

Abstract
Background
Alternative splicing (AS) is an omnipresent regulatory mechanism of gene expression that enables the generation of diverse splice isoforms from a single gene. Recently, AS events have gained considerable momentum in the pathogenesis of inflammatory bowel disease (IBD).
Methods
Our review has summarized the complex process of RNA splicing, and firstly highlighted the potential involved molecules that target aberrant splicing events in IBD. The quantitative transcriptome analyses such as microarrays, next‐generation sequencing (NGS) for AS events in IBD have been also discussed.
Results
Available evidence suggests that some abnormal splicing RNAs can lead to multiple intestinal disorders during the onset of IBD as well as the progression to colitis‐associated cancer (CAC), including gut microbiota perturbations, intestinal barrier dysfunctions, innate/adaptive immune dysregulations, pro‐fibrosis activation and some other risk factors. Moreover, current data show that the advanced technologies, including microarrays and NGS, have been pioneeringly employed to screen the AS candidates and elucidate the potential regulatory mechanisms of IBD. Besides, other biotechnological progresses such as the applications of third‐generation sequencing (TGS), single‐cell RNA sequencing (scRNA‐seq) and spatial transcriptomics (ST), will be desired with great expectations.
Conclusions
To our knowledge, the current review is the first one to evaluate the potential regulatory mechanisms of AS events in IBD. The expanding list of aberrantly spliced genes in IBD along with the developed technologies provide us new clues to how IBD develops, and how these important AS events can be explored for future treatment.
Keywords: alternative splicing, basic mechanisms, biotechnological progresses, inflammatory bowel disease, perspectives
Our review have summarised aberrant splicing events and mechanisms in the pathogenesis of IBD.
The progresses of AS detection in IBD patients are systematically described, and challenges of technological innovation are also raised.
These AS‐related genes provide us novel clues how IBD develops, and how these important AS events can be further elucidated for future treatment.
1. INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic inflammatory‐mediated disorder, which is clinically characterised by ulcerative colitis (UC), Crohn's disease (CD) and some other conditions. 1 In clinical practice, the main features of gastrointestinal tract inflammation in IBD patients include episodes of diarrhoea, bloody stools, abdominal pain, weight loss, fever and other immune‐related extraintestinal manifestations. 1 , 2 , 3 As the disease progresses, most of IBD patients may experience some more severe complications, including strictures, abscesses, fistulas, perforation and others, which may cause significant morbidity. Notably, long‐term colitis in patients with IBD can also lead to colon cancer called colitis‐associated cancer (CAC). 4 , 5 IBD is now considered as an unignorable life‐threatening disease in people of all ages, including children, adults, pregnant and geriatric populations, and often significantly reduces the quality of life. More depressingly, the global morbidity of IBD has increased rapidly over the past 20 years from developing and recently developed countries. 6 , 7 , 8 , 9 , 10 Over the past decades, considerable progresses have been made to greatly improve our knowledge of the pathophysiology of this disease. It is thought that IBD results from the individual's genetic susceptibility, defects in barrier function and an aberrant and continuing immune response to intestinal microbial flora. 1 , 11 Besides, other factors, including autophagy, reactive oxygen species production, endoplasmic reticulum stress and metabolic pathways associating with cellular homeostasis are also closely correlated with the development of IBD. 12 , 13 Recent studies using multi‐sampling and data integration combined with other novel techniques, such as whole‐genome sequencing, RNA sequencing, single nucleotide polymorphism arrays, genome‐wide methylation analysis and meta‐analyse, can help us better explain the cause of IBD. 14 , 15 , 16 , 17 However, we still have not fully elucidated this complex pathogenetic process. Therefore, it is an urgent need for investigators to reveal the detail pathogenesis of IBD and prevent the development of this costly and disturbing disease.
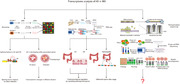
Alternative splicing (AS) is an omnipresent regulatory mechanism of gene expression that enables the generation of diverse splice isoforms from a single gene, 18 which contributes to the diversity of proteomes in >90% of human genes. During the constitutive splicing, each intron will be removed and the remained exons can be joined together to produce a mature mRNA. Compared with the constitutive splice, AS is far more complex. The process is performed by the spliceosome, which is a big complex consisting of 5 ribonucleoproteins (RNPs) involving the small nuclear RNA U1, U2, U4, U5, U6 and multiple auxiliary proteins cooperating to precisely recognise the splicing sites and catalyse the two splicing reaction steps. 18 , 19 First of all, the splicing process starts with the identification of the 5′ splicing site by the snRNP U1 and the combination of the splicing factor 1 (SF1) with the branch point 3 and of the U2 auxiliary factor (U2AF) heterodimer with the 3′ terminal AG and polypyrimidine tract. This assembly contributes to the E complex formation, which can be transformed to an ATP‐reliant, pre‐spliceosome A complex after replacing SF1 with the U2 snRNP at the branch site. Subsequently, the recruitment of U4/U6–U5 tri‐snRNP complex causes the B complex formation. These changes of complex contain the release of U1 and U4 and the formation of intron lariat which is known as the result of the first splicing catalytic step. Finally, the excised intron lariat is degraded, and U2, U5 and U6 snRNPs can be released to produce mature mRNA, which is known as the result of the second splicing catalytic step. 19 , 20 In addition, the splicing of pre‐mRNA is also regulated by splicing regulatory factors that serve as regulatory proteins and bind to the position which are able to enhance or silence the splicing process. There are four binding domains termed exonic splicing silencers, intronic splicing silencers, exonic splicing enhancers and intronic splicing enhancers. 21 Two primary families of splicing regulatory factors are characterised by heterogeneous nuclear RNPs (hnRNPs) or Ser/Arg‐rich proteins (SRs), which exert either inhibitory or activating effects on the recognition and usage of binding site (Figure 1A). 22 , 23 As shown in Figure 1B, the main AS patterns are divided into seven types: exon skipping (also called cassette exon), alternative 3′splice site selection, alternative 5′splice site selection, alternative first exon, alternative last exon, intron retention and mutually exclusive exons. 18 , 21
FIGURE 1.
The process and diverse patterns of AS. (A) The splicing process undergoes sequential phosphodiester transfer reactions, which is catalysed by spliceosomes, including five snRNPs U1, U2, U4, U5 and U6 as well as splicing factors. The snRNAs cooperate to recognise the splice sites and catalyse the two steps of splicing reaction. (B) The diverse patterns of AS include exon skipping, alternative 3′SS selection, alternative 5′SS selection, alternative first exon, alternative last exon, intron retention and mutually exclusive exons.
In normal circumstances, AS can be heavily regulated. However, like most pathophysiological processes, AS is open to faults, which can change the functions of proteins and lead to a vast repertoire of diseases. For instance, AS has been proved to account for multiple neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease and spinal muscular atrophy. 24 , 25 ‘Angiogenesis’ is defined as the constitution of new capillary blood vessels from pre‐existing micro vasculatures and regulated by a variety of factors. 26 , 27 In 2019, Bowler et al. 28 uncovered the potential AS mechanisms in angiogenesis and summarised the alternative spliced isoforms of essential genes that were involved in the process of angiogenesis. Defects in AS are frequently found and closely associated with the occurrence of human tumours, and abnormal changes of AS can also affect the cancer progression. 29 , 30 Recently, a study from Zhou et al. 31 preliminarily discussed all splicing defects both in adults and paediatric IBD. Our review have summarised the complex process of RNA splicing, and first highlighted involved molecules that target aberrant splicing events from a view of the basic mechanisms of IBD. The expanding list of aberrantly spliced genes in IBD along with the developed technologies provide us novel clues to how IBD develops, and how these important AS events can be further elucidated for future treatment.
2. HOW AS EVENTS PARTICIPATE IN THE PATHOGENESIS OF IBD?
2.1. AS and intestinal microbiota
The mammalian gastrointestinal tract can be considered as a suitable habitat for an enormous and interconnected community of microorganisms. The complex aggregation of microbes in gut, including fungi, viruses, protozoans and bacteria, is termed as intestinal microbiota. A great deal of clinical and experimental data have confirmed that alterations in microbial communities can promote the intestinal damage and play a primary role in the occurrence, progression and treatment of IBD. 32 , 33 The gut microbiota composition in IBD patients has been reported to be remarkably distinct from that of healthy individuals. 34 , 35 Moreover, the gut microbiota‐originated metabolites, including bile acids, short‐chain fatty acids, tryptophan metabolites and signals from microbial metabolites, can help opportunistic pathogens colonise and invade to the gut, increase the risk of dysregulated host responses and finally lead to the initiation of IBD. 36 , 37 , 38
Notably, several studies have found that defects in RNA splicing are also linked to the dysbiosis of intestinal microbiota and contribute partly to the pathogenesis of IBD. Intestinal epithelia express two myosin light chain kinase (MLCK) splice variants (the full‐length and shorter isoform MLCK2). In IBD patients, the pro‐inflammatory cytokine tumour necrosis factor (TNF)‐like 1A (TL1A) was demonstrated to activate the phosphatidylinositol 3‐kinase/protein kinase B (AKT) signals and up‐regulate the MLCK splice variant (MLCK2), which might induce the MLCK‐mediated terminal web contraction, and invoke bacterial internalisation (Figure 2A). 39 Cao et al. 40 showed that Enterotoxigenic Bacteroides fragilis (ETBF, a subtype of B fragilis) was closely relevant to the occurrence of IBD and CAC. ETBF‐infected cells could significantly down‐regulate miR‐149‐3p depending on the METTL14‐mediated N6‐methyladenosine methylation, which further increased the PHD finger protein 5A (PHF5A, a splicing modulator interacting with SF3b complex) expression, and promoted the RNA AS of Lysine acetyltransferase 2A (KAT2A) in CRC cells. The exon 8 of KAT2A was less frequently skipped after ETBF‐treatment and directly bound to the superoxide dismutase 2 (SOD2) promoter region, thus transactivating SOD2 and leading to cell proliferation (Figure 2B). 40 Heterochromatin Protein 1γ (HP1γ) is a protein which can safeguard the RNA splicing accuracy in the intestinal epithelium and reduce the impact of naturally occurring non‐canonical spicing events (spicing noise). In UC patients and mice, the HP1γ gene inactivation was proved to broadly increase splicing noise, lead to more opportunities of lamin A mRNA splice variants (progerin) and finally result in gut homeostasis rupture and trigger the IBD‐like traits (Figure 2C). 41
FIGURE 2.
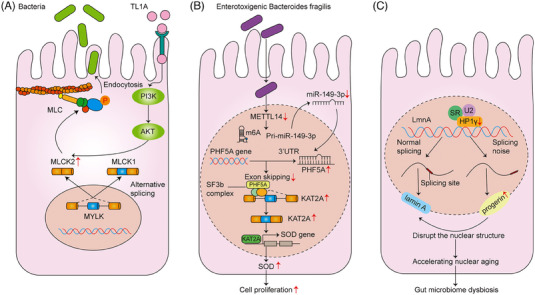
AS and intestinal microbiota. (A) The pro‐inflammatory cytokine TL1A activates the PI3/AKT pathway and up‐regulates the MLCK splice variant (MLCK2), which finally leads to MLCK‐mediated terminal web contraction, and invokes bacterial internalisation. (B) ETBF‐infected epithelial cells can down‐regulate miR‐149‐3p depending on the METTL14‐mediated N6‐methyladenosine methylation, increase the PHF5A expression and promote the RNA alternative splicing of KAT2A. The exon 8 of KAT2A is less frequently skipped after ETBF‐treatment, and can directly binds to the SOD2 promoter region, thus transactivating SOD2 and leading to cell proliferation. (C) The HP1γ gene inactivation is proved to broadly increase splicing noise, lead to more opportunities of lamin A mRNA splice variants (progerin), and finally result in gut homeostasis rupture and trigger the IBD‐like traits.
2.2. AS and intestinal epithelial barrier function
The gastrointestinal mucosa constitutes an essential barrier that serves as nutrient and fluid absorption as well as secretion. Defending the barrier integrity plays an essential role in the regulation of immune system. The healthy mucosal barrier comprises the mucus layer, epithelial cells and junctional complexes. 42 The mucus layer is considered as the first‐line physical defence that consists of highly glycosylated mucin proteins and limits exposure to all threats, including intestinal chemical and biological pathogens, to epithelial cells. Intestinal epithelial cells (IECs) are the central barrier, which can form a physiochemical protection that separates the microbes and antigens from the host's internal milieu. These IECs are derived from a pool of pluripotent stem cells at the bottom of the crypts and ultimately differentiated into goblet cells, Paneth cells, microfold cells, absorptive enterocytes and enteroendocrine cells. Together with the mucus layer and cellular immune system, IECs are crucial and associate with each other via a range of intercellular junctions, including adherens junctions (AJs), tight junctions and desmosomes. 43 , 44 Over the past decade, there have been increasing recognitions that either defects or breakdowns of the intestinal epithelial barrier function have been observed in many intestinal disorders such as IBD. 45 , 46
Of note, a weakened intestinal epithelial barrier caused by abnormal AS events is also closely associated with the susceptibility to IBD. As early as 2007, receptor protein‐tyrosine phosphatase sigma (PTPRS) has been proved as a susceptibility gene for IBD by the animal and genetic studies. E‐cadherin and β‐catenin are two essential AJ proteins that maintain the barrier defence in the gut and also serve as the colonic substrates for protein tyrosine phosphatases (PTPs). In human IBD, three SNPs (rs17130, rs886936 and rs8100586) that flanked exon 8 in the PTPRS gene were found to result in potential alternate splicing of exon 9 and meB, which could entirely remove the third immunoglobulin like domain of PTPσ and alter the ligand binding or recognition for E‐cadherin and β‐catenin. Subsequently, the E‐cadherin and β‐catenin phosphorylation caused the redistribution of E‐cadherin and cell disassembly, which finally led to the decomposition of AJ (Figure 3A). 47 Kindlin‐1 is a focal adhesion protein that contributes to the activation of integrin receptors. Previous data have found that there are two Kindlin‐1 transcripts (5.8 and 4.9 kb) in murine and human colon, which can correspondingly encode the 43 kDa kindlin‐1 and full‐length 74 kDa protein isoform, respectively. In 2007, Kern et al. 48 reported that the Kindlin‐1 short isoform might impair the interactions with ras homolog gene family member A (RhoA), cause the epithelial disconnection and defect of intestinal barrier and seem to be an event sequence in the pathogenesis of Kindler syndrome‐linked colitis (Figure 3B). Besides, the regulation of intestinal epithelial permeability and barrier loss is identified to require the MLCK and myosin regulatory light chain (MLC) phosphorylation. 49 Myosin phosphatase target subunit 1 (MYPT1) is a housekeeping gene, and IECs can express both isoforms (full length and variant 2 of MYPT1). Compared with the full‐length isoform, the variant 2 of MYPT1 could reduce the binding affinity for the myosin light chain phosphatase (MLCP). Thus, the dominant variant 2 might increase the MLCK activity and MLC phosphorylation and subsequently trigger the perijunctional actomyosin ring (PAMR) contraction‐mediated switch of intestinal epithelial permeability in IBD (Figure 3C). 50 In 2017, Mager et al. 51 also discussed that the novel importance of epithelial splicing regulator protein 1 (ESRP1, an AS regulator) in the colitis and altered colorectal cancer development. In humans, ESRP1 expression was significantly reduced in the biopsies taken from patients with IBD, and the low level of ESRP1 was closely related with a poorer outcome for CRC patients. Mechanistically, G protein‐coupled receptor 137 (GPR137) was identified as a newly splicing target of ESRP1, and a low level of ESRP1 as well as an elevated level of the long version of GPR137 could differently mediate the Wnt/β‐catenin signalling pathway, thus impairing intestinal barrier integrity and increasing the susceptibility to colitis or CAC (Figure 3D). 51
FIGURE 3.
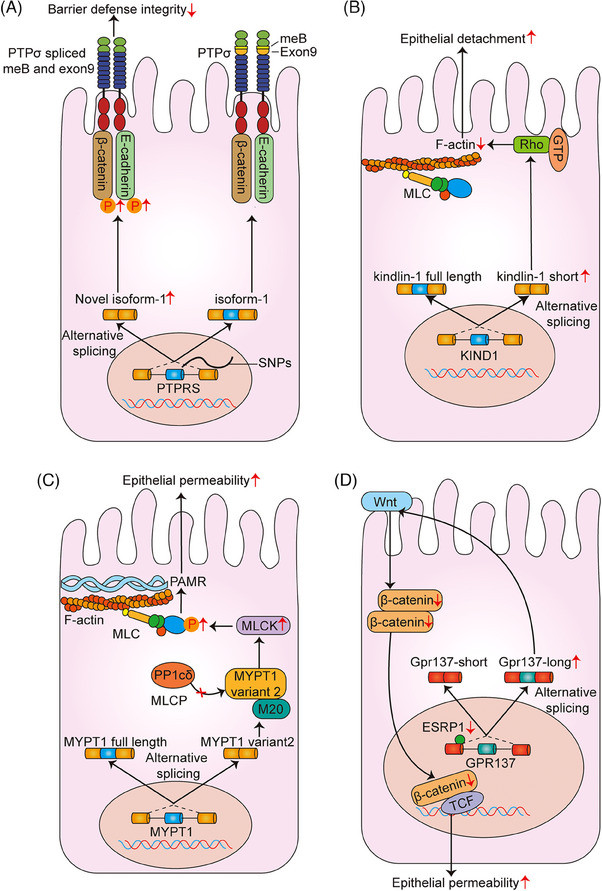
AS and intestinal epithelial barrier function. (A) Three SNPs (rs17130, rs886936 and rs8100586) that flanked exon 8 in the PTPRS gene induce alternative splicing of exon 9 and meB. Remove of the third immunoglobulin like domain of PTPσ alters the ligand binding or recognition for E‐cadherin and β‐catenin, which lead to the decomposition of adherens junction. (B) The Kindlin‐1 short isoform impairs the interactions with RhoA, and causes the epithelial detachment and defect of intestinal barrier. (C) The variant 2 of MYPT1 lacks the binding affinity to the catalytic substrate of MLCP, increase the MLCK activity and MLC phosphorylation, and subsequently triggers the PAMR contraction‐mediated switch of intestinal epithelial permeability. (D) An elevated level of the long version of GPR137 differently mediates the Wnt/β‐catenin signalling pathway, thus impairing intestinal barrier integrity and increasing the susceptibility to colitis or CAC.
2.3. AS and innate immune
The pathogenesis of IBD remains elusive, but IBD appears to be related to underlying excessive immune responses against the microorganisms of the intestinal flora, including the innate and adaptive immunity. 52 The innate immune response is non‐specific, quick and represents the first line of defence against pathogens within a short time. In the case of innate immune response cells, a large variety of different types, including neutrophils, macrophages, monocytes, myeloid‐derived suppressor cells, innate lymphoid cells, IECs, dendritic cells and natural killer cells, are critical and play a pivotal role in the active phase of IBD. 53
New insights into the molecular mechanisms of IBD indicate that AS events can also induce an abnormal innate immunity and contribute to the risk of this disease, which have received a particular attention in recent years. Nucleotide‐binding oligomerisation domain 1 (NOD1), which contains three domains, including the C‐terminal region comprising various numbers of leucine‐rich repeat (LRR) domains, nucleotide‐binding domain and the N‐terminal caspase activation and recruitment domain (CARD), is an intracellular pattern recognition protein. Among them, the LRR domain plays a key role in achieving bacterial sensing. The downstream receptor‐interactional serine/threonine kinase (RICK), a caspase‐recruitment domain‐containing kinase, plays as a crucial medium of NOD1 and NOD2 signals. Once sensing the specific muropeptide, RICK can interact with the CARD of NOD protein and serves as a bridge between IκB kinase (IKK) and TGF‐β‐activated kinase 1 (TAK1) complex in the NF‐κB signalling pathway. Subsequently, TAK1 complex will activate the IKK complex, thus resulting in the NF‐κB activation and impairing the anti‐microbial response. 54 , 55 , 56 , 57 , 58 In 2005, Girardin et al. 59 identified the existence of several NOD1 splicing variants, and only the full‐length molecule could trigger the NF‐κB activation upon stimulation and activated the host responses to bacterial infection. In terms of IBD, three NOD1 splicing variants were up‐regulated by inflammatory stimuli, which could block the NF‐κB pathway induced by the full‐length molecule to favour the development of IBD (Figure 4A). 59 In the last two decades, the adherent‐invasive Escherichia coli (AIEC) has been implicated in the pathophysiology of IBD. Adhesion of AIEC to IECs mainly depends on FimH and its binding receptor glycoprotein 2 (GP2) on the apical cell membrane of intestinal L or M cells. 60 , 61 An interesting study from Derer et al. 62 reported that GP2‐splicing variant 4 (GP2#4) rather than variant 2 was specially expressed in intestinal L or M cells, which could be induced by TNF‐α. Initially, elevated GP2#4 performed as a particular receptor for FimH‐positive bacteria to induce translocation of AIEC to the below Peyer's patches and activate protective immune responses. Afterwards, the IBD‐related serum GP2 autoantibodies were generated and inhibited the FimH binding to GP2#4, thereby leading to a corresponding reinforced attachment of flagellated bacteria to other intestinal epithelium, impairing mucosal immune and exacerbating intestinal inflammation (Figure 4B). 62 BCL‐Gonad (BCL‐G) is a unique and conserved member of the BCL‐2 family and initially considered as a pro‐apoptotic gene in humans. 63 In healthy gut tissue, both human BCL‐G splice variants (BCL‐GL, long; BCL‐GS short) were found to be over‐expressed in IECs. Compared with the non‐IBD individuals, increased tissue expressions of Th1 cytokines (IFN‐γ and TNF‐α) could strongly suppress the BCL‐GS expression, which differentially regulated the inflammatory chemokines (CCL5 and CCL20) and thereby might drive the pathophysiology of IBD (Figure 4C). 64
FIGURE 4.
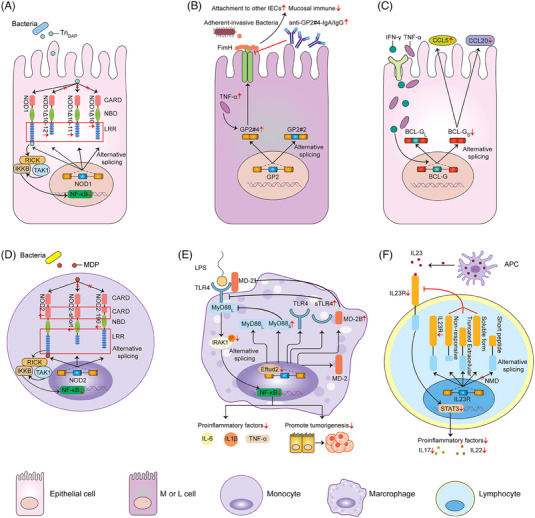
AS and innate immune. (A) In IBD, only other three splicing variants but not the full‐length of NOD1 are up‐regulated by inflammatory stimuli, thus blocking the NF‐κB pathway upon stimulation and inactivating the host responses to bacterial infection. (B) GP2‐splicing variant 4 (GP2#4) rather than variant 2 induced by TNF‐α performs as a specific receptor for FimH‐positive bacteria to induce translocation of AIEC to the below Peyer's patches and activate protective immune responses. On the contrary, serum GP2 autoantibodies inhibit the FimH binding to GP2#4, thereby leading to a corresponding reinforced attachment of flagellated bacteria to other intestinal epithelium, impairing mucosal immune and exacerbating intestinal inflammation. (C) In healthy gut tissue, both human BCL‐G splice variants (BCL‐GL and BCL‐GS) are over‐expressed in IECs. In IBD patients, increased tissue expressions of Th1 cytokines (IFN‐γ and TNF‐α) can strongly suppress the BCL‐GS expression, which differentially regulates the inflammatory chemokines (CCL5 and CCL20). (D) In the PBMCs of IBD patients, both the full‐length and alternatively spliced variants (NOD2‐short and NOD2‐190) are synchronously down‐regulated, but only the former is active and responsive to the NF‐κB. (E) Eftud2 deletion in macrophage deregulates the mRNA splicing of MyD88/TLR4/MD‐2. Alternatively spliced forms of MyD88s, MD‐2B and sTLR4 may contribute to the inhibition of the TLR4 signalling, NF‐κB activation as well as the release of some pro‐inflammatory mediators. (F) The changes of different splice variants of IL23R mediate the STAT3 pathway via the ligand‐binding interaction.
Similar to the function of NOD1, NOD2 is also identified as a crucial intracellular recognition receptor of pathogens, the ligand of which is muramyl dipeptide (MDP). Once bound to MDP, NOD2 can activate the NF‐κB signals, which brings out an up‐regulation of pro‐inflammatory cytokines. 65 , 66 In 2007, Leung et al. found that extensive AS targeting to the LRR domain and N‐terminal encoded the full‐length and alternatively spliced forms of NOD2 (NOD2‐short and NOD2‐190). In the peripheral blood mononuclear cells (PBMC) of IBD patients, both the full‐length and alternatively spliced variants were synchronously down‐regulated, but only the former was active and responsive to the NF‐κB. Thus, mutated variants of NOD2 might represent a novel mechanism in which the intracellular recognisation of bacterial peptidoglycan by the full length of NOD2 was significantly suppressed or altered (Figure 4D). 67 Elongation factor Tu GTP binding domain containing 2 (Eftud2) is a crucial component of the U5 snRNP that modulates AS to possibly regulate innate immune response in C. elegans and mouse macrophage. 68 Using an established mouse CAC model by azoxymethane (AOM)/dextran sulphate sodium (DSS), Lv et al. 69 first demonstrated that Eftud2 was constantly over‐expressed in the colonic tissue samples as well as infiltrating macrophages. Oppositely, in myeloid‐specific Eftud2(−/−) mice, Eftud2 deletion in macrophages was found to deregulate the mRNA splicing of MyD88/TLR4/MD‐2. The alternatively spliced forms of MyD88s, MD‐2B and sTLR4 might contribute to the inhibition of toll‐like receptor 4 (TLR4) signalling, NF‐κB activation as well as the release of some pro‐inflammatory mediators (Figure 4E). 69 Studies in recent years have identified the significance of IL‐23/IL‐23R signalling in regulating innate immune response by Th17 cells, and its downstream signal transducer and activator of transcription 3 (STAT3), janus‐kinase 2 and IL17RA have been also reported in IBD. 70 , 71 An interesting study from Kan et al. 72 identified a series of newly spliced variants of IL23R, and discovered four diverse premature termination forms of IL‐23Ra. These changes might regulate the function of IL‐23R through influencing the ligand‐binding interaction and perhaps therefore represented an inherent protective mechanism against the pathogenesis of IBD (Figure 4F). 72
2.4. AS and adaptive immune
In contrast to the innate immunity, adaptive immune responses involved in the IBD development are more time‐consuming, precise and complex. Once initiated by signals from microorganisms and damaged tissue, antigen‐presenting cells (APC) present antigens to T or B lymphocytes, and aggressive T or B cells via their productions of IgG antibodies will initiate a state of chronic inflammation response. 73 , 74 Recently, immunologists have also observed that AS events may participate into the different steps of adaptive immune response in IBD, such as activating the Th lymphocytes (Th1, Th2, Th17 and Th22 cells) and suppressing the activity of regulatory T (Treg) cells. CD44 is a widely expressed cell surface glycoprotein and transmembrane adhesion molecule that functions in many processes such as haematopoiesis, lymphocyte activation and tumour progression. Diverse CD44 isoforms are generated from AS of up to 10 separate exons (v1–v10). 75 , 76 As early as 1995 and 1996, the descriptions of alternatively spliced CD44 species were proposed between the normal, inflammatory and neoplastic lesions by Rosenberg and Yoshida, respectively, which first unravelled the mystery of CD44 variants in IBD patients. 77 , 78 In the study of Rosenberg et al., 77 the epithelial expressions of CD44v3 and CD44v6 were found to be significantly up‐regulated in biopsy samples of UC patients, but not in colonic CD patients. The possible mechanisms might be that these two CD44 isoforms (CD44v3 and CD44V6) could increase the lamina propria lymphocyte adhesion in colonic tissues of UC patients. 79 , 80 Similarly, the up‐regulation of CD44v6 was also confirmed in the lesions of inflamed IBD colonic epithelium as a means of assessing the disease activity, 81 whereas another report from Pfister et al. 82 found that CD44v6 was deceased on CD4+ lamina propria T cells in the mucosa of IBD patients. At the same time, another interesting variant CD44v7 began to be discovered in autoimmune disease and IBD. 83 , 84 On the one hand, CD44v7 appeared to endow lamina propria mononuclear cells with downstream contra‐apoptotic signals that might led to resistance to apoptosis and sustenance of the chronic colitis. 85 On the other hand, the expression of CD44v7 isoform on macrophages was proved to be indispensable for provoking the chronic colonic inflammation in the mice gut. CD44v7 deletion in macrophages of recipient mice might link to the down‐regulation of STAT3‐activating and forkhead box P3 (Foxp3)‐counteracting IL‐6, which would cause decreased numbers of phospho‐STAT3‐containing lymphocytes as well as elevated counts of Foxp3+ T‐cells in the gut (Figure 5A). 86 TL1A as well as its functional receptor (death‐domain receptor 3, DR3) have been considered as the key members of TNF/tumour necrosis factor receptor superfamilies of proteins. Once APC‐derived TL1A is bound to the lymphocytic DR3, TL1A–DR3 interaction exerts pleiotropic effects on different adaptive immune cells, including Treg and helper T cells, to influence cell proliferation, maintenance and differentiation. 87 , 88 It was confirmed that chronic colonic inflammation linked to the AS of DR3. Predominant expression of the transmembrane form of the receptor DR3 (tmDR3) in preference to the soluble form on lymphocytes could trigger the costimulatory signals that significantly amplified the IFN‐γ secretion and connected to the pathogenesis of Th1‐associated inflammation (Figure 5B). 89 In normal physiologically relevant conditions, T lymphocytes exist in the intestine epithelium (also known as intraepithelial lymphocytes, IELs). In cases of IBD, evidence shows that cytotoxic T lymphocytes are present in increased numbers and can promote the cryptal apoptosis and mucosal damage by releasing cytotoxic products, including granzymes and perforin, and the interaction of Fas ligand (Fas L) with a transmembrane death‐signalling receptor Fas. 90 , 91 Two studies in 2000 and 2001 all proved that T cell‐restricted intracellular antigen (TIA‐1) might serve on a mediator of alternative pre‐mRNA splicing, and could generate a mRNA isoform that coded for the membrane‐bound form of FAS receptor. 92 , 93 By immunohistochemical analyses from the IBD mucosal biopsy specimens and normal controls, Mitomi et al. 91 demonstrated that TIA‐1 + IELs were significantly elevated as compared with healthy individuals, and thereby leading to more cryptal apoptosis and abscesses in the destructive inflammatory condition of IBD (Figure 5C). Carcinoembryonic antigen‐related cell adhesion molecule 1 (CEACAM1), that is belonged to the carcinoembryonic antigen (CEA) family, is considered as a cell‐cell adhesion receptor and holds a complex role in inflammation and tumours. 94 , 95 The alternative CEACAM1 variants in human differ in the length of cytoplasmic tail and variable membrane distant Ig‐like domains. The long variants contain two intracellular immunoreceptor tyrosine‐based inhibitory motifs (ITIM) that negatively regulate the T cell activation, while the short isoforms lack the ITIM sequences that serves as costimulatory receptors. 96 In IBD, CEACAM1 was confirmed as a newly, non‐CD28‐associated co‐inhibitory receptor that mediated suppression of the T‐cell receptor–CD3 complex in a cell autonomous manner, and subsequently blocked the progression of intestinal inflammation. 97 In 2006, in the murine colitis model, Nagaishi et al. 96 revealed that over‐expression of the CEACAM1 isoform (CEACAM1‐4L) specifically in T lymphocytes might result in T‐cell inhibition ex vivo. In the study of Chen et al., CEACAM1‐L and CEACAM1‐S, were found to be up‐regulated after the protein–protein interaction. Owing to the two phosphorylated ITIMs, CEACAM1‐L could inhibit TCR–CD3 complex induced signal cascade, which depended upon the Src homology domain phosphatases 1 (SHP‐1) activity. Then the inhibitory role of CEACAM1‐L was mediated by the phosphorylation of C‐Jun N‐terminal kinase (JNK) and extracellular response kinase (ERK) pathways, thus leading to inhibition of Th1 pathways and cytokines secretion. 95 , 96 , 98 (Figure 5D). Interleukin‐6 (IL‐6) is a both pro‐ and anti‐inflammatory mediator and exerts multiple functions when largely induced during infection, inflammation and cancer. 99 A growing body of evidence show that two models of IL‐6 activation are presented and plays a dual role in the process of IBD as well as CAC. On one hand, the classic IL‐6 activation through membrane‐bound IL‐6 receptors (IL‐6Rs) seems to play a protective role that the regenerative and anti‐apoptotic response of IECs to the damage induced by DSS are observed. On the other hand, a soluble IL‐6R (sIL‐6R)‐conducted cell signal (IL‐6 trans‐signalling) promotes pro‐inflammatory pathway by activating the immune systems, including recruiting mononuclear cells, inhibiting T‐cell apoptosis and suppressing Treg differentiation. 100 , 101 , 102 , 103 , 104 , 105 It is interesting that two types of IL‐6 receptor subunit (IL‐6R or sIL‐6R; gp130 or sgp130) are also generated by differential corresponding pre‐mRNA splicing. 106 , 107 Recently, it was demonstrated that blockade of IL‐6 trans‐signalling by anti‐IL‐6R antibodies or recombinant sgp130 protein bound to the Fc region of human IgG1 might not only partly ameliorate the development of IBD but also the CAC progression (Figure 5E). 108 Treg cells are key regulators of inflammation and as well as in the maintenance of immune tolerance and homeostasis. 109 Forkhead box P3 (FOXP3, as a primary transcription factor of Treg cells) is necessary for the development of Treg cells. AS events consequently allow a single FOXP3 gene to produce different isoforms, including FOXP3fl (the full‐length FOXP3), FOXP3Δ2 (FOXP3 lacking exon 2) and FOXP3Δ2Δ7 (FOXP3 lacking exon 2 and 7), which exert multiple or even opposing functions. 110 , 111 In patients suffering CD, the pro‐inflammatory cytokine IL‐1β was found to promote abnormal patterns of FOXP3 splicing with an elevated proportion of FOXP3Δ2Δ7, which could favour the differentiation of naïve T cells into Th17 cells, and contribute to IL‐17 production and disease severity (Figure 5F). 112 Speckled Protein 140 (SP140) is a nuclear protein that is belonged to the speckled protein (SP) family, and its loss‐of‐function mutations is associated with multiple sclerosis (MS), CD and chronic lymphocytic leukaemia. 113 , 114 A study from Fraschilla et al. 115 demonstrated that Sp140−/− mice might harbour altered microbiota and exhibit more severe colitis. Mechanistically, a causal variant of rs28445040‐T was found to alter the splicing of the 7th exon of SP140 gene, which produced a transcript lacking the exon 7 (SP140Δ7) and decreased the full‐length transcript expression. These changes subsequently reduced the SP140 protein expression in lymphoblastoid cells and inhibited the NF‐κB activity in B cells. GO analysis implied that differentially expressed genes (DEGs) after SP140 silencing were enriched in regulation of inflammatory response, cell–cell adhesion and cytokine production, thereby leading to the progression of IBD (Figure 5G). 116 CD28 is a 44‐kDa homodimeric glycoprotein expressed on the surface of the majority of T cells. Previous data have revealed that CD28 is a key co‐stimulatory molecular, and ligation of CD28 with ligands (CD80/CD86) may play an essential role in naïve T cell activation. 117 In 2022, the existence of AS isoforms of CD28 was reported. Among these IBD‐related isoforms, full‐length CD28 was demonstrated to show a higher binding affinity with CD80/CD86, while both CD28i and CD28Δex2 were confirmed as loss‐of‐function spliced products that might decrease the disease risk through bringing out anergy of effector T cells, inducing tolerance, and inactivation to intestinal antigens and allergens (Figure 5H). 118
FIGURE 5.
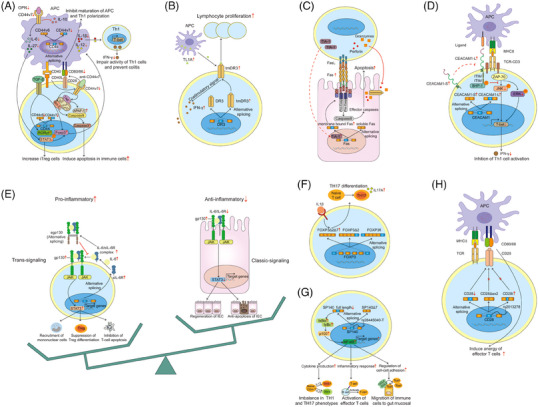
AS and adaptive immune. (A) In intestinal macrophage and T cells, blockade of CD44v6 and CD44v7 induces apoptosis in immune cells and prevent the chronic inflammation by distinct pathways. (B) Predominant expression of the tmDR3 in preference to the soluble form on lymphocytes can trigger the costimulatory signals, amplify the IFN‐γ secretion and connect to the pathogenesis of Th1‐associated inflammation. (C) In CTLs, TIA‐1 acts as an AS regulator, and generates a membrane‐bound form of FAS receptor, which can lead to more cryptal apoptosis and abscesses. (D) In mouse intestinal T cells, owing to the two phosphorylated ITIMs, up‐regulated CEACAM1‐L and CEACAM1‐S after ligand interaction mediate the TCR–CD3 pathway, and result in the inhibition of Th1 differentiation and secretion. (E) Classic IL‐6 activation via IL‐6Rs seems to play a protective role, while sIL‐6R‐mediated cell signal (IL‐6 trans‐signalling) exerts pro‐inflammatory effects. (F) IL‐1β promotes abnormal patterns of FOXP3 splicing with an up‐regulated proportion of FOXP3Δ2Δ7, which can favour the differentiation of naïve T cells into Th17 cells, and contribute to IL‐17 production and disease severity. (G) SP140Δ7 altered by rs28445040‐T inhibits the NF‐κB activity in B cells, and is involved in regulation of cytokine production, inflammatory response and cell‐cell adhesion. (H) Both CD28i and CD28Δex2 associated with ligands are confirmed as loss‐of‐function splicing isoform products that can reduce disease risk by inducing anergy of effector T cells.
2.5. AS and fibrosis
Chronic inflammation is a prerequisite for CD, but progression to strictures is predominantly driven by intestinal fibrosis. Investigations on the mechanism of fibrosis involve the excessive accumulation of extracellular matrix and expansion of mesenchymal cells, such as myofibroblasts, fibroblasts and smooth muscle cells. 119 , 120 , 121 Fibroblasts synergistically bind to fibronectin (FN) through integrin α5β1 and syndecan‐4 (recognising the 10th FN‐III domain (III10) and the 13th FN‐III domain (III13) of FN, respectively), which can activate the RhoA signals, and induce the assembly of actin stress fibre and cell spreading. 122 , 123 Tenascin‐C (TN‐C), which is a extracellular matrix glycoprotein and contains an alternatively spliced FNIII repeats A1‐D, can presumably bind to the 13th FN‐III domain and syndecan‐4, interfere with the FN/syndecan‐4 interaction. 122 , 124 In CD patients, though TN‐C was significantly induced in inflamed lesions of the colonic mucosa, many meprinβ‐positive leukocytes that appeared throughout the aberrant and inflamed intestinal tissue was able to cut the spliced N‐terminal of TN‐C at two distinct cleavage sites (the 7th constant FN‐III repeat and the alternative FN‐III repeat D). For this reason, the reactivated RhoA signalling might partly drive the FN‐mediated sustained fibroblasts activation, thereby progression of fibrosis in the pathogenesis of CD (Figure 6A). 125 Insulin‐like growth factor‐I (IGF‐I) that is induced by the fibrogenic cytokine TGF‐1, is highly expressed in all layers of intestinal in CD patients. 126 This gene can be alternatively spliced into IGF‐IEa, IGF‐IEb and IGF‐IEc variants in humans. Among these variants, the IGF‐IEa splice variant was confirmed to encode pro‐IGF‐IEa producing mature IGF‐I, result in smooth muscle hyperplasia and excessive collagen I productions. 127 Simultaneously, another up‐regulated variant IGF‐IEc might significantly increase the phosphorylated levels of myocyte enhancer factor 2 C (MEF2C) and extracellular signal‐related kinase (Erk5), which controlled the transcription of smooth muscle‐particular proteins, such as smoothelin, α‐smooth muscle actin and γ‐smooth muscle actin. These elevated molecules were all essential participants in the smooth muscle cell hypertrophy, which contributed to the formation of intestinal stricture (Figure 6B). 128
FIGURE 6.
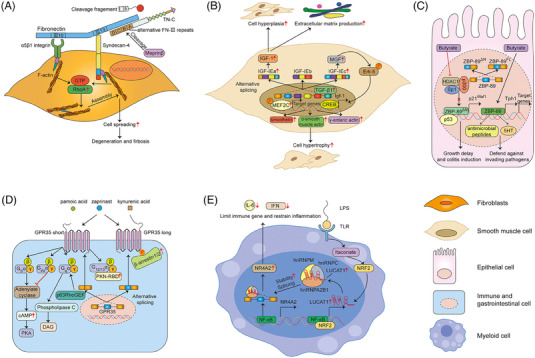
AS regulates fibrosis and other risk factors in the pathogenesis of IBD. (A)TN‐C that contains an alternatively spliced FNIII repeats A1–D interferes the FN/syndecan‐4 interaction, resulting in actin stress fibre assembly and cell spreading. (B) The splicing isoforms of IGF‐I induced by TGF‐1 are involved in cell hyperplasia, cell hypertrophy and extracellular matrix production in smooth muscle cell. (C) Two ZBP‐89 splice isoforms (ZBP‐89FL and ZBP‐89DN) regulate biological functions in epithelial cells. ZBP‐89FL tends to protect against chronic colitis, while ZBP‐89DN renders the colonic mucosa more susceptible to colitis. (D) Two distinct variants of GPR35 (GPR35 long and short) implicate in gut‐related diseases, and these two variants differed only in the length of their extracellular N‐termini by 31 amino acids. The short isoform can activate different major G proteins, while the presence of GRP35 long plays a positive modulator for arrestin recruitment. (E) LUCAT1 controls the splicing and stability of anti‐inflammatory NR4A2, thereby contributing to the suppressing effects of interferons and inflammatory mediators.
2.6. AS and other risk factors
In addition to the potential mechanisms discussed above, recent investigations also have demonstrated that AS events indeed participate into the IBD development in several different manners. For instance, ZBP‐89 (Zfp148, ZNF148, BFCOL1, BERF1), is a zinc finger transcription factor, that can bind to GC‐rich DNA elements in promoters, and involves the cell growth and death regulation. Two ZBP‐89 splice isoforms (ZBP‐89FL and ZBP‐89DN) have been identified and co‐expressed in gastrointestinal cell lines and tissues, which may regulate diverse biological functions in colitis. ZBP‐89FL was found to directly bind to the TPH1 promoter, which encoded the rate‐limiting enzyme in 5HT biosynthesis, and subsequently generated optimal amounts of 5HT or other antimicrobial peptides in response to bacterial infections. This data implied that the expression of ZBP‐89FL tended to protect against chronic colitis. 129 , 130 The other ZBP‐89 spliced isoform (ZBP‐89DN), which retained its zinc‐finger domain, could interact directly with p53 protein, while loss of amino terminal residues 1−127 of the full‐length protein might interfere the butyrate‐mediated p21waf1 activation by interacting with p300. As a consequence, ZBP‐89DN/DN mice might experience growth delay, decreased viability and rendered the colonic mucosa more susceptible to DSS colitis (Figure 6C). 131 Emerging evidence shows that the mechanisms that G protein‐coupled receptor 35 (GPR35) modulates the pathological processes of gastrointestinal inflammation through the mucosal healing of colonic epithelium, 132 immune system 133 and intestinal homeostasis. 134 , 135 GPR35, is a receptor for lysophosphatidic acid, and over‐expressed in IECs and some specific subtypes of immune cells. Single‐nucleotide polymorphisms also confirmed GPR35 as a key susceptibility gene for IBD. 136 Recently, investigators also elaborated on the two distinct variants of GPR35 (GPR35 long and short) implicated in gut‐related diseases, and these two variants differed only in the length of their extracellular N‐termini by 31 amino acids. The short isoform could activate different major G proteins, while the presence of GRP35 long played a positive modulator for arrestin recruitment (Figure 6D). 137 As mentioned above, FN is an adhesive glycoprotein existing in the extracellular matrix, and the heterogeneity of FN subunits derives mainly from AS of a primary precursor mRNA at three distinct sites termed EDB, EDA and IIICS. 138 , 139 In 2015, Bootz et al. 140 found that the alternatively spliced EDA domain of FN could not be virtually detected in most adult normal organs, while it was strongly stained in the sub‐mucosa and certain structures in the muscularis mucosa around blood vessels within the specimens of IBD patients and mice models of colitis. This means that exploring specific antibodies to the EDA domain of FN may provide us a prospective therapeutical option for the treatment of IBD conditions. 140 It is known that glucocorticoids (GC) exert an established immunosuppressive effect and are widely applied in the moderate‐to‐severe IBD treatment. The response to GC is mainly mediated through glucocorticoid receptor (hGR), of which two isoforms hGRα and hGRβ exist. 141 , 142 In 2005, Towers et al. 143 evaluated the hGRα and hGRβ expressions in CD patients and looked for a potential link between these two receptors and their response to GC treatment. The data implied that the over‐expression of hGRα mRNA in active CD patients was independent on steroid‐resistant or steroid‐responsive. But the augmented expressions of hGRβ were connected with GC resistant during the active phase of UC and CD patients. 143 However, in 2007 Hausmann and coworkers published the controversial data that neither of the GC isoforms associated with the GC sensitivity, which denied its predictive value for efficacy of steroid treatment. 144
In addition to the molecules mentioned above in which their potential regulatory mechanisms in the pathogenesis of IBD have been fully elucidated, there are still many well‐established positive or negative regulatory genes encoded by different variants but lacking of detailed elaborations for their possible pro‐ or anti‐inflammatory mechanisms. For example, some newly reported molecules, such as caspase‐associated recruitment domain 8 (CARD8), neurokinin‐1 receptor (NK‐1R), orosomucoid 1‐like protein 3 (ORMDL3), protein tyrosine phosphatase non‐receptor type 2 (PTPN2), IL‐15 receptor alpha (IL‐15Rα) are all positional and functional candidate genes for IBD, and variants of splicing of these genes have been addressed by numerous reports. 145 , 146 , 147 , 148 , 149 NK‐1R, a principal receptor of pro‐inflammatory neuropeptide substance P (SP), has been proved to play a vital role in rodent models of chronic colitis and in UC as well as CD patients. 150 , 151 In 2015, Gillespie et al. first examined the truncated (tr‐NK‐1R) and full‐length (fl‐NK‐1R) receptor expressions in colonic tissues from patients of quiescent colitis, high‐grade dysplasia (HGD) and CAC. The data implied that enhance of total NK‐1R protein in HGD and CAC was attributable to an elevation of tr‐NK‐1R mRNA, strongly suggesting an essential role of tr‐NK‐1R during the colitis‐to‐CAC malignant transformation. The tr‐NK‐1R variant could be therefore served as a diagnostic biomarker to distinguish patients at risk of neoplasia or associated cancer. 146 Nuclear receptor subfamily 4, group A, member 2 (NR4A2) is a nuclear receptor involving in modulating target gene transcription and regulating distinctive physiological processes. 152 , 153 In IBD, NR4A2 was considered as a negative regulator of immune response, and deletion of NR4A2 in T cells was confirmed to attenuate Tregs induction and led to aberrant increase of Th1 cells, which might partly exacerbate the colonic inflammation. 154 Likewise, NR4A2 is also a spliced gene, 155 , 156 and a recent study from Vierbuchen identified NR4A2 as a downstream mediator and binding protein of nuclear long non‐coding RNA LUCAT1‐dependent immune gene suppression. LUCAT1 was induced to control the splicing and stability of anti‐inflammatory NR4A2, thereby contributing to the suppressing effects of interferons and inflammatory mediators (Figure 6E). 157
3. TRANSCRIPTOMIC ANALYSIS OF AS EVENTS IN THE PATHOGENESIS OF IBD
As described above, we have systematically summarised all single splicing molecules that may cause AS events in the pathogenesis of IBD within the last two decades. However, we shall take a comprehensive and profound view of this problem. The main reason is that both UC and CD are highly complex disease processes that result from the integration of multiple and incompletely identified pathogenic elements. Therefore, transcriptomics has emerged as a powerful approach that provides us a highly sensitive and robust examination for multiple attributes such as strandedness, sequence composition, splicing factors, AS and alternative transcription start/stop sites (Figure 7). 158 Since the introduction of microarrays as the mainstream technology of the last decade, researchers have provided dozens of valuable datasets for identifying DEGs in large IBD samples readily available. 159 , 160 In 2011, Häsler et al. 161 first used the commercial and custom‐designed microarrays to investigate the gene expressing profile of pre‐mRNA splicing factors, and identified intron retention as an exemplary splicing event that was possibly linked to the IBD aetiology. A number of population studies have demonstrated that patients with long duration of UC present a relatively higher risk of developing CAC, in comparison with those patients with a shorter‐time inflammation. 162 , 163 One of proposed mechanisms may due to accumulated genetic abnormalities when IECs are persistently and chronically exposed to long‐term inflammation. 164 From this, using the Affymetrix Human Transcriptome Array 2.0, a similar study from the Asia Pacific region had been conducted to screen the transcriptome profiling, including DEGs and AS events, in inflamed colonic biopsies of long‐ and short‐duration UC patients. Totally, 640 DEGs and 3560 genes with differential splicing were identified. 165
FIGURE 7.
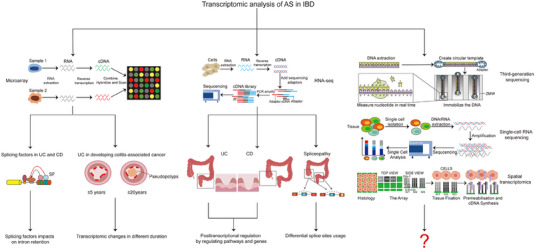
Transcriptomic analysis of AS events in the pathogenesis of IBD. Microarrays and NGS have emerged as powerful approaches that provide investigators highly sensitive and robust examinations for AS events in IBD. Notably, newly technologies, such as TGS, scRNA‐seq and ST, are also desired with great expectations.
RNA‐seq, the current next‐generation sequencing (NGS) approach, is expected to a superior technology to microarrays, which can provide global and digital rather than analogue information on transcripts and their corresponding isoforms. In the last years, it has merged as a revolutionising and mainstream approach and used to interrogate the transcriptome in various illnesses, as well as some chronic immune‐mediated disorders. 166 , 167 , 168 From this, genome‐wide analyses of gene expression alternations and AS signatures in patients with different subtypes of IBD are also performed by RNA‐seq. For example, a recent pioneering study by Li et al. presented an interesting genomic landscape of AS signatures in UC patients based on RNA‐seq data from two cohorts, and found that skipped exon (SE) and alternative first exon (AFE) were the two most remarkably enriched AS events during the UC development. In addition, they also performed a combined mRNA‐seq experiment between four UC patients and four healthy individuals and discovered that the immune response‐associated pathways and cell chemotaxis were significantly enriched in UC‐related AS events. 169 Coincidentally, another study from the same research team employing a well‐established public NCBI GEO dataset (GSE66207) identified a total of 2980 important AS events in CD patients, in comparison with controls. To validate the reliability in the GSE66207 dataset, authors also analysed the RNA‐seq data focused on a Chinese cohort and demonstrated 1715 significantly AS events were involved. Interestingly, the results from public or validation RNA‐seq dataset all suggested a strong similarity that SE and AFE were the two most common types of AS events in patients with CD. 170 In 2021, a transcriptomic research using RNA‐seq was carried out to present the whole mRNA sequencing profiles of 124 biopsies obtained from 34 young donors with UC or CD. In this study, a newly definition of ‘spliceopathy’ was first supported by Berger et al., 171 and the meaningful results implied that tissue location might be the largest contributor to variability in gene expression and splicing of IBD patients.
4. FUTURE PERSPECTIVES
With research progresses, many investigators have now noticed the significance of abnormal AS events accompanied by the occurrence and progression of diverse diseases, which are able to produce multiple different isoforms and diversify protein products. For instance, our recent published data using the long‐read sequencing technology first investigated the potential spicing events in CRC. Among the newly identified splicing isoforms, tissue inhibitor of metalloproteinase‐1 Δ4‐5 transcript (TIMP Δ4‐5) was significantly down‐regulated in CRC tissues, while the full length of TIMP (TIMP‐FL) possibly served as an oncogenic transcript and promoted the CRC growth. 172 Recently, AS investigations has also gained considerable momentum during the initiation and development of IBD. Though this study has described the regulatory mechanisms of AS, as well as its detection in patients with IBD and CAC, there are still some relative limitations. First, in addition to coding RNAs, non‐coding RNAs, including circular RNAs, microRNAs, long non‐coding RNAs and small nuclear RNAs, are also inextricably linked to AS. 173 , 174 , 175 These multiple different types of ncRNAs, which are generated by AS of precursor messenger RNAs, have been also implicated in the initiation, progression and therapy resistance or acted as regulatory molecules in various types of diseases through AS. 176 , 177 But it is a pity that there are not still any reports focusing on the non‐coding RNAs through AS events to elucidate the occurrence of IBD. Second, the majority of literature mainly reported the novel splicing molecules or elucidate the potential mechanisms of AS in IBD patients or experimental models, whereas no validated AS‐related biomarkers of IBD have been fully investigated or used in clinic. Third, the revolution of genome‐wide analyses of gene expression alternations and AS events is continuing after the successful applications of microarrays and NGS technology. Third‐generation sequencing (TGS) is proved to be a newly and improved sequencing technology, which can access in‐depth the splicing regulation, enhance the RNA isoforms’ characterisation and predict more comprehensive gene expression diversity. 178 , 179 In addition, traditional methods determine the DEGs owing to the analyses of whole‐tissue samples, but the contribution of individual cell populations are unknown. Single‐cell RNA sequencing (scRNA‐seq) including genomics, transcriptomics, proteomics, epigenomics and metabolomics sequencing, can successfully resolve this problem and directly measure molecular signatures in thousands to millions of individual cells, which provides us an opportunity to uncover the mysteries underlying cellular populations. 180 Since its introduction in 2009, 181 studies based on scRNA‐seq are rapidly increasing and have discovered more and more profound information about health and diseases. 182 , 183 The scRNA‐seq technology is also employed by many investigators to characterise cell‐type‐specific transcriptional heterogeneity in IBD. 184 , 185 , 186 , 187 Besides, a prevailing ‘spatial transcriptomics’ (ST) has been developed that allows spatially resolved, high‐dimensional assessment of gene transcription. Researches are able to obtain more high‐quality RNA‐sequencing data with three‐dimensional positional information from tissues sections. 182 , 188 , 189 More interestingly, scRNA‐seq and ST are not contradictory. On the contrary, these two current technologies can be integrated, and newly integrative computational methods are also studied in depth to propose ways to effectively capture more useful signatures in biomedical researches. 190 , 191 , 192 , 193 These three newly developed technologies can help investigators better understand the complex pathogenesis of IBD. It is believed that these currently developed technologies have opened a door for us to broaden the scope of the complex IBD pathogenesis, and more significant IBD‐related AS events and variants will be revealed and studied in the near future (Figure 7) . At last, this review presents all current data concerning the pathogenic role of AS during the IBD progression (Table 1). This may help us to further understand the molecular mechanisms of IBD, and develop and find new therapeutic methods or targets for IBD treatment, such as the development of an IL‐6R‐specific mAb, optimised version of sgp130Fc, some other small‐molecule splicing modulators and splice site‐switching anti‐sense oligonucleotides. 104 , 194 Of course, though great efforts have been made, further studies are still required to address the associations between AS and IBD in more detail.
TABLE 1.
Overview of AS‐related genes and variants modulated in IBD.
| Gene | Locus | Spliceosome | Splice variant | Sample | Source | Up/Down‐regulation | Function | References |
|---|---|---|---|---|---|---|---|---|
| MYLK | 3q21.1 | N/A |
MLCK1 MLCK2 |
Colonic mucosal samples with UC and CD, DSS‐induced model sample | Human and mice | Up‐regulation | Pro‐inflammatory | 39 |
| KAT2A | 17q21.2 | SF3b complex | KAT2A | Colonic biopsies from patients with IBD and CRC | Human | Up‐regulation | Pro‐inflammatory | 40 |
| LmnA | 1q22 | U2 and SR | Progerin | Colonic biopsies from healthy controls and patients with IBD, Cbx3 mouse model | Human and mice | Up‐regulation | Pro‐inflammatory | 41 |
| PTPRS | 19p13.3 | N/A |
Isoform2 Isoform3 Novel isoform1 |
DSS‐induced model sample | Mice | Up‐regulation | Pro‐inflammatory | 47 |
| KIND1 | 20p12.3 | N/A | Kindlin‐1 short | Intestinal biopsies from KS patients | Human | Up‐regulation | Pro‐inflammatory | 48 |
| MYPT1 | 12q21.2‐q21.31 | N/A | MYPT1 variant2 | Colonic biopsies from mouse sample | Mice | N/A | Pro‐inflammatory | 50 |
| GPR137 | 11q13.1 | ESRP1 |
Gpr137‐short Gpr137‐long |
Tumour, adenoma and normal tissue from CRC patient, AOM/DSS model sample | Human and mice | Up‐regulation | Pro‐inflammatory | 51 |
| NOD1 | 7p14.3 | N/A |
Nod1Δ10 Nod1Δ10‐11 Nod1Δ10‐12 |
Human epithelial cells | Human | Up‐regulation | Pro‐inflammatory | 59 |
| GP‐2 | 16p12.3 | N/A |
GP2#2 GP2#4 |
Ileal and colonic biopsies from IBD patients | Human | Up‐regulation | Pro‐inflammatory | 62 |
| BCL‐G | 12p13.2 | N/A |
BCL‐Gs BCL‐GL |
Colonic biopsies from non‐IBD and IBD patients | Human | Down‐regulation | Pro‐inflammatory | 64 |
| NOD2 | 16q12.1 | N/A |
NOD2‐190 NOD2‐short |
Peripheral blood mononuclear cells (PBMC) from healthy donors | Human | Up‐regulation | Pro‐inflammatory | 67 |
|
MyD88 TLR4 MD‐2 |
3p22.2 9q33.1 8q21.11 |
Eftud2 |
MyD88s sTLR4 MD‐2s |
DSS‐induced model sample | Mice | Up‐regulation | Anti‐inflammatory | 69 |
| IL23R | 1p31.3 | N/A |
Short peptide Soluble form Truncated Extracellular Non‐responsive |
PBMC from healthy donors | Human | Down‐regulation | Anti‐inflammatory | 72 |
| CD44 | 11p13 | N/A |
CD44v7 CD44v6 |
Transgenic mice model, colonic biopsies from patient with IBD, | Human and mice | Down‐regulation | Anti‐inflammatory | 84 , 86 |
| DR3 | 1p36.31 | N/A | tmDR3 | Ileitis murine model | Mice | Up‐regulation | Pro‐inflammatory | 89 |
| Fas | 10q23.31 | N/A | Membrane bound Fas | Colonic biopsies from patient with IBD | Human | Up‐regulation | Anti‐inflammatory | 91 |
| CEACAM1 | 19q13.2 | N/A |
CEACAM1‐L CEACAM1‐S |
Jurkat‐T cell transfection model | Human | Up‐regulation | Anti‐inflammatory | 96 |
| IL‐6R | 1q21.3 | N/A | sIL‐6R | AOM/DSS‐induced model sample | Mice | Up‐regulation | Pro‐inflammatory | 100 , 101 , 102 , 103 , 104 , 105 , 106 , 107 , 108 |
| FOXP3 | Xp11.23 | N/A |
FOXP3Δ2 FOXP3Δ2Δ7 |
Colonic biopsies from CD patients | Human | Up‐regulation | Pro‐inflammatory | 112 |
| SP140 | 2q37.1 | N/A | SP140Δ7 | Lymphoblastoid cell line | Fetal bovine | Down‐regulation | Pro‐inflammatory | 116 |
| CD28 | 2q33.2 | N/A |
CD28Δex2 CD28i |
Jurkat‐T cell transfection model | Up‐regulation | Anti‐inflammatory | 118 | |
| TN‐C | 9q33.1 | N/A | FNIIID | Transfected plasmid, chicken embryo | Chicken | N/A | Fibrosis | 125 |
| Igf‐1 | 12q23.2 | N/A |
IGF‐IEa IGF‐IEb IGF‐IEc |
Intestinal muscle cell from patient with CD and healthy donors | Human | Up‐regulation | Cell hyperplasia, hypertrophy | 128 |
| ZBP‐89 | 3q21.2 | N/A |
ZBP‐89ΔN ZBP‐89FL |
DSS‐induced model sample | Mice | Up‐regulation | Pro‐inflammatory | 131 |
| GPR35 | 2q37.3 | N/A |
GPR35 short GPR35 long |
Transfected plasmid | N/A | N/A | Mediate intracellular pathways | 137 |
| EDA | Xq13.1 | N/A | EDA | DSS‐induced model sample | Mice | Up‐regulation | Pro‐inflammatory | 140 |
| GR | 5q31.3 | N/A |
GRα GRβ |
Colonic tissue from CD patients and healthy donors | Human | Up‐regulation | Steroid resistance | 143 |
| CARD8 | 19q13.33 | N/A | T60, T54, T51, T48, T47 | Lymphoblastoid cell lines of CD patients | Human | N/A | N/A | 145 |
| NK‐1R | 2p12 | N/A |
tr‐NK‐1R fl‐NK‐1R |
Colonic tissue from UC patients | Human | Up‐regulation | Malignant transformation | 146 |
| ORMDL3 | 17q21.1 | N/A | ORMDL3 V1 | Hela cells, HEK293 cells, HL60 cells | Human | N/A | N/A | 147 |
| PTPN2 | 18q11.21 | N/A |
PTPN2‐001 PTPN2‐002 PTPN2‐003 |
Human peripheral blood mononuclear cells and mouse peritoneal macrophages | Human and mice | N/A | N/A | 148 |
| IL15RA | 10p15.1 | N/A |
Variant1 Variant2 Variant3 |
Colon and duodenum biopsy specimens from healthy individuals | Human | N/A | N/A | 149 |
| NR4A2 | 2q24.1 |
hnRNPC hnRNPM hnRNPA2B1 |
N/A | PBMC from donors | Human | Up‐regulation | Anti‐inflammatory | 157 |
AUTHOR CONTRIBUTIONS
Chunfang Xu, Airong Wu and Qiaoming Zhi designed the study. Chentao Zou, Xinquan Zan and Zhenyu Jia collected all literature and wrote the manuscript. Lu Zheng, Yijie Gu, Fei Liu and Ye Han provided the technical and writing supports. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
The authors agree to the publication of all the data involved in this article. No data from other entities are used in this study.
ACKNOWLEDGEMENTS
This study was supported by grants from the National Science Foundation of China (81902805) and the Suzhou Gusu Medical Youth Talent (GSWS2019032).
Zou C, Zan X, Jia Z, et al. Crosstalk between alternative splicing and inflammatory bowel disease: Basic mechanisms, biotechnological progresses and future perspectives. Clin Transl Med. 2023;13:e1479. 10.1002/ctm2.1479
Chentao Zou, Xinquan Zan and Zhenyu Jia contributed equally to this work.
Contributor Information
Chunfang Xu, Email: xcf601@163.com.
Airong Wu, Email: arwu@suda.edu.cn.
Qiaoming Zhi, Email: strexboy@163.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. 2019;2019:7247238. 10.1155/2019/7247238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307‐317. 10.1038/nature10209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427‐434. [DOI] [PubMed] [Google Scholar]
- 4. Hisamatsu T, Kanai T, Mikami Y, Yoneno K, Matsuoka K, Hibi T. Immune aspects of the pathogenesis of inflammatory bowel disease. Pharmacol Ther. 2013;137(3):283‐297. 10.1016/j.pharmthera.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 5. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573‐621. 10.1146/annurev-immunol-030409-101225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population‐based studies. Lancet. 2017;390(10114):2769‐2778. 10.1016/S0140-6736(17)32448-0 [DOI] [PubMed] [Google Scholar]
- 7. Korzenik JR, Podolsky DK. Evolving knowledge and therapy of inflammatory bowel disease. Nat Rev Drug Discov. 2006;5(3):197‐209. [DOI] [PubMed] [Google Scholar]
- 8. Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(1):S3‐S9. Suppl. [DOI] [PubMed] [Google Scholar]
- 9. Flanagan A, Allsopp SM, O'Connor SA, et al. High incidence of inflammatory bowel disease in Northern Australia: a prospective community population‐based Australian incidence study in the Mackay‐Isaac‐Whitsunday region. Intern Med J. 2022. 10.1111/imj.15941 [DOI] [PubMed] [Google Scholar]
- 10. Aniwan S, Santiago P, Loftus EV, Park SH. The epidemiology of inflammatory bowel disease in Asia and Asian immigrants to Western countries. United Eur Gastroenterol J. 2022;10(10):1063‐1076. 10.1002/ueg2.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee SH, Kwon JE, Cho M‐L. Immunological pathogenesis of inflammatory bowel disease. Intest Res. 2018;16(1):26‐42. 10.5217/ir.2018.16.1.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Momozawa Y, Mni M, Nakamura K, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43(1):43‐47. 10.1038/ng.733 [DOI] [PubMed] [Google Scholar]
- 13. Monteleone G, Fina D, Caruso R, Pallone F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr Opin Gastroenterol. 2006;22(4):361‐364. [DOI] [PubMed] [Google Scholar]
- 14. Singh S, Murad MH, Fumery M, Dulai PS, Sandborn WJ. First‐ and second‐line pharmacotherapies for patients with moderate to severely active ulcerative colitis: an updated network meta‐analysis. Clin Gastroenterol Hepatol. 2020;18(10). 10.1016/j.cgh.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Markus B, Birk OS, Geiger D. Integration of SNP genotyping confidence scores in IBD inference. Bioinformatics. 2011;27(20):2880‐2887. 10.1093/bioinformatics/btr486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooke J, Zhang H, Greger L, et al. Mucosal genome‐wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(11):2128‐2137. 10.1002/ibd.22942 [DOI] [PubMed] [Google Scholar]
- 17. Rajamäki K, Taira A, Katainen R, et al. Genetic and epigenetic characteristics of inflammatory bowel disease‐associated colorectal cancer. Gastroenterology. 2021;161(2):592‐607. 10.1053/j.gastro.2021.04.042 [DOI] [PubMed] [Google Scholar]
- 18. Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10(11):741‐754. 10.1038/nrm2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153‐165. 10.1038/nrm3525 [DOI] [PubMed] [Google Scholar]
- 20. Kim E, Goren A, Ast G. Alternative splicing: current perspectives. Bioessays. 2008;30(1):38‐47. [DOI] [PubMed] [Google Scholar]
- 21. Jin Y, Dong H, Shi Y, Bian L. Mutually exclusive alternative splicing of pre‐mRNAs. Wiley Interdiscip Rev RNA. 2018;9(3):e1468. 10.1002/wrna.1468 [DOI] [PubMed] [Google Scholar]
- 22. Keren H, Lev‐Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11(5):345‐355. 10.1038/nrg2776 [DOI] [PubMed] [Google Scholar]
- 23. Yang Q, Zhao J, Zhang W, Chen D, Wang Y. Aberrant alternative splicing in breast cancer. J Mol Cell Biol. 2019;11(10):920‐929. 10.1093/jmcb/mjz033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mills JD, Janitz M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol Aging. 2012;33(5):1012. 10.1016/j.neurobiolaging.2011.10.030. e11‐1012.e24. [DOI] [PubMed] [Google Scholar]
- 25. Li D, McIntosh CS, Mastaglia FL, Wilton SD, MT Aung‐Htut. Neurodegenerative diseases: a hotbed for splicing defects and the potential therapies. Transl Neurodegener. 2021;10(1):16. 10.1186/s40035-021-00240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kontos CD, Annex BH. Angiogenesis. Curr Atheroscler Rep. 1999;1(2):165‐171. [DOI] [PubMed] [Google Scholar]
- 27. Flournoy J, Ashkanani S, Chen Y. Mechanical regulation of signal transduction in angiogenesis. Front Cell Dev Biol. 2022;10:933474. 10.3389/fcell.2022.933474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bowler E, Oltean S. Alternative splicing in angiogenesis. Int J Mol Sci. 2019;20(9). 10.3390/ijms20092067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peng Q, Zhou Y, Oyang L, et al. Impacts and mechanisms of alternative mRNA splicing in cancer metabolism, immune response, and therapeutics. Mol Ther. 2022;30(3):1018‐1035. 10.1016/j.ymthe.2021.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan Y‐J, Huo F‐C, Kang M‐J, Liu B‐W, Wu M‐D, Pei D‐S. Alternative splicing of HSPA12A pre‐RNA by SRSF11 contributes to metastasis potential of colorectal cancer. Clin Transl Med. 2022;12(11):e1113. 10.1002/ctm2.1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou J, Zhang Q, Zhao Y, et al. The regulatory role of alternative splicing in inflammatory bowel disease. Front Immunol. 2023;14:1095267. 10.3389/fimmu.2023.1095267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1). 10.1007/s12328-017-0813-5 [DOI] [PubMed] [Google Scholar]
- 33. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573‐584. 10.1038/nrgastro.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004. 10.1038/nmicrobiol.2017.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khan I, Ullah N, Zha L, et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): cause or consequence? IBD treatment targeting the gut microbiome. Pathogens. 2019;8(3). 10.3390/pathogens8030126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lavelle A, Sokol H. Gut microbiota‐derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223‐237. 10.1038/s41575-019-0258-z [DOI] [PubMed] [Google Scholar]
- 37. Li G, Lin J, Zhang C, et al. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes. 2021;13(1):1968257. 10.1080/19490976.2021.1968257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flannigan KL, Nieves KM, Szczepanski HE, et al. The Pregnane X receptor and indole‐3‐propionic acid shape the intestinal mesenchyme to restrain inflammation and fibrosis. Cell Mol Gastroenterol Hepatol. 2023;15(3):765‐795. 10.1016/j.jcmgh.2022.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pai Y‐C, Weng L‐T, Wei S‐C, et al. Gut microbial transcytosis induced by tumor necrosis factor‐like 1A‐dependent activation of a myosin light chain kinase splice variant contributes to IBD. J Crohns Colitis. 2020;15(2):258‐272. 10.1093/ecco-jcc/jjaa165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cao Y, Wang Z, Yan Y, et al. Enterotoxigenic bacteroidesfragilis promotes intestinal inflammation and malignancy by inhibiting exosome‐packaged miR‐149‐3p. Gastroenterology. 2021;161(5). 10.1053/j.gastro.2021.08.003 [DOI] [PubMed] [Google Scholar]
- 41. Mata‐Garrido J, Xiang Y, Chang‐Marchand Y, et al. The Heterochromatin protein 1 is a regulator in RNA splicing precision deficient in ulcerative colitis. Nat Commun. 2022;13(1):6834. 10.1038/s41467-022-34556-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4(1):33‐46. 10.1016/j.jcmgh.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merga Y, Campbell BJ, Rhodes JM. Mucosal barrier, bacteria and inflammatory bowel disease: possibilities for therapy. Dig Dis. 2014;32(4):475‐483. 10.1159/000358156 [DOI] [PubMed] [Google Scholar]
- 44. Vancamelbeke M, Vermeire S. The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol. 2017;11(9):821‐834. 10.1080/17474124.2017.1343143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mehandru S, Colombel J‐F. The intestinal barrier, an arbitrator turned provocateur in IBD. Nat Rev Gastroenterol Hepatol. 2021;18(2):83‐84. 10.1038/s41575-020-00399-w [DOI] [PubMed] [Google Scholar]
- 46. Stolfi C, Maresca C, Monteleone G, Laudisi F. Implication of intestinal barrier dysfunction in gut dysbiosis and diseases. Biomedicines. 2022;10(2). 10.3390/biomedicines10020289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muise AM, Walters T, Wine E, et al. Protein‐tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol. 2007;17(14):1212‐1218. [DOI] [PubMed] [Google Scholar]
- 48. Kern JS, Herz C, Haan E, et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin‐1 isoforms. J Pathol. 2007;213(4):462‐470. [DOI] [PubMed] [Google Scholar]
- 49. He WQ, Wang J, Sheng JY, Zha JM, Graham WV, Turner JR. Contributions of myosin light chain kinase to regulation of epithelial paracellular permeability and mucosal homeostasis. Int J Mol Sci. 2020;21(3). 10.3390/ijms21030993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zha J‐M, Li H‐S, Wang Y‐T, Lin Q, Tao M, He W‐Q. Characterization of isoform expression and subcellular distribution of MYPT1 in intestinal epithelial cells. Gene. 2016;588(1):1‐6. 10.1016/j.gene.2016.04.048 [DOI] [PubMed] [Google Scholar]
- 51. Mager LF, Koelzer VH, Stuber R, et al. The ESRP1‐GPR137 axis contributes to intestinal pathogenesis. eLife. 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kałużna A, Olczyk P, Komosińska‐Vassev K. The role of innate and adaptive immune cells in the pathogenesis and development of the inflammatory response in ulcerative colitis. J Clin Med. 2022;11(2). 10.3390/jcm11020400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saez A, Herrero‐Fernandez B, Gomez‐Bris R, Sánchez‐Martinez H, Gonzalez‐Granado JM. Pathophysiology of inflammatory bowel disease: innate immune system. Int J Mol Sci. 2023;24(2). 10.3390/ijms24021526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32(6):597‐608. 10.1042/bsr20120055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaparakis‐Liaskos M. The intracellular location, mechanisms and outcomes of NOD1 signaling. Cytokine. 2015;74(2):207‐212. 10.1016/j.cyto.2015.02.018 [DOI] [PubMed] [Google Scholar]
- 56. Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. NOD‐like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth Factor Rev. 2011;22(5‐6):257‐276. 10.1016/j.cytogfr.2011.09.003 [DOI] [PubMed] [Google Scholar]
- 57. Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod‐induced NF‐kappaB activation. Embo J. 2008;27(2):373‐383. 10.1038/sj.emboj.7601962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898‐908. 10.1016/j.immuni.2014.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Girardin SE, Jéhanno M, Mengin‐Lecreulx D, Sansonetti PJ, Alzari PM, Philpott DJ. Identification of the critical residues involved in peptidoglycan detection by Nod1. J Biol Chem. 2005;280(46):38648‐38656. [DOI] [PubMed] [Google Scholar]
- 60. Ohno H, Hase K. Glycoprotein 2 (GP2): grabbing the FimH bacteria into M cells for mucosal immunity. Gut Microbes. 2010;1(6):407‐410. 10.4161/gmic.1.6.14078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Palmela C, Chevarin C, Xu Z, et al. Adherent‐invasive Escherichia coli in inflammatory bowel disease. Gut. 2018;67(3):574‐587. 10.1136/gutjnl-2017-314903 [DOI] [PubMed] [Google Scholar]
- 62. Derer S, Brethack A‐K, Pietsch C, et al. Inflammatory bowel disease‐associated GP2 autoantibodies inhibit mucosal immune response to adherent‐invasive bacteria. Inflamm Bowel Dis. 2020;26(12):1856‐1868. 10.1093/ibd/izaa069 [DOI] [PubMed] [Google Scholar]
- 63. Hartman ML, Czyz MBCL‐G. 20 years of research on a non‐typical protein from the BCL‐2 family. Cell Death Differ. 2023;30(6):1437‐1446. 10.1038/s41418-023-01158-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Woznicki JA, Flood P, Bustamante‐Garrido M, et al. Human BCL‐G regulates secretion of inflammatory chemokines but is dispensable for induction of apoptosis by IFN‐γ and TNF‐α in intestinal epithelial cells. Cell Death Dis. 2020;11(1):68. 10.1038/s41419-020-2263-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial‐induced inflammation. Cell Microbiol. 2003;5(9):581‐592. [DOI] [PubMed] [Google Scholar]
- 66. Kufer TA, Banks DJ, Philpott DJ. Innate immune sensing of microbes by Nod proteins. Ann N Y Acad Sci. 2006;1072:19‐27. [DOI] [PubMed] [Google Scholar]
- 67. Leung E, Hong J, Fraser A, Krissansen GW. Splicing of NOD2 (CARD15) RNA transcripts. Mol Immunol. 2007;44(4):284‐294. [DOI] [PubMed] [Google Scholar]
- 68. De Arras L, Laws R, Leach SM, et al. Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics. 2014;197(2):485‐496. 10.1534/genetics.113.160499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lv Z, Wang Z, Luo L, et al. Spliceosome protein Eftud2 promotes colitis‐associated tumorigenesis by modulating inflammatory response of macrophage. Mucosal Immunol. 2019;12(5):1164‐1173. 10.1038/s41385-019-0184-y [DOI] [PubMed] [Google Scholar]
- 70. Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL‐17F in inflammatory bowel disease (IBD): upregulated colonic IL‐17F expression in active Crohn's disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2008;14(4):437‐445. [DOI] [PubMed] [Google Scholar]
- 71. Neurath MF. IL‐23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019;45:1‐8. 10.1016/j.cytogfr.2018.12.002 [DOI] [PubMed] [Google Scholar]
- 72. Kan SH, Mancini G, Gallagher G. Identification and characterization of multiple splice forms of the human interleukin‐23 receptor alpha chain in mitogen‐activated leukocytes. Genes Immun. 2008;9(7):631‐639. 10.1038/gene.2008.64 [DOI] [PubMed] [Google Scholar]
- 73. Blumberg RS. Inflammation in the intestinal tract: pathogenesis and treatment. Dig Dis. 2009;27(4):455‐464. 10.1159/000235851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lin R, Wu W, Chen H, et al. GPR65 promotes intestinal mucosal Th1 and Th17 cell differentiation and gut inflammation through downregulating NUAK2. Clin Transl Med. 2022;12(3):e771. 10.1002/ctm2.771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hudson DL, Speight PM, Watt FM. Altered expression of CD44 isoforms in squamous‐cell carcinomas and cell lines derived from them. Int J Cancer. 1996;66(4):457‐463. 10.1002/(sici)1097-0215(19960516)66:4<457::Aid‐ijc8>3.0.Co;2‐v [DOI] [PubMed] [Google Scholar]
- 76. Vela E, Roca X, Isamat M. Identification of novel splice variants of the human CD44 gene. Biochem Biophys Res Commun. 2006;343(1):167‐170. 10.1016/j.bbrc.2006.02.128 [DOI] [PubMed] [Google Scholar]
- 77. Rosenberg WM, Prince C, Kaklamanis L, et al. Increased expression of CD44v6 and CD44v3 in ulcerative colitis but not colonic Crohn's disease. Lancet. 1995;345(8959):1205‐1209. 10.1016/s0140-6736(95)91991-0 [DOI] [PubMed] [Google Scholar]
- 78. Yoshida K, Sugino T, Bolodeoku J, et al. Detection of exfoliated carcinoma cells in colonic luminal washings by identification of deranged patterns of expression of the CD44 gene. J Clin Pathol. 1996;49(4):300‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Macdonald DC, Leir SH, Brooks C, Sanders E, Lackie P, Rosenberg W. CD44 isoform expression on colonic epithelium mediates lamina propria lymphocyte adhesion and is controlled by Th1 and Th2 cytokines. Eur J Gastroenterol Hepatol. 2003;15(10):1101‐1110. 10.1097/00042737-200310000-00007 [DOI] [PubMed] [Google Scholar]
- 80. Johnson P, Ruffell B. CD44 and its role in inflammation and inflammatory diseases. Inflamm Allergy Drug Targets. 2009;8(3):208‐220. 10.2174/187152809788680994 [DOI] [PubMed] [Google Scholar]
- 81. Camacho FI, Muñoz C, Sánchez‐Verde L, Sáez AI, Alcántara M, Rodríguez R. CD44v6 expression in inflammatory bowel disease is associated with activity detected by endoscopy and pathological features. Histopathology. 1999;35(2):144‐149. [DOI] [PubMed] [Google Scholar]
- 82. Pfister K, Wittig BM, Mueller‐Molaian I, Remberger K, Zeitz M, Stallmach A. Decreased CD44v6 expression in lamina propria lymphocytes of patients with inflammatory bowel disease. Exp Mol Pathol. 2001;71(3):186‐193. [DOI] [PubMed] [Google Scholar]
- 83. Wittig B, Seiter S, Schmidt DS, Zuber M, Neurath M, Zöller M. CD44 variant isoforms on blood leukocytes in chronic inflammatory bowel disease and other systemic autoimmune diseases. Lab Invest. 1999;79(6):747‐759. [PubMed] [Google Scholar]
- 84. Wittig BM, Stallmach A, Zeitz M, Günthert U. Functional involvement of CD44 variant 7 in gut immune response. Pathobiology. 2002;70(3):184‐189. [DOI] [PubMed] [Google Scholar]
- 85. Hoffmann U, Heilmann K, Hayford C, et al. CD44v7 ligation downregulates the inflammatory immune response in Crohn's disease patients by apoptosis induction in mononuclear cells from the lamina propria. Cell Death Differ. 2007;14(8):1542‐1551. [DOI] [PubMed] [Google Scholar]
- 86. Wittig BM, Sabat R, Holzlöhner P, et al. Absence of specific alternatively spliced exon of CD44 in macrophages prevents colitis. Mucosal Immunol. 2018;11(3):846‐860. 10.1038/mi.2017.98 [DOI] [PubMed] [Google Scholar]
- 87. Aiba Y, Nakamura M. The role of TL1A and DR3 in autoimmune and inflammatory diseases. Mediat Inflamm. 2013;2013:258164. 10.1155/2013/258164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Valatas V, Kolios G, Bamias G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co‐stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front Immunol. 2019;10:583. 10.3389/fimmu.2019.00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bamias G, Mishina M, Nyce M, et al. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci USA. 2006;103(22):8441‐8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Souza HSP, Tortori CJA, Castelo‐Branco MTL, et al. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: evidence of altered expression of FasL and perforin cytotoxic pathways. Int J Colorectal Dis. 2005;20(3):277‐286. [DOI] [PubMed] [Google Scholar]
- 91. Mitomi H, Ohkura Y, Yokoyama K, et al. Contribution of TIA‐1+ and granzyme B+ cytotoxic T lymphocytes to cryptal apoptosis and ulceration in active inflammatory bowel disease. Pathol Res Pract. 2007;203(10):717‐723. [DOI] [PubMed] [Google Scholar]
- 92. Förch P, Puig O, Kedersha N, et al. The apoptosis‐promoting factor TIA‐1 is a regulator of alternative pre‐mRNA splicing. Mol Cell. 2000;6(5):1089‐1098. [DOI] [PubMed] [Google Scholar]
- 93. Förch P, Valcárcel J. Molecular mechanisms of gene expression regulation by the apoptosis‐promoting protein TIA‐1. Apoptosis. 2001;6(6):463‐468. [DOI] [PubMed] [Google Scholar]
- 94. Ling Y, Wang J, Wang L, Hou J, Qian P. Xiang‐dong W. Roles of CEACAM1 in cell communication and signaling of lung cancer and other diseases. Cancer Metastasis Rev. 2015;34(2):347‐357. 10.1007/s10555-015-9569-x [DOI] [PubMed] [Google Scholar]
- 95. Kelleher M, Singh R, O'Driscoll CM, Melgar S. Carcinoembryonic antigen (CEACAM) family members and inflammatory bowel disease. Cytokine Growth Factor Rev. 2019;47:21‐31. 10.1016/j.cytogfr.2019.05.008 [DOI] [PubMed] [Google Scholar]
- 96. Nagaishi T, Iijima H, Nakajima A, Chen D, Blumberg RS. Role of CEACAM1 as a regulator of T cells. Ann N Y Acad Sci. 2006;1072:155‐175. 10.1196/annals.1326.004 [DOI] [PubMed] [Google Scholar]
- 97. Nagaishi T, Chen Z, Chen L, Iijima H, Nakajima A, Blumberg RS. CEACAM1 and the regulation of mucosal inflammation. Mucosal Immunol. 2008;1(1):S39‐42. 10.1038/mi.2008.50. Suppl 1. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen D, Iijima H, Nagaishi T, et al. Carcinoembryonic antigen‐related cellular adhesion molecule 1 isoforms alternatively inhibit and costimulate human T cell function. J Immunol. 2004;172(6):3535‐3543. 10.4049/jimmunol.172.6.3535 [DOI] [PubMed] [Google Scholar]
- 99. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10):a016295. 10.1101/cshperspect.a016295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rose‐John S, Scheller J, Elson G, Jones SA. Interleukin‐6 biology is coordinated by membrane‐bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80(2):227‐236. [DOI] [PubMed] [Google Scholar]
- 101. Atreya R, Neurath MF. Signaling molecules: the pathogenic role of the IL‐6/STAT‐3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr Drug Targets. 2008;9(5):369‐374. [DOI] [PubMed] [Google Scholar]
- 102. Rose‐John S, Mitsuyama K, Matsumoto S, Thaiss WM, Scheller J. Interleukin‐6 trans‐signaling and colonic cancer associated with inflammatory bowel disease. Curr Pharm Des. 2009;15(18):2095‐2103. [DOI] [PubMed] [Google Scholar]
- 103. Neurath MF, Finotto S. IL‐6 signaling in autoimmunity, chronic inflammation and inflammation‐associated cancer. Cytokine Growth Factor Rev. 2011;22(2):83‐89. 10.1016/j.cytogfr.2011.02.003 [DOI] [PubMed] [Google Scholar]
- 104. Chalaris A, Schmidt‐Arras D, Yamamoto K, Rose‐John S. Interleukin‐6 trans‐signaling and colonic cancer associated with inflammatory bowel disease. Dig Dis. 2012;30(5):492‐499. 10.1159/000341698 [DOI] [PubMed] [Google Scholar]
- 105. Matsumoto S, Hara T, Mitsuyama K, et al. Essential roles of IL‐6 trans‐signaling in colonic epithelial cells, induced by the IL‐6/soluble‐IL‐6 receptor derived from lamina propria macrophages, on the development of colitis‐associated premalignant cancer in a murine model. J Immunol. 2010;184(3):1543‐1551. 10.4049/jimmunol.0801217 [DOI] [PubMed] [Google Scholar]
- 106. Sommer J, Garbers C, Wolf J, et al. Alternative intronic polyadenylation generates the interleukin‐6 trans‐signaling inhibitor sgp130‐E10. J Biol Chem. 2014;289(32):22140‐22150. 10.1074/jbc.M114.560938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wolf J, Waetzig GH, Reinheimer TM, Scheller J, Rose‐John S, Garbers C. A soluble form of the interleukin‐6 family signal transducer gp130 is dimerized via a C‐terminal disulfide bridge resulting from alternative mRNA splicing. Biochem Biophys Res Commun. 2016;470(4):870‐876. 10.1016/j.bbrc.2016.01.127 [DOI] [PubMed] [Google Scholar]
- 108. Moriasi C, Subramaniam D, Awasthi S, Ramalingam S, Anant S. Prevention of colitis‐associated cancer: natural compounds that target the IL‐6 soluble receptor. Anticancer Agents Med Chem. 2012;12(10):1221‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Göschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to Treg‐based therapies. Semin Immunopathol. 2019;41(3):301‐314. 10.1007/s00281-019-00741-8 [DOI] [PubMed] [Google Scholar]
- 110. Kristensen B, Hegedüs L, Madsen HO, Smith TJ, Nielsen CH. Altered balance between self‐reactive T helper (Th)17 cells and Th10 cells and between full‐length forkhead box protein 3 (FoxP3) and FoxP3 splice variants in Hashimoto's thyroiditis. Clin Exp Immunol. 2015;180(1):58‐69. 10.1111/cei.12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mailer RK. IPEX as a consequence of alternatively spliced FOXP3. Front Pediatr. 2020;8:594375. 10.3389/fped.2020.594375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mailer RKW, Joly A‐L, Liu S, Elias S, Tegner J, Andersson J. IL‐1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep. 2015;5:14674. 10.1038/srep14674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fraschilla I, Jeffrey KL. The Speckled Protein (SP) family: immunity's chromatin readers. Trends Immunol. 2020;41(7):572‐585. 10.1016/j.it.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tanagala KKK, Morin‐Baxter J, Carvajal R, et al. SP140 inhibits STAT1 signaling, induces IFN‐γ in tumor‐associated macrophages, and is a predictive biomarker of immunotherapy response. J Immunother Cancer. 2022;10(12). 10.1136/jitc-2022-005088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fraschilla I, Amatullah H, Rahman R‐U, Jeffrey KL. Immune chromatin reader SP140 regulates microbiota and risk for inflammatory bowel disease. Cell Host Microbe. 2022;30(10). 10.1016/j.chom.2022.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Karaky M, Fedetz M, Potenciano V, et al. SP140 regulates the expression of immune‐related genes associated with multiple sclerosis and other autoimmune diseases by NF‐κB inhibition. Hum Mol Genet. 2018;27(23):4012‐4023. 10.1093/hmg/ddy284 [DOI] [PubMed] [Google Scholar]
- 117. Thompson CB, Lindsten T, Ledbetter JA, et al. CD28 activation pathway regulates the production of multiple T‐cell‐derived lymphokines/cytokines. Proc Natl Acad Sci USA. 1989;86(4):1333‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hitomi Y, Aiba Y, Ueno K, et al. rs2013278 in the multiple immunological‐trait susceptibility locus CD28 regulates the production of non‐functional splicing isoforms. Hum Genomics. 2022;16(1):46. 10.1186/s40246-022-00419-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li J, Mao R, Kurada S, et al. Pathogenesis of fibrostenosing Crohn's disease. Transl Res. 2019;209:39‐54. 10.1016/j.trsl.2019.03.005 [DOI] [PubMed] [Google Scholar]
- 120. Alfredsson J, Wick MJ. Mechanism of fibrosis and stricture formation in Crohn's disease. Scand J Immunol. 2020;92(6):e12990. 10.1111/sji.12990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hayashi Y, Nakase H. The molecular mechanisms of intestinal inflammation and fibrosis in Crohn's disease. Front Physiol. 2022;13:845078. 10.3389/fphys.2022.845078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yoshida T, Akatsuka T, Imanaka‐Yoshida K. Tenascin‐C and integrins in cancer. Cell Adh Migr. 2015;9(1‐2). 10.1080/19336918.2015.1008332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Missirlis D, Haraszti T, Kessler H, Spatz JP. Fibronectin promotes directional persistence in fibroblast migration through interactions with both its cell‐binding and heparin‐binding domains. Sci Rep. 2017;7(1):3711. 10.1038/s41598-017-03701-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bhattacharyya S, Midwood KS, Varga J. Tenascin‐C in fibrosis in multiple organs: translational implications. Semin Cell Dev Biol. 2022;128:130‐136. 10.1016/j.semcdb.2022.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ambort D, Brellier F, Becker‐Pauly C, et al. Specific processing of tenascin‐C by the metalloprotease meprinbeta neutralizes its inhibition of cell spreading. Matrix Biol. 2010;29(1):31‐42. 10.1016/j.matbio.2009.08.007 [DOI] [PubMed] [Google Scholar]
- 126. Zimmermann EM, Li L, Hou YT, Mohapatra NK, Pucilowska JB. Insulin‐like growth factor I and insulin‐like growth factor binding protein 5 in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G1022‐G1029. [DOI] [PubMed] [Google Scholar]
- 127. Hazelgrove KB, Flynn RS, Qiao L‐Y, Grider JR, Kuemmerle JF. Endogenous IGF‐I and alpha v beta3 integrin ligands regulate increased smooth muscle growth in TNBS‐induced colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296(6):G1230‐G1237. 10.1152/ajpgi.90508.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li C, Vu K, Hazelgrove K, Kuemmerle JF. Increased IGF‐IEc expression and mechano‐growth factor production in intestinal muscle of fibrostenotic Crohn's disease and smooth muscle hypertrophy. Am J Physiol Gastrointest Liver Physiol. 2015;309(11):G888‐G899. 10.1152/ajpgi.00414.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bai L, Kao JY, Law DJ, Merchant JL. Recruitment of ataxia‐telangiectasia mutated to the p21(waf1) promoter by ZBP‐89 plays a role in mucosal protection. Gastroenterology. 2006;131(3):841‐852. [DOI] [PubMed] [Google Scholar]
- 130. Essien BE, Grasberger H, Romain RD, et al. ZBP‐89 regulates expression of tryptophan hydroxylase I and mucosal defense against Salmonella typhimurium in mice. Gastroenterology. 2013;144(7). 10.1053/j.gastro.2013.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Law DJ, Labut EM, Adams RD, Merchant JL. An isoform of ZBP‐89 predisposes the colon to colitis. Nucleic Acids Res. 2006;34(5):1342‐1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tsukahara T, Hamouda N, Utsumi D, Matsumoto K, Amagase K, Kato S. G protein‐coupled receptor 35 contributes to mucosal repair in mice via migration of colonic epithelial cells. Pharmacol Res. 2017;123:27‐39. 10.1016/j.phrs.2017.06.009 [DOI] [PubMed] [Google Scholar]
- 133. Kaya B, Doñas C, Wuggenig P, et al. Lysophosphatidic acid‐mediated GPR35 signaling in CX3CR1+ macrophages regulates intestinal homeostasis. Cell Rep. 2020;32(5):107979. 10.1016/j.celrep.2020.107979 [DOI] [PubMed] [Google Scholar]
- 134. Boleij A, Fathi P, Dalton W, et al. G‐protein coupled receptor 35 (GPR35) regulates the colonic epithelial cell response to enterotoxigenic Bacteroides fragilis. Commun Biol. 2021;4(1):585. 10.1038/s42003-021-02014-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kaya B, Melhem H, Niess JH. GPR35 in intestinal diseases: from risk gene to function. Front Immunol. 2021;12:717392. 10.3389/fimmu.2021.717392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xiao Y, Wang X‐Q, Yu Y, et al. Comprehensive mutation screening for 10 genes in Chinese patients suffering very early onset inflammatory bowel disease. World J Gastroenterol. 2016;22(24):5578‐5588. 10.3748/wjg.v22.i24.5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Schihada H, Klompstra TM, Humphrys LJ, et al. Isoforms of GPR35 have distinct extracellular N‐termini that allosterically modify receptor‐transducer coupling and mediate intracellular pathway bias. J Biol Chem. 2022;298(9):102328. 10.1016/j.jbc.2022.102328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K. Modulation of cell‐adhesive activity of fibronectin by the alternatively spliced EDA segment. J Cell Biol. 1997;139(1):295‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kumra H, Reinhardt DP. Fibronectin‐targeted drug delivery in cancer. Adv Drug Deliv Rev. 2016;97:101‐110. 10.1016/j.addr.2015.11.014 [DOI] [PubMed] [Google Scholar]
- 140. Bootz F, Schmid AS, Neri D. Alternatively spliced EDA domain of fibronectin is a target for pharmacodelivery applications in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(8):1908‐1917. 10.1097/MIB.0000000000000440 [DOI] [PubMed] [Google Scholar]
- 141. Mwinyi J, Wenger C, Eloranta JJ, Kullak‐Ublick GA. Glucocorticoid receptor gene haplotype structure and steroid therapy outcome in IBD patients. World J Gastroenterol. 2010;16(31):3888‐3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sidoroff M, Kolho K‐L. Glucocorticoid sensitivity in inflammatory bowel disease. Ann Med. 2012;44(6):578‐587. 10.3109/07853890.2011.590521 [DOI] [PubMed] [Google Scholar]
- 143. Towers R, Naftali T, Gabay G, Carlebach M, Klein A, Novis B. High levels of glucocorticoid receptors in patients with active Crohn's disease may predict steroid resistance. Clin Exp Immunol. 2005;141(2):357‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hausmann M, Herfarth H, Schölmerich J, Rogler G. Glucocorticoid receptor isoform expression does not predict steroid treatment response in IBD. Gut. 2007;56(9):1328‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bagnall RD, Roberts RG, Mirza MM, Torigoe T, Prescott NJ, Mathew CG. Novel isoforms of the CARD8 (TUCAN) gene evade a nonsense mutation. Eur J Hum Genet. 2008;16(5):619‐625. 10.1038/sj.ejhg.5201996 [DOI] [PubMed] [Google Scholar]
- 146. Gillespie E, Leeman SE, Watts LA, et al. Truncated neurokinin‐1 receptor is increased in colonic epithelial cells from patients with colitis‐associated cancer. Proc Natl Acad Sci USA. 2011;108(42):17420‐17425. 10.1073/pnas.1114275108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Jin R, Yuan W‐X, Xu H‐G, Ren W, Zhuang L‐L, Zhou G‐P. Characterization of a novel isoform of the human ORMDL3 gene. Cell Tissue Res. 2011;346(2):203‐208. 10.1007/s00441-011-1261-z [DOI] [PubMed] [Google Scholar]
- 148. Bussières‐Marmen S, Hutchins AP, Schirbel A, et al. Characterization of PTPN2 and its use as a biomarker. Methods. 2014;65(2):239‐246. 10.1016/j.ymeth.2013.08.020 [DOI] [PubMed] [Google Scholar]
- 149. Escudero‐Hernández C, Martínez‐Abad B, Ruipérez V, Garrote JA, Arranz E. New IL‐15 receptor‐α splicing variants identified in intestinal epithelial Caco‐2 cells. Innate Immun. 2017;23(1):44‐53. 10.1177/1753425916674263 [DOI] [PubMed] [Google Scholar]
- 150. Goode T, O'Connell J, Anton P, et al. Neurokinin‐1 receptor expression in inflammatory bowel disease: molecular quantitation and localisation. Gut. 2000;47(3):387‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Koon HW, Pothoulakis C. Immunomodulatory properties of substance P: the gastrointestinal system as a model. Ann N Y Acad Sci. 2006;1088:23‐40. [DOI] [PubMed] [Google Scholar]
- 152. Herring JA, Elison WS, Tessem JS. Function of Nr4a orphan nuclear receptors in proliferation, apoptosis and fuel utilization across tissues. Cells. 2019;8(11). 10.3390/cells8111373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wan PK‐T, Siu MK‐Y, Leung TH‐Y, Mo X‐T, Chan KK‐L, Ngan HY‐S. Role of Nurr1 in carcinogenesis and tumor immunology: a state of the art review. Cancers (Basel). 2020;12(10). 10.3390/cancers12103044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sekiya T, Kashiwagi I, Inoue N, et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat Commun. 2011;2:269. 10.1038/ncomms1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xu P‐y, Le W‐d. Novel splicing variant of the human orphan nuclear receptor Nurr1 gene. Chin Med J (Engl). 2004;117(6):899‐902. [PubMed] [Google Scholar]
- 156. Liu H, Liu H, Li T, et al. NR4A2 genetic variation and Parkinson's disease: evidence from a systematic review and meta‐analysis. Neurosci Lett. 2017;650:25‐32. 10.1016/j.neulet.2017.01.062 [DOI] [PubMed] [Google Scholar]
- 157. Vierbuchen T, Agarwal S, Johnson JL, et al. The lncRNA LUCAT1 is elevated in inflammatory disease and restrains inflammation by regulating the splicing and stability of NR4A2. Proc Natl Acad Sci USA. 2023;120(1):e2213715120. 10.1073/pnas.2213715120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Rahman H, Vikram P, Hammami Z, Singh RK. Recent advances in date palm genomics: a comprehensive review. Front Genet. 2022;13:959266. 10.3389/fgene.2022.959266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Yamamoto‐Furusho JK, Fonseca‐Camarillo G. Genetic markers associated with clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(11):2683‐2695. 10.1097/MIB.0000000000000500 [DOI] [PubMed] [Google Scholar]
- 160. Wan D, Wang S, Xu Z, et al. PRKAR2A‐derived circular RNAs promote the malignant transformation of colitis and distinguish patients with colitis‐associated colorectal cancer. Clin Transl Med. 2022;12(2):e683. 10.1002/ctm2.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Häsler R, Kerick M, Mah N, et al. Alterations of pre‐mRNA splicing in human inflammatory bowel disease. Eur J Cell Biol. 2011;90(6‐7):603‐611. 10.1016/j.ejcb.2010.11.010 [DOI] [PubMed] [Google Scholar]
- 162. Olén O, Erichsen R, Sachs MC, et al. Colorectal cancer in ulcerative colitis: a Scandinavian population‐based cohort study. Lancet. 2020;395(10218):123‐131. 10.1016/S0140-6736(19)32545-0 [DOI] [PubMed] [Google Scholar]
- 163. Bopanna S, Ananthakrishnan AN, Kedia S, Yajnik V, Ahuja V. Risk of colorectal cancer in Asian patients with ulcerative colitis: a systematic review and meta‐analysis. Lancet Gastroenterol Hepatol. 2017;2(4):269‐276. 10.1016/S2468-1253(17)30004-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Arai N, Kudo T, Tokita K, et al. Expression of oncogenic molecules in pediatric ulcerative colitis. Digestion. 2022;103(2):150‐158. 10.1159/000519559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Low END, Mokhtar NM, Wong Z, Raja Ali RA. Colonic mucosal transcriptomic changes in patients with long‐duration ulcerative colitis revealed colitis‐associated cancer pathways. J Crohns Colitis. 2019;13(6):755‐763. 10.1093/ecco-jcc/jjz002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Beigh MM. Next‐generation sequencing: the translational medicine approach from “bench to bedside to population”. Medicines (Basel). 2016;3(2). 10.3390/medicines3020014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang F, Wu L, Yin L, Shi H, Gu Y, Xing N. Combined treatment with anti‐PSMA CAR NK‐92 cell and anti‐PD‐L1 monoclonal antibody enhances the antitumour efficacy against castration‐resistant prostate cancer. Clin Transl Med. 2022;12(6):e901. 10.1002/ctm2.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kinugasa H, Hiraoka S, Nouso K, et al. Liquid biopsy for patients with IBD‐associated neoplasia. BMC Cancer. 2020;20(1):1188. 10.1186/s12885-020-07699-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Li D, Tan Y. Dysregulation of alternative splicing is associated with the pathogenesis of ulcerative colitis. Biomed Eng Online. 2021;20(1):121. 10.1186/s12938-021-00959-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li D, Liang Y, Lu J, Tan Y. An alternative splicing signature in human Crohn's disease. BMC Gastroenterol. 2021;21(1):420. 10.1186/s12876-021-02001-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Berger K, Somineni H, Prince J, Kugathasan S, Gibson G. Altered splicing associated with the pathology of inflammatory bowel disease. Hum Genomics. 2021;15(1):47. 10.1186/s40246-021-00347-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Sun Q, Han Y, He J, et al. Long‐read sequencing reveals the landscape of aberrant alternative splicing and novel therapeutic target in colorectal cancer. Genome Med. 2023;15(1):76. 10.1186/s13073-023-01226-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Eger N, Schoppe L, Schuster S, Laufs U, Boeckel J‐N. Circular RNA splicing. Adv Exp Med Biol. 2018;1087:41‐52. 10.1007/978-981-13-1426-1_4 [DOI] [PubMed] [Google Scholar]
- 174. Xu B, Meng Y, Jin Y. RNA structures in alternative splicing and back‐splicing. Wiley Interdiscip Rev RNA. 2021;12(1):e1626. 10.1002/wrna.1626 [DOI] [PubMed] [Google Scholar]
- 175. Ouyang J, Zhong Y, Zhang Y, et al. Long non‐coding RNAs are involved in alternative splicing and promote cancer progression. Br J Cancer. 2022;126(8):1113‐1124. 10.1038/s41416-021-01600-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wang X, Hua J, Li J, et al. Mechanisms of non‐coding RNA‐modulated alternative splicing in cancer. RNA Biol. 2022;19(1):541‐547. 10.1080/15476286.2022.2062846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Chen QW, Cai QQ, Yang Y, et al. LncRNA BC promotes lung adenocarcinoma progression by modulating IMPAD1 alternative splicing. Clin Transl Med. 2023;13(1):e1129. 10.1002/ctm2.1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Żmieńko A, Satyr A. [Nanopore sequencing and its application in biology]. Postepy Biochem. 2020;66(3):193‐204. 10.18388/pb.2020_328 [DOI] [PubMed] [Google Scholar]
- 179. Allemand E, Ango F. Analysis of splicing regulation by third‐generation sequencing. Methods Mol Biol. 2022;2537:81‐95. 10.1007/978-1-0716-2521-7_6 [DOI] [PubMed] [Google Scholar]
- 180. Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single‐cell RNA sequencing technologies and applications: a brief overview. Clin Transl Med. 2022;12(3):e694. 10.1002/ctm2.694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Tang F, Barbacioru C, Wang Y, et al. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377‐382. 10.1038/nmeth.1315 [DOI] [PubMed] [Google Scholar]
- 182. Williams CG, Lee HJ, Asatsuma T, Vento‐Tormo R, Haque A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022;14(1):68. 10.1186/s13073-022-01075-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Liu S, Zhao Y, Lu S, et al. Single‐cell transcriptomics reveals a mechanosensitive injury signaling pathway in early diabetic nephropathy. Genome Med. 2023;15(1):2. 10.1186/s13073-022-01145-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Martin JC, Chang C, Boschetti G, et al. Single‐cell analysis of Crohn's disease lesions identifies a pathogenic cellular module associated with resistance to Anti‐TNF therapy. Cell. 2019;178(6). 10.1016/j.cell.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Boland BS, He Z, Tsai MS, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single‐cell analyses. Sci Immunol. 2020;5(50). 10.1126/sciimmunol.abb4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Devlin JC, Axelrad J, Hine AM, et al. Single‐cell transcriptional survey of ileal‐anal pouch immune cells from ulcerative colitis patients. Gastroenterology. 2021;160(5):1679‐1693. 10.1053/j.gastro.2020.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Kong L, Pokatayev V, Lefkovith A, et al. The landscape of immune dysregulation in Crohn's disease revealed through single‐cell transcriptomic profiling in the ileum and colon. Immunity. 2023;56(2). 10.1016/j.immuni.2023.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Vickovic S, Eraslan G, Salmén F, et al. High‐definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16(10):987‐990. 10.1038/s41592-019-0548-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Saul D, Kosinsky RL. Spatial transcriptomics herald a new era of transcriptome research. Clin Transl Med. 2023;13(5):e1264. 10.1002/ctm2.1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Moncada R, Barkley D, Wagner F, et al. Integrating microarray‐based spatial transcriptomics and single‐cell RNA‐seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38(3):333‐342. 10.1038/s41587-019-0392-8 [DOI] [PubMed] [Google Scholar]
- 191. Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single‐cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet. 2021;22(10):627‐644. 10.1038/s41576-021-00370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Ye C, Zhu J, Wang J, et al. Single‐cell and spatial transcriptomics reveal the fibrosis‐related immune landscape of biliary atresia. Clin Transl Med. 2022;12(11):e1070. 10.1002/ctm2.1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ji K, Chen L, Wang X, et al. Integrating single‐cell RNA sequencing with spatial transcriptomics reveals an immune landscape of human myometrium during labour. Clin Transl Med. 2023;13(4):e1234. 10.1002/ctm2.1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Bonnal SC, López‐Oreja I, Valcárcel J. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat Rev Clin Oncol. 2020;17(8):457‐474. 10.1038/s41571-020-0350-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.