

Abstract

Borane cluster-based porous covalent networks, named activated borane (ActB), were prepared by cothermolysis of decaborane(14) (nido-B10H14) and selected hydrocarbons (toluene, ActB-Tol; cyclohexane, ActB-cyHx; and n-hexane, ActB-nHx) under anaerobic conditions. These amorphous solid powders exhibit different textural and Lewis acid (LA) properties that vary depending on the nature of the constituent organic linker. For ActB-Tol, its LA strength even approaches that of the commonly used molecular LA, B(C6F5)3. Most notably, ActBs can act as heterogeneous LA catalysts in hydrosilylation/deoxygenation reactions with various carbonyl substrates as well as in the gas-phase dehydration of ethanol. These studies reveal the potential of ActBs in catalytic applications, showing (a) the possibility for tuning catalytic reaction outcomes (selectivity) in hydrosilylation/deoxygenation reactions by changing the material’s composition and (b) the very high activity toward ethanol dehydration that exceeds the commonly used γ-Al2O3 by achieving a stable conversion of ∼93% with a selectivity for ethylene production of ∼78% during a 17 h continuous period on stream at 240 °C.
Keywords: activated borane, Lewis acid catalyst, heterogeneous catalysis, deoxygenation reaction, ethanol dehydration, Gutmann−Beckett method, solid-state NMR
Introduction
Lewis acid-catalyzed reactions are numerous and of considerable significance.1 In chemical and petrochemical industries, it is well established that heterogeneous catalysts offer an important advantage over their homogeneous counterparts that they allow the design of continuous production processes. For this and other advantageous reasons, significant resources have, in recent decades, been allocated to the development of novel acid-based heterogeneous catalysts.
Traditionally, heterogeneous catalysts such as inorganic oxides, aluminosilicates, sulfonated zirconia, zeolites, and alumina halides have all contained metals as the Lewis acid centers. Alternatively, the incorporation of boron into zeolites and amorphous silica has been thoroughly studied and found to promote the oxidative dehydrogenation of alkanes to alkenes (ODH).2 Interestingly, however, isolated tricoordinated BO3 sites in MCM-22 have been shown to be inactive.3 Instead, an amorphous oligomeric boron oxide/hydroxide layer was shown to be active in ODH, the activity and selectivity being beneficially influenced by the ability of the catalyst to form radicals.4 Mixed boria–alumina oxides represent another family of boron-containing inorganic catalysts. These materials can be described as mildly acidic solids and are used in alcohol dehydration,5 Beckmann rearrangement,6 and phenyl glyoxal conversion.7
The traditional inorganic catalysts described above are very robust and effective even at very high temperatures. However, the set of reactions catalyzed by these materials is different from those catalyzed by molecular tricoordinated boron catalysts, which can, e.g., utilize “frustrated Lewis pair” (FLP) reactivity.8 The first examples of such tricoordinated boron molecular catalysts were based on boron halides BX3 and perfluoroalkyl boranes; however, such compounds are difficult to handle and/or are thermolabile. Since the 1980s, B(C6F5)3 (commonly referred to as “BCF”) has become the homogeneous catalyst of choice for many applications because of the strong electron-withdrawing effect, bulkiness of the pentafluorophenyl substituents, and the general ease and convenience of its use.9 Indeed, the versatility of BCF provided the impetus to its heterogenization by linking it to nonreactive solid supports. Thus, silica,10 alumina,11 graphene oxide,12 organic polymers,13 and other substrates were modified with -B(C6F5)2 acid centers. However, these heterogenized catalysts are laborious to synthesize, and their catalytic activity is in most cases significantly lower than that of the homogeneous BCF counterpart.
Recently, borane clusters (usually represented by derivatives of icosahedral carborane C2B10H12) have been proposed as a viable alternative to the commonly used polyfluorinated aryl substituents at the tricoordinated boron atom sites of BCF.14 Kondo and co-workers have recently reported the catalytic dehydration of ethanol to ethylene by hydrogenated borophene, a 2D material with an empirical formula H1B1.15 These HB sheets likely possess Brønsted sites represented by protons on the edges of the 2D structure, but the precise nature of the catalytically active sites in these materials is still unknown. This development prompted us to cast a wider net in the borane cluster field and test the boron acid centers found in activated borane (abbreviated hereafter as ActB)16 for heterogeneous catalytic activity. ActB is a porous polymer that is formed by cothermolysis of decaborane(14) (nido-B10H14) and organic solvents such as toluene or hexane. We have shown that the structure of ActB is most probably composed of closo borane clusters linked together by organic linking moieties.17
Here, in this paper, we present an improved strategy for the synthesis of ActB and present evidence for the catalytic activity of its Lewis acidic boron sites as well as a comparison of its performance compared with contemporary catalysts. The nature of the Lewis acid sites in ActB was characterized by the Gutmann–Beckett method (31P solid-state NMR after the adsorption of Et3PO) and by temperature-programmed desorption of ammonia. Furthermore, we showcase the utility of ActB for hydrosilylation/deoxygenation reactions of various carbonyl compounds and also in the dehydration of ethanol into ethylene. In the latter reaction, ActB exerted a higher catalytic activity than commonly used alumina and excellent catalyst stability even after 17 h on stream.
Results and Discussion
Synthesis of Activated Borane
Building on our previous findings,16 we have improved the synthesis of ActB and, by using an Ar-filled glovebox, a stainless steel autoclave (Berghof BR-300), and a Schlenk line, we have prepared pristine samples of ActB devoid of B–OH groups (see below IR and ssNMR). In order to compare the effect of aromatic, cyclic aliphatic, and acyclic aliphatic linking molecules, we have prepared ActBs by heating nido-B10H14 in toluene, cyclohexane, and n-hexane to 250 °C in a high-pressure reactor for 24 h (Scheme 1). The crude dark solids were Soxhlet-extracted with the same solvent as that used for the synthesis to remove all unreacted and partially unreacted impurities. The residual solvent was removed by heating to 100 °C under vacuum for 4 h. For details on synthesis, see the SI. The samples are denoted as ActB-Tol (toluene), ActB-cyHx (cyclohexane), and ActB-nHx (n-hexane).
Scheme 1. Synthesis of ActB Materials.

Characterization
All prepared ActBs are solids lacking long-range order. ActB-Tol and ActB-cyHx are black powders, whereas ActB-nHx is brown-colored. This crude observation is consistent with the materials’ UV–vis spectra, in which all ActBs absorb strongly below 1000 nm, with the absorption edge for ActB-nHx being shifted to lower wavelengths in comparison with ActB-Tol and ActB-cyHx. This translates to optical band gaps of 1.19, 1.20, and 1.34 eV for ActB-Tol, ActB-cyHx, and ActB-nHx, respectively, as derived from Tauc plots using the Kubelka–Munk-transformed reflectance spectra (for details, see the SI). The elemental analysis of ActBs was done by the standard CHN analysis, and the content of boron was determined by ICP-MS (see the SI for details). ICP-MS was also used to check for the presence of metal traces; from the comparison with blank samples, we did not observe elevated amounts of metals, which could be responsible for catalytic activity (such as Pd, Ni, Cu, Cr, Fe, Zn, Cr, etc.). Scanning electron microscopy (SEM) (Figure 1) shows larger particles of irregular shape for ActB-Tol and ActB-cyHx, while ActB-nHx forms spherical particles approximately 1 μm in diameter.
Figure 1.

SEM images of ActB-Tol (left), ActB-cyHx (middle), and ActB-nHx (right). For additional images, see the SI.
The Ar adsorption isotherms for all ActBs correspond well to those typical for microporous materials (Figure 2). ActB-Tol and ActB-cyHx contain pore size maxima between 0.9 and 1.6 nm, while ActB-nHx contains pore size maxima between 1.0 and 2.0 nm (Figure S4). The extent of porosity and specific surface area, however, varies greatly between the derivatives, with the clear trend ActB-Tol > ActB-cyHx > ActB-nHx (see Table 1). The adsorption of CO2 follows a similar pattern, with the highest adsorption achieved for ActB-Tol and a lower adsorption achieved for ActB-cyHx and ActB-nHx. In this case, however, no significant difference between the latter two was observed. To confirm the batch-to-batch reproducibility of the synthesis of ActB-Tol, we compared the adsorption isotherms of Ar and pore size distribution for individual batches (see Figures S5 and S6 in the SI).
Figure 2.

Adsorption isotherms of Ar at 87 K (top) and CO2 at 195 K (bottom) for ActB-Tol, ActB-cyHx, and ActB-nHx.
Table 1. Specific Surface Areas, Pore Diameters, and Pore Volumes for ActB-Tol, ActB-cyHx, and ActB-nHx.
| sample | SBET (m2g–1)a | pore size (nm)b | Vpore (cm3g–1)c |
|---|---|---|---|
| ActB-Tol | 784 | 0.9–1.6 | 0.47 |
| ActB-cyHx | 608 | 0.9–1.5 | 0.30 |
| ActB-nHx | 160 | 1.0–2.0 | 0.10 |
BET specific surface area.
Pore size maxima calculated by the MDFT method.
Total pore volume at p/p0 = 0.99.
The FTIR spectra (Figure 3) for these species contain peaks corresponding to both constituent moieties—borane cluster and organic linker. Most notably, the peaks at approximately 2900 and 2550 cm–1 correspond to C–H and B–H stretchings, respectively. In all cases, the absence of any peak at around 1900 cm–1 suggests the absence of μ-H (B–H–B bridges) in the structure. As expected, the peak at approximately 1600 cm–1, which can be attributed to the aromatic stretching vibration, is present only in ActB-Tol. A significant difference from the original ActB spectrum published earlier16 is the absence of the BO–H stretching peak over 3000 cm–1, which can be explained by the different preparations and measurement procedures used that avoid oxygen contamination. To study the effect of humid air, we exposed ActBs for 1 min to air and remeasured FTIR, which in all cases revealed a quick water adsorption onto the material, manifested by the appearance of the O–H stretching band at around 3200 cm–1.
Figure 3.

FTIR (ATR-Si) spectra of ActB-Tol (top), ActB-cyHx (middle), and ActB-nHx (bottom); red lines correspond to samples exposed to air for 1 min.
The thermal stabilities of ActBs were studied by TGA coupled with MS (Figures S10–S19). ActB-Tol showed the lowest stability with the onset of its main degradation step (Tonset) at around 245 °C, compared to the higher limits measured for ActB-cyHx and ActB-nHx at 319 and 322 °C, respectively. For a detailed discussion, see the SI.
The 1H MAS NMR spectrum of ActB-Tol (Figure 4, left column) reveals two broad signals at 1.64 and 6.5 ppm, which can be attributed to the methyl group and aromatic protons of toluene, respectively, overlapped with signals from borane clusters.16 The 1H MAS NMR spectra of ActB-cyHx and ActB-nHx samples show a broad and asymmetric dominant peak at 1.14 and 0.65 ppm, respectively, assigned to aliphatic protons overlapped with signals from borane clusters. Interestingly, both spectra contain a broad shoulder at about 6.5 and 6.2 ppm, respectively, which indicate the presence of unsaturated (−CH=) species, suggesting that partial dehydrogenation of cyclohexane and n-hexane occurs during the synthesis (further confirmed by 13C CP/MAS NMR; see below). In all cases, signals from open-face “bridging” μ-hydrogens of nido-B10H14 are not detected (the expected range of signals between −4 and 0 ppm),18 confirming the transformation of open nido-borane clusters into closed cluster derivatives and/or full substitution of the μ-hydrogens16 indicated by FTIR. The 13C CP/MAS NMR spectrum of ActB-Tol (Figure 4, middle column) exhibits three major peaks at 19.3, 126.6, and 134.6 ppm and minor peaks at 0.5 and 29.4 ppm. The major peaks are assigned, respectively, to the methyl carbon and the nonequivalent groups of aromatic carbons from immobilized toluene. The minor peak at 0.5 ppm was attributed to carbon atoms bonded to the borane cluster forming nodal points, similar to the bonding system that was observed for boron carbide systems.19 The second minor broad peak at 29.4 ppm, ranging from about −10 to 70 ppm, can be assigned to methyl groups directly connected to borane clusters and/or carbons incorporated into these clusters possibly forming a substituted carborane cluster.20 The 13C CP/MAS NMR spectra of ActB-cyHx and ActB-nHx display a series of partially overlapping signals ranging from about 0 to 40 ppm that correspond well with aliphatic carbons (Figure 4, middle column). The presence of signals around 133 ppm confirms the formation of unsaturated (−CH=) species. Furthermore, the distribution of chemical shifts points to a wide range of chemical environments typical of solids lacking a long-range order. In addition, the signal at around 0.7 ppm on the respective spectra of ActB-cyHx and ActB-nHx indicates carbon atoms forming nodal points similarly to what was observed for ActB-Tol.
Figure 4.

1H MAS NMR (left column), 13C CP/MAS NMR (middle column), and 11B MAS NMR (right column) spectra of ActB-Tol, ActB-cyHx, and ActB-nHx samples.
All 11B MAS NMR spectra (Figure 4, right column) possess two broad composite signals with maxima at around −7.3 and 1.5 ppm (3.5 ppm in the case of ActB-Tol) corresponding to the borane cluster molecules substituted at different positions forming a variety of chemical environments.16 Even 2D 11B 3Q/MAS NMR spectra did not reveal additional signals and clearly confirmed the structure lacking a long-range order (Figure S8).
Lewis Acid Sites
The Gutmann–Beckett method is commonly used to assess the relative acidity of molecular Lewis acid-based catalysts. It uses the sensitivity of the 31P NMR chemical shift (δP) in Et3PO (TEPO) to interactions with Lewis acids.21 As a result, the δP of TEPO can be shifted from 48 to over 90 ppm depending on the strength of the acid–base interaction. In recent years, the Gutmann–Beckett method was also used for porous solid acids using ssNMR. However, in this case, the NMR lines are broadened not only because of the measurement in solid state but also because of the varying chemical environments within the pores.22
From a comparison of all 31P MAS NMR spectra (Figure 5) of the investigated samples, it is evident that TEPO interacts only with ActB-Tol and ActB-cyHx and forms LA-TEPO adducts. Interestingly, the signals are very narrow (half-width of dominant signals is ca. 1.3 ± 0.2 kHz), displaying similar sharpness as zeolite-Y-TEPO adduct and much higher sharpness than γ-alumina or micromesoporous zirconosilicates.22 Therefore, the LA strength can be compared: BCF > ActB-Tol > ActB-cyHx. Given that ActBs do not contain any perfluorinated electron-withdrawing substituents, it points to a remarkable Lewis acidity of ActB-Tol. On the other hand, only one sharp signal, with almost unchanged position in comparison with pristine TEPO, was detected in the 31P MAS NMR spectrum of ActB-nHx. This indicates the presence of TEPO molecules in the investigated system but without any detectable chemical interaction.
Figure 5.

31P MAS ssNMR spectra from top: BCF+TEPO adduct, TEPO, ActB-Tol+TEPO, ActB-cyHx+TEPO, and ActB-nHx+TEPO.
11B 3Q/MAS NMR spectra after the adsorption
of TEPO
(Figure 6) display
one specific, relatively sharp signal with 11B δiso = 1 ppm (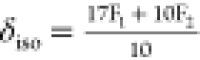 , where F1 = 1.3 ± 0.5 ppm
and F2 = 0.5 ± 0.5 ppm) in ActB-Tol+TEPO
and ActB-cyHx+TEPO systems. The shape of the signal and 11B NMR shift
suggest an almost ideal tetracoordinated geometry of affected boron
atoms.23 This clearly confirms the Lewis
acidity of ActB-Tol and ActB-cyHx by the formation of tetracoordinated
borons upon reaction with the Lewis base (TEPO). It should be noted
that only a small fraction of borons in ActBs are LA
sites, and therefore, the peaks are relatively small in comparison
with the rest of the spectra. In 13C CP/MAS NMR spectra, ActB-TEPO samples contain additional signals corresponding
to TEPO and toluene, which come from the TEPO treatment; otherwise,
the spectra are unchanged in comparison with as-prepared ActBs, thus confirming the preserved disordered structure (see Figure S9).
, where F1 = 1.3 ± 0.5 ppm
and F2 = 0.5 ± 0.5 ppm) in ActB-Tol+TEPO
and ActB-cyHx+TEPO systems. The shape of the signal and 11B NMR shift
suggest an almost ideal tetracoordinated geometry of affected boron
atoms.23 This clearly confirms the Lewis
acidity of ActB-Tol and ActB-cyHx by the formation of tetracoordinated
borons upon reaction with the Lewis base (TEPO). It should be noted
that only a small fraction of borons in ActBs are LA
sites, and therefore, the peaks are relatively small in comparison
with the rest of the spectra. In 13C CP/MAS NMR spectra, ActB-TEPO samples contain additional signals corresponding
to TEPO and toluene, which come from the TEPO treatment; otherwise,
the spectra are unchanged in comparison with as-prepared ActBs, thus confirming the preserved disordered structure (see Figure S9).
Figure 6.

11B 3Q/MAS NMR spectra of ActB-Tol+TEPO (a), ActB-cyHx+TEPO (b), and ActB-nHx+TEPO (c). The newly detected signal is highlighted by gray boxes.
To quantify the number of acid sites, we employed temperature-programmed desorption of ammonia (TPD). All three samples provided a similar pattern in which ammonia desorbs first at ∼120 °C (weakly adsorbed NH3) and then at ∼420 °C (strongly chemisorbed NH3; Figure 7 and Table 2). The fraction of the weakly adsorbed NH3 out of the total number of NH3 molecules adsorbed is very uniform for all three samples and ranges from 7 to 8%. Of note are the high ammonia desorption temperatures (varying from 403 °C for ActB-cyHx to 438 °C for ActB-Tol) that lie within the range of the strong Brønsted acid sites present in HZSM-5 zeolite (400–470 °C)24 and much higher than for the acid sites in amorphous aluminosilicates (250–300 °C).25 Importantly, ActB-Tol possesses the strongest acid sites according to ammonia TPD, which is in line with 31P MAS NMR after TEPO adsorption; see above.
Figure 7.

TPD curves for ActB-Tol (left), ActB-cyHx (middle), and ActB-nHx (right).
Table 2. Summary of Ammonia TPDa.
| sample | weakly adsorbed NH3 (mmol g–1) | chemisorbed NH3 (mmol g–1) | Tdes of chemisorbed NH3 (°C) |
|---|---|---|---|
| ActB-Tol | 0.30 | 3.47 | 438 |
| ActB-cyHx | 0.60 | 8.10 | 403 |
| ActB-nHx | 0.66 | 7.31 | 415 |
The amounts of adsorbed NH3 were estimated from the areas under the TPD curves.
The most significant difference between the ActBs samples is the total number of NH3 molecules adsorbed (Table 2). While ActB-Tol exhibited 3.77 mmol g–1 adsorbed NH3 in total, ActB-nHx and ActB-cyHx provided more than twice as much, with the maximum observed for ActB-cyHx (8.70 mmol g–1). These results indicate that ActB-cyHx and ActB-nHx contain a significantly higher number of acid sites in comparison to ActB-Tol. Interestingly, ActB-nHx contains a high number of acid sites according to NH3 TPD but virtually none according to 31P MAS NMR after TEPO adsorption. We assume that these differences originate in the different size and basicity of the probe molecules. Nevertheless, all obtained values are very high, suggesting that there might be multiple NH3 molecules adsorbed to each available LA site and/or strongly bound NH3 in other areas within the framework not only to the LA centers.
To analyze the ability of ActBs to adsorb medium-sized molecules, pyridine and benzene were preadsorbed onto each sample at RT followed by TGA measurement. The difference between the adsorption of neutral molecules (benzene) and basic molecules (pyridine) can be used as a proxy for the estimation of the number of accessible acid sites to medium-sized molecules. Thus, the amount of adsorbed pyridine followed the trend ActB-Tol > ActB-cyHx > ActB-nHx. Based on these data, we can assume that the number of LA sites accessible to medium-sized molecules is between 1 and 1.5 mmol g–1 for ActB-Tol and ActB-cyHx and yet probably only around 0.5 mmol g–1 for ActB-nHx (see Table 3). This correlates well with the specific surface area of the ActBs materials as determined by Ar adsorption and the Gutmann–Beckett method utilizing TEPO adsorption but not complementary with the ammonia TPD (for a detailed discussion, see the SI).
Table 3. Summary of Quantification of Probe Molecular Adsorption.
| sample | adsorbed benzene (mmol g–1) | adsorbed pyridine (mmol g–1) | chemisorbed NH3 (mmol g–1) | Δpyridine–benzene (mmol g–1)a |
|---|---|---|---|---|
| ActB-Tol | 0.8 | 2.1 | 3.47 | 1.3 |
| ActB-cyHx | 0.3 | 1.3 | 8.10 | 1.0 |
| ActB-nHx | 0.3 | 0.7 | 7.31 | 0.4 |
The difference between absorbed benzene and absorbed pyridine.
The origin of the LA boron atoms probably lies in the solvothermal thermolysis of open clusters. It has been observed earlier (e.g., see the work of J. Kennedy26) that open borane clusters subject to high energy (e.g., thermolysis, photolysis, and electron bombardment) form clusters with different number of vertices. We made similar observations during the thermal treatment of nido-B10H14 in benzene at 200 °C where we detected substituted B18H22 clusters.17 This change in the number of boron vertices is probably accompanied by the formation of highly reactive lower boranes that are the seeds for LA centers.
Catalytic Activity
Hydrosilylation and Deoxygenation Reactions
Lewis acidic boron molecules, e.g., BCF, are known to activate the Si–H bond.27 This can be utilized in hydrosilylation or reductive deoxygenation of carbonyl compounds in mild conditions.28 Such reactions cannot be catalyzed by conventional heterogeneous inorganic LA catalysts, e.g., zeolites, which are, on the other hand, known to promote hydrodeoxygenation (HDO) processes during biomass processing.29 These reactions, however, typically require high temperatures and pressures of hydrogen gas, as well as the presence of metal dopants, which are responsible for the hydrogenation reactivity. In addition, hydrosilylation reactions, related to those described herein, were only reported using solid-supported metal-based catalysts in several literature reports.30 For this reason, we endeavored to test the new ActBs described here for their ability to catalyze the reduction of selected substrates with silanes. Initial testing was carried out in batch reactions with an exclusion of air and moisture, typically employing 1 mmol of the corresponding substrate, 20 mg of the ActB catalyst, 2 mL of solvent, and an excess of Et3SiH (or other silanes).
Benzophenone (1) as a model substrate was cleanly converted over both ActB-Tol and ActB-cyHx materials to the corresponding deoxygenated product, diphenylmethane (2), by Et3SiH in toluene (Scheme 2), whereas the silyl ether intermediate 3 was only observed when using the ActB-nHx material at the conditions applied. Using 1.5 equiv of Et3SiH with ActB-Tol gave incomplete conversion, while 3 equiv of Et3SiH afforded full conversion, and the product was subsequently isolated by column chromatography in an 88% yield. Expectedly, the siloxane (Et3Si)2O was identified as the only other product of deoxygenation. The kinetic profiles at 60 and 100 °C show that ActB-Tol is slightly more active than ActB-cyHx, while ActB-nHx exhibits only poor activity in this deoxygenation reaction.
Scheme 2. Deoxygenation of Benzophenone (1) Performed Using Different ActB Catalysts, Yields Determined by GC (Isolated Yield in Parentheses).

1 mol % B(C6F5)3 (BCF) used as a catalyst (reaction time 2 h). kinetic profiles (bottom) of the deoxygenation of 1 catalyzed by ActBs at 60 or 100 °C. Conversion of 1 determined by GC.
These differences in performance are consistent with the observed relative LA strengths of the ActB materials, where ActB-Tol contained the most strongly acidic sites and ActB-nHx contained the weakest or least accessible LA sites. To assess the relevance of these results, we performed a comparison test with the BCF catalyst. Thus, the exact same deoxygenation reaction was done but in the presence of 1 mol % of BCF as a catalyst. Here, the BCF converted 1 in 2 h, but in contrast to the ActB-Tol and ActB-cyHx catalysts, BCF gave predominantly the silylated product 3 (84%) and only a small amount of 2. We also tested the pristine nido-B10H14 as well as a sample prepared by thermolysis of nido-B10H14 without solvent at the same conditions as applied for the synthesis of ActB materials (dark brown powder without detectable porosity measured by Ar adsorption at 87 K). Neither of these, however, exhibited any catalytic activity in the above transformation (for details, see the SI).
Next, we employed acetophenone (4) as a substrate for ActB catalytic assessment. In general, the reaction proceeded more slowly than for benzophenone, and the selectivity was greatly affected by conditions; see Scheme 3. ActB-Tol displayed a clear preference for deoxygenation under all tested conditions, with no detectable amounts of the silyl ether product 5. Significant formation of styrene (6) was observed, accompanied by ethylbenzene (7). The relative amount of 7 was maximized at higher reaction temperatures (100 °C being the optimum) when an excess of silane was used (Scheme 3b), which also ensured full conversion of the starting material. ActB-cyHx, in contrast, gave predominantly hydrosilylation product 5 irrespective of the amount of silane used. Prolonged reaction times with less available silane generally increased the amount of styrene formed. In contrast, ActB-nHx was much less active but gave almost exclusively the hydrosilylation product 5.
Scheme 3. Hydrosilylation/deoxygenation of Acetophenone (4): (a) Screening of Different ActB Materials Using 1.5 or 3 equiv of Silane and Toluene as the Solvent; (b) Reaction Temperature Optimization Using ActB-Tol in Toluene; and (c) Variation of Reaction Conditions in THF Solvent. Yields Determined by GC.

The styrene formation has been previously noted as a minor product in reactions of 4 catalyzed by BCF;31 its formation was significantly affected when 60 bar of H2 was applied combined with the presence of molecular sieves.32 In our hands, a control reaction employing BCF as a catalyst yielded only a mixture of ethylbenzene 7 (70%) and silyl ether 5 (25%), while no styrene was detected. Contrarily, the ActB-Tol catalyst under adjusted conditions (THF solvent, 60 °C; see Scheme 3c) afforded styrene (6) as the major product with a 65% selectivity (at an 89% conversion of 4). It should be noted that the formation of olefins from ketones on acidic zeolite catalysts at temperatures above 250 °C yielding mixture of hydrocarbons is well-recognized.33
Reaction conditions were optimized for various silane hydrides, solvents, reaction times, temperatures, and type of ActB catalyst. The results can be summarized as follows: (a) best conversions were achieved with secondary silanes (Et2SiH2 or Ph2SiH2), yet it was still comparable with cheaper Et3SiH; (b) by means of conversion of acetophenone 4, toluene appears to be the solvent of choice followed by THF; (c) selectivity is significantly affected by solvent and catalyst type: ActB-Tol showed increased selectivity toward the formation of styrene, whereas ActB-cyHx yields predominantly silyl ether 5; (d) excess of silane favors the formation of symmetrical ether 8; and (e) moderate temperature favors the formation of ethers 5 and 8, whereas increased temperature deoxygenation products 6 and 7. For a reaction overview, see Scheme 3, and for complete results, see the SI.
The effect of exposure of the ActB-Tol catalyst to air for a limited time (1 h) was studied. Interestingly, the activity does not seem to be affected much, which highlights the relative robustness of the ActB catalysts. Additionally, we tested the variability of catalytic properties of different batches of ActB-Tol in the hydrosilylation/deoxygenation of 1 and 4. Results summarized in Table S16 confirmed the good reproducibility of activity as well as selectivity.
In order to uncover the reaction mechanism of acetophenone transformation, we performed additional experiments employing products 5 and 6 and the related rac-1-phenylethanol (9) (Scheme 4). Silyl ether 5 is a plausible intermediate in the formation of both 6 and 7, while 6 is the preferred product in the absence of silane as shown in (reaction b) in Scheme 4. When 3 equiv of Et3SiH was applied (reaction a) in (Scheme 4), 7 becomes the dominant product, as expected. Styrene (6), when exposed to the standard reaction conditions as depicted in (reaction c), is not converted to any other molecular products even in the presence of the silane. Alcohol 9 is also completely converted to a mixture of products 5–7 under the same conditions (reaction d). The formation of styrene under these conditions, and especially without the presence of silane (reaction e), caught our attention as this is formally a dehydration reaction.34 This transformation was further studied in the context of the technologically important dehydration of ethanol to ethylene; see below.
Scheme 4. Reactions Involved in the Mechanism of 4-Hydrosilylation/Deoxygenation by ActB-Tol: (a) Deoxygenation of Silyl Ether 5 in the Presence of Silane, (b) without Silane, (c) Attempted Reaction of Styrene (6), (d) Reaction of Alcohol 9 in the Presence of Silane, and (e) without Silane. Conditions: 60 °C, 22 h, Toluene Solvent (1 mmol Substrate, 20 mg of ActB). GC Yields Given in Parentheses.

ActB-Tol and ActB-cyHx catalysts were further tested in the hydrosilylation/deoxygenation reaction of various other carbonyl-containing substrates (Scheme 5), namely, benzaldehyde (10), trans-chalcone (13), benzil (16), cyclohexanone (22), and 2-heptanone (27). It should be noted that in all cases, the reaction conversion was higher than 90%, often yielding a mixture of products, the composition of which is strongly dependent on the conditions used.
Scheme 5. Hydrosilylation/Deoxygenation of (a) Benzaldehyde (10), (b) Trans-chalcone (13), (c) Benzil (16), (d) Cyclohexanone (22), and (e) 2-Heptanone (27), using ActB-Tol or ActB-cyHx as the Catalysts at Various Conditions (Silane Stoichiometry, Temperature, Time) as Indicated. All Reactions Performed in Toluene. Yields Determined by GC.

While each substrate behaves uniquely, it is evident that it follows similar trends as in the case of acetophenone: with ActB-Tol, higher temperature and excess of Et3SiH favoring deoxygenation products or the formation of ethers, while ActB-cyHx affording predominantly hydrosilylated products. The formation of hydrosilylated products was obtained in much higher selectivities, which could be because of the milder conditions used. For full details of the optimization procedures and discussion of each substrate, see the SI.
Flow Conditions
On the basis of the batch experiments, ActB-Tol and ActB-cyHx catalysts were chosen for further investigation under flow conditions using a microfluidics-based flow reactor (X-Cube) in continuous flow mode. CatCart cartridges were loaded with the catalysts, and the catalyst bed was washed continuously with toluene solutions of the reactants and the resulting reaction mixtures were analyzed by GC. Based on the results from the preparative batch experiments described above, two reactions with high selectivity were chosen to test using flow conditions—the benzophenone (1) deoxygenation catalyzed by ActB-Tol and trans-chalcone (13) 1,4-hydrosilylation catalyzed by ActB-cyHx. In both cases, the catalysts were able to operate in the flow mode, giving initially full conversion of the substrate to the corresponding product when running with a flow rate of 0.1 mL min–1. Gradual loss of activity was observed for the ActB-Tol catalyst, while the ActB-cyHx also started losing its activity after ca. 2 h on stream (Figure 8). The same behavior was observed even when the flow rate was increased to 0.2 mL min–1. The leaching of boron moieties into the reaction mixture during the flow experiments was monitored by ICP-MS. The boron concentration in samples taken between 30 min and 2 h on stream was identical to the starting reaction mixture. For this reason, we conclude that the structure of ActBs is stable but its catalytic centers are gradually deactivated under hydrosilylation/deoxygenation reaction conditions.
Figure 8.

Time-dependence of the catalytic activity of ActB-Tol in benzophenone (1) deoxygenation and ActB-cyHx in trans-chalcone (13) 1,4-hydrosilylation under flow conditions (X-Cube, 100 °C, flow rate: 0.1 mL min–1—solid lines—or 0.2 mL min–1—segmented line, concentration of substrates 0.5 mM mL–1).
Dehydration of Ethanol
As noted above, the reaction of 1-phenylethanol in the absence of silane produced mostly the products of dehydration (styrene and ether (8)). These results gave us reason to test ActBs for the industrially relevant gas-phase dehydration of ethanol and compare catalytic properties with traditional solid acid catalysts, namely, zeolite HZSM-524b,24c,25a,35 and γ-alumina.25b,36 To validate the need for strong LA sites present in ActBs, we performed a blank experiment: impregnated activated charcoal with boric acid in B: C molar ratio is similar to ActBs.
Ethanol dehydration was performed within the temperature range of 170 and 240 °C; see Figure 9. The conversion decreases in the following order: HZSM-5>ActB-cyHx>ActB-Tol ≈ ActB-nHx ≈ γ-Al2O3, with ethylene yields following a similar pattern. Thus, ActB-cyHx achieved conversions over 90% and a ca. 80% yield of ethylene, while the control-activated charcoal–boric acid material was inactive throughout the whole temperature range.
Figure 9.

Ethanol conversion (top) and ethylene yield (bottom) during ethanol dehydration at 170, 190, 210, and 240 °C. Weight hour space velocity (WHSV) was kept for all measurements at 2.2 g g–1 h–1.
The major products of the dehydration reactions were ethylene and diethyl ether, the ratio of which is dependent on the temperature. In this regard, ActBs behave similarly to many conventional catalysts. Only in the case of HZSM-5 were butenes observed as products of ethylene dimerization, which is in line with the literature.24b In contrast to γ-Al2O3 and HZSM-5, all three ActBs produced only small amounts of ethane (ActB-cyHx provided up to 3.6% ethane at 240 °C). We assume that ethane is formed via ethylene hydrogenation, with the hydrogen needed for this step coming from the B–H bonds in ActBs. This hypothesis is in agreement with the following observations: (i) ethane yield progressively decreased with time in all cases, (ii) addition of H2 gas to the reactant flow did not change the amount of formed ethane, and (iii) significant decrease in the intensity of B–H absorption bands in IR spectra (∼2550 cm–1) after the catalytic experiments; see Figure S33.
From all of the ActBs, ActB-cyHx showed the most promising catalytic performance in ethanol dehydration. This behavior can be related to the high number of acidic sites as evidenced by ammonia TPD analysis and the materials’ porosity. ActB-Tol exhibited a similar porosity but a significantly lower number of acidic sites.
In an effort to probe the limits of the stability of ActBs, a 17 h time-on-stream experiment at 240 °C was conducted (Figure 10). Importantly, all ActBs proved to be stable, with ActB-cyHx exerting a stable conversion of ∼93% and an ethylene selectivity of ∼78%. HZSM-5, in particular, has been shown to suffer from ethylene oligomerization,35b coking, and deactivation.37 In line with the literature, HZSM-5 proved to be unstable with ethanol conversion decreasing from 98 to 76% and ethylene yield dropping from 99 to 62%. For a detailed discussion, all ethanol conversions, selectivities, and carbon balances, see the SI.
Figure 10.
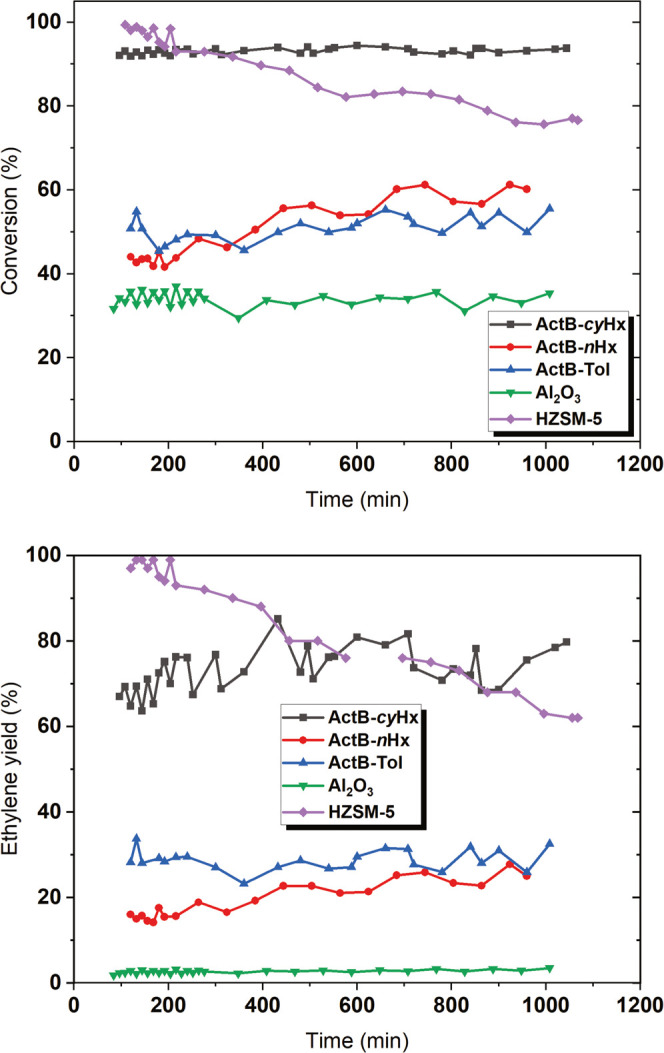
Ethanol conversion (top) and ethylene yield (bottom) during ethanol dehydration at 240 °C overnight (stability test). WHSV was kept at 4.4 g g–1 h–1 except for HZSM-5 due to its high activity. WHSV was set to 17.6 g g–1 h–1; for details, see the SI.
Conclusions
We have demonstrated that controlled cothermolyses of decaborane with aromatic or aliphatic hydrocarbons in strictly anaerobic conditions give highly porous and halide-free solid materials, ActBs, containing high numbers of Lewis acidic (LA) sites in their structures. These sites give these new materials a level of catalytic activity that approaches that of the well-established molecular borane LA B(C6F5)3 but with the added benefit of being a heterogeneous catalyst, allowing the use in flow reactors. The strength and number of LA sites in ActBs seem to be controllable by the nature of the hydrocarbon linkers incorporated into the material. We proved ActBs to be valuable heterogeneous catalysts in batch as well as flow conditions for hydrosilylation/deoxygenation reactions of various carbonyl substrates using silanes as the reductants. More importantly, not only can ActBs be used for analogous reactions as catalyzed by B(C6F5)3 but they also show some unique reactivity, e.g., the formation of styrene from acetophenone. In general, the toluene-based ActB-Tol preferred deoxygenation pathways in comparison to the other two catalysts with aliphatic linkers, which were, in most cases, more selective toward the silylation products.
The potential for dehydration reactions was studied in detail in ethanol dehydration in the gas phase, which revealed the capability of ActB catalysts to operate efficiently under flow conditions at 240 °C with excellent stability over 17 h on stream. The activity and selectivity for ethylene exceed those of commonly used γ-alumina catalysts. Further investigation of the effects of the catalytic material composition on its LA and consequently catalytic properties as well as utilization in other types of catalytic reactions is currently under way.
Acknowledgments
This work was supported by the Czech Science Foundation (No. GA 23-05818S), Research Infrastructure NanoEnviCz, the Ministry of Education, Youth and Sports of the Czech Republic under Project no. LM2023066, and the Ministry of Education, Youth and Sports of the Czech Republic and The European Union—European Structural and Investments Funds within the project Pro-NanoEnviCz II (Project No. CZ.02.1.01/0.0/0.0/18_046/0015586); the ssNMR measurement was supported by No. GA 20-01233S; and the catalytic testing of dehydration of ethanol was supported by the Grant Agency of Masaryk University (GAMU) No. MUNI/J/0007/2021. The authors are grateful to Dr. Jan Šubrt for SEM measurements, Slavomír Adamec for ICP-MS measurements, and Dr. Francesco Walenszus (3P Instruments GmbH) for TPD measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c04011.
Full experimental details of syntheses and catalytic experiments, additional characterization data for the ActB materials, detailed results of catalytic investigations (product distribution, characterization data) including an extended discussion. The authors have cited additional references within the Supporting Information.38 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Corma A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. 10.1021/cr00035a006. [DOI] [Google Scholar]; b Busca G. Acid Catalysts in Industrial Hydrocarbon Chemistry. Chem. Rev. 2007, 107, 5366–5410. 10.1021/cr068042e. [DOI] [PubMed] [Google Scholar]
- a Gao B.; Qiu B.; Zheng M.; Liu Z.; Lu W.-D.; Wang Q.; Xu J.; Deng F.; Lu A.-H. Dynamic Self-Dispersion of Aggregated Boron Clusters into Stable Oligomeric Boron Species on MFI Zeolite Nanosheets under Oxidative Dehydrogenation of Propane. ACS Catal. 2022, 12, 7368–7376. 10.1021/acscatal.2c01622. [DOI] [Google Scholar]; b Qiu B.; Lu W.-D.; Gao X.-Q.; Sheng J.; Yan B.; Ji M.; Lu A.-H. Borosilicate zeolite enriched in defect boron sites boosting the low-temperature oxidative dehydrogenation of propane. J. Catal. 2022, 408, 133–141. 10.1016/j.jcat.2022.02.017. [DOI] [Google Scholar]
- Altvater N. R.; Dorn R. W.; Cendejas M. C.; McDermott W. P.; Thomas B.; Rossini A. J.; Hermans I. B-MWW Zeolite: The Case Against Single-Site Catalysis. Angew. Chem., Int. Ed. 2020, 59, 6546–6550. 10.1002/anie.201914696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendejas M. C.; Dorn R. W.; McDermott W. P.; Lebrón-Rodríguez E. A.; Mark L. O.; Rossini A. J.; Hermans I. Controlled Grafting Synthesis of Silica-Supported Boron for Oxidative Dehydrogenation Catalysis. J. Phys. Chem. C 2021, 125, 12636–12649. 10.1021/acs.jpcc.1c01899. [DOI] [Google Scholar]
- a de Farias A. M. D.; Esteves A. M. L.; Ziarelli F.; Caldarelli S.; Fraga M. A.; Appel L. G. Boria modified alumina probed by methanol dehydration and IR spectroscopy. Appl. Surf. Sci. 2004, 227, 132–138. 10.1016/j.apsusc.2003.11.052. [DOI] [Google Scholar]; b Chaichana E.; Boonsinvarothai N.; Chitpong N.; Jongsomjit B. Catalytic dehydration of ethanol to ethylene and diethyl ether over alumina catalysts containing different phases with boron modification. J. Porous Mater. 2019, 26, 599–610. 10.1007/s10934-018-0663-7. [DOI] [Google Scholar]; c Delmastro A.; Gozzelino G.; Mazza D.; Vallino M.; Busca G.; Lorenzelli V. Characterization of microporous amorphous alumina–boria. J. Chem. Soc., Faraday Trans. 1992, 88, 2065–2070. 10.1039/FT9928802065. [DOI] [Google Scholar]
- a Forni L.; Fornasari G.; Tosi C.; Trifirò F.; Vaccari A.; Dumeignil F.; Grimblot J. Non-conventional sol–gel synthesis for the production of boron-alumina catalyst applied to the vapour phase Beckmann rearrangement. Appl. Catal., A 2003, 248, 47–57. 10.1016/S0926-860X(03)00147-9. [DOI] [Google Scholar]; b Kröcher O.; Elsener M. Hydrolysis and oxidation of gaseous HCN over heterogeneous catalysts. Appl. Catal., B 2009, 92, 75–89. 10.1016/j.apcatb.2009.07.021. [DOI] [Google Scholar]; c Curtin T.; McMonagle J. B.; Hodnett B. K. Influence of boria loading on the acidity of B2O3/Al2O3 catalysts for the conversion of cyclohexanone oxime to caprolactam. Appl. Catal., A 1992, 93, 91–101. 10.1016/0926-860X(92)80296-O. [DOI] [Google Scholar]
- Yang W.; Kim K. D.; O’Dell L. A.; Wang L.; Xu H.; Ruan M.; Wang W.; Ryoo R.; Jiang Y.; Huang J. Brønsted acid sites formation through penta-coordinated aluminum species on alumina-boria for phenylglyoxal conversion. J. Catal. 2022, 416, 375–386. 10.1016/j.jcat.2022.11.012. [DOI] [Google Scholar]
- a Lam J.; Szkop K. M.; Mosaferi E.; Stephan D. W. FLP catalysis: main group hydrogenations of organic unsaturated substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. 10.1039/C8CS00277K. [DOI] [PubMed] [Google Scholar]; b Stephan D. W. Diverse Uses of the Reaction of Frustrated Lewis Pair (FLP) with Hydrogen. J. Am. Chem. Soc. 2021, 143, 20002–20014. 10.1021/jacs.1c10845. [DOI] [PubMed] [Google Scholar]
- a Piers W. E.; Chivers T. Pentafluorophenylboranes: from obscurity to applications. Chem. Soc. Rev. 1997, 26, 345–354. 10.1039/cs9972600345. [DOI] [Google Scholar]; b Lawson J. R.; Melen R. L. Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. 10.1021/acs.inorgchem.6b02911. [DOI] [PubMed] [Google Scholar]
- Wanglee Y.-J.; Hu J.; White R. E.; Lee M.-Y.; Stewart S. M.; Perrotin P.; Scott S. L. Borane-Induced Dehydration of Silica and the Ensuing Water-Catalyzed Grafting of B(C6F5)3 To Give a Supported, Single-Site Lewis Acid, ≡SiOB(C6F5)2. J. Am. Chem. Soc. 2012, 134, 355–366. 10.1021/ja207838j. [DOI] [PubMed] [Google Scholar]
- Tian J.; Wang S.; Feng Y.; Li J.; Collins S. Borane-functionalized oxide supports: development of active supported metallocene catalysts at low aluminoxane loading. J. Mol. Catal. A: Chem. 1999, 144, 137–150. 10.1016/S1381-1169(98)00341-0. [DOI] [Google Scholar]
- Correa S. A.; Diaz-Droguett D. E.; Galland G. B.; Maraschin T. G.; De Sousa Basso N.; Dogan F.; Rojas R. S. Modification of rGO by B(C6F5)3 to generated single-site Lewis Acid rGO-O-B(C6F5)2 as co activator of nickel complex, to produce highly disperse rGO-PE nanocomposite. Appl. Catal., A 2019, 580, 149–157. 10.1016/j.apcata.2019.05.004. [DOI] [Google Scholar]
- a Horton T. A. R.; Wang M.; Shaver M. P. Polymeric frustrated Lewis pairs in CO2/cyclic ether coupling catalysis. Chem. Sci. 2022, 13, 3845–3850. 10.1039/D2SC00894G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yolsal U.; Horton T. A. R.; Wang M.; Shaver M. P. Cyclic Ether Triggers for Polymeric Frustrated Lewis Pair Gels. J. Am. Chem. Soc. 2021, 143, 12980–12984. 10.1021/jacs.1c06408. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vidal F.; McQuade J.; Lalancette R.; Jäkle F. ROMP-Boranes as Moisture-Tolerant and Recyclable Lewis Acid Organocatalysts. J. Am. Chem. Soc. 2020, 142, 14427–14431. 10.1021/jacs.0c05454. [DOI] [PubMed] [Google Scholar]; d Chen L.; Liu R.; Yan Q. Polymer Meets Frustrated Lewis Pair: Second-Generation CO2-Responsive Nanosystem for Sustainable CO2 Conversion. Angew. Chem., Int. Ed. 2018, 57, 9336–9340. 10.1002/anie.201804034. [DOI] [PubMed] [Google Scholar]; e Yolsal U.; Horton T. A. R.; Wang M.; Shaver M. P. Polymer-supported Lewis acids and bases: Synthesis and applications. Prog. Polym. Sci. 2020, 111, 101313 10.1016/j.progpolymsci.2020.101313. [DOI] [Google Scholar]
- a Zhang C.; Wang J.; Su W.; Lin Z.; Ye Q. Synthesis, Characterization, and Density Functional Theory Studies of Three-Dimensional Inorganic Analogues of 9,10-Diboraanthracene—A New Class of Lewis Superacids. J. Am. Chem. Soc. 2021, 143, 8552–8558. 10.1021/jacs.1c03057. [DOI] [PubMed] [Google Scholar]; b Akram M. O.; Tidwell J. R.; Dutton J. L.; Martin C. D. Tris(ortho-carboranyl)borane: An Isolable, Halogen-Free, Lewis Superacid. Angew. Chem., Int. Ed. 2022, 61, e202212073 10.1002/anie.202212073. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yruegas S.; Axtell J. C.; Kirlikovali K. O.; Spokoyny A. M.; Martin C. D. Synthesis of 9-borafluorene analogues featuring a three-dimensional 1,1′-bis(o-carborane) backbone. Chem. Commun. 2019, 55, 2892–2895. 10.1039/C8CC10087J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fujino A.; Ito S.-i.; Goto T.; Ishibiki R.; Kondo J. N.; Fujitani T.; Nakamura J.; Hosono H.; Kondo T. Hydrogenated Borophene Shows Catalytic Activity as Solid Acid. ACS Omega 2019, 4, 14100–14104. 10.1021/acsomega.9b02020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li Q.; Kolluru V. S. C.; Rahn M. S.; Schwenker E.; Li S.; Hennig R. G.; Darancet P.; Chan M. K. Y.; Hersam M. C. Synthesis of borophane polymorphs through hydrogenation of borophene. Science 2021, 371, 1143–1148. 10.1126/science.abg1874. [DOI] [PubMed] [Google Scholar]
- Bůžek D.; Škoch K.; Ondrušová S.; Kloda M.; Bavol D.; Mahun A.; Kobera L.; Lang K.; Londesborough M. G. S.; Demel J. ″Activated Borane″ - A Porous Borane Cluster Network as an Effective Adsorbent for Removing Organic Pollutants. Chem.—Eur. J. 2022, 28, e202201885 10.1002/chem.202201885. [DOI] [PubMed] [Google Scholar]
- Demel J.; Kloda M.; Lang K.; Škoch K.; Hynek J.; Opravil A.; Novotný M.; Bould J.; Ehn M.; Londesborough M. G. S. Direct Phenylation of nido-B10H14. J. Org. Chem. 2022, 87, 10034–10043. 10.1021/acs.joc.2c00997. [DOI] [PubMed] [Google Scholar]
- Hermanek S. Boron-11 NMR spectra of boranes, main-group heteroboranes, and substituted derivatives. Factors influencing chemical shifts of skeletal atoms. Chem. Rev. 1992, 92, 325–362. 10.1021/cr00010a007. [DOI] [Google Scholar]
- a Harazono T.; Hiroyama Y.; Watanabe T. Solid State NMR of 11B and 13C in Boron Carbide, B12C3 and 11B Enriched B12C3. Bull. Chem. Soc. Jpn. 1996, 69, 2419–2423. 10.1246/bcsj.69.2419. [DOI] [Google Scholar]; b Mauri F.; Vast N.; Pickard C. J. Atomic Structure of Icosahedral B4C Boron Carbide from a First Principles Analysis of NMR Spectra. Phys. Rev. Lett. 2001, 87, 085506 10.1103/PhysRevLett.87.085506. [DOI] [PubMed] [Google Scholar]
- Diaz M.; Jaballas J.; Arias J.; Lee H.; Onak T. 13C NMR Studies on Carboranes and Derivatives: Experimental/Calculational Correlations. J. Am. Chem. Soc. 1996, 118, 4405–4410. 10.1021/ja954089y. [DOI] [Google Scholar]
- Beckett M. A.; Strickland G. C.; Holland J. R.; Sukumar Varma K. A convenient n.m.r. method for the measurement of Lewis acidity at boron centres: correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity. Polymer 1996, 37, 4629–4631. 10.1016/0032-3861(96)00323-0. [DOI] [Google Scholar]
- a Lang S.; Benz M.; Obenaus U.; Himmelmann R.; Hunger M. Novel Approach for the Characterization of Lewis Acidic Solid Catalysts by Solid-State NMR Spectroscopy. ChemCatChem. 2016, 8, 2031–2036. 10.1002/cctc.201600372. [DOI] [Google Scholar]; b Wiper P. V.; Amelse J.; Mafra L. Multinuclear solid-state NMR characterization of the Brønsted/Lewis acid properties in the BP HAMS-1B (H-[B]-ZSM-5) borosilicate molecular sieve using adsorbed TMPO and TBPO probe molecules. J. Catal. 2014, 316, 240–250. 10.1016/j.jcat.2014.05.017. [DOI] [Google Scholar]; c Hradsky D.; Machac P.; Skoda D.; Leonova L.; Sazama P.; Pastvova J.; Kaucky D.; Vsiansky D.; Moravec Z.; Styskalik A. Catalytic performance of micro-mesoporous zirconosilicates prepared by non-hydrolytic sol-gel in ethanol-acetaldehyde conversion to butadiene and related reactions. Appl. Catal., A 2023, 652, 119037 10.1016/j.apcata.2023.119037. [DOI] [Google Scholar]
- a Britovsek G. J. P.; Ugolotti J.; White A. J. P. From B(C6F5)3 to B(OC6F5)3: Synthesis of (C6F5)2BOC6F5 and C6F5B(OC6F5)2 and Their Relative Lewis Acidity. Organometallics 2005, 24, 1685–1691. 10.1021/om049091p. [DOI] [Google Scholar]; b Beringhelli T.; Donghi D.; Maggioni D.; D’Alfonso G. Solution structure, dynamics and speciation of perfluoroaryl boranes through 1H, 11B and 19F NMR spectroscopy. Coord. Chem. Rev. 2008, 252, 2292–2313. 10.1016/j.ccr.2008.01.018. [DOI] [Google Scholar]; c Lewiński J.; Kubicki D.. NMR Spectroscopy, Heteronuclei, B, Al, Ga, In, Tl. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon J. C.; Tranter G. E.; Koppenaal D. W., Eds.; Academic Press: Oxford, 2017; pp 318–329. [Google Scholar]
- a Zhang X.; Wang R.; Yang X.; Zhang F. Comparison of four catalysts in the catalytic dehydration of ethanol to ethylene. Microporous Mesoporous Mater. 2008, 116, 210–215. 10.1016/j.micromeso.2008.04.004. [DOI] [Google Scholar]; b Xin H.; Li X.; Fang Y.; Yi X.; Hu W.; Chu Y.; Zhang F.; Zheng A.; Zhang H.; Li X. Catalytic dehydration of ethanol over post-treated ZSM-5 zeolites. J. Catal. 2014, 312, 204–215. 10.1016/j.jcat.2014.02.003. [DOI] [Google Scholar]; c Bi J.; Guo X.; Liu M.; Wang X. High effective dehydration of bio-ethanol into ethylene over nanoscale HZSM-5 zeolite catalysts. Catal. Today 2010, 149, 143–147. 10.1016/j.cattod.2009.04.016. [DOI] [Google Scholar]
- a Li Y.; Yang Q.; Yang J.; Li C. Mesoporous aluminosilicates synthesized with single molecular precursor (sec-BuO)2AlOSi(OEt)3 as aluminum source. Microporous Mesoporous Mater. 2006, 91, 85–91. 10.1016/j.micromeso.2005.11.021. [DOI] [Google Scholar]; b Styskalik A.; Kordoghli I.; Poleunis C.; Delcorte A.; Moravec Z.; Simonikova L.; Kanicky V.; Aprile C.; Fusaro L.; Debecker D. P. Hybrid mesoporous aluminosilicate catalysts obtained by non-hydrolytic sol–gel for ethanol dehydration. J. Mater. Chem. A 2020, 8, 23526–23542. 10.1039/D0TA07016E. [DOI] [Google Scholar]
- Bould J.; Clegg W.; Teat S. J.; Barton L.; Rath N. P.; Thornton-Pett M.; Kennedy J. D. An approach to megalo-boranes. Mixed and multiple cluster fusions involving iridaborane and platinaborane cluster compounds. Crystal structure determinations by conventional and synchrotron methods. Inorg. Chim. Acta 1999, 289, 95–124. 10.1016/S0020-1693(99)00071-7. [DOI] [Google Scholar]
- a Parks D. J.; Blackwell J. M.; Piers W. E. Studies on the Mechanism of B(C6F5)3-Catalyzed Hydrosilation of Carbonyl Functions. J. Org. Chem. 2000, 65, 3090–3098. 10.1021/jo991828a. [DOI] [PubMed] [Google Scholar]; b Piers W. E.; Marwitz A. J. V.; Mercier L. G. Mechanistic Aspects of Bond Activation with Perfluoroarylboranes. Inorg. Chem. 2011, 50, 12252–12262. 10.1021/ic2006474. [DOI] [PubMed] [Google Scholar]; c Rendler S.; Oestreich M. Conclusive Evidence for an SN2-Si Mechanism in the B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Compounds: Implications for the Related Hydrogenation. Angew. Chem., Int. Ed. 2008, 47, 5997–6000. 10.1002/anie.200801675. [DOI] [PubMed] [Google Scholar]; d Oestreich M.; Hermeke J.; Mohr J. A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. 10.1039/C4CS00451E. [DOI] [PubMed] [Google Scholar]
- a Parks D. J.; Piers W. E. Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters. J. Am. Chem. Soc. 1996, 118, 9440–9441. 10.1021/ja961536g. [DOI] [Google Scholar]; b Fang H.; Oestreich M. Defunctionalisation catalysed by boron Lewis acids. Chem. Sci. 2020, 11, 12604–12615. 10.1039/D0SC03712E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prabhudesai V. S.; Gurrala L.; Vinu R. Catalytic Hydrodeoxygenation of Lignin-Derived Oxygenates: Catalysis, Mechanism, and Effect of Process Conditions. Energy Fuels 2022, 36, 1155–1188. 10.1021/acs.energyfuels.1c02640. [DOI] [Google Scholar]; b Kumar A.; Jindal M.; Maharana S.; Thallada B. Lignin Biorefinery: New Horizons in Catalytic Hydrodeoxygenation for the Production of Chemicals. Energy Fuels 2021, 35, 16965–16994. 10.1021/acs.energyfuels.1c01651. [DOI] [Google Scholar]; c Perego C.; Bosetti A.; Ricci M.; Millini R. Zeolite Materials for Biomass Conversion to Biofuel. Energy Fuels 2017, 31, 7721–7733. 10.1021/acs.energyfuels.7b01057. [DOI] [Google Scholar]; d Shi Y.; Xing E.; Wu K.; Wang J.; Yang M.; Wu Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. 10.1039/C7CY00574A. [DOI] [Google Scholar]; e Tang X.; Ding W.; Li H. Improved hydrodeoxygenation of bio-oil model compounds with polymethylhydrosiloxane by Brønsted acidic zeolites. Fuel 2021, 290, 119883 10.1016/j.fuel.2020.119883. [DOI] [Google Scholar]; f Witsuthammakul A.; Sooknoi T. Selective hydrodeoxygenation of bio-oil derived products: ketones to olefins. Catal. Sci. Technol. 2015, 5, 3639–3648. 10.1039/C5CY00367A. [DOI] [Google Scholar]
- a Christensen D. B.; Mortensen R. L.; Kramer S.; Kegnæs S. Study of CoCu Alloy Nanoparticles Supported on MOF-Derived Carbon for Hydrosilylation of Ketones. Catal. Lett. 2020, 150, 1537–1545. 10.1007/s10562-019-03065-2. [DOI] [Google Scholar]; b Le Roux E.; De Mallmann A.; Merle N.; Taoufik M.; Anwander R. Immobilization of Heteroleptic Bis(oxazoline) Zinc Catalysts on SBA-15 for Asymmetric Hydrosilylation. Organometallics 2015, 34, 5146–5154. 10.1021/acs.organomet.5b00714. [DOI] [Google Scholar]; c Lázaro G.; Fernández-Alvarez F. J.; Iglesias M.; Horna C.; Vispe E.; Sancho R.; Lahoz F. J.; Iglesias M.; Pérez-Torrente J. J.; Oro L. A. Heterogeneous catalysts based on supported Rh–NHC complexes: synthesis of high molecular weight poly(silyl ether)s by catalytic hydrosilylation. Catal. Sci. Technol. 2014, 4, 62–70. 10.1039/C3CY00598D. [DOI] [Google Scholar]
- Keess S.; Simonneau A.; Oestreich M. Direct and Transfer Hydrosilylation Reactions Catalyzed by Fully or Partially Fluorinated Triarylboranes: A Systematic Study. Organometallics 2015, 34, 790–799. 10.1021/om501284a. [DOI] [Google Scholar]
- Mahdi T.; Stephan D. W. Facile Protocol for Catalytic Frustrated Lewis Pair Hydrogenation and Reductive Deoxygenation of Ketones and Aldehydes. Angew. Chem., Int. Ed. 2015, 54, 8511–8514. 10.1002/anie.201503087. [DOI] [PubMed] [Google Scholar]
- Chang C. D.; Silvestri A. J. The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts. J. Catal. 1977, 47, 249–259. 10.1016/0021-9517(77)90172-5. [DOI] [Google Scholar]
- a Korstanje T. J.; Jastrzebski J. T. B. H.; Klein Gebbink R. J. M. Catalytic Dehydration of Benzylic Alcohols to Styrenes by Rhenium Complexes. ChemSusChem 2010, 3, 695–697. 10.1002/cssc.201000055. [DOI] [PubMed] [Google Scholar]; b Bertero N. M.; Trasarti A. F.; Apesteguía C. R.; Marchi A. J. Liquid-phase dehydration of 1-phenylethanol on solid acids: Influence of catalyst acidity and pore structure. Appl. Catal., A 2013, 458, 28–38. 10.1016/j.apcata.2013.03.018. [DOI] [Google Scholar]
- a Styskalik A.; Vykoukal V.; Fusaro L.; Aprile C.; Debecker D. P. Mildly acidic aluminosilicate catalysts for stable performance in ethanol dehydration. Appl. Catal., B 2020, 271, 118926 10.1016/j.apcatb.2020.118926. [DOI] [Google Scholar]; b Wang Z.; O’Dell L. A.; Zeng X.; Liu C.; Zhao S.; Zhang W.; Gaborieau M.; Jiang Y.; Huang J. Insight into Three-Coordinate Aluminum Species on Ethanol-to-Olefin Conversion over ZSM-5 Zeolites. Angew. Chem., Int. Ed. 2019, 58, 18061–18068. 10.1002/anie.201910987. [DOI] [PubMed] [Google Scholar]
- a Phung T. K.; Proietti Hernández L.; Lagazzo A.; Busca G. Dehydration of ethanol over zeolites, silica alumina and alumina: Lewis acidity, Brønsted acidity and confinement effects. Appl. Catal., A 2015, 493, 77–89. 10.1016/j.apcata.2014.12.047. [DOI] [Google Scholar]; b Phung T. K.; Lagazzo A.; Rivero Crespo M. Á.; Sánchez Escribano V.; Busca G. A study of commercial transition aluminas and of their catalytic activity in the dehydration of ethanol. J. Catal. 2014, 311, 102–113. 10.1016/j.jcat.2013.11.010. [DOI] [Google Scholar]
- Neelakandeswari N.; Karvembu R.; Dharmaraj N. Mesoporous Nickel–Aluminosilicate Nanocomposite: A Solid Acid Catalyst for Ether Synthesis. J. Nanosci. Nanotechnol. 2013, 13, 2853–2863. 10.1166/jnn.2013.7419. [DOI] [PubMed] [Google Scholar]
- a Amoureux J.-P.; Fernandez C.; Steuernagel S. ZFiltering in MQMAS NMR. J. Magn. Reson., Ser. A 1996, 123, 116–118. 10.1006/jmra.1996.0221. [DOI] [PubMed] [Google Scholar]; b Equbal A.; Bjerring M.; Madhu P. K.; Nielsen N. C. Improving spectral resolution in biological solid-state NMR using phase-alternated rCW heteronuclear decoupling. Chem. Phys. Lett. 2015, 635, 339–344. 10.1016/j.cplett.2015.07.008. [DOI] [Google Scholar]; c Brus J. Heating of samples induced by fast magic-angle spinning. Solid State Nucl. Magn. Reson. 2000, 16, 151–160. 10.1016/S0926-2040(00)00061-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.