

Abstract
Background:
Sirtuin 3 (SIRT3) is a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase that confers resilience to cellular stress by promoting mitochondrial activity. Mitochondrial dysfunction is a major driver of inflammation during sepsis. We hypothesize that Sirt3 expression improves survival in polymicrobial sepsis by mitigating the inflammatory response.
Materials and Methods:
Sirt3 knockout (S3KO) and wild-type (WT) mice underwent cecal ligation and puncture (CLP) or sham surgery. mRNA expression was quantified using quantitative polymerase chain reaction (qPCR) and protein expression was quantified using enzyme-linked immunosorbent assay (ELISA). Spectrophotometric assays were used to quantify serum markers of organ dysfunction. For in vitro studies, bone marrow-derived macrophages (BMDMs) were harvested from S3KO and WT mice and treated with lipopolysaccharide (LPS).
Results:
After CLP, hepatic Sirt3 levels decreased from baseline by nine hours and remained depressed at 24 hours. Peak serum interleukin-6 (IL-6) protein levels were higher in S3KO mice. In LPS-treated BMDMs, IL-6 mRNA levels peaked earlier in S3KO cells, although peak levels were comparable to WT. Although S3KO mice had decreased median survival after CLP compared with WT, there was no difference in five-day survival or organ dysfunction.
Conclusions:
Although S3KO mice initially had increased inflammation and mortality, this difference abated with time, and overall survival was comparable between the groups. This pattern is consistent with the timeline of sepsis-induced Sirt3 downregulation in WT mice, and suggests that Sirt3 downregulation occurring in sepsis is at least partially responsible for the initial hyperinflammatory response and subsequent mortality. Our data support upregulation of Sirt3 as a promising therapeutic strategy for further research in sepsis.
Keywords: anti-inflammatory, dysregulated host response, mitochondrial activity, sirtuin
Despite international campaigns to improve the early identification and supportive care of sepsis, there are approximately 11 million sepsis-related deaths worldwide every year.1 The morbidity and mortality associated with sepsis is not, however, due solely to the underlying infectious agent. Instead, it is a product of the dysregulated host response that causes inflammation, oxidative stress, mitochondrial dysfunction, and cellular apoptosis.2–6 Therefore, to improve outcomes in septic shock, it is imperative that we discover new strategies to modulate the inflammatory response.
One group of proteins that may mitigate the dysfunctional host response to sepsis are sirtuins. Sirtuins are a highly conserved group of nicotinamide adenine dinucleotide (NAD)-dependent deacetylases that confer resilience to cellular stress.7–9 Sirtuin 1 has been studied extensively in sepsis, but the importance of sirtuin 3 (Sirt3) has only recently begun to be elucidated.10–12 Sirt3 is a mitochondrial sirtuin the expression of which in sepsis is known to improve mitochondrial function13,14 and reduce tissue oxidative stress.15,16 Enhancing Sirt3 can therefore prevent inflammation17,18 and cellular apoptosis.19 Indeed, increased Sirt3 expression may decrease sepsis-induced organ dysfunction in multiple organs including kidney,19,20 heart,21,22 and brain.14
Although many benefits have been postulated for Sirt3 expression in a polymicrobial sepsis model, the overall effects of Sirt3 on morbidity and mortality in vivo remain unknown. In this study, we demonstrate that Sirt3 expression is necessary to mitigate the inflammatory response to sepsis and improve early survival. Furthermore, we provide evidence that the anti-inflammatory effects of Sirt3 may be mediated via its expression in macrophages.
Materials and Methods
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committees at the University of Pennsylvania and The Ohio State University. Sirt3 knockout (S3KO) mice were originally generated by Lombard et al.23 and were backcrossed on a C57BL/6J background for 10 generations prior to use. Studies were performed on S3KO mice and their wild-type (WT) littermates.
We performed cecal ligation and puncture (CLP) on eight to 12-week-old mice as described by Rittirsch et al.24 Briefly, we anesthetized the animals using 2% isoflurane prior to making a midline abdominal incision. We ligated the cecum with 4-0 silk suture just below the ileocecal valve and punctured the cecum twice with a 23-gauge needle. The abdominal incision was closed in two layers with 4-0 vicryl suture. Control animals underwent sham surgery, which consisted of midline abdominal incision under anesthesia with manipulation of the cecum without ligation or puncture, followed by closure of the abdominal incision with 4-0 vicryl suture.
After surgery, all animals received 0.1 mg/kg subcutaneous buprenorphine, followed by a lower subcutaneous dose (0.05 mg/kg) every 12 hours for the first 48 hours post-operatively. Subcutaneous normal saline (75 mcL/g) was administered every 12 hours starting immediately after CLP. Antibiotic agents (ceftriaxone, 25 mg/kg; metronidazole, 12.5 mg/kg) were administered 12 hours after CLP and every 12 hours subsequently for the duration of the experiment. After surgery, animals were monitored at 12 hours and every six hours thereafter. Glucose was measured via AlphaTRAK rodent glucometer (Abbott Laboratories, Abbott Park, IL). Animals were weighed daily. A cohort of animals was euthanized at 36 hours or five days, and serum was collected for quantification of interleukin-6 (IL-6) and markers of organ dysfunction. Kidney and liver tissue were also harvested at these time points and either snap-frozen or used immediately for mitochondrial function testing. Five-day survival studies were carried out on a separate cohort of mice. To minimize animal discomfort, mice were euthanized when they reached predetermined end points of euthanasia (Supplementary Table S1). After five days all surviving animals were euthanized.
Bone marrow-derived macrophages
We harvested bone marrow from eight to 12-week-old S3KO and WT mice. After euthanasia, the bilateral lower extremities were prepped and sterilized with alcohol. Both femurs and tibias were harvested using sterile technique. Marrow was extracted with RPMI +1% P/S, filtered through a 100 mcm filter, and centrifuged at 300g for 10 minutes at 4°C. The cells were resuspended in growth media (400 mL RPMI, 1% P/S, 20% fetal bovine serum [FBS], 10 mcg macrophage colony-stimulating factor [MCSF]), plated, and incubated at 37°C for seven days. Media was changed on day 4. Cells were then washed with phosphate buffered saline (PBS), incubated for five minutes with 1 mL 0.25% trypsin, and resuspended in growth media without MCSF. Cells were centrifuged at 300g for 10 minutes at 4°C and then resuspended in growth media and counted. In a six-well plate, 0.5 × 10^6 cells in 1 mL growth media were added per well and allowed to incubate overnight at 37°C. Lipopolysaccharide (LPS) was added for a concentration of 1 mcg/mL and allowed to incubate for either four or eight hours. Cells were then collected for RNA extraction.
mRNA expression
Sirt3, IL6, and SOD2 mRNA levels were measured using quantitative polymerase chain reaction (qPCR). Liver and kidney RNA was extracted from frozen tissue and bone marrow-derived macrophages (BMDMs). RNA was extracted from fresh cells with ethanol precipitation. One microgram of RNA was used to create cDNA using a High-Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Waltham, MA) according to manufacturer's recommendations. Quantitative PCR was performed with SYBR green using an Applied Biosystems 7900HT system. Cycle threshold (CT) was normalized to β-actin to calculate ΔCT, and individual sample ΔCTs were compared with the average sham ΔCT at each timepoint (for in vivo) or untreated ΔCT (for in vitro) to calculate ΔΔCT. Relative mRNA levels were expressed as fold change, calculated as 2-ΔΔCT for each sample. Primers used: β-actin (F: GGCTGTATTCCCCTCCATCG, R: CCAGTTGGTAACAATGCCATGT), IL6 (F: TAGTCCTTCCTACCCCAATTTCC, R: TTGGTCCTTAGCCACTCCTTC), SOD2 (F: GCCTGCACTGAAGTTCAATG, R: ATCTGTAAGCGACCTTGCTC), Sirt3 (F: GCTGCTTCTGCGGCTCTATAC, R: GAAGGACCTTCGACAGACCGT).
Protein expression
Serum blood urea nitrogen (BUN) was quantified using point of care veterinary cartridges (i-STAT, Abbott Point of Care, Inc., Princeton, NJ). Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were quantified spectrophotometrically using commercially available kits (Pointe Scientific kit A7561150 and A7526150, Canton, MI). Serum IL-6 was quantified using enzyme-linked immunosorbent assay (ELISA; Invitrogen BMS603HS, Thermo Fisher Scientific, Waltham, MA). Each sample was run in duplicate with known standards and according to the manufacturer's guidelines.
Mitochondrial content
Citrate synthase was used to quantify mitochondrial content. Kidney or liver tissue (100 mcg) was suspended in assay buffer (0.1 mM 5,5-dithio-bis-(2-nitrobenzoic acid) [DTNB] in 1 M Tris buffer (pH 8.0), 0.3 mM acetyl coenzyme A, and 0.05% Triton X-100), and 1 mM oxaloacetate was added. Citrate synthase activity was then determined spectrophotometrically at 37°C with absorbance at 412 nm.25
Mitochondrial isolation
Liver and kidney tissue was harvested at 36 hours and five days post-surgery for mitochondrial isolation. Immediately after the mice were euthanized, 4–6 g of liver and kidney tissue were immersed and homogenized separately in ice-cold mitochondrial isolation buffer (MIB; 210 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM ethylenediaminetetraacetic acid [EDTA], 0.5% fatty acid-free bovine serum albumin [BSA], potassium hydroxide [KOH] to pH 7.2). Differential centrifugation was performed as previously described.26 The mitochondrial pellet was then resuspended in MIB.
High-resolution respirometry
Freshly isolated mitochondria (0.15 mg) were suspended in respiration medium B (RM-B; 110 mM mannitol, 0.5 mM ethylenegloltetraacetic acid [EGTA], 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 60 mM K lactobionate, 0.3 mM dithiothreitol [DTT], 1% fatty acid-free bovine serum albumin [BSA], adjusted to pH 7.1 with KOH). Oxygen consumption was measured using high resolution respirometry at 37°C (Oxygraph-2k Oroboros Instruments, Innsbruck, Austria). After stabilization, real-time oxygen concentration and flux data were continuously collected (DatLab software 4.3, Oroboros Instruments, Innsbruck, Austria). A standard substrate/inhibitor titration (SUIT) protocol was used to analyze respiratory function as a factor of rate of oxygen consumption. Basal respiration was recorded. For rates of fatty acid oxidation (FAO), 0.04 mM palmitoyl carnitine and 0.1 mM malate were added, followed by 1 mM adenosine diphosphate (ADP). For activity of individual complexes, complex I (CI)-dependent respiration was induced by adding 10 mM glutamate, 5 mM malate, and 1 mM ADP. Complex II (CII)-dependent respiration was then determined by adding 0.5 mcM rotenone (a CI inhibitor) and CII substrate succinate (10 mM). Complex III (CIII) inhibitor antimycin A (5 mcM) was then added and complex IV (CIV) substrates tetramethyl-para-phenylenediamine (TMPD; 0.5 mM) and ascorbate (2 mM) were added to measure CIV-dependent respiration.
Statistical analysis
Results are expressed as mean ± standard error. Comparisons between groups were performed using Student t-test, Mann-Whitney test, analysis of variance (ANOVA), or Kruskal-Wallis test. Mean survival was analyzed using a Kaplan-Meier curve with a Gehan-Breslow-Wilcoxon test, and 48 hour and five-day survival were analyzed using Fisher exact test. Statistical significance was designated as p ≤ 0.05. All statistical analysis was performed using Prism 7 (GraphPad Software, Inc).
Results
Sepsis induces a functional knockdown of Sirt3 in WT mice
We first evaluated the timeline of Sirt3 expression changes during sepsis in WT mice. Compared with sham mice, hepatic Sirt3 gene expression was downregulated by nine hours in mice subjected to CLP (77% decrease, p < 0.01), and remained decreased at 24 hours (75% decrease, p < 0.01; Fig. 1A). In contrast, there was no significant difference in Sirt3 expression in the kidney at three, nine, or 24 hours after CLP induction (Fig. 1B). Levels of Sirt3 mRNA were consistently downregulated in S3KO mouse tissue (data not shown).
FIG. 1.

Sirt3 and IL-6 expression over time in wild-type (WT) mice after cecal ligation and puncture (CLP). Fold change is expressed as change from average sham normalized cycle threshold (ΔCT) values at each time point. A: Changes in liver Sirt3 mRNA over time in WT sham versus CLP mice. B: Changes in kidney Sirt3 mRNA over time in WT sham versus CLP mice. C: Serum interleukin-6 (IL-6) protein levels in pg/mL in WT mice and Sirt3 knockout (S3KO) mice before CLP and three hours, six hours, 12 hours, 36 hours, and five days after CLP. *p < 0.05; **p < 0.01.
Deleting Sirt3 is proinflammatory in polymicrobial sepsis
Serum IL-6 protein levels increased after CLP and peaked around six hours. Peak serum IL-6 levels were higher in S3KO compared with WT mice (169.5 ± 30.1 ng/mL vs. 84.0 ± 16.6 ng/mL; p = 0.03) and remained elevated in S3KO mice at 12 hours (101.7 ± 21.8 ng/mL vs. 54.9 ± 5.5 ng/mL; p = 0.05). By 36 hours, there was no difference between S3KO and WT mice as serum IL-6 levels returned to baseline (Fig. 1C).
Sirt3 may increase hepatic mitochondrial activity in polymicrobial sepsis
We next aimed to determine whether the increased inflammation in septic S3KO mice could be related to changes in mitochondrial function. Sepsis caused a general decrease in hepatic and renal mitochondrial function in WT mice at 36 hours and five days after CLP. In the S3KO mice, the respiratory activity of complexes I and IV in liver tissue were increased at 36 hours post-CLP compared with WT (180.6 ± 12.5 vs. 140.2 ± 8.6 nmol O2/mg/min; p = 0.02 and 1741.1 ± 60.1 vs. 1,496.5 ± 96.4 nmol O2/mg/min; p = 0.05, respectively; Fig. 2A). By day 5, however, this difference had dissipated, and hepatic mitochondrial complex function was comparable between the septic WT and S3KO groups (Fig. 2B). There was no difference in renal mitochondrial complex activity between the S3KO and WT mice at 36 hours or five days (Fig. 2C and 2D). Sirt3 deletion had no effect on renal or hepatic fatty acid oxidation (Fig. 2A–2D) or mitochondrial content (Fig. 2E and 2F) at 36 hours or five days post-CLP.
FIG. 2.

Hepatic and renal mitochondrial content and function after cecal ligation and puncture (CLP) or sham surgery in wild-type (WT) and Sirt3 knockout (S3KO) mice. Respiratory function is determined by rate of oxygen consumption (nmol O2/mg/min). Rate of oxygen consumption was measured for fatty acid oxidation (FAO) and mitochondrial CI (complex I), CII (complex II), and CIV (complex IV). A: Oxygen consumption in hepatic tissue at 36 hours after CLP. B: Oxygen consumption in hepatic tissue at five days after CLP. C: Oxygen consumption in renal tissue at 36 hours after CLP. D: Oxygen consumption in renal tissue at five days after CLP. Mitochondrial content is measured by citrate synthase activity at 36 hours and five days in liver (E) and kidney (F). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Sirt3 deletion does not affect morbidity, but does decrease median survival
After CLP, 48-hour survival in S3KO mice was lower than WT (55.0% vs. 94.1%; p < 0.01). Five-day survival trended lower in S3KO mice (50.0% vs. 64.7%; p = 0.0952), however, this did not reach statistical significance. Median survival in S3KOs was 84 hours, whereas median survival for WT mice was not reached (Fig. 3A). Body temperature and glucose both decreased after CLP and nadired at 18 hours, however, there was no difference in either between the WT and S3KO mice (Fig. 3B and 3C). The S3KO mice weighed less at baseline compared with WT (25.6 ± 0.5 g vs. 27.0 ± 0.4 g; p = 0.04), but by day 5 post-CLP, both S3KO and WT mice had similar weights (23.1 ± 0.6 g vs. 23.1 ± 0.9 g; p = 0.98; Fig. 3D). Sirt3 deletion also did not affect serum markers of kidney or liver dysfunction at 36 hours or five days after CLP (Fig. 4A–4D).
FIG. 3.

Physiologic changes over time in wild-type (WT) versus Sirt3 knockout (S3KO) mice after cecal ligation and puncture (CLP). (A) Five-day survival, p value calculated using Gehan-Breslow-Wilcoxon test. (B) Temperature (C°) over time in the five days after CLP. (C) Glucose (mg/dL) over time in the five days after CLP. D: Daily weight (g) after CLP. *p < 0.05.
FIG. 4.

Markers of organ dysfunction in wild-type (WT) versus Sirt3 knockout (S3KO) mice after cecal ligation and puncture (CLP). A: Serum blood urea nitrogen (BUN; mg/mL) in WT versus S3KO mice at 36 hours after CLP. B: Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT; U/L) in WT versus S3KO mice at 36 hours after CLP. C: Serum BUN (mg/mL) in WT versus S3KO mice at five days after CLP. D: Serum AST and ALT (U/L) in WT versus S3KO mice at five days after CLP.
Sirt3 deletion promotes inflammation in LPS-stimulated BMDMs
Bone marrow-derived macrophages are known to play a vital role in the immune response to sepsis, and our previous work suggests that the inflammatory response to sepsis may be due in large part to expression of sirtuin proteins within myeloid cells.27 We therefore questioned whether Sirt3 deletion within BMDMs was sufficient to promote the BMDM hyperinflammatory response. Compared with baseline levels in untreated BMDMs, Sirt3 mRNA expression in LPS-treated BMDMs decreased by 66% in the first four hours (p = 0.019) but returned to baseline by eight hours (Fig. 5A). Although there was no difference between WT and S3KO cells in terms of the maximum IL6 mRNA expression observed, the IL6 expression peaked earlier in S3KO cells compared with WT (4 vs. 8 hours; Fig. 5B). LPS-treated S3KO BMDMs had an increase in SOD2 mRNA compared with WT at four hours (12.4-fold increase vs. 5.8-fold increase; p < 0.01). SOD2 expression in both WT and S3KO continued to increase at 8 hours but was not different between the groups (Fig. 5C).
FIG. 5.
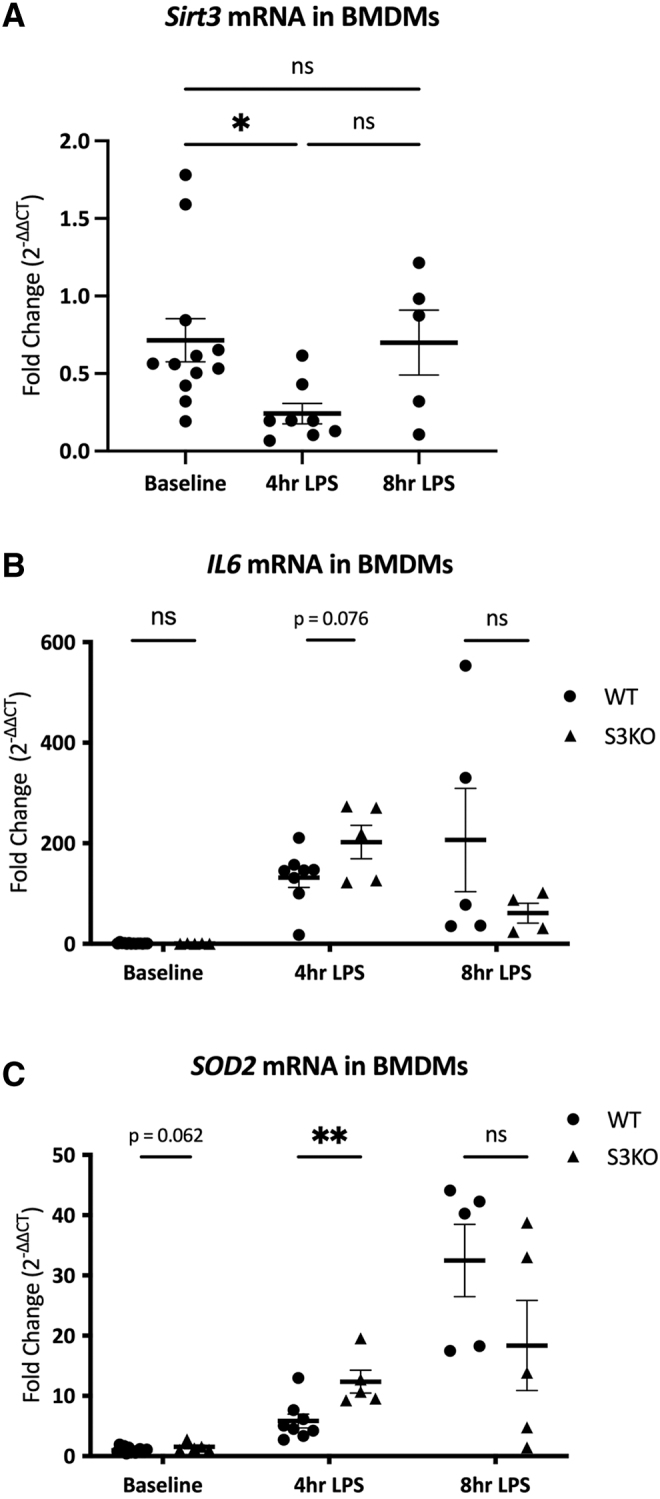
mRNA Expression over time in lipopolysaccharide (LPS)-treated bone marrow-derived macrophages (BMDMs): fold change is expressed as change from average untreated wild-type (WT) normalized cycle threshold (ΔCT) values at each time point. A: Changes in Sirt3 mRNA over time in untreated versus LPS-treated WT BMDMs. B: Changes in IL6 mRNA in untreated versus LPS-treated WT and S3KO BMDMs over time. C: Changes in SOD2 mRNA in untreated vs. LPS-treated WT and S3KO BMDMs over time. *p < 0.05; **p < 0.01.
Discussion
Sepsis remains a highly morbid and fatal disease process, in part because of the dysregulated host response that induces inflammation, oxidative damage, and mitochondrial dysfunction.2–6 Although the use of sepsis protocols and antibiotic agents have improved outcomes, further advancements will require finding ways to modulate this dysregulated host response.1 Sirt3 is generally thought to promote mitochondrial function and inhibit oxidative stress13–16; therefore, we evaluated its role on inflammation and survival in polymicrobial sepsis.
Although it has been shown previously that Sirt3 deletion upregulates proinflammatory cytokines in myeloid and non-myeloid cells during sepsis,18,19 we are the first to demonstrate a temporal link, with IL6 expression peaking earlier in S3KO myeloid cells compared with WT cells. This is consistent with our finding that WT cells become functional SIRT3 knockdown cells a few hours after a septic insult. Together these data support that a natural downregulation of SIRT3 contributes to the lethal hyperinflammatory response known to occur early during sepsis.28
Oxidative stress is another marker of cellular stress and is closely linked with inflammation.15,18 Superoxide has been shown to activate the inflammatory mediator nuclear factor kappa B (NFkB), which stimulates production of SOD2 as a compensatory mechanism.18,29–32 We found that LPS treatment was associated with increased expression of the antioxidant SOD2 in BMDMs. Similar findings in a murine model of sepsis by Bloise et al.33 confirmed that this upregulation of SOD2 correlated with an increase in superoxide production. Therefore, our results likely represent an overall increase in cellular oxidative stress with LPS treatment. The earlier increase in SOD2 expression in S3KO cells compared with WT that we identified is also consistent with our inflammatory cytokine findings and supports the theory that Sirt3 deletion contributes to early oxidative stress. Future studies will need to further examine the role of SIRT3 on early oxidative stress by studying lipid peroxidation in the tissues of septic mice with downregulated SIRT3 expression as well as those overexpressing SIRT3.
The S3KO mice had a shorter median survival after CLP compared with WT mice. The increase in early mortality, but lack of difference in five-day mortality, is consistent with our finding that WT animals become functional Sirt3 knockdown animals several hours after septic insult. We expect that because S3KO animals have an earlier increase in hyperinflammation and oxidative stress, they have increased mortality early on. This early survival advantage among the WT mice appears to be lost between days 3 and 5, however, consistent with the delayed Sirt3 downregulation in the WT mice and subsequent increases in inflammation and oxidative stress.
There is currently conflicting evidence regarding the effects of sepsis on mitochondrial activity, with some studies showing increased respiration in response to infection.34,35 This is believed to be due in part to increased mitochondrial uncoupling in response to cellular stress, which promotes reactive oxygen species (ROS) production.36,37 Whereas Sirt3 deletion is generally thought to decrease mitochondrial respiration,18 we found increased hepatic mitochondrial oxygen consumption in S3KO mice. Given our findings of increased SOD2 in S3KO mice, this increased oxygen consumption most likely reflects an increase in mitochondrial uncoupling and subsequent complex-dependent ROS production, as opposed to improved adenosine triphosphatase (ATP) production.36,37
We saw no other significant Sirt3-dependent effects on mitochondrial activity or serum markers of organ function in sepsis at 36 hours or five days. Of note, because serum markers of organ dysfunction tend to be a delayed sign of tissue damage, this lack of difference in serum marker of organ dysfunction is likely due to a survival bias. Although 36 hours was chosen as a timepoint to capture the delayed presentation of elevated AST, ALT, and BUN, we found that 25% of the S3KO mice, but none of the WT mice, had died by the 36-hour timepoint. It may therefore be beneficial for future studies on SIRT3 in early sepsis to focus on tissue oxidative stress as a surrogate for morbidity.
Our study had several limitations. We primarily measured the expression of inflammatory cytokines and Sirt3 using mRNA. Although our mRNA findings were supported by our studies of serum IL-6 protein levels, further experiments are needed to confirm that the changes in mRNA do, in fact, translate to changes in tissue protein expression. Additionally, because sepsis appears to create a functional knockdown of Sirt3 in WT mice, the results of our study may underestimate the true effects of Sirt3 during sepsis. Further studies comparing septic responses of S3KO and WT mice to mice overexpressing Sirt3 will be necessary to better elucidate the role of Sirt3 in polymicrobial sepsis.
Conclusions
In this study, we demonstrated that Sirt3 is naturally downregulated in sepsis, and that this downregulation promotes a hyperinflammatory response, contributing to increased mortality. Furthermore, our in vitro work established that Sirt3 expression in BMDMs influences their inflammatory potential, and therefore BMDMs may be particularly important in modulating the Sirt3-dependent response to sepsis. Sirt3 is a promising therapeutic target that could help ameliorate the harmful inflammatory response in early sepsis and improve survival.
Supplementary Material
Authors' Contributions
Investigation: Labiner, Sims. Formal analysis: Labiner, Sas, Sims. Writing: Labiner, Sas, Sims. Editing: Labiner, Sas, Sims. Conceptualization: Baur, Sims. Methodology: Baur, Sims.
Funding Information
Supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number T32AI106704-07. The research reported in this publication and the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
The authors have no conflicts of interest to disclose.
Supplementary Material
References
- 1. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020;395(10219):200–211; doi: 10.1016/S0140-6736(19)32989-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 2017;43(3):304–377; doi: 10.1007/s00134-017-4683-6 [DOI] [PubMed] [Google Scholar]
- 3. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016;315(8):801–810; doi: 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sair M, Etherington PJ, Peter Winlove C, et al. Tissue oxygenation and perfusion in patients with systemic sepsis. Crit Care Med 2001;29(7):1343–1349; doi: 10.1097/00003246-200107000-00008 [DOI] [PubMed] [Google Scholar]
- 5. Singer M, De Santis V, Vitale D, et al. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 2004;364(9433):545–548; doi: 10.1016/S0140-6736(04)16815-3 [DOI] [PubMed] [Google Scholar]
- 6. Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 1999;27(7):1230–1251; doi: 10.1097/00003246-199907000-00002 [DOI] [PubMed] [Google Scholar]
- 7. Imai S, Armstrong CM, Kaeberlein M, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000;403(6771):795–800; doi: 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 8. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999;13(19):2570–2580; doi: 10.1101/gad.13.19.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gallo CM, Smith DL Jr., Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol Cell Biol 2004;24(3):1301–1312; doi: 10.1128/mcb.24.3.1301-1312.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 2014;25(3):138–145; doi: 10.1016/j.tem.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakagawa T, Guarente L. Sirtuins at a glance. J Cell Sci 2011;124(Pt 6):833–838; doi: 10.1242/jcs.081067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu S, Gao Y, Zhang Q, et al. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxid Med Cell Longev 2016;2016:7296092; doi: 10.1155/2016/7296092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu TF, Vachharajani V, Millet P, et al. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 2015;290(1):396–408; doi: 10.1074/jbc.M114.566349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun F, Si Y, Bao H, et al. Regulation of Sirtuin 3-mediated deacetylation of cyclophilin d attenuated cognitive dysfunction induced by sepsis-associated encephalopathy in mice. Cell Mol Neurobiol 2017;37(8):1457–1464; doi: 10.1007/s10571-017-0476-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu J, Deng Z, Sun M, et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation. Lab Invest 2020;100(4):643–656; doi: 10.1038/s41374-019-0332-8 [DOI] [PubMed] [Google Scholar]
- 16. Xin T, Lu C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY) 2020;12(16):16224–16237; doi: 10.18632/aging.103644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeng H, He X, Tuo QH, et al. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2alpha/Notch3 pathways. Sci Rep 2016;6:20931; doi: 10.1038/srep20931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurundkar D, Kurundkar AR, Bone NB, et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 2019;4(1):e120722; doi: 10.1172/jci.insight.120722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao WY, Zhang L, Sui MX, et al. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci Rep 2016;6:33201; doi: 10.1038/srep33201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao W, Zhang L, Chen R, et al. SIRT3 protects against acute kidney injury via AMPK/mTOR-regulated autophagy. Front Physiol 2018;9:1526; doi: 10.3389/fphys.2018.01526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y, Zhang S, Rong J, et al. Sirt3 is a novel target to treat sepsis induced myocardial dysfunction by acetylated modulation of critical enzymes within cardiac tricarboxylic acid cycle. Pharmacol Res 2020;159:104887; doi: 10.1016/j.phrs.2020.104887 [DOI] [PubMed] [Google Scholar]
- 22. Koentges C, Cimolai MC, Pfeil K, et al. Impaired SIRT3 activity mediates cardiac dysfunction in endotoxemia by calpain-dependent disruption of ATP synthesis. J Mol Cell Cardiol 2019;133:138–147; doi: 10.1016/j.yjmcc.2019.06.008 [DOI] [PubMed] [Google Scholar]
- 23. Lombard DB, Alt FW, Cheng HL, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007;27(24):8807–8814; doi: 10.1128/MCB.01636-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rittirsch D, Huber-Lang MS, Flierl MA, et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 2009;4(1):31–36; doi: 10.1038/nprot.2008.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Srere PA, Matsuoka Y. Inhibition of rat citrate synthase by acetoacetyl CoA and NADH. Biochem Med 1972;6(3):262–266; doi: 10.1016/0006-2944(72)90047-6 [DOI] [PubMed] [Google Scholar]
- 26. Wang H, Guan Y, Karamercan MA, et al. Resveratrol rescues kidney mitochondrial function following hemorrhagic shock. Shock 2015;44(2):173–180; doi: 10.1097/SHK.0000000000000390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Labiner HE, Sas KM, Baur JA, et al. Sirt1 deletion increases inflammation and mortality in sepsis. J Trauma Acute Care Surg 2022;93(5):672–678; doi: 10.1097/TA.0000000000003751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X, Buechler NL, Woodruff AG, et al. Sirtuins and immuno-metabolism of sepsis. Int J Mol Sci 2018;19(9):2738; doi: 10.3390/ijms19092738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;552(Pt 2):335–344; doi: 10.1113/jphysiol.2003.049478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lingappan K. NF-kappaB in oxidative stress. Curr Opin Toxicol 2018;7:81–86; doi: 10.1016/j.cotox.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yao X, Carlson D, Sun Y, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS One 2015;10(10):e0139416; doi: 10.1371/journal.pone.0139416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011;472(7344):476–480; doi: 10.1038/nature09973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bloise FF, Santos AT, de Brito J, et al. Sepsis impairs thyroid hormone signaling and mitochondrial function in the mouse diaphragm. Thyroid 2020;30(7):1079–1090; doi: 10.1089/thy.2019.0124 [DOI] [PubMed] [Google Scholar]
- 34. Kohoutova M, Dejmek J, Tuma Z, et al. Variability of mitochondrial respiration in relation to sepsis-induced multiple organ dysfunction. Physiol Res 2018;67(Suppl 4):S577–S592; doi: 10.33549/physiolres.934050 [DOI] [PubMed] [Google Scholar]
- 35. Preau S, Vodovar D, Jung B, et al. Energetic dysfunction in sepsis: a narrative review. Ann Intensive Care 2021;11(1):104; doi: 10.1186/s13613-021-00893-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berry BJ, Trewin AJ, Amitrano AM, et al. Use the protonmotive force: mitochondrial uncoupling and reactive oxygen species. J Mol Biol 2018;430(21):3873–3891; doi: 10.1016/j.jmb.2018.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X, Fang P, Mai J, et al. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol 2013;6:19; doi: 10.1186/1756-8722-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.