

Abstract
Cancer-associated fibroblasts (CAFs) are a major cell type in the stroma of solid tumors and can exert both tumor-promoting and tumor-restraining functions. CAF heterogeneity is frequently observed in pancreatic ductal adenocarcinoma (PDAC), a tumor characterized by a dense and hypoxic stroma that features myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs) that are thought to have opposing roles in tumor progression. While CAF heterogeneity can be driven in part by tumor cell-produced cytokines, other determinants shaping CAF identity and function are largely unknown. In vivo, we found that iCAFs displayed a hypoxic gene expression and biochemical profile and were enriched in hypoxic regions of PDAC tumors, while myCAFs were excluded from these regions. Hypoxia led fibroblasts to acquire an inflammatory gene expression signature and synergized with cancer cell-derived cytokines to promote an iCAF phenotype in a HIF-1α dependent fashion. Furthermore, HIF-1α stabilization was sufficient to induce an iCAF phenotype in stromal cells introduced into PDAC organoid co-cultures and to promote PDAC tumor growth. These findings indicate hypoxia-induced HIF-1α as a regulator of CAF heterogeneity and promoter of tumor progression in PDAC.
Statement of Significance
Hypoxia in the tumor microenvironment of pancreatic cancer potentiates the cytokine-induced inflammatory cancer-associated fibroblast phenotype and promotes tumor growth.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive tumor and projected to become the second-leading cause of cancer-related mortality by 2030 in the United States (1). A significant barrier to the delivery of effective therapy for PDAC is the desmoplastic stroma that can constitute up to 90% of the tumor volume (1). The prominent desmoplastic response observed in PDAC is characterized by a fibrotic and inflammatory stromal milieu which is produced primarily by cancer-associated fibroblasts (CAFs) and plays a role in both supporting tumor cell growth and promoting therapeutic resistance (2). The basal activity of CAFs to produce extracellular matrix is not sufficient to mediate these effects, as depletion of CAF-derived collagen promotes PDAC growth and reduces survival in mouse models (3). Thus, CAFs can have either tumor-promoting or tumor-suppressing properties within the pancreatic tumor microenvironment (TME).
Transcriptionally and functionally heterogeneous subsets of CAFs have been identified in mouse and human PDAC (4–7). Myofibroblastic CAFs (myCAFs) are marked by expression of alpha smooth muscle actin (αSMA), produce extracellular matrix and are thought to restrain tumor growth (8). Inflammatory CAFs (iCAFs) express only low levels of αSMA, produce a variety of growth factors and inflammatory cytokines such as IL6 and can directly and indirectly promote tumor growth (9). Other, cancer-associated phenotypes of fibroblasts have also been reported, including antigen-presenting CAFs (apCAFs) marked by MHC-II expression (5). Heterogeneity within the CAF population has been suggested to be established in part by growth factor and cytokine gradients within the TME including the local accumulation of tumor-derived TGFβ and IL1/TNFα (10), indicating that spatial differences in the accumulation of different CAF subpopulations exist. However, whether the metabolic conditions present in the pancreatic TME also contribute to regulating CAF heterogeneity is less well explored.
Understanding regulators of CAF heterogeneity has clinical implications: while PDAC patients with high amounts of myCAFs in tumors had improved overall survival, they responded poorly to anti-PD-L1 therapy in retrospective studies (6,8). In contrast, iCAFs are associated with poor response to chemotherapy in patients (11), and iCAF-derived factors including IL6 are directly involved in PDAC progression in mouse models (12–14). Thus, a better understanding of the determinants of CAF heterogeneity may facilitate the development of therapies selectively targeting tumor promoting CAFs.
The TME of PDAC is characterized by nutrient depletion and hypoxia as a result of increased cancer cell demand and impaired vascularization (15,16). Hypoxia results in stabilization of the transcription factor HIF-1α which mediates cellular adaptation to low oxygen tension (17). In cancer cells, this adaptive response promotes epithelial-mesenchymal transition and angiogenesis, and a hypoxia gene expression signature is associated with poor prognosis of PDAC patients (18). In the stroma, hypoxia is known to promote lysyl oxidase expression to increase collagen crosslinking and tumor stiffness (19). Hypoxia is associated with an inflammatory fibroblast expression signature in genomic studies of human PDAC and has been shown to promote a secretory phenotype in CAFs while conversely, reducing αSMA expression (10,20,21). These data suggest that hypoxia could influence the CAF phenotype, but whether hypoxia and HIF-1α are causatively involved in the generation of distinct CAF subsets in PDAC is not understood. Here, we report the ability of hypoxia to synergize with cancer cell-derived cytokines in activating HIF-1α to promote the iCAF phenotype and tumor growth in PDAC.
Materials and Methods
Mouse experiments
All animal experiments described adhered to policies and practices approved by Memorial Sloan Kettering Cancer Center’s Institutional Animal Care and Use Committee (IACUC) and were conducted as per NIH guidelines for animal welfare (Protocol Number 11–03-007, Animal Welfare Assurance Number FW00004998). Mouse experiments were performed as previously described (22). The maximal tumor size/burden permitted by the IACUC (Tumor burden may not exceed 10% of the weight of the mouse which is equivalent to a tumor volume of 2.5 cm3 for a 25 g mouse) was not exceeded. Mice were maintained under specific pathogen-free conditions and housed at 4–5 mice per cage at a 12-hour light/dark cycle at a relative humidity of 30% to 70% and room temperature of 22.2 ± 1.1°C, and were allowed access to food and water ad libitum. Mice were maintained in individually ventilated polysulfone cages with a stainless-steel wire bar lid and filter top on autoclaved aspen chip bedding. Mice were fed a closed-formula, natural-ingredient, γ-irradiated diet (5053 - PicoLab® Rodent Diet 20, Purina LabDiet) which was surface decontaminated using “flash” sterilization (100°C for 1 minute). Mice were provided reverse-osmosis acidified (pH 2.5 to 2.8, with hydrochloric acid) water. Cage bottoms were changed weekly, whereas the wire bar lid, filter top and water bottle were changed biweekly.
Orthotopic organoid injection model
Orthotopic injections were performed as previously described (23). Organoids derived from pancreatic tumors of KPC (KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre) in a C57BL/6 background were used. Syngeneic C57BL/6 mice were anesthetized with isoflurane and an incision was made in the left abdominal side. Organoids were dissociated from cultures with TrypLE (Thermo Fisher) and resuspended in 30 μL growth factor reduced Matrigel (Corning). Approximately 1×105 cells were injected per recipient mouse into the tail region of the pancreas using a Hamilton Syringe. Successful injection was verified by the appearance of a fluid bubble without signs of intraperitoneal leakage. The abdominal wall was sutured with absorbable Vicryl sutures (Ethicon), and the skin was closed with wound clips (CellPoint Scientific Inc.). Mice were monitored for tumor development by ultrasound five weeks after injection and once/week afterwards using a Vevo 2100 System with a MS250 13–24MHz scan head (VisualSonics). When tumors were approximately 500 mm3 in size, 60 mg/kg body weight of pimonidazole (Hypoxyprobe) in 0.9% saline was injected i.p. one hour before euthanasia. Tumors were collected, and half of the tumor was allocated for 10% formalin fixation for histological analysis, and the other half was used to generate single cell suspensions for flow cytometry analysis.
Subcutaneous co-injection model
Subcutaneous injections were performed as described (22). 2×105 KPC cells alone or together with 1×106 PSCs were resuspended in 100 μL PBS and injected subcutaneously into the flanks of 8–10 weeks old female syngeneic C57BL/6 mice (JAX, 000664). At the beginning of each experiment, and at the onset of treatment with monoclonal antibodies, mice were randomly assigned to the different groups. No estimation of sample size was performed before the experiments. Treatment with monoclonal antibodies was started one week after tumor inoculation and performed as described (23). Mice were injected intraperitoneally every three days over two weeks with anti-IL-6 (200 μg; MP5–20F3, BioXCell), anti-VEGFR2 (800 μg; DC101, BioXCell), or IgG1 isotype control (800 μg; MOPC-21, BioXCell). Mice were monitored daily, and tumor volume was measured by calipers. Measurements were taken in two dimensions, and tumor volume was calculated as length × width2 × π/6. At the end of the experiment, mice were euthanized with CO2, and tumors were collected and aliquoted for 10% formalin fixation for immunohistochemistry and digestion for flow cytometry analysis.
Immunostaining of mouse PDAC tumors
Automated multiplex IF was conducted using the Leica Bond BX staining system. Paraffin-embedded tissues were sectioned at 5 μm and baked at 58°C for 1 hr. Slides were loaded in Leica Bond and immunofluorescence staining was performed as follows. Samples were pretreated with EDTA-based epitope retrieval ER2 solution (AR9640, Leica) for 20 min at 95°C. The quadruplex-plex antibody staining and detection was conducted sequentially. The primary antibodies against PDPN (0.05 μg/ml, hamster, DSHB, 8.1.1, RRID:AB_531893), PIMO (0.12 μg/ml, mouse, Hydroxyprobe Inc. MAB1, RRID:AB_2801307), SMA (0.1 μg/ml, rabbit, Abcam, ab5694, RRID:AB_2223021), HIF-1α (0.5 μg/ml, rabbit, Novus, NB100, RRID:AB_350071) were used. For the rabbit antibody, Leica Bond Polymer anti-rabbit HRP was used, for the hamster antibody and the mouse antibody, rabbit anti-Hamster (Novex, A18891) and rabbit anti-mouse (Abcam, ab133469) secondary antibodies were used as linkers before the application of the Leica Bond Polymer anti-rabbit HRP. After that, Alexa Fluor tyramide signal amplification reagents (Life Technologies, B40953, B40958) or CF dye tyramide conjugates (Biotium, 92174, 96053) were used for detection. After each round of IF staining, Epitope retrieval was performed for denaturization of primary and secondary antibodies before another primary antibody was applied. After the run was finished, slides were washed in PBS and incubated in 5 μg/ml 4’,6-diamidino-2-phenylindole (DAPI) (Sigma Aldrich) in PBS for 5 min, rinsed in PBS, and mounted in Mowiol 4–88 (Calbiochem). Slides were kept overnight at −20°C before imaging. Immunohistochemistry was performed by Histowiz (NY) using the following antibodies: SMA (ab5694, RRID:AB_2223021), CD31 (ab28364, RRID:AB_726362).
Imaging and analysis
Images from tissue sections of PDAC tumors were acquired with a Mirax Slide Scanner at 40x magnification. Images were analyzed in ImageJ (RRID:SCR_003070). Pimonidazole+ regions were located in each tissue section. Within each region, the number of PDPN+ only pixels, SMA+ only pixels, and double positive pixels were quantified. Thresholds were set manually for each channel and kept consistent for each image. Two sections per tumor were analyzed. Live images from PSC monocultures were acquired with a Leica SP5 Inverted confocal microscope with cells placed in an environmental chamber. IHC stained slides were scanned by Histowiz, and images were analyzed in ImageJ. Thresholds were set manually set and kept consistent for each image.
Cell culture
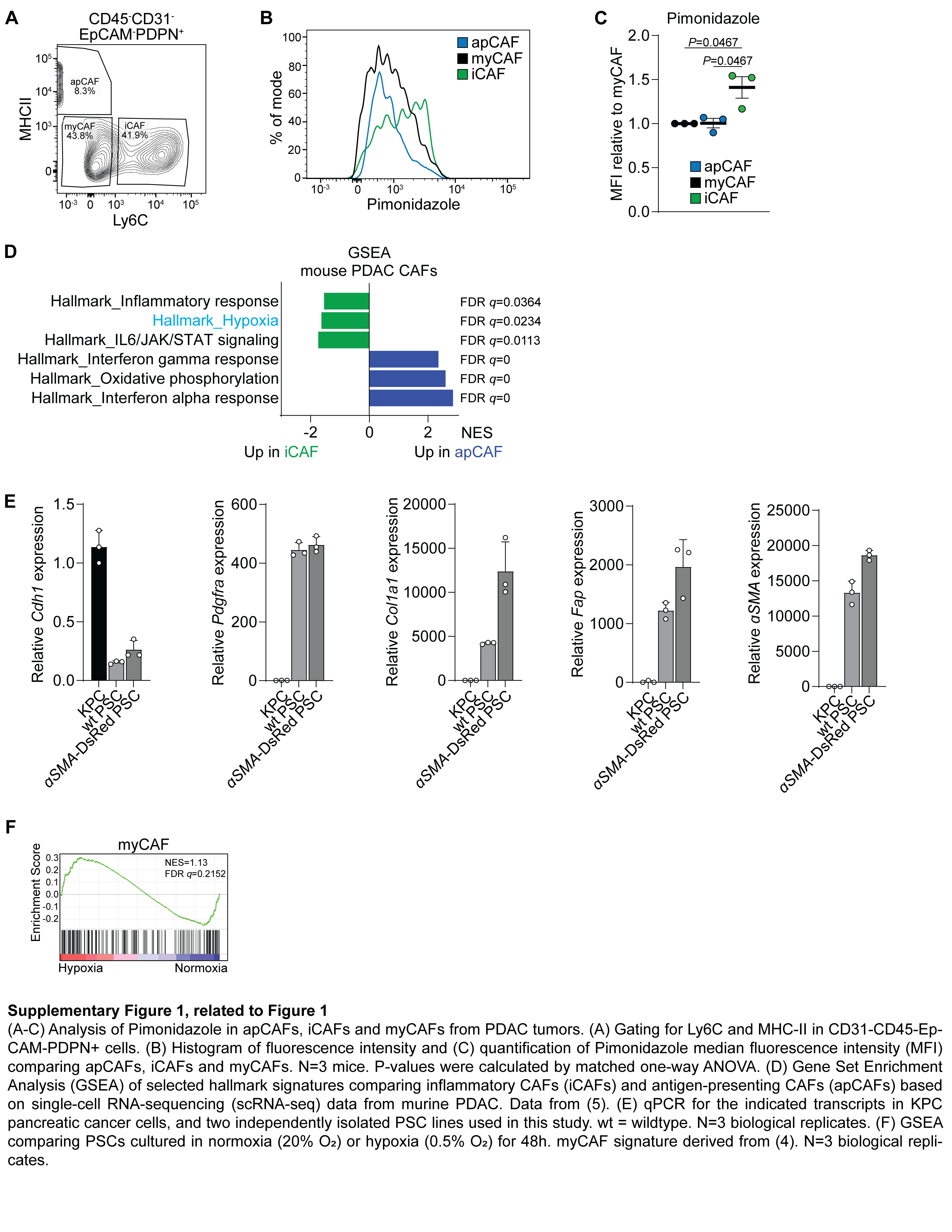
293T cells were obtained from ATCC (CRL-3216). PSCs were isolated from either wildtype C57BL/6 mice or αSMA-DsRed mice (24) by differential centrifugation as previously described (25) and immortalized by spontaneous outgrowth. KPC (KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre) mouse PDAC cells and organoids were described before (23). All cells were cultured at 37°C in 5% CO2 and 20% O2 and were maintained in DMEM supplemented with 10% FBS (Gemini), 100 U/ml penicillin and 100 μg/ml streptomycin (1% P/S). For hypoxia experiments, cells were cultured in a hypoxia glove box (Coy) set at 0.5% O2, 37°C and 5% CO2 for 48h. Cells were verified as mycoplasma-free by the MycoAlert Mycoplasma Detection Kit (Lonza). Cells were treated with 2 ng/mL murine IL1α (211–11A, Peprotech) and TNFα (315–01A, Peprotech) as indicated (“cytokines”). Cells were also treated with MLN120B (IKKβ inhibitor, 10 μM), AZD1480 (JAK inhibitor, 2 μM), anti-LIF (4 μg/mL, AF449, R&D), CoCl2 (100 μM, Sigma)
Organoid culture
Organoids were derived from pancreatic tumors of KPC (KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre) mice in a C57BL/6 background and described before (23). Organoids were cultured 24-well plates in growth factor reduced (GFR) Matrigel (Corning) in Advanced DMEM/F12 supplemented with the following: 1% P/S, 2 mM glutamine, 1X B27 supplement (12634–028, Invitrogen), 50 ng/ml murine EGF (PMG8043, Peprotech), 100 ng/ml murine Noggin (250–38; Peprotech), 100 ng/ml human FGF10 (100–26; Peprotech), 10 nM human Leu-Gastrin I (G9145, Sigma), 1.25 mM N-acetylcysteine (A9165; Sigma), 10 mM nicotinamide (N0636; Sigma), and R-spondin1 conditioned media (10% final). Organoids were passaged with every 3–4 days. For PSC co-culture, confluent wells of organoids were dissociated with 1x TrypLE (12604013, Thermo Fisher) and plated at a splitting ratio of 1:5 (approximately 1×104 cells) together with 8×104 αSMA-DsRed expressing PSCs in GFR Matrigel. Co-cultures were cultured with DMEM supplemented with 10% FBS (Gemini) and 1% P/S in 20% O2 and 5% CO2. For experiments in hypoxia, co-cultures were placed in a hypoxia glove box (Coy) set at 0.5% O2 for the last 48h of the experiment. For measurement of organoid growth in co-cultures, organoids expressing Luciferase were used. Cultures were treated with Luciferin, lysed and luminescence was measured.
MEMIC experiments
MEMICs were fabricated and used as described in detail previously (26). In brief, MEMICs were 3D printed in a 12-well format, and coverslips were glued at the bottom and the top to create inner and outer chambers. For each condition tested, one well was prepared without the coverslip on the top to create a control well without gradients. MEMICs were washed with water, UV-sterilized, washed twice with PBS and once with complete media before cell seeding. A 85 μL cell suspension containing 2×104 PSCs was filled in the inner chamber. For the open wells, 1.5 mL of a 1×105/mL cell suspension was added to the entire well. Cells were allowed to settle for 1h, and 1.5 mL media was added in the outer chamber in wells plated with cells in the inner chamber. The next day, cells were mock-treated or treated with cytokines. The gradient was allowed to form for 48h, and cells were either imaged live or fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100, blocked with 2.5% bovine serum albumin in PBS, and stained for 1h with an anti-GFP antibody (A10262, Invitrogen, RRID:AB_2534023). Wells were washed three times with PBS and incubated with an anti-chicken Alexa Fluor 488 coupled secondary antibody (A11039, Invitrogen) and Hoechst for nuclear staining for 30 min before being washed three times with PBS. Wells were imaged using BZ-X800 microscope from Keyence (20x magnification) and stitched using the BZ-X800 analysis software. Images were processed using custom MATLAB (RRID:SCR_001622) scripts. GFP/UnaG and DsRed fluorescence intensities were quantified and plotted according to their distance to the opening of the well. For per cell fluorescence quantification, images were segmented using nuclear staining and dilated to include adjacent cytoplasmic areas creating a mask for each cell. Then total fluorescence was integrated for each cell using these masks. Image analysis code is available upon request.
Ectopic gene expression and CRISPR/Cas9 mediated gene deletion
Guide RNAs targeting murine Hif1a and Vhl were designed using GuideScan (http://www.guidescan.com/) and cloned into pLentiCRISPRv2 (RRID:Addgene_127644). The following guide sequences were used: TCGTTAGGCCCAGTGAGAAA (Hif1a sg1), CAAGATGTGAGCTCACATTG (Hif1a sg2), CCGATCTTACCACCGGGCAC (Vhl sg1), GGCTCGTACCTCGGTAGCTG (Vhl sg2). Rosa26 targeting guides (Ctrl sg) were described before (27). To create IL6 and hypoxia reporters, a Gibson assembly-based modular assembly platform (GMAP) was used (28). HRE-dUnaG from pLenti-HRE-dUnaG (RRID:Addgene_124372), and a PGK driven hygromycin selection cassette from MSCV Luciferase PGK-hygro (RRID:Addgene_18782) were amplified using primers containing overhangs with the homology sites for GMAP cloning and inserted into a lentiviral vector (LV 1–5; Addgene, 68411). IL6-EGFP from pmIL-6promoterEGFP (RRID:Addgene_112896), and a PGK driven blasticidin selection cassette from pMSCV-Blasticidin (RRID:Addgene_75085) were amplified for GMAP similarly and inserted into LV 1–5. Lentiviral particles were produced in 293T cells by using psPAX2 (RRID:Addgene_12260) and pCMV-VSV-G (RRID:Addgene_8454) packaging plasmids. Viral supernatant was collected after 48h, passed through a 0.45 μm nylon filter and used to transduce PSCs in the presence of 8 μg/mL polybrene (Sigma) overnight. Cells were subjected to puromycin (2 μg/mL, Sigma), hygromycin (250 μg/mL) or blasticidin (10 μg/mL, Invivogen) antibiotic selection the following day. Polyclonal cell populations were used for the experiments.
Western blot
Lysates were generated by incubating cells in RIPA buffer (Millipore). Cytoplasmic and nuclear fractions were generated as previously described (29). 20–30 μg of cleared lysate were analyzed by SDS-PAGE as previously described (27). The following primary antibodies were used: Vinculin (1:5000 dilution, Sigma, V9131, RRID:AB_477629), β-Actin (1:5000; Sigma, A5441, RRID:AB_476744), HIF-1α (1:1000, 10006421, Cayman, RRID:AB_409037), VHL (1:200, sc-5575, Santa Cruz, RRID:AB_2241850), LDHA (1:1000, 2012S, Cell Signaling, RRID:AB_2137173), GFP (1:1000, 11814460001, Sigma, RRID:AB_390913), phospho-Stat3 Tyr709 (1:1000, 9145S, Cell Signaling, RRID:AB_2491009) or Stat3 (1:1000, 9139S, Cell Signaling), p65 (1:1000, 8242S, Cell Signaling, RRID:AB_331757), Lamin A/C (1:1000, 4777, Cell Signaling, RRID:AB_10545756). The following secondary antibodies were used: anti-rabbit HRP (1:5000, NA934V, GE, RRID:AB_11112914) and anti-mouse HRP (1:5000, NA931, GE, RRID:AB_772210).
ELISA
Quantification of IL6 in PSC conditioned media was performed with the Mouse IL-6 ELISA Kit (ab100712, Abcam). VEGF was quantified with the Mouse VEGF Quantikine ELISA Kit (MMV00, R&D).
Flow cytometry
For analysis of PSC monocultures, cells were trypsinized, washed, stained with DAPI and analyzed on an LSRFortessa II (BD). Live cells (DAPI-) were analyzed for EGFP fluorescence. For organoid/PSC co-cultures, Matrigel was digested with Dispase (Corning), and cells and organoids were dissociated mechanically by pipetting up and down at least 30 times. PSCs were analyzed by gating for DAPI- and DsRed+ cells followed by analysis of EGFP fluorescence intensity. For analysis of CAFs from PDAC tumors arising from orthotopic injection of KPC organoids, tumors were minced and resuspended in 5 mL DMEM with 800 μg/mL Dispase (Sigma), 500 μg/mL Collagenase P (Sigma), 100 ug/mL Liberase TL (Roche), 100 μg/mL DNaseI (Sigma), 100 μg/mL Hyaluronidase (Sigma). Samples were then transferred to C-tubes and processed using program 37C_m_TDK1_2 on a gentleMACS Octo dissociator with heaters (Miltenyi Biotec). Dissociated tissue was passaged through a 40 μm cell strainer and centrifuged at 1500 rpm × 5 minutes. Red blood cells were lysed with ACK lysis buffer (A1049201, Thermo Fisher) for 1 minute, and tubes were filled up with PBS. Samples were centrifuged and resuspended in FACS buffer (PBS supplemented with 2% FBS) and stained with Ghost Dye Violet 510 (1:1000, Tonbo Biosciences) on ice for 10 min for discrimination of viable and non-viable cells. Samples were blocked with anti-CD16/32 (FC block, 1:100, Biolegend, RRID:AB_2612550) for 15 minutes on ice and then incubated with the following antibodies (all from Biolegend) in Brilliant stain buffer (Thermo Fisher) for 30 minutes on ice: CD326-FITC (G8.8, 1:50), CD45-BV711 (30-F11, 1:200, RRID:AB_2564590), CD31-PE/Cy7 (390, 1:200, RRID:AB_2795050), PDPN-APC/Cy7 (8.1.1, 1:100, RRID:AB_2629804), Ly6C-BV421 (HK1.4, 1:200, RRID:AB_2562178), MHCII-BV785 (M5/114.15.2, 1:200, RRID:AB_2565977). Samples were washed in FACS buffer and fixed and permeabilized with the Foxp3 / Transcription Factor Staining Buffer Set (00–5523-00, Thermo Fisher) according to the manufacturer’s instructions. Samples were stained with anti-pimonidazole antibody (4.3.11.3, 1:50, Hypoxyprobe, RRID:AB_2801307) in permeabilization buffer at 4°C over night. Samples were incubated with anti-mouse Alexa Fluor 647 (1:400, Thermo Fisher, RRID:AB_2535804) in permeabilization buffer for 15 min at room temperature. Samples were resuspended in FACS buffer and analyzed on an LSRFortessa II by gating for Ghost Dye-, CD45-, CD31, CD326-, PDPN+ cells comparing Ly6C- with Ly6C+ cells or iCAFs (Ly6C+, MHCII-), apCAFs (Ly6C-, MHCII+) and myCAFs (Ly6C-, MHCII-). Compensation was performed with UltraComp eBeads (01–2222-42, Thermo Fisher). Data were analyzed with FlowJo software (RRID:SCR_008520). For analysis of αSMA expression, fixed and permeabilized samples were stained with Alexa Fluor 488 coupled anti-SMA antibody (1A4, 1:200, Thermo Fisher, RRID:AB_2574461).
Quantification of gene expression
RNA isolation and analysis was done as previously described (22). Total RNA was isolated from fibroblasts with Trizol (Life Technologies) according to the manufacturer’s instructions, and 1 μg RNA was used for cDNA synthesis using iScript (Bio-Rad). Quantitative real-time PCR (qPCR) analysis was performed in technical triplicates using 1:20 diluted cDNAs and 0.1 μM forward and reverse primers together with Power SYBR Green (Life Technologies) in a QuantStudio 7 Flex (Applied Biosystems). Gene expression was quantified in Microsoft Excel 365 (RRID:SCR_016137) as relative expression ratio using primer efficiencies calculated by a relative standard curve. The geometric mean of the endogenous control genes Actb and Rplp0 was used as reference sample. Primer pairs are as follows: TACCACCATGTACCCAGGCA (Actb FW), CTCAGGAGGAGCAATGATCTTGAT (Actb RV), AGATTCGGGATATGCTGTTGGC (Rplp0 FW), TCGGGTCCTAGACCAGTGTTC (Rplp0 RV), CCATCATGCGTCTGGACTT (αSMA FW), GGCAGTAGTCACGAAGGAATAG (αSMA RV), CTTCCATCCAGTTGCCTTCT (IL6 FW), CTCCGACTTGTGAAGTGGTATAG (IL6 RV), CATTGTCAAGTACAGTCCACACT (Ldha FW), TTCCAATTACTCGGTTTTTGGGA (Ldha RV). ATTTCGCTTCGGGACTAGC (Socs3 FW), AACTTGCTGTGGGTGACCAT (Socs3 RV), CTGCTCTCCCTCTTTCCTTTC (Lif FW), ACATTCCCACAGGGTACATTC (Lif RV).
RNA sequencing
Total RNA was isolated with Trizol as above, and libraries were prepared from polyA-selected mRNA using the TruSeq RNAsample preparation kit v2 (Illumina) according to the manufacturer’s instructions. Libraries were sequenced using an Illumina HiSeq 4000 generating 150 bp paired-end reads. An average of 58 million reads per sample was retrieved. Adaptor sequences were removed from fastq files with Trimmomatic v.0.36 (RRID:SCR_011848), and trimmed reads were mapped to the mus musculus GRCm38 reference genome using the STAR aligner v.2.5.2b (RRID:SCR_004463). Aligned features were counted with featureCounts from the Subread package v.1.5.2 and differential expression was determined using DESeq2 v3.10 (RRID:SCR_000154) from Bioconductor in R v4.1.0.
Gene set enrichment analysis (GSEA)
GSEA was performed using a pre-ranked gene list based on the log2 fold change comparing three Ctrl sg samples cultured in Normoxia against three Ctrl sg samples cultured in Hypoxia for 48h, or comparing three Ctrl sg samples cultured in Hypoxia against three Hif1a sg7 samples cultured in Hypoxia. GSEA 4.3.0 (RRID:SCR_003199) was used with 1000 permutations and mouse gene symbols remapped to human orthologs v7.5 (MSigDB). Enrichment of the iCAF signature (4) or Hallmark signatures (MSigDB) was analyzed.
Statistics
A student’s t-test was applied to compare one variable between two groups. One-way ANOVA was applied to compare one variable between three or more groups. Two-way ANOVA was applied to compare two independent variables between two groups. Correction for multiple comparisons was done using the Holm-Sidak method. Statistical analysis was done in GraphPad Prism 9 (RRID:SCR_002798). Most graphs show the mean + SD with individual datapoints, unless indicated otherwise in the figure legends. One tumor in the anti-VEGFR2 group in Fig. 6L was identified as an outlier (ROUT test) and excluded from the analysis.
Data availability
RNA-sequencing data have been deposited in GEO (GSE221761). Other expression data analyzed in this study were obtained from NCBI dbGaP (phs001840.v1.p1). All other data are available from the corresponding authors upon reasonable request.
Results
To investigate factors regulating CAF heterogeneity in PDAC, we analyzed publicly available single cell RNA (scRNA)-sequencing data from human PDAC patients (5). Single sample gene set enrichment analysis (GSEA) revealed enrichment of an inflammatory response signature in iCAFs and a collagen formation signature in myCAFs (Fig. 1A), as reported (5). Using these data, we found that an oxidative phosphorylation signature was enriched in myCAFs (Fig. 1A), consistent with our previous work showing that mitochondrial oxidative metabolism is required for proline biosynthesis and for collagen production (27). Conversely, a hypoxic gene expression signature was enriched in iCAFs (Fig. 1A). To confirm this finding, we used a murine orthotopic PDAC organoid transplantation model which closely recapitulates key features of human PDAC (30). PDAC organoids derived from the KPC (KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre) mouse model (31) were injected orthotopically into the pancreas of syngeneic C57BL/6 mice. Once tumors reached ~500 mm3, pimonidazole, a hypoxia indicator (32), was injected intraperitoneally one hour before euthanasia (Fig. 1B). Half of each tumor was digested, and CAFs (gated for CD31−CD45−EpCAM−PDPN+ cells) were counterstained for Ly6C as an iCAF surface marker (10) and analyzed for pimonidazole accumulation by flow cytometry (Fig. 1C–E). Consistent with the gene expression data, this analysis revealed that Ly6C+ CAFs had accumulated higher amounts of pimonidazole than Ly6C− CAFs (Fig 1D, E). In a separate experiment, we also stained for MHC-II (Sup. Fig. 1A), an apCAF surface marker (10). Consistent with the previous experiment, iCAFs (MHC-II−Ly6C+) accumulated significantly more pimonidazole compared to apCAFs (MHC-II+Ly6C−) and myCAFs (MHC-II−Ly6C−) (Sup. Fig. 1B, C). In addition, a hypoxia gene expression signature was enriched in iCAFs compared to apCAFs in scRNA-sequencing data from murine PDAC tumors (5) (Sup. Fig. 1D). Next, we analyzed the other halves of the PDAC tumors for the presence of pimonidazole, the general CAF marker PDPN and the myCAF marker αSMA by immunofluorescence (Fig. 1F). Strikingly, the vast majority of αSMA+ cells was located outside pimonidazole+ areas (Fig. 1F). In turn, 80% of the PDPN+ areas within pimonidazole+ regions stained negative for αSMA (Fig.1F, G).
Figure 1: A hypoxic signature is enriched in inflammatory fibroblasts in PDAC.
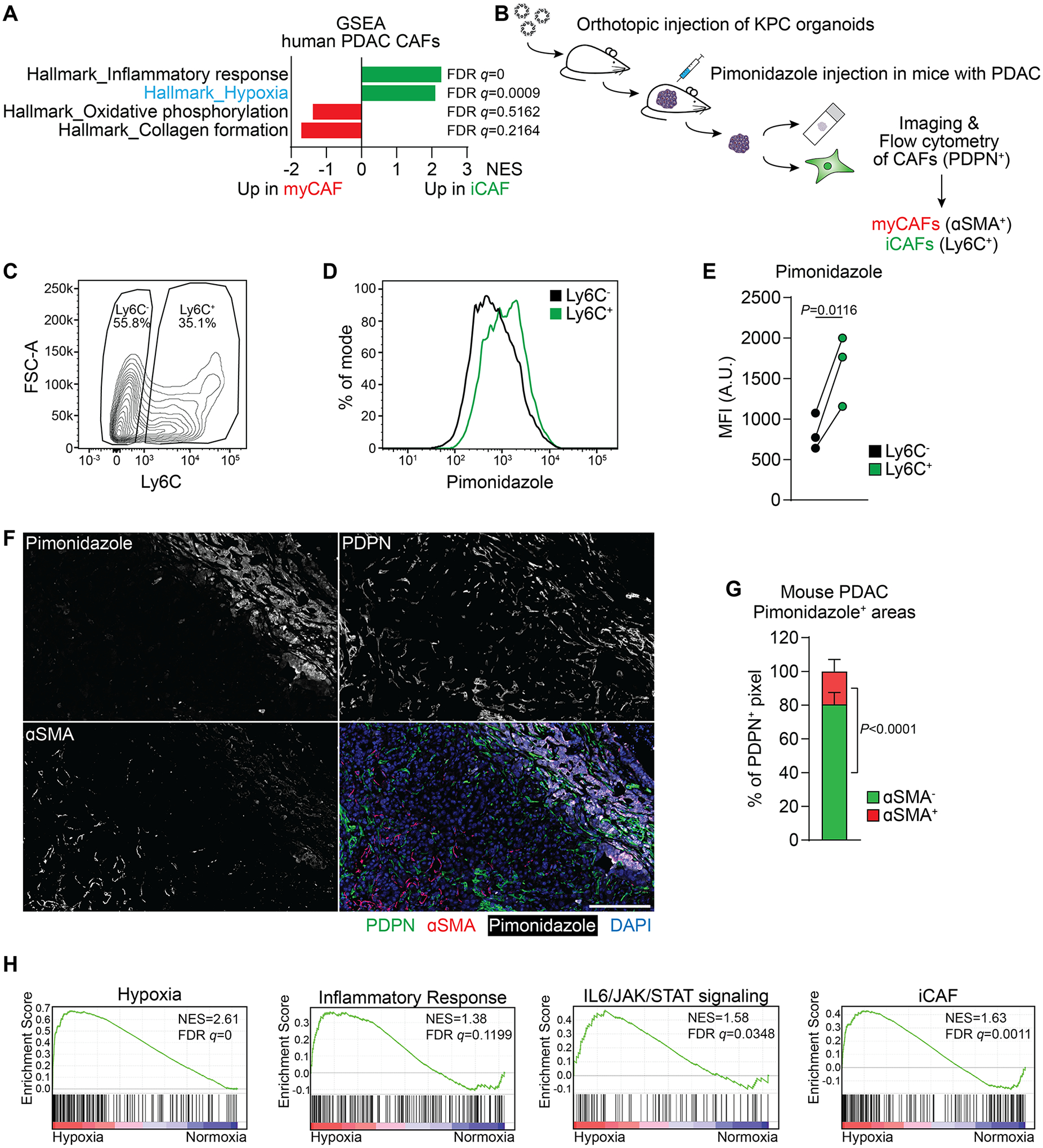
(A) Single sample Gene Set Enrichment Analysis (ssGSEA) of selected hallmark signatures in myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs) based on single-cell RNA-sequencing (scRNA-seq) data from human PDAC. Data from (5).
(B) Schematic of experimental workflow to analyze Pimonidazole enrichment and localization in mouse PDAC tumors arising from orthotopic transplantation of KPC organoids.
(C-E) Analysis of Pimonidazole in Ly6C+ and Ly6C− cells among live, CD31−CD45−EpCAM−PDPN+ cells in PDAC tumors. (C) Gating for Ly6C in PDPN+ cells. (D) Histogram of fluorescence intensity and (E) quantification of Pimonidazole median fluorescence intensity (MFI) comparing Ly6C+ and Ly6C− cells. A.U. = arbitrary units. N=3 mice. P-value was calculated by ratio paired t-test.
(F, G) Immunofluorescence staining of Pimonidazole, PDPN and αSMA in mouse PDAC tumors. (F) Representative image. Nuclei are labeled with DAPI. Scale bar = 500 μm. (G) Quantification of αSMA− and αSMA+ pixel among PDPN+ pixel within Pimonidazole-stained regions. N=8 sections from 4 mice. Data represent mean+SD. P-value was calculated by ratio paired t-test.
(H) GSEA comparing PSCs cultured in normoxia (20% O2) or hypoxia (0.5% O2) for 48h. iCAF signature derived from (4). Other signatures represent Hallmark signatures from MSigDB. N=3 biological replicates.
The above data indicate a significant positive correlation between hypoxia and the iCAF phenotype in PDAC. To test the hypothesis that hypoxia causally promotes acquisition of an iCAF state in fibroblasts, we used immortalized pancreatic stellate cells (PSCs) which contribute to CAFs in PDAC (33). After confirming their fibroblast nature (Sup. Fig. 1E), PSCs were cultured for 48 hours in normoxic (20% O2) or hypoxic (0.5% O2) conditions and their transcriptome was interrogated by RNA-sequencing. While there was no significant change in the myCAF signature (Sup. Fig. 1F), hypoxic culture conditions resulted in enrichment of an iCAF signature in PSCs, as well as an inflammatory response signature and IL6/JAK/STAT signaling (Fig. 1H).
The above data suggest a role of hypoxia in promoting an iCAF-like state. IL1 and TNFα have been identified as major cytokines secreted by pancreatic cancer cells that are capable of inducing an iCAF phenotype in PDAC (10). To assess the role of hypoxia in regulating CAF heterogeneity in relation to known inducers of the iCAF state, we treated PSCs with a combination of IL1 and TNFα (hereafter “cytokines”) to maximize cytokine signaling known to promote an iCAF phenotype. Hypoxia resulted in induction of the iCAF marker IL6 and repression of the myCAF marker αSMA (encoded by Acta2, hereafter αSMA) (Sup. Fig. 2A). When we cultured cytokine-treated PSCs in hypoxia there was a significant increase in IL6 expression and a further decrease in αSMA mRNA levels (Sup. Fig. 2A). To monitor acquisition of an iCAF state in PSCs by orthogonal methods, we developed a reporter system in which EGFP expression is driven by the murine IL6 promoter region (Fig. 2A). Responsiveness of the reporter to cytokine treatment was confirmed (Fig. 2A, B). Hypoxia was sufficient to increase the IL6-EGFP reporter signal to a similar level as did cytokine treatment, and culture of cytokine-treated cells in hypoxia further increased the reporter signal to more than 15-fold above mock-treated cells cultured in normoxia (Fig. 2A, B). Similarly, hypoxia increased IL6 protein levels, and in combination with cytokine treatment led to a synergistic upregulation of IL6 in the media (Fig. 2C). Next, we combined the iCAF reporter with a myCAF reporter in which DsRed expression is driven by the murine αSMA promoter region (24). Consistent with the prior observation that PSCs in monoculture on plastic display characteristics of myCAFs (4,10), high expression of αSMA-DsRed was detected in untreated PSCs cultured in normoxia, while IL6-EGFP was undetectable (Fig. 2D). Cytokine treatment reduced αSMA-DsRed levels, and in combination with hypoxia resulted in suppression of the αSMA-DsRed signal (Fig. 2D). In contrast, both cytokines and hypoxia increased IL6-EGFP levels individually to similar levels and when combined led to marked accumulation of the IL6-EGFP signal (Fig. 2D), indicating a switch in the expression of myCAF and iCAF markers under these conditions.
Figure 2: Hypoxia potentiates the cytokine-induced inflammatory fibroblast phenotype.
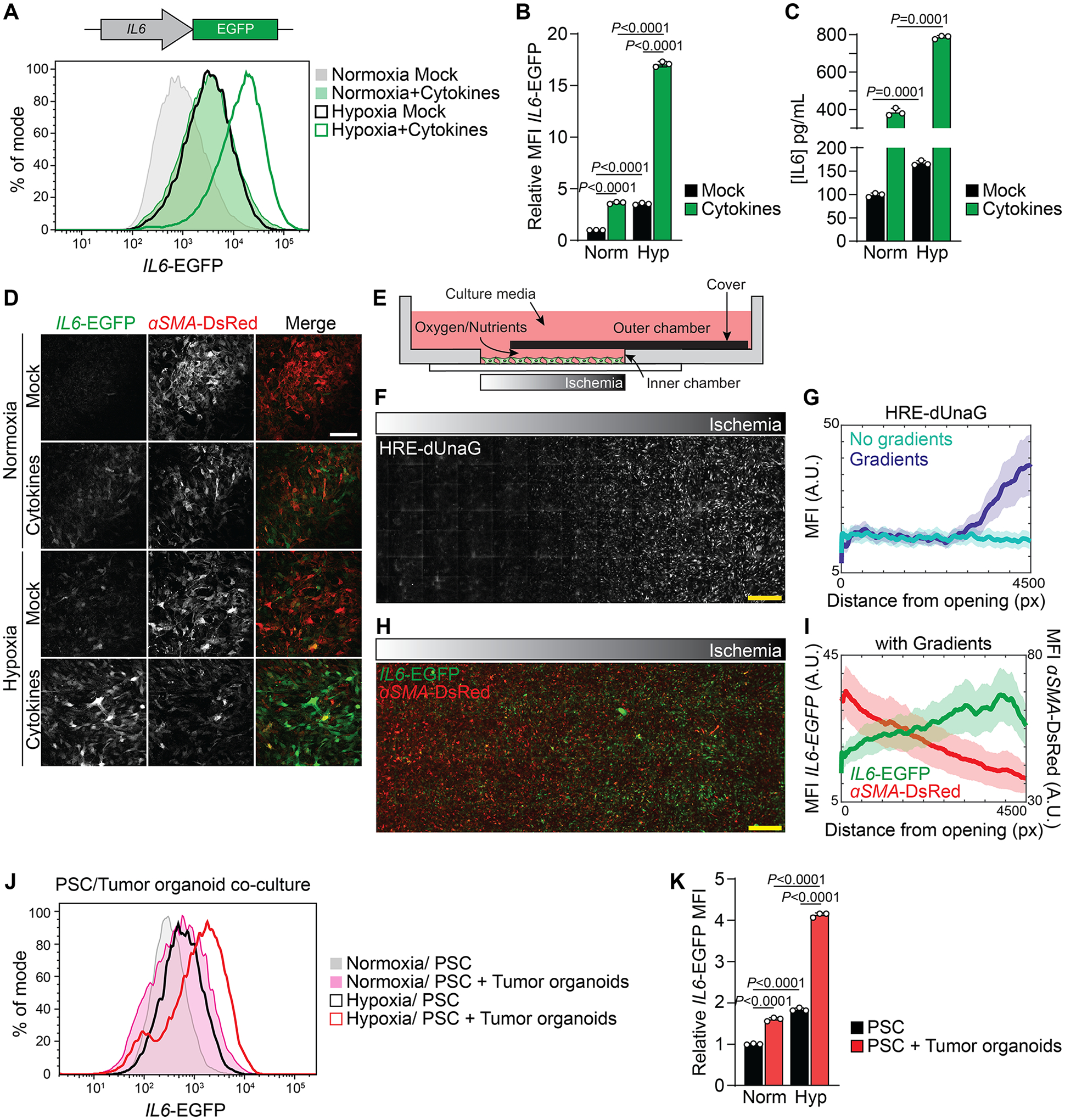
(A, B) Fluorescence intensity of IL6-EGFP expressing PSCs cultured in normoxia or hypoxia and mock-treated or treated with cytokines (IL1/TNFα) for 48h. (A) Histogram of IL6-EGFP fluorescence intensity. (B) Quantification of the relative MFI of IL6-EGFP. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(C) Quantification of IL6 levels in media conditioned by PSCs cultured in normoxia or hypoxia and mock-treated or treated with cytokines for 48h. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(D) Representative images of IL6-EGFP and αSMA-DsRed expressing PSCs cultured in normoxia or hypoxia and mock-treated or treated with cytokines for 48h. Scale bar = 200 μm.
(E-I) MEMIC experiment. (E) Schematic of the MEMIC, adapted from (26,35). PSCs expressing IL6-EGFP were plated in the inner chamber and treated with cytokines the next day. Gradients were allowed to form for 48h. (F, H) Representative images of MEMIC experiments with PSCs expressing HRE-dUnaG (F) or IL6-EGFP and αSMA-DsRed (H), treated with cytokines and cultured in the MEMIC for 48h. Scale bar = 500 μm. (G, I) Quantification of HRE-dUnaG (G) or IL6-EGFP and αSMA-DsRed (I) fluorescence intensity with increasing distance from the oxygen-rich opening. A.U. = arbitrary units, px = pixel.
(J, K) PSC/Tumor organoid co-culture experiment. PSCs expressing IL6-EGFP and αSMA-DsRed were cultured alone or together with KPC organoids in Matrigel for five days. In the last 48h, part of the cultures was incubated in hypoxia. (J) Histogram of IL6-EGFP fluorescence intensity in PSCs. (K) Quantification of the relative MFI of IL6-EGFP in PSCs. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
In tumors, there are gradients of oxygen and nutrient availability (34). To better model these gradients, we cultured PSCs together with cytokines in a metabolic microenvironment chamber (MEMIC) which allows the establishment of oxygen and nutrient gradients within the same culture well (Fig. 2E) (26,35). Using PSCs expressing the hypoxia reporter HRE-dUnaG (36), we confirmed establishment of an oxygen gradient along the MEMIC (Fig. 2F, G; Sup. Fig. 2B). While αSMA-DsRed reporter levels gradually declined along the gradient, the IL6-EGFP reporter signal increased towards ischemic regions (Fig. 2H, I; Sup. Fig. 2C–F), indicating that PSCs lose myCAF but acquire iCAF markers in ischemic conditions.
While PSCs cultured on plastic are considered myCAFs, PSCs cultured in Matrigel become quiescent and can acquire an iCAF state when co-cultured with PDAC organoids in Matrigel (4), a process dependent on organoid-derived cytokines (10). Consistent with this, we observed higher levels of IL6-EGFP but lower levels of αSMA-DsRed in PSCs co-cultured with KPC organoids in Matrigel for five days (Sup. Fig. 2G–J). Next, co-cultures of PSCs and KPC organoids were placed in hypoxia for the last 48h of the culture period. Hypoxia was sufficient to elevate expression of IL6-EGFP in PSCs to similar levels as did organoid co-culture, and exposure of co-cultures to hypoxia further elevated IL6-EGFP reporter levels in PSCs (Fig. 2J, K).
The above data suggest that hypoxia can induce an iCAF-like state in PSCs and that hypoxia potentiates the ability of cancer cell-secreted cytokines to promote an iCAF phenotype in PSCs while suppressing expression of myCAF markers. Next, we sought to determine the underlying mechanism. Among factors known to induce the iCAF state (10), we found a significant upregulation of Lif expression after culturing PSCs for 4h in hypoxia, while the iCAF marker IL6 was induced only after 24h exposure to hypoxia (Sup. Fig. 3A). To test whether Lif expression plays a role in promoting an iCAF-like state in hypoxia, we cultured PSCs for 24h in hypoxia in the presence of an anti-LIF neutralizing antibody. Blocking LIF reduced the IL6-EGFP reporter signal induced by hypoxic culture, while in turn, rescuing downregulation of the αSMA-DsRed reporter in hypoxia (Sup. Fig. 3B). IL1α-induced LIF has been shown to promote the iCAF state by activating JAK/STAT signaling in PSCs (10). Hypoxia increased activation of STAT3 and expression of the JAK/STAT target Socs3, peaking at 4–24h after exposure to hypoxia (Sup. Fig. 3C, D). STAT3 activation and Socs3 expression were reduced in the presence of an anti-LIF neutralizing antibody (Sup. Fig. 3E, F). Consistent with the role of JAK/STAT signaling in regulating the iCAF state, we found that the JAK inhibitor AZD1480 reduced IL6 expression induced by hypoxia, while rescuing hypoxia-mediated repression of αSMA (Sup. Fig. 3G, H). These data implicate a role for LIF and JAK/STAT signaling in the acquisition of an iCAF-like state in PSCs following exposure to hypoxia.
Previous analysis of transcription factor activity in human PDAC scRNA-sequencing data (5) indicated that in addition to the known iCAF regulators STAT3 and REL (NF-κB), HIF-1α activity is enriched in iCAFs (Sup. Fig. 3I). To test whether HIF-1α is active in CAFs accumulating in hypoxic regions in PDAC, we stained tumors from pimonidazole-injected mice (Fig. 1B) with a HIF-1α antibody. Indeed, nuclear HIF-1α was found in PDPN+αSMA− CAFs in pimonidazole+ regions, but not in PDPN+αSMA+ CAFs accumulating outside of pimonidazole+ regions (Fig. 3A). Hif1a is not transcribed basally in resting fibroblasts and its transcription is induced by growth factor and/or cytokine stimulation (37). Even when transcription is induced, fibroblasts like other cells do not accumulate HIF-1α protein due to the oxygen-dependent degradation by VHL (38). Like fibroblasts, PSCs accumulated little HIF-1α under hypoxia, however, when stimulated by cytokines under hypoxic conditions HIF-1α was upregulated synergistically over time, and we observed increased expression of the HIF-1α target LDHA compared to hypoxia alone (Fig. 3B). Hif1a transcription has been shown to be induced by NF-κB signaling as a result of cytokine stimulation (37,39). To test the role of NF-κB signaling in hypoxia and cytokine-induced HIF-1α upregulation, we treated PSCs with the IKKβ inhibitor (IKKβi) MLN120B. Cytokine stimulation resulted in nuclear accumulation of the NF-κB subunit p65, which was inhibited by IKKβi (Sup. Fig. 3J). While hypoxia did not alter Hif1a mRNA expression, Hif1a transcript and protein were induced by cytokine treatment in hypoxic PSCs in an NF-κB dependent fashion (Fig. 3C; Sup. Fig. 3J). In contrast, hypoxia reduced Hif2a mRNA expression, and Hif2a did not respond to additional stimulation by cytokines (Sup. Fig. 3K). Higher levels of HIF-1α were also found in PSCs co-treated with cytokines and cobalt chloride (CoCl2), a known inducer of HIF-1α stabilization and signaling (38), compared to CoCl2 treatment alone (Sup. Fig. 3L). While CoCl2 treatment alone increased levels of the IL6-EGFP reporter, combined treatment with CoCl2 and cytokines elevated the IL6-EGFP signal even more (Sup. Fig. 3L–N).
Figure 3: HIF-1α mediates the hypoxia-induced inflammatory phenotype in fibroblasts.
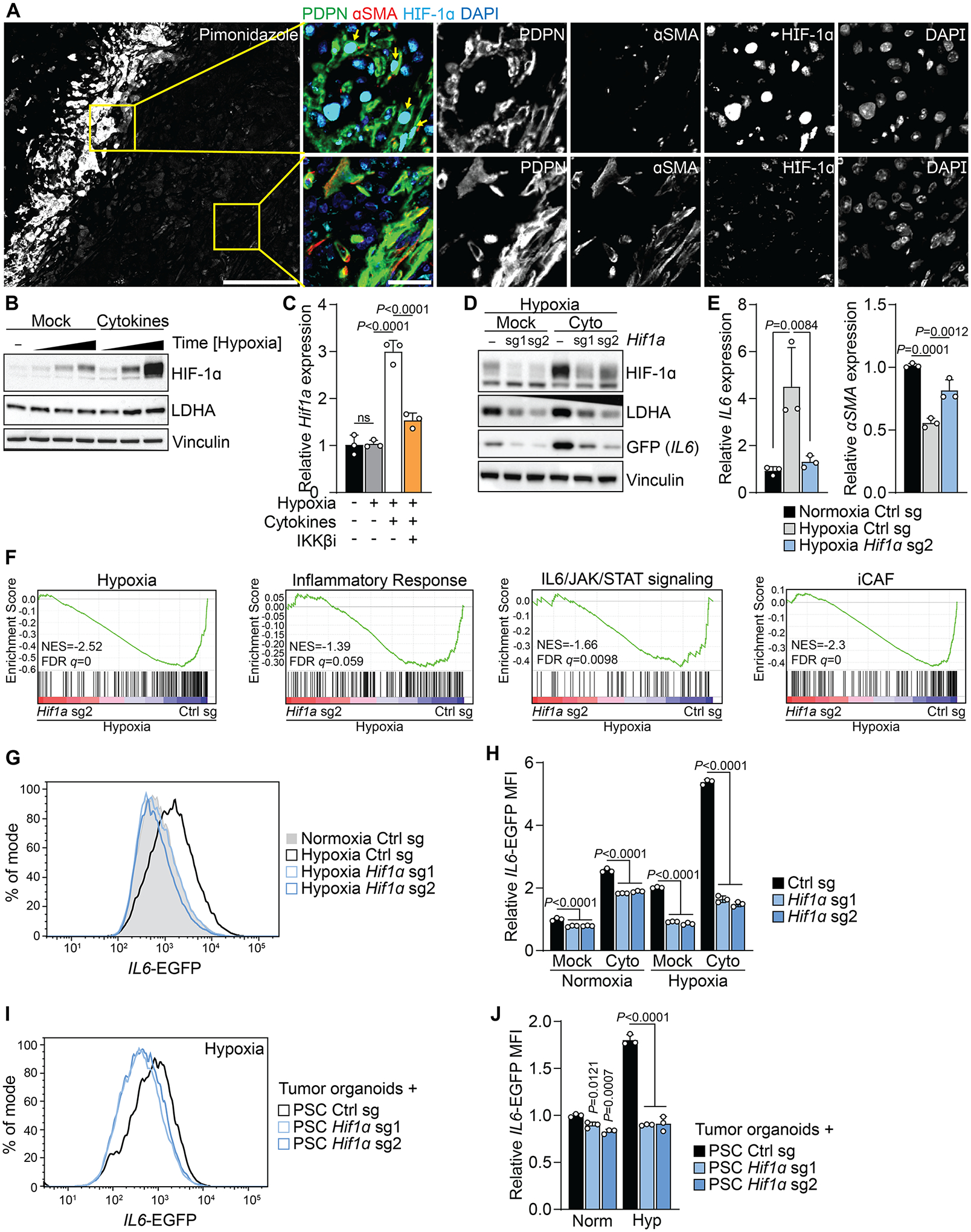
(A) Immunofluorescence staining of Pimonidazole, PDPN, αSMA and HIF-1a in mouse PDAC tumors. Representative image. Nuclei are labeled with DAPI. Scale bar = 500 μm. Yellow arrows indicate PDPN+HIF-1a+ cells.
(B) Western blot of PSCs cultured in Normoxia or in hypoxia in the presence or absence of cytokines for 15 min, 1 h or 4 h. Cytokine treatment and hypoxia were started at the same time. Representative experiments are shown. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(C) qPCR for Hif1a in PSCs cultured in normoxia or hypoxia for 4h and treated with cytokines in the presence or absence of the IKKβ inhibitor MLN120B.
(D) Western Blot of PSCs expressing IL6-EGFP and control or Hif1a sgRNA and cultured in Hypoxia. Cells were mock-treated or treated with cytokines for 48h. Representative experiment.
(E) qPCR for the indicated transcripts in PSCs expressing control or Hif1a sgRNA and cultured in normoxia or hypoxia for 48h. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(F) GSEA comparing PSCs expressing control or Hif1a sgRNA and cultured in hypoxia for 48h. iCAF signature derived from (4). Other signatures represent Hallmark signatures from MSigDB. N=3 biological replicates.
(G, H) Fluorescence intensity of PSCs expressing IL6-EGFP and control or Hif1a sgRNA cultured in Normoxia or Hypoxia and mock-treated or treated with cytokines for 48h. (G) Histogram of IL6-EGFP fluorescence intensity in mock-treated cells. (H) Quantification of the relative MFI of IL6-EGFP. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(I, J) Fluorescence intensity of PSCs expressing IL6-EGFP and control or Hif1a sgRNA co-cultured with KPC organoids for five days. In the last 48h, part of the cultures were incubated in Hypoxia. (I) Histogram of IL6-EGFP fluorescence intensity in PSCs cultured with organoids in Hypoxia. (J) Quantification of relative MFI of IL6-EGFP in PSCs. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
The above data suggested a role of HIF-1α in the hypoxia-mediated regulation of the iCAF state. To investigate this possibility, we expressed Hif1a sgRNAs which reduced protein levels of HIF-1α and its target LDHA in mock-treated as well as cytokine-treated PSCs in hypoxia (Fig. 3D). Hypoxic induction of IL6 and Ldha as well as repression of αSMA in PSCs was dependent on HIF-1α (Fig. 3E, Fig. 3O). In addition, Hif1a sgRNA reduced IL6 accumulation in the media of PSCs cultured in hypoxia (Sup. Fig. 3P). On a global gene expression level, the iCAF signature as well as inflammatory response, IL6/JAK/STAT signaling signatures in hypoxic PSCs were dependent on HIF-1α (Fig. 3F). The expression of the myCAF signature was unchanged in HIF-1α deleted PSCs in hypoxia (Sup. Fig. 3Q). Furthermore, the hypoxia-induced increase in IL6-EGFP fluorescence required HIF-1α (Fig. 3G, H). Given the further upregulation of HIF-1α in hypoxic cells by cytokine treatment, we also analyzed Hif1a sgRNA expressing PSCs in the presence of cytokines. Hif1a sgRNA prevented the synergistic accumulation of IL6-EGFP in cytokine-treated PSCs cultured in hypoxia (Fig. 3D, H). Similar results were obtained in Hif1a sgRNA expressing PSCs treated with CoCl2 (Sup. Fig. 3R, S). Moreover, the hypoxia-induced upregulation of IL6-EGFP reporter levels in PSCs co-cultured with KPC organoids without addition of exogenous cytokines beyond those produced by organoid cultures was also dependent on HIF-1α (Fig. 3I, J).
Next, we investigated whether HIF-1α stabilization can be sufficient to shift fibroblasts towards an iCAF-like state. To induce HIF-1α accumulation under normoxic conditions, we deleted Vhl, which targets hydroxylated HIF-1α for proteasomal degradation (38) (Fig. 4A). Vhl-deleted PSCs displayed higher expression of IL6 and Ldha mRNA, while αSMA expression was reduced (Fig. 4B). We also detected higher levels of IL6 in spent media of Vhl-deleted PSCs (Fig. 4C). Vhl deletion alone increased IL6-EGFP levels more than cytokine treatment, and when combined, Vhl deletion and cytokines elevated IL6-EGFP reporter signals ten-fold (Fig. 4A, D, E). Vhl deletion also promoted the IL6-EGFP signal in PSCs co-cultured with KPC organoids without addition of exogenous cytokines (Fig. 4F, G).
Figure 4: HIF-1α stabilization in fibroblasts can be sufficient to promote an inflammatory phenotype and tumor growth.
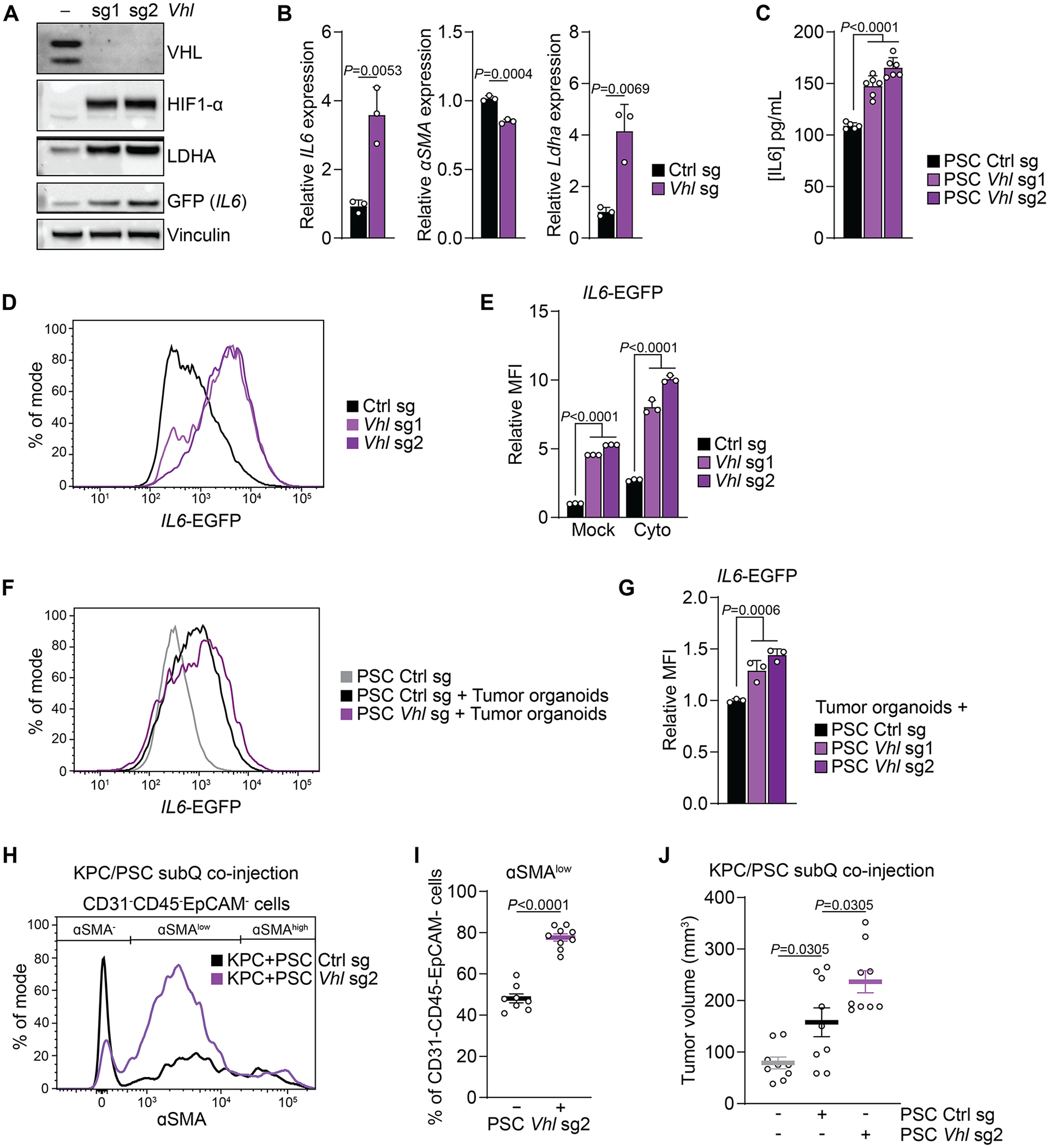
(A) Western blot of PSCs expressing control or Vhl sgRNAs and cultured in normoxia. A representative experiment is shown.
(B) qPCR for the indicated transcripts in PSCs expressing control or Vhl sgRNA cultured in normoxia. N=3 biological replicates. Data represent mean+SD. P-values were calculated by Student’s t-test.
(C) Quantification of IL6 levels in media conditioned by PSCs cultured in normoxia or hypoxia and mock-treated or treated with cytokines for 48h. N=6 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(D, E) Fluorescence intensity of PSCs expressing IL6-EGFP and control or Vhl sgRNA cultured in normoxia and mock-treated or treated with cytokines for 48h. (D) Histogram of IL6-EGFP fluorescence intensity in mock-treated cells. (E) Quantification of the relative MFI of IL6-EGFP. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(F, G) Fluorescence intensity of PSCs expressing IL6-EGFP and control or Vhl sgRNA co-cultured with KPC organoids for five days in normoxia. (F) Histogram of IL6-EGFP fluorescence intensity in PSCs. (G) Quantification of the relative MFI of IL6-EGFP in PSCs. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(H-J) Subcutaneous co-injection of KPC cells alone or together with PSCs expressing control of Vhl sgRNA. (H) aSMA fluorescence intensity of CD31-CD45-EpCAM- cells. (I) Quantification of CD31-CD45-EpCAM- cells expressing low levels of aSMA. N=8 (Ctrl) or 9 (Vhl sg2) biological replicates. Data represent mean+SD. P-values were calculated by Student’s t-test. (J) Tumor volume. N=9 biological replicates. Data represent mean+/−SEM. P-values were calculated by one-way ANOVA.
The above data indicate that Vhl deletion induces an iCAF-like state in PSCs. To test this in vivo, we used a subcutaneous allograft model. Tumors formed by KPC pancreatic cancer cells co-injected with Vhl-deleted compared to control PSCs had a lower abundance of αSMA+ CAFs (Sup. Fig. 4A, B). Within the αSMA+ population, a higher proportion of cells that expressed only low levels of αSMA, a characteristic of iCAFs, was found in tumors formed by KPC cells co-injected with Vhl-deleted PSCs (Fig. 4H, I). Given that iCAFs are associated with tumor growth (10), we sought to test whether Vhl deletion in PSCs would increase their ability to promote tumor growth in the same model. Co-injection of PSCs together with KPC pancreatic cancer cells promoted tumor growth compared to KPC cells alone (Fig. 4J), as reported before (40). Notably, co-injection of Vhl-deleted PSCs increased tumor growth significantly more than did control PSCs (Fig. 4J).
Our data indicate that Vhl deletion-induced HIF-1α stabilization in PSCs can be sufficient to promote an iCAF-like state and tumor growth. Given that VHL also targets HIF-2α for degradation, we tested whether the Vhl deletion-induced effects in PSCs were HIF-1α dependent. To this end, we generated Vhl/Hif1a double-knockout PSCs (Fig. 5A). Hif1a deletion rescued IL6 and Ldha induction as well as αSMA repression in Vhl-deleted PSCs (Fig. 5A, B). Similarly, upregulation of the IL6-EGFP reporter and accumulation of IL6 protein in Vhl-deleted PSCs was abolished by Hif1a deletion (Fig. 5C, D). To investigate the role of HIF-1α in Vhl deletion-mediated tumor support of PSCs, we co-injected KPC cells with control, Vhl-deleted, and Vhl/Hif1a double-knockout PSCs. While Vhl-deleted PSCs promoted tumor growth compared to control PSCs, this effect was not observed upon injection Vhl/Hif1a double-knockout PSCs (Fig. 5E).
Figure 5: Vhl-deletion promotes in PSC promotes an inflammatory phenotype and tumor growth via HIF-1α.
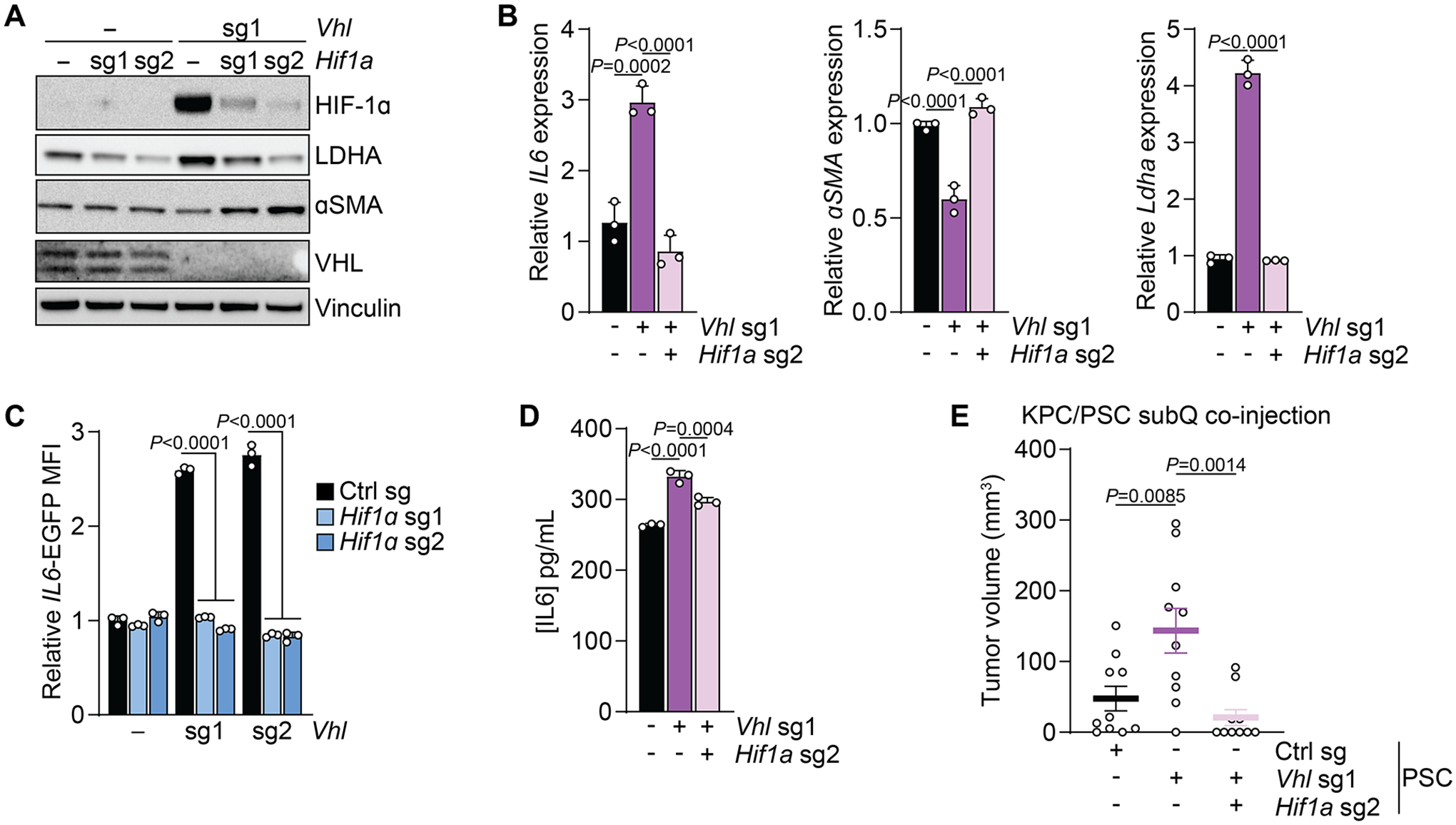
(A) Western blot of PSCs expressing control or Hif1a sgRNAs in the presence or absence of Vhl deletion. Representative experiment.
(B) qPCR for the indicated transcripts in PSCs expressing control, Vhl sgRNA, or Vhl and Hif1a sgRNAs cultured in normoxia. N=3 biological replicates. Data represent mean+SD. P-values were calculated by Student’s t-test.
(C) Quantification of fluorescence intensity of PSCs expressing control, Vhl sgRNA, or Vhl and Hif1a sgRNAs cultured in normoxia. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(D) Quantification of IL6 levels in media conditioned by PSCs expressing control, Vhl sgRNA, or Vhl and Hif1a sgRNAs cultured in normoxia. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(E) Volume of tumors arising from subcutaneous co-injection of KPC cells alone or together with PSCs expressing control, Vhl sgRNA, or Vhl and Hif1a sgRNAs. N=10 biological replicates. Data represent mean+/−SEM. P-values were calculated by one-way ANOVA.
Next, we sought to understand how HIF-1α stabilization in PSCs promotes tumor growth. In a co-culture model, Vhl deletion in PSCs added only a minimal benefit to organoid growth promoted by co-cultured PSCs (Sup. Fig. 4C), indicating the relevance of the in vivo context for tumor support by PSC HIF-1α. Given the HIF-1α dependent expression of IL6 described above, and the critical role of IL6 in PDAC progression (14), we hypothesized that PSC-derived IL6 is involved in tumor growth induced by PSC HIF-1α. To test this, we first co-injected with KPC cells and Vhl-deleted PSCs into the flanks of wildtype mice, and one week later treated the mice with an anti-IL6 monoclonal antibody or IgG control every three days for two weeks (Sup. Fig. 4D). No significant reduction in tumor growth following anti-IL6 treatment was observed (Sup. Fig. 4D).
Next, we sought to identify factors beyond IL6 that could mediate PSC HIF-1α-promoted tumor growth. Analysis of signaling molecule activity in human PDAC scRNA-sequencing data (5) indicated that in addition to IL6, the HIF-1α target VEGFA is among the highest enriched signaling molecules in iCAFs (Fig. 6A). Both Vegfa mRNA and VEGF protein accumulated in PSC cultured in hypoxia and further increased upon cytokine treatment (Fig. 6B, C). Upregulation of Vegfa mRNA and VEGF protein in hypoxia was HIF-1α dependent (Fig. 6D, E). Vegfa and VEGF were also upregulated by Vhl deletion in PSCs cultured in normoxia in a HIF-1α dependent fashion (Fig. 6F–H). Consistent with these findings, an angiogenesis expression signature was upregulated in hypoxic PSCs in a HIF-1α dependent fashion (Sup. Fig. 4E), indicating a role for PSC HIF-1α in remodeling the tumor vasculature. While no difference was observed in the abundance of CD31+ endothelial cells in tumors arising from co-injection of Vhl-deleted compared to control PSCs (Sup. Fig. 4F, G), there was a significant correlation between the number of CD31+ cells and tumor weight at endpoint (Fig. 6I). Thus, we sought to test the role of VEGF signaling in PSC HIF-1α mediated tumor growth. Mice co-injected with KPC and Vhl-deleted PSCs were treated with an anti-VEGFR2 monoclonal antibody or IgG control for two weeks (Sup. Fig. 4D). Consistent with the previous experiment, the number of CD31+ cells correlated with tumor weight at endpoint (Sup. Fig. 4H). Anti-VEGFR2 treatment reduced the abundance of CD31+ cells (Fig. 6J, K) and slowed down tumor growth that was promoted by Vhl-deleted PSCs (Fig. 6L). Taken together, these data indicate hypoxia-induced HIF-1α as a regulator of the iCAF state and promotor of tumor growth in PDAC at least in part via VEGF secretion.
Figure 6: VEGF derived from hypoxia induced iCAFs promotes tumor growth.
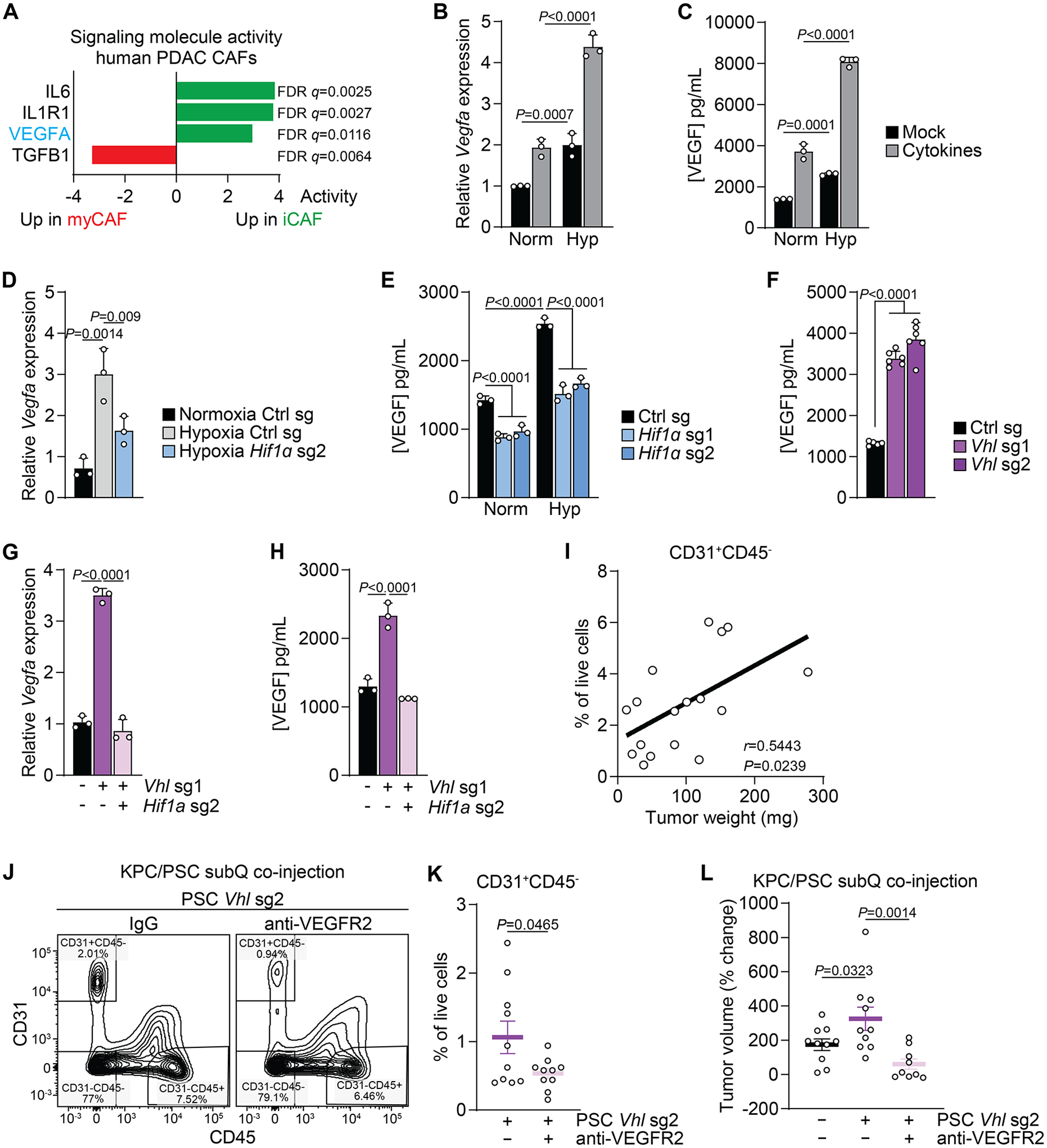
(A) Activity of a selected set of signaling molecules factors in myCAFs and iCAFs based on scRNA-seq data from human PDAC. Data from (5).
(B, C) Expression of Vefga mRNA (B) and protein (C) in PSCs cultured in normoxia or hypoxia and mock-treated or treated with cytokines for 48h. N=3 biological replicates. Data represent mean+SD. P-values were calculated by two-way ANOVA.
(D, E) Expression of Vefga mRNA (D) and protein (E) in PSCs expressing control of Hif1a sgRNAs cultured in normoxia or hypoxia for 48h. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA (D) or two-way ANOVA (E).
(F) Quantification of VEGF in media conditioned by PSCs expressing control or Vhl sgRNA. N=6 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(G, H) Expression of Vefga mRNA (G) and protein (H) in PSCs expressing control, Vhl sgRNA, or Vhl and Hif1a sgRNAs cultured in normoxia. N=3 biological replicates. Data represent mean+SD. P-values were calculated by one-way ANOVA.
(I) Correlation of the number of CD31+CD45- cells with tumor weight in tumors arising from subcutaneous co-injection of KPC cells together with PSCs expressing control or Vhl sgRNA.
(J-L) Subcutaneous co-injection of KPC cells together with PSCs expressing Vhl sgRNA and treated with an anti-VEGFR2 monoclonal antibody or IgG control. Gating for (J) and quantification of (K) CD31+CD45- cells among live cells. (L) Tumor volume as % of change since onset of treatment.
Discussion
Hypoxia is has long been recognized as a characteristic of the PDAC TME and is associated with poor outcomes of PDAC patients which is at least in part due to its influence on the cancer cell state (15,18). Whether hypoxia also affects the stromal cell state in the PDAC TME and their influence on tumor progression is less well understood. Here, we show that hypoxia-induced HIF-1α signaling shifts pancreatic CAFs towards acquisition of an iCAF-like state. While hypoxia by itself induces an inflammatory program, this is likely a hybrid state in which cells also display some myCAF features, consistent with upregulation of the iCAF signature in hypoxia while the myCAF signature was unchanged. Our observations that hypoxia potentiates the effects of cytokines secreted by PDAC cells suggests that when both hypoxia and cytokines are present, there is a switch from a myCAF to an iCAF state. This is consistent with our observations that iCAFs accumulate biochemical markers of hypoxia and that αSMA-positive CAFs are largely absent from hypoxic regions in murine PDAC. Furthermore, iCAFs display a hypoxic gene expression profile in human PDAC patients. The selective accumulation of hypoxia markers in iCAFs has also been confirmed in a separate study (41). These data provide evidence for hypoxia as an environmental regulator of fibroblast heterogeneity in PDAC. Given that hypoxia and fibroblast heterogeneity is a feature of the microenvironment of many solid tumors (9), the findings of this study could be relevant to a wide range of solid tumors.
The idea that hypoxia promotes an inflammatory response has been supported by several studies. In mice, short term exposure to hypoxia is sufficient to promote accumulation of inflammatory cells in several tissues and increases serum levels of various cytokines (42). In addition to being observed in tumors, hypoxia is also a feature of wounds, and fibroblast heterogeneity has been observed in wound healing (43). Thus, our observations further support the idea that cancer cell metabolism can co-opt the normal stromal regenerative response to support tumor growth (44).
Our data suggest that the hypoxia-induced shift of PSCs towards an iCAF-like state is mediated in part by upregulation of LIF in hypoxia, which activates JAK/STAT signaling in a cell-autonomous fashion. LIF is also induced in PSCs by IL1a via NF-κB, and PSC-derived LIF promotes PDAC progression (10,45). Together, these data indicate a role for hypoxia in PDAC progression via the induction of LIF in the tumor stroma, by promoting the inflammatory fibroblast state, as well as by acting non-cell autonomously on cancer cells.
Hypoxia also potentiates the ability of cytokines to promote acquisition of an iCAF phenotype in PSCs in a HIF-1α-dependent fashion. Elevated HIF-1α levels in hypoxic PSCs stimulated with cytokines results from increased Hif1α transcription as a consequence of cytokine induced NF-κB signaling, consistent with prior reports (37,39). Furthermore, cytokine signaling and HIF-1α cooperate to activate HIF-1α transcriptional activity by co-binding of STAT3 to promoter regions of HIF-1α target genes (46), further supporting the idea that cytokine signaling can cooperate with hypoxic signaling to influence the cell state. In addition, hypoxia can promote inflammatory cytokine production in cancer cells (47), suggesting the possibility of a feed forward mechanism resulting in autocrine cytokine signaling that might influence CAFs. This is supported by observations that PDAC cells produce higher levels of the iCAF inducer IL1α in hypoxic culture conditions (41). While in this study we considered tumor cells as a major source of cytokine that synergize with hypoxia to alter the CAF state, it is possible that other stromal cell types in the TME are involved in this process. Hypoxia is known to induce secretion of IL1 and TNFα in macrophages (42). Interestingly, tumor-associated macrophages accumulate in hypoxic PDAC regions, resulting in polarization into distinct subtypes (35), similar to our observations in CAFs. Taken together, these findings argue that hypoxia can results in significant remodeling of the TME, altogether promoting a phenotypic change in CAFs towards a more inflammatory state via both cell autonomous and non-cell autonomous mechanisms.
We provide evidence that HIF-1α stabilization in PSCs is sufficient to promote tumor growth in vivo in an allograft co-injection model. While Vhl-deleted PSCs secrete more IL6, blocking IL6 was insufficient to alleviate the tumor promoting effect of these cells. IL6 is critical for PDAC progression (14), however IL6 blockade or whole-body deletion of IL6 did not impact progression or overall survival (13,48). Responsiveness to IL6 appears to be dependent on p53 status in the tumor compartment (49), and consistent with this notion, the PDAC cells used in this study were p53 mutant. Thus, in the model systems used here, IL6 acts primarily as a marker for iCAFs, rather than as the effector of PSC-induced tumor growth. Instead, we found VEGF as PSC-derived, HIF-1α regulated molecule that is highly active in iCAFs, and implicate VEGF signaling as a mediator of the tumor promoting effects of PSC HIF-1α. Our data support a role for CAF-derived VEGF in modulating endothelial cells and the angiogenic response in tumors, which is consistent with prior findings in breast cancer (20). Overall, these findings support the idea that HIF-1α functions as tumor promoter in CAFs, but as tumor suppressor in cancer cells (50).
In this work, we identify a role for HIF-1α in promoting an inflammatory state in CAFs and to support tumor growth. Interestingly, HIF-2α but not HIF-1α expression in αSMA+ myCAFs has been shown to accelerate PDAC progression by establishing an immunosuppressive TME (51). Consistently, we detected nuclear HIF-1α only in αSMA− CAFs in hypoxic PDAC regions, but not in αSMA+ CAFs outside hypoxic areas. Together, these data suggest that αSMA+ myCAFs rely on HIF-2α, while αSMA−/αSMALow iCAFs depend on HIF-1α. To support this idea, future studies will be required that target HIF proteins in iCAFs in autochthonous tumor models, for example by using a dual recombinase approach including a Cre driver that is highly expressed in the iCAF population, such as Pdgfra or Fap (5,48). Understanding mechanism behind the differential dependence of CAF populations on HIF proteins will be an important area of investigation, and might involve selective modulation of cellular metabolism (52). Furthermore, the exclusion of αSMA+ CAFs from pimonidazole+ hypoxic PDAC regions reported in this study indicates that HIF-2α might be activated in myCAFs via oxygen-independent mechanisms. These might involve reactive oxygen species (ROS), as myofibroblast induction by TGFβ has been shown to depend on ROS (53).
Collectively, these data argue that hypoxic signaling via HIF-1α or HIF-2α alters the CAF phenotype to promote PDAC tumor growth by remodeling other stromal cell types including endothelial cells and macrophages. Given the presence of hypoxia and CAF heterogeneity in most solid tumors (9), targeting hypoxic signaling in the tumor stroma might be a generalizable strategy to impair cancer progression. Small molecules that can selectively target HIF-1α or HIF-2α are in clinical development, but their application will need to be considered in light of the tumor suppressive role of HIF-1α in the tumor epithelium (50). Selective targeting of HIF proteins in the tumor stroma could involve the use of chimeric antigen receptor T-cells (CAR-T), in which CAR expression is driven by a hypoxia response element (HRE) and targeted against a surface molecule highly expressed in CAFs, such as fibroblast activation protein (FAP). Hypoxia-sensing CAR-T cells and targeting FAP with CAR-T have proven safe in mouse models of cancer (54,55).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank the members of the Thompson laboratory for helpful discussions. We are thankful to Tullia Lindsten for help with planning of and protocol preparation for mouse experiments, and to Natalya N. Pavlova for help with GMAP. We also thank Elisa De Stanchina, Inna Kudos and Janelle Simon for help with orthotopic organoid injections into the pancreas, and Wenfei Kang and Eric Rosiek of the MSKCC Molecular Cytology Core for help with immunofluorescence staining, microscopy and image analysis. S.S. received support from the NCI (5K99CA259224) and the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center at MSKCC. S.S. is also supported by a Hirshberg Foundation Seed Grant, the AGA Research Foundation’s Caroline Craig Augustyn & Damian Augustyn Award in Digestive Cancer, and an ACS-IRG Pilot Grant from the University of Chicago Comprehensive Cancer Center. K.M.T. was supported by the Jane Coffin Childs Memorial Fund for Medical Research and a Shulamit Katzman Endowed Postdoctoral Research Fellowship. This work was supported by MSKCC’s David Rubenstein Center for Pancreatic Research Pilot Project (to S.W.L.) and NIH grant P01CA013106 (to S.W.L.). S.W.L. is the Geoffrey Beene Chair for Cancer Biology at MSKCC. C.C.F. receives support from the National Cancer Institute at the NIH (DP2 CA250005), the American Cancer Society (RSG-21-179-01-TBE) and the Pew Charitable Trust (00034121). C.B.T. was supported by the NCI (R01CA201318). This work used core facilities at MSKCC that were supported by the cancer center support grant (P30CA008748).
Footnotes
Conflict of interest:
C.B.T. is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. He is also a former member of the Board of Directors and stockholder of Merck and Charles River Laboratories. He holds patents related to cellular metabolism. S.W.L. is a consultant and holds equity in Blueprint Medicines, ORIC Pharmaceuticals, Mirimus, Inc., PMV Pharmaceuticals, Faeth Therapeutics, and Constellation Pharmaceuticals. All other authors do not declare any conflict of interest.
References
- 1.Orth M, Metzger P, Gerum S, Mayerle J, Schneider G, Belka C, et al. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol. Radiation Oncology; 2019;14:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helms E, Onate MK, Sherman MH. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020;10:648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell [Internet]. Elsevier Inc.; 2021;39:548–565.e6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33667385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med [Internet]. 2017;214:579–96. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2019;CD-19–0644. [DOI] [PubMed] [Google Scholar]
- 7.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. J Clin Oncol. 2019;37:e15739–e15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biffi G, Tuveson DA. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev. 2021;101:147–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicolas AM, Pesic M, Engel E, Ziegler PK, Diefenhardt M, Kennel KB, et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell. 2022;40:168–184.e13. [DOI] [PubMed] [Google Scholar]
- 12.Lee BY, Hogg EKJ, Below CR, Kononov A, Blanco-Gomez A, Heider F, et al. Heterocellular OSM-OSMR signalling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat Commun [Internet]. 2021;12:7336. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34921158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut [Internet]. 2018;67:320–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27797936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys [Internet]. 2000;48:919–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11072146 [DOI] [PubMed] [Google Scholar]
- 16.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol Cell [Internet]. Elsevier Inc.; 2010;40:294–309. Available from: 10.1016/j.molcel.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, et al. Targeting the LOX / hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat Rev Cancer [Internet]. Nature Publishing Group; 2014;14:430–9. Available from: 10.1038/nrc3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal. 2019;12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madsen CD, Pedersen JT, Venning FA, Singh LB, Moeendarbary E, Charras G, et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep [Internet]. 2015;16:1394–408. Available from: http://embor.embopress.org/cgi/doi/10.15252/embr.201540107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwörer S, Pavlova NN, Cimino FV, King B, Cai X, Sizemore GM, et al. Fibroblast pyruvate carboxylase is required for collagen production in the tumour microenvironment. Nat Metab [Internet]. 2021;3:1484–99. Available from: https://www.nature.com/articles/s42255-021-00480-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell [Internet]. Elsevier Inc.; 2020;1–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32234521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lebleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jesnowski R, Fürst D, Ringel J, Chen Y, Schrödel A, Kleeff J, et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and N-acetylcysteine. Lab Investig. 2005;85:1276–91. [DOI] [PubMed] [Google Scholar]
- 26.Janská L, Anandi L, Kirchberger NC, Marinkovic ZS, Schachtner LT, Guzelsoy G, et al. The MEMIC is an ex vivo system to model the complexity of the tumor microenvironment. DMM Dis Model Mech. 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwörer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, et al. Proline biosynthesis is a vent for TGFβ‐induced mitochondrial redox stress. EMBO J [Internet]. 2020;39:e103334. Available from: https://onlinelibrary.wiley.com/doi/abs/10.15252/embj.2019103334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee DY, et al. A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci Rep. Nature Publishing Group; 2016;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King B, Araki J, Palm W, Thompson CB. Yap / Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 2020;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boj SF, Hwang C Il, Baker LA, Chio IIC, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 32.Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC, et al. Pimonidazole: A Novel Hypoxia Marker for Complementary Study of Tumor Hypoxia and Cell Proliferation in Cervical Carcinoma. Gynecol Oncol [Internet]. 1998;71:270–7. Available from: http://www.sciencedirect.com/science/article/pii/S0090825898951630 [DOI] [PubMed] [Google Scholar]
- 33.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discov. 2022;12:484–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–82. [DOI] [PubMed] [Google Scholar]
- 35.Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci [Internet]. 2017;114:2934–9. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1700600114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erapaneedi R, Belousov VV, Schäfers M, Kiefer F. A novel family of fluorescent hypoxia sensors reveal strong heterogeneity in tumor hypoxia at the cellular level. EMBO J. 2016;35:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. [DOI] [PubMed] [Google Scholar]
- 39.Van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1αa by NF-κB. Biochem J. 2008;412:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwörer S, Pavlova NN, Cimino FV, King B, Cai X, Sizemore GM, et al. Fibroblast pyruvate carboxylase is required for collagen production in the tumor microenvironment. Nat Metab. 2021;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mello AM, Ngodup T, Lee Y, Donahue KL, Li J, Rao A, et al. Hypoxia promotes an inflammatory phenotype of fibroblasts in pancreatic cancer. Oncogenesis. Springer Nature; 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med [Internet]. 2011;364:656–65. Available from: https://portlandpress.com/biochemist/article/39/4/34/475/Hypoxia-and-inflammation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21. [DOI] [PubMed] [Google Scholar]
- 44.Schwörer S, Vardhana SA, Thompson CB. Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab [Internet]. 2019;29:576–91. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413119300154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature [Internet]. Springer US; 2019;569:131–5. Available from: 10.1038/s41586-019-1130-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawlus MR, Wang L, Hu CJ. STAT3 and HIF1α cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene. 2014;33:1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lappano R, Talia M, Cirillo F, Rigiracciolo DC, Scordamaglia D, Guzzi R, et al. The IL1β-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J Exp Clin Cancer Res. Journal of Experimental & Clinical Cancer Research; 2020;39:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov American Association for Cancer Research Inc.; 2022;12:1580–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wörmann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Görgülü K, et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology [Internet]. Elsevier, Inc; 2016;151:180–193.e12. Available from: 10.1053/j.gastro.2016.03.010 [DOI] [PubMed] [Google Scholar]
- 50.Tiwari A, Tashiro K, Dixit A, Soni A, Vogel K, Hall B, et al. Loss of HIF1A From Pancreatic Cancer Cells Increases Expression of PPP1R1B and Degradation of p53 to Promote Invasion and Metastasis. Gastroenterology. 2020;159:1882–1897.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D, et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology. 2022;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu C-J, Wang L-Y, Chodosh LA, Keith B, Simon MC. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol Cell Biol. 2003;23:9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J Biol Chem [Internet]. 2013;288:770–7. Available from: http://www.jbc.org/lookup/doi/10.1074/jbc.M112.431973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Reports Med [Internet]. ElsevierCompany.; 2021;2:100227. Available from: 10.1016/j.xcrm.2021.100227 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data have been deposited in GEO (GSE221761). Other expression data analyzed in this study were obtained from NCBI dbGaP (phs001840.v1.p1). All other data are available from the corresponding authors upon reasonable request.