

Abstract
The cognitive abilities of humans are distinctive among primates, but their molecular, cellular, and circuit substrates are poorly understood. We used comparative single-nucleus transcriptomics to analyze samples of the middle temporal gyrus (MTG) from adult humans, chimpanzees, gorillas, rhesus macaques, and common marmosets to understand human-specific features of the neocortex. Human, chimpanzee, and gorilla MTG showed highly similar cell-type composition and laminar organization as well as a large shift in proportions of deep-layer intratelencephalic-projecting neurons compared with macaque and marmoset MTG. Microglia, astrocytes, and oligodendrocytes had more-divergent expression across species compared with neurons or oligodendrocyte precursor cells, and neuronal expression diverged more rapidly on the human lineage. Only a few hundred genes showed human-specific patterning, suggesting that relatively few cellular and molecular changes distinctively define adult human cortical structure.
Print Summary:
Introduction:
The cerebral cortex is involved in complex cognitive functions such as language. Although the diversity and organization of cortical cell types has been extensively studied in several mammalian species, human cortical specializations that may underlie our unique cognitive abilities remain poorly understood.
Rationale:
Single-nucleus RNA sequencing (snRNA-seq) offers a relatively unbiased characterization of cellular diversity of brain regions. Comparative transcriptomic analysis enables the identification of molecular and cellular features that are conserved and specialized but is often limited by the number of species analyzed. We applied deep transcriptomic profiling of the cerebral cortex of humans and four non-human primate (NHP) species to identify homologous cell types and human specializations.
Results:
We generated snRNA-seq data from human, chimpanzee, gorilla, rhesus macaque, and marmoset (over 570,000 nuclei in total) to build a cellular classification of a language-associated region of cortex (middle temporal gyrus, MTG) in each species and a consensus primate taxonomy. Cell type proportions and distributions across cortical layers are highly conserved among great apes, while marmoset has higher proportions of L5/6 IT Car3 and L5 ET excitatory neurons and Chandelier inhibitory neurons. This strongly points to other cellular features driving human-specific cortical evolution. Profiling gorillas enabled discrimination of which human and chimpanzee expression differences are specialized in humans. We discovered that chimpanzee neurons have more similar gene expression profiles to gorilla than human neurons, despite chimpanzees and humans sharing a more recent common ancestor. In contrast, glial expression changes were consistent with evolutionary distances and were more rapid than neuronal expression changes in all species. Thus, our data support faster divergence of neuronal but not glial expression on the human lineage. For all primate species, many differentially expressed genes (DEGs) were specific to one or a few cell types and were significantly enriched in molecular pathways related to synaptic connectivity and signaling. Hundreds of genes had human-specific differences in transcript isoform usage, and these genes were largely distinct from DEGs. We leveraged published data sets to link human-specific DEGs to regions of the genome with human-accelerated mutations or deletions (HARs and hCONDELs). This led to the surprising discovery that a large fraction of human-specific DEGs (15-40%), and particularly those associated with synaptic connections and signaling, were near these genomic regions that are under adaptive selection.
Conclusion:
Our study finds that MTG cell types are largely conserved across approximately 40 million years of primate evolution, and the composition and spatial positioning of cell types is shared among great apes. In each species, hundreds of genes exhibit cell type-specific expression changes, particularly in pathways related to neuronal and glial communication. Human-specific DEGs are enriched near likely adaptive genomic changes and are poised to contribute to human-specialized cortical function.
One Sentence Summary:
Human specializations in cortical expression are poised to alter circuit wiring and are linked to adaptive genomic changes.
Graphical Abstract
Divergent gene expression in the primate neocortex. (A) Proportions of neuronal subclasses are conserved across species, except for increased proportions of three subclasses (asterisks) in marmoset. Among great apes, neuronal gene expression has evolved faster on the human lineage, and glial expression has diverged faster than neurons in all species. (B) Many human-specific DEGs are associated with circuit function and are linked to potentially adaptive changes in gene regulation.
Humans have unique cognitive abilities compared to non-human primates (NHPs), including chimpanzees, our closest evolutionary cousins. For example, humans have the capacity for vocal learning that requires a highly interconnected set of brain regions, including the middle temporal gyrus (MTG) region of the neocortex that integrates multimodal sensory information and is critical for visual and auditory language comprehension (1, 2). Human MTG is larger and more connected to other language-associated cortical areas compared to chimpanzees and other NHPs (3–5). These gross anatomical changes may be accompanied by changes in the molecular programs of cortical neurons and non-neuronal cells. Indeed, previous work has identified hundreds of genes with up- or down-regulated expression in the cortex of humans compared to chimpanzees and other primates (6–9) but have been limited to comparing broad populations of cells or have lacked another great ape species to study changes specific to the human lineage.
Single nucleus RNA-sequencing has enabled generation of high-resolution transcriptomic taxonomies of cell types in neocortex and other brain regions. Comparative analyses have established homologous cell types across mammals, including human and NHPs, and identified conserved and specialized features: cellular proportions (10), spatial distributions (11), and transcriptomic and epigenomic profiles (12). In this study, we profiled over 570,000 single nuclei using RNA-sequencing from MTG of 5 species: human, two great apes (chimpanzee and gorilla), a cercopithecid monkey (rhesus macaque), and a platyrrhine monkey (common marmoset). Based on a recently published mammalian phylogeny (13), this represents approximately 38 million years of evolution since these primate species shared a last common ancestor, and encompasses the relatively recent divergence of the human lineage from that of chimpanzees at 6 million years ago.
We defined cell type taxonomies for each species and a consensus taxonomy of 57 homologous cell types that were conserved across these haplorhine primates. This enabled comparison of the cellular architecture of cortex in humans to a representative sample of non-human primates at unprecedented resolution to disentangle evolutionary changes in cellular composition from gene expression profiles. Including gorillas, whose ancestry branched from that leading to humans and other great apes approximately 7 million years ago, enabled testing for faster evolution on the human lineage and inference of differences between human and chimpanzee that are derived (novel) in humans. Including two phylogenetically diverse monkey species enabled identification of cellular specializations that humans share with other great apes that may contribute to our enhanced cognitive abilities. Finally, establishing putative links between HARs and hCONDELs and human expression specializations by leveraging recently generated datasets of the in vitro activity of HARs (14) and cell type-specific chromatin folding (15, 16) identifies a subset of changes that may be adaptive.
Within-species cell type taxonomies
MTG cortical samples were collected from post-mortem adult male and female human, chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), rhesus macaque (Macaca mulatta), and common marmoset (Callithrix jacchus) individuals for single nucleus RNA-sequencing (Fig. 1B). MTG was identified in each species using gross anatomical landmarks. Layer dissections for human, chimpanzee, and gorilla datasets were identified and sampled as previously described (17). MTG slabs were sectioned, stained with fluorescent Nissl, and layers were microdissected and processed separately for nuclear isolation.
Fig. 1. Transcriptomic cell type taxonomies of human and NHP MTG.
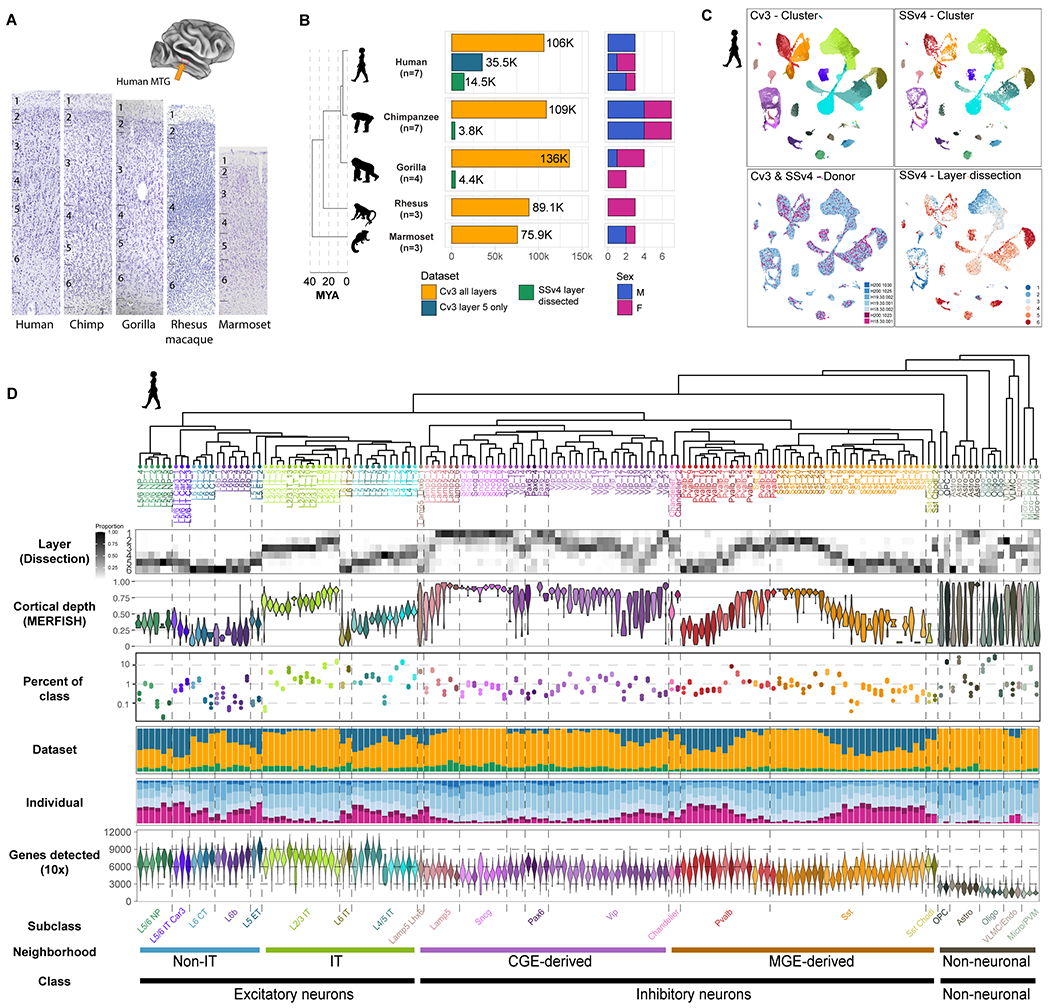
(A) Approximate MTG region dissected from human brain (inset). Representative Nissl-stained cross-sections of MTG in the five species profiled. (B) Phylogeny of species (left; MYA, millions of years ago) and barplots of QC-passing nuclei (center) and sampled individuals (right) for each dataset. (C) UMAP plots of single nuclei from human MTG integrated across individuals and RNA-seq technologies and colored by cluster, individual id, and dissected layer. (D) Human taxonomy dendrogram based on Cv3 cluster median expression. Heatmap of laminar distributions estimated from SSv4 layer dissections. Violin plots of the relative cortical depth (pia to white matter) of cells grouped by type based on in situ measurement of marker expression in human MTG. Dot plot of cell type abundance represented as a proportion of class (excitatory, inhibitory, glia). Error bars denote standard deviation across Cv3 individuals (L5 only dissection excluded). Barplots indicate the proportion of each cluster that is composed of Cv3 all layers, Cv3 layer 5 only, and SSv4 layer dissected datasets. Barplots indicating the proportion of each cluster that is composed of each individual. Violin plots showing the number of unique genes detected from Cv3 datasets.
For humans, single nuclei from 7 individuals contributed to three RNA-seq datasets: a Chromium 10x v3 (Cv3) dataset sampled from all 6 cortical layers (n = 107k nuclei); a Cv3 dataset sampled from micro-dissected layer 5 to capture rare excitatory neuron types (n = 36k); and our previously characterized (17) SMARTseq v4 (SSv4) dataset of six micro-dissected layers (n = 14.5k). Chimpanzee (n = 7 individuals) datasets included Cv3 across layers (n = 109k nuclei) and SSv4 layer dissections (n = 3.9k), and gorilla (n = 4) datasets included Cv3 (n = 136k) and SSv4 (n = 4.4k). Macaque (n = 3) and marmoset (n = 3) datasets included Cv3 from all layers (n = 89.7k and 76.9k nuclei, respectively). All nuclei preparations were stained for the pan-neuronal marker NeuN and FACS-purified to enrich for neurons over non-neuronal cells. Samples containing 90% NeuN+ (neurons) and 10% NeuN- (non-neuronal cells) nuclei were used for library preparations and sequencing. Nuclei from Cv3 experiments were sequenced to a saturation target of 60%, resulting in approximately 120k reads per nucleus. Nuclei from SSv4 experiments were sequenced to a target of 500k reads per nucleus.
Each species was independently analyzed to generate a ‘within-species’ taxonomy of cell types. First, datasets were annotated with cell subclass labels from our published human MTG and primary motor (M1) taxonomies (12, 17) using Seurat (18). Cell types were grouped into five neighborhoods – intratelencephalic (IT)-projecting and non-IT-projecting excitatory neurons, CGE- and MGE-derived interneurons, and non-neuronal cells – that were analyzed separately. High-quality nuclei were normalized using SCTransform (19) and integrated across individuals and data modalities using canonical correlation analysis. Human nuclei were well-mixed across the three datasets and across individuals (Fig. 1C), and similar mixing was observed for the other species (Figs. S1, S2). The integrated space was clustered into small ‘metacells’ that were merged into 151 clusters (Fig. 1D, S3) that included nuclei from all datasets and individuals. Cell types had robust gene detection (neuronal, median 3000 to 9000 genes; non-neuronal, 1500 to 3000) and were often rare (less than 1-2% of the cell class) and restricted to one or two layers (Table S1). Single nuclei from the other four species were clustered using identical parameters, resulting in 109 clusters in chimpanzees (Fig. S1A), 116 in gorillas (Fig. S1B), 120 in macaques (Fig. S2A), and 104 in marmosets (Fig. S2B). Significantly, human had the most cell type diversity (151 clusters), although the number of cell types could have been driven by technical factors: sampled individuals (only female macaques), tissue dissections (additional layer 5 sampling for humans), RNA-seq method (SSv4 included for great apes), and genome annotation quality.
Species cell types were hierarchically organized into dendrograms based on transcriptomic similarity (Fig. 1D, S1, S2, S3) and grouped into three major cell classes: excitatory (glutamatergic) neurons, inhibitory (GABAergic) neurons, and non-neuronal cells. Each of the three major classes were further divided into cell neighborhoods and subclasses based on integrated analysis of marker gene expression, layer dissections, and comparison to published cortical cell types (12). In total, we identified 24 conserved subclasses (18 neuronal, 6 non-neuronal) (Fig. S4A) that were used as a prefix for cell type labels. Inhibitory neurons comprised five CGE-derived subclasses (Lamp5 Lhx6, Lamp5, Vip, Pax6, and Sncg) expressing the marker ADARB2 and four MGE-derived subclasses (Chandelier, Pvalb, Sst, and Sst Chodl) expressing LHX6. Excitatory neurons include five intratelencephalically (IT)-projecting subclasses (L2/3 IT, L4 IT, L5 IT, L6 IT, and L5/6 IT Car3) and four deep layer non-IT-projecting subclasses (L5 ET, L5/6 NP, L6b, and L6 CT). Non-neuronal cells were grouped into six subclasses: astrocytes, oligodendrocyte precursor cells (OPCs), oligodendrocytes, microglia and perivascular macrophages (Micro/PVM), endothelial cells, and vascular and leptomeningeal cells (VLMCs).
This human MTG taxonomy provided substantially higher cell type resolution than our previously published human cortical taxonomies (12, 17) due to increased sampling (155k vs. 15-85k nuclei; Fig. S3). Furthermore, the in situ spatial distributions of cell types were characterized using MERFISH and are included as a gallery of human MTG sections (Supplementary Media 1) and summarized by cortical depth (Fig. 1D). All cell types matched one-to-one or one-to-many, and diversity was particularly expanded for non-neuronal subclasses and several neuronal subclasses and types: L5/6 NP (6 types), L6 CT (4), L2/3 IT FREM3 (8), and SST CALB1 (9). The FREM3 subtypes had a graded distribution across layers 2 and 3, consistent with spatial variation in FREM3 neuron morphology and electrophysiology (20).
Divergent abundances of cell types
Neuronal subclass frequencies were estimated as a proportion of excitatory and inhibitory neuron classes based on snRNA-seq sampling to account for species differences in the ratio of excitatory to inhibitory neurons (E:I ratio) (Fig. S4B) (12). Subclass proportions were highly consistent across individuals within each species and varied significantly (one-way ANOVA, P < 0.05) across species (Fig. 2A). Post-hoc pairwise t-tests between humans and each NHP identified up to 5-fold more L5/6 IT Car3, L5 ET, and PVALB-expressing chandelier interneurons in marmosets. Interestingly, L2/3 IT neurons had similar proportions in MTG, in contrast to the 50% expansion of L2/3 IT neurons in humans versus marmosets in M1 (12).
Fig. 2. Great ape specialization of L5/6 IT Car3 neuron proportions.
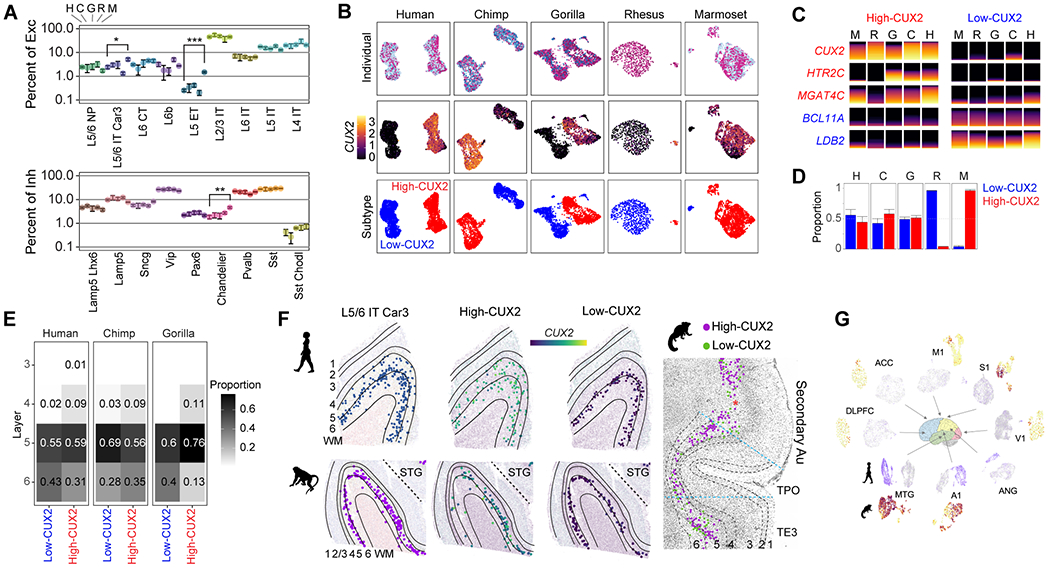
(A) Average subclass proportions of neuronal classes (whiskers, standard deviation across individuals). Significant differences in proportions compared to human (two-sided t-tests; Benjamini-Hochberg corrected * P < 0.05, ** P < 0.01, *** P < 0.001). (B) UMAPs of L5/6 IT Car3 neurons labeled by individual, CUX2 expression, and subtype. (C) Conserved marker gene expression of High-CUX2 and Low-CUX2 subtypes. (D) Average proportions of L5/6 IT Car3 neuron subtypes (whiskers, standard deviation across individuals). (E) Laminar distributions of subtypes across great apes estimated based on layer dissections. (F) L5/6 IT Car3 neurons labeled in situ based on marker expression in human and macaque MTG and in the matching regions in marmoset cortex (TPO and TE3). Low-CUX2 neurons are enriched at the layer 5/6 border in human and macaque. Red asterisk, area of low probe density due to a technical artifact. STG, superior temporal gyrus. Au, auditory cortex. (G) UMAPs of CUX2 expression in L5/6 IT Car3 neurons from matched cortical regions in humans (purple, high expression) and marmosets (dark orange, high expression).
Among L5/6 IT Car3 neurons, two distinct subtypes expressed CUX2 at high or low levels in all species (Fig. 2B). HTR2C and MGAT4C were additional conserved markers of the High-CUX2 subtype, and BCL11A and LDB2 were markers of the Low-CUX2 subtype (Fig. 2C). Subtype proportions were balanced in great apes, mostly Low-CUX2 in macaques, and mostly High-CUX2 in marmosets (Fig. 2D). Low-CUX2 neurons were consistently enriched in deeper layers than High-CUX2 in all three great apes (Fig. 2E). In human and macaque MTG, in situ labeling of marker genes using MERFISH (Fig. 2F) validated that the Low-CUX2 subtype was enriched at the border of layers 5 and 6, and the High-CUX2 subtype extended from upper layer 6 through layer 5. In macaque MTG, the proportion of High-CUX2 neurons varied along the gyrus (Fig. 2F) with little on the ventral side consistent with the snRNA-seq data and more on the dorsal side. In marmoset, in situ labeling of marker genes using RNAscope showed that High-CUX2 neurons were enriched in MTG (TPO and TE3) consistent with snRNA-seq data and in adjacent secondary auditory regions (Fig. 2F) Intriguingly, based on snRNA-seq data we collected from 7 additional regions of the human cortex (21), Low-CUX2 neurons were more common in many regions, and High-CUX2 neurons were enriched in temporal (MTG and primary auditory, A1) and parietal cortex (angular gyrus, ANG and primary somatosensory, S1) (Fig. 2G). Similarly, snRNA-seq data collected from 6 additional regions of the marmoset cortex (10, 12, 22) revealed that the High-CUX2 subtype was most enriched in temporal areas (MTG and A1) and less enriched in S1 (Fig. 2G).
Primate specializations of cell type expression
Next, we compared the transcriptomic similarity of subclasses across primates. For each species, gene markers were defined that could reliably predict the subclass identities of cells and were filtered to include one-to-one orthologs (Table S2). Non-neuronal subclasses expressed hundreds of markers and demonstrated greater distinction than neuronal subclasses that had 50-100 markers. Each subclass had a similar number of markers in all species (Fig. 3A), but only 10-20% had strongly conserved specificity (Fig. 3A,B). To compare the global expression profile of subclasses across primates, we correlated normalized median expression of variable genes between each species pair for each cell subclass (excluding undersampled endothelial cells and VLMCs) (Fig. 3C). Surprisingly, glial cells (except OPCs) had greater expression changes between species compared to neurons. Expression similarity decreased with evolutionary distance between human and NHPs at a similar rate across neuronal subclasses and OPCs and faster in oligodendrocytes, astrocytes, and particularly microglia (Fig. 3D). Glial expression remained significantly more divergent across species after normalizing for increased variation within species (Fig. S4C). Strikingly, chimpanzee neuronal subclasses were significantly more similar to gorilla than to human (Fig. 3E), despite a more recent common ancestor with humans (6 versus 7 million years ago). This was consistent with faster evolution of neurons on the human lineage since the divergence with chimpanzees. In contrast, there was no evidence for faster divergence among non-neuronal cells on the human lineage (Fig. 3E) or on the lineage leading to great apes (Fig. S4D).
Fig. 3. Rapid divergence of neuronal expression on the human lineage.
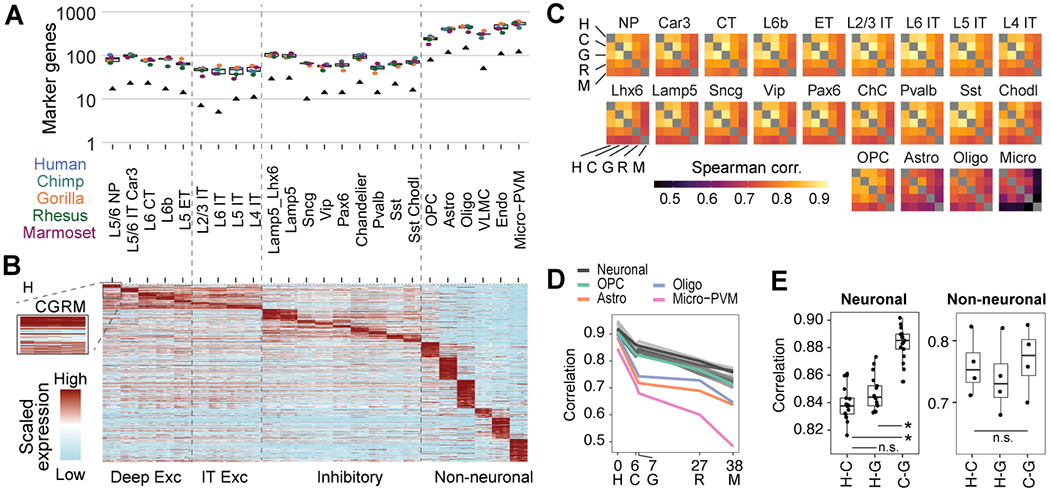
(A) Boxplot showing the distribution of subclass marker genes across species. Points denote the number of species markers and black triangles denote the number of conserved markers. (B) Expression heatmap for 924 conserved markers shown in A. Expression is row-scaled for each subclass for each species. (C) Heatmaps showing Spearman correlations of subclass median expression for all variable genes between species. (D) Line graph of subclass correlations from D for humans compared to each NHP as a function of the evolutionary distance (millions of years ago) of the most recent common ancestor (13). The zero point denotes the median correlation between human individuals for each subclass. (E) Boxplots of great ape pairwise correlations for neuronal and non-neuronal subclasses from C. ANOVA and post-hoc two-sided t-tests; Benjamini-Hochberg corrected *P < 0.05. Non-neuronal ANOVA was not significant.
In addition to evolutionary changes in expression levels, there may be changes in transcript isoform usage. We quantified isoform expression using full-length transcript information from SSv4 RNA-seq data acquired from great apes. For each cell subclass, we identified differentially expressed genes (DEGs; Table S3) and genes with at least moderately high expression that strongly switched isoform usage between each pair of species (Table S4). Notably, there was little overlap between genes with differential expression and isoform usage for L2/3 IT neurons (Fig. S4E). Genes with a human-specialized switch in isoform expression included BCAR1, INO80B, and SBNO1 (Fig. S4F). BCAR1 is a scaffold protein that is a component of the netrin signaling pathway and involved in axon guidance (23). INO80B (24) and SBNO1 are involved in chromatin remodeling, and SBNO1 contributes to brain axis development in zebrafish (25) and is a risk gene for intellectual disability (26). Interestingly, the predominant isoform of INO80B in human L2/3 IT neurons includes a retained intron (Fig. S4G) that may suppress transcription of this gene (27) and contribute to human specializations.
Finally, we quantified the conservation of gene expression patterns across cell types between human and NHPs. As expected, expression differences increased with evolutionary distance (Fig. S4H), and 75% of genes were conserved in all species (r > 0.9 in great apes; r > 0.65 in marmoset). 651 genes had highly divergent expression (r < 0.25), often in only a single species (Fig. S4I), such as FAM177B that was exclusively expressed in human microglia (Fig. S4J). Interestingly, a few genes had fixed derived expression in the great ape lineage. For instance, MEPE is a secreted calcium-binding phosphoprotein that was restricted to PVALB-expressing interneurons in great apes (Fig. S4J), and prolactin receptor (PRLR) had enriched expression in SST-expressing interneurons and L5/6 IT Car3 neurons in great apes as compared to CGE-derived interneurons in macaque and marmoset, potentially altering hormonal modulation of these neurons.
Human specializations of glial cells
Since glial cells exhibited the most divergent gene expression changes across species (Fig. 3C,D), we next aimed to uncover their specialized transcriptional programs in humans compared to other great apes. For astrocytes, we found more human DEGs (1189) than chimpanzee (787) or gorilla (617) DEGs (Fig. 4A,B; Fig. S5A; Table S3), and three times more highly divergent (>10-fold) human DEGs (Fig. 4A). Human astrocyte DEGs were significantly enriched in synaptic signaling and protein translation pathways based on enrichment analyses using Gene Ontology (GO) (Fig. 4C) and Synaptic GO (SynGO) (28) (Fig. 4D; Fig. S5B) databases. To study synapse-related astrocytic gene programs, we used a molecular database of astrocyte cell-surface molecules enriched at astrocyte-neuron junctions from an in vivo proteomic labeling approach in the mouse cortex (29). Among genes encoding 118 proteins that were robustly enriched in perisynaptic astrocytic processes, 24 genes (20%) were differentially expressed in human astrocytes compared to chimpanzee and gorilla astrocytes (Fig. 4E), and 47 genes (40%) had conserved expression across great apes (Fig. S5C,D). Interestingly, neuroligins and neurexins, ligand-receptor pairs that play a key role in astrocytic morphology and synaptic development (30), showed divergent expression patterns across great ape species (Fig. 4F,G). Other cell-adhesion gene families with well-known functions in astrocytic morphological and synaptic development also had multiple members among human astrocyte DEGs, including ephrins and their cognate receptors (EFNA5, EPHA6), clustered protocadherins (PCDH9), and teneurins (TENM2, TENM3, TENM4) (Fig. S5D,F).
Fig. 4. Human cortical astrocytes have specialized molecular features.
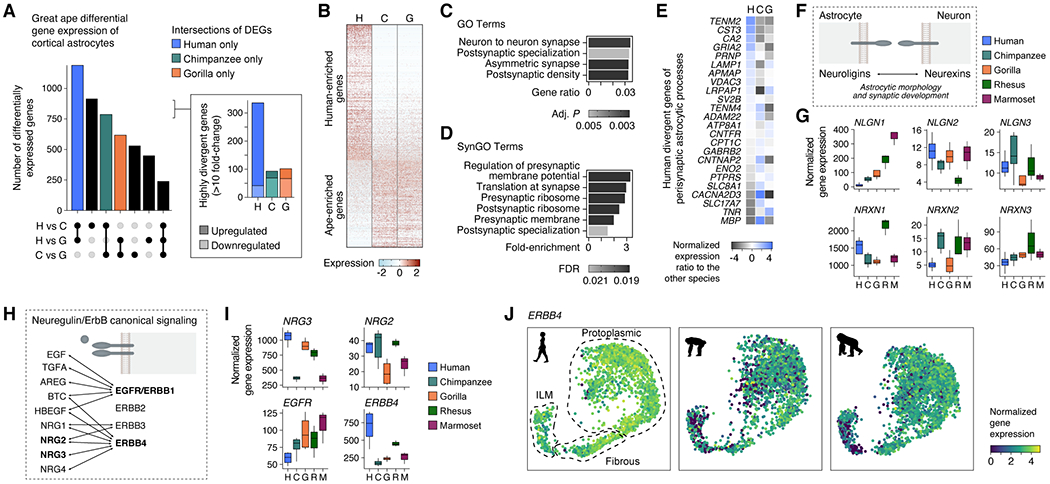
(A) Upset plot showing the number of DEGs in cortical astrocytes for pairwise comparisons between great ape species. Inset shows the number of highly divergent genes (fold-change > 10). (B) Heatmap showing row-scaled expression of human versus chimpanzee and gorilla DEGs. (C,D) Significantly enriched GO (C) and SynGO (D) terms in the union of astrocyte DEGs from the pairwise comparison between human and chimpanzee and the pairwise comparison between human and gorilla (FDR < 0.05). (E) Heatmap showing human DEGs (FDR < 0.01, normalized gene count > 5) of the proteome of perisynaptic astrocytic processes (Takano et al., 2020). (F) Schematic illustrating the trans-cellular interaction of astrocytic neuroligins and neuronal neurexins that is known to play a role in astrocytic morphology and synaptic development. (G) Box plots showing differential gene expression of neuroligins and neurexins in astrocytes across primate species. (H) Schematic illustrating ligand-receptor interactions of the neuregulin/ErbB signaling pathway. (I) Box plots showing differential expression of the ligands NRG2 and NRG3 and the receptors EGFR and ERBB4 in astrocytes across primate species. (J) Gene expression patterns of ERBB4 across astrocyte subtypes and great ape species. ILM, interlaminar.
In addition to cell-adhesion programs, we explored other cell-surface or secreted ligands and receptors that contribute to astrocyte function. We found that several astrocyte-secreted synaptogenic molecules such as Osteonectin (SPARC) and Hevin (SPARCL1) and ECM-related proteins (Brevican, BCAN; Neurocan, NCAN; and Phosphacan, PTPRZ1) were up-regulated in human astrocytes (Fig. S5E,F). Of note, four members of the neuregulin/ErbB signaling pathway showed differential gene expression in great ape astrocytes, with two receptors (EGFR and ERBB4) displaying expression changes in opposite directions (Figs. 5I,J). Interestingly, up-regulation of human ERBB4 expression was higher in protoplasmic and fibrous astrocytes than interlaminar astrocytes (Figs. 3J, S5G), demonstrating that transcriptional specializations can occur in a subtype-specific fashion. Finally, glutamate AMPA receptor subunits (GRIA1, GRIA2, GRIA4) had more than 3-fold greater expression in human astrocytes, suggesting a human-specific responsiveness of astrocytes to glutamate (Fig. S5H).
Fig. 5. Divergent expression across conserved cell types.
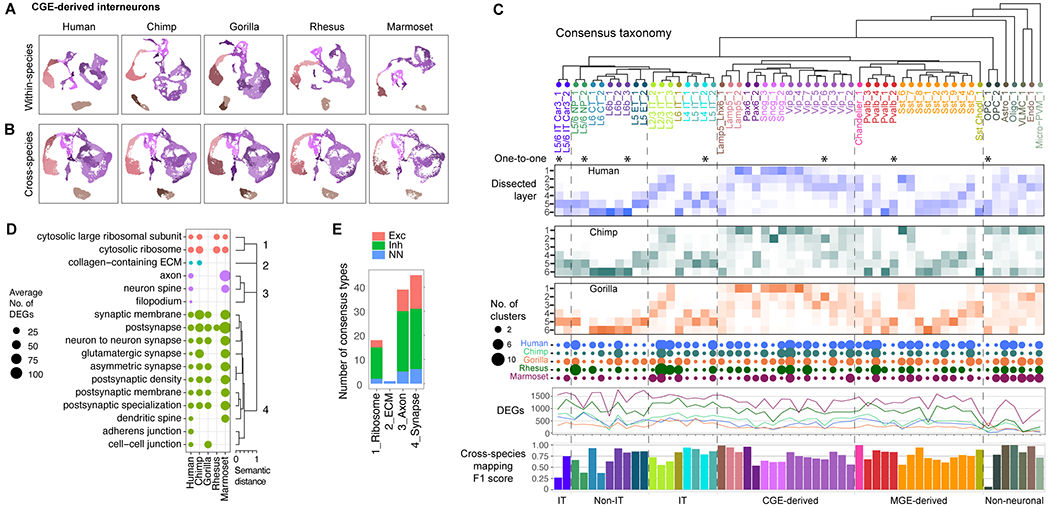
(A) UMAPs of CGE-derived interneuron expression generated for each species and colored by within-species cell types. (B) UMAPs of CGE interneuron expression integrated across the five species and with the same coloring as A. (C) Consensus taxonomy of 57 homologous cell types identified in all five species (*, one-to-one match across all species). For great ape species, heatmaps denote the proportion of nuclei dissected from layers 1 through 6 for each type. Dot plot denotes the number of within-species clusters that are associated with each consensus type. Line plots show the number of hDEGs per consensus type with fold-change (FC) > 1.4 for each species (colors as in dot plot). Barplots show the average classification accuracy (F1 score) across the five species using scPoli (84) (Fig. S12). (D) Summary of GO enrichment analysis of species DEGs. Cellular component terms are shown that were significantly enriched for hDEGs in at least one consensus type and form four distinct groups of similar GO terms. (E) Summary of the number of consensus types that express hDEGs that were enriched for at least one term in the four semantic GO groups.
We next examined gene expression changes in microglia, which also play critical roles in cortical circuit formation (31, 32). Recent comparative spatial transcriptomic data indicate that microglia-neuron contacts are more prevalent in human cortical circuits compared to mice, particularly in superficial layers (33). We reasoned that evolutionary changes in microglial connectivity could be mediated by fine-tuning expression of cell-surface ligands and receptors. Indeed, we found that human microglia have more DEGs (328) than chimpanzee (175) or gorilla (164) microglia (Fig. S6A–C), and human DEGs were significantly overrepresented in GO and SynGO terms related to synaptic compartments (Fig. S6D,E,F). Among the human microglia DEGs were several disease-associated genes, including SNCA (encoding alpha-synuclein) and TMEM163 implicated in neurodegenerative disorders (34–36), and Kalirin (KALRN) associated with neurodevelopmental and neuropsychiatric disorders (37) (Fig. S6G). We also corroborated the human-specific up-regulation of FOXP2 and CACNA1D that was recently reported in the dorsolateral prefrontal cortex (9).
Oligodendrocytes also showed human specializations, including DEGs involved in myelin organization and cell adhesion (e.g., CNTNAP2, LAMA2) (Fig. S6). Notably, unlike astrocytes and microglia, human and chimpanzee oligodendrocytes had similar numbers of DEGs, although humans had more up-regulated, highly divergent DEGs (Fig. S6I). In summary, these findings support faster divergence of glial expression in the human lineage that parallels neuronal divergence and likely impacts interactions between glia and neurons.
Consensus cell type conservation and divergence
To further investigate the canonical architecture of primate MTG, we built a transcriptomic taxonomy of high resolution consensus cell types. Starting with CGE-derived interneurons, we integrated single nucleus expression profiles across the five species based on conserved co-expression using Seurat (18). Within-species cell types remained distinct, and nuclei were well integrated (Fig. 5A,B; Fig. S7), particularly for humans and chimpanzees (Fig. S12A). Similar results were observed for the other cell neighborhoods (Figs. S8,S9,S10,S11). Separate pairwise alignments between human and NHPs confirmed that cell type homologies were better resolved in more closely related species (Fig. S12B). We also found that excitatory neurons were less well integrated than inhibitory neurons, and this finding was consistent with greater species specializations of excitatory types.
We established homologous cell types between all pairs of species using MetaNeighbor, a statistical framework (38, 39) that identified cell types that could be reliably discriminated (AUROC >0.6) from nearest neighbors in one species based on training data from the other species or that were reciprocal best matches. Pairwise cell type homologies were integrated to define 57 consensus types that included cell types identified in the five species, and a dendrogram was constructed based on transcriptomic similarities (Fig. 5C). The robustness of the 57 homologous types across species was confirmed using a complementary approach to consensus clustering, scArches (40) (Fig. S12C–H). Classification accuracy varied across consensus types (Fig. 5C) and with nearly perfect classification performance across species (average F1 score > 0.95) for distinct interneuron types (Lamp5 Lhx6, Pax6_1, and Chandelier cells) and non-neuronal types (astrocytes, oligodendrocytes, and endothelial cells). The rare OPC_1 subtype (5% of OPCs) had the lowest classification accuracy and somewhat ambiguous homology across species (Fig. S11) and may represent different subpopulations of OPCs across species. Eight consensus types represented one-to-one matches across all species, and the majority of types represented multiple matches of between two and ten within-species types. Differential sampling of nuclei across species due to differences in dissections or cell type proportions likely contributed to the number of cell types mapping to a consensus type. For example, in human MTG, more nuclei were sampled from layer 5 and more subtypes of the layer 5-enriched SST_3 consensus type were identified. Thus, there was a conserved set of cell types in primate MTG with transcriptomic specializations of subtypes, but no distinct novel types in any species. Laminar distributions of types were remarkably conserved across the great apes, except Sst Chodl_1 was present in more superficial layers of gorilla MTG (Fig. 5C), although more sampling of this rare type is needed for validation.
Previous work reported the lack of transcript and protein expression of tyrosine hydroxylase (TH), a key enzyme in the dopamine synthesis pathway, in the neocortex of non-human African great apes including chimpanzee and gorilla (41, 42). Recent transcriptomic profiling of chimpanzee prefrontal cortex suggests that this represents loss of dopamine signaling in a conserved cell type rather than loss of a homologous type (9). In MTG, we identified 9 consensus SST-expressing interneuron types present in all five primates (Fig. 5E) that had robust sets of conserved and species-specific markers (Fig. S13A; Table S5). The Sst_1 consensus type was distinct from most other MGE-derived interneurons (Fig. S13B) and expressed TH in human, macaque, and marmoset neurons but not chimpanzee or gorilla neurons (Fig. S13C–E). Conserved (e.g. NCAM2, PTPRK, UNC5D, and CNTNAP5) and species-specific genes were enriched in pathways for connectivity and signaling (Fig. S13F–J). Sst_1 was the rarest type in all primates (Fig. S13K) and varied from 0.3% of SST-expressing interneurons in gorillas and macaques to 1-3% in humans, chimpanzees, and marmosets. Interestingly, the majority of TH-expressing neurons belonged to different interneuron subclasses in humans (SST), macaques (PVALB), and marmosets (VIP) (Fig. S13L), and this was confirmed by in situ labeling of TH-expressing neurons in human and macaque MTG (Fig. S13M). Dopamine receptor expression varied across primates but did not track with predicted differences in local dopamine production (Fig. S13N–O). This is likely because subcortical regions provide the majority of dopaminergic input to the neocortex and mask the effects of evolutionary changes in local input.
We tested for changes in proportions of neuronal consensus types across primates using a Bayesian model (scCODA) that accounted for the compositional nature of the data (Fig. S14A) (43). We found that the higher E:I ratio in marmosets (Fig. S4B) was driven by increased proportions of most excitatory types, and the lower E:I ratio in macaques was driven by increased proportions of particularly Sst and Vip interneuron types and by decreased proportions of L2/3 IT_2, L2/3 IT_3, and L5/6 IT Car3_2 excitatory types. There were smaller changes among the great apes, except for an increased proportion of L5/6 NP_2 neurons in humans and chimpanzees.
Next, we identified species-specialized genes by comparing consensus cell type expression for each species to all other primates. Human consensus types had a broad range (fewer than 100 to over 1000) of statistically significant DEGs (Fig. 5C; Table S7) that represented 1-8% of expressed genes (Fig. S14B). Excitatory types in deep layers (IT and non-IT) had the most human-specific DEGs (hDEGs), including L5/6 NP_2, L6 CT_1, and both subtypes of L5/6 IT Car3 neurons (Fig. 5C). Surprisingly, non-neuronal types had the fewest hDEGs despite having the lowest correlated expression between species (Fig. 2D,E). Two factors contributed to this apparent inconsistency. First, non-neuronal cells expressed fewer transcripts than neurons, and the number of hDEGs as a proportion of median expressed genes was similar for non-neuronal and some neuronal types (Fig. S14B). Second, non-neuronal cells were more variable across individuals than neurons (Fig. S4C), and there was reduced power to detect smaller expression changes. Indeed, non-neuronal and neuronal types with fewer hDEGs had larger median fold-changes that were statistically significant despite high inter-individual variation (Fig. S14B).
Strikingly, many species DEGs were restricted to one or a few cell types, particularly for great apes (Fig. S14C). The cell type specificity of DEGs was not simply a result of expression changes in marker genes, but also selective changes in broadly expressed genes (Fig. S14D). hDEGs had a median 4-fold change in expression, while a few metabolism-related genes changed expression by 20-fold or more in most cell types (Fig. S14E). The same genes were often differentially expressed in multiple species (Fig. S14F) but in different cell types, and highly divergent (>10-fold) genes were usually found across all species. In situ measurement of two hDEGs, COL11A1 and DACH1, validated enriched expression in human Chandelier and L5/6 IT Car3 neurons, respectively (Fig. S14G). Species DEGs were enriched in four major pathways: ribosomal processing, extracellular matrix (ECM), axon structure, and the synapse (Fig. 5D,E). Ribosomal processing was primarily associated with interneurons in humans and all cell types in chimpanzees, macaques, and marmosets. Intriguingly, ECM-associated DEGs, including several laminin genes, were specific to the VLMC_1 consensus type in humans, chimpanzees, and marmosets (Fig. S14H) and have potential to alter the blood brain barrier as shown in a mouse model of pericyte dysfunction (44). Hundreds of axonal and synaptic genes were differentially expressed in most cell types in all species, and this suggests extensive molecular remodeling of connectivity and signaling during primate evolution.
Enrichment of HARs and hCONDELs near human differentially expressed genes
Genes may change expression between species due to neutral or adaptive evolution. To investigate which hDEGs may be under positive selection, we linked hDEGs to human-specific genomic sequence changes. Because hDEGs are differentially expressed in only one or a few consensus cell types, expression changes are likely caused by sequence modifications to regulatory regions that can alter transcription in select cell types. We examined three previously identified classes of genomic regions that have changed along the human lineage: (1) human accelerated regions (HARs) that are highly conserved across mammals and have higher substitution rates in the human lineage (14); (2) human conserved deletions (hCONDELs) that are highly conserved across mammals and deleted in humans (45, 46); and (3) human ancestor quickly evolved regions (HAQERs) that are the fastest evolved regions in the human genome (47). Strikingly, we find that HARs and hCONDELs are significantly (FDR < 0.05) enriched near hDEGs in many consensus cell types (Fig. 6A; Fig. S15A, B). The proportion of hDEGs near HARs and hCONDELs is highest for non-neuronal consensus types such as VLMCs (VLMC_1), microglia (Micro-PVM_1), and oligodendrocytes (Oligo_1), likely due to the larger intronic and flanking intergenic regions of hDEGs in these cell types (Fig. S15D). We find some enrichment of HARs and hCONDELs near NHP-specific DEGs (Fig. S16), and this is not surprising because accelerated genomic regions in different primate lineages cluster near similar genes (48).
Fig. 6. HARs and hCONDELs are enriched near hDEGs.
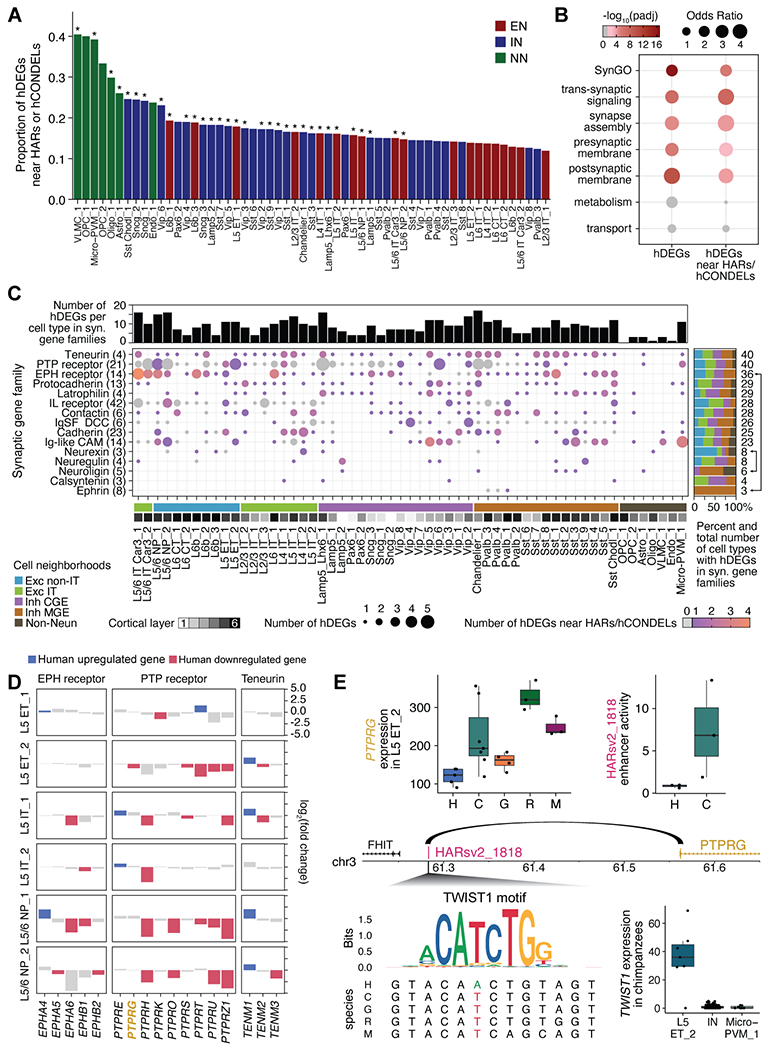
(A) Asterisks indicate HARs and hCONDELs that are enriched near hDEGs in specific consensus cell types at 5% FDR. EN: excitatory neuron, IN: inhibitory neuron, NN: non-neuronal. (B) hDEGs, as well as hDEGs near HARs or hCONDELs, are enriched for genes annotated in specific SynGO terms (28). Non-significant associations are in gray. (C) Synaptic gene families with highly divergent expression patterns. Brackets on the far right highlight examples of synaptic gene families with trans-synaptic interactions that show human differential gene expression in cell types known to form canonical cortical circuits. (D) Patterns of expression change between humans and NHPs for three highly-divergent families in consensus types of L5 excitatory neurons. (E) PTPRG has decreased expression in human L5 ET_2 (each point represents normalized pseudobulk gene expression per individual). Its promoter interacts with the intergenic region containing HARsv2_1818 (16), which has decreased enhancer activity in human SH-SY5Y cells (14). A base pair change in the human HARsv2_1818 sequence removes a potential binding site for TWIST1, which is highly expressed in L5 ET_2.
In contrast, HAQERs are not enriched near hDEGs in any consensus cell type (Fig. S15C). Unlike HARs and hCONDELs, HAQERs need not be conserved across other species and potentially include genomic regions that were previously non-functional but that acquire new functions in humans. Therefore, we tested if HAQERs were enriched near genes with differential expression between humans and chimpanzees, without regard for their expression in other primates, and found significant enrichment for the OPC and L5/6 IT Car3 subclasses (Fig. S17). HARs and hCONDELs are also enriched near DEGs between humans and chimpanzees in multiple cell subclasses, reflecting the enrichment of HARs and hCONDELs near hDEGs.
Since hDEGs are highly enriched for synaptic genes (Fig. 5D), we asked whether a subset of hDEGs that are potentially adaptive (i.e. near HARs or hCONDELs) are associated with specialized localizations or molecular functions of the synapse by performing gene set enrichment analysis using SynGO (28). We found a significant enrichment of hDEGs among SynGO genes compared to all expressed genes (P < 10−16) and a further enrichment of hDEGs near HARs and hCONDELs among SynGO genes compared to all hDEGs (P < 10−5) (Fig. 6B; Fig. S18; Table S8). Among the most enriched SynGO terms were synapse assembly, synaptic membrane organization, and trans-synaptic signaling. Other SynGO terms were not enriched, including synaptic transport, metabolism, cytoskeleton, and vesicle exocytosis machinery (Fig. 6B; Fig. S18; Fig. S19; Table S8). We also found a significant enrichment of hDEGs, and those near HARs and hCONDELs, within gene families encoding synaptic adhesion molecules (P < 10−6) (Fig. 6C; Fig. S20; Fig. S21; Table S8).
We next examined how synaptic genetic programs have changed expression in specific human consensus types. Some gene families (neurexins, interleukin receptors, FLRT proteins, and Trk receptors) mainly changed in excitatory types in deep cortical layers, while other families (neuroligins, protocadherins, latrophilins, and Ig superfamily DCC receptors) primarily changed in inhibitory types (Fig. 6C; Fig. S20). Interestingly, Pvalb interneurons and deep layer excitatory neurons are known to establish specific microcircuits in deeper cortical layers (49), and those types show complementary expression changes in ephrin ligands and receptors, respectively (Fig. 6C; Fig. S20B). Moreover, although teneurins, PTP receptors, and EPH receptors include hDEGs in almost all consensus types (Fig. 6C), specific family members are hDEGs only in a subset of types. For instance, 13 genes within these families (EPHA3, EPHA4, EPHA5, EPHA7, EPHB6, PTPRF, PTPRG, PTPRK, PTPRQ, PTPRS, PTPRT, PTPRU, TENM3) changed expression in only 1-2 consensus types within the 14 consensus types of L5-6 excitatory neurons (Fig. 6D; Fig. S20B; Fig. S22A,B). Similarly, several genes (CDH1, CDH2, CDH24, EFNA5, EFNB2, IGSF9B, LGI1, LGI2, LRFN5, SLITRK4) that only diverged in expression in inhibitory interneurons also showed selective changes in only 1-2 consensus types (Fig. S20B; Fig. S22C). Taken together, our data highlight human specializations of synaptic gene programs that are highly localized to specific cell types and may underlie differences in synaptic connectivity in specific microcircuits.
We leveraged existing data to identify human-specific sequence changes in regulatory regions linked to hDEGs that may drive differential expression in select cell types. For example, PTPRG is a member of the PTP receptor family that acts as presynaptic organizers for synapse assembly (50). Genetic variants in PTPRG have been associated with neuropsychiatric disorders, and Ptprg mutant mice show memory deficits, supporting an important role for PTPRG in cognitive function (51–54). We found that PTPRG was widely expressed across cell types (Fig. S23A) and had lower expression in humans compared to NHPs in four consensus types: one excitatory neuron type (L5 ET_2), microglia (Micro-PVM_1), and two inhibitory neuron types (Vip_2 and Vip_6) (Fig. 6E; Fig. S23B). PTPRG is located near HARsv2_1818 (chr3:61283266-61283416, hg38), which has decreased enhancer activity from the human sequence compared to the orthologous chimpanzee sequence (14). Intriguingly, a 5 kb genomic interval that includes HARsv2_1818 has been shown to interact with the promoter of PTPRG, specifically in excitatory neurons, but not in inhibitory neurons or microglia (15, 16). This raises the possibility that decreased enhancer activity from HARsv2_1818 in humans may have decreased PTPRG expression specifically in the excitatory neuron consensus type L5 ET_2 and that separate regulatory mechanisms may decrease PTPRG expression in microglia and specific inhibitory neuron consensus types. In support of this hypothesis, there is a base pair substitution in the human HARsv2_1818 sequence that removes a binding site for TWIST1, a basic helix-loop-helix transcription factor. We find that TWIST1 is expressed predominantly in L5 ET_2 compared to microglia or inhibitory neuron consensus types (Fig. 6E; Fig. S23C), further suggesting that human-specific sequence changes in HARsv2_1818 may specifically decrease PTPRG expression in L5 ET_2. We extended this analysis to link 112 HARs to 92 hDEGs in neurons using existing data (15, 16), and we posit that genomic interaction data from specific cell types may reveal additional genes that may be regulated by human-specific sequence changes.
Discussion:
Transcriptomic profiling of over 570,000 nuclei from the MTG region of primate neocortex reveals a remarkably conserved cellular architecture across humans and four NHPs: chimpanzees, gorillas, macaques, and marmosets. Humans and the other great apes have nearly identical proportions and laminar distributions of consensus types, while marmosets are the most distinct with markedly increased proportions of L5 ET and L5/6 IT Car3 excitatory neurons and Chandelier interneurons. Great apes have equal proportions of two major subtypes of L5/6 IT Car3 neurons that have high or low CUX2 expression and distinct positions in layers 5 and 6, and marmosets have mostly High-CUX2 neurons. Unlike in primates, L5/6 IT Car3 neurons in mice express markers of both subtypes and are transcriptomically homogeneous across the cortex although they project to diverse cortical targets including proximal areas and homotypic areas in the contralateral hemisphere (55). Remarkably, High-CUX2 neurons are selectively enriched in language-related regions in the human temporal and parietal cortex (MTG, A1, and ANG)(21), and these neurons may have distinct connectivity and contribute to the functional specializations of these regions.
Cell type expression differences are more pronounced than proportion differences and mostly parallel evolutionary distances. One notable exception is that neuronal expression diverged more rapidly in the human lineage (56) compared to chimpanzee and gorilla lineages. In all primates, evolutionary expression changes are significantly accelerated in microglia, astrocytes, and oligodendrocytes compared to neurons and OPCs, even after accounting for higher glial expression variability between individuals. In addition, human glia express more highly divergent genes than chimpanzee or gorilla glia, suggesting faster divergence of human microglia and astrocytes (9) as well as oligodendrocytes (8) among great apes. Deeper sampling of cells and individuals will be needed to disentangle the genetic and environmental effects driving these glial specializations and whether expression differences represent changes in cell types or cell states. Finally, human-specific changes in transcription also involve substantial switching of isoform usage in genes that, surprisingly, often have conserved expression levels. This highlights the importance of profiling full-length transcripts in molecular studies of cellular diversity to identify a more comprehensive set of genes with potentially functional changes.
Humans and NHPs have hundreds of DEGs that are specific to one or a few consensus types and are enriched in molecular pathways related to ribosomal processing, cell connectivity, and synaptic function. Human-specific changes in synaptic gene expression are complex, and distinct families of genes are differentially expressed in select neuronal and non-neuronal types. For example, ephrin molecules specifically differ in PVALB inhibitory cell types, while their cognate receptors (EPH receptors) are changing prominently in deep layer excitatory neurons. Importantly, ephrin/EPH receptor signaling has been shown to promote synaptogenesis in the mouse developing cortex (57, 58). Since PVALB interneurons and excitatory neurons form selective patterns of connectivity in a cell type-specific fashion (49), the differential expression of ephrins and EPH receptors in these cells observed in primate species could reflect species differences in the formation of inhibitory microcircuits involving specific subtypes of PVALB interneurons and excitatory neurons.
Of note, a substantial proportion of synaptic cell-adhesion genes showed down-regulated expression in human neurons, particularly in gene families encoding PTP receptors, including PTPRG, and EPH receptors (Fig. 6D). Some studies have proposed roles in synapse elimination for members of highly-divergent synaptic families, including Pcdh10, ephrin-B1, and ephrin-A2 (59, 60). In such a case, reduced expression of negative regulators of synaptic assembly in human neurons could lead to an enhanced ability to form synaptic connections, potentially underlying the greater number of synapses per neuron observed in the human cortex compared to NHPs (61). Notably these molecular and morphological specializations of human cortical neurons may be linked to macroscale anatomical changes since the number of synapses per neuron increases predictably with brain size across human and non-human primates (62). This highlights the need to sample a phylogenetically broader set of mammals, particularly large-brained, non-primate species, to help differentiate between cellular features that result from brain scaling versus human-specialized cognitive capacities, such as language.
Emerging evidence demonstrates the critical role that non-neuronal cell types play in cortical development, network function, and behavior (63–68). Previous molecular assays have identified a role for ErbB4-mediated signaling in astrogenesis, astrocyte-neuron communication, and astrocyte-induced neuronal remodeling, potentially through both paracrine and autocrine signaling (69–71). Interestingly, we observed changes in expression of ERBB4 receptor and its cognate ligands NRG2 and NRG3 in human astrocytes compared to chimpanzees and gorillas. Altogether, these findings point towards finely regulated molecular specializations underlying neuronal and glial communication in the human cortex. Our data also serves as a resource for future investigation of human-enriched astrocyte and microglia gene programs associated with disease.
Cell type-specific evolutionary changes in gene expression are likely driven by sequence changes to regulatory regions that can be active with high spatial and temporal precision. This is supported by prior studies of genome sequence evolution in humans and other species that estimated that more than 80% of adaptive sequence change is likely regulatory (45, 72, 73). Indeed, we find that previously identified genomic regions that have human-specific sequence changes, such as HARs and hCONDELs, are enriched near hDEGs. Intriguingly, this association is observed for both neuronal and non-neuronal consensus types. In addition to well-described changes in the number and function of neurons in the human brain (74), many non-neuronal cell types also undergo comparable changes in the human lineage (75, 76). Moreover, hDEGs, including those near HARs and hCONDELs, have been found to play critical roles in synapse establishment, elimination, and maintenance when expressed by neuronal and non-neuronal cells (77). Associating genomic regions with signatures of selection in humans to hDEGs provides a framework to link regulatory sequence changes to human-specific cellular and circuit-level phenotypes via expression changes in select cell types.
Methods:
Tissue specimens from primate species
Human postmortem tissue specimens.
De-identified postmortem adult human brain tissue was obtained after receiving permission from the deceased’s next-of-kin. Tissue collection was performed in accordance with the provisions of the United States Uniform Anatomical Gift Act of 2006 described in the California Health and Safety Code section 7150 (effective 1/1/2008) and other applicable state and federal laws and regulations. The Western Institutional Review Board reviewed tissue collection procedures and determined that they did not constitute human subjects research requiring institutional review board (IRB) review.
Male and female individuals 18-68 years of age with no known history of neuropsychiatric or neurological conditions were considered for inclusion in the study. Routine serological screening for infectious disease (HIV, Hepatitis B, and Hepatitis C) was conducted using individual blood samples and individuals testing positive for infectious disease were excluded from the study. Specimens were screened for RNA quality and samples with average RNA integrity (RIN) values ≥7.0 were considered for inclusion in the study. Postmortem brain specimens were processed as previously described (17) (dx.doi.org/10.17504/protocols.io.bf4ajqse). Briefly, coronal brain slabs were cut at 1 cm intervals, frozen in dry-ice cooled isopentane, and transferred to vacuum-sealed bags for storage at −80°C until the time of further use. To isolate the MTG, tissue slabs were briefly transferred to −20°C and the region of interest was removed and subdivided into smaller blocks on a custom temperature controlled cold table. Tissue blocks were stored at −80°C in vacuum-sealed bags until later use.
Chimpanzee and gorilla tissue specimens.
Chimpanzee tissue was obtained from the National Chimpanzee Brain Resource (supported by NIH grant NS092988). Gorilla samples were collected postmortem following naturally occurring death or euthanasia of the animals for medical conditions at various zoos. Gorilla and chimpanzee brains were divided into 2 cm coronal slabs, flash frozen using dry-ice cooled isopentane, liquid nitrogen, or a −80°C freezer, and then stored in freezer bags at −80°C. Tissues from the MTG were removed from appropriate slabs which were maintained on dry-ice during dissection and were shipped to the Allen Institute overnight on dry-ice.
Macaque tissue specimens.
Macaque tissue samples were obtained from the University of Washington National Primate Resource Center under a protocol approved by the University of Washington Institutional Animal Care and Use Committee. Immediately following euthanasia, macaque brains were removed and transported to the Allen Institute in artificial cerebral spinal fluid equilibrated with 95% O2 and 5% CO2. Upon arrival at the Allen Institute, brains were divided down the midline and each hemisphere was subdivided coronally into 0.5 cm slabs. Slabs were flash frozen in dry-ice cooled isopentane, transferred to vacuum-sealed bags, and stored at −80°C. MTG was removed from brain slabs as described above for human tissues.
Marmoset tissue specimens.
Marmoset experiments were approved by and in accordance with Massachusetts Institute of Technology IACUC protocol number 051705020. Adult marmosets (1.5–2.5 years old, 3 individuals) were deeply sedated by intramuscular injection of ketamine (20–40 mg kg−1) or alfaxalone (5–10 mg kg−1), followed by intravenous injection of sodium pentobarbital (10–30 mg kg−1). When the pedal with-drawal reflex was eliminated and/or the respiratory rate was diminished, animals were trans-cardially perfused with ice-cold sucrose-HEPES buffer (78). Whole brains were rapidly extracted into fresh buffer on ice. Sixteen 2-mm coronal blocking cuts were rapidly made using a custom-designed marmoset brain matrix. Slabs were transferred to a dish with ice-cold dissection buffer (78). All regions were dissected using a marmoset atlas as reference (79), and were snap-frozen in liquid nitrogen or dry ice-cooled isopentane, and stored in individual microcentrifuge tubes at −80 °C.
Temporal lobe dissections targeted area TE3 and TPO on the lateral temporal surface. Though a true homology to catarhine MTG may not exist in marmosets, these areas in marmoset form part of the temporal lobe association cortex. Moreover, on the basis of tract tracing connectivity studies (80), TE3 and TPO participate in the ‘default mode network,’ a functionally coupled network of higher-order association cortex that includes MTG in other species (81). Cortical area DLPFC targeted the dorsolateral surface of PFC, approximately 2.5-3 mm from the frontal pole. ACC/PFCm included medial frontal cortex anterior to the genu of the corpus callosum. M1 dissections were stained with fluorescent Nissl and targeted the hand/trunk region. S1 like sampled all primary somatosensory areas (A3,A1/2). A1 dissections targeted primary auditory area but likely include some rostral and caudal parabelt cortex. V1 dissections were collected on the dorsal bank of the calcarine sulcus approximately 4-6 mm from the posterior pole.
Tissue processing and single nucleus RNA-sequencing
SMART-seq v4 nucleus isolation and sorting (human, chimpanzee, gorilla).
Vibratome sections of MTG blocks were stained with fluorescent Nissl permitting microdissection of individual cortical layers as previously described (dx.doi.org/10.17504/protocols.io.bq6ymzfw). Nucleus isolation was performed as described (dx.doi.org/10.17504/protocols.io.ztqf6mw). Briefly, single nucleus suspensions were stained with DAPI (4’,6-diamidino-2-phenylindole dihydrochloride, ThermoFisher Scientific, D1306) at a concentration of 0.1μg/ml. Controls were incubated with mouse IgG1k-PE Isotype control (BD Biosciences, 555749, 1:250 dilution) or DAPI alone. To discriminate between neuronal and non-neuronal nuclei, samples were stained with mouse anti-NeuN conjugated to PE (FCMAB317PE, EMD Millipore) at a dilution of 1:500. Single-nucleus sorting was carried out on either a BD FACSAria II SORP or BD FACSAria Fusion instrument (BD Biosciences) using a 130 μm nozzle and BD Diva software v8.0. A standard gating strategy based on DAPI and NeuN staining was applied to all samples as previously described (17). Doublet discrimination gates were used to exclude nuclei multiplets. Individual nuclei were sorted into 96-well plates, briefly centrifuged at 1000 rpm, and stored at −80°C.
SMART-seq v4 RNA-sequencing.
The SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara #634894) was used per the manufacturer’s instructions. Standard controls were processed with each batch of experimental samples as previously described. After reverse transcription, cDNA was amplified with 21 PCR cycles. The NexteraXT DNA Library Preparation (Illumina FC-131-1096) kit with NexteraXT Index Kit V2 Sets A-D (FC-131-2001, 2002, 2003, or 2004) was used for sequencing library preparation. Libraries were sequenced on an Illumina HiSeq 2500 instrument (Illumina HiSeq 2500 System, RRID:SCR_016383) using Illumina High Output V4 chemistry. The following instrumentation software was used during data generation workflow; SoftMax Pro v6.5; VWorks v11.3.0.1195 and v13.1.0.1366; Hamilton Run Time Control v4.4.0.7740; Fragment Analyzer v1.2.0.11; Mantis Control Software v3.9.7.19.
SMART-seq v4 gene expression quantification.
For human, raw read (fastq) files were aligned to the GRCh38 genome sequence (Genome Reference Consortium, 2011) with the RefSeq transcriptome version GRCh38.p2 (RefSeq, RRID:SCR_003496, current as of 4/13/2015) and updated by removing duplicate Entrez gene entries from the gtf reference file for STAR processing, as previously described (17). For chimpanzee and gorilla, the Clint_PTRv2 and Susie3 NCBI reference genomes were used for alignment, respectively. For alignment, Illumina sequencing adapters were clipped from the reads using the fastqMCF program (from ea-utils). After clipping, the paired-end reads were mapped using Spliced Transcripts Alignment to a Reference (STAR v2.7.3a, RRID:SCR_015899) using default settings. Reads that did not map to the genome were then aligned to synthetic construct (i.e. ERCC) sequences and the E. coli genome (version ASM584v2). Quantification was performed using summerizeOverlaps from the R package GenomicAlignments v1.18.0. Expression levels were calculated as counts per million (CPM) of exonic plus intronic reads.
10x RNA-seq (human, chimpanzee, gorilla, and macaque).
Nucleus isolation for 10x Chromium snRNAseq was conducted as described (dx.doi.org/10.17504/protocols.io.y6rfzd6). Gating was as described for SSv4 above. NeuN+ and NeuN- nuclei were sorted into separate tubes and were pooled at a defined ratio (90% NeuN+, 10% NeuN-) after sorting. Sorted samples were centrifuged, frozen in a solution of 1X PBS, 1% BSA, 10% DMSO, and 0.5% RNAsin Plus RNase inhibitor (Promega, N2611), and stored at −80°C until the time of 10x chip loading. Immediately before loading on the 10x Chromium instrument, frozen nuclei were thawed at 37°C, washed, and quantified for loading as described (dx.doi.org/10.17504/protocols.io.nx3dfqn). Samples were processed using the 10x Chromium Single-Cell 3’ Reagent Kit v3 following the manufacturer’s protocol. Gene expression was quantified using the default 10x Cell Ranger v3 (Cell Ranger, RRID:SCR_017344) pipeline. Reference genomes included the modified genome annotation described above for SMART-seq v4 quantification (human), Clint_PTRv2 (chimpanzee), Susie3 (gorilla), and Mmul_10 (rhesus macaque). Introns were annotated as “mRNA”, and intronic reads were included in expression quantification.
10x RNA-seq (marmoset).
Unsorted single-nucleus suspensions from frozen marmoset samples were generated as in (10). GEM generation and library preparation followed the manufacturer’s protocol (10X Chromium single-cell 3′ v.3, protocol version #CG000183_ ChromiumSingleCell3′_v3_UG_Rev-A). Raw sequencing reads were aligned to the CJ1700 reference. Reads that mapped to exons or introns were assigned to annotated genes.
RNA-sequencing processing and clustering
Cell type label transfer.
Human MTG and M1 reference taxonomy subclass labels (12, 21) were transferred to nuclei in the current MTG dataset using Seurat’s label transfer (3000 high variance genes using the ‘vst’ method then filtered through exclusion list). For human label mapping to other species, higher variance genes were included from a list of orthologous genes (14,870 genes; downloaded from NCBI Homologene (https://www.ncbi.nlm.nih.gov/homologene) in November 2019; RRID SCR_002924). This was carried out for each species and RNA-seq modality dataset; for example, human-Cv3 and human-SSv4 were labeled independently. Each dataset was subdivided into 5 neighborhoods – IT and Non-IT excitatory neurons, CGE- and MGE-derived interneurons, and non-neuronal cells – based on marker genes and transferred subclass labels from published studies of human and mouse cortical cell types and cluster grouping relationships in a reduced dimensional gene expression space. MTG and M1 subclass labels were highly consistent for all neighborhoods and species (adjusted Rand index 0.88 to 0.99), and a final set of labels was manually curated using additional information, such as layer dissections.
Filtering low-quality nuclei.
SSv4 nuclei were included for analysis if they passed all QC criteria:
> 30% cDNA longer than 400 base pairs
> 500,000 reads aligned to exonic or intronic sequence
> 40% of total reads aligned
> 50% unique reads
> 0.7 TA nucleotide ratio
QC was then performed at the neighborhood level. Neighborhoods were integrated together across all species and modality; for example, deep excitatory neurons from human-Cv3, human-SSv4, Chimp-Cv3, etc. datasets were integrated using Seurat integration functions with 2000 high variance genes from the orthologous gene list. Integrated neighborhoods were Louvain clustered into over 100 meta cells, and Low-quality meta cells were removed from the dataset based on relatively low UMI or gene counts (included glia and neurons with greater than 500 and 1000 genes detected, respectively), predicted doublets using DoubletFinder(82) and default parameters (included nuclei with doublet scores under 0.3), and/or subclass label prediction metrics within the neighborhood (ie excitatory labeled nuclei that clustered with majority inhibitory or non-neuronal nuclei).
RNA-seq clustering.
Nuclei were normalized using SCTransform (19), and neighborhoods were integrated together within a species and across individuals and modalities by identifying mutual nearest neighbor anchors and applying canonical correlation analysis as implemented in Seurat (18). For example, deep excitatory neurons from human-Cv3 were split by individual and integrated with the human-SSv4 deep excitatory neurons. Integrated neighborhoods were Louvain clustered into over 100 meta cells. Meta cells were then merged with their nearest neighboring meta cell until merging criteria were sufficed, a split and merge approach that has been previously described (12). The remaining clusters underwent further QC to exclude Low-quality and outlier populations. These exclusion criteria were based on irregular groupings of metadata features that resided within a cluster.
Robustness tests of cell subclasses using MetaNeighbor
MetaNeighbor v1.12 (38, 39) was used to provide a measure of neuronal and non-neuronal subclass and cluster replicability within and across species. We subset snRNA-seq datasets from each species to the list of common orthologs before further analysis. For each assessment, we identified highly variable genes using the get_variable_genes function from MetaNeighbor. In order to identify homologous cell types, we used the MetaNeighborUS function, with the fast_version and one_vs_best parameters set to TRUE. The one_vs_best parameter identifies highly specific cross-dataset matches by reporting the performance of the closest neighboring cell type over the second closest as a match for the training cell type, and the results are reported as the relative classification specificity (AUROC). This step identified highly replicable cell types within each species and across each species pair. All 24 subclasses are highly replicable within and across species (one_vs_best AUROC of 0.96 within species and 0.93 across species in Fig. S4A).
Defining cross-species consensus cell types
While cell type clusters are highly replicable within each species (one_vs_best AUROC of 0.93 for neurons and 0.87 for non-neurons), multiple transcriptionally similar clusters mapped to each other across each species pair (average cross-species one_vs_best AUROC of 0.76). To build a consensus cell type taxonomy across species, we defined a cross-species cluster as a group of clusters that are either reciprocal best hits or clusters with AUROC > 0.6 in the one_vs_best mode in at least one pair of species. This lower threshold (AUROC>0.6) reflects the high difficulty/specificity of testing only against the best performing other cell type. We identified 86 cross-species clusters, each containing clusters from at least two primates. Any unmapped clusters were assigned to one of the 86 cross-species clusters based on their transcriptional similarity. For each unmapped cluster, the top 10 of their closest neighbors are identified using MetaNeighborUS one_vs_all cluster replicability scores, and the unmapped cluster is assigned to the cross-species cluster in which a strict majority of its nearest neighbors belong. For clusters with no hits, this is repeated using the top 20 closest neighbors, still requiring a strict majority to assign a cross-species type. 594 clusters present in five primates (i.e., union) mapped to 86 cross-species clusters, with 493 clusters present across 57 consensus cross-species clusters shared by all five primates (Table S6). This is described in more detail in our companion manuscript (83). Additional sampling of species and developmental time points will be needed to distinguish between transcriptomic specializations of conserved cell types and the emergence of closely related but novel cell types. In this study, the 101 clusters with initial homologies across fewer than 5 species were assigned to the most similar of the 57 consensus types.
An alternative approach for consensus clustering was used to assess the robustness of homologous cell type clusters identified by MetaNeighbor. For each of the five cell type neighborhoods (non-neuronal, MGE- and CGE-derived interneurons, IT- and non-IT-projecting excitatory neurons), we built a reference with four primate datasets and used the fifth primate dataset as query for cell type annotation using scArches(40). We built each reference dataset using 2000 highly variable genes, trained a model on the reference using scPoli (84), and mapped the query cells onto the reference data (Fig. S12). scPoli learns a set of cell-type prototypes from the latent cell representation of the reference data (Fig. S12C,D). The cells in the query dataset are annotated based on their closest cell-type prototype in the reference data (Fig. S12E), and the classification uncertainty estimated by euclidean distance from this prototype (Fig. S12F). Query cells typically mapped to cell type prototypes identified in the reference data with low label transfer uncertainty, highlighting the robustness of the primate MTG consensus taxonomy. Cell type labels predicted by scPoli were largely consistent with the consensus cell types identified by MetaNeighbor (overall classification accuracy with scPoli = 0.74, average cell type classification accuracy = 0.68), although the classification accuracy varied with cell type neighborhood (ranging from 0.91 across glial cell types to 0.69 across IT-type excitatory neurons).
Cell type taxonomy generation
For each species, a taxonomy was built using the final set of clusters and was annotated using subclass mapping scores, dendrogram relationships, marker gene expression, and inferred laminar distributions. Within-species taxonomy dendrograms were generated using build_dend function from scrattch_hicat R package. A matrix of cluster median log2(cpm + 1) expression across the 3000 High-variance genes for Cv3 nuclei from a given species were used as input. The cross-species dendrogram was generated with a similar workflow but was downsampled to a maximum of 100 nuclei per cross-species cluster per species. The 3000 High-variance genes used for dendrogram construction were identified from the downsampled matrix containing Cv3 nuclei from all five species. We generated the complete cross-species cluster dendrogram using average-linkage hierarchical clustering with (1 - average MetaNeighborUS one_vs_all cluster replicability scores) for each pair of 86 cross-species clusters as a measure of distance between cell types.
Cell type comparisons across species
Differential gene expression.
To identify subclass marker genes within a species, Cv3 datasets from each species were downsampled to a maximum of 100 nuclei per cluster per individual. Differentially expressed marker genes were then identified using the FindAllMarkers function from Seurat, using the Wilcoxon sum rank test on log-normalized matrices with a maximum of 500 nuclei per group (subclass vs. all other nuclei as background). Statistical thresholds for markers are indicated in their respective figures. To identify species marker genes across subclasses and consensus cell types, Cv3 datasets from each species were downsampled to a maximum of 50 nuclei per cluster per individual. Downsampled counts matrices were then grouped into pseudo-bulk replicates (species, individual, subclass/consensus types) and the counts were summed per replicate. DESeq2 functionality was then used to perform a differential expression analysis between species pairs (or comparisons of interest) for each subclass/consensus type using the Wald test statistic.
Expression correlations.
Subclasses were compared between each pair of species using Spearman correlations on subclass median log2(cpm + 1) expression of orthologous genes that had a median value greater than 0 in both species. These Spearman correlations were then visualized as heatmaps and also compared to the human-centric evolutionary distance from each species in Figure 2. Similarly, subclasses were compared across individuals within each species, and the average Spearman correlation of all pairwise comparisons of individuals was calculated. Within species correlations were performed on orthologous genes with median values greater than 0 in all donors for a given subclass. Nuclei were downsampled to a maximum of 100 nuclei per subclass per donor for comparisons.
Taxonomy comparisons.
To assess homologies between clusters from taxonomies of different species or different studies, we constructed Euclidean distance heatmaps that were anchored on one side by the taxonomies’ dendrogram. The heatmaps display the cluster labels of a single taxonomy on either end, and the heatmap values represent the Euclidean distance between cluster centroids in the reduced dimensional space using 30-50 principal components from a PC analysis. In the case of cross-species comparisons, the reduced space was derived from Cv3 data. The -log(Euclidean distance) is plotted, with smaller values indicating more similar transcriptomic profiles.
Estimating differential isoform usage between great apes.
We used Smart-seq single-nucleus RNA-seq data from humans (~14,500 cells), chimpanzees (~3,500 cells), and gorillas (~4,300 cells) to assess isoform switching between the species for each cell subclass. The RNA-seq reads were mapped to each species’ genome using STAR as described above. The isoforms were quantified using RSEM on a common set of annotated transcripts (TransMap V5 downloaded from the UCSC browser, RRID:SCR_005780) by aggregating reads from cells in each cell subtype using a pseudo-bulk method:
Aggregated reads from cells in each subclass
Mapped reads to the human, chimpanzee, or gorilla reference genome with STAR 2.7.7a using default parameters
Transformed genomic coordinates into transcriptomic coordinates using STAR parameter: --quantMode TranscriptomeSAM
Quantified isoform and gene expression using RSEM v1.3.3 parameters (RSEM, RRID:SCR_013027): --bam --seed 12345 --paired-end --forward-prob 0.5 --single-cell-prior --calc-ci
The isoform proportion metric (isoP) was defined as the isoform expression (transcripts per million, TPM) normalized by the total expression of the gene the isoform belongs to. To focus on highly expressed genes, we considered only isoforms originating from the top 50% (ranked by gene expression) of genes for each species. To control the variability of isoP values, we derived the 80% confidence intervals by comparing the isoP-values of different donors for each species using the following procedure:
The isoP values (ranging from 0 to 1) for donor 1 are binned into 10 bins of size 0.1.
The isoforms in each bin are sorted by the isoP values in donor 2.
The lower and upper bounds of the 80% isoP confidence interval are defined as 10% and 90% percentile of this sorted list.
The procedure was repeated, switching donors 1 and 2, and the isoP confidence interval bounds values from the two calculations were averaged.
The isoform switching between species was considered significant for isoforms whose confidence intervals were non-overlapping. We defined cross-species isoform switches as those that involved a major isoform in one of the species (i.e., isoP > 0.5) and report them in Table S4. A subset of isoforms with strong cross-species switching were identified that had isoP > 0.7 in one species, isoP < 0.1 in the other species, and >3-fold change in proportions between the species.
Identifying changes in cell type proportions across species.
Cell type proportions are compositional, where the gain or loss of one population necessarily affects the proportions of the others, so we used scCODA (43) to determine which changes in cell class, subclass, and cluster proportions across species were statistically significant. We focused these analyses on neuronal populations since these were deeply sampled in all five species based on sorting of nuclei with NeuN immunostaining. The proportion of each neuronal class, subclass, and cluster was estimated using a Bayesian approach where proportion differences across individuals were used to estimate the posterior. All compositional and categorical analyses require a reference population to describe differences with respect to and, because we were uncertain which populations should be unchanged, we iteratively used each cell type and each species as a reference when computing abundance changes. To account for sex differences, we included it as a covariate when testing for abundance changes. We report the effect size of each species and sex for each cell subclass and used a mean inclusion probability cutoff of 0.7 for calling a population credibly different.
In situ profiling of gene expression
MERFISH data collection.
Human postmortem frozen brain tissue was embedded in Optimum Cutting Temperature medium (VWR,25608-930) and sectioned on a Leica cryostat at −17 C at 10 um onto Vizgen MERSCOPE coverslips (VIZGEN 2040003). These sections were then processed for MERSCOPE imaging according to the manufacturer’s instructions. Briefly: sections were allowed to adhere to these coverslips at room temperature for 10 min prior to a 1 min wash in nuclease-free phosphate buffered saline (PBS) and fixation for 15 min in 4% paraformaldehyde in PBS. Fixation was followed by 3×5 minute washes in PBS prior to a 1 min wash in 70% ethanol. Fixed sections were then stored in 70% ethanol at 4 C prior to use and for up to one month. Human sections were photobleached using a 150W LED array for 72 h at 4 C prior to hybridization then washed in 5 ml Sample Prep Wash Buffer (VIZGEN 20300001) in a 5 cm petri dish. Sections were then incubated in 5 ml Formamide Wash Buffer (VIZGEN 20300002) at 37 C for 30 min. Sections were hybridized by placing 50 ul of VIZGEN-supplied Gene Panel Mix onto the section, covering with parafilm and incubating at 37 C for 36-48 h in a humidified hybridization oven.
Following hybridization, sections were washed twice in 5 ml Formamide Wash Buffer for 30 min at 47 C. Sections were then embedded in acrylamide by polymerizing VIZGEN Embedding Premix (VIZGEN 20300004) according to the manufacturer’s instructions. Sections were embedded by inverting sections onto 110 ul of Embedding Premix and 10% Ammonium Persulfate (Sigma A3678) and TEMED (BioRad 161-0800) solution applied to a Gel Slick (Lonza 50640) treated 2×3 glass slide. The coverslips were pressed gently onto the acrylamide solution and allowed to polymerize for 1.5 h. Following embedding, sections were cleared for 24-48 h with a mixture of VIZGEN Clearing Solution (VIZGEN 20300003) and Proteinase K (New England Biolabs P8107S) according to the Manufacturer’s instructions. Following clearing, sections were washed twice for 5 min in Sample Prep Wash Buffer (PN 20300001). VIZGEN DAPI and PolyT Stain (PN 20300021) was applied to each section for 15 min followed by a 10 min wash in Formamide Wash Buffer. Formamide Wash Buffer was removed and replaced with Sample Prep Wash Buffer during MERSCOPE set up. 100 ul of RNAse Inhibitor (New England BioLabs M0314L) was added to 250 ul of Imaging Buffer Activator (PN 203000015) and this mixture was added via the cartridge activation port to a pre-thawed and mixed MERSCOPE Imaging cartridge (VIZGEN PN1040004). 15 ml mineral oil (Millipore-Sigma m5904-6X500ML) was added to the activation port and the MERSCOPE fluidics system was primed according to VIZGEN instructions. The flow chamber was assembled with the hybridized and cleared section coverslip according to VIZGEN specifications and the imaging session was initiated after collection of a 10X mosaic DAPI image and selection of the imaging area. For specimens that passed the minimum count threshold, imaging was initiated and processing completed according to VIZGEN proprietary protocol.
The 140 gene Human cortical panel was selected using a combination of manual and algorithmic based strategies requiring a reference single cell/nucleus RNA-seq data set from the same tissue, in this case the human MTG snRNAseq dataset and resulting taxonomy(Hodge and Bakken 2019). First, an initial set of High-confidence marker genes are selected through a combination of literature search and analysis of the reference data. These genes are used as input for a greedy algorithm (detailed below). Second, the reference RNA-seq data set is filtered to only include genes compatible with mFISH. Retained genes need to be 1) long enough to allow probe design (> 960 base pairs); 2) expressed highly enough to be detected (FPKM >= 10), but not so high as to overcrowd the signal of other genes in a cell (FPKM < 500); 3) expressed with low expression in off-target cells (FPKM < 50 in non-neuronal cells); and 4) differentially expressed between cell types (top 500 remaining genes by marker score20). To more evenly sample each cell type, the reference data set is also filtered to include a maximum of 50 cells per cluster.
The spatial distribution of human MTG cell types was estimated from several sections from two donors. For each section, we made two manual annotations: a parallelogram spanning pia to white matter that selected cells from all cortical layers and a line segment from pia to white matter along the local cortical column axis. The cortical depth was calculated as the projection of the coordinates of the selected cells onto the cortical column axis. Annotations were done in napari using a notebook: https://github.com/AllenInstitute/Great_Ape_MTG/blob/master/cell_type_mapping/Great_apes_subsetting_cortical_depth.ipynb.
Cell type mapping in MERSCOPE data.
Any genes not matched across both the MERSCOPE gene panel and the snRNASeq mapping taxonomy were filtered from the snRNASeq dataset. We calculated the mean gene expression for each gene in each snRNAseq cluster. We assigned MERSCOPE cells to snRNAseq clusters by finding the nearest cluster to the mean expression vectors of the snRNASeq clusters using the cosine distance. All scripts and data used are available at: https://github.com/AllenInstitute/Great_Ape_MTG.
The main step of gene selection uses a greedy algorithm to iteratively add genes to the initial set. To do this, each cell in the filtered reference data set is mapped to a cell type by taking the Pearson correlation of its expression levels with each cluster median using the initial gene set of size n, and the cluster corresponding to the maximum value is defined as the “mapped cluster”. The “mapping distance” is then defined as the average cluster distance between the mapped cluster and the originally assigned cluster for each cell. In this case a weighted cluster distance, defined as one minus the Pearson correlation between cluster medians calculated across all filtered genes, is used to penalize cases where cells are mapped to very different types, but an unweighted distance, defined as the fraction of cells that do not map to their assigned cluster, could also be used. This mapping step is repeated for every possible n+1 gene set in the filtered reference data set, and the set with minimum cluster distance is retained as the new gene set. These steps are repeated using the new get set (of size n+1) until a gene panel of the desired size is attained. Code for reproducing this gene selection strategy is available as part of the mfishtools R library (https://github.com/AllenInstitute/mfishtools).
We used our 140 gene MERFISH panel designed to identify human cortical cell types to map every type described in this updated human MTG taxonomy to determine cell type locations within cortex and confirm cell type proportions. All cell type locations are provided for reference in graphical format as localized in a representative human MTG section H19.30.001.Cx46.MTG.02.02.007.5 (Supplementary Media 1).
RNAscope.
Fresh-frozen human postmortem brain tissues were sectioned at 16-25 μm onto Superfrost Plus glass slides (Fisher Scientific). Sections were dried for 20 minutes on dry ice and then vacuum sealed and stored at −80°C until use. The RNAscope multiplex fluorescent V2 kit was used per the manufacturer’s instructions for fresh-frozen tissue sections (ACD Bio), except that slides were fixed 60 minutes in 4% paraformaldehyde in 1X PBS at 4°C and treated with protease for 15 minutes. Sections were imaged using a 40X oil immersion lens on a Nikon TiE fluorescence microscope equipped with NIS-Elements Advanced Research imaging software (v4.20, RRID:SCR_014329). Positive cells were called by manual assessment of RNA spots for each gene. Cells were called positive for a gene if they contained ≥ 5 RNA spots for that gene. High versus low expression of CUX2 was determined by measuring fluorescence intensity for that gene in ImageJ. Lipofuscin autofluorescence was distinguished from RNA spot signal based on the broad fluorescence spectrum and larger size of lipofuscin granules. Staining for each probe combination was repeated with similar results on at least 2 separate individuals and on at least 2 sections per individual. Images were assessed with the FIJI distribution of ImageJ v1.52p and with NIS-Elements v4.20. RNAscope probes used were CUX2 (ACD Bio, 425581-C3), LDB2 (1003951-C2), and SMYD1 (493951-C2).
Fresh-frozen marmoset brain tissue was sectioned and processed for RNAscope staining as described above for human. Sections were imaged with a 10X lens on a Nikon TiE fluorescence microscope to collect large overview images and smaller regions of tissue were re-imaged using a 40X oil immersion lens. Images were assessed as above for human except that lipofuscin autofluorescence was not apparent in marmoset tissues. RNAscope probes used were CUX2 (ACD Bio, 554631-C2), NTNG2 (ACD Bio, custom probe targeting base pairs 1894-2819 of XM_035261022.2), and MGAT4C (custom probe targeting base pairs 704-1799 of XM_035257223.2). Staining for this probe combination was repeated on 3 sections from one individual. On all sections, an area of probe signal dropout was noted at the same location in the secondary auditory cortex that we attribute to a potential imaging or experimental artifact. All 3 probes had reduced signal intensity in this area and the area is marked in the figure panel displaying the RNAscope data (Fig. 2F) with a red asterisk.
Analysis of great ape species pairwise comparison for glial cells.
We used 10x single nucleus RNA-seq data for the comparison of normalized gene expression across species. Significant differential gene expression in pairwise comparisons of glial cells (astrocytes, microglia, oligodendrocytes) across great ape species was determined at log2 fold-change > 0.5 and FDR < 0.01. Among DEGs from great ape pairwise comparisons, species-specific highly divergent genes were identified as having >10 fold change in expression in a given species relative to the other two great ape species, and with a threshold of gene expression of normalized gene counts >5 in at least one species. GO enrichment analysis was performed using the Bioconductor package ‘clusterProfiler’ (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html), and the Fisher’s exact test was used for SynGO enrichment analysis (https://www.syngoportal.org/). GO and SynGO analyses were performed on the union of DEGs from the pairwise comparison between human and chimpanzee and the pairwise comparison between human and gorilla to increase power to detect significant GO terms. GO terms under biological process, molecular function, and cellular component categories were considered in the analysis. Significance for enriched terms was determined at 5% FDR. All MTG expressed genes in the consensus cell types (astrocytes, microglia, oligodendrocytes) were considered as the background gene set in the respective analyses. Gene expression change in glial cell types shown in heatmaps (Figures 3E; S5A,B,G; S6E,F) is calculated as the log2 ratio of normalized expression counts in a given species relative to the other two great ape species. To analyze astrocyte genes associated with perisynaptic astrocytic processes, a list of genes encoding proteins enriched at astrocyte-neuron junctions was used from a proteomic study in the mouse cortex (29). To analyze microglia genes associated with intercellular communication and signaling, a list of genes predicted to act as the ligand-receptor interactome of microglia-neuron communication was used from a recent study in the mouse cortex (85).
Enrichment of HARs and hCONDELs near hDEGs.
The set of HARs used in our analysis was obtained from (14), and the set of HAQERs used in our analysis was obtained from (47). The set of hCONDELs was obtained from (45, 46), and only hCONDELs that could be mapped to a syntenic orthologous location in hg38 were retained (1175 total) (86). We assigned intronic HARs/HAQERs/hCONDELs to the genes they are intronic to and intergenic HARs/HAQERs/hCONDELs to the closest upstream and downstream genes (Table S8) using Ensembl GRCh38 annotations obtained in May 2021 and Ensembl Pan_tro_3.0, gorGor4, and Mmul_10 annotations obtained in January 2023. With respect to the human annotations, 63.2% of HARs, 53.7% of HAQERs, and 59.4% of hCONDELs are intronic. For 83.2% of the 1165 intergenic HARs, at least one of their assigned genes is within 100kb. For 90.7% of the 732 intergenic HAQERs, at least one of their assigned genes is within 100kb. For 85.5% of the 477 intergenic hCONDELs, at least one of their assigned genes is within 100kb. The proportion of intronic and intergenic HARs and hCONDELs is similar for the chimpanzee, gorilla, and macaque annotations. We considered HARs/HAQERs/hCONDELs to be enriched near DEGs in a specific cell type if they are significant at 5% FDR for both of the following tests (87): (1) Are DEGs enriched for genes near HARs, HAQERs, and/or hCONDELs (Fisher’s exact test)? We set the background as expressed genes, which adjusts for the fact that HARs, HAQERs, and hCONDELs are known to be enriched near neural genes. (2) Are HARs, HAQERs, and/or hCONDELs more likely to fall near DEGs than expected by chance? We assign each gene a regulatory domain that comprises the genomic interval containing the gene, along with the upstream and downstream intergenic regions that extend to the nearest flanking genes, with an upper bound of 5 Mb in total size. We then ask whether HARs/HAQERs/hCONDELs are enriched within the regulatory domains of DEGs using the binomial test. This takes into account differences in genomic structure between genes, under the assumption that HARs/HAQERs/hCONDELs will be more likely to fall within the regulatory domains of genes with large intronic or flanking regions by chance.
SynGO and synaptic gene family enrichment.
To analyze the association between synaptic terms and human divergent gene expression patterns, we used an expert-curated database of GO annotations of synapse-related terms known as SynGO (28). To test whether hDEGs and hDEGs near HARs/hCONDELs are enriched in SynGO terms, we used Fisher’s exact test. We focused on SynGO terms within the first and second hierarchical levels of SynGO that broadly comprise the entire range of Cellular Components (CC) and Biological Processes (BP) terms, allowing for the visualization of enrichment patterns across a wide range of synaptic localizations and processes (Fig. S18). We grouped SynGO terms into two levels based on their hierarchical organization in SynGO (https://www.syngoportal.org/), corresponding to the following reference codes: 11 terms within level 1 (A1, B1, C1, D1, E1, F1, G1, H1, I1, J1, K1), and 71 terms within level 2 (A2-3, B2-11, C2, D2-11, E2, F2-10, G2-7, H2-4, I2-15, J2-11, K2-6). For synaptic gene families, we examined 15 functionally related categories: (1) families of cell-adhesion and synaptic-adhesion molecules, (2) families of ligand-receptor complexes involved in growth factor signaling, (3) families of other cell-surface receptors and ligands, (4) families of other G protein-coupled receptors (GPCRs) and their ligands (including orphan GPCRs), (5) families of ligand-receptor complexes involved in neuropeptidergic signaling and related GPCRs and ligands, (6) families of neurotransmitter-gated receptors and other ligand-gated receptors (including glutamate ionotropic receptors), (7) Ras GTPase superfamily, (8) families of Ras GAP and GEF signaling molecules, (9) families of other regulatory molecules and structural scaffolding proteins, (10) families related to other signaling complexes including intracellular kinases and phosphatases, (11) families related to the extracellular matrix (ECM) and proteoglycan families, (12) families related to cytoskeletal composition and organization and other related proteins, (13) families involved in synaptic vesicle exocytosis and other membrane fusion components, (14) families of proteases and peptidases, (15) families of voltage-gated ion channels and other gated ion channels and solute transporters. For each of these, we assembled a comprehensive list based on HGNC reference and a previously-curated catalog of synaptic molecules (88) (Table S8). Significance was determined at 5% FDR. “All” genes are genes that are expressed and can be assessed for differential expression by DESeq2 in at least one consensus type.
Supplementary Material
Table S1: Within-species cluster proportions across SMARTseq cortical layer dissections. Estimates of laminar distributions of clusters in human, chimpanzee, and gorilla based on the relative proportions of nuclei dissected from individual cortical layers.
Table S2: Within-species subclass marker genes. Prior to DEG analysis, datasets were downsampled to a maximum of 100 nuclei per individual per cluster (finer curation resolution than subclass) in the Cv3 only samples. DEG analysis was performed on log-normalized gene expression matrices. The gene list was generated by implementing Seurat’s FindAllMarkers function using the Wilcoxon sum rank test on a maximum of 500 nuclei per group (subclass versus all other cell types) within each species. Genes with Bonferroni adjusted p-values less than 0.05 were retained. This list is not filtered for orthologous genes and reflects the species-specific gene symbol.
Table S3: Subclass differential gene expression across great apes. Gene list of pairwise differential expression comparing human, chimpanzee, and gorilla for each subclass. The gene list was generated by implementing a pseudobulk approach across individuals with DESeq2 using Ward’s test on downsampled datasets with a maximum 50 nuclei per cluster per individual per species for Cv3 nuclei using one-to-one orthologous genes. Genes with Bonferroni adjusted p-values less than 0.05 were retained.
Table S4: Subclass isoform switching across great apes. Relative isoform abundances were compared between human, chimpanzee, and gorilla for one-to-one orthologous genes with at least moderate expression. Gene expression and isoform proportions are reported for two individuals from each species that had SSv4 data. The column ‘Large_prop_change’ indicates if one species expresses a larger proportion (> 0.7, > 3-fold change on average across individuals) of a transcript than the other species (proportion < 0.1).
Table S5: SST consensus type markers across species. List of marker genes that are differentially expressed in each of 9 SST-expressing consensus types relative to the other types. Markers were independently generated for each species. The gene list was generated by implementing the Wilcoxon sum rank test with Seurat’s FindAllMarkers function on downsampled datasets with a maximum 100 nuclei per cross-species cluster per individual for Cv3 nuclei.
Table S6: Consensus type cluster membership. 86 cross-species clusters included clusters from two or more species and were merged to give a final set of 57 consensus types with homologous clusters from all 5 primates.
Table S7: Consensus type species-specific expression. Gene list of differential gene expression comparing each species to the other four species for each consensus type. The gene list was generated by implementing a pseudobulk approach across individuals with DESeq2 using Ward’s test on downsampled datasets with a maximum 50 nuclei per cluster per individual per species for Cv3 nuclei using one-to-one orthologous genes. Genes with Bonferroni adjusted p-values less than 0.05 were retained.
Table S8. Analysis of hDEGs near HARs or hCONDELs. Lists of hCONDEL coordinates, hDEGs in consensus types that are near HARs or hCONDELs, potential HAR-hDEG interactions from existing PLAC-Seq datasets, and manually curated synapse-related gene families. This table also includes statistics on the enrichment of hDEGs, and hDEGs near HARs or hCONDELs, for SynGO and for manually curated lists of synapse-related gene families.
Galleries of the spatial distributions of human MTG cell types. A representative human MTG section indicating spatial locations of 151 cell types grouped by subclass. Upper left images of each subclass show all types within the subtype. Note that for the rare types PVALB_3, PVALB_8, and SST_4, a mapped cell did not occur within the example section or was obscured by other cells in the section. For these 3 types, the cell locations were approximated based on laminar positions in other sections. L1: Cortical Layer 1; L2: Cortical Layer 2; L3: Cortical Layer 3; L4: Cortical Layer 4; L5: Cortical Layer 5; L6: Cortical Layer 6; WM: White Matter.
Acknowledgements:
We thank the tissue procurement, tissue processing and facilities teams at the Allen Institute for Brain Science for assistance with the transport and processing of postmortem and neurosurgical brain specimens; the technology team at the Allen Institute for assistance with data management; Cassandra Sobieski for assistance with RNAscope data generation; M. Vawter, J. Davis and the San Diego Medical Examiner’s Office for assistance with postmortem tissue donations. We thank M. Hawrylycz for improvements to taxonomy visualizations. This publication was supported by and coordinated through BICCN. This publication is part of the Human Cell Atlas- www.humancellatlas.org/publications/. Research reported in this publication was supported by the National Institute Of Mental Health of the National Institutes of Health under Award Numbers U01MH114812 and U19MH114830. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank the founder of the Allen Institute, Paul G. Allen, for his vision, encouragement and support.
Funding:
Allen Discovery Center for Human Brain Evolution (CAW, DEA, JHS, MB)
Dutch Research Council (NWO) Applied and Engineering Sciences (AES) grant 3DOMICS 17126 (BPL, JE, TH, TK)
Dutch Research Council (NWO) Gravitation Program BRAINSCAPES 024.004.012 (BPL, SB, TH)
EMBO Postdoctoral Fellowship (ALTF 336-2022) (DEA)
Helen Hay Whitney Fellowship (JHS)
Nancy and Buster Alvord Endowment (CDK)
NARSAD Young Investigator Award (MC)
National Institutes of Health grant F32MH114501 (MC)
National Institutes of Health grant HG011641 (CCS, WDH)
National Institutes of Health grant NS092988 (CCS, WDH)
National Institutes of Health grant R01 HG009318 (AD)
National Institutes of Health grant R01LM012736 (HS, JGi)
National Institutes of Health grant R01MH113005 (HS, JGi)
National Institutes of Health grant U01MH114812 (AMY, AT, CR, DB, DM, ESL, JGo, KSi, KSm, KW, MT, NLJ, RDH, SD, SL, TEB, TP)
National Institutes of Health grant U01MH114819 (FMK, GF, SAM)
National Institutes of Health grant U19MH114821 (HS, JGi)
National Institutes of Health grant U19MH114830 (AG, AT, DB, DM, JGo, KSm, KW, MT, TP)
NSF EF-2021785 (CCS, WDH)
Y. Eva Tan Postdoctoral Fellowship (JHS)
Footnotes
Competing interests: From April 11, 2022, N.L.J. is an employee of Genentech.
Data and materials availability:
Raw sequence data were produced as part of the BRAIN Initiative Cell Census Network (BICCN: RRID:SCR_015820) are available for download from the Neuroscience Multi-omics Archive (RRID:SCR_016152; https://assets.nemoarchive.org/dat-net1412) and the Brain Cell Data Center (RRID:SCR_017266; https://biccn.org/data). Code for analysis and generation of figures is available for download from https://github.com/AllenInstitute/Great_Ape_MTG. Visualization and analysis tools for integrated species comparison are available using Cytosplore Viewer (RRID SCR_018330; https://viewer.cytosplore.org/). These tools allow comparison of gene expression in consensus clusters across species, as well as species-specific clusters and to calculate differential expression within and among species. The following publicly available datasets were used for analysis: Synaptic Gene Ontology (SynGO) and orthologous genes across species from NCBI Homologene (downloaded November 2019). MTG human SMARTseq v4 data (https://portal.brain-map.org/atlases-and-data/rnaseq/human-mtg-smart-seq, https://assets.nemoarchive.org/dat-swzf4kc).
References:
- 1.Papeo L, Agostini B, Lingnau A, The Large-Scale Organization of Gestures and Words in the Middle Temporal Gyrus. J. Neurosci 39, 5966–5974 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Özyürek A, Hearing and seeing meaning in speech and gesture: insights from brain and behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozlenkov A, Vermunt MW, Apontes P, Li J, Hao K, Sherwood CC, Hof PR, Ely JJ, Wegner M, Mukamel EA, Creyghton MP, Koonin EV, Dracheva S, Evolution of regulatory signatures in primate cortical neurons at cell-type resolution. Proc. Natl. Acad. Sci. U. S. A 117, 28422–28432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roumazeilles L, Eichert N, Bryant KL, Folloni D, Sallet J, Vijayakumar S, Foxley S, Tendler BC, Jbabdi S, Reveley C, Verhagen L, Dershowitz LB, Guthrie M, Flach E, Miller KL, Mars RB, Longitudinal connections and the organization of the temporal cortex in macaques, great apes, and humans. PLoS Biol. 18, e3000810 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ardesch DJ, Scholtens LH, Li L, Preuss TM, Rilling JK, van den Heuvel MP, Evolutionary expansion of connectivity between multimodal association areas in the human brain compared with chimpanzees. Proc. Natl. Acad. Sci. U. S. A 116, 7101–7106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khrameeva E, Kurochkin I, Han D, Guijarro P, Kanton S, Santel M, Qian Z, Rong S, Mazin P, Sabirov M, Bulat M, Efimova O, Tkachev A, Guo S, Sherwood CC, Camp JG, Pääbo S, Treutlein B, Khaitovich P, Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 30, 776–789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Z, Han D, Efimova O, Guijarro P, Yu Q, Oleksiak A, Jiang S, Anokhin K, Velichkovsky B, Grünewald S, Khaitovich P, Comprehensive transcriptome analysis of neocortical layers in humans, chimpanzees and macaques. Nat. Neurosci 20, 886–895 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Berto S, Mendizabal I, Usui N, Toriumi K, Chatterjee P, Douglas C, Tamminga CA, Preuss TM, Yi SV, Konopka G, Accelerated evolution of oligodendrocytes in the human brain. Proc. Natl. Acad. Sci. U. S. A 116, 24334–24342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma S, Skarica M, Li Q, Xu C, Risgaard RD, Tebbenkamp ATN, Mato-Blanco X, Kovner R, Krsnik Ž, de Martin X, Luria V, Martí-Pérez X, Liang D, Karger A, Schmidt DK, Gomez-Sanchez Z, Qi C, Gobeske KT, Pochareddy S, Debnath A, Hottman CJ, Spurrier J, Teo L, Boghdadi AG, Homman-Ludiye J, Ely JJ, Daadi EW, Mi D, Daadi M, Marín O, Hof PR, Rasin M-R, Bourne J, Sherwood CC, Santpere G, Girgenti MJ, Strittmatter SM, Sousa AMM, Sestan N, Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science, eabo7257 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krienen FM, Goldman M, Zhang Q, Del Rosario RCH, Florio M, Machold R, Saunders A, Levandowski K, Zaniewski H, Schuman B, Wu C, Lutservitz A, Mullally CD, Reed N, Bien E, Bortolin L, Fernandez-Otero M, Lin JD, Wysoker A, Nemesh J, Kulp D, Burns M, Tkachev V, Smith R, Walsh CA, Dimidschstein J, Rudy B, Kean LS, Berretta S, Fishell G, Feng G, McCarroll SA, Innovations present in the primate interneuron repertoire. Nature. 586, 262–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang R, Xia C, Zhang M, He J, Close J, Long B, Miller J, Lein E, Zhuang X, Conservation and divergence in cortical cellular organization between human and mouse revealed by single-cell transcriptome imaging. bioRxiv (2021), p. 2021.11.01.466826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, Crow M, Hodge RD, Krienen FM, Sorensen SA, Eggermont J, Yao Z, Aevermann BD, Aldridge AI, Bartlett A, Bertagnolli D, Casper T, Castanon RG, Crichton K, Daigle TL, Dalley R, Dee N, Dembrow N, Diep D, Ding S-L, Dong W, Fang R, Fischer S, Goldman M, Goldy J, Graybuck LT, Herb BR, Hou X, Kancherla J, Kroll M, Lathia K, van Lew B, Li YE, Liu CS, Liu H, Lucero JD, Mahurkar A, McMillen D, Miller JA, Moussa M, Nery JR, Nicovich PR, Niu S-Y, Orvis J, Osteen JK, Owen S, Palmer CR, Pham T, Plongthongkum N, Poirion O, Reed NM, Rimorin C, Rivkin A, Romanow WJ, Sedeño-Cortés AE, Siletti K, Somasundaram S, Sulc J, Tieu M, Torkelson A, Tung H, Wang X, Xie F, Yanny AM, Zhang R, Ament SA, Behrens MM, Bravo HC, Chun J, Dobin A, Gillis J, Hertzano R, Hof PR, Höllt T, Horwitz GD, Keene CD, Kharchenko PV, Ko AL, Lelieveldt BP, Luo C, Mukamel EA, Pinto-Duarte A, Preissl S, Regev A, Ren B, Scheuermann RH, Smith K, Spain WJ, White OR, Koch C, Hawrylycz M, Tasic B, Macosko EZ, McCarroll SA, Ting JT, Zeng H, Zhang K, Feng G, Ecker JR, Linnarsson S, Lein ES, Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 598, 111–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foley NM, Mason VC, Harris AJ, Bredemeyer KR, Damas J, Lewin HA, Eizirik E, Gatesy J, Karlsson EK, Lindblad-Toh K, Consortium Z, Springer MS, Murphy WJ, Andrews G, Armstrong JC, Bianchi M, Birren BW, Bredemeyer KR, Breit AM, Christmas MJ, Clawson H, Damas J, Palma FD, Diekhans M, Dong MX, Eizirik E, Fan K, Fanter C, Foley NM, Forsberg-Nilsson K, Garcia CJ, Gatesy J, Gazal S, Genereux DP, Goodman L, Grimshaw J, Halsey MK, Harris AJ, Hickey G, Hiller M, Hindle AG, Hubley RM, Hughes GM, Johnson J, Juan D, Kaplow IM, Karlsson EK, Keough KC, Kirilenko B, Koepfli K-P, Korstian JM, Kowalczyk A, Kozyrev SV, Lawler AJ, Lawless C, Lehmann T, Levesque DL, Lewin HA, Li X, Lind A, Lindblad-Toh K, Mackay-Smith A, Marinescu VD, Marques-Bonet T, Mason VC, Meadows JRS, Meyer WK, Moore JE, Moreira LR, Moreno-Santillan DD, Morrill KM, Muntané G, Murphy WJ, Navarro A, Nweeia M, Ortmann S, Osmanski A, Paten B, Paulat NS, Pfenning AR, Phan BN, Pollard KS, Pratt HE, Ray DA, Reilly SK, Rosen JR, Ruf I, Ryan L, Ryder OA, Sabeti PC, Schäffer DE, Serres A, Shapiro B, Smit AFA, Springer M, Srinivasan C, Steiner C, Storer JM, Sullivan KAM, Sullivan PF, Sundström E, Supple MA, Swofford R, Talbot J-E, Teeling E, Turner-Maier J, Valenzuela A, Wagner F, Wallerman O, Wang C, Wang J, Weng Z, Wilder AP, Wirthlin ME, Xue JR, Zhang X, A genomic timescale for placental mammal evolution. Science. 380, eabl8189 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girskis KM, Stergachis AB, DeGennaro EM, Doan RN, Qian X, Johnson MB, Wang PP, Sejourne GM, Nagy MA, Pollina EA, Sousa AMM, Shin T, Kenny CJ, Scotellaro JL, Debo BM, Gonzalez DM, Rento LM, Yeh RC, Song JHT, Beaudin M, Fan J, Kharchenko PV, Sestan N, Greenberg ME, Walsh CA, Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron. 109, 3239–3251.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O’Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK, Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song M, Pebworth M-P, Yang X, Abnousi A, Fan C, Wen J, Rosen JD, Choudhary MNK, Cui X, Jones IR, Bergenholtz S, Eze UC, Juric I, Li B, Maliskova L, Lee J, Liu W, Pollen AA, Li Y, Wang T, Hu M, Kriegstein AR, Shen Y, Cell-type-specific 3D epigenomes in the developing human cortex. Nature. 587, 644–649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding S-L, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, Conserved cell types with divergent features in human versus mouse cortex. Nature. 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell. 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berg J, Sorensen SA, Ting JT, Miller JA, Chartrand T, Buchin A, Bakken TE, Budzillo A, Dee N, Ding S-L, Gouwens NW, Hodge RD, Kalmbach B, Lee C, Lee BR, Alfiler L, Baker K, Barkan E, Beller A, Berry K, Bertagnolli D, Bickley K, Bomben J, Braun T, Brouner K, Casper T, Chong P, Crichton K, Dalley R, de Frates R, Desta T, Lee SD, D’Orazi F, Dotson N, Egdorf T, Enstrom R, Farrell C, Feng D, Fong O, Furdan S, Galakhova AA, Gamlin C, Gary A, Glandon A, Goldy J, Gorham M, Goriounova NA, Gratiy S, Graybuck L, Gu H, Hadley K, Hansen N, Heistek TS, Henry AM, Heyer DB, Hill D, Hill C, Hupp M, Jarsky T, Kebede S, Keene L, Kim L, Kim M-H, Kroll M, Latimer C, Levi BP, Link KE, Mallory M, Mann R, Marshall D, Maxwell M, McGraw M, McMillen D, Melief E, Mertens EJ, Mezei L, Mihut N, Mok S, Molnar G, Mukora A, Ng L, Ngo K, Nicovich PR, Nyhus J, Olah G, Oldre A, Omstead V, Ozsvar A, Park D, Peng H, Pham T, Pom CA, Potekhina L, Rajanbabu R, Ransford S, Reid D, Rimorin C, Ruiz A, Sandman D, Sulc J, Sunkin SM, Szafer A, Szemenyei V, Thomsen ER, Tieu M, Torkelson A, Trinh J, Tung H, Wakeman W, Waleboer F, Ward K, Wilbers R, Williams G, Yao Z, Yoon J-G, Anastassiou C, Arkhipov A, Barzo P, Bernard A, Cobbs C, de Witt Hamer PC, Ellenbogen RG, Esposito L, Ferreira M, Gwinn RP, Hawrylycz MJ, Hof PR, Idema S, Jones AR, Keene CD, Ko AL, Murphy GJ, Ng L, Ojemann JG, Patel AP, Phillips JW, Silbergeld DL, Smith K, Tasic B, Yuste R, Segev I, de Kock CPJ, Mansvelder HD, Tamas G, Zeng H, Koch C, Lein ES, Human neocortical expansion involves glutamatergic neuron diversification. Nature. 598, 151–158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorstad NL, Close J, Johansen N, Yanny AM, Barkan ER, Travaglini KJ, Bertagnolli D, Campos J, Casper T, Crichton K, Dee N, Ding S-L, Gelfand E, Goldy J, Hirschstein D, Kroll M, Kunst M, Lathia K, Long B, Martin N, McMillen D, Pham T, Rimorin C, Ruiz A, Shapovalova N, Shehata S, Siletti K, Somasundaram S, Sulc J, Tieu M, Torkelson A, Tung H, Ward K, Callaway EM, Hof PR, Dirk Keene C, Levi BP, Linnarsson S, Mitra PP, Smith K, Hodge RD, Bakken TE, Lein ES, Transcriptomic cytoarchitecture reveals principles of human neocortex organization. bioRxiv (2022), p. 2022.11.06.515349. [DOI] [PubMed] [Google Scholar]
- 22.Krienen FM, Levandowski KM, Zaniewski H, del Rosario RCH, Schroeder ME, Goldman M, Lutservitz A, Zhang Q, Li KX, Beja-Glasser VF, Sharma J, Shin TW, Mauermann A, Wysoker A, Nemesh J, Kashin S, Vergara J, Chelini G, Dimidschstein J, Berretta S, Boyden E, McCarroll SA, Feng G, A marmoset brain cell census reveals persistent influence of developmental origin on neurons. bioRxiv (2022), p. 2022.10.18.512442. [Google Scholar]
- 23.Liu G, Li W, Gao X, Li X, Jürgensen C, Park H-T, Shin N-Y, Yu J, He M-L, Hanks SK, Wu JY, Guan K-L, Rao Y, p130CAS is required for netrin signaling and commissural axon guidance. J. Neurosci 27, 957–968 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayala R, Willhoft O, Aramayo RJ, Wilkinson M, McCormack EA, Ocloo L, Wigley DB, Zhang X, Structure and regulation of the human INO80-nucleosome complex. Nature. 556, 391–395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takano A, Zochi R, Hibi M, Terashima T, Katsuyama Y, Function of strawberry notch family genes in the zebrafish brain development. Kobe J. Med. Sci 56, E220–30 (2011). [PubMed] [Google Scholar]
- 26.Bulayeva K, Lesch K-P, Bulayev O, Walsh C, Glatt S, Gurgenova F, Omarova J, Berdichevets I, Thompson PM, Genomic structural variants are linked with intellectual disability. J. Neural Transm 122, 1289–1301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ, Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 24, 1774–1786 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, Hussain NK, Imig C, de Jong APH, Jung H, Kohansalnodehi M, Kramarz B, Lipstein N, Lovering RC, MacGillavry H, Mariano V, Mi H, Ninov M, Osumi-Sutherland D, Pielot R, Smalla K-H, Tang H, Tashman K, Toonen RFG, Verpelli C, Reig-Viader R, Watanabe K, van Weering J, Achsel T, Ashrafi G, Asi N, Brown TC, De Camilli P, Feuermann M, Foulger RE, Gaudet P, Joglekar A, Kanellopoulos A, Malenka R, Nicoll RA, Pulido C, de Juan-Sanz J, Sheng M, Südhof TC, Tilgner HU, Bagni C, Bayés À, Biederer T, Brose N, Chua JJE, Dieterich DC, Gundelfinger ED, Hoogenraad C, Huganir RL, Jahn R, Kaeser PS, Kim E, Kreutz MR, McPherson PS, Neale BM, O’Connor V, Posthuma D, Ryan TA, Sala C, Feng G, Hyman SE, Thomas PD, Smit AB, Verhage M, SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron. 103, 217–234.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano T, Wallace JT, Baldwin KT, Purkey AM, Uezu A, Courtland JL, Soderblom EJ, Shimogori T, Maness PF, Eroglu C, Soderling SH, Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature. 588, 296–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji R-R, Eroglu C, Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature. 551, 192–197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammond TR, Robinton D, Stevens B, Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev. Biol 34, 523–544 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Thion MS, Ginhoux F, Garel S, Microglia and early brain development: An intimate journey. Science. 362, 185–189 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Fang R, Xia C, Close JL, Zhang M, He J, Huang Z, Halpern AR, Long B, Miller JA, Lein ES, Zhuang X, Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science. 377, 56–62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fellner L, Jellinger KA, Wenning GK, Stefanova N, Glial dysfunction in the pathogenesis of α-synucleinopathies: emerging concepts. Acta Neuropathol. 121, 675–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brettschneider J, Del Tredici K, Lee VM-Y, Trojanowski JQ, Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci 16, 109–120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kia DA, Zhang D, Guelfi S, Manzoni C, Hubbard L, Reynolds RH, Botía J, Ryten M, Ferrari R, Lewis PA, Williams N, Trabzuni D, Hardy J, Wood NW, United Kingdom Brain Expression Consortium (UKBEC) and the International Parkinson’s Disease Genomics Consortium (IPDGC), Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets. JAMA Neurol. 78, 464–472 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paskus JD, Herring BE, Roche KW, Kalirin and Trio: RhoGEFs in Synaptic Transmission, Plasticity, and Complex Brain Disorders. Trends Neurosci. 43, 505–518 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J, Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun 9, 884 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer S, Crow M, Harris BD, Gillis J, Scaling up reproducible research for single-cell transcriptomics using MetaNeighbor. Nat. Protoc 16, 4031–4067 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lotfollahi M, Naghipourfar M, Luecken MD, Khajavi M, Büttner M, Wagenstetter M, Avsec Ž, Gayoso A, Yosef N, Interlandi M, Rybakov S, Misharin AV, Theis FJ, Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol 40, 121–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sousa AMM, Zhu Y, Raghanti MA, Kitchen RR, Onorati M, Tebbenkamp ATN, Stutz B, Meyer KA, Li M, Kawasawa YI, Liu F, Perez RG, Mele M, Carvalho T, Skarica M, Gulden FO, Pletikos M, Shibata A, Stephenson AR, Edler MK, Ely JJ, Elsworth JD, Horvath TL, Hof PR, Hyde TM, Kleinman JE, Weinberger DR, Reimers M, Lifton RP, Mane SM, Noonan JP, State MW, Lein ES, Knowles JA, Marques-Bonet T, Sherwood CC, Gerstein MB, Sestan N, Molecular and cellular reorganization of neural circuits in the human lineage. Science. 358, 1027–1032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghanti MA, Spocter MA, Stimpson CD, Erwin JM, Bonar CJ, Allman JM, Hof PR, Sherwood CC, Species-specific distributions of tyrosine hydroxylase-immunoreactive neurons in the prefrontal cortex of anthropoid primates. Neuroscience. 158, 1551–1559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Büttner M, Ostner J, Müller CL, Theis FJ, Schubert B, scCODA is a Bayesian model for compositional single-cell data analysis. Nat. Commun 12, 6876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H, The extracellular matrix protein laminin α2 regulates the maturation and function of the blood-brain barrier. J. Neurosci 34, 15260–15280 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, Wenger AM, Bejerano G, Kingsley DM, Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 471, 216–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJP, Dougherty ML, Munson KM, Hastie AR, Diekhans M, Hormozdiari F, Lorusso N, Hoekzema K, Qiu R, Clark K, Raja A, Welch AE, Sorensen M, Baker C, Fulton RS, Armstrong J, Graves-Lindsay TA, Denli AM, Hoppe ER, Hsieh P, Hill CM, Pang AWC, Lee J, Lam ET, Dutcher SK, Gage FH, Warren WC, Shendure J, Haussler D, Schneider VA, Cao H, Ventura M, Wilson RK, Paten B, Pollen A, Eichler EE, High-resolution comparative analysis of great ape genomes. Science. 360 (2018), doi: 10.1126/science.aar6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangan RJ, Alsina FC, Mosti F, Sotelo-Fonseca JE, Snellings DA, Au EH, Carvalho J, Sathyan L, Johnson GD, Reddy TE, Silver DL, Lowe CB, Adaptive sequence divergence forged new neurodevelopmental enhancers in humans. Cell. 185, 4587–4603.e23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kostka D, Holloway AK, Pollard KS, Developmental Loci Harbor Clusters of Accelerated Regions That Evolved Independently in Ape Lineages. Mol. Biol. Evol 35, 2034–2045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, Sohal VS, Pyramidal Neurons in Prefrontal Cortex Receive Subtype-Specific Forms of Excitation and Inhibition. Neuron. 81, 61–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi H, Craig AM, Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 36, 522–534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kranz TM, Harroch S, Manor O, Lichtenberg P, Friedlander Y, Seandel M, Harkavy-Friedman J, Walsh-Messinger J, Dolgalev I, Heguy A, Chao MV, Malaspina D, De novo mutations from sporadic schizophrenia cases highlight important signaling genes in an independent sample. Schizophr. Res 166, 119–124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kranz TM, Berns A, Shields J, Rothman K, Walsh-Messinger J, Goetz RR, Chao MV, Malaspina D, Phenotypically distinct subtypes of psychosis accompany novel or rare variants in four different signaling genes. EBioMedicine. 6, 206–214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cressant A, Dubreuil V, Kong J, Kranz TM, Lazarini F, Launay J-M, Callebert J, Sap J, Malaspina D, Granon S, Harroch S, Loss-of-function of PTPR γ and ζ, observed in sporadic schizophrenia, causes brain region-specific deregulation of monoamine levels and altered behavior in mice. Psychopharmacology . 234, 575–587 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Wellcome Trust Case Control Consortium, Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661–678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng H, Xie P, Liu L, Kuang X, Wang Y, Qu L, Gong H, Jiang S, Li A, Ruan Z, Ding L, Yao Z, Chen C, Chen M, Daigle TL, Dalley R, Ding Z, Duan Y, Feiner A, He P, Hill C, Hirokawa KE, Hong G, Huang L, Kebede S, Kuo H-C, Larsen R, Lesnar P, Li L, Li Q, Li X, Li Y, Li Y, Liu A, Lu D, Mok S, Ng L, Nguyen TN, Ouyang Q, Pan J, Shen E, Song Y, Sunkin SM, Tasic B, Veldman MB, Wakeman W, Wan W, Wang P, Wang Q, Wang T, Wang Y, Xiong F, Xiong W, Xu W, Ye M, Yin L, Yu Y, Yuan J, Yuan J, Yun Z, Zeng S, Zhang S, Zhao S, Zhao Z, Zhou Z, Huang ZJ, Esposito L, Hawrylycz MJ, Sorensen SA, Yang XW, Zheng Y, Gu Z, Xie W, Koch C, Luo Q, Harris JA, Wang Y, Zeng H, Morphological diversity of single neurons in molecularly defined cell types. Nature. 598, 174–181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Somel M, Rohlfs R, Liu X, Transcriptomic insights into human brain evolution: acceleration, neutrality, heterochrony. Curr. Opin. Genet. Dev 29, 110–119 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME, EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 103, 945–956 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Margolis SS, Salogiannis J, Lipton DM, Mandel-Brehm C, Wills ZP, Mardinly AR, Hu L, Greer PL, Bikoff JB, Ho H-YH, Soskis MJ, Sahin M, Greenberg ME, EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 143, 442–455 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai N-P, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, Huber KM, Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 151, 1581–1594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen AQ, Sutley S, Koeppen J, Mina K, Woodruff S, Hanna S, Vengala A, Hickmott PW, Obenaus A, Ethell IM, Astrocytic Ephrin-B1 Controls Excitatory-Inhibitory Balance in Developing Hippocampus. J. Neurosci 40, 6854–6871 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elston GN, Benavides-Piccione R, DeFelipe J, The pyramidal cell in cognition: a comparative study in human and monkey. J. Neurosci 21, RC163 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sherwood CC, Miller SB, Karl M, Stimpson CD, Phillips KA, Jacobs B, Hof PR, Raghanti MA, Smaers JB, Invariant Synapse Density and Neuronal Connectivity Scaling in Primate Neocortical Evolution. Cereb. Cortex. 30, 5604–5615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oliveira JF, Sardinha VM, Guerra-Gomes S, Araque A, Sousa N, Do stars govern our actions? Astrocyte involvement in rodent behavior. Trends Neurosci. 38, 535–549 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Favuzzi E, Huang S, Saldi GA, Binan L, Ibrahim LA, Fernández-Otero M, Cao Y, Zeine A, Sefah A, Zheng K, Xu Q, Khlestova E, Farhi SL, Bonneau R, Datta SR, Stevens B, Fishell G, GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. 184, 4048–4063.e32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poskanzer KE, Yuste R, Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. U. S. A 113, E2675–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mederos S, Sánchez-Puelles C, Esparza J, Valero M, Ponomarenko A, Perea G, GABAergic signaling to astrocytes in the prefrontal cortex sustains goal-directed behaviors. Nat. Neurosci 24, 82–92 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Nagai J, Yu X, Papouin T, Cheong E, Freeman MR, Monk KR, Hastings MH, Haydon PG, Rowitch D, Shaham S, Khakh BS, Behaviorally consequential astrocytic regulation of neural circuits. Neuron. 109, 576–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kofuji P, Araque A, Astrocytes and Behavior. Annu. Rev. Neurosci 44, 49–67 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharif A, Duhem-Tonnelle V, Allet C, Baroncini M, Loyens A, Kerr-Conte J, Collier F, Blond S, Ojeda SR, Junier M-P, Prevot V, Differential erbB signaling in astrocytes from the cerebral cortex and the hypothalamus of the human brain. Glia. 57, 362–379 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G, Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 127, 185–197 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Sandau US, Mungenast AE, McCarthy J, Biederer T, Corfas G, Ojeda SR, The synaptic cell adhesion molecule, SynCAM1, mediates astrocyte-to-astrocyte and astrocyte-to-GnRH neuron adhesiveness in the mouse hypothalamus. Endocrinology. 152, 2353–2363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carroll SB, Evo-Devo and an Expanding Evolutionary Synthesis: A Genetic Theory of Morphological Evolution. Cell. 134, 25–36 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, Birney E, Searle S, Schmutz J, Grimwood J, Dickson MC, Myers RM, Miller CT, Summers BR, Knecht AK, Brady SD, Zhang H, Pollen AA, Howes T, Amemiya C, Lander ES, Di Palma F, Lindblad-Toh K, Kingsley DM, The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 484, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geschwind DH, Rakic P, Cortical evolution: judge the brain by its cover. Neuron. 80, 633–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oberheim NA, Goldman SA, Nedergaard M, Heterogeneity of astrocytic form and function. Methods Mol. Biol 814, 23–45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, Balic A, Giladi A, Sheban F, Dutertre C-A, Pfeifle C, Peri F, Raffo-Romero A, Vizioli J, Matiasek K, Scheiwe C, Meckel S, Mätz-Rensing K, van der Meer F, Thormodsson FR, Stadelmann C, Zilkha N, Kimchi T, Ginhoux F, Ulitsky I, Erny D, Amit I, Prinz M, Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell. 179, 1609–1622.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Tan CX, Eroglu C, Cell adhesion molecules regulating astrocyte-neuron interactions. Curr. Opin. Neurobiol 69, 170–177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA, Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell. 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paxinos G, Watson C, Petrides M, Rosa M, Tokuno H, The Marmoset Brain in Stereotaxic Coordinates (Elsevier Science, 2011). [Google Scholar]
- 80.Buckner RL, Margulies DS, Macroscale cortical organization and a default-like apex transmodal network in the marmoset monkey. Nat. Commun 10, 1976 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buckner RL, Krienen FM, The evolution of distributed association networks in the human brain. Trends Cogn. Sci 17, 648–665 (2013). [DOI] [PubMed] [Google Scholar]
- 82.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suresh H, Crow M, Jorstad N, Hodge R, Lein E, Dobin A, Bakken T, Gillis J, Conserved coexpression at single cell resolution across primate brains. bioRxiv (2022), p. 2022.09.20.508736. [Google Scholar]
- 84.De Donno C, Hediyeh-Zadeh S, Wagenstetter M, Moinfar AA, Zappia L, Lotfollahi M, Theis FJ, Population-level integration of single-cell datasets enables multi-scale analysis across samples. bioRxiv (2022), p. 2022.11.28.517803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stogsdill JA, Kim K, Binan L, Farhi SL, Levin JZ, Arlotta P, Pyramidal neuron subtype diversity governs microglia states in the neocortex. Nature (2022), doi: 10.1038/s41586-022-05056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turakhia Y, Chen HI, Marcovitz A, Bejerano G, A fully-automated method discovers loss of mouse-lethal and human-monogenic disease genes in 58 mammals. Nucleic Acids Res. 48, e91–e91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G, GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Földy C, Darmanis S, Aoto J, Malenka RC, Quake SR, Südhof TC, Single-cell RNAseq reveals cell adhesion molecule profiles in electrophysiologically defined neurons. Proc. Natl. Acad. Sci. U. S. A 113, E5222–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Within-species cluster proportions across SMARTseq cortical layer dissections. Estimates of laminar distributions of clusters in human, chimpanzee, and gorilla based on the relative proportions of nuclei dissected from individual cortical layers.
Table S2: Within-species subclass marker genes. Prior to DEG analysis, datasets were downsampled to a maximum of 100 nuclei per individual per cluster (finer curation resolution than subclass) in the Cv3 only samples. DEG analysis was performed on log-normalized gene expression matrices. The gene list was generated by implementing Seurat’s FindAllMarkers function using the Wilcoxon sum rank test on a maximum of 500 nuclei per group (subclass versus all other cell types) within each species. Genes with Bonferroni adjusted p-values less than 0.05 were retained. This list is not filtered for orthologous genes and reflects the species-specific gene symbol.
Table S3: Subclass differential gene expression across great apes. Gene list of pairwise differential expression comparing human, chimpanzee, and gorilla for each subclass. The gene list was generated by implementing a pseudobulk approach across individuals with DESeq2 using Ward’s test on downsampled datasets with a maximum 50 nuclei per cluster per individual per species for Cv3 nuclei using one-to-one orthologous genes. Genes with Bonferroni adjusted p-values less than 0.05 were retained.
Table S4: Subclass isoform switching across great apes. Relative isoform abundances were compared between human, chimpanzee, and gorilla for one-to-one orthologous genes with at least moderate expression. Gene expression and isoform proportions are reported for two individuals from each species that had SSv4 data. The column ‘Large_prop_change’ indicates if one species expresses a larger proportion (> 0.7, > 3-fold change on average across individuals) of a transcript than the other species (proportion < 0.1).
Table S5: SST consensus type markers across species. List of marker genes that are differentially expressed in each of 9 SST-expressing consensus types relative to the other types. Markers were independently generated for each species. The gene list was generated by implementing the Wilcoxon sum rank test with Seurat’s FindAllMarkers function on downsampled datasets with a maximum 100 nuclei per cross-species cluster per individual for Cv3 nuclei.
Table S6: Consensus type cluster membership. 86 cross-species clusters included clusters from two or more species and were merged to give a final set of 57 consensus types with homologous clusters from all 5 primates.
Table S7: Consensus type species-specific expression. Gene list of differential gene expression comparing each species to the other four species for each consensus type. The gene list was generated by implementing a pseudobulk approach across individuals with DESeq2 using Ward’s test on downsampled datasets with a maximum 50 nuclei per cluster per individual per species for Cv3 nuclei using one-to-one orthologous genes. Genes with Bonferroni adjusted p-values less than 0.05 were retained.
Table S8. Analysis of hDEGs near HARs or hCONDELs. Lists of hCONDEL coordinates, hDEGs in consensus types that are near HARs or hCONDELs, potential HAR-hDEG interactions from existing PLAC-Seq datasets, and manually curated synapse-related gene families. This table also includes statistics on the enrichment of hDEGs, and hDEGs near HARs or hCONDELs, for SynGO and for manually curated lists of synapse-related gene families.
Galleries of the spatial distributions of human MTG cell types. A representative human MTG section indicating spatial locations of 151 cell types grouped by subclass. Upper left images of each subclass show all types within the subtype. Note that for the rare types PVALB_3, PVALB_8, and SST_4, a mapped cell did not occur within the example section or was obscured by other cells in the section. For these 3 types, the cell locations were approximated based on laminar positions in other sections. L1: Cortical Layer 1; L2: Cortical Layer 2; L3: Cortical Layer 3; L4: Cortical Layer 4; L5: Cortical Layer 5; L6: Cortical Layer 6; WM: White Matter.
Data Availability Statement
Raw sequence data were produced as part of the BRAIN Initiative Cell Census Network (BICCN: RRID:SCR_015820) are available for download from the Neuroscience Multi-omics Archive (RRID:SCR_016152; https://assets.nemoarchive.org/dat-net1412) and the Brain Cell Data Center (RRID:SCR_017266; https://biccn.org/data). Code for analysis and generation of figures is available for download from https://github.com/AllenInstitute/Great_Ape_MTG. Visualization and analysis tools for integrated species comparison are available using Cytosplore Viewer (RRID SCR_018330; https://viewer.cytosplore.org/). These tools allow comparison of gene expression in consensus clusters across species, as well as species-specific clusters and to calculate differential expression within and among species. The following publicly available datasets were used for analysis: Synaptic Gene Ontology (SynGO) and orthologous genes across species from NCBI Homologene (downloaded November 2019). MTG human SMARTseq v4 data (https://portal.brain-map.org/atlases-and-data/rnaseq/human-mtg-smart-seq, https://assets.nemoarchive.org/dat-swzf4kc).