

Abstract
Genetic variation around the LRRK2 gene affects risk for both familial and sporadic Parkinson’s disease (PD). LRRK2 levels have become an appealing target for potential PD therapeutics with LRRK2 antisense oligonucleotides (ASOs) now moving toward clinical trials. However, LRRK2 has been suggested to play a fundamental role in peripheral immunity, and it is currently unknown if targeting increased LRRK2 levels in peripheral immune cells will be beneficial or deleterious. Here it was observed that G2019S macrophages exhibited increased stimulation-dependent lysosomal tubule formation (LTF) and MHC-II trafficking from the perinuclear lysosome to the plasma membrane in an mTOR-dependent manner with concomitant increases in pro-inflammatory cytokine release. Both ASO-mediated knockdown of mutant Lrrk2 and LRRK2 kinase inhibition ameliorated this phenotype and decreased these immune responses in control cells. Given the critical role of antigen presentation, lysosomal function, and cytokine release in macrophages, it is likely LRRK2-targeting therapies with systemic activity may have therapeutic value with regard to mutant LRRK2, but deleterious effects on the peripheral immune system, such as altered pathogen control in these cells, should be considered when reducing levels of non-mutant LRRK2.
Keywords: MT: Oligonucleotides: Therapies and Applications, antisense oligonucleotide, LRRK2, antigen presentation, lysosomal tubules, immune response
Graphical abstract
Tansey and colleagues identified a hyperinflammatory phenotype in macrophages associated with the Parkinson’s disease-associated G2019S Lrrk2 mutation. This was ameliorated with the use of Lrrk2-targeting ASOs and Lrrk2 kinase inhibitors. Furthermore, a novel Lrrk2-mediated mechanism was unveiled, demonstrating that Lrrk2 modulates antigen presentation via lysosomal tubule formation.
Introduction
Parkinson’s disease (PD) is a common progressive neurodegenerative disease, affecting about 1%–2% of the population older than 65 years.1 The prevalence of PD is expected to increase 2-fold by 2030.2 In addition, it is estimated that projected total economic burden will surpass $79 billion by 2037,3 highlighting the need for interventions that could delay disease progression. The fact that there are currently no disease-modifying drugs for people with PD indicates that knowledge gaps still need to be closed to identify ways to cure or prevent this disease.
The most prevalent LRRK2 mutation, G2019S, resides in the kinase domain and causes a 2- to 3-fold increase in kinase activity.4,5 Furthermore, increased LRRK2 mRNA and protein levels have been observed in B cells, T cells, non-classical CD16+ monocytes6 and neutrophils7 of patients with sporadic PD compared with age-matched healthy controls. LRRK2 levels have therefore become an appealing target for potential PD therapeutics, with LRRK2 antisense oligonucleotides (ASOs) now in clinical trials. Indeed, in a preclinical mouse model, administration of Lrrk2 ASOs to the brain reduces LRRK2 protein levels and fibril-induced α-synuclein inclusions.8 Similarly, the use of an ASO that blocks splicing of LRRK2 exon 41, which encodes part of the kinase domain, reverses aberrant endoplasmic reticulum (ER) calcium levels and mitophagy defects in PD patient-derived cell lines harboring the LRRK2 G2019S mutation.9,10 The administration of Lrrk2 ASOs have also been shown to rescue aberrant Rab10 phosphorylation levels and autophagic processing in the brains of mice expressing human G2019S-LRRK2.11 Although these early studies show promising results with LRRK2 ASO in the brain, relatively little is known about the potential effects of targeting LRRK2 levels in the periphery, where it is highly expressed in immune cells.6,12
It has been shown that LRRK2 levels increase in immune cells upon immune cell activation.6,13 However, whether LRRK2 expression increases in peripheral immune cells to dampen or promote inflammation is still unknown. Of note, complete abolition of LRRK2 kinase activity in the peripheral immune system leads to deleterious effects in Lrrk2-knockout (KO) models, with increased risk for infection and decreased pathogen control.12,14,15,16 This immune dysfunction may be mediated by lysosomal defects, as it has recently been demonstrated that a mouse Lrrk2-KO macrophage cell line displays vacuolization and lipofuscin autofluorescence upon lysosomal overload stress.17 On the basis of this, it can be inferred that LRRK2 may modulate lysosomal function in peripheral immune cells and increases in expression to regulate inflammation.
LRRK2 was first shown to colocalize to endosomal-autophagic structures and LC3 and p62-positive puncta in human embryonic kidney 293 (HEK293) cells transfected with a bacterial artificial chromosome (BAC)-YPet-LRRK2-WT (wild-type) construct.18 It was subsequently demonstrated that upon Toll-like receptor 4 (TLR4) stimulation with lipopolysaccharide (LPS), LRRK2 levels increase and the protein is recruited to endolysosomal membranes whereby it regulates the autophagy pathway in RAW264.7 macrophage cells.19 Such data suggest that LRRK2 may increase in response to inflammatory stimuli to mediate lysosomal function. Interestingly, LRRK2 has recently been shown to mediate tubulation and vesicle sorting from lysosomes in astrocytes.20 Bonet-Ponce and colleagues demonstrated that upon lysosomal membrane rupturing, LRRK2 is recruited to the lysosomal membrane whereby it mediates the formation of lysosomal tubules and release of lysosomal contents, with G2019S-Lrrk2 expression significantly increasing the formation of these tubules in astrocytes. Interestingly, it is known that lysosomal tubulation is usually observed in macrophages and other professional phagocytes undergoing immune activation.21 Lysosomal tubules are crucial for two immune-related functions upon immune activation; phagocytosis and antigen presentation.21,22 This is intriguing as LRRK2 has been heavily implicated in modulating phagocytosis23 and LRRK2 expression is also positively correlated with HLA-DR expression in human monocytes,6,13 suggesting a potential role of LRRK2 in antigen presentation. It is therefore possible that LRRK2 mediates inflammatory responses, such as antigen presentation and phagocytosis, via lysosomal tubulation.
The aim of this study is 2-fold: to assess the effects of Lrrk2 knockdown via ASOs on immune cell responses and lysosomal function and to test the hypothesis that LRRK2 mediates inflammatory responses, specifically antigen presentation, via the formation of lysosomal tubules. Here, we use Lrrk2 ASOs and Lrrk2 kinase inhibitors to investigate a novel mechanism that may link the role of LRRK2 at the lysosome to its role in inflammation and antigen presentation. As both strategies are being evaluated in clinical trials with PD patients,8 our inclusion of Lrrk2 knockdown and targeting of its kinase activity provides vital information on the potentially deleterious effects of reduced LRRK2 activity in peripheral immune cells. We investigated this in BAC transgenic mouse strains overexpressing either mouse WT Lrrk2 (WTOE) or mutant G2019S-Lrrk224,25 and B6 controls. BAC overexpressers were chosen for the purpose of this study for the potential to mimic the increase in LRRK2 expression in peripheral immune cells that is seen in sporadic PD patients.6 Peritoneal macrophages (pMacs) were isolated from these mice to examine the effects of Lrrk2 over-expression and mutation effects on macrophage cell function. pMacs are not a homogeneous population of cells, but rather a mix of small pMacs (SPMs) and large pMacs (LPMs). LPMs are resident to the peritoneal cavity and are traditionally thought of as anti-inflammatory, phagocytic, and responsible for the presentation of exogenous antigens.26 SPMs, on the other hand, are generated from bone-marrow-derived myeloid precursors which migrate to the peritoneal cavity in response to infection, inflammatory stimuli, or thioglycolate, and present a pro-inflammatory functional profile.26 We observe an increase in stimulation-dependent antigen presentation, and cytokine release in pMacs from G2019S BAC mice relative toWTOE and B6 controls. We also observe alterations in lysosomal function, with increased pan-cathepsin activity and degradative capacity of lysosomes in G2019S pMacs early in the inflammatory response. Knockdown of Lrrk2, as well as Lrrk2 kinase inhibition, successfully ameliorate these phenotypes, and NanoString nCounter-based transcriptomic profiling suggest altered vesicular trafficking, lysosomal positioning, and autophagy activity may underlie the effects of Lrrk2 knockdown on antigen presentation. Indeed, it was observed that G2019S pMacs exhibited increased stimulation-dependent lysosomal tubule formation (LTF) and MHC-II trafficking from the perinuclear lysosome to the plasma membrane in an mTOR-dependent manner.
Results
G2019S BAC transgenic pMacs exhibit increased antigen presentation and cytokine release
LRRK2 protein expression has previously been shown to increase in immune cells from both murine preclinical models and patient cells in response to inflammatory stimuli.6,13,27,28,29 To determine if Lrrk2 increases in response to such stimulus in pMacs, Lrrk2 protein levels were assessed in pMacs treated with 100 U IFNγ for 18 h. An increase in Lrrk2 levels was observed in all three genotypes upon IFNγ treatment (Figures 1A and 1B). Furthermore, significantly more Lrrk2 is present in WTOE and G2019S pMacs relative to B6 pMacs in both vehicle- and IFNγ-treated pMacs, with no significant difference seen between the two BAC models. Furthermore, we observed that IFNγ treatment caused a significant increase in phosphorylated Lrrk2 in all genotypes, indicative of increased kinase activity levels,30 with significantly higher levels of phosphorylated Lrrk2 observed in G2019S pMacs relative to the other genotypes (Figures S1A and S1B). Co-treatment with 100 nM of the LRRK2 kinase inhibitor, PF-06685360 (PF360), significantly reduced phosphorylated Lrrk2 levels in all genotypes and prevented stimulation-dependent increase (Figure S1B).
Figure 1.

G2019S BAC transgenic pMacs exhibit increased antigen presentation and cytokine release
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were stimulated with 100 U IFNγ for 18 h. (A and B) Lrrk2 protein levels were assessed and normalized to total protein levels and quantified. Representative western blots are shown. (C and D) Surface MHC-II geometric mean of fluorescence intensity (GMFI) GMFI was quantified on LPMs and SPMs using flow cytometry. (E–G) Levels of the cytokines TNF, IL-4, and IL-10 in medium were assessed, normalized to total protein levels and quantified. (H) Cells were co-treated with 100 nM PF360 or 50 nM bafilomycin A1 (baf), and YAe MFI was quantified on LPMs using flow cytometry. Bars represent mean ± SEM (n = 6–10). One- or two-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
As pMacs are not a homogeneous population of cells, as previously discussed, we used flow-cytometry-based methods to immunophenotype LPMs and SPMs, as the two populations can be distinguished on the basis of CD11b expression (Figure S1C).26 When assessing LPM and SPM count, it was observed that no differences were seen in LPM count between genotype nor treatment (Figure S1D), although a significant upregulation of SPMs was observed in WTOE pMacs upon IFNγ treatment relative to G2019S and B6 pMacs (Figure S1E). As bone-marrow-derived myeloid precursors are known to differentiate into SPMs upon inflammation, it may be, therefore, that WTOE bone-marrow-derived myeloid precursors present in these cultures have increased propensities to differentiate in such conditions.
To begin to investigate the effects of Lrrk2 over-expression and mutation on pMac function, we assessed surface MHC-II expression on both LPMs and SPMs as a measure of antigen presentation. Although no significant differences were observed between genotypes regarding MHC-II+ LPM count (Figure S1F), it was observed that G2019S-expressing LPMs express significantly more MHC-II in vehicle-treated LPMs, with all three genotypes increasing surface MHC-II expression with IFNγ treatment (Figure 1C). In SPMs, no differences between genotypes were observed in vehicle treatment, but increased surface MHC-II expression was observed in IFNγ-treated G2019S-expressing SPMs relative to the other genotypes (Figure 1D). Regarding MHC-II+ SPM count, increased counts were observed in IFNγ-treated WTOE pMacs (Figure S1G), but when calculated as a percentage of total SPMs, this difference did not persist (Figure S1H), suggesting that this observation was due to more SPMs overall as opposed to more MHC-II+ SPMs.
To determine whether changes in surface MHC-II levels were due to changes in transcription and translation, total MHC-II levels in pMac lysates were quantified. A significant increase in MHC-II protein levels was seen in all genotypes upon IFNγ treatment (Figures S1I and S1J). However, no significant differences were observed between genotypes, suggesting the increased MHC-II surface expression seen in G2019S pMacs is not driven by increased transcription and translation but likely due to altered trafficking to the plasma membrane.
To see if alterations in surface MHC-II expression were accompanied by changes in cytokine release, media from vehicle- and IFNγ-treated cells were collected and cytokine levels quantified. Increased levels of the pro-inflammatory TNF (Figure 1E) and the anti-inflammatory IL-4 were observed in media from IFNγ-treated G2019S pMacs (Figure 1F). As well, increased levels of the anti-inflammatory IL-10 were also observed in media from G2019S pMacs when treated with vehicle relative to media from WTOE and B6 pMacs. In both WTOE and G2019S pMacs, IFNγ treatment caused a reduction in IL-10 in the media compared with vehicle treatment, but no change was observed in B6 pMacs (Figure 1G). No significant differences were observed in other cytokines measured (Figures S1K–S1N).
The Eα: YAe model is used to monitor the antigen presentation capabilities of cells by incubating an endogenous peptide (Eα 52–68) which is subsequently phagocytosed, transported to the lysosome, loaded onto an MHC-II complex at the lysosome and transported back to the plasma membrane for antigen presentation (Figure S2). This Eα peptide-loaded MHC-II can subsequently be detected using flow cytometry using the YAe antibody.26,31 This model allows us to measure antigen presentation of a peptide directly and acts as a measure of the whole antigen presentation pathway, from uptake to peptide loading to presentation. It was observed here that YAe median fluorescence intensity (MFI) was significantly increased in IFNγ-treated G2019S-expressing LPMs relative to the other genotypes (Figure 1H). When co-treated with PF360, this phenotype was ameliorated and no significant differences between genotypes observed. Antigen presentation and pathogen sensing requires protease action and sufficient lysosomal function in order to occur,32 which is why lysosomotropic agents have been shown to decrease peptide-loaded MHC-II surface expression in antigen-presenting cells.33 Indeed, when co-treated with the vacuolar H+ ATPase (V-ATPase) inhibitor bafilomycin A1, YAe MFI significantly decreased in all three genotypes. Because of this observation and the crucial role of the lysosome in antigen presentation, we next sought to probe the effects of Lrrk2 over-expression and mutation on lysosomal function in these pMacs.
Lrrk2 kinase activity modulates lysosomal function early in the inflammatory response in cells engaging in antigen presentation
The immune response to an inflammatory stimulus is a dynamic process with peaks in different cellular activities occurring at different times. Although LRRK2 levels have been shown to increase in response to inflammatory stimuli, reports typically measure LRRK2 levels at end time point, neglecting to show changes in LRRK2 levels over time during the inflammatory response. We therefore wanted to examine how LRRK2 levels and phosphorylation change over time in response to IFNγ. Regarding total Lrrk2 levels, a significant, steady increase in Lrrk2 levels were seen over the 18 h IFNγ treatment with G2019S and WTOE-expressing pMacs consistently expressing increased levels relative to B6 pMacs (Figures 2A and 2B). Regarding phosphorylated Lrrk2, a similar pattern was observed, with levels of pLrrk2 at S935 increasing over the 18 h IFNγ treatment in all genotypes, with increased levels observed in G2019S pMacs relative to the other genotypes at the 18 h time point (Figure 2C).
Figure 2.

Lrrk2 kinase activity modulates lysosomal function early in the inflammatory response in cells engaging in antigen presentation
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were stimulated with 100 U IFNγ and harvested at 2, 6, or 18 h. (A–C) Lrrk2 protein and phosphorylated protein levels were assessed and normalized to total protein/total Lrrk2 levels and quantified. Representative western blots are shown. pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were stimulated with 100 U IFNγ and harvested at 2, 6, or 18 h after 1 h treatment with 100 nM PF360 immediately prior to harvesting. (D) pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were subject to one of two treatment conditions. All cells were stimulated with 100 U IFNγ and harvested at 2, 6, or 18 h. In the first treatment condition, cells were treated for 1 h with 100 nM PF360 immediately prior to harvesting. In the second treatment condition, cells were treated ± 100 nM PF360 from the beginning of the 18 h IFNγ treatment. For this first treatment condition with 1 h of Lrrk2 kinase inhibition, (E) DQ-BSA MFI was quantified in LPMs using flow cytometry, (F) BMV109 MFI was quantified in LPMs using flow cytometry, and (G and H) surface MHC-II GMFI was quantified in LPMs and SPMs using flow cytometry. For the second treatment condition with Lrrk2 kinase inhibition from the start of the 18 h IFNγ treatment, (I) BMV109 MFI was quantified in LPMs using flow cytometry, (J) DQ-Red BSA MFI was quantified in LPMs using flow cytometry, and (J and K) surface MHC-II GMFI was quantified in LPMs and SPMs using flow cytometry. Bars represent mean ± SEM (n = 8–10). Three-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
Next, pMacs were plated in the presence of IFNγ and collected at 2, 6, or 18 h into the treatment and measures of lysosomal activity were recorded using flow cytometry. To understand the role of Lrrk2 kinase activity over this time course, we applied one of two experimental designs (Figure 2D); PF360 or vehicle was either applied for 1 h prior to collection at each time point, or PF360 or vehicle was present from the start of the IFNγ treatment when cells were plated. In the conditions in which PF360 or vehicle was present for the 1 h prior to collection, it was observed that lysosomal degradation, as measured by DQ Red BSA fluorescence, peaked at 6 h into the IFNγ-treated LPMs in vehicle conditions for all 3 genotypes (Figure 2E). G2019S pMacs treated with vehicle exhibited even greater lysosomal degradation at this 6 h time point than WTOE and B6 pMacs. Interestingly, the 1 h treatment with PF360 prior to collection significantly reduced this 6 h peak in lysosomal degradation in all 3 genotypes. Similarly, cathepsin activity was measured using the pan-cathepsin probe BMV109, and a significant peak in cathepsin activity was observed at the 6 h time point in vehicle-treated pMacs, independent of genotype, relative to the other time points (Figure 2F). Interestingly, G2019S-expressing LPMs had significantly increased cathepsin activity at this 6 h time point relative to the other genotypes. Treatment of PF360 for 1 h prior to collection could decrease this 6 h peak in all genotypes, mirroring what was seen in the DQ Red BSA measurements. However, it was also observed that this PF360 treatment significantly increased cathepsin activity at the 2 h time point in all 3 genotypes.
It seems, therefore, that in cells treated with a vehicle, there is a peak in cathepsin activity at 6 h post IFNγ treatment. However, upon loss of total Lrrk2 kinase activity, this peak shifts, appearing earlier at 2 h. It seems therefore that a loss of Lrrk2 dysregulates the orchestrated response to IFNγ, at least regarding lysosomal cathepsin activity levels and protein degradation. Interestingly, when measuring surface MHC-II expression on LPMs and SPMs, although effects of genotype were observed at the 18 h time point, no significant effects of 1 h PF360 treatment were observed at any time point (Figures 2G and 2H).
Keeping in mind that the role of the lysosome occurs earlier in the inflammatory response than the end product of antigen presentation,32 we repeated these experiments with PF360 present from the start of the 18 h IFNγ treatment. Interestingly, in this condition, the increased surface MHC-II expression in G2019S LPMs and SPMs at 18 h was ameliorated (Figures 2I and 2J). Surface MHC-II expression was also significantly decreased at the 18 h time point in B6 and WTOE LPMs treated with PF360 from the start of the IFNγ response (Figure 2G). Similar changes in DQ Red BSA and BMV109 fluorescence were observed in PF360-treated pMacs in this experimental design that were seen in the 1 h PF360 condition (Figures 2K and 2L). Collectively, such data suggest that Lrrk2 kinase activity modulates antigen presentation earlier in the inflammatory response, as PF360 was unable to ameliorate the G2019S-mediated increase in MHC-II on LPMs when present for 1 h prior to collection at the 18 h time point, but could decrease surface MHC-II expression when present from the start of the inflammatory response. Given what is understood about the role of the lysosome in antigen presentation pathways, and the fact that we see significant effects of Lrrk2 kinase inhibition on lysosomal activity at the 6 h time point, we hypothesized that Lrrk2 may regulate antigen presentation via lysosomal activity early in the inflammatory response (Figure S3).
G2019S pMac antigen presentation and lysosomal phenotypes are rescued by knockdown of Lrrk2
It is currently unknown whether targeting increased LRRK2 levels in peripheral immune cells with LRRK2-targeting ASOs will be beneficial or deleterious to immune cell function, therefore these flow-cytometry-based assays were repeated in pMacs nucleofected with Lrrk2-targeting ASO (Ionis) or control ASO (Ionis). Ionis provided 3 Lrrk2-targeting ASOs which were used to nucleofect B6 pMacs at a concentration of 1 μg per reaction (2 × 106 cells per reaction); it was observed that ASO 3 significantly reduced total Lrrk2 levels in pMacs relative to control ASO at both the protein and the mRNA level (Figures S4A and S4B). Moreover, the control ASO did not significantly reduce Lrrk2 protein levels relative to non-nucleofected cells. When pMacs from BAC mice and B6 controls were nucleofected with this Lrrk2 ASO, a significant reduction in Lrrk2 protein levels were observed in all genotypes relative to control ASO (Figures 3A and 3B). Furthermore, in Lrrk2 ASO-nucleofected cells, 18 h treatment with IFNγ was unable to increase Lrrk2 levels, as seen in control ASOs.
Figure 3.

G2019S pMac antigen presentation and lysosomal phenotypes are rescued by knockdown of LRRK2
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were nucleofected with a Lrrk2-targeting ASO or control ASO and stimulated with 100 U IFNγ and harvested at 2, 6, or 18 h. (A and B) Lrrk2 protein levels normalized to β-actin levels and quantified. (C) YAe MFI was quantified on LPMs using flow cytometry. (D) Surface MHC-II GMFI was quantified in LPMs using flow cytometry. (E) DQ-BSA MFI was quantified in LPMs using flow cytometry. (F) BMV109 MFI was quantified in LPMs using flow cytometry. Bars represent mean ± SEM (n = 8–10). Two- or three-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
To ensure that nucleofection did not have adverse effects on inflammatory responses that may confound the interpretation of results, pMacs from BAC mice and B6 controls were nucleofected with control ASO or Lrrk2 ASO, and surface MHC-II expression at baseline and in response to IFNγ was quantified using flow cytometry alongside non-nucleofected control cells. A significant main effect of treatment was observed, with IFNγ treatment significantly increasing surface MHC-II expression in both nucleofected and non-nucleofected cells (Figure S4C). No significant differences were observed between non-nucleofected cells and those nucleofected with control ASO. However, a significant reduction in surface MHC-II expression was observed in IFNγ stimulated cells nucleofected with Lrrk2 ASO relative to non-nucleofected and control ASO treatments. Collectively, these data suggest that nucleofection with a control ASO does not significantly reduce Lrrk2 protein levels (Figure S4A) or significantly modify immune responses in macrophages relative to non-nucleofected cells. Therefore, all experiments discussed here on directly compare the effects of Lrrk2 knockdown via ASO relative to control ASO, with no non-nucleofected control cells.
Using the Eα: YAe model, antigen presentation was assessed in nucleofected pMacs after 18 h of IFNγ treatment. It was seen that in control ASO conditions increased YAe MFI was observed in G2019S-expressing LPMs relative to other genotypes as previously shown (Figure 3C). Interestingly, nucleofection with the Lrrk2-targeting ASO significantly reduced this YAe MFI in G2019S-expressing LPMs. Furthermore, knockdown of Lrrk2 also significantly decreased YAe MFI in B6 pMacs. This was supported by flow-cytometry-based surface MHC-II expression analysis in LPMs (Figure 3D).
In order to assess the effects of Lrrk2 knockdown on lysosomal function, lysosomal degradation and cathepsin activity was measured using flow cytometry as before. Similar to what was observed in experiments assessing effects of Lrrk2 kinase inhibition, Lrrk2 ASO treatment caused a significant reduction in both lysosomal protein degradation (Figure 3E) and cathepsin activity (Figure 3F) at the 6 h time point in G2019S and WTOE LPMs. All genotypes had a peak in lysosomal degradation at 6 h time point when treated with control ASO, but treatment with Lrrk2 ASO caused the peak to appear earlier at the 2 h time point instead (Figure 3E), the same effect observed with Lrrk2 kinase inhibition. Collectively, therefore, these data demonstrate that ASO-mediated knockdown of Lrrk2 phenocopies the effects of Lrrk2 kinase inhibition, suggesting the effects of Lrrk2 ASO on the phenotypes observed here are driven by a loss of Lrrk2 kinase activity.
Lrrk2 kinase inhibition and knockdown via ASO reduces trafficking of MHC-II to the plasma membrane
As knockdown of Lrrk2 and Lrrk2 kinase inhibition reduces both YAe MFI and MHC-II levels using flow cytometry suggests there is decreased transport of peptide-loaded MHC-II-complexes from the lysosome to the cell surface. It can therefore be hypothesized that an increase in intracellular MHC-II expression would be observed in conditions in which Lrrk2 is knocked down, or its kinase activity inhibited. To test this, using pMacs treated with the LRRK2 kinase inhibitor, PF360, or nucleofected with a control or Lrrk2 ASO, we co-stained for extracellular MHC-II (exMHCII) vs. intracellular MHC-II (icMHCII) and monitored expression via fluorescent microscopy. It was observed that G2019S pMacs exhibited increased exMHCII:icMHCII ratio relative to other genotypes, which was ameliorated upon LRRK2 kinase inhibition (Figures 4A and 4B), with this treatment decreasing exMHCII:icMHCII ratio in all genotypes. Such data suggest that Lrrk2 kinase inhibition reduces the transport of icMHCII to the cell surface to engage in antigen presentation. Indeed, it was observed that, under certain conditions, perinuclear clustering of icMHCII could be observed (Figures 4A and 4C). When the percentage of cells exhibiting this perinuclear icMHCII was quantified, it was observed that G2019S-expressing pMacs had a significant decrease in cells with perinuclear icMHCII relative to WTOE and B6 pMacs, with a significant decrease also observed in WTOE relative to B6 pMacs (Figure 4C). Furthermore, PF360 treatment significantly increased the percentage of cells with perinuclear icMHCII, further supporting a role of Lrrk2 in transport of MHC-II to the cell surface. A similar effect of Lrrk2 ASO was also observed (Figure 4E).
Figure 4.

Lrrk2 kinase inhibition and knockdown via ASO reduces trafficking of MHC-II to the plasma membrane
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were stimulated with 100 U IFNγ for 18 h and stained for intracellular and extracellular MHC-II MFI, and exMHCII:icMHCII ratio was quantified. White arrows indicate perinuclear clustering of icMHCII. Scale bars, 30 μM (A and B). Percentage of cells with perinuclear clustering was quantified (C). pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were nucleofected with a Lrrk2-targeting ASO or control ASO and stimulated with 100 U IFNγ for 18 h and stained for intracellular and extracellular MHC-II MFI, and exMHCII:icMHCII ratio was quantified. White arrows indicate perinuclear clustering of icMHCII. Scale bars, 30 μM (D and E). Bars represent mean ± SEM (n = 6). two-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
Lrrk2 knockdown via ASO and kinase inhibition alters cytokine release from pMacs
To see if alterations in antigen presentation and lysosomal function in Lrrk2 ASO or Lrrk2 kinase inhibitor treated cells were accompanied by changes in cytokine release, media from vehicle- and IFNγ-treated cells were collected and cytokine levels quantified. It was observed that both ASO-mediated Lrrk2 knockdown and Lrrk2 kinase inhibition was able to significantly reduce IFNγ-dependent IL-10 release in G2019S-expressing pMacs (Figures 5A and 5B), suggesting that IL-10 release from stimulated pMacs may be dependent on Lrrk2, specifically its kinase activity. This same observation was made regarding TNF release from G2019S pMacs (Figures 5C and 5D). A significant reduction in IL-12p70 release in G2019S pMacs nucleofected with the Lrrk2-targeting ASO (Figure 5E). On the other hand, although ASO-mediated Lrrk2 knockdown was capable of decreasing IL-4 release in IFNγ-treated G2019S-expressing pMacs (Figure 5F), Lrrk2 kinase inhibition was unable to do so (Figure S5A). No significant effects of ASO treatment or Lrrk2 kinase inhibition on cytokine release were observed in other cytokines measured (Figures S5B–S5E).
Figure 5.

LRRK2 knockdown via antisense oligonucleotide and kinase inhibition alters cytokine release from pMacs
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice were nucleofected with a Lrrk2-targeting ASO or control ASO and stimulated with 100 U IFNγ, or were plated with 100 U IFNγ ± 100 nM of Pf360 and media collected after 18 h. Cytokine levels of IL-10 (A and B), TNF (C and D), IL12p70 (E), and IL-4 (F) were quantified and normalized to live cell count. Bars represent mean ± SEM (n = 8–10). Two-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
ASO-mediated Lrrk2 knockdown alters critical immune pathways in G2019S and WTOE pMacs
In order to further probe the effects of G2019S-Lrrk2 and Lrrk2 ASO treatment on lysosomal pathways, NanoString-based mRNA expression analysis was performed using a custom code set of 250 genes located in lysosomal and immune-related pathways (Sup.file.1). pMacs from WTOE- and G2019S-Lrrk2 mice were nucleofected with control or Lrrk2-targeting ASO and plated in the presence or absence of IFNγ for 18 h prior to harvesting of RNA (Figure 6A). Lrrk2 mRNA was significantly reduced in G2019S pMacs nucleofected with Lrrk2-targeting ASO relative to control ASO in both vehicle- and IFNγ-treated conditions (Figure 6B). Although Lrrk2 mRNA significantly decreased in vehicle-treated WTOE pMacs nucleofected with Lrrk2-targeting ASO relative to control ASO, this failed to reach significance in IFNγ-treated WTOE pMacs.
Figure 6.

ASO-mediated Lrrk2 knockdown alters critical autophagy and cytokine signaling pathways in G2019S and WTOE pMacs
Transcriptomic analysis from WTOE or G2019S vehicle and IFNγ-treated pMacs nucleofected with control or Lrrk2 ASO. (A) Normalized log2(count) value for Lrrk2 mRNA was plotted. Bars represent mean ± SEM (n = 6). Two-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying the same letter are significantly different (p < 0.05). (B and C) Significant control ASO:Lrrk2 ASO DEGs in G2019S pMacs were counted and compared across vehicle and IFNγ treatments. (D and E) Significant control ASO:Lrrk2 ASO DEGs in WTOE pMacs were counted and compared across vehicle and IFNγ treatments. Volcano plots show proteins with fold change > 1.5 and adjusted p ≤ 0.05. (F) ShinyGO version 0.76.3 was used to identify pathways in which significant DEGs were associated with. (G) Heatmaps show DEGs seen only in G2019S pMacs treated with IFNγ. (H) ShinyGO version 0.76.3 was used to identify pathways in which significant DEGs were associated with. (I) Heatmaps show DEGs seen only in WTOE pMacs treated with IFNγ.
A principal-component analysis (PCA) map was generated from the resulting dataset which demonstrated that groups in our dataset primarily cluster on the basis of vehicle or IFNγ treatment (Figure 6C). Within these treatment clusters, there is distinct clusters on the basis of genotype and, furthermore, within these genotype clusters, there is distinct clustering on the basis of control ASO or Lrrk2 ASO nucleofection.
It was observed that an increase in number of control ASO:Lrrk2 ASO differentially expressed genes (DEGs) was observed in IFNγ-treated G2019S pMacs relative to those seen in vehicle-treated conditions (Figures 6B and 6C). A similar observation was made in WTOE pMacs (Figures 6D and 6E). As Lrrk2 protein levels increase in response to IFNγ, it follows that knockdown of Lrrk2 would have a more significant biological impact in these conditions in which Lrrk2 levels are increased.
Using ShinyGO analysis to identify pathways in which these DEGs were situated in, the control ASO:Lrrk2 ASO DEGs identified in G2019S and WTOE pMacs treated with IFNγ were found in critical immune-related pathways such as cytokine-cytokine receptor interaction, TNF production, and responses to bacteria (Figures 6F–6I). It was also noted that critical autophagy-related genes, such as sqstm1, ctss, becn1, and Atp6v1a were up-regulated when Lrrk2 levels were decreased as it has previously been described that mTOR inhibition, and therefore increased expression of autophagy-related genes and increased autophagic flux, decreases antigen presenting capabilities of AP cells.34
NanoString-based transcriptome analysis reveals genotype differences in a treatment specific manner and reveals differential response to IFNγ by G2019S pMacs
Next, we wanted to identify key cellular pathways that were altered in G2019S pMacs relative to WTOE pMacs in control ASO conditions, in both vehicle and IFNγ-treated conditions; that is, the effects of increased kinase activity levels in a context-dependent manner. Although a degree of overlap was seen regarding WTOE:G2019S DEGs identified in both vehicle and IFNγ treatments, a number of DEGs were identified as novel to each of these treatment conditions (Figures S6A–S6D). ShinyGO pathway analysis showed that WTOE:G2019S DEGs seen in both treatment conditions were identified in pathways such as glucosylceramide catabolic processing, lysosomal pH, and lysosomal/vacuole organization (Figure S6E). A number of these DEGs have previously been identified as substrates or interactors of Lrrk235,36,37 and have previously been implicated in PD38,39,40 (Figure S6F). These DEGs were down-regulated in G2019S pMacs relative to WTOE pMacs, indicating a potential downregulation of these pathways in G2019S pMacs. WTOE:G2019S DEGs seen in vehicle treatment only were identified in pathways related to cytokine production, signal transduction, and cellular communication (Figures S6G and S6H). Interestingly, WTOE:G2019S DEGs seen in IFNγ treatment only were most identified in pathways associated with vesicle transport and Rab and Ras signal transduction (Figures S6I and S6J). These DEGs were down-regulated in G2019S pMacs relative to WTOE pMacs, indicating a potential downregulation of these pathways in G2019S pMacs. Again, many of these DEGs have previously been identified as substrates of Lrrk237 and have previously been implicated in PD as well as other neurodegenerative diseases.41,42,43
NanoString-based transcriptome analysis reveals differential response to IFNγ by G2019S pMacs
We were also interested to understand how increased Lrrk2 kinase activity may modulate macrophages responses to an inflammatory stimulus, and if vehicle:IFNγ DEGs in G2019S pMacs differed from those in WTOE pMacs. There was a vast degree of overlap between vehicle:IFNγ DEGs in the two genotypes, but 32 DEGs were identified in G2019S pMacs that were not found in WTOE pMacs (Figures S7A–S7D). These 32 DEGs were identified in pathways related to autophagy, immune cell activation and regulation of phosphorylation (Figure S7E). Most interestingly, the pathway termed “regulator of neuronal death” was significantly enriched by these DEGs. DEGs identified in this pathway include Lgmn, Bad, Casp2, Tlr4, Grn, IL18, Cln3, and Gba, with the majority of these genes being down-regulated upon IFNγ treatment in G2019S pMacs (Figure S7F).
Vesicular trafficking and lysosomal positioning pathways are associated with the response to IFNγ in G2019S pMacs nucleofected with Lrrk2 ASO
We also wanted to understand how Lrrk2 knockdown affects the response to IFNγ in G2019S pMacs, and begin to unveil a mechanism of action regarding the capabilities of Lrrk2 ASO treatment to ameliorate antigen presentation and lysosomal phenotypes observed in G2019S pMacs here. When comparing vehicle:IFNγ DEGs in control ASO and Lrrk2 ASO treated G2019S pMacs, we found that, although many of the DEGs persisted in the Lrrk2 ASO condition, suggesting no effect of Lrrk2 knockdown on these DEGs, 15 DEGs were novel to control ASO conditions and 34 DEGs were seen only in Lrrk2 ASO treated cells (Figures 7A–7C). Interestingly, a number of the vehicle:IFNγ DEGs in control ASO-treated G2019S pMacs only included those that enriched the “regulator of neuronal death” pathway previously described (Figure 7D). The fact that these DEGs are only seen in control ASO conditions and not in Lrrk2 ASO conditions suggests that the knockdown of Lrrk2 sufficiently prevents these changes in gene expression in response to IFNγ in G2019S pMacs. Interestingly, vehicle:IFNγ DEGs identified in Lrrk2 ASO-treated G2019S pMacs were identified in pathways related to acute inflammatory responses to antigen, lysosome localization, and exocytosis (Figure 7E). A number of the genes identified in exocytosis and lysosome positioning pathways have specifically been identified to play a role in the release of secretory lysosomes, exocytosis of lysosomes and transportation of lysosomal content to the plasma membrane, including Snap23, Abca1, Tmem55b, Mtor, Rab3a and Rab3d21,34,44,45,46,47 (Figure 7F), and these were all down-regulated in response to IFNγ in G2019S pMacs treated with Lrrk2 ASO. Interestingly, LRRK2 has recently been shown to mediate tubulation and vesicle sorting from lysosomes.20,48 with G2019S-Lrrk2 expression significantly increasing the formation of these tubules from lysosomes. In the context of myeloid cells, it is known that lysosomal tubulation is usually observed in cells undergoing immune activation, and these tubules are crucial for both; phagocytosis and antigen presentation.34,49 Collectively, therefore, we hypothesized that increased lysosomal activity and antigen presentation observed in G2019S pMacs here may be due to increased LTF and that knockdown of Lrrk2 with ASO may ameliorate these phenotypes by reducing LTF.
Figure 7.

Vesicular trafficking and lysosomal positioning pathways are associated with the response to IFNγ in G2019S pMacs nucleofected with Lrrk2 ASO
Transcriptomic analysis from Lrrk2 ASO nucleofected G2019S pMacs treated with vehicle or IFNγ (A). Volcano plot shows proteins with fold change > 1.5 and adjusted p ≤ 0.05. (B and C) Significant DEGs were counted and compared across genotypes. (D) Heatmaps show DEGs seen only in control ASO nucleofected G2019S pMacs. (E) ShinyGO version 0.76.3 was used to identify pathways in which significant DEGs were associated with. (F) Heatmaps show DEGs seen only in G2019S pMacs nucleofected with Lrrk2 ASO.
Lrrk2 modulates antigen presentation via LTF
In order to determine if alterations in antigen presentation in G2019S pMacs were due to altered LTF, pMacs were loaded with dextran Alexa Fluor 546 for 1 h, followed by a 2 h pulse period to ensure dextran was fully loaded into lysosomal compartments and treated with 100 U IFNγ to induce LTF. Live cells were imaged after 2 h at which LTF would have occurred.34 When quantifying the percentage of cells with lysosomal tubules (defined as >2 μM), it was observed that IFNγ-treated G2019S pMacs exhibited significantly increased percentage of cells with tubular lysosomes relative to B6 controls and WTOE pMacs (Figures 8A–8C and S7A). It is known that tubular lysosomes that form in immune cells for the purpose of phagocytosis and antigen presentation are dependent on mTOR activity, whereas tubular lysosomes that form for the purpose of autophagy are not.50 Therefore, to differentiate between these two functions, cells were co-treated with the mTOR inhibitor Torin1, a significant reduction in cells with tubular lysosomes was observed in all genotypes (Figure 8B). When this was quantified as a percentage of cells with Torin1-dependent tubular lysosomes, the significant increase in G2019S pMacs relative to other genotypes persisted (Figure S7B), suggesting that the tubular lysosomes quantified here, and those that are increased in G2019S pMacs are mTOR dependent and therefore involved in antigen presentation. When cells were co-treated with the LRRK2 kinase inhibitor, PF360, a significant reduction was also observed in all genotypes, suggesting LTF in macrophages in dependent on LRRK2 kinase activity. Similarly, when cells were nucleofected with a Lrrk2 ASO, a significant reduction in LTF was observed relative to control ASOs (Figures 8C and S7C).
Figure 8.

LRRK2 modulates antigen presentation via lysosomal tubule formation
pMacs from 10- to 12-week-old male B6, WTOE, or G2019S mice treated with 0.5 mg/mL Dextran Alexa Fluor 546 for 1 h, followed by a 2 h pulse period to ensure loading into lysosomes, and then treated with 100 U IFNγ ± 100 nM Torin1 or 100 nM PF360 for 2 h to stimulate LFT. (A) Cells were imaged live, and (B) percentage of cells with tubular lysosomes quantified. Filled white arrows indicate pMacs with tubular structures, empty arrow heads indicate pMacs with punctate dextran. Dotted lines indicate masks of cells on the basis of bright-field images. Scale bars, 10 μM. (C) Cells were nucleofected with 1 μg of Control ASO or Lrrk2-targeting ASO and percentage of cells with tubular lysosomes quantified. (D) pMacs were treated with 100 U IFNγ ± 10 μM nocodazole or 100 nM Torin1 for 18 h, and YAe MFI in LPMs was quantified using flow cytometry. (E–G) pMacs were treated with 100 U IFNγ ± 100 nM PF360 and protein lysate assessed for mTOR, and s60k protein levels and normalized to β-actin levels and quantified. Representative western blots are shown. Bars represent mean ± SEM (n = 4–6). Two-way ANOVA, Bonferroni post hoc test, groups sharing the same letters are not significantly different (p > 0.05), and groups displaying different letters are significantly different (p < 0.05).
Fluorescently labeled dextrans have been shown to enter the cell by both macropinocytosis51 and micropinocytosis.52 It has recently been demonstrated that LRRK2 and Rab10 coordinate micropinocytosis in human and mouse phagocytic cells.53 In order to ensure that differences in dextran-labeled tubules were not due to differences in uptake between genotypes in our study, cells were loaded with 20 μg of dextran for 1 h, fixed, and imaged with no pulse phase and dextran-fluorescence quantified. As well, to ensure other endocytic pathways that may affect uptake of dextran54 are not significantly different between genotypes, cells were incubated with Alexa Fluor 594-transferrin and uptake measured via fluorescent microscopy. No significant effects of genotype or LRRK2 kinase inhibition on transferrin or dextran uptake were observed (Figures S7D–S7G), although a significant reduction in transferrin uptake was observed upon IFNγ treatment.
To further explore the role of LTF in the G2019S-associated phenotypes observed here, we repeated the YAe antigen presentation assay described before with co-treatments with compounds known to inhibit various steps of the antigen presenting process that depends on LTF. Both the previously described Lrrk2-mediated tubulation20 and antigen presentation via tubules are dependent on microtubules;55 co-treatment of IFNγ-treated pMacs with the microtubule destabilizer nocodazole significantly reduced YAe MFI in LPMs, indicating decreased antigen presenting abilities of these cells, in all three genotypes and ameliorating the G2019S-dependent increase observed in vehicle-treated cells (Figure 8D). Such observations suggest that the G2019S-dependent increase in antigen presentation in LPMs is dependent on microtubules. mTOR inhibition has also been shown to decrease LTF-dependent antigen presentation in dendritic cells and macrophages.34 Treatment with the mTOR inhibitor Torin1 also decreased YAe MFI in LPMs in all genotypes, ameliorating any effects of the G2019S mutation relative to the other two genotypes (Figure 8D). As previously discussed, presentation and pathogen sensing requires protease action and sufficient lysosomal function in order to occur,32 and treatment with the vATPase-A1 pump inhibitor bafilomycin A1 significantly reduced YAe MFI in these cells (Figure 1H).
We next wanted to determine if the trafficking of MHC-II to the plasma membrane was dependent on mTOR-dependent LTF. We found that Torin1 co-treatment induced the same effects as Lrrk2 kinase inhibition and knockdown on exMHCII:icMHCII ratio and nuclear clustering (Figures S9A–S9C). This suggests that the LTF-dependent MHC-II antigen presentation phenotype observed is mTOR dependent. This is further supported by the fact that mTOR was a significant DEG observed in the Lrrk2 ASO NanoString-based transcriptomics in G2019S pMacs (Figure 7F), with mTOR expression decreasing in response to IFNγ only in cells treated with the Lrrk2 ASO.
Inhibition of mTOR is a key trigger for autophagy. Therefore, we wondered whether the observed decrease in LTF in cells treated with Lrrk2 kinase inhibitor was accompanied by an up-regulation in autophagic flux. Furthermore, we hypothesized that the increased stimulation-dependent LFT observed in G2019S cells would be accompanied by a decrease in autophagic flux. Indeed, when quantifying LC3-II flux (calculated as signal difference between conditions with and without bafilomycin A1), it was observed that all genotypes exhibited decreased autophagic flux upon IFNγ treatment, with this effect exacerbated in G2019S pMacs (Figures S9D and S9E). Treatment with either the LRRK2 kinase inhibitor, PF360, or the mTOR inhibitor, Torin1, ameliorated this stimulation-dependent effect on LC3 flux in all genotypes. Collectively, such data suggest that there is a delicate balance between prioritizing autophagic flux or LTF in macrophages and that both Lrrk2 and mTOR are key components in regulating and maintaining this balance.
Given this, we wanted to assess whether G2019S pMacs exhibited an increase in mTOR expression and/or activity levels which may be driving LTF in these cells. Indeed, we saw a significant increase in mTOR protein levels in G2019S pMacs in a stimulation-dependent manner, and this was ameliorated with the knockdown of Lrrk2 via ASO (Figures 8E and 8F). Furthermore, we saw a significant increase in phosphorylated s6k levels (p70S6 kinase 1), a downstream target of mTOR signaling, in G2019S pMacs upon IFNγ stimulation which was ameliorated with the knockdown of Lrrk2 via ASO (Figures 8F and 8G). As well, we observed an increase in RILPL1 (Rab-interacting lysosomal protein-like 1 expression), a modulator of lysosomal positioning and a previously identified interactor of LRRK2,56 in G2019S pMacs upon IFNγ stimulation, which was ameliorated with the knockdown of Lrrk2 via ASO (Figures S9F and S9G). Collectively, these data suggest that the knockdown of Lrrk2 in G2019S pMacs ameliorates LTF-dependent antigen presentation by modulating mTOR levels and activity.
Discussion
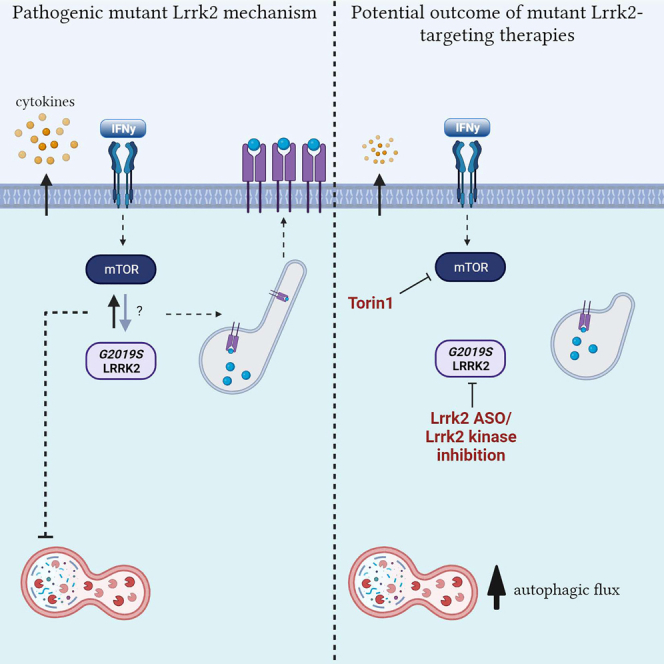
Recent advances in understanding LRRK2 function at the lysosome have suggested a potential link for the role of LRRK2 on the regulation of lysosomal function to that of immune cell function and modulation of inflammatory responses. Our data demonstrate increased antigen presentation, cytokine release, and lysosomal activity in pMacs from G2019S mice which were successfully ameliorated with knockdown of Lrrk2 via ASO or treatment with a LRRK2 kinase inhibitor. Our findings suggest that increased antigen presentation in mutant Lrrk2 cells is intrinsically linked to the alterations in lysosomal function observed, with inhibition of lysosomal function via bafilomycin A1 causing decreased antigen presentation in these cells. Our LRRK2 kinase inhibitor findings also demonstrate that the role of Lrrk2 in antigen presentation occurs early on in the response to IFNγ, and is accompanied by alterations in lysosomal peak activity during this response. Furthermore, tour LRRK2 kinase inhibitor findings suggest the effects of Lrrk2 on the reported phenotypes appear to be specifically mediated through its kinase activity. Furthermore, it was demonstrated that increased LTF is the underlying mechanism for the phenotypes observed in G2019S pMacs, in a potentially mTOR-dependent manner, with Lrrk2 ASO and kinase inhibition ameliorating this.
We found that release of three cytokines were consistently elevated in G2019S pMac cultures: IL-10, IL-4, and TNF. It has recently been shown that RAW264.7 cells expressing T1348N-LRRK2, an artificial P loop-null mutation that disrupts GTP binding,57,58 produce significantly less IL-10 relative to WT cells in response to LPS and zymosan.59 IL-10 is produced by macrophages and is critical in limiting immune-mediated pathology,60 and it was therefore suggested by Nazish et al. that there may be a neuroprotective role of LRRK2 in immune signaling through altered IL-10 secretion. Indeed, IL-4, which is also critical in resolving inflammation,61 was increased in G2019S pMac cultures in this study. Although a pro-inflammatory cytokine, the fact that TNF was also increased in G2019S pMac cultures in this study supports the hypothesis of LRRK2 being protective in the immune system given the fact that mice lacking TNF and/or TNF receptors have been reported to exhibit increased susceptibility to infection with increased bacterial load and increased inducible nitric oxide species (iNOS) production.62 In agreement with the hypothesis that LRRK2 may play a protective role in immune signaling, Lrrk2 has been shown to be required for efficient control of certain pathogens; LRRK2 has been implicated in the control of the enteric pathogen Salmonella typhimurium via NLRC4 inflammasome regulation in macrophages from Lrrk2-KO mice.14,15 This is supported by the observation that G2019S knock-in mice, controlled S. typhimurium infection better, with reduced bacterial growth and longer survival during sepsis; an effect which was dependent on myeloid cells.63 Furthermore, Paneth cells from Lrrk2-KO mice are more susceptible to infection from Listeria monocytogenes, with a loss of Lrrk2 causing decreased levels of lysozyme, an antimicrobial enzyme responsible for the degradation and lysis of bacteria.16 It is possible that increased LTF and therefore antigen presentation in G2019S pMacs is the underlying mechanism of the role of Lrrk2 in pathogen control and its protective role in the immune system. However, other reports describe a deleterious role of Lrrk2 in pathogen control. For example, animals with reovirus-induced encephalitis that expressed the G2019S mutation exhibited increased mortality, increased reactive oxygen species and higher concentrations of α-synuclein in the brain.63 Furthermore, loss of LRRK2 enhances Mycobacterium tuberculosis (Mtb) control and decreases bacterial burdens in both primary mouse macrophages and human iPSC-derived macrophages.64 It seems therefore that the role of Lrrk2 in pathogen control may be pathogen specific as well as cell and tissue type specific. S. typhimurium and L. monocytogenes are both food-borne pathogens that enter the body through the gut, whereas reovirus and Mtb are airborne that enter through the lungs. Indeed, macrophages in the body are heterogeneous, showing specific transcription factors and markers and therefore different functions in the body.65 It is possible therefore that the role Lrrk2 plays is different between different macrophages subtypes, and may also be sex and age dependent.63,66,67 In summary, we have described a role of LRRK2 in LTF which can be disrupted by ASO knockdown, but further research is required to determine if this is seen in other antigen-presenting cell types and whether such phenotypes alter pathogen control in these cells.
As well as pathogen and infection control, the role of LRRK2 in immune responses and inflammation has also been discussed in the context of inflammation in the gut and Crohn’s disease (CD).68 In 2008, a genome-wide association study (GWAS) implicated LRRK2 in CD, and this was further confirmed in a subsequent study in populations with European descent.5,7 The newly identified N2081D variant is associated with increased risk for both CD and PD while the N551K and R1398H variants are associated with reduced risk for both diseases.2,63 Interestingly, the N2081D variant results in increased kinase activity, while the R1398H variant that falls in the ROC domain has been shown to deactivate LRRK2 by increasing GTPase activity.2 This suggests that PD and CD pathogenesis maybe closely linked to specific LRRK2 functions directly related to the enzymatic domains of LRRK2, and therefore targeting this may be beneficial for the dysregulated inflammatory responses seen in both of these diseases. Indeed, much like immune cells from PD patients, LRRK2 expression is up-regulated with IFNγ stimulation in immune cells from CD patients.69 Furthermore, LRRK2 mRNA levels are increased in inflamed CD intestinal tissue relative to uninflamed tissue from the same patient.69 Collectively, such data suggest targeting increased LRRK2 levels and enzymatic activity may be beneficial in CD. However, the M2397T variant associated with sporadic CD has been show to affect LRRK2 protein levels by significantly decreasing the half-life of the protein,7,61,62 suggesting that a significant reduction in LRRK2 levels also increases risk for gut inflammation. It seems, therefore, that there is a sensitive balance between LRRK2 levels and inflammation, at least in the gut, with both increases and decreases in LRRK2 levels and activity increasing risk for inflammation. Such observations need to be taken into consideration when targeting LRRK2 levels and activity for potential therapeutics.
The use of both kinase inhibitors and knockdown of total Lrrk2 in this study also sheds light on the role of the kinase domain vs. other enzymatic and protein-protein interaction domains of Lrrk2. For example, it was observed that both 18 h Lrrk2 kinase inhibition and Lrrk2 ASO treatment were capable of ameliorating G2019S-associated increases in antigen presentation and LTF, suggesting a role of the Lrrk2 kinase domain in these functions. Lrrk2 has recently been implicated in a function termed lysosomal tubulation/sorting driven by LRRK2 (LYTL).20 Bonet-Ponce and colleagues reported that LRRK2 recruits JIP4 to lysosomes in a kinase-dependent manner via the phosphorylation of RAB35 and RAB10, promoting the formation of tubules in response to lysosomal membrane damage. Rab35 was indeed identified in the NanoString nCounter analysis in this study with Rab35 mRNA being down-regulated in G2019S IFNγ-treated pMacs relative to WTOE. Such a downregulation may be a compensatory mechanism for increased Rab35 phosphorylation. Additional research is needed to determine if similar Lrrk2 substrates are involved in LYTL and LTF; although similar, tubular lysosomes are known to play a role in multiple functions and therefore different interacting partners and characteristics of these tubular lysosomes are likely.48
It is interesting that, although IL-10 and TNF production was decreased by Lrrk2 kinase inhibition in this study, IL-4 release was unaffected by Lrrk2 kinase inhibition and was only ameliorated upon knockdown of Lrrk2, suggesting a kinase-independent role of Lrrk2 in the release of IL-4 from pMacs. Cytokine secretion pathways are often adapted to suit specific cytokines, their function, and cell type. Macrophages lack granules, which enable rapid release of the cytokines upon cell activation, and instead cytokines must be synthesized after cell activation and secreted.70 Three canonical transport pathways for cytokine secretion have been identified to date; direct transport to the cell surface from the trans-Golgi network, via the recycling endosome, and during phagocytosis where cytokine is routed from the recycling endosome to the phagocytic cup.70 Notably, TNF and IL-10 have been detailed to be processed and released via these canonical pathways, whereas much less is known about the non-classical secretory pathways for cytokine release thought to be used by cytokines such as IL-1β, IL-18, and IL-4.70 It is therefore possible that Lrrk2 mediates these canonical pathways via kinase-activity, which is plausible given the numerous reports identifying Rabs, known for regulating protein transport, vesicle trafficking, and membrane fusion, as bona fide kinase substrates of LRRK2.37,71 Additional research is required to understand the role of LRRK2 in non-canonical pathways of cytokine release and the requirement of LRRK2 GTPase activity and protein-protein interaction domains in this role.
LRRK2 has previously been shown to coordinate macropinocytosis via Rab10 recruitment to macropinosomes, which are MHC-II and Cd11b positive, and induce subsequent CL5-stimulated Akt signaling and bone-marrow-derived macrophage chemotaxis.53 Although Lrrk2 kinase inhibition decreased chemotaxis in these cells, increased surface receptor recycling was seen with Lrrk2 kinase inhibition. It has previously been described that mature macropinosomes will fuse with tubular lysosomes that mediate their contents to the cell surface.53 It may be, therefore, that LRRK2 plays multiple roles in the processing and trafficking of MHC-II to the cell surface to engage in antigen presentation, thus the role of LRRK2 in macropinocytosis could be a potential confound in this study with multiple interpretations of the data. For example, increased surface MHC-II expression on LPMs in this study may be due to increased surface receptor recycling as opposed to increased antigen presentation due to increased LTF; however, Liu et al.53 observed an increase in surface receptor recycling during Lrrk2 kinase inhibition, so therefore we would not expect this mechanism to be involved in G2019S pMacs which exhibit increased kinase activity. Furthermore, Liu et al.53 looked at MHC-II expression, as opposed to MHC-II complexes loaded with a peptide for antigen presentation; the use of the YAe assays in this study and the fact we can quantify antigen presenting MHC-II complexes leads us to conclude that the mechanism we are measuring here is indeed antigen presentation via LTF. Furthermore, no differences were observed in the uptake levels of dextran or transferrin via micropinocytosis or receptor-mediated endocytosis between genotypes, with no effects of Lrrk2 kinase inhibition, suggesting these mechanisms are unlikely playing a role in the phenotypes observed. However, it will still be of interest to future studies to unveil how macropinocytosis and LTF for antigen presentation interact and how LRRK2 may be implicated in this.
Interestingly, we observed different pathways altered and DEGs between WTOE and G2019S pMacs depending on whether cells were stimulated with IFNγ or not. This observation suggests that there is a differential role of LRRK2 in macrophages depending on the inflammatory status of the cell. As well, these data, and the fact that Lrrk2 kinase inhibitors had different effects on the phenotypes described here depending on when in the IFNγ-response it is administered supports the hypothesis that LRRK2 may behave as a “date-hub.” The “date-hub” hypothesis describes two types of “hubs,” one of which are “date hubs,” which bind their different partners at different times or locations. The potential for LRRK2 behaving as a “date-hub” has been discussed in the literature72 and may explain the differences in DEGs and effects of kinase inhibition described here, and may also explain the discrepancies reported regarding conflicting results.14 However, despite being discussed in the literature, no study has yet been completed to definitively show whether the LRRK2 interactome varies in a cell-type- or time-dependent manner.
Intriguingly, Rab8a, Rab8b, Rab35, and Rab5b were identified as DEGs between WTOE and G2019S pMacs, only when treated with IFNγ. Such data are in agreement with previous literature that reports concomitant increases in LRRK2 and pRab10 in human monocytes upon IFNγ stimulation13 and that LRRK2 is recruited to membranes upon LPS stimulation.18,19 However, perhaps counterintuitively, mRNA of these Rabs are down-regulated in G2019S pMacs relative to WTOE. Further exploration is necessary regarding how these findings translate to the protein level for LRRK2 substrates. It may be, however, that mRNA levels are down-regulated in G2019S pMacs as a compensatory mechanism to compensate for increased phosphorylation of Rabs by Lrrk2.37,71
It was noted that many of the DEGs identified were down-regulated in G2019S pMacs relative to WTOE, as well as DEGs identified in IFNγ relative to vehicle-treated G2019S cells. It may be, therefore, that in G2019S pMacs, in particular those treated with IFNγ, exhibit an overall downregulation of gene expression. TFEB is a master regulator of autophagy- and lysosome-related genes, is known to regulate cytokine production in immune cells,69 and is inhibited by mTOR activity.73 Interestingly, mTOR is required for LPS-induced lysosome tubulation and presentation of MHC-II in macrophages and dendritic cells, with mTOR inhibition decreasing lysosomal tubulation.34 It therefore follows that in this study, one potential mechanism of action for the downregulation of gene expression in G2019S pMacs in response to IFNγ may be increased mTOR activity, leading to increased TFEB inhibition and decreased transcription, with concomitant increases in LTF. In support of this mechanism, we found that G2019S pMacs exhibited increased mTOR levels and mTOR activity upon immune stimulation, leading to increased LTF and decreased autophagic flux, all of which could be ameliorated by inhibition of mTOR activity via Torin1 treatment.
It is known that mTOR inhibition is a key trigger for autophagy.74 In healthy individuals, mTOR signaling is responsible for maintaining a balance between protein synthesis, autophagy, and nutrient usage and storage processes. This balance is crucial for the cell, as its dysregulation leads to cancer, obesity, and diabetes.75 The lysosome surface serves as a platform to assemble major signaling hubs like mTOR, as well as AMPK, GSK3, and the inflammasome; these molecular assemblies integrate and facilitate cross-talk between signals and ultimately enable responses such as autophagy, membrane repair, and microbe clearance.76 Here we have shown a phenotype present in G2019S pMacs that increases LTF while decreasing autophagic flux. Collectively, such observations suggest LRRK2 may be a modulator of lysosomal responses in immune cells, with increased LRRK2 kinase activity favoring the formation of lysosomal tubules, and therefore pathogen control, over other lysosome-associated functions such as autophagic flux.
ASOs have been suggested as a potential therapeutic for LRRK2-PD, with many hypothesizing that targeting the increased LRRK2 levels and kinase activity will be beneficial to pathology. Although ASO-mediated knockdown of Lrrk2 in this study did ameliorate the G2019S phenotypes described, it also decreased many of the functional readouts in both B6 and WTOE pMacs, and the consequences of these effects on key immune functions such as infection control will need to be considered carefully. We have already discussed that alterations in pathogen control need to be explored regarding Lrrk2 ASOs and LTF in G2019S pMacs, and it therefore follows that this could have significant consequences on the use of Lrrk2-targeting approaches for therapies. If LRRK2 plays a critical role in pathogen and infection control in a pathogen- or tissue-specific manner, it may mean that LRRK2-targeting therapies may need to be limited to mutant LRRK2 carriers and/or may need to be delivered in a compartment-targeted manner, as opposed to systemically to avoid deleterious effects in tissues and cell types where LRRK2 is crucial for immune responses. Indeed, the LRRK2 ASO currently being tested in the clinic is delivered via intrathecal injection directly into the cerebral spinal fluid, thus limiting systemic exposure (NCT03976349).
Materials and methods
Animals
BAC transgenic mouse strains overexpressing either mouse mutant G2019S-Lrrk2 (G2019S) or mouse WT Lrrk2 (WTOE) have previously been characterised24,25 and were maintained in the McKnight Brain Institute vivarium (University of Florida [UF]) at 22°C at 60%–70% humidity and animals were kept in a 12 h light/dark cycle. C57BL/6 littermate controls were used for all studies, with G2019S/WTOE and C57BL/6 controls cohoused. All animal procedures were approved by the UF Institutional Animal Care and Use Committee and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996. Male mice were aged to 8–10 weeks and sacrificed via cervical dislocation.
Harvesting and culturing of pMacs and ex-vivo stimulation of non-nucleofected cells
pMacs were harvested from mice which had received a 1 mL intraperitoneal administration of 3% Brewer thioglycolate broth 72 h prior collection. Mice also received sustained-release buprenorphine every 48 h for pain relief. Mice were sacrificed via cervical dislocation and abdomen sprayed with 70% ethanol. Skin of the abdomen was split along the midline, taking care to avoid puncturing or cutting the abdominal cavity. Ten milliliters of cold RPMI media (11875119; Gibco) was injected into the peritoneal cavity using a 27G needle. After gentle massaging of the peritoneal cavity, as much fluid was withdrawn as possible from the peritoneal cavity using a 25G needle and 10 mL syringe. Aspirated fluid was passed through a 70 μM nylon filter onto 50 mL falcon and pre-wet with 5 mL HBSS−/− (Hank’s balanced salt solution). Filters were then washed twice with 5 mL HBSS−/− and then tubes spun at 400 × g for 5 min at 4°C. Supernatant was aspirated and pellet resuspended in 3 mL pre-warmed growth media (RPMI, 10% fetal bovine serum [FBS], 1% penicillin-streptomycin). Cells were counted and viability was recorded using trypan blue exclusion on an automated cell counter (Countess; Thermo Fisher Scientific). Volume growth medium was adjusted so that cells were plated at 5 × 105/mL in 6-, 24-, or 96-well plates depending on the intended assay. Cells were incubated at 37°C, 5% CO2 for a minimum of 2 h to allow macrophages to adhere. Wells were washed twice with sterile PBS to remove non-adherent cells and new, pre-warmed growth media added. For cells requiring ex vivo stimulation, 100 U IFNγ (R&D Systems) or vehicle (H2O) was added for 18 h. For co-treatments, a final concentration of 10 μM nocodazole (Sigma-Aldrich), 40 nM bafilomycin A1 (Sigma-Aldrich), 100 nM Torin1 (Calbiochem) and 100 nM PF360 (MedChem) was used.
Nucleofection and plating of pMacs
pMacs were harvested from mice as previously described. Once passed through 70 μM nylon filter, tubes spun at 90 × g for 10 min at 4°C. Cells were resuspended and counted as previously described. Cells were aliquoted into 50 mL falcons with 1 × 106 cells per nucleofection reaction. Cells were spun at 90 × g for 10 min at 4°C. Supernatant was carefully aspirated so not to disturb the cell pellet, and cells resuspended in nucleofection buffer (acclimated to room temperature; P2 Primary Cell 4D-Nucleofector X Kit L, V4XP-2024; Lonza) containing 1 μM Lrrk2 or control ASO per 100 μL (sequences are detailed in Table S1), to a final concentration of 1 × 106 cells per 100 μL. 100 μL of cells were transferred to each Nucleocuvette, which was then placed into a 4D-Nucleofector X Unit (Lonza) and pulsed using the CM 138 pulse code. After nucleofection, 400 μL of growth media (acclimated in incubator 1 h prior) was added to each Nucleocuvette and cells transferred to plates pre-coated with poly-D-lysine (Sigma-Aldrich). Cells were left to incubate for 24 h, after which medium was aspirated, cells were washed, and assays were started as previously described.
Flow cytometry
One hour prior to collection, pan-cathepsin probe BMV109 (Vergent Bioscience) and DQ Red BSA (Invitrogen) were added to each well at a final concentration of 1 μM and 10 μg/mL, respectively, and cells were incubated at 37°C for 1 h. Cells were then washed 3 times in sterile PBS, harvested, and transferred to a V-bottom 96-well plate (CLS3896-48EA; Sigma-Aldrich) and centrifuged at 300 × g for 5 min at 4°C. Cells were resuspended in 50 μL of PBS containing diluted fluorophore-conjugated antibodies (see Table S2) and incubated in the dark at 4°C for 20 min. Cells were centrifuged at 300 × g for 5 min at 4°C and washed twice in PBS. Cells were fixed in 50 μL 1% paraformaldehyde (PFA) at 4°C in the dark for 30 min. Cells were centrifuged at 300 × g for 5 min and resuspended in 200 μL fluorescence-activated cell sorting (FACS) buffer (PBS, 0.5 mM EDTA, 0.1% sodium azide). Cells were taken for flow cytometry on a Macs Quant Analyzer (Miltenyi Biotec) or BD LSR Fortessa Cell Analyzer. A minimum of 100,000 events were captured per sample and data were analyzed using FlowJo version 10.6.2 software (BD Biosciences). When validating flow cytometry panels and antibodies, fluorescence minus one controls (FMOCs) were used to set gates and isotype controls were used to ensure antibody-specific binding.
Ea(52–68) uptake assay
MHC II Ea chain (Ea) (52–68) peptide (AnaSpec) was reconstituted in sterile distilled H2O to a final concentrate of 1 mg/mL. Once pMacs had adhered to plates, 5 μg per well was added in growth medium. Cells were incubated for 18 h and taken forward for flow cytometry.
Cytokine release measurements via Mesoscale discovery electrochemiluminescence
V-PLEX mouse pro-inflammatory panel 1 kit (K15048D Merck Sharpe & Dohme [MSD]) was used to quantify cytokines in conditioned medium from pMacs. Medium was diluted 1:1 with MSD kit diluent and incubated at room temperature in the provided MSD plate with capture antibodies for 2 h as per the manufacturer’s instructions. Plates were then washed 3 times with PBS with 0.1% Tween 20 and detection antibodies conjugated with electrochemiluminescent labels were added and incubated at room temperature for another 2 h. After 3 washes with PBS containing 0.05% Tween 20, MSD read buffer was diluted to 2× and added, and the plates were loaded into the QuickPlex MSD instrument for quantification. Results were normalized to total live cell counts as measured using flow cytometry.
Immunoblotting
Medium was aspirated and cells were washed in PBS and lysed in RIPA buffer (50 mM Tris [pH 8], 150 mM NaCl, 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS). Cell lysates were then centrifuged at 10,000 × g for 10 min at 4°C. 6X Laemmli sample buffer was added (12% SDS, 30% β-mercaptoethanol, 60% glycerol, 0.012% bromophenol blue, and 375 mM Tris [pH 6.8]), and samples were reduced and denatured at 95°C for 5 min. Samples were loaded into 4%–20% Criterion Tris-HCl polyacrylamide gels (Bio-Rad) alongside Precision plus protein dual-color ladder (Bio-Rad) to determine target protein molecular weight. Electrophoresis was performed at 100 V for ∼60 min, and proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using a Trans-Blot Turbo Transfer System (Bio-Rad) which uses Trans-Blot Turbo Midi PVDF transfer packs (Bio-Rad) in accordance to manufacturer’s instructions. Prior to blocking, total protein was measured using Revert total protein stain (Licor) and imaged on the Odyssey FC imaging system (Licor). Membranes were then blocked in 5% non-fat milk in TBS/0.1% Tween 20 (TBS-T) for 1 h at room temperate and subsequently incubated with primary antibody (see Table S3) in blocking solution overnight at 4°C. Membranes were washed with TBS-T (3 × 5 min) and incubated in horseradish peroxidase (HRP)-conjugated secondary antibody (1:1,000) (Bio-Rad) in blocking solution for 1 h. Membranes were washed in TBS-T (3 × 5 min) and developed using SuperSignal West Femto/Pico (Thermo Fisher Scientific). Membranes were imaged using the Odyssey FC imaging system and quantified using Image Studio Lite version 5.2 (Licor).
NanoString-based mRNA expression analysis of lysosomal and immune-related genes
RNA from approximately 2-4 x 106 cells was isolated. RNase Easy mini kit (Qiagen) was used according to the manufacturer’s instructions. Briefly, 10 μL β-mercaptoethanol was added to every 1 mL of RLT buffer, and 350 μL was added to each well and cells were homogenized manually with a Mini Cell Scraper. Cell lysate was transferred to an RNase-free Eppendorf, and an equal volume of 70% ethanol was added to each sample. Samples were loaded into supplier columns and centrifuged at 11,000 x g for 30 s and flow through discarded. 350 μL RW1 buffer was added to each column and centrifuged at 11,000 x g for 15 s and flow through discarded. RPE buffer was added to columns, centrifuged at 11,000 x g for 30 s, repeated, and 30 μL RNase-free water was added and RNA eluted. RNA concentration was quantified and 260/230 and 260/280 recorded using a spectrophotometer. RIN values were assessed to ensure RNA integrity using Agilent RNA 6000 Nano Kit (Agilent) and Agilent 2100 Bioanalyzer.
Approximately 100 ng total RNA was hybridized to a custom panel for profiling 250 mouse genes within lysosomal, autophagy, and inflammatory pathways (Table S4) in a final volume of 15 μL at 65°C for 22 h according to the manufacturer’s protocol (NanoString Technologies, Inc., Seattle, WA). Gene expression profiling was measured on the NanoString nCounter system. Hybridized samples were processed on the NanoString nCounter Preparation Station using the high-sensitivity protocol, in which excess Capture and Reporter Probes were removed and probe-transcript complexes were immobilized on a streptavidin-coated cartridge and data collected on an nCounter digital analyzer (NanoString), following the manufacturer’s instructions.
Background level was determined by mean counts of eight negative control probes plus two SDs. Samples that contain <50% of probes above background, or that have imaging or positive control linearity flags, were excluded from further analysis. Probes that have raw counts below background in all samples were excluded from differential expression analysis to avoid false positive results. Data were normalized by geometric mean of housekeeping genes. All statistical analyses were performed on log2-transformed normalized counts.
Pre-processing and normalization of the raw counts was performed with nSolver Analysis Software version 4.0 (www.nanostring.com). The 6 spiked-in RNA positive control and the 8 negative controls present in the panel were used to confirm the quality of the run. Data were analyzed using either ROSALIND (https://rosalind.onramp.bio/), with a HyperScale architecture developed by ROSALIND, Inc. (San Diego, CA), or in nSolver Analysis Software version 4.0. Fold changes and p values are calculated using the optimal method, as described in the nCounter Advanced Analysis 2.0 User Manual. p value adjustment is performed using the Benjamini-Hochberg method of estimating false discovery rates (FDRs). Heatmaps of DEGs were constructed using nSolver Analysis Software version 4.0. Identified DEGs were copied into ShinyGO version 0.76.3 to identify pathways in which these genes are functional. Functional enrichment could not be carried out, because of a pre-enriched custom code set for lysosomal and inflammatory genes. Therefore, the number of DEGs present in identified pathways is reported, and enrichment p values are not reported.
Dextran-AF488 labeling of lysosomal tubules and microscopy
pMacs were pulsed with 0.5 mg/mL dextran Alexa Fluor 488 or 564 (Invitrogen) for 1 h, followed by a pulse period of 2 h to ensure loading of dextran into lysosomes. Cells were then chased in growth medium containing 100 U IFNγ or vehicle for 2 h to induce LTF. Cells were imaged live using either a Leica THUNDER imager or Zeiss Confocal LSM800 (ICBR, UF) at 60/63× magnification. Image analysis was performed using CellProfiler version 4.2.5.
Intracellular and extracellular MHC-II immunostaining and microscopy
Methods for intracellular and extracellular MHC-II immunostaining were adapted from Hennies et al.33 Cells were washed 3 times with DPBS+/+ and incubated with 25 μg/mL APC-MHC-II (BioLegend) in DPBS+/+ containing FcR-blocking reagent for 30 min at room temperature. Cells were then washed 3 times with DPBS+/+ and fixed by incubation in 4% PFA for 10 min at room temperature and then washed 3 times with DPBS+/+. Fixed cells were permeabilized with permeabilization buffer (eBiosciences) on ice for 15 min. PE-610-MHC-II (25 μg/mL; BioLegend) was spiked in and cells incubated for 30 min at room temperature. Cells were then washed 3 times in DPBS+/+ and incubated in 1 μg/mL DAPI (Life Technologies) for 10 min at room temperature in DPBS+/+. Cells were imaged using a Leica THUNDER imager at 60× magnification. Image analysis was performed using CellProfiler version 4.2.5. Using the IdentifyPrimaryObject module in CellProfiler, icMHC was identified. From this, intensity distribution was measured via the CellProfiler MeasureObjectIntensityDistribution module. Here, objects were segmented into bins and each bin intensity measured. icMHC was considered perinuclear if the innermost bin had greater intensity than the outermost bin. The percentage of cells with perinuclear icMHC was averaged across images per animal for each treatment condition. A total of 6 images were collected per animals for each treatment.
Dextran and transferrin labeling
pMacs were pulsed with 0.5 mg/mL dextran or transferrin Alexa Fluor 488 (Invitrogen) for 1 h at 37°C. Cells were washed 3 times with DPBS+/+, fixed in 4% PFA for 10 min at room temperature, and incubated in 1 μg/mL DAPI (Life Technologies) for 10 min at room temperature in DPBS+/+. Cells were imaged on an EVOS M7000 at 20× magnification. Image analysis was performed using CellProfiler version 4.2.5.
Statistics and data analysis
Data and statistical analyses were performed using IBM SPSS Statistics 27 or GraphPad Prism 9. For assessing differences between groups, data were analyzed by either 1-way or 2-way ANOVA or Student’s t test. In instances when data did not fit parametric assumptions, Kruskal-Wallis non-parametric ANOVA was used. Post hoc tests following ANOVAs were conducted using Tukey HSD or Bonferroni correction. Two-tailed levels of significance were used, and p values <0.05 were considered to indicate statistical significance. Graphs are depicted by means ± SEM.
Data and code availability
All data are available in the article. Correspondence and requests for materials should be addressed to M.G.T.
Acknowledgments
We thank members of the Tansey lab for useful discussions and edits of the manuscript. We thank Biogen/Ionis for supplying all Lrrk2 and control ASOs. We thank the UF Interdisciplinary Center for Biotechnology Research (UF | ICBR) for use of flow cytometry facilities and advice. Partial funding for this work was derived from awards from the Bright Focus Fellowship (grant A2021017F to R.L.W.), a Moonshot Award from the Fixel Institute for Neurological Diseases (R.L.W.), NIH/NINDS (National Institute of Neurological Disorders and Stroke) grant RF1NS128800 (M.G.T.), and NIH National Institute on Aging grant 1RF1AG057247 (M.G.T.).
Author contributions
R.L.W. was responsible for experimental design, performing experiments, plotting and analyzing data, data interpretation, and drafting and editing manuscript. J.R.M., H.A.S., and D.A.G. were responsible for optimizing Lrrk2 ASOs. H.A.S. was responsible for running all MSD-experiments. H.K. and W.D.H. were responsible for providing Lrrk2 and controls ASOs. M.G.T. was responsible for funding acquisition, supervision, and project administration. All authors participated in editing the manuscript.
Declaration of interests
W.D.H. is an employee of Biogen, and H.K. is an employee of Ionis Pharmaceuticals, where ASOs are currently under development for neurological indications.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.102064.
Supplemental information
Custom panel for profiling 250 mouse genes within lysosomal, autophagy, and inflammatory pathways.
References
- 1.Von Campenhausen S., Bornschein B., Wick R., Bötzel K., Sampaio C., Poewe W., Oertel W., Siebert U., Berger K., Dodel R. Prevalence and incidence of Parkinson's disease in Europe. Eur. Neuropsychopharmacol. 2005;15:473–490. doi: 10.1016/j.euroneuro.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Marras C., Beck J.C., Bower J.H., Roberts E., Ritz B., Ross G.W., Abbott R.D., Savica R., Van Den Eeden S.K., Willis A.W., et al. Prevalence of Parkinson's disease across North America. NPJ Parkinsons Dis. 2018;4:21. doi: 10.1038/s41531-018-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang W., Hamilton J.L., Kopil C., Beck J.C., Tanner C.M., Albin R.L., Ray Dorsey E., Dahodwala N., Cintina I., Hogan P., Thompson T. Current and projected future economic burden of Parkinson's disease in the U.S. NPJ Parkinsons Dis. 2020;6:15. doi: 10.1038/s41531-020-0117-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., Van Der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 5.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook D.A., Kannarkat G.T., Cintron A.F., Butkovich L.M., Fraser K.B., Chang J., Grigoryan N., Factor S.A., West A.B., Boss J.M., Tansey M.G. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinsons Dis. 2017;3:11. doi: 10.1038/s41531-017-0010-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atashrazm F., Hammond D., Perera G., Bolliger M.F., Matar E., Halliday G.M., Schüle B., Lewis S.J.G., Nichols R.J., Dzamko N. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson's disease patients. Mov. Disord. 2019;34:406–415. doi: 10.1002/mds.27601. [DOI] [PubMed] [Google Scholar]
- 8.Zhao H.T., John N., Delic V., Ikeda-Lee K., Kim A., Weihofen A., Swayze E.E., Kordasiewicz H.B., West A.B., Volpicelli-Daley L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Ther. Nucleic Acids. 2017;8:508–519. doi: 10.1016/j.omtn.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korecka J.A., Thomas R., Christensen D.P., Hinrich A.J., Ferrari E.J., Levy S.A., Hastings M.L., Hallett P.J., Isacson O. Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson's disease patient fibroblasts. Hum. Mol. Genet. 2019;28:3232–3243. doi: 10.1093/hmg/ddz126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korecka J.A., Talbot S., Osborn T.M., De Leeuw S.M., Levy S.A., Ferrari E.J., Moskites A., Atkinson E., Jodelka F.M., Hinrich A.J., et al. Neurite Collapse and Altered ER Ca2+ Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem Cell Rep. 2019;12:29–41. doi: 10.1016/j.stemcr.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korecka J.A., Thomas R., Hinrich A.J., Moskites A.M., Macbain Z.K., Hallett P.J., Isacson O., Hastings M.L. Splice-Switching Antisense Oligonucleotides Reduce LRRK2 Kinase Activity in Human LRRK2 Transgenic Mice. Mol. Ther. Nucleic Acids. 2020;21:623–635. doi: 10.1016/j.omtn.2020.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardet A., Benita Y., Li C., Sands B.E., Ballester I., Stevens C., Korzenik J.R., Rioux J.D., Daly M.J., Xavier R.J., Podolsky D.K. LRRK2 Is Involved in the IFN- Response and Host Response to Pathogens. J. Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallings R.L., Hughes L.P., Staley H.A., Simon Z.D., McFarland N.R., Alcalay R.N., Garrido A., Martí M.J., Sarró E.T., Dzamko N., Tansey M.G. WHOPPA Enables Parallel Assessment of Leucine-Rich Repeat Kinase 2 and Glucocerebrosidase Enzymatic Activity in Parkinson's Disease Monocytes. Front. Cell. Neurosci. 2022;16 doi: 10.3389/fncel.2022.892899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallings R.L., Tansey M.G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 2019;47:1581–1595. doi: 10.1042/BST20180463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W., Liu X., Li Y., Zhao J., Liu Z., Hu Z., Wang Y., Yao Y., Miller A.W., Su B., et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 2017;214:3051–3066. doi: 10.1084/jem.20170014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q., Pan Y., Yan R., Zeng B., Wang H., Zhang X., Li W., Wei H., Liu Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015;16:918–926. doi: 10.1038/ni.3233. [DOI] [PubMed] [Google Scholar]
- 17.Eguchi T., Kuwahara T., Sakurai M., Komori T., Fujimoto T., Ito G., Yoshimura S.I., Harada A., Fukuda M., Koike M., Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA. 2018;115:E9115–E9124. doi: 10.1073/pnas.1812196115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alegre-Abarrategui J., Christian H., Lufino M.M.P., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schapansky J., Nardozzi J.D., Felizia F., Lavoie M.J. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet. 2014;23:4201–4214. doi: 10.1093/hmg/ddu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonet-Ponce L., Beilina A., Williamson C.D., Lindberg E., Kluss J.H., Saez-Atienzar S., Landeck N., Kumaran R., Mamais A., Bleck C.K.E., et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020;6:eabb2454. doi: 10.1126/sciadv.abb2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hipolito V.E., Ospina-Escobar E., Botelho R.J. Lysosome remodelling and adaptation during phagocyte activation. Cell Microbiol. 2018;20 doi: 10.1111/cmi.12824. [DOI] [PubMed] [Google Scholar]
- 22.Mantegazza A.R., Zajac A.L., Twelvetrees A., Holzbaur E.L.F., Amigorena S., Marks M.S. TLR-dependent phagosome tubulation in dendritic cells promotes phagosome cross-talk to optimize MHC-II antigen presentation. Proc. Natl. Acad. Sci. USA. 2014;111:15508–15513. doi: 10.1073/pnas.1412998111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K.S., Marcogliese P.C., Yang J., Callaghan S.M., Resende V., Abdel-Messih E., Marras C., Visanji N.P., Huang J., Schlossmacher M.G., et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc. Natl. Acad. Sci. USA. 2018;115:E5164–E5173. doi: 10.1073/pnas.1718946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X., Patel J.C., Wang J., Avshalumov M.V., Nicholson C., Buxbaum J.D., Elder G.A., Rice M.E., Yue Z. Enhanced Striatal Dopamine Transmission and Motor Performance with LRRK2 Overexpression in Mice Is Eliminated by Familial Parkinson's Disease Mutation G2019S. J. Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yue Z. Genetic mouse models for understanding LRRK2 biology, pathology and pre-clinical application. Parkinsonism Relat. Disorders. 2012;18:S180–S182. doi: 10.1016/S1353-8020(11)70056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudensky A.Y., Rath S., Preston-Hurlburt P., Murphy D.B., Janeway C.A. On the complexity of self. Nature. 1991;353:660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- 27.Hakimi M., Selvanantham T., Swinton E., Padmore R.F., Tong Y., Kabbach G., Venderova K., Girardin S.E., Bulman D.E., Scherzer C.R., et al. Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural. Transm. 2011;118:795–808. doi: 10.1007/s00702-011-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moehle M.S., Webber P.J., Tse T., Sukar N., Standaert D.G., DeSilva T.M., Cowell R.M., West A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozina E., Sadasivan S., Jiao Y., Dou Y., Ma Z., Tan H., Kodali K., Shaw T., Peng J., Smeyne R.J. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. 2018;141:1753–1769. doi: 10.1093/brain/awy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng Z., Zhang S., Bustos D., Kleinheinz T., Le Pichon C.E., Dominguez S.L., Solanoy H.O., Drummond J., Zhang X., Ding X., et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 2012;4:164ra161. doi: 10.1126/scitranslmed.3004485. [DOI] [PubMed] [Google Scholar]
- 31.Itano A.A., McSorley S.J., Reinhardt R.L., Ehst B.D., Ingulli E., Rudensky A.Y., Jenkins M.K. Distinct Dendritic Cell Populations Sequentially Present Antigen to CD4 T Cells and Stimulate Different Aspects of Cell-Mediated Immunity. Immunity. 2003;19:47–57. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- 32.Watts C. The endosome-lysosome pathway and information generation in the immune system. Biochim. Biophys. Acta. 2012;1824:14–21. doi: 10.1016/j.bbapap.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hennies C.M., Lehn M.A., Janssen E.M. Quantitating MHC class II trafficking in primary dendritic cells using imaging flow cytometry. J. Immunol. Methods. 2015;423:18–28. doi: 10.1016/j.jim.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saric A., Hipolito V.E.B., Kay J.G., Canton J., Antonescu C.N., Botelho R.J. mTOR controls lysosome tubulation and antigen presentation in macrophages and dendritic cells. Mol. Biol. Cell. 2016;27:321–333. doi: 10.1091/mbc.E15-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mir R., Tonelli F., Lis P., Macartney T., Polinski N.K., Martinez T.N., Chou M.-Y., Howden A.J.M., König T., Hotzy C., et al. The Parkinson's disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 2018;475:1861–1883. doi: 10.1042/BCJ20180248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallings R., Connor-Robson N., Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet. 2019;28:2696–2710. doi: 10.1093/hmg/ddz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steger M., Tonelli F., Ito G., Davies P., Trost M., Vetter M., Wachter S., Lorentzen E., Duddy G., Wilson S., et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5:e12813. doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimprich A., Benet-Pagès A., Struhal W., Graf E., Eck S.H., Offman M.N., Haubenberger D., Spielberger S., Schulte E.C., Lichtner P., et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A., et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nalls M.A., Pankratz N., Lill C.M., Do C.B., Hernandez D.G., Saad M., Destefano A.L., Kara E., Bras J., Sharma M., et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 42.Cruts M., Gijselinck I., van der Zee J., Engelborghs S., Wils H., Pirici D., Rademakers R., Vandenberghe R., Dermaut B., Martin J.J., et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 43.Benussi L., Binetti G., Sina E., Gigola L., Bettecken T., Meitinger T., Ghidoni R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging. 2008;29:427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 44.Escrevente C., Bento-Lopes L., Ramalho J.S., Barral D.C. Rab11 is required for lysosome exocytosis through the interaction with Rab3a, Sec15 and GRAB. J. Cell Sci. 2021;134 doi: 10.1242/jcs.246694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roh K., Noh J., Kim Y., Jang Y., Kim J., Choi H., Lee Y., Ji M., Kang D., Kim M.-S., et al. Lysosomal control of senescence and inflammation through cholesterol partitioning. Nat. Metab. 2023;5:398–413. doi: 10.1038/s42255-023-00747-5. [DOI] [PubMed] [Google Scholar]
- 46.Kinoshita D., Sakurai C., Morita M., Tsunematsu M., Hori N., Hatsuzawa K. Syntaxin 11 regulates the stimulus-dependent transport of Toll-like receptor 4 to the plasma membrane by cooperating with SNAP-23 in macrophages. Mol. Biol. Cell. 2019;30:1085–1097. doi: 10.1091/mbc.E18-10-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araki M., Kontani K. Regulation of lysosomal positioning via TMEM55B phosphorylation. J. Biochem. 2021;169:507–509. doi: 10.1093/jb/mvab013. [DOI] [PubMed] [Google Scholar]
- 48.Bonet-Ponce L., Cookson M.R. The endoplasmic reticulum contributes to lysosomal tubulation/sorting driven by LRRK2. Mol. Biol. Cell. 2022;33:ar124. doi: 10.1091/mbc.E22-04-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suresh B., Saminathan A., Chakraborty K., Zajac M., Cui C., Becker L., Krishnan Y. Tubular lysosomes harbor active ion gradients and poise macrophages for phagocytosis. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2113174118. e2113174118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohnert K.A., Johnson A.E. Branching Off: New Insight Into Lysosomes as Tubular Organelles. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.863922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frost B., Jacks R.L., Diamond M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J.T.H., Kerr M.C., Karunaratne S., Jeanes A., Yap A.S., Teasdale R.D. The SNX-PX-BAR Family in Macropinocytosis: The Regulation of Macropinosome Formation by SNX-PX-BAR Proteins. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z., Xu E., Zhao H.T., Cole T., West A.B. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. 2020;39:e104862. doi: 10.15252/embj.2020104862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L., Wan T., Wan M., Liu B., Cheng R., Zhang R. The effect of the size of fluorescent dextran on its endocytic pathway. Cell Biol. Int. 2015;39:531–539. doi: 10.1002/cbin.10424. [DOI] [PubMed] [Google Scholar]
- 55.Vyas J.M., Kim Y.M., Artavanis-Tsakonas K., Love J.C., Van der Veen A.G., Ploegh H.L. Tubulation of class II MHC compartments is microtubule dependent and involves multiple endolysosomal membrane proteins in primary dendritic cells. J. Immunol. 2007;178:7199–7210. doi: 10.4049/jimmunol.178.11.7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ito K., Araki M., Katai Y., Nishimura Y., Imotani S., Inoue H., Ito G., Tomita T. Pathogenic LRRK2 compromises the subcellular distribution of lysosomes in a Rab12-RILPL1 -dependent manner. Faseb. J. 2023;37:e22930. doi: 10.1096/fj.202200780RR. [DOI] [PubMed] [Google Scholar]
- 57.Biosa A., Trancikova A., Civiero L., Glauser L., Bubacco L., Greggio E., Moore D.J. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson's disease-associated LRRK2. Hum. Mol. Genet. 2013;22:1140–1156. doi: 10.1093/hmg/dds522. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen A.P.T., Moore D.J. Understanding the GTPase Activity of LRRK2: Regulation, Function, and Neurotoxicity. Adv. Neurobiol. 2017;14:71–88. doi: 10.1007/978-3-319-49969-7_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nazish I., Arber C., Piers T.M., Warner T.T., Hardy J.A., Lewis P.A., Pocock J.M., Bandopadhyay R. Abrogation of LRRK2 dependent Rab10 phosphorylation with TLR4 activation and alterations in evoked cytokine release in immune cells. Neurochem. Int. 2021;147 doi: 10.1016/j.neuint.2021.105070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanin D.E., Prendergast C.T., Mountford A.P. IL-10 Production in Macrophages Is Regulated by a TLR-Driven CREB-Mediated Mechanism That Is Linked to Genes Involved in Cell Metabolism. J. Immunol. 2015;195:1218–1232. doi: 10.4049/jimmunol.1500146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daseke M.J., 2nd, Tenkorang-Impraim M.A.A., Ma Y., Chalise U., Konfrst S.R., Garrett M.R., DeLeon-Pennell K.Y., Lindsey M.L. Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. J. Mol. Cell. Cardiol. 2020;145:112–121. doi: 10.1016/j.yjmcc.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X., Körner H., Liu X. Susceptibility to Intracellular Infections: Contributions of TNF to Immune Defense. Front. Microbiol. 2020;11:1643. doi: 10.3389/fmicb.2020.01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shutinoski B., Hakimi M., Harmsen I.E., Lunn M., Rocha J., Lengacher N., Zhou Y.Y., Khan J., Nguyen A., Hake-Volling Q., et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 2019;11:eaas9292. doi: 10.1126/scitranslmed.aas9292. [DOI] [PubMed] [Google Scholar]
- 64.Härtlova A., Herbst S., Peltier J., Rodgers A., Bilkei-Gorzo O., Fearns A., Dill B.D., Lee H., Flynn R., Cowley S.A., et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 2018;37 doi: 10.15252/embj.201798694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park M.D., Silvin A., Ginhoux F., Merad M. Macrophages in health and disease. Cell. 2022;185:4259–4279. doi: 10.1016/j.cell.2022.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herbst S., Gutierrez M.G. LRRK2 in Infection: Friend or Foe? ACS Infect. Dis. 2019;5:809–815. doi: 10.1021/acsinfecdis.9b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tong Y., Giaime E., Yamaguchi H., Ichimura T., Liu Y., Si H., Cai H., Bonventre J.V., Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012;7:2. doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herrick M.K., Tansey M.G. Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Parkinsons Dis. 2021;7:26. doi: 10.1038/s41531-021-00170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brady O.A., Martina J.A., Puertollano R. Emerging roles for TFEB in the immune response and inflammation. Autophagy. 2018;14:181–189. doi: 10.1080/15548627.2017.1313943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murray R.Z., Stow J.L. Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Front. Immunol. 2014;5:538. doi: 10.3389/fimmu.2014.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steger M., Diez F., Dhekne H.S., Lis P., Nirujogi R.S., Karayel O., Tonelli F., Martinez T.N., Lorentzen E., Pfeffer S.R., et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. 2017;6:e31012. doi: 10.7554/eLife.31012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manzoni C. The LRRK2-macroautophagy axis and its relevance to Parkinson's disease. Biochem. Soc. Trans. 2017;45:155–162. doi: 10.1042/BST20160265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J., Kundu M., Viollet B., Guan K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laplante M., Sabatini D.M. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Inpanathan S., Botelho R.J. The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Biol. 2019;7:113. doi: 10.3389/fcell.2019.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Custom panel for profiling 250 mouse genes within lysosomal, autophagy, and inflammatory pathways.
Data Availability Statement
All data are available in the article. Correspondence and requests for materials should be addressed to M.G.T.