

SUMMARY
Accurate chromosome segregation relies on kinetochores carrying out multiple functions, including establishing and maintaining microtubule attachments, forming precise bioriented attachments between sister chromatids, and activating the spindle assembly checkpoint. Central to these processes is the highly conserved Ndc80 complex. This kinetochore subcomplex interacts directly with microtubules, but also serves as a critical platform for recruiting kinetochore-associated factors and as a key substrate for error correction kinases. The precise manner in which these kinetochore factors interact, and regulate each other’s function, remains unknown – considerably hindering our understanding of how Ndc80 complex-dependent processes function together to orchestrate accurate chromosome segregation. Here, we aimed to uncover the role of Nuf2’s CH domain, a component of the Ndc80 complex, in ensuring accurate chromosome segregation. Through extensive mutational analysis, we identified a conserved “interaction hub” comprising two segments in Nuf2’s CH domain, forming the binding site for Mps1 within the yeast Ndc80 complex. Intriguingly, the interaction between Mps1 and the Ndc80 complex seems to be subject to regulation by competitive binding with other factors. Mutants disrupting this interaction hub exhibit defects in spindle assembly checkpoint function and severe chromosome segregation errors. Significantly, specifically restoring Mps1-Ndc80 complex association rescues these defects. Our findings shed light on the intricate regulation of Ndc80 complex-dependent functions and highlight the essential role of Mps1 in kinetochore biorientation and accurate chromosome segregation.
INTRODUCTION
The precise segregation of duplicated chromosomes into daughter cells is a fundamental process during cell division. This segregation relies on the interactions between microtubules and kinetochores, which are large protein complexes that assemble on the centromeres of each chromosome (reviewed in1). Accurate chromosome segregation relies on several critical functions performed by kinetochores. First, they must establish and maintain attachments to the tips of microtubules, whose dynamic growing and shrinking generates the forces to physically move chromosomes within the cell. Second, kinetochores on each pair of replicated sister chromatids must form bioriented attachments to microtubules originating from opposite spindle poles. Since initial kinetochore-microtubule attachments are randomly established, these attachments are often erroneous and must be detected and corrected to prevent mis-segregation. Third, kinetochores must activate the spindle assembly checkpoint when microtubule attachments are absent, thereby halting the cell cycle until these errors are corrected (reviewed in2,3). The precise mechanisms by which kinetochores coordinate and regulate these diverse functions remain important and open questions in the field.
At the core of these essential cellular functions lies the Ndc80 complex, a highly conserved subcomplex of the kinetochore. This complex is a heterotetrameric, rod-like assembly, comprised of four distinct proteins: Ndc80, Nuf2, Spc24, and Spc254–9. The complex engages with microtubules through multiple domains, including the globular CH-domains located at the N-terminal end of the Ndc80:Nuf2 complex, which provides a positively charged interaction surface for microtubules7,10,11. Furthermore, the unstructured, flexible N-terminal tail of the Ndc80 protein harbors an additional microtubule-binding element7,11–14. The role of Ndc80’s CH domains in microtubule binding is well-established both in vitro and in vivo7,10,14,15. In contrast, the function of Nuf2’s CH domain has remained somewhat enigmatic7,15. Structural studies, for instance, indicate that this domain does not make direct contact with the microtubule6,9,16. Nevertheless, its high degree of conservation suggests that it likely serves vital functions.
Besides its direct microtubule binding capability, the Ndc80 complex serves as a critical “landing pad” for numerous microtubule-associated proteins, as well as a substrate for various kinases. To enhance the ability to form load-bearing attachments and to track with dynamic microtubule tips, the Ndc80 complex associates with the Dam1 complex in yeast10,17–22 and the functionally analogous Ska complex in metazoans23–26. The Ndc80 protein itself serves as a critical phosphorylation target for the error correction kinases, namely Ipl1 and Mps17,11–13,27–38. Additionally, the Ndc80 complex directly interacts with additional factors implicated in correcting erroneous attachments, including Stu2/chTOG39–41. Lastly, the Ndc80 complex assumes a pivotal role in triggering the spindle assembly checkpoint by recruiting the most upstream component, the kinase Mps133–35,37,42–48. A significant barrier to understanding how these diverse Ndc80 complex-dependent functions are coordinated lies in our limited understanding of how these different factors interact with the Ndc80 complex and the degree to which they influence each other’s functions.
In this study, our aim was to elucidate the role of Nuf2’s CH domain in facilitating chromosome segregation fidelity. Through extensive mutational analysis, we identified a conserved “interaction hub” formed by two segments of Nuf2’s CH domain, its N-terminal loop and G-helix. We demonstrate that this hub constitutes a key portion of the Mps1 binding interface within the yeast Ndc80 complex. This same region of Nuf2 associates with additional factors, including the Dam1 complex, suggesting that Mps1’s kinetochore localization may be regulated by competitive binding. Mutants that disrupt this interaction hub exhibit defects in spindle assembly checkpoint function and severe chromosome segregation errors. Remarkably, Mps1 appears to be the critical factor that binds to this region, since the cellular defects can be rescued by specifically restoring Mps1-Ndc80 complex association. Our work sheds light on the manner in which numerous Ndc80 complex-dependent functions are regulated and demonstrates the essential role of Mps1 in kinetochore biorientation and chromosome segregation.
RESULTS
Identification of a Nuf2 patch that is essential for cell viability
For this study, we wished to determine the role that Nuf2’s CH domain plays in the establishment of correctly bioriented sister chromatids. To identify residues that are important for its function, we first looked for spatial patterns of amino acid conservation within Nuf2’s CH domain. Here, we aligned Nuf2 sequences from various fungal species and then mapped the resulting conservation scores onto a previously determined Ndc80 complex structure49 (PDB: 5TCS). This analysis revealed a region of conserved residues in close spatial proximity, which is centered on Nuf2’s so called “G-helix” as well as its amino-terminal “loop” (Fig 1A & Fig S1A). These residues, especially F8, P9, R118, S124, N128 and R131, appear highly conserved in fungal and metazoan Nuf2 sequences (Fig 1A & Fig S1A–B). To examine the functional importance of these residues, we generated strains containing an auxin-inducible degron of the endogenous NUF2, and ectopically expressed mutant nuf2 alleles to examine their phenotype in the presence of auxin. We focused on mutating residues that showed strong conservation, and assayed effects on cell viability using a spot dilution assay. We found that many of the mutations resulted in severe cell viability defects, and the most penetrant phenotypes were associated with mutations within the Nuf2 G-helix (nuf2S124D, nuf2A125D, nuf2N128A and nuf2R131E) as well as N-terminal loop (nuf2F8A P9A; Fig 1B & Fig S1D). Importantly, these mutations do not appear to affect Nuf2 expression levels (Fig S1C). A summary of all mutants and associated viability phenotypes can be found in Fig 1C and Fig S1D. Based on the severity of viability defects, amount of conservation, and degree of spatial proximity, we chose to focus on mutations nuf2F8A P9A, nuf2S124D, and nuf2N128A for further analysis.
Figure 1. Mutation of conserved Nuf2 residues affects cell viability.

(A) Cartoon of the heterotetrameric Ndc80 complex (Ndc80, Nuf2, Spc24, Spc25). CH domains are represented as ovals; coiled-coil domains and the N-terminal tail of Ndc80 are shown as wavy lines. Structure shows S. cerevisiae Nuf2 from49 (5TCS), with other members of the Ndc80 complex not shown. Left: Conservation of Nuf2 among 12 fungal species (listed in Fig S1), viewed using ChimeraX, on a scale of −2.5 to +2.5, with the most conserved residues in red and least conserved in blue. Right: Conserved residues used throughout this work are highlighted (F8 and P9 in the N-terminal loop; S124 and N128 in the G-helix).
(B) Yeast cell viability assay with a subset of nuf2 mutant alleles. Strains carry NUF2-AID and an ectopic copy of NUF2–3HA (No covering allele, “None”, M1889; NUF2WT, M2038; nuf2F8A P9A, M2042; nuf2S124A, M2040; nuf2S124D, M2041; nuf2N128A, M2414). Cells were serially diluted five-fold and spotted onto plates containing DMSO (control), 250 μM auxin (to degrade endogenous Nuf2-AID protein), or 250 μM auxin + 6.5 μg/mL benomyl.
(C) Structure of Nuf2 (5TCS), illustrating the range of cell viability phenotypes of mutant alleles observed on plates containing benomyl (data shown in Fig S1D). Nuf2 mutants are listed in groups in decreasing shades of purple, from the strongest phenotype (death on auxin only) to the weakest (slight sickness on auxin + benomyl).
Spindle assembly checkpoint function is perturbed in nuf2 patch mutants
To begin understanding how these nuf2 CH domain mutants result in cell viability defects, we first compared the growth phenotypes to previously described mutations in the CH domain of Nuf2’s binding partner, Ndc80. We found that nuf2 mutants, including nuf2F8A P9A and nuf2N128A, display a considerably more severe viability defect than ndc80 mutants which perturb microtubule binding7,10 (i.e. ndc80K204E and ndc80K122E K204E; Fig 2A and Fig S2). Furthermore, while ndc80K122E K204E mutants arrest in mitosis10, likely by activating the spindle assembly checkpoint, the nuf2 mutants did not induce a specific cell cycle arrest (Fig 2B). These observations suggest that these nuf2 CH domain mutations affect a kinetochore function beyond just microtubule binding. Given the lack of cell cycle arrest, we next assessed spindle assembly checkpoint (SAC) function by examining the localization of the SAC component, Bub1, which is recruited to kinetochores upon SAC activation50–53. For this assay, CDC20-AID cells were arrested in metaphase by the addition of auxin. Once arrested in metaphase, cells were treated with nocodazole, and co-localization of Bub1-GFP with the kinetochore marker Mtw1-mCherry was monitored. Approximately 72 ± 6% of NUF2WT cells showed Bub1-GFP recruitment to the kinetochore upon nocodazole treatment, consistent with prior observations51,53. In contrast, nuf2F8A P9A and nuf2N128A showed a dramatic decrease in the number of cells with Bub1 at kinetochores (14 ± 5.5% and 14 ± 3.5%, respectively; Fig 2C), consistent with defects in spindle assembly checkpoint function. We also note that Mtw1-mCherry distribution appeared normal in the nuf2 mutants, suggesting that these mutations do not cause gross defects in kinetochore organization (Fig 2C).
Figure 2. Lethal nuf2 mutant alleles fail to trigger the spindle assembly checkpoint.

(A) Yeast cell viability assay comparing nuf2 mutants with previously characterized ndc80 alleles. Strains carry either NUF2-AID or NDC80-AID at their endogenous loci, and ectopic copies of NUF2–3HA (None, M1889; NUF2WT, M2038; nuf2F8A P9A, M2042; nuf2N128A, M2414) or NDC80–3HA (None, M4182; NDC80WT, M692; ndc80K204E, M693; ndc80K122E K204E, M694). Cells were serially diluted five-fold and spotted onto plates containing DMSO, 250 μM auxin or 250 μM auxin + 6.5 μg/mL benomyl.
(B) Comparison of yeast cells expressing nuf2 mutant alleles to those expressing an ndc80 mutant allele that induces a metaphase arrest. Exponentially growing strains with NDC80-AID or NUF2-AID and ectopic copies of either ndc80K122E K204E-3HA (M694) or NUF2–3HA (NUF2WT, M2933; nuf2F8A P9A, M2935; nuf2S124D, M2936; nuf2N128A, M2945) were treated with 500 μM auxin for 2.5 hours to degrade endogenous Ndc80-AID or Nuf2-AID protein prior to fixation. Left: Representative micrographs. Right: Quantitation of cells showing the percentage of large-budded, mononucleate cells in ndc80 or nuf2 mutants, with error bars indicating the standard deviation among three replicates (n ≥ 100 cells for each replicate). Significance was determined by a two-tailed unpaired t test (**; P ≤ 0.01).
(C) Bub1-GFP assay for SAC activity. Exponentially growing NUF2-AID CDC20-AID strains (NUF2WT, M2792; nuf2F8A P9A, M2794; nuf2S124D, M2795; nuf2N128A, M2801) were treated with 500 μM auxin for 2 hours to arrest cells in metaphase, followed by 10 μM nocodazole for 1 hour to activate the SAC. Strains carry BUB1-GFP (SAC) and MTW1-mCherry (kinetochore) alleles. Left: Representative micrographs. Right: Quantitation of percent of large-budded, mononucleate cells with Bub1-GFP localized at the kinetochore, with error bars indicating the standard deviation among three replicates (n=66–112 cells for each replicate). Significance was determined by a two-tailed unpaired t test (***; P ≤ 0.001). Only cells with a single Mtw1 signal, indicating collapse of the spindle, were analyzed.
Mutants within Nuf2’s patch result in substantial chromosome segregation defects
While these nuf2 CH domain mutants display spindle assembly checkpoint defects, the lethal phenotypes observed cannot be explained by this limitation alone since the SAC is not essential for cell viability in yeast (Fig 3A). This discrepancy led us to examine sister kinetochore biorientation and chromosome segregation defects as a potential cause of the lethality. To monitor biorientation, we induced a metaphase arrest by depleting Cdc20 (using a methionine-repressible CDC20 allele) in cells that also carry a fluorescently marked centromere of chromosome III54. Under these conditions, opposing spindle pulling forces cause bioriented sister chromatids to separate, appearing as two distinct GFP puncta49. The frequency of biorientation in NUF2WT cells was similar to those typically observed in this assay (44 ± 6%, Fig 3B). However, in nuf2 mutant cells, we observed a marked reduction in biorientation of CEN III (nuf2F8A P9A, 10 ± 1.5%; nuf2S124D, 6.6 ± 1.5%; and nuf2N128A, 18 ± 1.5%). Furthermore, we noted a significant portion of nuf2 mutant cells bypassing the arrest induced by Cdc20 depletion (Fig S3A). In these nuf2 mutants, spindle elongation led exclusively to instances of sister chromatid non-disjunction and unequal DNA masses (Fig S3B–C). These findings can be attributed to substantial deficiencies in biorientation, where many or all pairs of sister chromatids form syntelic attachments to the mitotic spindle, consequently lacking the necessary forces to restrict spindle elongation.
Figure 3. Inviable nuf2 mutants display a loss of sister chromatid biorientation and mis-segregation of chromosomes.

(A) Comparison of nuf2 mutant cell viability relative to the mad2 SAC mutant. Strains carrying a NUF2-AID and ectopic copies of NUF2–3HA (NUF2WT, M2038; nuf2F8A P9A, M2042; nuf2N128A, M2414) are shown in comparison to mad2Δ (M35). Cells were serially diluted five-fold and spotted onto plates containing DMSO, 250 μM auxin, 6.5 μg/mL benomyl or both auxin and benomyl.
(B) Assay for sister chromatid biorientation in nuf2 mutants. Exponentially growing cells carrying NUF2-AID and ectopic copies of NUF2–3HA (NUF2WT, M4245; nuf2F8A P9A, M4251; nuf2S124D, M4249; nuf2N128A, M4253), pMET-CDC20, and CEN III marked with GFP (CEN III:lacO LacI-GFP) were arrested with 1 μg/mL alpha factor for 3 hours, followed by release into fresh media containing methionine (to induce metaphase arrest via pMET-CDC20) and 500 μM auxin (to degrade endogenous Nuf2-AID) for 1.5 hours. Left: Representative micrographs. Right: Quantitation of percent of large-budded, mononucleate cells displaying biorientation of the chromosome-marked GFP signal, with error bars indicating the standard deviation among three replicates (n= 71–137 cells for each replicate). Significance was determined by a two-tailed unpaired t test (** P ≤ 0.01).
(C) Chromosome segregation assay in nuf2 mutants. Exponentially growing cells carrying NUF2-AID and ectopic copies of NUF2–3HA (NUF2WT, M4469; nuf2F8A P9A, M4475; nuf2N128A, M4479), as well as CEN IV marked with GFP (CEN IV:lacO LacI-GFP) and SPC110-mCherry (spindle pole marker) were arrested with 1 μg/mL alpha factor for 3 hours, followed by release into fresh media containing 500 μM auxin for 2 hours. Left: Representative micrographs. Right: Quantitation of percent of binucleate cells displaying correct segregation of the chromosome-marked GFP signal, with error bars indicating the standard deviation among three replicates (n = 75–151 cells for each replicate). Significance was determined by a two-tailed unpaired t test (****; P ≤ 0.0001).
To examine chromosome segregation fidelity in these nuf2 mutants, cells again carrying a fluorescently marked centromere (now of chromosome IV) were released from a G1 arrest, and chromosome segregation was examined upon anaphase onset. Cells expressing nuf2F8A P9A and nuf2N128A displayed surprisingly high errors in segregating chromosome IV (NUF2WT 96 ± 4.6% correct segregation compared to nuf2F8A P9A 4 ± 3.1%, nuf2N128A 14 ± 3.5%, Fig 3C and Fig S3D). Together, these observations show that the region of Nuf2’s CH domain, consisting of its G-helix and N-terminal loop, are required for spindle assembly checkpoint function and essential for biorientation and accurate chromosome segregation.
Mps1-kinetochore localization perturbed in nuf2 patch mutants
The observed defects in both SAC function and biorientation led us to consider the idea that decreased recruitment of the Mps1 kinase to the kinetochore may be the cause of the observed phenotypes. Mps1 lies at the apex of spindle assembly checkpoint signaling. Beyond this, its activity is important in the establishment of correctly bioriented kinetochore attachments32–37,44,45, and Nuf2 and Ndc80 have been implicated as the binding site for Mps130,42,43,55. We therefore used an established kinetochore co-IP assay to examine the levels of Mps1 associated with the kinetochore in NUF2WT relative to nuf2 mutants31,56. Kinetochores were immunoprecipitated via anti-Flag pulldown of the kinetochore component Dsn1-Flag, and co-purification of Mps1-3V5 was determined by western blot. As expected, NUF2-AID cells (treated with auxin 2 hours prior to harvesting) expressing NUF2WT showed Mps1 co-purification with kinetochores. In contrast, nuf2F8A P9A, nuf2S124D, and nuf2N128A mutants displayed nearly undetectable Mps1 binding but preserved Ndc80 association (Fig 4A). We propose this region of Nuf2 forms a key part of the Mps1 binding interface to the yeast Ndc80 complex. These results are also consistent with Yu and colleagues who made a similar observation for human Mps1 binding to Ndc80 complex43, suggesting this is a remarkably conserved interface.
Figure 4. Mps1 kinetochore recruitment is lost in nuf2 mutants, and an mps1 mutant partially phenocopies a nuf2 mutant for chromosome segregation defects.

(A) Detection of Mps1-3V5 association with the kinetochore via immunoprecipitation. Exponentially growing cultures expressing DSN1–6His-3Flag (No tag, M2630) as well as MPS1–3V5, NUF2-AID and ectopic copies of NUF2–3HA (NUF2WT, M3494; nuf2F8A P9A, M3500; nuf2S124D, M3498; nuf2N128A, M3504), were treated with auxin 2 hours prior to harvest. Kinetochores were purified from lysates by anti-Flag immunoprecipitation (IP) and analyzed by immunoblotting.
(B) Cartoon of the Mps1 protein and a conservation histogram for amino acids 126 to 190. The region from 151 to 171 is shown in detail, with amino acids mutated in this work marked by an *. Numbering is according to the S. cerevisiae sequence.
(C) Complementation of the temperature sensitive mps1-1 allele (M56) by ectopically expressed MPS1 (MPS1WT, M4407; mps1Δ151−200, M4412; mps1Δ201−300, M4413; mps1RLR>EEE, M4949). Cells were serially diluted five-fold, spotted onto plates, and grown at mps1-1 permissive (23°C) and non-permissive (37°C) temperatures.
(D) Chromosome segregation assay in mps1 mutants. Exponentially growing cells carrying the mps1-1 temperature sensitive allele and ectopic copies of MPS1 (MPS1WT, M4714; mps1RLR>EEE, M4710, M4947, M4948), as well as CEN IV marked with GFP (CEN IV:lacO LacI-GFP) and SPC110-mCherry (spindle pole marker), were arrested with 1 μg/mL alpha factor for 3 hours, followed by release into fresh media at 37°C for 2 hours. Due to the difference in phenotypes between nuf2 mutants and mps1RLR>EEE, three independent strains were analyzed to ensure validity of the result. Left: Representative micrographs. Right: Quantitation of percent of binucleate cells displaying correct segregation of the chromosome-marked GFP signal, with error bars indicating the standard deviation among three to four replicates (n = 61–154 cells for each replicate). Significance was determined by a two-tailed unpaired t test (****; P ≤ 0.0001).
If the nuf2 mutant defects stem from reduced Mps1 binding, we hypothesized that an mps1 mutant at this interface would exhibit a similar phenotype. To identify potential interaction regions of Mps1 with the Ndc80 complex, we sought conserved regions within Mps1 outside its C-terminal kinase domain and identified two highly conserved patches encompassing residues 151–157 and 165–171 (Fig 4B). Earlier studies associated residues 151–200 with Mps1’s SAC and biorientation functions, while finding residues 201–300 are crucial for spindle pole body duplication57. To assess the functional significance of these conserved Mps1 segments, we initially examined cell viability. Here, we used a temperature sensitive allele, mps1-1, to disrupt the endogenous function, as an MPS1-AID allele did not fully disrupt cell viability in our strain background (data not shown). Consistent with previous observations, mps1Δ151−200 and mps1Δ201−300 mutants were inviable in this system57 (Fig 4C). While neither mps1Δ151−157 nor mps1Δ165−171 fully disrupted cell viability, combining these deletions or introducing point mutations spanning one or both regions resulted in complete inviability (Fig 4C and Fig S4A). The notion that this portion of Mps1 facilitates binding to the Ndc80 complex is well-supported by concurrent studies from Pleuger et al.58 and Zahm et al., employing biochemical and structural approaches to determine the Mps1-Ndc80 complex interface (personal communication S. Westermann and S. Harrison).
Based on the above observations we decided to further characterize the mps1R155E L165E R170E mutant (hereafter designated as “mps1RLR>EEE”), focusing on spindle pole body separation and chromosome segregation. Cells carrying a fluorescently marked centromere of chromosome IV were arrested in G1 at a permissive temperature. Upon release from this G1 arrest at a non-permissive temperature, we examined spindle pole body separation and chromosome segregation during anaphase. mps1RLR>EEE cells showed spindle pole body separation similar to that of MPS1WT, indicating this role of Mps159 remains functional in these mutants (Fig S4B). In contrast, mps1RLR>EEE mutants displayed a large degree of chromosome segregation errors (MPS1WT 99 ± 1.4% correct segregation, compared to mps1RLR>EEE 56 ± 4.3%, Fig 4D and Fig S4C). It is worth noting that the chromosome segregation defects observed in the mps1RLR>EEE mutant were not as pronounced as those observed in the nuf2 CH domain mutants (14% correct segregation for nuf2N128A, compared to 56% for mps1RLR>EEE, Fig 3C and Fig 4D). This discrepancy may be due to the mps1RLR>EEE mutant not completely disrupting Mps1’s ability to bind the Ndc80 complex. Alternatively, it is plausible that additional factors might bind this region of Nuf2’s CH domain, and the observed phenotypes could arise from the combination of these defects.
Dam1 complex recruitment to kinetochores is altered in nuf2 CH domain mutants
The inconsistency in observed chromosome segregation defects between the nuf2 and mps1 mutants prompted us to consider that the nuf2 mutants might disrupt binding to additional factors. Recent structural studies have suggested this region of the Ndc80 complex as a potential interaction site for the Dam1 complex19,20. To examine if the nuf2 CH domain mutants disrupt Dam1 recruitment to the kinetochore, we assessed the localization of a component of the Dam1 complex, Ask1-YFP, along the mitotic spindle in metaphase arrested cells. NUF2WT cells exhibited a characteristic bilobed distribution of Ask1 signals proximal to the spindle pole (marked by Spc110-mCherry), consistent with expected kinetochore localization. On the other hand, nuf2F8A P9A mutant cells displayed a substantial change in Ask1 localization, showing a dramatic decrease proximal to the spindle pole, and a noticeable increase in localization along the mitotic spindle (Fig 5A). These observations align with the nuf2F8A P9A mutant disrupting normal localization of the Dam1 complex to the kinetochore. If Dam1 complex localization is compromised in nuf2 mutants, we might anticipate synthetic lethality with hypomorphic alleles of Dam1 complex components. In line with this idea, we found that nuf2F8A, nuf2P9A, and nuf2I10A L11A display synthetic lethality and/or sickness with dad1-1 mutants (Fig 5B and Fig S5). Collectively, these findings support the notion that multiple factors, including Mps1 and the Dam1 complex, interact with this “interaction hub” of Nuf2’s CH domain, and the observed phenotypes are likely a result of the combined effects of these binding alterations. This also raises an intriguing possibility that competition for binding to the Ndc80 complex might contribute to regulating the localization and function of these factors.
Figure 5. Dam1 localization to the kinetochore is disrupted in nuf2 mutants.

(A) Ask1-YFP localization assay in NUF2WT and nuf2F8A P9A. Strains carry a NUF2-AID allele and ectopic copies of NUF2–3HA (NUF2WT, M4985; nuf2F8A P9A, M4987), as well as CDC20-AID, ASK1-YFP (Dam1 complex) and SPC110-mCherry (spindle pole marker). Left: Representative micrographs. Middle: Profile plots showing intensities of a line scan through the spindles of NUF2WT and nuf2F8A P9A cells. The Spc110-mCherry signal intensity (red) was plotted on the left y-axis, and the Ask1-YFP signal intensity (green) was plotted on the right y-axis. Right: Graph of YFP intensities, with each dot representing an individual spindle pole-proximal peak (n ≥ 100 cells, ≥ 200 peaks each for NUF2WT and nuf2F8A P9A, respectively). Significance was determined by a two-tailed unpaired t test (****; P < 0.0001).
(B) Cell viability assay assessing synthetic lethality of nuf2 mutant alleles with the temperature sensitive dad1-1 allele. Strains carry a NUF2-AID allele and ectopic copies of NUF2–3HA (NUF2WT DAD1, M2038; NUF2WT dad1-1, M4465; nuf2P9A DAD1, M2152; nuf2P9A dad1-1, M4926; nuf2I10A L11A DAD1, M3750; nuf2I10A L11A dad1-1, M4928). Cells were serially diluted five-fold, spotted onto plates containing DMSO or 250 μM auxin and grown at dad1-1 permissive (23°C) and semi-permissive (30°C) temperatures.
Ndc80 complex interaction hub mutants can be suppressed by modulating Mps1 levels
Considering that multiple factors seem to interact with this interaction hub, such as Mps1, the Dam1 complex, and potentially other factors, including the Aurora B kinase, Ipl1 (see Zahm et al., personal communication with S. Harrison), a challenging question arises: how do we determine the relative importance of each of these factors in promoting accurate chromosome segregation? Some insight into this question came from the following observations. We reasoned that if defects in Mps1-kinetochore association was the main cause of viability defects in our nuf2 mutants, perhaps modulating Mps1 levels at the kinetochore could suppress the associated defects. Prior research with human proteins demonstrated that the presence of the N-terminal tail of Ndc80 diminishes Mps1’s capacity to bind to the Ndc80 complex in vitro, likely due to competitive binding. Furthermore, this inhibition appears to be relieved by phosphorylation of the N-terminal tail43. If this idea is correct, and Ndc80’s N-terminal tail forms a ‘closed’ conformation to compete with Mps1’s ability to bind in cells, we reasoned that deletion of this tail should increase kinetochore-associated Mps1 (Fig 6A) and may suppress the nuf2 mutant growth defects. We constructed a NUF2-AID NDC80-AID ‘double AID’ strain to conduct these experiments, and found that this genetic background was far more sensitized, i.e. nuf2 mutants which show no detectable growth defect in a NUF2-AID ‘single AID’ background, now result in dramatic cell viability defects (Fig S6A). Consistent with the idea above, many of the nuf2 mutants at the described Mps1 binding interface showed prominent growth defects in the context of NDC80WT, however, many of these mutants were suppressed by ndc80ΔN-tail (Fig 6B and Fig S6B–C). These findings also raise the possibility that the N-terminus of Ndc80 associates directly with the interaction hub to regulate Mps1 localization.
Figure 6. Increasing the level of Mps1 suppresses the lethality of nuf2 mutants.
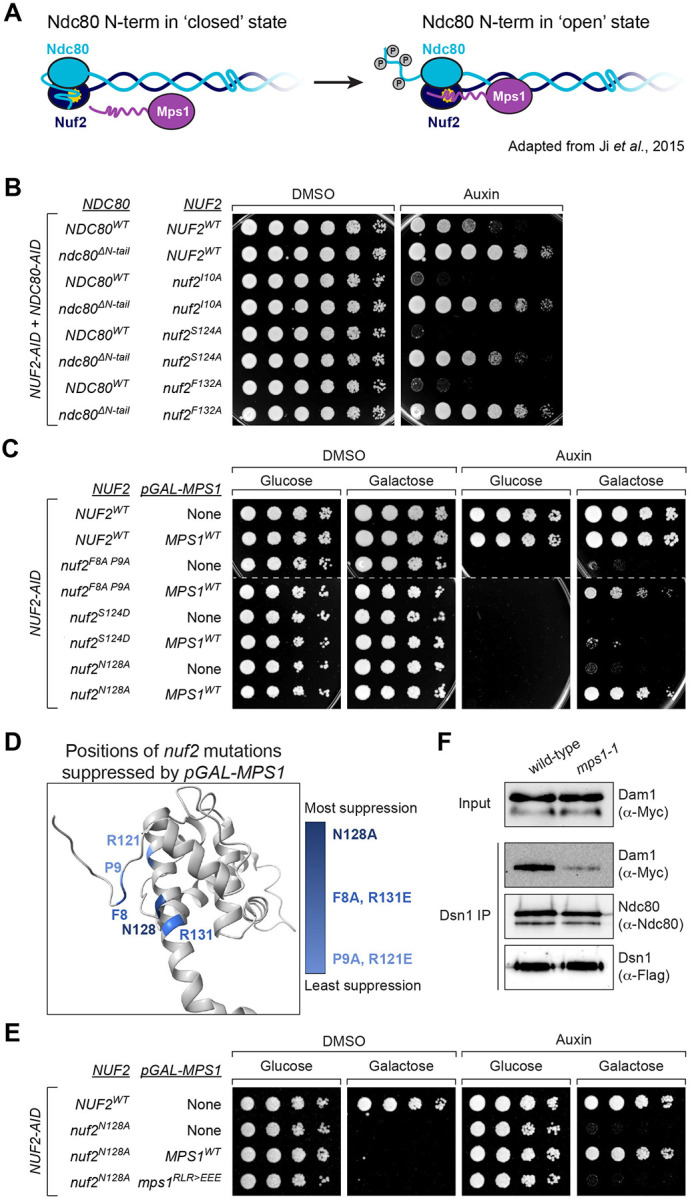
(A) Model illustrating two states of the Ndc80 N-terminal tail: “closed”, in which the N-terminal tail of Ndc80 associates with Nuf2, precluding Mps1 binding, and “open”, in which the tail is altered, likely via phosphorylation, to allow Mps1-Nuf2 association43.
(B) Yeast cell viability assay comparing nuf2 mutant phenotypes in NDC80WT and ndc80ΔN-tail strains. Each is a “double AID” strain, containing both NUF2-AID and NDC80-AID alleles, as well as ectopic copies of NUF2–3HA and NDC80–3HA (NUF2WT NDC80WT, M2998; NUF2WT ndc80ΔN-tail, M2999; nuf2I10A NDC80WT, M2654; nuf2I10A ndc80ΔN-tail, M2755; nuf2S124A NDC80WT, M2641; nuf2S124A ndc80ΔN-tail, M2742; nuf2F132A NDC80WT, M2841; nuf2F132A ndc80ΔN-tail, M2774). Cells were serially diluted five-fold and spotted onto plates containing DMSO or 250 μM auxin.
(C) Yeast cell viability assay to assess the effect of pGAL-driven overexpression of Mps1 in nuf2 mutants. Strains contain NUF2-AID and ectopic copies of NUF2–3HA, without or with an ectopic pGAL-MPS1 single integration vector allele, respectively (NUF2WT, M2038 and M3586; nuf2F8A P9A, M2042 and M3587; nuf2S124D, M2041 and M4619; nuf2N128A, M2414 and M3588). Cells were serially diluted five-fold and spotted onto plates containing either glucose (control) or galactose (induce over-expression of MPS1), as well as DMSO or 250 μM auxin.
(D) Structure of S. cerevisiae Nuf2 (5TCS) illustrating the residues with mutants that are suppressed by expression of pGAL-MPS1. Darker shades of blue indicate increasing strength of the suppression.
(E) Yeast cell viability assay, comparing pGAL-MPS1WT and pGAL-mps1RLR>EEE suppression of nuf2N128A. Strains contain NUF2-AID and an ectopic copy of NUF2–3HA (NUF2WT, M2038; nuf2N128A, M2414) or also contain a single, ectopic pGAL-MPS1 allele (nuf2N128A with MPS1WT, M3588; nuf2N128A with mps1RLR>EEE, M4946). Cells were serially diluted five-fold and spotted onto plates containing either glucose or galactose, as well as DMSO or 250 μM auxin.
(F) Detection of Dam1-9Myc association with the kinetochore via immunoprecipitation. Exponentially growing wild-type (M457) and mps1-1 (M469) cultures expressing DSN1–6His-3Flag, DAM1–9Myc and NDC80–3HA were shifted to 37°C for 2 hours prior to harvest. Kinetochores were purified from lysates by anti-Flag immunoprecipitation (IP) and analyzed by immunoblotting.
Another way to modulate Mps1 levels at the kinetochore is through overexpression. Compellingly, overexpression of Mps1 rescues the cell viability defects of both nuf2F8A P9A and nuf2N128A, as well as another nearby mutation (Fig 6C & Fig S6D–E). In fact, when we examine the nuf2 mutant alleles whose cell viability defects are suppressed by Mps1 overexpression, we find that the mutated residues spatially cluster on a surface of Nuf2 (Fig 6D). However, viability of nuf2S124D, the most penetrant single substitution nuf2 mutant we tested, was not suppressed by Mps1 overexpression (Fig 6C) even though it is close in spatial proximity to F8, P9 and N128 (Fig 1A & Fig 6D). We suggest the increased severity of nuf2S124D may be due to physical displacement of the N-term loop of Nuf2 from its G-helix, leading to a complete disruption of the Mps1 binding site. The finding that the overexpression of Mps1 can suppress the detrimental effects of mutations in this portion of the Ndc80 complex implies that these mutations result in lethality due to reduced association between Mps1 and the Ndc80 complex. However, it was plausible that the rescue of these mutants by Mps1 overexpression was solely due to the restoration of spindle assembly checkpoint function60–62. This does not appear to be the case, as Mps1 overexpression still suppresses nuf2 mutants even when the SAC is inactivated by deleting MAD3 (Fig S6F). Furthermore, this suppression appears specific to Mps1 as overexpression of Ipl1 does not rescue the cell viability of these nuf2 mutants (Fig S6G). Finally, the ability of Mps1 overexpression to suppress nuf2 mutant growth defects appeared to be dependent on the ability of Mps1 to interact with the Ndc80 complex. While the overexpression of MPS1WT effectively suppresses the cell viability defects of the nuf2 mutants, we found that the overexpression of mps1RLR>EEE or other mutations at the Ndc80 complex interface fail to achieve the same outcome (Fig 6E and Fig S6H–I). These findings indicate that overexpression of Mps1 alone is insufficient to suppress the nuf2 mutants, but instead requires Mps1’s interaction with the Ndc80 complex. Moreover, the observation that certain nuf2 mutants (e.g. nuf2F8A P9A and nuf2N128A) are susceptible to suppression by Mps1 overexpression suggests that these mutants disrupt, but do not entirely eliminate, the ability of Mps1 to bind the Ndc80 complex.
While modulating the levels of Mps1 at the kinetochore appears to suppress the nuf2 CH domain mutants, how do we reconcile the observation that these mutants also display reduced Dam1 complex recruitment (as described in Fig 5A)? A potential explanation is that the localization of the Dam1 complex to the kinetochore is regulated by Mps1 activity, also suggested by previous work31,36,63,64. To investigate this hypothesis, we used a kinetochore co-IP assay56 in which kinetochores were immunoprecipitated, and co-purification of the Dam1 complex was determined by western blot. In line with the notion above, kinetochores purified from mps1-1 cells lacking Mps153 exhibited reduced association with the Dam1 complex (Fig 6F). These findings potentially elucidate how modulating Mps1 levels at the kinetochore can mitigate the effects caused by nuf2 CH domain mutants, which disrupt the interaction hub and presumably lead to the loss of binding of multiple factors. Additionally, these results suggest an active role of Mps1 in recruiting a competing factor, the Dam1 complex, vying for the same kinetochore binding site.
Mps1 is the critical factor recruited to the Ndc80 complex interaction hub
Finally, while modulating Mps1 levels appears to suppress the cell viability defects of the nuf2 CH domain mutants, we wanted to more directly ask if the phenotypes of nuf2 mutants are due specifically to diminished Mps1-kinetochore association. Here, we asked if restoring Mps1 localization, using rapamycin induced dimerization of FRB and FKBP12, would rescue the nuf2 mutant phenotypes (Fig 7A). We generated strains containing NUF2-AID, NDC80-FKBP12 and MPS1-FRB at their endogenous loci, and either NUF2WT or nuf2N128A expressed from an ectopic locus. Since tethering Mps1-FRB to the C-terminus of Ndc80 constitutively activates the SAC, causing cell inviability65, these strains also included a deletion of MAD3. Compellingly, the tethering of Mps1 to Ndc80 effectively restored the viability of nuf2N128A mutants (Fig 7B). These data argue that Mps1 needs to be bound to the Ndc80 complex to carry out its kinetochore biorientation function. In summary, our findings establish a pivotal “interaction hub” within the Ndc80 complex that orchestrates SAC signaling and microtubule attachment. Moreover, it underscores the indispensable role played by the kinetochore-associated pool of Mps1 in ensuring precise chromosome segregation.
Figure 7. Tethering Mps1 to the kinetochore is sufficient to restore nuf2 mutant viability.

(A) Schematic of system to tether Mps1 to the kinetochore. In the presence of rapamycin, Mps1-FRB associates with Ndc80-FKBP12, artificially localizing Mps1 to the Ndc80 complex.
(B) Yeast cell viability assay using the rapamycin-induced tethering system. TOR1–1 fpr1Δ (alone, M1375), followed by strains that also contain MPS1-FRB and NDC80-FKBP12 tethering alleles (MAD3, M1463; mad3Δ, M4116) and NUF2-AID, with no nuf2 covering allele (“None”, M4841) or with ectopic copies of NUF2–3HA (NUF2WT, M4908; nuf2N128A, M4912), and finally a control nuf2N128A strain lacking the NDC80-FKBP12 allele (M4955). Cells were serially diluted five-fold and spotted onto plates containing DMSO, 50 ng/mL rapamycin, 250 μM auxin or 250 μM auxin plus 50 ng/mL rapamycin.
DISCUSSION
In this work, we set out to examine the role that Nuf2’s CH domain plays in the fidelity of chromosome segregation. Through extensive mutational analysis, we pinpointed a conserved “interaction hub” within Nuf2’s CH domain, formed by portions of its N-terminal loop and G-helix. Importantly, this patch serves as the binding site for Mps1 within the yeast Ndc80 complex, but also associates with numerous other factors. Consequently, mutants disrupting this hub exhibit defects in spindle assembly checkpoint function and display severe errors in chromosome segregation. The array of interactions occurring at this specific site on the Ndc80 complex appears critical for the functional regulation of Mps1’s association with the kinetochore. In fact, restoring Mps1-Ndc80 complex association completely rescues the cellular defects associated with mutations at this site. Overall, this work sheds light on mechanisms cells use to regulate Mps1 at the kinetochore, and also underscores the essential role of Mps1 in kinetochore biorientation and precise chromosome segregation.
Mps1 is the critical factor that associates with the Ndc80 complex’s interaction hub
Our work describes a previously uncharacterized region of Nuf2’s CH domain that is responsible for interacting with segments of various proteins. This list encompasses the N-terminus of the kinase Mps1, the C-terminus of the Dam1 protein, potentially the N-terminal tail of the Ndc80 protein, and the N-terminus of the kinase Ipl119,20,42,43 (this work as well as concurrent work by Zahm et al, Pleuger et al; personal communication S. Harrison and S. Westermann), among potentially other yet-to-be-identified factors. Notably, mutations affecting this interaction hub lead to cell inviability and substantial errors in kinetochore biorientation and chromosome segregation. Interestingly, our findings suggest that these severe phenotypes do not result from disrupting the binding of multiple factors to this site. Instead, our observation that specifically restoring Mps1-Ndc80 complex association, whether through overexpression or tethering via FRB-FKBP, implies that Mps1 might be the sole essential factor binding to this interaction hub. It remains possible that Mps1’s role may involve, in part, recruiting and/or regulating these additional factors when bound to the Ndc80 complex. Our observation that Mps1 appears to recruit the Dam1 complex aligns with this latter concept, and is consistent with previous work31,36,63,64. In any case, our identification of the Mps1 binding site on the Ndc80 complex highlights the crucial role that kinetochore-bound Mps1 plays in error correction, in addition to its well-studied function in spindle assembly checkpoint activation. This error correction function of Mps1 is essential for cell viability, revealing a more significant role than we previously recognized.
The activity of Mps1 at the kinetochore is subject to regulation at multiple levels
We propose the following working model, building upon the excellent work of others (reviewed in3,66), outlining the steps from the initial microtubule attachment to biorientation (Fig 8). Prior to initial attachment to spindle microtubules, the Ndc80 complex is in a ‘closed’ conformation with the N-terminal tail of Ndc80 folded back and occluding the association of other factors to this interaction hub43. Both error correction kinases, Ipl1 and Mps1, can phosphorylate the N-terminal tail7,11–13,27–31, which changes Ndc80 to an ‘open’ conformation43, allowing binding of Mps1. Kinetochore-associated Mps1 can now carry out its well-described function in spindle assembly checkpoint activation33–35,37,44,45. Importantly, this pool of Mps1 also appears essential for achieving kinetochore biorientation and ensuring accurate chromosome segregation. This is likely achieved through a tension-sensitive error correction mechanism31–37. However, the precise working mechanism of this process remains unclear (further discussion below). In part, this error correction function of Mps1 may involve recruiting the Dam1 complex31,36,63,64. Regardless, once attachments are correctly bioriented and no longer susceptible to error correction-mediated destabilization, the Dam1 complex establishes stable association with the Ndc80 complex, thereby displacing Mps1 from its binding site (Zahm et al., Pleuger et al.; personal communication S. Harrison and S. Westermann). In this manner, Mps1 seems to facilitate the recruitment of its own competitive inhibitor. Another mechanism through which Mps1 regulates its kinetochore localization is autophosphorylation58,67. Furthermore, competition between Mps1 and microtubules for binding to the Ndc80 complex is proposed as an additional means to displace Mps142,43, although Mps1 binding does not appear to be mutually exclusive to microtubule binding67. These cumulative events subsequently deactivate spindle assembly checkpoint signaling and also likely terminate Mps1’s participation in error correction. While this model provides insights into certain aspects of this process, several crucial questions remain.
Figure 8.
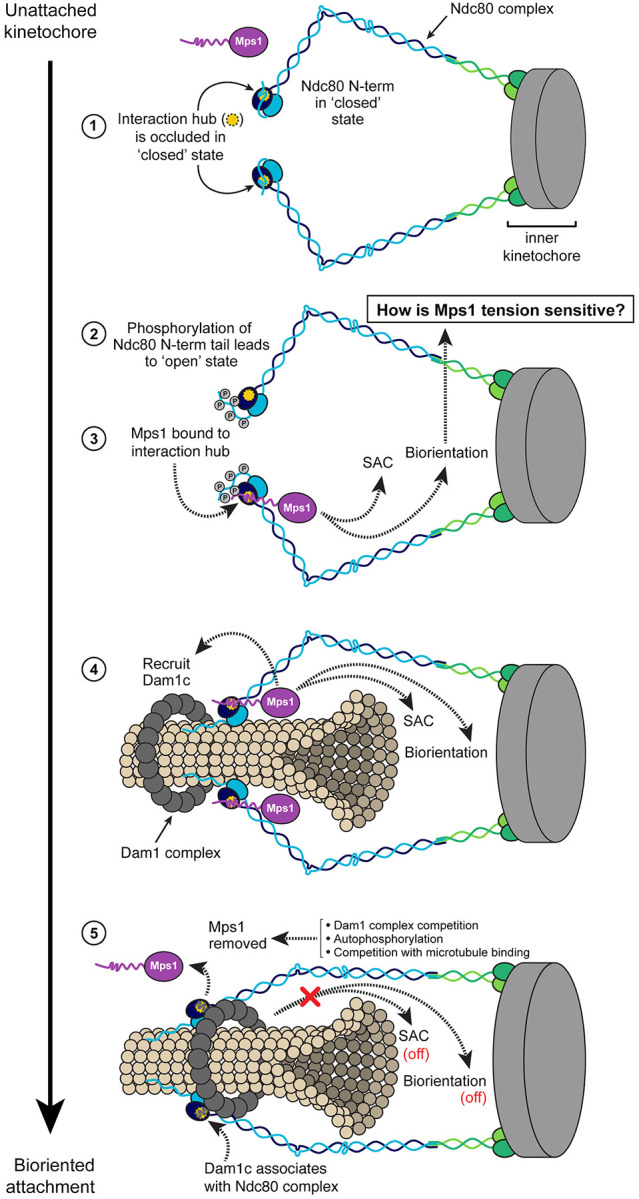
Model of the transition from unattached kinetochore to bioriented attachment, incorporating known protein-protein associations and events that may occur at the interaction hub on Nuf2.
- In the unattached kinetochore state, the N-terminus of Ndc80 folds over Nuf2 to occlude the interaction hub (“closed state”), precluding association of Mps1 with the kinetochore.
- Movement of the Ndc80 N-terminal tail away from the Nuf2 interaction hub (possibly via phosphorylation by Ipl1 or Mps1) leads to the “open state”, allowing access to other factors.
- Mps1 associates with the Ndc80 complex at the Nuf2 hub, triggering the SAC and participating in sister chromatid biorientation through an as yet undetermined mechanism that is likely tension sensitive.
- Mps1 also promotes association of the Dam1 complex with Ndc80 complex, which ultimately allows for stable kinetochore-microtubule interaction and the formation of bioriented attachments.
One key question is how does Mps1’s activity at kinetochores contribute to biorientation? Additionally, what mechanisms regulate these activities in response to tension, a critical signal that cells use to discern correct from incorrect attachments? It has been well-demonstrated that Mps1 phosphorylates sites on the N-terminal tail of Ndc80, leading to attachment destabilization. Notably, Mps1’s phosphorylation of Ndc80 is stimulated on kinetochores lacking tension31. However, disrupting the known Mps1 phosphorylation sites, even in conjunction with a spindle assembly checkpoint mutant, does not substantially impact cell viability30,31. Furthermore, the N-terminal tail of the Ndc80 protein is dispensable for viability in yeast30,68,69. These observations are in contrast to the nuf2 and mps1 mutants described here, which exhibit complete inviability. This discrepancy suggests that additional mechanisms must be involved in this process. Mps1 has been implicated in two other pathways that promote biorientation: the activation of the kinase Bub132,70 and the recruitment of pericentromeric localization of Sgo170–75. The interplay between these pathways, particularly in the context of our new mutants disrupting kinetochore-associated Mps1, requires further investigation. It’s worth noting that Bub1 and Sgo1 are non-essential proteins, and furthermore, the role of tension in these processes remains unclear.
Several key questions surround the regulation of the kinetochore-bound Mps1 pathway by tension. Given the proximity of Mps1 to the microtubule binding domains of the kinetochore, spatial separation of substrates from Mps1, as proposed for Aurora B76, seems unlikely. Instead, tension-dependent changes in substrate conformation, as recently demonstrated for Ipl177, provide a plausible mechanism by which tension suppresses phosphorylation of critical targets. Alternatively, Mps1’s kinase activity or localization could be influenced by a kinetochore factor, potentially a portion of the Ndc80 complex, and this regulation may be responsive to tension. For instance, the displacement of Nuf2’s N-terminal loop from its G-helix (similar to the nuf2S124D mutant) either via tension or another mechanism appears to be an intriguing way that cells could modulate Mps1 activity. This notion gains support from the observation that the assembly of the outer kinetochore is essential for generating most, if not all, cellular Mps1 activity55. Additionally, the interplay between Aurora B and Mps1 activities remains an open question.
In summary, our work identifies a critical “interaction hub” that governs essential kinetochore functions of the kinase Mps1. These functions encompass its broadly studied role in spindle assembly checkpoint signaling, but, notably, emphasize the significance of Mps1 in establishing kinetochore biorientation. Our findings clarify the mechanisms that govern kinetochore-associated Mps1 and how these mechanisms contribute to the fidelity of chromosome segregation.
METHODS
Yeast strains and media
All yeast strains used are listed in Table S1 and are isogenic in the W303 background78. Standard media and microbial techniques79 were used, as well as standard yeast genetic methods for strain construction80,81.
Single integration vectors (SIV) pM93 (HIS3), pM94 (URA3), pM95 (TRP1), and pM96 (LEU2) were provided by Leon Chan and were used in the construction of the NUF2, NDC80, MPS1 and IPL1 plasmids described below. The genomic NUF2-AID allele NUF2–3V5-IAA7:KanMX was constructed using pM66 (3V5-IAA7:KanMX plasmid; Leon Chan) as a template, and subsequent conversion to NUF2–3V5-IAA7:NATMX was performed as in82. A single integration LEU2 vector containing pNUF2-NUF2–3HA (pM604) was constructed by first tagging endogenous NUF2 with 3HA from pM9 (3HA:HIS3MX6)83, then using the pNUF2-NUF2–3HA strain as a template for plasmid construction. Mutants of NUF2 were constructed by mutagenizing pM604 as described in84,85. The NDC80–3V5-IAA7:KanMX is described in39. A TRP1 single integration vector containing pNDC80-NDC80–3HA (pM270) was constructed in a similar manner to NUF2–3HA above, and pNDC80-ndc80ΔN-tail-3HA (pM1342) was subsequently constructed using pM270 as a template for mutagenesis as in84,85.
The MPS1–3V5:KanMX allele was constructed using pM32 (V5:KanMX tagging vector; Leon Chan) as a template, and was marker converted to MPS1–3V5:HphMX as in82. pGAL10-MPS1 and pGAL10-IPl1 HIS3 single integration vectors were constructed by genomic PCR amplification of the respective ORFs, followed by Gibson assembly into pM93, downstream of the pGAL10 promoter86. The pGAL10 promoter was replaced with genomic PCR-amplified native MPS1 promoter to construct pM1595 (pMPS1-MPS1 HIS3 SIV), and subsequent cloning produced pM1718 (pMPS1-MPS1 URA3 SIV). Mutants of MPS1 were constructed in these plasmids by standard cloning or by mutagenizing pM1489, pM1595 and pM1718 as described in84,85. All plasmids and primers are listed in Table S2. Further details regarding plasmid and strain construction are available upon request.
Integration plasmids containing pGPD1-TIR1 (pM74 for integration at LEU2, pM76 for HIS3, and pM78 for TRP1) were provided by Leon Chan; the URA3 version was constructed from pM74 and the pM94 URA3 single integration vector. SPC110-mCherry:HphMX and ASK1-YFP:HIS3 were provided by Trisha Davis, and BUB1-GFP:KanMX, MTW1-mCherry:HphMX by Sue Biggins. Construction of pCUP1-GFP-LacI is described in87, CEN III::lacO:TRP1 is described in88, and CEN IV::lacO:TRP1 was provided by Andrew Murray. Strains containing the previously described pMET-CDC20 allele were provided by Frank Uhlmann89, and the CDC20-AID allele by Adele Marston. mps1-1 and dad1-1 temperature sensitive alleles were provided by Andrew Murray and Sue Biggins, respectively. The pGAL-MPS1-Myc:URA3 and mad2Δ alleles were provided by Andrew Murray, mad3Δ is described in90, and DSN1–6His-3Flag in56. TOR1–1, fpr1Δ, MPS1-FRB:KanMX, and NDC80-FKBP12:HISMX are described in65,91. The Dam1-9Myc allele was provided by Sue Biggins.
Auxin inducible degron
The auxin inducible degron (AID) system was used as described in92. Cells expressed C-terminal fusions of NUF2, NDC80, or CDC20 to the gene for an auxin responsive protein (IAA7) at each respective endogenous locus. Cells also expressed TIR1, which is required for auxin-induced degradation. For cell viability assays, 250 μM auxin (indole-3-acetic acid; Sigma Aldrich) dissolved in DMSO was top-plated on agar to induce degradation of the AID-tagged protein. Cells for microscopy were treated with 500 μM auxin in liquid media at the specified time prior to fixation. Auxin was added to liquid media 2 hours prior to harvesting cells for immunoprecipitation analysis.
Spot dilution yeast viability assays
Desired strains were grown overnight for two days on YPA plus 2% dextrose plates (YPAD). Equal amounts of each strain were then used for a 1:5 dilution series and spotted onto YPAD + DMSO, YPAD + Auxin, YPAD + Auxin and 6.5 μg/mL benomyl, YPA Gal (YPA + 2% galactose) + DMSO, or YPA Gal + Auxin. Plates were incubated at 23°C for 2 to 4 days, unless otherwise indicated for temperature sensitive alleles (30°C or 37°C, as noted in figure legends).
Cell fixation, imaging conditions and image analysis
Conditions for growing cells for each type of imaging are described in brief in the figure legends. For most experiments, cells were grown to log phase in standard YPAD liquid medium at room temperature (approximately 20°C). Where indicated, cells were treated with auxin (500 μM) or auxin and nocodazole (10 μM). In some cases, an α-factor arrest was used to synchronize cells (1 μg/mL for 3 hours at room temperature; using bar1-1 strains), followed by three washes in YPAD + 2% DMSO and resuspension in fresh YPAD media for the release. For the biorientation assay, cells were arrested with α-factor in synthetic liquid medium lacking methionine, then released into the pMET-CDC20 arrest in the presence of methionine at 30°C. 8 mM methionine was subsequently added every thirty minutes until harvest.
Fixation was performed in 3.7% formaldehyde in 100 mM phosphate buffer (pH 6.4) for 5 minutes. Cells were washed once with 100 mM phosphate (pH 6.4) and resuspended in 100 mM phosphate, 1.2 M sorbitol buffer (pH 7.5) and permeabilized with 1% Triton X-100 stained with 1 μg/mL DAPI (4’, 6-diamidino2-phenylindole; Molecular Probes). Cells were imaged with a DeltaVision Ultra microscope with a 60X objective (NA = 1.42), equipped with a sCMOS digital camera. Fifteen Z-stacks (0.3 micron apart) were acquired, and frames were deconvolved using standard settings. Image stacks were maximally projected. softWoRx image processing software was used for image acquisition and processing.
Quantitation of microscopy images was performed as indicated in the figure legends. For the Bub1-GFP assay for SAC activity, only mononucleate cells with a single Mtw1-mCherry signal were counted (indicating collapse of the spindle from nocodazole treatment). Note that nuf2 mutants often increased the number of cells with two Mtw1 signals, despite nocodazole treatment, and also displayed increased bypass of the CDC20-AID arrest (Fig S3A–C). Collectively, this reduced the number of cells in each field of view available for scoring the presence of Bub1-GFP at the kinetochore relative to NUF2WT. Numbers of cells counted are indicated in the figure legend. For the biorientation assay in Fig 3B, only mononucleate cells in metaphase were counted. For chromosome segregation assays in Fig 3C and Fig 4D, only binucleate cells that had undergone anaphase (approximate spindle length ≥ 4 μM) were counted.
Spindle length in Fig S3A was calculated by drawing a line in FIJI (Image J, NIH) that connected the edges of the Spc110-mCherry signals from the two poles. Spindle pole body separation in Fig S4B was calculated by the percentage of cells that displayed two Spc110-mCherry signals at 90 minutes following the temperature shift. Ask1-YFP intensities in Fig 5A were calculated by drawing a line in FIJI through the Spc110 spindle pole signals and performing plot profile analysis of the YFP signal across this line. The maximum peak intensity proximal to each pole was then determined (two signals for each cell, which were sometimes asymmetric, especially in the nuf2 mutant). Data points in the right graph in Fig 5 represent individual Ask1-YFP peak intensities, two for each cell.
Kinetochore immunoprecipitations
Native kinetochores were purified from asynchronously, exponentially growing S. cerevisiae cells containing Dsn1-6His-3Flag by immunoprecipitation with α-Flag, essentially as described in56. Cells were grown in standard YPAD medium. For strains containing NUF2-AID, cells were treated with 500 μM auxin 2 hours prior to harvest. For examining kinetochore enrichment of Dam1-9Myc in WT and mps1-1 strains, cells were shifted to 37°C for 2 hours prior to harvest. Protein lysates were prepared by mechanical disruption in the presence of lysis buffer using glass beads and a beadbeater (Biospec Products). Lysed cells were resuspended in buffer H (BH) (25 mM HEPES pH 8.0, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 0.1% NP-40, 15% glycerol with 150 mM KCl containing protease inhibitors (at 20 μg/mL final concentration for each of leupeptin, pepstatin A, chymostatin and 200 μM phenylmethylsulfonyl fluoride) and phosphatase inhibitors (0.1 mM Na-orthovanadate, 0.2 μM microcystin, 2 mM β-glycerophosphate, 1 mM Na pyrophosphate, 5 mM NaF) followed by centrifugation at 16,100 g for 30 min at 4°C to clarify the lysate. Dynabeads conjugated with α-Flag antibodies were incubated with extract for 3 hours with constant rotation, followed by three washes with BH containing protease inhibitors, phosphatase inhibitors, 2 mM dithiothreitol (DTT) and 150 mM KCl. Beads were further washed twice with BH containing 150 mM KCl and protease inhibitors. Associated proteins were eluted from the beads by boiling in 2x SDS sample buffer.
Immunoblotting
For immunoblot analysis in Fig S1C, cell lysates were prepared by mechanical disruption with glass beads in 2x SDS-PAGE sample buffer (0.4mM EDTA, 4% SDS, 125 mM Tris pH6.8, 20% glycerol, 2% bromophenol blue). Lysates for immunoprecipitation were described above. Standard procedures for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were followed as described in93,94. A nitrocellulose membrane (Bio-Rad) was used to transfer proteins from polyacrylamide gels. Commercial monoclonal antibodies used for immunoblotting were as follows: α-HA, 12CA5 (Roche, 1:2000), α-Flag, M2 (Sigma Aldrich, 1:3000), α-V5 (Invitrogen; 1:5000), α-Pgk1 (Abcam; 1:5000). The Ndc80 polyclonal antibody was a gift from Arshad Desai and was used at 1:10,000. Secondary antibodies used were a sheep anti-mouse and goat anti-rabbit conjugated to horseradish peroxidase (HRP, GE Biosciences; 1:10,000 dilution). Antibodies were detected using the SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific).
Statistics
GraphPad Prism version 10 was used for statistical analysis. Data normality was assumed for all experiments. Student’s t test was used for comparisons between strains, as indicated in the figure legends. In the text, mean ± standard deviation are reported.
Multiple sequence alignments
Fungal Nuf2 proteins were identified using a PSI-BLAST95 search on NCBI. Multiple sequence alignments of the entire proteins were generated with ClustalOmega default parameters and displayed in JalView 296. Species are listed in the figures and/or legends. Conservation scores shown on structures were generated in ChimeraX 1.5, using an entropy-based measure from AL2CO97.
Supplementary Material
ACKNOWLEDGEMENTS
The authors declare no competing financial interests. We are grateful to Stefan Westermann, Richard Pleuger, Steve Harrison and Jake Zahm for sharing data prior to publication. We thank Sue Biggins, Leon Chan, Trisha Davis, Adèle Marston, Michael Stewart, Andrew Murray, and Frank Uhlmann for providing plasmids and/or strains and Arshad Desai for providing antibodies. In addition, we would like to thank members of the Miller lab, Stefan Westermann and Steve Harrison for helpful discussions and critical reading of the manuscript. This work was supported by 5 For the Fight (to M.P.M), Pew Biomedical Scholars (to M.P.M), and NIH grant R35GM142749 (to M.P.M).
REFERENCES
- 1.Musacchio A., and Desai A. (2017). A Molecular View of Kinetochore Assembly and Function. Biology (Basel) 6, 5. 10.3390/biology6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monda J.K., and Cheeseman I.M. (2018). The kinetochore-microtubule interface at a glance. J Cell Sci 131, jcs214577. 10.1242/jcs.214577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McAinsh A.D., and Kops G.J.P.L. (2023). Principles and dynamics of spindle assembly checkpoint signalling. Nat Rev Mol Cell Biol 24, 543–559. 10.1038/s41580-023-00593-z. [DOI] [PubMed] [Google Scholar]
- 4.Wigge P.A., and Kilmartin J.V. (2001). The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol 152, 349–360. 10.1083/jcb.152.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCleland M.L., Gardner R.D., Kallio M.J., Daum J.R., Gorbsky G.J., Burke D.J., and Stukenberg P.T. (2003). The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev 17, 101–114. 10.1101/gad.1040903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alushin G.M., Ramey V.H., Pasqualato S., Ball D.A., Grigorieff N., Musacchio A., and Nogales E. (2010). The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467, 805–810. 10.1038/nature09423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciferri C., Pasqualato S., Screpanti E., Varetti G., Santaguida S., Dos Reis G., Maiolica A., Polka J., De Luca J.G., De Wulf P., et al. (2008). Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 133, 427–439. 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei R.R., Sorger P.K., and Harrison S.C. (2005). Molecular organization of the Ndc80 complex, an essential kinetochore component. Proc Natl Acad Sci U S A 102, 5363–5367. 10.1073/pnas.0501168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson-Kubalek E.M., Cheeseman I.M., Yoshioka C., Desai A., and Milligan R.A. (2008). Orientation and structure of the Ndc80 complex on the microtubule lattice. J Cell Biol 182, 1055–1061. 10.1083/jcb.200804170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lampert F., and Westermann S. (2011). A blueprint for kinetochores - new insights into the molecular mechanics of cell division. Nat Rev Mol Cell Biol 12, 407–412. 10.1038/nrm3133. [DOI] [PubMed] [Google Scholar]
- 11.Wei R.R., Al-Bassam J., and Harrison S.C. (2007). The Ndc80/HEC1 complex is a contact point for kinetochore-microtubule attachment. Nat Struct Mol Biol 14, 54–59. 10.1038/nsmb1186. [DOI] [PubMed] [Google Scholar]
- 12.Guimaraes G.J., Dong Y., McEwen B.F., and Deluca J.G. (2008). Kinetochore-microtubule attachment relies on the disordered N-terminal tail domain of Hec1. Curr Biol 18, 1778–1784. 10.1016/j.cub.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller S.A., Johnson M.L., and Stukenberg P.T. (2008). Kinetochore attachments require an interaction between unstructured tails on microtubules and Ndc80(Hec1). Curr Biol 18, 1785–1791. 10.1016/j.cub.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tooley J.G., Miller S.A., and Stukenberg P.T. (2011). The Ndc80 complex uses a tripartite attachment point to couple microtubule depolymerization to chromosome movement. Mol Biol Cell 22, 1217–1226. 10.1091/mbc.E10-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundin L.J.R., Guimaraes G.J., and Deluca J.G. (2011). The NDC80 complex proteins Nuf2 and Hec1 make distinct contributions to kinetochore-microtubule attachment in mitosis. Mol Biol Cell 22, 759–768. 10.1091/mbc.E10-08-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alushin G.M., Musinipally V., Matson D., Tooley J., Stukenberg P.T., and Nogales E. (2012). Multimodal microtubule binding by the Ndc80 kinetochore complex. Nat Struct Mol Biol 19, 1161–1167. 10.1038/nsmb.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tien J.F., Umbreit N.T., Gestaut D.R., Franck A.D., Cooper J., Wordeman L., Gonen T., Asbury C.L., and Davis T.N. (2010). Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J Cell Biol 189, 713–723. 10.1083/jcb.200910142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez A., Kim J.O., Umbreit N.T., Asbury C.L., Davis T.N., Miller M.P., and Biggins S. (2020). Cdk1 Phosphorylation of the Dam1 Complex Strengthens Kinetochore-Microtubule Attachments. Curr Biol 30, 4491–4499.e5. 10.1016/j.cub.2020.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zahm J.A., Jenni S., and Harrison S.C. (2023). Structure of the Ndc80 complex and its interactions at the yeast kinetochore-microtubule interface. Open Biol 13, 220378. 10.1098/rsob.220378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muir K.W., Batters C., Dendooven T., Yang J., Zhang Z., Burt A., and Barford D. (2023). Mechanism of outer kinetochore assembly on microtubules and its regulation by mitotic error correction. Preprint at bioRxiv, 10.1101/2023.07.20.549907 10.1101/2023.07.20.549907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J.O., Zelter A., Umbreit N.T., Bollozos A., Riffle M., Johnson R., MacCoss M.J., Asbury C.L., and Davis T.N. (2017). The Ndc80 complex bridges two Dam1 complex rings. Elife 6, e21069. 10.7554/eLife.21069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flores R.L., Peterson Z.E., Zelter A., Riffle M., Asbury C.L., and Davis T.N. (2022). Three interacting regions of the Ndc80 and Dam1 complexes support microtubule tip-coupling under load. J Cell Biol 221, e202107016. 10.1083/jcb.202107016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Hooff J.J.E., Snel B., and Kops G.J.P.L. (2017). Unique Phylogenetic Distributions of the Ska and Dam1 Complexes Support Functional Analogy and Suggest Multiple Parallel Displacements of Ska by Dam1. Genome Biol Evol 9, 1295–1303. 10.1093/gbe/evx088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huis In ‘t Veld P.J., Volkov V.A., Stender I.D., Musacchio A., and Dogterom M. (2019). Molecular determinants of the Ska-Ndc80 interaction and their influence on microtubule tracking and force-coupling. Elife 8, e49539. 10.7554/eLife.49539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helgeson L.A., Zelter A., Riffle M., MacCoss M.J., Asbury C.L., and Davis T.N. (2018). Human Ska complex and Ndc80 complex interact to form a load-bearing assembly that strengthens kinetochore-microtubule attachments. Proc Natl Acad Sci U S A 115, 2740–2745. 10.1073/pnas.1718553115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maciejowski J., Drechsler H., Grundner-Culemann K., Ballister E.R., Rodriguez-Rodriguez J.-A., Rodriguez-Bravo V., Jones M.J.K., Foley E., Lampson M.A., Daub H., et al. (2017). Mps1 Regulates Kinetochore-Microtubule Attachment Stability via the Ska Complex to Ensure Error-Free Chromosome Segregation. Dev Cell 41, 143–156.e6. 10.1016/j.devcel.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akiyoshi B., Nelson C.R., Ranish J.A., and Biggins S. (2009). Analysis of Ipl1-mediated phosphorylation of the Ndc80 kinetochore protein in Saccharomyces cerevisiae. Genetics 183, 1591–1595. 10.1534/genetics.109.109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheeseman I.M., Chappie J.S., Wilson-Kubalek E.M., and Desai A. (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997. 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 29.DeLuca J.G., Gall W.E., Ciferri C., Cimini D., Musacchio A., and Salmon E.D. (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982. 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 30.Kemmler S., Stach M., Knapp M., Ortiz J., Pfannstiel J., Ruppert T., and Lechner J. (2009). Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J 28, 1099–1110. 10.1038/emboj.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarangapani K.K., Koch L.B., Nelson C.R., Asbury C.L., and Biggins S. (2021). Kinetochore-bound Mps1 regulates kinetochore-microtubule attachments via Ndc80 phosphorylation. J Cell Biol 220, e202106130. 10.1083/jcb.202106130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benzi G., Camasses A., Atsunori Y., Katou Y., Shirahige K., and Piatti S. (2020). A common molecular mechanism underlies the role of Mps1 in chromosome biorientation and the spindle assembly checkpoint. EMBO Rep 21, e50257. 10.15252/embr.202050257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hewitt L., Tighe A., Santaguida S., White A.M., Jones C.D., Musacchio A., Green S., and Taylor S.S. (2010). Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol 190, 25–34. 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maciejowski J., George K.A., Terret M.-E., Zhang C., Shokat K.M., and Jallepalli P.V. (2010). Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol 190, 89–100. 10.1083/jcb.201001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maure J.-F., Kitamura E., and Tanaka T.U. (2007). Mps1 kinase promotes sisterkinetochore bi-orientation by a tension-dependent mechanism. Curr Biol 17, 2175–2182. 10.1016/j.cub.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer R.E., Kim S., Obeso D., Straight P.D., Winey M., and Dawson D.S. (2013). Mps1 and Ipl1/Aurora B act sequentially to correctly orient chromosomes on the meiotic spindle of budding yeast. Science 339, 1071–1074. 10.1126/science.1232518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santaguida S., Tighe A., D’Alise A.M., Taylor S.S., and Musacchio A. (2010). Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190, 73–87. 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayward D., Roberts E., and Gruneberg U. (2022). MPS1 localizes to end-on microtubule-attached kinetochores to promote microtubule release. Curr Biol 32, 5200–5208.e8. 10.1016/j.cub.2022.10.047. [DOI] [PubMed] [Google Scholar]
- 39.Miller M.P., Asbury C.L., and Biggins S. (2016). A TOG Protein Confers Tension Sensitivity to Kinetochore-Microtubule Attachments. Cell 165, 1428–1439. 10.1016/j.cell.2016.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahm J.A., Stewart M.G., Carrier J.S., Harrison S.C., and Miller M.P. (2021). Structural basis of Stu2 recruitment to yeast kinetochores. Elife 10, e65389. 10.7554/eLife.65389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herman J.A., Miller M.P., and Biggins S. (2020). chTOG is a conserved mitotic error correction factor. Elife 9, e61773. 10.7554/eLife.61773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiruma Y., Sacristan C., Pachis S.T., Adamopoulos A., Kuijt T., Ubbink M., von Castelmur E., Perrakis A., and Kops G.J.P.L. (2015). CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 348, 1264–1267. 10.1126/science.aaa4055. [DOI] [PubMed] [Google Scholar]
- 43.Ji Z., Gao H., and Yu H. (2015). CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 348, 1260–1264. 10.1126/science.aaa4029. [DOI] [PubMed] [Google Scholar]
- 44.Jelluma N., Brenkman A.B., van den Broek N.J.F., Cruijsen C.W.A., van Osch M.H.J., Lens S.M.A., Medema R.H., and Kops G.J.P.L. (2008). Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132, 233–246. 10.1016/j.cell.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 45.Jones M.H., Huneycutt B.J., Pearson C.G., Zhang C., Morgan G., Shokat K., Bloom K., and Winey M. (2005). Chemical genetics reveals a role for Mps1 kinase in kinetochore attachment during mitosis. Curr Biol 15, 160–165. 10.1016/j.cub.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Yamagishi Y., Yang C.-H., Tanno Y., and Watanabe Y. (2012). MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat Cell Biol 14, 746–752. 10.1038/ncb2515. [DOI] [PubMed] [Google Scholar]
- 47.Yuan I., Leontiou I., Amin P., May K.M., Soper Ní Chafraidh S., Zlámalová E., and Hardwick K.G. (2017). Generation of a Spindle Checkpoint Arrest from Synthetic Signaling Assemblies. Curr Biol 27, 137–143. 10.1016/j.cub.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayward D., Bancroft J., Mangat D., Alfonso-Pérez T., Dugdale S., McCarthy J., Barr F.A., and Gruneberg U. (2019). Checkpoint signaling and error correction require regulation of the MPS1 T-loop by PP2A-B56. J Cell Biol 218, 3188–3199. 10.1083/jcb.201905026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valverde R., Ingram J., and Harrison S.C. (2016). Conserved Tetramer Junction in the Kinetochore Ndc80 Complex. Cell Rep 17, 1915–1922. 10.1016/j.celrep.2016.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roberts B.T., Farr K.A., and Hoyt M.A. (1994). The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol Cell Biol 14, 8282–8291. 10.1128/mcb.14.12.8282-8291.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillett E.S., Espelin C.W., and Sorger P.K. (2004). Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J Cell Biol 164, 535–546. 10.1083/jcb.200308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krenn V., Wehenkel A., Li X., Santaguida S., and Musacchio A. (2012). Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. J Cell Biol 196, 451–467. 10.1083/jcb.201110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.London N., Ceto S., Ranish J.A., and Biggins S. (2012). Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol 22, 900–906. 10.1016/j.cub.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Straight A.F., Belmont A.S., Robinett C.C., and Murray A.W. (1996). GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol 6, 1599–1608. 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- 55.Kuijt T.E.F., Lambers M.L.A., Weterings S., Ponsioen B., Bolhaqueiro A.C.F., Staijen D.H.M., and Kops G.J.P.L. (2020). A Biosensor for the Mitotic Kinase MPS1 Reveals Spatiotemporal Activity Dynamics and Regulation. Curr Biol 30, 3862–3870.e6. 10.1016/j.cub.2020.07.062. [DOI] [PubMed] [Google Scholar]
- 56.Akiyoshi B., Sarangapani K.K., Powers A.F., Nelson C.R., Reichow S.L., Arellano-Santoyo H., Gonen T., Ranish J.A., Asbury C.L., and Biggins S. (2010). Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature 468, 576–579. 10.1038/nature09594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Araki Y., Gombos L., Migueleti S.P.S., Sivashanmugam L., Antony C., and Schiebel E. (2010). N-terminal regions of Mps1 kinase determine functional bifurcation. J Cell Biol 189, 41–56. 10.1083/jcb.200910027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pleuger R., Cozma C., Hohoff S., Denkhaus C., Dudziak A., Kaschani F., Musacchio A., Vetter I.R., and Westermann S. (2023). Ipl1-controlled attachment maturation regulates Mps1 association with its kinetochore receptor. Preprint at bioRxiv, 10.1101/2023.10.30.564738 10.1101/2023.10.30.564738. [DOI] [PubMed] [Google Scholar]
- 59.Schutz A.R., and Winey M. (1998). New alleles of the yeast MPS1 gene reveal multiple requirements in spindle pole body duplication. Mol Biol Cell 9, 759–774. 10.1091/mbc.9.4.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hardwick K.G., Weiss E., Luca F.C., Winey M., and Murray A.W. (1996). Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 273, 953–956. 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- 61.Palframan W.J., Meehl J.B., Jaspersen S.L., Winey M., and Murray A.W. (2006). Anaphase inactivation of the spindle checkpoint. Science 313, 680–684. 10.1126/science.1127205. [DOI] [PubMed] [Google Scholar]
- 62.Fraschini R., Beretta A., Lucchini G., and Piatti S. (2001). Role of the kinetochore protein Ndc10 in mitotic checkpoint activation in Saccharomyces cerevisiae. Mol Genet Genomics 266, 115–125. 10.1007/s004380100533. [DOI] [PubMed] [Google Scholar]
- 63.Shimogawa M.M., Graczyk B., Gardner M.K., Francis S.E., White E.A., Ess M., Molk J.N., Ruse C., Niessen S., Yates J.R., et al. (2006). Mps1 phosphorylation of Dam1 couples kinetochores to microtubule plus ends at metaphase. Curr Biol 16, 1489–1501. 10.1016/j.cub.2006.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer R.E., Brown J., Beck L., and Dawson D.S. (2018). Mps1 promotes chromosome meiotic chromosome biorientation through Dam1. Mol Biol Cell 29, 479–489. 10.1091/mbc.E17-08-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aravamudhan P., Goldfarb A.A., and Joglekar A.P. (2015). The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat Cell Biol 17, 868–879. 10.1038/ncb3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pachis S.T., and Kops G.J.P.L. (2018). Leader of the SAC: molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol 8, 180109. 10.1098/rsob.180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koch L.B., Opoku K.N., Deng Y., Barber A., Littleton A.J., London N., Biggins S., and Asbury C.L. (2019). Autophosphorylation is sufficient to release Mps1 kinase from native kinetochores. Proc Natl Acad Sci U S A 116, 17355–17360. 10.1073/pnas.1901653116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lampert F., Hornung P., and Westermann S. (2010). The Dam1 complex confers microtubule plus end-tracking activity to the Ndc80 kinetochore complex. J Cell Biol 189, 641–649. 10.1083/jcb.200912021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Demirel P.B., Keyes B.E., Chaterjee M., Remington C.E., and Burke D.J. (2012). A redundant function for the N-terminal tail of Ndc80 in kinetochore-microtubule interaction in Saccharomyces cerevisiae. Genetics 192, 753–756. 10.1534/genetics.112.143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Storchová Z., Becker J.S., Talarek N., Kögelsberger S., and Pellman D. (2011). Bub1, Sgo1, and Mps1 mediate a distinct pathway for chromosome biorientation in budding yeast. Mol Biol Cell 22, 1473–1485. 10.1091/mbc.E10-08-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fernius J., and Hardwick K.G. (2007). Bub1 kinase targets Sgo1 to ensure efficient chromosome biorientation in budding yeast mitosis. PLoS Genet 3, e213. 10.1371/journal.pgen.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Breit C., Bange T., Petrovic A., Weir J.R., Müller F., Vogt D., and Musacchio A. (2015). Role of Intrinsic and Extrinsic Factors in the Regulation of the Mitotic Checkpoint Kinase Bub1. PLoS One 10, e0144673. 10.1371/journal.pone.0144673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nerusheva O.O., Galander S., Fernius J., Kelly D., and Marston A.L. (2014). Tension-dependent removal of pericentromeric shugoshin is an indicator of sister chromosome biorientation. Genes Dev 28, 1291–1309. 10.1101/gad.240291.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peplowska K., Wallek A.U., and Storchova Z. (2014). Sgo1 regulates both condensin and Ipl1/Aurora B to promote chromosome biorientation. PLoS Genet 10, e1004411. 10.1371/journal.pgen.1004411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Indjeian V.B., Stern B.M., and Murray A.W. (2005). The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science 307, 130–133. 10.1126/science.1101366. [DOI] [PubMed] [Google Scholar]
- 76.Liu D., Vader G., Vromans M.J.M., Lampson M.A., and Lens S.M.A. (2009). Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 323, 1350–1353. 10.1126/science.1167000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Regt A.K., Clark C.J., Asbury C.L., and Biggins S. (2022). Tension can directly suppress Aurora B kinase-triggered release of kinetochore-microtubule attachments. Nat Commun 13, 2152. 10.1038/s41467-022-29542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thomas B.J., and Rothstein R. (1989). Elevated recombination rates in transcriptionally active DNA. Cell 56, 619–630. 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 79.Sherman F., Fink G., and Lawrence C. (1974). Methods in Yeast Genetics: Laboratory Manual (Cold Spring Harbor Laboratory; ). [Google Scholar]
- 80.Rothstein R. (1991). Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol 194, 281–301. 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 81.Sherman F. (1991). Getting started with yeast. Methods Enzymol 194, 3–21. 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 82.Lorenz A. (2015). New cassettes for single-step drug resistance and prototrophic marker switching in fission yeast. Yeast 32, 703–710. 10.1002/yea.3097. [DOI] [PubMed] [Google Scholar]
- 83.Longtine M.S., McKenzie A., Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., and Pringle J.R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. . [DOI] [PubMed] [Google Scholar]
- 84.Liu H., and Naismith J.H. (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91. 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tseng W.-C., Lin J.-W., Wei T.-Y., and Fang T.-Y. (2008). A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal Biochem 375, 376–378. 10.1016/j.ab.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 86.Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., and Smith H.O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 87.Biggins S., Severin F.F., Bhalla N., Sassoon I., Hyman A.A., and Murray A.W. (1999). The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev 13, 532–544. 10.1101/gad.13.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shonn M.A., Murray A.L., and Murray A.W. (2003). Spindle checkpoint component Mad2 contributes to biorientation of homologous chromosomes. Curr Biol 13, 1979–1984. 10.1016/j.cub.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 89.Uhlmann F., Wernic D., Poupart M.A., Koonin E.V., and Nasmyth K. (2000). Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103, 375–386. 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 90.Pinsky B.A., Kung C., Shokat K.M., and Biggins S. (2006). The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol 8, 78–83. 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- 91.Haruki H., Nishikawa J., and Laemmli U.K. (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31, 925–932. 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 92.Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., and Kanemaki M. (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922. 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 93.Burnette W.N. (1981). “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112, 195–203. 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- 94.Towbin H., Staehelin T., and Gordon J. (1992). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology 24, 145–149. [PubMed] [Google Scholar]
- 95.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., and Lipman D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., and Barton G.J. (2009). Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pei J., and Grishin N.V. (2001). AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712. 10.1093/bioinformatics/17.8.700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.