

Abstract
An NAD-malic enzyme was purified to homogeneity from Bradyrhizobium japonicum A1017, and its molecular characteristics were surveyed. The enzyme exhibited native and subunit molecular masses of 388 and 85 kDa, respectively, suggesting that it exists as a homotetramer, and was activated by metabolic intermediates in glycolysis. The role of the enzyme in bacteroids’ carbon metabolism is discussed.
Malic enzymes (MEs [EC 1.1.38–40]) catalyze an anaplerotic reaction supplying pyruvate from malate and are supposed to function in continuous operation of the trichloroacetic acid cycle. MEs have been isolated from various sources, and their characteristics have been surveyed extensively by many researchers (9, 13, 14, 16, 20).
In legume nodules, MEs in the bacteroids are believed to play a unique physiological role in supporting the nitrogenase reaction (5, 17). Clear genetic evidences for the significance of the MEs for the nitrogenase reaction of bacteroids was recently reported by Driscoll and Finan (6, 7), who showed that an NAD-ME-defective mutant of Rhizobium meliloti was Nod+ and Fix−, and conversely, that an NADP-ME-defective mutant was Nod+ and Fix+. These data indicate that the NAD-ME reaction is a principal step in support of the nitrogenase reaction.
In Bradyrhizobium japonicum, however, genetic evidence for the roles of NAD-ME and NADP-ME in bacteroids remains obscure. Only partially purified samples of NAD-ME and NADP-ME have been available for characterization of biochemical activity (4, 19). In a previous study, we purified NADP-ME from B. japonicum A1017 to homogeneity, and its molecular characteristics were surveyed (3). In the present study, NAD-ME was first purified to homogeneity from B. japonicum A1017, and the purified protein was characterized biochemically to elucidate its physiological role in nodule bacteroids. The N-terminal amino acid sequence of NAD-ME was also analyzed to prepare DNA probes for gene cloning.
B. japonicum A1017 was supplied from a stock culture of the Institute of Agrobiological Sciences (Tsukuba, Japan) and was cultured in a liquid medium (10 liters) containing CaSO4 · 2H2O (0.1 g), MgSO4 · 7H2O (0.1 g), KH2PO4 (0.3 g), K2HPO4 (0.7 g), NaCl (0.2 g), sucrose (1 g), yeast extract (1 g), Polypepton (5 g), and DL-ME (1 g) per liter. After 5 days at 24°C with continuous aeration, the cells were harvested by centrifugation (12,000 × g for 30 min).
NAD-ME activity was determined spectrophotometrically at 340 nm according to the method described by Ochoa (15). The standard reaction mixture contained 50 mM HEPES, 6 mM l-malate, 20 mM KCl, 2 mM MnCl2, 0.75 mM EDTA, and 1 mM NAD+ at pH 7.3. One unit of the enzyme was defined as the amount of enzyme that catalyzed the reduction of 1 nmol of NAD+ per min under a standard assay system. Amounts of protein were determined as reported by Bradford with bovine serum albumin as the standard (2).
A summary of the purification steps is shown in Table 1. The harvested cells were resuspended in buffer A (50 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM MgSO4, 5 mM dithiothreitol, 5% glycerol) and broken by sonication (Branson model 250/450) at an output of 10% for 3 h at 4°C. After an ammonium sulfate precipitation (50 to 60%) and dialysis, the protein solution was loaded onto a MonoQ column (HR10/10; Pharmacia) with buffer B (50 mM Tris-HCl [pH 7.5], 50 mM KCl, 2 mM MgCl2, 3 mM MnCl2, 5 mM dithiothreitol) and eluted with a linear gradient of KCl from 50 mM to 1 M. The active fractions were applied to an HW55 column (Tosoh Co., Tokyo, Japan) for gel filtration. Buffer C (50 mM Tris-HCl [pH 7.5], 2 mM MgCl2, 3 mM MnCl2, 5 mM dithiothreitol, 10 mM KCl) was used to equilibrate the column and to elute the enzyme. For the final purification step, a 5′AMP-Sepharose 4B column (80 mm by 10 mm; Pharmacia) was used with buffer D (50 mM Tris-HCl [pH 7.5]). The enzyme fraction was eluted with an increasing NAD+ gradient from 0 to 5 mM. The enzyme was eluted as a single peak.
TABLE 1.
Purification of NAD-ME from B. japonicum A1017
| Purification step | Amt of protein (mg) | Total activity (U) | Sp act (U/mg) | Purification factor (fold) | Recovery (%) |
|---|---|---|---|---|---|
| Crude extract | 438 | 65.8 | 0.15 | 1.00 | 100 |
| 50–60% saturation ppt | 14.6 | 31.5 | 2.16 | 14.4 | 47.9 |
| MonoQ column | 1.20 | 9.12 | 7.60 | 50.7 | 13.9 |
| HW55 column | 0.037 | 10.6 | 285 | 1,902 | 16.1 |
| 5′-AMP-Sepharose column | 0.020 | 6.02 | 301 | 2,007 | 9.15 |
Approximately 20 μg of purified NAD-ME with a specific enzyme activity of 301 nmol of NAD+ reduction/min/mg of protein was obtained from 6 g (wet weight) of B. japonicum A1017 cells. The purified enzyme was shown to be homogeneous by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1), which was performed according to the methods described by Laemmli (11).
FIG. 1.
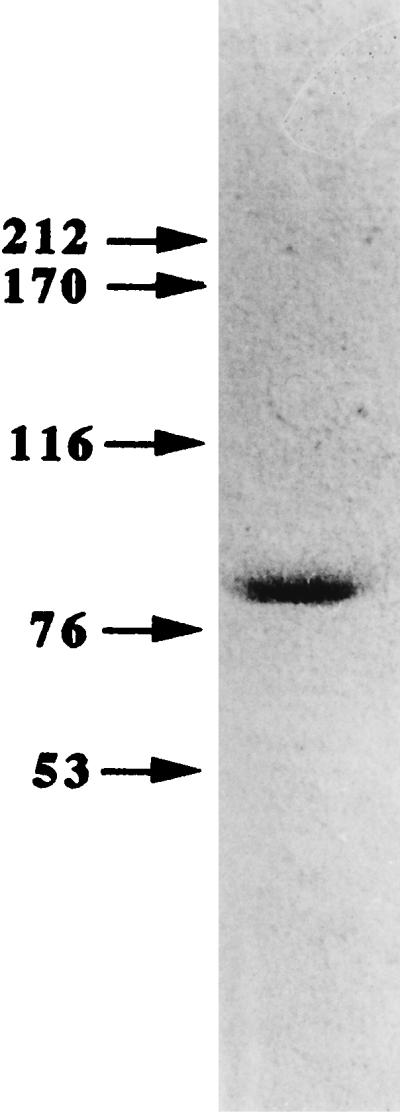
SDS-PAGE of NAD-ME from B. japonicum A1017. The numbers on the left indicate molecular masses (in kilodaltons). The purified enzyme (0.5 μg) was loaded onto the gel, and protein in the gel was detected by silver staining (3).
The molecular mass of the enzyme was estimated by high-performance liquid chromatography (Superose 6 HR10/30; Pharmacia) with the following standard proteins: cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), and apoferritin (445 kDa). The molecular mass of native NAD-ME was estimated to be 388,000 Da in 0.05 M phosphate buffer and 0.15 M NaCl (pH 7.2). By SDS-PAGE assays with standard proteins from Pharmacia’s High Molecular Weight kit, NAD-ME was determined to be composed of a single subunit of approximately 85,000 Da, suggesting that the NAD-ME is a tetramer. Similar oligometric structures have been observed in NAD-MEs from other bacteria. The molecular masses of subunits of NAD-ME of Bacillus stearothermophilus (10) and of Escherichia coli (21) were reported to be 50,000 and 52,000, respectively, and their native forms showed much higher values (200,000 and 203,000, respectively), suggesting their tetrameric structure.
The N-terminal amino acid sequence of the purified protein was identified as MEEQLDLQLKPKVAEFLN by the automated Millipore Prosequencer (Waters, Milford, Mass.). No similarity was detected by a BLAST search with the terminal amino acid sequences of other MEs (1).
The Kms of NAD-ME against malate and NAD+ were calculated to be 0.15 and 0.36 mM, respectively, by the Lineweaver-Burk plot (data not shown). All enzyme assays were done in triplicate. The concentrations of l-malate used were 0.05 to 20 mM. The assay system contained 2 mM Mn2+ and 1 mM NAD+ for assaying at maximum activity. The Km obtained was significantly smaller than those of other B. japonicum strains (4, 5, 19), suggesting that B. japonicum A1017 may express a different type of NAD-ME. In Streptococcus bovis (0.63 mM [9]) and E. coli (0.4 mM [21]), the value of NAD-ME for l-malate were similar to that of B. japonicum A1017. In contrast, the affinity for NAD+ of NAD-ME from B. japonicum A1017 was lower than those of other bacterial NAD-MEs (9, 21). The maximum enzyme activity was obtained at 0.8 mM NAD+ but continued to increase slightly (2 to 5%) until the NAD+ concentration attained 2 mM. When the reaction mixture contained NADP+ (1 mM) instead of NAD+ (1 mM), the enzyme activity decreased to 17% of the value under standard assay conditions.
A divalent cation was strictly required for NAD-ME activity. The stimulating effects on enzyme activity by various divalent cations were analyzed at various concentrations (0 to 10 mM). The most stimulating effect was obtained by adding Mn2+ to the assay system with 6 mM l-malate, and the enzyme activity attained its plateau at 2 mM Mn2+. When the assay system contained Mg2+, the maximum effect was observed at 5 mM, although the activity was 50% of the value when the assay system contained 2 mM Mn2+. Other divalent cations (Cu2+, Ca2+, Fe2+, and Zn2+) showed much less stimulating effects. The addition of 1 mM EDTA did not affect NAD-ME activity when 2 mM MnCl2 was also added to the assay system with 6 mM l-malate.
Most metabolic intermediates involved in the citric acid cycle performed as inhibitors of NAD-ME, except succinate, amino-n-butyrate, and hydroxy-n-butyrate (Tables 2 and 3). Similar inhibition of NAD-ME activity by fumarate at a concentration of 1 mM was reported for Ascaris sp. (14). Malonate and pyruvate inhibited the activity by approximately 60% at 5 mM. ATP and AMP inhibited the activity at high concentrations (>1.5 mM). Coenzyme A (CoA), NADH, and NADPH were strong inhibitors of NAD-ME, but acetyl-CoA had no obvious effect on NAD-ME activity (Table 3).
TABLE 2.
Effects of metabolic intermediates on NAD-ME activity from B. japonicum A1017a
| Compound | Relative activity (%) at the following concn:
|
|
|---|---|---|
| 1 mM | 5 mM | |
| Control | 100 | 100 |
| Succinate | 103 | 121 |
| 2-Ketoglutarate | 83 | 73 |
| dl-Isocitrate | 65 | 64 |
| Malonate | 94 | 41 |
| Tartarate | 95 | 78 |
| Hydroxypyruvate | 110 | |
| γ-Amino-n-butyrate | 121 | 121 |
| Hydroxy-n-butyrate | 134 | 121 |
| Pyruvate | 76 | 38 |
| l-Glutarate | 112 | 107 |
| l-Aspartate | 97 | 48 |
| d-Fructose-6-phosphate | 191 | 211 |
| a-d-glucose-1-phosphate | 189 | 170 |
| d-Glucose-6-phosphate | 242 | 268 |
| d-Fructose-1,6-diphosphate | 155 | 209 |
| Uridine-5′-diphosphoglucose | 160 | 175 |
| Adenosine-5′-diphosphoglucose | 123 | 161 |
| AMP | 82 | 65 |
| Phosphoenolpyruvate | 70 | 70 |
The enzyme was equilibrated in assay mixtures containing 50 mM HEPES, 6 mM l-malate, 20 mM KCl, 2 mM MnCl2, and 0.75 mM EDTA at pH 7.3. The enzyme reaction was initiated by the addition of 1 mM NAD+. The results are percentages of the control values (293 ± 8 nmol min−1 mg protein−1) and were determined from the means of triplicate measurements for each sample.
TABLE 3.
Effects of metabolic intermediates on NAD-ME activity from B. japonicum A1017
| Compound | Concn (mM) | Relative activity (%) |
|---|---|---|
| Control | 100 | |
| Fumarate | 1.0 | 72 |
| 3.0 | 66 | |
| 5.0 | 73 | |
| 50 | 36 | |
| 100 | 18 | |
| ATP | 0.5 | 112 |
| 1.0 | 120 | |
| 1.5 | 102 | |
| 2.0 | 97 | |
| 3.0 | 61 | |
| 5.0 | 28 | |
| 3-Acetylpyridine adenine dinucleotide | 0.25 | 58 |
| 1.0 | 106 | |
| 5.0 | 165 | |
| CoA | 0.1 | 78 |
| 0.5 | 78 | |
| 1.0 | 66 | |
| 5.0 | 56 | |
| Acetyl-CoA | 0.05 | 103 |
| 0.1 | 107 | |
| 1.0 | 90 | |
| NADH | 0.05 | 63 |
| 0.1 | 51 | |
| NADPH | 0.05 | 58 |
| 0.1 | 56 |
Interestingly, glutamate and various metabolic intermediates in glycolysis stimulated NAD-ME activity (Table 2). Especially, d-glucose-6-phosphate, d-fructose-6-phosphate, and d-fructose-1,6-diphosphate greatly stimulated enzyme activity even at a concentration of 1 mM. No contaminating enzyme activities that cause NADH formation with sugar phosphates were observed in the purified enzyme. To our knowledge, this is the first report of such significant activation of NAD-ME. This activation was not observed in the case of NADP-ME of B. japonicum A1017 (3).
We previously reported low NADH-to-NAD+ and high NADPH-to-NADP+ ratios in bacteroids of B. japonicum (18), suggesting that the condition is inhibitory for pyruvate formation of NADP-ME. The data concerning the characteristics of NAD-ME that were observed, such as no inhibition by acetyl-CoA and significant activation by metabolic intermediates in glycolysis, suggested that NAD-ME can operate to supply pyruvate from malate continuously to form acetyl-CoA in order to maintain citric acid cycle operation, lipid biosynthesis, and polysaccharide formation through gluconeogenesis in B. japonicum bacteroids when the supply of photosynthate is high enough. However, in R. meliloti, acetyl-CoA inhibited the activity of NAD-ME, and no effect on NADP-ME activity was observed (8). This suggests that the physiological roles of NAD-ME and NADP-ME are different in R. meliloti and B. japonicum.
Acknowledgments
This work was supported by a grant from the Ministry of Agriculture and Fisheries, Tokyo, Japan.
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Chen F, Okabe Y, Osano K, Tajima S. Purification and characterization of the NADP-malic enzyme from Bradyrhizobium japonicum A1017. Biosci Biotech Biochem. 1997;61:384–386. doi: 10.1271/bbb.61.384. [DOI] [PubMed] [Google Scholar]
- 4.Copeland L, Quinnell R G, Day D A. Malic enzyme activity in bacteroids from soybean nodules. J Gen Microbiol. 1989;135:2005–2011. [Google Scholar]
- 5.Day D A, Quinnell R G, Bergersen F J. An hypothesis on the role of malic enzyme in symbiotic nitrogen fixation in soybean nodules. In: Graham P H, Sadowsky M J, Vance C P, editors. Symbiotic nitrogen fixation. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1994. pp. 159–164. [Google Scholar]
- 6.Driscoll B T, Finan T M. NAD+-dependent malic enzyme of Rhizobium meliloti is required for symbiotic nitrogen fixation. Mol Microbiol. 1993;7:856–873. doi: 10.1111/j.1365-2958.1993.tb01177.x. [DOI] [PubMed] [Google Scholar]
- 7.Driscoll B T, Finan T M. NADP+-dependent malic enzyme of Rhizobium meliloti. J Bacteriol. 1996;178:2224–2231. doi: 10.1128/jb.178.8.2224-2231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Driscoll B T, Finan T M. Properties of NAD+- and NADP+-dependent malic enzymes of Rhizobium (Sinorhizobium) meliloti and differential expression of their genes in nitrogen-fixing bacteroids. Microbiology. 1997;143:489–498. doi: 10.1099/00221287-143-2-489. [DOI] [PubMed] [Google Scholar]
- 9.Kawai S, Suzuki H, Yamamoto K, Inui M, Yukawa H, Kumagai H. Purification and characterization of a malic enzyme from the ruminal bacterium Streptococcus bovis ATCC 15352 and cloning and sequencing of its gene. Appl Environ Microbiol. 1996;62:2692–2700. doi: 10.1128/aem.62.8.2692-2700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi K, Doi S, Negoro S, Urabe I, Okada H. Structure and properties of malic enzyme from Bacillus stearothermophilus. J Biol Chem. 1989;264:3200–3205. [PubMed] [Google Scholar]
- 11.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 12.Lai C, Harris B G, Cook P F. Mechanism of activation of the NAD-malic enzyme from Ascaris ssum by fumarate. Arch Biochem Biophys. 1992;299:214–219. doi: 10.1016/0003-9861(92)90266-y. [DOI] [PubMed] [Google Scholar]
- 13.Murai T, Tokushige M, Nagai J, Katsuki H. Physiological functions of NAD- and NADP-linked malic enzymes in Escherichia coli. Biochem Biophys Res Commun. 1971;43:875–881. doi: 10.1016/0006-291x(71)90698-x. [DOI] [PubMed] [Google Scholar]
- 14.Nagel W O, Sauer L A. Mitochondrial malic enzymes. J Biol Chem. 1982;257:12405–12411. [PubMed] [Google Scholar]
- 15.Ochoa S. Malate dehydrogenase from pig heart. Methods Enzymol. 1955;1:735. [Google Scholar]
- 16.Spina J, Jr, Bright H J, Rosenbloom J. Purification and properties of L-malic enzyme from Escherichia coli. Biochemistry. 1970;9:3794–3801. [Google Scholar]
- 17.Tajima S, Kimura I, Kouzai K, Kasai T. Succinate degradation through the critric acid cycle in Bradyrhizobium japonicum J501 bacteroids under low oxygen concentration. Agric Biol Chem. 1990;54:891–897. [Google Scholar]
- 18.Tajima S, Kouzai K. Nucleotide pools in soybean nodule tissues. A survey of NAD(P)/NAD(P)H ratios and energy charge. Plant Cell Physiol. 1989;30:589–593. [Google Scholar]
- 19.Tomaszewska B, Werner D. Purification and properties of NAD- and NADP-depedent malic enzymes from Bradyrhizobium japonicum bacteroids. J Plant Physiol. 1995;146:591–595. [Google Scholar]
- 20.Winning B M, Bourguignon J, Leaver C J. Plant mitochondrial NAD+-dependent malic enzyme. J Biol Chem. 1994;269:4780–4786. [PubMed] [Google Scholar]
- 21.Yamaguchi M, Tokushige M, Katsuki H. Studies on regulatory functions of malic enzymes. J Biochem. 1973;73:169–180. [PubMed] [Google Scholar]