

Abstract
The ability to manipulate the chemical composition of proteins and peptides has been central to the development of improved polypeptide-based therapeutics and has enabled researchers to address fundamental biological questions that would otherwise be out of reach. Protein ligation, in which two or more polypeptides are covalently linked, is a powerful strategy for generating semisynthetic products and for controlling polypeptide topology. However, specialized tools are required to efficiently forge a peptide bond in a chemoselective manner with fast kinetics and high yield. Fortunately, nature has addressed this challenge by evolving enzymatic mechanisms that can join polypeptides using a diverse set of chemical reactions. Here, we summarize how such nature-inspired protein ligation strategies have been repurposed as chemical biology tools that afford enhanced control over polypeptide composition.
Graphical Abstract
Introduction
Proteins and peptides carry out a vast number of biological functions that are ultimately dictated by their chemical composition. Owing to their involvement in most biological processes and the ever-increasing market for protein and peptide therapeutics1, there is a great interest in strategies that can control polypeptide composition at levels that stretch beyond the linear, ribosomal assembly of genetically encoded natural amino acids. To this end, the ligation of two or more protein-based substrates is a facile approach that enables researchers to flexibly tailor the composition of proteins in various ways. Protein ligation strategies pave the way for protein semisynthesis, in which recombinantly produced proteins are fused with synthetic peptides, thereby expanding the chemical space that is available for (re)defining the primary structure of proteins. These strategies have been used to install a wide range of chemical probes into proteins2, site-specifically incorporate post-translational modifications (PTMs) and conjugate cytotoxic drugs to antibodies3,4. In addition, protein ligation can be leveraged for segmental isotope labelling of proteins, thereby reducing the complexity of protein NMR spectra, which allows the structural characterization of protein domains that are otherwise inaccessible5. Ligation-based strategies have also proved useful for manipulating protein topology, as exemplified by the cyclization of polypeptides through intramolecular reactions, which can be used to generate stable and therapeutically relevant peptide libraries6. Moreover, polypeptide topologies can also be engineered through isopeptide bond formation between amino acid side chains. Evidently, the motivation for performing protein ligations can vary significantly, and it is therefore critical to choose a ligation platform that is best suited for the desired outcome (Fig. 1).
Fig. 1 |. An overview of the enzymatic toolbox for protein ligation-mediated polypeptide engineering covered in this Review.
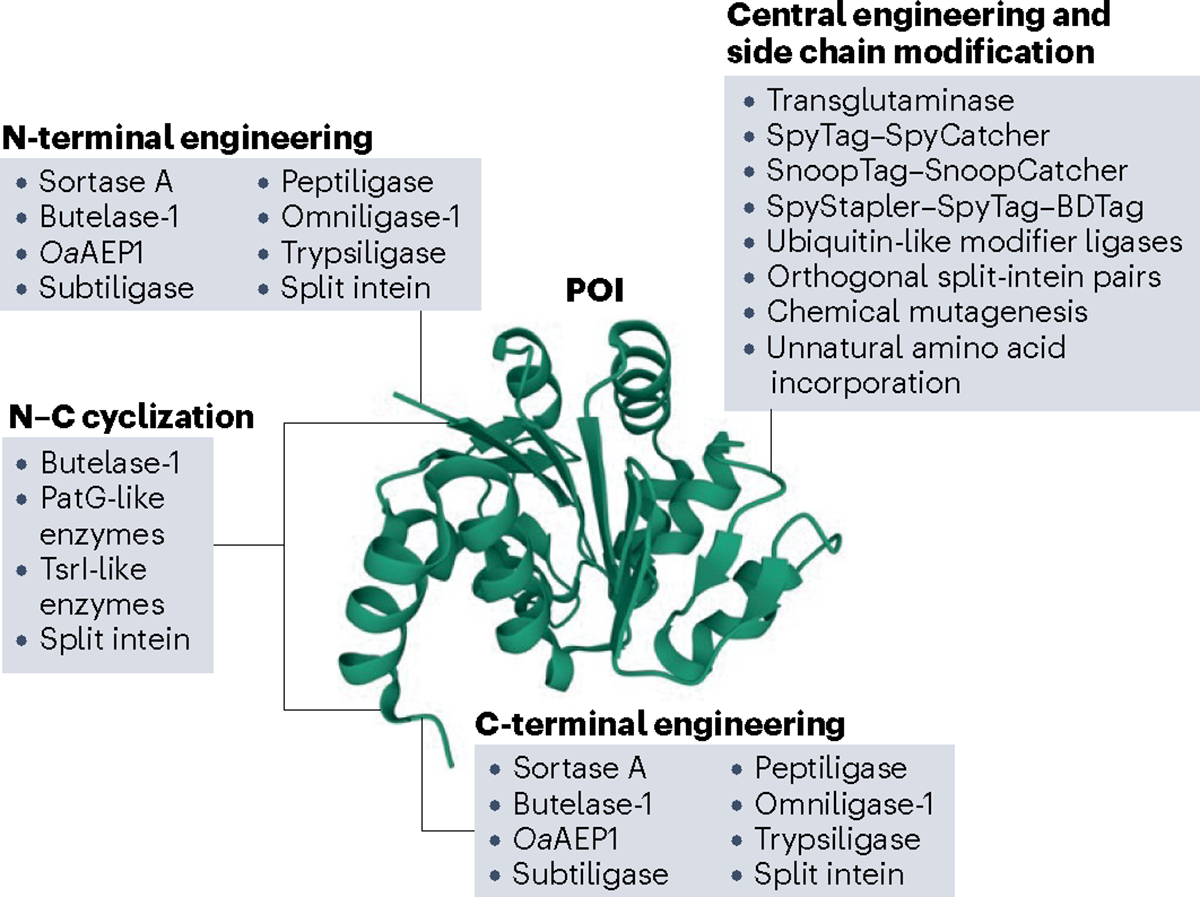
There are multiple enzyme-based platforms available for modifying a protein of interest (POI). As all ligation strategies are not created equal, they have distinct advantages and disadvantages. Different enzymes allow access to different segments of the POI. These enzymes also differ in the mechanism by which they catalyse amide bond formation, as N–C cyclization requires intramolecular ligation, whereas N- and C-terminal engineering is achieved by intermolecular reactions. Other platforms modify side chains by generating isopeptide bonds. Two fundamental questions for any protein ligation endeavour are thus, where in the POI the modifications will be introduced and what the nature of the resulting bond should be. Human DJ-1 is shown as a model protein, Protein Data Bank (PDB): 1PDV.
Successful ligation of polypeptides requires an efficient, chemoselective reaction that creates a specific amide bond, despite the presence of the ensemble of functional groups found in proteins. Chemical methods for protein ligation, of which native chemical ligation (NCL) and its extension expressed protein ligation (EPL), are the most common (Box 1), represent valuable platforms for generating semisynthetic, site-specifically modified proteins. However, these chemical approaches are ultimately limited by their biocompatibility and requirement for high concentrations of reactants. Alternatively, semisynthetic proteins, which are composed of recombinant and synthetic pieces, can be generated through non-ligation-based platforms, such as genetic code expansion (Box 1), bioorthogonal conjugation to reactive side chains3,7 and post-translational mutagenesis8,9, although all of these have inherent limitations. To develop novel strategies for protein ligation, inspiration has been drawn from natural enzyme-based approaches. Enzymes have long been known to be able to perform ‘reverse proteolysis’ and join two protein segments together10. Since this discovery, the toolbox for enzyme-mediated protein ligation has markedly expanded and now includes several powerful platforms that allow precise ligation of peptides and proteins under complex and dynamic conditions, such as living cells, at low reactant concentrations2,3.
Box 1. Alternative strategies for generating semisynthetic polypeptides.
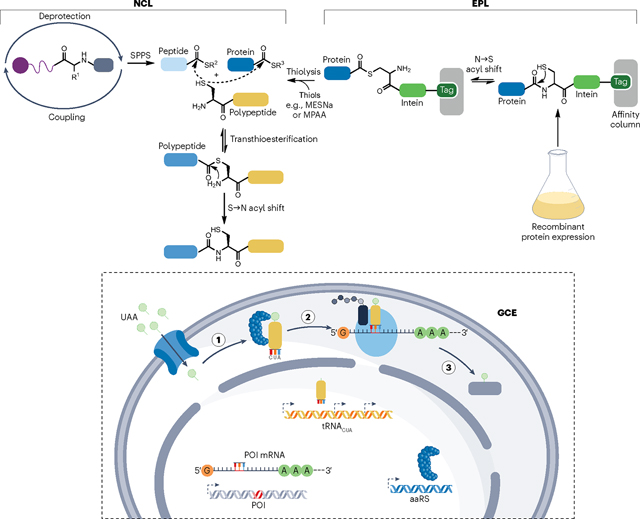
Native chemical ligation (NCL) joins unprotected polypeptides through condensation of a C-terminal thioester and an N-terminal cysteine by a mechanism that is reminiscent of intein-based ligation207. NCL is performed under aqueous conditions at neutral pH and has been instrumental for incorporating synthetic moieties into polypeptides. However, to routinely generate larger, semisynthetic proteins, at least one fragment must first be recombinantly produced. Although there are several routes for producing recombinant proteins with N-terminal cysteines2, facile production of C-terminal thioesters requires assistance from nature. By fusing a protein of interest (POI) to an intein variant that arrests splicing at the thioester intermediate level208, an NCL-suitable α-thioester can be released through thiolysis. The expressed protein ligation (EPL) strategy overcomes the size limitation for C-terminal modification and has been widely used since its conception209. Importantly, the use of multiple sequential ligation steps, including both NCL-based and EPL-based reactions, further increases the size of the semisynthetic product and allows internal residues to be modified210–213. Nevertheless, the applicability of NCL and EPL is still restricted by the fact that millimolar reactant concentrations are needed to drive the rate-limiting transthioesterification step to promote spontaneous amide bond formation over thioester hydrolysis. This prevents these ligation strategies from being used in cells where there are high concentrations of thiols.
Synthetic moieties can also be introduced site specifically into proteins by forcing the translational machinery to accommodate unnatural amino acids (UAAs) through genetic code expansion (GCE). This strategy classically overwrites the natural decoding of an mRNA triplet, often the amber codon, by ectopic expression of an orthogonal aminoacyl-tRNA synthetase and a modified tRNA that recognizes the specific triplet within the mRNA of interest214. GCE has the advantage that it can readily target the entire protein sequence, whereas ligation-based strategies require multiple ligations to extend beyond the termini. Conversely, although ligation of synthetic peptides unlocks the door to the entire chemical space available through solid-phase peptide synthesis, GCE requires an aminoacyl-tRNA synthetase and tRNA pair for each UAA. Furthermore, it remains challenging to install multiple UAAs by GCE, despite novel approaches relying on quadruplet codons215 or even entirely synthetic genomes216. Thus, NCL, EPL and GCE are invaluable platforms for protein manipulation that are constantly undergoing further developments and aptly complement enzyme-based strategies. MESNa, sodium 2-mercaptoethane sulfonate; MPAA, 4-mercaptophenylacetic acid; aaRS, aminoacyl-tRNA synthetase; SPPS, solid-phase peptide synthesis.
In this Review, we summarize the different classes of enzymes, which are grouped on the basis of the mechanism by which they facilitate amide bond formation, that are available to researchers for the generation of semisynthetic proteins in vitro and in cells. We highlight how these enzymes vary in key aspects of protein ligation to inform the selection of which strategy is best suited for a given task. These differences include how traceless the ligation is, that is, how substantial the ‘ligation scar’ that remains in the final product is, the selectivity and efficiency of the reaction, the type of bond that is created (native peptide bond or isopeptide bond), the synthetic availability of the reactants, and whether the platform is bioorthogonal. Moreover, we present selected examples of applications of these strategies together with additional sources for in-depth descriptions of each system. Finally, we emphasize how engineering of the naturally occurring enzymes has been critical for moving the field towards more efficient and expansive protein ligation.
Transpeptidase-based methods
Transpeptidases are a family of enzymes isolated from bacteria or plants that catalyse nucleophilic carbonyl substitution and transamination11. Transpeptidases perform their biochemical functions by initially cleaving an amide bond within a recognition sequence of the acyl donor using a catalytic cysteine residue. This generates an activated thioester intermediate that can then be attacked by an amine donor, restoring an amide bond and completing the transpeptidase cycle. Transpeptidases possess promiscuous enzymatic activities, including the cleavage of the D-alanyl–D-alanine bond and the subsequent crosslinkage of bacterial cell wall peptidoglycan (Fig. 2a) as well as peptide or protein macrocyclization. Consequently, transpeptidases are versatile scaffolds for developing protein ligases, and two major types of transpeptidases are widely used for protein ligation: sortases and asparaginyl endopeptidases (AEPs).
Fig. 2 |. Biochemistry of protein and peptide ligations mediated by sortase and butelase-1.
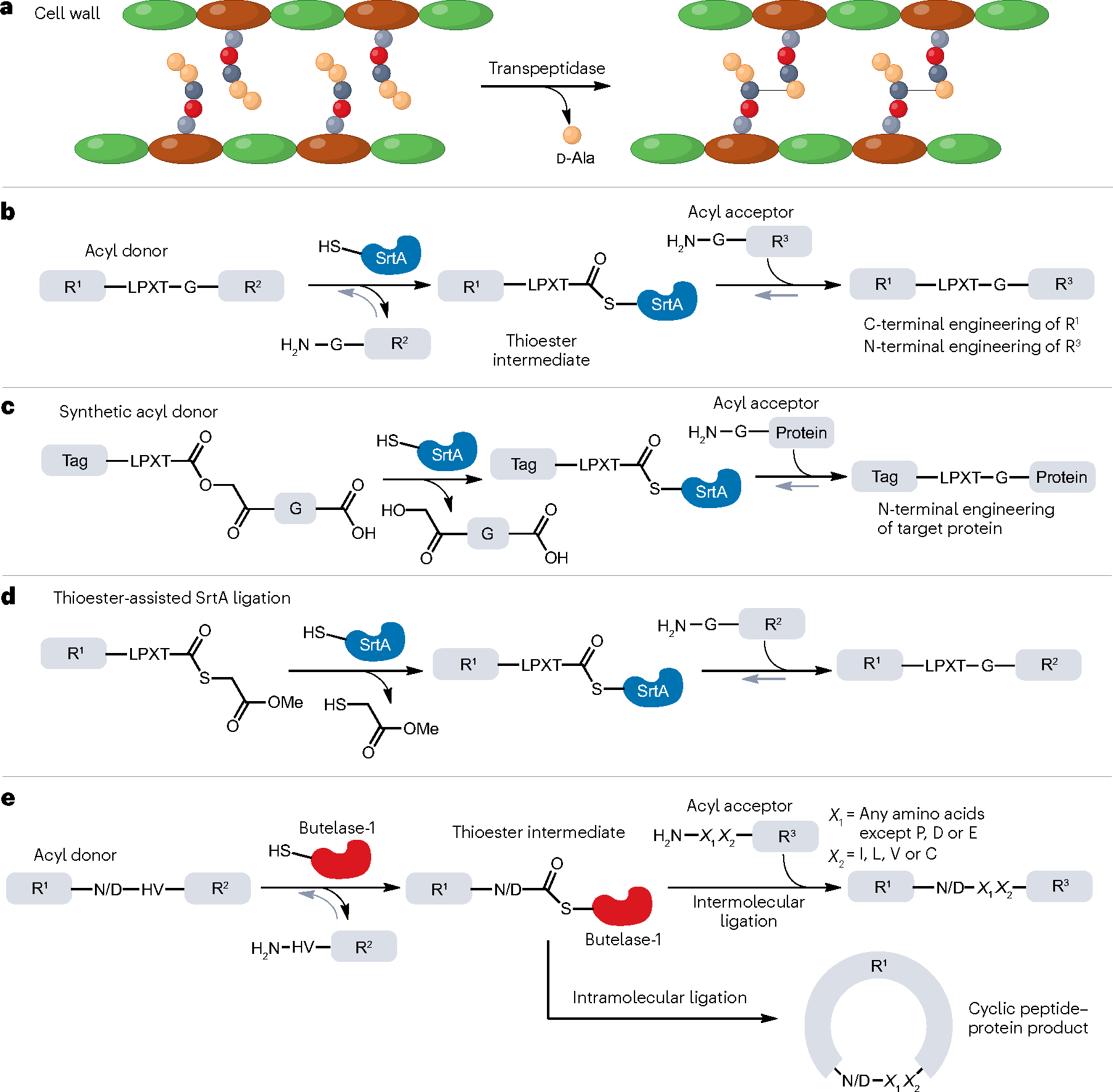
a, The biological function of transpeptidase-mediated peptide ligation is to covalently modify the cell wall of bacteria and plants. Shown here, the crosslinking of peptidoglycan by the d-alanyl–d-alanine-transpeptidase. b, Biochemical mechanism of sortase A (SrtA)-catalysed reversible ligation and its application in protein N- and C-terminal engineering (sortagging). The LPXT-G sequence is recognized and cleaved by the enzyme SrtA. R1 and R2 are peptide or protein sequences. c, Irreversible protein–peptide ligations mediated by SrtA with rationally designed synthetic or semisynthetic acyl donors, where the ‘G’ residue in the recognition sequence ‘LPXT-G’ is replaced by synthetic moieties. ‘Tag’ could be synthetic fluorescent molecules or specific peptide sequences for imaging, enrichment and tracking of the products after ligation. R1 and R2 are peptide or protein sequences. d, As in panel c, irreversible SrtA-based ligation can be achieved using thioesters to modify the N-terminal part of the protein of interest. e, Biochemistry and application of protein ligation by butelase-1, which proceeds through formation of a key thioester intermediate. The N/D–HV sequence can be recognized and cleaved by butelase-1 to generate a thioester intermediate, which can be resolved to yield a linear or cyclic product. R1, R2 and R3 are peptide or protein sequences. X1 and X2 are specific amino acid residues that can be recognized by butelase-1 in this ligation reaction: X1 can be any amino acid except P, D or E; X2 can be I, L, V or C.
Sortases
Sortases are a highly ubiquitous group of transpeptidases found in Gram-positive bacteria that use their enzymatic activity to anchor various surface proteins to the peptidoglycan of the cell wall12. Of these, sortase A (SrtA), isolated from Staphylococcus aureus, has found widespread use for protein ligation13. In SrtA-mediated ligation, the catalytic cysteine attacks the scissile bond between threonine and glycine within a conserved amino acid sequence (R1-LPXTG-R2) to form a key thioester intermediate (R1-LPXT-SrtA) and releases the by-product NH2-GR2 (Fig. 2b). The activated thioester then undergoes aminolysis by the N-terminal α-amine group of a donor (NH2-GR3), to generate a new protein sequence (R1-LPXTG-R3). This makes SrtA useful for both N-terminal and C-terminal protein modification. As the N-terminal residue of the amine donor must be glycine, the original LPXTG motif is regenerated in the ligated product. This enzymatic process, also referred to as the sortagging reaction, is Ca2+-dependent owing to an allosteric Ca2+ binding site that stabilizes an otherwise disordered loop in SrtA14. Recombinant SrtA can be purified with high yields from Escherichia coli (typically >40 mg l−1), cementing its utility as a chemical biology tool.
However, wild-type SrtA has relatively poor catalytic efficiency, even in the presence of Ca2+, and the reaction requires high enzyme concentrations (0.1–1.0 molar ratios of SrtA:substrate) as well as long ligation times (more than 20 h). Moreover, the reversibility of SrtA-mediated transpeptidation restricts the overall yield of the ligated product, as the product (R1-LPXT-GR3) acts as a substrate for the reverse enzymatic cleavage and ligation (Fig. 2b). It has been shown that the addition of excess amine donor (NH2-GR3) or removal of the glycine by-product (NH2-GR2) can shift the equilibrium towards an increased yield of the desired product15,16. The yield can be further improved by using engineered depsipeptide substrates that mimic the glycine amide structure and release hydroxyacetyl by-products that cannot re-attack the thioester intermediate17 (Fig. 2c). Although this strategy has only been successfully applied to N-terminal protein modification18, an alternative approach has recently been developed in which SrtA uses a thioester substrate in an irreversible ligation reaction that is practical for both N-terminal and C-terminal protein modification19 (Fig. 2c). Moreover, thioester-assisted ligation displays improved sequence tolerance, leaving only a single glycine residue as a ‘scar’ at the ligation site (Fig. 2d). Finally, StrA has also been engineered, for example, through directed evolution or rational design, to enhance its catalytic efficiency20,21 and remove its Ca2+ dependency22, thus improving its utility for protein ligation in living cells, which usually contain low concentrations of Ca2+.
The chemoselectivity of SrtA is attributed to its recognition of the LPXTG motif, which serves as a conjugation tag for protein ligation. Even though this enzymatic ligation is not ‘traceless’, the modest size of the ‘ligation scar’ (LPXTG) can in some cases be introduced without remarkably affecting the functions of the ligated product protein substrates15. Still, several studies have used directed evolution of SrtA to broaden its substrate preference, thus increasing the flexibility of sortase-mediated protein ligation. By applying yeast display selection20,23, error-prone PCR-based construction of random mutation libraries and structure-guided saturated mutagenesis24, the catalytic efficiency of SrtA has been improved by increasing the substrate binding affinity for the mutants eSrtA20,23 and 5M-D124G-Y187L-E189R (with three mutations relative to eSrtA)24.
SrtA accepts any amine donor with an N-terminal glycine and therefore has limited selectivity in complex biological microenvironments. The combination of this low acyl donor selectivity and the requirement of high Ca2+ concentrations means that SrtA is mostly used in simpler systems with a single amine donor (with a N-terminal polyglycine sequence), including in vitro protein ligation25, protein lipidation to facilitate membrane insertion26, enzyme immobilization27 and the homogeneous generation of antibody–drug conjugates28. Nevertheless, there are a few examples of SrtA-mediated ligation in eukaryotic cells29, including a platform in which genetic code expansion facilitates sumoylation of an internal residue by SrtA30.
Asparaginyl endopeptidases
AEPs are a subtype of cysteine proteases that cleave proteins at C-terminal asparagine or aspartic acid residues, forming a thioester intermediate that then undergoes hydrolysis or nucleophilic attack (for example, by a peptide α-amine) to yield the ligation product31. The result of AEP-mediated catalysis (hydrolysis or ligation) is therefore dependent on the concentration of nucleophiles in the microenvironment. In plants, AEPs play important roles in the maturation of seed storage proteins in the low pH environment of storage vacuoles32. To facilitate this, plant AEPs are expressed as zymogens that require low pH-induced autoactivation through the cleavage of N-terminal and C-terminal pro-domains33. The following examples, butelase-1 and OaAEP1, are two representative AEPs that are widely applied in protein and/or peptide engineering.
Butelase-1.
Butelase-1 is a unique cysteine transpeptidase isolated from Clitoria ternatea seeds that acts as a cyclase in the biosynthesis of cyclotides, a family of cyclic, cysteine-rich peptides in plants. It exhibits little hydrolase activity but instead cleaves an Asn or Asp(Asx)–His–Val motif between Asx and His to form a reactive thioacyl–enzyme intermediate that can then be intercepted by the N-terminal α-amine of a peptide to eventually form a stable amide bond34 (Fig. 2e). An important in vitro application of butelase-1 is to produce cyclic proteins and/or peptides through an intramolecular reaction driven by a nucleophilic attack by the N-terminal α-amine of the substrate on the thioester intermediate35 (Fig. 2e). In comparison with wild-type SrtA [kcat/KM (catalytic constant/Michaelis constant) ≈200 l mol−1 s−1]20,24, butelase-1 has a much higher catalytic activity, with kcat/KM values as high as 1,340,000 l mol−1 s−1 for medium-sized peptides. As a result, butelase-1-mediated ligations require only ~0.005 molar equivalents of the enzyme. Butelase-1 also possesses incredible cyclization rates that are >10,000 times faster than those of sortases36. Moreover, butelase-1 displays high catalytic promiscuity with negligible N-terminal sequence requirements for the acyl acceptor (Fig. 2e). This flexibility is highlighted by the fact that substrates consisting of d-amino acids (except for the P1 Asx residue) can also be ligated efficiently by butelase-1 (ref. 37), thus enabling the efficient synthesis of d-amino-acid-containing peptide macrocycles38. Introduction of d-amino acids into cyclic peptides may largely improve the stability and pharmacokinetics of peptide drugs39, making this an intriguing application of butelase-1-mediated ligation. This has been exemplified in the cyclization of sunflower trypsin inhibitor, the conotoxin MrlA and the antimicrobial θ-defensin38.Conversely, the high sequence tolerance of butelase-1 prevents it from being used for chemoselective ligation in complex microenvironments.
The biochemical machinery of butelase-1 provides a route for the synthesis of protein thioesters, thereby enabling tandem chemoenzymatic ligations (for example, via NCL)40. Notably, another advantage of butelase-1 is that it does not rely on cofactors and is thus not limited by their availability, as in the case of the Ca2+-dependent activity of SrtA. These advantages have enabled the use of butelase-1 for a number of applications besides cyclization, including engineering of the bacterial cell surface41, production of peptide dendrimers42 and high-yielding, N-terminal protein labelling using thiodepsipeptides as the acyl acceptor43. Importantly, SrtA and butelase-1 are orthogonal, and have been used for dual labelling of antibodies in one-pot reactions as well as C-to-C fusion of proteins44. However, the use of butelase-1 is not without drawbacks. Noticeably, the heterologous expression of recombinant butelase-1 has not been very successful so far, and most studies have been performed using the natural, plant-derived enzyme45. As a plant protein possessing three pairs of disulfide bonds, recombinant butelase-1 purified from E. coli or Pichia pastorishas exhibits undesirably low catalytic efficiency and yield, and thus the optimization of high-yield preparation of butelase-1 with excellent catalytic efficiency is still in demand46. This also means that engineering of butelase-1 has been limited. Similar to SrtA, butelase-1 suffers from the intrinsic reversibility of the transpeptidase reaction, which lowers the product yield, and an excess of substrate is required to reach yields of >50%. These obstacles largely limit the potential biotechnological applications of butelase-1 (ref.16).
OaAEP1.
In the search for butelase-1-like transpeptidases, the genomes of cyclotide-producing plants such as the Rubiaceae, Violaceae, Fabaceae, Solanaceae and Cucurbitaceae families have been mined to identify enzymes that can recognize and transform cyclotide pre-cursors containing a conserved C-terminal Asp or Asn residue. Among the identified AEPs, a promising alternative to butelase-1 is OaAEP1, isolated from the plant Oldenlandia affinis16. Although OaAEP1 is a less-active homologue of butelase-1, it is amenable to recombinant expression in E. coli, albeit at relatively modest levels (<2 mg l−1)47. OaAEP1 can catalyse cyclization of a diverse range of substrates without the assistance of any cofactors and, similar to butelase-1, OaAEP1 and its mutants (for example, E371V) have been widely used for macrocycle synthesis48 as well as for protein modification, such as site-specific sequential protein labelling49. OaAEP1 can also be used for ligating peptide–nucleic acid conjugates to proteins, thereby allowing erasable imaging of membrane proteins that rely on the sequential hybridization and removal of a fluorescent probe50. OaAEP1 therefore represents an important platform for further evolution of AEP-based ligation strategies31.
Protease-based methods
The major biological function of proteases is to cleave target protein substrates rather than facilitate transamination as seen for trans-peptidases. Proteases can be classified into broad groups on the basis of the nucleophilic residue that attacks the scissile bond in the substrate (serine proteases, cysteine proteases, threonine proteases, aspartic proteases, glutamic proteases, metalloproteases and asparagine peptide lyases)51. For protein ligation, serine proteases are of particular interest, as they can be engineered to catalyse protein and/or peptide ligation by favouring the ‘reverse proteolysis’ reaction. Here, the enzyme–substrate complex is resolved through aminolysis rather than hydrolysis, as the α-amine of a peptide donor serves as a nucleophile52. Most of these peptide ligases are derived from subtilisins (such as subtiligase, peptiligase and omniligase-1), which are secretory proteases found in soil bacteria53. These bona fide protein ligases accept a larger range of recognition motifs than the aforementioned transpeptidases, which makes them powerful and versatile tools for protein modification in vitro, but their lack of specificity does limit their potential for modifying proteins in complex microenvironments.
Subtiligases
The subtilase family of enzymes possesses an Asp–Ser–His catalytic triad and is the second largest serine protease family characterized to date, with more than 200 members identified. Subtilases are widespread and are found in eubacteria, archaebacteria, eukaryotes and even viruses54.
Among all the subtilases, subtilisins, which are secretory proteins from soil bacteria with typical molecular weights of ~27 kDa, are well studied as protein ligase scaffolds55. One successful example is subtilisin BPN’, isolated from Bacillus amyloliquefaciens. For most natural subtilisins, their peptide bond hydrolysis activity strongly dominates over the reverse peptide ligation activity. However, the introduction of two mutations (S221C and P225A) in subtilisin BPN’ tunes the engineered enzyme (subtiligase) to efficiently catalyse ligation of a C-terminal peptide ester acyl donor and an N-terminal α-amine of a peptide or protein using Ca2+ as a cofactor56,57. The S221C mutation converts the enzyme from a serine into a cysteine protease that is able to form a thioacyl–enzyme tetrahedral intermediate instead of the original oxyester, thereby generating an intermediate that is more prone to aminolysis. The P225A mutation enhances the peptide ligase activity by two orders of magnitude by negating the steric crowding within the active site58.
Unlike SrtA and butelase-1, subtiligase does not require a specific recognition motif at the substrate termini to catalyse ligation. Nevertheless, the residues on both sides of the ligation site greatly influence the catalytic performance of subtiligase, and the target is therefore usually modified to increase the yield. Notably, a protein and/or peptide ester substrate is needed in subtiligase-mediated ligation to serve as the acyl donor (Fig. 3a), necessitating that the N-terminal ligation partner is either entirely synthetic or has its C terminus functionalized. Subtiligase also requires a large excess of acyl acceptor to suppress hydrolysis. Moreover, subtiligases are typically expressed as pre-pro-proteins, in which the pre-sequence serves as a signal peptide for secretion and the pro-domain is required for folding of the functional mature enzyme before its autocatalytic removal59.
Fig. 3 |. Biochemistry of protease-based protein and peptide ligations.
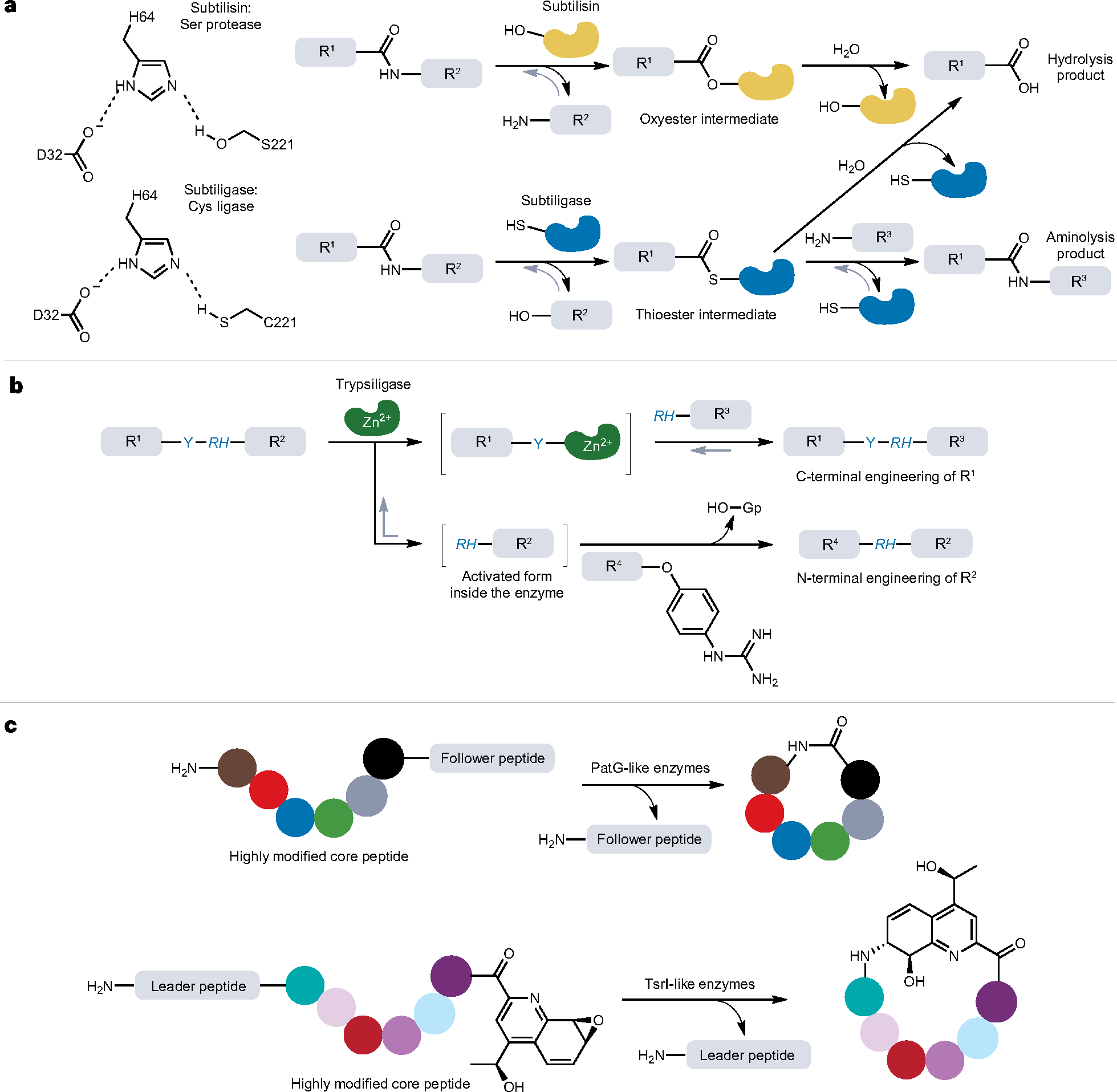
a, Biochemical mechanisms of subtilisin and subtiligase-mediated protein and peptide ligation. The catalytic triad of subtilisin is Asp–His–Ser (D32–H64–S221) and that of subtiligases is Asp–His–Cys (D32–H64–C221). For subtiligase, aminolysis is favoured over hydrolysis, thereby enabling the enzyme to support protein ligation. b, Trypsiligase-mediated protein ligation and its applications. The Y–RH sequence can be recognized and cleaved by trypsiligase. Guanidinophenyl (Gp) is a good leaving group that activates the C terminus of acyl donor, R4. R1–R4 denotes peptide or protein sequences throughout (a,b). c, Macrolactonization catalysed by protease-derived macrocyclases, PatG and TsrI, involved in microbial biosynthetic pathways. These enzymes catalyse not only the removal of signal peptides (such as leader or follower peptides) but also the intramolecular peptide bond formation between the N and C termini, as in the case of peptide macrolactonization. The coloured circles represent individual amino acid residues after post-translational modifications.
After years of optimization and screening, functionally enhanced subtiligase variants have been successfully generated60. In one variant obtained through directed evolution61, the pro-domain and calcium loop were deleted to circumvent the need for autocatalytic cleavage to generate the mature enzyme as well as the Ca2+ dependency. Similarly, a subtiligase variant termed stabiligase has been generated via the introduction of five stabilizing mutations (M50F, N76D, N109S, K213R and N218S) that enable protein ligation in the presence of denaturants such as SDS and guanidinium hydrochloride62,63.
Owing to their high promiscuity, subtiligases have been widely used in protein chemistry. One important application is subtiligase-mediated EPL, in which the thioester substrate is usually synthesized from an intein-tagged recombinant protein through thiolysis64. Classical EPL is performed via NCL, meaning that an N-terminal cysteine in the amine donor is required. However, in subtiligase-mediated EPL, the cysteine residue is no longer needed because the transthioesterification and subsequent S→N-acyl shift of NCL are not required for the enzymatic ligation. This strategy has been used to efficiently produce C-terminally phosphorylated tumour suppressor protein PTEN (phosphatase and tensin homologue)65. A proteomics-based characterization of the specificity of 36 subtiligase variants has identified mutants with distinct reactivities that enable orthogonal N-terminal labelling of proteins with different N-terminal sequences63. By combining four of these mutants with broad N-terminal specificity together, the promiscuity of subtiligase-mediated labelling was exploited to enrich, and hence map, the cellular N-terminome.
Peptiligase and omniligase-1.
In the pursuit of improved peptide ligases, two-point mutations were introduced into an already heavily engineered subtiligase scaffold66. The resulting ligase, termed peptiligase, contains 18 stabilizing mutations as well as deletions of the pre-domain, pro-domain and calcium-binding domain. Peptiligase can catalyse ligations to extremely high yields (>98% yield in less than 1 h) using only a slight excess of one of the reagents, for example, 1.1–1.5 equivalents of acyl acceptor. Compared with other commonly used peptide ligases, peptiligase is thermostable (TM = 66 °C) and functions well in the presence of organic solvents (up to 50 vol% N, N-dimethyl-formamide) as well as denaturants (2 M urea or guanidinium chloride), making it a particularly useful tool for the ligation of poorly soluble or folded proteins or peptides.
To gain a broader acyl acceptor substrate scope, peptiligase has been further engineered via site-directed mutagenesis67. One resulting enzyme is omniligase-1, which is useful for chemo-enzymatic peptide synthesis as well as for protein semisynthesis. Furthermore, omniligase-1 can catalyse the formation of head-to-tail macrocyclic products using substrates that are >300 residues long, and it has been applied in the gram-scale synthesis of cyclic peptides, making it viable for industrial-scale protein ligation68,69. In summary, both omniligase-1 and peptiligase-mediated coupling reactions are scalable and can be used as a versatile stand-alone technology, as well as in combination with chemical or intein-based protein ligation methodologies67.
Trypsiligase
Trypsin is a serine protease belonging to the PA (proteases of mixed nucleophile, superfamily A) clan superfamily and is one of the most widely used enzymes in proteomics research70. Trypsin is found in the digestive system of many vertebrates, where it hydrolyses proteins by cleaving peptide bonds at the carboxyl side of unmodified lysine and arginine residues71. Although the potential use of trypsin variants for peptide and protein synthesis has been known for decades71, extensive protein engineering was required before trypsin-derived ligases became an integral part of the protein semisynthesis toolbox.
One of the most successful and widely used trypsin variants is trypsiligase, a rationally designed quadruple mutant (K60E, N143H, E151H and D189K) of anionic rat trypsin II (ref. 72). Trypsiligase has a high ligation rate and specifically cleaves the tripeptide motif Y–RH, followed by transpeptidation of the acyl to an α-amine with an N-terminal RH motif (Fig. 3b). This means that trypsiligase catalyses N-terminal modification that only leaves a small, two-residue (RH) ‘ligation scar’. However, as with all transpeptidase reactions, trypsiligase-catalysed ligation suffers from lower yields caused by the reversibility of the reaction and competing hydrolysis of the acyl– enzyme intermediate. Additionally, trypsiligase adopts a zymogenlike conformation, meaning that both the Y–RH tripeptide motif and its cofactor Zn2+ are required to induce its ligation activity73. This unique biochemistry minimizes proteolytic side reactions, enabling trypsiligase to mediate N-terminal labelling with a substrate mimetic as the acyl donor74 (Fig. 3b), as well as C-terminal labelling with synthetic moieties75, including click handles that allow further derivatization73. Intriguingly, only 0.5% of all proteins in the SwissProt database contain the Y–RH recognition motif despite its small size45, thereby allowing Y–RH to serve as a useful tag for site-specific modification of either the N-terminal or C-terminal region of target proteins in trypsiligase-mediated ligations (Fig. 3b).
Macrocyclases from microbial biosynthetic pathways
In the biosynthetic pathways of microbial polypeptide metabolites, some macrocyclases have evolved dual functions and perform both proteolysis and macrolactonization76–79. These enzymes have been discovered in the biosynthesis of cyanobactins, a family of ribosomal cyclic peptides produced by cyanobacteria80.
One particularly well-studied example is PatG, a macrocyclase involved in the biosynthesis of patellamide. The PatG-like macrocyclases contain an Asp–His–Ser catalytic triad, enabling them to catalyse proteolytic cleavage of a C-terminal recognition sequence, termed the ‘follower peptide’, in tandem with peptide macrocyclization81 (Fig. 3c). In contrast to other protease-derived protein ligases, PatG-like enzymes have a narrower substrate scope, limited to peptides that have been post-translationally modified by heterocyclization of cysteine, serine or threonine to form thiazole, thiazoline or oxazoline residues. Although this equips PatG with a high degree of specificity, it also necessitates the synthesis of non-standard proteogenic substrates for ligation82. Structural biology studies indicate that PatG enzymes contain a conserved helix–loop–helix insertion that may prevent the acyl–enzyme intermediate from being attacked by a water molecule81, thus preventing hydrolysed by-products. Interestingly, deletion of this segment results in a PatG variant with maintained protease activity that no longer catalyses peptide macrocyclization83,84. Although useful in producing peptide macrocycles, further engineering and optimization are required to make PatG a more widely applied molecular tool, as illustrated by efforts that circumvent the need for a C-terminal proline/thiazoline (for example, PagGmac-Cys275Ala)85,86.
Another subtype of dual-functional macrocyclases are TsrI-like enzymes, which are involved in the biosynthetic pathway of bicyclic thiopeptides and play essential roles in the construction of the molecular structure of thiostrepton. TsrI belongs to the α/β-hydrolase fold enzyme family and possesses a Ser–His–Asp catalytic triad. Similar to PatG, TsrI catalyses proteolytic cleavage followed by macrocylization. However, unlike PatG, TsrI-mediated macrocylization occurs through a unique epoxide ring-opening reaction following cleavage of the N-terminal leader peptide87 (Fig. 3c). Although previous studies have shown that TsrI-like enzymes can tolerate amino acid substitution in the sequences of the leader and core peptides, this family of enzymes has not been commonly used in protein or peptide chemistry as they require highly modified substrates87.
Transglutaminase-mediated methods
Unlike the enzymes mentioned previously that specifically catalyse ligations between the N-terminal α-amino groups and C-terminal carboxyl groups of peptides or proteins, transglutaminases (TGMs) are a type of naturally occurring protein ligase that non-specifically catalyses isopeptide bond formation using the side chain amide group of glutamine residues88. An aminolysis reaction between the glutamine γ-carboxamide and the ε-amino group of lysine results in the formation of covalent crosslinks that bind the proteins together (Fig. 4). The food industry employs TGMs to crosslink pieces of meat; hence, TGMs are also referred to as ‘meat glue’89. TGMs are found in most domains of life including animals, plants and microorganisms. Mammalian TGMs (for example, TGM2) require Ca2+ as a cofactor, whereas those isolated from bacteria (such as Streptomyces mobaraensis) are calcium-independent enzymes. TGMs have a very broad natural substrate scope ranging from cytoplasmic proteins to histones90. Because TGMs do not have any sequence selectivity, they have limited use for in vivo and in cellulo applications for protein ligation91.
Fig. 4 |. Biochemistry of the isopeptide bond formation mediated by transglutaminases.
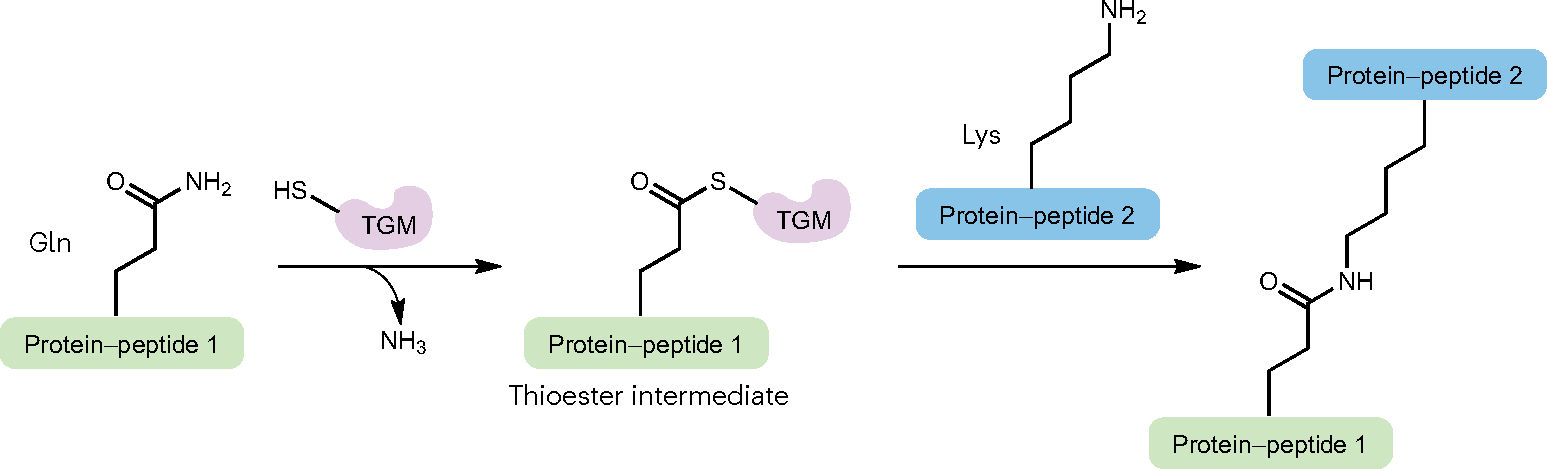
The isopeptide bond is formed between Gln and Lys residues of the protein or peptide substrates. A Cys in transglutaminases (TGMs) is the catalytic residue for this reaction, which proceeds through a reactive thioester intermediate. Ammonia is the by-product of this enzymatic ligation.
Although TGMs are unable to catalyse the formation of peptide bonds, constructing isopeptide bonds can be useful for producing side chain-conjugated proteins. For example, S. mobaraensis TGM is a versatile tool for manufacturing antibody–drug conjugates, such as human IgG1 derivatives92. Here, either lysine or glutamine residues within the antibody are used to create the conjugate, and the drug molecule is designed with a carboxamide or primary amine as the reactive group to target the complementary residue (or residues).
Molecular superglue-mediated methods
The emergence of ‘molecular superglue’ techniques has provided new protein and peptide ligation strategies, such as the SpyTag–SpyCatcher and SnoopTag–SnoopCatcher reactive pairs93,94. These techniques rely on the formation of an isopeptide bond between aspartate (or glutamate) and lysine residues by spontaneous condensation95. The reaction was initially discovered in Gram-positive bacteria (for example, Streptococcus pyogenes) where it serves to stabilize extracellular proteins such as Spy0128 (refs. 96,97). In the invasive strains of S. pyogenes, the second immunoglobulin-like collagen adhesion domain (CnaB2) from the fibronectin-binding protein FbaB contains a single isopeptide bond that is autocatalytically formed, stabilizing the protein and extending its half-life and durability. To take advantage of this unique biochemical arrangement, CnaB2 has been split and engineered to produce a 13-amino acid peptide tag (SpyTag) and a 138-amino acid protein partner (SpyCatcher). The SpyTag and SpyCatcher associate with nanomolar affinity and are able to ligate two protein segments in vitro or in vivo93 (Fig. 5a). Recently, the SpyTag–SpyCatcher pair was used to enforce asymmetry on nucleosome core particles, which represent the fundamental unit of chromatin, by covalently linking two different variants of a given histone subtype (in this case, H3) during in vitro reconstitution, thus generating a more physiologically relevant form of the complex98. Moreover, SpyTag–SpyCatcher has been used to overcome the low catalytic activity of SrtA by covalently linking the enzyme to its substrate in the so-called ‘proximity-based sortase-mediated ligation’99.
Fig. 5 |. Molecular superglue-mediated isopeptide bond formation.
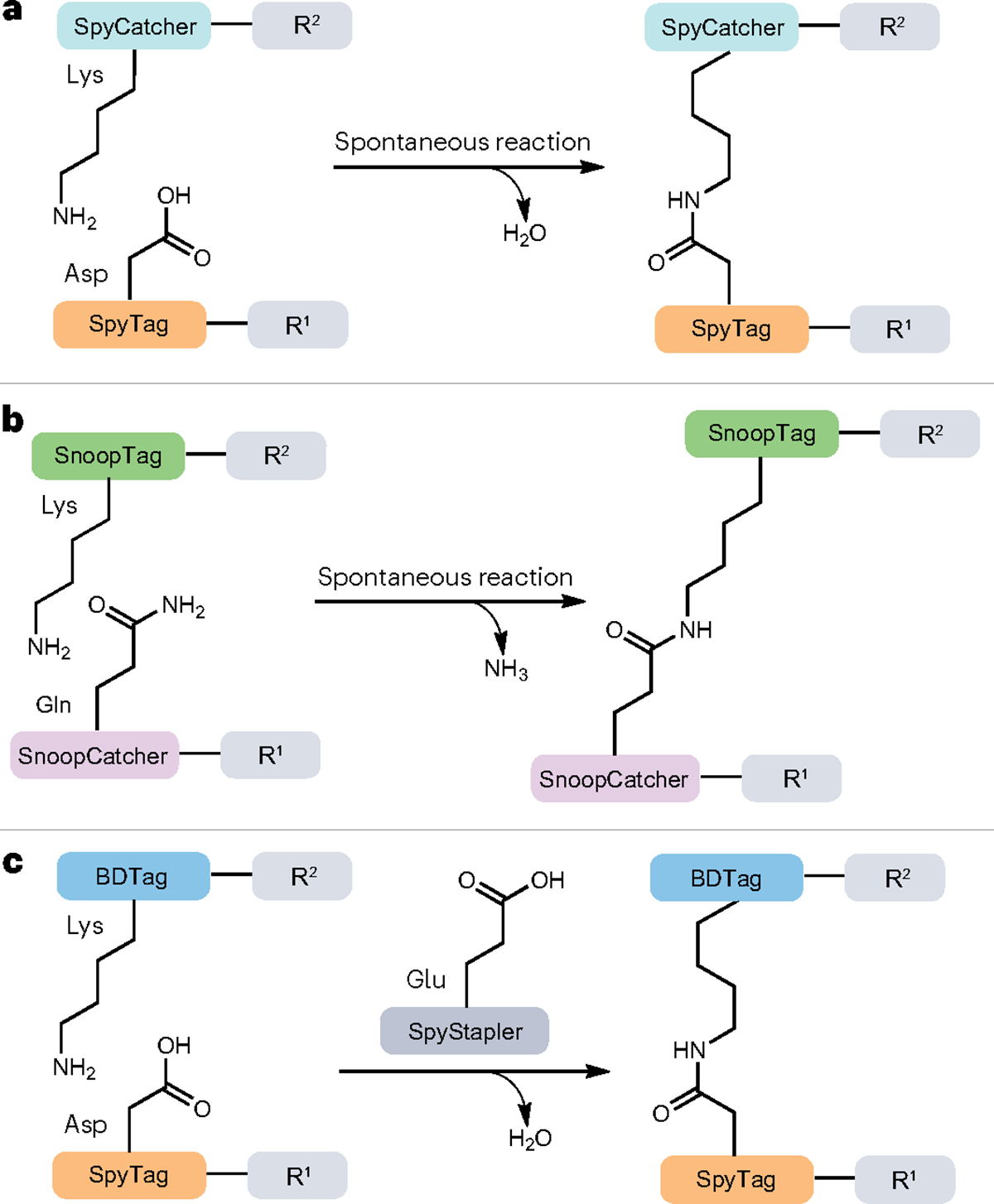
a–c, Spontaneous ligation reactions mediated by SpyCatcher–SpyTag (a), SnoopCatcher–SnoopTag (b) and SpyStapler–SpyTag–BDTag (c) (also known as ‘peptide–peptide staplers’) systems. In the SpyStapler–SpyTag–BDTag system, the second immunoglobulin-like collagen adhesin domain (CnaB2) is divided into three components by further splitting SpyCatcher to yield an intrinsically disordered protein, SpyStapler, which can form a stably folded structure in the presence of SpyTag and BDTag to facilitate the formation of an isopeptide bond between them. R1 and R2 stand for peptide–protein sequences. Water (a,c) and ammonia (b) are the by-products of this type of enzymatic ligation.
A similar splitting and engineering strategy has been used for the adhesion protein RrgA from S. pneumoniae, yielding the peptide SnoopTag (12 amino acids) and its protein partner SnoopCatcher (112 amino acids) (Fig. 5b). The condensation of SnoopTag and Snoop-Catcher occurs in near quantitative yields and does not crossreact with SpyTag–SpyCatcher, enabling bioorthogonal protein labelling using these protein pairs94,100. In a similar Tag–Catcher system, the CnaB protein from Streptococcus dysgalactiae has been split and engineered into the SdyTag–SdyCatcher pair, a homologue of SpyTag–SpyCatcher101.
The molecular superglue principle has been further developed to generate ‘peptide–peptide staplers’, an alternative approach for efficient protein ligation both in vivo and in vitro102. In this unique ligation reaction, the CnaB2 protein is divided into three components by further splitting the SpyCatcher piece at a solvent-exposed second loop region to yield SpyStapler and BDTag. Although SpyStapler is intrinsically disordered on its own, it forms a stably folded structure in the presence of SpyTag and BDTag, and a glutamate residue in SpyStapler is critical for isopeptide bond formation between SpyTag and BDTag. (Fig. 5c). By expanding the Tag–Catcher platform with a third component, the level of spatiotemporal control over covalent coupling may be improved by controlling the localization or expression of SpyStapler102. Although these systems are far from traceless and involve fairly large tags, they are promising bioorthogonal protein ligation tools for generating intramolecularly and intermolecularly crosslinked structures. As such, these techniques have been employed for tracking the dynamics of a membrane protein during cell division in E. coli103, increasing the thermostability of luciferase by cyclization104 and creating artificial protein structures of different topologies105.
Ubiquitin ligase-based ligation strategies
Ubiquitin (Ub) is a small protein of 76 amino acids, which is highly conserved from yeast to humans, that can be covalently linked to the lysine residues of target proteins to signal their degradation by the 26S proteasome or to modify their function or localization106. A set of three enzymes (E1, E2 and E3) catalyses the ligation of Ub to the ε-amino group of lysine (Fig. 6A) to generate a branched protein structure through an isopeptide bond. The Ub-ligating enzymes are referred to as E3s and operate in conjunction with an E1 Ub-activating enzyme and an E2 Ub-conjugating enzyme107. E1 enzymes require ATP as a cofactor to activate Ub and generate a thioester complex that is subsequently transferred to an E2 enzyme. Most commonly, the E2 enzyme then interacts with an E3 enzyme, enabling Ub to be transferred to the target protein substrate. This process can occur multiple times on a single substrate to eventually generate a polyubiquitylated product108. The E3 ligases are often multisubunit protein complexes, and they control the specificity of ubiquitylation by directing the PTM to specific substrate proteins.
Fig. 6 |. Ubiquitin and ubiquitin-like modifier ligase-mediated isopeptide bond formation and its applications.
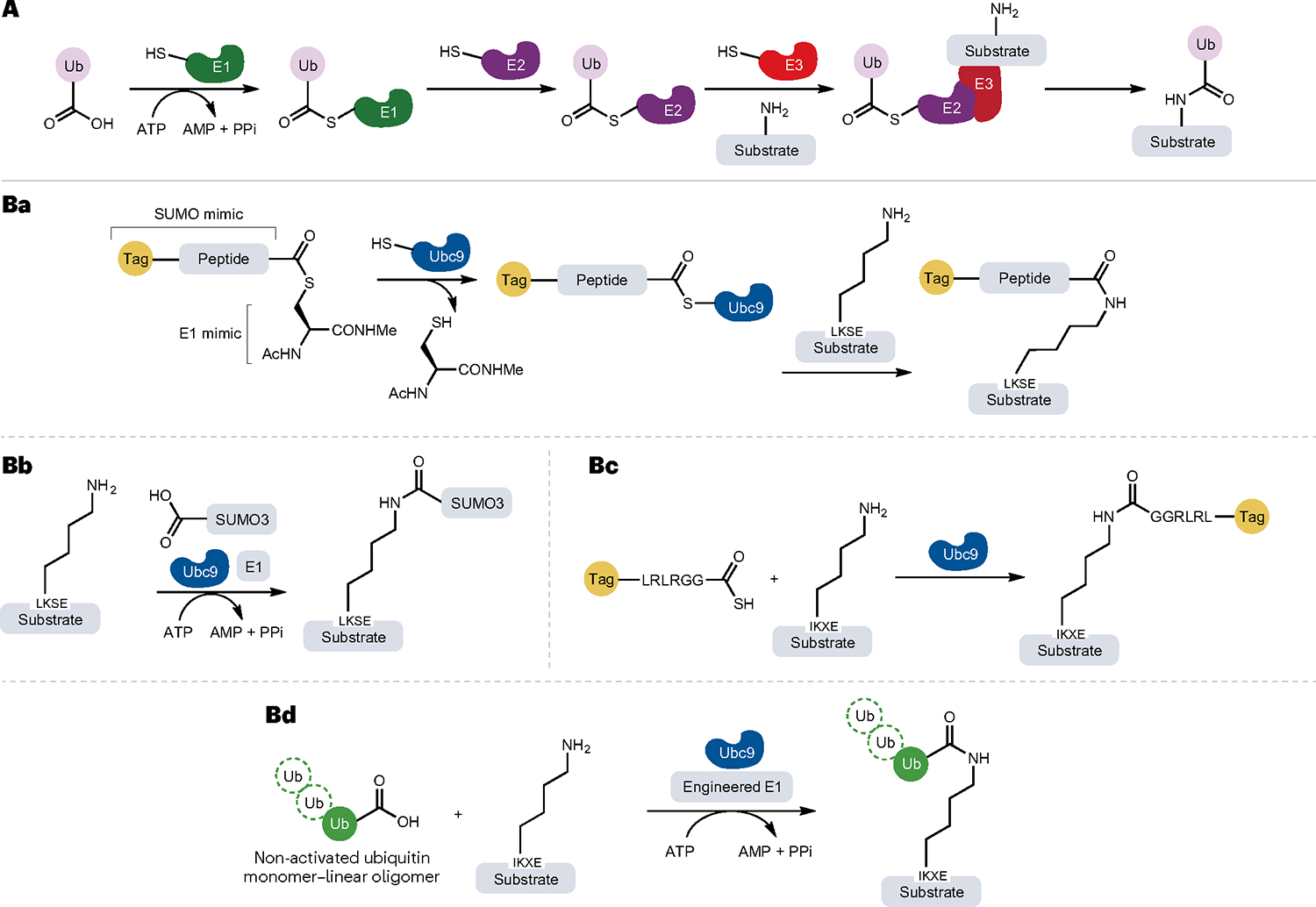
A, General mechanism of protein ubiquitylation (isopeptide bond formation) catalysed by E1, E2 and E3 enzymes. Ba,Bc, Site-specific modification and ubiquitylation of target proteins containing synthetic or genetically encoded tags by the LACE platform (lysine acylation using conjugating enzymes). A minimal genetically encoded tag (LKSE or IKXE) in the substrate proteins or peptides acts as acyl acceptor, whereas a peptide sequence (LRLRGG) mimicking the C terminus of ubiquitin (Ub) functions as an acyl donor when functionalized as a thioester. Thus, the thioester acyl donor mimics the structure of the small Ub-like modifier (SUMO)–E1 complex. Using thioesters and Ub-conjugating enzyme E2 (Ubc9), proteins can be modified with chemical probes and even small proteins. Bb, Non-activated SUMO3 can be site specifically installed at the LACE tag by the combined action of Ubc9 and an E1 enzyme in the presence of the cofactor ATP, which is hydrolysed to AMP and two molecules of phosphate (PPi). Bd, The development of a chimeric E1 enzyme enables conjugation of non-activated Ub. Ac, acetyl group; Me, methyl group.
The unique biochemical features of ubiquitylation have been used to turn Ub into a degradation tag for selectively downregulating cellular levels of target proteins. Such proteolysis targeting chimeras (PRO-TACs) have emerged as a powerful tool for studies of protein function as well as for drug development. However, as protein ubiquitylation plays essential roles in cellular signal transduction, especially protein degradation, E3 Ub ligases have not found use in protein semisynthesis or engineering owing to the risk of off-target activity that could perturb cellular function. In addition to Ub, there are multiple Ub-like (UBL) modifiers that are structurally similar to Ub and possess a conserved C-terminal glycine residue, which facilitates the condensation with lysine residues in substrate proteins109. Of the eight families of UBL modifiers that are conjugated to protein substrates109, only the machinery installing SUMO (small UBL modifier) has served as a starting point for the development of protein ligation strategies so far110,111.
The E2 conjugase of SUMO, Ubc9, has been used for the site-specific attachment of biochemical probes, one-pot dual labelling in combination with either SpyTag–SpyCatcher or the sortase variant SrtA7M and conjugation of wild-type Ub and ISG15 to recombinant target proteins112. This strategy, termed lysine acylation using conjugating enzymes (LACE), bypasses the need for E1 and E3 enzymes and enables isopeptide bond formation using just Ubc9. In this reaction, a short genetically encoded tag (LKSE or IKXE) is required in the substrate proteins or peptides to act as an acyl acceptor, whereas the acyl donors are thioesters possessing a C-terminal LRLRGG sequence that can be further activated by Ubc9 (Fig. 6Ba). In this way, lysine acylation using conjugating enzymes permits site-specific modification of internal lysine residues through the introduction of a small ‘ligation scar’. Additionally, Ubc9 can also use the non-activated SUMO3 as a substrate in combination with the E1 enzyme and ATP cofactor112 (Fig. 6Bb). Because the loading of the thioester onto Ubc9 is rate-limiting, a recent method used an engineered E1 enzyme to speed up the formation of the Ubc9–thioester intermediate113 (Fig. 6Bc). Importantly, this strategy also circumvents the need for synthetic thioesters, thus enabling conjugation of non-activated Ub (Fig. 6Bd).
Intein-based methods and applications
Although the field of transpeptidase-mediated and protein ligase-mediated protein ligation is rapidly evolving, these strategies have found limited use in complex biological systems. The specificity of these enzymes is dictated by recognition sequences that are retained as ‘ligation scars’ in the product, preventing protein ligation from being simultaneously highly specific and traceless.
Inteins (intervening proteins) can overcome these issues by relying on high-affinity, protein–protein interactions rather than primary sequence motifs for nearly traceless and chemoselective ligation. Additionally, intein-mediated ligation is irreversible and can thus reach high yields. More than 1,000 intein-encoding genes have now been identified in unicellular organisms from all three domains of life: Bacteria, Archaea and Eukarya114,115. Inteins self-excise from their host proteins, thereby ligating the flanking polypeptides (exteins) together through a native peptide bond (Fig. 7a). Inteins can therefore be viewed as single-turnover enzymes that break two amide bonds to form a single new bond116. Contiguous inteins are produced as single polypeptide chains and can control the reassembly, and hence function, of the naturally or artificially split proteins in which they are embedded. Importantly, inteins themselves can be split into two discrete fragments that facilitate protein trans-splicing (PTS) upon association (Fig. 7b). Such split inteins are powerful tools for the bioorthogonal production of semisynthetic, site-specifically modified proteins through fragment condensation. Here, we focus on major developments in intein technology that have moved the field towards control of the chemical composition of proteins in complex biological systems. Other parts of the intein toolbox have been excellently reviewed elsewhere2,117,118.
Fig. 7 |. Mechanism of intein-mediated protein ligation.
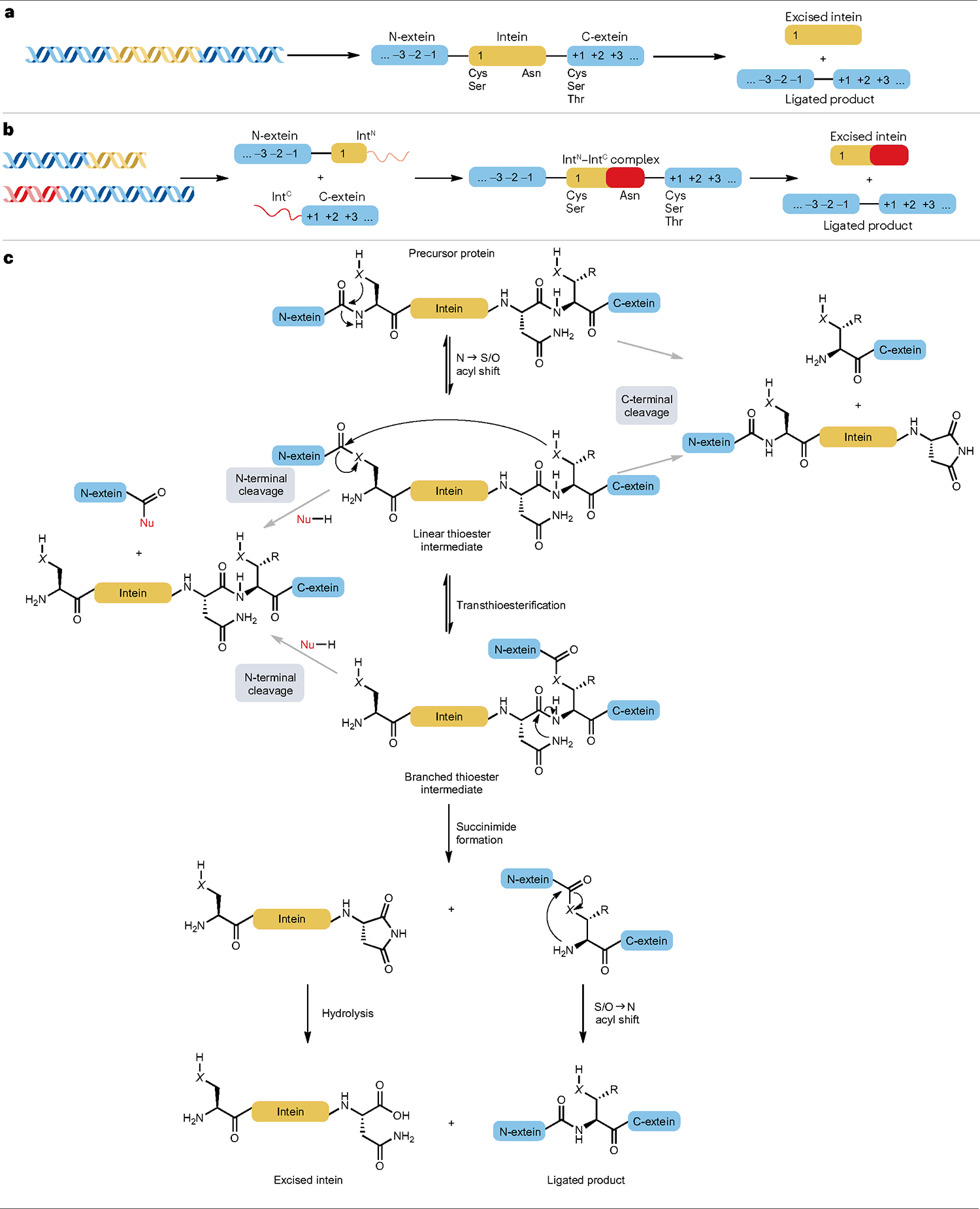
a, Contiguous inteins are produced as single proteins flanked by two extein sequences. The numbering highlights the nomenclature used to denote key amino acid positions in inteins and exteins together with the type of amino acids found at the indicated positions. b, Split inteins are generated as discrete polypeptides that associate to facilitate ligation of the flanking extein sequences. Split inteins contain disordered regions that fold into a splicing-competent complex upon association to carry out protein trans-splicing. The canonical intein split sites yield larger N-terminal split-intein fragments (IntN) and smaller C-terminal split-intein fragments (IntC). c, Mechanism of intein-based splicing. Efficient splicing competes with premature cleavage reactions in which the thioester intermediates are intercepted by a nucleophile (Nu-H, usually H2O). X designates oxygen for inteins with a Ser residue at position 1 or sulfur for inteins that use Cys as their nucleophile.
The mechanism of intein-mediated protein ligation
Inteins catalyse protein splicing through a series of nucleophilic attacks that are supported by conformational changes in intein structure119,120. Although the precise splicing mechanism may vary, most inteins use a four-step reaction sequence that relies on a few conserved residues (Fig. 7a–c). First, the N-terminal nucleophile of the intein (Cys or Ser) attacks the carbonyl carbon of the adjacent amide bond, inducing a reversible NS/O acyl shift that generates a linear intermediate. To favour (thio)ester formation, inteins may distort the scissile bond and/or increase the nucleophilicity of the position 1 Cys or Ser121–124. Second, the N-extein is transferred to the C-extein through a trans(thio)esterification step mediated by the +1 nucleophile of the C-extein, resulting in a branched intermediate. The +1 nucleophile, which is the N-terminal residue of the C-extein, can be Cys, Ser or Thr (Fig. 7a). Third, the irreversible cyclization of a conserved, C-terminal Asn resolves the branched intermediate and excises the intein as a succinimide125. This is the rate-limiting step of protein splicing, which is accelerated by conformational changes associated with the formation of the branched intermediate126–129. Lastly, the exteins undergo a spontaneous S/O→N acyl shift to restore a native peptide bond and complete protein ligation. Along its reaction path, protein splicing competes with side reactions that lead to N-terminal or C-terminal cleavage through hydrolysis of the (thio)ester intermediates130. Inteins generally facilitate efficient protein splicing when situated within their native extein contexts, but the risk of side reactions increases when the coordination of the reaction steps is perturbed by, for example, splicing of non-native exteins126,131. Thus, as discussed subsequently, increasing the extein tolerance of inteins will decrease the level of side product formed during splicing.
PTS follows the same overall mechanism as canonical inteins but depends on initial association of the split-intein fragments IntN and IntC (Fig. 7b). The split fragments are largely disordered and can refold into a functional intein by a two-step, association–collapse sequence into an intertwined, stable complex132–134. Split inteins can either be naturally occurring or artificially split versions of contiguous inteins, but the natural split inteins often display superior properties and associate more strongly with KD values in the low nanomolar range132,135, whereas the KD value of artificially split inteins can be in the low micromolar range136. Furthermore, the kinetics of split-intein association and the subsequent folding are fast and do not limit the rate of PTS137. Consequently, efficient PTS can be achieved using even low concentrations of the reactants.
The ever-expanding intein toolbox
Despite the great potential of intein-based protein modification, its application comes with a set of challenges, including poor intein fragment solubilities, slow splicing rates, strong extein dependencies and the size of split inteins making them practically inaccessible to solid-phase peptide synthesis (SPPS). Although these issues remain relevant, tremendous strides have been made to lower these barriers through protein engineering and continued characterization of new naturally occurring intein pairs.
The capacity to carry out efficient protein splicing is considered the only evolutionary pressure exerted on inteins. Because natural selection has only occurred within their native extein context, inteins are often highly specialized, and many contiguous inteins splice inefficiently with half-lives of several hours when tasked with ligating non-native extein sequences120,126. The first naturally split intein to be identified, the Ssp DnaE intein from Synechocystis sp. PCC6803, splices with a half-life of 75 min at 30 °C138, which is considerably slower than many biological processes. However, nature has also developed ultrafast inteins, including the well-characterized Npu DnaE intein, identified from Nostoc punctiforme PCC73102, which splices with a half-life of 63 s at 37 °C139. Fast inteins are widespread in cyanobacteria138 and have enabled the generation of a consensus fast (Cfa) intein that splices with an improved half-life of 20 s at 30 oC and display enhanced protein stability140. Additionally, even faster intein pairs have been identified from metagenomic data, including Gp41–1, which remains the fastest intein to date with a half-life of ~5 s at 37 °C141. Examination of data from a saline lake in Antarctica identified the AceL–TerL intein, which splices efficiently at 8 °C (t1/2 ~ 7 min)142, paving the way for efficient protein labelling at low temperatures.
Traceless PTS requires inteins to accommodate the sequences of any protein of interest (POI) as exteins. However, inteins are not inherently promiscuous, as the +1 nucleophile is essential for the initial (thio) esterification step that generates the linear (thio)ester intermediate. Furthermore, the splicing efficiency depends on the nature of the C-terminal and N-terminal residues in the N-extein and C-extein, respectively127,130,143, and it has often been necessary to insert three to five residues of the native extein sequences into target proteins to promote splicing. In particular, splicing is highly sensitive to the identity of the residues at the +2 and +3 positions. Owing to this extein dependency, protein engineering has been used to alter the extein preference of the chimeric Npu DnaEN + Ssp DnaEC pair144, Ssp DnaB (yielding the M86 intein)145 and the Pho RadA intein146. Furthermore, the splicing capacity of the widely used DnaE inteins depends strongly on the presence of a large hydrophobic +2 residue147–149, a requirement that can be alleviated by introducing three mutations that increase the promiscuity of both Npu DnaE and Cfa150. Finally, different intein pairs will naturally have varying extein preferences, as they have adapted to different host proteins146,151, suggesting that the continuous characterization of new inteins will further increase the extein tolerance of the intein toolbox at large.
A key application of PTS is the generation of semisynthetic proteins by fusing one split intein to a truncated protein and ligating synthetic cargo to the other (Fig. 8a). Hence, the size of the intein–synthetic cargo fusion should ideally be compatible with standard SPPS. Most naturally split inteins are split at a canonical site that produces IntN and IntC fragments of approximately 100 and 35 residues, respectively2. This means that the IntN is well beyond the reach of SPPS, whereas IntC is near the feasible limit of SPPS, leaving little room for the addition of synthetic cargo. Thus, it has been a priority to search for novel split sites that reduce intein fragment size while preserving efficient PTS kinetics. The feasibility of N-terminal modification, in which IntN carries the synthetic cargo (Fig. 8a), has been increased by identifying atypically split inteins from metagenomic data142,152,153. The IntN of these inteins is significantly shorter, with the shortest being only 15 residues153. Interestingly, a consensus atypical split intein (Cat) was found to have a high C-extein tolerance134, highlighting that the location of the split site may be an underappreciated strategy for relaxing extein dependency. The IntN size has been further reduced to 11–12 residues using artificial split sites154,155; however, this does compromise splicing efficiency156. Moreover, artificially split-intein pairs often suffer from decreased binding affinities, requiring intein fragments to be fused to a pair of high-affinity interacting modules to achieve efficient PTS136,157. To support C-terminal modification, IntC fragments have been shortened to only five and six residues without a significant loss of splicing efficiency156,158. Recently, Thompson et al.159 developed a one-pot strategy termed transpeptidase-assisted intein ligation (TAIL) for making semisynthetic proteins in which SrtA-mediated ligation is combined with PTS (Fig. 8b). By using SrtA for assembling an active split intein, TAIL reduces the size of the synthetic intein to seven residues and is applicable to N-terminal and C-terminal protein modification.
Fig. 8 |. Split intein-based strategies for generating semisynthetic proteins.
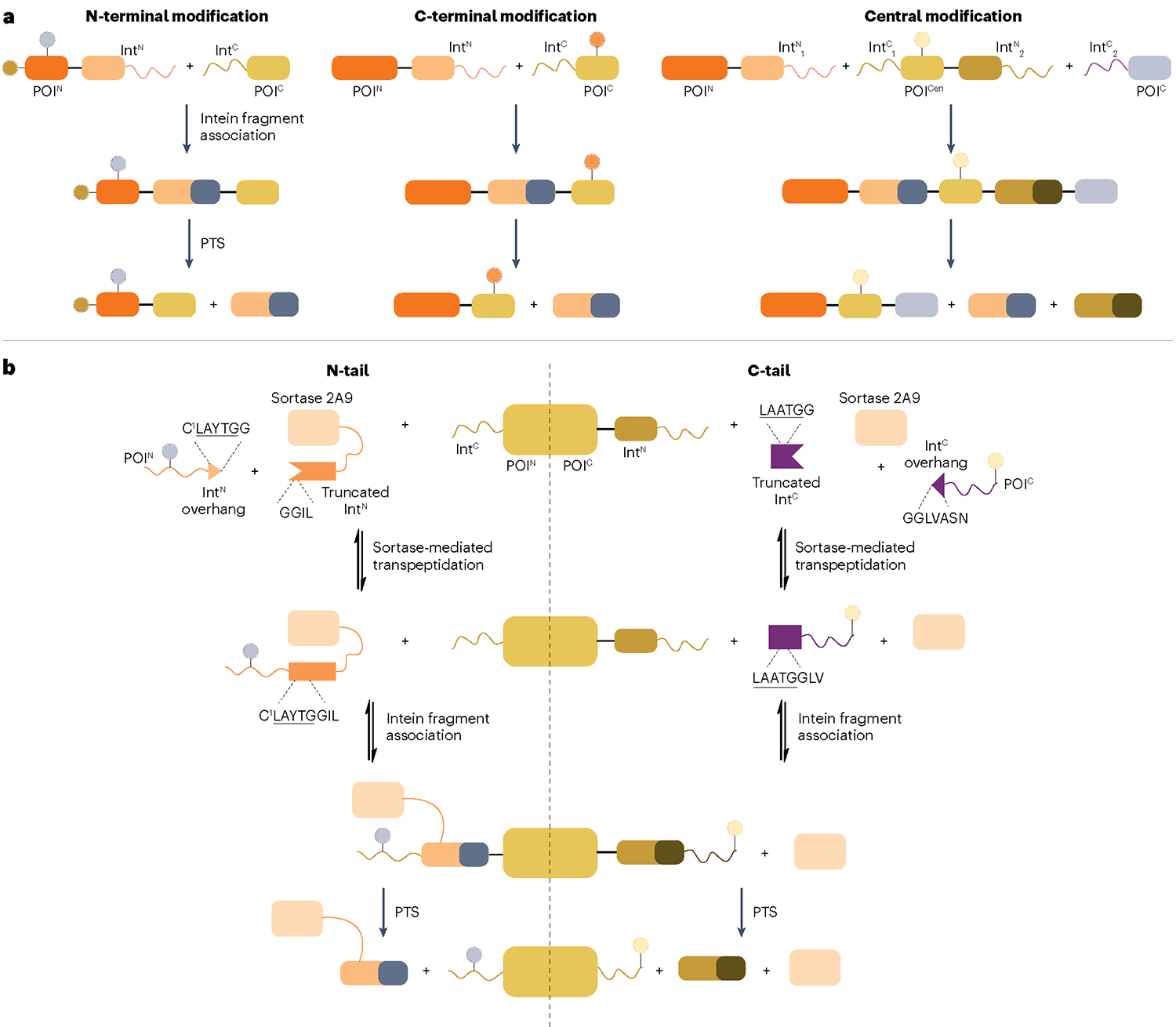
a, Proteins can be site-specifically modified at their termini by splitting them into two separate pieces, each fused to a split-intein fragment. For N-terminal modification, a short, synthetic N-terminal segment of the protein of interest (POI) (POIN) is fused to the N-terminal split intein fragment (IntN), whereas the recombinantly expressed C-terminal POI fragment (POIC) is fused to the C-terminal split-intein fragment (IntC). Upon association and protein trans-splicing (PTS), an N-terminally modified POI is produced. Similarly, C-terminal modification is performed by making the IntC–POIC complex synthetically. To modify a central segment of a POI (POICen), orthogonal intein pairs are used in a tandem protein splicing scheme in which the POICen is synthetic and fused to intein fragments at both termini. IntN1 and IntC1 as well as IntN2 and IntC2 represent two pairs of orthogonal split inteins. b, Transpeptidase-assisted intein ligation (TAIL) enables N-terminal (N-tail) and C-terminal (C-tail) modification of proteins by combining sortase- and intein-mediated protein ligation. Split inteins are further split into small overhangs (IntN overhang and IntC overhang) containing sortase recognition sequences LAYTG or LAATG and truncated, inactive inteins. The reversible, sortase-mediated transpeptidation step generates active split inteins carrying synthetic cargo, which associate with their split-intein partner for protein semisynthesis through irreversible PTS. Note that sortase and the truncated IntN are fused in N-tail to increase the reaction rate and thereby suppress premature cleavage of IntC–POIN complex.
Conditional protein splicing
The spontaneous nature of protein splicing makes naturally occurring inteins unattractive for examining biological processes that require strict temporal and/or spatial control. Therefore, several conditional protein splicing (CPS) techniques have been developed, which allow control of intein activity by extrinsic cues, that can be grouped based on the nature of the trigger (Fig. 9).
Fig. 9 |. Strategies for conditional protein splicing (CPS).
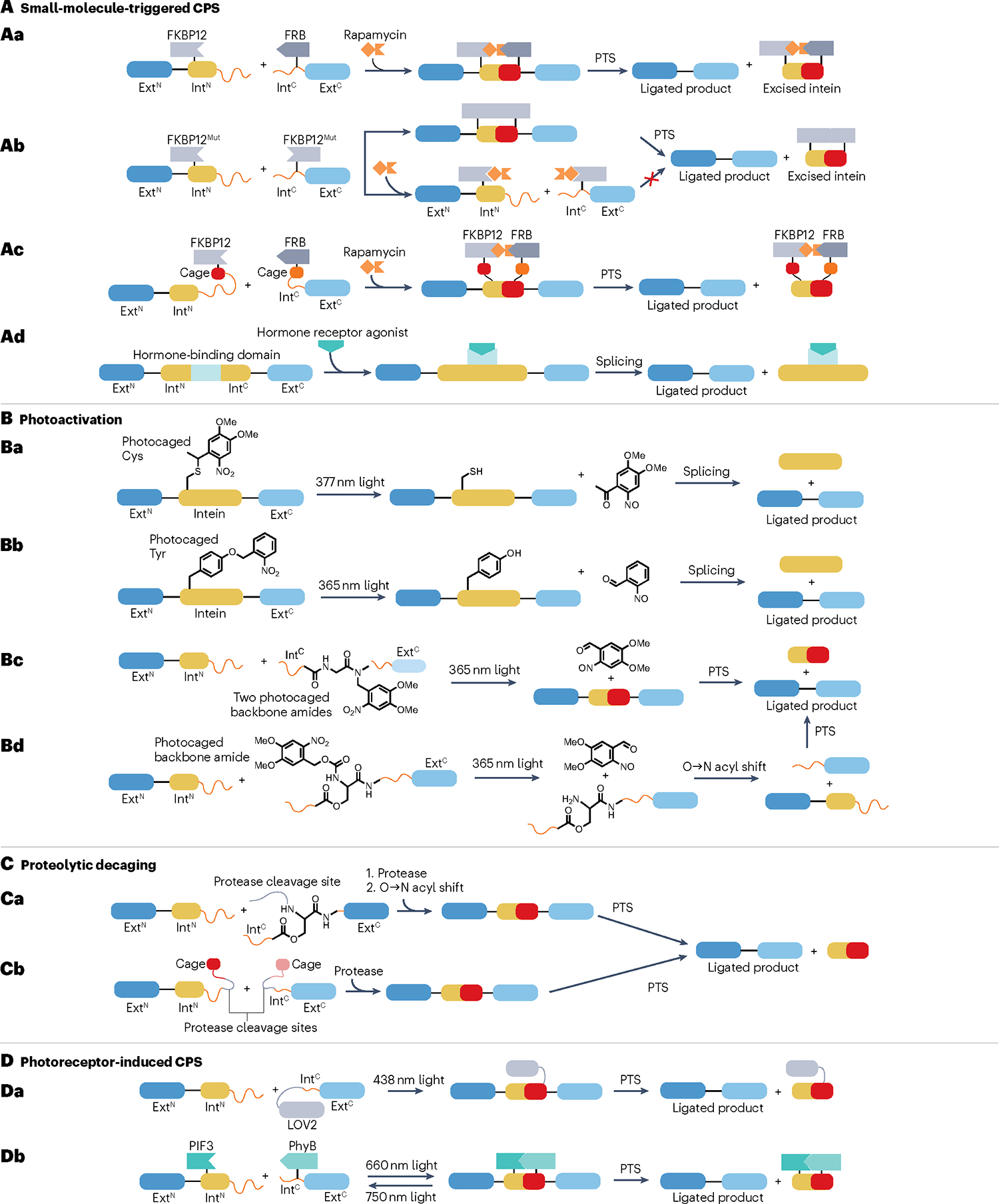
A, Small-molecule-triggered conditional protein splicing (CPS). Aa, The association of split inteins is controlled through the rapamycin-dependent interaction of the 12-kDa FK506-binding protein (FKBP12) and FKBP12-rapamycin binding (FRB) domain. By linking the N-terminal extein (ExtN) to a fusion of the N-terminal split intein (IntN) and FKBP12 protein, protein trans-splicing (PTS) occurs when rapamycin brings the construct into proximity of a fusion of the C-terminal split intein (IntC), FRB and the C-terminal extein (ExtC). Ab, Using a homodimerizing mutant of FKBP12, rapamycin can turn PTS off by disrupting the interaction between the FKBP12 domains. Ac, PTS is inhibited by the introduction of ‘cages’ that prevent split-intein association by binding to their cognate intein fragments. As rapamycin brings the constructs together, these cages are displaced upon binding of the split-intein fragments, thus resulting in PTS. Ad, Contiguous inteins carrying insertions of binding domains from hormone receptors can be triggered by the hormones 4-hydroxy tamoxifen and oestrogen. B, Photoactivated CPS. Splicing can be controlled by installing photoremovable protecting groups on side chains of either Cys (Ba) or Tyr (Bb) residues that are key for splicing. Bc, Insertion of photoremovable protecting groups on the backbone amides of two Gly residues prevents proper folding of the split-intein fragment. Upon removal of these photocages, the split-intein refolds and associates with its intein partner to facilitate PTS. Bd, An O-acyl linkage on a backbone amide introduces a kink in the protein that prevents PTS. The protecting group can be removed by light, inducing an O→N acyl shift to restore the backbone conformation and allow splicing to occur. C, CPS through proteolytic decaging. Ca, A kink introduced into the intein backbone can be resolved by removing the protecting group through proteolysis. The subsequent O→N acyl shift occurs similarly to that in panel Bd. Cb, Inhibitory cages (as in panel Ac) are fused to the intein fragments through linkers that contain a protease cleavage site, and proteolytic removal of these cages therefore triggers PTS. D, Photoreceptor-mediated CPS. Da, The IntC is fused to the light, oxygen or voltage domain 2 (LOV2), which undergoes conformational changes upon illumination to facilitate PTS. Db, The interaction between transcription factor phytochrome-interacting factor 3 (PIF3) and phytochrome B (PhyB) can be modulated by light at different wavelengths, thereby enabling CPS to be turned on and off.
CPS can be induced using small molecules that promote binding, and hence splicing, of weakly associating split inteins. The first example of such proximity-induced CPS was based on fusing IntN and IntC of the Sce VMA intein from Saccharomyces cerevisiae to the 12-kDa FK506-binding protein (FKBP12) and FKBP12-rapamycin binding domain, respectively160 (Fig. 9Aa). The resulting rapamycin-dependent system has been used for in vitro kinase activation161, three-piece ligation148, in cellulo splicing162–164 and CPS in live Drosophila melanogaster165. Additionally, rapamycin can also act as an off switch in a system that relies on a homodimerizing mutant of FKBP12 (ref. 166) (Fig. 9Ab). Contiguous intein-based CPS can similarly be achieved by coupling the conformational changes of hormone receptors upon ligand binding to splicing. Insertion of the ligand binding domain of the human oestrogen receptor into the Mtu RecA intein enables 4-hydroxy tamoxifen to trigger splicing in yeast167, and this system has been further optimized for CPS in human cells168–171 (Fig. 9Ac). Noticeably, this intein has been shown to improve the genome-editing specificity of Cas9 by restricting Cas9 activity to the window of 4-HT treatment, thereby minimizing off-target cleavage171. Moreover, conceptually similar CPS systems that rely on the thyroid hormone receptor have been developed, enabling oestrogen to act as both an on switch and an off switch172.
PTS can also be controlled using photosensitive strategies. In the most straightforward of such strategies, PTS is inhibited by a photocaged nucleophile analogue at position 1 (Fig. 9Ba), making splicing contingent on light-induced deprotection173,174. Similarly, splicing can be obstructed by incorporation of ortho-nitrobenzyl-tyrosine at the position of a conserved Phe residue in the Gp41–1 and M86 inteins175,176 (Fig. 9Bb). Backbone-modifying strategies also allow photoinducible PTS, as two photocaged glycine residues can prevent an IntC fragment from attaining its splicing-competent conformation before protecting group removal177 (Fig. 9Bc). Similarly, a kink can be introduced in the intein backbone using an O-acyl linkage, and PTS is induced by deprotection of a photocleavable α-amino group and a subsequent O→N acyl migration that restores the native intein structure178,179 (Fig. 9Bd). By substituting the α-amino-protecting group to a protease cleavage site, this strategy also enables CPS to be triggered by proteolysis178 (Fig. 9Ca). Protease-regulated splicing can also be realized by fusing each split intein to a segment of their cognate intein partner using a protease cleavage site-containing linker. The resulting ‘caged’, inactive inteins are only able to associate and splice, following their proteolytic liberation180 (Fig. 9Cb). These auto-inhibited inteins have also been combined with the proximity-induced FKBP12–FRP system to allow rapamycin-triggered splicing181 (Fig. 9Ac).
Light-triggered CPS can also rely on conformational changes in protein domains rather than removal of photolabile groups (Fig. 9D). The light, oxygen or voltage domain 2 of a photoreceptor from Avena sativa undergoes a flavin-dependent conformational change upon illumination, which has been used to generate trans CPS systems using split inteins (Fig. 9Da) as well as cis-CPS systems in mammalian cells182,183. Furthermore, a photosensitive, proximity-induced CPS platform has been developed by fusing Sce VMA split fragments to phytochrome B and transcription factor phytochrome-interacting factor 3 from Arabidopsis thaliana. When the phycocyanobilin chromophore is available, these proteins associate upon illumination at 660 nm, whereas 750-nm light leads to their dissociation, thereby enabling light to act as an on–off switch of PTS activity184 (Fig. 9Db).
In vivo applications of protein splicing
Intein-mediated protein ligation has found use for the in vitro production of bioconjugates117, isotopic labelling of protein segments for NMR studies151 and bypassing the packaging constraints of adeno-associated virus-based delivery185. In addition, split inteins are powerful tools for N–C cyclization of polypeptides through intramolecular splicing186, a reaction that is used in split-intein circular ligation of peptides and proteins (SICLOPPS) to make genetically encoded libraries of cyclic polypeptides6,187. Although these applications constitute key aspects of intein technology, the ability to tailor protein composition in complex microenvironments by generating semisynthetic proteins with temporal and/or spatial control still represents a key goal in protein ligation. Here, we focus on selected examples that have advanced the field of intein-mediated protein ligation in cellular contexts. However, more examples exist and have been reviewed elsewhere2,3,117.
To modify proteins in cells, one split intein carrying a synthetic cargo is delivered to cells expressing a POI fused to the other split-intein fragment (Fig. 10Aa). Thus, PTS will result in a full-length protein containing the modification (or modifications) defined by the delivered, semisynthetic cargo. Using this general strategy, in cellulo protein semi-synthesis has been used to install protein tags188, biotin150,159, fluorophores157,159,189–191 and quantum dots192 into various proteins, allowing them to be probed within their native cellular environment.
Fig. 10 |. Cellular protein manipulation strategies using inteins.
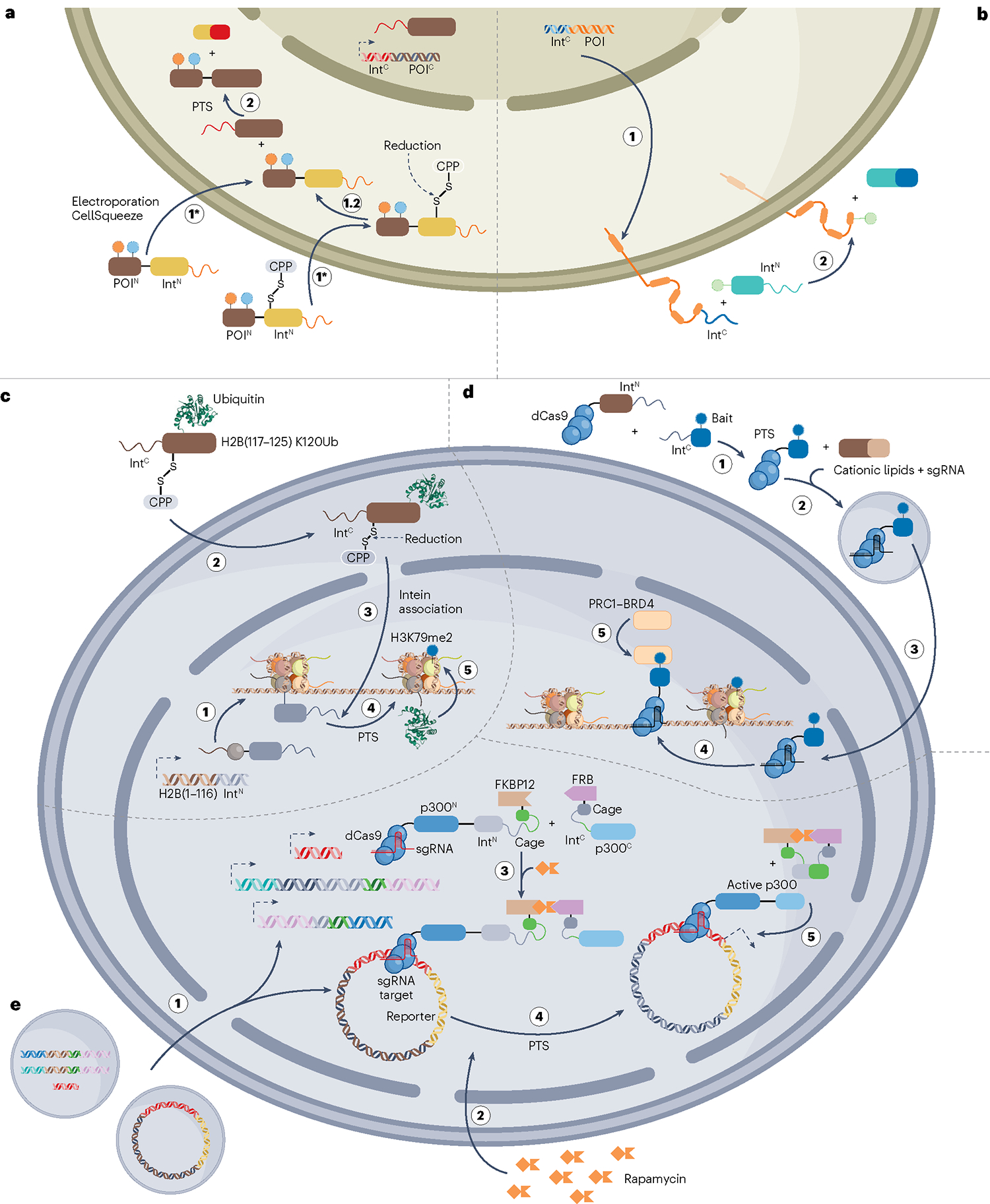
a, Split-intein-based modification of proteins of interest (POIs) in vivo. To modify intracellular POIs at their N terminus using protein trans-splicing (PTS), two constructs are designed: (1) the larger, C-terminal segment of the POI (POIC) fused to the C-terminal split-intein fragment (IntC) and (2) the synthetic, N-terminal segment of the POI (POIN) carrying the modification (or modifications) of choice fused to the N-terminal split-intein fragment (IntN). The cells are transfected to recombinantly express the IntC–POIC construct, whereas the semisynthetic piece can be delivered by electroporation, CellSqueeze or conjugation of the cargo to a cell-penetrating peptide (CPP). For CPP-based delivery, the CPP is often conjugated to IntN through a disulfide bond that is reduced in the cytoplasm upon cell entry. The modified POI is then generated by PTS as the IntN–POIN and IntC–POIC constructs associate within the cell. Although this figure only depicts N-terminal POI modification, this strategy is also applicable to C-terminal modification (c). b, PTS-based modification of the extracellular part of membrane proteins can be achieved using a strategy similar to that in panel a. However, as splicing occurs at the cell surface, such modifications are more straightforward than those targeting intracellular proteins. c, Site-specific modification of histones. To install ubiquitin (Ub) at lysine 120 of histone H2B, cells are transfected with a plasmid encoding a truncated segment of H2B (residues 1–116) fused to IntN. By delivering the missing piece of H2B (residues 117–125) carrying the K120Ub modification fused to IntC using a CPP, the PTM is site specifically installed in the full-length H2B protein. The control of H2B composition afforded by this strategy highlighted that H2B K120Ub stimulates writing of another histone PTM, namely, H3K79me2. d, Locus-specific recruitment of epigenetic regulators. PTS was used to fuse a synthetic bait moiety to catalytically dead Cas9 (dCas9) in the culture medium. The semisynthetic dCas9 was then delivered to the cell together with a single guide RNA (sgRNA) using cationic lipid transfection. In the nucleus, the sgRNA guides the dCas9–bait complex to the DNA sequences of interest, leading to specific recruitment of either of the epigenetic modulators polycomb repressive complex 1 (PRC1) or bromodomain-containing protein 4 (BRD4) depending on the choice of synthetic bait. e, Genome-specific activation of the histone acetyltransferase p300 by conditional protein splicing. p300 was split into inactive N-terminal (p300N) and C-terminal (p300C) fragments. The p300N fragment was fused to dCas9 and a caged, N-terminal split-intein fragment linked to the 12-kDa FK506-binding protein (FKBP12) domain. Similarly, p300C was fused to the cognate, caged IntC linked to the FKBP12-rapamycin binding (FRB) domain. In cells that express these two constructs together with sgRNAs of interest, p300 activity can be reconstituted at specific genomic regions by rapamycin-triggered conditional protein splicing. This was used to selectively enhance transcription of a reporter gene that was targeted by the sgRNA.
The extracellular part of membrane proteins can also be modified using PTS189,193 (Fig. 10a,b). Cysteine-containing inteins are redox-sensitive, which complicates PTS in oxidizing environments, such as the extracellular space. Therefore, an artificially split, cysteinefree intein has been developed by engineering a split intein from an Aeromonas bacteriophage136. One drawback of PTS-based modification is that it is limited to modification of protein termini; to modify central protein segments, two (or more) orthogonal intein pairs are required to perform tandem PTS148,194,195 (Fig. 8a). For this, a central, synthetic segment of a POI is fused to an N-terminal IntC1 and a C-terminal IntN2, thus enabling reconstitution of the full-length protein through PTS with the N-terminal part of the POI fused to IntN1 and the C-terminal protein segment fused to IntC2. Tandem PTS was recently extended to eukaryotic cells by modifying central residues in membrane proteins in Xenopus laevis oocytes and green fluorescent protein in HEK293 cells196. Noticeably, a library of 15 mutually orthogonal intein pairs has recently been established, thereby expanding the selection of inteins that are suitable for tandem PTS197.
Intein-based protein semisynthesis in cells has mostly been leveraged for proof-of-principle studies. The field of histone PTMs is an exception to this, as PTS is finding increased use for tailoring the chemical composition of chromatin. Chromatin, which is the physiologically relevant form of DNA in eukaryotes, is a complex comprising histone proteins bound to genomic DNA, and the compaction state of chromatin is key for the regulation of all DNA-templated events, including replication and transcription. Histones can be extensively modified, and the combined pattern of histone PTMs, the so-called histone code198, is read in a way that directly impacts chromatin compaction and hence cellular function. To uncover a ‘rosetta stone’ for the histone code, it is thus crucial to be able to control histone composition in cells to probe the role of individual PTMs and combinations thereof.
In the first example of split-intein-based, chromatin modification in the nucleus, Ub was installed at position K120 in histone H2b, following the general strategy outlined in Fig. 10a. A C-terminally truncated version of H2B fused to IntN (H2B (1–116)–IntN) was first expressed in cells. Nuclei were isolated from these cells and incubated with the cognate IntC fused to the synthetic, missing piece of H2B carrying the PTM (IntC–H2B(117–125)–K120Ub). Upon protein splicing, full-length H2B carrying K120Ub was generated, which showed that the site-specific installation of this mark promotes H3K79 methylation199,200 (Fig. 10c). This proved that ligation-based protein semisynthesis can be used to dissect the relationship between histone chromatin composition and function, and a similar strategy has also been adopted for modifying the N terminus of histone H3 (refs. 150,201). Importantly, intein-driven chromatin modification can be extended from in nucleo to living cells by conjugating the semisynthetic cargo to a cell-penetrating peptide199 or by delivering it using electroporation202 (Fig. 10a,c).
One drawback of these studies is that chromatin is modified across the entire genome, whereas endogenous chromatin is organized into spatially discrete regions. In a step towards achieving genome specificity, inteins have been used to modify dead Cas9 (dCas9) in the cell medium with synthetic ligands that recruit epigenetic modifiers. Subsequently, cationic lipid-mediated transfection was used to deliver the semisynthetic dCas9 into the cells, thereby recruiting the target enzymes to the DNA sequences of interest203 (Fig. 10d). In addition, the specificity of dCas9 has been combined with a rapamycin-induced CPS platform. The histone acetyltransferase p300 was split at a site that resulted in two non-functional pieces that were each fused to one member of a split-intein pair. Furthermore, the N-terminal p300 fragment was fused to dCas9 to direct the construct to the loci of interest. By adding rapamycin to cells expressing these constructs, it was possible to reconstitute functional p300 to drive transcription at the reporter sequence targeted by dCas9 (ref. 181) (Fig. 10e). Going forward, it is interesting to see whether CPS-based, genome-targeted strategies in combination with in situ protein semisynthesis will enable site-specific histone modification in a temporally and spatially controlled reaction.
Summary and outlook
Nature has provided multiple routes for enzymatically forming amide bonds between two distinct polypeptides. The past decades have seen impressive advances towards repurposing protein ligation strategies for unprecedented control over protein composition. At the forefront of these efforts have been the continued discovery of novel enzymes as well as the improvement of existing enzymes through protein engineering. With this expanded toolbox at hand, many proteins are now amenable substrates for protein ligation. In this Review, we summarize the mechanisms of various enzyme-mediated protein ligation technologies and use this knowledge to highlight the advantages and disadvantages that come with each approach. In particular, there is a general juxtaposition between the need for a chemoselective and site-selective reaction and the pursuit of traceless ligation. This is exemplified by butelase-1 facilitating efficient and promiscuous ligation that leaves as little as a single residue behind in the ligated product, whereas the more selective SrtA-mediated ligation creates a pentapeptide ‘scar’. A similar choice between selectivity and tracelessness is seen for the isopeptide-generating enzymes, as TGM2-based modification is highly promiscuous and traceless, whereas the molecular superglues are highly specific but require the POI to be fused to large tags. Moreover, additional features of the different ligation strategies, including catalytic efficiency, enzyme availability and reaction conditions, should all be taken into consideration before committing to a protein ligation strategy.
The manipulation of protein composition in cellular environments remains a major goal in protein ligation, with the applicability of many enzymes restricted by their lack of chemoselectivity and/or need for high reactant concentrations or chemically modified reactants to drive reversible reactions towards higher yields. Moving forward, these issues could partly be mitigated by developing systems in which protein ligation is temporally and spatially controlled by exogenous triggers, thereby tightly regulating an otherwise promiscuous protein modification reaction. Currently, only SrtA, inteins and molecular superglues have been applied in cells, whereas intein-based strategies remain the only feasible route for general in situ protein semisynthesis, which is still considered a technically demanding feat. One obstacle that reduces the efficiency of in vivo ligations is the cell membrane itself, as delivery of the synthetic cargo is a considerable challenge. Fortunately, much progress is being made in cellular delivery techniques204, including the use of nanoparticle technology205. As these delivery platforms become more routine, we expect the entry barrier for in vivo protein ligation to become lower. Considering the recent maturation of intein-based techniques, the field seems primed for a shift from method development towards probing the role of protein composition and modification in complex biological processes. Nevertheless, there is still room for continued methodological development, as the generation of semisynthetic integral membrane proteins with high yields remains out of reach. It is similarly unfeasible to use inteins to install multiple modifications within a protein segment that are located too far apart in the primary sequence to be covered by a single synthetic piece, highlighting that no one ligation platform is likely to be able to solve all protein modification challenges.
The emergence of novel platforms that harness the benefits of two (or more) protein manipulation techniques thus constitutes an intriguing development in the field. The recently developed TAIL strategy159 combines the advantageous features of two different ligation strategies, by exploiting the short, and hence synthetically accessible, recognition sequence of SrtA and the irreversibility of intein-based PTS. Similarly, the ability of genetic code expansion to site specifically install unnatural amino acids, which allow for bioorthogonal chemical reactions, at any position along the entire protein sequence has been combined with SrtA-mediated protein ligation to modify proteins with Ub and SUMO30. This method was recently expanded further to use OaAEP1-based ligation to modify internal UAAs introduced by genetic code expansion with biophysical probes and Ub206. Thus, continued innovation that allows the best of two (or more) protein engineering platforms to be merged is likely to improve our control of protein composition in the future. Although protein ligation is still not a straightforward task, there is now a diverse set of well-established platforms available. This paves the way for researchers to aptly transcend the chemical space afforded to proteins by the genetic code in pursuit of semisynthetic proteins as well as novel protein and peptide conjugates.
Acknowledgements
The authors thank M. Luo for critical reading of the manuscript. Work in the David Laboratory is supported by the Josie Robertson Foundation, the Pershing Square Sohn Cancer Research Alliance, the NIH (CCSG core grant P30 CA008748, MSK SPORE P50 CA192937, R21 DA044767 and R35 GM138386), the Parker Institute for Cancer Immunotherapy and the Anna Fuller Trust. In addition, the David Laboratory is supported by the William H. Goodwin and Alice Goodwin Commonwealth Foundation for Cancer Research and by the Center for Experimental Therapeutics at MSKCC. R.P. is supported by the Novo Nordisk Foundation grant NNF20OC0061064. The Zheng laboratory is supported by OSUCCC startup funds.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information Nature Reviews Chemistry thanks Ashraf Brik, Anne Conibear and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Usmani SS et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS ONE 12, e0181748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson RE & Muir TW Chemoenzymatic semisynthesis of proteins. Chem. Rev. 120, 3051–3126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conibear AC Deciphering protein post-translational modifications using chemical biology tools. Nat. Rev. Chem. 4, 674–695 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Drago JZ, Modi S & Chandarlapaty S Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muona M, Aranko AS, Raulinaitis V & Iwaï H Segmental isotopic labeling of multi-domain and fusion proteins by protein trans-splicing in vivo and in vitro. Nat. Protoc. 5, 574–587 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Sohrabi C, Foster A & Tavassoli A Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 4, 90–101 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Sornay C, Vaur V, Wagner A & Chaubet G An overview of chemo- and site-selectivity aspects in the chemical conjugation of proteins. R. Soc. Open Sci. 9, 211563 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalker JM & Davis BG Chemical mutagenesis: selective post-expression interconversion of protein amino acid residues. Curr. Opin. Chem. Biol. 14, 781–789 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Wright TH et al. Posttranslational mutagenesis: a chemical strategy for exploring protein side-chain diversity. Science 354, aag1465–3 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Bergmann M & Fraenkel-Conrat H The enzymatic synthesis of peptide bonds. J. Biol. Chem. 124, 1–6 (1938). [Google Scholar]
- 11.Aliashkevich A & Cava F LD-transpeptidases: the great unknown among the peptidoglycan cross-linkers. FEBS J. 289, 4718–4730 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Spirig T, Weiner EM & Clubb RT Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pishesha N, Ingram JR & Ploegh HL Sortase A: a model for transpeptidation and its biological applications. Annu. Rev. Cell Dev. Biol. 34, 163–188 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Ilangovan U, Ton-That H, Iwahara J, Schneewind O & Clubb RT Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl Acad. Sci. USA 98, 6056–6061 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao H, Hart SA, Schink A & Pollok BA Sortase-mediated protein ligation: a new method for protein engineering. J. Am. Chem. Soc. 126, 2670–2671 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Schmidt M, Toplak A, Quaedflieg PJ & Nuijens T Enzyme-mediated ligation technologies for peptides and proteins. Curr. Opin. Chem. Biol. 38, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Williamson DJ, Fascione MA, Webb ME & Turnbull WB Efficient N-terminal labeling of proteins by use of sortase. Angew. Chem. Int. Ed. 51, 9377–9380 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Williamson DJ, Webb ME & Turnbull WB Depsipeptide substrates for sortase-mediated N-terminal protein ligation. Nat. Protoc. 9, 253–262 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Zuo C et al. Thioester-assisted sortase-A-mediated ligation. Angew. Chem. Int. Ed. 61, e202201887 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Dorr BM, Ham HO, An C, Chaikof EL & Liu DR Reprogramming the specificity of sortase enzymes. Proc. Natl Acad. Sci. USA 111, 13343–13348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freund C & Schwarzer D Engineered sortases in peptide and protein chemistry. ChemBioChem 22, 1347–1356 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirakawa H, Ishikawa S & Nagamune T Ca2+-independent sortase-A exhibits high selective protein ligation activity in the cytoplasm of Escherichia coli. Biotechnol. J. 10, 1487–1492 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Podracky CJ et al. Laboratory evolution of a sortase enzyme that modifies amyloid-β protein. Nat. Chem. Biol. 17, 317–325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L et al. Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 6, 31899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warden-Rothman R, Caturegli I, Popik V & Tsourkas A Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Anal. Chem. 85, 11090–11097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wöll S, Bachran C, Schiller S, Swee LK & Scherließ R Sortase-A mediated chemoenzymatic lipidation of single-domain antibodies for cell membrane engineering. Eur. J. Pharm. Biopharm. 153, 121–129 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Fauser J, Savitskiy S, Fottner M, Trauschke V & Gulen B Sortase-mediated quantifiable enzyme immobilization on magnetic nanoparticles. Bioconjug. Chem. 31, 1883–1892 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Gébleux R, Briendl M, Grawunder U & Beerli RR in Enzyme-Mediated Ligation Methods (eds Nuijens T. & Schmidt M.) 1–13 (Springer, 2019). [DOI] [PubMed] [Google Scholar]
- 29.Strijbis K, Spooner E & Ploegh HL Protein ligation in living cells using sortase. Traffic 13, 780–789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fottner M et al. Site-specific ubiquitylation and SUMOylation using genetic-code expansion and sortase. Nat. Chem. Biol. 15, 276–284 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Tang TMS & Luk LYP Asparaginyl endopeptidases: enzymology, applications and limitations. Org. Biomol. Chem. 19, 5048–5062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruis (Fred) D. & Selinger DA. & Curran JM. & Jung R. Redundant proteolytic mechanisms process seed storage proteins in the absence of seed-type members of the vacuolar processing enzyme family of cysteine proteases. Plant Cell 14, 2863–2882 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James AM et al. The macrocyclizing protease butelase 1 remains autocatalytic and reveals the structural basis for ligase activity. Plant J. 98, 988–999 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Nguyen GKT et al. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 10, 732–738 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Nguyen GKT et al. Butelase 1: a versatile ligase for peptide and protein macrocyclization. J. Am. Chem. Soc. 137, 15398–15401 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Nguyen GKT et al. Butelase-mediated cyclization and ligation of peptides and proteins. Nat. Protoc. 11, 1977–1988 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hemu X, Zhang X, Bi X, Liu C-F & Tam JP in Enzyme-Mediated Ligation Methods (eds Nuijens T. & Schmidt M.) 83–109 (Springer, 2019). [Google Scholar]
- 38.Nguyen GKT, Hemu X, Quek J-P & Tam JP Butelase-mediated macrocyclization of D-amino-acid-containing peptides. Angew. Chem. Int. Ed. 55, 12802–12806 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Lee AC, Harris JL, Khanna KK & Hong J-H A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 20, 2383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao Y, Nguyen GKT, Tam JP & Liu C-F Butelase-mediated synthesis of protein thioesters and its application for tandem chemoenzymatic ligation. Chem. Commun. 51, 17289–17292 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Bi X et al. Enzymatic engineering of live bacterial cell surfaces using butelase 1. Angew. Chem. Int. Ed. 56, 7822–7825 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Cao Y, Nguyen GKT, Chuah S, Tam JP & Liu C-F Butelase-mediated ligation as an efficient bioconjugation method for the synthesis of peptide dendrimers. Bioconjug. Chem. 27, 2592–2596 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Nguyen GKT, Cao Y, Wang W, Liu CF & Tam JP Site-specific N-terminal labeling of peptides and proteins using butelase 1 and thiodepsipeptide. Angew. Chem. Int. Ed. 54, 15694–15698 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Harmand TJ et al. One-pot dual labeling of IgG 1 and preparation of C-to-C fusion proteins through a combination of sortase A and butelase 1. Bioconjug. Chem. 29, 3245–3249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nuijens T, Toplak A, Schmidt M, Ricci A & Cabri W Natural occurring and engineered enzymes for peptide ligation and cyclization. Front. Chem. 7, 829 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao J et al. Enzymatic properties of recombinant ligase butelase-1 and its application in cyclizing food-derived angiotensin I-converting enzyme inhibitory peptides. J. Agric. Food Chem. 69, 5976–5985 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Yang R et al. Engineering a catalytically efficient recombinant protein ligase. J. Am. Chem. Soc. 139, 5351–5358 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Harris KS et al. Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase. Nat. Commun. 6, 10199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rehm FBH et al. Site-specific sequential protein labeling catalyzed by a single recombinant ligase. J. Am. Chem. Soc. 141, 17388–17393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Z et al. Oa AEP1-mediated PNA–protein conjugation enables erasable imaging of membrane protein. Chem. Commun. 58, 8448–8451 (2021). [DOI] [PubMed] [Google Scholar]
- 51.López-Otín C & Bond JS Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goettig P Reversed proteolysis — proteases as peptide ligases. Catalysts 11, 33 (2021). [Google Scholar]
- 53.Razzaq A et al. Microbial proteases applications. Front. Bioeng. Biotechnol. 7, 110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siezen RJ & Leunissen JAM Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6, 501–523 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao MB, Tanksale AM, Ghatge MS & Deshpande VV Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 62, 597–635 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carter P, Nilsson B, Burnier JP, Burdick D & Wells JA Engineering subtilisin BPN′ for site-specific proteolysis. Proteins Struct. Funct. Bioinform. 6, 240–248 (1989). [DOI] [PubMed] [Google Scholar]
- 57.Ballinger MD, Tom J & Wells JA Designing subtilisin BPN’ to cleave substrates containing dibasic residues. Biochemistry 34, 13312–13319 (1995). [DOI] [PubMed] [Google Scholar]
- 58.Abrahmsen L et al. Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry 30, 4151–4159 (1991). [DOI] [PubMed] [Google Scholar]
- 59.Weeks AM & Wells JA Subtiligase-catalyzed peptide ligation. Chem. Rev. 120, 3127–3160 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Shane A & Wells JA Selection for improved subtiligases by phage display. Proc. Natl Acad. Sci. USA 96, 9497–9502 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strausberg SL et al. Directed evolution of a subtilisin with calcium-independent stability. Biotechnology (N Y) 13, 669–673 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Chang TK, Jackson DY, Burnier JP & Wells JA Subtiligase: a tool for semisynthesis of proteins. Proc. Natl Acad. Sci. USA 91, 12544–12548 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weeks AM & Wells JA Engineering peptide ligase specificity by proteomic identification of ligation sites. Nat. Chem. Biol. 14, 50–57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henager SH et al. Enzyme-catalyzed expressed protein ligation. Nat. Methods 13, 925–927 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henager SH, Henriquez S, Dempsey DR & Cole PA Analysis of site-specific phosphorylation of PTEN by using enzyme-catalyzed expressed protein ligation. ChemBioChem 21, 64–68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toplak A, Nuijens T, Quaedflieg PJLM, Wu B & Janssen DB Peptiligase, an enzyme for efficient chemoenzymatic peptide synthesis and cyclization in water. Adv. Synth. Catal. 358, 2140–2147 (2016). [Google Scholar]
- 67.Toplak A et al. From thiol-subtilisin to omniligase: design and structure of a broadly applicable peptide ligase. Comput. Struct. Biotechnol. J. 19, 1277–1287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt M & Nuijens T in Enzyme-Mediated Ligation Methods (eds Nuijens T. & Schmidt M.) 43–61 (Springer, 2019). [Google Scholar]
- 69.Schmidt M et al. Omniligase-1: a powerful tool for peptide head-to-tail cyclization. Adv. Synth. Catal. 359, 2050–2055 (2017). [Google Scholar]
- 70.Simpson RJ Fragmentation of protein using trypsin. Cold Spring Harb. Protoc. 2006, pdb.prot4550 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Huber R & Bode W Structural basis of the activation and action of trypsin. Acc. Chem. Res. 11, 114–122 (1978). [Google Scholar]
- 72.Liebscher S, Mathea S, Aumüller T, Pech A & Bordusa F Trypsiligase-catalyzed labeling of proteins on living cells. ChemBioChem 22, 1201–1204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meyer C, Liebscher S & Bordusa F Selective coupling of click anchors to proteins via trypsiligase. Bioconjug. Chem. 27, 47–53 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Liebscher S et al. N-terminal protein modification by substrate-activated reverse proteolysis. Angew. Chem. Int. Ed. 53, 3024–3028 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Liebscher S et al. Derivatization of antibody fab fragments: a designer enzyme for native protein modification. ChemBioChem 15, 1096–1100 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Kopp F & Marahiel MA Macrocyclization strategies in polyketide and nonribosomal peptide biosynthesis. Nat. Prod. Rep. 24, 735–749 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Arnison PG et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jonathan RC, Paola E, Patrick SC & Nair KS Characterization of the macrocyclase involved in the biosynthesis of RiPP cyclic peptides in plants. Proc. Natl Acad. Sci. USA 114, 6551–6556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ongpipattanakul C & Nair SK Biosynthetic proteases that catalyze the macrocyclization of ribosomally synthesized linear peptides. Biochemistry 57, 3201–3209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sivonen K, Leikoski N, Fewer DP & Jokela J Cyanobactins — ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 86, 1213–1225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koehnke J et al. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat. Struct. Mol. Biol. 19, 767–772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Houssen WE in Enzyme-Mediated Ligation Methods (eds Nuijens T. & Schmidt M.) 193–210 (Springer, 2019). [Google Scholar]
- 83.Agarwal V, Pierce E, McIntosh J, Schmidt EW & Nair SK Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem. Biol. 19, 1411–1422 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Czekster CM, Ludewig H, McMahon SA & Naismith JH Characterization of a dual function macrocyclase enables design and use of efficient macrocyclization substrates. Nat. Commun. 8, 1045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sarkar S, Gu W & Schmidt EW Expanding the chemical space of synthetic cyclic peptides using a promiscuous macrocyclase from prenylagaramide biosynthesis. ACS Catal. 10, 7146–7153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oueis E, Stevenson H, Jaspars M, Westwood NJ & Naismith JH Bypassing the proline/thiazoline requirement of the macrocyclase PatG. Chem. Commun. 53, 12274–12277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qingfei Z et al. An α/β-hydrolase fold protein in the biosynthesis of thiostrepton exhibits a dual activity for endopeptidyl hydrolysis and epoxide ring opening/macrocyclization. Proc. Natl Acad. Sci. USA 113, 14318–14323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savoca MP, Tonoli E, Atobatele AG & Verderio EAM Biocatalysis by transglutaminases: a review of biotechnological applications. Micromachines 9, 562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Griffin M, Casadio R & Bergamini CM Transglutaminases: nature’s biological glues. Biochem. J. 368, 377–396 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Farrelly LA et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rachel NM & Pelletier JN Biotechnological applications of transglutaminases. Biomolecules 3, 870–888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dickgiesser S, Deweid L, Kellner R, Kolmar H & Rasche N in Enzyme-Mediated Ligation Methods (eds Nuijens T. & Schmidt M.) 135–149 (Springer, 2019). [Google Scholar]
- 93.Bijan Z et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl Acad. Sci. USA 109, E690–E697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gianluca V et al. Programmable polyproteams built using twin peptide superglues. Proc. Natl Acad. Sci. USA 113, 1202–1207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Veggiani G, Zakeri B & Howarth M Superglue from bacteria: unbreakable bridges for protein nanotechnology. Trends Biotechnol. 32, 506–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kang HJ, Coulibaly F, Clow F, Proft T & Baker EN Stabilizing isopeptide bonds revealed in Gram-positive bacterial pilus structure. Science 318, 1625–1628 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Kang HJ & Baker EN Intramolecular isopeptide bonds: protein crosslinks built for stress? Trends Biochem. Sci. 36, 229–237 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Lukasak BJ et al. A genetically encoded approach for breaking chromatin symmetry. ACS Cent. Sci. 8, 176–183 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang HH, Altun B, Nwe K & Tsourkas A Proximity-based sortase-mediated ligation. Angew. Chem. Int. Ed. 56, 5349–5352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brune KD et al. Dual plug-and-display synthetic assembly using orthogonal reactive proteins for twin antigen immunization. Bioconjug. Chem. 28, 1544–1551 (2017). [DOI] [PubMed] [Google Scholar]
- 101.Tan LL, Hoon SS & Wong FT Kinetic controlled Tag–Catcher interactions for directed covalent protein assembly. PLoS ONE 11, e0165074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu X-L, Liu Y, Liu D, Sun F & Zhang W-B An intrinsically disordered peptide–peptide stapler for highly efficient protein ligation both in vivo and in vitro. J. Am. Chem. Soc. 140, 17474–17483 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Keeble AH et al. Evolving accelerated amidation by SpyTag/SpyCatcher to analyze membrane dynamics. Angew. Chem. Int. Ed. 56, 16521–16525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Si M, Xu Q, Jiang L & Huang H SpyTag/SpyCatcher cyclization enhances the thermostability of firefly luciferase. PLoS ONE 11, e0162318 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang W-B, Sun F, Tirrell DA & Arnold FH Controlling macromolecular topology with genetically encoded SpyTag–SpyCatcher chemistry. J. Am. Chem. Soc. 135, 13988–13997 (2013). [DOI] [PubMed] [Google Scholar]
- 106.Bedford L, Lowe J, Dick LR, Mayer RJ & Brownell JE Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scheffner M, Nuber U & Huibregtse JM Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83 (1995). [DOI] [PubMed] [Google Scholar]
- 108.Li W & Ye Y Polyubiquitin chains: functions, structures, and mechanisms. Cell. Mol. Life Sci. 65, 2397–2406 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cappadocia L & Lima CD Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem. Rev. 118, 889–918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao B et al. Protein engineering in the ubiquitin system: tools for discovery and beyond. Pharmacol. Rev. 72, 380–413 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fottner M & Lang K Decorating proteins with LACE. Nat. Chem. 12, 980–982 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Hofmann R, Akimoto G, Wucherpfennig TG, Zeymer C & Bode JW Lysine acylation using conjugating enzymes for site-specific modification and ubiquitination of recombinant proteins. Nat. Chem. 12, 1008–1015 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Akimoto G, Fernandes AP & Bode JW Site-specific protein ubiquitylation using an engineered, chimeric E1 activating enzyme and E2 SUMO conjugating enzyme Ubc9. ACS Cent. Sci. 8, 275–281 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Green CM, Novikova O & Belfort M The dynamic intein landscape of eukaryotes. Mob. DNA 9, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Novikova O et al. Intein clustering suggests functional importance in different domains of life. Mol. Biol. Evol. 33, 783–799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paulus H Inteins as enzymes. Bioorg. Chem. 29, 119–129 (2001). [DOI] [PubMed] [Google Scholar]
- 117.Shah NH & Muir TW Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Novikova O, Topilina N & Belfort M Enigmatic distribution, evolution, and function of inteins. J. Biol. Chem. 289, 14490–14497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eryilma E, Shah NH, Muir TW & Cowburn D Structural and dynamical features of inteins and implications on protein splicing. J. Biol. Chem. 289, 14506–14511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mills KV, Dorval DM & Lewandowski KT Kinetic analysis of the individual steps of protein splicing for the Pyrococcus abyssi PolII intein. J. Biol. Chem. 280, 2714–2720 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Callahan BP, Topilina NI, Stanger MJ, Van Roey P & Belfort M Structure of catalytically competent intein caught in a redox trap with functional and evolutionary implications. Nat. Struct. Mol. Biol. 18, 630–633 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dearden AK et al. A conserved threonine spring-loads precursor for intein splicing. Protein Sci. 22, 557–563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Romanelli A, Shekhtman A, Cowburn D & Muir TW Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein–intein junction. Proc. Natl Acad. Sci. USA 101, 6397–6402 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du Z, Zheng Y, Patterson M, Liu Y & Wang C pKa coupling at the intein active site: implications for the coordination mechanism of protein splicing with a conserved aspartate. J. Am. Chem. Soc. 133, 10275–10282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu MQ et al. Protein splicing: an analysis of the branched intermediate and its resolution by succinimide formation. EMBO J. 13, 5517–5522 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Frutos S, Goger M, Giovani B, Cowburn D & Muir TW Branched intermediate formation stimulates peptide bond cleavage in protein splicing. Nat. Chem. Biol. 6, 527–533 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shah NH, Eryilmaz E, Cowburn D & Muir TW Extein residues play an intimate role in the rate-limiting step of protein trans-splicing. J. Am. Chem. Soc. 135, 5839–5847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Z et al. Structure of the branched intermediate in protein splicing. Proc. Natl Acad. Sci. USA 111, 8422–8427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu Q et al. Conserved residues that modulate protein trans-splicing of Npu DnaE split intein. Biochem. J. 461, 247–255 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Chong S et al. Protein splicing involving the Saccharomyces cerevisiae VMA intein. J. Biol. Chem. 271, 22159–22168 (1996). [DOI] [PubMed] [Google Scholar]
- 131.Mills KV, Johnson MA & Perler FB Protein splicing: how inteins escape from precursor proteins. J. Biol. Chem. 289, 14498–14505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shah NH, Eryilmaz E, Cowburn D & Muir TW Naturally split inteins assemble through a ‘capture and collapse’ mechanism. J. Am. Chem. Soc. 135, 18673–18681 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zheng Y, Wu Q, Wang C, Xu MQ & Liu Y Mutual synergistic protein folding in split intein. Biosci. Rep. 32, 433–442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stevens AJ, Sekar G, Gramespacher JA, Cowburn D & Muir TW An atypical mechanism of split intein molecular recognition and folding. J. Am. Chem. Soc. 140, 11791–11799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shah NH, Vila-Perelló M & Muir TW Kinetic control of one-pot trans-splicing reactions by using a wild-type and designed split intein. Angew. Chem. Int. Ed. 50, 6511–6515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bhagawati M et al. A mesophilic cysteine-less split intein for protein trans-splicing applications under oxidizing conditions. Proc. Natl Acad. Sci. USA 116, 22164–22172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martin DD, Xu MQ & Evans TC Characterization of a naturally occurring trans-splicing intein from Synechocystis sp. PCC6803. Biochemistry 40, 1393–1402 (2001). [DOI] [PubMed] [Google Scholar]
- 138.Shah NH, Dann GP, Vila-Perelló M, Liu Z & Muir TW Ultrafast protein splicing is common among cyanobacterial split inteins: implications for protein engineering. J. Am. Chem. Soc. 134, 11338–11341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zettler J, Schütz V & Mootz HD The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 583, 909–914 (2009). [DOI] [PubMed] [Google Scholar]
- 140.Stevens AJ et al. Design of a split intein with exceptional protein splicing activity. J. Am. Chem. Soc. 138, 2162–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Carvajal-Vallejos P, Pallissé R, Mootz HD & Schmidt SR Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources. J. Biol. Chem. 287, 28686–28696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thiel IV, Volkmann G, Pietrokovski S & Mootz HD An atypical naturally split intein engineered for highly efficient protein labeling. Angew. Chem. Int. Ed. 53, 1306–1310 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Amitai G, Callahan BP, Stanger MJ, Belfort G & Belfort M Modulation of intein activity by its neighboring extein substrates. Proc. Natl Acad. Sci. USA 106, 11005–11010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lockless SW & Muir TW Traceless protein splicing utilizing evolved split inteins. Proc. Natl Acad. Sci. USA 106, 10999–11004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Appleby-Tagoe JH et al. Highly efficient and more general cis- and trans-splicing inteins through sequential directed evolution. J. Biol. Chem. 286, 34440–34447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oeemig JS, Zhou D, Kajander T, Wlodawer A & Iwaï H NMR and crystal structures of the Pyrococcus horikoshii RadA intein guide a strategy for engineering a highly efficient and promiscuous intein. J. Mol. Biol. 421, 85–99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cheriyan M, Pedamallu CS, Tori K & Perler F Faster protein splicing with the nostoc punctiforme DnaE intein using non-native extein residues. J. Biol. Chem. 288, 6202–6211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shi J & Muir TW Development of a tandem protein trans-splicing system based on native and engineered split inteins. J. Am. Chem. Soc. 127, 6198–6206 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Iwai H, Züger S, Jin J & Tam PH Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 580, 1853–1858 (2006). [DOI] [PubMed] [Google Scholar]
- 150.Stevens AJ et al. A promiscuous split intein with expanded protein engineering applications. Proc. Natl Acad. Sci. USA 114, 8538–8543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Züger S & Iwai H Intein-based biosynthetic incorporation of unlabeled protein tags into isotopically labeled proteins for NMR studies. Nat. Biotechnol. 23, 736–740 (2005). [DOI] [PubMed] [Google Scholar]
- 152.Bachmann AL & Mootz HD An unprecedented combination of serine and cysteine nucleophiles in a split intein with an atypical split site. J. Biol. Chem. 290, 28792–28804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Neugebauer M, Böcker JK, Matern JCJ, Pietrokovski S & Mootz HD Development of a screening system for inteins active in protein splicing based on intein insertion into the LacZα-peptide. Biol. Chem. 398, 57–67 (2017). [DOI] [PubMed] [Google Scholar]
- 154.Sun W, Yang J & Liu XQ Synthetic two-piece and three-piece split inteins for protein trans-splicing. J. Biol. Chem. 279, 35281–35286 (2004). [DOI] [PubMed] [Google Scholar]
- 155.Lin Y et al. Protein trans-splicing of multiple atypical split inteins engineered from natural inteins. PLoS ONE 8, e59516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ludwig C, Pfeiff M, Linne U & Mootz HD Ligation of a synthetic peptide to the N terminus of a recombinant protein using semisynthetic protein trans-splicing. Angew. Chem. Int. Ed. 45, 5218–5221 (2006). [DOI] [PubMed] [Google Scholar]
- 157.Braner M, Kollmannsperger A, Wieneke R & Tampé R ‘Traceless’ tracing of proteins — high-affinity trans-splicing directed by a minimal interaction pair. Chem. Sci. 7, 2646–2652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Appleby JH, Zhou K, Volkmann G & Liu XQ Novel split intein for trans-splicing synthetic peptide onto C terminus of protein. J. Biol. Chem. 284, 6194–6199 (2009). [DOI] [PubMed] [Google Scholar]
- 159.Thompson RE, Stevens AJ & Muir TW Protein engineering through tandem transamidation. Nat. Chem. 11, 737–743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mootz HD & Muir TW Protein splicing triggered by a small molecule. J. Am. Chem. Soc. 124, 9044–9045 (2002). [DOI] [PubMed] [Google Scholar]
- 161.Mootz HD, Blum ES & Muir TW Activation of an autoregulated protein kinase by conditional protein splicing. Angew. Chem. Int. Ed. 43, 5189–5192 (2004). [DOI] [PubMed] [Google Scholar]
- 162.Sonntag T & Mootz HD An intein-cassette integration approach used for the generation of a split TEV protease activated by conditional protein splicing. Mol. Biosyst. 7, 2031–2039 (2011). [DOI] [PubMed] [Google Scholar]
- 163.Mootz HD, Blum ES, Tyszkiewicz AB & Muir TW Conditional protein splicing: a new tool to control protein structure and function in vitro and in vivo. J. Am. Chem. Soc. 125, 10561–10569 (2003). [DOI] [PubMed] [Google Scholar]
- 164.Alford SC, O’Sullivan C, Obst J, Christie J & Howard PL Conditional protein splicing of α-sarcin in live cells. Mol. Biosyst. 10, 831–837 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Schwartz EC, Saez L, Young MW & Muir TW Post-translational enzyme activation in an animal via optimized conditional protein splicing. Nat. Chem. Biol. 3, 50–54 (2007). [DOI] [PubMed] [Google Scholar]
- 166.Brenzel S & Mootz HD Design of an intein that can be inhibited with a small molecule ligand. J. Am. Chem. Soc. 127, 4176–4177 (2005). [DOI] [PubMed] [Google Scholar]
- 167.Buskirk AR, Ong YC, Gartner ZJ & Liu DR Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc. Natl Acad. Sci. USA 101, 10505–10510 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hartley PD & Madhani HD Mechanisms that specify promoter nucleosome location and identity. Cell 137, 445–458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yuen CM, Rodda SJ, Vokes SA, McMahon AP & Liu DR Control of transcription factor activity and osteoblast differentiation in mammalian cells using an evolved small-molecule-dependent intein. J. Am. Chem. Soc. 128, 8939–8946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Peck SH, Chen I & Liu DR Directed evolution of a small-molecule-triggered intein with improved splicing properties in mammalian cells. Chem. Biol. 18, 619–630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Davis KM, Pattanayak V, Thompson DB, Zuris JA & Liu DR Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 11, 316–318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Skretas G & Wood DW Regulation of protein activity with small-molecule-controlled inteins. Protein Expr. Purif. 14, 523–532 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cook SN et al. Photochemically lnitiated protein splicing. Angew. Chem. Int. Ed. Engl. 34, 1629–1630 (1995). [Google Scholar]
- 174.Ren W, Ji A & Ai HW Light activation of protein splicing with a photocaged fast intein. J. Am. Chem. Soc. 137, 2155–2158 (2015). [DOI] [PubMed] [Google Scholar]
- 175.Böcker JK, Dörner W & Mootz HD Light-control of the ultra-fast Gp41–1 split intein with preserved stability of a genetically encoded photo-caged amino acid in bacterial cells. Chem. Commun. 55, 1287–1290 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Böcker JK, Friedel K, Matern JCJ, Bachmann AL & Mootz HD Generation of a genetically encoded, photoactivatable intein for the controlled production of cyclic peptides. Angew. Chem. Int. Ed. 54, 2116–2120 (2015). [DOI] [PubMed] [Google Scholar]
- 177.Berrade L, Kwon Y & Camarero JA Photomodulation of protein trans-splicing through backbone photocaging of the DnaE split intein. ChemBioChem 11, 1368–1372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vila-Perelló M, Hori Y, Ribó M & Muir TW Activation of protein splicing by protease- or light-triggered O to N acyl migration. Angew. Chem. Int. Ed. 47, 7764–7767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jung D et al. Photo-triggered fluorescent labelling of recombinant proteins in live cells. Chem. Commun. 51, 9670–9673 (2015). [DOI] [PubMed] [Google Scholar]
- 180.Gramespacher JA, Stevens AJ, Nguyen DP, Chin JW & Muir TW Intein zymogens: conditional assembly and splicing of split inteins via targeted proteolysis. J. Am. Chem. Soc. 139, 8074–8077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gramespacher JA, Burton AJ, Guerra LF & Muir TW Proximity induced splicing utilizing caged split inteins. J. Am. Chem. Soc. 141, 13708–13712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wong S, Mosabbir AA & Truong K An engineered split intein for photoactivated protein trans-splicing. PLoS ONE 10, e0135965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jones DC, Mistry IN & Tavassoli A Post-translational control of protein function with light using a LOV-intein fusion protein. Mol. Biosyst. 12, 1388–1393 (2016). [DOI] [PubMed] [Google Scholar]
- 184.Tyszkiewicz AB & Muir TW Activation of protein splicing with light in yeast. Nat. Methods 5, 303–305 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Truong DJJ et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 43, 6450–6458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Scott CP, Abel-Santos E, Wall M, Wahnon DC & Benkovic SJ Production of cyclic peptides and proteins in vivo. Proc. Natl Acad. Sci. USA 96, 13638–13643 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tavassoli A & Benkovic SJ Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli. Nat. Protoc. 2, 1126–1133 (2007). [DOI] [PubMed] [Google Scholar]
- 188.Giriat I & Muir TW Protein semi-synthesis in living cells. J. Am. Chem. Soc. 125, 7180–7181 (2003). [DOI] [PubMed] [Google Scholar]
- 189.Volkmann G & Liu XQ Protein C-terminal labeling and biotinylation using synthetic peptide and split-intein. PLoS ONE 4, e8381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Borra R, Dong D, Elnagar AY, Woldemariam GA & Camarero JA In-cell fluorescence activation and labeling of proteins mediated by FRET-quenched split inteins. J. Am. Chem. Soc. 134, 6344–6353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bhagawati M et al. In cellulo protein semi-synthesis from endogenous and exogenous fragments using the ultra-fast split Gp41–1 intein. Angew. Chem. Int. Ed. 59, 21007–21015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Charalambous A, Antoniades I, Christodoulou N & Skourides PA Split-inteins for simultaneous, site-specific conjugation of quantum dots to multiple protein targets in vivo. J. Nanobiotechnol. 9, 1–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ray DM, Flood JR & David Y Harnessing split-inteins as a tool for the selective modification of surface receptors in live cells. ChemBioChem 24, e202200487 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Otomo T, Ito N, Kyogoku Y & Yamazaki T NMR observation of selected segments in a larger protein: central-segment isotope labeling through intein-mediated ligation. Biochemistry 38, 16040–16044 (1999). [DOI] [PubMed] [Google Scholar]
- 195.Busche AEL et al. Segmental isotopic labeling of a central domain in a multidomain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Ed. 48, 6128–6131 (2009). [DOI] [PubMed] [Google Scholar]
- 196.Khoo KK et al. Chemical modification of proteins by insertion of synthetic peptides using tandem protein trans-splicing. Nat. Commun. 11, 2284 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pinto F, Thornton EL & Wang B An expanded library of orthogonal split inteins enables modular multi-peptide assemblies. Nat. Commun. 11, 1529 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 199.David Y, Vila-Perelló M, Verma S & Muir TW Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat. Chem. 7, 394–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Holt MT et al. Identification of a functional hotspot on ubiquitin required for stimulation of methyltransferase activity on chromatin. Proc. Natl Acad. Sci. USA 112, 10365–10370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Burton AJ et al. In situ chromatin interactomics using a chemical bait and trap approach. Nat. Chem. 12, 520–527 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Burton AJ, Haugbro M, Parisi E & Muir TW Live-cell protein engineering with an ultra-short split intein. Proc. Natl Acad. Sci. USA 117, 12041–12049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liszczak GP et al. Genomic targeting of epigenetic probes using a chemically tailored Cas9 system. Proc. Natl Acad. Sci. USA 114, 681–686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Morshedi Rad D et al. A comprehensive review on intracellular delivery. Adv. Mater. 33, 2005363 (2021). [DOI] [PubMed] [Google Scholar]
- 205.Mitchell MJ et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fottner M et al. Site-specific protein labeling and generation of defined ubiquitin–protein conjugates using an asparaginyl endopeptidase. J. Am. Chem. Soc. 144, 13118–13126 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dawson PE, Muir TW, Clark-Lewis I & Kent SBH Synthesis of proteins by native chemical ligation. Science 266, 776–779 (1994). [DOI] [PubMed] [Google Scholar]
- 208.Chong S et al. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 192, 271–281 (1997). [DOI] [PubMed] [Google Scholar]
- 209.Muir TW, Sondhi D & Cole PA Expressed protein ligation: a general method for protein engineering. Proc. Natl Acad. Sci. USA 95, 6705–6710 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kumar KSA, Spasser L, Erlich LA, Bavikar SN & Brik A Total chemical synthesis of di-ubiquitin chains. Angew. Chem. Int. Ed. 49, 9126–9131 (2010). [DOI] [PubMed] [Google Scholar]
- 211.Tashiro K, Mohapatra J, Brautigam CA & Liszczak G A Protein semisynthesis-based strategy to investigate the functional impact of linker histone serine ADP-ribosylation. ACS Chem. Biol. 17, 810–815 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schwagerus S, Reimann O, Despres C, Smet-Nocca C & Hackenberger CPR Semi-synthesis of a tag-free O-GlcNAcylated tau protein by sequential chemoselective ligation. J. Pept. Sci. 22, 327–333 (2016). [DOI] [PubMed] [Google Scholar]
- 213.Kulkarni SS, Sayers J, Premdjee B & Payne RJ Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nat. Rev. Chem. 2, 0122 (2018). [Google Scholar]
- 214.Chin JW Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem. 83, 379–408 (2014). [DOI] [PubMed] [Google Scholar]
- 215.Neumann H, Wang K, Davis L, Garcia-Alai M & Chin JW Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444 (2010). [DOI] [PubMed] [Google Scholar]
- 216.Fredens J et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]