

Abstract
Background
A randomized, phase II, placebo-controlled, and blinded clinical trial (NCT01062425) was conducted to determine the efficacy of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, versus placebo in combination with radiation and temozolomide in newly diagnosed glioblastoma.
Methods
Patients with newly diagnosed glioblastoma were randomly assigned 2:1 to receive (1) cediranib (20 mg) in combination with radiation and temozolomide; (2) placebo in combination with radiation and temozolomide. The primary endpoint was 6-month progression-free survival (PFS) based on blinded, independent radiographic assessment of postcontrast T1-weighted and noncontrast T2-weighted MRI brain scans and was tested using a 1-sided Z test for 2 proportions. Adverse events (AEs) were evaluated per CTCAE version 4.
Results
One hundred and fifty-eight patients were randomized, out of which 9 were ineligible and 12 were not evaluable for the primary endpoint, leaving 137 eligible and evaluable. 6-month PFS was 46.6% in the cediranib arm versus 24.5% in the placebo arm (P = .005). There was no significant difference in overall survival between the 2 arms. There was more grade ≥ 3 AEs in the cediranib arm than in the placebo arm (P = .02).
Conclusions
This study met its primary endpoint of prolongation of 6-month PFS with cediranib in combination with radiation and temozolomide versus placebo in combination with radiation and temozolomide. There was no difference in overall survival between the 2 arms.
Keywords: angiogenesis, cediranib, glioblastoma, vascular endothelial growth factor
Key Points.
Cediranib is safe and feasible in newly diagnosed glioblastoma subjects.
Cediranib improved progression-free survival (PFS) but not overall survival.
Disease progression is challenging to define with VEGF-targeting therapies.
Importance of the Study.
This randomized, placebo-controlled, and double-blind study confirmed the safety and benefit of an oral, pan-VEGF receptor tyrosine inhibitor, and cediranib in prolonging progression-free survival, the primary study endpoint when added to radiation and temozolomide in newly diagnosed glioblastoma. However, there was no overall survival benefit of cediranib and defining disease progression is confounded by the anti-permeability effects of a VEGF-targeting drug. These results with cediranib are consistent with those observed with bevacizumab in newly diagnosed glioblastoma.
Glioblastoma, IDH-wildtype (glioblastoma), the most common primary malignant brain tumor in adults, causes significant neurological morbidity and is associated with poor survival.1,2 Microvascular proliferation, a histopathological hallmark of glioblastoma, is a consequence of the high expression levels of proangiogenic cytokines, particularly of vascular endothelial growth factor (VEGF) and signaling via its endothelial tyrosine kinase receptor VEGFR2.3–6 Levels of VEGF and its receptor correlate with the histological grade of gliomas, with the highest levels present in glioblastoma.7,8Thus, novel anti-VEGF agents, such as monoclonal antibodies and tyrosine kinase inhibitors are attractive therapeutic strategies in glioblastoma.9 The US Food and Drug Administration approved bevacizumab, an anti-VEGF monoclonal antibody, as a monotherapy for recurrent glioblastoma in 2017 based on the radiographic response rates observed in 2 phase II trials.10–12 However, 2 subsequent randomized trials of bevacizumab versus placebo in combination with radiation and temozolomide demonstrated no survival benefit in newly diagnosed glioblastoma.13,14
Cediranib, an orally available pan-VEGFR tyrosine kinase inhibitor has a sub-nanomolar IC50 for VEGF receptors with additional activity against c-Kit and lower potency against PDGFRß.15 Based on a half-life of 22 h it can be administered once daily. In a phase II study of cediranib (45 mg/day) for patients with recurrent glioblastoma, 8/30 (27%) subjects achieved a partial radiographic response.16,17 Subsequently, this phase II, randomized, blinded, and placebo-controlled study (ClinicalTrials.gov identifier NCT01062425) was conducted to investigate the efficacy of cediranib, in combination with temozolomide and fractionated radiation in newly diagnosed glioblastoma.
Patients and Methods
Patients
Patients with newly diagnosed glioblastoma were the target population for this clinical trial. Inclusion criteria included age ≥ 18 years, pathological diagnosis of glioblastoma, and Karnofsky Performance Status (KPS) ≥ 70. Exclusion criteria included any prior anti-VEGF therapy, prior treatment with temozolomide, or prior treatment with radiation to the head or neck (except for T1 glottic cancer). All patients were required to sign an informed consent form approved by the Institutional Review Board of the enrolling institution.
The study was performed in accordance with the Declaration of Helsinki and the International Conference on Harmonization/Good Clinical Practice.
Study Design
The study was a phase II, comparative, randomized, and multicenter trial. Patients were stratified by Recursive Partitioning Analysis (RPA), class III versus IV versus V and by the promoter methylation status of 06-methylguanine-DNA-methyltransferase (MGMT), methylated versus unmethylated versus invalid, prior to being randomized to receive cediranib (20 mg) versus placebo in combination with temozolomide (75 mg/m2/daily) and radiation (60 Gy in 2 Gy fractions) daily for 42 days followed by cediranib (20 mg) versus placebo in combination with temozolomide (150–200 mg/m2 for 5 consecutive days in 28-day cycles) for up 12 cycles maximum. Patient randomization was performed at the time of registration, with a 2:1 allocation ratio in favor of the cediranib arm, based on the permuted block design using the method described by Zelen.18 The primary endpoint of this study is 6-month progression-free survival (PFS) using MacDonald criteria.19 Secondary endpoints of this study included overall survival (OS), PFS, treatment-related toxicity, and the association between MGMT methylation status and clinical outcomes.
The sample size calculation would address whether the addition of cediranib to concurrent chemoradiation and standard temozolomide would improve the 6-month PFS rate in patients with glioblastoma. The null hypothesis was that the 6-month PFS rates for both arms were 50%, and the alternative hypothesis was that patients receiving the experimental regimen would have a 6-month PFS rate of 66%. With 150 eligible patients, there would be an 80% statistical power to detect the 16% absolute increase in 6-month PFS at a significance level of 0.15, using a 1-sided Z test for 2 proportions.20 Guarding against up to a 47% rate for patients who were retrospectively found ineligible or did not get randomized due to early disease progression, patient refusal, insufficient tissues, or other reasons, 283 patients were required to be enrolled in order to have 150 eligible and randomized patients.
Imaging reviews of disease progression by 6 months were first performed locally. The central radiology review of progression was performed by a team of reviewers including 2 readers and an adjudicator, per MacDonald criteria. The reviewers were blinded to treatment assignment and clinical information (except for neurologic function and KPS). If there was a disagreement between the 2 readers, the adjudicator provided the final determination of the progression status. Out of 34 patients judged by local sites to have disease progression by 6 months, 30 cases (88%) were confirmed by central reviews. However, among 83 patients who were judged to be progression-free by 6 months by local assessments, only 49 cases (59%) were confirmed by central reviews; the central reviews identified 34 progressors who were judged to be progression-free by 6 months per local reads.
OS was measured from the date of randomization to the date of death, or otherwise, the last follow-up date on which the patient was reported alive. PFS was measured from the date of randomization to the date of first progression, or death, or otherwise, the last follow-up date on which the patient was reported alive without progression. OS and PFS rates were estimated using the Kaplan–Meier method,21 and differences between the 2 treatment arms were tested using the log-rank test.22 Multivariate analyses for OS and PFS were performed using the Cox proportional hazards model23 with the stratification factors included as covariates, to assess the adjusted treatment effects. Toxicities were measured using CTCAE version 4. Differences in the observed severe toxicities (grade 3+) between the 2 arms were tested using the Chi-square test. The log-rank test was used to assess the effect of MGMT methylation status on OS and PFS, both overall (combining 2 treatment arms) and within each treatment arm. The Cox proportional hazards model was used to adjust for stratification factors (RPA class and MGMT methylation status). The proportional hazards assumption was verified using testing and graphical methods. For all the secondary endpoints, a 2-sided test with a significance level of 0.05 was used to declare statistical significance.
Results
Summary of Patient Enrollment
This study opened to accrual on February 26, 2010. Accrual was completed on May 9, 2012, with a total of 261 patients enrolled. One hundred and fifty-eight patients (60.5%) were randomized, and 9 (5.7%) of them were subsequently found ineligible. Reasons for not being randomized and ineligibility are noted in Figure 1. Therefore, for the statistical analyses, there were 52 and 97 eligible and randomized patients in the placebo and cediranib arm, respectively. Table 1 shows the distributions of pretreatment characteristics by treatment arm for all the eligible and randomized patients. The distributions of the stratification factors, MGMT methylation status, and RPA class appear balanced between the 2 treatment arms.
Figure 1.

CONSORT diagram.
Table 1.
Patient and tumor characteristics for all eligible patients in RTOG 0837
| Placebo | Cediranib | Total | ||||
|---|---|---|---|---|---|---|
| Patient or Tumor Characteristic | n | % | n | % | n | % |
| Age (years) | ||||||
| Median | 59 | 61 | 60 | |||
| Min–max | 37–82 | 27–83 | 27–83 | |||
| Q1–Q3 | 51–67 | 54–65 | 53–66 | |||
| ≤ 49 | 11 | 21.2 | 16 | 16.5 | 27 | 18.1 |
| 50–59 | 17 | 32.7 | 27 | 27.8 | 44 | 29.5 |
| 60–69 | 15 | 28.8 | 38 | 39.2 | 53 | 35.6 |
| ≥ 70 | 9 | 17.3 | 16 | 16.5 | 25 | 16.8 |
| Gender | ||||||
| Male | 28 | 53.8 | 53 | 54.6 | 81 | 54.4 |
| Female | 24 | 46.2 | 44 | 45.4 | 68 | 45.6 |
| Race | ||||||
| Black or African American | 3 | 5.8 | 4 | 4.1 | 7 | 4.7 |
| White | 48 | 92.3 | 93 | 95.9 | 141 | 94.6 |
| Unknown or not reported | 1 | 1.9 | 0 | 0.0 | 1 | 0.7 |
| Ethnicity | ||||||
| Hispanic or Latino | 1 | 1.9 | 9 | 9.3 | 10 | 6.7 |
| Not Hispanic or Latino | 48 | 92.3 | 87 | 89.7 | 135 | 90.6 |
| Unknown (individuals not reporting ethnicity) | 3 | 5.8 | 1 | 1.0 | 4 | 2.7 |
| KPS | ||||||
| 70–80 | 25 | 48.1 | 42 | 43.3 | 67 | 45.0 |
| 90–100 | 27 | 51.9 | 55 | 56.7 | 82 | 55.0 |
| Surgery | ||||||
| Subtotal | 26 | 50.0 | 33 | 34.0 | 59 | 39.6 |
| Total (gross) | 24 | 46.2 | 64 | 66.0 | 88 | 59.1 |
| Other | 2 | 3.8 | 0 | 0.0 | 2 | 1.3 |
| Neurologic Function | ||||||
| No symptoms | 11 | 21.2 | 29 | 29.9 | 40 | 26.8 |
| Minor symptoms | 26 | 50.0 | 49 | 50.5 | 75 | 50.3 |
| Moderate symptoms | 15 | 28.8 | 19 | 19.6 | 34 | 22.8 |
| MGMT status | ||||||
| Methylated | 18 | 34.6 | 36 | 37.1 | 54 | 36.2 |
| Unmethylated | 31 | 59.6 | 57 | 58.8 | 88 | 59.1 |
| Invalid | 3 | 5.8 | 4 | 4.1 | 7 | 4.7 |
| RPA class at randomization | ||||||
| III | 8 | 15.4 | 11 | 11.3 | 19 | 12.8 |
| IV | 34 | 65.4 | 68 | 70.1 | 102 | 68.5 |
| V | 10 | 19.2 | 18 | 18.6 | 28 | 18.8 |
Treatment Adverse Events
Information on the adverse events (AEs) is presented by the treatment arm for all eligible and randomized patients who received protocol treatment. Table 2 lists the summary, overall and by system organ class, of the highest-grade AEs regardless of relationship to protocol treatment. Overall, there were 5 patients (9.6%) in the placebo arm, and 11 (12.0%) in the cediranib arm with reported grade 5 AEs. Of these 11 grade 5 AEs, 4/11 (3 infectious and 1 neurological) grade 5 events were deemed possibly, probably, or definitely related to cediranib. The proportions of patients with grade ≥ 3 AEs were summarized and compared between the treatment arms. Out of all eligible and randomized patients who received protocol treatment, 35 (67.3%) from the placebo arm and 77 (83.7%) from the cediranib arm had reported grade ≥ 3 AEs regardless of relationship to protocol treatment, resulting in a P-value of .02.
Table 2.
Distribution of RTOG 0837 patients by highest grade adverse event by system organ class for all reported adverse events without regard to attribution
| Placebo (n = 52) | Cediranib (n = 92) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| System Organ Class | n and (%) of Patients by Grade | n and (%) of Patients by Grade | ||||||||
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 | |
| Overall highest grade | 2 | 12 | 19 | 11 | 5 | 1 | 13 | 44 | 22 | 11 |
| (3.8) | (23.1) | (36.5) | (21.2) | (9.6) | (1.1) | (14.1) | (47.8) | (23.9) | (12.0) | |
| Blood and lymphatic system disorders | 16 | 2 | 5 | 2 | 0 | 19 | 7 | 7 | 3 | 0 |
| (30.8) | (3.8) | (9.6) | (3.8) | (0.0) | (20.7) | (7.6) | (7.6) | (3.3) | (0.0) | |
| Cardiac disorders | 2 | 2 | 0 | 0 | 0 | 7 | 1 | 0 | 1 | 0 |
| (3.8) | (3.8) | (0.0) | (0.0) | (0.0) | (7.6) | (1.1) | (0.0) | (1.1) | (0.0) | |
| Ear and labyrinth disorders | 8 | 2 | 0 | 0 | 0 | 7 | 4 | 0 | 0 | 0 |
| (15.4) | (3.8) | (0.0) | (0.0) | (0.0) | (7.6) | (4.3) | (0.0) | (0.0) | (0.0) | |
| Endocrine disorders | 2 | 1 | 0 | 0 | 0 | 7 | 3 | 1 | 0 | 0 |
| (3.8) | (1.9) | (0.0) | (0.0) | (0.0) | (7.6) | (3.3) | (1.1) | (0.0) | (0.0) | |
| Eye disorders | 6 | 1 | 0 | 0 | 0 | 21 | 1 | 0 | 0 | 0 |
| (11.5) | (1.9) | (0.0) | (0.0) | (0.0) | (22.8) | (1.1) | (0.0) | (0.0) | (0.0) | |
| Gastrointestinal disorders | 17 | 15 | 2 | 0 | 1 | 18 | 49 | 16 | 0 | 0 |
| (32.7) | (28.8) | (3.8) | (0.0) | (1.9) | (19.6) | (53.3) | (17.4) | (0.0) | (0.0) | |
| General disorders and administration site conditions | 13 | 25 | 7 | 0 | 2 | 16 | 47 | 16 | 0 | 2 |
| (25.0) | (48.1) | (13.5) | (0.0) | (3.8) | (17.4) | (51.1) | (17.4) | (0.0) | (2.2) | |
| Hepatobiliary disorders | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| (0.0) | (0.0) | (0.0) | (0.0) | (0.0) | (1.1) | (0.0) | (1.1) | (0.0) | (0.0) | |
| Immune system disorders | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| (0.0) | (1.9) | (0.0) | (0.0) | (0.0) | (1.1) | (1.1) | (0.0) | (0.0) | (0.0) | |
| Infections and infestations | 2 | 10 | 6 | 2 | 0 | 3 | 16 | 9 | 0 | 4 |
| (3.8) | (19.2) | (11.5) | (3.8) | (0.0) | (3.3) | (17.4) | (9.8) | (0.0) | (4.3) | |
| Injury, poisoning, and procedural complications | 13 | 2 | 1 | 0 | 0 | 18 | 8 | 1 | 0 | 0 |
| (25.0) | (3.8) | (1.9) | (0.0) | (0.0) | (19.6) | (8.7) | (1.1) | (0.0) | (0.0) | |
| Investigations | 8 | 10 | 10 | 8 | 0 | 24 | 24 | 16 | 21 | 0 |
| (15.4) | (19.2) | (19.2) | (15.4) | (0.0) | (26.1) | (26.1) | (17.4) | (22.8) | (0.0) | |
| Metabolism and nutrition disorders | 15 | 10 | 11 | 2 | 0 | 22 | 28 | 18 | 6 | 0 |
| (28.8) | (19.2) | (21.2) | (3.8) | (0.0) | (23.9) | (30.4) | (19.6) | (6.5) | (0.0) | |
| Musculoskeletal and connective tissue disorders | 7 | 3 | 9 | 0 | 0 | 15 | 21 | 11 | 0 | 0 |
| (13.5) | (5.8) | (17.3) | (0.0) | (0.0) | (16.3) | (22.8) | (12.0) | (0.0) | (0.0) | |
| Neoplasms benign, malignant, and unspecified (incl cysts and polyps) | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 2 |
| (0.0) | (0.0) | (0.0) | (0.0) | (1.9) | (1.1) | (0.0) | (0.0) | (0.0) | (2.2) | |
| Nervous system disorders | 14 | 13 | 14 | 1 | 0 | 20 | 29 | 18 | 1 | 3 |
| (26.9) | (25.0) | (26.9) | (1.9) | (0.0) | (21.7) | (31.5) | (19.6) | (1.1) | (3.3) | |
| Psychiatric disorders | 9 | 12 | 2 | 1 | 0 | 17 | 18 | 2 | 0 | 0 |
| (17.3) | (23.1) | (3.8) | (1.9) | (0.0) | (18.5) | (19.6) | (2.2) | (0.0) | (0.0) | |
| Renal and urinary disorders | 5 | 5 | 1 | 0 | 0 | 15 | 6 | 1 | 0 | 0 |
| (9.6) | (9.6) | (1.9) | (0.0) | (0.0) | (16.3) | (6.5) | (1.1) | (0.0) | (0.0) | |
| Reproductive system and breast disorders | 2 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 |
| (3.8) | (0.0) | (0.0) | (0.0) | (0.0) | (2.2) | (1.1) | (0.0) | (0.0) | (0.0) | |
| Respiratory, thoracic, and mediastinal disorders | 13 | 1 | 0 | 0 | 1 | 24 | 7 | 5 | 2 | 0 |
| (25.0) | (1.9) | (0.0) | (0.0) | (1.9) | (26.1) | (7.6) | (5.4) | (2.2) | (0.0) | |
| Skin and subcutaneous tissue disorders | 19 | 15 | 1 | 0 | 0 | 33 | 27 | 5 | 0 | 0 |
| (36.5) | (28.8) | (1.9) | (0.0) | (0.0) | (35.9) | (29.3) | (5.4) | (0.0) | (0.0) | |
| Social circumstances | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| (0.0) | (0.0) | (0.0) | (0.0) | (0.0) | (1.1) | (0.0) | (0.0) | (0.0) | (0.0) | |
| Surgical and medical procedures | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| (0.0) | (0.0) | (0.0) | (0.0) | (0.0) | (0.0) | (1.1) | (1.1) | (0.0) | (0.0) | |
| Vascular disorders | 3 | 8 | 6 | 1 | 0 | 5 | 23 | 17 | 2 | 0 |
| (5.8) | (15.4) | (11.5) | (1.9) | (0.0) | (5.4) | (25.0) | (18.5) | (2.2) | (0.0) | |
Test for the Primary Endpoint
The median follow-up time for all eligible patients who were still alive at the time of the analyses was 36.7 months, with a range of 2.2–53.9 months. Out of the 149 eligible and randomized patients for both arms, 137 (91.9%) were evaluable for 6-month PFS. Three patients from the placebo arm and 9 patients from the cediranib arm were not evaluable for the primary endpoint due to withdrawal before 6 months, scan not evaluable, or no protocol treatment given. Based on central radiology reviews on the primary endpoint, the 6-month PFS rate for the cediranib arm was 46.6%, as compared to 24.5% for the placebo arm, resulting in a P-value of .005 by 1-sided Z test for 2 proportions. This suggested that the experimental regimen significantly improved 6-month PFS for this patient population, as compared to the treatment of the placebo arm.
Results for the Secondary Endpoints
The median survival time (MST) was 14.5 months (95% CI: 12.3–19.7 months) for the cediranib arm, and 13.8 months (95% CI: 9.6–18.9 months) for the placebo arm, with a hazard ratio (HR) of 0.87 (95% CI: 0.60–1.24; P-value = .44). The median PFS time was 6.2 months (95% CI: 4.5–8.1 months) for the cediranib arm, and 2.7 months (95% CI: 2.5–3.7 months) for the placebo arm. The corresponding HR for PFS was 0.67 (95% CI: 0.47–0.95) with a P-value of .03. The Kaplan–Meier curves on OS and PFS by treatment arm are demonstrated in Figures 2 and 3, respectively.
Figure 2.

Overall survival by treatment arm.
Figure 3.

Progression-free survival by treatment arm.
Tables 3 and 4 show the results of Cox proportional hazards models for OS and PFS. After adjusting for the stratification factors, the HR of the cediranib effect on OS was 0.91 (95% CI: 0.62–1.34) with a P-value of 0.65 and the HR of the cediranib effect on PFS was 0.67 (95% CI: 0.46–0.97) with a P-value of 0.04.
Table 3.
Cox proportional hazards model for overall survival
| Variable (Bolded value has unfavorable outcome) |
P-value | Hazard Ratio (95% CI) |
|---|---|---|
| Assigned treatment (Placebo vs. Cediranib) | 0.648 | 0.91 (0.62, 1.34) |
| MGMT (Methylated vs. Unmethylated) | < 0.001 | 2.11 (1.40, 3.17) |
| RPA (RPA III vs. IV) | 0.014 | 2.22 (1.18, 4.20) |
| RPA (RPA III vs. V) | < 0.001 | 5.72 (2.74, 11.97) |
Table 4.
Cox proportional hazards model for PFS
| Variable (Bolded Value has Unfavorable Outcome) |
P-value | Hazard Ratio (95% CI) |
|---|---|---|
| Assigned treatment (Placebo vs. Cediranib) | 0.036 | 0.67 (0.46, 0.97) |
| MGMT (methylated vs. Unmethylated) | 0.950 | 0.99 (0.68, 1.44) |
| RPA (RPA III vs. IV) | 0.458 | 1.23 (0.71, 2.13) |
| RPA (RPA III vs. V) | 0.010 | 2.39 (1.23, 4.63) |
The 6-month PFS rates were also compared between the treatment arms by gender. For males, the 6-month PFS rates for the cediranib and placebo arm were 55.3% (95% CI: 40.1–69.8%) and 26.9% (95% CI: 11.6–47.8%), respectively. For females, the 6-month PFS rates for the cediranib and placebo arm were 36.6% (95% CI: 22.1–53.1%) and 21.7% (95% CI: 7.5–43.7%), respectively.
Results of the Analyses on the MGMT Methylation Status
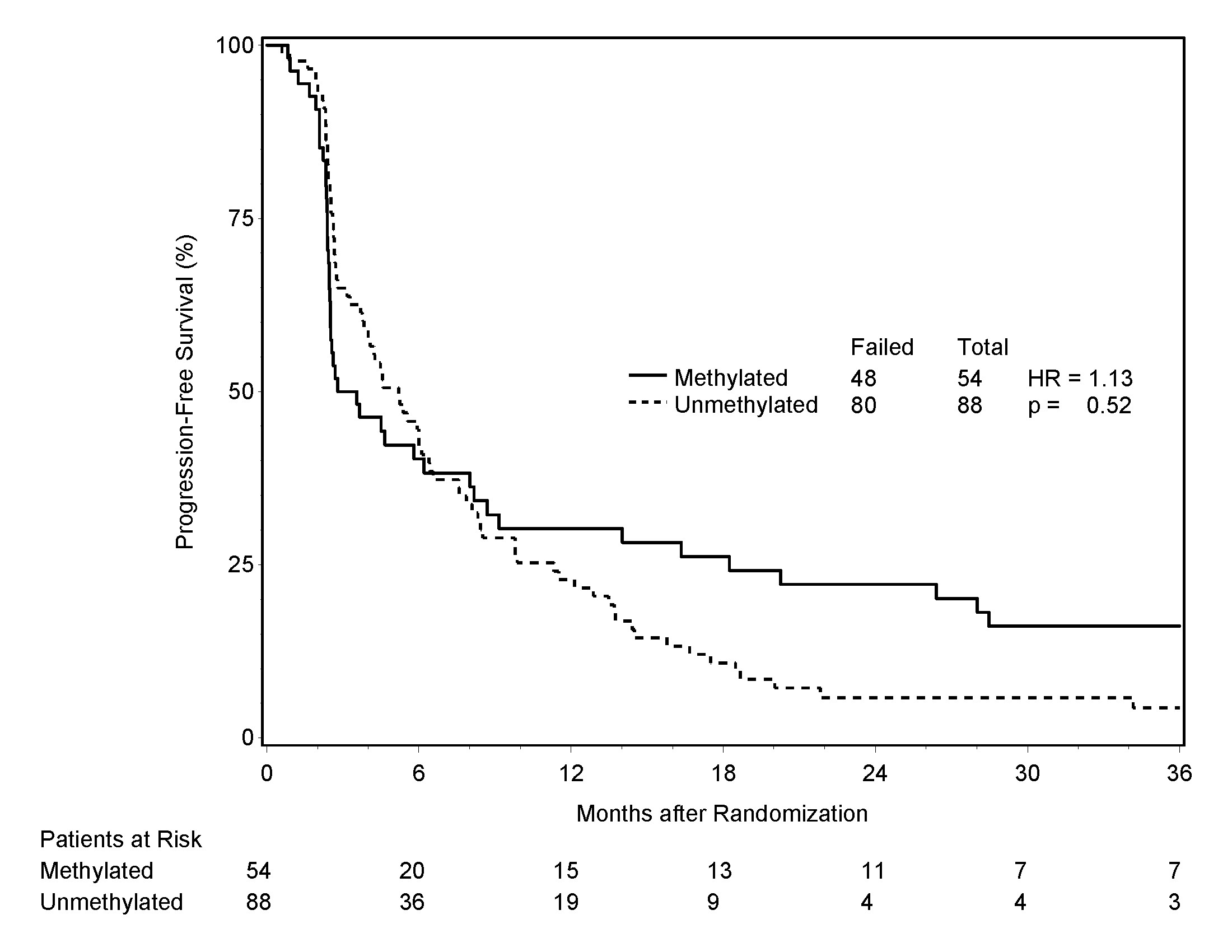
OS and PFS rates by MGMT methylation status were assessed for all the eligible and randomized patients from both arms. The MST was 13.0 months (95% CI: 10.9–15.4 months) for unmethylated patients, and 29.3 months (95% CI: 17.1–36.2 months) for methylated patients, with a P-value < .001 and the HR was 2.13 (95% CI: 1.44–3.15). The median PFS time was 5.2 months (95% CI: 3.8–6.4 months) for unmethylated patients, and 3.2 months (95% CI: 2.5–8.0 months) for methylated patients, with a P-value of.52 and an HR of 1.13 (95% CI: 0.78–1.63). The Kaplan–Meier curves on OS and PFS by MGMT methylation status for patients from both arms are demonstrated in Supplementary Figures S1 and S2, respectively online only.
OS and PFS rates by MGMT methylation status were assessed for patients in the placebo arm. The MST was 13.4 months (95% CI: 10.7–19.7 months) for unmethylated patients, and 17.7 months (95% CI: 2.4–39.6 months) for methylated patients, with a P-value = .20 and an HR of 1.56 (95% CI: 0.79–3.06). The median PFS time was 3.2 months (95% CI: 2.6–6.1 months) for unmethylated patients, and 2.4 months (95% CI: 2.1–2.7 months) for methylated patients, with a P-value = .054, and an HR of 0.55 (95% CI: 0.30–1.02).
Similar analyses were performed for patients in the cediranib arm. The MST was 13.0 months (95% CI: 10.3–14.5 months) for unmethylated patients, and 30.0 months (95% CI: 18.9–40.1 months) for methylated patients, with an HR of 2.59 (95% CI: 1.58–4.25; P-value < .001). The median PFS time was 6.0 months (95% CI: 4.1–7.9 months) for unmethylated patients, and 6.2 months (95% CI: 2.5–16.3 months) for methylated patients, with an HR of 1.51 (95% CI: 0.96–2.40; P-value = .07). As an exploratory analysis, we examined the prognostic value of MGMT methylation status for patients in the cediranib arm, adjusting for RPA class in the Cox proportional hazards model: the HR for MGMT unmethylated versus methylated for OS and PFS were 2.91 (95% CI: 1.74–4.86) and 1.46 (95% CI: 0.91–2.35), respectively.
Discussion
Anti-angiogenic therapy is a beneficial component in the treatment of multiple solid tumors given the importance of adequate blood supply for tumor growth and metastasis.3 Despite promising preclinical data and early clinical trials, anti-angiogenic agents have failed to show an overall survival benefit in randomized controlled trials of patients with glioblastoma.13,14,24,25 In particular, agents targeting VEGF appear to prolong PFS, possibly improve quality of life in some patients, and decrease steroid usage, yet the trials to date have demonstrated no improvement in overall survival. Moreover, it remains unclear whether the extension of “progression”-free survival observed in multiple randomized trials of anti-VEGF therapies represents a delay of tumor growth versus an artefactual effect of this class of agents. The anti-permeability effects of VEGF inhibitors reduce contrast extravasation through tumor blood vessels complicating the interpretation of postcontrast CT and MRI studies.
The addition of cediranib to standard concurrent chemoradiation and adjuvant temozolomide in this trial significantly improved the 6-month PFS rate in patients with newly diagnosed glioblastoma. However, OS was not significantly improved, and the rate of severe toxicities was significantly increased.
The prognostic value of MGMT methylation status for patients in the cediranib arm reflected a similar pattern, implying no significant biologic effect.
Although the primary endpoint of NRG Oncology/RTOG 0837, PFS at 6 months, was met, the anti-permeability effects of cediranib may confound the interpretation of postcontrast brain CT and MRI scans and raise questions about the validity of this endpoint in assessing the utility of VEGF inhibitors. However, no alternative imaging endpoint has been validated as a method of identifying tumor progression in this setting. Development of cediranib is ongoing in glioblastoma and other solid tumors, including ovarian carcinoma. Preclinical models demonstrate that the hypoxia that results from inhibition of angiogenesis disrupts homologous recombination repair mechanisms rendering cancer cells susceptible to inhibition of poly (ADP-ribose) polymerase (PARP) even in the absence of BRCA 1/2 mutations.26,27 Cediranib in combination with PARP inhibition is being studied in a recurrent glioblastoma trial (NCT02974621).
The results of NRG Oncology/RTOG 0837 are consistent with the results observed in 4 randomized, controlled trials of bevacizumab or cediranib in newly diagnosed and recurrent glioblastoma.13,14,24,25 The lack of an overall survival benefit with VEGF inhibitors with different mechanisms of action (VEGF-A ligand sequestration, VEGF receptor tyrosine kinase receptor inhibition) in both the newly diagnosed and recurrent disease settings raises the question of whether these drugs have any role as single agents in the treatment of glioblastoma patients. While the confounding impact of patient crossover from the placebo arms to ultimately receive anti-VEGF therapy exists for all these studies the precise impact of such crossover on overall survival cannot be defined. Moreover, there are no predictive markers to identify glioblastoma subpopulations most likely to benefit from anti-VEGF therapy. While the anti-permeability effects of VEGF inhibitors confound the interpretation of postcontrast brain imaging studies this biological effect has the benefit of reducing brain edema as demonstrated by the steroid-sparing impact of these agents. While neuropsychological and quality of life outcomes are conflicting in these prior studies there is at least some indication of benefit. Future studies are needed to identify predictive biospecimen or imaging markers of glioblastoma subpopulations most likely to benefit from anti-VEGF therapies and to identify the molecular mechanisms involved in the development of resistance to these drugs. The latter will inform future clinical trials of combinations of anti-VEGF agents with inhibitors of other proangiogenic signal transduction pathways.
Supplementary Material
{kind=link}
{kind=link}
Contributor Information
Tracy T Batchelor, Department of Neurology, Brigham and Women’s Hospital, Boston, Massachusetts, USA.
Minhee Won, Department of Statistics, NRG Oncology Statistics and Data Management Center, Philadelphia, Pennsylvania, USA.
Arnab Chakravarti, Department of Radiation Oncology, Wexner Medical Center, Ohio State University Comprehensive Cancer Center, Columbus, Ohio, USA.
Costas G Hadjipanayis, Department of Neuro-Oncology, Neurosurgery, University of Pittsburgh Medical Center, Pittsburg, Pennsylvania, USA.
Wenyin Shi, Department of Radiation Oncology, Thomas Jefferson University Hospital, Philadelphia, Pennsylvania, USA.
Lynn S Ashby, Department of Neurology, Barrow Neurological Institute, Phoenix, Arizona, USA.
Volker W Stieber, Department of Radiation Oncology, Novant Health Forsyth Medical Center, Winston-Salem, North Carolina, USA.
H Ian Robins, Department of Medicine, School of Medicine and Public Health, University of Wisconsin, Madison, Wisconsin, USA.
Heidi J Gray, Department of Obstetrics and Gynecology, University of Washington Medical Center, Seattle, Washington, USA.
Alfredo Voloschin, Department of Neuro-Oncology, Orlando Health Cancer Institute, Orlando, Florida, USA.
John B Fiveash, Department of Radiation Oncology, University of Alabama at Birmingham Medical Center, Birmingham, Alabama, USA.
Clifford G Robinson, Department of Radiation Oncology, Washington University, St. Louis, Missouri, USA.
UshaSree Chamarthy, Department of Medical Oncology/Hematology, Sparrow HH Cancer Center, Lansing, Michigan, USA.
Young Kwok, Department of Radiation Oncology, University of Maryland Medical Systems, Baltimore, Maryland, USA.
Terrence P Cescon, Department of Hematology, Reading Hospital, Reading, Pennsylvania, USA.
Anand K Sharma, Department of Radiation Oncology, Medical University of South Carolina, Charleston, South Carolina, USA.
Rekha Chaudhary, Department of Hematology Oncology, University of Cincinnati, Cincinnati, Ohio, USA.
Mei-Yin Polley, Department of Statistics, NRG Oncology Statistics and Data Management Center, Philadelphia, Pennsylvania, USA; Department of Statistics, University of Chicago, Chicago, Illinois, USA.
Minesh P Mehta, Department of Radiation Oncology, Miami Cancer Institute, Miami, Florida, USA (M.P.M.).
Funding
This work was supported by National Cancer Institute (U10CA180868, U10CA180822); and AstraZeneca.
Conflict of interest statement
Dr Chakravarti, Ashby, Robins, Gray, Voloschin, Chamarthy, Kwok, Cescon, Sharma, Chaudhary, Polley and Ms Won have nothing to disclose. Dr Batchelor reports grants from AstraZeneca and Pfizer for support of clinical trials; personal fees from Merck for lectures, NXDC, Amgen, Roche, Oxigene, Proximagen/Usher, Genomicare, and Champions Biotechnology for scientific advisory board outside the submitted work; other potentially influencing activities, which are CME lectures Oakstone Medical Publishing, Oncology Audio Digest, Research To Practice, and Imedex, Editorial Board of UpToDate, Inc, and consulting in Jiahui Health, Consulting. Dr Hadjipanayis reports royalties from NX Development Corporation, and consulting in Synaptive Medical. Dr Shi reports grants from Regeneron for research funding; grants and personal fees from Novocure for research funding and consulting; personal fees from Brainlab, and Varian for consulting. Dr Stieber reports personal fees from Novocure for the speaker’s bureau. Dr Fiveash reports grants and others from Varian for consulting research and educational contracts. Dr Robinson reports grants and personal fees from Varian for MRA, grants for personal projects and consulting; grants from Elekta for personal projects; and equity from Radialogica. Dr Mehta reports personal fees from Karyopharm, Tocagen, AstraZeneca, Blue Earth, Celgene, and Abbvie for consulting, and Oncoceutics for the board of directors.
Authorship statement
T.T.B. contributed to experimental design, implementation of the study, analysis, and interpretation of data, writing and approval of the manuscript, and principal investigator of the study. M.W. contributed to the experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. A.C. contributed to the experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. C.G.H. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. W.S. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. L.S.A. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. V.W.S. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. H.I.R. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. H.J.G. contributed to the experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. A.V. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. J.B.F. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. C.G.R. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. U.C. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. Y.K. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. T.P.C. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. A.K.S. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. L.P. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. M.Y.P. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript. M.P.M. contributed to experimental design, implementation of study, analysis, and interpretation of data, writing and approval of the manuscript.
References
- 1. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. Lyon (France): International Agency for Research on Cancer; 2021. (WHO classification of tumours series, 5th ed.; vol. 6) [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 3. Jain RK, di Tomaso E, Duda DG, et al. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. [DOI] [PubMed] [Google Scholar]
- 4. Holash J, Maisonpierre PC, Compton D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284(5422):1994–1998. [DOI] [PubMed] [Google Scholar]
- 5. Shweiki D, Itin A, Soffer D, Keshet E.. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359(6398):843–845. [DOI] [PubMed] [Google Scholar]
- 6. Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A.. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367(6463):576–579. [DOI] [PubMed] [Google Scholar]
- 7. Samoto K, Ikezaki K, Ono M, et al. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res. 1995;55(5):1189–1193. [PubMed] [Google Scholar]
- 8. Schmidt NO, Westphal M, Hagel C, et al. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84(1):10–18. [DOI] [PubMed] [Google Scholar]
- 9. Jain RK, Duda DG, Clark JW, Loeffler JS.. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3(1):24–40. [DOI] [PubMed] [Google Scholar]
- 10. Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–4740. [DOI] [PubMed] [Google Scholar]
- 11. Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen MH, Shen YL, Keegan P, Pazdur R.. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14(11):1131–1138. [DOI] [PubMed] [Google Scholar]
- 13. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722. [DOI] [PubMed] [Google Scholar]
- 14. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28(17):2817–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. [DOI] [PubMed] [Google Scholar]
- 18. Zelen M. The randomization and stratification of patients to clinical trials. J Chronic Dis. 1974;27(7-8):365–375. [DOI] [PubMed] [Google Scholar]
- 19. Macdonald DR, Cascino TL, ScholdSC, Jr, Cairncross JG.. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. [DOI] [PubMed] [Google Scholar]
- 20. Ott L. An Introduction to Statistical Methods and Data Analysis. Belmont, Calif.: Duxbury Press; 1993. [Google Scholar]
- 21. Kaplan EL, Meier P.. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 22. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50(3):163–170. [PubMed] [Google Scholar]
- 23. Cox DR. Regression models and life-tables. J R Stat Soc Series B 1972;34:187–202. [Google Scholar]
- 24. Wick W, Gorlia T, Bendszus M, et al. Lomustine and Bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377(20):1954–1963. [DOI] [PubMed] [Google Scholar]
- 25. Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31(26):3212–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein TJ, Glazer PM.. The tumor microenvironment and DNA repair. Semin Radiat Oncol. 2010;20(4):282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim JJ, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ.. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16(4):343–53.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.