

Abstract
Background
Cystic fibrosis (CF) is a common life‐shortening genetic condition caused by a variant in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. A class II CFTR variant F508del is the commonest CF‐causing variant (found in up to 90% of people with CF (pwCF)). The F508del variant lacks meaningful CFTR function ‐ faulty protein is degraded before reaching the cell membrane, where it needs to be to effect transepithelial salt transport. Corrective therapy could benefit many pwCF. This review evaluates single correctors (monotherapy) and any combination of correctors (most commonly lumacaftor, tezacaftor, elexacaftor, VX‐659, VX‐440 or VX‐152) and a potentiator (e.g. ivacaftor) (dual and triple therapies).
Objectives
To evaluate the effects of CFTR correctors (with or without potentiators) on clinically important benefits and harms in pwCF of any age with class II CFTR mutations (most commonly F508del).
Search methods
We searched the Cochrane CF Trials Register (28 November 2022), reference lists of relevant articles and online trials registries (3 December 2022).
Selection criteria
Randomised controlled trials (RCTs) (parallel design) comparing CFTR correctors to control in pwCF with class II mutations.
Data collection and analysis
Two authors independently extracted data, assessed risk of bias and judged evidence certainty (GRADE); we contacted investigators for additional data.
Main results
We included 34 RCTs (4781 participants), lasting between 1 day and 48 weeks; an extension of two lumacaftor‐ivacaftor studies provided additional 96‐week safety data (1029 participants). We assessed eight monotherapy RCTs (344 participants) (4PBA, CPX, lumacaftor, cavosonstat and FDL169), 16 dual‐therapy RCTs (2627 participants) (lumacaftor‐ivacaftor or tezacaftor‐ivacaftor) and 11 triple‐therapy RCTs (1804 participants) (elexacaftor‐tezacaftor‐ivacaftor/deutivacaftor; VX‐659‐tezacaftor‐ivacaftor/deutivacaftor; VX‐440‐tezacaftor‐ivacaftor; VX‐152‐tezacaftor‐ivacaftor). Participants in 21 RCTs had the genotype F508del/F508del, in seven RCTs they had F508del/minimal function (MF), in one RCT F508del/gating genotypes, in one RCT either F508del/F508del genotypes or F508del/residual function genotypes, in one RCT either F508del/gating or F508del/residual function genotypes, and in three RCTs either F508del/F508del genotypes or F508del/MF genotypes.
Risk of bias judgements varied across different comparisons. Results from 16 RCTs may not be applicable to all pwCF due to age limits (e.g. adults only) or non‐standard designs (converting from monotherapy to combination therapy).
Monotherapy
Investigators reported no deaths or clinically relevant improvements in quality of life (QoL). There was insufficient evidence to determine effects on lung function.
No placebo‐controlled monotherapy RCT demonstrated differences in mild, moderate or severe adverse effects (AEs); the clinical relevance of these events is difficult to assess due to their variety and few participants (all F508del/F508del).
Dual therapy
In a tezacaftor‐ivacaftor group there was one death (deemed unrelated to the study drug). QoL scores (respiratory domain) favoured both lumacaftor‐ivacaftor and tezacaftor‐ivacaftor therapy compared to placebo at all time points (moderate‐certainty evidence). At six months, relative change in forced expiratory volume in one second (FEV1) % predicted improved with all dual combination therapies compared to placebo (high‐ to moderate‐certainty evidence).
More pwCF reported early transient breathlessness with lumacaftor‐ivacaftor (odds ratio (OR) 2.05, 99% confidence interval (CI) 1.10 to 3.83; I2 = 0%; 2 studies, 739 participants; high‐certainty evidence). Over 120 weeks (initial study period and follow‐up), systolic blood pressure rose by 5.1 mmHg and diastolic blood pressure by 4.1 mmHg with twice‐daily 400 mg lumacaftor‐ivacaftor (80 participants). The tezacaftor‐ivacaftor RCTs did not report these adverse effects.
Pulmonary exacerbation rates decreased in pwCF receiving additional therapies to ivacaftor compared to placebo (all moderate‐certainty evidence): lumacaftor 600 mg (hazard ratio (HR) 0.70, 95% CI 0.57 to 0.87; I2 = 0%; 2 studies, 739 participants); lumacaftor 400 mg (HR 0.61, 95% CI 0.49 to 0.76; I2 = 0%; 2 studies, 740 participants); and tezacaftor (HR 0.64, 95% CI 0.46 to 0.89; 1 study, 506 participants).
Triple therapy
No study reported any deaths (high‐certainty evidence). All other evidence was low‐ to moderate‐certainty. QoL respiratory domain scores probably improved with triple therapy compared to control at six months (six studies). There was probably a greater relative and absolute change in FEV1 % predicted with triple therapy (four studies each across all combinations). The absolute change in FEV1 % predicted was probably greater for F508del/MF participants taking elexacaftor‐tezacaftor‐ivacaftor compared to placebo (mean difference 14.30, 95% CI 12.76 to 15.84; 1 study, 403 participants; moderate‐certainty evidence), with similar results for other drug combinations and genotypes. There was little or no difference in adverse events between triple therapy and control (10 studies). No study reported time to next pulmonary exacerbation, but fewer F508del/F508del participants experienced a pulmonary exacerbation with elexacaftor‐tezacaftor‐ivacaftor at four weeks (OR 0.17, 99% CI 0.06 to 0.45; 1 study, 175 participants) and 24 weeks (OR 0.29, 95% CI 0.14 to 0.60; 1 study, 405 participants); similar results were seen across other triple therapy and genotype combinations.
Authors' conclusions
There is insufficient evidence of clinically important effects from corrector monotherapy in pwCF with F508del/F508del.
Additional data in this review reduced the evidence for efficacy of dual therapy; these agents can no longer be considered as standard therapy. Their use may be appropriate in exceptional circumstances (e.g. if triple therapy is not tolerated or due to age). Both dual therapies (lumacaftor‐ivacaftor, tezacaftor‐ivacaftor) result in similar small improvements in QoL and respiratory function with lower pulmonary exacerbation rates. While the effect sizes for QoL and FEV1 still favour treatment, they have reduced compared to our previous findings. Lumacaftor‐ivacaftor was associated with an increase in early transient shortness of breath and longer‐term increases in blood pressure (not observed for tezacaftor‐ivacaftor). Tezacaftor‐ivacaftor has a better safety profile, although data are lacking in children under 12 years. In this population, lumacaftor‐ivacaftor had an important impact on respiratory function with no apparent immediate safety concerns, but this should be balanced against the blood pressure increase and shortness of breath seen in longer‐term adult data when considering lumacaftor‐ivacaftor.
Data from triple therapy trials demonstrate improvements in several key outcomes, including FEV1 and QoL. There is probably little or no difference in adverse events for triple therapy (elexacaftor‐tezacaftor‐ivacaftor/deutivacaftor; VX‐659‐tezacaftor‐ivacaftor/deutivacaftor; VX‐440‐tezacaftor‐ivacaftor; VX‐152‐tezacaftor‐ivacaftor) in pwCF with one or two F508del variants aged 12 years or older (moderate‐certainty evidence). Further RCTs are required in children under 12 years and those with more severe lung disease.
Keywords: Adult, Child, Humans, Aminophenols, Aminophenols/adverse effects, Cystic Fibrosis, Cystic Fibrosis/drug therapy, Cystic Fibrosis/genetics, Cystic Fibrosis Transmembrane Conductance Regulator, Cystic Fibrosis Transmembrane Conductance Regulator/genetics, Dyspnea, Dyspnea/drug therapy, Mutation
Plain language summary
CFTR correctors, a therapy for cystic fibrosis targeted at specific variants (most commonly F508del)
Review question
How do drugs (or drug combinations) for correcting the basic defect in the most common cystic fibrosis (CF)‐causing gene variant (F508del) impact on outcomes important to people with CF (pwCF), e.g. survival, quality of life (QoL), lung function and safety?
Key messages
There is a lack of evidence to support monotherapy and limited evidence to support dual therapy for pwCF who have the gene variant F508del. We found important differences across key outcomes, with fewer unwanted side effects in the triple therapy studies when compared to one dual therapy drug combination (lumacaftor plus ivacaftor).
More research is needed to assess triple therapy combinations in children and to monitor their safety profiles over the longer term.
How would drugs (or drug combinations) correct the basic defect in the most common CF‐causing gene variant (F508del)?
The CF gene makes a protein that helps salts move across cells in many parts of the body. Over 80% of pwCF have at least one copy of the genetic variant F508del, meaning they make a full length of this protein, but it cannot move through the cell correctly. Laboratory experiments suggest that if this protein reaches the cell wall, it may be able to function, restore salt movement and correct the chronic problems that pwCF experience.
What did we want to find out?
How do drugs (or drug combinations) used to treat people of any age who have the genetic variant F508del impact their lives? Are these drugs associated with any unwanted side effects?
What did we do?
We searched for and included studies that looked at drugs (and drug combinations) used to treat pwCF who had at least one copy of the most common CF‐causing gene variant (F508del) compared to control medications.
We compared and summarised the results of the studies and rated our confidence in the evidence, based on factors such as study methods and sizes.
What did we find?
We included 34 studies that involved 4781 pwCF and which lasted between 1 day and 48 weeks; all studies compared an active drug treatment to a placebo (drug containing no active treatment). We do not have enough information to decide if 14 studies should be included or not and eight studies have not yet been completed.
Eight studies looked at treatment with a single drug (monotherapy). These studies did not report any deaths or clinically relevant improvements in quality of life (QoL) scores. There was not enough evidence to show an effect on lung function. All studies reported side effects; there were a wide range of side effects each reported by a small number of participants in the studies.
There were 16 studies assessing a combination of two drugs (dual therapy), either tezacaftor plus ivacaftor or lumacaftor plus ivacaftor. One participant taking tezacaftor plus ivacaftor died, but this was not thought to be related to the treatment. Both dual therapies resulted in improvements in QoL and lung function; rates of pulmonary exacerbations (a flare‐up of symptoms) were also lower. Neither dual therapy was linked to severe side effects, although people starting treatment with lumacaftor plus ivacaftor experienced shortness of breath for one to two weeks; this usually stopped without further treatment. More concerningly, in longer studies some people taking lumacaftor plus ivacaftor experienced a rise in blood pressure. Two people (out of over 500) even stopped lumacaftor plus ivacaftor treatment because of high blood pressure. These side effects were not reported for tezacaftor plus ivacaftor.
There were 11 studies assessing different combinations of three drugs (triple therapy). The combinations were based on tezacaftor with ivacaftor (or deutivacaftor, which is a similar but chemically altered version of ivacaftor) and investigators then added either elexacaftor, VX‐659, VX‐440 or VX‐152 and compared these to either triple placebo, or tezacaftor with ivacaftor and one placebo. Some studies split groups by treatment and also by different genetic variants. No study reported any deaths. Triple therapies improved QoL scores and lung function in all comparisons, with no difference in the number or severity of side effects. Fewer people with two copies of F508del taking elexacaftor plus tezacaftor plus ivacaftor experienced a pulmonary exacerbation than those taking the control treatment.
What are the limitations of the evidence?
Our certainty in the evidence ranged from uncertain to very certain, but we were quite certain of the evidence for most results. While studies generally provided few details about their design, so we could not make clear judgements on potential biases, we had fewer concerns with the larger, more recent studies. Some of our findings are based on studies that were too small to show important effects and for 16 studies the results may not be applicable to all pwCF due to them only including people of certain ages (i.e. only adults or only children). Also, one study had an unusual design where people were given monotherapy and then later a dual therapy.
How up‐to‐date is this evidence?
This review updates our earlier review. We last looked for evidence on 28 November 2022.
Summary of findings
Summary of findings 1. Summary of findings ‐ monotherapy: lumacaftor compared to placebo for cystic fibrosis.
| Lumacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: lumacaftor Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Lumacaftor | |||||
|
Survival Follow‐up: 14 to 28 days |
No deaths reported. | No deaths reported. | NA | 147 (2 studies) |
⊕⊕⊝⊝ lowa | — |
|
Quality of life ‐ total score Follow‐up: 14 to 28 days |
Outcome not reported. | NA | A higher score indicates a better outcome. | |||
|
Quality of life ‐ CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 14 to 28 days |
There was a significant decrease in the CFQ‐R respiratory domain in the 50 mg lumacaftor group compared to placebo. No differences were found in the other dose groups (25 mg, 100 mg, 200 mg) compared to placebo. | NA | 85 (1 study) |
⊕⊕⊝⊝ lowa | A higher score indicates a better outcome. | |
|
FEV1 % predicted: relative change from baseline Follow‐up: 14 to 28 days |
Outcome not reported. | NA | — | |||
|
FEV1 % predicted: absolute change from baseline Follow‐up: 14 to 28 days |
The mean change from baseline was 1.7% predicted. | The mean change from baseline was 1.90% predicted lower (4.13 lower to 0.33 higher). | NA | 61 (1 study) |
⊕⊕⊕⊝ moderateb | — |
|
Adverse events Follow‐up: 14 to 28 days |
There were no significant differences between groups in terms of participants experiencing any specific adverse event. In 1 of the studies, 1 participant from each of the 4 different dosage lumacaftor arms (4 participants in total) discontinued the study drug due to respiratory adverse effects. No participants discontinued from the placebo group. |
NA | 115 (2 studies) |
⊕⊝⊝⊝ very lowa,b,c | — | |
|
Time to first pulmonary exacerbation Follow‐up: 14 to 28 days |
Outcome not reported (see comment). | NA | Time to first pulmonary exacerbation was not reported. There was no significant difference between groups in the number of participants experiencing pulmonary exacerbations. | |||
| *The basis for the assumed risk is the mean placebo group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; EQ‐5D‐3L: 5‐Dimension‐3 Level FEV1: forced expiratory volume at one second; MD: mean difference; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded twice due to risk of bias: in one study, data were selectively reported and the presentation of data often did not allow for inclusion in analysis. There are also incomplete outcome data in the study, with participants unaccounted for in the analysis.
bDowngraded once due to indirectness: the design of the study means that monotherapy treatment was measured for only 14 days before a combination therapy phase was started.
cDowngraded once due to imprecision: few events occurred, therefore CIs for occurrence of specific events are very wide.
Summary of findings 2. Summary of findings ‐ monotherapy: cavosonstat compared to placebo for cystic fibrosis.
| Cavosonstat compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: cavosonstat 200 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Cavosonstat | |||||
|
Survival Follow‐up: 28 days |
No deaths reported. | No deaths reported. | NA | 26 (1 study) |
⊕⊝⊝⊝ very lowa,b,c | — |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | A higher score indicates a better outcome. | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 28 days |
The mean absolute change from baseline in CFQ‐R respiratory domain was ‐4.6 points in the placebo group. | The mean absolute change from baseline in CFQ‐R respiratory domain was 3.80 higher (11.30 lower to 18.90 higher) in the cavosonstat group than the placebo group. | NA | 24 (1 study) |
⊕⊝⊝⊝ very lowa,b,c,d | A higher score indicates a better outcome. |
|
FEV1 % predicted: relative change from baseline Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
FEV1 % predicted: absolute change from baseline Follow‐up: 28 days |
There were no treatment‐related changes in FEV1 (% predicted) compared to placebo. | NA | 26 (1 study) |
⊕⊝⊝⊝ very lowa,b,c | A graphical figure of change from baseline in FEV1 (% predicted) is provided, but numerical data cannot be extracted to include in analysis due to overlapping lines. | |
|
Adverse events: occurring in at least 10% of cavosonstat‐treated participants Follow‐up: 28 days |
There was no significant difference between groups in terms of cough, pulmonary exacerbation, chest discomfort and fatigue. | NA | 26 (1 study) |
⊕⊝⊝⊝ very lowa,b,c,e | — | |
|
Time to first pulmonary exacerbation Follow‐up: NA |
Outcome not reported. | NA | — | |||
| *The basis for the assumed risk is the control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume in 1 second; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to potential risk of bias: unclear details related to methodological design, high risk of attrition bias and some unbalanced baseline characteristics.
bDowngraded once due to indirectness: only adults were recruited into the study, therefore the results are not applicable to children.
cDowngraded once due to imprecision: a single study with a small sample size.
dDowngraded once due to imprecision: wide CIs around the result.
eDowngraded once due to imprecision: very wide CIs around the results (due to small event numbers).
Summary of findings 3. Summary of findings ‐ dual therapy: lumacaftor plus ivacaftor (once daily) compared with placebo for cystic fibrosis (short term).
| Lumacaftor plus ivacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: lumacaftor (600 mg once daily or 400 mg once daily) plus ivacaftor (250 mg twice daily) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Lumacaftor plus ivacaftor | |||||
|
Survival Follow‐up: 6 months |
No deaths reported. | No deaths reported. | NA | 1108 (2 studies) |
⊕⊕⊕⊕ high | — |
|
Quality of life ‐ (EuroQol) EQ‐5D‐3L Index Score (total score): absolute change from baseline Follow‐up: 6 months |
The mean absolute change from baseline ranged from 0.0006 to 0.0017 points. | The mean absolute change from baseline was 0.00 points higher (0.01 lower to 0.01 higher). | NA | 1061 (2 studies) |
⊕⊕⊕⊝ moderatea | A higher score indicates a better outcome. |
|
Quality of life ‐ CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 6 months |
The mean absolute change from baseline ranged from ‐6.1 to 2.81 points. | The mean absolute change from baseline was 2.83 points higher (0.91 higher to 4.74). | NA | 1139 (3 studies) |
⊕⊕⊕⊝ moderatea | A higher score indicates a better outcome. There was also a significant difference between groups at 28 days, MD 3.70 points (95% CI 1.81 to 5.58). |
|
FEV1 % predicted: relative change from baseline Follow‐up: 6 months |
The mean relative change from baseline ranged from ‐0.54% to 0%. | The mean relative change from baseline was 5.12% higher (3.57% higher to 6.67% higher). | NA | 1134 (3 studies) |
⊕⊕⊕⊕ high | — |
|
FEV1 % predicted: absolute change from baseline Follow‐up: 6 months |
The mean absolute change from baseline ranged from ‐4.0 to ‐0.15% predicted. | The mean absolute change from baseline was 3.08% predicted higher (2.20 higher to 3.97 higher). | NA | 1134 (3 studies) |
⊕⊕⊕⊝ moderatea | There was also a significant difference between groups at 28 days, MD 2.37% predicted (95% CI 1.52 to 3.22). |
|
Adverse events Follow‐up: 6 months |
Cough was significantly more common in the placebo group compared to the lumacaftor‐ivacaftor group. Dyspnoea was significantly more common in the lumacaftor‐ivacaftor group compared to the placebo group (OR 2.05, 99% CI 1.10 to 3.83; I2 = 0%; 2 studies, 739 participants). There were no significant differences between groups in terms of the number of participants experiencing adverse events, serious adverse events or other adverse events. |
NA | 1108 (2 studies) |
⊕⊕⊕⊕ high | Long‐term open‐label follow‐up data from the 2 studies showed a significant rise in blood pressure (mean (SE) increase in systolic blood pressure of 5.1 (1.5) mm Hg and in diastolic blood pressure of 4.1 (1.2) mm Hg) in participants allocated a 400 mg twice‐daily dose. | |
|
Time to first pulmonary exacerbation Follow‐up: 6 months |
Time to first pulmonary exacerbation was significantly longer in both the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group (HR 0.70, 95% CI 0.57 to 0.87; I2 = 0%; 2 studies, 739 participants); and the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group (HR 0.61, 95% CI 0.49 to 0.76; I2 = 0%; 2 studies, 740 participants). | NA | 1108 (2 studies) |
⊕⊕⊕⊝ moderatea | Presentation of data did not allow an analysis of the lumacaftor doses pooled. Please note: the control participants are the same participants in both the reported summary statistics as both studies had 3 arms. |
|
| *The basis for the assumed risk is the mean placebo group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; EQ‐5D‐3L: 5‐Dimension‐3 Level; EuroQol: Euro Quality of Life Scale; FEV1: forced expiratory volume at one second; HR: hazard ratio; MD: mean difference; mmHg: millimetres of mercury; NA: not applicable; OR: odds ratio; SE: standard error. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to risk of bias from selective reporting: data contributing to analyses were extrapolated from published graphs or estimated. We have requested confirmation of the exact data from the study investigators. Any unpublished information we receive will be included in a future update and this judgement will be reconsidered.
Summary of findings 4. Summary of findings ‐ dual therapy: lumacaftor plus ivacaftor (twice daily) compared with placebo for cystic fibrosis (short term).
| Lumacaftor plus ivacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Placebo | Lumacaftor plus ivacaftor | |||||
| Assumed risk | Corresponding risk | |||||
|
Survival Follow‐up: 24 weeks |
No deaths reported. | No deaths reported. | NA | 204 (1 study) | ⊕⊕⊕⊝ moderatea | — |
|
Quality of life ‐ total score Follow‐up: 24 weeks |
Outcome not reported. | NA | A higher score indicates a better outcome. | |||
|
Quality of life ‐ CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 24 weeks |
See comment. | The mean change in the CFQ‐R respiratory domain was 2.50 points higher in the lumacaftor‐ivacaftor group compared to the placebo group, ranging from 0.10 lower to 5.10 higher. | NA | 204 (1 study) | ⊕⊕⊝⊝ lowa,b | A higher score indicates a better outcome. Data were analysed via a MMRM. Results provided by this model can be interpreted as a treatment effect averaged from each study visit until week 24. |
|
FEV1 % predicted: relative change from baseline Follow‐up: 24 weeks |
Outcome not reported. | NA | Relative change from baseline in FEV1 was listed in the methods of the study but no numerical results were presented. If numerical data become available at a later date, they will be included in an update of this review. |
|||
|
FEV1 % predicted: absolute change from baseline Follow‐up: 24 weeks |
See comment. | The mean change in FEV1 % predicted was 2.40 higher in the lumacaftor‐ivacaftor group compared to the placebo group, ranging from 0.40 higher to 4.40 higher. | NA | 204 (1 study) | ⊕⊕⊝⊝ lowa,b | Data were analysed via a MMRM. Results provided by this model can be interpreted as a treatment effect averaged from each study visit until week 24. |
|
Adverse events Follow‐up: 24 weeks |
There was no significant difference between the groups in terms of productive cough, nasal congestion, oropharyngeal pain, upper abdominal pain, rhinorrhoea, increased sputum, cough, pyrexia, headache, upper respiratory tract infection, abdominal pain, nausea, vomiting, fatigue and respiratory events (such as wheezing, dyspnoea, asthma and chest discomfort). | NA | 204 (1 study) | ⊕⊕⊝⊝ lowb,c | — | |
|
Time to first pulmonary exacerbation Follow‐up: 24 weeks |
Outcome not reported. | NA | Time to first pulmonary exacerbation was listed in the methods of the study but no numerical results were presented. If numerical data become available at a later date, they will be included in an update of this review. |
|||
| *The basis for the assumed risk is the control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; MMRM: mixed model for repeated measures; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to indirectness: children aged 6 to 11 years were recruited in this study, therefore the results are not applicable to other age groups.
bDowngraded once due to risk of bias from selective reporting: limited data available, which are adjusted for all visits. Further graphical data were available in the publication but could not be accurately extracted. We have requested confirmation of the exact data from the study investigators. Any unpublished information we receive will be included in a future update and this judgement will be reconsidered.
cDowngraded once due to imprecision: few events occurred, therefore the CIs for occurrence of specific events are very wide.
Summary of findings 5. Summary of findings ‐ dual therapy: lumacaftor plus ivacaftor compared with placebo for cystic fibrosis (immediate term).
| Lumacaftor plus ivacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: lumacaftor (200 mg) plus ivacaftor (150 mg or 250 mg twice daily)a Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Lumacaftor plus ivacaftora | |||||
|
Survival Follow‐up: 21 days1 |
No deaths reported. | No deaths reported. | NA | 62 (1 study) |
⊕⊕⊕⊝ moderateb | — |
|
Quality of life: total score Follow‐up: 21 days1 |
Outcome not reported. | NA | — | |||
|
Quality of life: respiratory domain Follow‐up: 21 days1 |
Outcome not reported. | NA | — | |||
|
FEV1 % predicted: relative change from baseline Follow‐up: 21 days1 |
Outcome not reported. | NA | — | |||
|
FEV1 % predicted: absolute change from baseline Follow‐up: 21 days1 |
The mean change from baseline was 0.3. | The mean change from baseline was 1.57% predicted higher (‐2.13 lower to 5.27 higher). | NA | 59 (1 study) |
⊕⊕⊕⊝ moderateb | — |
|
Adverse events Follow‐up: 21 days1 |
There were no significant differences between groups in terms of participants experiencing: cough, oropharyngeal pain, nasal congestion, dizziness, a prolonged prothrombin time and upper respiratory tract infection. | NA | 61 (1 study) |
⊕⊕⊝⊝ lowb,c | — | |
|
Time to first pulmonary exacerbation Follow‐up: 21 days1 |
Outcome not reported (see comment). | NA | Time to first pulmonary exacerbation was not reported. There was no significant difference between groups in the number of participants experiencing pulmonary exacerbations. | |||
| *The basis for the assumed risk is the mean placebo group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume at 1 second; NA: not applicable | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe design of the study was 14 days of lumacaftor monotherapy (200 mg once daily), then a dose of ivacaftor (150 mg or 250 mg once daily) was added on for 7 days of combination therapy. Results presented in this table are from the combination treatment period only.
bDowngraded once due to indirectness: the design of the study means that combination treatment was measured for only 7 days and the prior lumacaftor monotherapy phase (see footnote 1) may have influenced results of the combination phase.
cDowngraded once due to imprecision: few events occurred, therefore the CIs for occurrence of specific events are very wide.
Summary of findings 6. Summary of findings ‐ dual therapy: tezacaftor plus ivacaftor compared with placebo or ivacaftor alone for cystic fibrosis.
| Tezacaftor plus ivacaftor compared with placebo or ivacaftor alone for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: tezacaftor (100 mg once daily or 50 mg twice daily) plus ivacaftor (150 mg twice daily) Comparison: placebo (i.e. tezacaftor placebo) or ivacaftor (150 mg twice daily) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or ivacaftor alone | Tezacaftor plus ivacaftor | |||||
|
Survival Follow‐up: up to 24 weeks |
No deaths reported. | 1 death occurred but was not deemed to be related to the study drug. | NA | 1132 (9 studies) | ⊕⊕⊕⊝ lowa,b | No other study in this comparison reported any deaths. |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: up to 24 weeks |
The mean absolute change from baseline in CFQ‐R respiratory domain score in the tezacaftor‐ivacaftor group was 2.88 points higher (2.48 higher to 3.29 higher) than the placebo group. | NA | 946 (5 studies) | ⊕⊕⊝⊝ lowa,c | A higher score indicates a better outcome. The difference in absolute change from baseline was calculated by least‐squares regression, hence the assumed risk is not presented. The mean absolute change from baseline in CFQ‐R respiratory domain score in the tezacaftor once daily plus ivacaftor group was also significantly higher than the placebo group at 4 weeks (MD 5.10, 95% CI 2.99 to 7.21); this was also true at 3 months in the tezacaftor 50 mg twice daily plus ivacaftor group (MD 10.20, 95% CI ‐0.06 to 20.46). |
|
|
FEV1 % predicted: relative change from baseline Follow‐up: up to 24 weeks |
The mean relative change from baseline in FEV1 % predicted in the tezacaftor‐ivacaftor group was 0.92% higher (0.72% higher to 1.11% higher) than the placebo group. | NA | 944 (5 studies) | ⊕⊕⊝⊝ lowa,c | The difference in relative change from baseline was calculated by least‐squares regression, hence the assumed risk is not presented. At 1 month there was no difference in relative change in FEV1 % predicted in the 100 mg tezacaftor once daily group compared to placebo (MD 3.72, 95% CI ‐7.77 to 15.21; 1 trial; 18 participants). 1 study comparing twice‐daily treatment with tezacaftor 50 mg plus ivacaftor 150 mg to placebo reported that at 3 months there was no difference between groups (MD 1.60%, 95% CI ‐7.63 to 10.83; 1 study; 11 participants). |
|
|
FEV1 % predicted: absolute change from baseline Follow‐up: up to 24 weeks |
The mean absolute change from baseline in FEV1 % predicted in the tezacaftor plus ivacaftor group was 0.39% predicted higher (0.27% higher to 0.52% higher) than the placebo group. | NA | 944 (5 studies) | ⊕⊕⊝⊝ lowa,c | The difference in absolute change from baseline was calculated by least‐squares regression, hence the assumed risk is not presented. At 1 month the mean absolute change from baseline in FEV1 % predicted in the tezacaftor‐ivacaftor group was significantly higher than the placebo group (MD 3.54 % predicted, 95% CI 2.39 to 4.69; 3 studies, 556 participants; I² = 0%). One trial comparing twice‐daily treatment with 50 mg tezacaftor plus ivacaftor 150 mg to placebo reported that at 3 months there was no difference between groups (MD 1.00% predicted, 95% CI ‐3.35 to 5.35; 1 trial, 11 participants). |
|
|
Adverse events: most commonly occurring events (occurring in at least 10% of participants) Follow‐up: up to 24 weeks |
The most commonly occurring adverse events in both groups were cough and pulmonary exacerbation. There were no significant differences between groups (99% confidence intervals) in the number of participants experiencing cough, pulmonary exacerbation, headache, nasal congestion or nasopharyngitis, increased sputum, haemoptysis, pyrexia, oropharyngeal pain, nausea or fatigue. |
NA | 1057 (8 studies) | ⊕⊕⊕⊝ moderatea | — | |
|
Time to first pulmonary exacerbation Follow‐up: up to 24 weeks |
The hazard ratio for pulmonary exacerbation in the tezacaftor plus ivacaftor group, as compared with the placebo group, was 0.64 (95% CI 0.46 to 0.89). | NA | 506 (1 study) |
⊕⊕⊕⊝ moderatea | A hazard ratio below 1 favours the tezacaftor‐ivacaftor group. | |
| *The basis for the assumed risk is the control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; MD: mean difference; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to indirectness: 8 out of the 9 trials in this outcome included children over the age of 12 and adults; only 1 trial included children under the age of 12, therefore the results may not be applicable to younger children.
bDowngraded once due to risk of bias across the trials contributing to this outcome: 3 of the trials were at unclear risk of bias from randomisation and allocation concealment and there was concern around unbalanced treatment group sizes and baseline characteristics (Donaldson 2018; Munck 2020; NCT02070744). Four of the trials were at high risk of bias due to incomplete outcome data (McKone 2021; Munck 2020; NCT02070744; NCT02508207).
cDowngraded once due to risk of bias across the included trials for this outcome: 2 trials were at unclear or high risk of bias across all domains; 3 trials were at risk of bias from incomplete outcome reporting; and 2 were at unclear risk of bias around the randomisation and allocation concealment process.
Summary of findings 7. Summary of findings ‐ triple therapy: VX‐659‐tezacaftor‐ivacaftor/deutivacaftor compared to control for cystic fibrosis.
| VX‐659 plus tezacaftor plus ivacaftor or deutivacaftor compared with control for cystic fibrosis | ||||||
|
Patient or population: adults with cystic fibrosis and either F508del/MF or F508del/F508del genotype Settings: outpatients Intervention: VX‐659 (80 mg once daily, 120 mg twice daily, 240 mg once daily or 400 mg once daily) plus tezacaftor 100 mg once per day plus ivacaftor 150 mg twice daily or deutivacaftor 150 mg once daily Comparison: F508del/MF participants: triple placebo; F508del/F508del participants: placebo plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Triple placebo or placebo‐tezacaftor‐ivacaftor | VX‐659 plus tezacaftor plus ivacaftor or deutivacaftor | |||||
|
Survival Follow‐up: up to 24 weeks |
No deaths reported. | No deaths reported. | NA | 634 (4 studies) | ⊕⊕⊕⊝ moderatea | — |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: up to 24 weeks |
Mean (SD) absolute change in CFQ‐R score was ‐1.5 points (15.16) in the control group. | Mean absolute change in CFQ‐R score in the VX‐659‐ tezacaftor‐ivacaftor group was 20.10 points higher (17.19 higher to 23.01 higher) in the intervention group. | NA | 382 (1 study) | ⊕⊕⊕⊝ moderatea | A higher score indicates a better outcome. This outcome was only reported at up to 6 months in 1 study at a dose of VX‐659 240 mg once daily; 3 further studies reported at up to 1 month as follows. Davies 2018b reported an improvement in the VX‐659 80 mg once daily plus tezacaftor plus ivacaftor group compared to placebo (MD 10.00, 95% CI 0.29 to 19.71; 1 study, 21 participants with F508del/MF genotype). There was also an improvement in the VX‐659 240 mg once daily plus tezacaftor plus ivacaftor group compared to placebo (MD 16.13 points, 95% CI 13.02 to 19.24; 2 studies; 412 participants) (Davies 2018b; NCT03447249). NCT03460990 reported an improvement with VX‐659 240 mg once daily plus tezacaftor 100 mg once daily compared to placebo (MD 13.50, 95% CI 8.79 to 18.21; 1 study, 111 participants with F508del/F508del genotype). Davies 2018b reported further doses compared to the controls for different genotypes at up to 1 month: VX‐659 400 mg once daily plus tezacaftor plus ivacaftor (MD 7.90, 95% CI ‐0.58 to 16.38; 32 participants with F508del/MF genotype and MD 18.10, 95% CI 10.85 to 25.35; 29 participants with F508del/F508del genotype); and VX‐659 400 mg once daily plus tezacaftor plus deutivacaftor (MD 20.30, 95% CI 7.05 to 33.55; 25 participants with F508del/MF genotype and MD 20.30, 95% CI 7.05 to 33.55; 25 participants with F508del/MF genotype). |
|
FEV1 (% predicted): relative change from baseline Follow‐up: up to 24 weeks |
Outcome not reported at up to 6 months. | 1 study reported at up to 1 month and found improvements in the relative change from baseline in FEV1 % predicted across all dose levels and genotypes when compared to placebo. | ||||
|
FEV1 (% predicted): absolute change from baseline Follow‐up: up to 24 weeks |
Mean (SD) absolute change in FEV1 % predicted was ‐0.8% (8.27) in the control group. |
Mean absolute change in FEV1 % predicted was 14.20% higher (12.54 higher to 15.86 higher) in the intervention group. | NA | 382 (1 study) | ⊕⊕⊕⊝ moderatea | This outcome was only reported for VX‐659 240 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily versus triple placebo in participants with the F508del/MF genotype at the 6‐month time point. The same trial also reported at 1 month (MD 14.00% predicted, 95% CI 12.34 to 15.66) (NCT03447249). 2 further trials reported at up to 1 month. Davies 2018b found a greater absolute change in FEV1 % predicted with VX‐659 120 mg twice daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily compared to placebo (MD 10.00% predicted, 95% CI 3.04 to 16.96; 1 study, 12 participants with F508del/MF genotype). NCT03460990 reported a greater absolute change in FEV1 % predicted with VX‐659 240 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily compared to tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (MD 9.90% predicted, 95% CI 7.41 to 12.39; 1 study, 111 participants with F508del/F508del genotype). Keating 2018 (n = 117) found a significant improvement in the absolute change in FEV1 (L) at all dose levels and genotypes for the interventions compared to control. |
|
Adverse events Follow‐up: up to 24 weeks |
There was no difference in the number of participants experiencing at least 1 adverse event between the intervention and placebo groups at any dose or for any genotype. There was also no statistical difference versus placebo relating to the severity of adverse events across all doses and genotype groups. |
NA | 634 (4 studies) | ⊕⊕⊕⊝ moderatea | — | |
| Time to first pulmonary exacerbation | Outcome not reported. | NA | Keating 2018 (n = 117) did report that there was no difference in the number of courses of antibiotics required or the number of pulmonary exacerbations between groups at all dose levels and genotypes for the interventions compared to control. | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; MD: mean difference; MF: minimal function; NA: not applicable; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to indirectness or lack of applicability: data do not include children under the age of 12 and the results may not be applicable to younger children.
bDowngraded once for imprecision due to the very small sample size.
Summary of findings 8. Summary of findings ‐ triple therapy: elexacaftor‐tezacaftor‐ivacaftor/deutivacaftor compared to control for cystic fibrosis.
| Elexacaftor plus tezacaftor plus ivacaftor or deutivacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults with cystic fibrosis and either F508del/MF or F508del/F508del genotype Settings: outpatients Intervention: elexacaftor (50 mg once daily, 100 mg once daily or 200 mg once daily) plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily or deutivacaftor 150 mg once daily Comparison: F508del/MF participants: triple placebo; F508del/F508del participants: placebo tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Triple placebo or placebo tezacaftor plus ivacaftor | Elexacaftor plus tezacaftor plus ivacaftor or VX‐561 | |||||
|
Survival Follow‐up: 4 weeks to 24 weeks |
No deaths reported. | No deaths reported. | NA | 603 (3 studies) | ⊕⊕⊕⊕ high | — |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 4 weeks to 24 weeks |
A significant improvement in the elexacaftor plus tezacaftor plus ivacaftor or deutivacaftor groups in the CFQ‐R respiratory domain was observed compared to control versus placebo across all dose levels and both genotypes. | NA | 599 (3 studies) | ⊕⊕⊕⊝ moderatea | A higher score indicates a better outcome. | |
|
FEV1 (% predicted): relative change from baseline Follow‐up: 4 weeks to 24 weeks |
A significant improvement in the relative change from baseline in FEV1 % predicted in the elexacaftor plus tezacaftor plus ivacaftor groups was observed across all dose levels and genotypes when compared to control groups. | NA | 603 (3 studies) | ⊕⊕⊕⊝ moderatea | — | |
|
FEV1 (% predicted): absolute change from baseline Follow‐up: 24 weeks |
A significant improvement in the absolute change from baseline in FEV1 % predicted in the elexacaftor plus tezacaftor plus ivacaftor groups compared to control groups was observed in 2 studies including participants with F508del/MF genotype (MD 14.30% predicted, 95% CI 12.76 to 15.84; 1 study, 403 participants) and F508del/F508del genotype (MD 10.20% predicted, 95% CI 8.26 to 12.14; 1 study, 175 participants). | NA | 578 (2 studies) | ⊕⊕⊕⊝ moderatea | 1 study (n = 123) reported a significant improvement in the absolute change from baseline in FEV1 (L) favouring the intervention across all dose levels and genotypes for the interventions compared to control. | |
|
Adverse events Follow‐up: 4 weeks to 24 weeks |
There was no significant difference in the number of participants experiencing at least 1 adverse event between the intervention and placebo groups at any dose or for any genotype. There was also no statistical difference versus placebo relating to the severity of adverse events across all doses and genotype groups. | NA | 603 (3 studies) | ⊕⊕⊕⊝ moderatea | — | |
|
Time to first pulmonary exacerbation Follow‐up: N/A |
Outcome not reported; see comments. | N/A | 1 study (403 participants with genotype F508del/MF genotype) reported that fewer participants in the intervention group needed additional antibiotics for an exacerbation over the 6‐month reporting period (OR 0.29, 95% CI 0.14 to 0.60). 1 further study (175 participants with genotype with F508del/F508del) reported that fewer participants in the intervention group experienced an exacerbation at 1 month than in the control group (OR 0.17, 99% CI 0.06 to 0.45). |
|||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; MD: mean difference; MF: minimal function: NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to indirectness or lack of applicability: data do not include children under the age of 12 and those with more severe disease.
Summary of findings 9. Summary of findings ‐ triple therapy: elexacaftor‐tezacaftor‐ivacaftor compared to control (ivacaftor alone or tezacaftor plus ivacaftor) for cystic fibrosis.
| Elexacaftor plus tezacaftor plus ivacaftor compared to control (ivacaftor alone or tezacaftor plus ivacaftor) for cystic fibrosis | ||||||
|
Patient or population: adults and children aged 12 years and over with cystic fibrosis and either F508del/gating variant or F508del/residual function genotype Settings: outpatients Intervention: elexacaftor 200 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours Comparison: ivacaftor 150 mg every 12 hours or tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Ivacaftor alone or tezacaftor plus ivacaftor | Elexacaftor plus tezacaftor plus ivacaftor | |||||
|
Survival Follow‐up: 8 weeks |
No deaths reported. | No deaths reported. | NA | 258 (1 study) | ⊕⊕⊕⊝ moderatea | — |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: NA |
Outcome not reported | NA | — | |||
|
FEV1 (% predicted): relative change from baseline Follow‐up: NA |
Outcome not reported | NA | — | |||
|
FEV1 (% predicted): absolute change from baseline Follow‐up: 8 weeks |
Mean (SD) absolute change from baseline in FEV1 was 0.2% (5.10) in the control group. | Mean absolute change in FEV1 % predicted was 3.50% higher (2.24% higher to 4.76 higher). | NA | 258 (1 study) | ⊕⊕⊕⊝ moderatea | — |
|
Adverse events: number of participants with treatment‐emergent adverse events Follow‐up: 8 weeks |
659 per 1000 | 667 per 1000 (506 to 797) |
OR 1.04 (0.53 to 2.04) | 258 (1 study) | ⊕⊕⊕⊝ moderatea | — |
|
Time to first pulmonary exacerbation Follow‐up: NA |
Outcome not reported | NA | — | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; MD: mean difference; NA: not applicable; OR: odds ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to indirectness: data do not include children under the age of 12.
Summary of findings 10. Summary of findings ‐ triple therapy: VX‐440‐tezacaftor‐ivacaftor compared to placebo for cystic fibrosis.
| VX‐440 plus tezacaftor plus ivacaftor compared with placebo for cystic fibrosis | ||||||
|
Patient or population: adults with cystic fibrosis and with the F508del/MF genotype Settings: outpatients Intervention: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) Comparison: placebo or placebo plus tezacaftor | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | VX‐440 plus tezacaftor plus ivacaftor | |||||
|
Survival Follow‐up: up to 4 weeks |
No deaths reported. | No deaths reported. | NA | 55 (1 study)a |
⊕⊕⊝⊝ lowb,c | — |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: up to 4 weeks |
The mean difference in absolute change from baseline in CFQ‐R respiratory domain was 18.50 points (95% CI 8.99 to 28.01) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo group (29 participants). The mean difference in absolute change from baseline in CFQ‐R respiratory domain was 20.10 points (95% CI 13.76 to 26.44) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo plus tezacaftor group (26 participants). |
NA | 55 (1 study)a |
⊕⊕⊝⊝ lowb,c | A higher score indicates a better outcome. | |
|
FEV1 (% predicted): relative change from baseline Follow‐up: up to 4 weeks |
The mean difference in relative change from baseline in FEV1 (% predicted) was 19.10% (95% CI 10.62 to 27.58) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo group (29 participants). The mean difference in relative change from baseline in FEV1 (% predicted) was 20.00% (95% CI 12.59 to 27.41) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo plus tezacaftor group (26 participants). |
NA | 55 (1 study)a |
⊕⊕⊝⊝ lowb,c | — | |
|
FEV1 (% predicted): absolute change from baseline Follow‐up: up to 4 weeks |
The mean difference in absolute change from baseline in FEV1 (% predicted) was 10.60% (95% CI 5.93 to 15.27) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo group (29 participants). The mean difference in absolute change from baseline in FEV1 (% predicted) was 12.00% (95% CI 7.64 to 16.36) in the VX‐440 plus tezacaftor plus ivacaftor group compared to the placebo plus tezacaftor group (26 participants). |
NA | 55 (1 study)a |
⊕⊕⊝⊝ lowb,c | — | |
|
Adverse events Follow‐up: up to 4 weeks |
There was no difference in the number of participants with severe adverse events between either the treatment group compared to placebo (OR 3.48, 99% CI 0.06 to 212.67) (29 participants) or the treatment group compared to placebo plus tezacaftor (OR 0.11, 99% CI 0.00 to 3.34) (26 participants). | NA | 55 (1 study)a |
⊕⊕⊝⊝ lowb,c | — | |
|
Time to first pulmonary exacerbation Follow‐up: NA |
Outcome not reported | NA | — | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; NA: not applicable; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe single study was in two parts. The first part (29 participants) compared the triple therapy to placebo. The second part (26 participants) compared to placebo plus tezacaftor (NCT02951182). bDowngraded once due to indirectness or lack of applicability: data do not include children under the age of 12 and those with more severe disease. Also short‐term data only. cDowngraded once for imprecision due to small sample size.
Summary of findings 11. Summary of findings ‐ triple therapy: VX‐152‐tezacaftor‐ivacaftor compared to placebo for cystic fibrosis.
| VX‐152 plus tezacaftor plus ivacaftor compared with control for cystic fibrosis | ||||||
|
Patient or population: adults with cystic fibrosis and with the F508del/MF genotype Settings: outpatients Intervention: VX‐152 (100 mg, 200 mg or 300 mg twice daily) plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily Comparison: placebo or placebo plus tezacaftor plus ivacaftor | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or placebo plus tezacaftor plus ivacaftor | VX‐152 plus tezacaftor plus ivacaftor | |||||
|
Survival Follow‐up: 15 days |
No deaths reported. | NA | 76 (1 study)a | ⊕⊕⊝⊝ lowb,c | — | |
|
Quality of life: total score Follow‐up: NA |
Outcome not reported. | NA | — | |||
|
Quality of life: CFQ‐R respiratory domain: absolute change from baseline Follow‐up: 15 days |
The results favoured the VX‐152 100 mg plus tezacaftor plus ivacaftor group compared to the placebo group, MD 14.20 points (95% CI 0.98 to 27.42) (14 participants). The results favoured the VX‐152 200 mg plus tezacaftor plus ivacaftor group compared to the placebo group, MD 29.40 points (95% CI 16.97 to 41.83) (18 participants). The results favoured the VX‐152 300 mg plus tezacaftor plus ivacaftor group compared to the placebo group, MD 26.20 points (95% CI 13.71 to 38.69) (18 participants). There was no difference between VX‐152 200 mg twice daily plus tezacaftor plus ivacaftor compared to the control of placebo plus tezacaftor plus ivacaftor, MD 4.80 (95% CI ‐6.53 to 16.13) (14 participants). Results favoured the VX‐152 300 mg twice daily group plus tezacaftor plus ivacaftor compared to placebo plus tezacaftor plus ivacaftor MD 11.30 (95% CI 3.08 to 19.52) (28 participants). |
NA | 76 (1 study)a | ⊕⊕⊝⊝ lowb,c | — | |
|
FEV1 (% predicted): relative change from baseline Follow‐up: 15 days |
The mean relative change from baseline in FEV1 % predicted was greater across all treatment groups compared to either placebo or placebo plus tezacaftor plus ivacaftor. VX‐152 100 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 12.60% (95% CI 3.48 to 21.72) (14 participants). VX‐152 200 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 21.30% (95% CI 12.73 to 29.87) (18 participants). VX‐152 300 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 17.10% (95% CI 8.05 to 26.15) (18 participants). VX‐152 200 mg plus tezacaftor plus ivacaftor group compared to placebo plus tezacaftor plus ivacaftor, MD 14.60% (95% CI 1.88 to 27.32) (14 participants). VX‐152 300 mg plus tezacaftor plus ivacaftor group compared to placebo plus tezacaftor plus ivacaftor, MD 13.50 (95% CI 6.57 to 20.43) (28 participants). |
NA | 76 (1 study)a | ⊕⊕⊝⊝ lowb,c | — | |
|
FEV1 (% predicted): absolute change from baseline Follow‐up: 15 days |
The mean absolute change from baseline in FEV1 % predicted was greater across all treatment groups compared to either placebo or placebo plus tezacaftor plus ivacaftor. VX‐152 100 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 6.5% (95% CI 1.62 to 11.38) (14 participants). VX‐152 200 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 10.50% (95% CI 5.92 to 15.08) (18 participants). VX‐152 300 mg plus tezacaftor plus ivacaftor group compared to placebo, MD 8.80% (95% CI 3.98 to 13.62) (18 participants). VX‐152 200 mg plus tezacaftor plus ivacaftor group compared to placebo plus tezacaftor plus ivacaftor, MD 8.30% (95% CI 1.15 to 15.45) (14 participants). VX‐152 300 mg plus tezacaftor plus ivacaftor group compared to placebo plus tezacaftor plus ivacaftor, MD 8.70% (95% CI 4.50 to 12.90) (28 participants). |
NA | 76 (1 study)a | ⊕⊕⊝⊝ lowb,c | — | |
|
Adverse events Follow‐up: 15 days |
There was no difference in the number of adverse events at the 100 mg (14 participants) or 200 mg dose (18 participants) compared to placebo and 200 mg plus tezacaftor plus ivacaftor (14 participants) or 300 mg plus tezacaftor plus ivacaftor compared to placebo plus tezacaftor plus ivacaftor (28 participants). In the 300 mg group compared to placebo (18 participants), participants in both groups reported treatment‐emergent adverse events. |
NA | 76 (1 study)a | ⊕⊕⊝⊝ lowb,c | — | |
|
Time to first pulmonary exacerbation Follow‐up: NA |
Outcome not reported. | NA | — | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CI: confidence interval; FEV1: forced expiratory volume at 1 second; NA: not applicable; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe included trial was split into two parts. The first part compared to placebo and the second part compared to placebo plus tezacaftor/ivacaftor. The total number of participants was 76, but each of the arms had smaller numbers of participants, ranging from 14 to 28.
bDowngraded once due to indirectness or lack of applicability: data do not include children under the age of 18 and those with more severe disease. Also short‐term data only.
cDowngraded once due potential of risk of bias: high risk of attrition bias and 4 participants with FEV1 < 40% despite the inclusion criteria of FEV1 ≥ 40% and ≤ 90% of predicted normal for age, sex and height at the screening visit.
Background
Description of the condition
Cystic fibrosis (CF) is the most common inherited life‐shortening illness with a prevalence of 1 in 2000 at birth in Northern Europeans (Bobadilla 2002) and varying prevalence in other populations depending on ethnic composition. The affected gene codes for a protein called the cystic fibrosis transmembrane conductance regulator (CFTR) (Riordan 1989; Southern 1997). CFTR protein is transported to the outer cell membrane, where it has a role in the transport of salts (anions, chloride and bicarbonate) in and out of the cell (Rogan 2011). This role is important in all epithelial cells, particularly those lining the airways, pancreatic ducts, sweat gland, bile ducts in the liver and vas deferens.
In the lungs of people with CF (pwCF), defective salt transport leads to a reduction in airway surface liquid volume. This, in turn, leads to compromised mucociliary clearance, which makes the airway susceptible to infection, which initiates a cycle of inflammation, chronic infection and progressive lung damage. Eventually this causes respiratory failure, which is the commonest cause of premature death for pwCF. In addition to the airway problems, the abnormal transepithelial salt transport can lead to complications in other organs. This can result in malnutrition and diabetes (through pancreatic damage), salt depletion (through excess loss in sweat) and subfertility.
Over 2000 variants have been identified in the CFTR gene. These variants are classified according to the impact they have on the synthesis, processing or function of the CFTR gene (CFMD 2013). Classes of CFTR variant are described in more detail in Table 12 (Rowntree 2003; Southern 2007). Most CFTR variants are associated with a complete loss of CFTR protein and result in a classical CF phenotype. Some CFTR variants are associated with residual function and these tend to be associated with less severe phenotype, e.g. individuals may be pancreatic sufficient and not require pancreatic replacement therapy.
1. Classes of mutations affecting CFTR production, structure and function.
| Class | Example mutation | Impact on CFTR structure and function |
| I | G542X | Synthesis of CFTR is critically impaired, and no functional protein is produced. This is due to the presence of a premature stop codon in the nucleotide sequence. Individuals have minimal CFTR function. |
| II | F508del | A full length of CFTR is produced, but this is structurally abnormal and destroyed by the cell before it reaches the cell membrane. This is called a defect in the intracellular trafficking pathway. Small amounts of CFTR do reach the cell membrane; however, here they display defective ion transport, demonstrating that the F508del mutation is more than just a processing defect. Individuals have minimal CFTR function. |
| III | G551D | CFTR is produced and embedded in the cell membrane, but the chloride channel does not respond ('switch on') to normal stimulation from the cell. This means there is no meaningful ion transport across the protein. Individuals have minimal CFTR function. |
| IV | R347P | CFTR is transported to the outer cell membrane, and responds to normal stimulation, but functions at a low level because chloride ions do not cross the channel appropriately. Individuals have some residual CFTR function. |
| V | A455E | Normal CFTR is produced, but the amount of protein is reduced. Individuals have some residual CFTR function. |
CFTR: cystic fibrosis transmembrane regulator
The commonest CF‐causing variant, F508del (also known as ΔF508 or delta F508), is found in the majority of pwCF (up to 80% to 90% of some populations, e.g. pwCF from a Northern European heritage). For pwCF with F508del, a full length of protein is transcribed but recognised as misfolded by the cell and is degraded before reaching the cell membrane, where it needs to be positioned to effect transepithelial salt transport. Hence, this is a severe variant associated with no meaningful CFTR function. This type of variant is called a class II mutation (or trafficking defect) and much research has explored masking the molecular defect, bypassing the cellular mechanisms and enabling the F508del protein to traffic to the cell membrane, where it may have some normal salt transport capability.
Description of the intervention
Increasing understanding of how different variants affect the production, structure and function of CFTR has led to the concept of mutation‐specific therapies (Table 12). For class II variants where a full length of protein is produced, recognised as abnormal by the cell and degraded before reaching the cell membrane, scientists have recognised that certain laboratory manoeuvres can affect this process, e.g. reducing cell temperature and the trafficking defect can be overcome (Colledge 1995). In such circumstances, the F508del protein may reach the cell membrane, where it has some ability to transport salt. This has led to the search for molecules that can overcome the F508del trafficking defect and these drugs have been called 'correctors'.
Two distinct scientific approaches have resulted in the recognition of candidate drugs with this mode of action (Amaral 2007):
testing of compounds known to affect CFTR or other ion channels (either pharmaceutical drugs or chemicals that occur naturally in plants, herbs, fruits or food components);
high throughput screening, which involves testing large numbers of diverse chemicals, on laboratory cell lines, to identify which of these may overcome the intracellular trafficking defect.
These approaches have resulted in the identification of small molecules that may be taken orally (Rubenstein 1997; Van Goor 2011).
Since our initial review in 2018, there has been considerable evolution in this field, with the development of further combination agents that address the molecular challenge of correcting the class II variants; these agents are all correctors (e.g. tezacaftor, VX‐659, elexacaftor), which are given in combination with the same potentiator ivacaftor. A separate review is available for potentiators (Skilton 2019). This method of administering combined drugs has been accommodated in this updated version of our Cochrane Review and interventions are presented as monotherapy, dual therapy and, more recently since the 2020 update, triple therapy. Given the close relationship of these approaches and to illustrate the development of the field, we have evaluated all of these therapies in this single updated Cochrane Review rather than considering these in separate reviews. Dependent on how the field develops, we may review this decision for future updates of this review.
The drugs names, company codes used throughout this review and presumed mechanism of action have been summarised in Table 13.
2. Drug names based on company codes with presumed mechanism of action.
| Company | Code during product development | Name of drug | Mechanism of action |
| Vertex | VX‐121 | vanzacaftor | Potentiator |
| VX‐445 | elexacaftor | Corrector | |
| VX‐561 | deutivacaftor | Potentiator | |
| VX‐661 | tezacaftor | Corrector | |
| VX‐770 | ivacaftor | Potentiator | |
| VX‐809 | lumacaftor | Corrector | |
| Proteostasis Therapeutics | PTI‐428 | nesolicaftor | Amplifier |
| PTI‐801 | posenacaftor | Corrector | |
| PTI‐808 | dirocaftor | Potentiator |
How the intervention might work
Correction of the basic CF defect may lead to normalisation of airway surface liquid, and correction of mucociliary clearance, reducing the susceptibility to airway infection and inflammation.
In addition to correctors, other drugs that aim to treat the CFTR defect are also under investigation. These include potentiators for class III and IV variants, which enhance the function of mutated CFTR protein embedded in the cell membrane by increasing the time the CFTR salt channel remains open, and therapies for class I variant, which act to prevent structural abnormalities of CFTR that occur when premature stop codons terminate protein synthesis. Cochrane Reviews assessing these interventions have been published (Aslam 2023; Skilton 2019).
While correctors are successful at facilitating the F508del protein to reach the cell membrane, it still has suboptimal function. Our initial version of this Cochrane Review suggested that CFTR correctors would need to be combined with other agents (potentiators) to achieve a clinical benefit for pwCF who have a class II variant (e.g. F508del) (Southern 2018). For the purposes of this Cochrane Review, we consider combination therapies for class II variants as 'correctors'. We appreciate that these therapies contain single agents with distinct molecular properties, e.g. the dual therapy tezacaftor‐ivacaftor is a combination of a corrector (tezacaftor) and a potentiator (ivacaftor), but together we class this as a corrector therapy. The likely mechanism of action for each agent is described in Table 13.
Why it is important to do this review
CFTR correctors are novel therapies and it is important that randomised controlled trials (RCTs) are conducted and critically appraised to provide clear evidence assessing their benefits and harms. It is important that funding bodies have a clear evidence base on which to assess new therapies for CF that aim to correct the basic defect. In addition, critical appraisal of studies will help inform future study design.
New therapies that correct the F508del mutation will have a positive impact on an important proportion of the CF population (Southern 1997). Given the number of pwCF who will be prescribed this treatment, there will be an important healthcare cost. Experience from other licensed agents that correct the underlying CF defect suggests that these costs may be considerable (NICE 2016).
This review aims to collate evidence from RCTs that have evaluated the benefits and harms of CFTR correctors in pwCF and class II CFTR variants. This is an updated version of the review (Southern 2018; Southern 2020).
Objectives
To evaluate the effects of CFTR correctors (with or without potentiators) on clinically important benefits and harms in people with CF of any age with class II CFTR mutations (most commonly F508del).
Methods
Criteria for considering studies for this review
Types of studies
We have included RCTs of parallel design (published or unpublished). We have not included quasi‐RCTs. We have not included cross‐over studies as we do not feel this study design is appropriate given that the intervention aims to correct the underlying defect of CF and if the intervention is effective it will have an important impact on the course of the disease. This has been established from the data from trials examining ivacaftor for people with class III mutations.
Types of participants
We have included RCTs involving children or adults with CF, as confirmed either by the presence of two disease‐causing variants (at least one class II variant), or by a combination of positive sweat test and recognised clinical features of CF. We have included RCTs that include participants with any level of disease severity. RCTs limited to pwCF who are homozygous for a class II variant are analysed separately.
Types of interventions
A CFTR corrector is defined as a drug that aims to increase the amount of CFTR expressed at the epithelial cell apical membrane, by reducing or preventing degradation of CFTR by normal intracellular mechanisms. The main variant targeted by this approach is F508del. As this review focuses on small molecule therapies that correct the intracellular trafficking defect of variants, such as F508del, interventions that target DNA correction (e.g. antisense technology) are not included.
We have included RCTs comparing CFTR correctors with either placebo or another intervention. We have also included RCTs in which CFTR correctors are administered alongside another class of drug that also aims to improve CFTR function (e.g. potentiators).
Types of outcome measures
We assessed the following outcome measures.
Primary outcomes
Survival
-
Quality of life (QoL) (measured using validated quantitative scales or scores (e.g. Cystic Fibrosis Questionnaire‐Revised (CFQ‐R) (Quittner 2009))
Total QoL score
Different sub‐domains that may be reported
-
Physiological measures of lung function (L or per cent (%) predicted for age, sex and height)
Forced expiratory flow rate at one second (FEV1) (relative change from baseline)
FEV1 absolute values (and change from baseline)
Forced vital capacity (FVC) (absolute values and change from baseline)
Lung clearance index (LCI) (post hoc change)
Other relevant physiological measures of lung function
Secondary outcomes
-
Adverse effects (AEs)
Graded by review authors as mild (therapy does not need to be discontinued)
Graded by review authors as moderate (therapy is discontinued, and the adverse effect ceases)
Graded by review authors as severe (life‐threatening or debilitating, or which persists even after treatment is discontinued)
Other adverse effects of therapy (of any severity) that are not classifiable according to these categories
-
Hospitalisation
Number of days
Number of episodes
Time to next hospitalisation
School or work attendance (i.e. number of days missed)
-
Extra courses of antibiotics (measured as time to the next course of antibiotics and the total number of courses of antibiotics)
Oral
Intravenous
Inhaled
Sweat chloride (change from baseline) as a measure of CFTR function
-
Radiological measures of lung disease (assessed using any scoring system)
Chest radiograph scores
Computerised tomogram (CT) score
-
Acquisition of respiratory pathogens
Pseudomonas aeruginosa
Staphylococcus aureus
Haemophilus influenzae
Other pathogen clinically relevant in CF
-
Eradication of respiratory pathogens (as defined by study authors)
P aeruginosa
S aureus
H influenzae
Other pathogen clinically relevant in CF
-
Nutrition and growth (measured as relative change from baseline) (including z scores or centiles)
Weight
Body mass index (BMI)
Height
With regards to exacerbations, investigators of different studies do not use consistent definitions of exacerbations in CF, and sometimes investigators do not explicitly define what they consider to be an exacerbation for their study. Therefore, in order to incorporate data for exacerbations from the different included studies, we have used a broad definition of an exacerbation, such that we consider an exacerbation to be an increase in symptoms, the need for antibiotics or hospital admission, or any combination of these. We report these events under secondary outcome 4.
Search methods for identification of studies
We searched for all relevant published and unpublished studies without restrictions on language (we did not exclude studies reported in a language other than English), year or publication status.
Electronic searches
We identified relevant studies from the Cochrane Cystic Fibrosis Trials Register using the terms: 'drugs that correct defects in CFTR transcription, translation or processing'. Relevant studies have been tagged with these terms for indexing purposes in the Cystic Fibrosis Trials Register.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work was identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cystic Fibrosis and Genetic Disorders Group website.
Date of the most recent search: 28 November 2022.
We also searched the following trial registries and registers:
US National Institutes of Health Ongoing Trials Register Clinicaltrials.gov (www.clinicaltrials.gov);
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (trialsearch.who.int/);
European Medicines Agency (www.clinicaltrialsregister.eu/ctr-search/search).
Date of the most recent search: 3 December 2022.
For details of our search strategies, please see Appendix 1.
Searching other resources
We screened the bibliographies of included studies and any relevant systematic reviews identified for further references to potentially relevant studies. We also contacted authors of included studies, leaders in the field and companies known to be developing and investigating CFTR correctors, to identify any studies that may have been missed by this search. We recorded response rates from this contact process below (Results of the search).
Data collection and analysis
Selection of studies
Two authors (MH and JM) independently assessed the suitability of each potential study identified by the search. If disagreement arose on the suitability of a study for inclusion in the review, we attempted to reach a consensus by discussion, failing which, a third author arbitrated.
Data extraction and management
Two authors (MH and JM) independently extracted relevant data from each included study. If disagreement arose on data extraction, we attempted to reach a consensus by discussion, failing which, a third author (KWS) arbitrated. Four authors (MH, JM, SP and SN) entered the data into RevMan for analysis.
If studies had reported data on our primary outcome (survival), we planned to report these as a binary outcome or a time‐to‐event outcome. We planned to extract QoL scores as relative change from baseline ((measurement at end of treatment ‐ measurement at baseline) / measurement at baseline) x 100). We extracted data presented as post‐treatment values or change from baseline when this was not possible.
With regards to the secondary outcome 'Extra courses of antibiotics', we planned to extract data as time to the next course of antibiotics and the total number of courses of antibiotics. We noted whether episodes of pulmonary exacerbations were physician‐defined or protocol‐defined. If studies reported baseline and post‐treatment sweat chloride concentration values, we calculated the relative change from baseline values ((measurement at end of treatment ‐ measurement at baseline) / measurement at baseline) x 100).
We reported data as immediate (up to and including one month), short‐term (over one month and up to six months) and longer‐term (over six months). The exception to this is one study that presents data for lumacaftor monotherapy for 14 days and then adds ivacaftor for the final seven days of the study; in this case we present data at 14 and 21 days (Boyle 2014). If a study we include in future updates of the review reports multiple time points within each of these ranges, we will report the later time point.
We attempted to extract the most precise data possible for each outcome; we preferred to extract data from tables in the papers. Where papers presented data only graphically, three authors (JM, SP and SJN) estimated the relevant data from graphs and compared estimations for accuracy.
We have not combined data from studies evaluating distinct agents or combinations of agents, as they have different mechanisms of actions. Also, where RCTs have recruited pwCF with different genotypes and presented these results separately, we have analysed the data by genotype.
Assessment of risk of bias in included studies
Two authors (MH and JM) assessed the risk of bias for each study using the Cochrane risk of bias tool (Higgins 2017). This includes assessment of the following methodological aspects of the included studies:
procedure for randomisation (selection bias);
allocation concealment (selection bias);
masking (blinding) of the intervention from participants, clinicians and trial personnel evaluating outcomes (performance bias);
missing outcome data (attrition bias);
selective outcome reporting (reporting bias);
other sources of bias (e.g. the influence of funding sources or industry on trial characteristics and presented results).
We also assessed whether all participants were included in an intention‐to‐treat analysis, regardless of whether they completed the treatment schedule or not. If disagreement arose on the assessment of risk of bias of a study, we attempted to reach a consensus by discussion, failing which, a third author (KWS) arbitrated.
Measures of treatment effect
For binary outcomes, we calculated a pooled estimate of the treatment effect for each outcome using the pooled odds ratio (OR) and 95% confidence intervals (CIs) or 99% CIs for analysis of separate adverse events.
For continuous outcomes, we calculated the mean change from baseline for each group or the mean post‐intervention values and standard deviation (SD) for each group. We converted standard errors (SEs) to SDs. We produced a pooled estimate of treatment effect by calculating the mean difference (MD) and 95% CIs.
In future updates of this review, if different trials present data for the same outcomes in different forms (e.g. absolute values of lung function measures, or change in these measures from a baseline), we will combine these in a meta‐analysis where appropriate.
Where the studies did not report change data, but instead presented absolute post‐treatment data without baseline data (so it was not possible to calculate change data), we planned to use absolute post‐treatment data instead of change from baseline. However, if the report presented baseline and post‐treatment data for any outcome, we calculated SDs for the change from baseline, for example if the CI was available. If there was not enough information available to calculate the SDs for the changes, we planned to impute them from other trials in the review, where data were available and trials were similar (i.e. when they used the same measurement scale, had the same degree of measurement error and had the same time periods between baseline and final value measurement). If neither of these methods were possible, we planned to calculate a change‐from‐baseline SD, making use of an imputed correlation coefficient (methods described in section 23.2.7.3 in the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2022)).
Where time‐to‐event data were reported (e.g. survival time, time to next hospitalisation, time to first exacerbation), we reported a hazard ratio (HR) and 95% CIs. Where HRs and 95% CIs were not reported for a time‐to‐event outcome, we assessed whether any reported data, including graphical data (e.g. Kaplan‐Meier curves), could be used to indirectly estimate HRs and 95% CIs via the published methods (Parmar 1998; Williamson 2002).
When reporting on outcomes, we used the following subheadings to describe the time points: immediate (up to and including one month); short term (over one month and up to six months); and longer term (over six months).
Unit of analysis issues
We included results from RCTs of parallel design in which individual study participants are randomised. We have not included cross‐over studies as we do not feel this study design is appropriate given that the intervention aims to correct the underlying defect. If the intervention is effective, it will have an important impact on the course of the disease. This has been established from the data from trials examining ivacaftor for people with class III mutations (Skilton 2019).
In one included study, continuous outcomes were analysed via a mixed‐model repeated‐measures (MMRM) analysis based on the average effect across the measured time points (Ratjen 2017). Such an analysis is longitudinal and uses all available data at every visit and allows adjustment for covariates such as the baseline measurement of the outcome. All analyses were also adjusted for baseline weight (less than 25 kg versus 25 kg or over) and baseline FEV1 (% predicted ‐ less than 90% versus 90% or above). Results provided by this model can be interpreted as treatment effect averaged from each study visit until week 24. Within this review, results are entered into the analysis via generic inverse variance and are not pooled with other studies, due to the different approaches to analysis.
For the studies of triple therapies we included combinations with VX‐561 (deuterated ivacaftor), as we considered it to be a sufficiently similar intervention to standard ivacaftor. We pooled the data for those participants with F508del/MF genotypes as these pwCF were in the groups that tested the ascending doses and VX‐561. Only one dose was tested in pwCF with the F508del/F508del genotype and VX‐561 was not tested in this group. Pooling data from studies of differing genotypes (participants with one or two class II variants) was not considered appropriate.
Dealing with missing data
In order to allow an intention‐to‐treat analysis, we extracted data on the number of participants with each outcome event, by allocated treated group, irrespective of compliance and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment or follow‐up. If any data were missing or unclear, we contacted the primary investigators for clarification.
As stated above, where HRs and 95% CIs were not reported for a time‐to‐event outcome, we assessed whether any reported data, including graphical data (e.g. Kaplan‐Meier curves) could be used to indirectly estimate HRs and 95% CIs via the published methods (Parmar 1998; Williamson 2002).
Assessment of heterogeneity
We assessed heterogeneity through visual examination of the combined data presented in the forest plots, and by considering overlap of study‐specific CIs, and the I² statistic (Higgins 2003) together with Chi² values (Deeks 2022). The I² statistic reflects the likelihood that variation of results across studies is due to heterogeneity rather than chance, and we interpreted this statistic using the following simple classification:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
In order to identify selective outcome reporting, where possible, we have compared outcomes described in the study protocol with those reported in the publication. We have requested protocols for specific studies from the primary investigators and recorded the proportion of protocols that were available to us. If a protocol was not available, we searched for information about outcomes from trial registry databases. We also compared outcomes listed in the 'Methods' section of the final paper with those presented in the 'Results' section. If the published papers reported negative findings either only partially, or not at all, we contacted primary investigators for these data.
We would have assessed publication bias by constructing and assessing the symmetry of a funnel plot. This would have been possible if we included more than 10 studies in a meta‐analysis in the review. We would have plotted the number of participants in the study against a measure of treatment effect. If the funnel plot was asymmetrical, we would consider whether this was due to publication bias, or whether methodology or small sample size caused the results of certain studies to show exaggerated treatment effects.
Data synthesis
As we intended to assess different CFTR correctors within this review, we assumed that there would not be a single common true effect. We also anticipated that participants in each study would vary due to different eligibility criteria. Therefore, regardless of I² value, we intended to use a random‐effects model to analyse data from studies.
As the review progressed, we included a number of early‐phase studies of interventions (which were ultimately not taken forward) in addition to large Phase 3 studies of combination therapies; therefore, we felt it more appropriate to employ separate comparisons within the review. As only a relatively small number of studies were included in each comparison (and when meta‐analysis was undertaken), we considered it more appropriate to employ a fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
We would have investigated any heterogeneity that we identified using subgroup analyses of potential confounding factors, if sufficient numbers (at least 10 studies included in a meta‐analysis) were available. For this review, these confounding factors would be:
age (children (defined as younger than 18 years of age) versus adults);
gender;
different variant classes (Table 12).
As we did not seek individual participant data from study investigators and such information was not available within published reports, we did not undertake a subgroup analysis on the basis of disease severity. We may incorporate such an analysis in future updates of this review.
Sensitivity analysis
In future updates of this review, if sufficient data are available, we will examine the impact of bias on the results by comparing meta‐analyses including and excluding studies with concerns of high risk of selection or reporting bias due to issues relating to randomisation, allocation concealment or masking of interventions from participants or study personnel.
Summary of findings and assessment of the certainty of the evidence
In a post hoc change from protocol, we have presented 11 summary of findings tables (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11).
We have presented two tables under the comparison of 'Monotherapy compared to control'. In one table we compare lumacaftor monotherapy to placebo (Table 1); we have presented lumacaftor results only rather than other correctors in the table for this comparison due to the relevance of this particular treatment at the time of writing (NICE 2016). In a further table we present results for cavosonstat compared to placebo (Table 2). We have not presented other monotherapy treatments in the summary of findings tables as interventions have not been taken forward on larger, more representative populations in Phase 3 studies.
We have presented four tables under the comparison of 'Dual therapy (correctors plus potentiators) compared to control':
lumacaftor (600 mg once daily or 400 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, reporting short‐term results (one month to six months) (Table 3);
lumacaftor (200 mg twice daily) and ivacaftor (250 mg twice daily) versus placebo, reporting immediate‐term results (up to one month (Table 4);
lumacaftor (200 mg) plus ivacaftor (150 mg or 250 mg twice daily) versus placebo, reporting immediate‐term results (up to one month) (Table 5).
tezacaftor (100 mg once daily) and ivacaftor (150 mg twice daily) versus placebo or ivacaftor (150 mg twice daily alone) (one month to six months) (Table 6).
We have presented tables separately for lumacaftor plus ivacaftor under this comparison due to the differences in doses, measurement times and approaches to analysis.
We have presented five tables for the comparison of 'Triple therapy (correctors plus potentiators) compared to control':
VX‐659 (80 mg once daily, 120 mg twice daily, 240 mg once daily or 400 mg once daily) plus tezacaftor 100 mg once per day plus ivacaftor 150 mg twice daily or VX‐561 150 mg once daily compared to triple placebo (for F508del/MF participants) or placebo tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (for F508del/F508del participants) (Table 7);
elexacaftor (50 mg once daily, 100 mg once daily or 200 mg once daily) plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily or VX‐561 150 mg once daily compared to triple placebo (for F508del/MF participants) or placebo tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (for F508del/F508del participants) (Table 8);
elexacaftor 200 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours compared to ivacaftor 150 mg every 12 hours or tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours (Table 9);
VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo in adults with cystic fibrosis and with the F508del/MF genotype (Table 10);
VX‐152 (100 mg, 200 mg or 300 mg twice daily) plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily versus placebo in adults with cystic fibrosis and with the F508del/MF genotype (Table 11).
We reported the following outcomes in all tables (chosen based on relevance to clinicians and consumers):
survival;
QoL (total score);
QoL (respiratory domain);
FEV1 % predicted (relative and absolute change);
AEs; and
time to first pulmonary exacerbation.
For clarity in the tables, we do not present adverse events according to the subdomains in Effects of interventions; instead we have inserted a general statement about the summary of findings for these outcomes and graded the evidence based on all of the subdomains combined.
We determined the certainty of the evidence using the GRADE approach, and downgraded evidence in the presence of a high risk of bias in at least one study, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results or high probability of publication bias. We downgraded the evidence by one level if we considered the limitation to be serious and by two levels if very serious.
Results
Description of studies
For full information on the characteristics of studies, please see Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification; Characteristics of ongoing studies.
Results of the search
Previously (last update 2020), the search of the specified databases identified 190 unique references corresponding to 75 studies (Figure 1). Two included trials were identified through communication with authors, before publication (Heijerman 2019; Middleton 2019). In the most recent update of this Cochrane Review, a search of the Cochrane Cystic Fibrosis Trials Register identified 44 studies and a search of the ongoing trials databases identified a further 237 studies (Figure 2).
1.

Study flow diagram for searches up to and including 2020.
2.
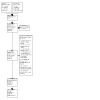
Study flow diagram for the search for the 2023 update of this review.
There are 34 studies (175 references) that meet the eligibility criteria for inclusion in this review (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; Horsley 2017; Keating 2018; McCarty 2002; McKone 2021; Middleton 2019; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Rubenstein 1998; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021; Zeitlin 2002). The results from two of these studies were jointly reported in 20 papers (TRAFFIC 2015; TRANSPORT 2015); a further paper reports two separate studies of the same intervention ‐ a Phase 1 study (Davies 2018a) and a Phase 2 study (Davies 2018b).
We excluded 35 studies (67 references) (Berkers 2014; Berkers 2021; Chadwick 1998; Chilvers 2021; Chmiel 2021; Drevinek 2017; ISRCTN14081521; Krivec 2021; Leonard 2012; Marsh 2022; Meijer 2016; NCT00945347; NCT01899105; NCT02323100; NCT03447262; NCT03525574; NCT03537651; NCT03601637; NCT03633526; NCT03691779; NCT03756922; NCT04043806; NCT04058210; NCT04058366; NCT04183790; NCT04235140; NCT04362761; NCT04537793; NCT04545515; NCT05535959; Nick 2014; Rowe 2017; Rubenstein 2006; Sumner 2014; Ziady 2015).
We have listed 14 studies (32 references ‐ one full paper reports two separate studies) as completed and awaiting classification, to be assessed for inclusion or exclusion at the next update (ALBATROSS; Downey 2019; Downey 2020; FLAMINGO; Hunt 2017; NCT03768089; Uluer 2023a; Uluer 2023b; NCT03969888; NCT04353817; PELICAN; Rio‐CF; Taylor‐Cousar 2019; Wainwright 2019).
We identified eight relevant ongoing studies (12 references), which we will assess for inclusion once completed (Jain 2018; NCT02589236; NCT02718495; NCT03258424; NCT04853368; NCT05033080; NCT05076149; NCT05274269).
Included studies
Study design
The 34 included studies ranged from Phase 1 to Phase 4 RCTs, and all employed a parallel study design (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; Horsley 2017; Keating 2018; McCarty 2002; McKone 2021; Middleton 2019; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Rubenstein 1998; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021; Zeitlin 2002). The PROGRESS study was an extension study of the TRAFFIC and TRANSPORT studies included in the review (TRAFFIC 2015; TRANSPORT 2015), but with participants in the control group from the initial trials randomised to receive the active treatment at one of two doses (PROGRESS 2017).
A total of 4781 randomised participants were included in this review (participants in the PROGRESS study have only been counted in their original studies and not in this extension study). Study sample sizes ranged from 12 participants (Davies 2018a) to 563 participants (TRANSPORT 2015). One study was composed of three cohorts ‐ cohort 1 (n = 62), cohort 2 (n = 109) and cohort 3 (n = 15); any reference to this study is to participants randomised to cohort 1 only, since data for the placebo participants from cohorts 2 and 3 were pooled, undoing the effects of randomisation and rendering them ineligible for inclusion in this review (Boyle 2014). In the Phase 2 study of tezacaftor‐ivacaftor, only data from the heterozygous population are included (n = 18), as the placebo groups in the homozygous arms of the trial were pooled (Donaldson 2018).
Two triple therapy studies included both run‐in and washout periods where participants received 100 mg once daily tezacaftor plus 150 mg twice daily ivacaftor; in between these periods investigators in one study assessed either 600 mg VX‐440 twice daily plus 50 mg tezacaftor once daily plus 300 mg ivacaftor twice daily or matched placebo for VX‐440 and the same dose of tezacaftor‐ivacaftor (NCT02951182). In the second study, both 200 mg VX‐152 and 300 mg VX‐152 combination therapies were compared to tezacaftor plus ivacaftor control groups (NCT02951195).
The duration of the included studies ranged from a single day (Phase 1 single‐dose testing) (McCarty 2002) to 48 weeks (Stahl 2021) with an extension of two studies to 96 weeks (PROGRESS 2017).
Two studies were undertaken at single centres (Rubenstein 1998; Zeitlin 2002), but the remaining studies were conducted at multiple centres, ranging from four (McCarty 2002) to 191 sites (PROGRESS 2017). Eight studies were conducted in the USA only (Donaldson 2014; Donaldson 2017; McCarty 2002; NCT02070744; NCT02508207; NCT02951195; Rubenstein 1998; Zeitlin 2002), one in Germany only (Stahl 2021), two in the UK only (Davies 2018a; Davies 2018b), one in Australia only (NCT02730208), six in North America and Europe (Clancy 2012; Donaldson 2018; Heijerman 2019; Ratjen 2017; Schwarz 2021; Taylor‐Cousar 2017), four in Europe and Australia (Davies 2021; Horsley 2017; Sutharsan 2022; Wilson 2021) and the remainder across North America, Europe and Australia (Barry 2021; Boyle 2014; Keating 2018; McKone 2021; Middleton 2019; Munck 2020; NCT02951182; NCT03447249; NCT03460990; PROGRESS 2017; TRAFFIC 2015; TRANSPORT 2015).
Full texts were available for 24 studies (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2017; Donaldson 2018; Heijerman 2019; Keating 2018; McCarty 2002; McKone 2021; Middleton 2019; Munck 2020; PROGRESS 2017; Ratjen 2017; Rubenstein 1998; Schwarz 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021; Zeitlin 2002). One study was available only as two conference abstracts (Horsley 2017); one study was reported in two conference abstracts and an online summary on Clinicaltrials.gov (Donaldson 2014); a further study was reported as a conference abstract and an online summary (Stahl 2021); and seven studies were reported as online summaries only (NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990).
One Phase 2 study of triple combination therapy indicated that there had been a corresponding Phase 1 study, but it was conducted in "healthy volunteers"; the paper does not state if this means people who do not have CF or people who do have CF but are in a good state of health (Keating 2018). The publication does not include any data for the Phase 1 study, although a continuation into a Phase 2 study implies that the safety profile was considered acceptable during the study period. It was not explicitly stated whether any AEs or safety concerns were observed in the Phase 1 study, nor does it state the dose tested or whether elexacaftor was tested in triple combination or as an individual agent for the purposes of the Phase 1 study (Keating 2018).
Participants
One study recruited pwCF with one F508del variant (the other variant was classified as residual function (RF) (ivacaftor responsive)) (Donaldson 2018). One study recruited a number of pwCF with two F508del variant copies and a number of pwCF with one F508del copy and a RF variant (Davies 2021). One study recruited pwCF with F508del/gating genotypes (McKone 2021). One study recruited participants with F508del/gating and F508del/RF genotypes (Barry 2021). Five studies recruited a number of pwCF with two F508del variant copies and a number of pwCF with one F508del copy and a minimal function (MF) (non‐ivacaftor responsive) variant (Davies 2018a; Davies 2018b; Keating 2018; NCT02951182; NCT02951195). Three studies recruited pwCF with F508del/MF genotypes (Middleton 2019; Munck 2020; NCT03447249). The remaining 23 studies recruited participants who had F508del/F508del genotypes.
Only one study recruited children between the ages of two to five years (Stahl 2021), two studies recruited children between the ages of six to 11 years (Davies 2021; Ratjen 2017), 14 studies recruited adolescents and adults (Barry 2021; McKone 2021; Munck 2020; NCT02730208; NCT03447249; NCT03460990; PROGRESS 2017; Rubenstein 1998; Schwarz 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021) and the remaining 17 studies recruited only adults.
Interventions
Single included studies examined the effects of 4‐phenylbutyrate (4PBA) (Rubenstein 1998; Zeitlin 2002), 8‐cyclopentyl‐1, 3‐dipropylxanthine (CPX) (McCarty 2002), N6022 (Donaldson 2014), cavosonstat (N91115) (Donaldson 2017), lumacaftor monotherapy (Boyle 2014; Clancy 2012) and FDL169 monotherapy (Horsley 2017). Seven studies assessed lumacaftor‐ivacaftor dual combination therapy (Boyle 2014; PROGRESS 2017; Ratjen 2017; Stahl 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021) and nine studies assessed tezacaftor‐ivacaftor dual combination therapy (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017). Four studies assessed the triple combination therapy of VX‐659‐tezacaftor‐ivacaftor (Davies 2018a; Davies 2018b; NCT03447249; NCT03460990), five studies assessed the triple combination therapy of elexacaftor‐tezacaftor‐ivacaftor (Barry 2021; Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022), one study assessed the triple combination therapy of VX‐440‐tezacaftor‐ivacaftor (NCT02951182) and one study the triple combination therapy of VX‐152‐tezacaftor‐ivacaftor (NCT02951195).
Monotherapy
Eight studies (n = 344) report on monotherapy (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
Two studies compared 4PBA to placebo (Rubenstein 1998; Zeitlin 2002). In the earlier study, participants received either 19 g 4PBA (split into three daily doses) or placebo for one week (Rubenstein 1998). The subsequent Phase 2 study examined the effects 4PBA given at either 20 g (n = 6), 30 g (n = 6) or 40 g (n = 3), given in three daily doses for one week (Zeitlin 2002).
One study compared escalating doses of CPX to placebo (McCarty 2002). Participants were randomised to receive single doses of either placebo (n = 8) or 1 mg (n = 4), 3 mg (n = 4), 10 mg (n = 4), 30 mg (n = 4), 100 mg (n = 5), 300 mg (n = 4) or 1000 mg (n = 4) CPX.
One study compared sequential ascending doses of N6022 to placebo (Donaldson 2014). Participants were randomised to receive placebo (n = 19) or the active drug (intravenous solution of N6022 in normal saline) at a dose of either 5 mg (n = 10), 10 mg (n = 9), 20 mg (n = 9) or 40 mg (n = 19). Both treatments were administered by infusion pump over one to eight minutes once per day for seven days.
The study of cavosonstat included both healthy volunteers and pwCF (Donaldson 2017). Those with CF (only these participants provide data for the review) were randomised to receive 50 mg placebo (n = 12) or cavosonstat at different doses (50 mg (n = 12), 100 mg (n = 13), or 200 mg (n = 14)) twice daily for 28 days.
Two included studies compared lumacaftor monotherapy (Boyle 2014; Clancy 2012). One study (n = 64) compared 200 mg lumacaftor once daily for 14 days to placebo; then from day 15, participants took a combination of lumacaftor and ivacaftor twice daily until day 21, thus contributing to two sections of this review (Boyle 2014). The second study of lumacaftor monotherapy used escalating doses of 25 mg (n = 18), 50 mg (n = 18), 100 mg (n = 17) and 200 mg (n = 19), to placebo (n = 17) for 28 days (Clancy 2012).
One study compared FDL169 at doses of 400 mg (n = 6), 600 mg (n = 6) and 800 mg (n = 8), each taken three times daily, versus placebo (n = 7) for 28 days (Horsley 2017).
Dual therapy
A total of 16 studies reported on dual therapy (n = 2627; including 62 participants from one study which also reported on monotherapy (Boyle 2014)).
Lumacaftor‐ivacaftor combination therapy
Seven studies have evaluated lumacaftor‐ivacaftor combination therapy (Boyle 2014; PROGRESS 2017; Ratjen 2017; Stahl 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
In one cohort of a Phase 2 study, participants received 200 mg lumacaftor once daily for 14 days, followed by seven days of 200 mg lumacaftor once daily plus either 150 mg (n = 20) or 250 mg (n = 21) of ivacaftor twice daily (day 15 to 21), or placebo (Boyle 2014). In one Phase 2 placebo‐controlled study with 51 participants, children weighing less than 14 kg at screening received 100 mg twice‐daily lumacaftor plus 125 mg twice‐daily ivacaftor, and children weighing at least 14 kg at screening received 150 mg twice‐daily lumacaftor plus 188 mg twice‐daily ivacaftor (Stahl 2021).
In one Phase 3 study, children received either a combination of lumacaftor 200 mg plus ivacaftor 250 mg every 12 hours or placebo for 24 weeks (Ratjen 2017).
Two Phase 3, three‐arm studies (TRAFFIC and TRANSPORT) also compared lumacaftor‐ivacaftor combination therapy to placebo. In these studies, two separate doses of lumacaftor (600 mg once daily and 400 mg twice daily) were combined with twice‐daily 250 mg of ivacaftor. The placebo group received lumacaftor‐matched placebo every 12 hours in combination with ivacaftor‐matched placebo every 12 hours (TRAFFIC 2015; TRANSPORT 2015). A long‐term extension study (96 weeks) randomised those in the placebo groups of the TRAFFIC and TRANSPORT studies to one of the two lumacaftor‐ivacaftor combination doses; those already receiving an active treatment continued with their existing treatment (PROGRESS 2017).
In one Phase 4 placebo‐controlled study with 70 participants, participants received 400 mg twice‐daily lumacaftor plus 250 mg twice‐daily ivacaftor (Wilson 2021).
Tezacaftor‐ivacaftor combination therapy
Nine studies evaluated tezacaftor‐ivacaftor combination therapy (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017).
One Phase 2 study included a dose‐escalation arm, a comparison of various doses of tezacaftor‐ivacaftor in people with the F508del/F508del genotype, and a comparison of tezacaftor‐ivacaftor against ivacaftor alone in people with one F508del variant and one G551D variant (Donaldson 2018).
A further 12‐week Phase 2 study (40 participants homozygous for the F508del variant) had two different arms; the first arm compared 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to a matched placebo, and the second arm compared two 50 mg once‐daily tablets of tezacaftor plus 150 mg twice‐daily ivacaftor to a matched placebo (NCT02070744).
One Phase 3 study compared a combination of 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to a matched placebo for 24 weeks (Taylor‐Cousar 2017).
Five studies (497 participants) compared once‐daily tezacaftor 100 mg plus twice‐daily ivacaftor 150 mg to either a matched placebo or twice‐daily ivacaftor 150 mg (McKone 2021; Munck 2020; NCT02508207; NCT02730208; Schwarz 2021). Two of these studies were Phase 2 studies (NCT02508207; NCT02730208), two were Phase 3 studies (McKone 2021; Munck 2020) and one was a Phase 3b study (Schwarz 2021).
One Phase 3 study (67 participants) randomised participants 4:1 to tezacaftor‐ivacaftor or a blinding group (placebo for homozygous F508del participants and ivacaftor for F508del/residual function participants); doses were weight‐dependent. Participants homozygous for the F508del variant and randomised to the tezacaftor‐ivacaftor arm who weighed less than 40 kg on day 1 received 50 mg once‐daily tezacaftor plus 75 mg twice‐daily ivacaftor, while those weighing at least 40 kg on day 1 received 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor. Participants who were homozygous for the F508del variant and randomised to the blinding group received a matched placebo. Participants in the intervention group who were heterozygous (F508del/residual function) and randomised to tezacaftor‐ivacaftor who weighed less than 40 kg on day 1 received 50 mg once‐daily tezacaftor plus 75 mg twice‐daily ivacaftor and an ivacaftor‐matching placebo in the morning; while those weighing at least 40 kg on day 1 received 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor and an ivacaftor‐matching placebo in the morning. Participants who were heterozygous (F508del/residual function) and randomised to the ivacaftor blinding group also received a weigh‐dependent dose: those weighing less than 40 kg on day 1 received a tezacaftor‐ivacaftor‐matching placebo in the morning and 75 mg twice‐daily ivacaftor, while those weighing at least 40 kg on day 1 received a tezacaftor‐ivacaftor‐matching placebo in the morning plus 150 mg twice‐daily ivacaftor (Davies 2021).
Triple therapy
In total, 11 studies (n = 1872) reported on triple therapy (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
VX‐659‐tezacaftor‐ivacaftor
Four studies evaluated triple therapy of VX‐659‐tezacaftor‐ivacaftor (Davies 2018a; Davies 2018b; NCT03447249; NCT03460990).
A 14‐day Phase 1 study compared a single dose of 120 mg VX‐659 taken twice daily in combination with tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily in individuals with a compound heterozygous genotype of F508del/MF (Davies 2018a).
The subsequent four‐week Phase 2 study compared three different doses of VX‐659 in combination with tezacaftor‐ivacaftor to a single placebo group (n = 10) in participants with genotype of F508del/MF variant (n = 53) as follows: VX‐659 80 mg and tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 11); VX‐659 240 mg and tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 20); VX‐659 400 mg and tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 22). These groups taking one of the previously stated doses of the test intervention regimen had a four‐day washout period taking the tezacaftor‐ivacaftor preparation only (same doses). This study also compared once daily VX‐659 400 mg plus tezacaftor 100 mg plus deutivacaftor (deuterated ivacaftor) 150 mg to placebo in another group of participants with genotype F508del/MF variant (n = 25) for four weeks. In a further arm of the study, 29 participants with the F508del/F508del variant were randomised to either VX‐659 400 mg plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 18) or to placebo plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (this dual therapy combination is currently considered the standard of care in individuals with this genotype) (n = 11). This cohort (n = 29) had a four‐week run‐in period taking the same dose of tezacaftor‐ivacaftor only before starting the triple therapy combination for four weeks. Once the intervention period was over, these participants had a further four‐week washout period of taking the same dose of tezacaftor‐ivacaftor as a dual combination (Davies 2018b).
One Phase 3 study (385 participants with the genotype F508del/MF) compared 240 mg once‐daily VX‐659 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to a matched placebo for 24 weeks (NCT03447249). Another Phase 3 study (116 participants with F508del/F508del) compared 240 mg once‐daily VX‐659 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor for four weeks (NCT03460990).
VX‐440‐tezacaftor‐ivacaftor
One Phase 2 study (73 participants F508del/MF and F508del/F508del) was split into two parts. Participants in the first part had the F508del/MF genotype and participants in the second part had the F508del/F508del genotype. In the first part, there were three interventional cohorts in the initial four‐week phase of the study: participants in one cohort received 200 mg VX‐440 twice daily plus 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily; participants in the second cohort received 200 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor, and finally participants in the third cohort received 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor. Each cohort was compared against a placebo group. In the second part of the study, participants received 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor in a four‐week run‐in period, and then participants in the treatment group received 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor for four weeks, and participants in the control group received placebo matched to VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor for four weeks. Participants in both cohorts then took 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor for four weeks in a washout period (NCT02951182).
VX‐152‐tezacaftor‐ivacaftor
One Phase 2 study (76 participants, F508del/MF and F508del/F508del) was split into two parts. Participants in the first part had the F508del/MF genotype and participants in the second part had the F508del/F508del genotype. In the first part of this study, there were three cohorts in the initial two‐week phase. Participants in one cohort received 100 mg twice‐daily VX‐152 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor; participants in the second cohort each received 200 mg twice‐daily VX‐152 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor; and, finally, participants in the third cohort received 300 mg twice‐daily VX‐152 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor. Each interventional cohort was compared against a placebo group. In the second part of this study, participants were split into four different cohorts. Participants in all four cohorts took part in a run‐in period, and each received 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for four weeks. In the first cohort, participants then received 200 mg twice‐daily VX‐152 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor for two weeks. The results of these participants were compared to the second cohort, who received placebo matched to VX‐152 plus 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for two weeks. Participants in the third cohort received 300 mg VX‐152 twice daily plus 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for four weeks in the treatment period. The results of these participants were compared to the fourth cohort, who received placebo matched to VX‐152 plus 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for four weeks. Participants in all four cohorts then received 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for two weeks in a washout period (NCT02951195).
Elexacaftor‐tezacaftor‐ivacaftor
Five studies examined the triple combination of elexacaftor in combination with tezacaftor and ivacaftor (Barry 2021; Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022). The studies included different doses of this combination, as outlined below.
One Phase 2 study compared three different doses of elexacaftor to placebo in 65 participants with F508del/MF for four weeks, followed by a one‐week washout period of tezacaftor‐ivacaftor or dual placebo (Keating 2018). The intervention doses were as follows:
elexacaftor 50 mg and tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 10);
elexacaftor 100 mg and tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (n = 22); and
elexacaftor 200 mg and tezacaftor 200 mg once daily plus ivacaftor 150 mg twice daily (n = 21).
The same study also compared once‐daily elexacaftor 200 mg plus tezacaftor 100 mg plus deutivacaftor 150 mg to triple placebo in a group of 29 participants with F508del/MF; these participants had no run‐in or washout period. A further group of 29 participants with F508del/F508del had a four‐week run‐in period of once‐daily tezacaftor 100 mg plus ivacaftor 150 mg, followed by the intervention period of once‐daily elexacaftor 200 mg or equivalent placebo while continuing with tezacaftor‐ivacaftor at the same doses; this was followed by a four‐week washout period where all participants took just tezacaftor‐ivacaftor at the previous doses (Keating 2018).
One Phase 3 study compared 200 mg once‐daily elexacaftor plus 100 mg once‐daily tezacaftor and 150 mg twice‐daily ivacaftor (n = 55) versus placebo once daily plus tezacaftor 100 mg once daily and ivacaftor 150 mg twice daily over four weeks in 52 participants with a F508del/F508del genotype (Heijerman 2019). Another Phase 3 study compared 200 mg once‐daily elexacaftor plus 100 mg once‐daily tezacaftor and 150 mg twice‐daily ivacaftor (n = 200) versus triple placebo (n = 203) in participants with a F508del/MF genotype for six months (Middleton 2019). The second variant in these participants was a MF mutation, so the trial can be considered to reflect the impact of the triple therapy on the single F508del variant. It should be considered that some of the MF variants may also have been responsive to triple therapy, but we considered it appropriate to consider this group collectively.
A further Phase 3 study compared 200 mg once‐daily elexacaftor plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to either 150 mg twice‐daily ivacaftor monotherapy or 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor in 271 participants with the genotypes F508del/gating variant and F508del/residual function variant for eight weeks (Barry 2021).
One Phase 3b study (176 participants, F508del/F508del genotype) compared 200 mg once‐daily elexacaftor plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor for 24 weeks (Sutharsan 2022).
Outcomes
All included studies (monotherapy, dual therapy or triple therapy) reported on survival. There were 26 studies reporting QoL, all of which utilised the respiratory domain of the CFQ‐R (Barry 2021; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2017; Donaldson 2018; Heijerman 2019; Horsley 2017; Keating 2018; McKone 2021; Middleton 2019; Munck 2020; NCT02070744; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Lung function using FEV1 was reported in 30 studies (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; Horsley 2017; Keating 2018; McCarty 2002; McKone 2021; Middleton 2019; Munck 2020; NCT02070744; NCT02508207; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Three studies additionally reported LCI (Davies 2021; Ratjen 2017; Stahl 2021).
Reporting of the pre‐specified secondary outcomes in this review varied across studies. All included studies monitored the AEs of therapy, but the manner in which these safety outcomes were analysed and reported varied considerably. In 17 studies, investigators reported outcomes relating to pulmonary exacerbations (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; Munck 2020; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Rubenstein 1998; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015) (please see our definition of exacerbations above (Types of outcome measures)). For monotherapy, two studies stated that exacerbations were physician‐defined (Boyle 2014; Clancy 2012), one study referred to them as 'infective exacerbations' (Donaldson 2017), in two studies it was unclear whether exacerbations were protocol‐ or physician‐defined (Donaldson 2014; Horsley 2017) and in two studies pulmonary exacerbation was not included as an outcome (Rubenstein 1998; Zeitlin 2002). For dual therapy, two studies defined exacerbations as episodes requiring antibiotics or hospitalisation (TRAFFIC 2015; TRANSPORT 2015), for one study, exacerbations were physician‐defined (Boyle 2014) and for four studies, it is unclear whether exacerbations are protocol‐ or physician‐defined (Donaldson 2018; PROGRESS 2017; Ratjen 2017; Taylor‐Cousar 2017). One study defined pulmonary exacerbation "as the treatment with new or changed antibiotics therapy (intravenous, inhaled, oral) for greater than or equal to 4 sinopulmonary signs/symptoms" (Munck 2020). For triple therapy, six trials defined exacerbations as those which were infective in nature or required antibiotics (Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT03447249). One study specifically reported on rates of hospitalisation (Middleton 2019). Changes in sweat chloride, as a marker of CFTR function, were reported by 26 studies (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Heijerman 2019; Horsley 2017; Keating 2018; McKone 2021; Middleton 2019; NCT02070744; NCT02508207; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Ratjen 2017; Rubenstein 1998; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; Zeitlin 2002, Munck 2020). Three studies reported radiological outcomes (NCT02730208; Ratjen 2017; Stahl 2021). Four studies reported microbiological outcomes (NCT02730208; Stahl 2021; Taylor‐Cousar 2017; Zeitlin 2002) and 12 studies reported body mass index (BMI) (Davies 2021; Heijerman 2019; Munck 2020; NCT02070744; NCT03447249; PROGRESS 2017; Ratjen 2017; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
Funding sources
Pharmaceutical companies primarily funded 27 studies (Barry 2021; Davies 2018a; Davies 2018b; Donaldson 2014; Donaldson 2017; Heijerman 2019; Horsley 2017; Keating 2018; McCarty 2002; McKone 2021; Middleton 2019; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Five studies were funded jointly by pharmaceutical companies and other sources (Boyle 2014; Clancy 2012; Davies 2021; Donaldson 2018; Schwarz 2021). Two studies were not funded by pharmaceutical companies at all: one was funded by the Cystic Fibrosis Foundation (CFF) (Zeitlin 2002), and one jointly by the CFF and the NIH (Rubenstein 1998).
Further information about the studies is presented in the tables (Characteristics of included studies).
Excluded studies
We excluded a total of 35 studies. Of these, nine studies were of cross‐over design (Berkers 2014; Berkers 2021; Leonard 2012; Marsh 2022; NCT00945347; NCT01899105; NCT05535959; Nick 2014; Rowe 2017). In 15 studies, participants were not randomised to different study arms (Chilvers 2021; NCT03447262; NCT03525574; NCT03537651; NCT03601637; NCT03633526; NCT03691779; NCT04043806; NCT04058366; NCT04183790; NCT04235140; NCT04362761; NCT04537793; NCT04545515; Rubenstein 2006). Two studies comprised different treatment arms, but participants were not randomised (Chadwick 1998; Krivec 2021). One study was an expanded‐access study and not an RCT (NCT04058210). One study was a pre‐clinical laboratory study (Ziady 2015). One study was of general gene therapy and not a mutation‐specific therapy (Sumner 2014). In two studies, the intervention was not considered to be a corrector for type II variants (Chmiel 2021; Drevinek 2017). One study did not look at any corrector as an intervention (ISRCTN14081521). Two studies have been terminated with no data available (either published or on the trials database) (Meijer 2016; NCT02323100). In the final study, the initial phases were not randomised and also involved healthy participants; phase 4 of the trial is due to be randomised and enrol pwCF, however the trial has been suspended for "business reasons" (NCT03756922).
Studies awaiting classification
There are 14 studies awaiting classification due to a lack of information allowing us to make a decision on their inclusion or exclusion (ALBATROSS; Downey 2019; Downey 2020; FLAMINGO; Hunt 2017; NCT03768089; Uluer 2023a; Uluer 2023b; NCT03969888; NCT04353817; PELICAN; Rio‐CF; Taylor‐Cousar 2019; Wainwright 2019). For further details on each of these studies, please see the table Characteristics of studies awaiting classification.
Five studies describe monotherapy exclusively (ALBATROSS; FLAMINGO; Hunt 2017; Rio‐CF; Uluer 2023a), one study describes both monotherapy and dual therapies (NCT03969888), one study exclusively describes dual therapy (Wainwright 2019), one study describes both dual and triple therapy combinations (Downey 2019) and six studies describe triple therapy exclusively (Downey 2020; NCT03768089; NCT04353817; PELICAN; Taylor‐Cousar 2019; Uluer 2023b).
All 14 are RCTs of parallel design with either two or three or four arms. The duration of studies varied from 14 days (Downey 2019) to 72 weeks (Wainwright 2019). Participants ranged in age, with 12 studies recruiting participants aged 18 years and over (ALBATROSS; Downey 2019; Downey 2020; FLAMINGO; Hunt 2017; NCT03768089; NCT03969888; PELICAN; Rio‐CF; Taylor‐Cousar 2019; Uluer 2023a; Uluer 2023b), one study recruiting participants aged 12 years and over (Wainwright 2019) and one study recruiting children aged six to 11 years of age (NCT04353817). The genotype of participants also varied with eight studies recruiting participants with F508del/F508del (Downey 2019; Downey 2020; FLAMINGO; Hunt 2017; NCT03969888; PELICAN; Rio‐CF; Wainwright 2019), two studies recruiting participants with F508del/MF (NCT03768089; NCT04353817), one study recruiting participants with both the genotypes F508del/F508del and F508del/MF (Uluer 2023b), one study recruiting participants with F508del/class III variant (ALBATROSS), and one study recruiting participants with at least one copy of G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P or G1349D (Uluer 2023a). One study did not state the genotype of participants (Taylor‐Cousar 2019).
There are five placebo‐controlled monotherapy studies evaluating GLPG2222 (ALBATROSS; FLAMINGO), riociguat (Rio‐CF), sildenafil (Hunt 2017) and deutivacaftor (Uluer 2023a). One three‐arm study compares ABBV‐3067 versus ABBV‐3067 plus ABBV‐2222 versus placebo and so falls into the monotherapy and dual therapy categories (NCT03969888). Two further studies describe dual therapies: one two‐arm study compares tezacaftor‐ivacaftor versus placebo (Wainwright 2019), while a three‐arm study compares a dual therapy of posenacaftor (PTI‐801) with dirocaftor (PTI‐808) versus a triple therapy of posenacaftor plus dirocaftor plus nesolicaftor (PTI‐428) versus placebo, thus falling into both the dual therapy and triple therapy comparisons (Downey 2019). Triple therapy is also described in a further six studies: dirocaftor plus posenacaftor plus nesolicaftor versus placebo (Downey 2020); nesolicaftor plus tezacaftor‐ivacaftor versus placebo plus tezacaftor‐ivacaftor (Taylor‐Cousar 2019); GLPG‐2737 plus lumacaftor‐ivacaftor versus placebo plus lumacaftor‐ivacaftor (PELICAN); elexacaftor‐tezacaftor‐ivacaftor versus placebo (NCT04353817); vanzacaftor‐tezacaftor‐ivacaftor versus placebo (NCT03768089); and vanzacaftor plus tezacaftor and deutivacaftor versus placebo (Uluer 2023b).
Ongoing studies
We have listed eight studies as ongoing (Jain 2018; NCT02589236; NCT02718495; NCT03258424; NCT04853368; NCT05033080; NCT05076149; NCT05274269).
Monotherapy
Three studies are currently evaluating different monotherapy correctors (Jain 2018; NCT02718495; NCT03258424).
Two placebo‐controlled studies are evaluating nesolicaftor, a particular type of CFTR corrector called an amplifier, which augments the actions of other CFTR modulators (NCT02718495; NCT03258424). The first study expects to recruit 56 adults with CF (mutation not specified) and is being conducted at 29 centres across North America and Europe (NCT02718495). The 28‐day intervention is comparing ascending dose treatment (doses not stated) and outcome measures include AEs, lung function, pharmacokinetics, sweat chloride, weight and QoL (NCT02718495). The second study is a Phase 1 RCT being run at two centres in the UK (NCT03258424). It plans to recruit 16 adults with CF who are already receiving ivacaftor for 14 days of treatment, but dose levels of nesolicaftor are not stated. Outcome measures include AEs, pharmacokinetic outcomes, sweat chloride, lung function and weight (NCT03258424).
The final study is evaluating posenacaftor alone and also in combination with nesolicaftor compared to placebo in a Phase 1 study in adults with CF (homozygous F508del in three cohorts and heterozygous F508del in one cohort) who have a baseline FEV1 of 40% to 90% and who are currently receiving lumacaftor‐ivacaftor as background therapy (Jain 2018). It is a multicentre UK‐based study with an estimated enrolment of 32 participants and a treatment duration of 14 days with a follow‐up visit at 21 days. There are four arms, two 14‐day arms comparing different doses of combined dirocaftor and posenacaftor to placebo, one 14‐day arm comparing combined dirocaftor, posenacaftor and nesolicaftor to placebo and one arm comparing dirocaftor, posenacaftor and nesolicaftor to placebo for seven days followed by dirocaftor and posenacaftor versus placebo for a further seven days (no washout period). Outcomes include AEs, pharmacokinetics, lung function, sweat chloride, nutritional outcomes and QoL (Jain 2018).
Triple therapy
Five ongoing studies are evaluating triple therapies (NCT02589236; NCT04853368; NCT05033080; NCT05076149; NCT05274269).
One ongoing Phase 2, placebo‐controlled, parallel RCT (n = 138) is evaluating the efficacy of adding cavosonstat to pre‐existing lumacaftor‐ivacaftor therapy for 12 weeks in adults with the genotype F508del/F508del (NCT02589236). There are three arms (the doses of lumacaftor and ivacaftor are not stated in any arm), lumacaftor with ivacaftor and cavosonstat 200 mg twice daily versus lumacaftor with ivacaftor and cavosonstat 400 mg twice daily versus lumacaftor with ivacaftor and matched placebo. The primary outcome is the absolute change from baseline in FEV1 % predicted. Secondary outcomes are the relative change from baseline in FEV1 % predicted, the absolute change from baseline in sweat chloride, the absolute change from baseline in CFQ‐R respiratory domain, the absolute change from baseline in BMI, the absolute change from baseline in "patient global impression of change (PGIC)", the incidence of treatment‐emergent AEs, pharmacological measures and number of pulmonary exacerbations (NCT02589236).
One ongoing Phase 2 RCT (n = 90) is evaluating the oral treatments galicaftor plus navocaftor plus ABBC‐119 or galicaftor plus navocaftor plus ABBV‐576 combination therapies in four arms: (NCT04853368):
Arm 1: participants with the genotype F508del/508del receive oral capsules of galicaftor plus navocaftor dual combination for 28 days followed by galicaftor plus navocaftor plus ABBV‐119 triple combination for 28 days.
Arm 2: participants with the genotype F508del/MF receive galicaftor plus navocaftor plus ABBV‐119 triple combination therapy for 28 days.
Arm 3: participants with the genotype F508del/MF receive placebo for 28 days.
Arm 4: participants with the genotype F508del/508del or F508del/MF receive galicaftor plus navocaftor plus ABBV‐576 triple combination therapy for 28 days.
The exact doses for each arm are not available from the trial registry entry (clinicaltrials.gov). Participants in Arm 4 must be receiving stable elexacaftor plus tezacaftor plus ivacaftor treatment, while participants in Arms 1, 2 and 3 must not be receiving this treatment. The primary outcomes for Arms 1 and 2 are absolute change from baseline in FEV1 % predicted up to 29 days, and the primary outcome in Arm 3 is absolute change in sweat chloride up to 29 days. Secondary outcomes for Arms 1 and 2 are absolute change from baseline in sweat chloride, absolute change from baseline in FVC, absolute change from baseline in forced expiratory flow (FEF)25-75, relative changes from baseline in FEV1 % predicted, relative changes from baseline in FVC, relative changes from baseline in FEF25-75 and absolute change in the CFQ‐R respiratory domain score. The secondary outcome measure for cohort 3 is absolute change from baseline in FEV1 % predicted (NCT04853368).
One ongoing Phase 3, parallel RCT is evaluating vanzacaftor in triple combination therapy over 24 weeks (NCT05033080). Participants are aged 12 years and older with the genotype F508del/MF. Participants receive vanzacaftor plus tezacaftor plus deutivacaftor and participants or elexacaftor‐tezacaftor‐ivacaftor. The primary outcome is the absolute change from baseline in FEV1 % predicted; secondary outcomes are absolute change from baseline in sweat chloride, the proportion of participants with sweat chloride less than 60 mmol/L and proportion of participants with sweat chloride less than 30 mmol/L (NCT05033080).
A further 24‐week study is a Phase 3, double‐blind RCT evaluating vanzacaftor triple combination therapy in participants aged 12 years and older (NCT05076149). Participants must have one of the following genotypes: F508del/F508del, heterozygous for F508del and a gating variant, F508del and a residual function variant, at least one other triple combination responsive CFTR gene variant identified as responsive to elexacaftor‐tezacaftor‐ivacaftor and no F508del variant. Participants receive either vanzacaftor‐tezacaftor‐deutivacaftor or elexacaftor‐tezacaftor‐ivacaftor. The primary outcome is absolute change from baseline in FEV1 % predicted; secondary outcomes are absolute change from baseline in sweat chloride, the proportion of participants with sweat chloride less than 60 mmol/L and the proportion of participants with sweat chloride less than 30 mmol/L (NCT05076149).
The final ongoing study is a Phase 3, double‐blind, placebo‐controlled RCT of parallel design evaluating elexacaftor plus tezacaftor plus ivacaftor in participants with CF aged six years and older who have a non‐F508del elexacaftor‐tezacaftor‐ivacaftor responsive CFTR variant. Participants receive elexacaftor‐tezacaftor‐ivacaftor in the morning and ivacaftor in the evening or a matched placebo at both time points. Outcomes are measured at 24 weeks, except AEs, which are measured at 28 weeks. The primary outcome measure of the study is the absolute change in FEV1 % predicted; secondary outcome measures are the absolute change in sweat chloride, absolute change in CFQ‐R respiratory domain score, absolute change in BMI, absolute change in weight, number of pulmonary exacerbations and the number of participants with AEs and serious AEs (NCT05274269).
Risk of bias in included studies
We have summarised our risk of bias judgements in the figures (Figure 3; Figure 4).
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
4.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Sequence generation
We judged 24 studies to have a low risk of bias for sequence generation (Barry 2021; Boyle 2014; Davies 2018a; Davies 2018b; Davies 2021; Heijerman 2019; Keating 2018; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Of these, one study used a computer‐generated randomisation schedule developed by an independent party (Boyle 2014), and the others randomised participants via an interactive web response system (Barry 2021; Davies 2018a; Davies 2018b; Davies 2021; Heijerman 2019; Keating 2018; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). As none of the remaining 10 included studies reported details of random sequence generation, we have judged the risk of bias as unclear (Clancy 2012; Donaldson 2014; Donaldson 2017; Donaldson 2018; Horsley 2017; McCarty 2002; Munck 2020; NCT02070744; Rubenstein 1998; Zeitlin 2002).
Allocation concealment
We judged 24 studies to have a low risk of bias due to allocation concealment (Barry 2021; Boyle 2014; Davies 2018a; Davies 2018b; Davies 2021; Heijerman 2019; Keating 2018; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). In the Phase 2 lumacaftor‐ivacaftor study, site pharmacists dispensed drugs on the basis of an interactive voice response system, making it unlikely that participants or study personnel would have been aware of group assignments prior to recruitment into the study (Boyle 2014). The remaining studies also employed an interactive web response system to allocate participants to treatment groups (Barry 2021; Davies 2018a; Davies 2018b; Davies 2021; Heijerman 2019; Keating 2018; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Methods to conceal group allocation were not reported by the remaining 10 studies, who also failed to report on random sequence generation, so we judged these as having an unclear risk of bias (Clancy 2012; Donaldson 2014; Donaldson 2017; Donaldson 2018; Horsley 2017; McCarty 2002; Munck 2020; NCT02070744; Rubenstein 1998; Zeitlin 2002).
Blinding
We judged 27 studies to have a low risk of performance bias (Barry 2021; Boyle 2014; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; Keating 2018; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). We judged six studies to have an unclear risk of performance bias (Clancy 2012; Horsley 2017; McCarty 2002; Munck 2020; NCT02070744; Rubenstein 1998) and we judged one study to have a high risk of performance bias (Zeitlin 2002).
With regards to detection bias, we judged 23 studies to have a low risk (Barry 2021; Boyle 2014; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; McKone 2021; Middleton 2019; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021) and 11 studies to have an unclear risk (Clancy 2012; Davies 2018a; Davies 2018b; Horsley 2017; Keating 2018; McCarty 2002; Munck 2020; NCT02070744; Ratjen 2017; Rubenstein 1998; Zeitlin 2002).
Two monotherapy studies had a low risk of both performance and detection bias (Boyle 2014; Donaldson 2014). In the Boyle study, drug doses were prepared by an independent unmasked pharmacist and dispensed by site pharmacists who were masked to treatment assignment. Site investigators and the study sponsor were also masked to treatment assignment and to sweat chloride levels ‐ data that could have potentially disclosed treatment assignment. Participant blinding was maintained by placebo, which was matched to intervention by the quantity of tablets and by size, colour, coating and packaging (Boyle 2014). In the earlier Donaldson study, participants, caregivers, investigators and outcome assessors were double‐blinded via intravenous administration of placebo (saline) using the same volume as the active drug groups (Donaldson 2014).
Four studies had an unclear risk of performance and detection bias (Clancy 2012; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002). In the pilot 4PBA, CPX, FDL169 and the lumacaftor monotherapy studies, there was insufficient information about how participant, study personnel or outcome assessor blinding was maintained and so we judged these four studies to have an unclear risk of performance and detection bias (Clancy 2012; Horsley 2017; McCarty 2002; Rubenstein 1998).
Participants from the three intervention groups (20 g, 30 g and 40 g) in the Phase 2 4PBA study had different dosing schedules and were given a different number of tablets. Therefore we judged this study to have a high risk of performance bias. Also in this study, there were insufficient data on blinding of outcome assessors and we therefore judged it to have an unclear risk of detection bias (Zeitlin 2002).
We judged 12 dual therapy studies to have a low risk of both performance and detection bias. In two Phase 3 lumacaftor‐ivacaftor studies and the extension study, the participants and study team remained blinded to the treatment assignments and the placebo was matched in appearance and packaging to the active intervention. The online protocol further stated that all site personnel, including the investigator, the site monitor and the study team would remain blinded to treatment group (PROGRESS 2017; TRAFFIC 2015; TRANSPORT 2015). In the Phase 4 lumacaftor‐ivacaftor study, the participants and study team were blinded in the trial, and there was a clear statement regarding the exceptions to the blinding procedure in the protocol (Wilson 2021). In one paediatric lumacaftor‐ivacaftor study, double‐blinding was achieved by using placebo tablets visually identical to the test product and all site personnel were also blinded to allocation (Stahl 2021). Six tezacaftor‐ivacaftor dual combination studies made use of matched placebo and employed double‐blinding to reduce performance bias (Davies 2021; Donaldson 2018; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017). In one tezacaftor‐ivacaftor study, in which an active control was used, all participants and the study team were blinded (McKone 2021).
In the earlier paediatric lumacaftor‐ivacaftor study, there was a low risk of performance bias since double‐blinding was achieved by using placebo tablets visually identical to the test product; however, there was an unclear risk of detection bias since the blinding of outcome assessors was not discussed (Ratjen 2017).
Two tezacaftor‐ivacaftor studies provided insufficient information about blinding of participants, study personnel or outcome assessors, so we judged these studies to have an unclear risk of performance and detection bias (Munck 2020; NCT02070744).
We judged six triple therapy studies to have low risks of both performance and detection bias since all participants, study personnel and outcome assessors were blinded to the treatment codes, and the protocols clearly identified when individuals could be unblinded (Barry 2021; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
We judged five triple combination studies to have a low risk of performance bias, as the participants and site personnel were blinded to allocation. However, we considered them to have an unclear risk of detection bias, as although all authors were blinded to allocation, no mention is made of other outcome assessors (e.g. clinicians who were not authors, but were involved in seeing participants and measuring outcomes of interest) and whether there was a possibility of them knowing the allocated intervention (Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019).
Incomplete outcome data
We judged 20 studies to have a low risk of bias due to incomplete outcome data (Barry 2021; Davies 2018a; Davies 2018b; Donaldson 2014; Donaldson 2018; Heijerman 2019; Horsley 2017; Keating 2018; McCarty 2002; Middleton 2019; NCT02730208; NCT02951182; PROGRESS 2017; Ratjen 2017; Rubenstein 1998; Schwarz 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015).
We judged two studies to have an unclear risk of attrition bias (Boyle 2014; Zeitlin 2002). In the Phase 2 lumacaftor‐ivacaftor study, one out of 62 participants (1.6%) withdrew due to an AE, demonstrating a low withdrawal rate. However, in the analysis the investigators only included participants for whom data were available. Although these participants were excluded because of insufficient data rather than for unfavourable characteristics, e.g. AEs, we judged this study as having an unclear risk of attrition bias because we are not sure how these exclusions would have affected the balance between groups in baseline characteristics (Boyle 2014). In the Phase 2 4PBA study, all 19 randomised participants completed the final study visit, but risk of attrition bias was unclear because there was no report of how many of these participants were included in the analysis (Zeitlin 2002). We approached the primary author to clarify this, but did not receive any additional information.
We judged 12 studies in total to have a high risk of attrition bias (Clancy 2012; Davies 2021; Donaldson 2017; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02951195; NCT03447249; NCT03460990; Stahl 2021; Wilson 2021). In the study of lumacaftor monotherapy, although there was a low withdrawal rate, with only four out of 89 (5%) participants withdrawing from the study due to AEs, data for a number of outcomes were excluded from the analysis (Clancy 2012). A total of 42 participants were excluded from reports of AEs; nine participants were excluded from reports on change from baseline in sweat chloride concentration (see figure 1b in the full‐text article) and four participants were excluded from the information on CFQ‐R domain scores. Our concerns were due firstly to the high level of excluded participant data and secondly to the lack of reasons for the exclusion of these participant data. The study's lead investigator was approached for clarification, but we have received no response to date (Clancy 2012). In the cavosonstat study, two out of 51 participants are unaccounted for in the final analysis (Donaldson 2017). We judged the Phase 4 lumacaftor‐ivacaftor trial to have a high risk of attrition bias as a number of outcomes (including outcomes relevant to our review) reported data for fewer participants than the number assigned to that group (Wilson 2021). For example, only 30 out of 34 participants in the intervention group were analysed for both absolute and relative change from baseline in FEV1 % predicted at week 24 with no explanation given for the missing participants.
In the paediatric lumacaftor‐ivacaftor study for the outcome absolute change from baseline in magnetic resonance imaging (MRI) global chest score at week 48, investigators only analysed 32 out of 35 participants in the intervention group, and 15 out of 16 participants in the placebo group. No explanation was given for these missing participants (Stahl 2021).
We judged five tezacaftor‐ivacaftor dual therapy studies to have a high risk of attrition bias. Davies 2021 based the mean, SD and P values on fewer participants than had been randomised to each group, with no explanation offered as to why this was the case. For example, the P value for the change in sweat chloride was based on 48 participants, with no explanation offered as to why this number was lower than the 54 participants in the intervention group (Davies 2021). There were also fewer participants analysed across a range of outcomes in the other tezacaftor‐ivacaftor studies, with no further explanations offered (McKone 2021; Munck 2020; NCT02070744; NCT02508207).
We judged one Phase 2 VX‐152 study to have a high risk of attrition bias due to the fact that 4 out of 46 participants enrolled in Part 2 of the study discontinued during the run‐in period, and were not randomised to the treatment period, without any further explanation given (NCT02951195).
We judged two Phase 3 VX‐659 triple combination therapy studies to have a high risk of attrition bias. In the first study, five participants who were included in the run‐in period were not dosed in the treatment period, but no further information or explanation was provided (NCT03460990). In the second study, it is unclear why three of the participants enrolled in the study were not dosed in the treatment period; furthermore, five participants in the placebo group did not complete the regimen, with the reason for three of these listed as "other" with no further explanation. It was also unclear why the "safety set" for both the placebo and treatment groups were 189 and 193 participants respectively, though 190 and 192 initially started each regimen (NCT03447249).
Selective reporting
Where study protocols were not available, or there were missing outcome data, we approached the studies' primary authors for additional information.
We judged nine studies to have a low risk of reporting bias (Boyle 2014; Donaldson 2017; Donaldson 2018; Heijerman 2019; Middleton 2019; PROGRESS 2017; McCarty 2002; Rubenstein 1998; Stahl 2021). For the Phase 2 lumacaftor‐ivacaftor study, the protocol was not available, but outcomes were presented on the NIH trials registry and we did not identify any missing outcomes in the publication (Boyle 2014). For the pilot 4PBA and CPX studies, protocols were not available and planned outcomes were not listed in ongoing online trials databases (McCarty 2002; Rubenstein 1998). So, we compared the outcomes reported in the 'Methods' sections to the outcomes reported in the 'Results' sections of the publications and again did not identify any missing outcomes (McCarty 2002; Rubenstein 1998). For the extension study of TRAFFIC and TRANSPORT, we compared the list of outcomes provided on the NIH trials registry to the results reported in the published paper; all listed outcomes were reported (PROGRESS 2017). Two Phase 3 triple combination studies and the paediatric lumacaftor‐ivacaftor study stated and reported outcomes in both the protocol and results (Heijerman 2019; Middleton 2019; Stahl 2021).
In total, we judged 19 studies to have an unclear risk of reporting bias (Barry 2021; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Horsley 2017; Keating 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Schwarz 2021; Sutharsan 2022; Wilson 2021). For two monotherapy studies only limited results were available from the NIH trials registry and it was unclear if all relevant information has been made available (Donaldson 2014; Horsley 2017). Seven studies were of dual therapy and each stated in their methods that they would measure 12‐lead electrocardiogram (ECG) or vital signs, or both; however, none of the studies report data or information for these outcomes in their results or supplements (Davies 2021; McKone 2021; Munck 2020; NCT02508207; NCT02730208; Schwarz 2021; Wilson 2021). We have been unable to access the trial protocol for one dual therapy study, however we have contacted the trial authors for further information (NCT02070744). Nine studies were of triple combination therapy and each stated in their methods that they would measure 12‐lead ECG and vital signs; however, none of the studies report data or information for these outcomes in their results or supplements (Barry 2021; Davies 2018a; Davies 2018b; Keating 2018; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
We judged six studies to have a high risk of bias from selective outcome reporting (Clancy 2012; Ratjen 2017; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Zeitlin 2002). The protocol for the lumacaftor study was not available, but the planned outcomes were listed on the National Institutes for Health (NIH) trials registry; comparing these outcomes to those reported in the 'Results' section of the published paper, we ascertained that no data were reported for FEF25-75% or FVC at 28 days (Clancy 2012). The study protocol for the Phase 2 4PBA study was not available and planned outcomes were not listed in ongoing online trials databases (Zeitlin 2002). We compared the outcomes reported in the 'Methods' section of the paper to the outcomes reported in the 'Results' section and identified that data were not reported for the change from baseline in FEV1 or microbiology scores at day seven (Zeitlin 2002).
In the two Phase 3 lumacaftor‐ivacaftor studies, pre‐specified outcomes were reported on the NIH trials registry (TRAFFIC 2015; TRANSPORT 2015). In both studies, data for the absolute change from baseline in FEV1 and relative change from baseline in FEV1 were combined at 16 and 24 weeks. This was not pre‐specified and the primary author was contacted for clarification. Furthermore, we needed to extrapolate some results from graphical figures and some additional data for outcomes not reported in the final paper were only reported in ClinicalTrials.gov. Also, the investigators state that they measured FVC (which was not listed as an endpoint) and do not report this in the joint paper (TRAFFIC 2015; TRANSPORT 2015).
In the paediatric lumacaftor‐ivacaftor combination study, several outcomes, including LCI5.0, weight, height and time to first pulmonary exacerbation, which were listed in the methods of the full publication and also on the ClinicalTrials.gov entry for this study, were not reported in the 'Results' section of the paper (Ratjen 2017).
In the tezacaftor‐ivacaftor combination study, a number of outcomes were recorded according to the study protocol, but were not reported in the published paper (Taylor‐Cousar 2017). These outcomes were the CF respiratory symptom diary, duration of daily physical activity (number of minutes), the Pittsburgh Sleep Quality Index (PSQI), SF‐12 health survey, sputum microbiology, the time to first exacerbation and the number of days with an exacerbation, the time to first hospitalisation and the number of days hospitalised with exacerbation, the number of exacerbations requiring intravenous (IV) therapy, the time to the first IV therapy and the number of days on IV therapy.
Other potential sources of bias
We judged there to be a low risk of other bias due to no significant difference between baseline characteristics in six studies (PROGRESS 2017; Rubenstein 1998; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Zeitlin 2002) and due to well‐matched baseline characteristics in a further nine studies (Boyle 2014; Clancy 2012; Donaldson 2014; Ratjen 2017; Davies 2018a; Davies 2018b; Keating 2018; Heijerman 2019; McKone 2021; Middleton 2019; Wilson 2021). Furthermore, in both the TRAFFIC and TRANSPORT studies, adherence to treatment was high with similar compliance rates across the different treatment groups (TRAFFIC 2015; TRANSPORT 2015).
In 16 studies, there was insufficient detail about baseline characteristics or an apparent imbalance in baseline characteristics, leading to an unclear risk of bias (Barry 2021; Davies 2021; Donaldson 2017; Donaldson 2018; Horsley 2017; McCarty 2002; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT03447249; NCT03460990; Schwarz 2021; Stahl 2021; Sutharsan 2022).
We judged there to be a high risk of other bias in one study as results from four participants with an FEV1 below 40% were analysed, despite the inclusion criteria of the study stating that participants must have an FEV1 of greater than or equal to 40% and less than or equal to 90% of predicted normal for age, sex and height at the screening visit (NCT02951195).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11
As described above, we identified three types of intervention relevant for this review. The first group of studies examined single agents (monotherapy) that aim to correct the F508del trafficking defect (commonly referred to as "correctors"). The second and third groups of studies examined a combination of various correctors with ivacaftor (a drug known to potentiate the function of the CFTR in the membrane). As these interventions have different potential mechanisms of action, we present the results separately for 'Monotherapy compared to control', 'Dual therapy (correctors plus potentiators) compared to control' and 'Triple therapy (correctors plus potentiators) compared to control'. Results are summarised for all doses reported separately and for treatment doses combined where appropriate. In the summary of findings tables, the certainty of the evidence has been graded for pre‐defined outcomes (see above) and definitions of these gradings provided (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11).
Monotherapy compared to control
Eight studies with 344 participants contributed to this comparison (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
Two studies (n = 37) compared 4PBA to placebo (Rubenstein 1998; Zeitlin 2002), one study (n = 66) compared N6022 to placebo (Donaldson 2014), one study (n = 37) compared CPX to placebo (McCarty 2002) and two studies (n = 151) compared varying doses of lumacaftor alone to placebo (Boyle 2014; Clancy 2012). One study (n = 51) compared cavosonstat 200 mg (twice daily) to placebo; we only present the 200 mg dose comparison (n = 26) from this early‐phase study as this is the only dose that is being studied further and other doses are not relevant to current clinical practice (Donaldson 2017). Participants in one study (n = 62) received lumacaftor monotherapy for 14 days followed by combination therapy with ivacaftor for seven days, therefore this study contributes to both comparisons in this review (Boyle 2014). One study (n = 27) compared FDL169 at doses of 400 mg (n = 6), 600 mg (n = 6) and 800 mg (n = 8) to placebo (n = 7). All different dose levels were compared to the same placebo group of seven participants (Horsley 2017).
With regards to pulmonary exacerbations, please see our definition above (Types of outcome measures). For monotherapy, two studies stated that exacerbations were physician‐defined (Boyle 2014; Clancy 2012), one study referred to them as 'infective exacerbations' (Donaldson 2017), in two studies it was unclear whether exacerbations were protocol‐ or physician‐defined (Donaldson 2014; Horsley 2017) and in two studies pulmonary exacerbation was not included as an outcome (Rubenstein 1998; Zeitlin 2002).
Important results for the drugs lumacaftor and cavosonstat within this comparison are summarised in the tables (Table 1; Table 2). We have assessed the following outcomes using the GRADE criteria in each of the tables and indicated our findings in the relevant text below.
survival;
QoL (total score);
QoL (respiratory domain);
FEV1 % predicted (relative and absolute change);
AEs; and
time to first pulmonary exacerbation.
For the comparison of lumacaftor versus placebo, we judged the certainty of the evidence to be of moderate to very low certainty; we downgraded the evidence due to serious concerns over risk of bias, due to indirectness related to the design of the studies and due to imprecision where few events occurred and CIs around the result were very wide (Table 1). For the comparison of cavosonstat versus placebo, we judged the certainty of the evidence to be very low; we downgraded the evidence due to concerns over risk of bias, due to indirectness as the results are not applicable to children and due to imprecision as only a single study with a small sample size contributed evidence for some outcomes so the CIs around the result were wide (Table 2).
We have not presented other monotherapy treatments in the summary of findings tables as interventions have not been taken forward on larger, more representative populations in Phase 3 studies.
Primary outcomes
1. Survival
No deaths were reported during any of the included studies (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
2. QoL
a. Total QoL score
Data for this outcome were not reported by any study (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
b. Different sub‐domains
i. Immediate term (up to and including one month)
Lumacaftor versus placebo
The study by Clancy (n = 89) reported on the change from baseline scores for all CFQ‐R domains at 28 days (Table 14). We have presented these absolute change from baseline scores as we were unable to calculate the relative change from baseline in CFQ‐R scores since baseline CFQ‐R scores were not reported. Furthermore, no SDs or CIs were reported to allow calculation of SDs for entry into the analysis (Clancy 2012).
3. Change from baseline CFQ‐R domain scores at 28 days (Clancy 2012).
| Lumacaftor | Placebo | ||||
| Domain | 25 mg (n = 17) | 50 mg (n = 17) | 100 mg (n = 16) | 200 mg (n =18) | (n = 17) |
| Body | ‐0.21 | ‐1.63 | 2.61 | 0.06 | ‐1.34 |
| Digestion | 2.28 | ‐0.72 | 0.25 | 2.58 | 4.62 |
| Eating | ‐3.66 | ‐7.27* | 3.24 | ‐2.58 | 2.11 |
| Emotion | ‐3.22 | ‐1.36 | 3.49 | ‐2.62 | 4.86 |
| Health perceptions | ‐2.84 | ‐6.97* | ‐0.44 | ‐1.9 | 5.03 |
| Physical | ‐5.97 | ‐7.38* | ‐3.46 | ‐0.98 | 1.23 |
| Respiratory | ‐5.22 | ‐6.32* | ‐1.29 | 2.22 | 4.53 |
| Role | ‐5.94* | ‐4.6 | 1.1 | ‐6.53* | 2.21 |
| Social | 0 | ‐1.01 | 0.47 | ‐2.64 | ‐0.55 |
| Treatment burden | 4.19 | ‐5.96* | 1.42 | ‐0.68 | 2.46 |
| Vitality | ‐4.65 | ‐7.23* | ‐1.52 | 0.73 | ‐2.18 |
| Weight | 5.41 | 2.18 | 8.83 | ‐4.19 | 0.3 |
*Significant results versus placebo are highlighted by stars.
Participants in the 25 mg group reported significantly lower CFQ‐R scores for the role domain (MD ‐8.15) and respiratory domain (MD ‐9.75) compared to participants in the placebo group. Participants in the 50 mg lumacaftor group reported significantly lower CFQ‐R scores for the eating domain (MD ‐9.4), health perceptions domain (MD ‐12.0), respiratory domain (MD ‐10.85) and treatment burden domain (MD ‐8.42) compared to participants assigned to placebo. Participants in the 200 mg group reported significantly lower CFQ‐R scores for the role domain (P < 0.05) compared to participants in the placebo group (Clancy 2012).
Cavosonstat versus placebo
Donaldson 2017 (n = 51) also reported data for both the respiratory and eating domains of the CFQ‐R at 28 days, but neither result showed any difference between cavosonstat and placebo groups at up to one month (Analysis 2.1; Analysis 2.2).
2.1. Analysis.

Comparison 2: Cavosonstat (N91115) (200 mg twice daily) versus placebo, Outcome 1: CFQR respiratory domain: absolute change from baseline
2.2. Analysis.

Comparison 2: Cavosonstat (N91115) (200 mg twice daily) versus placebo, Outcome 2: CFQR eating domain: absolute change from baseline
FDL169 versus placebo
Horsley reported the change from baseline at up to one month for the CFQ‐R respiratory domain (Horsley 2017); this favoured the 400 mg group (n = 6) compared to placebo (n = 7), MD 5.09 (95% CI ‐2.72 to 12.90) (Analysis 4.1); there was no difference between the 600 mg group (n = 6) and placebo, MD ‐4.33 (95% CI ‐12.01 to 3.35) (Analysis 5.1); and favoured the 800 mg group (n = 8) over placebo, MD 8.84 (95% CI 1.40 to 16.28) (Analysis 6.1)
4.1. Analysis.

Comparison 4: FDL169 (400 mg three times daily) versus placebo, Outcome 1: Mean change in CFQ‐R respiratory domain
5.1. Analysis.

Comparison 5: FDL169 (600 mg three times daily) versus placebo, Outcome 1: Mean change in CFQ‐R respiratory domain
6.1. Analysis.

Comparison 6: FDL169 (800 mg three times daily) versus placebo, Outcome 1: Mean change in CFQ‐R respiratory domain
ii. Short term (over one month and up to and including six months)
Data for this outcome were not reported by any study (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
3. Physiological measures of lung function
a. FEV1 (relative change from baseline)
i. Immediate term (up to and including one month)
Lumacaftor versus placebo
The study by Clancy (n = 89) reported the mean relative change from baseline in FEV1 % predicted after 28 days of treatment with escalating doses of lumacaftor, but did not present the corresponding SDs, precluding analysis (Clancy 2012). No differences were reported between the placebo group and the different lumacaftor dose groups: 25 mg, MD ‐2.53% predicted; 50 mg, MD ‐2.22% predicted; 100 mg, MD 0.25% predicted; and 200 mg, MD 0.40% predicted. No SDs or CIs were reported to allow calculation of SDs for entry into the analysis (Clancy 2012).
Cavosonstat versus placebo
Donaldson (n = 51) presents data for cavosonstat versus placebo pictorially in the graph (supplementary tables), but overlapping SD lines render these data difficult to extract. The paper reports that no treatment‐related changes in FEV1 were seen with cavosonstat compared to placebo at up to one month (Donaldson 2017).
N6022 versus placebo
The study by Donaldson (n = 66) reported the mean relative change from baseline in FEV1 % predicted after seven days of treatment with sequential ascending doses of N6022 (5 mg, 10 mg, 20 mg or 40 mg per day) (Donaldson 2014). The paper reports no significant differences between the placebo group and any of the N6022 dose groups at up to one month (Analysis 3.1).
3.1. Analysis.

Comparison 3: N6022 versus placebo, Outcome 1: FEV1 % predicted (relative change from baseline at up to 1 month)
ii. Short term (over one month and up to and including six months)
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
b. FEV1 (absolute values)
i. Immediate term (up to and including one month)
Lumacaftor versus placebo
The Phase 2 study reported on the absolute change from baseline in FEV1 % predicted after lumacaftor monotherapy (day 14) (Boyle 2014); there was no difference between treatment groups at up to one month (MD ‐1.90, 95% CI ‐4.13 to 0.33; 1 study, 61 participants; moderate‐certainty evidence; Analysis 1.1).
1.1. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 1: FEV1 % predicted (absolute change from baseline)
Cavosonstat versus placebo
As previously stated, Donaldson (n = 51) reported that no treatment‐related changes in FEV1 were seen with cavosonstat compared to placebo (Donaldson 2017) (low‐certainty evidence).
FDL169 versus placebo
This study reported the absolute change from baseline in FEV1 % predicted at up to one month (Horsley 2017); there was a greater increase in the 400 mg (n = 6) group than placebo (n = 7), MD 4.68 (95% CI 0.12 to 9.24) (Analysis 4.2); but no difference between the 600 mg group (n = 6) and placebo, MD 2.80 (95% CI ‐1.82 to 7.42) (Analysis 5.2) or between the 800 mg group (n = 8) and placebo, MD 0.68 (95% CI ‐3.80 to 5.16) (Analysis 6.2).
4.2. Analysis.

Comparison 4: FDL169 (400 mg three times daily) versus placebo, Outcome 2: FEV1 % predicted absolute change (% points)
5.2. Analysis.

Comparison 5: FDL169 (600 mg three times daily) versus placebo, Outcome 2: FEV1 % predicted absolute change (% points)
6.2. Analysis.

Comparison 6: FDL169 (800 mg three times daily) versus placebo, Outcome 2: FEV1 % predicted absolute change (% points)
ii. Short term (over one month and up to and including six months)
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
c. FVC
Seven studies did not report data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
i. Immediate term (up to and including one month)
Cavosonstat versus placebo
Similarly to the measurement of FEV1, Donaldson (n = 51) reported that no treatment‐related changes in FVC were seen with cavosonstat compared to placebo (Donaldson 2017).
Secondary outcomes
1. Adverse effects
All included studies reported on the AEs of therapy (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002). The extent and type of AE reporting varied between studies.
a. Mild (therapy does not need to be discontinued)
In Phase 2 trials of potential correctors (CPX, 4PBA, N6022, lumacaftor and cavosonstat), there was a lack of evidence to show any significant increase in AE reporting compared to placebo (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002). However, a large number of events were reported and it is difficult to assess the clinical relevance of these events with the small number of participants in the trials. Further details are given below.
Lumacaftor versus placebo
We present the AE events occurring in more than one participant in any dose group in the lumacaftor study by Clancy 2012 in the additional tables (Table 15). We have combined the total number of participants with AEs occurring in the 100 mg and 200 mg lumacaftor groups and compared this to the number of participants experiencing AEs in the placebo group (Analysis 1.2). AE data for participants receiving a lower dose (25 mg or 50 mg of lumacaftor) were not included as there was a lack of evidence of efficacy. The most commonly reported side effect was cough; there was no difference in the number of participants who reported cough between the participants assigned to either 100 mg or 200 mg lumacaftor and those assigned to placebo (OR 1.28, 99% CI 0.28 to 5.92; Analysis 1.2) (Clancy 2012).
4. Frequency of adverse effects occurring in more than one participant in any VX‐809 treatment group (Clancy 2012).
|
Placebo n (%) |
Lumacaftor n (%) |
Total n (%) |
||||
| Adverse effect n (%) | (n = 17) | 25 mg (n = 18) | 50 mg (n = 18) | 100 mg (n = 17) | 200 mg (n = 18) | (n = 45)* |
| Cough | 7 (41.2) | 10 (55.6) | 6 (33.3) | 7 (41.2) | 10 (52.6) | 40 (88.9) |
| Headache | 3 (17.6) | 4 (22.2) | 5 (27.8) | 2 (11.8) | 5 (26.3) | 19 (42.2) |
| Rales | 1 (5.9) | 6 (33.3) | 2 (11.1) | 3 (17.6) | 3 (15.8) | 15 (33.3) |
| Productive cough | 3 (17.6) | 2 (11.1) | 0 (0.0) | 4 (23.5) | 6 (31.6) | 15 (17.8) |
| Dyspnoea | 1 (5.9) | 5 (27.8) | 3 (16.7) | 2 (11.8) | 4 (21.1) | 15 (33.3) |
| Pulmonary exacerbation* | 2 (11.8) | 4 (22.2) | 2 (11.1) | 2 (11.8) | 4 (21.1) | 14 (31.1) |
| Fatigue | 2 (11.8) | 3 (16.7) | 3 (16.7) | 2 (11.8) | 3 (15.8) | 13 (28.9) |
| Fever | 2 (11.8) | 2 (11.1) | 1 (5.6) | 1 (5.9) | 5 (26.3) | 11 (24.4) |
| Nasal congestion | 3 (17.6) | 2 (11.1) | 1 (5.6) | 2 (11.8) | 2 (10.5) | 10 (22.2) |
| Wheezing | 3 (17.6) | 1 (5.6) | 4 (22.2) | 1 (5.9) | 0 (0.0) | 9 (20.0) |
| Diarrhoea | 3 (17.6) | 3 (16.7) | 1 (5.6) | 2 (11.8) | 0 (0.0) | 9 (20) |
| Oropharyngeal pain | 3 (17.6) | 0 (0.0) | 3 (16.7) | 0 (0.0) | 2 (10.5) | 8 (17.8) |
| Upper respiratory tract infection | 1 (5.9) | 2 (11.1) | 1 (5.6 | 3 (17.6) | 0 (0.0) | 7 (15.6) |
| Sinus congestion | 2 (11.8) | 1 (5.6) | 2 (11.1) | 0 (0.0) | 1 (5.3) | 6 (13.3) |
| Respiration abnormal | 0 (0.0) | 1 (5.6) | 1 (5.6) | 0 (0.0) | 4 (21.1) | 6 (13.3) |
| Haemoptysis | 2 (11.8) | 1 (5.6) | 1 (5.6) | 0 (0.0) | 2 (10.5) | 6 (13.3) |
| Constipation | 0 (0.0) | 2 (11.1) | 2 (11.1) | 1 (5.9) | 1 (5.3) | 6 (13.3) |
| Abdominal pain | 1 (5.9) | 3 (16.7) | 1 (5.6) | 0 (0.0) | 1 (5.3) | 6 (13.3) |
| Myalgia | 1 (5.9) | 0 (0.0) | 3 (16.7) | 0 (0.0) | 1 (5.3) | 5 (11.1) |
| Post‐tussive vomiting | 0 (0.0) | 0 (0.0) | 2 (11.1) | 1 (5.9) | 1 (5.3) | 4 (8.9) |
| Nausea | 0 (0.0) | 3 (16.7) | 0 (0.0) | 0 (0.0) | 1 (5.3) | 4 (8.9) |
| Nasopharyngitis | 0 (0.0) | 1 (5.6) | 0 (0.0) | 1 (5.9) | 2 (10.5) | 4 (8.9) |
| Dizziness | 0 (0.0) | 1 (5.6) | 0 (0.0) | 2 (11.8) | 1 (5.3) | 4 (8.9) |
| Back pain | 0 (0.0) | 2 (11.1) | 1 (5.6) | 0 (0.0) | 1 (5.3) | 4 (8.9) |
| Abdominal pain upper | 1 (5.9) | 0 (0.0) | 0 (0.0) | 1 (5.9) | 2 (10.5) | 4 (8.9) |
| Sputum abnormal | 0 (0.0) | 2 (11.1) | 0 (0.0) | 0 (0.0) | 1 (5.3) | 3 (6.7) |
| Epistaxis | 1 (5.9) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (10.5) | 3 (6.7) |
| C‐reactive protein increased | 0 (0.0) | 1 (5.6) | 0 (0.0) | 2 (11.8) | 0 (0.0) | 3 (6.7) |
| Paranasal sinus hypersecretion | 0 (0.0) | 2 (11.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (4.4) |
| Lung hyperinflation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.8) | 0 (0.0) | 2 (4.4) |
*Unclear why the total number of participants in the study is shown to be 45. The author has been contacted for clarification.
1.2. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 2: Adverse effects: 100 mg and 200 mg lumacaftor groups (combined data) versus placebo at up to 1 month
Data for 14 days of lumacaftor monotherapy (200 mg once daily) demonstrated no significant differences between participants treated with lumacaftor therapy and placebo in the number of participants experiencing cough, oropharyngeal pain, nasal congestion, dizziness, a prolonged prothrombin time and upper respiratory tract infection (very low‐certainty evidence; Analysis 1.3) (Boyle 2014).
1.3. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 3: Adverse effects: 200 mg lumacaftor group versus placebo at up to 1 month
Cavosonstat versus placebo
In the cavosonstat study, there was no difference in cough, pulmonary exacerbation, chest discomfort or fatigue in the treatment group compared to placebo (very low‐certainty evidence; Analysis 2.3) (Donaldson 2017). All AEs observed in this study were reported to be 'mild or moderate' in severity.
2.3. Analysis.

Comparison 2: Cavosonstat (N91115) (200 mg twice daily) versus placebo, Outcome 3: Adverse events occurring in > 10% of participants at up to 1 month
N6022 versus placebo
The numbers of Grade 1 (mild) AEs across all N6022 doses and placebo were reported (Donaldson 2014). There was no difference between any of the N6022 doses and placebo in terms of the number of mild AEs; specific events were not reported (Analysis 3.2).
3.2. Analysis.

Comparison 3: N6022 versus placebo, Outcome 2: Treatment‐emergent adverse events (mild) at up to 1 month
CPX versus placebo
Participants received a single dose of the assigned CPX dose level (1 mg, 3 mg, 10 mg, 30 mg, 100 mg, 300 mg or 1000 mg) (McCarty 2002). AEs were recorded on the day of dosing (day one), day two and followed up one week post‐dosing. AEs that occurred in more than 3% of participants are shown in the additional tables (Table 16). Combined data from all CPX groups versus placebo demonstrated that the following events were less common in the placebo group: abdominal pain, OR 0.45 (99% CI 0.01 to 24.92); asthenia, OR 0.65 (99% CI 0.01 to 39.69); headache, OR 0.33 (99% CI 0.01 to 17.72); pain, OR 0.45 (99% CI 0.01 to 24.92); diarrhoea, OR 0.65 (99% CI 0.01 to 39.69); lung disease, OR 0.45 (99% CI 0.01 to 24.92); and rhinitis, OR 0.45 (99% CI 0.01 to 24.92) (Analysis 7.1). Dizziness was more common amongst participants in the placebo group, OR 9.33 (99% CI 0.32 to 268.92) (Analysis 7.1). The difference between CPX groups (combined data) and placebo was not statistically significant for any AE (McCarty 2002).
5. Frequency of occurrence of adverse effects occurring in more than 3% of participants in any CPX treatment group in McCarty 2002.
| Placebo | CPX | |||||||
| Adverse effects, n | (n = 8) | 1 mg (n = 4) | 3 mg (n = 4) | 10 mg (n = 4) | 30 mg (n = 4) | 100 mg (n = 5) | 300 mg (n = 4) | 1000 mg (n = 4) |
| Abdominal pain | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 |
| Asthenia | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Headache | 0 | 0 | 0 | 2 | 1 | 0 | 1 | 0 |
| Pain | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 0 |
| Diarrhoea | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| Dizziness | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Lung disease | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 |
| Rhinitis | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 2 |
CPX: 8‐cyclopentyl‐1, 3‐dipropylxanthine
7.1. Analysis.

Comparison 7: CPX versus placebo, Outcome 1: Adverse events occurring in more than 3% of participants in all treatment groups (combined data) versus placebo at up to 1 month
4PBA versus placebo
In the pilot 4PBA study (n = 18) (Rubenstein 1998), the differences between groups in the number of participants who reported episodes of bad taste in their mouth and diarrhoea were not statistically significant, OR 0.44 (99% CI 0.01 to 13.44) and OR 3.35 (99% CI 0.04 to 267.31), respectively (Analysis 8.1).
8.1. Analysis.

Comparison 8: 4PBA versus placebo, Outcome 1: Adverse events at up to 1 month
In the Phase 2 4PBA study (n = 19), participants randomised to the 20 g cohort reported episodes of transient nausea, headache, sleepiness and body odour after the initial dose; the transient nausea, sleepiness and headache resolved with a dose of Tylenol® (acetaminophen). No numerical data were reported regarding AEs, therefore we are not able to analyse any data for this study (Zeitlin 2002).
b. Moderate (therapy is discontinued, and the adverse effect ceases)
None of the participants in the Phase 2 lumacaftor‐ivacaftor study, the pilot 4PBA study or the CPX study required study drug interruption due to the AEs of therapy (Boyle 2014; McCarty 2002; Rubenstein 1998).
Lumacaftor versus placebo
There were no differences in the number of AEs requiring study drug discontinuation up to day 28 between any lumacaftor dose and placebo (Analysis 1.4) (Clancy 2012).
1.4. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 4: Adverse effects requiring study drug discontinuation at up to 1 month
Cavosonstat versus placebo
In the cavosonstat study, there was no difference in cough, pulmonary exacerbation, chest discomfort or fatigue in the treatment group compared to placebo and all AEs observed in this study were reported to be 'mild or moderate' in severity (very low‐certainty evidence; Analysis 2.3) (Donaldson 2017).
N6022 versus placebo
The number of Grade 2 (moderate) AEs across all N6022 doses and placebo were reported (Donaldson 2014). There were no differences between any of the N6022 doses and placebo in terms of the number of Grade 2 AEs; specific events were not reported (Analysis 3.3).
3.3. Analysis.

Comparison 3: N6022 versus placebo, Outcome 3: Treatment‐emergent adverse events (moderate) at up to 1 month
4PBA versus placebo
In the Phase 4 4PBA study, participants who were discontinued from a particular study dose were assigned a reduced dose and this is discussed under severe AEs (Zeitlin 2002).
c. Severe (life‐threatening or debilitating, or which persists even after treatment is discontinued)
None of the participants from the CPX study or the cavosonstat study required study drug termination (Donaldson 2017; Rubenstein 1998).
Lumacaftor versus placebo
In the Clancy study, AEs in eight participants were considered severe: fatigue (n = 1); sinus congestion (n = 1); musculoskeletal discomfort (n = 1); cough (n = 2); and pulmonary exacerbation (n = 3). It is not stated to which arm these participants were randomised. Four out of 89 participants (5%) ‐ one participant from each of the lumacaftor arms ‐ discontinued the study drug due to respiratory AEs. No participants discontinued from the placebo group (Clancy 2012).
N6022 versus placebo
The number of Grade 3 or above (serious or life‐threatening) AEs across all N6022 doses and placebo were reported (Donaldson 2014). There were no differences between any of the N6022 doses and placebo in terms of the number of Grade 3 or above AEs (Analysis 3.4). The events were as follows: one participant with appendicitis in the 5 mg/day N6022 group and three participants with a pulmonary exacerbation of CF, one each in the placebo, 5 mg/day and 40 mg/day N6022 groups.
3.4. Analysis.

Comparison 3: N6022 versus placebo, Outcome 4: Treatment‐emergent adverse events (serious or severe) at up to 1 month
4PBA versus placebo
None of the participants from the pilot 4PBA study required study drug termination (McCarty 2002). In the Phase 2 4PBA study, none of the participants in the 20 g group required study drug termination (Zeitlin 2002). "Several" participants (exact number not stated) in the 30 g group reported episodes of transient nausea, headache, sleepiness and transient visual disturbances after the initial dose. Two participants from the 30 g cohort required dose reduction to 20 g due to headache (n = 1) and for an unknown reason (n = 1). One participant who started in this group had to discontinue medication after developing acute distal intestinal obstruction syndrome on day two, but was replaced by another participant. The three participants assigned 40 g of 4PBA reported episodes of nausea, headache and visual disturbances and one participant reported cramp in hands and fingers. One participant tolerated the dose whilst splitting the 40 g into six daily doses, one participant had a dose reduction to 30 g daily and another participant in this group was discontinued from the study due to intolerable symptoms (nausea, headache and visual disturbances). The 40 g cohort was terminated early following analysis of the data by the safety monitoring committee (Zeitlin 2002).
d. Other adverse effects of therapy (of any severity) that are not classifiable according to these categories
Lumacaftor versus placebo
Two studies also reported on the number of participants who experienced episodes of pulmonary exacerbations described as AEs (Boyle 2014; Clancy 2012). Results are presented in the analyses and described below (see 'Extra courses of antibiotics') (Analysis 1.2; Analysis 1.3).
N6022 versus placebo
Donaldson reported on "none serious" AEs on each dose of N6022 and placebo (Donaldson 2014). Due to the small numbers of participants experiencing different AEs, these results are not entered into analysis and are reported in the additional tables (Table 17).
6. Adverse events (non‐serious) reported in Donaldson 2014 (N6022 versus placebo).
| Placebo | N6022 | Total | ||||
| Adverse events, n | (n = 19) | 5 mg (n = 10) | 10 mg (n = 9) | 20 mg (n = 9) | 40 mg (n = 19) | (n = 66) |
| Lymphadenopathy | 1 | 0 | 0 | 0 | 0 | 1 |
| Chest tightness | 1 | 2 | 0 | 0 | 2 | 5 |
| Atrioventricular block second degree | 0 | 0 | 1 | 0 | 0 | 1 |
| Nodal rhythm | 0 | 0 | 0 | 1 | 0 | 1 |
| Supraventricular extrasystoles | 0 | 0 | 0 | 1 | 0 | 1 |
| Supraventricular tachycardia | 0 | 0 | 1 | 0 | 0 | 1 |
| Ventricular extrasystoles | 1 | 0 | 0 | 0 | 0 | 1 |
| Ventricular tachycardia | 0 | 0 | 0 | 1 | 0 | 1 |
| Diarrhoea | 2 | 0 | 1 | 0 | 0 | 3 |
| Nausea | 1 | 1 | 0 | 0 | 1 | 3 |
| Vomiting | 0 | 0 | 0 | 0 | 2 | 2 |
| Flatulence | 0 | 0 | 1 | 0 | 0 | 1 |
| Parosmia | 0 | 0 | 0 | 2 | 0 | 2 |
| Night sweats | 0 | 0 | 2 | 0 | 0 | 2 |
| Fatigue | 1 | 1 | 0 | 0 | 2 | 4 |
| Pyrexia | 0 | 1 | 0 | 0 | 2 | 3 |
| Infective pulmonary exacerbations of CF | 1 | 1 | 0 | 0 | 1 | 3 |
| Upper respiratory tract infection | 1 | 0 | 0 | 0 | 0 | 1 |
| Headache | 1 | 1 | 1 | 2 | 1 | 6 |
| Cough | 7 | 3 | 1 | 3 | 2 | 16 |
| Increased bronchial secretion | 3 | 2 | 2 | 2 | 1 | 10 |
| Nasal congestion | 1 | 3 | 0 | 0 | 1 | 5 |
| Rales | 0 | 3 | 0 | 1 | 0 | 4 |
| Total participants with at least one adverse event, n (%) | 18 (95%) | 9 (90%) | 9 (100%) | 9 (100%) | 15 (79%) | 60 (91%) |
CF: cystic fibrosis
FDL169 versus placebo
Horsley reported the number of participants experiencing at least one AE, and the number of 'serious' AEs; AEs were not categorised under mild, moderate or severe (Horsley 2017). No difference was found in the number of participants experiencing at least one AE between any tested dose level of FDL169 and placebo (n = 7); for the 400 mg group (n = 6), OR 6.67 (99% CI 0.21 to 207.87) (Analysis 4.3); for the 600 mg group (n = 6), OR 0.06 (99% CI 0.00 to 4.00) (Analysis 5.3); and for the 800 mg group (n = 8), OR 21.86 (99% CI 0.34 to 1419.86) (Analysis 6.3). Similarly, there was no statistical difference observed in the occurrence of any particular AE or of serious AEs (Analysis 4.3; Analysis 5.3; Analysis 6.3).
4.3. Analysis.

Comparison 4: FDL169 (400 mg three times daily) versus placebo, Outcome 3: Adverse events at up to 1 month
5.3. Analysis.

Comparison 5: FDL169 (600 mg three times daily) versus placebo, Outcome 3: Adverse events at up to 1 month
6.3. Analysis.

Comparison 6: FDL169 (800 mg three times daily) versus placebo, Outcome 3: Adverse events at up to 1 month
2. Hospitalisation
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
3. School or work attendance
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
4. Extra courses of antibiotics
a. Time to the next course of antibiotics
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
b. Total number of courses of antibiotics
i. Immediate term (up to and including one month)
Lumacaftor versus placebo
In the lumacaftor study, pulmonary exacerbations were physician‐defined and there was no difference in the frequency of participants who developed pulmonary exacerbations between those in the lumacaftor groups and the placebo group (OR 1.50, 99% CI 0.16 to 14.31; 1 study, 53 participants; Analysis 1.2) (Clancy 2012).
In the Boyle study, it was unclear whether the reported exacerbations were protocol‐defined or physician‐defined (Boyle 2014). At day 14, exacerbations were more common in participants receiving 200 mg lumacaftor once daily in comparison to participants receiving placebo; however, the difference between groups was not significant (OR 2.72, 99% CI 0.05 to 156.17; 1 study, 62 participants; Analysis 1.3).
FDL169 versus placebo
From the published abstract for this Phase 1 study, it is unclear whether exacerbations were physician‐ or protocol‐defined. A total of three participants across all groups were reported to have had an infective respiratory exacerbation; no participants in the 400 mg group (n = 6), one participant in the 600 mg group (n = 6), one participant in the 800 mg group (n = 8) and one participant in the placebo group (n = 7) (Horsley 2017).
5. Sweat chloride (change from baseline) as a measure of CFTR function
All included studies reported on sweat chloride concentration (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
i. Immediate (up to one month)
Lumacaftor versus placebo
In the Clancy study (n = 89), data at seven days demonstrated small reductions in the change from baseline in sweat chloride concentration compared to placebo for the participants taking 25 mg lumacaftor, MD 1.7 mmol/L; 50 mg lumacaftor, MD ‐1.5 mmol/L; 100 mg lumacaftor, MD ‐0.1mmol/L; and 200 mg lumacaftor, MD ‐4.4 mmol/L (Clancy 2012). No SDs or CIs were reported to allow the inclusion of these results into the analysis (Clancy 2012). At 28 days, participants in the 25 mg lumacaftor group demonstrated a marginal increase in sweat chloride concentration compared to placebo, MD 0.1 mmol/L, and those in the 50 mg lumacaftor group demonstrated a decreased sweat chloride concentration compared to placebo, MD ‐4.61 mmol/L (Clancy 2012). These differences were not statistically significant and no SDs or CIs were reported for inclusion of these results into the analysis (Clancy 2012). Data at one month demonstrated statistically significant reductions in sweat chloride concentration compared to placebo for participants in the once‐daily 100 mg lumacaftor group, MD ‐6.13 mmol/L (95% CI ‐12.25 to ‐0.01) and once‐daily 200 mg lumacaftor group, MD ‐8.21 (95% CI ‐14.30 to ‐2.12) (Analysis 1.5) (Clancy 2012).
1.5. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 5: Sweat chloride concentration (change from baseline at up to 1 month) [mmol/L]
Boyle reported that at day 14, there was a small reduction in sweat chloride concentration reported in participants taking 200 mg lumacaftor once daily compared to placebo, which was not statistically significant (MD ‐2.75 mmol/L, 95% CI ‐7.65 to 2.15; Analysis 1.6) (Boyle 2014). Results for up to 21 days (monotherapy and combination therapy) are reported under 'Correctors plus potentiators in combination therapy compared to placebo'.
1.6. Analysis.

Comparison 1: Lumacaftor versus placebo, Outcome 6: Sweat chloride concentration (change from baseline)
Cavosonstat versus placebo
There was no difference in sweat chloride concentration between cavosonstat and placebo at up to one month (MD ‐3.30 mmol/L, 95% CI ‐9.13 to 2.53; 1 study, 24 participants; Analysis 2.4) (Donaldson 2017).
2.4. Analysis.

Comparison 2: Cavosonstat (N91115) (200 mg twice daily) versus placebo, Outcome 4: Sweat chloride
CPX versus placebo
In the CPX study (n = 37), McCarty reported post‐treatment sweat chloride concentration values at the end of treatment on day one (McCarty 2002). The baseline sweat chloride values in the CPX group and the placebo group appear to have been pooled. By calculating the values for relative change from baseline, we have assumed that the baseline sweat chloride value represents the baseline sweat chloride concentration value for each arm. At the end of treatment on day one, there were no differences in sweat chloride concentration between the placebo group and the 1 mg CPX group, MD 12.8%; the 3 mg CPX group, MD 7.5%; the 10 mg CPX group, MD 11.3%; the 30 mg CPX group, MD 5.4%; the 100 mg CPX group, MD 5.1%; the 300 mg CPX group, MD 14.7%; and the 1000 mg CPX group, MD ‐8.2%. No SDs or CIs were reported to allow calculation of SDs for entry into the analysis (McCarty 2002).
4BPA versus placebo
In the pilot 4PBA study by Rubenstein (n = 18), there was no difference in sweat chloride concentration at one week between participants in the 4PBA group and the placebo group (P = 0.387). Data were plotted on a graph and could not be extracted with accuracy (Rubenstein 1998).
The Phase 2 4PBA study by Zeitlin reported post‐treatment sweat chloride concentration values at day two, day three, day four and day seven; we calculated the relative change from baseline values at each time point. There was no difference in sweat chloride concentration between the 20 g 4PBA group and the placebo group after two days of treatment, MD ‐7.8%; three days, MD ‐4.9%; four days, MD ‐3.3% and seven days, MD ‐8.7%. Furthermore, there was no difference in sweat chloride concentration between the 30 g 4PBA group and the placebo group after two days of treatment, MD ‐25.9%; three days, MD 0.5%; four days, MD ‐6.4% and seven days, MD ‐3.9% (Zeitlin 2002). No SDs or CIs were reported to allow calculation of SDs for entry into the analysis. Due to insufficient reporting of data by both 4PBA studies, we were unable to include the results in the analysis (Rubenstein 1998; Zeitlin 2002).
FDL169 versus placebo
Horsley reported the absolute change in sweat chloride (mmol/L) at 28 days (Horsley 2017). There was no difference between the 400 mg group (n = 6) and placebo (n = 7) (MD 2.47, 95% CI ‐4.47 to 9.41; Analysis 4.4) or between the 800 mg group (n = 8) and placebo (MD 3.48, 95% CI ‐3.35 to 10.31; Analysis 6.4), but there was a greater drop in sweat chloride in the placebo group than the 600 mg group (n = 6) (MD 8.07, 95% CI 0.98 to 15.16; Analysis 5.4).
4.4. Analysis.

Comparison 4: FDL169 (400 mg three times daily) versus placebo, Outcome 4: Sweat chloride change from baseline (mmol/L)
6.4. Analysis.

Comparison 6: FDL169 (800 mg three times daily) versus placebo, Outcome 4: Sweat chloride change from baseline (mmol/L)
5.4. Analysis.

Comparison 5: FDL169 (600 mg three times daily) versus placebo, Outcome 4: Sweat chloride change from baseline (mmol/L)
6. Radiological measures of lung disease
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
7. Acquisition of respiratory pathogens
No study reported data for the acquisition of S aureus, H influenzae or any other clinically relevant pathogens except P aeruginosa (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
a. P aeruginosa
This was a pre‐defined outcome of interest in the Phase 1/2 4PBA study, but no study results were reported (Zeitlin 2002). The other studies did not report data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998).
8. Eradication of respiratory pathogens
No study reported data for this outcome (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017McCarty 2002; Rubenstein 1998; Zeitlin 2002).
9. Nutrition and growth
No study reported data for this outcome, either in terms of weight, BMI or height (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002).
Dual therapy (correctors plus potentiators) compared to control
We included 15 studies with 2627 participants, which contributed to the efficacy results in this comparison (of which 62 participants also contributed to the monotherapy comparison) (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). A further study contributed additional safety data to this comparison (see below) (PROGRESS 2017).
Lumacaftor plus ivacaftor versus placebo
Six studies (1495 participants) compared lumacaftor plus ivacaftor to placebo (Boyle 2014; Ratjen 2017; Stahl 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
One study (62 participants) compared lumacaftor 200 mg once daily plus 150 mg or 250 mg twice‐daily ivacaftor to placebo (Boyle 2014). Participants in this study received lumacaftor monotherapy for 14 days followed by combination therapy with ivacaftor for seven days, therefore this study contributes to both comparisons of this review (Boyle 2014).
The paediatric combination study (204 children) compared 200 mg lumacaftor twice daily plus 250 mg ivacaftor twice daily to placebo for six months (Ratjen 2017). Primary and secondary outcomes of this study were analysed via a mixed model for repeated measures (MMRM); further details of this analysis approach are provided in the tables (Characteristics of included studies). Results provided by this model can be interpreted as a treatment effect averaged from each study visit until six months.
A further paediatric study (51 participants) compared lumacaftor 100 mg plus ivacaftor 125 mg twice daily to placebo in participants weighing less than 14 kg at screening. For those participants weighing greater than or equal to 14 kg at screening, investigators compared lumacaftor 150 mg plus ivacaftor 188 mg twice daily to placebo (Stahl 2021). The results of this study have not been combined with other comparisons of lumacaftor plus ivacaftor versus placebo, due to the fact that it was unclear as to the number of participants weighing less than, or greater than or equal to, 14 kg at screening. This study has not been included in a summary of findings table.
One study (70 participants) compared 400 mg lumacaftor plus 250 mg ivacaftor twice daily to placebo (Wilson 2021).
Two three‐arm studies (1108 participants) compared 600 mg once‐daily lumacaftor plus 250 mg twice‐daily ivacaftor to 400 mg twice‐daily lumacaftor plus 250 mg twice‐daily ivacaftor and to placebo (TRAFFIC 2015; TRANSPORT 2015). The PROGRESS study was an extension to the TRAFFIC and TRANSPORT studies (TRAFFIC 2015; TRANSPORT 2015), in which participants from the original placebo groups were randomised to one of the two interventions (PROGRESS 2017). Due to the overlap of participants in these three studies, we have not included efficacy data for the PROGRESS study under a comparison of lumacaftor (plus ivacaftor) doses. We have included safety data from this study as these are important longer‐term results for participants on this intervention; results for the PROGRESS study are presented in the tables for information (PROGRESS 2017; Table 18; Table 19).
7. Adverse events with an incidence of ≥ 0.20 events per patient‐year in Konstan 2017.
| Event | Lumacaftor 400 mg twice daily/ivacaftor 250 twice daily (n = 340) | Placebo transitioned to lumacaftor 400 mg twice daily/ivacaftor 250 mg twice daily (n = 176) | Lumacaftor 600 mg once daily/ivacaftor 250 mg twice daily (n = 335) | Placebo transitioned to lumacaftor 600 mg once daily/ivacaftor 250 mg twice daily (n = 178) |
| Total exposure in patient‐years | 570 | 290 | 570 | 300 |
| Infective pulmonary exacerbation | 0.980 | 1.035 | 1.157 | 1.080 |
| Cough | 0.510 | 0.573 | 0.627 | 0.609 |
| Haemoptysis | 0.266 | 0.200 | 0.235 | 0.239 |
| Increased sputum | 0.208 | 0.207 | 0.224 | 0.175 |
| Nasopharyngitis | 0.194 | 0.169 | Not reported | Not reported |
| Headache | 0.140 | 0.107 | 0.129 | 0.101 |
| Dyspnoea | 0.124 | 0.166 | 0.117 | 0.128 |
| Pyrexia | 0.114 | 0.152 | 0.148 | 0.148 |
| Upper respiratory tract infection | 0.129 | 0.131 | Not reported | Not reported |
| Diarrhoea | 0.093 | 0.145 | 0.111 | 0.101 |
| Abnormal respiration | 0.077 | 0.128 | 0.088 | 0.145 |
| Nausea | 0.072 | 0.104 | Not reported | Not reported |
| Fatigue | 0.084 | 0.090 | Not reported | Not reported |
| Abdominal pain | 0.087 | 0.066 | 0.087 | 0.084 |
| Oropharyngeal pain | Not reported | Not reported | 0.101 | 0.081 |
| Nasal congestion | Not reported | Not reported | 0.104 | 0.091 |
| Rhinitis | Not reported | Not reported | 0.064 | 0.030 |
| Any adverse event: n (%) | 333 (97.9) | 176 (100) | 331 (98.8) | 177 (99.4) |
| Any serious adverse event: n (%) | 143 (42.1) | 89 (50.6) | 156 (46.6) | 77 (43.3) |
| Any treatment‐emergent respiratory event: n (%) | 99 (29) | 67 (38) | 102 (30) | 67 (38) |
8. Secondary efficacy outcomes reported in Konstan 2017.
| Outcome | Lumacaftor 400 mg twice daily/ivacaftor 250 mg twice daily (n = 340) | Placebo transitioned to lumacaftor 400 mg twice daily/ivacaftor 250 mg twice daily (n = 176) | Lumacaftor 600 mg once daily/ivacaftor 250 mg twice daily (n = 335) | Placebo transitioned to lumacaftor 600 mg once daily/ivacaftor 250 mg twice daily (n = 178) |
| FEV1 (% predicted):1 Week 72 |
0.5 (95% CI ‐0.4 to 1.5) P = 0.2806 |
1.5 (95% CI 0.2 to 2.9) P = 0.0254 |
1.2 (95% CI 0.3 to 2.2) P = 0.0127 |
1.9 (95% CI 0.6 to 3.2) P = 0.0037 |
| FEV1 (% predicted):1 Week 96 |
0.5 (95% CI ‐0.7 to 1.6) P = 0.4231 |
0.8 (95% CI ‐0.8 to 2.3) P = 0.3495 |
0.0 (95% CI ‐1.1 to 1.1) P = 0.9682 |
1.6 (95% CI ‐0.1 to 3.2) P = 0.0632 |
| FEV1 (% predicted):2 Week 72 |
0.9 (95% CI 0.0 to 1.9) P = 0.0500 |
1.9 (95% CI 0.6 to 3.2) P = 0.0040 |
1.7 (95% CI 0.8 to 2.7) P = 0.0003 |
2.2 (95% CI 1.0 to 3.5) P = 0.0005 |
| FEV1 (% predicted):2 Week 96 |
1.1 (95% CI 0.0 to 2.2) P = 0.0535 |
1.1 (95% CI ‐0.5 to 2.6) P = 0.1696 |
0.7 (95% CI ‐0.4 to 1.8) P = 0.1966 |
2.0 (95% CI 0.4 to 3.6) P = 0.0149 |
| FEV1 (% predicted):1 Relative change Week 72 |
1.4 (95% CI ‐0.3 to 3.2) P = 0.1074 |
2.6 (95% CI 0.2 to 5.0) P = 0.0332 |
2.4 (95% CI 0.6 to 4.1) P = 0.0080 |
3.8 (95% CI 1.4 to 6.1) P = 0.0017 |
| FEV1 (% predicted):1 Relative change Week 96 |
1.2 (95% CI ‐0.8 to 3·3) P = 0·2372 |
1.1 (95% CI ‐1.7 to 3.9) P = 0·4415 |
0.1 (95% CI ‐1.9 to 2.1) P = 0.9297 |
3.6 (95% CI 0.6 to 6.6) P = 0.0172 |
| BMI Week 72 |
0.69 (95% CI 0.56 to 0.81) P < 0.0001 |
0.62 (95% CI 0.45 to 0.79) P < 0.0001 |
0.72 (95% CI 0.60 to 0.84) P < 0.0001 |
0.52 (95% CI 0.36 to 0.69) P < 0.0001 |
| BMI Week 96 |
0.96 (95% CI 0.81 to 1.11) P < 0.0001 |
0.76 (95% CI 0.56 to 0.97) P < 0.0001 |
0.81 (95% CI 0.66 to 0.95) P < 0.0001 |
0.55 (95% CI 0.34 to 0.76) P < 0.0001 |
| CFQ‐R respiratory domain Week 72 |
5.7 (95% CI 3.8 to 7.5) P < 0.0001 |
3.3 (95% CI 0.7 to 5.9) P = 0.0124 |
3.2 (95% CI 1.4 to 5.1) P = 0.0007 |
3.3 (95% CI 0.7 to 5.8) P = 0.0116 |
| CFQ‐R respiratory domain Week 96 |
3.5 (95% CI 1.3 to 5.8) P = 0.0018 |
0.5 (95% CI ‐2.7 to 3.6) P = 0.7665 |
1.1 (95% CI ‐1.1 to 3.2) P = 0.3339 |
2.0 (95% CI ‐1.1 to 5.1) P = 0.2033 |
| Pulmonary exacerbations: events per patient year | 0.65 (95% CI 0.56 to 0.75) | 0.69 (95% CI 0.56 to 0.85) | 0.80 (95% CI 0.70 to 0.92) | 0.76 (95% CI 0.62 to 0.93) |
| Pulmonary exacerbations: events requiring hospital admission per patient year | 0.24 (95% CI 0.19 to 0.29) | 0.30 (95% CI 0.22 to 0.40) | 0.31 (95% CI 0.25 to 0.38) | 0.35 (95% CI 0.26 to 0.47) |
| Pulmonary exacerbations: events requiring IV antibiotics per patient year | 0.32 (95% CI 0.26 to 0.38) | 0.37 (95% CI 0.29 to 0.49) | 0.38 (95% CI 0.32 to 0.46) | 0.42 (95% CI 0.33 to 0.54) |
BMI: body mass index CFQ‐R: cystic fibrosis questionnaire‐revised CI: confidence interval FEV1: forced expiratory volume at one second IV: intravenous
Unless otherwise stated, all outcomes reported are the mean (95% CI) absolute change from baseline. P values correspond to the within‐group change compared to baseline.
1Calculated using Wang‐Hankinson equations. 2Calculated using Global Lungs Initiative equations.
Tezacaftor plus ivacaftor versus placebo or versus ivacaftor alone
Nine studies (1132 participants) compared tezacaftor plus ivacaftor to placebo or to ivacaftor alone (i.e. using ivacaftor as placebo) (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Taylor‐Cousar 2017; Schwarz 2021).
One study (510 participants) compared a combination of tezacaftor 100 mg plus ivacaftor 150 mg every 12 hours to a matched placebo for six months (Taylor‐Cousar 2017) and one study (18 participants) compared tezacaftor (100 mg per day) plus ivacaftor (150 mg twice daily) against control (150 mg twice‐daily ivacaftor alone) in people with one F508del mutation and one G551D mutation (Donaldson 2018).
Five studies (497 participants) compared tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily to either a matched placebo or ivacaftor 150 mg twice daily alone (McKone 2021; Munck 2020; NCT02508207; NCT02730208; Schwarz 2021). Duration of studies ranged from 29 days (NCT02508207) to 72 weeks (NCT02730208).
One eight‐week study (67 participants) compared tezacaftor plus ivacaftor to either a matched placebo or ivacaftor. We present these data narratively, as the study presents the control group as two separate analyses (either placebo or ivacaftor alone) (Davies 2021).
A further 12‐week study (40 participants) compared two different active interventions to placebo: one arm compared tezacaftor 50 mg every 12 hours plus ivacaftor 150 mg every 12 hours to a matched placebo; and the second arm compared two 50 mg tablets of tezacaftor once daily plus ivacaftor 150 mg every 12 hours to a matched placebo. We report both arms in the same category of tezacaftor 50 mg twice daily plus ivacaftor 150 mg twice daily versus either placebo or ivacaftor 150 mg twice daily alone (NCT02070744).
We have not combined data from studies evaluating distinct agents or combinations of agents, as they have different mechanisms of actions. For each separate comparison we have assessed the following outcomes using the GRADE criteria, presented these gradings in individual tables and indicated our findings in the relevant text below.
survival;
QoL (total score);
QoL (respiratory domain);
FEV1 % predicted (relative and absolute change);
AEs; and
time to first pulmonary exacerbation.
For the comparison of lumacaftor (600 mg once daily or 400 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, reporting short‐term results (one month to six months), we judged the certainty of the evidence to be high to moderate; we downgraded the evidence due to risk of bias from selective reporting where data contributing to analyses were extrapolated from published graphs or estimated (Table 3).
For the comparison of lumacaftor (200 mg twice daily) and ivacaftor (250 mg twice daily) versus placebo, reporting immediate‐term results (up to one month), we judged the certainty of the evidence to be moderate to low; we downgraded evidence due to risk of bias from selective reporting where data contributing to analyses were extrapolated from published graphs or estimated, due to indirectness as results are applicable only to children between the ages of six to 11 years and due to imprecision where few events occurred and CIs around the result were very wide (Table 4).
For the comparison of lumacaftor (200 mg) plus ivacaftor (150 mg or 250 mg twice daily) versus placebo, reporting immediate‐term results (up to one month), we judged the certainty of the evidence to be moderate to low; we downgraded the evidence due to indirectness related to the design of the study and due to imprecision where few events occurred and CIs around the result were very wide (Table 5).
For the comparison of tezacaftor (100 mg once daily) and ivacaftor (150 mg twice daily) versus placebo or ivacaftor (150 mg twice daily alone) we judged the certainty of the evidence to be low to moderate; we downgraded the evidence due to indirectness as results are not applicable to children under the age of 12 and some results are not applicable to individuals homozygous for F508del (Table 6).
Primary outcomes
1. Survival
Only one study reported deaths. There was one death in the tezacaftor‐ivacaftor group due to “postinfluenza sepsis and multiple organ dysfunction syndrome”, but it was deemed not to be related to the study drug (Schwarz 2021).
No other study in this comparison reported any deaths (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
2. QoL
A total of 11 studies reported on QoL (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; Ratjen 2017; Schwarz 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
a. Total QoL score
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
Two studies reported QoL according to the Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score at six months (TRAFFIC 2015; TRANSPORT 2015). This information was not reported in the primary journal article, but is available from the study record on ClinicalTrials.gov (TRAFFIC 2015; TRANSPORT 2015).
There was no difference in the absolute change from baseline of EQ‐5D‐3L index score between the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily group and placebo group (MD 0.00, 95% CI ‐0.01 to 0.02; Analysis 10.1), or the lumacaftor 400 mg twice daily plus 250 mg ivacaftor twice daily group and placebo group (MD 0.00, 95% CI ‐0.01 to 0.02; Analysis 11.1), or when the two lumacaftor doses were pooled at six months (MD 0.00, 95% CI ‐0.01 to 0.01; moderate‐certainty evidence; Analysis 12.1).
10.1. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 1: Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline)
11.1. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 1: Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline)
12.1. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 1: Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline)
b. QoL sub‐domains
i. Immediate term (up to and including one month)
Lumacaftor plus ivacaftor versus placebo
In the TRAFFIC and TRANSPORT studies (1108 participants), at 28 days participants in the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily group experienced a greater absolute improvement from baseline in the CFQ‐R respiratory domain compared to the placebo group (MD 3.32, 95% CI 1.13 to 5.51; Analysis 10.2). There was also a difference demonstrated with the lumacaftor 400 mg twice daily plus 250 mg ivacaftor twice daily group at 28 days (MD 4.13, 95% CI 1.94 to 6.31; Analysis 11.2) (TRAFFIC 2015; TRANSPORT 2015). There was also a greater absolute improvement from baseline in the CFQ‐R respiratory domain compared to the placebo group when the two lumacaftor doses were pooled (MD 3.70, 95% CI 1.81 to 5.58; I2 = 82%; 2 studies, 1108 participants; Analysis 12.2). After examination of the two studies in this analysis, we are unable to identify potential causes for this level of heterogeneity.
10.2. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 2: Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline)
11.2. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 2: Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline)
12.2. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 2: Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline)
Tezacaftor plus ivacaftor versus control
Taylor‐Cousar reported on the CFQ‐R respiratory domain at up to one month (Taylor‐Cousar 2017) and found a greater improvement in the treatment group (MD 5.10, 95% CI 2.99 to 7.21; 1 study, 504 participants; Analysis 18.2).
18.2. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 2: CFQ‐R physical functioning domain (absolute change from baseline)
Donaldson (18 participants) presents the within‐group change from baseline to Day 28 for the CFQ‐R respiratory domain and at the end of the study the difference in treatment effect between tezacaftor‐ivacaftor and placebo was 6.81 points (P = 0.2451) (Donaldson 2018). These data were extrapolated and we have requested confirmation of the exact data from the study investigators. We will include any unpublished information we receive in a future update.
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
At six months, this statistically significant improvement was maintained in both the 600 mg lumacaftor and 400 mg lumacaftor groups (MD 3.04, 95% CI 0.76 to 5.32; Analysis 10.2 and MD 2.50, 95% CI 0.30 to 4.70; I2 = 0%; 3 studies, 783 participants; Analysis 11.2, respectively). The difference was also maintained when both of these doses were pooled together (MD 2.83, 95% CI 0.91 to 4.74; I2 = 0%; 3 studies, 1139 participants; Analysis 12.2) (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). These data from the TRAFFIC and TRANSPORT studies were extrapolated and we have requested confirmation of the exact data from the study investigators. We will include any unpublished information we receive in a future update.
The EQ‐5D‐3L visual analogue scale (VAS) domain score was also reported at six months in two studies (TRAFFIC 2015; TRANSPORT 2015). Participants in both the lumacaftor 600 mg group and the lumacaftor 400 mg group experienced greater absolute improvements from baseline in the EQ‐5D‐3L VAS domain compared to the placebo group (MD 2.24, 95% CI 0.18 to 4.31; Analysis 10.3 and MD 2.30, 95% CI 0.25 to 4.36; Analysis 11.3, respectively). There was also a greater absolute improvement from baseline in the EQ‐5D‐3L VAS domain compared to the placebo group when the two lumacaftor doses were pooled (MD 2.28, 95% CI 0.50 to 4.06; Analysis 12.3). This information was not reported in the primary journal article, but is available from the study record on ClinicalTrials.gov (TRAFFIC 2015; TRANSPORT 2015). We have requested immediate‐term data for this domain from the study investigators and will include any unpublished information we receive in a future update.
10.3. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 3: Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline)
11.3. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 3: Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline)
12.3. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 3: Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline)
The paediatric combination study also reported the absolute change from baseline (up to and including 24 weeks) of the CFQ‐R respiratory domain (Ratjen 2017). The change within the lumacaftor plus ivacaftor group was higher compared to the placebo group, but this difference did not reach statistical significance (MD 2.50, 95% CI ‐0.10 to 5.10; low‐certainty evidence; Analysis 15.1). Additional results at earlier time points (day 15, week 4 and week 16) were published graphically in the full study publication, but the graphical plots were too small to allow for accurate extraction of data (Ratjen 2017). Numerical data for these time points have been requested from the study investigators and we will include any unpublished information we receive in a future update.
15.1. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 1: Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline)
Investigators in this study also list the absolute change in Treatment Satisfaction Questionnaire for Medication (TSQM) domains as a secondary outcome of the study, but do not present results for this outcome (Ratjen 2017). Numerical data for this outcome have also been requested from the study investigators and any unpublished information we receive will be included in a future update.
Tezacaftor plus ivacaftor versus control
One study (11 participants) comparing two active arms and placebo reported on the absolute change from baseline in CFQ‐R respiratory domain score (NCT02070744). In the first active arm, participants received tezacaftor 50 mg every 12 hours plus ivacaftor 150 mg every 12 hours; at three months, there was a greater change in score for the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor group (MD 10.20, 95% CI ‐0.06 to 20.46; Analysis 17.1).
17.1. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 1: Absolute change from baseline in CFQ‐R respiratory domain score
Five studies (946 participants; including the second arm of NCT02070744 in which participants received two tezacaftor 50 mg tablets once daily plus ivacaftor 150 mg every 12 hours) reported data for the CFQ‐R respiratory domain at up to six months (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021; Taylor‐Cousar 2017) and found a greater increase in QoL score for tezacaftor‐ivacaftor (MD 2.88, 95% CI 2.48 to 3.29; I2 = 36%; 5 studies, 946 participants; low‐certainty evidence; Analysis 18.1).
18.1. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 1: CFQ‐R respiratory domain (absolute change from baseline)
One study also examined the other domains of the CFQ‐R questionnaire at six months (Taylor‐Cousar 2017). In five out of 12 domains (including the respiratory symptoms domain already reported above) there was a greater improvement in the active intervention group compared to placebo: physical functioning (MD 3.80, 95% CI 1.90 to 5.70; Analysis 18.2); treatment burden (MD 3.40, 95% CI 1.60 to 5.20; Analysis 18.3); health perceptions (MD 3.20, 95% CI 1.20 to 5.20; Analysis 18.4); and vitality (MD 2.30, 95% CI 0.10 to 4.50; Analysis 18.5). In the remaining seven domains (social functioning, role functioning, eating problems, emotional functioning, weight, digestive symptoms and body image), there was no difference between active treatment and placebo groups (Analysis 18.6; Analysis 18.7; Analysis 18.8; Analysis 18.9; Analysis 18.10; Analysis 18.11; Analysis 18.12).
18.3. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 3: CFQ‐R treatment burden domain (absolute change from baseline)
18.4. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 4: CFQ‐R health perceptions domain (absolute change from baseline)
18.5. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 5: CFQ‐R vitality domain (absolute change from baseline)
18.6. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 6: CFQ‐R social functioning domain (absolute change from baseline)
18.7. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 7: CFQ‐R role functioning domain (absolute change from baseline)
18.8. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 8: CFQ‐R eating problems domain (absolute change from baseline)
18.9. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 9: CFQ‐R emotional functioning (absolute change from baseline)
18.10. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 10: CFQ‐R weight domain (absolute change from baseline)
18.11. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 11: CFQ‐R digestive symptoms domain(absolute change from baseline)
18.12. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 12: CFQ‐R body image domain (absolute change from baseline)
One study (67 participants) reported the mean within‐group change from baseline in the CFQ‐R respiratory domain score at week eight; for the tezacaftor‐ivacaftor group this was 2.30 points (95% CI ‐0.10 to 4.60; P = 0.0546), compared to a mean within‐group change of 9.20 (SD 23.10) points in the placebo group, and 2.80 (SD 9.60) points in the ivacaftor group (Davies 2021).
3. Physiological measures of lung function
a. FEV1 (relative change from baseline)
i. Immediate term (up to and including one month)
Lumacaftor plus ivacaftor versus placebo
We have requested immediate‐term data for this domain from the investigators of two studies (TRAFFIC 2015; TRANSPORT 2015). We will include any unpublished information we receive in a future update.
Tezacaftor plus ivacaftor versus control
There was no difference between tezacaftor plus ivacaftor compared to ivacaftor alone at one month (MD 3.72, 95% CI ‐7.77 to 15.21; 1 study, 18 participants; Analysis 18.13) (Donaldson 2018).
18.13. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 13: FEV1 % predicted (relative change from baseline)
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
At six months, participants in the TRAFFIC and TRANSPORT studies in the the lumacaftor 600 mg group experienced greater relative changes from baseline in FEV1 (% predicted) compared to the placebo group (MD 5.63, 95% CI 3.80 to 7.47; 2 studies, 720 participants; Analysis 10.4). This was also true for the lumacaftor 400 mg group, when data were pooled with the participants from the Wilson study (MD 4.69, 95% CI 2.91 to 6.46; I2 = 0%; 3 studies, 777 participants; Analysis 11.4) (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
10.4. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 4: FEV1 % predicted (relative change from baseline)
11.4. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 4: FEV1 % predicted (relative change from baseline)
There was also a greater relative change from baseline in FEV1 % predicted compared to the placebo group when the two lumacaftor doses were pooled (MD 5.12, 95% CI 3.57 to 6.67; I2 = 0%; 3 studies, 1134 participants; high‐certainty evidence; Analysis 12.4).
12.4. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 4: FEV1 % predicted (relative change from baseline)
Tezacaftor plus ivacaftor versus control
One arm of one study reported on the relative change from baseline in FEV1 % predicted at three months (NCT02070744). We found no difference between the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor group compared to either placebo or 150 mg twice‐daily ivacaftor (MD 1.60, 95% CI ‐7.63 to 10.83; 1 study, 11 participants; Analysis 17.2).
17.2. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 2: Relative change from baseline in % predicted FEV1
Five studies (including the second arm of the NCT02070744 trial) reported at up to six months (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021; Taylor‐Cousar 2017). Data showed a greater relative change from baseline in FEV1 % predicted in the 100 mg tezacaftor once daily plus 150 mg twice‐daily ivacaftor group compared to control (either placebo or 150 mg twice‐daily ivacaftor) (MD 0.92, 95% CI 0.72 to 1.11; I2 = 94%; 5 studies, 944 participants; low‐certainty evidence; Analysis 18.13). We investigated this very high level of heterogeneity and when we split the results by genotype of participants, McKone 2021 and Munck 2020 included participants heterozygous for F508del, while NCT02070744, Schwarz 2021 and Taylor‐Cousar 2017 included participants homozygous for F508del); results from both groups showed no heterogeneity (I2 = 0%).
b. FEV1 absolute values
i. Immediate term (up to and including one month)
Lumacaftor plus ivacaftor versus placebo
In two studies, participants in both the lumacaftor 600 mg group and the lumacaftor 400 mg group experienced greater absolute changes from baseline in FEV1 % predicted at 28 days compared to the placebo group (MD 2.32, 95% CI 1.34 to 3.31; 2 studies, 739 participants; Analysis 10.5 and MD 2.42, 95% CI 1.43 to 3.40; 2 studies, 740 participants; Analysis 11.5, respectively) (TRAFFIC 2015; TRANSPORT 2015). There was also a greater absolute change from baseline in FEV1 % predicted compared to the placebo group when the two lumacaftor doses were pooled (MD 2.37, 95% CI 1.52 to 3.22; Analysis 12.5). These data were extrapolated and we have requested confirmation of the exact data from the study investigators. Any unpublished information we receive will be included in a future update.
10.5. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 5: FEV1 % predicted (absolute change from baseline)
11.5. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 5: FEV1 % predicted (absolute change from baseline)
12.5. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 5: FEV1 % predicted (absolute change from baseline)
The Phase 2 lumacaftor‐ivacaftor study (62 participants) reported on the absolute change from baseline in FEV1 % predicted after lumacaftor monotherapy (day 14) and lumacaftor (200 mg daily) and ivacaftor (150 mg or 250 mg twice daily) combination therapy (day 21) (Boyle 2014). Results for lumacaftor monotherapy are discussed above (see 'Correctors (monotherapy) compared to placebo').
Small, but statistically non‐significant improvements in FEV1 % predicted were reported at day 21 for participants treated with 200 mg lumacaftor once daily (day 1 to 21) and either 150 mg ivacaftor twice daily (day 15 to 21) (MD 2.80, 95% CI ‐1.39 to 6.99; Analysis 13.1) or 250 mg ivacaftor twice daily (day 15 to 21) (MD 0.20, 95% CI ‐4.20 to 4.60; Analysis 14.1), respectively, and when ivacaftor doses were combined (MD 1.57, 95% CI ‐2.13 to 5.27; moderate‐certainty evidence; Analysis 16.1) (Boyle 2014).
13.1. Analysis.

Comparison 13: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (150 mg twice daily) for days 15 to 21 versus placebo, Outcome 1: FEV1 % predicted (absolute change from baseline)
14.1. Analysis.

Comparison 14: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (250 mg twice daily) for days 15 to 21 versus placebo, Outcome 1: FEV1 % predicted (absolute change from baseline)
16.1. Analysis.

Comparison 16: Lumacaftor (200 mg once daily monotherapy for 14 days) plus ivacaftor (150 mg or 250 mg twice daily for days 15 to 21) for 21 days, Outcome 1: FEV1 % predicted (absolute change from baseline)
Tezacaftor plus ivacaftor versus control
At one month, there was a greater absolute improvement from baseline in FEV1 % predicted compared to the control groups in the three tezacaftor‐ivacaftor studies (MD 3.54, 95% CI 2.39 to 4.69; 3 studies, 556 participants; Analysis 18.14) (Taylor‐Cousar 2017; Donaldson 2018; NCT02508207).
18.14. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 14: FEV1 % predicted (absolute change from baseline)
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
In the TRAFFIC and TRANSPORT studies (1108 participants), at six months the difference in the absolute change from baseline in FEV1 % predicted was maintained in the lumacaftor 600 mg group (MD 3.34, 95% CI 2.30 to 4.38; 2 studies, 720 participants; Analysis 10.5). This was also true when these two studies were combined with a third (n = 70) comparing lumacaftor 400 mg to placebo (MD 2.83, 95% CI 1.81 to 3.84; 3 studies, 777 participants; Analysis 11.5) (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). There was also a greater absolute change from baseline in FEV1 % predicted compared to the placebo group when the two lumacaftor doses were pooled (MD 3.08, 95% CI 2.20 to 3.97; I2 = 0%; 3 studies, 1134 participants; moderate‐certainty evidence; Analysis 12.5).
In the paediatric combination study, investigators reported a greater absolute change from baseline in FEV1 % predicted in the lumacaftor plus ivacaftor group compared to the placebo group up to and including six months (MD 2.40, 95% CI 0.40 to 4.40; 1 study, 204 participants; low‐certainty evidence; Analysis 15.2) (Ratjen 2017). Additional results at earlier time points (day 15, week 4 and week 16) were published graphically in the study report, but the graphical plots were too small to allow for accurate extraction of data (Ratjen 2017). We have requested numerical data for these time points from the study investigators and will include any unpublished information we receive in a future update. Investigators in this study also reported early post‐drug dose declines in FEV1 % predicted at day one in the lumacaftor plus ivacaftor group (Ratjen 2017). A markedly smaller decline was observed post‐dose at day 15, and no decline was observed by four months. These data are not available for all participants, so are not entered into analysis for this review; instead these results are presented in the additional tables (Table 20).
15.2. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 2: FEV1 % predicted (absolute change from baseline)
9. Acute changes in FEV1 (% predicted) following study drug administration in Ratjen 2017.
|
Lumacaftor plus ivacaftor Mean (SD) |
Placebo Mean (SD) |
|
| Day 1, ≤ 2 hours post dose | n = 91 ‐5.5 (8.2) |
n = 97 ‐0.1 (5.1) |
| Day 1, 4 to 6 hours post dose | n = 92 ‐7.7 (7.3) |
n = 96 ‐1.4 (7.1) |
| Day 1, 24 hours post dose | n = 38 ‐4.1 (10.1) |
n = 44 ‐1.7 (6.8) |
| Day 15, ≤ 2 hours post dose | n = 88 ‐1.4 (7.0) |
n = 87 0.9 (5.5) |
| Day 15, 4 to 6 hours post dose | n = 86 ‐1.3 (6.4) |
n = 87 0.1 (5.2) |
| Week 16, ≤ 2 hours post dose | n = 33 1.7 (4.8) |
n = 42 0.8 (5.8) |
| Week 16, 4 to 6 hours post dose | n = 33 0.5 (7.4) |
n = 42 0.6 (7.1) |
| Week 24, ≤ 2 hours post dose | n = 25 0.3 (4.1) |
n = 23 0.0 (3.4) |
| Week 24, 4 to 6 hours post dose | n = 24 ‐2.8 (4.0) |
n = 24 0.1 (4.3) |
SD: standard deviation
Tezacaftor plus ivacaftor versus control
At three months, one arm of one study showed no difference in the absolute change from baseline in FEV1 % predicted between the intervention and control groups when tezacaftor was given at 50 mg twice a day (MD 1.00 % predicted, 95% CI ‐3.35 to 5.35; 1 study, 11 participants; Analysis 17.3) (NCT02070744).
17.3. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 3: Absolute change from baseline in % predicted FEV1
Five studies (n = 944; including the second arm of the NCT02070744 trial) reported the absolute change from baseline in FEV1 % predicted at up to six months (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021; Taylor‐Cousar 2017). There was a greater change in the 100 mg tezacaftor plus 150 mg twice daily ivacaftor group compared to control (either placebo or 150 mg twice‐daily ivacaftor) (MD 0.39% predicted, 95% CI 0.27 to 0.52; I2 = 95%; 5 studies, 944 participants; low‐certainty evidence; Analysis 18.14). Similar to the investigation of heterogeneity for the earlier outcome of relative change in FEV1 % predicted, spitting by genotype resulted in no heterogeneity between the two studies recruiting participants heterozygous for F508del (McKone 2021; Munck 2020). However, there was still heterogeneity (I2 = 47%) between the three studies recruiting participants homozygous for F508del (NCT02070744; Schwarz 2021; Taylor‐Cousar 2017); removing Taylor‐Cousar 2017 (n = 504) from the analysis reduced heterogeneity between the two smaller studies to zero (NCT02070744, n = 28; Schwarz 2021, n = 97).
In the Davies study, through week eight, the mean within‐group change from baseline in FEV1 % predicted for the tezacaftor‐ivacaftor group was 2.80 % predicted (95% CI 1.00 to 4.60; P = 0.0024), compared to a mean (SD) within‐group change of ‐3.70 (6.10) % predicted in the placebo group, and ‐0.40 (6.00) % predicted in the ivacaftor group (Davies 2021).
c. FVC (absolute values and change from baseline)
No study in this comparison reported data for this outcome (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). However, the protocol of the TRAFFIC and TRANSPORT studies states that FVC data were collected (although not considered as an outcome). We have requested any recorded data relevant to this outcome from the study investigators and will include any unpublished information we receive in a future update.
d. LCI
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
Only one study (n = 204) reported this outcome and its primary outcome was LCI2.5, i.e. the number of lung volume turnovers required to reach 2.5% of starting tracer gas concentration (Ratjen 2017). There was a significantly larger reduction in LCI2.5 in the lumacaftor plus ivacaftor group compared to the placebo group up to and including six months (MD ‐1.10, 95% CI ‐1.40 to ‐0.80; Analysis 15.3). Additional results at earlier time points (day 15, one month and four months) were published graphically in the full study report, but the graphical plots were too small to allow for accurate extraction of data (Ratjen 2017). We have requested numerical data for these time points from the study investigators and will include any unpublished information we receive in a future update.
15.3. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 3: LCI2.5 (absolute change from baseline)
The study investigators also list LCI5.0 as a secondary outcome of the study, i.e. number of lung volume turnovers required to reach 5% of starting tracer gas concentration (Ratjen 2017). However, results for this outcome are not presented. We have requested numerical data for these time points from the study investigators and will include any unpublished information we receive in a future update.
Tezacaftor plus ivacaftor versus control
At week eight, the Davies study reported that the mean within‐group change from baseline in LCI₂.₅ for the tezacaftor‐ivacaftor group was ‐0.51 (95% CI ‐0.74 to ‐0.29; P < 0.0001), compared to a mean (SD) within‐group change of 0.10 (1.16) in the placebo group and ‐0.61 (0.88) in the ivacaftor group (Davies 2021). Furthermore, in the same study, at week eight, the mean within‐group change from baseline in LCI₅․₀ for the tezacaftor‐ivacaftor group was ‐0.30 (95% CI ‐0.39 to ‐0.20; P < 0.0001), compared to a mean (SD) within‐group change of 0.08 (0.36) in the placebo group, and ‐0.48 (0.51) in the ivacaftor group (Davies 2021).
ii. Longer term (over six months)
Lumacaftor plus ivacaftor versus placebo
One study found a slightly greater change from baseline in LCI2.5 at over six months in the lumacaftor‐ivacaftor group compared to placebo (MD ‐0.69, 95% CI ‐1.35 to ‐0.03; 1 study, 51 participants; Analysis 9.1) (Stahl 2021).
9.1. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 1: Absolute change from baseline in lung clearance index 2.5 (LCI2.5)
Secondary outcomes
1. Adverse events
All studies examining combination therapies reported AEs (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). The type and extent of AE reporting was not consistent across studies, making comparison between different treatment regimens and interventions a challenge. For the Phase 3 studies, a common AE was defined by the researchers as one that occurred in more than 10% of participants. For nine studies we have used the definition of a common AE as being an event that occurred in more than 10% of participants (Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017).
Lumacaftor plus ivacaftor versus placebo
Three studies reported no differences in the number of participants experiencing AEs during the study, either by lumacaftor dose or when lumacaftor doses were pooled: for 600 mg lumacaftor, OR 1.00 (99% CI 0.37 to 2.71; 2 studies, 739 participants; Analysis 10.6); for 400 mg lumacaftor, OR 0.66 (99% CI 0.28 to 1.60; 3 studies, 808 participants; Analysis 11.6), and for combined doses, OR 0.76 (99% CI 0.34 to 1.71; I2 = 0%; 3 studies, 1178 participants; Analysis 12.6) (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
10.6. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 6: Adverse events by end of study (at 6 months)
11.6. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 6: Adverse events by end of study (at 6 months)
12.6. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 6: Adverse events by end of study (at 6 months)
In the paediatric lumacaftor‐ivacaftor study, the overall rate of reporting of AEs was lower than for the TRAFFIC and TRANSPORT studies with a similar profile, including increased reporting of chest tightness on starting the lumacaftor‐ivacaftor intervention compared to placebo (Ratjen 2017). The study also reported no significant difference between the lumacaftor plus ivacaftor group compared to placebo in the number of participants experiencing AEs during the study (OR 0.60, 99% CI 0.14 to 2.58; 1 study, 204 participants; low‐certainty evidence; Analysis 15.4).
15.4. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 4: Treatment‐emergent adverse events with incidence > 10% in any treatment group (at 6 months)
Stahl 2021 reported no differences between groups in the number of serious AEs (OR 1.75, 99% CI 0.19 to 16.29; 1 study, 51 participants; Analysis 9.2) and other (not including serious) AEs (OR 0.41, 99% CI 0.01 to 23.66; 1 study, 51 participants; Analysis 9.2).
9.2. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 2: Most common adverse events (occurring in at least 10% of participants in either group)
Tezacaftor plus ivacaftor versus control
For the tezacaftor‐ivacaftor studies, we present the most common AEs that occurred in at least 10% of participants in either study (moderate‐certainty evidence; Analysis 18.16). There were two studies that reported less commonly occuring AEs and these are presented in the original study reports, none of which showed any difference between groups (Donaldson 2018; Taylor‐Cousar 2017).
18.16. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 16: Most common adverse events (occurring in at least 10% of participants in either group)
a. Mild (therapy does not need to be discontinued)
Lumacaftor plus ivacaftor versus placebo
Boyle reported data for lumacaftor‐ivacaftor combination therapy at 21 days (day 14 to 21) (Boyle 2014). The combined analysis showed no differences between participants treated with lumacaftor‐ivacaftor combination therapy and placebo in the number of participants experiencing cough, oropharyngeal pain, nasal congestion, dizziness, a prolonged prothrombin time and upper respiratory tract infection (Analysis 13.2; Analysis 14.2; Analysis 16.2) (low‐certainty evidence).
13.2. Analysis.

Comparison 13: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (150 mg twice daily) for days 15 to 21 versus placebo, Outcome 2: Adverse events occurring in 10% or more participants (from days 15 to 21)
14.2. Analysis.

Comparison 14: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (250 mg twice daily) for days 15 to 21 versus placebo, Outcome 2: Adverse events occurring in 10% or more participants (from days 15 to 21)
16.2. Analysis.

Comparison 16: Lumacaftor (200 mg once daily monotherapy for 14 days) plus ivacaftor (150 mg or 250 mg twice daily for days 15 to 21) for 21 days, Outcome 2: Adverse events occurring in 10% or more participants (from days 15 to 21)
For participants in the three studies receiving the lumacaftor‐ivacaftor therapy, the most regularly reported AEs were respiratory in nature (e.g. chest tightness) (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). In TRAFFIC and TRANSPORT, most respiratory AEs occurred shortly after starting lumacaftor‐ivacaftor therapy and for those who continued with the intervention they were reported to be transient in nature. Dyspnoea was more common in the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group compared to placebo, OR 2.05 (99% CI 1.10 to 3.83) (Analysis 10.6) and when lumacaftor doses were combined, OR 1.78 (99% CI 1.02 to 3.08) (Analysis 12.6). Cough was less common in the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group compared to placebo, OR 0.59 (99% CI 0.40 to 0.87) (Analysis 11.6) and when lumacaftor doses were combined, OR 0.65 (99% CI 0.46 to 0.91) (Analysis 12.6). Infective pulmonary exacerbations were less common in the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group compared to placebo, OR 0.60 (99% CI 0.42 to 0.87) (Analysis 11.6) and when lumacaftor doses were combined, OR 0.64 (99% CI 0.46 to 0.88) (Analysis 12.6).
There were no differences between lumacaftor 600 mg once daily, lumacaftor 400 mg twice daily plus 250 mg ivacaftor twice daily or lumacaftor doses combined compared to placebo in terms of the number of participants experiencing other AEs: headache, haemoptysis, diarrhoea, abnormal respiration, increased sputum, nasopharyngitis, oropharyngeal pain, abdominal pain, fatigue, nausea, pyrexia, nasal congestion, upper respiratory tract infection (Analysis 10.6; Analysis 11.6, Analysis 12.6).
In the TRAFFIC and TRANSPORT studies, in seven participants receiving lumacaftor‐ivacaftor therapy abnormal liver function (elevated liver enzyme) results led to a temporary discontinuation of the intervention, after which liver function improved (to baseline in six participants). Treatment was re‐started in six of these participants; in one participant, abnormal liver function was associated with hepatitis E infection (TRAFFIC 2015; TRANSPORT 2015).
The paediatric lumacaftor‐ivacaftor study reported the number of treatment‐emergent AEs with an incidence over 10% (Ratjen 2017). Productive cough, nasal congestion, oropharyngeal pain, upper abdominal pain, rhinorrhoea and increased sputum were observed more frequently in the lumacaftor‐ivacaftor group compared to the placebo group, but there was no statistically significant difference between the groups (Analysis 15.4). There were also no differences between the groups in terms of cough, pyrexia, headache, upper respiratory tract infection, abdominal pain, nausea, vomiting, fatigue and respiratory events (such as wheezing, dyspnoea, asthma and chest discomfort) (Analysis 15.4).
The Stahl 2021 study did not report any differences between any of the groups in the following AEs: abdominal pain, upper abdominal pain, constipation, diarrhoea, pale faeces, vomiting, pyrexia, positive carrier status for bacterial disease, gastroenteritis, infective pulmonary exacerbation of CF, nasopharyngitis, rhinitis, upper respiratory tract infection, lung infiltration, positive test for P aeruginosa, headache, cough, dyspnoea, epistaxis, nasal congestion, nasal polyps and productive cough (Analysis 9.2).
Tezacaftor plus ivacaftor versus control
There were no differences between tezacaftor‐ivacaftor and control groups (99% CIs) in the number of participants (eight studies, 1057 participants) experiencing the following AEs (moderate‐certainty evidence): cough, pulmonary exacerbation, headache, nasal congestion or nasopharyngitis, increased sputum, haemoptysis, pyrexia, oropharyngeal pain, nausea or fatigue, upper abdominal pain, dyspnoea, lung infection with P aeruginosa, respiratory tract infection (either upper or lower), gastroenteritis, influenza, pharyngitis, sunburn, positive bacterial test, migraine, arthralgia, renal impairment, acute and chronic sinusitis, pneumothorax, tinnitus, diarrhoea, dyspepsia, non‐cardiac chest pain, muscle strain, decreased pulmonary function test, decrease in weight, increased alanine aminotransferase, increased heart rate, abnormal breath sounds, decrease in FEV1, decreased vitamin D, decreased oxygen saturation, iron deficiency, back pain, muscle spasms, musculoskeletal pain, hyposmia, sinus headache, testicular pain, amenorrhoea, abnormal respiration, lower respiratory tract congestion, discoloured sputum, epistaxis, nasal oedema, paranasal sinus hypersecretion, productive cough, sinus congestion, rales and rash (Analysis 17.4; Analysis 18.16) (Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017). Taylor‐Cousar specified respiratory compromise on initiation as an AE in light of the reports from the TRAFFIC and TRANSPORT studies, but there was no increased reporting of this event (Taylor‐Cousar 2017).
17.4. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group)
The most common AEs occurring in at least 10% of participants in the tezacaftor‐ivacaftor group in the Davies study were cough (14.8%), headache (14.8%) and productive cough (13.0%). Of the AEs in the intervention group in this study, most were considered not related (38.9%) or unlikely related (18.5%) to the drug (Davies 2021).
b. Moderate (therapy is discontinued, and the adverse effect ceases)
Lumacaftor plus ivacaftor versus placebo
None of the participants in the Phase 2 lumacaftor‐ivacaftor study required study drug interruption for the AEs of therapy (Boyle 2014). It was not stated in the TRAFFIC and TRANSPORT studies whether study drug interruption for the AEs of therapy was required for any participants (TRAFFIC 2015; TRANSPORT 2015). The combined safety data demonstrated similar rates of serious AE reporting for participants receiving placebo (28.6%) and those receiving the lumacaftor‐ivacaftor combination therapy (17.3% to 22.8%); however, the characteristics of these events were different (Analysis 10.6; Analysis 11.6; Analysis 12.6). Although it was not stated whether study drug interruption was required in the Wilson study, two out of 34 participants in the intervention group discontinued treatment due to AEs, whereas no participants in the control group discontinued treatment (Wilson 2021).
In three studies, 14 participants on lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily and 19 participants on lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily discontinued the study due to AEs. In total, 33 of 772 (4.3%) of participants receiving lumacaftor‐ivacaftor discontinued compared to six of 406 (1.5%) participants receiving placebo (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). The differences in the discontinuation rates in the treatment groups were not statistically significant compared to placebo at the 1% statistical significance level to allow for multiple analyses related to AEs: 600 mg once‐daily lumacaftor plus 250 mg twice‐daily ivacaftor versus placebo (OR 2.38, 99% CI 0.67 to 8.50; Analysis 10.6); 400 mg twice‐daily lumacaftor plus 250 mg twice‐daily ivacaftor versus placebo (OR 3.11, 99% CI 0.96 to 10.11; Analysis 11.6); and 600 mg once‐daily or 400 mg twice‐daily lumacaftor plus 250 mg twice‐daily ivacaftor versus placebo (OR 2.81, 99% CI 0.92 to 8.57; Analysis 12.6).
In the same studies, 84 participants on lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily, 79 participants on lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily and 115 participants on placebo experienced at least one serious AE, defined as "death, life threatening adverse experience, in‐patient hospitalization/prolongation of hospitalization, persistent/significant disability or incapacity, congenital anomaly/birth defect, important medical event" (TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). The following AEs, which occurred on more than one occasion, were reported to have resulted in discontinuation of the lumacaftor‐ivacaftor therapy: elevated serum creatinine kinase level (n = 4), haemoptysis (n = 3), bronchospasm (n = 2), dyspnoea (n = 2), pulmonary exacerbation (n = 2) (see below) and rash (n = 2). One participant developed hypertension and discontinued the study (not included in the initial reports of this study); other reasons for discontinuation were not recorded (TRAFFIC 2015; TRANSPORT 2015).
There was no difference in the number of AEs between participants on lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily compared to placebo (OR 0.73, 99% CI 0.47 to 1.13; Analysis 10.6) and fewer participants experienced serious AEs on lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily compared to placebo (OR 0.62, 99% CI 0.40 to 0.95; Analysis 11.6); this was also true when lumacaftor doses were combined (OR 0.69, 99% CI 0.48 to 1.00; Analysis 12.6).
The paediatric study of lumacaftor‐ivacaftor did not report a difference between groups in the number of serious AEs (OR 1.18, 99% CI 0.50 to 2.78; Analysis 15.4). There were 13 participants who had serious AEs in the lumacaftor plus ivacaftor group; these were considered to be treatment‐related in two participants (one drug interaction and one obstructive airways disorder). In the placebo group, 11 participants had serious AEs; these were considered to be treatment‐related in three participants (one distal intestinal obstruction syndrome, two elevated aminotransferases) (Ratjen 2017). In this study, six out of 103 participants discontinued, three due to AEs. One participant discontinued due to an early respiratory event, a second due to persistently abnormal liver function tests and the reasons for the remaining four who discontinued were not recorded (Ratjen 2017).
In the longer‐term follow‐up study to the TRAFFIC and TRANSPORT studies (PROGRESS), in which participants were randomised to two different lumacaftor‐ivacaftor dose regimens, the paper reported that 7% of the participants withdrew because of AEs during the 96‐week study period; in one participant this was due to hypertension (PROGRESS 2017).
It is not clear if any participants in the Stahl study discontinued treatment due to AEs. Information on the trial registry entry states that one participant did not complete the study in the intervention group; however, the published paper states that no AE led to the discontinuation of treatment (Stahl 2021).
Tezacaftor plus ivacaftor versus control
In the four placebo‐controlled studies evaluating 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor studies, 10 out of 460 participants receiving tezacaftor‐ivacaftor discontinued compared to 12 out of 465 participants in the control groups (McKone 2021; Munck 2020; Schwarz 2021; Taylor‐Cousar 2017). Reasons for discontinuation in the tezacaftor‐ivacaftor group included abdominal pain (n = 2), raised serum creatinine phosphokinase (n = 1), raised liver enzymes (n = 1), a generalised tonic‐clonic seizure (n = 1), adenocarcinoma of the colon (n = 1), malaise (n = 1) and multiple organ dysfunction syndrome and sepsis in the setting of influenza infection (n = 1). It is unclear as to the number of participants who interrupted and discontinued treatment in the two studies comparing 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to placebo (NCT02508207; NCT02730208).
Across the 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor studies, we found no differences between the number of participants experiencing treatment‐emergent AEs and serious AEs when compared to control (OR 0.79, 99% CI 0.49 to 1.26; I2 = 63%; 6 studies, 1000 participants; Analysis 18.16 and OR 0.61, 99% CI 0.38 to 0.98; I2 = 0%; 6 studies, 1000 participants; Analysis 18.16) (McKone 2021; Munck 2020; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017). Regarding treatment‐emergent AEs, removing the results from NCT02508207 reduced I2 to 0%; we are unable to explain this heterogeneity.
All participants in the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor study experienced treatment‐emergent AEs, and there was no difference between groups in the number of participants who experienced serious AEs (OR 0.49, 99% CI 0.08 to 3.02; Analysis 17.4). It is unclear as to the number of participants who interrupted and discontinued treatment (NCT02070744).
No participants in the tezacaftor‐ivacaftor group had AEs that led to either treatment interruption or discontinuation in the Davies study. There were no participants in either the ivacaftor or placebo group who discontinued treatment after receiving at least one dose (Davies 2021).
c. Severe (life‐threatening or debilitating, or which persists even after treatment is discontinued)
Lumacaftor plus ivacaftor versus placebo
There were no AEs reported in trials evaluating lumacaftor‐ivacaftor, which in our assessment were life‐threatening or debilitating. When treatments were discontinued, the reported AEs resolved (Boyle 2014; Ratjen 2017; Stahl 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
Tezacaftor plus ivacaftor versus control
Two studies (n = 608) comparing 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to placebo reported two life‐threatening AEs in participants in the tezacaftor‐ivacaftor treatment groups: one due to haemoptysis and one due to postinfluenza sepsis and multiple organ dysfunction syndrome (Schwarz 2021; Taylor‐Cousar 2017).
There were no life‐threatening events recorded in four studies (Davies 2021; McKone 2021; Munck 2020; NCT02508207) and it is unclear whether any of the serious AEs recorded in two studies were deemed to be life‐threatening (NCT02070744; NCT02730208).
d. Other adverse effects of therapy (of any severity) that are not classifiable according to these categories
All studies reported on the number of participants who experienced episodes of pulmonary exacerbations described as AEs (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). We present these results in the analyses (Analysis 9.2; Analysis 10.6; Analysis 11.6; Analysis 12.6; Analysis 13.2; Analysis 14.2; Analysis 16.2; Analysis 15.4; Analysis 17.4; Analysis 18.16; Analysis 18.19) and they are also described below (see 'Extra courses of antibiotics').
18.19. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 19: Number of pulmonary exacerbations
Investigators in Davies 2021 reported that 3 out of 54 participants in the tezacaftor‐ivacaftor group, 2 out of 10 participants in the placebo group and no participants in the ivacaftor group experienced an infective pulmonary exacerbation; these data are not presented in the analysis (Davies 2021).
Lumacaftor plus ivacaftor versus placebo
After a second participant on lumacaftor‐ivacaftor was withdrawn with hypertension, the researchers for the TRAFFIC and TRANSPORT and PROGRESS studies reported the blood pressure measurements for the participants over the total study period (PROGRESS 2017; TRAFFIC 2015; TRANSPORT 2015). Average blood pressure data were presented from participants over the total 120‐week study period of TRAFFIC and TRANSPORT and PROGRESS for participants who continued on lumacaftor‐ivacaftor, but only for those who received the 400 mg twice daily dose, as this was the dose for which the company received a product licence (PROGRESS 2017). There was a significant mean (SE) increase in systolic blood pressure of 5.1 (1.5) mm Hg and in diastolic blood pressure of 4.1 (1.2) mm Hg (n = 80) (PROGRESS 2017).
2. Hospitalisation
Two lumacaftor‐ivacaftor studies reported data for this outcome (TRAFFIC 2015; TRANSPORT 2015), as well as the tezacaftor‐ivacaftor study (Taylor‐Cousar 2017). The remaining studies in this comparison did not report on hospitalisation (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Wilson 2021).
ii. Short term (over one month and up to and including six months)
Tezacaftor plus ivacaftor versus control
The rate of pulmonary exacerbations that led to hospitalisation or treatment with intravenous antibiotic agents (or both) was lower in the tezacaftor‐ivacaftor group than in the placebo group (0.29 versus 0.54 events per year; rate ratio 0.53, 95% CI 0.34 to 0.82) (Taylor‐Cousar 2017).
iii. Long term (over six months)
Lumacaftor plus ivacaftor versus placebo
Exacerbations were protocol‐defined in the TRAFFIC and TRANSPORT studies as exacerbations leading to hospitalisation or treatment with intravenous antibiotics (TRAFFIC 2015; TRANSPORT 2015). We present information relating to events leading to hospitalisation here and information relating to all pulmonary exacerbations below (see 'Extra courses of antibiotics').
The study publications reported on the rate of events per participant leading to hospitalisation over 48 weeks, graphically pooled across both studies (TRAFFIC 2015; TRANSPORT 2015). We estimate that the event rate per participant over 48 weeks in the placebo group was 0.45. The corresponding event rate in the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group was 0.27 (equal to a 39% decrease compared to the placebo group, P = 0.003). In the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group the event rate was 0.18 (equal to a 61% decrease compared to the placebo group, P < 0.001). The presentation of data did not allow us to estimate the rate over the two lumacaftor doses combined. These data were extrapolated and we have requested confirmation of the exact data from the study investigators. Any unpublished information we receive will be included in a future update.
3. School or work attendance
No study reported data for this outcome (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
4. Extra courses of antibiotics
With regards to pulmonary exacerbations, please see our definition above (Types of outcome measures). For this comparison, three studies defined exacerbations as episodes requiring antibiotics or hospitalisation (Munck 2020; TRAFFIC 2015; TRANSPORT 2015); in one study, exacerbations were physician‐defined (Boyle 2014); for four studies, it is unclear whether exacerbations are protocol‐ or physician‐defined (Donaldson 2018; PROGRESS 2017; Ratjen 2017; Taylor‐Cousar 2017).
a. Time to the next course of antibiotics
The paediatric combination study listed the time to first pulmonary exacerbation as a secondary outcome of the study, but did not present results (Ratjen 2017). We have requested numerical data for this outcome from the trial investigators and we will include any unpublished information we receive in a future update of the review.
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
In the TRAFFIC and TRANSPORT studies, when compared to placebo the time to first pulmonary exacerbation was longer in both in the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group (HR 0.70, 95% CI 0.57 to 0.87; 2 studies, 739 participants; moderate‐certainty evidence; Analysis 10.7) and the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group (HR 0.61, 95% CI 0.49 to 0.76; 2 studies, 740 participants; moderate‐certainty evidence; Analysis 11.7). Similarly, the rate of exacerbations was lower in both the active intervention groups compared to placebo; the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group (rate ratio 0.70, 95% CI 0.57 to 0.87; Analysis 10.8) and the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group (rate ratio 0.61, 95% CI 0.49 to 0.76; Analysis 11.8). This information was not reported in the primary journal article, but is available from the study record on ClinicalTrials.gov (TRAFFIC 2015; TRANSPORT 2015). Information regarding time to first pulmonary exacerbation was reported only as a hazard ratio and P value; the SE used in this analysis was estimated using the methods of Parmar (Parmar 1998).
10.7. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 7: Time to first pulmonary exacerbation
11.7. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 7: Time to first pulmonary exacerbation
10.8. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 8: Rate of exacerbations
11.8. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 8: Rate of exacerbations
Tezacaftor plus ivacaftor versus control
The time to first pulmonary exacerbation was longer in the tezacaftor‐ivacaftor group compared to the placebo group (HR 0.64, 95% CI 0.46 to 0.89; 1 study, 506 participants; moderate‐certainty evidence; Analysis 18.17) (Taylor‐Cousar 2017).
18.17. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 17: Time to first pulmonary exacerbation
The published paper of the Munck study showed a Kaplan‐Meier plot of time to first pulmonary exacerbation, though the trials registry stated that as a result of “less than 50% of events, time‐to‐first event data was not estimated” (Munck 2020). The number of participants with at least one pulmonary exacerbation was recorded, and there was no statistically significant difference between groups (OR 0.90, 99% CI 0.36 to 2.30; 1 study, 168 participants; Analysis 18.18).
18.18. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 18: Number of participants with at least 1 pulmonary exacerbation
b. Total number of courses of antibiotics
ii. Immediate term (up to one month)
Data from the Phase 2 lumacaftor‐ivacaftor study on the number of exacerbations were reported at day 21 (Boyle 2014). These data demonstrated no differences between treatment groups from participants receiving 200 mg lumacaftor once daily plus either 150 mg or 250 mg of ivacaftor twice daily or ivacaftor doses combined compared to placebo (OR 2.22, 99% CI 0.08 to 58.11; Analysis 13.2, OR 1.05, 99% CI 0.03 to 44.10; Analysis 14.2 and OR 1.62, 99% CI 0.08 to 34.55; Analysis 16.2, respectively).
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
In two studies, both the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily and 400 mg twice daily plus 250 mg ivacaftor twice daily groups experienced fewer pulmonary exacerbations than the placebo group (OR 0.66, 99% CI 0.45 to 0.97; 2 studies, 739 participants; Analysis 10.6 and OR 0.60, 99% CI 0.42 to 0.87; 3 studies, 808 participants; Analysis 11.6, respectively) (TRAFFIC 2015; TRANSPORT 2015). This difference was also observed for the two lumacaftor doses combined compared to placebo (OR 0.64, 99% CI 0.46 to 0.88; 3 studies, 1178 participants; Analysis 12.6).
The presentation of data did not allow us to estimate the time to first pulmonary exacerbation or rate of exacerbations over the two lumacaftor doses combined. We extrapolated these data and we have requested confirmation of the exact data from the study investigators; we will include any unpublished information we receive in a future update.
There were no differences between treatment groups in the number of pulmonary exacerbations experienced in the paediatric combination study (OR 1.11, 99% CI 0.55 to 2.25; 1 study, 204 participants; Analysis 15.4) (Ratjen 2017).
Tezacaftor plus ivacaftor versus control
The larger study (n = 510) also reported the rate of pulmonary exacerbations that led to hospitalisation or treatment with intravenous antibiotic agents (or both); see secondary outcome 'Hospitalisation' above for further details (Taylor‐Cousar 2017).
There was no difference between groups in the Munck study for the number of pulmonary exacerbations (OR 0.97, 99% CI 0.40 to 2.39; 1 study, 168 participants; Analysis 18.19). There was also no difference between groups measuring the annualised rate of pulmonary exacerbation events ratio (Munck 2020).
ii. Long term (over six months)
The TRAFFIC and TRANSPORT studies also reported the rate of events per participant leading to intravenous antibiotic treatment over 48 weeks graphically pooled across the two studies (TRAFFIC 2015; TRANSPORT 2015). These data were extrapolated and we have requested confirmation of the exact data from the study investigators. Any unpublished information we receive will be included in a future update.
We estimate that the event rate per participant over 48 weeks in the placebo group was 0.58. The corresponding event rate in the lumacaftor 600 mg once daily plus ivacaftor 250 mg twice daily group was 0.32 (equal to a 45% decrease compared to the placebo group, P < 0.001) and in the lumacaftor 400 mg twice daily plus ivacaftor 250 mg twice daily group was 0.18 (equal to a 56% decrease compared to the placebo group, P < 0.001).
5. Sweat chloride (change from baseline) as a measure of CFTR function
Five studies did not report this outcome (NCT02730208; Schwarz 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
ii. Immediate term (up to one month)
Lumacaftor plus ivacaftor versus placebo
Boyle reported that following lumacaftor (day 1 to 21) and ivacaftor (day 15 to 21) combination therapy, data at 21 days demonstrated reductions in sweat chloride concentration in the 150 mg ivacaftor group (MD ‐5.00 mmol/L, 95% CI ‐11.61 to 1.61; Analysis 13.3) and 250 mg ivacaftor group (MD ‐10.90 mmol/L, 95% CI ‐17.60 to ‐4.20; Analysis 14.3), the latter of which was statistically significant (Boyle 2014). There was also a statistically significant reduction in sweat chloride concentration when ivacaftor doses were combined (MD ‐7.95, 95% CI ‐13.81 to ‐2.09; Analysis 16.3).
13.3. Analysis.

Comparison 13: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (150 mg twice daily) for days 15 to 21 versus placebo, Outcome 3: Sweat chloride concentration (mmol/L) (change from baseline)
14.3. Analysis.

Comparison 14: Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (250 mg twice daily) for days 15 to 21 versus placebo, Outcome 3: Sweat chloride concentration (mmol/L) (change from baseline)
16.3. Analysis.

Comparison 16: Lumacaftor (200 mg once daily monotherapy for 14 days) plus ivacaftor (150 mg or 250 mg twice daily for days 15 to 21) for 21 days, Outcome 3: Sweat chloride concentration (mmol/L) (change from baseline)
Tezacaftor plus ivacaftor versus control
Data from three studies showed a reduction in sweat chloride concentration in the tezacaftor‐ivacaftor groups at one month compared to control groups (MD ‐9.14 mmol/L, 95% CI ‐10.93 to ‐7.34; 3 studies, 552 participants; Analysis 18.20) (Donaldson 2018; NCT02508207; Taylor‐Cousar 2017).
18.20. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 20: Sweat chloride (change from baseline)
ii. Short term (over one month and up to and including six months)
The paediatric combination study reported a greater absolute reduction in sweat chloride concentration from baseline in the lumacaftor plus ivacaftor group compared to the placebo group, up to and including four weeks (MD ‐20.80, 95% CI ‐23.40 to ‐18.20; Analysis 15.5). Additional results (at time points day 15, four months and six months) were published graphically in the study publication, but the graphical plots were too small to allow for accurate extraction of data (Ratjen 2017). We have requested numerical data for these time points from the study investigators and will include any unpublished information we receive in a future update.
15.5. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 5: Sweat chloride concentration (absolute change from baseline)
Tezacaftor plus ivacaftor versus control
At week eight, Davies 2021 reported that the mean within‐group change from baseline in sweat chloride concentration for the tezacaftor‐ivacaftor group was ‐12.30 mmol/L (95% CI ‐15.30 to ‐9.30; P < 0.0001), compared to ‐1.00 mmol/L (12.30) in the placebo group, and ‐1.00 mmol/L (9.00) in the ivacaftor group.
At three months, the results from the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor study also demonstrated a difference between the groups, favouring tezacaftor‐ivacaftor (MD ‐13.50, 95% CI ‐19.47 to ‐7.53; 1 study, 10 participants; Analysis 17.5) (NCT02070744).
17.5. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 5: Absolute change from baseline in sweat chloride
The reduction in sweat chloride concentration in the 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor groups seen at one month compared to the control groups (MD ‐9.14 mmol/L, 95% CI ‐10.93 to ‐7.34; I2 = 0%; 3 studies, 552 participants; Analysis 18.20) (Donaldson 2014; NCT02508207; Taylor‐Cousar 2017) was maintained at six months (MD ‐6.38 mmol/L, 95% CI ‐6.89 to ‐5.86; I2 = 93%; 4 studies, 837 participants; Analysis 18.20) (McKone 2021; Munck 2020; NCT02070744; Taylor‐Cousar 2017).
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus control
Stahl 2021 reported a greater reduction in sweat chloride in the lumacaftor‐ivacaftor group compared to placebo at over six months (MD ‐26.40, 95% CI ‐34.57 to ‐18.23; 1 study, 49 participants; Analysis 9.3).
9.3. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 3: Absolute change from baseline in sweat chloride
6. Radiological measures of lung disease (assessed using any scoring system)
Twelve studies did not report data for this outcome (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; Schwarz 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
ii. Short term (over one month and up to and including six months)
A substudy was performed for the Ratjen study (lumacaftor 200 mg twice daily plus ivacaftor 250 mg twice daily) and investigators reported data for CT scans and Brody score for 10 children, seven on active treatment and three on placebo (Ratjen 2017). There was no difference between groups in the change in overall Brody score at up to 24 weeks (MD ‐16.70, 95% CI ‐36.05 to 2.65; 1 study, 10 participants; Analysis 15.6), or in the mean change in the bronchiectasis component of the Brody score (MD ‐2.40, 95% CI ‐4.96 to 0.16; 1 study, 10 participants; Analysis 15.7) or in the mean change in the air trapping component of the Brody score (MD ‐6.60, 95% CI ‐20.77 to 7.57; 1 study, 10 participants; Analysis 15.8).
15.6. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 6: CT Brody score (mean change)
15.7. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 7: CT Brody score bronchiectasis score (mean change)
15.8. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 8: CT Brody score air trapping score (mean change)
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus placebo
Stahl 2021 found no difference in the absolute change from baseline in MRI global chest score at over six months between the lumacaftor‐ivacaftor and placebo groups (MD ‐1.40, 95% CI ‐5.24 to 2.44; 1 study, 47 participants; Analysis 9.4).
9.4. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 4: Absolute change from baseline in MRI global chest score
Tezacaftor plus ivacaftor versus placebo
NCT02730208 compared 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor to placebo and demonstrated no difference between the tezacaftor‐ivacaftor and placebo groups for the absolute change from baseline in total Brody/CF‐CT score at over six months (MD ‐1.48, 95% CI ‐7.25 to 4.29; 1 study, 41 participants; Analysis 18.21).
18.21. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 21: Absolute change in total Brody/CF‐CT score from baseline
7. Acquisition of respiratory pathogens
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus placebo
Stahl 2021 reported that 1 out of 35 participants in the lumacaftor‐ivacaftor group tested positive for P aeruginosa compared to 2 out of 16 participants in the placebo group. However, when analysed, the difference between groups was not statistically significant (OR 0.21, 99% CI 0.01 to 5.36; Analysis 9.2).
Tezacaftor plus ivacaftor versus control
One study looking at 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor reported that 2 out of 20 participants in the tezacaftor‐ivacaftor group had a pseudomonal lung infection compared to no participants in the placebo group at over six months (OR 5.81, 99% CI 0.10 to 341.36; 1 study, 41 participants; Analysis 18.22) (NCT02730208).
18.22. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 22: Lung infection (pseudomonal)
8. Eradication of respiratory pathogens
No study reported data for this outcome (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
9. Nutrition and growth
a. Weight
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
Three studies reported data for this outcome (Ratjen 2017; TRAFFIC 2015; TRANSPORT 2015).
Two studies presented results for the absolute change from baseline in weight (kg) at six months (TRAFFIC 2015; TRANSPORT 2015). Participants in both the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily group and the 400 mg twice daily plus 250 mg ivacaftor twice daily group experienced a greater absolute weight gain from baseline compared to the placebo group (MD 0.80 kg, 95% CI 0.42 to 1.18; 2 studies, 725 participants; Analysis 10.9 and MD 0.65 kg, 95% CI 0.27 to 1.03; 2 studies, 723 participants; Analysis 11.9, respectively). There was also a greater absolute weight gain from baseline compared to the placebo group when the two lumacaftor doses were pooled (MD 0.72 kg, 95% CI 0.39 to 1.05; I2 = 75%; 2 studies, 1081 participants; Analysis 12.7). After examination of the two studies in this analysis, we are unable to identify potential causes for this level of heterogeneity.
10.9. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 9: Weight (kg) (absolute change from baseline)
11.9. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 9: Weight (kg) (absolute change from baseline)
12.7. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 7: Weight (kg) (absolute change from baseline)
The paediatric combination study listed absolute change in weight and absolute change in weight‐for‐age z score as secondary outcomes of the study, but did not present any results (Ratjen 2017). We have requested numerical data from the study investigators and will include any unpublished information we receive in a future update.
Tezacaftor plus ivacaftor versus control
Neither the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor study (NCT02070744) nor the 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor studies (Munck 2020; NCT02070744) found a difference in the change from baseline in body weight between intervention and control groups at three months (MD ‐0.60, 95% CI ‐2.05 to 0.85; 1 study, 11 participants; Analysis 17.6 and MD ‐0.04, 95% CI ‐0.55 to 0.46; I2 = 0%; 2 studies, 195 participants; Analysis 18.23, respectively).
17.6. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 6: Absolute change from baseline in body weight (kg)
18.23. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 23: Absolute change from baseline in body weight
In the Davies study, at week eight, the mean (SD) within‐group change from baseline in weight for the tezacaftor‐ivacaftor group was 0.30 kg (0.80) compared to 0.60 kg (1.00) in the placebo group and 0.50 kg (0.90) in the ivacaftor group (Davies 2021). Davies also reported the mean (SD) within‐group changes from baseline in weight z score: for the tezacaftor‐ivacaftor group ‐0.04 (0.17) compared to ‐0.02 (0.15) in the placebo group and 0.03 (0.23) in the ivacaftor group (Davies 2021).
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus placebo
Stahl 2021 found no difference between groups for the absolute change from baseline in weight‐for‐age z score at over six months (MD 0.20, 95% CI ‐0.01 to 0.41; 1 study, 51 participants; Analysis 9.5).
9.5. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 5: Absolute change from baseline in weight‐for‐age z score
b. BMI
Eight studies reported on the absolute change from baseline in BMI (Davies 2021; Munck 2020; NCT02070744; Ratjen 2017; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021); two studies additionally reported absolute change in BMI‐for‐age z score (Ratjen 2017; Stahl 2021), and two studies reported absolute change in BMI z score (Davies 2021; Munck 2020). One study reported the relative change from baseline in BMI (Wilson 2021).
i. Immediate term (up to and including one month)
Lumacaftor plus ivacaftor versus placebo
At one month, there was no difference in the absolute change in BMI from baseline between the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily and placebo groups (MD 0.01, 95% CI ‐0.07 to 0.09; 2 studies, 739 participants; Analysis 10.10) or the lumacaftor 400 mg twice daily plus 250 mg ivacaftor twice daily and placebo groups (MD 0.02, 95% CI ‐0.06 to 0.10; 2 studies, 740 participants; Analysis 11.10). There was also no difference in absolute change in BMI from baseline compared to the placebo group when the two lumacaftor doses were pooled (MD 0.02, 95% CI ‐0.05 to 0.08; I2 = 0%; 2 studies, 1108 participants; Analysis 12.8).
10.10. Analysis.

Comparison 10: Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 10: BMI (absolute change from baseline)
11.10. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 10: BMI (absolute change from baseline)
12.8. Analysis.

Comparison 12: Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 8: BMI (absolute change from baseline)
Tezacaftor plus ivacaftor versus control
There was no difference between tezacaftor‐ivacaftor and placebo in terms of change from baseline in BMI in the Taylor‐Cousar 2017 study at one month (MD ‐0.03, 95% CI ‐0.13 to 0.07; 1 study, 504 participants; Analysis 18.24).
18.24. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 24: BMI (change from baseline)
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
Despite no immediate differences between treatment groups, at six months participants in both the lumacaftor 600 mg once daily plus 250 mg ivacaftor twice daily and 400 mg twice daily plus 250 mg ivacaftor twice daily groups experienced a greater absolute change in BMI from baseline compared to the placebo group (MD 0.29, 95% CI 0.16 to 0.43; I2 = 0%; 2 studies, 725 participants; Analysis 10.10 and MD 0.25, 95% CI 0.12 to 0.39; I² = 31%; 3 studies, 786 participants; Analysis 11.10, respectively). When both lumacaftor doses were pooled, there was also a greater absolute change in BMI from baseline compared to the placebo group (MD 0.27, 95% CI 0.16 to 0.38; I² = 53%; 3 studies, 1144 participants; Analysis 12.8). After examination of the studies in this analysis, we are unable to identify potential causes for this level of heterogeneity.
At six months, Rajten reported no difference between groups in the absolute change in BMI or the absolute change in BMI‐for‐age z score (MD 0.10, 95% CI ‐0.10 to 0.30; 1 study, 204 participants; Analysis 15.9 and MD 0.00, 95% CI ‐0.10 to 0.10; 1 study, 204 participants; Analysis 15.10, respectively). Additional results for BMI at earlier time points (day 15, one month and four months) were published graphically in the full paper, but the graphical plots were too small to allow for accurate extraction of data (Ratjen 2017). We have requested numerical data for these time points from the study investigators and will include any unpublished information we receive in a future update.
15.9. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 9: BMI (absolute change from baseline)
15.10. Analysis.

Comparison 15: Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 10: BMI‐for‐age z score (absolute change from baseline)
At six months, Wilson 2021 reported no difference between study groups in the relative change from baseline in BMI (MD 1.00, 95% CI ‐1.14 to 3.14; 1 study, 63 participants; Analysis 11.11).
11.11. Analysis.

Comparison 11: Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo, Outcome 11: BMI (relative change from baseline)
Tezacaftor plus ivacaftor versus control
At three months, there was no difference between the 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor and the placebo groups in the absolute change from baseline in BMI (MD ‐0.16, 95% CI ‐0.66 to 0.34; 1 study, 11 participants; Analysis 17.7) (NCT02070744).
17.7. Analysis.

Comparison 17: Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 7: Absolute change from baseline BMI
There was no difference between tezacaftor‐ivacaftor and placebo in terms of change from baseline in BMI (MD 0.02, 95% CI ‐0.09 to 0.13; I2 = 0%; 3 studies, 699 participants; Analysis 18.24) (Munck 2020; NCT02070744; Taylor‐Cousar 2017) and in absolute change from baseline in BMI z score (MD ‐0.05, 95% CI ‐0.20 to 0.10; 1 study, 47 participants; Analysis 18.25) (Munck 2020).
18.25. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 25: Absolute change from baseline in BMI z score
In the Davies study, at week eight, the mean (SD) within‐group change from baseline in BMI for the tezacaftor‐ivacaftor group was ‐0.04 kg/m² (0.43) compared to 0.02 kg/m² (0.41) in the placebo group and 0.11 kg/m² (0.53) in the ivacaftor group (Davies 2021). Davies also reported that the mean (SD) within‐group changes from baseline in BMI z score for the tezacaftor‐ivacaftor group were ‐0.08 (0.27) compared to ‐0.05 (0.22) in the placebo group and 0.08 (0.37) in the ivacaftor group (Davies 2021).
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus control
Stahl 2021 found a greater absolute change from baseline in BMI‐for‐age z score in the lumacaftor‐ivacaftor group compared to the placebo group at 48 weeks (MD 0.44, 95% CI 0.09 to 0.79; 1 study, 51 participants; Analysis 9.6).
9.6. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 6: Absolute change from baseline in BMI‐for‐age z score
c. Height
This outcome was not reported by 11 studies (Boyle 2014; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021).
ii. Short term (over one month and up to and including six months)
Lumacaftor plus ivacaftor versus placebo
One study listed the absolute change in height and absolute change in height‐for‐age z score as a secondary outcome of the study, but did not present results for this outcome (Ratjen 2017). We have requested numerical data for this outcome from the study investigators and will include any unpublished information we receive in a future update.
Tezacaftor plus ivacaftor versus control
At week eight, Davies reported the mean (SD) within‐group changes from baseline in height were 0.90 cm (0.70) for the tezacaftor‐ivacaftor group compared to 1.20 cm (0.50) in the placebo group and 0.90 cm (0.40) in the ivacaftor group (Davies 2021). Additionally, the mean (SD) within‐group changes from baseline in height z score for the tezacaftor‐ivacaftor group were 0.01 (0.13) compared to 0.04 (0.08) in the placebo group and ‐0.01 (0.07) in the ivacaftor group (Davies 2021).
iii. Longer term (over six months)
Lumacaftor plus ivacaftor versus placebo
At over six months, Stahl 2021 found no difference between the intervention and control groups for absolute change from baseline in stature‐for‐age z score (MD ‐0.01, 95% CI ‐0.19 to 0.17; 1 study, 51 participants; Analysis 9.7).
9.7. Analysis.

Comparison 9: Lumacaftor plus ivacaftor versus placebo, Outcome 7: Absolute change from baseline in stature‐for‐age z score
Triple therapy (correctors plus potentiators) compared to control
A total of 11 studies (1872 participants) contributed to the efficacy and safety results in this comparison (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022). The comparisons are as follows; the exact different doses employed and further study details are described above (Included studies).
Four studies (634 participants) compared VX‐659‐tezacaftor‐ivacaftor to placebo‐tezacaftor‐ivacaftor or triple placebo in participants with the genotype F508del/MF or F508del/F508del (Davies 2018a; Davies 2018b; NCT03447249; NCT03460990). Please see Table 7.
Four studies (808 participants) compared elexacaftor‐tezacaftor‐ivacaftor to placebo‐tezacaftor‐ivacaftor or triple placebo in participants with the genotype F508del/MF or F508del/F508del (Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022). Please see Table 8.
One study (271 participants) compared elexacaftor‐tezacaftor‐ivacaftor to either ivacaftor or tezacaftor‐ivacaftor in participants with the genotypes F508del/gating variant and F508del/residual function variant (Barry 2021). Please see Table 9.
One two‐part study (73 participants) compared VX‐440‐tezacaftor‐ivacaftor to triple placebo in participants with the genotypes F508del/MF and F508del/F508del (NCT02951182). For this study, we have narratively described some results and reported other results in the analyses. The participants receiving active treatment were split into three cohorts in the first part; participants in cohort 1A received 200 mg VX‐440 every 12 hours plus 100 mg tezacaftor once daily plus 150 mg ivacaftor every 12 hours for four weeks; participants in cohort 1B (low dose) received 200 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 150 mg ivacaftor every 12 hours for four weeks; and participants in cohort 1B (high dose) received 600 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 300 mg ivacaftor every 12 hours for four weeks. We have analysed the high‐dose data in the review, and narratively described the pooled results from cohorts 1A and 1B (low dose). Participants in the second cohort received 200 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor, and finally participants in the third cohort received 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor. Each cohort was compared against a placebo group. In the second part of the study, participants received 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor in a four‐week run‐in period, and then participants in the treatment group received 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor for four weeks, and participants in the control group received placebo matched to VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor for four weeks. Participants in both cohorts then took 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor for four weeks in a washout period. Please see Table 10.
One two‐part study (76 participants) compared VX‐152‐tezacaftor‐ivacaftor to triple placebo in participants with the genotypes F508del/MF and F508del/F508del (NCT02951195). Please see Table 11.
Important results for this comparison are summarised in the tables (Table 7; Table 8; Table 9; Table 10; Table 11). We have assessed the following outcomes using the GRADE criteria in each of the tables and indicated our findings in the relevant text below:
survival;
QoL (total score);
QoL (respiratory domain);
FEV1 % predicted (relative and absolute change);
AEs; and
time to first pulmonary exacerbation.
For the comparisons examined, we judged the certainty of the evidence to range from low to high, but to be mostly moderate. We downgraded the evidence due to indirectness as results are not applicable to children under the age of 12 and those with more severe disease. We have presented the findings for the comparison of VX‐659 (80 mg once daily, 120 mg twice daily, 240 mg once daily or 400 mg once daily) plus tezacaftor 100 mg once per day plus ivacaftor 150 mg twice daily or deutivacaftor 150 mg once daily compared to triple placebo (for F508del/MF participants) or placebo tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (for F508del/F508del participants) in one table (Table 7). We have presented the findings for the comparison of elexacaftor (50 mg once daily, 100 mg once daily or 200 mg once daily) plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily or deutivacaftor 150 mg once daily compared to triple placebo (for F508del/MF participants) or placebo tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (for F508del/F508del participants) in a separate table (Table 8).
Primary outcomes
1. Survival
None of the included studies reported any deaths (certainty evidence of evidence ranges across comparisons from low to high) (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
2. QoL
a. Total QoL score
None of the triple combination therapy studies reported on total QoL score (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
b. QoL sub‐domains
i. Immediate term (up to and including one month)
Eight studies reported on QoL subdomains at one month (Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990).
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
Two studies (502 participants) reported the absolute change in the respiratory domain of the CFQ‐R score after one month of treatment (Davies 2018b; NCT03447249). Results favoured the intervention over placebo at the 80 mg dose (MD 10.00, 95% CI 0.29 to 19.71; 1 study, 21 participants; moderate‐certainty evidence; Analysis 19.1) (Davies 2018b) and also for the 240 mg dose (MD 16.13, 95% CI 13.02 to 19.24; I2 = 88%; 2 studies, 412 participants; moderate‐certainty evidence; Analysis 21.1) (Davies 2018b; NCT03447249). Investigating this high level of heterogeneity, we note that Davies 2018b was a four‐arm phase 2 trial from which we are reporting just one active arm and the placebo arm (n = 30) compared to NCT03447249, which was a larger phase 3 trial (n = 382); this may explain the differences seen between the study results. We found no difference between the 400 mg group and the placebo group (MD 7.90, 95% CI ‐0.58 to 16.38; 1 study, 32 participants; moderate‐certainty evidence; Analysis 23.1) (Davies 2018b).
19.1. Analysis.

Comparison 19: VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
21.1. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
23.1. Analysis.

Comparison 23: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
Participants with F508del/F508del
There were improvements in the CFQ‐R respiratory domain at doses of VX‐659 240 mg (MD 13.50, 95% CI 8.79 to 18.21; 1 study, 111 participants; moderate‐certainty evidence; Analysis 22.1) (NCT03460990) and VX‐659 400 mg with tezacaftor‐ivacaftor compared to tezacaftor‐ivacaftor‐placebo (MD 18.10, 95% CI 10.85 to 25.35; 1 study, 29 participants; moderate‐certainty evidence; Analysis 24.1) (Davies 2018b).
22.1. Analysis.

Comparison 22: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 1: Quality of life: absolute change in CFQ‐R respiratory domain
24.1. Analysis.

Comparison 24: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
VX‐659 plus tezacaftor plus deutivacaftor
One study tested this combination in a group of participants with F508del/MF, but only at a dose of VX‐659 400 mg (Davies 2018b). Results showed an improvement with the active intervention versus placebo in the respiratory domain (MD 20.30, 95% CI 7.05 to 33.55; 1 study, 25 participants; moderate‐certainty evidence; Analysis 25.1).
25.1. Analysis.

Comparison 25: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
In Keating 2018, elexacaftor showed greater improvements versus placebo in the CFQ‐R respiratory domain at one month at the 50 mg dose (MD 17.20, 95% CI 4.44 to 29.96; 1 study 22 participants; Analysis 26.1) and at the 100 mg dose (MD 14.50, 95% CI 3.72 to 25.28; 1 study, 34 participants; Analysis 27.1). Two studies reported data for the 200 mg dose at one month (Keating 2018; Middleton 2019) and pooled data showed a greater improvement in the CFQ‐R respiratory domain compared to triple placebo (MD 19.15, 95% CI 16.12 to 22.19; I² = 71%; 2 studies, 436 participants; moderate‐certainty evidence; Analysis 28.1). We investigated this heterogeneity between studies and noted that the phase 3 Middleton 2019 study (n = 403) was larger than the phase 1/2 Keating 2018 study (n = 33); also, the participants in Middleton 2019 were slightly younger on average than in Keating 2018. These differences may explain the heterogeneity observed.
26.1. Analysis.

Comparison 26: Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
27.1. Analysis.

Comparison 27: Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory sub‐domain
28.1. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 1: Quality of life: CFQ‐R respiratory domain (change from baseline)
Participants with F508del/F508del
At one month, data from two studies (Keating 2018; Heijerman 2019) showed an improvement in QoL with the elexacaftor 200 mg group compared to tezacaftor‐ivacaftor‐placebo (MD 17.78, 95% CI 12.90 to 22.66; I² = 0%; 2 studies, 135 participants; moderate‐certainty evidence; Analysis 29.1).
29.1. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 1: Quality of life: CFQ‐R respiratory domain (change from baseline)
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
This regimen was only tested at a dose of elexacaftor of 200 mg in one study (Keating 2018). Results showed a greater improvement in the respiratory domain of CFQ‐R compared to placebo (MD 12.80, 95% CI 0.93 to 24.67; 1 study, 29 participants; moderate‐certainty evidence; Analysis 30.1).
30.1. Analysis.

Comparison 30: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 1: Quality of life: change in CFQ‐R respiratory domain
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
This regimen was tested in one study at doses of VX‐152 100 mg twice daily, VX‐152 200 mg twice daily and VX‐152 300 mg twice daily (NCT02951195). Results favoured the intervention group at all doses when compared to placebo: 100 mg twice daily (MD 14.20, 95% CI 0.98 to 27.42; 1 study, 14 participants; low‐certainty evidence; Analysis 32.1); 200 mg twice daily (MD 29.40, 95% CI 16.97 to 41.83; 1 study, 18 participants; low‐certainty evidence; Analysis 33.1); and 300 mg twice daily (MD 26.20, 95% CI 13.71 to 38.69; 1 study, 18 participants; low‐certainty evidence; Analysis 34.1). Compared to the control of placebo plus tezacaftor plus ivacaftor, there was no difference found for VX‐152 200 mg twice daily (MD 4.80, 95% CI ‐6.53 to 16.13; 1 study, 14 participants; low‐certainty evidence; Analysis 35.1), and results favoured the VX‐152 300 mg twice daily group (MD 11.30, 95% CI 3.08 to 19.52; 1 study, 28 participants; low‐certainty evidence; Analysis 36.1).
32.1. Analysis.

Comparison 32: VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
33.1. Analysis.

Comparison 33: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
34.1. Analysis.

Comparison 34: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 1: CFQ‐R Respiratory domain score (absolute change from baseline)
35.1. Analysis.

Comparison 35: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
36.1. Analysis.

Comparison 36: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
Only one study tested this regimen at doses of VX‐440 200 mg twice daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily, 200 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor, and 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor (NCT02951182). Results for the first two dosing schedules were pooled, and there was a greater improvement from baseline in the intervention group, least squares MD 16.1 (95% CI 5.40 to 26.80). For the final dosing schedule, results favoured the intervention group when compared to placebo (MD 18.50, 95% CI 8.99 to 28.01; 1 study, 29 participants; low‐certainty evidence; Analysis 37.1) and tezacaftor plus ivacaftor respectively (MD 20.10, 95% CI 13.76 to 26.44; 1 study, 26 participants; Analysis 38.1).
37.1. Analysis.

Comparison 37: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo, Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
38.1. Analysis.

Comparison 38: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily), Outcome 1: CFQ‐R respiratory domain score (absolute change from baseline)
ii. Short term (over one month and up to and including six months)
VX‐659 plus tezacaftor plus ivacaftor versus placebo
Participants with F508del/MF
At six months, there was a greater improvement in the QoL respiratory domain with VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) compared to placebo (MD 20.10, 95% CI 17.19 to 23.01; 1 study, 382 participants; moderate‐certainty evidence; Analysis 21.1) (NCT03447249)
Elexacaftor plus tezacaftor plus ivacaftor versus triple placebo
Participants with F508del/MF
At six months, results favoured elexacaftor 200 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily compared to triple placebo (MD 20.20, 95% CI 16.19 to 24.21; 1 study, 403 participants; moderate‐certainty evidence; Analysis 28.1) (Middleton 2019).
Elexacaftor plus tezacaftor plus ivacaftor versus either ivacaftor or tezacaftor plus ivacaftor
Participants with F508del/gating and F508del/residual function
One study compared elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (Barry 2021); results showed a greater improvement in the CFQ‐R respiratory domain score with the elexacaftor arm compared to control (MD 8.70, 95% CI 5.34 to 12.06; 1 study, 258 participants; moderate‐certainty evidence; Analysis 31.1).
31.1. Analysis.

Comparison 31: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/gating or F508del/residual function), Outcome 1: Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline)
Elexacaftor plus tezacaftor plus ivacaftor versus placebo plus tezacaftor plus ivacaftor
Participants with F508del/F508del
The greater improvement in CFQ‐R respiratory domain score seen at one month was maintained through to six months (MD 15.90, 95% CI 11.74 to 20.06; 1 study, 175 participants; moderate‐certainty evidence; Analysis 29.1) (Sutharsan 2022).
3. Physiological measures of lung function
a. FEV1 (relative change from baseline)
i. Immediate term (up to and including one month)
Four studies reported on the relative change from baseline in FEV1 % predicted at one month (Davies 2018b; Keating 2018; NCT02951182; NCT02951195).
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
One study reported a greater relative change from baseline in FEV1 % predicted with the active intervention compared to placebo (n = 10) for the 80 mg dose (MD 18.36%, 95% CI 3.63 to 33.09; 1 study, 21 participants; Analysis 19.2), the 240 mg dose (MD 20.17%, 95% CI 8.73 to 31.61; 1 study, 30 participants; Analysis 21.2) and the 400 mg dose (MD 23.85%, 95% CI 14.52 to 33.18; 1 study, 32 participants; Analysis 23.2) (Davies 2018b).
19.2. Analysis.

Comparison 19: VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
21.2. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
23.2. Analysis.

Comparison 23: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
Participants with F508del/F508del
The same study reported on a cohort homozygous for F508del and found a greater relative change in FEV1 % predicted with VX‐659 400 mg (n = 18) than with a placebo added to the tezacaftor‐ivacaftor intervention (MD 15.99%, 95% CI 8.61 to 23.37; 1 study, 29 participants; Analysis 24.2) (Davies 2018b).
24.2. Analysis.

Comparison 24: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 2: FEV1 % predicted (relative change from baseline)
VX‐659 plus tezacaftor plus deutivacaftor
Participants with F508del/MF
This regimen was only assessed at a dose of VX‐659 400 mg in participants with F508del/MF and results showed a greater relative change from baseline in FEV1 % predictedcompared to placebo (MD 33.05%, 95% CI 22.05 to 44.05; 1 study, 25 participants; Analysis 25.2) (Davies 2018b).
25.2. Analysis.

Comparison 25: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Results showed greater relative improvements in FEV1 % predicted from baseline compared to placebo (n = 12) at each dose of elexacaftor: at 50 mg (MD 19.00%, 95% CI 7.08 to 30.92; 1 study, 22 participants; moderate‐certainty evidence; Analysis 26.2); at 100 mg (MD 13.50%, 95% CI 3.28 to 23.72; 1 study, 34 participants; moderate‐certainty evidence; Analysis 27.2); and at 200 mg (MD 25.90%, 95% CI 15.57 to 36.23; 1 study, 33 participants; moderate‐certainty evidence; Analysis 28.2) (Keating 2018).
26.2. Analysis.

Comparison 26: Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
27.2. Analysis.

Comparison 27: Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
28.2. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
Participants with F508del/F508del
The same study reported on participants homozygous for F508del and found a greater relative change from baseline in FEV1 % predicted in the elexacaftor 200 mg group compared to the tezacaftor‐ivacaftor‐placebo group (MD 17.80%, 95% CI 6.66 to 28.94; 1 study, 28 participants; moderate‐certainty evidence; Analysis 29.2) (Keating 2018).
29.2. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 2: FEV1 % predicted (relative change from baseline)
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
Only the elexacaftor 200 mg dose was tested in this group and results again showed a greater improvement in FEV1 % predicted from baseline in the test intervention group compared to placebo (MD 18.30%, 95% CI 7.64 to 28.96; 1 study, 29 participants; moderate‐certainty evidence; Analysis 30.2) (Keating 2018).
30.2. Analysis.

Comparison 30: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 2: FEV1 % predicted (relative change from baseline)
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
One study reported that the low‐dose pooled data (cohort 1A: 200 mg VX‐440 every 12 hours plus 100 mg tezacaftor once daily plus 150 mg ivacaftor every 12 hours for 4 weeks; cohort 1B (low dose): 200 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 150 mg ivacaftor every 12 hours for four weeks) showed improvements in the relative change from baseline in FEV1 % predicted at one month, least squares MD 14.80 (95% CI 5.30 to 24.20) (NCT02951182); the high‐dose data from this study also showed greater improvement with treatment compared to placebo (MD 19.10, 95% CI 10.62 to 27.58; 1 study, 29 participants; low‐certainty evidence; Analysis 37.2) and tezacaftor plus ivacaftor respectively (MD 20.00, 95% CI 12.59 to 27.41; 1 study, 26 participants; low‐certainty evidence; Analysis 38.2) (NCT02951182).
37.2. Analysis.

Comparison 37: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo, Outcome 2: FEV1 % predicted (relative change from baseline)
38.2. Analysis.

Comparison 38: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily), Outcome 2: FEV1 % predicted (relative change from baseline)
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
Only one study tested this regimen of VX‐152 plus tezacaftor plus ivacaftor (NCT02951195). Results favoured the intervention group at all doses when compared to placebo: 100 mg twice daily (MD 12.60, 95% CI 3.48 to 21.72; 1 study, 14 participants; low‐certainty evidence; Analysis 32.2); 200 mg twice daily (MD 21.30, 95% CI 12.73 to 29.87; 1 study, 18 participants; low‐certainty evidence; Analysis 33.2); and 300 mg twice daily (MD 17.10, 95% CI 8.05 to 26.15; 1 study, 18 participants; low‐certainty evidence; Analysis 34.2). Results also favoured the intervention group at all doses when compared to tezacaftor plus ivacaftor: 200 mg twice daily (MD 14.60, 95% CI 1.88 to 27.32; 1 study, 14 participants; low‐certainty evidence; Analysis 35.2); and 300 mg twice daily (MD 13.50, 95% CI 6.57 to 20.43; 1 study, 28 participants; low‐certainty evidence; Analysis 36.2).
32.2. Analysis.

Comparison 32: VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 2: FEV1 % predicted (relative change from baseline)
33.2. Analysis.

Comparison 33: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 2: FEV1 % predicted (relative change from baseline)
34.2. Analysis.

Comparison 34: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 2: FEV1 % predicted (relative change from baseline)
35.2. Analysis.

Comparison 35: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 2: FEV1 % predicted (relative change from baseline)
36.2. Analysis.

Comparison 36: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 2: FEV1 % predicted (relative change from baseline)
ii. Short term (over one month and up to and including six months)
None of the triple therapy trials reported over this time frame (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
b. FEV1 (absolute values and change from baseline)
All of the triple combination trials reported on the absolute change in FEV1; two studies reported in L at one month (Davies 2018b; Keating 2018) and nine studies reported in % predicted, two at two weeks (Davies 2018a; NCT02951195), three at one month (Heijerman 2019; NCT02951182; NCT03460990), one at two months (Barry 2021), one at six months (Sutharsan 2022) and two at both one and six months (Middleton 2019; NCT03447249).
i. Immediate term (up to and including one month)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
In both studies reporting absolute change from baseline in FEV1 % predicted (each with a different dose), the intervention improved FEV1 % predicted compared to control: VX‐659 120 mg twice daily (MD 10.00%, 95% CI 3.04 to 16.96; 1 study, 12 participants; Analysis 20.1) (Davies 2018a) and VX‐659 240 mg once daily (MD 14.00, 95% CI 12.34 to 15.66; 1 study, 382 participants; Analysis 21.3) (NCT03447249).
20.1. Analysis.

Comparison 20: VX‐659 (120 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 1: FEV1 % predicted (absolute change from baseline)
21.3. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 3: FEV1 % predicted (absolute change from baseline)
The second Davies study reported a greater absolute change in FEV1 (L) with the intervention regimen compared to placebo for all dose levels (Davies 2018b): 80 mg dose (MD 0.37 L, 95% CI 0.15 to 0.59; 1 study, 21 participants; moderate‐certainty evidence; Analysis 19.3); 240 mg (MD 0.42 L, 95% CI 0.20 to 0.64; 1 study, 30 participants; moderate‐certainty evidence; Analysis 21.4); and 400 mg (MD 0.52 L, 95% CI 0.34 to 0.70; 1 study, 32 participants; Analysis 23.3).
19.3. Analysis.

Comparison 19: VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
21.4. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 4: FEV1 L (absolute change from baseline)
23.3. Analysis.

Comparison 23: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
Participants with F508del/F508del
Two studies reported the absolute change in FEV1 at one month in participants with F508del/F508del; each employed a different dose of VX‐659, but both studies showed a greater improvement in the intervention group compared to the control group (Davies 2018b; NCT03460990). The study with the lower dose, VX‐659 240 mg, reported the absolute change from baseline in FEV1 % predicted (MD 9.90, 95% CI 7.41 to 12.39; 1 study, 111 participants; Analysis 22.2) (NCT03460990). The study with the higher dose, VX‐659 400 mg, reported the absolute change from baseline in FEV1 L (MD 0.35 L, 95% CI 0.19 to 0.51; 1 study, 29 participants; Analysis 24.3) (Davies 2018b).
22.2. Analysis.

Comparison 22: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 2: FEV1 % predicted (absolute change from baseline)
24.3. Analysis.

Comparison 24: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 3: FEV1 L (absolute change from baseline)
VX‐659 plus tezacaftor plus deutivacaftor
Only a dose of VX‐659 of 400 mg was tested in participants with the F508del/MF genotype and results showed a greater absolute change from baseline in FEV1 (L) in the intervention group compared to placebo (MD 0.68 L, 95% CI 0.45 to 0.91; 1 study, 25 participants; Analysis 25.3) (Davies 2018b).
25.3. Analysis.

Comparison 25: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
One study reported on the absolute change in FEV1 % predicted at one month in this population (Middleton 2019). Results showed that the triple therapy combination led to a greater absolute change in FEV1 % predicted compared to triple placebo (MD 13.80, 95% CI 12.18 to 15.42; 1 study, 403 participants; Analysis 28.3).
28.3. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 3: FEV1 % predicted (absolute change from baseline)
One study reported on the absolute change in FEV1 (L) from baseline at one month (Keating 2018). The triple therapy combination led to greater changes at all doses of the intervention compared to placebo (n = 12): 50 mg (n = 10) (MD 0.46 L, 95% CI 0.19 to 0.73; 1 study, 22 participants; Analysis 26.3); 100 mg (MD 0.38 L, 95% CI 0.20 to 0.56; 1 study, 34 participants; Analysis 27.3); and 200 mg (MD 0.57 L, 95% CI 0.36 to 0.78; 1 study, 33 participants; Analysis 28.4).
26.3. Analysis.

Comparison 26: Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
27.3. Analysis.

Comparison 27: Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
28.4. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 4: FEV1 L (absolute change from baseline)
Participants with F508del/F508del
One study reported on the absolute change in FEV1 % predicted at one month and showed that the elexacaftor‐tezacaftor‐ivacaftor combination led to a greater absolute change compared to tezacaftor‐ivacaftor‐placebo (MD 10.00% predicted, 95% CI 7.51 to 12.49; 1 study, 107 participants; Analysis 29.3) (Heijerman 2019).
29.3. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 3: FEV1 % predicted (absolute change from baseline)
A further study reported on the absolute change in FEV1 (L) at one month (Keating 2018). Results showed a greater absolute change in the elexacaftor 200 mg (plus tezacaftor and ivacaftor) group compared to the tezacaftor‐ivacaftor‐placebo group (MD 0.46 L, 95% CI 0.26 to 0.66; 1 study, 28 participants; moderate‐certainty evidence; Analysis 29.4).
29.4. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 4: FEV1 L (absolute change from baseline)
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
Keating only tested a dose of elexacaftor 200 mg in 21 participants with the F508del/MF genotype (Keating 2018); the results showed a greater absolute change in FEV1 (L) versus placebo (MD 0.44 L, 95% CI 0.25 to 0.63; 1 study, 29 participants; moderate‐certainty evidence; Analysis 30.3).
30.3. Analysis.

Comparison 30: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 3: FEV1 L (absolute change from baseline)
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
This regimen was tested in one study at doses of 100 mg twice‐daily VX‐152, 200 mg twice‐daily VX‐152 and 300 mg twice‐daily VX‐152 (NCT02951195). Results from the intervention group demonstrated a greater change from baseline in FEV1 % predicted compared to placebo at all dosage levels: 100 mg (MD 6.50% predicted, 95% CI 1.62 to 11.38; 1 study, 14 participants; Analysis 32.3); 200 mg (MD 10.50% predicted, 95% CI 5.92 to 15.08; 1 study, 18 participants; Analysis 33.3); and 300 mg (MD 8.80% predicted, 95% CI 3.98 to 13.62; 1 study, 18 participants; Analysis 34.3). Results from the intervention group also demonstrated a greater change from baseline in FEV1 % predicted compared to tezacaftor plus ivacaftor at all dosage levels: 200 mg (MD 8.30% predicted, 95% CI 1.15 to 15.45; 1 study, 14 participants; Analysis 35.3); and 300 mg (MD 8.70% predicted, 95% CI 4.50 to 12.90; 1 study, 28 participants; Analysis 36.3).
32.3. Analysis.

Comparison 32: VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 3: FEV1 % predicted (absolute change from baseline)
33.3. Analysis.

Comparison 33: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 3: FEV1% predicted (absolute change from baseline)
34.3. Analysis.

Comparison 34: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 3: FEV1 % predicted (absolute change from baseline)
35.3. Analysis.

Comparison 35: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 3: FEV1 % predicted (absolute change from baseline)
36.3. Analysis.

Comparison 36: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 3: FEV1 % predicted (absolute change from baseline)
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
In one study, both the low‐dose pooled arms (cohort 1A: 200 mg VX‐440 every 12 hours plus 100 mg tezacaftor once daily plus 150 mg ivacaftor every 12 hours for 4 weeks; cohort 1B (low dose): 200 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 150 mg ivacaftor every 12 hours for 4 weeks) and the high‐dose arm (cohort 1B (high dose): 600 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 300 mg ivacaftor every 12 hours for 4 weeks) showed improvements at one month in the absolute change from baseline in FEV1 % predicted when compared to placebo (NCT02951182) (least squares MD 8.60, 95% CI 3.50 to 13.80 and MD 10.60, 95% CI 5.93 to 15.27; 1 study, 29 participants; low‐certainty evidence; Analysis 37.3). Results also showed improvements in the 600 mg twice‐daily VX‐440 group when compared to tezacaftor plus ivacaftor (MD 12.00, 95% CI 7.64 to 16.36; 1 study, 26 participants; Analysis 38.3).
37.3. Analysis.

Comparison 37: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo, Outcome 3: FEV1 % predicted (absolute change from baseline)
38.3. Analysis.

Comparison 38: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily), Outcome 3: FEV1 % predicted (absolute change from baseline)
ii. Short term (over one month and up to and including six months)
Elexacaftor‐tezacaftor‐ivacaftor
Participants with F508del/MF
One triple therapy study reported the absolute change in FEV1 % predicted at six months for participants with F508del/MF (Middleton 2019). There was a greater change from baseline in the treatment group compared to the triple placebo group (MD 14.30% predicted, 95% CI 12.76 to 15.84; 1 study, 403 participants; moderate‐certainty evidence; Analysis 28.3).
Participants with F508del/F508del
One study looking at 200 mg once‐daily elexacaftor with tezacaftor and ivacaftor compared to placebo with tezacaftor and ivacaftor found there to be a greater absolute change from baseline in FEV1 % predicted at six months in the treatment group (MD 10.20% predicted, 95% CI 8.26 to 12.14; 1 study, 175 participants; Analysis 29.3) (Sutharsan 2022).
Participants with F508del/gating and F508del/residual function
One triple therapy study reported on the absolute change in FEV1 % predicted for participants with F508del/gating and F508del/residual function (Barry 2021). It found a slightly greater change from baseline in the treatment group compared to the control (MD 3.50% predicted, 95% CI 2.24 to 4.76; 1 study, 258 participants; moderate‐certainty evidence; Analysis 31.2).
31.2. Analysis.

Comparison 31: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/gating or F508del/residual function), Outcome 2: FEV1 % predicted (absolute change from baseline)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
The greater absolute change in FEV1 % predicted in the intervention group that was reported at one month was also seen at six months in the study looking at 240 mg once‐daily VX‐659 (MD 14.20% predicted, 95% CI 12.54 to 15.86; 1 study, 382 participants; moderate‐certainty evidence; Analysis 21.3) (NCT03447249).
c. FVC (absolute values and change from baseline)
No study in this comparison reported data for this outcome (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
d. LCI
No study in this comparison reported data for this outcome (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
Secondary outcomes
1. Adverse events
AEs were reported by all of the studies examining triple combination therapies, in one study over a period of two weeks (Davies 2018a), in four studies over a period of one month (Davies 2018b; Heijerman 2019; Keating 2018; NCT03460990), in one study over a period of eight weeks (Barry 2021), in one study at one month and at six months (Middleton 2019), and two studies at six months (NCT03447249; Sutharsan 2022). One study was split in two parts; in part 1, AEs were reported up to day 57 and in part 2, AEs were reported up to day 85. Part 2 of this study included a four‐week run‐in period, a four‐week treatment period and a four‐week washout period (NCT02951182). Another study was split in two parts; in part 1, AEs were reported up to day 43 and in part 2, AEs were reported up to day 71. Part 2 of this study included a four‐week run‐in period, a four‐week treatment period and a two‐week washout period (NCT02951195). All of the studies either reported AEs in terms of mild, moderate or severe or recorded the "most common AEs", which they defined as occurring in at least 5% of participants. We have set CIs for AEs at 99%, as per Measures of treatment effect in this review's methodology.
VX‐659 plus tezacaftor plus ivacaftor versus control
There was no statistically significant difference in the number of participants experiencing at least one AE between the test intervention and control at any dose or for any genotype.
Participants with F508del/MF
One Phase 1 study reported the number of participants experiencing an AE with a dose level of VX‐659 120 mg (Davies 2018a); there was no difference between groups in the number of participants experiencing at least one AE (OR 31.67, 99% CI 0.32 to 3111.29; 1 study, 12 participants; moderate‐certainty evidence; Analysis 20.2). One Phase 2 study reported on this outcome for each of three dose levels of VX‐659 and found no difference between intervention and placebo groups at any level (Davies 2018b): 80 mg (OR 1.11, 99% CI 0.02 to 51.19; 1 study, 21 participants; moderate‐certainty evidence; Analysis 19.4); 240 mg (OR 0.33, 99% CI 0.02 to 6.85; 1 study, 30 participants; moderate‐certainty evidence; Analysis 21.5); and 400 mg (OR 0.38, 99% CI 0.02 to 7.70; 1 study, 32 participants; moderate‐certainty evidence; Analysis 23.4).
20.2. Analysis.

Comparison 20: VX‐659 (120 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 2: Adverse events (at up to 1 month)
19.4. Analysis.

Comparison 19: VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 4: Adverse events (at 1 month)
21.5. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 5: Adverse events (at 1 month)
23.4. Analysis.

Comparison 23: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 4: Adverse events (at 1 month)
One Phase 3 study assessing a dose level of VX‐659 240 mg reported on the number of participants with treatment‐emergent AEs and found no difference between groups (OR 0.69, 99% CI 0.27 to 1.77; 1 study, 382 participants; moderate‐certainty evidence; Analysis 21.6) (NCT03447249).
21.6. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 6: Most common adverse events (occurring in at least 10% of participants in either group) (6 months)
All doses are once daily except for 120 mg, which was taken twice daily in the Phase 1 study (Davies 2018a).
Participants with F508del/F508del
Two studies with different dose levels reported on AEs in participants with this genotype (Davies 2018b; NCT03460990). At the VX‐659 240 mg dose level, investigators reported no difference in the number of participants with treatment‐emergent AEs (OR 1.32, 99% CI 0.49 to 3.56; 1 study, 111 participants; Analysis 22.3) (NCT03460990). At the 400 mg dose level, there was no difference between groups in the number experiencing any AE (OR 1.11, 99% CI 0.08 to 14.81; 1 study, 29 participants; Analysis 24.4).
22.3. Analysis.

Comparison 22: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 3: Most common adverse events (occurring in at least 10% of participants in either group)
24.4. Analysis.

Comparison 24: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 4: Adverse events (at 1 month)
VX‐659 plus tezacaftor plus deutivacaftor versus placebo
Participants with F508del/MF
For this cohort, there was no difference in the number of participants experiencing at least one AE between groups (OR 0.95, 99% CI 0.01 to 74.78; 1 study, 25 participants; moderate‐certainty evidence; Analysis 25.4) (Davies 2018b).
25.4. Analysis.

Comparison 25: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 4: Adverse events (at 1 month)
Elexacaftor plus tezacaftor plus ivacaftor versus control
Participants with F508del/MF
The Keating 2018 Phase 2 study reported that for the 50 mg cohort, every participant in both groups had an AE, therefore an OR was not estimable (Analysis 26.4). For the other doses in this study, there was no difference between groups in the number experiencing any AE: 100 mg (OR 0.57, 99% CI 0.01 to 42.46; 1 study, 34 participants; Analysis 27.4); and 200 mg (OR 0.21, 99% CI 0.00 to 11.62; 1 study, 33 participants; Analysis 28.5). The Middleton 2019 Phase 3 study of participants with this genotype reported no difference between the 200 mg elexacaftor and placebo groups in the total number of participants experiencing an AE at six months (OR 0.56, 99% CI 0.23 to 1.36; 1 study, 403 participants; Analysis 28.6).
26.4. Analysis.

Comparison 26: Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 4: Adverse events (at 1 month)
27.4. Analysis.

Comparison 27: Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 4: Adverse events at (1 month)
28.5. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 5: Adverse events (at up to 1 month)
28.6. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 6: Adverse events (at up to 6 months)
Participants with F508del/F508del
Data from two studies combined (Heijerman 2019; Keating 2018) showed no difference in the number of people experiencing any AE at one month (OR 0.94, 99% CI 0.46 to 1.96; I2 = 42%; 2 studies, 135 participants; Analysis 29.5). There was also no difference in the number of participants with treatment‐emergent AEs at six months (OR 0.67, 99% CI 0.18 to 2.53; 1 study, 176 participants; Analysis 29.6) (Sutharsan 2022).
29.5. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 5: Adverse events (at up to 1 month)
29.6. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 6: Most common adverse events (occurring in at least 10% of participants in either group)
Participants with F508del/gating and F508del/residual function
One study reporting on this comparison in participants with these genotypes found no difference in the number of participants with treatment‐emergent AEs between intervention and control groups (OR 1.04, 99% CI 0.53 to 2.04; 1 study, 258 participants; moderate‐certainty evidence; Analysis 31.3) (Barry 2021).
31.3. Analysis.

Comparison 31: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/gating or F508del/residual function), Outcome 3: Most common adverse events (occurring in at least 10% of participants in either group)
Elexacaftor plus tezacaftor plus deutivacaftor versus placebo
Participants with F508del/MF
At one month, Keating 2018 observed no difference in the number of AEs between the intervention and placebo groups (OR 1.36, 99% CI 0.05 to 38.84; 1 study, 29 participants; moderate‐certainty evidence; Analysis 30.4).
30.4. Analysis.

Comparison 30: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 4: Adverse events (at 1 month)
VX‐152 plus tezacaftor plus ivacaftor versus placebo
Participants with F508del/MF and F508del/F508del
NCT02951195 reported no difference in the number of AEs between intervention and placebo groups for the 100 mg dose of VX‐152 (OR 0.06, 99% CI 0.00 to 4.02; 1 study, 14 participants; Analysis 32.4) or for the 200 mg dose of VX‐152 (OR 0.13, 99% CI 0.00 to 7.63; 1 study, 18 participants; Analysis 33.4). At the 300 mg dose, every participant in both the intervention and placebo groups experienced treatment‐emergent AEs and therefore an odds ratio was not estimable (Analysis 34.4). There was also no difference in the number of AEs between intervention and tezacaftor plus ivacaftor control groups for the 200 mg dose of VX‐152 (OR 0.50, 99% CI 0.02 to 15.09; 1 study, 14 participants; Analysis 35.4) or for the 300 mg dose of VX‐152 (OR 3.80, 99% CI 0.21 to 67.89; 1 study, 28 participants; Analysis 36.4).
32.4. Analysis.

Comparison 32: VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group) (day 43)
33.4. Analysis.

Comparison 33: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group)
34.4. Analysis.

Comparison 34: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group)
35.4. Analysis.

Comparison 35: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group) (at day 71)
36.4. Analysis.

Comparison 36: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group) (at day 71)
VX‐440 plus tezacaftor plus ivacaftor versus placebo
Participants with F508del/MF and F508del/F508del
One study (n = 73) reported on this genotype for this treatment combination (NCT02951182). In both the treatment arms looking at 200 mg twice‐daily VX‐440 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor and 200 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor, all nine participants experienced treatment‐emergent AEs. This was compared to nine out of 11 participants in the placebo group. In the treatment arm looking at 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor, there was no difference found between the intervention and control groups for the number of participants with treatment‐emergent AEs (OR 1.11, 99% CI 0.08 to 14.81; low‐certainty evidence; Analysis 37.4). There was also no difference found between the 600 mg VX‐440 and the tezacaftor plus ivacaftor control groups for the number of participants with treatment‐emergent AEs (OR 0.22, 99% CI 0.00 to 11.73; 1 study, 26 participants; Analysis 38.4).
37.4. Analysis.

Comparison 37: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo, Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group) (at day 57)
38.4. Analysis.

Comparison 38: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily), Outcome 4: Most common adverse events (occurring in at least 10% of participants in either group) (at day 85)
a. Mild (therapy does not need to be discontinued)
We could not accurately record the number of mild AEs occurring in any of the triple therapy studies since they record the number of participants experiencing at least one AE, by the maximum severity, meaning that a participant may have had numerous 'mild' AEs and a single moderate or severe event, but we would only be aware of the single most severe event (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022).
In the studies where there is not yet any published paper, we could not determine the severity of any of the listed AEs (NCT02951182; NCT02951195; NCT03447249; NCT03460990).
b. Moderate (therapy is discontinued, and the adverse effect ceases)
Our definition of a moderate AE differed to that used in the studies, however the studies also reported the number of AEs that led to discontinuation of therapy. We therefore used this number to record the number of moderate AEs according to our definition.
In the studies where there is not yet any published paper, we could not determine whether the therapy was discontinued or not (NCT02951182; NCT02951195; NCT03447249; NCT03460990).
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
No participants in either the intervention or placebo groups were recorded as having a moderate AE in the dose groups VX‐659 80 mg, VX‐659 120 mg twice daily and VX‐659 400 mg, meaning that an OR was not calculable (moderate‐certainty evidence; Analysis 19.4; Analysis 20.2; Analysis 23.4). For the VX‐659 240 mg group, there was no difference in the number of moderate AEs between the intervention and placebo groups (OR 1.62, 99% CI 0.02 to 121.50; 1 study, 30 participants; moderate‐certainty evidence; Analysis 21.5) (Davies 2018b).
Participants with F508del/F508del
At one month, no participants in either the intervention or placebo groups were recorded as having a moderate AE at the dose VX‐659 400 mg, meaning that an OR was not calculable (moderate‐certainty evidence; Analysis 24.4) (Davies 2018b).
VX‐659 plus tezacaftor plus deutivacaftor
Participants with F508del/MF
No difference was observed in the number of moderate AEs between the intervention (n = 19) and placebo groups (n = 6) after one month of this treatment regimen (OR 1.86, 99% CI 0.03 to 119.25; 1 study, 25 participants; moderate‐certainty evidence; Analysis 25.4) (Davies 2018b).
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Keating 2018 reported that no participants in either the elexacaftor 50 mg or the elexacaftor 100 mg groups or the placebo groups experienced a moderate AE at one month, meaning that an OR was not calculable for the groups taking this dose (moderate‐certainty evidence; Analysis 26.4; Analysis 27.4). There were no differences in the number of moderate AEs experienced by the elexacaftor 200 mg (n = 21) and placebo groups (OR 3.21, 99% CI 0.05 to 193.04; 1 study, 33 participants; Analysis 28.5).
Participants with F508del/F508del
Two studies found no difference between the elexacaftor 200 mg and placebo groups in the number of participants experiencing moderate AEs at one month (OR 0.94, 99% CI 0.39 to 2.26; I2 = 0%; 2 studies, 135 participants; moderate‐certainty evidence; Analysis 29.5) (Heijerman 2019; Keating 2018).
At six months, Sutharsan 2022 found no difference between groups in the number of participants experiencing moderate AEs (OR 0.50, 99% CI 0.02 to 12.01; 1 study, 176 participants; moderate‐certainty evidence; Analysis 29.6).
Participants with F508del/gating and F508del/residual function
Barry 2021 compared elexacaftor‐tezacaftor‐ivacaftor to control and found no difference between groups in the number of participants experiencing moderate AEs for these genotypes (OR 0.47, 99% CI 0.02 to 11.28; 1 study, 258 participants; Analysis 31.3).
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
In the single study (29 participants) comparing elexacaftor plus tezacaftor plus deutivacaftor in participants with this genotype, no participants in either the intervention or placebo group experienced a moderate AE at one month, meaning an OR was not calculable for this group (moderate‐certainty evidence; Analysis 30.4) (Keating 2018).
c. Severe (life‐threatening or debilitating, or which persists even after stopping treatment)
Our definition of severe AEs was equivalent to the studies' definition of a serious AE, therefore we counted the number of participants reporting serious AEs.
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
Severe AE s also occurred at every dose level in both intervention and placebo groups for participants with this genotype (Davies 2018a; Davies 2018b; NCT03447249). Results showed no differences between groups at most dose levels at one month: VX‐659 80 mg (OR 0.23, 99% CI 0.01 to 5.92; 1 study, 21 participants; Analysis 19.4) (Davies 2018b); VX‐659 120 mg twice daily (OR 2.33, 99% CI 0.03 to 176.29; 1 study, 12 participants; Analysis 20.2) (Davies 2018a); VX‐659 240 mg (OR 0.58, 99% CI 0.06 to 5.75; 1 study, 30 participants; Analysis 21.5) (Davies 2018b); and VX‐659 400 mg (OR 0.11, 99% CI 0.00 to 2.67; 1 study, 32 participants; Analysis 23.4) (Davies 2018b). At six months, there were fewer participants with severe AEs in the VX‐659 250 mg treatment arm than the placebo group (OR 0.14, 99% CI 0.06 to 0.33; 1 study, 382 participants; Analysis 21.6) (NCT03447249).
Participants with F508del/F508del
Severe AEs occurred in the VX‐659 240 mg, VX‐659 400 mg and placebo groups, but there was no difference between groups: at eight weeks VX‐659 240 mg (OR 5.48, 99% CI 0.10 to 305.22; 1 study, 111 participants; Analysis 22.3) (NCT03460990); and at one month VX‐659 400 mg (OR 0.26, 99% CI 0.01 to 7.39; 1 study, 29 participants; Analysis 24.4) (Davies 2018b).
VX‐659 plus tezacaftor plus deutivacaftor
Participants with F508del/MF
No statistical difference was found between the VX‐659 400 mg and placebo groups for the number of severe AEs at one month (OR 0.12, 99% CI 0.01 to 2.04; 1 study, 25 participants; moderate‐certainty evidence; Analysis 25.4) (Davies 2018b).
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Keating 2018 reported on severe AEs in this comparison and genotype at one month and found no differences between the intervention and placebo groups across doses: elexacaftor 50 mg (OR 0.56, 99% CI 0.02 to 16.15; 1 study, 22 participants; moderate‐certainty evidence; Analysis 26.4); elexacaftor 100 mg (OR 0.50, 99% CI 0.03 to 7.92; 1 study, 34 participants; moderate‐certainty evidence; Analysis 27.4); elexacaftor 200 mg (OR 0.10, 99% CI 0.00 to 5.93; 1 study, 33 participants; moderate‐certainty evidence; Analysis 28.5). At six months, Middleton 2019 also reported no difference in the number of severe AEs between groups (OR 1.39, 99% CI 0.54 to 3.57; 1 study, 403 participants; moderate‐certainty evidence; Analysis 28.6).
Participants with F508del/F508del
Two studies compared elexacaftor 200 mg to placebo and found no difference in the number of severe AEs between groups at one month (OR 0.19, 99% CI 0.02 to 1.92; I2 = 0%; 2 studies, 135 participants; Analysis 29.5) (Heijerman 2019; Keating 2018). At six months, Sutharsan 2022 also found no difference in the number of severe AEs between the intervention and control groups (OR 0.32, 99% CI 0.08 to 1.31; 1 study, 176 participants; Analysis 29.6).
Participants with F508del/gating and F508del/residual function
At eight weeks, Barry 2021 found no difference in the number of severe AEs between the intervention and control groups for these genotypes (OR 0.41, 99% CI 0.10 to 1.72; 1 study, 258 participants; Analysis 31.3).
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
At one month, Keating 2018 found no difference between the groups in the number of severe AEs (OR 0.12, 99% CI 0.00 to 8.97; 1 study, 29 participants; moderate‐certainty evidence; Analysis 30.4).
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
One study evaluated 100 mg twice daily, 200 mg twice daily and 300 mg twice daily VX‐152 in conjunction with tezacaftor and ivacaftor (NCT02951195). At up to day 43, investigators found no difference in the incidence of severe AEs between groups for any dose level: 100 mg twice daily VX‐152 (OR 0.20, 99% CI 0.00 to 13.86; 1 study, 14 participants; Analysis 32.4); 200 mg twice daily VX‐152 (OR 0.12, 99% CI 0.00 to 8.19; 1 study, 18 participants; Analysis 33.4); and 300 mg twice daily VX‐152 (OR 0.33, 99% CI 0.01 to 10.34; 1 study, 18 participants) (Analysis 34.4). For the 200 mg twice daily and 300 mg twice daily VX‐152 combination therapy groups and the tezacaftor plus ivacaftor control groups, there were no participants who experienced serious AEs at up to day 71 (Analysis 35.4; Analysis 36.4).
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
One study assessed three different dosing schedules for this genotype, reporting at two months (NCT02951182). No participants in either of the two following dosing schedules experienced severe AEs: VX‐440 200 mg twice daily plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor; and VX‐440 200 mg twice daily plus 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor. No participants experienced severe AEs in the matched placebo group. The study found no difference in the number of participants with severe AEs between the treatment arm receiving VX‐440 600 mg twice daily plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor and the placebo group (OR 3.48, 99% CI 0.06 to 212.67; 1 study, 29 participants; Analysis 37.4). The study found no difference in the number of participants with severe AEs between the treatment arm receiving VX‐440 600 mg twice daily plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor and the 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor control group (OR 0.11, 99% CI 0.00 to 3.34; 1 study, 26 participants; Analysis 38.4).
2. Hospitalisation
ii. Short term (over one month and up to and including six months)
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Only one Phase 3 study reported this outcome (Middleton 2019); investigators found that at six months, elexacaftor 200 mg in combination with tezacaftor and ivacaftor reduced the odds of hospitalisation versus triple placebo (OR 0.29, 95% CI 0.14 to 0.60; 1 study, 403 participants; Analysis 28.7).
28.7. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 7: Hospitalisation
3. School or work attendance
No study reported data for this outcome (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
4. Extra courses of antibiotics
a. Time to the next course of antibiotics
No study reported data for this outcome (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
b. Total number of courses of antibiotics
As for the monotherapy and dual combination therapy sections above, under this outcome we report the occurrence of infective pulmonary exacerbations.
With regards to pulmonary exacerbations, please see our definition above (Types of outcome measures). For triple therapy, four studies defined exacerbations as those that were infective in nature or required antibiotics (Davies 2018a; Davies 2018b; Heijerman 2019; Middleton 2019). For one triple therapy study, it was unclear whether the exacerbations were protocol‐ or physician‐defined (Keating 2018).
i. Immediate term (up to one month)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
One 14‐day Phase 1 study found no difference in infective pulmonary exacerbations between the 120 mg twice‐daily VX‐659 group and the placebo group (OR 2.33, 95% CI 0.03 to 176.29; 1 study, 12 participants; Analysis 20.2) (Davies 2018a).
ii. Short term (over one month and up to and including six months)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
After one month, the Davies 2018b Phase 2 study found no difference between groups in the number of participants experiencing a pulmonary exacerbation at any dose level of VX‐659: 80 mg once daily (OR 1.50, 99% CI 0.10 to 21.90; 1 study, 21 participants; Analysis 19.4); 240 mg once daily (OR 0.71, 99% CI 0.05 to 9.48; 1 study, 30 participants; Analysis 21.5); and 400 mg once daily (OR 0.89, 99% CI 0.07 to 10.67; 1 study, 32 participants; Analysis 23.4).
The Phase 3 study reported the number of pulmonary exacerbations, defined as "treatment with new or changed antibiotic therapy (intravenous, inhaled, or oral) for greater than or equal to 4 sinopulmonary signs/symptoms" (NCT03447249). There were fewer participants in the intervention group experiencing pulmonary exacerbations at six months compared to the placebo group (OR 0.06, 99% CI 0.03 to 0.13; 1 study, 382 participants; Analysis 21.7).
21.7. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 7: Number of pulmonary exacerbations
Participants with F508del/F508del
Neither of the two studies assessing VX‐659 plus tezacaftor plus ivacaftor found a difference between groups in the number of pulmonary exacerbations either at the 240 mg dose level (OR 0.21, 99% CI 0.03 to 1.64; 1 study, 111 participants; Analysis 22.3) (NCT03460990) or at the 400 mg dose level (OR 1.03, 99% CI 0.11 to 9.34; 1 study, 29 participants; Analysis 24.4) (Davies 2018b).
VX‐659 plus tezacaftor plus deutivacaftor
Participants with F508del/MF
Davies 2018b reported no difference between the VX‐659 400 mg group and the placebo group in participants experiencing a pulmonary exacerbation at one month (OR 0.12, 99% CI 0.01 to 2.04; 1 study 25 participants; Analysis 25.4).
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
One study assessing this intervention reported on the number of participants experiencing exacerbations at up to day 43 and day 71 (NCT02951195). At up to day 43, there were no differences between treatment and control groups at any of the VX‐152 dose levels: 100 mg twice‐daily VX‐152 (OR 0.08, 99% CI 0.00 to 4.89; 1 study, 14 participants; Analysis 32.4); 200 mg twice‐daily VX‐152 (OR 0.11, 99% CI 0.00 to 2.92; 1 study, 18 participants; Analysis 33.4); and 300 mg twice‐daily VX‐152 (OR 0.11, 99% CI 0.00 to 2.92; 1 study, 18 participants; Analysis 34.4). At up to day 71, in part 2 of the study, there were no differences between the 200 mg twice‐daily VX‐152 and the 300 mg twice‐daily VX‐152 groups and the tezacaftor plus ivacaftor control groups (OR 1.42, 99% CI 0.02 to 122.55; 1 study, 14 participants; Analysis 35.4 and OR 0.42, 99% CI 0.04 to 4.45; 1 study, 28 participants; Analysis 36.4).
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Keating 2018 reported data at one month for these participants at three dose levels and found no difference between the elexacaftor‐tezacaftor‐ivacaftor groups and the placebo groups in the number of participants experiencing a pulmonary exacerbation at any dose level: 50 mg (OR 0.86, 99% CI 0.08 to 9.23; 1 study, 22 participants; Analysis 26.4); 100 mg (OR 0.59, 99% CI 0.08 to 4.57; 1 study, 34 participants; Analysis 27.4); and 200 mg (OR 0.21, 99% CI 0.02 to 2.52; 1 study, 33 participants; Analysis 28.5).
Middleton 2019 reported a significantly lower odds of having an exacerbation requiring antibiotics at 24 weeks (OR 0.29, 95% CI 0.14 to 0.60; 1 study, 403 participants; Analysis 28.8). However, as reported in the supplementary paper (complete data requested from the authors), the time to the next exacerbation was significantly shorter for participants on placebo (HR 0.34, 95% CI 0.22 to 0.52) (Middleton 2019).
28.8. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 8: Exacerbation (need for antibiotics)
Participants with F508del/F508del
At six months, Sutharsan 2022 reported fewer participants in the 200 mg group experiencing pulmonary exacerbations than in the control group (OR 0.17, 99% CI 0.06 to 0.45; 1 study, 175 participants; Analysis 29.6).
Participants with F508del/gating and F508del/RF
At eight weeks, Barry 2021 reported slightly fewer participants experiencing pulmonary exacerbations in the 200 mg elexacaftor group than in the control group (OR 0.20, 99% CI 0.05 to 0.87; 1 study, 258 participants; Analysis 31.3).
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
Two studies report data for this comparison and the combined data showed a significant difference in favour of elexacaftor‐tezacaftor‐ivacaftor (OR 0.15, 99% CI 0.02 to 1.00; I2 = 0%; 2 studies, 136 participants; Analysis 30.4).
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
NCT02951182 reported that a total of nine out of 36 participants across all dosing schedules of VX‐440 plus tezacaftor plus ivacaftor compared to two out of 11 participants in the placebo group experienced infective pulmonary exacerbations at up to day 57. For the 200 mg twice‐daily VX‐440 plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor group, there were six out of nine participants who experienced an infective pulmonary exacerbation. For the 200 mg twice‐daily VX‐440 plus 50 mg twice daily tezacaftor plus 150 mg twice daily ivacaftor group, there were no participants experiencing an infective pulmonary exacerbation. For the 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor dosing schedule, there was no difference found between the intervention and placebo groups (OR 0.90, 99% CI 0.07 to 12.00; 1 study, 29 participants; Analysis 37.4). At day 85, for the 600 mg twice‐daily VX‐440 plus 50 mg twice‐daily tezacaftor plus 300 mg twice‐daily ivacaftor dosing schedule, there was no difference found between the intervention and tezacaftor plus ivacaftor control groups (OR 0.88, 99% CI 0.03 to 22.76; 1 study, 26 participants; Analysis 38.4).
5. Sweat chloride (change from baseline) as a measure of CFTR function
i. Immediate term (up to one month)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
At two weeks, the Phase 1 study found that the 120 mg twice‐daily dose of VX‐659 reduced sweat chloride more than with placebo (MD ‐30.60 mmol/L, 95% CI ‐46.38 to ‐14.82; 1 study, 12 participants; Analysis 20.3) (Davies 2018a).
20.3. Analysis.

Comparison 20: VX‐659 (120 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 3: Sweat chloride (absolute change from baseline)
At one month, all active intervention groups showed a reduction in sweat chloride compared to placebo: at the 80 mg dose level (MD ‐48.60 mmol/L, 95% CI ‐60.94 to ‐36.26; 1 study, 21 participants; Analysis 19.5) (Davies 2018b); at the 240 mg dose level (MD ‐43.50 mmol/L, 95% CI ‐46.19 to ‐40.81; I2 = 0%; 2 studies, 412 participants; Analysis 21.8) (Davies 2018b; NCT03447249); and at the 400 mg dose level (MD ‐54.30 mmol/L, 95% CI ‐65.28 to ‐43.32; 1 study, 32 participants; Analysis 23.5) (Davies 2018b).
19.5. Analysis.

Comparison 19: VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
21.8. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 8: Sweat chloride (change from baseline) (mmol/L)
23.5. Analysis.

Comparison 23: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (absolute change from baseline)
Participants with F508del/F508del
Two studies reported a greater reduction in sweat chloride at one month; one study compared 240 mg VX‐659 to control (MD ‐48.70 mmol/L, 95% CI ‐53.83 to ‐43.57; 1 study, 111 participants; Analysis 22.4) (NCT03460990) and the second compared 400 mg to control (MD ‐45.20 mmol/L, 95% CI ‐52.18 to ‐38.22; 1 study, 29 participants; Analysis 24.5) (Davies 2018b).
22.4. Analysis.

Comparison 22: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 4: Sweat chloride (absolute change from baseline)
24.5. Analysis.

Comparison 24: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
VX‐659 plus ivacaftor plus deutivacaftor
Participants with F508del/MF
At one month, Davies 2018b reported a greater reduction in sweat chloride in the 400 mg compared to the placebo group (MD ‐36.80 mmol/L, 95% CI ‐48.74 to ‐24.86; 1 study, 25 participants; Analysis 25.5).
25.5. Analysis.

Comparison 25: VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
At one month, Keating 2018 found that all active intervention groups in this comparison showed a difference in the change in sweat chloride compared to placebo. Middleton 2019 also reported on the 200 mg dose level at this time point: 50 mg elexacaftor (MD ‐36.00 mmol/L, 95% CI ‐47.23 to ‐24.77; 1 study, 22 participants; Analysis 26.5); 100 mg elexacaftor (MD ‐31.00 mmol/L, 95% CI ‐40.41 to ‐21.59; 1 study, 34 participants; Analysis 27.5); and 200 mg (MD ‐40.96 mmol/L, 95% CI ‐43.60 to ‐38.33; I2 = 0%; 2 studies, 436 participants; Analysis 28.9).
26.5. Analysis.

Comparison 26: Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
27.5. Analysis.

Comparison 27: Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
28.9. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 9: Sweat chloride (absolute change from baseline)
Participants with F508del/F508del
At one month, two studies reported that triple therapy with elexacaftor 200 mg showed a greater decrease in sweat chloride than placebo (MD ‐44.32 mmol/L, 95% CI ‐48.80 to ‐39.84; I2 = 0%; 2 studies, 135 participants; Analysis 29.7) (Heijerman 2019; Keating 2018).
29.7. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 7: Sweat chloride (absolute change from baseline)
Elexacaftor plus tezacaftor plus deutivacaftor
Participants with F508del/MF
Keating 2018 also reported a decrease in sweat chloride at one month for this intervention compared to control (MD ‐34.60 mmol/L, 95% CI ‐45.15 to ‐24.05; 1 study, 29 participants; Analysis 30.5).
30.5. Analysis.

Comparison 30: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF), Outcome 5: Sweat chloride (change from baseline) (mmol/L)
VX‐152 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
NCT02951195 reported three different dose levels at two weeks and consistently found a reduction in sweat chloride when compared with the placebo groups: 100 mg VX‐152 (MD ‐19.40 mmol/L, 95% CI ‐29.45 to ‐9.35; 1 study, 14 participants; Analysis 32.5); 200 mg VX‐152 (MD ‐13.50 mmol/L, 95% CI ‐23.17 to ‐3.83; 1 study, 18 participants; Analysis 33.5); and 300 mg VX‐152 (MD ‐27.40 mmol/L, 95% CI ‐36.86 to ‐17.94; 1 study, 18 participants; Analysis 34.5). The same study also found a reduction in sweat chloride when compared to tezacaftor plus ivacaftor at two weeks for the 200 mg twice‐daily VX‐152 group (MD ‐24.80 mmol/L, 95% CI ‐35.37 to ‐14.23; 1 study, 14 participants; Analysis 35.5), and a reduction was also found at one month for the 300 mg twice‐daily VX‐152 group (MD ‐23.90 mmol/L, 95% CI ‐32.11 to ‐15.69; 1 study, 28 participants; Analysis 36.5).
32.5. Analysis.

Comparison 32: VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 5: Sweat chloride (absolute change from baseline)
33.5. Analysis.

Comparison 33: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 5: Sweat chloride (absolute change from baseline)
34.5. Analysis.

Comparison 34: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo, Outcome 5: Sweat chloride (absolute change from baseline)
35.5. Analysis.

Comparison 35: VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 5: Sweat chloride (absolute change from baseline)
36.5. Analysis.

Comparison 36: VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily), Outcome 5: Sweat chloride (absolute change from baseline)
VX‐440 plus tezacaftor plus ivacaftor
Participants with F508del/MF and F508del/F508del
The NCT02951182 study reported that the participants in the low‐dose pooled arm had a greater reduction in sweat chloride levels when compared to the placebo group, least squares MD ‐22.3 mmol/L (95% CI ‐32.1 to ‐12.4); our analysis of the 600 mg high‐dose arm had similar findings when compared to placebo (MD ‐34.70 mmol/L, 95% CI ‐43.54 to ‐25.86; 1 study, 29 participants; Analysis 37.5) and tezacaftor plus ivacaftor, respectively (MD ‐33.40 mmol/L, 95% CI ‐45.41 to ‐21.39; 1 study, 26 participants; Analysis 38.5).
37.5. Analysis.

Comparison 37: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo, Outcome 5: Sweat chloride (absolute change from baseline)
38.5. Analysis.

Comparison 38: VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily), Outcome 5: Sweat chloride (absolute change from baseline)
ii. Short term (over one month and up to and including six months)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
NCT03447249 reported that at six months and with a dose of 240 mg VX‐659, the reduction in sweat chloride was maintained when compared to placebo (MD ‐44.50 mmol/L, 95% CI ‐47.14 to ‐41.86; 1 study, 382 participants; Analysis 21.8).
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Middleton 2019 reported a greater reduction in sweat chloride in the treatment group compared to the placebo group at six months (MD ‐41.80 mmol/L, 95% CI ‐44.33 to ‐39.27; 1 study, 403 participants; Analysis 28.9).
Participants with F508del/F508del
At six months, Sutharsan 2022 also reported a reduction in sweat chloride when compared with the control group at a dose of 200 mg elexacaftor (MD ‐42.80 mmol/L, 95% CI ‐46.27 to ‐39.33; 1 study, 175 participants; Analysis 29.7).
Participants with F508del/gating and F508del/RF
Barry 2021 reported that at eight weeks, the sweat chloride levels were decreased in the intervention group for these genotypes when compared with the control group (MD ‐23.00 mmol/L, 95% CI ‐25.98 to ‐20.02; 1 study, 258 participants; Analysis 31.4).
31.4. Analysis.

Comparison 31: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/gating or F508del/residual function), Outcome 4: Sweat chloride (absolute change from baseline)
6. Radiological measures of lung disease
No study reported data for this outcome (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
7. Acquisition of respiratory pathogens
No study reported data on the acquisition of pre‐specified or any other clinically relevant pathogens (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
8. Eradication of respiratory pathogens
No study reported data on eradication of respiratory pathogens (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022).
9. Nutrition and growth
Three studies reported data on nutrition and growth parameters (Heijerman 2019; Middleton 2019; NCT03447249).
ii. Short term (over one month and up to six months)
Elexacaftor plus tezacaftor plus ivacaftor
Participants with F508del/MF
Middleton reported on the change from baseline in weight (kg) and BMI z score at six months (Middleton 2019). Results favoured triple therapy for both weight (MD 2.90 kg, 95% CI 2.40 to 3.40; 1 study, 403 participants; Analysis 28.10) and BMI z score (MD 0.30, 95% CI 0.17 to 0.43; 1 study, 403 participants; Analysis 28.11).
28.10. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 10: Weight (absolute change from baseline)
28.11. Analysis.

Comparison 28: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF), Outcome 11: BMI z score (absolute change from baseline)
Participants with F508del/F508del
The four‐week Phase 3 study reported a least squares mean for the change from baseline of both weight and BMI (Heijerman 2019). Investigators reported a greater increase in weight in the elexacaftor group (MD 1.6 kg, 95% CI 1.0 to 2.10; 1 study, 107 participants; Analysis 29.8). Similarly, there was a greater increase in BMI in the intervention group (MD 0.6 kg/m², 95% CI 0.41 to 0.79; 1 study, 107 participants; Analysis 29.9).
29.8. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 8: Weight (change from baseline)
29.9. Analysis.

Comparison 29: Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del), Outcome 9: BMI (change from baseline)
VX‐659 plus tezacaftor plus ivacaftor
Participants with F508del/MF
The Phase 3 study (n = 385) reported results for the absolute change from baseline in BMI, BMI z score and body weight (NCT03447249). Results favoured the intervention group at six months for each outcome: BMI (MD 1.11, 95% CI 0.92 to 1.30; 1 study, 382 participants; Analysis 21.10); BMI z score (MD 0.39, 95% CI 0.25 to 0.53; 1 study, 382 participants; Analysis 21.11); and body weight (MD 3.20 kg, 95% CI 2.65 to 3.75; 1 study, 382 participants; Analysis 21.9).
21.10. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 10: BMI (change from baseline)
21.11. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 11: BMI z score (absolute change from baseline)
21.9. Analysis.

Comparison 21: VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF), Outcome 9: Body weight (absolute change from baseline)
Discussion
Class II variants of the CFTR gene are defined as those that form a full length of protein, but the abnormal protein does not reach the cell membrane in any significant quantity. This is referred to as a trafficking defect. F508del, the most prevalent variant to cause CF, is a class II variant. A therapy that corrects the F508del trafficking defect would have a profound impact on the field of CF, providing a treatment option for the majority of pwCF.
Summary of main results
We identified 34 eligible RCTs evaluating correctors for pwCF and class II CFTR mutations (Barry 2021; Boyle 2014; Clancy 2012; Davies 2018a; Davies 2018b; Davies 2021; Donaldson 2014; Donaldson 2017; Donaldson 2018; Horsley 2017; Keating 2018; McCarty 2002; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; NCT02951182; NCT02951195; NCT03447249; NCT03460990; PROGRESS 2017; Ratjen 2017; Rubenstein 1998; Schwarz 2021; Stahl 2021; Sutharsan 2022; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021; Zeitlin 2002). Eight studies examined monotherapy with different correctors (Boyle 2014; Clancy 2012; Donaldson 2014; Donaldson 2017; Horsley 2017; McCarty 2002; Rubenstein 1998; Zeitlin 2002). In 16 studies (including the multi‐arm Boyle trial), investigators examined dual combination therapy of either lumacaftor‐ivacaftor or tezacaftor‐ivacaftor (Boyle 2014; Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; PROGRESS 2017; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). Six of these were well‐powered Phase 3 studies that enrolled pwCF (including children aged 6 to 11 years) with two copies of the F508del variant (F508del homozygotes) (Davies 2021; Ratjen 2017; Schwarz 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015). A total of 11 studies examined triple combination therapy, a combination of a novel corrector with tezacaftor and ivacaftor (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022); four of these were Phase 3 studies of the combination elexacaftor‐tezacaftor‐ivacaftor (Barry 2021; Heijerman 2019; Middleton 2019; Sutharsan 2022).
When considering the results reported below, please note that the generally accepted minimally important clinical difference (MCID) for FEV1 is 6%, as specified in the protocol for the included study TRAFFIC 2015, but there is no clear agreement on what constitutes a MCID for change in CFQ‐R score or LCI, although it has been suggested that an improvement of four points on the CFQ‐R scale could be considered a MCID (Quittner 2009; Ramsey 2011).
Monotherapy versus control
Early‐phase trials evaluated potential molecules 4PBA (Rubenstein 1998; Zeitlin 2002), CPX (McCarty 2002), N6022 (Donaldson 2014), cavosonstat (Donaldson 2017), lumacaftor (Boyle 2014; Clancy 2012) and FDL169 (Horsley 2017). No study reported on survival and there were only limited data available for QoL, which did not show any clinically relevant improvements. There was no significant impact on clinical outcomes (including sweat chloride) with either 4BPA, CPX or N6022 and Phase 3 studies of these drugs were not conducted. The study of FDL169 versus placebo reported an improvement in the absolute change in FEV1 % predicted at the 400 mg dose (MD 4.68 % predicted, 95% CI 0.12 to 9.24; Analysis 5.2). Though statistically significant, it is uncertain whether this improvement is clinically significant, meaning that based on the strength of relevant evidence, currently we can only say that this intervention may lead to a benefit for pwCF (Horsley 2017). There are no significant concerns about safety for any corrector at any dose when compared to placebo. In an early phase study of cavosonstat monotherapy, there was a reduction in sweat chloride of ‐4.1 mmol/L (P = 0.032) at the highest dose (200 mg); however, this reduction was not considered clinically significant. There was a modest improvement in sweat chloride with lumacaftor alone compared with placebo after one month (MD ‐8.21 mmol/L, 95% CI ‐14.30 to ‐2.12) (Clancy 2012), but not sufficient to warrant investigation of this agent as monotherapy in later phase studies. A difference was observed in the change from baseline of sweat chloride for the 600 mg dose of FDL169 versus placebo, in this case showing FDL169 to increase sweat chloride level versus placebo (MD 8.84 mmol/L, 95% CI 1.40 to 16.28; Analysis 5.4).
We have identified a further ongoing study of cavosonstat, which may potentially be eligible for inclusion in this review at a later date (NCT02589236) and one abstract stated that FDL169 will be studied in combination with a potentiator FDL176 (Horsley 2017).
Dual therapy versus control
Six studies with 1495 participants compared lumacaftor plus ivacaftor to placebo (Boyle 2014; Ratjen 2017; Stahl 2021; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021) and nine studies with 1132 participants compared tezacaftor plus ivacaftor to placebo or to ivacaftor alone (i.e. tezacaftor placebo) (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017). The efficacy outcomes (primary and secondary) for the tezacaftor‐ivacaftor Phase 3 study were similar to those reported with lumacaftor‐ivacaftor (Taylor‐Cousar 2017).
There was one death in the tezacaftor plus ivacaftor group of one study, and this was not deemed to be related to the study drug (Schwarz 2021). In participants allocated to the lumacaftor‐ivacaftor combination, combined trial data demonstrated no difference with regards to change in a generic measure of QoL (EQ‐5L‐3D), but there was an improvement in the respiratory domain of the CF‐specific QoL measure (CFQ‐R) at six months (MD 2.83, 95% CI 0.91 to 4.74; I2 = 0%; 3 studies, 1139 participants; moderate‐certainty evidence; Analysis 12.2). Data from five tezacaftor plus ivacaftor studies found differences in the respiratory domain of the CFQ‐R at six months in favour of the treatment group (MD 2.88, 95% CI 2.48 to 3.29; I2 = 36%; 5 studies, 946 participants; Analysis 18.1) (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021; Taylor‐Cousar 2017). Given the suggestion that the MCID for this measure is four points, and based on low‐ to moderate‐certainty evidence for these interventions and this outcome, lumacaftor‐ivacaftor and tezacaftor‐ivacaftor have not been associated with a clinically important increase in the CFQ‐R QoL measure. There were no differences between treatment groups found in the other tezacaftor‐ivacaftor studies (Davies 2021; NCT02070744).
With respect to respiratory function (primarily FEV1 % predicted), at six months there were differences in change from baseline in favour of the lumacaftor‐ivacaftor combination when the two lumacaftor doses were pooled over placebo in both relative change (MD 5.12, 95% CI 3.57 to 6.67; I2 = 0%; 3 studies, 1134 participants; high‐certainty evidence; Analysis 12.4; Table 3) and in absolute change (MD 3.08, 95% CI 2.20 to 3.97; I2 = 0%; 3 studies, 1134 participants; moderate‐certainty evidence; Analysis 12.5; Table 3). There was an improvement in absolute change in FEV1 values at one month in the pooled results of three studies looking at 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor versus either placebo or ivacaftor (MD 3.54, 95% CI 2.39 to 4.69; I2 = 0%; 3 studies, 556 participants; Analysis 18.14) (Donaldson 2018; NCT02508207; Taylor‐Cousar 2017). Data from the studies assessing this treatment regimen also showed a greater relative and absolute change from baseline in FEV1 % predicted at six months (MD 0.92, 95% CI 0.72 to 1.11; I2 = 94%; 5 studies, 944 participants; Analysis 18.13; and MD 0.39, 95% CI 0.27 to 0.52; I2 = 95%; 5 studies, 944 participants; Analysis 18.14) (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021; Taylor‐Cousar 2017). At three months, there were no differences found between treatment groups in the study looking at 50 mg twice‐daily tezacaftor plus 150 mg twice‐daily ivacaftor versus either placebo or ivacaftor (NCT02070744) and the paediatric Davies study also did not find any differences between groups (Davies 2021). Improvement in FEV1 is considered an important surrogate outcome measure for pwCF. The European Medicines Agency (EMA) has suggested that "as FEV1 is linked to mortality, any significant difference between placebo and active treatment is potentially clinically relevant" (EMA 2012); therefore, based on effect size and strength of evidence, both lumacaftor‐ivacaftor and tezacaftor‐ivacaftor have been associated with a clinically relevant improvement in FEV1 when compared to control. In the study protocol, the MCID in absolute change in FEV1 % predicted used to calculate the sample sizes for the TRAFFIC and TRANSPORT studies was 5% (TRAFFIC 2015; TRANSPORT 2015). This magnitude of improvement in respiratory function was not achieved with the lumacaftor‐ivacaftor combination in these studies. In a post hoc change to the protocol, the primary outcome for TRAFFIC and TRANSPORT was altered from absolute change from baseline in FEV1 % predicted at six months to an average of the FEV1 values at four and six months. The 200 mg twice‐daily lumacaftor plus 250 mg twice‐daily ivacaftor study on children aged 6 to 11 years reported the change in LCI as its primary outcome (Ratjen 2017); although this measure is a well‐validated research outcome assessing respiratory function, it is not yet routinely used in clinical practice. The children allocated to lumacaftor‐ivacaftor demonstrated a greater reduction in LCI compared to those receiving placebo (least squares MD ‐1.10, 95% CI ‐1.40 to ‐0.80; Analysis 15.3). Although this difference is statistically significant, it is difficult to assess the clinical relevance of this result for young pwCF as there are a lack of longer‐term data for this outcome. There were statistically significant improvements in LCI2.5 found in both the paediatric tezacaftor‐ivacaftor study (MD ‐0.51, 95% CI ‐0.74 to ‐0.29; P < 0.0001) (Davies 2021) and paediatric lumacaftor‐ivacaftor study (MD ‐0.69, 95% CI ‐1.35 to ‐0.03; Analysis 9.1) (Stahl 2021). In the paediatric Davies study, there was a greater improvement from baseline in LCI5.0 (MD ‐0.30, 95% CI ‐0.39 to ‐0.20; P < 0.0001) (Davies 2021). A study involving a subgroup of participants (n = 10; seven in the active treatment group and three in the placebo group) reported no improvement in chest CT scan score from baseline to 24 weeks; this was a secondary outcome in our review (Analysis 15.6; Analysis 15.7; Analysis 15.8; Ratjen 2017).
A number of secondary outcomes were reported in the included studies. Overall, the safety data reported for the lumacaftor‐ivacaftor combination were reassuring, but there was clear evidence of increased reports of early respiratory symptoms (OR 2.05, 99% CI 1.10 to 3.83; Analysis 10.6). The aetiology of this event is unclear and it was reported to settle after a few weeks if the intervention was continued (TRAFFIC 2015; TRANSPORT 2015). Two participants were withdrawn because of hypertension (one during the follow‐up study (PROGRESS 2017)). For participants (n = 80) receiving 400 mg twice a day of lumacaftor there was a significant mean (SE) increase in systolic blood pressure of 5.1 (1.5) mm Hg and in diastolic blood pressure of 4.1 (1.2) mm Hg (PROGRESS 2017). For children (aged 6 to 11 years) enrolled in the Phase 3 study of lumacaftor‐ivacaftor combination therapy, the safety profile reported was similar to the TRAFFIC and TRANSPORT studies, including transient early respiratory compromise and infrequent elevation in serum transaminases (liver enzymes) (Ratjen 2017). There were two life‐threatening AEs reported in the tezacaftor plus ivacaftor groups, not considered to be related to the trial drug (Schwarz 2021; Taylor‐Cousar 2017). There was no increased reporting of AEs for the tezacaftor‐ivacaftor studies, in particular the early transient dyspnoea reported with the lumacaftor‐ivacaftor combination.
In the TRAFFIC and TRANSPORT studies, pulmonary exacerbations were reported more frequently in participants allocated to placebo compared to those receiving the lumacaftor‐ivacaftor combination (Analysis 10.6; Analysis 11.6) (TRAFFIC 2015; TRANSPORT 2015); pulmonary exacerbations are challenging to record accurately, but important to pwCF. One tezacaftor‐ivacaftor study reported the time to first pulmonary exacerbation and found a decreased hazard ratio in the intervention group versus the placebo group (Taylor‐Cousar 2017). There was no difference found between groups in the number of participants with at least one pulmonary exacerbation, the number of pulmonary exacerbation events and the annualised rate of pulmonary exacerbation events ratio in one study looking at tezacaftor‐ivacaftor (Munck 2020). In participants allocated to the lumacaftor‐ivacaftor combination therapy, BMI improved after six months (Analysis 9.6; Analysis 10.10; Analysis 11.10). There was no difference found between study groups for the relative change from baseline in BMI (Analysis 11.11). Early phase studies of lumacaftor combined with ivacaftor demonstrated a greater magnitude of effect with a reduction in sweat chloride compared to lumacaftor monotherapy with the higher dose of ivacaftor (MD ‐10.9 mmol/L, 95% CI ‐17.6 to ‐4.2; 1 study, 34 participants; Analysis 14.3) (Boyle 2014). There was a reduction in sweat chloride in the lumacaftor‐ivacaftor group compared to placebo (Stahl 2021), and there were reductions in sweat chloride levels in all of the newly identified tezacaftor‐ivacaftor studies reporting on this outcome (Davies 2021; McKone 2021; Munck 2020; NCT02070744; NCT02508207). There was no difference found in the radiological measures of lung disease and the acquisition of respiratory pathogens between treatment and control groups in either of the newly identified tezacaftor‐ivacaftor or lumacaftor‐ivacaftor studies (NCT02730208; Stahl 2021).
Triple therapy versus control or dual therapy
A number of agents have been evaluated in combination with tezacaftor‐ivacaftor (triple therapy) in the 11 studies included in this comparison (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022). One triple combination, elexacaftor‐tezacaftor‐ivacaftor, was taken forward for Phase 3 studies for pwCF with one or two F508del variants (Barry 2021; Heijerman 2019; Middleton 2019; Sutharsan 2022). For pwCF with one F508del variant, this represented the first exposure to a variant‐specific therapy and the comparator was a placebo (Middleton 2019). In another study, the participants were heterozygous for the F508del variant and a gating or residual function variant, and the comparator was either ivacaftor or tezacaftor plus ivacaftor following a four‐week run‐in period (Barry 2021). For those with two F508del variants, the participants were already on tezacaftor‐ivacaftor and this was the comparator used for these studies (Heijerman 2019; Sutharsan 2022).
No deaths were reported in any of the included studies (Barry 2021; Davies 2018a; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022) (high‐certainty evidence).
In early phase studies, at one month, triple therapy combinations increased QoL scores across multiple doses for pwCF who have one or two F508del gene variants (Davies 2018a; Davies 2018b; Keating 2018; NCT02951182; NCT02951195; NCT03447249; NCT03460990); although this effect was not seen in the 400 mg trial of VX‐659 in people with the F508del/MF genotype (Davies 2018b). For pwCF with two F508del variants, over one month elexacaftor‐tezacaftor‐ivacaftor improved QoL respiratory domain scores compared to tezacaftor‐ivacaftor combination therapy (MD 17.78, 95% CI 12.90 to 22.66; I2 = 0%; 2 studies, 135 participants; moderate‐certainty evidence; Analysis 29.1) (Heijerman 2019; Keating 2018). At two months, elexacaftor‐tezacaftor‐ivacaftor improved QoL respiratory domain scores versus control for participants with F508del/gating and F508del/RF genotypes (MD 8.70, 95% CI 5.34 to 12.06; 1 study, 258 participants; moderate‐certainty evidence; Analysis 31.1) (Barry 2021). Similarly, at six months, elexacaftor‐tezacaftor‐ivacaftor improved QoL respiratory domain scores versus placebo in F508del/MF participants (MD 20.2, 95% CI 16.2 to 24.2; 1 study, 403 participants; moderate‐certainty evidence; Analysis 28.1) (Middleton 2019). At one month and six months, elexacaftor‐tezacaftor‐ivacaftor improved QoL respiratory domain scores versus tezacaftor‐ivacaftor in F508del/F508del participants (MD 17.40, 95% CI 11.94 to 22.86; 1 study, 107 participants; moderate‐certainty evidence ; Analysis 29.1) (Heijerman 2019) and (MD 15.90, 95% CI 11.74 to 20.06; 1 study, 175 participants; moderate‐certainty evidence; Analysis 29.1) (Sutharsan 2022).
For triple combinations including VX‐659, there was an improvement in both absolute and relative change in FEV1 from baseline at one month compared to placebo for pwCF with one or two copies of F508del (Davies 2018b; NCT02951182; NCT02951195; NCT03447249; NCT03460990). Similar results were found for the elexacaftor combination (Keating 2018) (moderate‐certainty evidence). For F508del/MF participants, at six months, the elexacaftor‐tezacaftor‐ivacaftor group demonstrated an improvement in absolute change in FEV1 % predicted compared to placebo (MD 14.30 % predicted, 95% CI 12.8 to 15.8; 1 study, 403 participants; moderate‐certainty evidence; Analysis 28.3) (Middleton 2019). For pwCF with two F508del variants, at one month elexacaftor‐tezacaftor‐ivacaftor improved absolute change in FEV1 % predicted (MD 10.0% predicted, 95% CI 7.5 to 12.5; 1 study, 107 participants; moderate‐certainty evidence; Analysis 29.3) (Heijerman 2019). At six months, the absolute change in FEV1 % predicted continued to improve (MD 10.20% predicted, 95% CI 8.26 to 12.14; 1 study, 175 participants; moderate‐certainty evidence; Analysis 29.3) (Sutharsan 2022). For pwCF with F508del/gating and F508del/RF genotypes, elexacaftor‐tezacaftor‐ivacaftor improved absolute change in FEV1 (MD 3.50% predicted, 95% CI 2.24 to 4.76; 1 study, 258 participants; moderate‐certainty evidence; Analysis 31.2) (Barry 2021).
There was no difference in the occurrence of AEs for any combination compared to control across genotype groups and there were no unexpected AEs related to the study drug (Davies 2018a; Davies 2018b; Keating 2018; NCT02951182; NCT02951195; NCT03447249; NCT03460990). Elexacaftor‐tezacaftor‐ivacaftor led to no difference in the number or severity of AEs compared to placebo or control (Barry 2021; Heijerman 2019; Middleton 2019; Sutharsan 2022). In one study, there were fewer participants with F508del/MF experiencing severe AEs in the VX‐659 240 mg treatment arm than the placebo group (NCT03447249). One study showed a longer time to the next pulmonary exacerbation in participants with F508del/MF taking elexacaftor‐tezacaftor‐ivacaftor compared to placebo over the six‐month study (Middleton 2019). There were fewer participants with F508del/gating and F508del/RF in the 200 mg elexacaftor group experiencing pulmonary exacerbations than in the control (Barry 2021) and there were also fewer participants, with F508del/MF, in the 240 mg once‐daily VX‐659 group experiencing pulmonary exacerbations at six months when compared to placebo (NCT03447249). Data from one study showed fewer exacerbations in F508del/F508del participants taking elexacaftor‐tezacaftor‐ivacaftor compared to control (MD 0.17, 99% CI 0.06 to 0.45; 1 study, 175 participants, moderate‐certainty evidence; Analysis 29.6) (Sutharsan 2022).
Reductions in sweat chloride were reported in both the early and the Phase 3 studies. In participants with two F508del variants, at one month, there was a greater mean reduction in sweat chloride (mmol/L) in the elexacaftor‐tezacaftor ivacaftor group compared to the tezacaftor‐ivacaftor group (MD ‐44.32 mmol/L, 95% CI ‐48.80 to ‐39.84; I2 = 0%; 2 studies, 135 participants; Analysis 29.7) (Heijerman 2019; Keating 2018). This difference was maintained at six months (MD ‐42.80 mmol/L, 95% CI ‐46.27 to ‐39.33; 1 study, 175 participants; Analysis 29.7) (Sutharsan 2022). For F508del/MF participants, the reduction was similar at one month (MD ‐40.96 mmol/L, 95% CI ‐43.60 to ‐38.33; I2 = 0%; 2 studies, 436 participants; Analysis 28.9) (Keating 2018; Middleton 2019) and at six months (MD ‐41.80 mmol/L, 95% CI ‐44.3 to ‐39.3; 1 study, 403 participants; Analysis 28.9) (Middleton 2019).
In three studies, triple therapy saw an improvement in measures of nutrition and growth versus control in participants with both F508del/F508del and F508del/MF genotypes (Heijerman 2019; Middleton 2019; NCT03447249). In homozygous participants, the intervention saw a greater increase in weight (MD 1.6 kg, 95% CI 1.0 to 2.10; 1 study, 107 participants; Analysis 29.8), as well as a greater increase in BMI (MD 0.6 kg/m², 95% CI 0.41 to 0.79; 1 study, 107 participants; Analysis 29.9) (Heijerman 2019). In the second study, heterozygous participants in the intervention group saw a greater increase in weight compared to the control group (MD 2.90 kg, 95% CI 2.40 to 3.40; 1 study, 403 participants; Analysis 28.10), and also a greater increase in BMI z score (MD 0.30, 95% CI 0.17 to 0.43; 1 study, 403 participants; Analysis 28.11) (Middleton 2019). For the 240 mg once daily VX‐659 study in participants with F508del/MF, there were improvements in BMI, BMI Z‐score and body weight found (NCT03447249).
Overall completeness and applicability of evidence
This review has examined evidence for efficacy and safety. We have not included outcomes related to cost‐effectiveness.
Monotherapy versus control
The single‐agent studies enrolled participants with two copies of the F508del variant. These studies have not been taken forward on larger, more representative populations into Phase 3 studies. New agents are currently being assessed in early‐phase studies: one study of cavosonstat including 51 adults aged over 18 with CF (Donaldson 2017) and a Phase 1 study of FDL169 including 27 participants aged 18 or over who were homozygous for F508del, early data from which were provided in a poster and conference abstract (Horsley 2017).
Dual therapy versus control
Phase 2 and the Phase 3 studies of both lumacaftor and tezacaftor combined with ivacaftor have examined this therapy for people with two copies of the F508del variant (F508del homozygotes), including one Phase 4 lumacaftor study (Donaldson 2018; NCT02070744; NCT02508207; NCT02730208; Ratjen 2017; Schwarz 2021; Stahl 2021; Taylor‐Cousar 2017; TRAFFIC 2015; TRANSPORT 2015; Wilson 2021). One Phase 3 tezacaftor study included participants who were heterozygous for the F508del variant and a gating variant (McKone 2021), and another included participants who were heterozygous for the F508del variant and a minimal function variant (Munck 2020). One paediatric Phase 3 tezacaftor study included participants who were homozygous for the F508del variant or heterozygous for the F508del variant and a residual function variant (Davies 2021). The Phase 2 study of tezacaftor‐ivacaftor additionally examined the impact of this therapy on adults with one F508del variant and the G551D variant (Donaldson 2018). One cross‐over study has examined pwCF with one F508del variant combined with an ivacaftor‐sensitive residual function mutation, to evaluate any potential additive impact of tezacaftor on the recognised ivacaftor benefit (Rowe 2017). We excluded this study because of concerns over study design, in particular carryover effects of an intervention (ivacaftor) that has been shown to correct the basic defect in CF.
The Phase 3 studies of both the lumacaftor and ivacaftor dual therapy are well‐powered and provide clear statistical evidence of improvement in clinical outcomes, even if these are limited in magnitude compared to the changes anticipated in the protocol and to those reported for individuals with G551D receiving ivacaftor (Ratjen 2017; TRAFFIC 2015; TRANSPORT 2015). These Phase 3 studies were conducted across a large number of CF centres in North America, Europe and Australia, and the results are applicable to pwCF who are homozygous for F508del in these regions with mild to moderate lung disease.
The Phase 3 studies of lumacaftor‐ivacaftor enrolled children and adults (age range 6 to 64 years) (Ratjen 2017; TRAFFIC 2015; TRANSPORT 2015). For lumacaftor‐ivacaftor, results were consistent across age groups, although for the 24‐week study of 6‐ to 11‐year olds the absolute change in FEV1 % predicted was less marked (MD 2.40, 95% CI 0.40 to 4.40; 1 study, 206 participants; low‐certainty evidence; Analysis 15.2) (Ratjen 2017).
Triple therapy versus control
The Phase 1 study by Davies, which was published as part of the same paper as the Phase 2 study, enrolled 12 pwCF with one copy of F508del and one MF variant (F508del/MF) and all were aged over 18 years; data on younger pwCF are required (Davies 2018a).
The Phase 2 trials of triple therapy enrolled pwCF homozygous for F508del (F508del/F508del) and also pwCF F508del/MF. Three studies did not include people under 18 years of age (Davies 2018b; Keating 2018; NCT02951195) and one study included participants aged 12 years and older (NCT02951182).
Two Phase 2 studies tested the triple therapy combination of VX‐659 or elexacaftor plus tezacaftor 100 mg once daily plus deutivacaftor, a deuterated form of ivacaftor that has a longer half‐life in the body than the typical ivacaftor formulation. Deutivacaftor is taken once daily at the same dose (150 mg), rather than the typical 150 mg twice per day with standard ivacaftor. This combination was only tested in a group of participants with the F508del/MF heterozygous genotype; it is only used in combination with the maximum tested doses of VX‐659 and elexacaftor. The studies do not state why a combination of deutivacaftor is tested in these trials, or why it is only tested in people with F508del/MF genotypes, or why it is only tested in combination with the maximum doses of the above medications (Davies 2018b; Keating 2018). One two‐part Phase 2 study compared VX‐440 plus tezacaftor plus ivacaftor to triple placebo for four weeks in part 1, and compared VX‐440 plus tezacaftor plus ivacaftor to tezacaftor plus ivacaftor for four weeks in part 2 (NCT02951182); the other two‐part Phase 2 study compared VX‐152 plus tezacaftor plus ivacaftor to triple placebo for two weeks in part 1, and compared VX‐152 plus tezacaftor plus ivacaftor to tezacaftor plus ivacaftor for two weeks in one cohort and four weeks in the second cohort (NCT02951195).
Three Phase 3 studies have tested elexacaftor‐tezacaftor‐ivacaftor versus placebo (for participants with F508del/MF) or versus tezacaftor‐ivacaftor (for participants with F508del/F508del). An additional Phase 3 study tested elexacaftor‐tezacaftor‐ivacaftor versus either ivacaftor (for participants with F508del‐gating genotypes) or tezacaftor‐ivacaftor (for participants with F508del‐residual function genotypes). The studies spanned three continents and included adults and children 12 years and older (Barry 2021; Heijerman 2019; Middleton 2019; Sutharsan 2022). The study on pwCF with either F508del/gating or F508del/residual function genotypes was for eight weeks (Barry 2021). The study on pwCF with one F508del variant was for six months (Middleton 2019), one study on participants with F508del/F508del was only one month in duration (Heijerman 2019) and the other study testing this regimen in participants with this genotype was for six months (Sutharsan 2022).
Two Phase 3 studies tested VX‐659 plus tezacaftor plus ivacaftor compared to either triple placebo (in participants with F508del/MF) or to tezacaftor‐ivacaftor (in participants with F508del/F508del). The studies included participants aged 12 years and older (NCT03447249; NCT03460990). The study on participants with F508del/MF was for six months (NCT03447249) and the study on participants with F508del/F508del was for eight weeks (NCT03460990).
Quality of the evidence
Reflecting the emerging field of variant‐specific therapy, studies included in this review were often difficult to appraise and interpret due to complex designs that incorporated several drug doses and genotype combinations.
Monotherapy versus control
Authors of some studies were only able to provide abstracts or posters (or both) as sources of data. For the early‐phase studies of 4PBA, CPX, N6022 and FDL169, relevant outcome data for this review were limited and the risk of bias for various domains was difficult to judge. Important results for the drugs lumacaftor and cavosonstat within this comparison are summarised in the tables (Table 1; Table 2). We have not presented other monotherapy treatments in the summary of findings tables as interventions have not yet been taken forward on larger, more representative populations in Phase 3 studies.
We judged the certainty of the evidence from a small study of lumacaftor monotherapy to be very low to moderate due to concerns over incomplete outcome data, selective reporting and limited outcome data resulting in wide CIs around effect sizes (Boyle 2014; Table 1).
The certainty of the evidence from a short‐term study of cavosonstat compared to placebo was low to very low due to concerns over unclear methodological design, indirectness (lack of applicability of results to children) and limited outcome data resulting in wide CIs around effect sizes (Donaldson 2017; Table 2).
Dual therapy versus control
We judged the certainty of the evidence from the three large multicentre RCTs of lumacaftor‐ivacaftor combination therapy to be moderate to high (PROGRESS 2017; TRAFFIC 2015; TRANSPORT 2015; Table 3). Not all outcomes were reported in the final study publications; some were available in the online supplement, some were extrapolated from graphical figures and others were available on the NIH database (ClinicalTrials.gov). Although the time point for assessment of the primary outcome changed post hoc from FEV1 % predicted at six months to an aggregate of four and six months (which represented a larger treatment effect), we did not judge this to reflect a high risk of bias. This was because the results at six months were also significant, and the amended protocol states that "This change was made during final review by senior management. It is important to note that this change was made based on theoretical considerations alone. No data analysis was used to support this change and, in fact, the spirometry data were maintained at the designated vendor and were not available to any Vertex personnel".
We judged the certainty of the evidence from an additional large multicentre RCT of lumacaftor‐ivacaftor combination therapy to be moderate to low (Ratjen 2017; Table 4). The study recruited children aged 6 to 11 years, so the results are not applicable to other age groups. Not all outcomes were reported in the final study report and additional data could not be extracted from graphical figures. Furthermore, the analysis approach taken within this review adjusted for earlier time points in the analysis at six months, therefore results should be interpreted as the treatment effect averaged from each study visit until six months.
We judged the certainty of the evidence from a small, very short‐term study of lumacaftor‐ivacaftor combination therapy to be very low to moderate, similar to the monotherapy lumacaftor study (see above). We had concerns over incomplete outcome data, selective reporting and limited outcome data resulting in wide CIs around effect sizes (Boyle 2014; Table 5).
We judged the evidence from the nine tezacaftor‐ivacaftor combination therapy studies to be of low to moderate certainty (Davies 2021; Donaldson 2018; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Taylor‐Cousar 2017; Table 6). Results may not be applicable to children under the age of 12 (as only one trial out of nine provided data for children aged under 12); also, some results are not applicable to individuals homozygous for F508del. Not all studies provide data for each outcome reported in Table 6; nine studies provide data for survival, five studies provide data for QoL and FEV1, eight studies provide AE data and one study provides data for the time to next pulmonary exacerbation. Furthermore, in the large tezacaftor‐ivacaftor combination study, a number of outcomes were recorded according to the study protocol, but not presented in the published study report (Taylor‐Cousar 2017).
Triple therapy versus control
The early phase triple therapy studies were complex because they evaluated a number of factors: different doses, different genotypes (F508del/F508del and F508del/MF), different correctors and different forms of ivacaftor. This resulted in eight different comparator groups. We judged the evidence for the comparison of VX‐659 plus tezacaftor plus ivacaftor or VX‐561 compared with control to be moderate‐ to high‐certainty (Davies 2018a; Davies 2018b; NCT03447249; NCT03460990; Table 7), but this intervention has not been taken beyond Phase 2 studies. Elexacaftor was selected for Phase 3 studies in combination with tezacaftor and ivacaftor.
The certainty of the Phase 3 studies of elexacaftor‐tezacaftor‐ivacaftor ranged from moderate to high. We downgraded the evidence for QoL, relative change from baseline in FEV1 % predicted, AEs and time to first pulmonary exacerbation due to indirectness or lack of applicability, as data do not include children under the age of 12 and those with more severe disease (Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022; Table 8).
We judged the evidence from the VX‐440 combination therapy study to be of moderate certainty for each outcome presented (NCT02951182; Table 10). We downgraded the evidence for indirectness as the study did not include children under the age of 12 and those with more severe disease; furthermore, this study only presented short‐term data. We additionally downgraded for imprecision (small sample size) (Table 10).
We judged the evidence from the VX‐152 combination therapy study to be low certainty for each outcome presented (NCT02951195; Table 11). We downgraded the evidence for indirectness ‐ similar to the VX‐440 combination therapy study, this study did not include children under the age of 18 and those with more severe disease, and only presented short‐term data. Additionally, the study included four participants with FEV1 less than 40% despite the inclusion criteria for the study stating that FEV1 should be at least 40% and up to and including 90% of predicted normal for age, sex and height at the screening visit, thus increasing the risk of bias (Table 11).
Potential biases in the review process
As outlined above, the review authors conducted comprehensive literature searches (Search methods for identification of studies; Appendix 1). For each iteration of the review, two authors individually applied the inclusion and exclusion criteria to the identified studies and excluded studies that were not relevant, with arbitration of a third author if required (KWS) (Data extraction and management). The analyses were undertaken by two review authors and checked for appropriateness by the review statistician (SN). This approach minimised the risks of bias in the review process.
None of the authors have received direct or indirect payments from the companies responsible for the development of any agents included in this review; however, KWS has previously attended and has organised educational events that have received financial support from Vertex, the company that has developed and is evaluating some of the agents included in this review (outside the time limits for declarations for Cochrane Conflict of Interest statements).
Not all results were reported in a format from which they could be accurately extracted, and we needed to extrapolate data for several outcomes from graphs and figures. We have contacted authors and are awaiting confirmation that these estimates are accurate. This review has assessed all available published study data and we contacted study authors for relevant unpublished information and individual participant data; to date we have not received any further data. We are not aware of any unpublished trials.
Definitions of various degrees of AEs and definitions of exacerbations differed between the review and the included studies; sometimes investigators did not specifically define what was recorded as an exacerbation. In order to incorporate data on exacerbations from different studies, we set a broad definition of what we would record as an exacerbation (Types of outcome measures). This broad definition, together with variation between defining and recording exacerbations in included studies, means that synthesising and reporting data on exacerbations could be viewed as a limitation of this review.
Seven out of 15 new studies were identified from a trials database (clinicaltrials.gov). Including data that have not yet been published in journals has allowed for the review to encompass a wide range of information. However, this is also a limitation of the review, as the data identified have not yet been peer‐reviewed. It is not yet clear if these data will be published in peer‐reviewed journals.
Agreements and disagreements with other studies or reviews
The National Institute for Health and Care Excellence (NICE) in the UK has undertaken a health technology appraisal for lumacaftor‐ivacaftor, which was published on 27 July 2016 (NICE 2016). The appraisal included the TRAFFIC, TRANSPORT and PROGRESS studies (PROGRESS 2017; TRAFFIC 2015; TRANSPORT 2015); the report concluded that the quality of these studies was generally good and that the results were generalisable to a UK population with mild‐moderate disease severity. The evidence review group (ERG) noted that there were significant effects on key outcomes compared with standard care alone, but it was unclear how clinically significant the effects were. AE data were recorded as per the published papers, but withdrawals due to hypertension and the overall increase in blood pressure in participants receiving 400 mg twice a day were not recorded. In addition, the ERG examined a detailed cost‐effectiveness assessment (including an estimate of incremental cost‐effectiveness ratio) and concluded, on that basis, that lumacaftor‐ivacaftor is not recommended, within its marketing authorisation, for treating CF in people 12 years and older who are homozygous for F508del mutation of the CFTR gene. These conclusions were reasonable at the time of publication, and in agreement with our earlier review, but new evidence outlined in this review now downgrades those recommendations, in our opinion. NICE (UK) have commissioned a further comprehensive appraisal of modulator therapy, the report of which is expected later in 2023.
An evaluation of the safety of lumacaftor and ivacaftor highlighted the finding of "transaminitis" (raised transaminases) in ivacaftor and combination studies (Talamo Guervara 2017). In addition, the review reported non‐congenital cataracts identified in pre‐clinical studies and in children taking ivacaftor and combined therapy. The review also highlighted that lumacaftor is a strong inducer of the liver enzyme, cytochrome P3A and the implications for co‐prescribing of drugs metabolised through this route.
A review and meta‐analysis published in December 2018 examined the efficacy and safety of dual therapy with CFTR correctors and potentiators (Wu 2018). It looked at pwCF with F508del/F508del (monotherapy not assessed). Two studies included in this Cochrane Review were not included in the Wu review, although they did meet the eligibility criteria (PROGRESS 2017; Taylor‐Cousar 2017). Also, for the Boyle study, we included data from cohort 1, but not cohorts 2 and 3 due to concerns over pooling the control group (Boyle 2014); the Wu review includes these data. Furthermore, we included heterozygous participants from the 2018 Donaldson tezacaftor‐ivacaftor study, due to other participants being pooled, which negated the effects of randomisation (Donaldson 2018). The Wu review includes all pooled and unpooled participants (including those not homozygous for F508del), impacting on potential bias by disrupting randomisation and blinding. We did not include pooled placebo data in our review, which explains the different number of participants. The Wu review presents a meta‐analysis of efficacy data for both lumacaftor‐ivacaftor and tezacaftor‐ivacaftor therapies, consistent with our data. Finally, Wu did not report the AE of hypertension found with lumacaftor‐ivacaftor therapy, which we reported. The strong conclusions of the Wu review are not supported by their meta‐analysis and overstate improvement in efficacy measures. In addition, the authors claim to demonstrate a dose‐response effect, but there is no evidence of this from the data presented. Some interpretation within the Wu review is based on observational and "experimental" studies not included in the review, rather than evidence from their meta‐analysis.
Another systematic review was published in March 2022 (Wang 2022). The review included pwCF with at least one F508del variant and after searching for RCTs on PubMed, Web of Science and the Cochrane Library, the authors included six RCTs, all of which were included in our Cochrane Review (Barry 2021; Davies 2018b; Heijerman 2019; Keating 2018; Middleton 2019; Sutharsan 2022). In contrast to our review, Wang did not include data from eligible trials on any trials registry. The authors of the Wang study assessed the risk of bias in all of the eligible studies using Cochrane methodology, but omitted a further domain used in our review entitled ‘other bias’. We used this section to comment on any additional uncertainties in the included studies. Wang’s assessments of bias differed from the Cochrane Review in a number of domains. Wang determined that each study had a low risk of bias for each domain, and did not give reasons for each justification. In contrast, our Cochrane Review found two studies to have unclear risks of both detection and reporting bias (Davies 2018b; Keating 2018) and two studies to have unclear risks of reporting bias (Barry 2021; Sutharsan 2022). Furthermore, the Wang review assessed only one of the groups from the Davies study (400 mg VX‐659‐tezacaftor‐ivacaftor) (Davies 2018b), thus it lacked significant amounts of data, including data involving deutivacaftor, when compared to our Cochrane Review. Similarly, Wang only reported data from the 200 mg elexacaftor‐tezacaftor‐ivacaftor group from the Keating study (Keating 2018). The Wang review assessed similar outcomes to this review, although it did not identify any increases in blood pressure across the studies. Nonetheless, the conclusions of the Wang review were very similar to those of this Cochrane Review, with both reviews commenting on the fact that all participants included in the reviews were aged 12 and over, and future evaluations of triple therapy combinations in younger pwCF are needed.
Authors' conclusions
Implications for practice.
There is a lack of evidence to support monotherapy with a corrector for people with cystic fibrosis (pwCF) who have two F508del variants (F508del/F508del). Future updates of this review will not include data from monotherapy studies, unless new agents emerge.
There are large dual therapy studies included in the current version of this review showing small differences in outcomes. Evidence for dual therapy combinations demonstrates that although the therapy is more effective than monotherapy, and some of the differences identified were statistically significant, these differences were small when compared to the triple therapy data.
Evidence to support dual therapy is reduced as more data become available. For example, we added six‐month data from four new studies (McKone 2021; Munck 2020; NCT02070744; Schwarz 2021) to the previously included study (Taylor‐Cousar 2017), which reduced the difference between once‐daily 100 mg tezacaftor plus twice‐daily 150 mg ivacaftor compared to placebo or 150 mg ivacaftor alone for the change in Cystic Fibrosis Questionnaire ‐ Revised (CFQ‐R) respiratory domain scores from mean difference (MD) 5.10 (95% confidence interval (CI) 3.20 to 7.00; 1 study, 504 participants) in the previous version of the review (Southern 2018) to MD 2.88 (95% CI 2.48 to 3.29; I2 = 36%; 5 studies, 946 participants; Analysis 18.1). Similarly, the MDs at six months for both the relative and absolute change from baseline in forced expiratory volume in one second (FEV1) % predicted were reduced: relative change decreased from MD 6.80 (95% CI 5.30 to 8.30; 1 study, 504 participants) (Southern 2018) to MD 0.92 (95% CI 0.72 to 1.11; I2 = 94%; 5 studies, 944 participants; Analysis 18.13), and absolute change decreased from MD 4.00 (95% CI 3.10 to 4.90; 1 study, 504 participants) (Southern 2018) to MD 0.39 (95% CI 0.27 to 0.52; I2 = 95%; 5 studies, 944 participants; Analysis 18.14).
New dual therapies have provided some limited evidence of treatment effect in children, with improvements seen in one measure of lung function, body mass index (BMI)‐for‐age z score and sweat chloride concentrations (Davies 2021; Stahl 2021).
In terms of the triple therapy studies, the new data for elexacaftor‐tezacaftor‐ivacaftor are consistent with previous studies in terms of the magnitude of improvement in efficacy outcomes. One of the newly identified studies examined participants with F508del/gating and F508del/residual function genotypes, allowing for the authors to conclude that the presence of one F508del allele is sufficient for improvements to be seen with this triple therapy combination in pwCF already established on a recognised modulator therapy (for example, ivacaftor) (Barry 2021). New correctors identified in this updated review, VX‐440 and VX‐152, demonstrated promising results in Phase 3 studies with improvements in outcomes that were comparable to triple therapy with elexacaftor‐tezacaftor‐ivacaftor (NCT02951182; NCT02951195).
In summary, there is a lack of evidence to support corrector monotherapy for pwCF. The certainty of the evidence to support dual therapy ranges from high to low, but treatment effect appears to decrease as the evidence base is increased and these agents can no longer be considered as standard therapy, except in exceptional circumstances (for example, if triple therapy is not tolerated or the individual is too young). Dual therapy is currently the only treatment option for children under six years of age and the results of this review highlight the importance of obtaining robust evidence for triple therapy in this age group; such trials are ongoing. There is moderate‐ to high‐certainty evidence of significant and clinically relevant differences in key outcomes, with improved safety profiles compared to lumacaftor‐ivacaftor. This evidence supports elexacaftor‐tezacaftor‐ivacaftor triple therapy for eligible pwCF.
We present changes to the implications for practice at this update in an additional table (Table 21).
10. Implications for practice: summary of changes in 2023 update.
| Previous implications for practice (2020) | Current implications for practice (2023) |
| There is a lack of evidence to support single agent corrector therapy (monotherapy) for people with CF who have two F508del genetic variants (F508del/F508del). | There is a lack of evidence to support single agent corrector therapy (monotherapy) for people with cystic fibrosis who have two F508del variants (F508del/F508del). |
| There is some evidence to support dual therapies (lumacaftor‐ivacaftor and tezacaftor‐ivacaftor) for people with CF with the genotype F508del/F508del. | New trial data have reduced the evidence supporting dual therapy. Key changes include:
|
| In children younger than 12 years of age, there are no data to assess tezacaftor‐ivacaftor. In a study of lumacaftor‐ivacaftor in children aged 6 to 11 years, there was some evidence of clinical efficacy (decreasing LCI value), but the clinical relevance of these changes is not clear. | New dual therapies have provided some limited evidence of treatment effect in children, with improvements seen in one measure of lung function, BMI‐for‐age z score and sweat chloride concentrations (Davies 2021; Stahl 2021). |
| The magnitude of the reported improvements in efficacy measures suggests the potential of elexacaftor‐tezacaftor‐ivacaftor to provide a significant intervention for people with CF with one or two F508del variants. | The new data for elexacaftor‐tezacaftor‐ivacaftor are consistent with previous studies in terms of the magnitude of improvement in efficacy outcomes. A newly identified study allowed for the conclusion that the presence of one F508del allele is sufficient for improvements to be seen with this triple therapy combination in people with CF already established on a recognised modulator therapy (for example, ivacaftor) (Barry 2021). New correctors identified (VX‐440 and VX‐152) have demonstrated promising results in Phase 3 studies (NCT02951182; NCT02951195). |
BMI: body mass index; CF: cystic fibrosis; CFQ‐R: Cystic Fibrosis Questionnaire ‐ Revised (Quittner 2009); CI: confidence interval; FEV₁: forced expiratory volume in 1 second; LCI: lung clearance index; MD: mean difference
Implications for research.
It is important that post‐market surveillance is undertaken for all agents that correct F508del and other class II variants. It is clear that lumacaftor‐ivacaftor is associated with adverse effects such as high blood pressure, some of which have necessitated the withdrawal of therapy. Tezacaftor‐ivacaftor appears to have a more favourable safety profile, however more data are required.
The current available data for the triple combination of elexacaftor‐tezacaftor‐ivacaftor for pwCF with one or two F508del variants is mainly limited to pwCF aged 12 years and older, although safety data are available for younger children. Studies are needed to evaluate the impact of elexacaftor‐tezacaftor‐ivacaftor on pwCF under 12 years of age and those with more severe lung disease. There is also a need for information from longer‐term monitoring of this intervention.
As outcomes such as disease‐specific quality of life measures and lung clearance index become more embedded within Phase 3 trials, it is important that the validity of these outcomes is determined to establish minimally important changes and outline these in the relevant study protocols.
As new variant‐specific therapies emerge, it is important that lessons learnt from this review are taken on board. Investigators should report clearly on methodological approaches to reduce the risk of bias, in particular with regards to random sequence generation, allocation concealment and blinding; they should also ensure that randomisation is maintained when analysing data. It is important that future studies examine and clearly report on outcomes relevant to pwCF and their families. Pharmaceutical companies must consider how they can evaluate any new benefits from these medications. In addition to the respiratory system, other systems in the body need to be evaluated in clinical trials, for example the endocrine and gastrointestinal systems. Moving forward, outcomes that focus on the frequency of drug administration may be incorporated into clinical trials.
As triple therapy is established, it is appropriate to investigate the withdrawal of other standard therapies such as nebulised mucoactive agents. This should be undertaken in a robust manner to ensure safety and good outcomes.
The cost‐effectiveness and accessibility of these medications should also be evaluated. Adverse events need to be reported fully and characterised clearly. Some events relate to the physiological impacts of triple therapy (for example, sinus pain, abdominal pain and testicular pain), whereas others are characterised as idiosyncratic changes (for example, mood changes, skin rashes and transaminitis). Safety reporting of these new agents needs to be comprehensive and clear.
What's new
| Date | Event | Description |
|---|---|---|
| 16 November 2023 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Cystic Fibrosis Trials Register and registry searches identified 103 references for inclusion in the review (one of these referred to two separate studies, which are listed as 'Awaiting assessment'). Included studies We have added 15 included studies (45 references) at this update; nine of these are dual therapy studies (Davies 2021; McKone 2021; Munck 2020; NCT02070744; NCT02508207; NCT02730208; Schwarz 2021; Stahl 2021; Wilson 2021) and six are triple therapy studies (Barry 2021; NCT02951182; NCT02951195; NCT03447249; NCT03460990; Sutharsan 2022). Of these, we have added 11 new references to five dual therapy studies previously listed as ongoing and now included (Davies 2021; McKone 2021; NCT02730208; Schwarz 2021; Stahl 2021); one dual therapy study was previously listed as ongoing and is now included, though no new references have been added (NCT02070744). We added three new references to two triple therapy studies previously listed as awaiting assessment and now included (Barry 2021; NCT03460990), and we added two references to a triple therapy study previously listed as excluded but which has now been included (Sutharsan 2022). Additionally, one new reference is to a previously included monotherapy study (Clancy 2012), two new references are to a previously included dual therapy study (Ratjen 2017), and nine new references are to four triple therapy studies previously listed as included (Davies 2018a; Davies 2018b; Heijerman 2019; Middleton 2019). Excluded studies There are 11 newly excluded studies (29 references) (Berkers 2021; Chilvers 2021; Chmiel 2021; ISRCTN14081521; Krivec 2021; Marsh 2022; Meijer 2016; NCT02323100; NCT03756922; NCT04058210; NCT05535959). The NCT02323100 study was previously listed as ongoing and is now excluded, and no new references were added at this update. We added three new references to a study previously listed as ongoing and now excluded (Meijer 2016), and we added two new references to an already excluded study (Berkers 2014). One previously excluded study (previously listed with a single reference to the ClinicalTrials.gov entry NCT04105972) has now been included ‐ see above (Sutharsan 2022). Ongoing studies We identified four new ongoing studies (eight references) (NCT04853368; NCT05033080; NCT05076149; NCT05274269). Six studies previously listed as ongoing have now been included (with a total of 11 additional references identified) (Davies 2021; McKone 2021; NCT02070744; NCT02730208; Schwarz 2021; Stahl 2021), and two have now been excluded (with a total of four additional references identified) (Meijer 2016; NCT02323100). Studies awaiting assessment There are two newly identified studies (four references) listed as awaiting assessment (Downey 2020; NCT03768089). Two studies that were previously listed as ongoing have been completed and are now listed as awaiting assessment (no new references were identified) (ALBATROSS; FLAMINGO). We added three references to studies already listed as awaiting assessment: one to a dual therapy study (NCT03969888), one to a triple therapy study (NCT04353817) and one reference that presents results from two separate studies (Uluer 2023a; Uluer 2023b). |
| 16 November 2023 | New citation required and conclusions have changed | A new author (Matthew Heneghan) has joined the review team. The addition of 14 studies to the meta‐analysis has changed the conclusions of the review. The evidence to support triple therapy with elexacaftor‐tezacaftor‐ivacaftor has been strengthened by the new data in this update, but the evidence for the dual cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy of lumacaftor‐ivacaftor for people with CF who have two copies of the F508del variant has been weakened. |
History
Protocol first published: Issue 2, 2014 Review first published: Issue 8, 2018
| Date | Event | Description |
|---|---|---|
| 16 December 2020 | New citation required and conclusions have changed | The title of the review has been changed to more accurately reflect its scope (previously 'Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis'). The inclusion of a new comparison of triple combination therapy has been added and changed the conclusions of our review. One author (Dr Sanjay Patel) has stepped down and a new author (Dr Jared Murphy) has joined the team. |
| 16 December 2020 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Cystic Fibrosis Trials Register and registry searches identified 65 new references. Included studies There were two new references to one included monotherapy study previously listed as ongoing (Horsley 2017). We identified one new reference to one already included dual combination study (Donaldson 2018), eight new references to a further dual combination study (Ratjen 2017) and seven references to another already included dual combination study (Taylor‐Cousar 2017). There were two new references to two further dual combination studies (TRAFFIC 2015; TRANSPORT 2015). Five new references describe three newly included triple combination studies, which were previously listed as ongoing (trial registry entries) (Davies 2018a; Davies 2018b; Keating 2018) and five references to two further new triple combination studies (Heijerman 2019; Middleton 2019). Excluded studies Two references were to a newly excluded study (Drevinek 2017) and eight references were additional references to already excluded studies (Chilvers 2021; Rowe 2017). Studies awaiting assessment Three references are to one new study of monotherapy (Rio‐CF). There was a single new reference to a dual therapy study (Wainwright 2019) and three references to a new study of dual therapy versus triple therapy versus placebo (Downey 2019). There were eight references to new triple therapy studies (Munck 2020; PELICAN; Taylor‐Cousar 2019; Uluer 2023b). Ongoing studies One reference was added to an already ongoing study of monotherapy (Meijer 2016) and four references were to two new ongoing studies of monotherapy (ALBATROSS; FLAMINGO). There was one reference to a new study looking at both monotherapy and dual combination therapy (Jain 2018) and four references to ongoing studies of dual therapy (Stahl 2021; Schwarz 2021). A search of ongoing trials registers identified 25 references: one additional reference to an already included study; 15 references that were excluded; seven references to studies awaiting assessment; and two references to ongoing studies. |
| 28 August 2018 | Amended | Wording in the Background section of the Abstract revised with regard to the populations where CF occurs. |
Acknowledgements
The review authors would like to acknowledge the help and support of Cochrane CF and Genetic Disorders, in particular Nikki Jahnke and Tracey Remmington. Toby Lasserson and Newton Opiyo, from the Cochrane Editorial Unit, provided valuable and precise editorial oversight. We would also like to acknowledge the study investigators, Professors/Doctors Elborn, Boyle, Clancy, McCarty, Ratjen, Rubenstein, Taylor‐Cousar, Davies, Drevinek and Zeitlin, who engaged constructively with this review and thank them for providing additional requested information.
The current review team would also like to thank Dr Sanjay Patel for his contributions to the protocol and the original version of this review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to Cochrane Cystic Fibrosis and Genetic Disorders. The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Search methods ‐ electronic searches
| Database/resource | Date searched | Search strategy |
| Cochrane CF Group's Trials Register (CF Trials Register) |
28 November 2022 | Searched using group allocated terms: drugs that correct defects in CFTR transcription, translation or processing Relevant studies have been tagged by the group with these terms for indexing purposes in the Group's Cystic Fibrosis Trials Register. |
| US National Institutes of Health database (clinicaltrials.gov/) |
3 December 2022 | [Advanced Search Form] Condition or disease: cystic fibrosis Other terms: VX OR corrector Study type: Interventional Studies (Clinical Trials) |
| WHO ICTRP (trialsearch.who.int/) |
3 December 2022 | Cystic fibrosis AND (VX OR corrector) |
| European Medicines Agency (www.clinicaltrialsregister.eu/) |
3 December 2022 | Cystic fibrosis AND (VX OR corrector) |
Data and analyses
Comparison 1. Lumacaftor versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 At up to 1 month | 1 | 61 | Mean Difference (IV, Fixed, 95% CI) | ‐1.90 [‐4.13, 0.33] |
| 1.2 Adverse effects: 100 mg and 200 mg lumacaftor groups (combined data) versus placebo at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.2.1 Cough | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.28 [0.28, 5.92] |
| 1.2.2 Headache | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.13 [0.16, 8.04] |
| 1.2.3 Rales | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.20 [0.18, 57.82] |
| 1.2.4 Productive cough | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.79 [0.27, 11.98] |
| 1.2.5 Dyspnoea | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.20 [0.18, 57.82] |
| 1.2.6 Pulmonary exacerbation | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.50 [0.16, 14.31] |
| 1.2.7 Fatigue | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.21 [0.12, 12.09] |
| 1.2.8 Fever | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.50 [0.16, 14.31] |
| 1.2.9 Nasal congestion | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.07, 4.93] |
| 1.2.10 Wheezing | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.13 [0.01, 2.91] |
| 1.2.11 Diarrhoea | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.27 [0.02, 3.31] |
| 1.2.12 Oropharyngeal pain | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.27 [0.02, 3.31] |
| 1.2.13 Upper respiratory tract infection | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.45 [0.07, 31.52] |
| 1.2.14 Sinus congestion | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.01, 5.55] |
| 1.2.15 Respiration abnormal | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.85 [0.10, 243.04] |
| 1.2.16 Haemoptysis | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.44 [0.03, 6.54] |
| 1.2.17 Constipation | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.54 [0.04, 147.25] |
| 1.2.18 Abdominal pain | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.46 [0.01, 18.95] |
| 1.2.19 Myalgia | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.46 [0.01, 18.95] |
| 1.2.20 Post‐tussive vomiting | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.54 [0.04, 147.25] |
| 1.2.21 Nausea | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.48 [0.02, 106.10] |
| 1.2.22 Nasopharyngitis | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.66 [0.07, 193.30] |
| 1.2.23 Dizziness | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.66 [0.07, 193.30] |
| 1.2.24 Back pain | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.48 [0.02, 106.10] |
| 1.2.25 Upper abdominal pain | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.45 [0.07, 31.52] |
| 1.2.26 Sputum abnormal | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.48 [0.02, 106.10] |
| 1.2.27 Epistaxis | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.94 [0.04, 24.27] |
| 1.2.28 C‐reactive protein increased | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.54 [0.04, 147.25] |
| 1.2.29 Paranasal sinus hypersecretion | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.2.30 Lung hyperinflation | 1 | 53 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.54 [0.04, 147.25] |
| 1.3 Adverse effects: 200 mg lumacaftor group versus placebo at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.3.1 Cough | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.43 [0.19, 60.73] |
| 1.3.2 Pulmonary exacerbation | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.72 [0.05, 156.17] |
| 1.3.3 Oropharyngeal pain | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.72 [0.05, 156.17] |
| 1.3.4 Nasal congestion | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.3.5 Dizziness | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.3.6 Prothrombin time prolonged | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.59 [0.02, 113.01] |
| 1.3.7 Upper respiratory tract infection | 1 | 62 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.4 Adverse effects requiring study drug discontinuation at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.4.1 25 mg lumacaftor | 1 | 35 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.11, 78.81] |
| 1.4.2 50 mg lumacaftor | 1 | 35 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.11, 78.81] |
| 1.4.3 100 mg lumacaftor | 1 | 34 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.18 [0.12, 83.76] |
| 1.4.4 200 mg lumacaftor | 1 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.11, 74.42] |
| 1.5 Sweat chloride concentration (change from baseline at up to 1 month) [mmol/L] | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.5.1 100 mg lumacaftor | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐6.13 [‐12.25, ‐0.01] |
| 1.5.2 200 mg lumacaftor | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | ‐8.21 [‐14.30, ‐2.12] |
| 1.6 Sweat chloride concentration (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 At up to 1 month | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐2.75 [‐7.65, 2.15] |
Comparison 2. Cavosonstat (N91115) (200 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 CFQR respiratory domain: absolute change from baseline | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1.1 At up to 1 month | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 3.80 [‐11.30, 18.90] |
| 2.2 CFQR eating domain: absolute change from baseline | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 At up to 1 month | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [‐2.75, 7.55] |
| 2.3 Adverse events occurring in > 10% of participants at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 2.3.1 Cough | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.05 [0.13, 8.17] |
| 2.3.2 Pulmonary exacerbation | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.26 [0.00, 20.03] |
| 2.3.3 Chest discomfort | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 308.20] |
| 2.3.4 Fatigue | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.13, 71.47] |
| 2.4 Sweat chloride | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.4.1 At up to 1 month | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐3.30 [‐9.13, 2.53] |
Comparison 3. N6022 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 FEV1 % predicted (relative change from baseline at up to 1 month) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1.1 5 mg/day N6022 | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐2.00 [‐5.31, 1.31] |
| 3.1.2 10 mg/day N6022 | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐4.73, 1.33] |
| 3.1.3 20 mg/day N6022 | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐2.20 [‐5.28, 0.88] |
| 3.1.4 40 mg/day N6022 | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | ‐1.00 [‐3.06, 1.06] |
| 3.2 Treatment‐emergent adverse events (mild) at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.2.1 5 mg/day N6022 | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.10, 2.30] |
| 3.2.2 10 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.28, 7.64] |
| 3.2.3 20 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.12, 2.88] |
| 3.2.4 40 mg/day N6022 | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.34, 4.59] |
| 3.3 Treatment‐emergent adverse events (moderate) at up to 1 month | 1 | 123 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.41, 2.04] |
| 3.3.1 5 mg/day N6022 | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.18, 4.90] |
| 3.3.2 10 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.20, 5.87] |
| 3.3.3 20 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.53, 13.85] |
| 3.3.4 40 mg/day N6022 | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.04, 1.48] |
| 3.4 Treatment‐emergent adverse events (serious or severe) at up to 1 month | 1 | 123 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.35, 5.41] |
| 3.4.1 5 mg/day N6022 | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.50 [0.35, 57.11] |
| 3.4.2 10 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.02, 17.51] |
| 3.4.3 20 mg/day N6022 | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.02, 17.51] |
| 3.4.4 40 mg/day N6022 | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.06, 17.25] |
Comparison 4. FDL169 (400 mg three times daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Mean change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 5.09 [‐2.72, 12.90] |
| 4.2 FEV1 % predicted absolute change (% points) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.2.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 4.68 [0.12, 9.24] |
| 4.3 Adverse events at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 4.3.1 Total number of participants experiencing at least 1 adverse event | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 6.67 [0.21, 207.87] |
| 4.3.2 Serious adverse events | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 4.3.3 Headache | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 6.00 [0.18, 196.26] |
| 4.3.4 Nasopharyngitis | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 4.3.5 Fatigue | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 4.3.6 Blood CK increase | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 4.3.7 Cough | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 4.3.8 Flatulence | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3.9 Respiratory exacerbation | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 4.3.10 Rhinorrhoea | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 4.3.11 Increased sputum | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3.12 Abdominal pain | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 4.3.13 Dry mouth | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 4.3.14 Lethargy | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.17 [0.00, 11.99] |
| 4.3.15 Myalgia | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3.16 Nasal congestion | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3.17 Nausea | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3.18 Oropharyngeal pain | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 4.3.19 Productive cough | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 4.3.20 Upper respiratory tract infection | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 4.4 Sweat chloride change from baseline (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.4.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 2.47 [‐4.47, 9.41] |
Comparison 5. FDL169 (600 mg three times daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Mean change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | ‐4.33 [‐12.01, 3.35] |
| 5.2 FEV1 % predicted absolute change (% points) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.2.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 2.80 [‐1.82, 7.42] |
| 5.3 Adverse events at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 5.3.1 Total number of participants experiencing at least 1 adverse event | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.06 [0.00, 4.00] |
| 5.3.2 Serious adverse events | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.04, 254.93] |
| 5.3.3 Headache | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 2.95] |
| 5.3.4 Nasopharyngitis | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.83 [0.02, 43.83] |
| 5.3.5 Fatigue | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 20.89] |
| 5.3.6 Blood CK increase | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 5.3.7 Cough | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.00, 8.63] |
| 5.3.8 Flatulence | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 5.3.9 Infective respiratory exacerbation | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 5.3.10 Rhinorrhoea | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 5.3.11 Increased sputum | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 8.33 [0.12, 599.51] |
| 5.3.12 Abdominal pain | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.20 [0.02, 63.12] |
| 5.3.13 Dry mouth | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 5.3.14 Lethargy | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.17 [0.00, 11.99] |
| 5.3.15 Myalgia | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 8.33 [0.12, 599.51] |
| 5.3.16 Nasal congestion | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 8.33 [0.12, 599.51] |
| 5.3.17 Nausea | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 5.3.18 Oropharyngeal pain | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 5.3.19 Productive cough | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.05, 349.57] |
| 5.3.20 Upper respiratory tract infection | 1 | 13 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.00, 28.33] |
| 5.4 Sweat chloride change from baseline (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.4.1 At up to 1 month | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 8.07 [0.98, 15.16] |
Comparison 6. FDL169 (800 mg three times daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Mean change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1.1 At up to 1 month | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | 8.84 [1.40, 16.28] |
| 6.2 FEV1 % predicted absolute change (% points) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.2.1 At up to 1 month | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.3 Adverse events at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.1 Total number of participants experiencing at least 1 adverse event | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.2 Serious adverse events | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.3 Headache | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.4 Nasopharyngitis | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.5 Fatigue | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.6 Blood CK increase | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.7 Cough | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.8 Flatulence | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.9 Infective respiratory exacerbation | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.10 Rhinorrhoea | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.11 Increased sputum | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.12 Abdominal pain | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.13 Dry mouth | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.14 Lethargy | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.15 Myalgia | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.16 Nasal congestion | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.17 Nausea | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.18 Oropharyngeal pain | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.19 Productive cough | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.3.20 Upper respiratory tract infection | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Totals not selected | |
| 6.4 Sweat chloride change from baseline (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.4.1 At up to 1 month | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 7. CPX versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Adverse events occurring in more than 3% of participants in all treatment groups (combined data) versus placebo at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 7.1.1 Abdominal pain | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.45 [0.01, 24.92] |
| 7.1.2 Asthenia | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.65 [0.01, 39.69] |
| 7.1.3 Headache | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.01, 17.72] |
| 7.1.4 Pain | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.45 [0.01, 24.92] |
| 7.1.5 Diarrhoea | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.65 [0.01, 39.69] |
| 7.1.6 Dizziness | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 9.33 [0.32, 268.92] |
| 7.1.7 Lung disease | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.45 [0.01, 24.92] |
| 7.1.8 Rhinitis | 1 | 37 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.45 [0.01, 24.92] |
Comparison 8. 4PBA versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Adverse events at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 8.1.1 Bad taste in mouth | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.44 [0.01, 13.44] |
| 8.1.2 Diarrhoea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.35 [0.04, 267.31] |
| 8.2 Participants requiring study drug termination or a reduced dosage at up to 1 month | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.2.1 30 g 4PBA | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
8.2. Analysis.

Comparison 8: 4PBA versus placebo, Outcome 2: Participants requiring study drug termination or a reduced dosage at up to 1 month
Comparison 9. Lumacaftor plus ivacaftor versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Absolute change from baseline in lung clearance index 2.5 (LCI2.5) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1.1 At week 48 | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.69 [‐1.35, ‐0.03] |
| 9.2 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 9.2.1 Number of serious adverse events | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.75 [0.19, 16.29] |
| 9.2.2 Other (not including serious) adverse events | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.41 [0.01, 23.66] |
| 9.2.3 Pseudomonas test positive | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.01, 5.36] |
| 9.2.4 Abdominal pain | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.75 [0.19, 16.29] |
| 9.2.5 Abdominal pain upper | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.01, 5.36] |
| 9.2.6 Constipation | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.95 [0.12, 289.70] |
| 9.2.7 Diarrhoea | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.94 [0.10, 38.55] |
| 9.2.8 Faeces pale | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 4.79] |
| 9.2.9 Vomiting | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.42 [0.03, 6.34] |
| 9.2.10 Pyrexia | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.90 [0.12, 6.72] |
| 9.2.11 Bacterial disease carrier | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 4.79] |
| 9.2.12 Gastroenteritis | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.05, 7.93] |
| 9.2.13 Infective pulmonary exacerbation of cystic fibrosis | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.64 [0.13, 3.12] |
| 9.2.14 Nasopharyngitis | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.69 [0.35, 8.15] |
| 9.2.15 Rhinitis | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.11, 3.04] |
| 9.2.16 Upper respiratory tract infection | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.13 [0.01, 2.80] |
| 9.2.17 Lung infiltration | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 4.79] |
| 9.2.18 Headache | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.05, 7.93] |
| 9.2.19 Cough | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.88 [0.16, 4.77] |
| 9.2.20 Dyspnoea | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.01, 5.36] |
| 9.2.21 Epistaxis | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.42 [0.03, 6.34] |
| 9.2.22 Nasal congestion | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.04 [0.00, 2.00] |
| 9.2.23 Nasal polyps | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.01, 5.36] |
| 9.2.24 Productive cough | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 4.79] |
| 9.3 Absolute change from baseline in sweat chloride | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.3.1 At week 48 | 1 | 49 | Mean Difference (IV, Fixed, 95% CI) | ‐26.40 [‐34.57, ‐18.23] |
| 9.4 Absolute change from baseline in MRI global chest score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.4.1 At week 48 | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | ‐1.40 [‐5.24, 2.44] |
| 9.5 Absolute change from baseline in weight‐for‐age z score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.5.1 At week 48 | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.01, 0.41] |
| 9.6 Absolute change from baseline in BMI‐for‐age z score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.6.1 At week 48 | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.44 [0.09, 0.79] |
| 9.7 Absolute change from baseline in stature‐for‐age z score | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.19, 0.17] |
| 9.7.1 At week 48 | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.19, 0.17] |
Comparison 10. Lumacaftor (600 mg once daily) plus ivacaftor (250 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1.1 At 6 months | 2 | 715 | Mean Difference (IV, Fixed, 95% CI) | 0.00 [‐0.01, 0.02] |
| 10.2 Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.2.1 At up to 1 month | 2 | 739 | Mean Difference (IV, Fixed, 95% CI) | 3.32 [1.13, 5.51] |
| 10.2.2 At 6 months | 2 | 725 | Mean Difference (IV, Fixed, 95% CI) | 3.04 [0.76, 5.32] |
| 10.3 Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.3.1 At 6 months | 2 | 712 | Mean Difference (IV, Fixed, 95% CI) | 2.24 [0.18, 4.31] |
| 10.4 FEV1 % predicted (relative change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.4.1 At 6 months | 2 | 720 | Mean Difference (IV, Fixed, 95% CI) | 5.63 [3.80, 7.47] |
| 10.5 FEV1 % predicted (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.5.1 At up to 1 month | 2 | 739 | Mean Difference (IV, Fixed, 95% CI) | 2.32 [1.34, 3.31] |
| 10.5.2 At 6 months | 2 | 720 | Mean Difference (IV, Fixed, 95% CI) | 3.34 [2.30, 4.38] |
| 10.6 Adverse events by end of study (at 6 months) | 2 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 10.6.1 Any adverse event | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.00 [0.37, 2.71] |
| 10.6.2 Discontinuation due to an adverse event | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.38 [0.67, 8.50] |
| 10.6.3 At least 1 serious adverse event | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.73 [0.47, 1.13] |
| 10.6.4 Infective pulmonary exacerbation | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.45, 0.97] |
| 10.6.5 Cough | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.72 [0.49, 1.08] |
| 10.6.6 Headache | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.00 [0.59, 1.68] |
| 10.6.7 Haemoptysis | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.60, 1.81] |
| 10.6.8 Diarrhoea | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.18 [0.61, 2.28] |
| 10.6.9 Abnormal respiration | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.91 [0.94, 3.88] |
| 10.6.10 Increased sputum | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.74 [0.44, 1.24] |
| 10.6.11 Dyspnoea | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.05 [1.10, 3.83] |
| 10.6.12 Nasopharyngitis | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.55 [0.27, 1.10] |
| 10.6.13 Oropharyngeal pain | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.52 [0.80, 2.89] |
| 10.6.14 Abdominal pain | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.80 [0.39, 1.62] |
| 10.6.15 Fatigue | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.51, 2.08] |
| 10.6.16 Nausea | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.51, 2.11] |
| 10.6.17 Pyrexia | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.54, 1.98] |
| 10.6.18 Nasal congestion | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.73 [0.39, 1.35] |
| 10.6.19 Upper respiratory tract infection | 2 | 739 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.21 [0.54, 2.70] |
| 10.7 Time to first pulmonary exacerbation | 2 | 739 | Hazard Ratio (IV, Fixed, 95% CI) | 0.70 [0.57, 0.87] |
| 10.8 Rate of exacerbations | 2 | 739 | Rate Ratio (IV, Fixed, 95% CI) | 0.70 [0.57, 0.87] |
| 10.9 Weight (kg) (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.9.1 At 6 months | 2 | 725 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [0.42, 1.18] |
| 10.10 BMI (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.10.1 At up to 1 month | 2 | 739 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.07, 0.09] |
| 10.10.2 At 6 months | 2 | 725 | Mean Difference (IV, Fixed, 95% CI) | 0.29 [0.16, 0.43] |
Comparison 11. Lumacaftor (400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1.1 At 6 months | 2 | 708 | Mean Difference (IV, Fixed, 95% CI) | 0.00 [‐0.01, 0.02] |
| 11.2 Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.2.1 At up to 1 month | 2 | 740 | Mean Difference (IV, Fixed, 95% CI) | 4.13 [1.94, 6.31] |
| 11.2.2 At 6 months | 3 | 783 | Mean Difference (IV, Fixed, 95% CI) | 2.50 [0.30, 4.70] |
| 11.3 Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.3.1 At 6 months | 2 | 710 | Mean Difference (IV, Fixed, 95% CI) | 2.30 [0.25, 4.36] |
| 11.4 FEV1 % predicted (relative change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.4.1 At 6 months | 3 | 777 | Mean Difference (IV, Fixed, 95% CI) | 4.69 [2.91, 6.46] |
| 11.5 FEV1 % predicted (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.5.1 At up to 1 month | 2 | 740 | Mean Difference (IV, Fixed, 95% CI) | 2.42 [1.43, 3.40] |
| 11.5.2 At 6 months | 3 | 777 | Mean Difference (IV, Fixed, 95% CI) | 2.83 [1.81, 3.84] |
| 11.6 Adverse events by end of study (at 6 months) | 3 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 11.6.1 Any adverse event | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.28, 1.60] |
| 11.6.2 Discontinuation due to an adverse event | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.11 [0.96, 10.11] |
| 11.6.3 At least 1 serious adverse event | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.62 [0.40, 0.95] |
| 11.6.4 Infective pulmonary exacerbation | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.60 [0.42, 0.87] |
| 11.6.5 Cough | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.59 [0.40, 0.87] |
| 11.6.6 Headache | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.96 [0.58, 1.61] |
| 11.6.7 Haemoptysis | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.60, 1.76] |
| 11.6.8 Diarrhoea | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.40 [0.76, 2.59] |
| 11.6.9 Abnormal respiration | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.23 [0.64, 2.39] |
| 11.6.10 Increased sputum | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.71 [0.43, 1.16] |
| 11.6.11 Dyspnoea | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.61 [0.87, 2.98] |
| 11.6.12 Nasopharyngitis | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.22 [0.69, 2.15] |
| 11.6.13 Oropharyngeal pain | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.72 [0.36, 1.46] |
| 11.6.14 Abdominal pain | 2 | 738 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.53, 2.01] |
| 11.6.15 Fatigue | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.23 [0.63, 2.40] |
| 11.6.16 Nausea | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.56 [0.85, 2.89] |
| 11.6.17 Pyrexia | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.54, 1.97] |
| 11.6.18 Nasal congestion | 2 | 738 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.51 [0.26, 1.02] |
| 11.6.19 Upper respiratory tract infection | 3 | 808 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.80 [0.91, 3.59] |
| 11.7 Time to first pulmonary exacerbation | 2 | 740 | Hazard Ratio (IV, Fixed, 95% CI) | 0.61 [0.49, 0.76] |
| 11.8 Rate of exacerbations | 2 | 740 | Rate Ratio (IV, Fixed, 95% CI) | 0.61 [0.49, 0.76] |
| 11.9 Weight (kg) (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.9.1 At 6 months | 2 | 723 | Mean Difference (IV, Fixed, 95% CI) | 0.65 [0.27, 1.03] |
| 11.10 BMI (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.10.1 At up to 1 month | 2 | 740 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.06, 0.10] |
| 11.10.2 At 6 months | 3 | 786 | Mean Difference (IV, Fixed, 95% CI) | 0.25 [0.12, 0.38] |
| 11.11 BMI (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.11.1 At 6 months | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | 1.00 [‐1.14, 3.14] |
Comparison 12. Lumacaftor (600 mg once daily or 400 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Quality of life ‐ Euro Quality of Life Scale (EuroQol) 5‐Dimension‐3 Level (EQ‐5D‐3L) Index Score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1.1 At 6 months | 2 | 1061 | Mean Difference (IV, Fixed, 95% CI) | 0.00 [‐0.01, 0.01] |
| 12.2 Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.2.1 At up to 1 month | 2 | 1108 | Mean Difference (IV, Fixed, 95% CI) | 3.70 [1.81, 5.58] |
| 12.2.2 At 6 months | 3 | 1139 | Mean Difference (IV, Fixed, 95% CI) | 2.83 [0.91, 4.74] |
| 12.3 Quality of life ‐ EQ‐5D‐3L VAS score (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.3.1 At 6 months | 2 | 1060 | Mean Difference (IV, Fixed, 95% CI) | 2.28 [0.50, 4.06] |
| 12.4 FEV1 % predicted (relative change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.4.1 At 6 months | 3 | 1134 | Mean Difference (IV, Fixed, 95% CI) | 5.12 [3.57, 6.67] |
| 12.5 FEV1 % predicted (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.5.1 At up to 1 month | 2 | 1108 | Mean Difference (IV, Fixed, 95% CI) | 2.37 [1.52, 3.22] |
| 12.5.2 At 6 months | 3 | 1134 | Mean Difference (IV, Fixed, 95% CI) | 3.08 [2.20, 3.97] |
| 12.6 Adverse events by end of study (at 6 months) | 3 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 12.6.1 Any adverse event | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.76 [0.34, 1.71] |
| 12.6.2 Discontinuation due to an adverse event | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.81 [0.92, 8.57] |
| 12.6.3 At least 1 serious adverse event | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.69 [0.48, 1.00] |
| 12.6.4 Infective pulmonary exacerbation | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.64 [0.46, 0.88] |
| 12.6.5 Cough | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.65 [0.46, 0.91] |
| 12.6.6 Headache | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.97 [0.62, 1.51] |
| 12.6.7 Haemoptysis | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.65, 1.66] |
| 12.6.8 Diarrhoea | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.27 [0.73, 2.21] |
| 12.6.9 Abnormal respiration | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.44 [0.80, 2.59] |
| 12.6.10 Increased sputum | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.72 [0.47, 1.10] |
| 12.6.11 Dyspnoea | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.78 [1.02, 3.08] |
| 12.6.12 Nasopharyngitis | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.88 [0.52, 1.49] |
| 12.6.13 Oropharyngeal pain | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.06 [0.60, 1.87] |
| 12.6.14 Abdominal pain | 2 | 1108 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.91 [0.51, 1.65] |
| 12.6.15 Fatigue | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.14 [0.63, 2.07] |
| 12.6.16 Nausea | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.29 [0.73, 2.27] |
| 12.6.17 Pyrexia | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.05 [0.60, 1.84] |
| 12.6.18 Nasal congestion | 2 | 1108 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.62 [0.36, 1.07] |
| 12.6.19 Upper respiratory tract infection | 3 | 1178 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.51 [0.79, 2.87] |
| 12.7 Weight (kg) (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.7.1 At 6 months | 2 | 1081 | Mean Difference (IV, Fixed, 95% CI) | 0.72 [0.39, 1.05] |
| 12.8 BMI (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.8.1 At up to 1 month | 2 | 1108 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.05, 0.08] |
| 12.8.2 At 6 months | 3 | 1144 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [0.16, 0.38] |
Comparison 13. Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (150 mg twice daily) for days 15 to 21 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 13.1.1 At 14 days (before addition of ivacaftor) | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐4.39, 0.79] |
| 13.1.2 At 21 days | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | 2.80 [‐1.39, 6.99] |
| 13.2 Adverse events occurring in 10% or more participants (from days 15 to 21) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 13.2.1 Cough | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.06 [0.14, 8.09] |
| 13.2.2 Pulmonary exacerbation | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.22 [0.08, 58.11] |
| 13.2.3 Oropharyngeal pain | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.02, 13.07] |
| 13.2.4 Nasal congestion | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.02, 13.07] |
| 13.2.5 Dizziness | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 13.2.6 Prothrombin time prolonged | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 13.2.7 Upper respiratory tract infection | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 13.3 Sweat chloride concentration (mmol/L) (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 13.3.1 At 14 days (before addition of ivacaftor) | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐8.58, 2.38] |
| 13.3.2 At 21 days | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐5.00 [‐11.61, 1.61] |
Comparison 14. Lumacaftor (200 mg once daily) for 21 days plus ivacaftor (250 mg twice daily) for days 15 to 21 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1.1 At 14 days (before addition of ivacaftor) | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | ‐2.00 [‐4.66, 0.66] |
| 14.1.2 At 21 days | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐4.20, 4.60] |
| 14.2 Adverse events occurring in 10% or more participants (from days 15 to 21) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 14.2.1 Cough | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.01, 4.52] |
| 14.2.2 Pulmonary exacerbation | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.05 [0.03, 44.10] |
| 14.2.3 Oropharyngeal pain | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.06 [0.07, 15.89] |
| 14.2.4 Nasal congestion | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.68 [0.14, 20.50] |
| 14.2.5 Dizziness | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.31 [0.05, 239.61] |
| 14.2.6 Prothrombin time prolonged | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 14.2.7 Upper respiratory tract infection | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 14.3 Sweat chloride concentration (mmol/L) (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.3.1 At 14 days (before addition of ivacaftor) | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐2.40 [‐8.00, 3.20] |
| 14.3.2 At 21 days | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐10.90 [‐17.60, ‐4.20] |
Comparison 15. Lumacaftor (200 mg twice daily) plus ivacaftor (250 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.1.1 At 6 months | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | 2.50 [‐0.10, 5.10] |
| 15.2 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.2.1 At 6 months | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [0.40, 4.40] |
| 15.3 LCI2.5 (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.3.1 At 6 months | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | ‐1.10 [‐1.40, ‐0.80] |
| 15.4 Treatment‐emergent adverse events with incidence > 10% in any treatment group (at 6 months) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.4.1 Any adverse event | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.14, 2.58] |
| 15.4.2 Any serious adverse event | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.50, 2.78] |
| 15.4.3 Pulmonary exacerbation | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.55, 2.25] |
| 15.4.4 Cough | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.53, 1.61] |
| 15.4.5 Productive cough | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.35 [1.27, 8.84] |
| 15.4.6 Nasal congestion | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.30 [0.94, 5.60] |
| 15.4.7 Oropharyngeal pain | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.66, 3.64] |
| 15.4.8 Pyrexia | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.33, 1.44] |
| 15.4.9 Upper abdominal pain | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.74, 5.08] |
| 15.4.10 Headache | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.60, 3.63] |
| 15.4.11 Upper respiratory tract infection | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.55, 3.15] |
| 15.4.12 Sputum increased | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.92 [1.28, 27.42] |
| 15.4.13 Abdominal pain | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.39, 2.46] |
| 15.4.14 Nausea | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.43, 2.83] |
| 15.4.15 Rhinorrhoea | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.06 [0.68, 6.27] |
| 15.4.16 Vomiting | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.39, 2.46] |
| 15.4.17 Fatigue | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.31, 1.98] |
| 15.4.18 Respiratory events | 1 | 204 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.71, 3.29] |
| 15.5 Sweat chloride concentration (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.5.1 At up to 1 month | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | ‐20.80 [‐23.40, ‐18.20] |
| 15.6 CT Brody score (mean change) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.6.1 At 6 months | 1 | 10 | Mean Difference (IV, Fixed, 95% CI) | ‐16.70 [‐36.05, 2.65] |
| 15.7 CT Brody score bronchiectasis score (mean change) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.7.1 At 6 months | 1 | 10 | Mean Difference (IV, Fixed, 95% CI) | ‐2.40 [‐4.96, 0.16] |
| 15.8 CT Brody score air trapping score (mean change) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.8.1 At 6 months | 1 | 10 | Mean Difference (IV, Fixed, 95% CI) | ‐6.60 [‐20.77, 7.57] |
| 15.9 BMI (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.9.1 At 6 months | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.10, 0.30] |
| 15.10 BMI‐for‐age z score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.10.1 At 6 months | 1 | 204 | Mean Difference (IV, Fixed, 95% CI) | 0.00 [‐0.10, 0.10] |
Comparison 16. Lumacaftor (200 mg once daily monotherapy for 14 days) plus ivacaftor (150 mg or 250 mg twice daily for days 15 to 21) for 21 days.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 16.1.1 At 21 days | 1 | 59 | Mean Difference (IV, Fixed, 95% CI) | 1.57 [‐2.13, 5.27] |
| 16.2 Adverse events occurring in 10% or more participants (from days 15 to 21) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 16.2.1 Cough | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.61 [0.09, 4.01] |
| 16.2.2 Pulmonary exacerbation | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.08, 34.55] |
| 16.2.3 Oropharyngeal pain | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.77 [0.07, 9.03] |
| 16.2.4 Nasal congestion | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.06 [0.10, 11.04] |
| 16.2.5 Dizziness | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.01 [0.08, 209.72] |
| 16.2.6 Prothrombin time prolonged | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.79 [0.05, 160.31] |
| 16.2.7 Upper respiratory tract infection | 1 | 61 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.79 [0.05, 160.31] |
| 16.3 Sweat chloride concentration (mmol/L) (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 16.3.1 At 21 days | 1 | 50 | Mean Difference (IV, Fixed, 95% CI) | ‐7.95 [‐13.81, ‐2.09] |
Comparison 17. Tezacaftor (50 mg twice daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Absolute change from baseline in CFQ‐R respiratory domain score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.1.1 At 3 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | 10.20 [‐0.06, 20.46] |
| 17.2 Relative change from baseline in % predicted FEV1 | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.2.1 At 3 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | 1.60 [‐7.63, 10.83] |
| 17.3 Absolute change from baseline in % predicted FEV1 | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.3.1 At 3 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | 1.00 [‐3.35, 5.35] |
| 17.4 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 17.4.1 Number of participants with treatment‐emergent adverse events | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 17.4.2 Number of participants with serious adverse events | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.49 [0.08, 3.02] |
| 17.4.3 Infective pulmonary exacerbation of cystic fibrosis | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.41 [0.08, 2.04] |
| 17.4.4 Chronic sinusitis | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.5 Pneumothorax | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.15 [0.00, 9.08] |
| 17.4.6 Tinnitus | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.7 Diarrhoea | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.23, 39.60] |
| 17.4.8 Nausea | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 305.28] |
| 17.4.9 Dyspepsia | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.80 [0.01, 45.09] |
| 17.4.10 Fatigue | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.80 [0.01, 45.09] |
| 17.4.11 Non‐cardiac chest pain | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.12 Nasopharyngitis | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 305.28] |
| 17.4.13 Acute sinusitis | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.14 Muscle strain | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.15 Pulmonary function test decreased | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.33 [0.11, 16.54] |
| 17.4.16 Weight decreased | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.85 [0.07, 51.01] |
| 17.4.17 Alanine aminotransferase increased | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.18 Aspartate aminotransferase increased | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.19 Heart rate increased | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.20 Breath sounds abnormal | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.21 Forced expiratory volume decreased | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.18 [0.01, 3.67] |
| 17.4.22 Vitamin D decreased | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.15 [0.00, 9.08] |
| 17.4.23 Oxygen saturation decreased | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.24 Iron deficiency | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.25 Back pain | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.55 [0.05, 6.48] |
| 17.4.26 Muscle spasms | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.27 Musculoskeletal pain | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.28 Headache | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.21 [0.15, 31.44] |
| 17.4.29 Hyposmia | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.30 Sinus headache | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.31 Testicular pain | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.32 Amenorrhoea | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.33 Cough | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.14, 4.40] |
| 17.4.34 Dyspnoea | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.82 [0.08, 8.20] |
| 17.4.35 Haemoptysis | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.83 [0.08, 8.96] |
| 17.4.36 Respiration abnormal | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.19 [0.15, 31.61] |
| 17.4.37 Nasal congestion | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.06, 5.59] |
| 17.4.38 Lower respiratory tract congestion | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.39 Sputum increased | 1 | 39 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.02, 3.06] |
| 17.4.40 Sputum discoloured | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.41 Epistaxis | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.42 Nasal oedema | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.43 Paranasal sinus hypersecretion | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.44 Productive cough | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.45 Sinus congestion | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.4.46 Rales | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.00, 20.63] |
| 17.4.47 Rash | 1 | 11 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.03, 265.75] |
| 17.5 Absolute change from baseline in sweat chloride | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.5.1 At 3 months | 1 | 10 | Mean Difference (IV, Fixed, 95% CI) | ‐13.50 [‐19.47, ‐7.53] |
| 17.6 Absolute change from baseline in body weight (kg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.6.1 At 3 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐2.05, 0.85] |
| 17.7 Absolute change from baseline BMI | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐0.66, 0.34] |
| 17.7.1 At 3 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐0.66, 0.34] |
Comparison 18. Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 CFQ‐R respiratory domain (absolute change from baseline) | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.1.1 At 1 month | 1 | 504 | Mean Difference (IV, Fixed, 95% CI) | 5.10 [2.99, 7.21] |
| 18.1.2 Up to and including 6 months | 5 | 946 | Mean Difference (IV, Fixed, 95% CI) | 2.88 [2.48, 3.29] |
| 18.2 CFQ‐R physical functioning domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.2.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 3.80 [1.90, 5.70] | |
| 18.3 CFQ‐R treatment burden domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.3.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 3.40 [1.60, 5.20] | |
| 18.4 CFQ‐R health perceptions domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.4.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 3.20 [1.20, 5.20] | |
| 18.5 CFQ‐R vitality domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.5.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 2.30 [0.10, 4.50] | |
| 18.6 CFQ‐R social functioning domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.6.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 1.50 [0.00, 3.00] | |
| 18.7 CFQ‐R role functioning domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.7.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 1.50 [‐0.30, 3.30] | |
| 18.8 CFQ‐R eating problems domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.8.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐0.60, 2.80] | |
| 18.9 CFQ‐R emotional functioning (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.9.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.00, 2.20] | |
| 18.10 CFQ‐R weight domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.10.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.50 [‐2.90, 3.90] | |
| 18.11 CFQ‐R digestive symptoms domain(absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.11.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐1.90, 1.70] | |
| 18.12 CFQ‐R body image domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.12.1 At 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | ‐0.50 [‐2.30, 1.30] | |
| 18.13 FEV1 % predicted (relative change from baseline) | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.13.1 At 1 month | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 3.72 [‐7.77, 15.21] |
| 18.13.2 Up to and including 6 months | 5 | 944 | Mean Difference (IV, Fixed, 95% CI) | 0.92 [0.72, 1.11] |
| 18.14 FEV1 % predicted (absolute change from baseline) | 7 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.14.1 At 1 month | 3 | 556 | Mean Difference (IV, Fixed, 95% CI) | 3.54 [2.39, 4.69] |
| 18.14.2 Up to and including 6 months | 5 | 944 | Mean Difference (IV, Fixed, 95% CI) | 0.39 [0.27, 0.52] |
| 18.15 Absolute change from baseline in mucociliary clearance | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐9.87, 9.07] |
| 18.15.1 At week 4 | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐9.87, 9.07] |
| 18.16 Most common adverse events (occurring in at least 10% of participants in either group) | 7 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 18.16.1 Number of participants with treatment‐emergent adverse events | 6 | 1000 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.79 [0.49, 1.26] |
| 18.16.2 Number of participants with serious adverse events | 6 | 1000 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.61 [0.38, 0.98] |
| 18.16.3 Cough | 7 | 1018 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.77 [0.53, 1.12] |
| 18.16.4 Pulmonary exacerbation | 7 | 1018 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.70 [0.48, 1.01] |
| 18.16.5 Headache | 4 | 665 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.09 [0.63, 1.91] |
| 18.16.6 Nasal congestion or nasopharyngitis | 5 | 833 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.15 [0.69, 1.94] |
| 18.16.7 Increased sputum | 4 | 815 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.77 [0.45, 1.33] |
| 18.16.8 Haemoptysis | 5 | 770 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.52, 1.66] |
| 18.16.9 Pyrexia | 3 | 568 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.48, 1.83] |
| 18.16.10 Oropharyngeal pain | 2 | 550 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.37, 1.63] |
| 18.16.11 Fatigue | 3 | 695 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.75 [0.38, 1.49] |
| 18.16.12 Nausea | 4 | 602 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.14 [0.53, 2.43] |
| 18.16.13 Abdominal pain upper | 1 | 97 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.73 [0.12, 4.48] |
| 18.16.14 Dyspnoea | 1 | 97 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.17, 5.21] |
| 18.16.15 Lung infection pseudomonal | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 18.16.16 Upper respiratory tract infection | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.38 [0.22, 26.07] |
| 18.16.17 Lower respiratory tract infection | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.68 [0.14, 20.50] |
| 18.16.18 Gastroenteritis | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.22 [0.08, 58.11] |
| 18.16.19 Influenza | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.22 [0.08, 58.11] |
| 18.16.20 Pharyngitis | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 18.16.21 Sunburn | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.01, 6.96] |
| 18.16.22 Bacterial test positive | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.06 [0.00, 2.81] |
| 18.16.23 Migraine | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 18.16.24 Arthralgia | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.13] |
| 18.16.25 Renal impairment | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.13] |
| 18.17 Time to first pulmonary exacerbation | 1 | Hazard Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 18.17.1 At 6 months | 1 | 506 | Hazard Ratio (IV, Fixed, 95% CI) | 0.64 [0.46, 0.89] |
| 18.18 Number of participants with at least 1 pulmonary exacerbation | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 18.18.1 At 3 months | 1 | 168 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.90 [0.36, 2.30] |
| 18.19 Number of pulmonary exacerbations | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 18.19.1 At 3 months | 1 | 168 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.97 [0.40, 2.39] |
| 18.20 Sweat chloride (change from baseline) | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.20.1 At 1 month | 3 | 552 | Mean Difference (IV, Fixed, 95% CI) | ‐9.14 [‐10.93, ‐7.34] |
| 18.20.2 Up to and including 6 months | 4 | 837 | Mean Difference (IV, Fixed, 95% CI) | ‐6.38 [‐6.89, ‐5.86] |
| 18.21 Absolute change in total Brody/CF‐CT score from baseline | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.21.1 At 18 months | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | ‐1.48 [‐7.25, 4.29] |
| 18.22 Lung infection (pseudomonal) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 18.22.1 Up to 19 months | 1 | 41 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.81 [0.10, 341.36] |
| 18.23 Absolute change from baseline in body weight | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.23.1 At 3 months | 2 | 195 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.55, 0.46] |
| 18.24 BMI (change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.24.1 At 1 month | 1 | 504 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.13, 0.07] |
| 18.24.2 Over 1 month and up to and including 6 months | 3 | 699 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.09, 0.13] |
| 18.25 Absolute change from baseline in BMI z score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.25.1 At 3 months | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 [‐0.20, 0.10] |
18.15. Analysis.

Comparison 18: Tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either placebo or ivacaftor (150 mg twice daily) alone, Outcome 15: Absolute change from baseline in mucociliary clearance
Comparison 19. VX‐659 (80 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.1.1 At 1 month | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | 10.00 [0.29, 19.71] |
| 19.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.2.1 At 1 month | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | 18.36 [3.63, 33.09] |
| 19.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.3.1 At 1 month | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | 0.37 [0.15, 0.59] |
| 19.4 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 19.4.1 Number of participants experiencing at least one adverse event | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.11 [0.02, 51.19] |
| 19.4.2 Number experiencing moderate adverse events | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.3 Number experiencing severe adverse events | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.01, 5.92] |
| 19.4.4 Infective pulmonary exacerbations | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.50 [0.10, 21.90] |
| 19.4.5 Cough | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.38 [0.13, 85.06] |
| 19.4.6 Headache | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.04, 233.35] |
| 19.4.7 Oropharyngeal pain | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.8 Increased sputum | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.53 [0.09, 351.89] |
| 19.4.9 Raised blood creatine phosphokinase | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.04, 233.35] |
| 19.4.10 Nasopharyngitis | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.00 [0.07, 58.76] |
| 19.4.11 Nasal congestion | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.12 Nausea | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.13 Pyrexia | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.90 [0.02, 41.46] |
| 19.4.14 Abnormal respiration | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.04, 233.35] |
| 19.4.15 Constipation | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.16 Diarrhoea | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.04, 233.35] |
| 19.4.17 Fatigue | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.53 [0.09, 351.89] |
| 19.4.18 Haemoptysis | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.53 [0.09, 351.89] |
| 19.4.19 Productive cough | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.53 [0.09, 351.89] |
| 19.4.20 URTI | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.28 [0.00, 21.45] |
| 19.4.21 Rash | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.22 Sinus congestion | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.23 Influenza | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.24 Pain | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.4.25 Rales | 1 | 21 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 19.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.5.1 At 1 month | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | ‐48.60 [‐60.94, ‐36.26] |
Comparison 20. VX‐659 (120 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 20.1.1 At up to 1 month | 1 | 12 | Mean Difference (IV, Fixed, 95% CI) | 10.00 [3.04, 16.96] |
| 20.2 Adverse events (at up to 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 20.2.1 Number of participants experiencing at least one adverse event | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 31.67 [0.32, 3111.29] |
| 20.2.2 Number experiencing moderate adverse events | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 20.2.3 Number experiencing severe adverse events | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.33 [0.03, 176.29] |
| 20.2.4 Infective respiratory exacerbations | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.33 [0.03, 176.29] |
| 20.2.5 Cough | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.33 [0.03, 176.29] |
| 20.2.6 Productive cough | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.33 [0.03, 176.29] |
| 20.2.7 Oral candidiasis | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.25 [0.00, 16.23] |
| 20.2.8 Abdominal discomfort | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.9 Raised blood creatine phosphokinase | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.10 Diarrhoea | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.11 Ear pain | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.12 Fatigue | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.13 Flatulence | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.14 Hypertension | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.15 Insomnia | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.16 Nausea | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.17 Photosensitivity reaction | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.18 Decreased pulmonary function tests | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.19 Increased sputum | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.20 Tinnitus | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.2.21 Viral URTI | 1 | 12 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.01, 112.68] |
| 20.3 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 20.3.1 At up to 1 month | 1 | 12 | Mean Difference (IV, Fixed, 95% CI) | ‐30.60 [‐46.38, ‐14.82] |
Comparison 21. VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus triple placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Quality of life: change in CFQ‐R respiratory domain | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.1.1 At 1 month | 2 | 412 | Mean Difference (IV, Fixed, 95% CI) | 16.13 [13.02, 19.24] |
| 21.1.2 At 6 months | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | 20.10 [17.19, 23.01] |
| 21.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.2.1 At 1 month | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | 20.17 [8.73, 31.61] |
| 21.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.3.1 At 1 month | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | 14.00 [12.34, 15.66] |
| 21.3.2 At 6 months | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | 14.20 [12.54, 15.86] |
| 21.4 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.4.1 At 1 month | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | 0.42 [0.20, 0.64] |
| 21.5 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 21.5.1 Number of participants experiencing at least one adverse event | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.02, 6.85] |
| 21.5.2 Number experiencing moderate adverse events | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.3 Number experiencing severe adverse events | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.06, 5.75] |
| 21.5.4 Infective pulmonary exacerbation | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.71 [0.05, 9.48] |
| 21.5.5 Cough | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.86 [0.19, 76.85] |
| 21.5.6 Headache | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.73 [0.11, 304.12] |
| 21.5.7 Oropharyngeal pain | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.20 [0.08, 234.41] |
| 21.5.8 Increased sputum | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.9 Raised blood creatine phosphokinase | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.10 Nasopharyngitis | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.47 [0.01, 20.94] |
| 21.5.11 Nasal congestion | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.12 Nausea | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.13 Pyrexia | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.47 [0.01, 20.94] |
| 21.5.14 Abnormal respiration | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.15 Constipation | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.16 Diarrhoea | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.17 Fatigue | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.84 [0.05, 173.42] |
| 21.5.18 Haemoptysis | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.19 Productive cough | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.62 [0.02, 121.50] |
| 21.5.20 URTI | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.15 [0.00, 11.69] |
| 21.5.21 Rash | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.22 Sinus congestion | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.23 Influenza | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.24 Pain | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.5.25 Rales | 1 | 30 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 21.6 Most common adverse events (occurring in at least 10% of participants in either group) (6 months) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 21.6.1 Number of participants with treatment‐emergent adverse events | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.69 [0.27, 1.77] |
| 21.6.2 Number of participants with serious adverse events | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.14 [0.06, 0.33] |
| 21.6.3 Infective pulmonary exacerbation of cystic fibrosis | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.07 [0.04, 0.15] |
| 21.6.4 Nausea | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.28 [0.10, 0.84] |
| 21.6.5 Fatigue | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.44 [0.15, 1.29] |
| 21.6.6 Upper respiratory tract infection | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.76 [1.91, 17.35] |
| 21.6.7 Nasopharyngitis | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.68 [0.71, 4.00] |
| 21.6.8 Headache | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.79 [0.38, 1.67] |
| 21.6.9 Cough | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.17, 0.57] |
| 21.6.10 Oropharyngeal pain | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.57 [0.60, 4.08] |
| 21.6.11 Sputum increased | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.67 [0.33, 1.36] |
| 21.6.12 Haemoptysis | 1 | 382 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.07, 0.82] |
| 21.7 Number of pulmonary exacerbations | 1 | Odds Ratio (IV, Fixed, 99% CI) | Subtotals only | |
| 21.7.1 At 6 months | 1 | 382 | Odds Ratio (IV, Fixed, 99% CI) | 0.06 [0.03, 0.13] |
| 21.8 Sweat chloride (change from baseline) (mmol/L) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.8.1 At 1 month | 2 | 412 | Mean Difference (IV, Fixed, 95% CI) | ‐43.50 [‐46.19, ‐40.81] |
| 21.8.2 At 6 months | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | ‐44.50 [‐47.14, ‐41.86] |
| 21.9 Body weight (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.9.1 At 6 months | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | 3.20 [2.65, 3.75] |
| 21.10 BMI (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.10.1 At 6 months | 1 | 382 | Mean Difference (IV, Fixed, 95% CI) | 1.11 [0.92, 1.30] |
| 21.11 BMI z score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.11.1 At 6 months | 1 | 117 | Mean Difference (IV, Fixed, 95% CI) | 0.39 [0.25, 0.53] |
Comparison 22. VX‐659 (240 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 22.1 Quality of life: absolute change in CFQ‐R respiratory domain | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | 13.50 [8.79, 18.21] |
| 22.1.1 At 1 month | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | 13.50 [8.79, 18.21] |
| 22.2 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 22.2.1 At 1 month | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | 9.90 [7.41, 12.39] |
| 22.3 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 22.3.1 Number of participants with treatment‐emergent adverse events | 1 | 111 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.32 [0.49, 3.56] |
| 22.3.2 Number of participants with serious adverse events | 1 | 111 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.48 [0.10, 305.22] |
| 22.3.3 Infective pulmonary exacerbations | 1 | 111 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.03, 1.64] |
| 22.3.4 Sputum increased | 1 | 111 | Odds Ratio (M‐H, Fixed, 99% CI) | 24.01 [0.55, 1043.88] |
| 22.4 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 22.4.1 At 1 month | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | ‐48.70 [‐53.83, ‐43.57] |
Comparison 23. VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 23.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 23.1.1 At 1 month | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 7.90 [‐0.58, 16.38] |
| 23.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 23.2.1 At 1 month | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 23.85 [14.52, 33.18] |
| 23.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 23.3.1 At 1 month | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 0.52 [0.34, 0.70] |
| 23.4 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 23.4.1 Number of participants experiencing at least one adverse event | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.02, 7.70] |
| 23.4.2 Number experiencing moderate adverse events | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.3 Number experiencing severe adverse events | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 2.67] |
| 23.4.4 Infective pulmonary exacerbation | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.89 [0.07, 10.67] |
| 23.4.5 Cough | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.00 [0.09, 42.91] |
| 23.4.6 Headache | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.11 [0.10, 269.76] |
| 23.4.7 Oropharyngeal pain | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.11 [0.10, 269.76] |
| 23.4.8 Increased sputum | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.77 [0.07, 209.36] |
| 23.4.9 Raised blood creatine phosphokinase | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.77 [0.07, 209.36] |
| 23.4.10 Nasopharyngitis | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.06, 33.22] |
| 23.4.11 Nasal congestion | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.12 Nausea | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.77 [0.07, 209.36] |
| 23.4.13 Pyrexia | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.43 [0.01, 18.86] |
| 23.4.14 Abnormal respiration | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.77 [0.07, 209.36] |
| 23.4.15 Constipation | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.77 [0.07, 209.36] |
| 23.4.16 Diarrhoea | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.47 [0.02, 109.79] |
| 23.4.17 Fatigue | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.18 Haemoptysis | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.19 Productive cough | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.20 URTI | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.90 [0.03, 24.89] |
| 23.4.21 Rash | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.22 Sinus congestion | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.23 Influenza | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.24 Pain | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.4.25 Rales | 1 | 32 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 23.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 23.5.1 At 1 month | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐54.30 [‐65.28, ‐43.32] |
Comparison 24. VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 24.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 24.1.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 18.10 [10.85, 25.35] |
| 24.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 24.2.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 15.99 [8.61, 23.37] |
| 24.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 24.3.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.35 [0.19, 0.51] |
| 24.4 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 24.4.1 Number of participants experiencing at least one adverse event | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.11 [0.08, 14.81] |
| 24.4.2 Number experiencing moderate adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.3 Number experiencing severe adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.26 [0.01, 7.39] |
| 24.4.4 Infective pulmonary exacerbation | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.03 [0.11, 9.34] |
| 24.4.5 Cough | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.29 [0.11, 15.47] |
| 24.4.6 Headache | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.19 [0.09, 289.65] |
| 24.4.7 Oropharyngeal pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 24.4.8 Increased sputum | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.00 [0.09, 46.89] |
| 24.4.9 Raised blood creatine phosphokinase | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.26 [0.01, 7.39] |
| 24.4.10 Nasopharyngitis | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.11 Nasal congestion | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 7.14 [0.13, 379.05] |
| 24.4.12 Nausea | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.56 [0.03, 9.16] |
| 24.4.13 Pyrexia | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.05, 34.62] |
| 24.4.14 Abnormal respiration | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.15 Constipation | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.16 Diarrhoea | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 24.4.17 Fatigue | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.19 [0.00, 14.26] |
| 24.4.18 Haemoptysis | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.97 [0.03, 148.00] |
| 24.4.19 Productive cough | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.97 [0.03, 148.00] |
| 24.4.20 URTI | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 24.4.21 Rash | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.22 Sinus congestion | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.23 Influenza | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.24 Pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.4.25 Rales | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 24.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 24.5.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐45.20 [‐52.18, ‐38.22] |
Comparison 25. VX‐659 (400 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 25.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 25.1.1 At 1 month | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 20.30 [7.05, 33.55] |
| 25.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 25.2.1 At 1 month | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 33.05 [22.05, 44.05] |
| 25.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 25.3.1 At 1 month | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.68 [0.45, 0.91] |
| 25.4 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 25.4.1 Number of participants experiencing at least one adverse event | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.95 [0.01, 74.78] |
| 25.4.2 Number experiencing moderate adverse events | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.3 Number experiencing severe adverse events | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.01, 2.04] |
| 25.4.4 Cough | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.04, 7.63] |
| 25.4.5 Infective respiratory exacerbation | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.01, 2.04] |
| 25.4.6 Headache | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.7 Oropharyngeal pain | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.8 Increased sputum | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.9 Raised blood creatine phosphokinase | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.10 Nasopharyngitis | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.11 Nasal congestion | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.12 Nausea | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.13 Pyrexia | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.76 [0.05, 161.94] |
| 25.4.14 Abnormal respiration | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.15 Constipation | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.16 Diarrhoea | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.17 Fatigue | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.18 Haemoptysis | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.19 Productive cough | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.20 URTI | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 25.4.21 Rash | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.22 Sinus congestion | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.23 Influenza | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.24 Pain | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.03, 119.25] |
| 25.4.25 Rales | 1 | 25 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.05 [0.00, 3.11] |
| 25.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 25.5.1 At 1 month | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐36.80 [‐48.74, ‐24.86] |
Comparison 26. Elexacaftor (50 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 26.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 26.1.1 At 1 month | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 17.20 [4.44, 29.96] |
| 26.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 26.2.1 At 1 month | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 19.00 [7.08, 30.92] |
| 26.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 26.3.1 At 1 month | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.46 [0.19, 0.73] |
| 26.4 Adverse events (at 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 26.4.1 Number of participants experiencing at least one adverse event | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.4.2 Number experiencing moderate adverse events | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.4.3 Number experiencing severe adverse events | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.56 [0.02, 16.15] |
| 26.4.4 Infective pulmonary exacerbation | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.86 [0.08, 9.23] |
| 26.4.5 Cough | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 7.33 [0.31, 173.32] |
| 26.4.6 Increased sputum | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.29 [0.11, 15.22] |
| 26.4.7 Haemoptysis | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.20 [0.00, 12.63] |
| 26.4.8 Pyrexia | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.37 [0.00, 28.22] |
| 26.4.9 Nausea | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.07, 21.63] |
| 26.4.10 Oropharyngeal pain | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.07, 21.63] |
| 26.4.11 Headache | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.75 [0.09, 80.30] |
| 26.4.12 Nasal congestion | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.07, 21.63] |
| 26.4.13 Nasopharyngitis | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.95 [0.05, 305.83] |
| 26.4.14 Increased blood creatine phosphokinase | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.4.15 Fatigue | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.01, 5.13] |
| 26.4.16 Elevated AST | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.4.17 Diarrhoea | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.22 [0.03, 55.87] |
| 26.4.18 Abnormal respiration | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.4.19 Rhinorrhoea | 1 | 22 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 26.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 26.5.1 At 1 month | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐36.00 [‐47.23, ‐24.77] |
Comparison 27. Elexacaftor (100 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 27.1 Quality of life: change in CFQ‐R respiratory sub‐domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 27.1.1 At 1 month | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 14.50 [3.72, 25.28] |
| 27.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 27.2.1 At 1 month | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 13.50 [3.28, 23.72] |
| 27.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 27.3.1 At 1 month | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.38 [0.20, 0.56] |
| 27.4 Adverse events at (1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 27.4.1 Number of participants experiencing at least one adverse event | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.57 [0.01, 42.46] |
| 27.4.2 Number experiencing moderate adverse events | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 27.4.3 Number experiencing severe adverse events | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.03, 7.92] |
| 27.4.4 Infective pulmonary exacerbation | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.59 [0.08, 4.57] |
| 27.4.5 Cough | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.24 [0.16, 64.50] |
| 27.4.6 Increased sputum | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.67 [0.07, 6.20] |
| 27.4.7 Haemoptysis | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.47 [0.14, 16.00] |
| 27.4.8 Pyrexia | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.24 [0.16, 64.50] |
| 27.4.9 Nausea | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.79 [0.06, 10.19] |
| 27.4.10 Oropharyngeal pain | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.03, 7.92] |
| 27.4.11 Headache | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.03, 7.92] |
| 27.4.12 Nasal congestion | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.03, 7.92] |
| 27.4.13 Nasopharyngitis | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 27.4.14 Increased blood creatine phosphokinase | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 27.4.15 Fatigue | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.04 [0.00, 2.24] |
| 27.4.16 Elevated AST | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 27.4.17 Diarrhoea | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.74 [0.08, 39.74] |
| 27.4.18 Abnormal respiration | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.74 [0.02, 129.18] |
| 27.4.19 Rhinorrhoea | 1 | 34 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.49 [0.08, 246.11] |
| 27.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 27.5.1 At 1 month | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐31.00 [‐40.41, ‐21.59] |
Comparison 28. Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus triple placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 28.1 Quality of life: CFQ‐R respiratory domain (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.1.1 At up to 1 month | 2 | 436 | Mean Difference (IV, Fixed, 95% CI) | 19.15 [16.12, 22.19] |
| 28.1.2 At up to 6 months | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | 20.20 [16.19, 24.21] |
| 28.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.2.1 At up to 1 month | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | 25.90 [15.57, 36.23] |
| 28.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.3.1 At up to 1 month | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | 13.80 [12.18, 15.42] |
| 28.3.2 At up to 6 months | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | 14.30 [12.76, 15.84] |
| 28.4 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.4.1 At up to 1 month | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | 0.57 [0.36, 0.78] |
| 28.5 Adverse events (at up to 1 month) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 28.5.1 Number of participants experiencing at least one adverse event | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.00, 11.62] |
| 28.5.2 Number experiencing moderate adverse events | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.21 [0.05, 193.04] |
| 28.5.3 Number experiencing severe adverse events | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 5.93] |
| 28.5.4 Infective pulmonary exacerbation | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.02, 2.52] |
| 28.5.5 Cough | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.50 [0.29, 104.32] |
| 28.5.6 Increased sputum | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.94 [0.11, 8.18] |
| 28.5.7 Haemoptysis | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.03, 8.36] |
| 28.5.8 Pyrexia | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.55 [0.01, 23.83] |
| 28.5.9 Nausea | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.25 [0.01, 6.84] |
| 28.5.10 Oropharyngeal pain | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.03, 8.36] |
| 28.5.11 Headache | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.03, 8.36] |
| 28.5.12 Nasal congestion | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.03, 8.36] |
| 28.5.13 Nasopharyngitis | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 6.43 [0.12, 336.06] |
| 28.5.14 Increased blood creatine phosphokinase | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.83 [0.02, 135.72] |
| 28.5.15 Fatigue | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.04 [0.00, 2.35] |
| 28.5.16 Elevated AST | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.83 [0.02, 135.72] |
| 28.5.17 Diarrhoea | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.18 [0.00, 13.28] |
| 28.5.18 Abnormal respiration | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.83 [0.02, 135.72] |
| 28.5.19 Rhinorrhoea | 1 | 33 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 28.6 Adverse events (at up to 6 months) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 28.6.1 Number with any adverse event | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.23, 1.36] |
| 28.6.2 Number with mild adverse events | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.90, 2.13] |
| 28.6.3 Number with moderate adverse events | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.42, 0.92] |
| 28.6.4 Number with severe adverse events | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.68, 2.85] |
| 28.6.5 Number with serious adverse events | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.36, 1.03] |
| 28.6.6 Adverse event leading to discontinuation of treatment | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.02 [0.24, 105.33] |
| 28.6.7 Infective pulmonary exacerbation of CF | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.20, 0.48] |
| 28.6.8 Sputum increased | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.63, 1.68] |
| 28.6.9 Headache | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.70, 2.03] |
| 28.6.10 Cough | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.20, 0.52] |
| 28.6.11 Diarrhoea | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.20, 0.52] |
| 28.6.12 Upper respiratory tract infection | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.59, 2.03] |
| 28.6.13 Nasopharyngitis | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.45, 1.51] |
| 28.6.14 Oropharyngeal pain | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.41, 1.44] |
| 28.6.15 Haemoptysis | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.17, 0.74] |
| 28.6.16 Fatigue | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.19, 0.95] |
| 28.7 Hospitalisation | 1 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 28.7.1 At up to 6 months | 1 | Rate Ratio (IV, Fixed, 95% CI) | 0.29 [0.14, 0.60] | |
| 28.8 Exacerbation (need for antibiotics) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 28.8.1 At up to 6 months | 1 | 403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.14, 0.60] |
| 28.9 Sweat chloride (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.9.1 At up to 1 month | 2 | 436 | Mean Difference (IV, Fixed, 95% CI) | ‐40.96 [‐43.60, ‐38.33] |
| 28.9.2 At up to 6 months | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | ‐41.80 [‐44.33, ‐39.27] |
| 28.10 Weight (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.10.1 At up to 6 months | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | 2.90 [2.40, 3.40] |
| 28.11 BMI z score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 28.11.1 At 6 months | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [0.17, 0.43] |
Comparison 29. Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor 150 mg twice daily versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/F508del).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 29.1 Quality of life: CFQ‐R respiratory domain (change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.1.1 At up to 1 month | 2 | 135 | Mean Difference (IV, Fixed, 95% CI) | 17.78 [12.90, 22.66] |
| 29.1.2 At 6 months | 1 | 175 | Mean Difference (IV, Fixed, 95% CI) | 15.90 [11.74, 20.06] |
| 29.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.2.1 At up to 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 17.80 [6.66, 28.94] |
| 29.3 FEV1 % predicted (absolute change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.3.1 At up to 1 month | 1 | 107 | Mean Difference (IV, Fixed, 95% CI) | 10.00 [7.51, 12.49] |
| 29.3.2 At 6 months | 1 | 175 | Mean Difference (IV, Fixed, 95% CI) | 10.20 [8.26, 12.14] |
| 29.4 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.4.1 At up to 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.46 [0.26, 0.66] |
| 29.5 Adverse events (at up to 1 month) | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 29.5.1 Number of participants experiencing at least one adverse event | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.46, 1.96] |
| 29.5.2 Number experiencing mild adverse events | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.49, 2.29] |
| 29.5.3 Number experiencing moderate adverse events | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.39, 2.26] |
| 29.5.4 Number experiencing severe adverse events | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.02, 1.92] |
| 29.5.5 Number experiencing serious adverse events | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.92 [0.17, 21.88] |
| 29.5.6 Adverse event leading to discontinuation of trial drug | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 29.5.7 Elevated transaminase | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.92 [0.17, 21.88] |
| 29.5.8 Rash | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.13, 6.95] |
| 29.5.9 Cough | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.90, 7.93] |
| 29.5.10 Nasopharyngitis | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.29, 5.33] |
| 29.5.11 Upper respiratory tract infection | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.96 [0.34, 11.19] |
| 29.5.12 Infective respiratory exacerbation | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.88 [0.18, 19.53] |
| 29.5.13 Headache | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.12, 1.88] |
| 29.5.14 Haemoptysis | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.17, 2.34] |
| 29.5.15 Pulmonary exacerbation | 1 | 107 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.22] |
| 29.5.16 Oropharyngeal pain | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.51 [0.44, 14.37] |
| 29.5.17 Increased sputum | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.24, 9.90] |
| 29.5.18 Pyrexia | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.09, 11.52] |
| 29.5.19 Nausea | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.02, 5.55] |
| 29.5.20 Nasal congestion | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 29.5.21 Increased blood creatine phosphokinase | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.86 [0.18, 80.99] |
| 29.5.22 Fatigue | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.86 [0.18, 80.99] |
| 29.5.23 Elevated AST | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.13, 61.89] |
| 29.5.24 Diarrhoea | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 29.5.25 Abnormal respiration | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.92 [0.08, 44.92] |
| 29.5.26 Rhinorrhoea | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.04, 30.00] |
| 29.6 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 29.6.1 Number of participants with treatment‐emergent adverse events | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.67 [0.18, 2.53] |
| 29.6.2 Number of participants with moderate adverse events | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.02, 12.01] |
| 29.6.3 Number of participants with serious adverse events | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.08, 1.31] |
| 29.6.4 Infective pulmonary exacerbations | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.17 [0.06, 0.45] |
| 29.6.5 Nasopharyngitis | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.40 [0.49, 3.97] |
| 29.6.6 Upper respiratory tract infection | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.92 [0.43, 8.52] |
| 29.6.7 Headache | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.57 [0.63, 3.91] |
| 29.6.8 Cough | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.41 [0.14, 1.16] |
| 29.6.9 Oropharyngeal pain | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.45, 6.22] |
| 29.6.10 Sputum increased | 1 | 175 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.19, 1.79] |
| 29.7 Sweat chloride (absolute change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.7.1 At up to 1 month | 2 | 135 | Mean Difference (IV, Fixed, 95% CI) | ‐44.32 [‐48.80, ‐39.84] |
| 29.7.2 At 6 months | 1 | 175 | Mean Difference (IV, Fixed, 95% CI) | ‐42.80 [‐46.27, ‐39.33] |
| 29.8 Weight (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.8.1 At up to 1 month | 1 | Mean Difference (IV, Fixed, 95% CI) | 1.60 [1.00, 2.20] | |
| 29.9 BMI (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 29.9.1 At up to 1 month | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [0.41, 0.79] |
Comparison 30. Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus deutivacaftor (150 mg once daily) versus placebo (F508del/MF).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 30.1 Quality of life: change in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 30.1.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 12.80 [0.93, 24.67] |
| 30.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 30.2.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 18.30 [7.64, 28.96] |
| 30.3 FEV1 L (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 30.3.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.44 [0.25, 0.63] |
| 30.4 Adverse events (at 1 month) | 2 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 30.4.1 Number of participants experiencing at least one adverse event | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.36 [0.05, 38.84] |
| 30.4.2 Number experiencing moderate adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 30.4.3 Number experiencing severe adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.00, 8.97] |
| 30.4.4 Infective pulmonary exacerbation | 2 | 136 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.15 [0.02, 1.00] |
| 30.4.5 Cough | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.94 [0.08, 11.23] |
| 30.4.6 Increased sputum | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.17 [0.05, 28.28] |
| 30.4.7 Nausea | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.18 [0.01, 2.57] |
| 30.4.8 Oropharyngeal pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.94 [0.08, 11.23] |
| 30.4.9 Nasal congestion | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.17 [0.05, 28.28] |
| 30.4.10 Productive cough | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.74 [0.03, 21.09] |
| 30.4.11 Chest pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.18 [0.04, 135.31] |
| 30.4.12 Paranasal sinus discomfort | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.18 [0.04, 135.31] |
| 30.4.13 Sinus congestion | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.18 [0.04, 135.31] |
| 30.4.14 URTI | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.18 [0.04, 135.31] |
| 30.4.15 Vomiting | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.06 [0.00, 3.85] |
| 30.5 Sweat chloride (change from baseline) (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 30.5.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐34.60 [‐45.15, ‐24.05] |
Comparison 31. Elexacaftor (200 mg once daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus either ivacaftor (150 mg twice daily) or tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) (F508del/gating or F508del/residual function).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 31.1 Quality of life ‐ CFQ‐R respiratory domain (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 31.1.1 At 2 months | 1 | 258 | Mean Difference (IV, Fixed, 95% CI) | 8.70 [5.34, 12.06] |
| 31.2 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 31.2.1 At 2 months | 1 | 258 | Mean Difference (IV, Fixed, 95% CI) | 3.50 [2.24, 4.76] |
| 31.3 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 31.3.1 Number of participants with treatment‐emergent adverse events | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.53, 2.04] |
| 31.3.2 Number of participants with moderate adverse events | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.47 [0.02, 11.28] |
| 31.3.3 Number of participants with serious adverse events | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.41 [0.10, 1.72] |
| 31.3.4 Infective pulmonary exacerbations | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.20 [0.05, 0.87] |
| 31.3.5 Headache | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.51 [0.18, 1.44] |
| 31.3.6 Cough | 1 | 258 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.14 [0.03, 0.72] |
| 31.4 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 31.4.1 At 2 months | 1 | 258 | Mean Difference (IV, Fixed, 95% CI) | ‐23.00 [‐25.98, ‐20.02] |
Comparison 32. VX‐152 (100 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 32.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 32.1.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 14.20 [0.98, 27.42] |
| 32.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 32.2.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 12.60 [3.48, 21.72] |
| 32.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 32.3.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 6.50 [1.62, 11.38] |
| 32.4 Most common adverse events (occurring in at least 10% of participants in either group) (day 43) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 32.4.1 Number of participants with treatment‐emergent adverse events | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.06 [0.00, 4.02] |
| 32.4.2 Number of participants with serious adverse events | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.20 [0.00, 13.86] |
| 32.4.3 Infective pulmonary exacerbations | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 4.89] |
| 32.4.4 Viral infection | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.5 Lymphadenopathy | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.6 Ear discomfort | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.7 Abdominal distension | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.8 Colitis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.9 Diarrhoea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 9.44 [0.13, 671.21] |
| 32.4.10 Nausea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.11 Vomiting | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.12 Fatigue | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.13 Vessel puncture site haemorrhage | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.14 Upper respiratory tract infection | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.15 Viral upper respiratory tract infection | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.16 Aspartate aminotransferase increased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.17 Blood alkaline phosphatase increased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.18 Gamma‐glutamyltransferase increased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.19 Hypoglycaemia | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.20 Musculoskeletal chest pain | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.21 Dizziness | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.40 [0.03, 72.18] |
| 32.4.22 Headache | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.23 Migraine | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.24 Anxiety | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.25 Cough | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.50 [0.10, 121.10] |
| 32.4.26 Dysphonia | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.27 Dyspnoea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.28 Haemoptysis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.29 Lower respiratory tract congestion | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.05, 391.56] |
| 32.4.30 Nasal congestion | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.31 Oropharyngeal pain | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.38 [0.00, 32.19] |
| 32.4.32 Sinus congestion | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.20 [0.00, 13.86] |
| 32.4.33 Sputum increased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.40 [0.03, 72.18] |
| 32.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 32.5.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | ‐19.40 [‐29.45, ‐9.35] |
Comparison 33. VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 33.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 33.1.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 29.40 [16.97, 41.83] |
| 33.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 33.2.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 21.30 [12.73, 29.87] |
| 33.3 FEV1% predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 33.3.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 10.50 [5.92, 15.08] |
| 33.4 Most common adverse events (occurring in at least 10% of participants in either group) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 33.4.1 Number of participants with treatment‐emergent adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.13 [0.00, 7.63] |
| 33.4.2 Number of participants with serious adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.00, 8.19] |
| 33.4.3 Infective pulmonary exacerbations | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 2.92] |
| 33.4.4 Lymphadenopathy | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.5 Ear discomfort | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.6 Colitis | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.7 Diarrhoea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 327.32] |
| 33.4.8 Faeces soft | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.9 Nausea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.10 Post‐tussive vomiting | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.11 Vomiting | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.12 Fatigue | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 327.32] |
| 33.4.13 Vessel puncture site haemorrhage | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.14 Seasonal allergy | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.15 Upper respiratory tract infection | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 33.4.16 Viral upper respiratory tract infection | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.17 Procedural haemorrhage | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.18 Aspartate aminotransferase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.19 Blood alkaline phosphatase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.20 Blood bilirubin increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.21 Gamma‐glutamyltransferase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.22 Dizziness | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.23 Headache | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.24 Migraine | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.25 Sinus headache | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.26 Anxiety | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.27 Insomnia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.28 Cough | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 7.00 [0.29, 171.64] |
| 33.4.29 Dysphonia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.30 Dyspnoea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.31 Dyspnoea exertional | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.32 Lower respiratory tract congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.33 Nasal congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.34 Oropharyngeal pain | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 33.4.35 Respiration abnormal | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.4.36 Sinus congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.00, 8.19] |
| 33.4.37 Sputum increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.75 [0.06, 53.76] |
| 33.4.38 Wheezing | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 33.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 33.5.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | ‐13.50 [‐23.17, ‐3.83] |
Comparison 34. VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 34.1 CFQ‐R Respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 34.1.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 26.20 [13.71, 38.69] |
| 34.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 34.2.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 17.10 [8.05, 26.15] |
| 34.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 34.3.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 8.80 [3.98, 13.62] |
| 34.4 Most common adverse events (occurring in at least 10% of participants in either group) | 2 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 34.4.1 Number of participants with treatment‐emergent adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 34.4.2 Number of participants with serious adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.01, 10.34] |
| 34.4.3 Infective pulmonary exacerbations | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 2.92] |
| 34.4.4 Anaemia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.5 Lymphadenopathy | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.6 Ear discomfort | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.7 Abdominal discomfort | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.8 Colitis | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.9 Diarrhoea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.10 Dyspepsia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.11 Flatulence | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.12 Nausea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.13 Vomiting | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.14 Fatigue | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.15 Thirst | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.16 Vessel puncture site haemorrhage | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.17 Chronic sinusitis | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.18 Upper respiratory tract infection | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.19 Viral upper respiratory tract infection | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 34.4.20 Procedural dizziness | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.21 Alanine aminotransferase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.22 Aspartate aminotransferase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 34.4.23 Blood alkaline phosphatase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 34.4.24 Gamma‐glutamyltransferase increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 34.4.25 Platelet count increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.26 Dizziness | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.27 Dysgeusia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.28 Headache | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 7.93 [0.13, 479.91] |
| 34.4.29 Migraine | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.30 Sinus headache | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.31 Anxiety | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.32 Chromaturia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.33 Breast discomfort | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.34 Breast tenderness | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.35 Cough | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.00 [0.11, 79.53] |
| 34.4.36 Dysphonia | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.37 Dyspnoea | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.38 Haemoptysis | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.39 Lower respiratory tract congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.40 Nasal congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.41 Oropharyngeal pain | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.24 [0.00, 19.06] |
| 34.4.42 Pulmonary pain | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.43 Respiration abnormal | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.00 [0.08, 327.32] |
| 34.4.44 Respiratory tract congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.4.45 Sinus congestion | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.12 [0.00, 8.19] |
| 34.4.46 Sputum increased | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.02, 37.18] |
| 34.4.47 Wheezing | 1 | 18 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.68 [0.03, 214.00] |
| 34.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 34.5.1 At day 15 | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | ‐27.40 [‐36.86, ‐17.94] |
Comparison 35. VX‐152 (200 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 35.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 35.1.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 4.80 [‐6.53, 16.13] |
| 35.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 35.2.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 14.60 [1.88, 27.32] |
| 35.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 35.3.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 8.30 [1.15, 15.45] |
| 35.4 Most common adverse events (occurring in at least 10% of participants in either group) (at day 71) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 35.4.1 Number of participants with treatment‐emergent adverse events | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.50 [0.02, 15.09] |
| 35.4.2 Number of participants with serious adverse events | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 35.4.3 Eye swelling | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.4 Nausea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.5 Fatigue | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.6 Gastroenteritis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.7 Infective pulmonary exacerbation of cystic fibrosis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.8 Nasopharyngitis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.9 Pneumonia | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.10 Incision site pain | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.11 Muscle strain | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.12 Weight decreased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.13 Arthralgia | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.14 Tendon pain | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.15 Headache | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.16 Bronchospasm | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.17 Cough | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.01, 18.70] |
| 35.4.18 Dyspnoea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.19 Haemoptysis | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.33 [0.01, 18.70] |
| 35.4.20 Lower respiratory tract congestion | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.21 Rhinorrhoea | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.22 Sputum discoloured | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.23 Sputum increased | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.4.24 Upper respiratory tract congestion | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.25 Wheezing | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.26 Erythema | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 9.97] |
| 35.4.27 Night sweats | 1 | 14 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.02, 122.55] |
| 35.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 35.5.1 At day 15 | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | ‐24.80 [‐35.37, ‐14.23] |
Comparison 36. VX‐152 (300 mg twice daily) plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily) versus placebo plus tezacaftor (100 mg once daily) plus ivacaftor (150 mg twice daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 36.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 36.1.1 At 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 11.30 [3.08, 19.52] |
| 36.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 36.2.1 Through 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 13.50 [6.57, 20.43] |
| 36.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 36.3.1 Through 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 8.70 [4.50, 12.90] |
| 36.4 Most common adverse events (occurring in at least 10% of participants in either group) (at day 71) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 36.4.1 Number of participants with treatment‐emergent adverse events | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.80 [0.21, 67.89] |
| 36.4.2 Number of participants with serious adverse events | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 36.4.3 Diarrhoea | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.84 [0.05, 163.01] |
| 36.4.4 Vomiting | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 7.90] |
| 36.4.5 Chest discomfort | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 7.90] |
| 36.4.6 Infective pulmonary exacerbation of cystic fibrosis | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.42 [0.04, 4.45] |
| 36.4.7 Nasopharyngitis | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 7.90] |
| 36.4.8 Headache | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.86 [0.07, 210.80] |
| 36.4.9 Chromaturia | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.86 [0.07, 210.80] |
| 36.4.10 Cough | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 6.29 [0.12, 326.72] |
| 36.4.11 Dyspnoea | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.63 [0.02, 18.50] |
| 36.4.12 Lower respiratory tract congestion | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 7.90] |
| 36.4.13 Productive cough | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.84 [0.05, 163.01] |
| 36.4.14 Sputum increased | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.40 [0.11, 50.56] |
| 36.4.15 Wheezing | 1 | 28 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 7.90] |
| 36.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 36.5.1 Through 1 month | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐23.90 [‐32.11, ‐15.69] |
Comparison 37. VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 37.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 37.1.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 18.50 [8.99, 28.01] |
| 37.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 37.2.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 19.10 [10.62, 27.58] |
| 37.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 37.3.1 Through 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 10.60 [5.93, 15.27] |
| 37.4 Most common adverse events (occurring in at least 10% of participants in either group) (at day 57) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 37.4.1 Number of participants with treatment‐emergent adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.11 [0.08, 14.81] |
| 37.4.2 Number of participants with serious adverse events | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 37.4.3 Infective pulmonary exacerbations | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.90 [0.07, 12.00] |
| 37.4.4 Diarrhoea | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.05, 34.62] |
| 37.4.5 Abdominal pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 37.4.6 Viral upper respiratory tract infection | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.05, 34.62] |
| 37.4.7 Upper respiratory tract infection | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.48 [0.06, 212.67] |
| 37.4.8 Alanine aminotransferase increased | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.05, 34.62] |
| 37.4.9 Aspartate aminotransferase increased | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.25 [0.05, 34.62] |
| 37.4.10 Dizziness | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 6.33] |
| 37.4.11 Cough | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.15 [0.01, 1.80] |
| 37.4.12 Sputum increased | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.16 [0.01, 3.74] |
| 37.4.13 Dyspnoea | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 6.33] |
| 37.4.14 Oropharyngeal pain | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 6.33] |
| 37.4.15 Respiration abnormal | 1 | 29 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 6.33] |
| 37.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 37.5.1 At 1 month | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐34.70 [‐43.54, ‐25.86] |
Comparison 38. VX‐440 (600 mg twice daily) plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily) versus placebo plus tezacaftor (50 mg twice daily) plus ivacaftor (300 mg twice daily).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 38.1 CFQ‐R respiratory domain score (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 38.1.1 At one month | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 20.10 [13.76, 26.44] |
| 38.2 FEV1 % predicted (relative change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 38.2.1 At 1 month | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 20.00 [12.59, 27.41] |
| 38.3 FEV1 % predicted (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 38.3.1 Through 1 month | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 12.00 [7.64, 16.36] |
| 38.4 Most common adverse events (occurring in at least 10% of participants in either group) (at day 85) | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 38.4.1 Number of participants with treatment‐emergent adverse events | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.00, 11.73] |
| 38.4.2 Number of participants with serious adverse events | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 3.34] |
| 38.4.3 Infective pulmonary exacerbation of cystic fibrosis | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.88 [0.03, 22.76] |
| 38.4.4 Adjustment disorder | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.5 Diarrhoea | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.55 [0.06, 197.65] |
| 38.4.6 Abdominal pain | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.7 Anorectal discomfort | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.8 Viral upper respiratory tract infection | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.11 [0.00, 3.34] |
| 38.4.9 Upper respiratory tract infection | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.76 [0.03, 112.52] |
| 38.4.10 Folliculitis | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.11 Pharyngitis | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.12 Alanine aminotransferase increased | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.76 [0.03, 112.52] |
| 38.4.13 Insomnia | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.09 [0.00, 7.17] |
| 38.4.14 Cough | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.61 [0.09, 249.50] |
| 38.4.15 Sputum increased | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.88 [0.03, 22.76] |
| 38.4.16 Nasal congestion | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.76 [0.03, 112.52] |
| 38.4.17 Lower respiratory tract congestion | 1 | 26 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.76 [0.03, 112.52] |
| 38.5 Sweat chloride (absolute change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 38.5.1 Through 1 month | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐33.40 [‐45.41, ‐21.39] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Barry 2021.
| Study characteristics | ||
| Methods | Phase 3, double‐blind RCT Parallel design Duration: 8 weeks (following ivacaftor or tezacaftor‐ivacaftor run‐in period of 4 weeks) Multicentre study (93 study locations) conducted across the USA, Australia, Belgium, Canada, Denmark, France, Germany, Ireland, Israel, Italy, Netherlands, Spain, UK |
|
| Participants | Inclusion criteria state aged 12 years and older with FEV1 40% to 90% predicted 258 participants in total: 132 participants received elexacaftor‐tezacaftor‐ivacaftor and 126 received active control Age, mean (SD): 37.7 (14.5) years Sex: 128 females, 130 males Genotype: F508del/gating variant or F508del/residual function Baseline FEV1 mean (SD) % predicted: 67.6 (16.0)% predicted |
|
| Interventions |
Intervention: triple therapy of elexacaftor‐tezacaftor‐ivacaftor; following an ivacaftor or tezacaftor‐ivacaftor run‐in period of 4 weeks, participants received elexacaftor 200 mg daily/tezacaftor 100 mg daily/ivacaftor 150 mg every 12 hours for 8 weeks Control: ivacaftor or tezacaftor‐ivacaftor; following an ivacaftor or tezacaftor‐ivacaftor run‐in period of 4 weeks, participants either received ivacaftor 150 mg every 12 hours or tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 8 weeks |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | A Phase 3 Study of VX‐445 Combination Therapy in Cystic Fibrosis Subjects Heterozygous for F508del and a Gating or Residual Function Mutation (F/G and F/RF Genotypes) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised (1:1) to the elexacaftor/tezacaftor/ivacaftor treatment arm or the control arm (ivacaftor or tezacaftor/ivacaftor) using an interactive web response system. The randomisation code list was described as being produced by Vertex Biometrics or a qualified randomisation vendor. The randomisation was stratified based on the comparator group, FEV1 % predicted (as determined during the run‐in period) and sweat chloride (also determined during the run‐in period). |
| Allocation concealment (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment and the randomisation code list was described as being produced by Vertex Biometrics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor, and the study team), and members of the Vertex study team were blinded to the treatment codes. Individuals who could be unblinded were outlined in protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor, and the study team), and members of the Vertex study team were blinded to the treatment codes. Individuals who could be unblinded were outlined in protocol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants analysed. Reasons given in supplementary materials as to why certain participants discontinued study at certain points. Appears to be missing data for exploratory endpoints, e.g. absolute change in BMI, though this does not raise the risk of bias. |
| Selective reporting (reporting bias) | Unclear risk | Methods state that ECGs and vital signs will be measured, however these have not been stated in the results, whether unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants generally well‐balanced, however it is unclear as to the extent to which the sponsors were involved in designing, writing up and publishing the report. |
Boyle 2014.
| Study characteristics | ||
| Methods | Phase 2, placebo‐controlled RCT with 3 different cohorts. Only Cohort 1 was included in this review (n = 62). (The following information will refer to cohort 1 only ‐ see 'Notes'). Parallel design Multicentre study conducted at 69 different sites in North America, Europe and Australia Duration: Cohort 1 lasted 21 days |
|
| Participants | Mutation: participants homozygous for F508del mutation Age: participants in Cohort 1 have a mean age of 29.1 years Gender split: 50% of participants are male Lung function: all participants in Cohort 1 have a FEV1 ≥ 40% of predicted normal for age, gender and height and a mean (range) predicted FEV1 of 66.9% (32.8 to 117.1) Sweat chloride levels: participants in Cohort 1 have a mean (range) level of 101.9 mmol/L (87.5 to 121.0) |
|
| Interventions |
Intervention 1: lumacaftor (also known as VX‐809, a CFTR corrector) alone Intervention 2: lumacaftor in combination with ivacaftor (also known as VX‐770, a CFTR potentiator) Intervention 3: placebo Cohort 1 (n = 62) Study drug participants: 200 mg lumacaftor once daily for 14 days; then from day 15, participants continue to take 200 mg of lumacaftor in addition to either 150 mg or 250 mg of ivacaftor twice daily until day 21 Placebo participants: placebo for 21 days |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals and the Cystic Fibrosis Foundation Therapeutics Development Network | |
| Notes | *Denotes outcomes relevant to this review. Only data from Cohort 1 were included in this review. This was because data for placebo participants from Cohorts 2 and 3 were pooled, although randomisation in these cohorts occurred separately. This meant that the effects of randomisation in these cohorts were undone. Data for participants in Cohorts 2 and 3 were excluded. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A random sequence was generated by a computer by an independent party. |
| Allocation concealment (selection bias) | Low risk | "Site pharmacists dispensed drugs on the basis of an interactive voice response system". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Drug doses were prepared by an independent unmasked pharmacist and dispensed by site pharmacists who were masked to treatment assignment. Participant blinding was maintained by placebo, which was matched to intervention by the quantity of tablets and by size, colour, coating and packaging. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Site investigators and the study sponsor were also masked to treatment assignment and to sweat chloride levels ‐ data that could have potentially disclosed treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Participant data were excluded from the analysis due to insufficient data, e.g. participants were excluded from the analysis of sweat chloride concentration if an insufficient amount of sweat was provided. We judged this trial as having an unclear risk of attrition bias because it was unclear how these exclusions would have affected the balance between groups in baseline characteristics. |
| Selective reporting (reporting bias) | Low risk | We compared the outcomes reported on the US NIH trials registry (www.clinicaltrials.gov) to the outcomes reported in the results of the published paper as the protocol was not available. No selective outcome reporting was identified. |
| Other bias | Low risk | Similar baseline characteristics. |
Clancy 2012.
| Study characteristics | ||
| Methods | Phase 2a placebo‐controlled RCT Parallel design Multicentre study conducted at 25 study locations across North America and Europe Duration: 28 days |
|
| Participants |
Inclusion criteria: FEV1 > 40% predicted 89 participants with a confirmed diagnosis of CF were randomised Baseline characteristics Age, median (range): 26 (18 to 54) years Sex: 60% of the participants were males Genotype: 88 out of 89 participants were homozygous for the F508del mutation Lung function, median (range) FEV1 % predicted: 71% predicted (34.2 to 126.3) Sweat chloride levels, median (range): 103.5 (66.0 to 129.0) mmol/L Nutritional status, BMI median (range): 22 (16 to 34) |
|
| Interventions |
Intervention 1: placebo (n = 17) Intervention 2: lumacaftor (VX‐809) 25 mg once daily (n = 18) Intervention 3: lumacaftor 50 mg once daily (n = 18) Intervention 4: lumacaftor 100 mg once daily (n = 17) Intervention 5: lumacaftor 200 mg once daily (n = 19) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals and grants from the NIH | |
| Notes | *Denotes outcomes relevant to this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | There was insufficient information on how participant or study personnel blinding was maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information on how outcome assessor blinding was maintained. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | In the adverse events table, the total number of participants shown (n = 45) is fewer than the total number of participants randomised (n = 89). In Figure 1B, the number of participants analysed for the outcome 'Change from baseline in sweat chloride' (n = 63) is fewer than the total number of participants randomised to the intervention (n = 72). Therefore, 9 participants have been unaccounted for. In the table of results of total CFQ‐R scores, 1 participant appears to be excluded from each of the treatment groups. |
| Selective reporting (reporting bias) | High risk | No results have been presented for FEF25-75% or FVC despite being stated as outcomes. |
| Other bias | Low risk | Baseline characteristics well matched except for the less severe lung disease in the 25 mg and placebo groups. |
Davies 2018a.
| Study characteristics | ||
| Methods | Phase 1, placebo‐controlled, double‐blind RCT Parallel design Multicentre study conducted at 9 sites across the UK Duration: 14 days |
|
| Participants | 12 participants with a confirmed CF diagnosis enrolled and randomised. All 12 participants completed the 14‐day trial period. Age: 18 years or older Genotype: F508del/MF Disease severity: body weight ≥ 35 kg and FEV1 % predicted 40% to 90% at screening Intervention group (n = 9) Age, mean (SD): 36.8 (9.9) years Sex: 8 males, 1 female FEV1 % predicted, mean (SD): 48.0 (12.7)% Sweat chloride, mean (SD): 107.7 (10.5) mmol/L Placebo group (n = 3) Age, mean (SD): 30.3 (5.1) years Sex: 3 males FEV1 % predicted, mean (SD): 44.9 (9.6)% Sweat chloride, mean (SD): 104.3 (4.9) mmol/L |
|
| Interventions |
Intervention: VX‐659 120 mg every 12 hours plus ivacaftor 150 mg every 12 hours plus tezacaftor 100 mg once daily . Control: triple placebo. |
|
| Outcomes |
|
|
| Funding source | Vertex Pharmaceutical supported the trial, who in turn received funding from the Cystic Fibrosis Foundation for development of VX‐659. Also grants from The National Institute for Health Research and NIH. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list made by Vertex Biostatistics or a randomisation vendor. Final list reviewed and approved by a designated unblinded statistician who is independent of the study team. Interactive web response system used to assign participants to treatment. |
| Allocation concealment (selection bias) | Low risk | Random allocation independent of study team. Use of interactive web response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants, site personnel and Vertex study team were blinded to allocation. Protocol sets out conditions when blinding could/should be broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | All authors were only allowed access to study data after they were unblinded. No mention is made of other outcome assessors (e.g. clinicians who were not authors, but were involved in seeing participants and measuring outcomes of interest) and whether there was a possibility of them knowing allocated intervention. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised are accounted for. |
| Selective reporting (reporting bias) | Unclear risk | States in methods that 12‐lead ECG and vital signs would be measured; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Low risk | Different groups of participants are balanced in baseline characteristics; no significant difference between them. Detail in paper and its supplement does not cause any concern about other sources of bias not previously mentioned. |
Davies 2018b.
| Study characteristics | ||
| Methods | Phase 2, placebo‐controlled, double‐blind RCT Parallel design with 3 arms, each arm comparing different doses to placebo Multicentre study conducted at 48 sites in the UK, US, Ireland and Israel Duration: 4 weeks active intervention (up to 12 weeks including run‐in and washout periods) |
|
| Participants | Eligible participants are aged 18 or older, with a confirmed CF diagnosis and a CFTR genotype of either F508del/MF or F508del/F508del Arm 1 63 participants enrolled and randomised; all with genotype F508del/MF Placebo (n = 10) Age, mean (SD): 26.6 (6.0) years Sex: 6 males, 4 females FEV1 % predicted, mean (SD): 53.9 (12.0)% Sweat chloride, mean (SD): 98.2 (13.3) mmol/L CFQ‐R score (respiratory domain), mean (SD): 98.2 (13.3) VX‐659 80 mg (n = 11) Age, mean (SD): 32.0 (11.7) years Sex: 4 males, 7 females FEV1 % predicted, mean (SD): 57.9 (10.8)% Sweat chloride, mean (SD): 102.7 (7.0) mmol/L CFQ‐R score (respiratory domain), mean (SD): 63.1 (18.5) VX‐659 240 mg (n = 20) Age, mean (SD): 31.4 (9.7) years Sex: 13 males, 7 females FEV1 % predicted, mean (SD): 58.0 (16.8)% Sweat chloride, mean (SD): 100.5 (9.0) mmol/L CFQ‐R score (respiratory domain), mean (SD): 64.4 (17.8) VX‐659 400 mg (n = 22) Age, mean (SD): 27.2 (6.6) years Sex: 10 males, 12 females FEV1 % predicted, mean (SD): 59.6 (15.4)% Sweat chloride, mean (SD): 100.7 (11.6) mmol/L CFQ‐R score (respiratory domain), mean (SD): 64.6 (20.7) Arm 2 29 participants enrolled and randomised all with the genotype F508del/F508del Placebo plus tezacaftor plus ivacaftor (n = 11) Age, mean (SD): 32.5 (7.5) years Sex: 7 males, 4 females FEV1 % predicted, mean (SD): 60.0 (12.6)% Sweat chloride, mean (SD): 96.6 (11.4) mmol/L CFQ‐R score (respiratory domain), mean (SD): 65.7 (17.4) VX‐659 400 mg plus tezacaftor plus ivacaftor (n = 18) Age, mean (SD): 33.4 (9.2) years Sex: 12 males, 6 females FEV1 % predicted, mean (SD): 58.6 (13.3)% Sweat chloride, mean (SD): 91.9 (11.6) mmol/L CFQ‐R score (respiratory domain), mean (SD): 68.5 (14.1) Arm 3 29 participants enrolled and randomised, all with genotype F508del/MF Triple placebo (n = 6) Age, mean (SD): 24.5 (5.3) years Sex: 3 males, 3 females FEV1 % predicted, mean (SD): 53.0 (12.3)% Sweat chloride, mean (SD): 96.6 (4.3) mmol/L CFQ‐R score (respiratory domain), mean (SD): 64.8 (24.0) VX‐659 400 mg plus tezacaftor plus VX‐561 (n = 19) Age, mean (SD): 32.5 (9.4) years Sex: 8 males, 11 females FEV1 % predicted, mean (SD): 59.8 (12.6)% Sweat chloride, mean (SD): 101.2 (9.5) mmol/L CFQ‐R score (respiratory domain), mean (SD): 69.3 (13.9) |
|
| Interventions |
Arm 1 (F508del/MF genotype): once‐daily VX‐659 80 mg or 240 mg or 400 mg plus tezacaftor 100 mg once‐daily plus ivacaftor 150 mg twice‐daily versus triple placebo for 4 weeks Arm 2 (F508del/F508del) : 4‐week run‐in of tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (standard of care) followed by the addition of once‐daily VX‐659 400 mg or matched placebo to existing regimen for 4 weeks followed by 4‐week washout period of tezacaftor 100 mg once daily plus ivacaftor 150 mg twice daily (standard of care) Arm 3 (F508del/MF): once‐daily VX‐659 400 mg plus tezacaftor 100 mg once daily plus VX‐561* 150 mg once daily versus triple placebo *Deuterated ivacaftor ‐ administered once daily instead of twice daily |
|
| Outcomes |
|
|
| Funding source | Vertex Pharmaceuticals supported the study, who in turn received funding from the Cystic Fibrosis Foundation for development of VX‐659. Also grants from the National Institute for Health Research and NIH. | |
| Notes | Different groups of participants are balanced in baseline characteristics; no significant difference between them. Detail in paper and its supplement does not cause any concern about other sources of bias not previously mentioned. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation code generated by Vertex Biometrics or a "qualified randomisation vendor". Randomisation stratified by FEV1 % predicted values (< 70 vs ≥ 70). |
| Allocation concealment (selection bias) | Low risk | Use of interactive web response system to allocate participants to groups. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants, site personnel and Vertex study team were blinded to allocation. Protocol sets out conditions when blinding could/should be broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | All authors were only allowed access to trial data after they were unblinded. No mention is made of other outcome assessors (e.g. clinicians who were not authors, but were involved in seeing participating patients and measuring outcomes of interest) and whether there was a possibility of them knowing the allocated intervention. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised are accounted for. |
| Selective reporting (reporting bias) | Unclear risk | States in methods that investigators would measure 12‐lead ECG and vital signs; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Low risk | Different groups of participants are balanced in baseline characteristics; no significant difference between them. Detail in paper and its supplement does not cause any concern about other sources of bias not previously mentioned. |
Davies 2021.
| Study characteristics | ||
| Methods | Phase 3, double‐blind RCT Parallel design Duration: 8 weeks |
|
| Participants |
Inclusion criteria Age, range: 6 to 11 years Genotype: F508del/F508del or F508del/RF Lung function, FEV1 % predicted ≥ 70%, adjusted for age, sex, height Screening LCI2.5 result ≥ 7.5 Able to swallow tablets Exclusion criteria Clinically significant cirrhosis with or without portal hypertension Colonisation with organisms associated with a more rapid decline in pulmonary status Solid organ or haematological transplantation Baseline characteristics 69 participants randomised, out of which 67 participants received study drug Age, mean (SD): 8.6 (1.7) years Sex: 37 females, 30 males Lung function, LCI2.5 mean (SD): 9.54 (1.97) |
|
| Interventions |
TEZ/IVA: participants with genotype F508del/F508del received tezacaftor/ivacaftor fixed‐dose combination in the morning and ivacaftor in the evening for 8 weeks. Participants with genotype F508del/RF received tezacaftor/ivacaftor fixed‐dose combination and placebo matched to ivacaftor in the morning and ivacaftor in the evening for 8 weeks Ivacaftor: participants with genotype F508del/RF received placebo matched to tezacaftor/ivacaftor fixed‐dose combination in the morning and ivacaftor in the morning and evening for 8 weeks Placebo: participants with genotype F508del/F508del received placebo matched to tezacaftor/ivacaftor fixed‐dose combination in the morning and placebo matched to ivacaftor in the evening for 8 weeks |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
Additional endpoints
|
|
| Funding source | Vertex Pharmaceuticals Incorporated and NIHR Respiratory Clinical Research Facility at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Interactive web or voice response system used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician (not a member of the study execution team). |
| Allocation concealment (selection bias) | Low risk | An interactive web or voice response system was used to assign participants to treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All study personnel were blinded and with a clear statement of the exceptions to this in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Clinicaltrials.gov states that masking was quadruple (participant, care provider, investigator, outcomes assessor), and the protocol states that all study personnel would be blinded, with a clear statement of the exceptions to this in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | All randomised participants analysed against outcomes, however according to the published paper, the mean and SD values, and P values were based on fewer participants than there were within the group (for example, the P value for the change in sweat chloride was based on 48 participants), with no explanations offered as to why these numbers were lower than the 54 participants in the intervention group. |
| Selective reporting (reporting bias) | Unclear risk | Unable to find exact values for "standard 12‐lead ECGs" and other parameters used to assess AEs. Although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Unclear risk | Baseline characteristics generally balanced. It is unclear as to the extent to which the funders were involved in designing, writing up and publishing the report. |
Donaldson 2014.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 7 days Multicentre: 17 sites in the USA |
|
| Participants | 66 participants Mean (SD) age: 29 (8) years Sex: 40 females and 26 males Disease severity: mean (SD) % predicted FEV1 70 (21)%, and mean (SD) sweat chloride 101 (11) mmol/L There were no significant differences among the treatment groups at baseline |
|
| Interventions |
Intervention: 4 sequential ascending doses of N6022 were assessed (5, 10, 20 and 40 mg/day) given by intravenous infusion once daily Control: placebo (normal saline) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Sponsored by Nivalis Therapeutics | |
| Notes | *Denotes outcomes relevant to this review 4 sequential ascending doses of N6022 were assessed (5, 10, 20 and 40 mg/day) followed by a confirmatory cohort of participants at the highest dose. An independent Data Monitoring Committee adjudicated dose escalation at the completion of each cohort after review of unblinded safety data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (participant, caregiver, investigator, outcomes assessor) achieved with IV administration of placebo (saline) using the same volume as the active drug groups. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind (participant, caregiver, investigator, outcomes assessor) achieved with IV administration of placebo (saline) using the same volume as the active drug groups. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants completed the 7 days of follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | No full‐text publication of the study available. Limited results (without any statistical analysis) available in the ongoing trials database (www.clinicaltrials.gov). Unclear if all relevant information has been made available. |
| Other bias | Low risk | Baseline characteristics across the 5 treatment groups seem fairly well‐balanced despite small numbers in each group. |
Donaldson 2017.
| Study characteristics | ||
| Methods | Phase 1, double‐blind RCT Parallel design Duration: 28 days treatment Multicentre (10 centres) |
|
| Participants | 51 adults with CF randomised Genotype: homozygous for the F508del mutation Age, mean (range): 50 mg cavosonstat twice daily group 31 (18 to 45) years; 100 mg cavosonstat twice daily group 27 (18 to 42) years; 200 mg cavosonstat twice daily group 27 (18 to 42) years Sex: 32 females, 19 males Lung function: FEV1 ≥ 40% predicted pre‐ or post‐bronchodilator value at screening Sweat chloride: ≥ 60 mEq/L |
|
| Interventions |
Intervention: cavosonstat 2x daily 50 mg, 100 mg or 200 mg Control: placebo 2x daily |
|
| Outcomes |
Primary outcome (no prespecified sample size)
Secondary outcomes (at 28 days)
|
|
| Funding source | Sponsored by Nivalis Therapeutics | |
| Notes | *Denotes an outcome relevant to this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description of method. |
| Allocation concealment (selection bias) | Unclear risk | No description of method. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Likely low risk as double‐blind and placebo‐controlled, but further information about this aspect of methodology is not described. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Likely low risk as double‐blind and placebo‐controlled, but further information about this aspect of methodology is not described. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2 participants unaccounted for in analysis. |
| Selective reporting (reporting bias) | Low risk | Likely low risk ‐ all outcomes reported, but they appear to have been measured at other time points that are not reported (7 days and 14 days). |
| Other bias | Unclear risk | "Approximately two‐thirds of CF subjects were female; however, there was a greater proportion of males in the 200 mg BID dose group. Other baseline characteristics were similar across the treatment groups." Unclear if this sex imbalance may have influenced the results. |
Donaldson 2018.
| Study characteristics | ||
| Methods | Double‐blind, controlled, Phase 2 RCT, which included a dose‐ranging arm Parallel design Duration: 28 days treatment followed by 28 days observation Multicentre: USA (19 sites), Canada (4 sites), Germany (8 sites), UK (5 sites) |
|
| Participants | Genotype: participants homozygous for F508del mutation, and heterozygous participants with 1 F508del mutation and 1 G551D mutation. Only the 18 heterozygous participants are included in the analysis of this review (this is because the placebo participants in the homozygous arms of the trial were pooled, and this was judged to negate the effects of randomisation). Active drug arm Age, mean (SD): 26.6 (7.0) years Sex: 6/14 (43%) participants female Lung function (FEV1 % predicted), mean (SD): 59.1 (16.6)% predicted Sweat chloride levels, mean (SD): 52.9 (19.6) Placebo arm Age, mean (SD): 34.5 (7.6) years Sex: 3/4 (75%) female Lung function (FEV1 % predicted), mean (SD): 62.6 (12.7)% predicted Sweat chloride levels, mean (SD): 56.7 (22.1) |
|
| Interventions |
Intervention: tezacaftor 100 mg/day and ivacaftor 150 mg Control: ivacaftor 150 mg (heterozygous arm only) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals and grants from the NIH | |
| Notes | *Denotes outcomes relevant to this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described. |
| Allocation concealment (selection bias) | Unclear risk | Method not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants included in analysis. |
| Selective reporting (reporting bias) | Low risk | All outcomes appear to have been reported. |
| Other bias | Unclear risk | Baseline characteristics of heterozygous participants somewhat imbalanced across groups (e.g. sex, age, FEV1). However, this imbalance may be due to small numbers (active drug arm n = 14 and placebo arm n = 4) and unclear if the imbalance has influenced results. |
Heijerman 2019.
| Study characteristics | ||
| Methods | Phase 3, double‐blind RCT Parallel design Multicentre study conducted at 44 sites in 4 countries (Belgium, the Netherlands, the UK and the USA) Duration: 4 weeks with a further 4‐week follow‐up period followed by participants being invited to a 96‐week open‐label extension study (VX17‐445‐105; NCT03525574) |
|
| Participants | Eligible participants were aged 12 or above with a confirmed diagnosis of CF and 2 copies of F508del, FEV1 % predicted 40% to 90% and in 'stable condition' as judged by study investigators 108 participants were randomised and 107 participants who received a dose of the study drug during the treatment period were included in analyses Intervention group (n = 55) Age, mean (SD): 28.8 (11.5) years Age distribution: ≥ 12 to < 18 years: 16 (29%); ≥ 18 years: 39 (71%) Sex: 31 females (56%) FEV1 % predicted, mean (SD): 61.6 (15.4) % predicted FEV1 % predicted, distribution: < 40%: 6 (11%); ≥ 40% to < 70%: 31 (56%); ≥ 70% to ≤ 90%: 18 (33%); > 90%: 0 BMI, mean (SD): 21.75 (3.19) kg/m² Sweat chloride concentration, mean (SD): 91.4 (11.0) mmol/L CFQ‐R respiratory domain score, mean (SD): 70.6 (16.2) CFTR modulator therapy: yes 32 (58%); no 23 (42%) Control group (n = 52) Age, mean (SD): 27.9 (10.8) years Age distribution: ≥ 12 to < 18 years: 14 (27%); ≥ 18 years: 38 (73%) Sex: 24 females (44%) FEV1 % predicted, mean (SD): 60.2 (14.4) % predicted FEV1 % predicted, distribution: < 40%: 4 (8%); ≥ 40% to < 70%: 34 (65%); ≥ 70% to ≤ 90%: 14 (27%); > 90%: 0 BMI, mean (SD): 21.88 (4.12) kg/m² Sweat chloride concentration, mean (SD): 90.0 (12.3) mmol/L CFQ‐R respiratory domain score, mean (SD): 72.6 (17.9) CFTR modulator therapy: yes 34 (65%); no 18 (35%) |
|
| Interventions | 4‐week run‐in period for both intervention and control groups: tezacaftor 100 mg daily, ivacaftor 150 mg every 12 hours Intervention (n = 55): elexacaftor 200 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours Control (n = 52): placebo matched to elexacaftor once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours |
|
| Outcomes | Outcomes measured at baseline, 2 weeks and 4 weeks Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals supported the study and were also involved in writing the study; however, the corresponding author had final responsibility for the decision to submit for publication. | |
| Notes | ClinicalTrials.gov: NCT03525548 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomly assigned in a 1:1 ratio by an interactive web response system. Randomisation was stratified by FEV1 % predicted. |
| Allocation concealment (selection bias) | Low risk | Study sites entered participant demographic information and stratification criteria into the interactive web response system, which established the treatment group and corresponding treatment kit numbers. Elexacaftor‐tezacaftor‐ivacaftor and tezacaftor‐ivacaftor regimens were distributed via identical treatment kits. An independent vendor, which was not involved in any other operational parts of this trial, operated and maintained the system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and people responsible for participants' care were all blinded to treatment allocation by use of identical treatment kits, which were matched in the appearance and number of tablets. Treatment kits were provided to participants in sealed blister cards only when the participants were on site. All participants, investigators and trial personnel were masked to treatment assignments during the conduct of this trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All investigators and trial personnel were masked to treatment assignments during the conduct of this trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 108 participants were randomised and 107 participants who received a dose of the study drug during the treatment period were included in analyses. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in protocol were reported. |
| Other bias | Unclear risk | None noted; well‐matched baseline characteristics. However, as it is stated that Vertex funded the study, it is unclear as to the extent to which they were involved in designing, writing up and publishing the report. |
Horsley 2017.
| Study characteristics | ||
| Methods | Phase 1b RCT Parallel design Multicentre: 14 centres in Australia, Czech Republic, Germany and the UK Duration: 28 days |
|
| Participants | 27 participants aged 18 or over, homozygous for F508del and with FEV1 % predicted at least 40% at baseline FDL169 400 mg (n = 6) Age, mean (range): 31.5 (18 to 56) years Sex: 3 males, 3 females FEV1 % predicted, mean (SD): 82.2 (22.5)% Sweat chloride, mean (SD): 96.9 (9.5) mmol/L CFQ‐R score (respiratory domain), mean (SD): 89.7 (8.1) FDL169 600 mg (n = 6) Age, mean (range): 37.7 (18 to 62) years Sex: 3 males, 3 females FEV1 % predicted, mean (SD): 59.3 (9.9)% Sweat chloride, mean (SD): 101.8 (10.1) mmol/L CFQ‐R score (respiratory domain), mean (SD): 74.0 (11.5) FDL169 800 mg (n = 8) Age, mean (range): 26.5 (21 to 37) years Sex: 4 males, 4 females FEV1 % predicted, mean (SD): 85.3 (13.5)% Sweat chloride, mean (SD): 98.5 (9.8) mmol/L CFQ‐R score (respiratory domain), mean (SD): 77.0 (18.8) Placebo (n = 7) Age, mean (range): 30.4 (20 to 51) years Sex: 2 males, 5 females FEV1 % predicted, mean (SD): 64.3 (21.5)% Sweat chloride, mean (SD): 104.4 (14.1) mmol/L CFQ‐R score (respiratory domain), mean (SD): 75.4 (11.6) |
|
| Interventions |
Cohort 1 (n = 15): FDL169 400 mg 3x daily (n = 6) versus FDL169 600 mg 3x daily (n = 6) versus placebo (n = 3) Cohort 2 (n = 12): FDL169 800 mg 3x daily (n = 8) versus placebo (n = 4) |
|
| Outcomes |
|
|
| Funding source | Flatley Discovery Lab was the sponsor for this Phase 1b study | |
| Notes | We also obtained a poster from the author, which was presented at a conference. A study is planned for 2019 to evaluate the corrector FDL169 in combination with potentiator FDL176 in individuals with CF homozygous for F508del. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States that participants were randomised, but does not state the method by which they were randomised. |
| Allocation concealment (selection bias) | Unclear risk | Does not state methods of allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Does not state who was and was not blinded during the study. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Does not state how outcome assessors were blinded during the study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised are accounted for. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported in the results. |
| Other bias | Unclear risk | As the only information available was as part of a poster and a full detailed publication has not been published; it is difficult to say with any certainty whether there are other sources of bias in the process of this study. |
Keating 2018.
| Study characteristics | ||
| Methods | Phase 2, double‐blind RCT Parallel design Multicentre: 38 sites in the USA, the Netherlands, Belgium and Australia Duration: 4 weeks intervention period (12 weeks for those arms with a 4‐week run‐in and 4‐week washout) Randomisation of participants was stratified by FEV1 % predicted being less than or equal to 70% versus greater than 70%, except for the first 10 F508del/MF participants who were not stratified. |
|
| Participants | All participants were aged 18 years or older, with CFTR genotype of either F508del/MF or F508del/F508del. They must have had FEV1 % predicted between 40% and 90% at screening, as well as stable disease. 123 participants underwent randomisation and received at least 1 dose of the intervention. Participants with F508del/MF (n = 65) Elexacaftor 50 mg 1x daily plus tezacaftor 100 mg 1x daily plus ivacaftor 150 mg every 12 hours (n = 10) Age, mean (SD): 27.1 (7.4) years Sex: 4 males, 6 females FEV1 % predicted, mean (SD): 56.4 (14.6)% Sweat chloride, mean (SD): 103.1 (7.8) mmol/L CFQ‐R score (respiratory domain), mean (SD): 62.8 (21.9) Elexacaftor 100 mg 1x daily plus tezacaftor 100 mg 1x daily plus ivacaftor 150 mg every 12 hours (n = 22) Age, mean (SD): 31.8 (8.3) years Sex: 15 males, 7 females FEV1 % predicted, mean (SD): 60.0 (15.5)% Sweat chloride, mean (SD): 103.6 (12.2) mmol/L CFQ‐R score (respiratory domain), mean (SD): 65.9 (13.4) Elexacaftor 200 mg 1x daily plus tezacaftor 100 mg 1x daily plus ivacaftor 150 mg every 12 hours (n = 21) Age, mean (SD): 33.3 (10.3) years Sex: 10 males, 11 females FEV1 % predicted, mean (SD): 59.4 (18.0)% Sweat chloride, mean (SD): 103.9 (9.7) mmol/L CFQ‐R score (respiratory domain), mean (SD): 61.1 (17.5) Triple placebo (n = 12) Age, mean (SD): 29.7 (7.5) years Sex: 10 males, 2 females FEV1 % predicted, mean (SD): 59.0 (14.9)% Sweat chloride, mean (SD): 103.1 (8.2) mmol/L CFQ‐R score (respiratory domain), mean (SD): 57.4 (14.1) Participants with F508del/F508del (n = 28)* Elexacaftor 200 mg plus tezacaftor plus ivacaftor (n = 21) Age, mean (SD): 29.9 (7.6) years Sex: 12 males, 9 females FEV1 % predicted, mean (SD): 60.0 (15.1)% Sweat chloride, mean (SD): 92.7 (11.1) mmol/L CFQ‐R score (respiratory domain), mean (SD): 71.2 (17.3) Placebo plus tezacaftor plus ivacaftor (n = 8) Age, mean (SD): 27.9 (8.0) years Sex: 6 males, 2 females FEV1 % predicted, mean (SD): 62.8 (13.2)% Sweat chloride, mean (SD): 99.5 (9.0) mmol/L CFQ‐R score (respiratory domain), mean (SD): 73.0 (22.3) *Had a 4‐week run‐in of tezacaftor‐ivacaftor and a further 4‐week post‐invention washout period of tezacaftor‐ivacaftor Participants with F508del/MF (n = 29) Elexacaftor 200 mg plus tezacaftor plus VX‐561 (n = 21) Age, mean (SD): 30.6 (9.5) years Sex: 11 males, 10 females FEV1 % predicted, mean (SD): 60.6 (17.5)% Sweat chloride, mean (SD): 100.8 (15.4) mmol/L CFQ‐R score (respiratory domain), mean (SD): 63.8 (18.2) Triple placebo (n = 8) Age, mean (SD): 27.8 (5.2) years Sex: 3 males, 5 females FEV1 % predicted, mean (SD): 60.7 (14.0)% Sweat chloride, mean (SD): 96.4 (1.5) mmol/L CFQ‐R score (respiratory domain), mean (SD): 43.8 (21.9) |
|
| Interventions |
Arm 1: 1x daily elexacaftor 50 mg or elexacaftor 100 mg or elexacaftor 200 mg plus tezacaftor 100 mg plus 2x daily (every 12 hours) ivacaftor 150 mg versus triple placebo Arm 2: 4‐week run in period of 1x daily tezacaftor 100 mg plus ivacaftor 150 mg for all participants; then 4‐week intervention period of 1x daily elexacaftor 200 mg and tezacaftor 100 mg plus 2x daily (every 12 hours) ivacaftor 150 mg versus matched placebo; then a 4‐week washout period of 1x daily tezacaftor 100 mg plus ivacaftor 150 mg for all participants Arm 3: 1x daily elexacaftor 200 mg and tezacaftor 100 mg and VX‐561 150 mg once daily versus triple placebo |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals, who received funding from the CF Foundation to develop elexacaftor. The NIH gave a grant to the University of Alabama at Birmingham. | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation code made by Vertex Biostatistics or a "qualified randomisation vendor". Randomisation stratified by FEV1 % predicted (less than or equal to 70% versus greater than 70%). |
| Allocation concealment (selection bias) | Low risk | Use of interactive web response system for allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants, site personnel and Vertex study team related to the study were blinded. A clear statement on when unblinding is necessary or permitted is provided in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | All authors were only allowed access to study data after they were unblinded. No mention is made of other outcome assessors (e.g. clinicians who were not authors, but were involved in seeing participants and measuring outcomes of interest) and whether there was a possibility of them knowing the allocated intervention. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised are accounted for. |
| Selective reporting (reporting bias) | Unclear risk | States in methods that 12‐lead ECG and vital signs would be measured; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Low risk | Different groups of participants are balanced in baseline characteristics; no significant difference between them. Detail in paper and its supplement does not cause any concern about other sources of bias not previously mentioned. |
McCarty 2002.
| Study characteristics | ||
| Methods | Phase 1, placebo‐controlled RCT Parallel design Multicentre, conducted at 4 sites in North America Duration: single‐dose assessment. Participants were monitored for 2 days and followed up at 1 week. |
|
| Participants | Mutation: all 37 participants were homozygous for the F508del mutation and were described as having mild CF. Age: 18 years or over; age range 18 to 38 years. Sex: 21 males and 16 females Lung function: participants were eligible if they had a baseline FEV1 ≥ 60% predicted and had not endured pulmonary colonisation by a drug resistant organism within 12 months of screening. |
|
| Interventions |
Intervention 1: placebo Intervention 2: CPX in the following escalating doses: 1 mg CPX 3 mg CPX 10 mg CPX 30 mg CPX 100 mg CPX 300 mg CPX 1000 mg CPX |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | SciClone Pharmaceuticals, and grants from the NIH | |
| Notes | *Denotes outcomes relevant to this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | There was insufficient information on how participant or study personnel blinding was maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information on how outcome assessor blinding was maintained. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No report of withdrawals and all originally randomised participants were included in the analysis. |
| Selective reporting (reporting bias) | Low risk | Protocol not available and outcomes not reported in the ongoing online database (www.clinicaltrials.gov). Reported results corresponded to outcomes listed in methods section. |
| Other bias | Unclear risk | Unclear whether baseline characteristics were well‐matched. |
McKone 2021.
| Study characteristics | ||
| Methods | Double‐blind, Phase 3 RCT Parallel design Multicentre study, conducted across the USA, Australia, Austria, Belgium, Canada, France, Germany, Ireland, Italy and the UK Duration of treatment: 4‐week ivacaftor run‐in period, 8‐week active comparator treatment period, 4‐week safety follow‐up period |
|
| Participants |
Inclusion criteria Age: 12 years and older Genotype: heterozygous for F508del‐CFTR mutation and a second CFTR allele with a gating defect that is clinically demonstrated to be ivacaftor‐responsive Lung function: FEV1 ≥ 40% and ≤ 90% of predicted Baseline characteristics 150 participants Age, mean (SD): 32.4 (12.2) years Sex: 66 females, 84 males |
|
| Interventions | Run‐in period: ivacaftor 150 mg tablet orally every 12 hours for 4 weeks VX‐661 + ivacaftor (active comparator period): VX‐661 100 mg and ivacaftor 150 mg fixed‐dose combination tablet orally once daily in the morning and ivacaftor 150 mg tablet orally once daily in the evening for 8 weeks Control (active comparator period): ivacaftor monotherapy 150 mg tablet orally every 12 hours as monotherapy for 8 weeks |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation codes were produced by the designated vendor. No Vertex biostatistician was unblinded to the actual randomisation list before the database lock. The randomisation procedures were provided in the protocol. Upon approval of the dummy randomisation, the external designated vendor generated the final randomisation list and provided it directly to the Interactive Web Response System vendor. |
| Allocation concealment (selection bias) | Low risk | An interactive web response system was used to assign participants to study treatment using a list of randomisation codes generated by a designated vendor. The study biostatistician was involved in developing the randomisation specifications and reviewing the dummy randomisation lists, and remained blinded to the final live unblinded randomisation lists and the actual treatment assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All participants, site personnel, including the investigator, the site monitor and the study team, were blinded. Protocol sets out conditions when blinding could/should be broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind study. All participants, site personnel, including the investigator, the site monitor and the study team, were blinded. Protocol sets out conditions when blinding could/should be broken. During the conduct of the study, the Vertex study team did not have access to the post‐dose spirometry data during the active comparator treatment period. During the conduct of the study, the Vertex study team did not have access to the post‐dose sweat chloride data; dummy data were used to develop statistical programs. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Out of 76 in the intervention group, only 74 were analysed for change in sweat chloride and 75 were analysed for PK parameters. Full set analysis was completed for 74 participants in the ivacaftor monotherapy group (75 started this regimen), and only 72 were analysed for absolute and relative change in FEV1 % predicted, 73 were analysed for change in CFQ‐R, 70 for change in sweat chloride and 68 for PK parameters. 143 of the 156 participants in the ivacaftor run‐in period were analysed for PK parameters. Within the ivacaftor monotherapy group, 6 participants ended treatment early and reasons were given for each, however no further detail than 'other' was given for 2 participants |
| Selective reporting (reporting bias) | Unclear risk | States in methods that 12‐lead ECG and vital signs would be measured; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Low risk | Different groups of participants are balanced in baseline characteristics. The paper states how Vertex Pharmaceuticals Incorporated participated in the study and publication. |
Middleton 2019.
| Study characteristics | ||
| Methods | Phase 3, double‐blind RCT Parallel design Multicentre study, conducted at 115 sites in 13 countries across North America, Europe and Australia Duration: 28‐day screening period followed by a 24‐week intervention period followed by a further 28‐day safety follow‐up, after which participants were invited to a 96‐week open‐label extension study in which all participants received active treatment (VX17‐445‐105; ClinicalTrials.gov number, NCT03525574) |
|
| Participants | Eligible participants were 12 years of age or older with CF and 1 copy of F508del and 1 copy of a MF allele, FEV1 % predicted 40% to 90% and in 'stable condition' as judged by study investigators. 405 participants randomised; 403 received at least 1 dose of intended treatment and were included in the analyses. Intervention group (n = 202) Age, mean (SD): 25.6 (9.7) years Age distribution: ≥ 12 to < 18 years: 56 (28.0%); ≥ 18 years: 144 (72.0%) Sex: 96 females (48.0%) FEV1 % predicted, mean (SD): 61.6 (15.0) % predicted FEV1 % predicted, distribution: < 40%: 18 (9.0%); ≥ 40% to < 70%: 114 (57.0%); ≥ 70% to ≤ 90%: 66 (33.0%); > 90%: 2 (1.0%) BMI, mean (SD): 21.49 (3.07) kg/m² Sweat chloride concentration, mean (SD): 102.3 (11.9) mmol/L CFQ‐R respiratory domain score, mean (SD): 68.3 (16.9) Control group (n = 201) Age, mean (SD): 26.8 (11.3) years Age distribution: ≥ 12 to < 18 years: 60 (29.6%); ≥ 18 years: 143 (70.4%) Sex: 98 females (48.3%) FEV1 % predicted, mean (SD): 61.3 (15.5) % predicted FEV1 % predicted, distribution: < 40%: 16 (7.9%); ≥ 40% to < 70%: 120 (59.1%); ≥ 70% to ≤ 90%: 62 (30.5%); > 90%: 5 (2.5%) BMI, mean (SD): 21.31 (3.14) kg/m² Sweat chloride concentration, mean (SD): 102.9 (9.8) mmol/L CFQ‐R respiratory domain score, mean (SD): 70.0 (17.8) |
|
| Interventions |
Intervention (n = 202): elexacaftor 200 mg once daily plus tezacaftor 100 mg once daily plus ivacaftor 150 mg every 12 hours Control (n = 201): triple‐matched placebo |
|
| Outcomes | Outcomes measured at weeks 4, 8, 12, 16 and 24 Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals supported the study and were also involved in writing the study; however, the first 2 authors and last 2 authors wrote the first draft of the manuscript and made final decisions regarding the content of the submitted manuscript. | |
| Notes | Eudract No. 2018‐000183‐28 403 participants underwent randomisation and received at least 1 dose, therefore included in analyses; 400 participants went on to the open‐label extension |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment in a 1:1 ratio stratified by FEV1 % predicted, age at screening and sex. The protocol states that the randomisation code list would be produced by Vertex Biometrics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | Interactive web response system used to assign participants to treatment. The pharmacist or designated study site staff maintained information regarding the dates and amounts of (1) study drug received; (2) study drug dispensed to the participants; and (3) study drug returned by the participants. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study drug and matched placebo administered orally and all participants received the same number of tablets each day to maintain the blind. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team), and members of the Vertex study team were blinded to the treatment codes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All site personnel (including the investigator, the site monitor and the study team), and members of the Vertex study team were blinded to the treatment codes. The interim analysis was performed by an external independent biostatistician who was not involved in the study, and the results were reviewed by the independent data monitoring committee. After the interim analysis, the study continued to completion and remained double‐blinded through week 24, apart from the planned unblinding of a limited Vertex team that was tasked with preparing regulatory submissions. To protect study integrity, members of the limited Vertex unblinded team were not involved in and did not influence the ongoing conduct of the study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 403/405 people who were randomised received at least 1 dose. They were all included in all applicable analyses. 3 discontinued in the intervention group (1 became pregnant and 2 due to AEs) after receiving at least 1 dose. All 400 remaining participants were enrolled into an open‐label extension study. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol are reported in either the primary publication or supplement. |
| Other bias | Unclear risk | None noted; well‐matched baseline characteristics. However, as it is stated that Vertex funded the study, it is unclear as to the extent to which they were involved in designing, writing up and publishing the report. |
Munck 2020.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled RCT Duration: 12 weeks Multicentre study, conducted across the USA, Australia, Austria, Canada, France, Israel, Spain Parallel design |
|
| Participants |
Inclusion criteria Aged over 12 years Diagnosed with CF with genotype F508del/MF FEV1 % predicted ≥ 40 % and ≤ 90% of predicted normal for age, sex and height at screening visit Baseline characteristics 168 participants Age, mean (SD): 26.1 (9.2) years Sex: 81 females and 87 males |
|
| Interventions |
Intervention: VX‐661 100 mg plus ivacaftor 150 mg fixed‐dose combination tablet administered orally in the morning and ivacaftor 150 mg film‐coated tablet administered orally in the evening up to Week 12 Control: placebo matched to VX‐661 plus ivacaftor 150 mg fixed‐dose combination tablet administered orally in the morning and placebo matched to ivacaftor 150 mg film‐coated tablet administered orally in the evening up to Week 12 |
|
| Outcomes |
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | There was insufficient information on how participant or study personnel blinding was maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information on how outcome assessor blinding was maintained. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Out of 83 in the intervention group, only 82 were analysed for absolute and relative change in FEV1 % predicted, change in BMI and absolute change in body weight. 79 were analysed for change in sweat chloride, and only 82 analysed for PK parameters (with sometimes fewer participants for individual measurements). Out of 85 in the placebo group, only 84 were analysed for change in sweat chloride. No further explanations were given for missing participants. Conflicting information between the clinicaltrials.gov website and the supplementary figures available in Munck's published paper. According to clinicaltrials.gov, 85 participants completed the placebo group treatment; however, according to the supplementary figures, only 84 participants completed the placebo treatment regimen. |
| Selective reporting (reporting bias) | Unclear risk | Unable to access trial protocol. Munck's paper states that it would measure ECGs and vital signs; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. Also, clinicaltrials.gov states that "Time‐to‐first pulmonary exacerbation was planned to be estimated using Kaplan‐Meier (KM) estimates. However, due to less than 50% of events, time‐to‐first event data was not estimated. Instead, number of participants with at least one pulmonary exacerbation event were collected and are reported"; however, there is a Kaplan‐Meier plot of time to first pulmonary exacerbation event in the supplementary figures of Munck's paper. |
| Other bias | Unclear risk | Well‐matched baseline characteristics. However, as it is stated that Vertex funded the study, it is unclear as to the extent to which they were involved in designing, writing up and publishing the report. |
NCT02070744.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled, Phase 2 RCT Parallel design Multicentre: conducted across 23 sites in the USA Sample size: 40 participants randomised (but 39 were analysed because 1 participant was randomised, but not treated) Duration: 12 weeks of treatment |
|
| Participants |
Inclusion criteria Age: 18 years or older Diagnosed with CF and homozygous for the F508del mutation Lung function: FEV1 ≥ 40% predicted and ≤ 90% predicted normal for age, sex and height Participants must have stable CF disease as judged by the investigator Baseline characteristics Age, mean (SD): 28.8 (8.3) years Sex: 14 females, 25 males |
|
| Interventions |
Intervention arm 1: VX‐661 50 mg tablet every 12 hours + ivacaftor 150 mg tablet every 12 hours for 12 weeks Control arm 1: VX‐661 matched placebo tablet every 12 hours + ivacaftor matched placebo tablet every 12 hours for 12 weeks Intervention arm 2: VX‐661 100 mg (2x 50 mg tablets) once daily + ivacaftor 150 mg table every 12 hours for 12 weeks Control arm 2: VX ‐661 (2x placebo‐matched tablets) once daily + ivacaftor matched placebo tablet every 12 hours for 12 weeks |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated ‐ unable to access study protocol. |
| Allocation concealment (selection bias) | Unclear risk | Not stated ‐ unable to access study protocol. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | States on clinicaltrials.gov that masking is triple (participant, care provider, investigator), although unable to access trial protocol to find exact method of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | States on clinicaltrials.gov that masking is triple (participant, care provider, investigator), although unable to access trial protocol to find exact method of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | One participant did not complete an intervention group, and the reason given for this was 'randomized, but not treated', with no other information given. There were only 36 participants analysed out of 39 participants for absolute change in sweat chloride. There were also only 20 out of 21 participants analysed for area under the concentration versus time curve from time 0 to 24 hours of VX‐661 and area under the concentration versus time curve from time 0 to 12 hours of ivacaftor. No reasons are given for missing participants. |
| Selective reporting (reporting bias) | Unclear risk | Unable to access study protocol to find initial outcomes set. |
| Other bias | Unclear risk | Characteristics of participants generally well balanced (however, there were 14 females and 25 males ‐ unable to determine if this imbalance impacted results). Unable to determine the extent to which the sponsor was involved in designing and conducting the study. |
NCT02508207.
| Study characteristics | ||
| Methods | Phase 2, placebo‐controlled RCT Parallel design Multicentre study, conducted across 7 sites in the USA Duration of treatment: 29 days. Safety follow‐up visit occurred 28 ± 7 days after the last dose. |
|
| Participants |
Inclusion criteria Diagnosed with CF and homozygous for F508del genotype Aged 18 years and older FEV1 % predicted ≥ 40% and ≤ 90% of predicted normal for age, sex and height at screening visit Baseline characteristics 34 participants randomised Age, mean (SD): 33.4 (9.5) years Sex: 19 females and 15 males |
|
| Interventions |
Intervention: participants received 100 mg tezacaftor/150 mg ivacaftor fixed‐dose combination tablet orally once daily in the morning followed by 150 mg ivacaftor tablet orally once daily in the evening for 29 days Control: participants received placebo matched to tezacaftor‐ivacaftor fixed‐dose combination tablet orally once daily in the morning followed by placebo matched to ivacaftor tablet orally once daily in the evening for 29 days |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment using a list of randomisation codes generated by a designated vendor. The only Vertex personnel involved in developing the randomisation specifications and reviewing the dummy randomisation code list was the study biostatistician who was blinded to the final randomisation code list and the actual treatment assignments. |
| Allocation concealment (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment using a list of randomisation codes generated by a designated vendor. The only Vertex personnel involved in developing the randomisation specifications and reviewing the dummy randomisation code list was the study biostatistician who was blinded to the final randomisation code list and the actual treatment assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. Blinding of treatment codes and applicable study data was maintained until the database was locked for the final analysis. The participants and all site personnel, including the investigator, the site monitor and the study team, were blinded with the exceptions to this clearly stated in the study protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. Blinding of treatment codes and applicable study data maintained until the database was locked for the final analysis. The participants and all site personnel, including the investigator, the site monitor and the study team, were blinded with the exceptions to this clearly stated in the study protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 23/27 participants in the intervention group were analysed for absolute change from baseline in small‐bowel area under the curve over 1‐minute mean pH increments at day 29 (and only 6/7 participants in the placebo group were analysed for this outcome) and absolute change from baseline in sweat chloride at day 29. Due to the lack of a published paper, it was unclear why there were fewer participants analysed for these outcomes. |
| Selective reporting (reporting bias) | Unclear risk | Methods stated that 12‐lead ECGs and vital signs would be measured; however, exact values for the parameters used to assess safety and tolerability were not available in the results, whether unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants generally well‐balanced. Unable to determine the extent to which the sponsor was involved in designing and conducting the study. |
NCT02730208.
| Study characteristics | ||
| Methods | Placebo‐controlled, double‐blind, Phase 2 RCT Parallel design Multicentre study, conducted across 7 sites in Australia 72‐week treatment duration Safety follow‐up 28 days after last dose |
|
| Participants |
Inclusion criteria Aged 12 years and older Homozygous for F508del CFTR mutation Stable CF disease as judged by the investigator FEV1 % predicted ≥ 70% of predicted normal for age, sex and height during screening Baseline characteristics 41 people randomised Age, mean (SD): 20.2 (8.4) years Sex: 21 females and 20 males |
|
| Interventions |
Intervention: participants received tezacaftor 100 mg/ivacaftor 150 mg fixed‐dose combination tablet orally once daily in the morning and ivacaftor 150 mg tablet orally once daily in the evening for 72 weeks Control: participants received placebo matched to tezacaftor/ivacaftor fixed‐dose combination tablet orally once daily in the morning and placebo matched to ivacaftor tablet orally once daily in the evening for 72 weeks |
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician who was not a member of the study execution team. |
| Allocation concealment (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician who was not a member of the study execution team. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. The participants and all site personnel, including the investigator, the site monitor and the study team, were blinded with the exceptions to this clearly stated in the study protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. The participants and all site personnel, including the investigator, the site monitor and the study team, were blinded with the exceptions to this clearly stated in the study protocol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants were analysed. |
| Selective reporting (reporting bias) | Unclear risk | Methods state that ECGs and vital signs would be measured; however, exact values for the parameters used to assess safety and tolerability were not available in the results, whether unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants were generally well‐balanced. Unable to determine the extent to which the sponsor was involved in designing and conducting the study. |
NCT02951182.
| Study characteristics | ||
| Methods | Phase 2 RCT Parallel design Multicentre study, conducted across the USA (22 sites), Australia (3 sites), Austria (1 site), Belgium (2 sites), Canada (2 sites), Denmark (1 site), Germany (3 sites), Italy (1 site), Spain (2 sites) and UK (3 sites) Duration of treatment: for part 1, treatment lasted 4 weeks. For part 2, there was a 4‐week run‐in period, 4‐week treatment period and 4‐week washout period. Outpatient safety follow‐up visit occurring approximately 28 days after the last dose of study drug for participants who complete study drug dosing and for participants who prematurely discontinued study drug dosing (certain exceptions for part 2 participants) |
|
| Participants |
Inclusion criteria Aged 12 years and older Participants must have an FEV1 ≥ 40% and ≤ 90% of predicted normal for age, sex and height at screening Genotype: F508del/MF with a mutation known or predicted not to be responsive to tezacaftor and/or ivacaftor, or F508del/F508del Baseline characteristics 73 participants randomised Age, range: 18 to 65 years Sex: 11 females and 62 males Genotype: participants in Part 1 were F508del/MF Genotype: participants in Part 2 were F508del/F508del |
|
| Interventions |
Control Part 1: placebo ‐ cohort 1A and 1B combined: placebo matched to VX‐440/tezacaftor/ivacaftor for 4 weeks Intervention Part 1 Cohort 1A: 200 mg VX‐440 every 12 hours plus 100 mg tezacaftor once daily plus 150 mg ivacaftor every 12 hours for 4 weeks Intervention Part 1 Cohort 1B (low dose): 200 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 150 mg ivacaftor every 12 hours for 4 weeks Intervention Part 1 Cohort 1B (high dose): 600 mg VX‐440 every 12 hours plus 50 mg tezacaftor every 12 hours plus 300 mg ivacaftor every 12 hours for 4 weeks Control Part 2 (tezacaftor/ivacaftor): following a 4‐week run‐in period on 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily, participants received placebo matched to VX‐440 and 50 mg tezacaftor twice daily plus 300 mg ivacaftor twice daily for 4 weeks in treatment period and 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for 4 weeks in washout period Intervention Part 2 (triple combination): following a 4‐week run‐in period on 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily, participants received 600 mg VX‐440 twice daily plus 50 mg tezacaftor once daily plus 300 mg ivacaftor twice daily for 4 weeks in treatment period and 100 mg tezacaftor once daily plus 150 mg ivacaftor twice daily for 4 weeks in washout period |
|
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | An interactive web response system was used to assign participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, placebo‐matched RCT. All participants, site personnel (including the investigator, the site monitor and the study team) and the Vertex study team were blinded to the treatment codes with the exceptions to this outlined in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind, placebo‐matched RCT. All participants, site personnel (including the investigator, the site monitor and the study team) and the Vertex study team were blinded to the treatment codes with the exceptions to this outlined in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not all of the participants who were randomised were analysed for Ctrough data, and there was no explanation as to why these participants were not included in the analysis. However, this was not an outcome for this review, so risk of bias is not increased. |
| Selective reporting (reporting bias) | Unclear risk | Methods stated that ECGs and vital signs would be measured to assess safety and tolerability; however, exact values for these measurements could not be found in the results, whether unremarkable or not. |
| Other bias | Unclear risk | There were 11 females and 62 males, and it was unclear whether this imbalance affected the results. The extent to which the sponsor was involved in designing and reporting the results from the study is also unclear. |
NCT02951195.
| Study characteristics | ||
| Methods | Phase 2 RCT Parallel design Multicentre study, conducted across 16 sites in the USA For part 1, the total study duration is approximately 10 weeks; participants received study drug for approximately 2 weeks For part 2, the total study duration is approximately 16 weeks for participants in Cohort 2A and 18 weeks for participants in Cohort 2B. Study drug administered for approximately 8 weeks in Cohort 2A and approximately 10 weeks in Cohort 2B Safety follow‐up 28 ± 7 days after last dose |
|
| Participants |
Inclusion criteria Aged 18 years and older FEV1 ≥ 40% and ≤ 90% of predicted normal for age, sex and height at the screening visit Genotype heterozygous F508del/MF mutation known or predicted not to respond to tezacaftor and/or ivacaftor, or F508del/F508del Baseline characteristics 76 participants randomised Age, range: 18 to 65 years Sex: 39 females and 37 males Cohorts 1A, 1B, 1C: heterozygous F508del/MF Cohorts 2A, 2B: F508del/F508del |
|
| Interventions |
Part 1 Placebo: participants received triple placebo matched to VX‐152/tezacaftor/ivacaftor triple combination for 2 weeks Part 1 Cohort 1A: participants received triple combination of VX‐152 100 mg every 12 hours/tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks Part 1 Cohort 1B: participants received triple combination of VX‐152 200 mg every 12 hours/tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks Part 1 Cohort 1C: participants received triple combination of VX‐152 300 mg every 12 hours/tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks Part 2 Cohort 2A Placebo: following a run‐in period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, participants received triple placebo matched to VX‐152 and tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks, then a washout period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks. Part 2 Cohort 2A: following a run‐in period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, participants received triple combination of VX‐152 200 mg every 12 hours/tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks, then a washout period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks Part 2 Cohort 2B Placebo: following a run‐in period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, participants received placebo matched to VX‐152 and tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, then a washout period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks Part 2 Cohort 2B: following a run‐in period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, participants received the triple combination of VX‐152 300 mg every 12 hours/tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 4 weeks, then a washout period on tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 2 weeks |
|
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biostatistics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biostatistics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind RCT. All participants, site personnel (including the investigator, the site monitor and the study team) and the Vertex study team were blinded to the treatment codes with the exceptions outlined in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind RCT. All participants, site personnel (including the investigator, the site monitor, and the study team) and the Vertex study team were blinded to the treatment codes with the exceptions outlined in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Appears to be missing data for Ctrough values, with no explanation offered as to why certain participants have been missed. However, this is not an outcome for this review, and so this does not increase the risk of bias. 4/46 participants enrolled in Part 2 discontinued during the run‐in period and were not randomised in the treatment period, without any further explanation given. |
| Selective reporting (reporting bias) | Unclear risk | Protocol stated that safety and tolerability measurements would be based on ECGs and vital signs, and there are no exact values for these in the results, whether they were unremarkable or not. |
| Other bias | High risk | There are 4 participants with FEV1 < 40% who appear to have been analysed, despite the inclusion criteria of the study stating that participants must have an FEV1 ≥ 40% and ≤ 90% of predicted normal for age, sex and height at the screening visit. The extent to which the sponsor was involved in this study is unclear due to the lack of a published paper. |
NCT03447249.
| Study characteristics | ||
| Methods | RCT, double‐blind, placebo‐controlled Parallel design Duration: 24 weeks |
|
| Participants |
Inclusion criteria Age: 12 years and older Genotype: F508del/MF Lung function: FEV1 40% to 90% predicted Baseline characteristics 385 participants randomised Age, mean (SD): 26.9 (9.9) years Sex: 214 males, 168 females FEV1 % predicted, mean (SD): 60.6 (14.9) % predicted |
|
| Interventions |
Intervention: VX‐659 240 mg/tezacaftor 100 mg/ivacaftor 150 mg as fixed‐dose combination tablets in the morning and ivacaftor 150 mg as mono tablet in the evening Control: matched placebo morning and evening |
|
| Outcomes |
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised (1:1) to the triple combination VX‐659/tezacaftor/ivacaftor arm or to the triple placebo arm. Randomisation was stratified by FEV1 % predicted determined during the screening period, age at the screening visit and sex. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | Participants were randomised (1:1) to the triple combination VX‐659/tezacaftor/ivacaftor arm or to the triple placebo arm. Randomisation was stratified by FEV1 % predicted determined during the screening period, age at the screening visit and sex. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who could be unblinded were listed in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who could be unblinded were listed in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 3 participants enrolled in the study were not dosed in the triple combination group (unclear why). 5 participants in the placebo group did not complete the regimen, and the reason for 3 of these participants is listed as 'other', with no further detail. Unclear as to why 'safety set' for the placebo and treatment group were 189 and 193 participants respectively, though 190 and 192 started each regimen. It is unclear as to the number of participants who would be evaluable for absolute change in BMI z score for participants 20 years of age and under at baseline (from baseline at week 24). The number of participants analysed for PK outcomes for each measurement was smaller than the overall number of participants in the treatment group, with no explanation as to why this was the case. |
| Selective reporting (reporting bias) | Unclear risk | States that it would measure ECG and vital signs; although these may have been measured, they are not stated in the results regardless of whether they were unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants were generally well‐balanced, although the extent to which the sponsor was involved in designing and reporting the study results is unclear. |
NCT03460990.
| Study characteristics | ||
| Methods | RCT, double‐blind, placebo‐controlled Parallel design Duration: 8 weeks |
|
| Participants |
Inclusion criteria Aged 12 years and older Diagnosed with CF and homozygous for F508del FEV1 40% to 90% predicted Baseline characteristics 116 participants randomised Age, mean (SD): 27.2 (9.3) years Sex: 59 females, 52 males FEV1 % predicted, mean (SD): 62.4 (14.8) % predicted |
|
| Interventions | Run‐in period of 4 weeks with tezacaftor/ivacaftor, then: Intervention 1: VX‐659 240 mg/tezacaftor 100 mg/ivacaftor 150 mg as fixed‐dose combination tablets in the morning and ivacaftor 150 mg as mono tablet in the evening Control: placebo/tezacaftor 100 mg/ivacaftor 150 mg as fixed‐dose combination tablets in the morning and ivacaftor 150 mg as mono tablet in the evening |
|
| Outcomes |
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | A Study of VX‐659 Combination Therapy in CF Subjects Homozygous for F508del (F/F) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised (1:1) to the triple combination VX‐659/tezacaftor/ivacaftor arm or to the tezacaftor/ivacaftor arm. Randomisation stratified by FEV1 % predicted determined during the tezacaftor/ivacaftor run‐in period and age at the screening visit. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | Participants randomised (1:1) to the triple combination VX‐659/tezacaftor/ivacaftor arm or to the tezacaftor/ivacaftor arm. Randomisation stratified by FEV1 % predicted determined during the tezacaftor/ivacaftor run‐in period and age at the screening visit. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who may be unblinded were listed in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind study. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who may be unblinded were listed in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | For certain Ctrough measurements, there appear to be fewer participants analysed at specified time points than the overall number of participants who were randomised, without any further explanation as to why this was the case. Additionally, 5 participants were included in the run‐in period but were not dosed in the triple combination treatment period, without any further information as to why this was the case. |
| Selective reporting (reporting bias) | Unclear risk | Protocol states that safety and tolerability assessments would be based on ECGs and vital signs. There are no exact values recorded for these in the results, whether they were unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants are generally well‐balanced, although it is unclear (due to the lack of a published paper) as to the extent to which the sponsor was involved in designing and reporting the study. |
PROGRESS 2017.
| Study characteristics | ||
| Methods | Phase 3 RCT Double‐blind rollover study (participants on active treatment continued their treatment, participants on placebo were randomised to 1 of the 2 active interventions) Parallel design Multicentre: 191 sites in 15 countries across North America, Australia and Europe Duration: 96 weeks |
|
| Participants |
Inclusion criteria Age: 12 years and older Sex: both males and females Confirmed diagnosis of CF Genotype: homozygous or heterozygous for the F508del mutation Participants have previously participated in TRAFFIC or TRANSPORT and completed 24 weeks of treatment Baseline characteristics 1030 participants enrolled in total. Across all groups, there were 505 females and 524 males. Breakdown of characteristics not given for interventions 1 and 2 (participants taking 600 mg lumacaftor once daily + 250 mg ivacaftor every 12 hours) Intervention 3: 400 mg lumacaftor every 12 hours + 250 mg ivacaftor every 12 hours (continued treatment) 340 participants Age, mean (SD): 25.1 (9.3) years Sex: 164 females, 176 males FEV1 % predicted, mean (SD): 60.4 (14.2) % predicted BMI, mean (SD): 21.4 (2.9) Pseudomonas positive:261 (77%) Intervention 4: placebo transitioned to lumacaftor 400 mg every 12 hours/ivacaftor 250 every 12 hours 176 participants Age, mean (SD): 24.9 (10.1) years Sex: 86 females, 90 males FEV1 % predicted, mean (SD): 60.2 (13.8) % predicted BMI, mean (SD): 20.9 (2.8) Pseudomonas positive: 126 (72%) |
|
| Interventions |
Intervention 1: 600 mg lumacaftor once daily + 250 mg ivacaftor every 12 hours (continued treatment) Intervention 2: 600 mg lumacaftor once daily + 250 mg ivacaftor every 12 hours (rolled over from placebo) Intervention 3: 400 mg lumacaftor every 12 hours + 250 mg ivacaftor every 12 hours (continued treatment) Intervention 4: 400 mg lumacaftor every 12 hours + 250 mg ivacaftor every 12 hours (rolled over from placebo) |
|
| Outcomes |
Primary outcome measure Treatment cohorts: safety of long‐term treatment based on AEs, clinical laboratory values (serum chemistry, haematology, coagulation studies, and urinalysis), standard digital ECGs, vital signs and pulse oximetry at 100 weeks Secondary outcome measures
|
|
| Funding source | Sponsored by Vertex Pharmaceuticals Inc. | |
| Notes | Long‐term extension of the TRAFFIC and TRANSPORT studies in which participants receiving an active treatment continued with this treatment and those receiving placebo were randomised to receive 1 of the 2 active treatments from the TRAFFIC and TRANSPORT studies. Additional analyses were conducted comparing participants receiving 400 mg lumacaftor every 12 hours + 250 mg ivacaftor every 12 hours to an observational registry cohort of matched controls. These analyses are not reported in this review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Only the placebo groups from the previous studies were randomised. Participants were randomly assigned (in a 1:1:1 ratio) to 1 of 3 study groups; the randomisation was established by an interactive web response system. Randomisation was stratified according to age (< 18 years versus ≥ 18 years), sex and pulmonary function (% predicted FEV1 at screening, < 70% versus ≥ 70%). |
| Allocation concealment (selection bias) | Low risk | Only the placebo groups from the previous studies were randomised. The randomisation was established by an interactive web response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. Interventions were matched in appearance and packaging. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all participants randomised who received at least 1 dose of study medication were included in analysis. Missing data were investigated in sensitivity analyses. |
| Selective reporting (reporting bias) | Low risk | All listed outcomes are reported in the results. |
| Other bias | Low risk | A 'rate of change' analysis showed that baseline characteristics across the groups were well‐balanced. |
Ratjen 2017.
| Study characteristics | ||
| Methods | Phase 3, placebo‐controlled RCT Parallel design Multicentre: 54 sites in 9 countries (USA, Australia, Belgium, Canada, Denmark, France, Germany, Sweden and the UK) Duration: 24 weeks |
|
| Participants |
Inclusion criteria Aged 6 to 11 years. Lung function: participants must have a FEV1 (% predicted) of 70% or more, and LCI2.5 of 7.5 or more Baseline characteristics 206 participants randomised (ivacaftor n = 104; placebo n = 102); 1 from each arm withdrew before the first dose leaving 204 participants in the analysis Genotype: all participants were homozygous for the F508del mutation Age, mean (SD): 8.8 (1.6) years Sex: 83 males and 121 females |
|
| Interventions |
Intervention: lumacaftor 200 mg every 12 hours in combination with ivacaftor 250 mg every 12 hours Control: matched placebo |
|
| Outcomes |
Primary outcome
Mean absolute change in LCI2.5 from baseline at all study visits up to and including week 24* Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals | |
| Notes | *Denotes outcomes relevant to this review Analyses were performed as the absolute change from baseline (including all measurements up to and including week 24, both on‐treatment measurements and measurements after treatment discontinuation) ‐ was based on a MMRM, adjusted for the baseline measurement of the outcome, baseline weight (less than 25 kg versus 25 kg or over and baseline FEV1 (% predicted) (less than 90% compared to 90% or more), with treatment‐by‐visit interaction as fixed‐effect, participant as a random‐effects. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Blocked randomisation was performed via an interactive web response system, stratified by baseline weight and FEV1 (% predicted). |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed centrally via the interactive web response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding was achieved by using placebo tablets visually identical to the test product. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not stated whether outcome assessment was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates were reported and an ITT approach was taken to analysis, with all randomised participants who received at least 1 dose of the study drug included in analysis (1 participant in each group was randomised but did not receive the study drug). |
| Selective reporting (reporting bias) | High risk | Several outcomes are listed in the methods (e.g. LCI5.0, time‐to‐first pulmonary exacerbation, absolute change in TSQM domains) but are not presented in the results. |
| Other bias | Low risk | Baseline characteristics were similar across the 2 groups. |
Rubenstein 1998.
| Study characteristics | ||
| Methods | A pilot, placebo‐controlled RCT Parallel design Single centre Duration: 1 week |
|
| Participants |
Inclusion criteria Aged 14 years or older Baseline characteristics 18 participants randomised (9 participants in each group) Age, mean (SD): intervention group: 22.3 (5.9) years; placebo group 24.8 (4.9) years Sex: intervention group 5 males and 4 females; placebo group 4 males and 5 females Genotype: homozygous for F508del mutation FVC % predicted, mean (SD): intervention group 73.4 (20.3) % predicted; placebo group: 65.5 (18.6) % predicted FEV1 % predicted, mean (SD): intervention group 57.8 (27.2) % predicted; placebo group 47.5 (22.1)% predicted |
|
| Interventions |
Intervention: sodium 4‐phenylbutyrate (also known as Buphenyl or 4PBA) 19 g, orally administered, in 3 daily doses of 6 g, 6 g and 7 g Control: placebo |
|
| Outcomes |
|
|
| Funding source | NIH and Cystic Fibrosis Foundation | |
| Notes | *Denotes outcomes relevant to this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study article states that "randomization and blinding were performed by the Johns Hopkins Hospital Investigational Drug Pharmacy" but exact method of randomisation has not been described. |
| Allocation concealment (selection bias) | Unclear risk | Study article states that "randomization and blinding were performed by the Johns Hopkins Hospital Investigational Drug Pharmacy" but exact method of allocation concealment has not been described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind. Study article states that "randomization and blinding were performed by the Johns Hopkins Hospital Investigational Drug Pharmacy" but exact method of blinding has not been described. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind. Study article states that "randomization and blinding were performed by the Johns Hopkins Hospital Investigational Drug Pharmacy" but exact method of blinding has not been described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data were analysed with 9 participants in each group, equivalent to the number originally randomised. |
| Selective reporting (reporting bias) | Low risk | Protocol not available and outcomes not presented in the ongoing trials database (www.clinicaltrials.gov/). Outcomes reported in the 'methods' were reported in the 'results' so selective reporting bias is low. |
| Other bias | Low risk | "... baseline characteristics between the groups were similar with respect to age, gender, pancreatic sufficiency, and baseline pulmonary function." |
Schwarz 2021.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled, 3‐part Phase 3b RCT Parallel design Multicentre: conducted across the USA (41 sites), France (7 sites) and Germany (5 sites) Duration: 56 days: screening period Day ‐28 through Day ‐1, treatment period Day 1 through Day 56 ± 5 days, safety follow‐up contact (28 days ± 7 days after the last dose of study drug) |
|
| Participants |
Inclusion criteria Aged 12 years and older Genotype: homozygous for F508del mutation Lung function: FEV1 ≥ 25% and ≤ 90% of predicted Baseline characteristics 97 participants Age, mean (SD): 33.8 (9.3) years 61 females, 36 males |
|
| Interventions |
Intervention: tezacaftor 100 mg/ivacaftor 150 mg fixed‐dose combination tablet orally once daily in the morning and ivacaftor 150 mg tablet orally once daily in the evening for 56 days Control: placebo matched to tezacaftor/ivacaftor fixed‐dose combination tablet orally once daily in the morning followed by placebo matched to ivacaftor tablet orally once daily in the evening for 56 days |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals Incorporated, National Institutes of Health and UK National Institute for Health Research (NIHR) Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield National Health Service (NHS) Foundation Trust and Imperial College London | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web response system was used for randomisation following a list of randomisation codes generated by a designated vendor. |
| Allocation concealment (selection bias) | Low risk | An Interactive web response system was used to assign participants to treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participants and site personnel, including the investigator, the site monitor and the study team were blinded and exceptions listed in the statistical analysis plan. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. All study personnel were blinded to participant treatment assignments, with the exceptions clearly stated in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants were included in analysis. There is a discrepancy between clinicaltrials.gov and the published paper ‐ according to clinicaltrials.gov, 1 person in the placebo group did not complete the treatment regimen, however in the published paper, it states that 2 did not. |
| Selective reporting (reporting bias) | Unclear risk | States that study would measure vital signs; although these may have been measured, they are not stated in the results or supplement regardless of whether they were unremarkable or not. |
| Other bias | Unclear risk | Baseline characteristics were generally balanced (however, there were 61 females compared to 36 males ‐ unclear how this imbalance may have affected results). States how Vertex Pharmaceuticals Incorporated participated in the study, however the extent to which support was provided by the other funders is unclear. |
Stahl 2021.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 48 weeks Location: Germany |
|
| Participants |
Baseline criteria 51 children with CF randomised Age, range: 2 to 5 years Sex: 18 females, 33 males Genotype: homozygous for F508del |
|
| Interventions |
Intervention: in placebo‐controlled period for 48 weeks, participants weighing less than 14 kg at screening received lumacaftor 100 mg/ivacaftor 125 mg fixed‐dose combination every 12 hours and participants weighing at least 14 kg at screening received lumacaftor 150 mg/ivacaftor 188 mg fixed‐dose combination every 12 hours Control: participants received placebo matched to lumacaftor/ivacaftor in placebo‐controlled period for 48 weeks |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | 2017‐003761‐99 (EudraCT Number) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An interactive web or voice response system was used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician who was not a member of the study execution team. |
| Allocation concealment (selection bias) | Low risk | An interactive web or voice response system was used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician who was not a member of the study execution team. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. The participants/caregivers and all site personnel, including the investigator and the study monitor, and the Vertex study team remained blinded to treatment assignments until the database was locked for Part 1 (i.e. database locked for data up to and including the Week 48 Visit) with the exceptions to this clearly stated in the trial protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. The participants/caregivers and all site personnel, including the investigator and the study monitor, and the Vertex study team remained blinded to treatment assignments until the database lock for Part 1 (i.e. database locked for data up to and including the Week 48 Visit) with the exceptions to this clearly stated in trial protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | For the primary outcome measured in the study, there were only 32/35 participants analysed in the intervention group, and 15/16 analysed in the placebo group (no detail given as to why there were missing participants). There was also conflicting information between clinicaltrials.gov and the published paper regarding the number of participants analysed for outcomes. |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the protocol are reported in the full trial paper. |
| Other bias | Unclear risk | Characteristics of participants were generally well‐balanced (however, there were 18 females and 33 males ‐ unable to determine if this imbalance impacted results). Unable to determine the extent to which the sponsor was involved in designing and conducting the study. |
Sutharsan 2022.
| Study characteristics | ||
| Methods | Phase 3b RCT Parallel design Multicentre study, conducted across Australia (6 sites), Belgium (4 sites), Germany (8 sites) and the UK (17 sites) 28‐day run‐in period, 24‐week treatment period, 4‐week safety follow‐up |
|
| Participants |
Inclusion criteria Aged 12 years and older Genotype: homozygous for F508del‐CFTR mutation FEV1 % predicted ≥ 40% and ≤ 90% of predicted mean for age, sex and height Baseline characteristics Age, mean (SD): 27.8 (11.4) years Sex: 88 females, 87 males CFQ‐R respiratory domain score, mean (SD): 72.2 (18.6) |
|
| Interventions | Following tezacaftor/ivacaftor run‐in period of 4 weeks, participants received either the intervention or control Intervention: elexacaftor 200 mg once daily/tezacaftor100 mg once daily/ivacaftor 150 mg every 12 hours for 24 weeks Control: tezacaftor 100 mg once daily/ivacaftor 150 mg every 12 hours for 24 weeks |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised (1:1) to the elexacaftor/tezacaftor/ivacaftor or tezacaftor/ivacaftor groups. Randomisation was stratified by FEV1 % predicted determined during the tezacaftor/ivacaftor run‐in period, age at the screening visit and whether the participant was receiving CFTR modulator treatment at the screening visit. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Allocation concealment (selection bias) | Low risk | Participants were randomised (1:1) to the elexacaftor/tezacaftor/ivacaftor or tezacaftor/ivacaftor groups. Randomisation was stratified by FEV1 % predicted determined during the tezacaftor/ivacaftor run‐in period, age at the screening visit and whether the participant was receiving CFTR modulator treatment at the screening visit. An interactive web response system assigned participants to treatment. The randomisation code list was produced by Vertex Biometrics or a qualified randomisation vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who may be unblinded were listed in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind study. All participants (and their parents/caregivers/companions), site personnel (including the investigator, the site monitor and the study team) and members of the Vertex study team were blinded to the treatment codes. Individuals who may be unblinded were listed in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants were analysed. |
| Selective reporting (reporting bias) | Unclear risk | Methods state that ECGs and vital signs would be measured; however, the exact values of the parameters used to measure safety and tolerability are not provided, whether unremarkable or not. |
| Other bias | Unclear risk | Characteristics of participants were generally well‐matched. Unclear as to the extent to which the sponsor was involved in designing the study and publishing the report. |
Taylor‐Cousar 2017.
| Study characteristics | ||
| Methods | Placebo‐controlled RCT Parallel design Duration: 24 weeks Multicentre in North America and Europe |
|
| Participants |
Inclusion criteria Aged 12 years and older Diagnosed with CF Baseline characteristics 510 participants were randomised Age: 23% were aged 12 to 18 years Sex: 49% female Genotype: homozygous for F508del Mean FEV1 at baseline: 60% (9.4% had baseline FEV1 < 40% predicted, 2% had baseline FEV1 > 90% predicted) Mean baseline sweat chloride: 100.5 Mean BMI: 21 |
|
| Interventions |
Intervention: 100 mg tezacaftor 1x daily and 150 mg ivacaftor 2x daily Control: placebo |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Funding source | Vertex Pharmaceuticals | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A statistician separate to study team produced a list of randomisation codes and allocations assigned via a web‐based interactive system (information provided in the online protocol). |
| Allocation concealment (selection bias) | Low risk | Web‐based interactive system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo ‐ all relevant people blinded (participants and study personnel). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ all relevant people blinded (participants and study personnel). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant who was randomised but did not receive the study intervention (due to a pulmonary exacerbation before the day 1 visit) was excluded from the efficacy and safety analyses. A further 5 participants who were randomised and received the study intervention were found to have an ineligible or unconfirmed CFTR genotype and excluded from the efficacy analyses. There were reasons given for the missing participants and so this is a low risk of bias. |
| Selective reporting (reporting bias) | High risk | The following outcomes were measured (according to the protocol), but not reported.
|
| Other bias | Low risk | Final manuscript written with the assistance of medical writers funded by the sponsor; however, this is unlikely to have introduced bias. |
TRAFFIC 2015.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled, Phase 3 RCT Parallel design Multicentre: 90 sites in North America, Australia and Europe Estimated sample size: 559 Duration: 24 weeks |
|
| Participants |
Inclusion criteria A confirmed diagnosis of CF and stable disease (as judged by the investigator) Lung function: FEV1 between ≥ 40% and ≤ 90% of predicted normal for age, sex and height Baseline characteristics 549 participants randomised Age, mean (range): intervention arm 1 24.7 (12 to 54) years; intervention arm 2 25.5 (12 to 57) years; placebo 25.0 (12 to 64 years) Sex: 295 (54%) males; 254 (46%) females Genotype: homozygous for the F508del mutation |
|
| Interventions |
Intervention 1 (n = 183): 600 mg of lumacaftor 1x daily and 250 mg of ivacaftor every 12 hours Intervention 2 (n = 182): 400 mg of lumacaftor every 12 hours and 250 mg of ivacaftor every 12 hours Placebo (n = 184): lumacaftor‐matched placebo every 12 hours in combination with ivacaftor‐matched placebo every 12 hours |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Sponsored by Vertex Pharmaceuticals Inc. | |
| Notes | Known as TRAFFIC study. The TRAFFIC and TRANSPORT studies were identical with the following exceptions: TRAFFIC included ambulatory ECG screening at days 1 and 15 in approximately 165 participants in the USA; TRANSPORT included additional pharmacokinetics assessments performed in approximately 28 adolescents in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomly assigned (in a 1:1:1 ratio) to 1 of 3 study groups; the randomisation was established by an interactive web response system. Randomisation was stratified according to age (< 18 years versus ≥ 18 years), sex and pulmonary function (% predicted FEV1 (% predicted) at screening, < 70% versus ≥ 70%). |
| Allocation concealment (selection bias) | Low risk | The randomisation was established by an interactive web response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. Placebo was matched in appearance and packaging. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Correct number of participants included in the analysis (i.e. those who received at least one dose of the study drug – ITT). Prior to first dose, 10 out of 559 participants withdrew: 2 withdrew from treatment arm 1; 5 withdrew from treatment arm 2; 3 withdrew from placebo group. Post first dose, 25 out of 549 participants withdrew (with reasons): 11 from treatment arm 1; 10 withdrew from treatment arm 2; 4 withdrew from placebo group. |
| Selective reporting (reporting bias) | High risk | Additional data available on www.clinicaltrials.gov for outcomes not reported in the final paper, such as:
Some results had to be extrapolated from graphical figures; we await confirmation from the study sponsor of the accuracy of the results. Investigators state that they measured FVC (which was not listed as an endpoint) and do not report this in the joint paper. |
| Other bias | Low risk | Adherence to study treatment was high and the mean compliance rate (determined by site personnel and ongoing study drug count) was similar across lumacaftor‐ivacaftor and placebo groups (99.1% versus 98.5%). |
TRANSPORT 2015.
| Study characteristics | ||
| Methods | Double‐blind, placebo‐controlled, Phase 3 RCT Parallel design Multicentre: 82 sites in North America, Australia and Europe Estimated sample size: 563 Duration: 24 weeks |
|
| Participants |
Inclusion criteria Confirmed diagnosis of CF and with stable disease (as judged by the investigator) Lung function: FEV1 (% predicted) between ≥ 40% and ≤ 90% of predicted normal for age, sex and height Baseline characteristics 559 participants randomised Age, mean (range): intervention arm 1 24.3 (12 to 48) years; intervention arm 2 25.0 (12 to 54) years; placebo 25.7 (12 to 55) years Sex: 268 (48%) males; 291 (52%) females Genotype: homozygous for the F508del mutation |
|
| Interventions |
Intervention 1: 600 mg of lumacaftor 1x daily and 250 mg of ivacaftor every 12 hours for 24 weeks Intervention 2: 400 mg of lumacaftor every 12 hours and 250 mg of ivacaftor every 12 hours for 24 weeks Placebo: placebo |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Sponsored by Vertex Pharmaceuticals Inc. | |
| Notes | Known as TRANSPORT study The TRAFFIC and TRANSPORT studies were identical with the following exceptions: TRAFFIC included ambulatory ECG screening at days 1 and 15 in approximately 165 participants in the USA; TRANSPORT included additional pharmacokinetics assessments performed in approximately 28 adolescents in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomly assigned (in a 1:1:1 ratio) to 1 of 3 study groups; the randomisation was established by an interactive web response system. Randomisation was stratified according to age (< 18 years versus ≥ 18 years), sex and pulmonary function (% predicted FEV1 (% predicted) at screening, < 70% versus ≥ 70%). |
| Allocation concealment (selection bias) | Low risk | The randomisation was established by an interactive web response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. Placebo was matched in appearance and packaging. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | These were double‐blind studies in which the participant and study team remained blinded to the treatment assignments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Correct number of participants included in the analysis (i.e. those who received at least 1 dose of the study drug – ITT). Prior to first dose, 4 out of 563 participants withdrew: 2 withdrew from treatment arm 1; 2 withdrew from treatment arm 2; none withdrew from placebo group. Post first dose, 29 out of 559 participants withdrew (with reasons): 9 from treatment arm 1; 15 withdrew from treatment arm 2; 5 withdrew from placebo group. |
| Selective reporting (reporting bias) | High risk | Additional data available on www.clinicaltrials.gov for outcomes not reported in the final paper, such as:
Some results had to be extrapolated from graphical figures; we await confirmation from the study sponsor of the accuracy of the results. Investigators state that they measured FVC (which was not listed as an endpoint) and do not report this in the joint paper. |
| Other bias | Low risk | Adherence to study treatment was high and the mean compliance rate (determined by site personnel and ongoing study drug count) was similar across lumacaftor‐ivacaftor and placebo groups (99.1% versus 98.5%). |
Wilson 2021.
| Study characteristics | ||
| Methods | Phase 4, placebo‐controlled RCT Parallel design Multicentre study, conducted across 13 sites in Australia and the UK Duration of treatment: 24 weeks. Safety follow‐up visit occurred 4 weeks after last study drug dose. |
|
| Participants |
Inclusion criteria Aged 12 years and older Genotype: homozygous for the F508del‐CFTR mutation FEV1 at least 40% and not greater than 90% of predicted Baseline characteristics Age, mean (SD): 25.5 (10.33) years Sex: 31 females, 39 males |
|
| Interventions |
Intervention: lumacaftor 400 mg/ivacaftor 250 mg fixed‐dose combination tablet orally every 12 hours for 24 weeks Control: placebo matched to lumacaftor/ivacaftor fixed‐dose combination tablet orally every 12 hours for 24 weeks |
|
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
|
| Funding source | Vertex Pharmaceuticals Incorporated | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Interactive web or voice response system used to assign participants to treatment. The randomisation code was produced by Vertex Biostatistics or a qualified randomisation vendor. The Vertex study biostatistician reviewed and approved the production of the final randomisation list, which was reviewed and approved by a designated unblinded biostatistician not a member of the study team. |
| Allocation concealment (selection bias) | Low risk | An interactive web or voice response system was used to assign participants to treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participants and all site personnel, including the investigator, the site monitor and the study team, were blinded. There is a clear statement of the exceptions to this in the protocol. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Matched placebo ‐ double‐blind RCT. The participants and all site personnel, including the investigator, the site monitor and the study team were blinded. There is a clear statement of the exceptions to this in the protocol. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A number of outcomes (including outcomes relevant to our review) reported data for fewer participants than the number assigned to that group. For example, only 30/34 participants in the intervention group were analysed for both absolute and relative change from baseline in FEV1 % predicted at week 24. No explanation was given for the missing participants. |
| Selective reporting (reporting bias) | Unclear risk | States in the protocol that safety and tolerability assessments would be based on a number of parameters, e.g. vital signs, although there are no exact values presented in results. |
| Other bias | Low risk | Baseline characteristics were generally well‐balanced (though there were 13 females compared to 21 males in the intervention group). The paper outlined the extent to which Vertex Pharmaceuticals Incorporated were involved (sponsor was involved in the study design and analysis and interpretation of the data, with collaboration from the authors. The sponsor helped develop the report with input, review and approval from the authors). |
Zeitlin 2002.
| Study characteristics | ||
| Methods | Phase 1/2, placebo‐controlled RCT Parallel design Single centre Duration: 1 week This study follows on from a pilot study (see above) (Rubenstein 1998) |
|
| Participants |
Baseline characteristics 19 participants were supposed to be randomised in a 3:1 ratio to either study drug or placebo. Randomisation to 40 g group discontinued due to safety reasons; therefore, 6 participants were allocated to the 20 g and 30 g groups, 3 to the 40 g group and 4 to the placebo group. It is unclear why only 4 participants were randomised to the placebo group. Age, mean: 28.5 years Sex: 12 males and 7 females Genotype: all participants were homozygous for the F508del mutation Lung function FEV1 % predicted, mean (SD): 63.7 (17.0) % predicted Nutritional status weight, mean (SD): 62.6 (17.0) kg |
|
| Interventions |
Control: placebo Intervention 2: 4‐phenylbutyrate (4PBA) 20 g Intervention 3: 4‐phenylbutyrate (4PBA) 30 g Intervention 4: 4‐phenylbutyrate (4PBA) 40 g All active interventions split into 3 daily doses |
|
| Outcomes |
|
|
| Funding source | Cystic Fibrosis Foundation | |
| Notes | *Denotes outcomes relevant to this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated as randomised but it is not clear how this was conducted. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Escalation to the next dose level was preceded by an examination of the safety profile of the preceding dose. Therefore, study personnel would have been aware of treatment allocation. Also, between the 3 intervention groups, participants received a different number of tablets and had different dosage schedules:
Therefore, it is unlikely that study personnel blinding and participant blinding was maintained throughout the study. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information on how outcome assessor blinding was maintained. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All 19 participants completed the final study visit, but it is unclear how many participants were used in the analysis. |
| Selective reporting (reporting bias) | High risk | Protocol not available. Pulmonary function or microbiology scores at day 7 were not reported. |
| Other bias | Low risk | "There were no significant differences in gender, baseline age, weight, or FEV1 (% predicted) among participants in the four groups." |
AE: adverse event BMI: body mass index CF: cystic fibrosis CFQ‐R: Cystic Fibrosis Questionnaire‐Revised CFRSD: Cystic Fibrosis Respiratory Symptom Diary CFTR: cystic fibrosis transmembrane conductance regulator CPX: 8‐cyclopentyl‐1, 3‐dipropylxanthine CT: computer tomography ECG: electrocardiogram EQ 5D 3L: EuroQol 3 Level FEF25-75%: forced expiratory flow FEV1: forced expiratory volume in one second FVC: forced vital capacity IRT: immunoreactive trypsinogen ITT: intention‐to‐treat IV: intravenous LCI: lung clearance index mEq/L: milliequivalents/L MF: minimal function MMRM: mixed‐effects model for repeated measurements NIH: National Institutes of Health PK: pharmacokinetic PSQI: Pittsburgh Sleep Quality Index QoL: quality of life RCT: randomised controlled trial RF: residual function SAE: serious adverse event SD: standard deviation SF12: short‐form 12 TSQM: Treatment Satisfaction Questionnaire for Medication VO2max: maximal oxygen consumption vs: versus
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Berkers 2014 | Cross‐over study with participants with gating defects and not a class II variant. |
| Berkers 2021 | Cross‐over assignment. |
| Chadwick 1998 | Investigators informed review authors that study was not randomised. |
| Chilvers 2021 | Participants not randomised; single‐group assignment. |
| Chmiel 2021 | Lenabasum is not a corrector. |
| Drevinek 2017 | QR‐010 is an anti‐sense oligonucleotide, which we did not consider to be a corrector. |
| ISRCTN14081521 | Not looking at any corrector as an intervention. |
| Krivec 2021 | Contacted author who stated that randomisation was not performed or ethically possible. |
| Leonard 2012 | Cross‐over design. |
| Marsh 2022 | Cross‐over assignment. |
| Meijer 2016 | The study has been terminated; no data available. |
| NCT00945347 | Cross‐over design. |
| NCT01899105 | Cross‐over design. |
| NCT02323100 | Study has been terminated due to funding ending. |
| NCT03447262 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03525574 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03537651 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03601637 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03633526 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03691779 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT03756922 | Initial phases not randomised and in healthy participants. Phase 4 due to be randomised and in people with CF, but trial suspended for "business reasons". |
| NCT04043806 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT04058210 | Not a randomised controlled trial. |
| NCT04058366 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT04183790 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT04235140 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT04362761 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT04537793 | Participants not randomised; sequential assignment; no masking, open‐label. |
| NCT04545515 | Participants not randomised; single‐group assignment; no masking, open‐label. |
| NCT05535959 | Cross‐over assignment. |
| Nick 2014 | Cross‐over study assessing CFTR mutations eligible for treatment with ivacaftor (not relevant to this review). |
| Rowe 2017 | Cross‐over design. |
| Rubenstein 2006 | Participants were not randomised. |
| Sumner 2014 | Gene therapy study, not a mutation‐specific therapy. |
| Ziady 2015 | Laboratory study conducted within cells donated by CF and non‐CF donors. Not a study of people with CF. |
CF: cystic fibrosis CFTR: cystic fibrosis transmembrane conductance regulator
Characteristics of studies awaiting classification [ordered by study ID]
ALBATROSS.
| Methods | Phase IIa, double‐blind, placebo‐controlled RCT Multicentre in Australia and Europe Duration: 4 weeks |
| Participants |
Inclusion criteria Aged 18 years or older Genotype: F508del/class III variant Disease status: baseline FEV1 % predicted 40% or greater On stable ivacaftor treatment for at least the past 4 weeks (during the study they also continued to take ivacaftor) Baseline characteristics 37 participants randomised GLPG2222 150 mg once daily group Age, median (range): 29.0 (19 to 42) years Sex: 12 males BMI, median (range): 23.95 (19.9 to 31.5) Sweat chloride, median (range): 38.8 (16 to 99) mmol/L FEV1 % predicted, median (range): 72.5 (41 to 113) % predicted GLPG2222 300 mg once daily group Age, median (range): 29.0 (18 to 35) years Sex: 6 males BMI, median (range): 22.00 (18.4 to 34.3) Sweat chloride, median (range): 48.8 (14 to 88) mmol/L FEV1 % predicted, median (range): 63.0 (37 to 99) % predicted Placebo group Age, median (range): 46.0 (19 to 53) years Sex: 3 males BMI, median (range): 25.30 (21.2 to 33.6) Sweat chloride, median (range): 32.5 (25 to 90) mmol/L FEV1 % predicted, median (range): 81.0 (37 to 97) % predicted |
| Interventions |
Intervention 1: GLPG2222 150 mg orally for 4 weeks Intervention 2: GLPG2222 300 mg orally for 4 weeks Control: placebo All participants were already on stable ivacaftor treatment for at least 4 weeks prior to this trial and continued taking their ivacaftor throughout this study period 30 participants took one of the GLPG2222 doses and 7 participants took placebo |
| Outcomes |
Primary outcomes
Secondary outcomes
Initial abstracts report AEs, PK and pharmacodynamic data, sweat chloride change and absolute change in FEV1 % predicted |
| Notes | Clinicaltrials.gov: NCT03045523 |
Downey 2019.
| Methods | RCT, double‐blind and placebo‐controlled Parallel design, 3 arms Duration: 14 days |
| Participants |
Inclusion criteria Adults (over 18 years of age) with CF homozygous for F508del and with FEV1 40% to 90% predicted |
| Interventions |
Intervention 1: dual therapy of PTI‐801 + PTI‐808 Intervention 2: triple therapy of PTI‐801 + PTI‐808 + PTI‐428 Control: placebo |
| Outcomes |
|
| Notes | Further details (full paper) not yet available |
Downey 2020.
| Methods | Randomised, double‐blind, placebo‐controlled, phase 2 clinical study. Treatment period 4 weeks followed by a 2‐week follow‐up visit. |
| Participants |
Inclusion criteria Diagnosed with CF and homozygous for the F508del CFTR mutation Aged ≥ 18 years FEV1 40% to 90% predicted |
| Interventions |
Intervention: combination of dirocaftor, posenacaftor and nesolicaftor (abstracts do not give dose levels) Control: placebo |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Initial data suggest a generally well‐tolerated safety profile and clinical benefit with dirocaftor, posenacaftor and nesolicaftor. In the preliminary analysis, mean absolute changes in FEV1 % predicted of 8% points (95% CI 3 to 12) (P ≤ 0.01, n = 11) versus placebo were observed following a treatment period of 4 weeks. Reduction in sweat chloride of ‐29 mmol/L (95% CI ‐42 to ‐16) (P < 0.0005, n = 10) versus placebo were observed at week 4. |
FLAMINGO.
| Methods | Phase 2, double‐blind, placebo‐controlled RCT Duration: 29 days Location: multicentre in North America and Europe |
| Participants |
Inclusion criteria Aged 18 years and over Diagnosed with CF with F508del/F508del genotype Disease status: FEV1 40% predicted or greater Not on concomitant CFTR modulator therapy within 4 weeks of study start Baseline characteristics 59 adults randomised (11 in placebo group) Age, median (range): placebo group 27.0 (21 to 58) years; GLPG2222 50 mg once daily group 26.0 (20 to 37) years; GLPG2222 100 mg once daily group 24.0 (18 to 35) years; GLPG2222 200 mg once daily group 2.0 (19 to 47) years; GLPG2222 400 mg once daily group 26.0 (19 to 59) years Sex: placebo group 7 males; GLPG2222 50 mg once daily group 7 males; GLPG2222 100 mg once daily group 4 males; GLPG2222 200 mg once daily group 7 males; GLPG2222 400 mg once daily group 9 males BMI, median (range): placebo group 22.20 (16.3 to 25.7); GLPG2222 50 mg once daily group 21.05 (18.7 to 25.1); GLPG2222 100 mg once daily group 20.75 (14.1 to 23.3); GLPG2222 200 mg once daily group 22.30 (18.5 to 26.8); GLPG2222 400 mg once daily group 22.40 (18.3 to 23.7) FEV1% predicted, median (range): placebo group 75.0 (43 to 92) % predicted; GLPG2222 50 mg once daily group 53.5 (35 to 80) % predicted; GLPG2222 100 mg once daily group 59.5 (39 to 113) % predicted; GLPG2222 200 mg once daily group 51.5 (40 to 88) % predicted; GLPG2222 400 mg once daily group 58.5 (38 to 92) % predicted Sweat chloride, median (range): placebo group 108.0 (91.5 to 114.5) mmol/L; GLPG2222 50 mg once daily group 105.5 (86.0 to 114.5) mmol/L; GLPG2222 100 mg once daily group 110.0 (96.0 to 113.0) mmol/L; GLPG2222 200 mg once daily group 102.8 (82.0 to 119.5) mmol/L; GLPG2222 400 mg once daily group 105.8 (100.0 to 123.0) mmol/L |
| Interventions | GLPG2222 once daily dose of 4 different doses taken once daily were tested (either 50, 100, 200, 400 mg) for 4 weeks Cohort A GLPG2222 50 mg tablet and 2x matching placebo tablets orally, 4x daily for 29 days GLPG2222 100 mg tablet and 2x matching placebo tablets orally, 4x daily for 29 days Cohort B GLPG2222 2x 100 mg tablets and 1x matching placebo tablet orally, 4x daily for 29 days GLPG2222 2x 150 mg tablets and GLPG2222 1x 100 mg tablet orally, 4x daily for 29 days |
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
| Notes | — |
Hunt 2017.
| Methods | RCT, placebo‐controlled (randomised 2:1 to sildenafil or placebo) Parallel design Duration: 4 weeks |
| Participants |
Baseline characteristics 18 adults homozygous for F508del Age, mean (SD): 28.7 (6.6) years Sex: 7/18 males (35%), 11/18 females (65%) FEV1 % predicted, mean (SD): 82.5% (12.9) % predicted BMI, mean (SD): 23.2 (6.6) kg/m² |
| Interventions |
Intervention: oral sildenafil 40 mg 3x daily Control: matching placebo All participants also on standard of care and lumacaftor/ivacaftor |
| Outcomes |
Routine laboratory tests were measured at baseline. |
| Notes | — |
NCT03768089.
| Methods | Double‐blind, placebo‐controlled RCT Sequential assignment/escalating dose |
| Participants |
Inclusion criteria Aged 18 years and over Healthy adults without CF and BMI 18 to 32 and weight over 50 kg and people with CF with F508del/MF genotype and FEV1 40% to 90% predicted and weight over 35 kg Only CF participants eligible for inclusion in review Baseline characteristics 114 adults randomised |
| Interventions |
Intervention: VX‐121 tablet plus 100 mg tezacaftor/150‐mg ivacaftor fixed‐dose combination tablet and ivacaftor 150 mg film‐coated tablet Control: triple placebo |
| Outcomes |
|
| Notes | A Phase 1/2 Study of VX‐121 in Healthy Subjects and in Subjects With Cystic Fibrosis |
NCT03969888.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design, 3 arms Duration: 29 days |
| Participants |
Inclusion criteria Adults (over 18 years of age) Diagnosed with CF and homozygous for F508del Lung function ≥ 40 % and ≤ 90 % of predicted normal for age, gender and height at screening |
| Interventions |
Experimental: Part 1: ABBV‐3067 plus placebo (participants will receive various dosing regimens for ABBV‐3067 plus placebo ABBV‐2222 taken orally depending on arm assignment) Experimental: Part 1: ABBV‐3067 plus ABBV‐2222 (participants will receive fixed dose of ABBV‐3067 plus various dosing regimens for ABBV‐2222 taken orally depending on arm assignment) Control: placebo (participants in Part 1 and Part 2 will receive placebo ABBV‐3067 plus placebo ABBV‐2222 taken orally) Experimental: Part 2: ABBV‐3067 plus ABBV‐2222 (participants will receive various dosing regimens for ABBV‐3067 plus a fixed dose of ABBV‐2222 taken orally depending on arm assignment) |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes | A Phase 2 Study of ABBV‐3067 Alone and in Combination with ABBV‐2222 in Cystic Fibrosis Subjects Who Are Homozygous for the F508del Mutation |
NCT04353817.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 24 weeks (safety 28 weeks) |
| Participants |
Inclusion criteria Aged 6 to 11 years Diagnosed with CF and genotype F508del/MF FEV1 over 70 % predicted Baseline characteristics 121 children randomised Age, mean (SD): 9.2 (1.7) years Sex: 70 females, 51 males LCI2.5 mean (SD): 10.01 (2.09) years |
| Interventions |
Intervention: participants weighing < 30 kg at screening received 100 mg once‐daily elexacaftor plus 50 once‐daily tezacaftor plus 75 mg twice‐daily ivacaftor and participants weighing ≥ 30 kg at screening received 200 mg once‐daily elexacaftor plus 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor in the treatment period for 24 weeks. Control: participants received placebo matched to elexacaftor plus tezacaftor plus ivacaftor and placebo matched to ivacaftor in the treatment period for 24 weeks. |
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Notes | A Study Evaluating Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Subjects 6 Through 11 Years of Age With Cystic Fibrosis and F/MF Genotypes |
PELICAN.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 28 days |
| Participants |
Inclusion criteria Aged 18 and over Diagnosed with CF and homozygous for F508del Previously treated with lumacaftor‐ivacaftor for at least 12 weeks before start of trial FEV1 at least 40% predicted Sweat chloride at least 60 mmol/L Baseline characteristics 22 adults randomised Age, median (range): intervention group 21.5 (19 to 42) years; placebo group 28.5 (19 to 38) years Sex: 9 males in intervention group, 5 males in placebo group FEV1 % predicted, median (range): intervention group 55.7 (37 to 86) % predicted; placebo group 68.8 (43 to 97) % predicted BMI, median (range): intervention group 21.1 (18 to 29); placebo group 20.3 (17 to 24) Sweat chloride concentration, median (range): intervention group 86.0 (61 to 113) mmol/L; placebo group 84.3 (72 to 109) mmol/L CFQ‐R respiratory domain score, median (range): intervention group 68.0 (39 to 100); placebo group 69.4 (44 to 89) |
| Interventions |
Intervention (n = 14): 75 mg twice‐daily GLPG‐2737 plus twice‐daily lumacaftor 400 mg/ivacaftor 250 mg Control (n = 8): placebo plus twice‐daily lumacaftor 400 mg/ivacaftor 250 mg |
| Outcomes |
Primary outcome measures:
Secondary outcome measures:
|
| Notes | — |
Rio‐CF.
| Methods | Placebo‐controlled, quadruple‐blind RCT. Randomised 2:1 (intervention n = 14, placebo n = 7). Phase 2 Parallel design Multicentre: USA (3 sites), Canada (1 site), Europe (6 sites) Duration: 28 days |
| Participants |
Inclusion criteria Diagnosed with CF and homozygous for F508del FEV1 % predicted 40% to 100% Baseline characteristics 21 adults randomised Age, mean (SD): total cohort 27.8 (6.9) years; intervention 27.1 (6.9) years; placebo 29.1 (7.2) years Sex: total cohort 16 males (76.2%); intervention 10 males (71.4%); placebo 6 males (85.7%) Sweat chloride, mean (SD): total cohort (n = 16) 95.53 (15.03) mmol/L; intervention (n = 9) 96.33 (17.28) mmol/L; placebo (n = 7) 94.50 (12.82) mmol/L |
| Interventions |
Intervention: riociguat 0.5 mg 3x daily for 14 days, then 1.0 mg 3 x daily for the next 14 days Control: matched placebo 3x daily |
| Outcomes |
Primary outcomes
Secondary outcomes
Outcomes measured at baseline, day 14 and day 28 |
| Notes | Author of the study was contacted and stated that further detail is awaited in a published manuscript. However, we note from clinicaltrials.gov that part 2 of this study has been terminated after no longer being deemed appropriate. |
Taylor‐Cousar 2019.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Escalating dose Duration: 28 days |
| Participants |
Inclusion criteria Aged 18 years and older Diagnosed with CF FEV1 40% to 90% predicted Participants had previously been taking tezacaftor/ivacaftor Baseline characteristics 40 adults randomised |
| Interventions |
Intervention: 2 different doses of PTI‐428 (doses not stated) plus tezacaftor/ivacaftor Control: placebo plus tezacaftor/ivacaftor |
| Outcomes |
|
| Notes | — |
Uluer 2023a.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 16 weeks |
| Participants |
Inclusion criteria Adults with CF with at least one copy of G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P or G1349D, already on ivacaftor Baseline characteristics 77 adults randomised |
| Interventions |
Intervention: 1 of 4 dose levels of deutivacaftor plus placebo Control: ivacaftor 150 mg film‐coated tablet plus placebo |
| Outcomes |
|
| Notes | A Phase 2 Study to Evaluate Efficacy and Safety of VX‐561 in Subjects Aged 18 Years and Older With Cystic Fibrosis Unclear whether this will include participants F508del ‐ waiting to see if appropriate for inclusion in this review. |
Uluer 2023b.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Sequential assignment Duration: 75 days |
| Participants |
Inclusion criteria Adults (aged 18 years and over) with CF either homozygous for F508del or with genotype F508del/MF and FEV1 40% to 90% predicted Baseline characteristics 87 adults randomised |
| Interventions |
F508del/MF group Intervention 1: vanzacaftor‐tezacaftor‐deutivacaftor Control: matched placebo F508del/F508del group Intervention 1: vanzacaftor‐ tezacaftor‐deutivacaftor Control: placebo‐tezacaftor‐ivacaftor |
| Outcomes |
|
| Notes | — |
Wainwright 2019.
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 72 weeks |
| Participants |
Inclusion criteria Aged 12 years and older Diagnosed with CF and homozygous for F508del FEV1 at least 70% predicted |
| Interventions |
Intervention: 100 mg once‐daily tezacaftor plus 150 mg twice‐daily ivacaftor Control: matched placebo |
| Outcomes |
|
| Notes | — |
AE: adverse event AUC: area under the curve BMI: body mass index CF: cystic fibrosis CT: computer tomography CFQ‐R: cystic fibrosis questionnaire ‐ revised CFTR: cystic fibrosis transmembrane conductance regulator ECG: electrocardiogram FEV1: forced expiratory volume in one second LCI: lung clearance index MF: minimal function PK: pharmacokinetic RCT: randomised controlled trial SAE: serious adverse event SD: standard deviation
Characteristics of ongoing studies [ordered by study ID]
Jain 2018.
| Study name | A phase 1 / 2, double‐blind, placebo‐controlled RCT designed to evaluate the safety, tolerability, and pharmacokinetics of PTI‐808, PTI‐801, and PTI‐428 combination therapy in people with CF Initial result evaluating third‐generation CFTR corrector PTI‐801 in people with CF |
| Methods | Phase 1, double‐blind RCT testing ascending doses of PTI‐801 Multicentre in the UK Duration: 14 days of treatment with follow‐up visit at 21 days |
| Participants |
Inclusion criteria Aged 18 years and older Diagnosed with CF clinically and by genetic testing Genotype: for cohorts 1, 2 and 4 F508del/F508del; for cohort 3 at least one copy of F508del Disease status: baseline FEV1 40% to 90% Baseline characteristics Estimated enrolment: 32 participants Participants were all currently receiving lumacaftor/ivacaftor as background therapy |
| Interventions |
Cohort 1: once‐daily PTI‐808 with PTI‐801 versus once‐daily PTI‐808 plus placebo for 14 days Cohort 2: once‐daily PTI‐808 with PTI‐801 versus once‐daily PTI‐808 plus placebo for 14 days Cohort 3: once‐daily PTI‐808 with PTI‐801 and PTI‐428 versus once‐daily placebos for 14 days Cohort 4: once‐daily PTI‐808 with PTI‐801 and PTI‐428 versus placebos once daily for 7 days immediately followed by PTI‐808 with PTI‐801 versus placebos for 7 days Multiple ascending doses of PTI‐801 versus placebo. Initial abstract does not provide detail on the actual doses, frequency, route of administration or details of placebo. |
| Outcomes |
Primary outcome
Secondary outcomes
Other outcomes
|
| Starting date | January 2018 |
| Contact information | Proteostasis Therapeutics Inc. |
| Notes | Estimated primary completion date: April 2019 Estimated study completion date: May 2019 |
NCT02589236.
| Study name | Study of Cavosonstat (N91115) in Patients With CF Homozygous for the F508del‐CFTR Mutation (SNO‐6) |
| Methods | Double‐blind, placebo‐controlled RCT Parallel design |
| Participants |
Inclusion criteria Aged 18 years and older FEV1: 40% to 85% predicted Participants must have been treated with lumacaftor‐ivacaftor for at least 8 weeks prior to day 1 |
| Interventions |
Intervention 1: lumacaftor‐ivacaftor‐cavosonstat 200 mg 2x daily Intervention 2: lumacaftor‐ivacaftor‐cavosonstat 400 mg 2x daily Control: lumacaftor‐ivacaftor‐matched placebo |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | November 2015 |
| Contact information | Principal Investigator: Scott Donaldson, MD, University of North Carolina, Chapel Hill Sponsors and collaborators: Nivalis Therapeutics, Inc. and Medidata Solutions |
| Notes | — |
NCT02718495.
| Study name | Study Assessing PTI‐428 Safety, Tolerability, and Pharmacokinetics in Subjects With Cystic Fibrosis |
| Methods | Quadruple‐blind, placebo‐controlled, 3‐arm, Phase 2 RCT Parallel design Multicentre: 29 centres in North America and Europe Sample size: expected to enrol 56 participants Duration: 28 days of treatment |
| Participants |
Inclusion criteria Aged 18 years and older Genotype: not specified Lung function: FEV1 40% to 90% predicted |
| Interventions | PTI‐428 versus placebo Part A has 2 groups: the 1st group will enrol adults with CF into a single ascending‐dose treatment group; the 2nd group will enrol adults with CF, including those on background treatment with lumacaftor‐ivacaftor (ORKAMBI®) and those not on a CFTR modulator into a multiple‐ascending dose treatment group Part B will enrol adults with CF currently on stable lumacaftor‐ivacaftor (ORKAMBI®) background therapy for a minimum of 3 months into a Phase 2 treatment group consisting of 2 cohorts Part C will enrol adults with CF, including those on background treatment with ivacaftor (KALYDECO®) and those not on a CFTR modulator, into a Phase 2 treatment group consisting of 3 cohorts |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | November 2017 |
| Contact information | Proteostasis Therapeutics, Inc. |
| Notes | — |
NCT03258424.
| Study name | Study Assessing PTI‐428 Safety, Tolerability, and Pharmacokinetics in Subjects With Cystic Fibrosis on KALYDECO® as Background Therapy |
| Methods | Phase 1 placebo‐controlled RCT Parallel design 2 centres Sample size: expected to enrol 16 participants Duration: 14 days of treatment |
| Participants |
Inclusion criteria Age: 18 years and older Genotype: no specific mutations specified as eligible or ineligible Taking ivacaftor Lung function: FEV1 40% to 90% predicted |
| Interventions |
Group 1: 1x daily dosing of PTI‐428 Group 2: placebo for 14 days All participants continue on ivacaftor |
| Outcomes |
Primary outcome
Secondary outcomes
Other outcomes
|
| Starting date | July 2017 |
| Contact information | Proteostasis Clinical Trials (pticlinicaltrials@proteostasis.com) |
| Notes | — |
NCT04853368.
| Study name | A Phase 2 Study of Galicaftor‐Navocaftor‐ABBV‐119 or Galicaftor‐Navocaftor‐ABBV‐576 Combination Therapies in Subjects With Cystic Fibrosis Who Are Homozygous or Heterozygous for the F508del Mutation |
| Methods | Phase 2 RCT Parallel design Multicentre: approximately 35 sites worldwide Participants will be allocated to 1 of the 4 treatment arms |
| Participants | Estimated sample size 90 participants Inclusion criteria Aged 18 years and older Sex: males and females Genotype: F508del/F508del or F508del/MF |
| Interventions | For all study arms, ABBV‐576, galicaftor, navocaftor will be given once daily and ABBV‐119 twice a day Arm 1: participants with the genotype F508del/F508del not receiving elexacaftor‐tezacaftor‐ivacaftor treatment will receive oral capsules of galicaftor/navocaftor dual combination for 28 days followed by galicaftor/navocaftor/ABBV‐119 triple combination for 28 days Arm 2: participants with genotype F508del/MF not receiving elexacaftor‐tezacaftor‐ivacaftor treatment will receive galicaftor/navocaftor/ABBV‐119 triple combination or placebo for 28 days Arm 3: participants with genotype F508del/MF not receiving elexacaftor‐tezacaftor‐ivacaftor treatment will receive galicaftor/navocaftor/ABBV‐119 triple combination or placebo for 28 days Arm 4: participants with either genotype homozygous or heterozygous for the F508del mutation and receiving stable elexacaftor‐tezacaftor‐ivacaftor treatment will receive galicaftor/navocaftor/ABBV‐576 triple combination therapy for 28 days |
| Outcomes |
Primary outcome measures Arms 1 and 2
Arm 3
Secondary outcome measures Arms 1 and 2
Arm 3
|
| Starting date | April 2021 |
| Contact information | AbbVie Ltd AbbVie House Vanwall Business Park Vanwall Road Maidenhead Berkshire SL6 4UB UK Telephone: +44 (0) 1628 561090 |
| Notes | — |
NCT05033080.
| Study name | A Phase 3, Randomized, Double‐blind, Controlled Study Evaluating the Efficacy and Safety of VX‐121 Combination Therapy in Subjects With Cystic Fibrosis (CF) Who Are Heterozygous for F508del and a Minimal Function Mutation (F/MF) |
| Methods | Double‐blind, placebo‐controlled RCT Parallel design Duration: 24 weeks Location: 139 locations across North America, Europe, Australia and New Zealand |
| Participants |
Inclusion criteria Aged 12 years and older Sex: males and females Genotype: F508del/MF. FEV1 ≥ 40% and ≤ 90% predicted mean for age, sex and height for participants receiving elexacaftor‐tezacaftor‐ivacaftor therapy; FEV1 ≥ 40% and ≤ 80% for participants not currently receiving elexacaftor‐tezacaftor‐ivacaftor therapy |
| Interventions |
Intervention: VX‐121‐tezacaftor‐deutivacaftor Control: elexacaftor‐tezacaftor‐ivacaftor therapy |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | 14 September 2021 |
| Contact information | Vertex Pharmaceuticals, Inc.(medicalinfo@vrtx.com) |
| Notes | — |
NCT05076149.
| Study name | A Phase 3, Randomized, Double‐blind, Controlled Study Evaluating the Efficacy and Safety of VX‐121 Combination Therapy in Subjects With Cystic Fibrosis Who Are Homozygous for F508del, Heterozygous for F508del and a Gating (F/G) or Residual Function (F/RF) Mutation, or Have At Least 1 Other Triple Combination Responsive CFTR Mutation and No F508del Mutation |
| Methods | Double‐blind, active‐controlled RCT Parallel design Duration: 24 weeks Location: 170 international locations |
| Participants |
Inclusion criteria Aged 12 years and over Have one of the following genotypes:
FEV1 ≥ 40% and ≤ 90% predicted mean for height, age and sex for those currently taking CFTR modulator therapy or ≥ 40% and ≤ 80% for participants not currently taking CFTR modulator therapy |
| Interventions |
Intervention: VX‐121/tezacaftor/deutivacaftor Active control: elexacaftor‐tezacaftor‐ivacaftor |
| Outcomes |
Primary outcome
Secondary outcomes Absolute change from baseline in sweat chloride at week 24 Proportion of participants with sweat chloride < 60 mmol/L at week 24 Proportion of participants with sweat chloride < 30 mmol/L at Week 24 |
| Starting date | 27 October 2021 |
| Contact information | Vertex Pharmaceuticals, Inc.(medicalinfo@vrtx.com) |
| Notes | — |
NCT05274269.
| Study name | A Phase 3 Double‐blind, Randomized, Placebo‐controlled Study Evaluating the Efficacy and Safety of ELX/TEZ/IVA in Cystic Fibrosis Subjects 6 Years of Age and Older With a Non‐F508del ELX/TEZ/IVA‐responsive CFTR Mutation |
| Methods | Phase 3, double‐blind, placebo‐controlled RCT Parallel design |
| Participants | Estimated sample size 270 participants Inclusion criteria Aged 6 years and older Sex: males and females Participants have a non‐F508del elexacaftor‐tezacaftor‐ivacaftor responsive CFTR variant |
| Interventions |
Intervention: elexacaftor‐tezacaftor‐ivacaftor in the morning and ivacaftor in the evening Control: placebo matched to elexacaftor‐tezacaftor‐ivacaftor in the morning and placebo matched to ivacaftor in the evening |
| Outcomes |
Primary outcome measure
Secondary outcome measures
|
| Starting date | March 2022 |
| Contact information | Contact: Medical information 617‐341‐6777 medicalinfo@vrtx.com |
| Notes | Sponsors and collaborators: Vertex Pharmaceuticals Incorporated |
AE: adverse event AUC: area under the curve BMI: body mass index CF: cystic fibrosis CFTR: cystic fibrosis transmembrane conductance regulator CFU: colony forming units CFQ‐R: Cystic Fibrosis Questionnaire‐Revised Cmax: maximum concentration CT: computer tomography ECG: electrocardiogram FEF: forced expiratory flow FEV1: forced expiratory volume in one second FVC: forced vital capacity LCI: lung clearance index MRI: magnetic resonance imaging P aeruginosa: Pseudomonas aeruginosa PK: pharmacokinetic RCT: randomised controlled trial SAE: serious adverse event TEZ/IVA: tezacaftor‐ivacaftor TSQM: Treatment Satisfaction Questionnaire for Medication
Differences between protocol and review
We added lung clearance index (LCI) as an outcome due to the increasing use of this outcome as a measure of lung function in the younger population.
We have added a statement to the Methods section that 99% confidence intervals will be used to analyse separate adverse events. This is the most appropriate statistical approach for considering adverse events individually.
Originally, we intended to combine all studies included in the review using a random‐effects approach to meta‐analysis. However, due to the substantial differences in the designs and interventions employed within the studies, we considered it more appropriate to make separate comparisons within the review, and where small numbers of studies of a similar design and intervention were pooled in meta‐analysis, a fixed‐effect approach was appropriate.
Upon identification and inclusion of new studies that examined triple combination therapies, we made the decision to analyse dual combination and triple combination therapy regimens separately.
In line with current Cochrane guidance, we have included summary of findings tables for all comparisons.
We have clarified the time points that data are reported and stated our intention that if a study which we include in future updates presents data at more than one time point within any of our stated time ranges, we will present the later data set from the study.
Contributions of authors
| Roles and responsibilities | |
| TASK | RESPONSIBILITY |
| Protocol stage: draft the protocol | IS, SP with comments from all |
| Review stage: select which trials to include (2 + 1 arbiter) | MH, JM, IS, SP (+ KWS) |
| Review stage: extract data from trials (2 people) | MH, JM, IS, SP |
| Review stage: enter data into RevMan | MH, JM, SP, SJN |
| Review stage: carry out the analysis | MH, JM, SP, SJN |
| Review stage: interpret the analysis | MH, JM, IS, SP, SJN |
| Review stage: draft the final review | MH, JM, IS, SP, KWS with comments from all |
| Update stage: update the review | MH, JM, IS, KWS and SJN |
Sources of support
Internal sources
No sources of support provided
External sources
-
National Institute for Health & Care Research, UK
This systematic review was supported by the National Institute for Health & Care Research, via Cochrane Infrastructure funding to Cochrane Cystic Fibrosis and Genetic Disorders.
Declarations of interest
Matthew Heneghan declares no potential conflict of interest.
Professor Kevin Southern declares no potential conflict of interest.
Dr Jared Murphy declares no potential conflict of interest.
Dr Ian Sinha is in receipt of a NIHR HTA grant for paediatric asthma and is a member of the NICE asthma committee; however, neither of these are related to cystic fibrosis or this review and thus do not constitute a potential conflict of interest.
Dr Sarah J Nevitt declares no potential conflict of interest.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Barry 2021 {published data only}
- 2018-002835-76. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-445 combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a gating or residual function mutation (F/G and F/RF genotypes). https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-002835-76/results (this version publication date 22 July 2021).
- Barry PJ, Mall MA, Alvarez A, Colombo C, Winter-de Groot KM, Fajac I, et al. Triple therapy for cystic fibrosis Phe508del-gating and - residual function genotypes. New England Journal of Medicine 2021;385(9):815-25. [CFGD REGISTER: BD293c] [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCTR2018-002835-76-IE. A phase 3 study of VX-445 combination therapy in cystic fibrosis (CF) subjects heterozygous for f508del and a gating or residual function mutation (F/G and F/RF genotypes). www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2018-002835-76-IE (date started 29 October 2019). [CFGD REGISTER: BD293b]
- NCT04058353. A phase 3 study of VX-445 combination therapy in cystic fibrosis (CF) subjects heterozygous for F508del and a gating or residual function mutation (F/G and F/RF genotypes). clinicaltrials.gov/ct2/show/NCT04058353 (first received 15 August 2019). [CFGD REGISTER: BD293a] [EUDRACT NUMBER: 2018-002835-76]
Boyle 2014 {published data only}2010‐020413‐90
- Boyle M, Bell SC, Konstan M, McColley S, Flume P, Kang L, et al. Lumacaftor, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in CF patients with the F508del-CFTR mutation: phase 2 interim analysis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S14. [ABSTRACT NO.: WS7.4] [CENTRAL: 921640] [CFGD REGISTER: BD169c] [Google Scholar]
- Boyle MP, Bell S, Konstan M, McColley SA, Kang L, Patel N, et al. The investigational CFTR corrector, VX-809 (lumacaftor) co-administered with the oral potentiator ivacaftor improved CFTR and lung function in F509-8DEL homozygous patients: phase II study results. Pediatric Pulmonology 2012;47 Suppl 35:315. [ABSTRACT NO.: 260] [CENTRAL: 921644] [CFGD REGISTER: BD169b] [Google Scholar]
- Boyle MP, Bell S, Konstan MW, McColley SA, Wisseh S, Spencer-Green G. VX-809, an investigational CFTR corrector, in combination with VX-770, an investigational CFTR potentiator, in subjects with CF and homozygous for the F508DEL-CFTR mutation. Pediatric Pulmonology 2011;46 Suppl 34:287. [ABSTRACT NO.: 212] [CENTRAL: 848840] [CFGD REGISTER: BD169a] [Google Scholar]
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet. Respiratory Medicine 2014;2(7):527-38. [CENTRAL: 994993] [CFGD REGISTER: BD169d] [EMBASE: 2014458184] [DOI] [PubMed] [Google Scholar]
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. Supplementary Appendix to "A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial.". Lancet. Respiratory Medicine 2014;2(7):527-38. [CENTRAL: 997711] [CFGD REGISTER: BD169e] [DOI] [PubMed] [Google Scholar]
- Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, et al. Lumacaftor/ivacaftor treatment of patients with cystic fibrosis heterozygous for F508del-CFTR. Annals of the American Thoracic Society 2017;14(2):213-9. [CFGD REGISTER: BD169f] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, et al. Lumacaftor/ivacaftor treatment of patients with cystic fibrosis heterozygous for F508del-CFTR. Annals of the American Thoracic Society 2017;14(2):213-9. Online supplement: data. [CFGD REGISTER: BD169h] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, et al. Lumacaftor/ivacaftor treatment of patients with cystic fibrosis heterozygous for F508del-CFTR. Annals of the American Thoracic Society 2017;14(2):213-9. Online supplement: disclosures. [CFGD REGISTER: BD169g] [DOI] [PMC free article] [PubMed] [Google Scholar]
Clancy 2012 {published data only}
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012;67(1):12-8. [CENTRAL: 806692] [CFGD REGISTER: BD166c] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Ballmann M, Boyle MP, DeBoeck C, et al. A phase II, randomized, placebo-controlled, clinical trial of four doses of VX-809 in CF patients homozygous for the F508del CFTR mutation. Pediatric Pulmonology 2010;45 Suppl 33(S33):298. [ABSTRACT NO.: 224] [CENTRAL: 848845] [CFGD REGISTER: BD166b] [Google Scholar]
- Clancy JP, Rowe SM, Liu B, Hathorne H, Dong Q, Wisseh S, et al. Variability of nasal potential difference measurements in clinical testing of CFTR modulators. Pediatric Pulmonology 2011;46 Suppl 34(S34):283. [ABSTRACT NO.: 202] [CENTRAL: 848842] [CFGD REGISTER: BD166d // BD165n] [Google Scholar]
- Clancy JP, Spencer-Green G, for the VX-809-101SG. Clinical evaluation of VX-809, a novel investigational oral F508del-CFTR corrector, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Journal of Cystic Fibrosis 2010;9 Suppl 1:S20. [ABSTRACT NO.: 73] [CENTRAL: 848846] [CFGD REGISTER: BD166a] [Google Scholar]
- NCT00865904. Study of VX-809 in cystic fibrosis subjects with the ∆F508-CFTR gene mutation. www.clinicaltrials.gov/ct2/show/NCT00865904 (first received 19 March 2009).
Davies 2018a {published data only}
- A study to evaluate safety and pharmacokinetics of VX-659 in healthy subjects and in adults with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT03029455 (first received 24 January 2017). [CFGD REGISTER: BD260a]
- Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, et al. VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. New England Journal of Medicine 2018;379(17):1599-611. [CENTRAL: CN-01650323] [CFGD REGISTER: BD260b // BD248d] [EMBASE: 624591628] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Davies 2018b {published data only}
- A phase 2, randomized, double-blind, controlled study to evaluate the safety and efficacy of VX-659 combination therapy in subjects aged 18 years and older with cystic fibrosis. www.clinicaltrialsregister.eu/ctr-search/trial/2016-003585-11/ (start date 1 September 2017).
- Colombo C, Tullis E, Davies JC, McKee C, DeSouza C, Waltz D, et al. Preliminary safety and efficacy of triple combination CFTR modulator regimens in CF. Italian Journal of Pediatrics 2018;44(Suppl 1):6. [ABSTRACT NO.: 03] [CFGD REGISTER: BD248b] [Google Scholar]
- Davies JC, Colombo C, Tullis E, Mckee C, Desouza C, Waltz D, et al. Preliminary safety and efficacy of triple combination CFTR modulator regimens in cystic fibrosis. Journal of Cystic Fibrosis 2018;17(Suppl 3):S3. [CENTRAL: CN-01730786] [CFGD REGISTER: BD248a] [EMBASE: 622930757] [Google Scholar]
- Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, et al. VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. New England Journal of Medicine 2018;379(17):1599-611. [CENTRAL: CN-01650323] [CFGD REGISTER: BD248d] [DOI: 10.1056/NEJMoa1807119] [EMBASE: 624591628] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03224351. A study evaluating the safety and efficacy of VX-659 combination therapy in subjects with cystic fibrosis. clinicaltrials.gov/show/NCT03224351 (first received 21 July 2017). [CFGD REGISTER: BD248e]
- Tullis E, Colombo C, Davies J, Wark P, McKee C, Desouza C, et al. Preliminary safety and efficacy of triple-combination CFTR modulator regimens. Respirology 2018;23(Suppl 1):33. [ABSTRACT NO.: TO 026] [CFGD REGISTER: BD248c] [Google Scholar]
Davies 2021 {published data only}
- 2016-004479-35. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in subjects aged 6 through 11 years with cystic fibrosis, homozygous or heterozygous for the F508del-CFTR mutation. www.clinicaltrialsregister.eu/ctr-search/trial/2016-004479-35 (first received 17 May 2018). [DOI] [PubMed]
- Davies JC, Sermet-Gaudelus I, Naehrlich L, Harris RS, Campbell D, Ahluwalia N, et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. Journal of Cystic Fibrosis 2021;20(1):68-77. [CFGD REGISTER: BD253b] [DOI] [PubMed] [Google Scholar]
- NCT03559062. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in subjects aged 6 through 11 years with cystic fibrosis, homozygous or heterozygous for the F508del-CFTR mutation. https://trialsearch.who.int/Trial2.aspx?TrialID=NCT03559062 (date of first enrolment 17 May 2018). [DOI] [PubMed]
- NCT03559062. A study to evaluate efficacy and safety of tez/iva in subjects aged 6 through 11 years with cystic fibrosis [A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in subjects aged 6 through 11 years with cystic fibrosis, homozygous or heterozygous for the F508del-CFTR mutation]. clinicaltrials.gov/ct2/show/NCT03559062 (first received 15 June 2018). [CFGD REGISTER: BD253a]
Donaldson 2014 {published data only}
- Donaldson SH, Shoemaker S, Mandagere A, Troha J. Novel modifiers of CFTR: emerging clinical experience with GSNOR inhibitors. Pediatric Pulmonology 2014;49 Suppl 38:154. [ABSTRACT NO.: S10.2] [CENTRAL: 1015872] [CFGD REGISTER: BD217b] [Google Scholar]
- Donaldson SH, Taylor-Cousar JL, Rosenbluth D, Zeitlin P, Chmiel J, Jain M, et al. Safety, tolerability, and pharmacokinetics of the intravenous S-nitrosoglutathione reductase inhibitor N6022: an ascending-dose study in subjects homozygous for the F508DEL-CFTR mutation. Pediatric Pulmonology 2014;49 Suppl 38:308. [ABSTRACT NO.: 258] [CENTRAL: 1012386] [CFGD REGISTER: BD217a] [Google Scholar]
- NCT01746784. Safety and pharmacokinetic study of N6022 in subjects with cystic fibrosis homozygous for the F508del CFTR mutation (SNO1) [A phase 1b, randomized, double-blind, placebo-controlled, dose escalation study of N6022 to evaluate safety and pharmacokinetics in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation (SNO1)]. clinicaltrials.gov/ct2/show/NCT01746784 (first received 11 December 2012). [CLINICALTRIALS.GOV: NCT01746784]
Donaldson 2017 {published data only}
- Donaldson SH, Solomon GM, Zeitlin PL, Flume PA, Casey A, McCoy K, et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. Journal of Cystic Fibrosis 2017;16(3):371-9. [CFGD REGISTER: BD226b] [DOI] [PubMed] [Google Scholar]
- Donaldson SH, Solomon GM, Zeitlin PL, Flume PA, Casey A, McCoy K, et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. Journal of Cystic Fibrosis 2017;16(3):371-9. Online supplementary tables and figures. [CFGD REGISTER: BD226c] [DOI] [PubMed] [Google Scholar]
- Donaldson SH. Safety and pharmacokinetics of N91115 in patients with cystic fibrosis homozygous for the F508DEL-CFTR mutation. Pediatric Pulmonology 2015;50 Suppl 41:293. [ABSTRACT NO.: 270] [CENTRAL: 1092200] [CFGD REGISTER: BD226a] [Google Scholar]
- NCT02275936. Study of N91115 in patients with cystic fibrosis homozygous F508del-CFTR mutation (SNO4) [A phase 1b, randomized, double-blind, placebo-controlled, parallel, group study of N91115 to evaluate safety and pharmacokinetics in patients with cystic fibrosis homozygous for the F508del-CFTR mutation]. clinicaltrials.gov/ct2/show/NCT02275936 (first received 27 October 2014).
Donaldson 2018 {published data only}
- Donaldson S, Pilewski J, Griese M, Dong Q, Lee PS, for theVX11-661-101SG. VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508Del-CFTR mutation: interim analysis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S14. [ABSTRACT NO.: WS7.3] [CENTRAL: 872941] [CFGD REGISTER: BD190a] [Google Scholar]
- Donaldson SH, Pilewski JM, Cooke J, Himes-Lekstrom J, VX11-661-101 SG. Addition of VX-661, an investigational CFTR corrector, to ivacaftor, a CFTR potentiator, in patients with CF and heterozygous for F508DEL/G551D-CFTR. Pediatric Pulmonology 2014;49 Suppl 38:308-9. [ABSTRACT NO.: 260] [CENTRAL: 1012385] [CFGD REGISTER: BD190c] [Google Scholar]
- Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. American Journal of Respiratory and Critical Care Medicine 2018;197(2):214-24. [CFGD REGISTER: BD190e] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. American Journal of Respiratory and Critical Care Medicine 2018;197(2):214-24. Online data supplement. [CFGD REGISTER: BD190f] [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCTR2011-003821-93-DE. A phase 2, multicenter, double blinded, placebo controlled study to evaluate safety, efficacy, pharmacokinetics, and pharmacodynamics of VX-661 monotherapy and vx-661/ivacaftor cotherapy in subjects with cystic fibrosis, homozygous or heterozygous for the f508del CFTR mutation. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2011-003821-93-DE (first received 2011). [CENTRAL: CN-01882341] [CFGD REGISTER: BD190g]
- NCT01531673. Study of VX-661 alone and in combination with ivacaftor in subjects homozygous or heterozygous to the F508del-Cystic Fibrosis Transmembrane Conductance Regulator(CFTR) mutation [A phase 2, multicenter, double-blinded, placebo controlled study to evaluate safety, efficacy, pharmacokinetics, and pharmacodynamics of VX-661 monotherapy and VX-661/ivacaftor cotherapy in subjects with cystic fibrosis, homozygous or heterozygous for the F508del-CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01531673 (first received 13 February 2012).
- Pilewski JM, Cooke J, Lekstrom-Himes J, Donaldson S, for the VX-661IG. VX-661 in combination with ivacaftor in patients with cystic fibrosis and the F508del-CFTR mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S1. [ABSTRACT NO.: WS01.4] [CENTRAL: 1081479] [CFGD REGISTER: BD190d] [Google Scholar]
- Pilewski JM, Donaldson SH, Cooke J, Lekstrom-Himes J. Phase 2 studies reveal additive effects of VX-661, an investigational CFTR corrector, and ivacaftor, a CFTR potentiator, in patients with CF who carry the F508Del-CFTR mutation. Pediatric Pulmonology 2014;49 Suppl 38:157-9. [CENTRAL: 1015871] [CFGD REGISTER: BD190b] [Google Scholar]
Heijerman 2019 {published data only}
- Department of Error. Erratum: Department of Error (The Lancet (2019) 394(10212) (1940–1948), (S0140673619325978), (10.1016/S0140-6736(19)32597-8)). Lancet 2020;395(10238):1694. [CENTRAL: CN-02164017] [CFGD REGISTER: BD268c] [EMBASE: 2006013175] [PMID: 32473673] [Google Scholar]
- EUCTR2018-000184-89-BE. A phase 3 study of VX-445 combination therapy in CF subjects homozygous for f508del (F/F). www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2018-000184-89-BE (date started 27 September 2018). [CFGD REGISTER: BD268g]
- Heijerman H, McKone E, Downey DG, Mall M, Ramsey B, Rowe S, et al. Phase 3 efficacy and safety of the ELX/TEZ/ iva triple combination in people with CF homozygous for the F508del mutation. Pediatric Pulmonology 2019;54 Suppl 2:347. [CENTRAL: CN-01990652] [CFGD REGISTER: BD268a] [EMBASE: 629389111] [Google Scholar]
- Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394(10212):1940-8. [CENTRAL: CN-02006814] [CFGD REGISTER: BD268b] [DOI: 10.1016/S0140-6736(19)32597-8] [EMBASE: 2003873327] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoor C, Van Brunt K, Daines C, Durieu I, Fajac I, Goralski J, et al. Impact of elexacaftor/tezacaftor/ivacaftor triple combination therapy on health-related quality of life in people with cystic fibrosis homozygous for F508DEL (F/F): results from a phase 3 clinical study. Thorax 2021;76(Suppl 1):A41-2. [CFGD REGISTER: BD268h] [Google Scholar]
- Majoor CJ, Van Brunt K, Daines C, Durieu I, Fajac I, Goralski JL, et al. Impact of elexacaftor/tezacaftor/ ivacaftor triple combination therapy on health-related quality of life in people with cystic fibrosis homozygous for F508del: results from a phase 3 clinical study. Pediatric Pulmonology 2020;55(Suppl 2):225. [CFGD REGISTER: BD268f] [Google Scholar]
- NCT03525548. A study of VX-445 combination therapy in CF subjects homozygous for F508del (F/F). clinicaltrials.gov/ct2/show/NCT03525548 (first received 15 May 2018). [EUDRACT NUMBER: 2018-000184-89]
Horsley 2017 {published and unpublished data}
- Horsley A, Burr L, Kotsimbos T, Ledson M, Schwarz C, Simmonds N, et al. Safety, pharmacokinetics and pharmacodynamics of the CFTR corrector FDL169. Journal of Cystic Fibrosis 2018;17(Suppl 3):S42. [CFGD REGISTER: BD250a] [Google Scholar]
- Horsley AR, Blaas S, Burr L, Caroll M, Downey DG, Drevinek P, et al. Novel CFTR corrector FDL169: safety, pharmacokinetics and pharmacodynamics. Journal of Cystic Fibrosis 2018;17 Suppl 3:S42. [CFGD REGISTER: BD250c] [Google Scholar]
- Horsley AR, Blaas S, Burr L, Carroll M, Downey DG, Drevinek P, et al. Novel CFTR corrector FDL169: safety, pharmacokinetics and pharmacodynamics. Pediatric Pulmonology 2018;53 (S2):252. [CFGD REGISTER: BD250b] [Google Scholar]
Keating 2018 {published data only}2017‐000797‐11
- Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. New England Journal of Medicine 2018;379(17):1612-20. [CENTRAL: CN-01650324] [CFGD REGISTER: BD259] [EMBASE: 624591484] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03227471. A study of VX-445 in healthy subjects and subjects with cystic fibrosis [A phase 1/2 study of VX-445 in healthy subjects and subjects with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT03227471 (first posted 24 July 2017).
McCarty 2002 {published data only}
- Ahrens RC, Standaert TA, Launspach J, Han SH, Teresi ME, Aitken ML, et al. Use of nasal potential difference and sweat chloride as outcome measures in multicenter clinical trials in subjects with cystic fibrosis. Pediatric Pulmonology 2002;33(2):142-50. [CENTRAL: 385693] [CFGD REGISTER: BD136d] [DOI] [PubMed] [Google Scholar]
- Aitken ML, Ahrens RC, Karlin DA, Konstan MW, McNamara SC, Regelman WE, et al. Safety of a phase I double-blind placebo-controlled dose escalation trial of oral CPX in adult CF patients. Pediatric Pulmonology 1998;26 Suppl 17:276. [CENTRAL: 385694] [CFGD REGISTER: BD136b] [Google Scholar]
- McCarty NA, Standaert TA, Teresi M, Tuthill C, Launspach J, Kelley TJ, et al. A phase I randomized, multicenter trial of CPX in adult subjects with mild cystic fibrosis. Pediatric Pulmonology 2002;33(2):90-8. [CENTRAL: 377220] [CFGD REGISTER: BD136c] [PMID: ] [DOI] [PubMed] [Google Scholar]
- McCarty NA, Weatherly MR, Kelley TJ, Konstan MW, Milgram LJ, Teresi M, et al. Multicenter phase I trial of CPX in adults patients with mild CF: results of nasal potential difference measurements. Pediatric Pulmonology 1998;26 Suppl 17:276. [CENTRAL: 291449] [CFGD REGISTER: BD136a] [Google Scholar]
McKone 2021 {published data only}
- 2014-004838-25. A phase 3, randomized, double-blind, ivacaftor-controlled, parallel-group study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the F508del-CFTR mutation and a second CFTR allele with a gating defect that is clinically demonstrated to be ivacaftor responsive. https://www.clinicaltrialsregister.eu/ctr-search/trial/2014-004838-25/DE (first received 17 May 2016).
- McKone EF, DiMango EA, Sutharsan S, Barto TL, Campbell D, Ahluwalia N, et al. A phase 3, randomized, double-blind, parallel-group study to evaluate tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for F508del-CFTR and a gating mutation. Journal of Cystic Fibrosis 2021;20(2):234-42. [CFGD REGISTER: BD288b] [DOI] [PubMed] [Google Scholar]
- NCT02412111. A phase 3 study of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, who have one F508del-CFTR mutation and a second mutation that has been demonstrated to be clinically responsive to ivacaftor [A phase 3, randomized, double-blind, ivacaftor-controlled, parallel-group study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the F508del-CFTR mutation and a second CFTR allele with a gating defect that Is clinically demonstrated to be ivacaftor responsive]. clinicaltrials.gov/ct2/show/NCT02412111 (first received 8 April 2015). [CFGD REGISTER: BD288a]
- NCT02412111. A phase 3, randomized, double-blind, ivacaftor-controlled, parallel-group study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the F508del-CFTR mutation and a second CFTR allele with a gating defect that is clinically demonstrated to be ivacaftor responsive. https://trialsearch.who.int/Trial2.aspx?TrialID=NCT02412111 (date of first enrolment June 2015).
Middleton 2019 {published data only}
- EUCTR2018-000183-28-SE. A phase 3 study of VX-445 combination therapy in subjects with cystic fibrosis heterozygous for the f508del mutation and a minimal function mutation (F/MF). www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2018-000183-28-SE (date started 27 August 2018). [CFGD REGISTER: BD267f]
- Fajac I, Van Brunt K, Daines C, Durieu I, Goralski J, Heijerman H, et al. Impact of elexacaftor/tezacaftor/ivacaftor triple combination therapy on health-related quality of life in people with cystic fibrosis heterozygous for F508DEL and a minimal function mutation (F/MF): results from a phase 3 clinical study. Thorax 2021;76(Suppl 1):A40-1. [CFGD REGISTER: BD267g] [Google Scholar]
- Fajac I, Van Brunt K, Daines C, Durieu I, Goralski J, Heijerman H, et al. Impact of elexacaftor/tezacaftor/ivacaftor triple combination therapy on health-related quality of life in people with cystic fibrosis heterozygous for F508del and a minimal function mutation: results from a Phase 3 clinical study. Journal of Cystic Fibrosis 2020;19:S118-9. [CENTRAL: CN-02140149] [CFGD REGISTER: BD267c] [EMBASE: 2006056615] [Google Scholar]
- Fajac I, Van Brunt K, Daines C, Durieu I, Goralski JL, Heijerman H, et al. Impact of elexacaftor/tezacaftor/ ivacaftor triple combination therapy on health-related quality of life in people with cystic fibrosis heterozygous for F508del and a minimal function mutation: results from a phase 3 clinical study. Pediatric Pulmonology 2020;55 Suppl 2:213. [CFGD REGISTER: BD267e] [Google Scholar]
- Jain R, Mall M, Drevinek P, Lands L, McKone E, Polineni D, et al. Phase 3 efficacy and safety of the ELX/TEZ/ iva triple combination in people with CF and F508del/minimal function genotypes. Pediatric Pulmonology 2019;54 Suppl:346-7. [CENTRAL: CN-01987255] [CFGD REGISTER: BD267a] [EMBASE: 629389084] [Google Scholar]
- Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single phe508del allele. New England Journal of Medicine 2019;381(19):1809-19. [CENTRAL: CN-02004607] [CFGD REGISTER: BD267b] [DOI: 10.1056/NEJMoa1908639] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03525444. A phase 3 study of VX-445 combination therapy in subjects with cystic fibrosis heterozygous for the f508del mutation and a minimal function mutation (F/MF) [A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-445 combination therapy in subjects with cystic fibrosis who are heterozygous for the f508del mutation and a minimal function mutation (F/MF)]. clinicaltrials.gov/show/NCT03525444 (first received 15 May 2018). [CENTRAL: CN-01659552] [CFGD REGISTER: BD267d] [EUDRACT NUMBER: 2018-000183-28]
Munck 2020 {published data only}
- 2014-004787-37. A phase 3, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the F508del-CFTR mutation and with a second CFTR mutation that is not likely to respond to VX-661 and/or ivacaftor therapy (F508del/NR). https://www.clinicaltrialsregister.eu/ctr-search/trial/2014-004787-37/results (first version publication date 1 March 2017).
- Munck A, Kerem E, Ellemunter H, Campbell D, Wang LT, Ahluwalia N, et al. Tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for minimal function CFTR mutations. Journal of Cystic Fibrosis 2020;19(6):962-8. [CENTRAL: CN-02142331] [CFGD REGISTER: BD274b] [DOI: 10.1016/j.jcf.2020.04.015] [EMBASE: 2006729203] [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT02516410. A study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the F508del-CFTR mutation. clinicaltrials.gov/ct2/show/NCT02516410 (first received 5 August 2015). [CFGD REGISTER: BD274a]
NCT02070744 {published data only}
- NCT02070744. Study to evaluate safety and efficacy of VX-661 in combination with ivacaftor in subjects with cystic fibrosis, homozygous for the F508del-CFTR mutation with an open-label expansion [A phase 2, randomized, multicenter, double blind, placebo controlled study to evaluate safety, efficacy, pharmacokinetics, and pharmacodynamics of VX-661 in combination with ivacaftor for 12 weeks in subjects with cystic fibrosis, homozygous for the F508del CFTR mutation with an open-label extension]. clinicaltrials.gov/ct2/show/NCT02070744 (first received 25 February 2014). [CLINICALTRIALS.GOV: NCT02070744]
NCT02508207 {published data only}
- NCT02508207. A phase 2 study to evaluate effects of VX-661/Ivacaftor on lung and extrapulmonary systems in subjects with cystic fibrosis, homozygous for the F508del-CFTR mutation. clinicaltrials.gov/ct2/show/NCT02508207 (first received 24 July 2015).
NCT02730208 {published data only}
- 2019-002189-11. A phase 2, randomized, placebo-controlled, double-blind study to evaluate the effect of VX-661 in combination with ivacaftor on chest imaging endpoints in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del-CFTR mutation. https://www.clinicaltrialsregister.eu/ctr-search/trial/2019-002189-11/results (this version publication date 7 November 2019).
- NCT02730208. A study to evaluate the effect of VX-661 in combination with ivacaftor on chest imaging endpoints in subjects with cystic fibrosis, homozygous for the F508del CFTR mutation [A phase 2, randomized, placebo-controlled, double-blind study to evaluate the effect of VX-661 in combination with ivacaftor on chest imaging endpoints in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del CFTR mutation]. clinicaltrials.gov/ct2/show/NCT02730208 (first received 6 April 2016).
NCT02951182 {published data only}
- 2016-000454-36. A phase 2, randomized, double-blind, controlled study to evaluate the safety and efficacy of VX-440 combination therapy in subjects aged 12 years and older with cystic fibrosis. www.clinicaltrialsregister.eu/ctr-search/trial/2016-000454-36/results (first published 1 December 2018). [CFGD REGISTER: BD305c]
- EUCTR2016-000454-36-AT. A study evaluating the safety and efficacy of VX-440 combination therapy in subjects with cystic fibrosis. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2016-000454-36-AT (first received 7 October 2016). [CFGD REGISTER: BD305b]
- NCT02951182. A study evaluating the safety and efficacy of VX-440 combination therapy in subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT02951182 (first received 1 November 2016). [CFGD REGISTER: BD305a]
NCT02951195 {published data only}
- NCT02951195. A study evaluating the safety of VX-152 combination therapy in adults with cystic fibrosis [A phase 2, randomized, double blind, controlled study to evaluate the safety of VX-152 combination therapy in adults with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02951195 (first received 1 November 2016). [CFGD REGISTER: BD297]
NCT03447249 {published data only}
- 2017-004132-11. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-659 combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a minimal function mutation (F/MF). www.clinicaltrialsregister.eu/ctr-search/trial/2017-004132-11 (first received 1 October 2018).
- NCT03447249. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-659 combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a minimal function mutation (F/MF). clinicaltrials.gov/ct2/show/NCT03447249 (first received 27 February 2018).
- NCT03447249. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-659 combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a minimal function mutation (F/MF). https://trialsearch.who.int/Trial2.aspx?TrialID=NCT03447249 (date of first enrolment 7 March 2018).
NCT03460990 {published data only}
- EUCTR2017-004133-82-GB. A study evaluating safety and efficacy of VX-659 combination therapy in subjects with cystic fibrosis. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2017-004133-82-GB (date started 1 May 2018). [CFGD REGISTER: BD294b]
- EUCTR2017-004133-82-IE. A study evaluating safety and efficacy of VX-659 combination therapy in subjects with cystic fibrosis. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2017-004133-82-IE (date started 1 May 2018). [CFGD REGISTER: BD294c]
- NCT03460990. A study of VX-659 combination therapy in CF subjects homozygous for F508del (F/F). clinicaltrials.gov/ct2/show/NCT03460990 (first received 9 March 2018). [CFGD REGISTER: BD294a] [EUDRACT NUMBER: 2017-004133-82]
PROGRESS 2017 {published data only}
- Konstan M, McKone E, Moss R, Marigowda G, Cooke J, Lubarsky B, et al. Evidence of reduction in annual rate of FEV1 decline and sustained benefits with lumacaftor and ivacaftor (LUM/IVA) in patients (pts) with cf homozygous for F508DEL-CFTR. Pediatric Pulmonology 2016;51 Suppl 45:260. [ABSTRACT NO.: 180] [CFGD REGISTER: BD213p // BD214p] [Google Scholar]
- Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet. Respiratory Medicine 2017;5(2):107-18. [CFGD REGISTER: BD213q // BD214q] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet. Respiratory Medicine 2017;5(2):107-18. Online supplementary appendix. [CFGD REGISTER: BD213r // BD214r] [DOI] [PubMed] [Google Scholar]
- NCT01931839. A phase 3 rollover study of lumacaftor in combination with ivacaftor in subjects 12 years and older with cystic fibrosis [A phase 3, rollover study to evaluate the safety and efficacy of long-term treatment with lumacaftor in combination with ivacaftor in subjects aged 12 Years and older with cystic fibrosis, homozygous or heterozygous for the F508del-CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01931839 (first received 29 August 2013). [CLINICALTRIALS.GOV: NCT01931839]
Ratjen 2017 {published data only}
- Anonymous. Corrections [Corrections: efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial (The Lancet Respiratory Medicine (2017) 5(7) (557-567)(S2213260017302151)(10.1016/S2213-2600(17)30215-1))]. Lancet Respiratory Medicine 2017 Aug;5(8):e28. [CENTRAL: CN-01473292] [CFGD REGISTER: BD233k] [EMBASE: 617478198] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Brody A, Nagle SK, Owen C, Marigowda G, Waltz D, Goldin J, et al. Effect of lumacaftor/ivacaftor on total, bronchiectasis, and air trapping computed tomography scores in children homozygous for F508DEL-CFTR: exploratory imaging substudy. Pediatric Pulmonology 2017;52 Suppl 47:286. [CFGD REGISTER: BD233d] [Google Scholar]
- Brody AS, Nagle S, Hug C, Marigowda G, Waltz D, Goldin J, et al. Effect of lumacaftor/ivacaftor on total, bronchiectasis and air trapping computed tomography (CT) scores in children homozygous for F508del-CFTR: exploratory imaging substudy. Thorax 2017;72(Suppl 3):A57. [CFGD REGISTER: BD233e] [Google Scholar]
- Brody AS, Nagle S, Hug C, Marigowda G, Waltz D, Goldin J, et al. Effect of lumacaftor/ivacaftor on total, bronchiectasis, and air trapping computed tomography (CT) scores in children homozygous for F508del-CFTR: exploratory imaging substudy. Thorax 2017;72(Suppl 3):A57. [CENTRAL: CN-01643820] [CFGD REGISTER: BD233e] [EMBASE: 619739057] [Google Scholar]
- Milla C, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M. Safety, tolerability, and pharmacodynamics of combination lumacaftor/ivacaftor therapy in patients aged 6-11 yrs with CF homozygous for the F508DEL-CFTR mutation. Pediatric Pulmonology 2016;51 Suppl:259. [CENTRAL: CN-01212610] [CFGD REGISTER: BD233j] [EMBASE: 612358598] [Google Scholar]
- NCT02514473. A study to evaluate the efficacy and safety of lumacaftor in combination with ivacaftor in subjects with CF, homozygous for the f508del-cftr mutation. clinicaltrials.gov/show/NCT02514473 (first received 3 August 2015). [CFGD REGISTER: BD233l]
- Nagle S, Brody AS, Woods J, Johnson KM, Wang L, Marigowda G, et al. Feasibility of ultrashort echo time (UTE) MRI to evaluate the effect of lumacaftor/ivacaftor therapy in children with cystic fibrosis (CF) homozygous for F508DEL. Thorax 2017;72(Suppl 3):A221. [CENTRAL: CN-01643817] [CFGD REGISTER: BD233g] [EMBASE: 619738999] [Google Scholar]
- Nagle SK, Brody A, Woods JC, Johnson KM, Wang L, Marigowda G, et al. Feasibility of ultrashort echo time MRI to evaluate the effect of lumacaftor/ ivacaftor therapy in children with cystic fibrosis homozygous for f508del. Pediatric Pulmonology 2017;52 Suppl 47:314-5. [CENTRAL: CN-01622615] [CFGD REGISTER: BD233f] [EMBASE: 619069874] [Google Scholar]
- Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet. Respiratory Medicine 2017;5(7):557-67. Online supplementary appendix. [CFGD REGISTER: BD233c] [DOI] [PubMed] [Google Scholar]
- Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous forF508del-CFTR: a randomised, placebo-controlled, phase 3 trial. Lancet Respiratory Medicine 2017;5(7):557-67. [CFGD REGISTER: BD233b] [DOI] [PubMed] [Google Scholar]
- Ratjen F, Tian S, Marigowda G, Hug C, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor/ivacaftor (LUM/IVA) in patients (pts) aged 6-11 years (yrs) with cystic fibrosis (CF) homozygous for F508del-CFTR: a randomized placebo (PBO)-controlled phase 3 trial. Journal of Cystic Fibrosis 2017;16 Suppl 1:S24. [ABSTRACT NO.: WS13.4] [CENTRAL: 1383249] [CFGD REGISTER: BD233a] [Google Scholar]
- Wainwright C, Brody A, Nagle S, Hug C, Marigowda G, Waltz D, et al. Effect of lumacaftor/ivacaftor on ct scores: exploratory imaging substudy. Respirology 2018;23(Suppl 1):57. [CENTRAL: CN-01920038] [CFGD REGISTER: BD233h] [EMBASE: 622091817] [Google Scholar]
- Wainwright C, Nagle S, Brody A, Woods J, Johnson K, Wang L, et al. Ultrashort echo time MRI can evaluate treatment effect of lumacaftor/Ivacaftor. Respirology 2018;23(Suppl 1):141. [CENTRAL: CN-01919971] [CFGD REGISTER: BD233i] [EMBASE: 622091399] [Google Scholar]
Rubenstein 1998 {published data only}
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. American Journal of Respiratory and Critical Care Medicine 1998;157(2):484-90. [CENTRAL: 201485] [CFGD REGISTER: BD146b] [EMBASE: 1998064104] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A randomized, double blind, placebo-controlled trial of sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Pediatric Pulmonology 1997;Suppl 14:272. [CENTRAL: 291563] [CFGD REGISTER: BD146a] [DOI] [PubMed] [Google Scholar]
Schwarz 2021 {published data only}
- 2017-000540-18. Phase 3b, randomized, double-blind, placebo-controlled, parallel group study to assess the safety, efficacy, and tolerability of tezacaftor/ivacaftor (TEZ/IVA) in an Orkambi-experienced population who are homozygous for the F508del-CFTR mutation. https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-000540-18/results (this version publication date 3 January 2020).
- EUCTR2017-000540-18-DE. Phase 3b, randomized, double-blind, placebo-controlled, parallel group study to assess the safety, efficacy, and tolerability of tezacaftor/ivacaftor (TEZ/IVA) in an Orkambi-experienced population who are homozygous for the F508del-CFTR mutation. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2017-000540-18-DE (first received 13 September 2017).
- NCT03150719. A study to evaluate safety, efficacy, and tolerability of TEZ/IVA in Orkambi® (lumacaftor/ivacaftor) - experienced subjects with cystic fibrosis (CF) [Phase 3b, randomized, double-blind, placebo-controlled, parallel group study to assess the safety, efficacy, and tolerability of tezacaftor/ivacaftor (TEZ/IVA) in an Orkambi-experienced population who are homozygous for the F508del CFTR mutation]. clinicaltrials.gov/ct2/show/NCT03150719 (first posted 12 May 2017). [CFGD REGISTER: BD273a]
- Schwarz C, Sutharsan S, Epaud R, Klingsberg R, Fischer R, Rowe SM, et al. Safety, efficacy, and tolerability of tezacaftor/ivacaftor in cystic fibrosis patients who previously discontinued lumacaftor/ivacaftor due to respiratory adverse events: a randomized, double-blind, placebo-controlled phase 3b study. Pneumologie 2019;73(Suppl 1):P13. [CENTRAL: CN-01960602] [CFGD REGISTER: BD273b] [DOI: 10.1055/s-0039-1678160] [EMBASE: 628475379] [DOI] [Google Scholar]
- Schwarz C, Sutharsan S, Epaud R, Klingsberg RC, Fischer R, Rowe SM, et al. Tezacaftor/ivacaftor in people with cystic fibrosis who stopped lumacaftor/ivacaftor due to respiratory adverse events. Journal of Cystic Fibrosis 2021;20(2):228-33. [CENTRAL: CN-02143375] [CFGD REGISTER: BD273c] [DOI: 10.1016/j.jcf.2020.06.001] [EMBASE: 2006829285] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stahl 2021 {published data only}
- EUCTR2017-003761-99-DE. A study of the effects of lumacaftor/ivacaftor on disease progression in subjects aged 2 through 5 years with cystic fibrosis, homozygous for f508del [An exploratory phase 2, 2-part, randomized, double blind, placebo controlled study with a long term, open label period to explore the impact of lumacaftor/ivacaftor on disease progression in subjects aged 2 through 5 years with cystic fibrosis, homozygous for f508del]. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2017-003761-99-DE (first received 2018). [CENTRAL: CN-01908852] [CFGD REGISTER: BD276b]
- NCT03625466. A study to explore the impact of lumacaftor/ivacaftor on disease progression in subjects aged 2 through 5 years with cystic fibrosis, homozygous for f508del [An exploratory phase 2, 2-part, randomized, double-blind, placebo-controlled study with a long-term, open-label period to explore the impact of lumacaftor/ivacaftor on disease progression in subjects aged 2 through 5 years with cystic fibrosis, homozygous for f508del]. clinicaltrials.gov/show/NCT03625466 (first received 10 August 2018). [CENTRAL: CN-01626171] [CFGD REGISTER: BD276a]
- Stahl M, Roehmel J, Eichinger M, Doellinger F, Naehrlich L, Kopp MV, et al. An exploratory study to determine the impact of lumacaftor/ivacaftor (LUM/IVA) on disease progression in children 2 through 5 years of age with cystic fibrosis homozygous for F508del-CFTR (F/F). Journal of Cystic Fibrosis 2021;20 Suppl 1:S22. [CFGD REGISTER: BD276d] [Google Scholar]
- Stahl M, Roehmel J, Eichinger M, Doellinger F, Naehrlich L, Kopp MV, et al. Long-term efficacy of lumacaftor/ivacaftor (LUM/IVA) in children aged 2 through 5 years with cystic fibrosis (CF) homozygous for the F508del-CFTR mutation (F/F): a phase 2, open-label extension study. Journal of Cystic Fibrosis 2022;21 Suppl 1:S32-3. [CFGD REGISTER: BD276c] [Google Scholar]
Sutharsan 2022 {published data only}
- EUCTR2019-001735-31-GB. A study to evaluate the safety and efficacy of VX-445 / tezacaftor / ivacaftor in patients suffering from cystic fibrosis. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2019-001735-31-GB (date started 13 September 2019). [CFGD REGISTER: BD292b]
- NCT04105972. A study evaluating the efficacy and safety of VX-445/tezacaftor/ivacaftor in cystic fibrosis subjects, homozygous for F508del. clinicaltrials.gov/ct2/show/NCT04105972 (first posted 26 September 2019). [CFGD REGISTER: BD292a]
- Sutharsan S, McKone EF, Downey DG, Duckers J, MacGregor G, Tullis E, et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. The Lancet. Respiratory Medicine 2022;10(3):267-77. [CFGD REGISTER: BD292c] [DOI: 10.1016/S2213-2600(21)00454-9] [DOI] [PubMed] [Google Scholar]
Taylor‐Cousar 2017 {published data only}
- Flume P, Lekstrom-Himes J, Fischer Biner R, Simard C, Downey DG, Zhou H, et al. A phase 3, open-label study of tezacaftor/ivacaftor (TEZ/IVA) therapy, interim analysis of pooled safety, and efficacy in patients homozygous for F508del-CFTR. Journal of Cystic Fibrosis 2018;17 Suppl 3:S64-5. [CFGD REGISTER: BD236d] [Google Scholar]
- Ingenito E, Nair N, Yi B, Lekstrom-Himes J, Elborn JS, Rowe SM. Retrospective analysis of physiological response patterns to tezacaftor/ivacaftor in patients with cystic fibrosis homozygous for F508DEL-CFTR or heterozygous for F508DEL-CFTR and a residual function mutation. Thorax 2018;73(Suppl 4):A42-3. [CENTRAL: CN-02002310] [CFGD REGISTER: BD236i // BD237h] [EMBASE: 627697348] [Google Scholar]
- NCT02347657. A randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of VX-661 in combination with ivacaftor. clinicaltrials.gov/show/NCT02347657 (first received 27 January 2015). [CFGD REGISTER: BD236k]
- Smith D, Flume P, Lekstrom-Himes J, Fischer Biner R, Simard C, Downey D, et al. Phase 3 interim analysis: tezacaftor/ivacaftor (TEZ/IVA) in patients homozygous for F508delcystic fibrosis transmembrane conductance regulator (CFTR). Respirology 2019;24(S1):30. [ABSTRACT NO.: TO017] [CENTRAL: CN-02002181] [CFGD REGISTER: BD236j] [EMBASE: 626940397] [Google Scholar]
- Sommerburg O, Yang Y, Rizio AA, Loop B, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor (TEZ/IVA) treatment in patients with cystic fibrosis and F508del/F508del-CFTR: patient-reported outcomes in a Phase 3, randomised, controlled trial (EVOLVE). Pneumologie 2019;73(Suppl 1):P13. [CENTRAL: CN-01960604] [CFGD REGISTER: BD236h] [DOI: 10.1055/s-0039-1678165] [EMBASE: 628475389] [DOI] [Google Scholar]
- Sutharsan S, Taylor-Cousar J, Lekstrom-Himes J, Wang L, Lu Y, Elborn JS. Efficacy and safety of tezacaftor/ivacaftor in patients aged >= 12 years with CF homozygous for F508del-CFTR: a randomized placebo (PBO)-controlled phase 3 trial. Pneumologie (Stuttgart, Germany) 2018;72(Suppl 1):S36. [CFGD REGISTER: BD236e] [Google Scholar]
- Taylor-Cousar JL, Elborn S. Advances in treating patients homozygous for F508del. Pediatric Pulmonology 2017;52 Suppl 47:173-5. [CFGD REGISTER: BD236c] [Google Scholar]
- Taylor-Cousar JL, Lekstrom-Himes J, Wang L, Lu Y, Elborn S. Efficacy and safety of tezacaftor/ ivacaftor in patients aged >=12 years with cf homozygous for f508del-cftr: a randomized placebo-controlled phase 3 trial. Pediatric Pulmonology 2017;52 Suppl 47:307. [CFGD REGISTER: BD236a] [Google Scholar]
- Taylor-Cousar JL, Munck A, McKone EF, Ent CK, Moeller A, Simard C, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. New England Journal of Medicine 2017;377(21):2013-23. [CFGD REGISTER: BD236b] [DOI: 10.1056/NEJMoa1709846] [DOI] [PubMed] [Google Scholar]
- Yang Y, Rizio A, Chuang C, Loop B, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor treatment in patients with cystic fibrosis and F508del/F508del-CFTR: patient-reported outcomes in a phase 3 randomized, controlled trial. Pediatric Pulmonology 2018;53(S2):264. [CFGD REGISTER: BD236f] [Google Scholar]
- Yang Y, Rizio AA, Chuang C-C, Loop B, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor (TEZ/IVA) treatment in patients with cystic fibrosis homozygous for F508DEL-CFTR: patient-reported outcomes in a phase 3 randomized, controlled trial (EVOLVE). Thorax 2018;73(Suppl 4):A42. [CENTRAL: CN-01936246] [CFGD REGISTER: BD236g] [DOI: 10.1136/thorax-2018-212555.74] [EMBASE: 627697250] [DOI] [Google Scholar]
TRAFFIC 2015 {published data only}
- De Boeck C. Long-term clinical effects of CFTR co-therapy with lumacaftor/ivacaftor. Pediatric Pulmonology 2015;50:135-7. [CENTRAL: 1163954] [CFGD REGISTER: BD213m/BD214m] [EMBASE: 72081237] [SYMPOSIUM SUMMARY: S9.1] [Google Scholar]
- De Boeck K, Elborn J, Ramsey B, Boyle MP, Konstan MW, Huang X, et al. Efficacy and safety of lumacaftor+ivacaftor combination therapy in patients with CF homozygous for F508DEL-CFTR by FEV1 subgroups. Pediatric Pulmonology 2015;50 Suppl 41:283. [ABSTRACT NO.: 245] [CENTRAL: 1092180] [CFGD REGISTER: BD213f/BD214f] [Google Scholar]
- Elborn J, Wainwright CE, Ramsey B, Huang X, Margowda G, Waltz D, et al. Effect of lumacaftor in combination with ivacaftor in patients with cystic fibrosis who are homozygous for F508-DEL-CFTR: the TRAFFIC Study. Pediatric Pulmonology 2014;49 Suppl 38:304. [ABSTRACT NO.: 249] [CENTRAL: 1012382] [CFGD REGISTER: BD213a] [Google Scholar]
- Elborn JS, Ramsey B, Boyle MP, Wainwright C, Konstan M, Huang X, et al. Lumacaftor in combination with ivacaftor in patients with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S1. [ABSTRACT NO.: WS01.3] [CENTRAL: 1077209] [CFGD REGISTER: BD213e/BD214d] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet. Respiratory Medicine 2016;4(8):617-26. [CENTRAL: 1157425] [CFGD REGISTER: BD213i/BD214i] [DOI: 10.1016/S2213-2600(16)30121-7] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet. Respiratory Medicine 2016;4(8):617-26. Online supplementary appendix. [CFGD REGISTER: BD213o/BD214o] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Wainwright CE, Konstan MW, Huang X, et al. Lumacaftor/ ivacaftor combination therapy in CF patients homozygous for F508del-CFTR with severe lung dysfunction. Journal of Cystic Fibrosis 2015;14 Suppl 1:S94. [ABSTRACT NO.: 143] [CENTRAL: 1077207] [CFGD REGISTER: BD213c/BD214d] [Google Scholar]
- Flume PA, Suthoff ED, Kosinski M, Marigowda G, Quittner AL. Measuring recovery in health-related quality of life during and after pulmonary exacerbations in patients with cystic fibrosis. Journal of Cystic Fibrosis 2019;18(5):737-42. [CENTRAL: CN-01742131] [CFGD REGISTER: BD213u // BD214u] [DOI: 10.1016/j.jcf.2018.12.004] [EMBASE: 2001400109] [DOI] [PubMed] [Google Scholar]
- Konstan M, McKone E, Moss R, Marigowda G, Cooke J, Lubarsky B, et al. Evidence of reduction in annual rate of FEV1 decline and sustained benefits with lumacaftor and ivacaftor (LUM/IVA) in patients (pts) with cf homozygous for F508DEL-CFTR. Pediatric Pulmonology 2016;51 Suppl 45:260. [ABSTRACT NO: 180] [CFGD REGISTER: BD213p // BD214p] [Google Scholar]
- Konstan MW, Ramsey BW, Elborn J, Boyle MP, Waltz D, Marigowda G, et al. Safety and efficacy of treatments with lumacaftor in combination with ivacaftor in patients with CF homozygous for F508DEL-CFTR. Pediatric Pulmonology 2015;50 Suppl 41:269. [ABSTRACT NO.: 211] [CENTRAL: 1092192] [CFGD REGISTER: BD213h/BD214h] [Google Scholar]
- McColley SA, Konstan MW, Ramsey BW, Elborn J, Boyle MP, Wainwright CE, et al. Association between changes in percent predicted FEV1 and incidence of pulmonary exacerbations, including those requiring hospitalization and/ or IV antibiotics, in patients with CF treated with lumacaftor in combination with ivacaftor. Pediatric Pulmonology 2015;50 Suppl 41:282. [ABSTRACT NO.: 241] [CENTRAL: 1092182] [CFGD REGISTER: BD213g/BD214g] [Google Scholar]
- McColley SA, Konstan MW, Ramsey BW, Stuart Elborn J, Boyle MP, Wainwright CE, et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. Journal of Cystic Fibrosis 2019;18(1):94-101. [CENTRAL: CN-01921335] [CFGD REGISTER: BD213t // BD214t] [EMBASE: 2001047042] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01807923. A study of lumacaftor in combination with ivacaftor in cystic fibrosis subjects aged 12 years and older who are homozygous for the F508del-CFTR mutation (TRAFFIC) [A phase 3, randomized, double blind, placebo controlled, parallel group study to evaluate the efficacy and safety of lumacaftor in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01807923 (first received 8 March 2013). [CLINICALTRIALS.GOV: NCT01807923]
- Seliger V, Bai Y, Volkova N, Tian S, Waltz. Prevalance of cataracts in a population of cystic fibrosis patients homozygous for the F508del mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S108. [ABSTRACT NO.: 196] [CENTRAL: 1077208] [CFGD REGISTER: BD213d/BD214c] [Google Scholar]
- Solem CT, Vera-Llonch M, Tai M, O’Callaghan L. Pulmonary exacerbations, lung dysfunction, and Eq-5d measures in adolescents and adults with cystic fibrosis and homozygous for the F508del-Cftr mutation. Value In Health 2016;19(3):PA116-7. [CFGD REGISTER: BD213s // BD214s] [DOI: 10.1016/j.jval.2016.03.461] [DOI] [Google Scholar]
- Sullivan JC, Accurso FJ, Marigowda G, Beusmans J, Geho D, Zhang E, et al. Combination lumacaftor/ivacaftor therapy improves inflammatory biomarkers in patients with cf homozygous for the f508DEL-CFTR mutations. Pediatric Pulmonology 2016;51 Suppl 45:274. [ABSTRACT NO.: 218] [CFGD REGISTER: BD213n/BD214n] [Google Scholar]
- Sullivan JC, Accurso FJ, Marigowda G, Beusmans J, Geho D, Zhang E, et al. Improvement in inflammatory biomarkers in patients (pts) wth cystic fibrosis (CF) homozygous for the F508del-CFTR mutation treated with lumacaftor (LUM) and ivacaftor (IVA). Journal of Cystic Fibrosis 2016;15 Suppl 1:S6. [ABSTRACT NO.: WS04.2] [CENTRAL: 1157463] [CFGD REGISTER: BD213k/BD214k] [Google Scholar]
- Suthoff ED, Kosinski M, Sikirica S, Quittner AL, Flume P. Impact of pulmonary exacerbations (PEx) on health-related quality of life (HRQoL) assessed by the Cystic Fibrosis Questionnaire-Revised (CFQ-R) in TRAFFIC and TRANSPORT. Journal of Cystic Fibrosis 2016;15 Suppl 1:S86. [ABSTRACT NO.: 138] [CENTRAL: 1157462] [CFGD REGISTER: BD213j/BD214j] [Google Scholar]
- Wainwright CE, Elborn JS, Ramsey B, Huang X, Marigowda G, Waltz D, et al. Effect of lumacaftor in combination with ivacaftor in patients with cf who are homozygous for F508-CFTR: phase 3 TRAFFIC and TRANSPORT studies. Pediatric Pulmonology 2014;49 Suppl 38:156. [ABSTRACT NO.: S10.3] [CENTRAL: 1383236] [CFGD REGISTER: BD213L/BD214L] [Google Scholar]
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. New England Journal of Medicine 2015;373(3):220-31. [CENTRAL: 1067433] [CFGD REGISTER: BD213b/BD214b] [5500133000000031] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
TRANSPORT 2015 {published data only}
- De Boeck C. Long-term clinical effects of CFTR co-therapy with lumacaftor/Ivacaftor. Pediatric Pulmonology 2015;50:135-7. [CENTRAL: 1163954] [CFGD REGISTER: BD214m/BD213m] [EMBASE: 72081237] [SYMPOSIUM SUMMARY: S9.1] [Google Scholar]
- De Boeck K, Elborn J, Ramsey B, Boyle MP, Konstan MW, Huang X, et al. Efficacy and safety of lumacaftor+ivacaftor combination therapy in patients with CF homozygous for F508DEL-CFTR by FEV1 subgroups. Pediatric Pulmonology 2015;50 Suppl 41:283. [ABSTRACT NO.: 245] [CENTRAL: 1092180] [CFGD REGISTER: BD214f/BD213f] [Google Scholar]
- Elborn JS, Ramsey B, Boyle MP, Wainwright C, Konstan M, Huang X, et al. Lumacaftor in combination with ivacaftor in patients with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S1. [ABSTRACT NO.: WS01.3] [CENTRAL: 1077209] [CFDG REGISTER: BD214d/BD213e] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet. Respiratory Medicine 2016;4(8):617-26. [CENTRAL: 1157425] [CFGD REGISTER: BD214i/BD213i] [DOI: 10.1016/S2213-2600(16)30121-7] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet. Respiratory Medicine 2016;4(8):617-26. Online supplementary appendix. [CFGD REGISTER: BD214o/BD213o] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborn JS, Ramsey BW, Boyle MP, Wainwright CE, Konstan MW, Huang X, et al. Lumacaftor/ ivacaftor combination therapy in CF patients homozygous for F508del-CFTR with severe lung dysfunction. Journal of Cystic Fibrosis 2015;14 Suppl 1:S94. [ABSTRACT NO.: 143] [CENTRAL: 1077207] [CFGD REGISTER: BD214d/BD213c] [Google Scholar]
- Flume PA, Suthoff ED, Kosinski M, Marigowda G, Quittner AL. Measuring recovery in health-related quality of life during and after pulmonary exacerbations in patients with cystic fibrosis. Journal of Cystic Fibrosis 2019;18(5):737-42. [CENTRAL: CN-01742131] [CFGD REGISTER: BD213u // BD214u] [DOI: 10.1016/j.jcf.2018.12.004] [EMBASE: 2001400109] [DOI] [PubMed] [Google Scholar]
- Konstan M, McKone E, Moss R, Marigowda G, Cooke J, Lubarsky B, et al. Evidence of reduction in annual rate of FEV1 decline and sustained benefits with lumacaftor and ivacaftor (LUM/IVA) in patients (pts) with cf homozygous for F508DEL-CFTR. Pediatric Pulmonology 2016;51 Suppl 45:260. [ABSTRACT NO: 180] [CFGD REGISTER: BD213p // BD214p] [Google Scholar]
- Konstan MW, Ramsey BW, Elborn J, Boyle MP, Waltz D, Marigowda G, et al. Safety and efficacy of treatments with lumacaftor in combination with ivacaftor in patients with CF homozygous for F508DEL-CFTR. Pediatric Pulmonology 2015;50 Suppl 41:269. [ABSTRACT NO.: 211] [CENTRAL: 1092192] [CFGD REGISTER: BD214h/BD213h] [Google Scholar]
- McColley SA, Konstan MW, Ramsey BW, Elborn J, Boyle MP, Wainwright CE, et al. Association between changes in percent predicted FEV1 and incidence of pulmonary exacerbations, including those requiring hospitalization and/ or IV antibiotics, in patients with CF treated with lumacaftor in combination with ivacaftor. Pediatric Pulmonology 2015;50 Suppl 41:282. [ABSTRACT NO.: 241] [CENTRAL: 1092182] [CFGD REGISTER: BD214g/BD213g] [Google Scholar]
- McColley SA, Konstan MW, Ramsey BW, Stuart Elborn J, Boyle MP, Wainwright CE, et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. Journal of Cystic Fibrosis 2019;18(1):94-101. [CENTRAL: CN-01921335] [CFGD REGISTER: BD213t // BD214t] [EMBASE: 2001047042] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01807949. A study of lumacaftor in combination with ivacaftor in cystic fibrosis subjects aged 12 years and older who are homozygous for the F508del-CFTR Mutation (TRANSPORT) [A phase 3, randomized, double blind, placebo controlled, parallel group study to evaluate the efficacy and safety of lumacaftor in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01807949 (first received 8 March 2013). [CLINICALTRIALS.GOV: NCT01807949]
- Ramsey B, Boyle MP, Elborn J, Huang X, Marigowda G, Waltz D, et al. Effect of lumacaftor in combination with ivacaftor in patients with cystic fibrosis who are homozygous for F508DEL-CFTR: TRANSPORT Study. Pediatric Pulmonology 2014;49 Suppl 38:305. [ABSTRACT NO.: 250] [CENTRAL: 1012383] [CFGD REGISTER: BD214a] [Google Scholar]
- Seliger V, Bai Y, Volkova N, Tian S, Waltz. Prevalance of cataracts in a population of cystic fibrosis patients homozygous for the F508del mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S108. [ABSTRACT NO.: 196] [CENTRAL: 1077208] [CFGD REGISTER: BD214c/BD213d] [Google Scholar]
- Solem CT, Vera-Llonch M, Tai M, O’Callaghan L. Pulmonary exacerbations, lung dysfunction, and Eq-5d measures in adolescents and adults with cystic fibrosis and homozygous for the F508del-Cftr mutation. Value In Health 2016;19(3):PA116-7. [CFGD REGISTER: BD213s // BD214s] [DOI: 10.1016/j.jval.2016.03.461] [DOI] [Google Scholar]
- Sullivan JC, Accurso FJ, Marigowda G, Beusmans J, Geho D, Zhang E, et al. Combination lumacaftor/ivacaftor therapy improves inflammatory biomarkers in patients with cf homozygous for the f508DEL-CFTR mutations. Pediatric Pulmonology 2016;51 Suppl 45:274. [ABSTRACT NO.: 218] [CFGD REGISTER: BD214n/BD213n] [Google Scholar]
- Sullivan JC, Accurso FJ, Marigowda G, Beusmans J, Geho D, Zhang E, et al. Improvement in inflammatory biomarkers in patients (pts) wth cystic fibrosis (CF) homozygous for the F508del-CFTR mutation treated with lumacaftor (LUM) and ivacaftor (IVA). Journal of Cystic Fibrosis 2016;15 Suppl 1:S6. [ABSTRACT NO.: WS04.2] [CENTRAL: 1157463] [CFGD REGISTER: BD214k/BD213k] [Google Scholar]
- Suthoff ED, Kosinski M, Sikirica S, Quittner AL, Flume P. Impact of pulmonary exacerbations (PEx) on health-related quality of life (HRQoL) assessed by the Cystic Fibrosis Questionnaire-Revised (CFQ-R) in TRAFFIC and TRANSPORT. Journal of Cystic Fibrosis 2016;15 Suppl 1:S86. [ABSTRACT NO.: 138] [CENTRAL: 1157462] [CFGD REGISTER: BD214j/BD213j] [Google Scholar]
- Wainwright CE, Elborn JS, Ramsey B, Huang X, Marigowda G, Waltz D, et al. Effect of lumacaftor in combination with ivacaftor in patients with CF who are homozygous for F508-CFTR: phase 3 TRAFFIC and TRANSPORT studies. Pediatric Pulmonology 2014;49 Suppl 38:156. [ABSTRACT NO.: S10.3] [CENTRAL: 1383236] [CFGD REGISTER: BD214L/BD213L] [Google Scholar]
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. New England Journal of Medicine 2015;373(3):220-31. [CENTRAL: 1067433] [CFGD REGISTER: BD214b/BD213b] [5500133000000031] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilson 2021 {published data only}
- 2016-000066-34. A phase 4, randomized, double-blind, placebo-controlled, parallel-design study of the effect of lumacaftor/ivacaftor combination therapy on exercise tolerance in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del-CFTR mutation. https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-000066-34/results (this version publication date 2 February 2019).
- EUCTR2016-000066-34-GB. A phase 4, randomized, double-blind, placebo-controlled, parallel-design study of the effect of lumacaftor/ivacaftor combination therapy on exercise tolerance in subjects aged 12 years and older with cystic fibrosis, homozygous for the F508del-CFTR mutation. https://trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2016-000066-34-GB (first received 25 July 2017).
- NCT02875366. A study of the effects of lumacaftor/ivacaftor (LUM/IVA) on exercise tolerance in subjects with cystic fibrosis (CF), homozygous for the f508del-cftr mutation. clinicaltrials.gov/show/NCT02875366 (first received 23 August 2016). [CFGD REGISTER: BD287a]
- Wilson J, You X, Ellis M, Urquhart DS, Jha L, Duncan M, et al. VO2max as an exercise tolerance endpoint in people with cystic fibrosis: lessons from a lumacaftor/ivacaftor trial. Journal of Cystic Fibrosis 2021;20(3):499-505. [CFGD REGISTER: BD287b] [DOI] [PubMed] [Google Scholar]
Zeitlin 2002 {published data only}
- Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee CK, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Molecular Therapy 2002;6(1):119-26. [CENTRAL: 409030] [CFGD REGISTER: BD148] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Berkers 2014 {published data only}
- Berkers G, Vijftigschild L, Bronsveld I, Arets H, Winter-de Groot K, Heijerman H, et al. A beta-2 agonist as a CFTR activator in CF; the ABBA study. Pediatric Pulmonology 2014;49 Suppl 38:299-300. [ABSTRACT NO.: 236] [CENTRAL: 1012380] [CFGD REGISTER: BD212a] [Google Scholar]
- EUCTR2014-000057-37-NL. Bronchodilcation as a CFTR activator in CF. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2014-000057-37-NL (first registered 12 February 2014). [CFGD REGISTER: BD212b]
- NTR4513. A B2-agonist as a CFTR activator in CF. http://www.who.int/trialsearch/Trial2.aspx?TrialID=NTR4513 (registered 14 April 2014). [CFGD REGISTER: BD212c]
Berkers 2021 {published data only}
- Berkers G, Meer R, Heijerman H, Beekman JM, Boj SF, Vries RGJ, et a. Lumacaftor/ivacaftor in people with cystic fibrosis with an A455E-CFTR mutation. Journal of Cystic Fibrosis 2021;20(5):761-7. [CFGD REGISTER: BD286b] [DOI] [PubMed] [Google Scholar]
- NCT03061331. Lumacaftor/ivacaftor combination therapy in subjects with CF WHO have an A455E CFTR mutation. clinicaltrials.gov/show/NCT03061331 (date first posted 23 February 2017). [CFGD REGISTER: BD286a]
Chadwick 1998 {published data only}
- Chadwick S, Browning JE, Stern M, Cheng SH, Gruenert DC, Geddes DM, et al. Nasal application of glycerol in DF508 cystic fibrosis patients. Pediatric Pulmonology 1998;Suppl 17:278. [ABSTRACT NO.: 275] [CENTRAL: 208568] [CFGD REGISTER: BD147] [Google Scholar]
Chilvers 2021 {published data only}
- Chilvers M, Davies JC, Ratjen F, Milla C, Owen CA, Tian S, et al. Long-term safety and efficacy of lumacaftor/ivacaftor therapy in patients aged 6-11 years with cystic fibrosis homozygous for the F508del-CFTR mutation (F/F). Journal of Cystic Fibrosis 2019;18 Suppl 1:S23. [ABSTRACT NO.: WS12-4] [CENTRAL: CN-02011205] [CFGD REGISTER: BD232c] [EMBASE: 2001976603] [Google Scholar]
- Chilvers M, Owen C, Marigowda G, Tian S, Solomon M, Black P, et al. Safety and efficacy of lumacaftor/ ivacaftor in patients aged =6 years with cf homozygous for F508DEL-CFTR (phase 3 extension study). Pediatric Pulmonology 2017;52 Suppl 47:319. [CFGD REGISTER: BD232b] [Google Scholar]
- Chilvers M, Tian S, Marigowda G, Bsharat M, Hug C, Solomon M, et al. An open-label extension (EXT) study of lumacaftor/ivacaftor (LIM/IVA) therapy in patients (pts) aged 6-11 years (yrs) with cystic fibrosis (CF) homozygous for F508del-CFTR. Journal of Cystic Fibrosis 2017;16 Suppl 1:S77. [ABSTRACT NO: 52] [CENTRAL: 1383248] [CFGD REGISTER: BD232a] [CLINICALTRIALS.GOV: NCT01897233] [Google Scholar]
- Chilvers MA, Davies JC, Milla C, Tian S, Han Z, Cornell AG, et al. Long-term safety and efficacy of lumacaftor-ivacaftor therapy in children aged 6-11 years with cystic fibrosis homozygous for the F508del-CFTR mutation: a phase 3, open-label, extension study. Lancet Respiratory Medicine 2021;9(7):721-32. [CFGD REGISTER: BD232d] [DOI] [PubMed] [Google Scholar]
- NCT01897233. Study of lumacaftor in combination with ivacaftor in subjects 6 through 11 years of age with cystic fibrosis, homozygous for the F508del-CFTR mutation [A phase 3, open-label study to evaluate the pharmacokinetics, safety, and tolerability of lumacaftor in combination with ivacaftor in subjects 6 through 11 years of age with cystic fibrosis, homozygous for the F508del-CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01897233 (first received 11 July 2013). [CLINICALTRIALS.GOV: NCT01897233]
Chmiel 2021 {published data only}
- Chmiel J, Elborn S, Constantine S, White B. A double-blind, placebo-controlled phase 2 study in adults with cystic fibrosis of anabasum, a selective cannabinoid receptor type 2 agonist. Pediatric Pulmonology 2017;52 Suppl 47:317. [CFGD REGISTER: IB118b] [Google Scholar]
- Chmiel J, Elborn S, Constantine S, White B. Recent advances in anti-inflammatory treatment. Pediatric Pulmonology 2017;52 Suppl 47:177-8. [CFGD REGISTER: IB118c] [Google Scholar]
- Chmiel JF, Flume P, Downey DG, Dozor AJ, Colombo C, Mazurek H, et al. Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. Journal of Cystic Fibrosis 2021;20(1):78-85. [CFGD REGISTER: IB118d] [DOI] [PubMed] [Google Scholar]
- NCT02465450. Safety, tolerability, pharmacokinetics, and efficacy of JBT-101 (lenabasum) in cystic fibrosis. clinicaltrials.gov/show/NCT02465450 (first received 8 June 2015). [CFGD REGISTER: IB118e]
- NCT02465450. Safety, tolerability, pharmacokinetics, and efficacy of JBT-101 (lenabasum) in cystic fibrosis. clinicaltrials.gov/show/NCT02465450 (first received 8 June 2015). [CFGD REGISTER: IB118a]
Drevinek 2017 {published data only (unpublished sought but not used)}
- Drevinek P, Pready N, Lamontagne N, Montgomery S, Henig N. QR-010 via inhalation is safe, well tolerated and achieves systemic concentrations in a single ascending dose study in subjects with cystic fibrosis homozygous for the F508del CFTR mutation. Journal of Cystic Fibrosis 2017;16 Suppl 1:S73-4. [CFGD REGISTER: BD244a] [Google Scholar]
- Elborn S, Bouisset F, Checcio T, Perquin J, Lamontagne N, Montgomery S, et al. A first-in-human, phase 1b, dose escalation study of QR-010, a novel antisense oligonucleotide administered in subjects with cystic fibrosis homozygous for the F508del CFTR mutation. Paediatric Pulmonology 2017;52 Suppl 47:289. [CFGD REGISTER: BD244b] [Google Scholar]
ISRCTN14081521 {published data only}
- ISRCTN14081521. Can people with cystic fibrosis safely stop taking some of their nebulised treatments if they are established on the new modulator therapy, Kaftrio? www.who.int/trialsearch/Trial2.aspx?TrialID=ISRCTN14081521 (date started February 2021). [CFGD REGISTER: BD289]
Krivec 2021 {published data only}
- Krivec U, Praprotnik M, Aldeco M, Lepej D, Kotnik Pirs A, Zver A, et al. Increased pulmonary interstitial fluid at CFTR modulator therapy start. Journal of Cystic Fibrosis 2021;20 Suppl 1:S55. [CFGD REGISTER: PI335] [Google Scholar]
Leonard 2012 {published data only}
- Leonard A, Dingemanse J, Lebecque P, Leal T. Oral miglustat in homozygous F508del CF patients. Journal of Cystic Fibrosis 2010;9 Suppl 1:S20. [ABSTRACT NO.: 75] [CENTRAL: 921950] [CFGD REGISTER: BD193a] [Google Scholar]
- Leonard A, Lebecque P, Dingemanse J, Leal T. A randomized placebo-controlled trial of miglustat in cystic fibrosis based on nasal potential difference. Journal of Cystic Fibrosis 2012;11:231-6. [CENTRAL: 840524] [CFGD REGISTER: BD193b] [PMID: ] [DOI] [PubMed] [Google Scholar]
Marsh 2022 {published data only}
- Lim G, Ng C, Yule A, Hoad C, Dellschaft NS, Stewart I, et al. An assessment of terminal ileum morphology using magnetic resonance imaging in people with cystic fibrosis. Journal of Cystic Fibrosis 2022;21 Suppl 1:S113. [CFGD REGISTER: BD312c] [Google Scholar]
- Marsh R, Hanson L, Ng C, Mitchell-Whyte M, Dellschaft N, Hoad C, et al. Effects of Symkevi (tezacaftor/ivacaftor) on the lung and gut microbiota in cystic fibrosis. Journal of Cystic Fibrosis 2022;21 Suppl 1:S97. [CFGD REGISTER: BD312d] [Google Scholar]
- NCT04006873. Gut imaging for function & transit in cystic fibrosis study 2. clinicaltrials.gov/show/NCT04006873 (first received 5 July 2019). [CFGD REGISTER: BD312a]
- Ng C, Dellschaft N, Hoad C, Marciani L, Mainz J, Hill T, et al. Effects of tezacaftor/ivacaftor on gut function and transit in cystic fibrosis: a randomized, double-blind, placebo-controlled, crossover trial. Journal of Cystic Fibrosis 2021;20(Suppl 2):S102. [CFGD REGISTER: BD312b] [Google Scholar]
Meijer 2016 {published data only}
- EUCTR2015-002911-13-FR. A Phase II, dose ranging, multicenter, double-blind, placebo controlled study to evaluate safety and efficacy of (R)-roscovitine in subjects with cystic fibrosis. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2015-002911-13-FR (date started 15 September 2015). [CFGD REGISTER: BD230e]
- Leven C, Schutz S, Audrezet M-P, Nowak E, Meijer L, Montier T. Non-linear pharmacokinetics of oral roscovitine (Seliciclib) in cystic fibrosis patients chronically infected with pseudomonas aeruginosa: a study on population pharmacokinetics with monte carlo simulations. Pharmaceutics 2020;12(11):1-16. [CFGD REGISTER: BD230f] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Hery-Arnaud G, Le Berre R, Nowak E, Le Roux L, Gueganton L, et al. ROSCO-CF, a safety and efficacy clinical trial of (r)-roscovitine in CF patients. Pediatric Pulmonology 2016;51 Suppl 45:269. [CFGD REGISTER: BD230b] [Google Scholar]
- Meijer L, Hery-Arnaud G, Le Berre R, Nowak E, Le Roux L, Gueganton L, et al. Rosco-CF, a safety and efficacy clinical trial of (R)-roscovitine in CF patients. Journal of Cystic Fibrosis 2016;15 Suppl 1:S42. [ABSTRACT NO.: ePS03.7] [CENTRAL: 1157464] [CFGD REGISTER: BD230a] [Google Scholar]
- Meijer L, Hery-Arnaud G, Le Berre R, Nowak E, Rault G, Mottier D. ROSCO-CF, a safety and efficacy clinical trial of (R)-roscovitine in cystic fibrosis patients. Journal of Cystic Fibrosis 2018;17 Suppl 3:S25. [CFGD REGISTER: BD230c] [Google Scholar]
- Meijer L, Hery-Arnaud G, Leven C, Nowak E, Hillion S, Renaudineau Y, et al. Safety and pharmacokinetics of Roscovitine (Seliciclib) in cystic fibrosis patients chronically infected with Pseudomonas aeruginosa, a randomized, placebo-controlled study. Journal of Cystic Fibrosis 2022;21(3):529-36. [CFGD REGISTER: BD230g] [DOI] [PubMed] [Google Scholar]
- NCT02649751. Evaluation of (R)-roscovitine safety and effects in subjects with cystic fibrosis, homozygous for the F508del-CFTR mutation (ROSCO-CF) [A phase II, dose ranging, multicenter, double-blind, placebo controlled study to evaluate safety and effects of (R)-Roscovitine in adults subjects with cystic fibrosis, carrying 2 cystic fibrosis causing mutations with at least one F508del-CFTR mutation and chronically infected with Pseudomonas aeruginosa, a study involving 36 CF patients (24 treated, 12 controls). ROSCO-CF]. clinicaltrials.gov/ct2/show/NCT02649751 (first received 7 January 2016). [CFGD REGISTER: BD230d]
NCT00945347 {published data only}
- NCT00945347. Does a nasal instillation of miglustat normalize the nasal potential difference in cystic fibrosis patients? [Does a nasal instillation of miglustat normalize the nasal potential difference in cystic fibrosis patients homozygous for the F508del mutation? A randomized, double blind placebo-controlled study]. clinicaltrials.gov/show/NCT00945347 (first posted 24 July 2009).
NCT01899105 {published data only}
- NCT01899105. A phase 1 study to investigate the food effect of lumacaftor in combination with ivacaftor [A phase 1, randomized, single-dose, open-label crossover study to investigate the effect of food on the relative bioavailability of 2 fixed-dose combinations of lumacaftor and ivacaftor tablet formulations in healthy adult subjects]. clinicaltrials.gov/show/NCT01899105 (first received 15 July 2013). [CLINICALTRIALS.GOV: NCT01899105]
NCT02323100 {published data only}
- NCT02323100. Glycerol phenylbutyrate corrector therapy for CF (cystic fibrosis) (GPBA) [A double blind, placebo controlled, dose escalation trial of glycerol phenylbutyrate corrector therapy for cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02323100 (first received 23 December 2014).
NCT03447262 {published data only}
- NCT03447262. A study evaluating the long term safety and efficacy of VX-659 combination therapy. clinicaltrials.gov/ct2/show/NCT03447262 (first received 27 February 2018).
NCT03525574 {published data only}
- NCT03525574. A study evaluating the long-term safety and efficacy of VX-445 combination therapy. clinicaltrials.gov/ct2/show/NCT03525574 (first received 15 May 2018).
NCT03537651 {published data only}
- NCT03537651. A study to evaluate the safety and efficacy of long-term treatment with TEZ/IVA in CF subjects with an F508del CFTR mutation. clinicaltrials.gov/ct2/show/NCT03537651 (first received 25 May 2018).
NCT03601637 {published data only}
- NCT03601637. Safety and pharmacokinetic study of lumacaftor/ivacaftor in subjects 1 to less than 2 years of age with cystic fibrosis, homozygous for F508del. clinicaltrials.gov/ct2/show/NCT03601637 (first received 26 July 2018).
NCT03633526 {published data only}
- NCT03633526. Evaluation of VX-659/TEZ/IVA in cystic fibrosis subjects 6 through 11 years of age. clinicaltrials.gov/ct2/show/NCT03633526 (first received 16 August 2018).
NCT03691779 {published data only}
- NCT03691779. Evaluation of VX 445/TEZ/IVA in cystic fibrosis subjects 6 through 11 years of age. clinicaltrials.gov/ct2/show/NCT03691779 (first received 2 October 2018).
NCT03756922 {published data only}
- NCT03756922. A DDI study of FDL169 and FDL176 in healthy subjects. www.clinicaltrials.gov/ct2/show/NCT03756922 (first received 28 November 2018).
NCT04043806 {published data only}
- NCT04043806. A study evaluating the long-term safety of VX-445 combination therapy. clinicaltrials.gov/ct2/show/NCT04043806 (first received 2 August 2019).
NCT04058210 {published data only}
- NCT04058210. VX-445/TEZ/IVA expanded access program for cystic fibrosis (CF) patients heterozygous for F508del mutation and a minimal function mutation (F/MF genotypes). clinicaltrials.gov/ct2/show/NCT04058210 (first received 15 August 2019).
NCT04058366 {published data only}
- NCT04058366. Study evaluating the long-term safety and efficacy of VX-445 combination therapy. clinicaltrials.gov/ct2/show/NCT04058366 (first received 15 August 2019).
NCT04183790 {published data only}
- NCT04183790. Evaluation of long-term safety and efficacy of VX-445 combination therapy in subjects with cystic fibrosis who are 6 years of age and older. clinicaltrials.gov/ct2/show/NCT04183790 (first received 3 December 2019).
NCT04235140 {published data only}
- NCT04235140. Long-term safety of lumacaftor/ivacaftor in subjects with cystic fibrosis who are homozygous for F508del and 12 to <24 months of age at treatment initiation. clinicaltrials.gov/ct2/show/NCT04235140 (first received 21 January 2020).
NCT04362761 {published data only}
- NCT04362761. A study evaluating the long-term safety of elexacaftor combination therapy. clinicaltrials.gov/ct2/show/NCT04362761 (first received 27 April 2020).
NCT04537793 {published data only}
- NCT04537793. Evaluation of ELX/TEZ/IVA in cystic fibrosis (CF) subjects 2 through 5 years. https://clinicaltrials.gov/study/NCT04537793 (first received 3 September 2020).
NCT04545515 {published data only}
- NCT04545515. A study evaluating the long-term safety and efficacy of elexacaftor/tezacaftor/ivacaftor in cystic fibrosis (CF) subjects 6 years and older and F/MF genotypes. clinicaltrials.gov/ct2/show/NCT04545515 (first received 11 September 2020).
NCT05535959 {published data only}
- NCT05535959. A study to evaluate the relative bioavailability of a fixed-dose combination tablet of vx-121/tezacaftor/deutivacaftor. clinicaltrials.gov/show/NCT05535959 (first received 10 September 2022). [CFGD REGISTER: BD314]
Nick 2014 {published data only}
- Nick JA, Rodman D, St Clair C, Jones MC, Li H, Higgins M, et al. Effect of ivacaftor in patients with cystic fibrosis, residual CFTR function, and FEV1 ≥40% of predicted, N-of-1 study. Pediatric Pulmonology 2014;49 Suppl 38:285. [ABSTRACT NO.: 196] [CENTRAL: 1012379] [CFGD REGISTER: BD211a] [Google Scholar]
- Nick JA, Rodman D, St Clair C, Jones MC, Li H, Higgins M. Utilization of an "n-of-1" study design to test the effect of ivacaftor in CF patients with residual CFTR function and FEV1 >40% of predicted. Pediatric Pulmonology 2014;49:188-9. [CENTRAL: 1008991] [CFGD REGISTER: BD211b] [EMBASE: 71616032] [Google Scholar]
Rowe 2017 {published data only}
- Chuang C, Rizio A, Loop B, Lekstrom-Himes J, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor treatment in patients heterozygous for f508del-cftr and a residual function mutation: patient reported outcomes in a phase 3 randomized, controlled trial. Paediatric Pulmonology 2018;53 Suppl 2:S264. [CFGD REGISTER: BD237e] [CTG: ] [DOI: 10.1002/ppul.24152] [DOI] [Google Scholar]
- Chuang C-C, Rizio AA, Loop B, Lekstrom-Himes J, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor (TEZ/IVA) treatment in patients heterozygous for F508DEL-CFTR and a residual function mutation: patient-reported outcomes in a phase 3 randomized, controlled trial (expand). Thorax 2018;73:A41. [CENTRAL: CN-01934562] [CFGD REGISTER: BD237f] [EMBASE: 627697165] [Google Scholar]
- Fischer R, Rizio AA, Loop B, Lekstrom-Himes J, You X, Kosinski M, et al. Effects of tezacaftor/ivacaftor (TEZ/IVA) treatment in patients heterozygous for F508del-CFTR and a residual function mutation: patient-reported outcomes in a Phase 3, randomised, controlled trial (EXPAND). Pneumologie 2019;73(Suppl 1):P13. [CENTRAL: CN-01960601] [CFGD REGISTER: BD237g] [DOI: 10.1055/s-0039-1678161] [EMBASE: 628475378] [DOI] [Google Scholar]
- Fischer R, Rowe SM, Davies JC, Nair N, Han L, Lekstrom-Himes J. Efficacy and safety of tezacaftor/ivacaftor in patients (Pts) aged >= 12 Years with CF heterozygous for F508del and a residual function mutation: a randomized, double-blind, placebo-controlled, crossover phase 3 study. Pneumologie 2018;72 Suppl 1:S35. [CFGD REGISTER: BD237d] [DOI: 10.1055/s-0037-1619210] [NCT: 02392234] [DOI] [Google Scholar]
- Ingenito E, Nair N, Yi B, Lekstrom-Himes J, Elborn JS, Rowe SM. Retrospective analysis of physiological response patterns to tezacaftor/ivacaftor in patients with cystic fibrosis homozygous for F508DEL-CFTR or heterozygous for F508DEL-CFTR and a residual function mutation. Thorax 2018;73(Suppl 4):A42-3. [CFGD REGISTER: BD237h] [DOI: 10.1136/thorax-2018-212555.75] [DOI] [Google Scholar]
- NCT02392234. A phase 3 study to evaluate the efficacy and safety of ivacaftor and VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the f508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutation [A phase 3, randomized, double-blind, placebo-controlled, crossover study to evaluate the efficacy and safety of ivacaftor and VX-661 in combination with ivacaftor in subjects aged 12 years and older with cystic fibrosis, heterozygous for the f508del-cftr mutation, and a second allele with a CFTR mutation predicted to have residual function]. clinicaltrials.gov/show/NCT02392234 (first received 18 March 2015). [CENTRAL: CN-02040467] [CFGD REGISTER: BD237i]
- Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. New England Journal of Medicine 2017;377(21):2024-35. [CFGD REGISTER: BD237c] [CLINICALTRIALS.GOV: NCT02392234] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe SM, Davies J. CFTR modulation with tezacaftor/ivacaftor in patients heterozygous for F508del and a residual function mutation. Pediatric Pulmonology 2017;52 Suppl 47:175-6. [CFGD REGISTER: BD237a] [CLINICALTRIALS.GOV: NCT02392234] [Google Scholar]
- Rowe SM, Davies JC, Nair N, Han L, Lekstrom-Himes J. Efficacy and safety of tezacaftor/ ivacaftor and ivacaftor in patients aged >=12 years with cf heterozygous for f508del and a residual function mutation: a randomized, double-blind, placebo-controlled, crossover phase 3 study. Pediatric Pulmonology 2017;52 Suppl 47:317. [CFGD REGISTER: BD237b] [CLINICALTRIALS.GOV: NCT02392234] [Google Scholar]
Rubenstein 2006 {published data only}
- Rubenstein RC, Propert KJ, Reenstra WW, Skotleski ML. A pilot trial of the combination of phenylbutyrate and genistein. Pediatric Pulmonology 2006;41 Suppl 29:294. [ABSTRACT NO.: 248] [CENTRAL: 593141] [CFGD REGISTER: BD149] [Google Scholar]
Sumner 2014 {published data only}
- Sumner Jones SG, Alton EW, Boyd A, Chang SH, Davies JC, Davies LA, et al. Molecular analyses of vector delivery and gene expression in a multidose trial of non-viral gene therapy in patients with CF. Pediatric Pulmonology 2014;49 Suppl 38:302. [ABSTRACT NO.: 243] [CENTRAL: 1012381] [CFGD REGISTER: BD210b] [Google Scholar]
- Waller MD, Harman KM, Boyd A, Chang SH, Gill DR, Griesenbach U, et al. Measurement of CFTR function in cystic fibrosis patients in response to multidose CFTR gene therapy. Pediatric Pulmonology 2014;49 Suppl 38:249. [ABSTRACT NO.: 97] [CENTRAL: 1012378] [CFGD REGISTER: BD210a] [Google Scholar]
Ziady 2015 {published data only}
- Ziady AG, Lin S, Heltshe SL, Kelley T, Muhlebach MS, Accurso FJ, et al. Protein expression in CF primary airway epithelia following treatment with VX-809 reveals significant changes in pkc-mediated signalling, proton and iron transport, and lipid metabolism. Pediatric Pulmonology 2015;50 Suppl 41:300. [ABSTRACT NO.: 290] [CENTRAL: 1092203] [CFGD REGISTER: BD225] [Google Scholar]
References to studies awaiting assessment
ALBATROSS {published data only (unpublished sought but not used)}BD247
- Bell S, De Boeck K, Drevinek P, Plant B, Barry P, Elborn S, et al. GLPG2222 in subjects with cystic fibrosis and the F508del/Class III mutation on stable treatment with ivacaftor: results from a phase II study (ALBATROSS). Journal of Cystic Fibrosis 2018;17 Suppl 3:S2. [ABSTRACT NO.: WS01.4] [CFGD REGISTER: BD247a] [Google Scholar]
- Bell S, De Boeck K, Drevinek P, Plant B, Barry P, Elborn S, et al. Results from a phase II study- ALBATROSS - evaluation of GLPG2222 in subjects with CF and the F508del/class III mutation on stable treatment with ivacaftor. Pediatric Pulmonology 2018;53 Suppl 2:249. [ABSTRACT NO.: 269] [CFGD REGISTER: BD247b] [Google Scholar]
- Bell SC, Barry PJ, De Boeck K, Drevinek P, Elborn JS, Plant BJ, et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. Journal of Cystic Fibrosis 2019;18(5):700-7. [CENTRAL: CN-01936893] [CFGD REGISTER: BD247c // BD254c] [DOI: 10.1016/j.jcf.2019.04.014] [EMBASE: 2001903987] [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT03045523. A study to evaluate GLPG2222 in ivacaftor-treated subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT03045523 (first received 7 February 2017). [STUDY NO.: GLPG2222-CL-201]
Downey 2019 {published data only}
- Downey D, Flume P, Jain M, Fajac I, Schwarz C, Pressler T, et al. Initial results evaluating combinations of the novel CFTR corrector PTI-801, potentiator PTI-808, and amplifier PTI-428 in cystic fibrosis subjects. Journal of Cystic Fibrosis 2019;18 Suppl 1:S10. [CENTRAL: CN-01990618] [CFGD REGISTER: BD269a] [EMBASE: 2001976571] [Google Scholar]
- Flume P, Downey DG, Jain M, Fajac I, Schwarz C, Pressler T, et al. Evaluation of novel CFTR modulator combinations of the corrector PTI-801, potentiator PTI-808, and amplifier PTI-428 in CF subjects. Pediatric Pulmonology 2019;54 Suppl 2:348. [CENTRAL: CN-01989480] [CFGD REGISTER: BD269c] [EMBASE: 629387551] [Google Scholar]
- Jain M, Pilewski J, Flume P, Taylor-Cousar J, Rowe S, Milla C, et al. Initial results evaluating the add-on effect of the novel CFTR corrector PTI-801 in cystic fibrosis subjects. Journal of Cystic Fibrosis 2019;18 Suppl 1:S10-1. [CENTRAL: CN-02007011] [CFGD REGISTER: BD269b] [EMBASE: 2001976532] [Google Scholar]
Downey 2020 {published data only}
- Downey DG, Fajac I, Flume P, O'Carroll M, Pressler T, Proesmans M, et al. Evaluation of combinations of the CFTR potentiator dirocaftor, corrector posenacaftor and amplifier nesolicaftor in CF subjects with two copies of the F508del mutation. Pediatric Pulmonology 2020;55 Suppl 2:204. [CFGD REGISTER: BD285b] [Google Scholar]
- Downey DG, Fajac I, Flume P, O'Carroll M, Pressler T, Proesmans M, et al. Evaluation of combinations of the CFTR potentiator dirocaftor, corrector posenacaftor and amplifier nesolicaftor in cystic fibrosis subjects with two copies of the F508del mutation. Journal of Cystic Fibrosis 2020;19 Suppl:S19. [CFGD REGISTER: BD285a] [Google Scholar]
FLAMINGO {published data only (unpublished sought but not used)}
- Bell SC, Barry PJ, De Boeck K, Drevinek P, Elborn JS, Plant BJ, et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. Journal of Cystic Fibrosis 2019;18(5):700-7. [CFGD REGISTER: BD247c // BD254c] [DOI: 10.1016/j.jcf.2019.04.014] [DOI] [PubMed] [Google Scholar]
- NCT03119649. A study to evaluate multiple doses of GLPG2222 in adult subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT03119649 (first received 18 April 2017). [STUDY NO.: GLPG2222-CL-202]
- Ent KC, Minic P, Verhulst S, Van Brackel E, Flume P, Boas S, et al. GLPG2222 in subjects with cystic fibrosis homozygous for F508del: results from a phase II study (FLAMINGO). Journal of Cystic Fibrosis 2018;17 Suppl 3:S42. [ABSTRACT NO.: EPS3.05] [CFGD REGISTER: BD254a] [Google Scholar]
- Ent KC, Minic P, Verhulst S, Van Brackel E, Flume P, Boas S, et al. GLPG2222 in subjects with cystic fibrosis homozygous for F508del: results from a phase II study (FLAMINGO). Pediatric Pulmonology 2018;53 Suppl 2:250. [ABSTRACT NO.: 271] [CFGD REGISTER: BD254b] [Google Scholar]
Hunt 2017 {published data only}
- Hunt K, St Clair C, Curran-Everett D, Solomon GM, Saavedra MT, Nick JA, et al. CFTR effects of oral sildenafil in combination with lumacaftor/ivacaftor in adults with CF. Pediatric Pulmonology 2017;52(Suppl 47):322. [CFGD REGISTER: IB117] [Google Scholar]
NCT03768089 {published data only}
- EudraCT 2018-000126-55. A phase 1/2 study of VX-121 in healthy subjects and in subjects with cystic fibrosis. www.clinicaltrialsregister.eu/ctr-search/search?query=2018-000126-55 (first received 15 March 2018). [CFGD REGISTER: BD295b]
- NCT03768089. Study of VX-121 in healthy subjects and in subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT03768089 (first received 7 December 2018). [CFGD REGISTER: BD295a] [EUDRACT NUMBER: 2018-000126-55]
NCT03969888 {published data only}
- EudraCT 2019-000750-63. A phase 2 study of ABBV-3067 alone and in combination with ABBV-2222 in cystic fibrosis subjects who are homozygous for the F508del mutation. www.clinicaltrialsregister.eu/ctr-search/search?query=2019-000750-63 (first received 31 October 2019). [CFGD REGISTER: BD296b]
- NCT03969888. A phase 2 study of ABBV-3067 alone and in combination with ABBV-2222. clinicaltrials.gov/show/nct03969888 (first received 31 May 2019). [CFGD REGISTER: BD296a]
NCT04353817 {published data only}
- EUCTR2019-003554-86-GB. A study evaluating efficacy and safety of elexacaftor/tezacaftor/ivacaftor in subjects 6 through 11 years old with cystic fibrosis and F/MF genotypes. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2019-003554-86-GB (date started 14 August 2020). [CFGD REGISTER: BD291b]
- NCT04353817. A study evaluating efficacy and safety of elexacaftor/tezacaftor/ivacaftor in subjects 6 through 11 years of age with cystic fibrosis and F/MF genotypes. clinicaltrials.gov/ct2/show/NCT04353817 (first received 20 April 2020). [CFGD REGISTER: BD291a]
PELICAN {published data only}
- EUCTR2017-002181-42-DE. GLPG2737 on top of Orkambi in subjects with cystic fibrosis [A phase IIa, randomized, double-blind, placebo-controlled study to evaluate GLPG2737 in Orkambi-treated subjects with cystic fibrosis homozygous for the F508del mutation]. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2017-002181-42-DE (first received 2017). [CENTRAL: CN-01887243] [CFGD REGISTER: BD272b]
- NCT03474042. GLPG2737 on top of Orkambi in subjects with cystic fibrosis [A phase IIa, randomized, double-blind, placebo-controlled study to evaluate GLPG2737 in orkambi-treated subjects with cystic fibrosis homozygous for the f508del mutation]. clinicaltrials.gov/show/NCT03474042 (first received 22 March 2018). [CFGD REGISTER: BD272a]
- Koningsbruggen-Rietschel S, Conrath K, Fischer R, Sutharsan S, Kempa A, Gleiber W, et al. GLPG2737 in lumacaftor/ivacaftor-treated CF subjects homozygous for the F508del mutation: a randomized phase 2A trial (PELICAN). Journal of Cystic Fibrosis 2020;19(2):292-8. [CENTRAL: CN-01999311] [CFGD REGISTER: BD272c] [EMBASE: 2003281625] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rio‐CF {published data only (unpublished sought but not used)}
- Derichs N, Taylor-Cousar J, Tullis E, Davies J, Nazareth D, Downey DG, et al. Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult phe508del homozygous cystic fibrosis patients: study design and rationale for the Rio-CF study. Journal of Cystic Fibrosis 2017;16 Suppl 1:S36. [CFGD REGISTER: BD246b] [Google Scholar]
- NCT02170025. Early signs of efficacy study with riociguat in adult homozygous delta F508 cystic fibrosis patients [Multi-center phase 2 study to assess the safety, tolerability and early signs of efficacy of tid orally administered BAY63-2521 in adult delta F508 homozygous cystic fibrosis patients]. clinicaltrials.gov/ct2/show/results/NCT02170025 (first received 23 June 2014).
- Taylor-Cousar JL, Tullis E, Derichs N, Davies JC, Nazareth D, Downey R, et al. Riociguat for the treatment of adult phe508del homozygous cystic fibrosis: efficacy data from the phase II Rio-CF study. Journal of Cystic Fibrosis 2018;17 Suppl 3:S67. [CFGD REGISTER: BD246a] [DOI] [PubMed] [Google Scholar]
Taylor‐Cousar 2019 {published data only}
- Taylor-Cousar J, Gifford AH, Flume P, Sawicki GS, Pilewski JM, Jain M, et al. Initial results evaluating the first-in-class CFTR amplifier, PTI-428, in subjects with CF on background treatment with tezacaftor/ ivacaftor. Pediatric Pulmonology 2019;54 Suppl 2:332. [CENTRAL: CN-01988388] [CFGD REGISTER: BD270] [EMBASE: 629388001] [Google Scholar]
Uluer 2023a {published data only}
- NCT03911713. A phase 2 study to evaluate efficacy and safety of VX-561 in subjects aged 18 years and older with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT03911713 (first received 11 April 2019). [EUDRACT NUMBER: 2018-003970-28]
- Uluer AZ, MacGregor G, Azevedo P, Indihar V, Keating C, Mall MA, et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: randomised, double-blind, controlled, phase 2 trials. Lancet Respiratory Medicine 2023 Feb 23 [Epub ahead of print]. [DOI: 10.1016/S2213-2600(22)00504-5] [DOI] [PubMed]
Uluer 2023b {published data only}
- EUCTR2018-002496-18-PT. A study to evaluate the safety and efficacy of VX-121 combination therapy in subjects with cystic fibrosis [A phase 2, randomized, double-blind, controlled study to evaluate the safety and efficacy of VX-121 combination therapy in subjects aged 18 years and older with cystic fibrosis]. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2018-002496-18-PT (first received 2019). [CENTRAL: CN-02068097] [CFGD REGISTER: BD277b]
- NCT03912233. A study to evaluate the safety and efficacy of VX-121 combination therapy in subjects with cystic fibrosis [A phase 2, randomized, double-blind, controlled study to evaluate the safety and efficacy of VX-121 combination therapy in subjects aged 18 years and older with cystic fibrosis]. clinicaltrials.gov/show/NCT03912233 (first received 11 April 2019). [CENTRAL: CN-01912167] [CFGD REGISTER: BD277a]
- Uluer AZ, MacGregor G, Azevedo P, Indihar V, Keating C, Mall MA, et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: randomised, double-blind, controlled, phase 2 trials. Lancet Respiratory Medicine 2023 Feb 23 [Epub ahead of print]. [DOI] [PubMed]
Wainwright 2019 {published data only}
- Wainwright C, Stick S, Goldin J, Lekstrom-Himes J, Wang L, Campbell D, et al. Change in low-dose chest computed tomography (CT) scores after 72 weeks of tezacaftor/ivacaftor (TEZ/IVA) in patients (pts) with cystic fibrosis and ppFEV1 >=70%: an exploratory phase 2 study. Journal of Cystic Fibrosis 2019;18 Suppl 1:S11-2. [CENTRAL: CN-02009338] [CFGD REGISTER: BD275] [EMBASE: 2001976227] [Google Scholar]
References to ongoing studies
Jain 2018 {published data only (unpublished sought but not used)}
- Jain M, Flume P, Escobar H, Taylor-Cousar JL, Pressler T, Liou TG, et al. Initial results evaluating third generation CFTR corrector PTI-801 in CF subjects. Paediatric Pulmonology 2018;53 Suppl 2:246. [CFGD REGISTER: BD257] [Google Scholar]
NCT02589236 {published data only}
- NCT02589236. Study of cavosonstat (N91115) in patients with CF homozygous for the F508del-CFTR mutation (SNO-6) [A phase 2, randomized, double-blind, placebo-controlled, parallel-group study of N91115 to evaluate efficacy and safety in patients with cystic fibrosis who are homozygous for the F508del-CFTR mutation treated with lumacaftor/ivacaftor]. clinicaltrials.gov/ct2/show/NCT02589236 (first received 28 October 2015).
NCT02718495 {published data only}
- NCT02718495. Study assessing PTI-428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis [A phase I/II, multi-center, randomized, placebo-controlled, study designed to assess the safety, tolerability, and pharmacokinetics of PTI-428 in subjects with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02718495 (first received 24 March 2016).
NCT03258424 {published data only}
- NCT03258424. Study assessing PTI-428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis on KALYDECO® as background therapy [A phase I, randomized, placebo-controlled, study designed to assess the safety, tolerability, and pharmacokinetics of PTI-428 in subjects with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT03258424 (first received 23 August 2017).
NCT04853368 {published data only}
- EUCTR2020-005805-25-HU. A study of galicaftor/navocaftor/abbv-119 combination therapy in subjects with cystic fibrosis. trialsearch.who.int/Trial2.aspx?TrialID=NCT04853368 (first received 19 April 2021). [CFGD REGISTER: BD313b]
- NCT04853368. Study to evaluate adverse events and change in disease activity with oral capsules of galicaftor/navocaftor/abbv-119 or galicaftor/navocaftor/abbv-576 combination therapies in adult participants with cystic fibrosis. clinicaltrials.gov/show/NCT04853368 (first received 21 April 2021). [CFGD REGISTER: BD313a]
NCT05033080 {published data only}
- EUCTR2021-000712-31-DE. A phase 3 study of VX-121 combination therapy in subjects with cystic fibrosis heterozygous for f508del and a minimal function mutation (F/MF). trialsearch.who.int/Trial2.aspx?TrialID=NCT05033080 (first received 26 August 2021). [CFGD REGISTER: BD290b]
- NCT05033080. A phase 3 study of VX-121 combination therapy in participants with cystic fibrosis (CF) heterozygous for F508del and a minimal function mutation (F/MF). www.clinicaltrials.gov/ct2/show/NCT05033080 (first received 2 September 2021). [CFGD REGISTER: BD290a]
NCT05076149 {published data only}
- EUCTR2021-000694-85-DK. A phase 3 study of VX-121 combination therapy in subjects with cystic fibrosis who are homozygous for f508del, heterozygous for f508del and a gating (F/G) or residual function (F/RF) mutation, or have at least 1 other triple combination responsive CFTR mutation and no f508del mutation. trialsearch.who.int/Trial2.aspx?TrialID=NCT05076149 (first received 29 September 2021). [CFGD REGISTER: BD311b]
- NCT05076149. A study of VX-121 combination therapy in participants with cystic fibrosis (CF) who are homozygous for F508del, heterozygous for F508del and a gating (F/G) or residual function (F/RF) mutation, or have at least 1 other triple combination responsive (TCR) CFTR mutation and no F508del mutation. www.clinicaltrials.gov/ct2/show/NCT05076149 (first received 13 October 2021). [CFGD REGISTER: BD311a]
NCT05274269 {published data only}
- EUCTR2021-005320-38-AT. Evaluation of efficacy and safety of ELX/TEZ/IVA in subjects without an f508del mutation. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2021-005320-38-AT (first received 5 January 2022). [CFGD REGISTER: BD306b]
- NCT05274269. Evaluation of efficacy and safety of elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA) in cystic fibrosis subjects without an f508del mutation. clinicaltrials.gov/show/NCT05274269 (first received 10 March 2022). [CFGD REGISTER: BD306a]
Additional references
Amaral 2007
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends in Pharmacological Sciences 2007;28(7):334-41. [DOI] [PubMed] [Google Scholar]
Aslam 2023
- Aslam AA, Sinha IP, Southern KW. Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2023, Issue 3. Art. No: CD012040. [DOI: 10.1002/14651858.CD012040.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bobadilla 2002
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations - correlation with incidence data and application to screening. Human Mutation 2002;19(6):575-606. [DOI] [PubMed] [Google Scholar]
CFMD 2013
- Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto, Canada. Cystic fibrosis mutation database. www.genet.sickkids.on.ca/app (accessed 23 September 2013).
Colledge 1995
- Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, et al. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nature Genetics 1995;10(4):445-52. [DOI] [PubMed] [Google Scholar]
Deeks 2022
- Deeks JJ, Higgins JP, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
EMA 2012
- European Medicines Agency. Report of the workshop on endpoints for cystic fibrosis clinical trials (EMA/769571/2012); November 2012. Available at www.ema.europa.eu/docs/en_GB/document_library/Report/2012/12/WC500136159.pdf.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 5.2.0 (updated June 2017). The Cochrane Collaboration, 2017. Available from training.cochrane.org/handbook/archive/v5.2.
Higgins 2022
- Higgins JP, Eldridge S, Li T, editor(s). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
NICE 2016
- Lumacaftor–ivacaftor for treating cystic fibrosis homozygous for the F508del mutation. www.nice.org.uk/guidance/ta398 (accessed 1 September 2020).
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815-34. [DOI] [PubMed] [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramsey 2011
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. New England Journal of Medicine 2011;365(18):1663-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Riordan 1989
- Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene – cloning and characterization of complementary. Science 1989;245(4922):1066-72. [DOI] [PubMed] [Google Scholar]
Rogan 2011
- Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011;139(6):1480-90. [DOI: 10.1378/chest.10-2077] [DOI] [PubMed] [Google Scholar]
Rowntree 2003
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Annals of Human Genetics 2003;67(5):471-85. [DOI] [PubMed] [Google Scholar]
Rubenstein 1997
- Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. Journal of Clinical Investigation 1997;100(10):2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Skilton 2019
- Skilton M, Krishan A, Patel S, Sinha IP, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2019, Issue 1. Art. No: CD009841. [DOI: 10.1002/14651858.CD009841.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Southern 1997
- Southern KW. Delta F508 in cystic fibrosis: willing but not able. Archives of Disease in Childhood 1997;76(3):278-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Southern 2007
- Southern KW. Cystic fibrosis and formes frustes of CFTR-related disease. Respiration: International Review of Thoracic Diseases 2007;74(3):241-51. [DOI] [PubMed] [Google Scholar]
Van Goor 2011
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America 2011;108(46):18843-8. [DOI: 10.1073/pnas.1105787108] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2022
- Wang Y, Ma B, Li W, Li P. Efficacy and safety of triple combination cystic fibrosis transmembrane conductance regulator modulators in patients with cystic fibrosis: a meta-analysis of randomized controlled trials. Frontiers in Pharmacology 2022;13:863280. [DOI: 10.3389/fphar.2022.863280] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Williamson 2002
- Williamson PR, Tudur Smith C, Hutton JL, Marson AG. Aggregate data meta-analysis with time-to-event outcomes. Statistics in Medicine 2002;21(11):3337-51. [DOI] [PubMed] [Google Scholar]
Wu 2018
- Wu HX, Zhu M, Xiong XF, Wei J, Zhuo KQ, Cheng DY. Efficacy and safety of CFTR corrector and potentiator combination therapy in patients with cystic fibrosis for the F508del-CFTR homozygous mutation: a systematic review and meta-analysis. Advances in Therapy 2018;36(2):451-61. [DOI: 10.1007/s12325-018-0860-4] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Southern 2018
- Southern KW, Patel S, Sinha IP, Nevitt SJ. Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 8. Art. No: CD010966. [DOI: 10.1002/14651858.CD010966.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Southern 2020
- Southern KW, Murphy J, Sinha IP, Nevitt SJ. Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane Database of Systematic Reviews 2020, Issue 12. Art. No: CD010966. [DOI: 10.1002/14651858.CD010966.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]