 kDA TRITC-dextran and ii) had permeabilities of 16.96 × 10−7 cm/s and 5.19 × 10−7 cm/s for the endosteal (n = 3) and perivascular niches (n = 5), respectively.
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.biomaterials.2021.121245
3.3. BMoaC supports maintenance of hematopoietic stem/progenitor cells
To complete the BMoaC model, we introduced hematopoietic stem/progenitor cells (HSPC; CD34+) into each of the niches. The HSPC were isolated from cord blood and cultured in each niche over a period of 7–14 days (Fig. 4A). Individual experiments were carried out with a single donor (fourteen donors total) and sample purity was assessed by flow cytometry. Prior to loading, the HSPC were on average 90.5% ± 6.4 CD34+ and 78.4% ± 8.6 CD34+/CD133+, which is consistent with a relatively pure stem/progenitor population; samples with purity below 80% CD34+ cells were not used (Fig. 4Bi, S4.). On average, 61.4 ± 25.7 and 42.4 ± 12.9 CD34+ cells were loaded into each individual endosteal and perivascular chamber, respectively (Fig. 4B(ii)). CD34+-derived cells were visible by brightfield in both niches as early as day 4 and proliferated within the device over the culture period (Fig. S4B). Small round cells could be clearly seen within the lumen of blood vessels in the perivascular niche (Fig. S4B, Day 12). CD34+-derived cells were also observed trafficking within the main chambers of the BMoaC and into the fluidic lines using time-lapse microscopy (Sup. Vid. 2). BMSC donor did not affect the proliferation or behavior of CD34+-derived cells. CD34+ cells loaded into the device without EC or stromal cells failed to proliferate after 7 days.
Fig. 4. BMoaC maintained a population of CD34+ HSPC.

(A) Experimental design. (B) (i) CD34+ cells isolated from human cord blood were 90.4% ± 6.37 CD34+ and 78.4% ± 8.57 CD34+/CD133+, n = 14. (ii) Approximately 40–60 CD34+ HSPC were loaded into each device, n = 21 devices. (C) BMoaC supported CD34+ blood vessels. (i,ii) Small, round CD34+ HSPC (arrows) were present along with Lin+ cells. Scale bars: 500 μm and 50 μm for low and high magnification images, respectively. (D) Cells isolated after 7, 10, and 14 days of culture in the BMoaC were analyzed for CD34/CD133 from both the endosteal and perivascular niches, biological replicates with n = 5. (E) Cells isolated after 7, 10, and 14 days of culture from either niche in the BMoaC consistently generated CFU-GEMM, biological replicates with n = 10–11. (F) Cells isolated after 10 and 14 days of culture within single niche (SN, endosteal or perivascular) controls produced CFU-GEMM; monolayer controls did not.
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.biomaterials.2021.121245
To analyze maintenance of CD34+ cells in the BMoaC, the tissue matrix was digested to release cells from the device and expression of CD34, CD133, and lineage (Lin; a mix of antibodies to CD3, CD14, CD16, CD19, CD20, and CD56) was measured with flow cytometry (Figs. S4D–E). These measurements were supported by qualitative observations made via confocal microscopy (Fig. 4C). In both niches, we observed clusters of Lin+ cells with kidney-shaped/multi-lobular nuclei and small, round CD34+ cells (both are distinguishable from CD34+ elongated EC) surrounded by a blood vessel (Fig. 4Ci, S4D, Sup. Vid. 3). BMoaC maintained both a population of Lin−/CD34+ cells and a differentiated population of Lin+/CD34− cells, as identified by flow cytometry (Fig. S4D). Both niches maintained a small CD34+/CD133+ population, as well as a CD34+/CD133− population, over the culture period (Fig. 4D). There was no significant change in either the CD34+/CD133+ or CD34+/CD133− cell population percentages after Day 7 (n = 5), though an upward trend in the absolute quantity of these stem cell-like populations in the perivascular niche was observed.
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.biomaterials.2021.121245
Methocult assays were performed with cells isolated from each niche of the BMoaC, to assess the colony forming unit (CFU) potential of cultured HSPC. We examined burst forming unit-erythroid (BFU-E); colony forming unit-granulocyte, macrophage (CFU-GM); and colony forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte (CFU-GEMM) (Fig. S5A). Cells isolated from the BMoaC after 7, 10, and 14 days of culture were capable of forming CFU-GEMM (Fig. 4E), as well as colonies representing granulocyte, erythroid, monocyte, and megakaryocyte lineages (Figs. S5B, D, F). The number of HSPC on each day and in each device may be estimated from the number of CFU-GEMM colonies and a dilution factor (see Methods for details). The number of cells capable of generating a CFU-GEMM colony was stable between Day 7 and Day 14 (7.36 ± 6.7 and 6.0 ± 5.6 cells per device, respectively; p > 0.05). SN and monolayer controls were performed to assess whether both niches were necessary to maintain the HSPC. Cells isolated from SN control devices generated GEMM colonies at Day 10 and Day 14 of culture (Fig. 4F), as well as other colonies (Figs. S5C, E, G). Monolayer controls had very limited CFU potential, including CFU-GEMM (Figs. 4F, S5C, E, G), despite hosting a CD45+ population containing a high percentage of stem/progenitor cells (CD34+/CD133+ and CD34+/CD133−) compared to SN controls (Fig. S5H).
BMoaC were incubated at 37 °C with 5% CO2 and ambient oxygen for 14 days. Mean O2 tension within each niche was determined using phosphorescence lifetime imaging microscopy (PhLIM) and the oxygen sensitive dye Oxyphor G4 [38,39] on days 7, 10 and 14 (Fig. S7). O2 tension at Day 14 (88.7% of ambient oxygen) was slightly lower than at Day 7 (97.5%) (Fig. S6) within both niches, consistent with enhanced cellular density and metabolism.
3.4. BMoaC supports CD45+ cell differentiation, expansion, and egress
In vivo, the bone marrow generates leukocytes of the innate immune system, including monocytes (CD14+) and neutrophils (CD66b+), and the BMoaC can replicate this differentiation of CD34+ cells into Lin+ cells over time. In the BMoaC, both niches contained CD14+ cells in extravascular spaces, within the blood vessels, and adjacent to the vessels (Fig. 5A). Next, we characterized egressed cells – or, cells that exit the tissue chamber and enter the fluidic lines. When collected on day 14 and assessed by flow cytometry, egressed cells were positive for the pan-immune cell marker CD45 (Fig. 5B) and contained a population of CD45+/CD66b+/CD33− cells, which is consistent with a mature neutrophil (Fig. 5C).
Fig. 5. BMoaC supported cellular differentiation and egress of cells from the myeloid lineage.

(A) After 10 days of culture, BMoaC contain cells expressing CD14 (Green) in both sides of the device and in the (i, ii) lumen of CD34+ blood vessels (red). Nuclei were counterstained with DAPI (blue). Scale bars: 500 μm and 50 μm for low and high magnification images, respectively. Cells that egressed from the bone marrow niche and into the adjacent fluidic line of devices were collected on Day 14 and analyzed by flow cytometry for (B) the leukocyte common antigen CD45 and (C) the myeloid marker CD33 and neutrophil marker CD66b. (D) BMoaC exposed to 0.5 M doxorubicin between days 8 and 10 of a 14 day culture produce significantly fewer egressed CD66b+/CD33− cells from the perivascular niche compared to control devices. **p < 0.01 by 2-way ANOVA followed by Sidak’s post hoc-analysis. (E) BMoaC exposed to 30 ng/mL G-CSF produced significantly more egressed CD66b+/CD33− cells from the perivascular niche compared to control devices. *p < 0.05, **p < 0.01 by 2-way ANOVA followed by Tukey’s post hoc-analysis.
Next, we tested the ability of the BMoaC, over 14 days of culture, to: i) mimic the neutropenic effect of chemotherapy, or ii) increase neutrophil production via G-CSF exposure. Monolayer controls and BMoaC were exposed to 0.5 M doxorubicin from Day 8–10 of culture (48 h). On Day 14, cells were collected from the device and analyzed via flow cytometry. Doxorubicin significantly reduced the viability of adherent and non-adherent cells in monolayer controls compared to the BMoaC, which demonstrated reduced viability only in egressed cells from the perivascular niche (Fig. S7A) (n = 3; 2-way ANOVA with a Sidak-post hoc comparison *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). In addition, the ratio of CD66b+/CD33− cells to CD66b−/CD33+ cells increased significantly in monolayer cultures following doxorubicin exposure, which suggests an enhanced sensitivity for myeloid progenitor cells (CD66b−/CD33+ cells) cultured in monolayer (Fig. S7B). Within the BMoaC, egressed CD66b+/CD33− cells from the perivascular niche decreased from ~250 cells/device for control devices to ~4 cells/device for treated devices (2-way ANOVA with Sidak post-hoc comparison, **p ≤ 0.01), consistent with a neutropenic response (Fig. 5D).
We followed the same protocol to analyze the impact of 30 ng/mL G-CSF on the BMoaC. There was a significant increase in the egress of CD45+ cells from the perivascular niche following treatment and from the perivascular versus endosteal niche under treated and untreated conditions (Fig. S7C). The ratio of CD66b+/CD33− cells to CD66b−/CD33+ cells increased in monolayer controls and in tissue resident cells of the BMoaC perivascular niche (Fig. S7D). The monolayer response is consistent with previous reports [44,45]. Following G-CSF exposure, we observed a decrease in the number of CD66b− /CD33+ cells within the BMoaC perivascular niche compared to the control (Fig. 5E), which was countered by an increase in the number of egressed CD66b−/CD33+ cells from the perivascular niche (p < 0.05, 2-way ANOVA, Sidak’s post hoc comparison). Treatment also increased the egress of CD66b+/CD33− and CD66b−/CD33+ cells from the BMoaC perivascular niche compared to the endosteal niche. The more mature population of CD66b+/CD33− cells within the BMoaC did not change following exposure to G-CSF (Fig. 5F).
3.5. Breast cancer cells migrate into BMoaC niches
The BMoaC design includes a third bottom chamber to observe the interaction between an additional tissue of interest (e.g., cancer cells) and the vascularized bone marrow niches. To demonstrate this feature, BMoaC were cultured for 4 days to allow for tissue development. Then, MDA-MB-231 expressing red fluorescent protein were loaded in a fibrin gel in the bottom chamber to simulate cancer cell invasion (Fig. 6A). As a control, cancer cells were also loaded into devices containing only fibrin in the hexagonal top chambers, which were cultured with media for 4 days prior to introduction of cancer cells. Next, BMoaC and fibrin controls were cultured for an additional 8–9 days and cancer cell migration into both chambers was observed (Fig. 6B and C). Cancer cells had similar rates of growth in the bottom chamber for both fibrin controls and BMoaC (Fig. S8A, n = 4 for fibrin controls, n = 6 for BMoaC). Growth patterns in the bottom chamber were not biased by soluble signaling from either the endosteal or perivascular niche (Fig. S8B). Fibrin control devices had minimal growth and migration into the hexagonal top chambers (n = 4). In contrast, MDA-MB-231 readily migrated into the adjacent BMoaC tissue chambers (n = 6) between days 4 and 8 (Fig. 6C), with no statistically significant differences between the endosteal and perivascular niches. When devices were stained for Ki-67, a marker of proliferation, we observed significantly more Ki-67+ cancer cells in both the endosteal (19.7 ± 26.1 vs 54.0 ± 17) and perivascular niches (8.0 ± 16.0 vs 66.7 ± 7.9) compared to the fibrin only controls (**p ≤ 0.01 and ***p ≤ 0.001, 2-way ANOVA with Tukey-post hoc comparison). There was no difference in the percentage of Ki-67+ cells in the bottom chamber of the fibrin control compared to the BMoaC (39.0 ± 15.4 vs 28.0 ± 9.7), consistent with the observed patterns of growth. These preliminary experiments replicate features of metastasis to the bone marrow and demonstrate the utility of the BMoaC to study the interaction of cancer cells with each niche of the bone marrow.
Fig. 6. Breast cancer cells migrated toward and proliferated in the endosteal and perivascular niches.

(A) Experimental design. Breast cancer cells were loaded into the bottom chamber of the BMoaC on Day 4 and cultured for an additional 8 days. (B) The triple negative breast cancer cell line MDA-MB-231 (green) proliferated over time and migrated into the bottom portion of both the endosteal and perivascular niches. EC and HSPC (red) were labeled with CD34 antibody and nuclei (blue) were counterstained with DAPI. Scale bar: 500 μm. The fluorescent area of cancer cells in each chamber of the device was quantified. (C) MDA-MB-231 cells migrated towards the endosteal and perivascular niches after 8 days of culture in the BMoaC. (D) MDA-MB-231 cancer cells in the BMoaC perivascular and endosteal niches versus fibrin only controls expressed significantly more Ki-67, suggesting active proliferation. Cancer cells in the BMoaC perivascular niche expressed more Ki-67 than those in the bottom chamber. *p < 0.05, **p < 0.01, ***p < 0.001 by 2-way ANOVA followed by Tukey’s post hoc-analysis.
4. Discussion
The ability to model human bone marrow in vitro and with high spatiotemporal resolution has the potential to impact how we study and understand an array of normal biological processes and diseases in the bone marrow. Our primary goal was to create a microphysiological mimetic of the bone marrow and demonstrate maintenance of HSPC, differentiation of the HSPC into primary lineages (i.e., hematopoiesis), and release (egress) of mature neutrophils in the platform. Since distinct niches are thought to exist in the bone marrow, we sought to physically separate the perivascular and endosteal niches, thus enabling niche-specific analyses of HSPC maintenance and function. To accomplish this goal, we created a model system with a dynamic, 3D, perfusable vascular network and separate, but adjacent, perivascular and endosteal niches. Our data demonstrate that both niches contribute to the microenvironment needed to maintain a small population of HSPC in vitro for up to two weeks. Furthermore, we observed generation of microvascular networks with physiological permeability and expression of soluble and membrane-bound proteins, differentiation of HSPC into both myeloid and erythroid lineages, egress of CD66b+/CD33− neutrophils into adjacent microfluidic lines, and a response to doxorubicin and G-CSF that is consistent with in vivo observations in human peripheral blood. Finally, our device incorporates an additional chamber that can be used to study the trafficking of tumor cells into the perivascular and endosteal niches; such experiments may provide insight into cancers that preferentially metastasize to bone marrow (including breast and prostate).
Maintaining, and in particular expanding, HSPC in vitro has proven challenging due to rapid differentiation and population outgrowth of cells that secrete soluble factors which inhibit HSPC expansion. Some success has been achieved in suspension and fed-batch culture systems that specifically inhibit these signals and/or stimulate the HSPC with factors such as stem regennin 1 (SR1), Notch-Delta like ligand, Angiopoietin-like 5, and IGFBP2R [46–48]. The primary goal of these systems is expansion of HSPC for clinical applications, including bone marrow transplantation; in contrast, our goal was to recreate the in vivo niche’s ability to maintain a small population of HSPC and observe egress of neutrophils into the microfluidic lines as a mimic of the systemic circulation. This stem cell niche is thought to be closely associated with the abluminal surface of the endothelial cell [3,4,49], and also thought to be a preferred location for metastatic breast cancer cells [1, 16,18]. Both of the in vitro niches in our platform contain an endothelial abluminal surface; however, our results demonstrate that both niches together are required to support a small but steady population of HSPC. Thus, it is likely that soluble factors derived from more than one stromal cell population are required to maintain HSPC.
Previous attempts to recreate human bone marrow using tissue engineering or “organ-on-a-chip” technologies are limited in number, but have successfully recreated several seminal features of bone marrow, including hematopoiesis, maintenance of HSPC, the potential for breast cancer cell metastasis, and drug response [19,20,50,51]. The studies by Torisawa and colleagues [19] were limited by the use of mouse HSPC and in vivo culture time within the mouse. Sieber and colleagues [51] demonstrated success using human cells, but did not include EC or any model of the vascular system. Marturano-Kruik and colleagues [50] presented a model of bone marrow that included a bone scaffold with EC and mesenchymal stem cells. This model demonstrated the formation of rudimentary blood vessels and the potential for studying the metastasis of breast cancer cells, but did not demonstrate hematopoiesis. Chou and colleagues [20] reported a model that recapitulates hematopoiesis, bone marrow drug toxicity, and features of a genetic disorder that impacts the bone marrow. However, this model was not able to demonstrate the functional maintenance of HSPC (i.e., lack of CFU-GEMM) and did not include an endosteal niche. In addition, all of the previous models are relatively large in size (3–7 mm), limiting the possibility of higher throughput applications. While it is difficult to recreate all features of bone marrow in a single model, our platform, which is < 1 mm, demonstrates: a 3D, perfusable vascular network; separate perivascular and endosteal niches; hematopoiesis; migration and egress of neutrophils; maintenance of HSPC, including the ability to form CFU-GEMM; and migration of breast cancer cells into the niche.
The two niches in our model demonstrated several similarities. For example, both niches were able to maintain a small population of CD34+/CD133+ cells when cultured together in the BMoaC for 14 days, mimicking the function of adult marrow (0.1%–5%) [52,53]. Tissues formed with both the hFOB and BMSC expressed hallmark proteins and markers – such as SCF, SDF-1/CXCL-12, and E-selectin/CD62E – that are constitutively expressed in native bone marrow [4,18]. In addition, by flow cytometry and immunofluorescence, both niches were able to stimulate the differentiation of CD34+ HSPC into Lin+ cells; further, these cells exited both niches and entered into the fluidic lines of the device, mimicking the in vivo process of leukocyte egress from the bone marrow into the circulation. However, the niches displayed differences in response to doxorubicin and G-CSF. The tissue resident and egressed cells from the BMoaC endosteal niche were relatively resistant to both drugs. In contrast, cells from the BMoaC perivascular niche demonstrated sensitivity to both drugs. The niches also displayed differences in the vascular network. For example, the two stromal populations differed in their vasculogenic potential; the hFOB formed robust vascular networks less reliably than the BMSC. The networks derived in the presence of hFOB had a significantly higher number of blood vessel endpoints—indicative of a less connected network—and also demonstrated a higher permeability to 70 kDa TRITC-dextran, consistent with the higher permeability of the sinusoidal blood vessels present in the bone marrow in vivo. These differential observations between the niches suggest that, in vivo, the less permeable vessels supported by the BMSC (and not the osteoblast) may play an important role in the differentiation and selective release of neutrophils into the systemic circulation. Finally, differences in CFU between the endosteal and perivascular SN compared to the BMoaC indicate that inter-niche communication may be needed to maintain a small steady population of HSPC in the bone marrow. The small steady number of CFU-GEMM in the BMoaC suggests that, when cultured together, these two niches maintain an erythro-myeloid progenitor state.
While the small volume of the tissue chambers in our model facilitates higher throughput applications and thus is a strength, we also understand that this feature limits the number and enhances the variability of HSPC that are initially seeded. In the Methocult assay, we seeded a constant number of cells from each device/experiment and, although we accounted for the dilution by differentiated cells, we observed a wide range of dilution factors. Thus, the small number of HSPC per chamber could account for the large variation in the number of CFU that we observed in the Methocult assay (Fig. 4 and Fig. S5). Future applications of our model must balance the need for throughput with the inherent variability in HSPC cell seeding density.
Besides providing a model of hematopoiesis, our microfluidic model of human bone marrow responded to exposure of doxorubicin and G-CSF by decreasing or increasing, respectively, the number of egressed cells from the device. Consistent with reports from other 3D models [54–56], the BMoaC appeared less sensitive to the cytotoxic effects of doxorubicin than the monolayer controls; in fact, treated and control BMoaC had similar numbers of viable cells. Importantly, we observed a decrease in the number of egressed CD66b−/CD33+ and CD66b+/CD33− cells from the perivascular niche compared to controls, consistent with a neutropenic response that occurs following chemotherapy in cancer patients. Similarly, when devices were stimulated with G-CSF, the perivascular niche released a higher number of CD66b+/CD33− cells, consistent with in vivo observations of healthy adults who received G-CSF [57,58].
The BMoaC provides an additional chamber to investigate cell migration and/or soluble mediator communication between the bone marrow and an alternate tissue type. Here, we used this chamber to examine interactions between breast cancer cells and bone marrow tissue. We observed MDA-MB-231, a triple negative breast cancer cell, migrate into the bone marrow but not into fibrin controls, consistent with the release of chemotactic factors from the bone marrow that attract cancer cells. Additionally, cells that migrated into the BMoaC had higher expression of the cell proliferation marker Ki-67 compared to cells in the bottom loading chamber, consistent with high Ki-67 expression in bone lesions [22,24]. To maintain the scope of our detailed initial model description, we did not explore the mechanism of communication between the bone marrow and cancer cells, which may include soluble mediators such as SDF-1/CXCL-12 or exosomes [59]. Thus, the BMoaC may be a relevant model for breast cancer cell metastasis, but could also be adapted for studies of hematological-based malignancies such as leukemia and myelodysplastic syndromes.
Finally, development of organ-on-a-chip models requires careful selection of which organ-specific features to include and which to exclude; accordingly, several features of the bone microenvironment are not included in our model. For example, innervation of the bone plays a central role in bone development and remodeling [60]. We chose to exclude this feature for simplicity and the fact that innervation is not likely to play a critical role in adult hematopoiesis. The mineralized bone present in vivo could impact the phenotype and function of the endosteal niche. We chose to model the endosteal niche with only the stromal cell (osteoblast) and a vascular network, and our results indicate this provides an adequate environment for hematopoiesis and drug response. Nonetheless, we acknowledge that both innervation and mineralized bone may be important features of the bone microenvironment for other applications, including accurate replication of a broader array of drug responses.
5. Conclusions
In this study, we present a novel microfluidic device that is capable of supporting two separate niches of human bone marrow while maintaining physiological proximity and communication. The microtissue model of human bone marrow displayed a perfusable, 3D vascular network and expression of ECM proteins and other biomarkers that are present in the native bone marrow. Importantly, CD34+ HSPC were cultured and maintained in the device for up to two weeks and were able to differentiate into mature neutrophils (CD66b+/CD33−) that could egress from the marrow and into adjacent microfluidic lines, thus providing a model of systemic neutrophil production. Finally, an adjacent chamber can be loaded at a later time point with an alternate cell of interest, in order to investigate its interaction and communication with the bone marrow. The platform offers high spatiotemporal resolution in a compact design providing an opportunity to investigate the stem cell niche and visualize cancer metastasis in a relatively high-throughput fashion; thus, the BMoaC may also be a useful tool for drug discovery or investigation of hematological diseases.
Supplementary Material
Acknowledgments
The authors would like to thank the Lewis Lab for use of the Invitrogen Attune NxT Flow Cytometer and Nuala Del Piccolo for editing the manuscript. The microfluidic device cartoon in the graphical abstract was created using Biorender.com. This work was supported by the National Institutes of Health F32CA210540-01A1 (D.E.G.); UH3TR000481 (S C G); and R21CA208519 (S C G., K.W., and D.L.).
Footnotes
Credit author statement
Drew E. Glaser: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – Reviewing & Editing, Visualization, Matthew B. Curtis: Investigation, Validation, Formal analysis, Peter A. Sariano: Investigation, Validation, Formal analysis, Zachary A. Rollins: Investigation, Validation, Formal analysis, Bhupinder S. Shergill: Investigation, Validation, Formal analysis, Aravind Anand: Investigation, Validation, Formal analysis, Alyssa M. Deely: Investigation, Validation, Formal analysis, Leif Anderson: Investigation, Validation, Formal analysis, Jeremy M. Lowen: Investigation, Validation, Formal analysis, Venktesh S. Shirure: Methodology, Software, Natalie R. Ng: Investigation, Formal analysis, Katherine Weilbacher: Conceptualization, Writing – review & editing, Daniel C. Link: Conceptualization, Writing – review & editing, Steven C. George: Conceptualization, Resources, Writing – original draft, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2021.121245.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
- [1].Ghajar CM, et al. , The perivascular niche regulates breast tumour dormancy, Nat. Cell Biol 15 (2013) 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schepers K, et al. , Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche, Cell Stem Cell 13 (2013) 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Itkin T, et al. , Distinct bone marrow blood vessels differentially regulate haematopoiesis, Nature 532 (2016) 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ding L, Saunders TL, Enikolopov G, Morrison SJ, Endothelial and perivascular cells maintain haematopoietic stem cells, Nature 481 (2012) 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marlow R, et al. , A novel model of dormancy for bone metastatic breast cancer cells, Cancer Res. 73 (2013) 6886–6899. [DOI] [PubMed] [Google Scholar]
- [6].Wang H, et al. , The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells, Cancer Cell 27 (2015) 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Levesque JP, Helwani FM, Winkler IG, The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization, Leukemia 24 (2010) 1979–1992. [DOI] [PubMed] [Google Scholar]
- [8].Boyerinas B, et al. , Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy, Blood 121 (2013) 4821–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Carlson P, et al. , Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy, Nat. Cell Biol 21 (2019) 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sugiyama T, Nagasawa T, Bone marrow niches for hematopoietic stem cells and immune cells, Inflamm. Allergy - Drug Targets 11 (2012) 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hwang PY, Brenot A, King AC, Longmore GD, George SC, Randomly distributed K14(+) breast tumor cells polarize to the leading edge and guide collective migration in response to chemical and mechanical environmental cues, Cancer Res. 79 (2019) 1899–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nilsson SK, et al. , Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells, Blood 106 (2005) 1232–1239. [DOI] [PubMed] [Google Scholar]
- [13].Price TT, et al. , Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone, Sci. Transl. Med 8 (2016) 340ra373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schweitzer KM, et al. , Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues, Am. J. Pathol 148 (1996) 165–175. [PMC free article] [PubMed] [Google Scholar]
- [15].Liu YF, et al. , ICAM-1 deficiency in the bone marrow niche impairs quiescence and repopulation of hematopoietic stem cells, Stem Cell Reports 11 (2018) 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mizuochi C, et al. , Intra-aortic clusters undergo endothelial to hematopoietic phenotypic transition during early embryogenesis, PLoS One 7 (2012), e35763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Winkler IG, et al. , Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance, Nat. Med 18 (2012) 1651–1657. [DOI] [PubMed] [Google Scholar]
- [18].Sipkins DA, et al. , In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment, Nature 435 (2005) 969–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Torisawa YS, et al. , Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro, Nat. Methods 11 (2014) 663–669. [DOI] [PubMed] [Google Scholar]
- [20].Chou DB, et al. , On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology, Nat Biomed Eng 4 (2020) 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ellis SL, et al. , The relationship between bone, hemopoietic stem cells, and vasculature, Blood 118 (2011) 1516–1524. [DOI] [PubMed] [Google Scholar]
- [22].Inari H, et al. , Clinicopathological and prognostic significance of Ki-67 immunohistochemical expression of distant metastatic lesions in patients with metastatic breast cancer, Breast Cancer 24 (2017) 748–755. [DOI] [PubMed] [Google Scholar]
- [23].Shirure VS, Lezia A, Tao A, Alonzo LF, George SC, Low levels of physiological interstitial flow eliminate morphogen gradients and guide angiogenesis, Angiogenesis 20 (2017) 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shirure VS, et al. , Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids, Lab Chip 18 (2018) 3687–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moya M, Tran D, George SC, An integrated in vitro model of perfused tumor and cardiac tissue, Stem Cell Res. Ther 4 (Suppl 1) (2013) S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pan H, et al. , 20-Year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years, N. Engl. J. Med 377 (2017) 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Labiano S, et al. , CD69 is a direct HIF-1alpha target gene in hypoxia as a mechanism enhancing expression on tumor-infiltrating T lymphocytes, OncoImmunology 6 (2017), e1283468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen X, et al. , Rapid anastomosis of endothelial progenitor cell-derived vessels with host vasculature is promoted by a high density of cotransplanted fibroblasts, Tissue Eng. 16 (2010) 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ingram DA, et al. , Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood, Blood 104 (2004) 2752–2760. [DOI] [PubMed] [Google Scholar]
- [30].Bi Y, et al. , Tumor-on-a-chip platform to interrogate the role of macrophages in tumor progression, Integr Biol (Camb) 12 (9) (2020) 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sewell-Loftin MK, Katz JB, George SC, Longmore GD, Micro-strains in the extracellular matrix induce angiogenesis, Lab Chip 20 (2020) 2776–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Weng KC, et al. , Human induced pluripotent stem-cardiac-endothelial-tumor-on-a-chip to assess anticancer efficacy and cardiotoxicity, Tissue Eng. C Methods 26 (2020) 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tjwa M, et al. , Fibrinolysis-independent role of plasmin and its activators in the haematopoietic recovery after myeloablation, J. Cell Mol. Med 13 (2009) 4587–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang X, et al. , Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels, Lab Chip 16 (2016) 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carrion B, Janson IA, Kong YP, Putnam AJ, A safe and efficient method to retrieve mesenchymal stem cells from three-dimensional fibrin gels, Tissue Eng. C Methods 20 (2014) 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Radtke S, et al. , Human multipotent hematopoietic progenitor cell expansion is neither supported in endothelial and endothelial/mesenchymal co-cultures nor in NSG mice, Sci. Rep 9 (2019) 12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Radtke S, Gorgens A, Liu B, Horn PA, Giebel B, Human mesenchymal and murine stromal cells support human lympho-myeloid progenitor expansion but not maintenance of multipotent haematopoietic stem and progenitor cells, Cell Cycle 15 (2016) 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Esipova TV, et al. , Two new “protected” oxyphors for biological oximetry: properties and application in tumor imaging, Anal. Chem 83 (2011) 8756–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lam SF, Shirure VS, Chu YE, Soetikno AG, George SC, Microfluidic device to attain high spatial and temporal control of oxygen, PLoS One 13 (2018), e0209574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Preibisch S, Saalfeld S, Tomancak P, Globally optimal stitching of tiled 3D microscopic image acquisitions, Bioinformatics 25 (2009) 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Moya ML, Hsu YH, Lee AP, Hughes CC, George SC, In vitro perfused human capillary networks, Tissue Eng. C Methods 19 (2013) 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cho H, Kim HY, Kang JY, Kim TS, How the capillary burst microvalve works, J. Colloid Interface Sci 306 (2007) 379–385. [DOI] [PubMed] [Google Scholar]
- [43].Nakatsu MN, et al. , Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1, Microvasc. Res 66 (2003) 102–112. [DOI] [PubMed] [Google Scholar]
- [44].Klein C, et al. , Ex vivo expansion of hematopoietic stem- and progenitor cells from cord blood in coculture with mesenchymal stroma cells from amnion, chorion, Wharton’s jelly, amniotic fluid, cord blood, and bone marrow, Tissue Eng. 19 (2013) 2577–2585. [DOI] [PubMed] [Google Scholar]
- [45].Bendall LJ, Bradstock KF, Csf G, From granulopoietic stimulant to bone marrow stem cell mobilizing agent, Cytokine Growth Factor Rev. 25 (2014) 355–367. [DOI] [PubMed] [Google Scholar]
- [46].Zhang CC, Kaba M, Iizuka S, Huynh H, Lodish HF, Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation, Blood 111 (2008) 3415–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Delaney C, et al. , Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution, Nat. Med 16 (2010) 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Csaszar E, et al. , Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling, Cell Stem Cell 10 (2012) 218–229. [DOI] [PubMed] [Google Scholar]
- [49].Chen JY, et al. , Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche, Nature 530 (2016) 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Marturano-Kruik A, et al. , Human bone perivascular niche-on-a-chip for studying metastatic colonization, Proc. Natl. Acad. Sci. U. S. A 115 (2018) 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sieber S, et al. , Bone marrow-on-a-chip: long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment, J Tissue Eng Regen Med 12 (2018) 479–489. [DOI] [PubMed] [Google Scholar]
- [52].Pang WW, et al. , Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 20012–20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE, Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 5320–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sobrino A, et al. , Mas receptor is involved in the estrogen-receptor induced nitric oxide-dependent vasorelaxation, Biochem. Pharmacol 129 (2017) 67–72. [DOI] [PubMed] [Google Scholar]
- [55].Perche F, Torchilin VP, Cancer cell spheroids as a model to evaluate chemotherapy protocols, Cancer Biol. Ther 13 (2012) 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Aljitawi OS, et al. , A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells, Leuk. Lymphoma 55 (2014) 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McCullough J, Clay M, Herr G, Smith J, Stroncek D, Effects of granulocyte-colony-stimulating factor on potential normal granulocyte donors, Transfusion 39 (1999) 1136–1140. [DOI] [PubMed] [Google Scholar]
- [58].Stroncek DF, et al. , Treatment of normal individuals with granulocyte-colony-stimulating factor: donor experiences and the effects on peripheral blood CD34+ cell counts and on the collection of peripheral blood stem cells, Transfusion 36 (1996) 601–610. [DOI] [PubMed] [Google Scholar]
- [59].Chen T, Moscvin M, Bianchi G, Exosomes in the pathogenesis and treatment of multiple myeloma in the context of the bone marrow microenvironment, Front Oncol 10 (2020) 608815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tomlinson RE, Christiansen BA, Giannone AA, Genetos DC, The role of nerves in skeletal development, adaptation, and aging, Front. Endocrinol 11 (2020) 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.