

Abstract
HIV‐1 reverse transcriptase (RT) remains a key target for HIV drug development. As successful management of the disease requires lifelong treatment, the emergence of resistance mutations is inevitable, making development of new RT inhibitors, which remain effective against resistant variants crucial. To this end, previous computationally guided drug design efforts have resulted in catechol diether compounds, which inhibit wildtype RT with picomolar affinities and appear to be promising preclinical candidates. To confirm that these compounds remain potent against Y181C, a widespread mutation conferring resistance to first generation inhibitors, they were screened against the HIV‐1 N119 clinical isolate, reported as a Y181C single mutant. In comparison to a molecular clone with the same mutation, N119 appears less susceptible to inhibition by our preclinical candidate compounds. A more detailed sequencing effort determined that N119 was misidentified and carries V106A in combination with Y181C. While both indolizine and naphthalene substituted catechol diethers are potent against the classical Y181C single mutant, the addition of V106A confers more resistance against the indolizine derivatives than the naphthalene derivatives. Crystal structures presented in this study highlight key features of the naphthyl group, which allow these compounds to remain potent in the double mutant, including stronger interactions with F227 and less reliance on V106 for stabilization of the ethoxy‐uracil ring, which makes critical hydrogen bonds with other residues in the binding pocket.
Keywords: drug resistance, HIV, macromolecular x‐ray crystallography, non‐nucleoside reverse transcriptase inhibitors, reverse transcriptase, structure‐guided drug design
1. INTRODUCTION
Human immunodeficiency virus (HIV) remains a major health issue worldwide. The most recent UNAIDS report estimates that approximately 40 million people have been infected with the virus. This includes 1.5 million new infections, the smallest annual decline in new infections since 2016. Of this population, 29 million people were accessing antiretroviral therapy, which resulted in viral suppression in 92% of cases. Inhibitors of HIV reverse transcriptase (RT), which is responsible for reverse transcribing the viral (+)‐sense RNA genome to dsDNA, were the first FDA‐approved anti‐HIV drugs and remain critical components of combinational therapies. These inhibitors fall into two categories, which inhibit the polymerase via two distinct mechanisms; nucleoside reverse transcriptase inhibitors (NRTIs) compete with incoming dNTPs and terminate the growing strand of DNA while non‐nucleotide reverse transcriptase inhibitors (NNRTIs) bind to an allosteric pocket 10 Å away from the active site, altering the rate‐limiting step in chemical catalysis (Holec et al., 2018; Sarafianos et al., 2009; Spence et al., 1995). RT is highly error prone and has no exonuclease activity, making an error every 4000 nucleotides and introducing approximately five errors in every complete genome replication (Preston et al., 1988). This leads to the rapid evolution of drug‐resistant mutants, particularly within the NNRTI binding pocket, as mutations there are less likely to reduce viral fitness than those in the enzyme's active site (De Luca, 2006).
As there is no cure for HIV and therapy must continue throughout a patient's life, it is important to design new inhibitors, which remain effective against emerging NNRTI‐resistant mutations (Phanuphak & Gulick, 2020). The Y181C mutation has proven particularly problematic, as it emerges rapidly after administration of first‐generation NNRTIs and has been identified in 10% or more of treatment‐naïve patient populations (Jourdain et al., 2004; Mbunkah et al., 2020; Nanfack et al., 2017; Zuo et al., 2020). Newer NNRTIs, including etravirine, rilpivirine, and doravirine, have been designed to remain active against this mutant, though the Y181C mutation is still identified in patient samples experiencing virologic failure after treatment with etravirine or rilpivirine (Alcaro et al., 2011; Molina et al., 2011). Additionally, rilpivirine must be dosed at low levels as it has low aqueous solubility and off‐target inhibition of the hERG ion channel causes dose‐limiting cardiac toxicity (Health Canada, 2011). While doravirine has minimal off‐target effects and remains potent against most common NNRTI‐resistant mutants, it is still susceptible to some mutations present at a low level within the population, including V106A and Y188L (Lai et al., 2014; Stanford University, 2022). It is important to continue development efforts so that a wider variety of NNRTIs are available to treat patients with a variety of resistance mutations.
Previous optimization of a micromolar hit from virtual screening resulted in the development of a class of cyanovinyl catechol diethers with picomolar affinity against wildtype RT (Bollini et al., 2011). As the cyanovinyl substituent was undesirable due to its potential to act as a Michael acceptor, subsequent efforts produced similarly potent compounds by replacing the cyanovinyl with either an indolizine or naphthalene ring (Lee et al., 2013, 2014). Lead naphthyl catechol diethers retain 2–5 nM EC50 values against Y181C (N119) and K103N/ Y181C (A17) viral isolates obtained from the HIV Reagent Program (Lee et al., 2014; Nunberg et al., 1991; Richman et al., 1991). However, lead indolizinyl catechol diethers seem to exhibit an interesting resistance profile; the Y181C single mutation results in approximately 1000‐fold lower potency, though low nanomolar potency is restored in the Y181C/K103N double mutant, which is usually associated with additional resistance (Lee et al., 2013). Earlier studies investigated this unexpected result by examining the antiviral efficacy, EC50, of Compound 1b, a lead indolizinyl catechol diether, against a Y181C molecular clone (Sasaki et al., 2019). Surprisingly, Compound 1b had 3.2 nM potency against the molecular clone, consistent with affinities observed in the naphthyl series, and no significant changes in binding in crystal structures (Sasaki et al., 2019).
In this study, we fully sequenced the N119 clinical isolate and determined that it is a V106A/Y181C double mutant rather than a Y181C single mutant. We have also confirmed that the V106A/Y181C double mutant confers more resistance to indolizinyl catechol diethers as compared with the naphthyl catechol diethers. To explain this difference, we have solved crystal structures of two representative indolizine compounds, 1a and 1b, and two representative naphthyl compounds, 2a and 2b, with both the single Y181C and double V106A/Y181C mutant RT. Our structural analysis suggests a possible mechanism for resistance in the V106A/Y181C mutant.
2. RESULTS
To explain the significant differences between EC50 measurements examining differing antiviral efficacies with the HIV‐1 N119 clinical isolate and the Y181C molecular clone, the HIV‐1 N119 isolate was sequenced. Representative sequencing results demonstrated two drug resistance mutations, V106A and Y181C revealing that a secondary mutation, V106A, was present as shown in (Figure 1).
FIGURE 1.
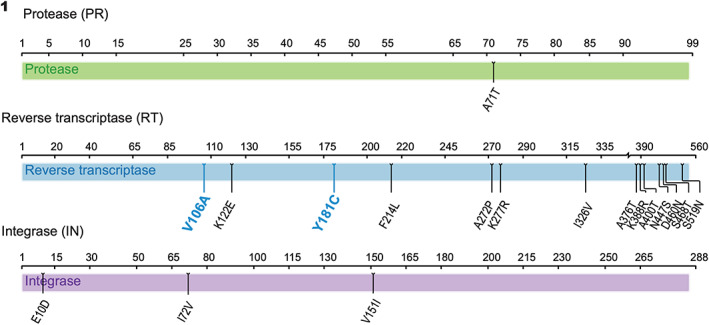
Viral genomic sequencing revealed two potential drug resistance mutations. Sequencing of HIV‐1 protease, reverse transcriptase, and integrase coding regions of viral stock HIV‐1 N119 nevirapine resistant variant. Consensus sequences from the assembled contigs covering HIV‐1 pol were uploaded onto HIVdb Program: Sequence Analysis interface on HIV Drug Resistance Database.
To confirm this additional mutation was responsible for the loss in potency at a molecular level, a recombinant version of RT containing the V106A/Y181C double mutant was expressed and purified. Two indolizine compounds, 1a and a difluorinated derivative, 1b, were studied along with the two corresponding naphthyl compounds, 2a and 2b (Figure 2a). Previous studies showed a nearly 1000 and 400‐fold decrease in potency for Compounds 1a and 1b, respectively, for the WT HIVIIIB versus mutant N119 strains of virus (Figure 2b) (Lee et al., 2013). Biochemical experiments using PicoGreen‐based assays with these WT and double mutant recombinant proteins were carried out to determine the IC50 values for all four compounds. This assay utilizes RT at an active site concentration of 20 nM, which limits determination of IC50 values to no lower than ~10 nM. Representative IC50 values for indolizine compounds, 1a and 1b, also showed a significant decrease in potency (33–47‐fold), in accord with the lower antiviral efficacy values, EC50s, observed in MT‐2 T cell assays (Figure 2b). Moreover, addition of the two fluorines on the indolizine and catechol rings of Compound 1b provides a boost in potency against the double mutant not observed in the cellular data (Figure 2b). As observed by previous cellular studies, representative naphthyl compounds, 2a and 2b, retained higher potency in the double mutant as compared with the indolizines. At the biochemical level, the substitution of two fluorines in Compound 2b versus 2a does not provide any additional boost in potency in IC50 values. However, these compounds are still slightly less potent (5–7‐fold) in the double mutant than in either WT or Y181C single mutant (Sasaki et al., 2019).
FIGURE 2.
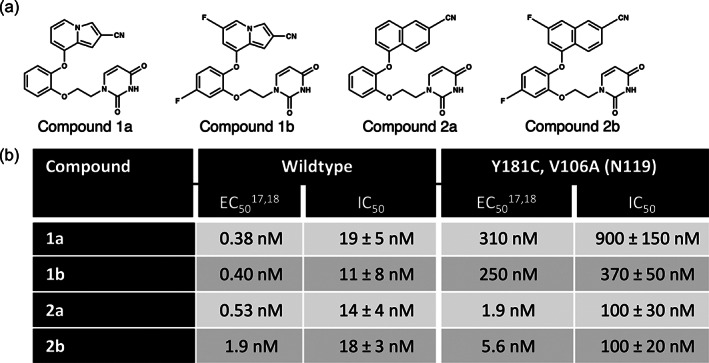
Biochemical assays with V106A/Y181C double mutant agree with cellular results obtained with the N119 variant. (a) Chemical structures of investigated compounds. (b) Affinity measurements of investigated compounds against wildtype and V106A/Y181C mutants. EC50s represent concentration of drug required decrease viral replication by 50% and IC50s represent concentration of drug required to inhibit half of the enzyme's activity. Reported errors represent standard deviations from three replicates.
To better understand the structural differences underlying the naphthyl derivatives ability to retain their potency in the double mutant, we determined the crystal structures with Compounds 1a, 1b, 2a, and 2b bound to V106A/Y181C RT. In addition, structures with Compounds 1a, 2a, and 2b bound to Y181C RT were solved to understand which changes in binding result from the single mutation and which result from their combination. Initial crystallization efforts for the double mutant involved soaking each compound into Apo V106A/Y181C RT52A crystals. While these soaked structures showed strong density for each ligand, B‐factors for the surrounding residues in the NNRTI binding pocket were abnormally high. To circumvent this issue, co‐crystal structures with V106A/Y181C RT were solved in two novel P21 unit cells; Compounds 1a and 2a crystallized in one, while Compound 2b crystallized in the other. The latter unit cell has been previously observed for K103N/Y181C cocrystals, though they diffracted to a lower resolution (Frey et al., 2015). These two unit cells are very similar, each containing two RT heterodimers in their asymmetric unit (ASU), with the major difference between them being the relative orientations of the two RT heterodimers (Figure S1). There is interpretable density for the ligand in both heterodimers, though the density is slightly more distinct in Assembly 1 (Figure S2).
All the structures present V106A/Y181C RT in a similar open conformation seen previously in NNRTI complexes with WT or Y181C RT. The connection and RNase H domains align well with the WT structures, while the thumb, palm, and finger domains are more variable, both between ASU mates and compared to WT (Figure S3). Of note, the palm domain of this double mutant shifts 3–4 Å downward toward the p51 subunit, bringing E169 in the p66 subunit close enough to hydrogen bond with K49 in the p51 subunit (Figure S4). This shift is observed in all four V106A/Y181C mutant structures, so it is likely not responsible for changes in compound affinity. For Compounds 1a and 2a, there is good agreement between the ligand placement in both copies of the protein (Figure S5A,B). However, in the Compound 2b structure, there are significant differences between the positions of both ligands and residues in the NNRTI binding pocket between the two copies in the ASU (Figure S5C).
2.1. Maintenance of key interactions in the Y181C single mutant
The Y181C mutation causes a loss of aryl edge‐to‐face interactions between Y181 and the catechol ring. As observed in our previous structure of Compound 1b in complex with the single Y181C mutant (Sasaki et al., 2019), all four compounds gain compensatory van der Waals interactions by varying degrees of catechol ring rotation toward V189 in both single and double mutant structures (Figure 3). As P95 is no longer interacting with Y181, it returns to the more common exo rotamer. However, interaction distances between the C6 substituent of the inhibitor's bicyclic ring and the nearest carbon of P95 remain relatively stable; the loop containing P95 moves into the pocket, such that the Cβ is in a similar position to the Cɣ in the WT (Figure 4). As a result of the catechol ring movement toward V189, the indolizine ring is also slightly shifted toward C181 in Y181C:1a and Y181C:1b structures (Figure 3a,b). This shift brings the C6 substituent of the indolizine ring into an ideal position for van der Waals interactions with the Cβ of P95 (Figure 4a,b). Since the catechol ring fluorine of Compound 2b made much closer contact with the Cβ of V189 than the equivalent substituent of Compound 1a or 1b in the WT structure, it did not have to shift as far to maintain the van der Waals interactions in the Y181C structure. As a result, the Compound 2b naphthyl group retains its WT positioning (Figure 3d).
FIGURE 3.
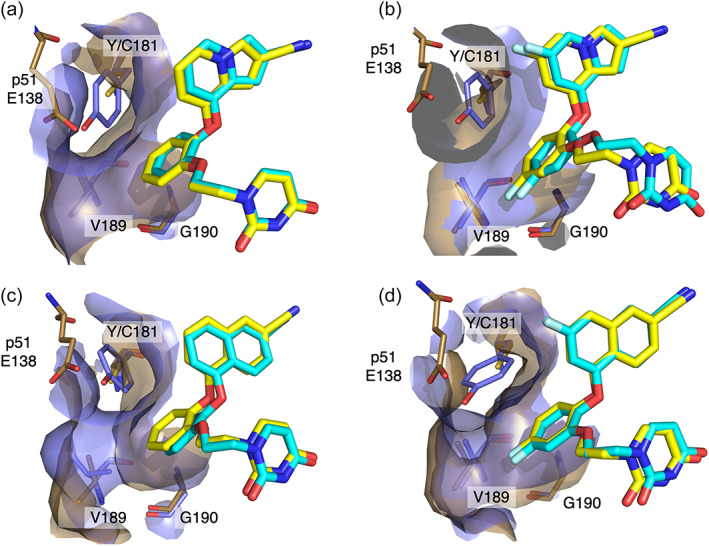
The catechol ring of the inhibitor shifts in the Y181C single mutant to increase contact with V189. (a) Superposition of Compound 1a bound to WT RT (protein in lilac, ligand in cyan; PDB 4MFB) and Y181C RT (protein in brown, ligand in yellow). (b) Superposition of Compound 1b bound to WT RT (protein in lilac, ligand in cyan; PDB: 6DTX) and Y181C RT (protein in brown, ligand in yellow; PDB: 6DTW). (c) Superposition of Compound 2a bound to WT RT (protein in lilac, ligand in cyan; PDB 4WE1) and Y181C RT (protein in brown, ligand in yellow). (d) Superposition of Compound 2b bound to WT RT (protein in lilac, ligand in cyan; PDB 5TW3) and Y181C RT (protein in brown, ligand in yellow).
FIGURE 4.
Interactions between P95 and the ligand C6 or C7 substituent are maintained in mutant structures. Interaction between the indolizine C6 substituent and V100 influences indolizine ring placement in double mutant structures. Van der Waals interactions made by the C6 substituents of (a) Compound 1a, (b) Compound 1b, or the C7 substituents of (c) Compound 2a, (d) Compound 2b with P95 and V100 in WT (ligand: cyan, protein: violet), Y181C (ligand: yellow, protein: sand), V106A/Y181C Assembly 1 (ligand: green, protein: deep teal) and V106A/Y181C Assembly 2 (ligand: purple, protein: pink). The C6/7 substituent of each ligand and the closest atom of Pro95 and V100 are illustrated as spheres. Interactions are illustrated as yellow dashed lines.
Compound 2a also benefits from van der Waals interactions between its catechol ring and the Cɣ of an unusual V189 rotamer in the WT structure. However, as the position of V189 in the Y181C:2a structure is rotated approximately 100° out of the pocket and into the rotamer observed in other WT and Y181C structures, Compound 2a must both rotate and twist its catechol ring much more than the other compounds to maintain contact with V189's Cβ, the closest carbon atom (Figure 3c). As a result, its naphthyl group must both rotate and twist approximately 20° to maintain key interactions at the top of the bicyclic ring with P95 and W229 and properly direct the cyano group into the tunnel. These movements appear to be well tolerated by the NNRTI binding pocket as Y188, F227, and W229 rotate slightly to maintain their respective stacking interactions with the ligand (Figure S6D).
Though the position of the bicyclic ring varies significantly in response to the Y181C mutation, the position of the uracil ring remains relatively constant. All four of these compounds have linkers in the anti‐anti‐gauche (aag) conformation in both WT and Y181C structures (Figure 5). This conformation was also observed in the earlier highly potent cyanovinyl compounds (Frey et al., 2014). Of note, the WT:2a structure deviates from this norm, particularly near the catechol ring, due to its interactions with V189 (Figure 5c). As the catechol rings of Compounds 1a and 2b do not have to shift much to maintain potency in the Y181C mutant, their uracil rings and ethoxy linkers directly superimpose in their respective WT and Y181C structures (Figure 5a,d). When the ethoxy linker returns to the conventional aag conformation in the Y181C:2a structure, consistent with V189 returning to its usual position, the uracil ring is in a similar position to that observed in the WT structure and the key hydrogen bond with the backbone of K102 is maintained (Figures 5c and 6c). In both Y181C:2a and Y181C:2b structures, the hydrogen bond between the uracil ring and the side chain of K102, observed in WT:2a and WT:2b structures, is not visible as there was insufficient density to model this residue in the mutant structures (Figure 6c,d). This lack of sidechain density for K102 is commonly observed in both WT and Y181C structures solved in this smaller unit cell and is likely due to changes in crystal contacts rather than a reflection of solution interactions (Figure S7). In contrast, Compound 1b maintains its linker in a strict aag conformation despite changes in its catechol ring position, causing the uracil ring to shift slightly in the Y181C structure as compared to the wildtype (Figure 5b). However, the uracil ring is rotated in the single mutant such that both hydrogen bonds are maintained (Sasaki et al., 2019) (Figure 6b).
FIGURE 5.
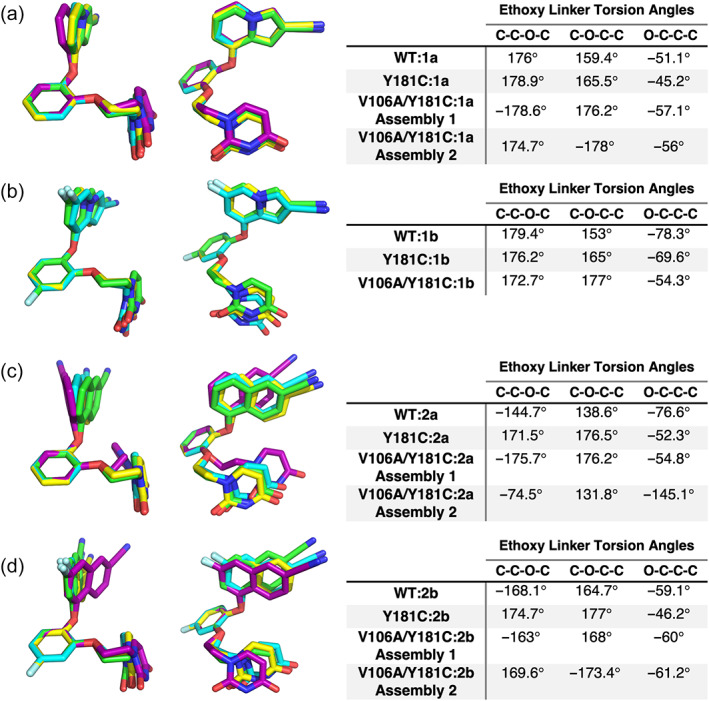
Ethoxy linker torsion angles of indolizine compounds adhere to the WT aag conformation more strictly than those of naphthyl compounds. Superposition of (a) Compound 1a, (b) Compound 1b, (c) Compound 2a, (d) Compound 2b in WT (cyan), Y181C (yellow), V106A/Y181C Assembly 1 (green), and V106A/Y181C Assembly 2 (purple) aligned by their catechol rings are shown from the side and front views. Tables listing each relevant torsion angle are provided to the right of each pair of structures.
FIGURE 6.
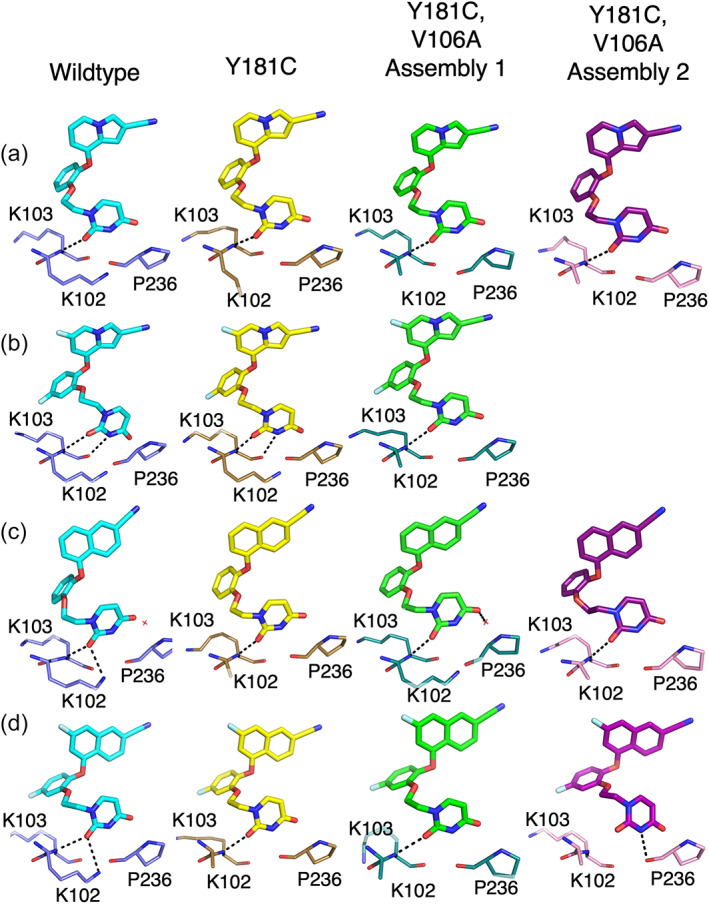
Changes in ethoxy linker conformation change the uracil ring orientation and hydrogen bonding partners in RT mutant structures. Hydrogen bonds made by (a) Compound 1a, (b) Compound 1b, (c) Compound 2a, (d) Compound 2b in WT (ligand: cyan, protein: violet), Y181C (ligand: yellow, protein: sand), V106A/Y181C Assembly 1 (ligand: green, protein: deep teal), and V106A/Y181C Assembly 2 (ligand: purple, protein: pink). Hydrogen bond interactions are illustrated as black dashed lines.
2.2. Differences in ligand binding in the Y181C/V106A double mutant
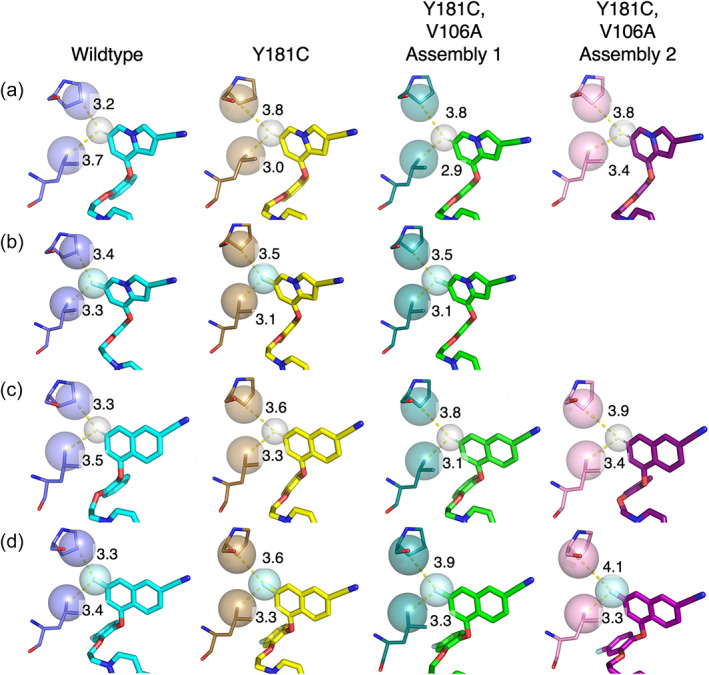
In all four single mutant structures, V106 interacts with several key points on the inhibitor. The Cɣ1 atom makes 3.3–3.7 Å contact with the oxygen of the indolizinyl or naphthyl‐substituted position of the catechol ring (Figure 7a). Additionally, the Cɣ2 atom makes 3.6–4 Å contact with the bottom oxygen of the catechol ring leading to the ethoxy linker and is placed 3.4–3.7 Å from the center of the uracil ring. Mutating this residue to alanine in the double mutant provides extra space both under the catechol ring and in the groove region where the uracil ring binds (Figure 7b,c).
FIGURE 7.
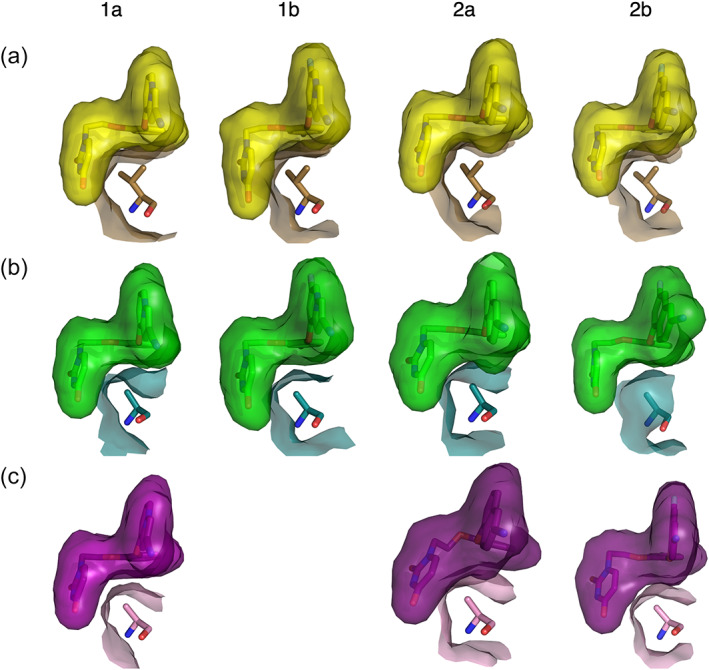
V106 makes several close interactions with the catechol ring, ethoxy linker, and uracil ring. Interaction surface between V/A106 and Compound 1a, Compound 1b, Compound 2a, or Compound 2b in (a) Y181C RT (ligand: yellow, protein: sand), (b) V106A/Y181C Assembly 1 (ligand: green, protein: deep teal), and (c) V106A/Y181C Assembly 2 (ligand: purple, protein: pink).
In addition to these short‐range effects, slight changes in the position of the A106 carbonyl impacts the conformation of the neighboring flexible loop containing K223, which projects into the tunnel to interact with the compounds' cyano group in wildtype and single Y181C mutant structures. Addition of the cyano group to form this interaction, either directly or mediated by a water molecule, is associated with a 1000‐fold increase in potency (Lee et al., 2013, 2014). In V106A/Y181C: 1a, 2a, and 2b structures, K223 is displaced by Q222, which projects closer to the bottom of the tunnel opening and may present a less favorable angle for the interaction (Figure 8; Figure S8). As this loop is surface exposed, this difference in positioning is likely due, at least in part, to changes in crystal contacts between the different space groups, though neither residue makes direct contact with the neighboring molecule (Figure S9). As both Y181C:1b and V106A/Y181C:1b structures were obtained from soaked crystals, it is likely that the modeled position of K223, which points away from the tunnel, is due to crystal contacts determined prior to ligand binding (Figures S8B and S9C). It is likely that Q222 is the residue found at the end of the tunnel in solution as it is commonly found where the region was exposed in a solvent channel, while K223 more commonly reached into the tunnel when adjacent residues are involved in crystal contacts (Figure S9).
FIGURE 8.
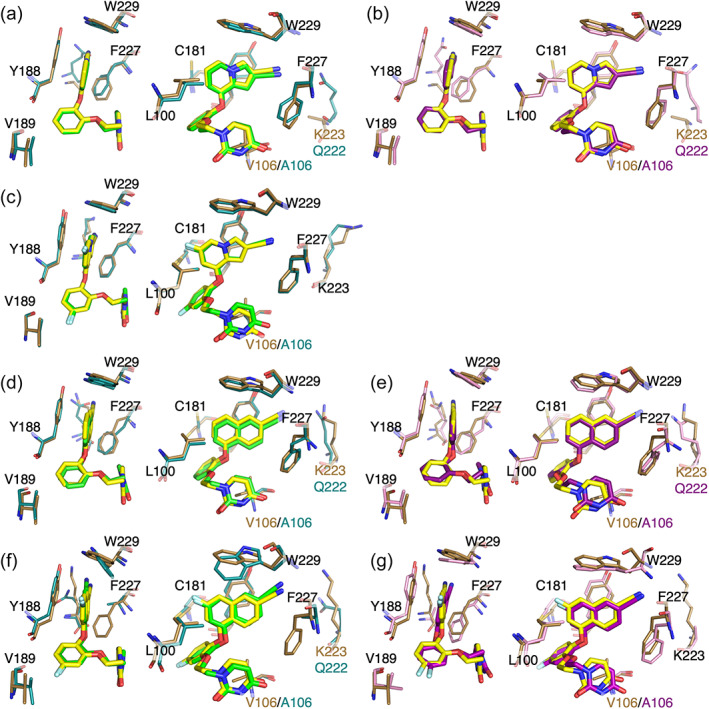
Key aromatic residues in the binding pocket shift to accommodate changes in naphthyl ring position in mutant structures. Superposition of Compound 1a bound to Y181C RT (protein in brown, ligand in yellow) and (a) V106A/Y181C Assembly 1 (protein in deep teal, ligand in green) or (b) V106A/Y181C Assembly 2 (protein in pink, ligand in purple). (c) Superposition of Compound 1b bound to Y181C RT (protein in brown, ligand in yellow) and V106A/Y181C (protein in deep teal, ligand in green). Superposition of Compound 2a bound to Y181C RT (protein in brown, ligand in yellow) and (d) V106A/Y181C Assembly 1 (protein in deep teal, ligand in green), or (e) V106A/Y181C Assembly 2 (protein in pink, ligand in purple). Superposition of Compound 2b bound to Y181C RT (protein in brown, ligand in yellow) and (f) V106A/Y181C Assembly 1 (protein in deep teal, ligand in green) or (g) V106A/Y181C Assembly 2 (protein in pink, ligand in purple). Both side (left) and front (right) views are shown for each superposition.
For Compound 1a, the indolizine ring shifts downward 0.5 Å in the V106A/Y181C structure relative to its position in the Y181C single mutant, allowing the indolizine ring to fill some of the available space provided by the V106A mutation. This downward shift pulls the indolizine ring farther away from Y188 and W229, key binding partners at the top of the binding pocket and places the cyano group lower in the tunnel region (Figure 8a,b). Additionally, while the aag conformation is maintained in the double mutant, the C—O—C—C torsion angle rotates 10°–15° relative to the single Y181C mutant, bringing the bottom catechol oxygen closer to the Cβ of A106 (Figure 5a). However, despite these changes, the uracil ring overlays well in WT, single, and double mutant structures, maintaining key hydrogen bonding interactions (Figure 6a).
The naphthyl of Compound 2a also shifts downward approximately 0.5 Å in the V106A/Y181C structure relative to its position in the Y181C single mutant. However, in this case, W229 rotates into the pocket such that the edge‐to‐face interaction with the top of the naphthyl group is maintained. Of note, F227 also rotates into the pocket to make a new edge‐to‐face interaction with the bottom edge of the naphthyl group (Figure 8d,e). Additionally, the cyano group of Compound 2a projects 1 Å farther into the tunnel than that of Compound 1a, allowing it to reach closer to Q222, increasing the strength of that interaction (Figure S10). While the downward shift is slightly less pronounced in Assembly 2, the naphthyl group rotates 10° into the pocket, toward its position in the wildtype structure, improving the angle for the interaction with Q222. This rotation is well tolerated as W229 rotates even farther into the pocket and Y188 makes a compensatory rotation to maintain the appropriate orientation for its face‐to‐face interaction with the naphthyl group (Figure 8e). Rotation of the naphthyl group also requires a compensatory reorientation of the catechol ring and complete repositioning of the ethoxy linker to correctly orient the uracil ring to maintain hydrogen bonding interactions with the backbone of K103 (Figures 5c and 6c). In contrast, in Assembly 1, the linker is found in the strict aag conformation observed in the WT and single mutant structures. While the sidechain of K102 is too far away from the uracil ring to form the hydrogen bond previously observed in the WT structure, a well‐ordered water, previously nearly 5 Å away from the uracil ring, moves within hydrogen bonding distance of the opposite carbonyl to compensate for the loss (Figure 6c). Though both partners are too disordered to be observed in Assembly 2, one or both may also interact with the ligand.
Compound 1b only shifts downward approximately 0.1 Å in the V106A/Y181C double mutant (Figure 8c). Farther downward movement is prevented by the 3.1 Å interaction between the fluorine in the C6 position of the indolizine ring and the Cδ1 of V100 (Figure 4b). If this fluorine substituent is modeled onto the V106A/Y181C:1a structure, it results in a 0.5 Å overlap with V100. Additionally, P95 shifts slightly into the pocket to maintain the ideal 3.5 Å interaction distance between the C6 fluorine and its Cβ observed in the single mutant. Since there is minimal change in position of the indolizine ring between the single and double mutant structures, the catechol rings almost directly superimpose between the two structures (Figure 8c). Although the aag conformation of the ethoxy linker is maintained in the double mutant, the uracil ring is rotated up 14° away from A106 and its hydrogen bonding partners (Figure 5b). This causes the loss of the hydrogen bond between the uracil nitrogen and the backbone of K103, which is likely responsible for the observed reduction in potency (Figure 6b).
Unlike the other compounds, Compound 2b binds to the V106A/Y181C double mutant in two distinct conformations. The conformation in Assembly 1 is similar to the conformation observed in the Y181C single mutant. V189 shifts about 0.5 Å deeper into the pocket, resulting in a compensatory 0.5 Å shift of the catechol ring into the pocket. This shift requires the naphthyl ring to rotate 7.5° toward the top of the pocket and approximately 10° toward the back of the pocket (Figure 8f). This rotation pulls the C7 fluorine substituent away from the Cβ of P95, increasing the interaction distance from 3.6 to 3.9 Å (Figure 4d). However, W229 rotates into the pocket, bringing its Cη 4.1 Å from the C7 fluorine. While this is a less favorable angle for its usual edge‐to‐face interaction with the upper edge of the naphthyl group, this rotation allows W229 to make a new edge‐to‐face interaction with Y183 (Figure 8f; Figure S11). If compensatory interactions with F227 similar to those observed in the V106A/Y181C:2a structure occur, they are likely much weaker as the naphthyl group is rotated away from the residue, toward the back of the pocket, and the residue is disordered (Figure 8f). As the shift in the catechol ring position is small, the conformation of the ethoxy uracil ring, and the hydrogen bond between the uracil ring carbonyl and the backbone of K103 are maintained in the double mutant structure (Figure 6d). However, the conformation of the compound in Assembly 2 significantly deviates from that observed in the Y181C single mutant. V189 rotates into the binding pocket, causing the catechol ring to rotate approximately 40° relative to the Y181C single mutant. This rotation causes a significant repositioning of both the naphthyl ring and ethoxy linker. As in the V106A/Y181C:2a structures, the naphthyl group shifts 0.5 Å downwards in the pocket. An 8° rotation of the naphthyl group into the binding pocket allows the C7 fluorine to fit between V100 and C181 without clashing (Figure 8g). Although P95 does not shift into the pocket to maintain its optimal interaction with the C7 fluorine on the naphthyl group, Y188, F227, and W229 rotate into the pocket to maximize their interactions with the shifted naphthyl group (Figures 4d and 8g). Though maintenance of the aag conformation of the ethoxy linker results in a repositioning of the uracil ring and weakening of the hydrogen bond to the backbone of K103, the new position optimizes the hydrogen bond between the uracil ring and the backbone carbonyl of P236 (Figure 6d).
3. DISCUSSION
The indolizine and naphthyl catechol diethers represent highly potent classes of NNRTIs with 0.3–2 nM affinities for WT HIV strains. Both classes of inhibitors remain potent against the common drug‐resistant Y181C mutant strain. However, only the naphthyl compounds retain this high level of potency against the V106A/Y181C double mutant identified in the HIV‐1 N119 mutant strain. While naphthyl compounds 2a and 2b have only a 3–4‐fold decrease in activity between WT and the double mutant, indolizine compounds 1a and 1b decrease in potency 500–1000‐fold. The structures presented here suggest a possible mechanism for the naphthyl compounds' retained activity in this mutant.
As the naphthyl group is larger than the indolizine group, it can make more extensive interactions with the aromatic residues lining the tunnel region, allowing the pocket to occupy a wider range of conformations. As the six‐membered naphthyl ring brings its edge within 4 Å of the phenylalanine ring, rotation into the pocket may be favored for optimizing interactions with F227 (Figure 9c,d). This proximity likely allows for the up to 20° rotation observed in naphthyl groups of both Compounds 2a and 2b as they accommodate optimal contacts between the catechol ring and V189 in both single and double mutant structures. In contrast, indolizines 1a and 1b have much less rotational flexibility in this region; any shifts necessary to accommodate catechol ring rotations are largely horizontal and vertical motions (Figure 8). As the edge of the five‐membered indolizine ring is nearly 5 Å from F227, their potency is likely more reliant on interactions with Y188 and W229 (Figure 9a,b). While F227 does rotate in the V106A/Y181C:1a structure, it likely does so to avoid a clash with the indolizine ring's new position rather than to optimize interactions with the ring (Figure 8a,b).
FIGURE 9.

The naphthyl ring can make closer interactions with surrounding aromatic residues in both WT and double mutant structures. Aromatic interaction partners of (a) Compound 1a, (b) Compound 1b, (c) Compound 2a, (d) Compound 2b in WT (ligand: cyan), V106A/Y181C Assembly 1 (ligand: green) and V106A/Y181C Assembly 2 (ligand: brown). Each atom in the aromatic residue is colored based on its distance from the closest atom in the indolizine ring of Compound 1a or 1b or the naphthyl ring of Compound 2a or 2b on a scale of 3.5–5 Å (scale bar shown below figures). For example, note that in panel (a) in the WT:1a structure, both W229 and Y188 are a dark purple, while W229 becomes more yellow in both V106A/Y181C:1a structures, indicating that the compound interacts with Y188 at a similar distance, but is farther away from W229.
Interactions with W229 are likely particularly crucial for indolizine compounds. The indolizine ring is placed slightly higher in the pocket than the naphthyl ring, reducing interaction distance from nearly 4 Å to less than 3.5 Å (Figure 9). While strong interactions with W229, which is highly conserved, are ideal in most cases, the reliance of the indolizine compounds on this interaction proves detrimental in the V106A/Y181C mutant. As W229 remains in a similar position in all observed structures, the downward shift in the Compound 1a structures increases this distance to 3.7 and 3.5 Å in assemblies 1 and 2, respectively. Despite the larger interaction distance in wildtype and single mutant structures, the naphthyl ring appears to make a stronger interaction with W229 than the indolizine ring. Although both Compounds 2a and 2b also shift downward in the V106A/Y181C double mutant, the tryptophan residue rotates down into the pocket to maintain an optimal interaction distance. A similar dependence of potency against mutant strains on W229 interaction strength has also been observed in the related 2‐naphthyl diether series (Duong et al., 2020).
The top of the molecule is not the only place that the naphthyl compounds have additional conformational flexibility. While aag is the preferred ethoxy‐linker conformation for both compounds, the first dihedral angle is more variable in the naphthyl compounds than in the indolizines. Since the ethoxy linker of indolizine compounds fits tightly in a groove between V106 and L100 in wildtype and single mutant structures, its conformation is restricted to fall between 176° and 180°. However, the ethoxy linker is not as tightly constrained in naphthyl compounds, allowing the first dihedral angle to vary between 170° and 215° (Figure 5). This additional flexibility may result from the need to maintain the uracil ring's hydrogen bonding interactions while accommodating the wider variety of catechol ring positions and orientations supported by the naphthyl ring's ability to rotate within the binding pocket.
Reliance on V106 to restrict the conformational flexibility of the ethoxy linker proves detrimental for indolizine compounds when the residue is replaced by a smaller alanine. While all observed conformations fall into the aag category, all have dihedral angles that fall outside the previously observed range, 182°, 175°, and 172° for Compound 1a, Assemblies 1 and 2, and Compound 1b, respectively (Figure 5). While these are minor rearrangements in this flexible linker, they contribute to the displacement of the uracil ring in the V106A/Y181C:1b, which results in the loss of the hydrogen bond between the uracil ring and the backbone of K103 (Figure 6f). As naphthyl compounds can tolerate a wider variety of linker conformations, this effect is less detrimental on their potency.
The V106A/Y181C mutant has also been identified in other passage experiments against nevirapine (Balzarini et al., 1993; Fujiwara et al., 1998), the first FDA‐approved NNRTI, and capravirine (Sato et al., 2006), a second generation NNRTI which failed to progress past Phase II clinical trials. This double mutant is reported to confer 600‐fold resistance against nevirapine (Sato et al., 2006). While 200‐fold decrease in nevirapine potency provided by the Y181C mutation alone confounds this result, the V106A mutation alone can also provide 60–150‐fold resistance (Stanford University, 2022), consistent with the fact that more than half of the compound's structure makes 4 Å or closer contact with V106 (Figure 10c). In contrast, the V106A/Y181C mutation was only found in viruses passaged at low capravirine concentrations and only confers five‐fold resistance to the drug (Sato et al., 2006). V106A, F227L, and V106A, L100I double mutants, however, can provide more than 100‐fold capravirine resistance. Though V106 is within 4 Å of three out of capravirine's four substituents, they all make additional stabilizing interactions with other residues in the binding pocket, including F227 and L100, so the virus requires additional mutations to fully destabilize the structure (Figure 10d).
FIGURE 10.
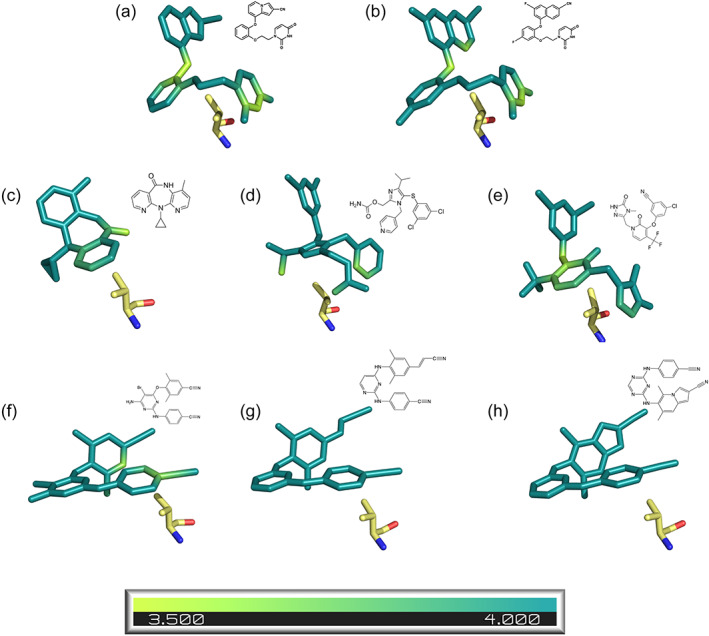
Inhibitors that make closer contact with V106 tend to have reduced potency in the V106A/181C double mutant. Wildtype structures with (a) Compound 1a (PDB: 4MFB), (b) Compound 2b (PDB: 5TW3), (c) Nevirapine (PDB: 1VRT), (d) Capravirine (PDB: 1EP4), (e) Doravirine (PDB: 4NCG), (f) Etravirine (PDB: 3MEC), (g) Rilpivirine (PDB: 2ZD1), and (h) Compound 3 (PDB: 5C24). V106 is shown in yellow for each structure. Ligands are colored by distance of each atom from V106 on a scale between 3.5 and 4 Å. The chemical structure of each compound is shown alongside the crystal structure.
Doravirine, the most recently approved NNRTI, also struggles against V106A, which emerges as a primary mutation and provides a 50‐fold reduction in potency alone (Lai et al., 2014). This effect increases to over 150‐fold resistance in combination with F227L (Martin et al., 2020). This is likely because doravirine's central pyridine ring sits directly on top of V106 and its triazole ring substituent sits against the back of the residue (Figure 10e). Other HIV cell culture experiments comparing the WT and V106A/Y181C variant found doravirine and efavirenz to be 3‐fold and 10‐fold, respectively, less effective with the double mutant.
Of the currently approved NNRTIs, rilpivirine and etravirine, both members of the diarylpyrimidine (DAPY) series, are the only ones to remain unaffected by V106 mutations (Stanford University, 2022). Of note, our most promising compounds, which replace rilpivirine's cyanovinyl group with either an indolizine or naphthyl ring have similar 0.5–10 nM potency against both wildtype and V106A/Y181C double mutant virus (Lee et al., 2015). This effectiveness against V106 mutations makes sense as this class of compounds makes very limited interactions with the residue. Etravirine makes glancing interactions with the sides of its two aryl rings (Figure 10f). In the rilpivirine structure, only one atom at the end of one of the aryl rings is within 4 Å of V106 and Compound 3, our lead indolizine DAPY, does not make any interactions within this threshold (Figure 10g,h).
This suggests a possible mechanism for the detrimental effects of the V106A mutation against a wider variety of NNRTIs. Increased contact of an inhibitor with V106 leads to an increase in susceptibility to resistance in mutant strains of the virus. This is particularly pronounced in compounds that rely on the residue to restrict the conformation of flexible regions to optimal geometries. However, in cases where the inhibitor only makes contact with the residue at the end of a substituent, an additional destabilizing mutation is often required for a significant reduction in potency. Taken together, the current studies provide valuable structural insight for further inhibitor design especially in considering variant strains of HIV that may be associated with drug resistance.
4. MATERIALS AND METHODS
4.1. Viral sequencing of clinical isolates
Viral aliquots underwent RNA extraction (Quick RNA Viral kit, Zymo Research, cat #Q1034), cDNA synthesis (SuperScript IV First Strand Synthesis System, Thermo Fisher, cat#18091050), and nested PCR as previously described (Ho et al., 2013). cDNA was first amplified using outer primers 5FLOut (5′‐GCCCCTAGGAAAAAGGGCTGTTGG‐3′) and 3INOut (5′‐AATCCTCATCCTGTCTACTTGCC‐3′) for 30 cycles of amplification by Platinum Taq HiFi polymerase. Two microliters of the outer PCR product were then amplified using inner PCR primers 5FLIn (5′‐TGCAGGGCCCCTAGGAAAAAGGGCTG‐3′) and 3INIn (5′‐CCACACAATCATCACCTGCC‐3′) for 35 cycles of amplification. Gel bands were visualized by 1% agarose gel electrophoresis, extracted, and sequenced using these primers: 3GagOut (5′‐TCTTTATCTAAGGGAACTGAAAAATATGCATC‐3′), 5FLIn (5′‐TGCAGGGCCCCTAGGAAAAAGGGCTG‐3′), 3FLOut (5′‐CCTTGCCCCTGCTTCTGTATTTCTGC‐3′), 5INIn (5′‐GAAAATTGAATTGGGCAAGTC‐3′), and 3INIn (5′‐CCACACAATCATCACCTGCC‐3′). Consensus sequences from the assembled contigs covering pol region were uploaded onto HIVdb Program: Sequence Analysis interface on HIV Drug Resistance Database.
4.2. Cloning, expression, and purification
Recombinant WT RT52A HIV‐1 RT was expressed and purified as previously described (Carter et al., 2023; Das et al., 2008). To produce double mutant V106A/Y181C RT52A construct for HIV‐1 RT, the V106A mutation was cloned into the p66 subunit of Y181C RT52A using Quikchange mutagenesis (Agilent) as previously described (Frey et al., 2015). This V106A/Y181C RT52A HIV‐1 RT was expressed and purified in the same manner as WT with slight modifications. To maximize formation of the correct p66/p51 heterodimer, transformed BL21(DE3) cells were induced early, at an OD600 of 0.4 with 1 mM of IPTG. Additionally, the heparin column gradient was modified to allow for cleaner separation between the p66/p51 heterodimer from any free p51 subunit. RT was loaded on a 5‐mL Heparin column (Cytiva) in Buffer C (50 mM Tris pH 8.0, 10% Glycerol, 0.5 mM TCEP) and eluted using a slow gradient from 28% to 55% Buffer D (50 mM Tris pH 8.0, 1M NaCl, 10% Glycerol, 2 mM DTT) over 16.2 column volumes. This protein was used for both biochemical and crystallographic experiments.
4.3. Biochemical assays
In vitro inhibition of both WT and V106A/Y181C RT was assayed using a PicoGreen‐based EnzChek Reverse Transcriptase Assay Kit (Thermo Fisher Scientific, Inc., E22064) as previously described (Carter et al., 2023). RT52A (20 nM active site concentration in the final reaction) was incubated at room temperature for 15 min with either test compounds at various concentrations or a DMSO control in a 96‐well plate (Greiner 655096). The reaction was initiated with annealed r(A)350 template and d(T)16 primer at a final concentration of 4 μg/mL in polymerization buffer (60 mM Tris pH 8.1, 60 mM KCl, 8 mM MgCl2, 13 mM DTT, 100 μM dTTP). After 30 min, the reaction was quenched with EDTA, and the PicoGreen reagent was added. PicoGreen fluorescence was measured with a plate reader (Molecular Devices Spectamax M5) with excitation and emission at 485 and 520 nm respectively. Each measurement was performed in triplicate and values were normalized to DMSO controls. The resulting curves were fit to the standard four‐variable dose–response curve in Prism (version 9.4.0) to determine the IC50. Since 20 nM enzyme is required for adequate signal to noise, we cannot accurately determine IC50 values below 10–20 nM.
4.4. Crystallization
Details of crystallographic methods can be found in the Supporting Information.
AUTHOR CONTRIBUTIONS
Klarissa Hollander: Investigation; formal analysis; visualization; writing—original draft; writing—review and editing. Albert H. Chan: Investigation; formal analysis. Kathleen M. Frey: Investigation; formal analysis. Olivia Hunker: Investigation. Joseph A. Ippolito: Investigation. Krasimir A. Spasov: Investigation. Yang‐Hui J. Yeh: Investigation; formal analysis. William L. Jorgensen: Funding acquisition; writing—review and editing. Ya‐Chi Ho: Funding acquisition; project administration; writing—review and editing. Karen S. Anderson: Conceptualization; funding acquisition; project administration; supervision; writing—review and editing.
Supporting information
Figure S1. Asymmetric units of V106A/Y181C structures form from different arrangements of p66/p51 heterodimers.
Figure S2. There is good quality density for the ligand in all solved structures.
Figure S3. Overall structure of V106A/Y181C RT is similar to that of WT RT.
Figure S4. Additional hydrogen bonding interactions between E169 and K49 in V106A/Y181C double mutant.
Figure S5. Ligand placement is similar in both RT heterodimers in each asymmetric unit.
Figure S6. Slight shifts within the binding pocket accommodate for the Y181C single mutation.
Figure S7. Crystal contacts influence the positioning of the side chain of K102.
Figure S8. Q222 commonly replaces K223 at the end of the tunnel in V106A/Y181C double mutant structures.
Figure S9. Crystal contacts influence whether Q222 or K223 point into the tunnel region of the binding pocket.
Figure S10. The cyano group of naphthyl compounds extends farther into the tunnel than those of indolizine compounds.
Figure S11. Close‐up view of edge‐to‐face interaction between Y183 and W229.
Table S1. Crystallographic statistics table.
Data S1. PDB validation reports for seven newly deposited structures.
Data S2. Results from deposition of N119 sequences to the Stanford HIV Database.
Tables. Supplementary methods.
ACKNOWLEDGMENTS
We express gratitude to the National Institutes of Health for funding this work (R01GM049551 and R01AI155072 to KSA; R01AI044616 to WLJ; T32 GM148333 to KH). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357, the National Synchrotron Light Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE‐AC02‐98CH10886. We thank the Yale Macromolecular X‐ray Crystallography Core Laboratory for their assistance with data set collection (NIH grant 1S10OD018007‐01).
Hollander K, Chan AH, Frey KM, Hunker O, Ippolito JA, Spasov KA, et al. Exploring novel HIV‐1 reverse transcriptase inhibitors with drug‐resistant mutants: A double mutant surprise. Protein Science. 2023;32(12):e4814. 10.1002/pro.4814
Review Editor: John Kuriyan
REFERENCES
- Alcaro S, Alteri C, Artese A, Ceccherini‐Silberstein F, Costa G, Ortuso F, et al. Docking analysis and resistance evaluation of clinically relevant mutations associated with the HIV‐1 non‐nucleoside reverse transcriptase inhibitors nevirapine, efavirenz and etravirine. ChemMedChem. 2011;6(12):2203–2213. [DOI] [PubMed] [Google Scholar]
- Balzarini J, Karlsson A, Pérez‐Pérez MJ, Camarasa MJ, Tarpley WG, De Clercq E. Treatment of human immunodeficiency virus type 1 (HIV‐1)‐infected cells with combinations of HIV‐1‐specific inhibitors results in a different resistance pattern than does treatment with single‐drug therapy. J Virol. 1993;67(9):5353–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollini M, Domaoal RA, Thakur VV, Gallardo‐Macias R, Spasov KA, Anderson KS, et al. Computationally‐guided optimization of a docking hit to yield catechol diethers as potent anti‐HIV agents. J Med Chem. 2011;54(24):8582–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ZJ, Hollander K, Spasov KA, Anderson KS, Jorgensen WL. Design, synthesis, and biological testing of biphenylmethyloxazole inhibitors targeting HIV‐1 reverse transcriptase. Bioorg Med Chem Lett. 2023;84:129216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, Shatkin AJ, et al. High‐resolution structures of HIV‐1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci. 2008;105(5):1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A. The impact of resistance on viral fitness and its clinical implications. In: Geretti A, editor. Antiretroviral resistance in clinical practice. London: Mediscript; 2006. [PubMed] [Google Scholar]
- Duong VN, Ippolito JA, Chan AH, Lee WG, Spasov KA, Jorgensen WL, et al. Structural investigation of 2‐naphthyl phenyl ether inhibitors bound to WT and Y181C reverse transcriptase highlights key features of the. Protein Sci. 2020;29(9):1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KM, Gray WT, Spasov KA, Bollini M, Gallardo‐Macias R, Jorgensen WL, et al. Structure‐based evaluation of C5 derivatives in the catechol diether series targeting HIV‐1 reverse transcriptase. Chem Biol Drug Des. 2014;83(5):541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KM, Puleo DE, Spasov KA, Bollini M, Jorgensen WL, Anderson KS. Structure‐based evaluation of non‐nucleoside inhibitors with improved potency and solubility that target HIV reverse transcriptase variants. J Med Chem. 2015;58(6):2737–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Sato A, El‐Farrash M, Miki S, Abe K, Isaka Y, et al. S‐1153 inhibits replication of known drug‐resistant strains of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 1998;42(6):1340–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health Canada . Summary basis of decision (SBD) EDURANT Rilpivirine hydrochloride, 25 mg tablet. Janssen Inc.: Health Canada. 2011.
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication‐competent noninduced proviruses in the latent reservoir increase barrier to HIV‐1 cure. Cell. 2013;155(3):540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holec AD, Mandal S, Prathipati PK, Destache CJ. Nucleotide reverse transcriptase inhibitors: a thorough review, present status and future perspective as HIV therapeutics. Curr HIV Res. 2018;15(6):411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain G, Ngo‐Giang‐Huong N, Le Coeur S, Bowonwatanuwong C, Kantipong P, Leechanachai P, et al. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine‐based antiretroviral therapy. N Engl J Med. 2004;351(3):229–240. [DOI] [PubMed] [Google Scholar]
- Lai M‐T, Feng M, Falgueyret J‐P, Tawa P, Witmer M, DiStefano D, et al. In vitro characterization of MK‐1439, a novel HIV‐1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2014;58(3):1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W‐G, Frey KM, Gallardo‐Macias R, Spasov KA, Bollini M, Anderson KS, et al. Picomolar inhibitors of HIV‐1 reverse transcriptase: design and crystallography of naphthyl phenyl ethers. ACS Med Chem Lett. 2014;5(11):1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W‐G, Frey KM, Gallardo‐Macias R, Spasov KA, Chan AH, Anderson KS, et al. Discovery and crystallography of bicyclic arylaminoazines as potent inhibitors of HIV‐1 reverse transcriptase. Bioorg Med Chem Lett. 2015;25(21):4824–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W‐G, Gallardo‐Macias R, Frey KM, Spasov KA, Bollini M, Anderson KS, et al. Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. J Am Chem Soc. 2013;135(44):16705–16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EA, Lai M‐T, Ngo W, Feng M, Graham D, Hazuda DJ, et al. Review of doravirine resistance patterns identified in participants during clinical development. J Acquir Immune Defic Syndr. 2020;85(5):635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbunkah HA, Bertagnolio S, Hamers RL, Hunt G, Inzaule S, Rinke de Wit TF, et al. Low‐abundance drug‐resistant HIV‐1 variants in antiretroviral drug‐naive individuals: a systematic review of detection methods, prevalence, and clinical impact. J Infect Dis. 2020;221(10):1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J‐M, Cahn P, Grinsztejn B, Lazzarin A, Mills A, Saag M, et al. Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment‐naive adults infected with HIV‐1 (ECHO): a phase 3 randomised double‐blind active‐controlled trial. Lancet. 2011;378(9787):238–246. [DOI] [PubMed] [Google Scholar]
- Nanfack AJ, Redd AD, Bimela JS, Ncham G, Achem E, Banin AN, et al. Multimethod longitudinal HIV drug resistance analysis in antiretroviral‐therapy‐naive patients. J Clin Microbiol. 2017;55(9):2785–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunberg JH, Schleif WA, Boots EJ, O'Brien JA, Quintero JC, Hoffman JM, et al. Viral resistance to human immunodeficiency virus type 1‐specific pyridinone reverse transcriptase inhibitors. J Virol. 1991;65(9):4887–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanuphak N, Gulick RM. HIV treatment and prevention 2019: current standards of care. Curr Opin HIV AIDS. 2020;15(1):4–12. [DOI] [PubMed] [Google Scholar]
- Preston BD, Poiesz BJ, Loeb LA. Fidelity of HIV‐1 reverse transcriptase. Science. 1988;242(2882):1168–1171. [DOI] [PubMed] [Google Scholar]
- Richman D, Shih CK, Lowy I, Rose J, Prodanovich P, Goff S, et al. Human immunodeficiency virus type 1 mutants resistant to nonnucleoside inhibitors of reverse transcriptase arise in tissue culture. Proc Natl Acad Sci. 1991;88(24):11241–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Marchand B, Das K, Himmel D, Parniak MA, Hughes SH, et al. Structure and function of HIV‐1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385(3):693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Gannam ZTK, Kudalkar SN, Frey KM, Lee W‐G, Spasov KA, et al. Molecular and cellular studies evaluating a potent 2‐cyanoindolizine catechol diether NNRTI targeting wildtype and Y181C mutant HIV‐1 reverse transcriptase. Bioorg Med Chem Lett. 2019;29(16):2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Hammond J, Alexander TN, Graham JP, Binford S, Sugita K‐I, et al. In vitro selection of mutations in human immunodeficiency virus type 1 reverse transcriptase that confer resistance to capravirine, a novel nonnucleoside reverse transcriptase inhibitor. Antiviral Res. 2006;70(2):66–74. [DOI] [PubMed] [Google Scholar]
- Spence R, Kati W, Anderson K, Johnson K. Mechanism of inhibition of HIV‐1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267(5200):988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford University . Major Non‐Nucleoside RT Inhibitor (NNRTI) Resistance Mutations. December 7, 2022. Stanford University.
- Zuo L, Liu K, Liu H, Hu Y, Zhang Z, Qin J, et al. Trend of HIV‐1 drug resistance in China: a systematic review and meta‐analysis of data accumulated over 17 years (2001–2017). eClinicalMedicine. 2020;18:100238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Asymmetric units of V106A/Y181C structures form from different arrangements of p66/p51 heterodimers.
Figure S2. There is good quality density for the ligand in all solved structures.
Figure S3. Overall structure of V106A/Y181C RT is similar to that of WT RT.
Figure S4. Additional hydrogen bonding interactions between E169 and K49 in V106A/Y181C double mutant.
Figure S5. Ligand placement is similar in both RT heterodimers in each asymmetric unit.
Figure S6. Slight shifts within the binding pocket accommodate for the Y181C single mutation.
Figure S7. Crystal contacts influence the positioning of the side chain of K102.
Figure S8. Q222 commonly replaces K223 at the end of the tunnel in V106A/Y181C double mutant structures.
Figure S9. Crystal contacts influence whether Q222 or K223 point into the tunnel region of the binding pocket.
Figure S10. The cyano group of naphthyl compounds extends farther into the tunnel than those of indolizine compounds.
Figure S11. Close‐up view of edge‐to‐face interaction between Y183 and W229.
Table S1. Crystallographic statistics table.
Data S1. PDB validation reports for seven newly deposited structures.
Data S2. Results from deposition of N119 sequences to the Stanford HIV Database.
Tables. Supplementary methods.