

Summary
Pulmonary fibrosis frequently occurs in rheumatic conditions, particularly systemic sclerosis-associated interstitial lung disease (SSc-ILD). The pathology involves cell transformation into interstitial structures and collagen accumulation. CD4+LAG3+T cells, known for immune inhibition, are relevant in autoimmunity. This study investigates CD4+LAG3+T cells in SSc-ILD. Clinical analysis revealed a correlation between CD4+LAG3+T cells and interleukin-6 (IL-6) and erythrocyte sedimentation rate (ESR). Using primary human lung fibroblasts (pHLFs) and murine bone marrow-derived macrophages (BMDMs), we showed that CD4+LAG3+T cells secreted TGF-β3 inhibits TGF-β1-induced mesenchymal transformation, modulates cellular function, and reduces collagen release. In mouse models, CD4+LAG3+T cells exhibited potential in alleviating bleomycin-induced pulmonary fibrosis. This study emphasizes CD4+LAG3+T cells’ therapeutic promise against fibrosis and proposes their role as biomarkers.
Subject areas: Health sciences, Rheumatology, Fibrosis
Graphical abstract
Highlights
-
•
Reduced CD4+LAG3+T cell in SSc-ILD correlate negatively with IL-6
-
•
CD4+LAG3+T cells block TGF-β1-induced mesenchymal transition in cells
-
•
CD4+LAG3+T cells hinder cell function and collagen secretion due to TGF-β1
-
•
CD4+LAG3+T cells ameliorate bleomycin-induced mouse pulmonary fibrosis
Health sciences; Rheumatology; Fibrosis
Introduction
Pulmonary fibrosis stands as a recurrent and intricate clinical ailment characterized by its multifaceted etiologies. The disease is invariably accompanied by a state of prolonged immune-mediated inflammation, heralding the activation of myofibroblasts. These myofibroblasts assume the mantle of principal effectors in the propagation of pulmonary fibrosis, primarily originating from lung fibroblasts and epithelial cells. They orchestrate the excessive secretion of collagen, a cumulatively aberrant deposition within the pulmonary milieu. The accrued collagen precipitates structural detriment to both the pulmonary mesenchymal and alveolar architecture, precipitating a cascade of events culminating in the erosion of lung ventilatory function, culminating inexorably in respiratory insufficiency.1,2
In the realm of clinical observation, numerous rheumatic disorders, encompassing systemic sclerosis, dermatomyositis, rheumatoid arthritis, and Sjogren’s syndrome, harbor the capacity to incite mesenchymal lung maladies. The natural progression of such conditions often gravitates toward the eventual emergence of pulmonary fibrosis, and at an advanced stage, even respiratory failure.3,4 The dearth of efficacious therapeutic modalities engenders formidable challenges for afflicted patients. Regulatory approval from the US Food and Drug Administration has been conferred upon agents like pirfenidone and nintedanib for the clinical management of pulmonary fibrosis.5,6 These agents operate by curtailing the transition of lung fibroblasts into myofibroblasts. Notably, the preponderant pro-inflammatory mediator instrumental in promoting myofibroblast transition is TGF-β1.7,8 This growth factor consortium orchestrates a spectrum of cellular differentiation processes inclusive of epithelial-mesenchymal transition, T cell differentiation, and macrophage polarization.9 Amongst these, TGF-β1 triggers the upregulation of markers associated with mesenchymal transition within lung fibroblasts, typified by α-smooth muscle actin (α-SMA) and fibronectin. These alterations are accompanied by pronounced changes in cytoskeletal architecture, heightened migratory and proliferative capacities, alongside an augmented propensity for collagen synthesis.10,11
Dissimilar to the pro-fibrotic attributes attributed to TGF-β1, TGF-β3 has garnered consensus in its propensity to counteract fibrosis and impede the undue accrual of extracellular matrix components.12 As propounded by Karamichos et al., the potential of TGF-β3 as an anti-fibrotic agent has been cogently established, manifesting its ability to ameliorate corneal fibrosis through a regressive course of action. This capacity renders TGF-β3 a potent candidate for the amelioration of fibroblast and matrix-mediated fibrotic elements, effectively assuming the role of a “rescue” modality.2 In parallel, the investigations of Xu et al. culminate in the efficacious application of TGF-β3 as a therapeutic intervention, effectively mitigating the severity of pulmonary fibrosis in murine models induced by radiation exposure.13 In the spectrum of cutaneous fibrotic phenomena, TGF-β3 emerges as a discerning agent, effectively attenuating scarring manifestations.14 Correspondingly, therapeutic intervention featuring TGF-β3 has been underscored as instrumental in abrogating the proliferative proclivities of smooth muscle cells.15 This phenomenon, reasonably inferred, may be rooted in the augmentation of G1 cell-cycle arrest, thereby ostensibly bolstering the process of DNA damage repair. Parallel instances of analogous import are extant within enterocyte crypt cells, bolstering the notion of TGF-β3’s pervasive influence.16 It hence follows that TGF-β3 assumes a central determinant role, intricately choreographing the dynamics of cellular transition and proliferation.
LAG3 stands as a sanguine target within the landscape of cancer therapeutics, accentuated by its pivotal stature as the second most salient target subsequent to PD1.17 Clinical evaluations pertinent to the efficacy of LAG3-targeted interventions are presently underway, a salient trajectory endorsed by the pervasive abnormal activation of the LAG3 pathway observed within neoplastic entities.18 Conversely, anomalies in both PD1 and LAG3 pathways are inextricably linked to the etiopathogenesis of autoimmune disorders.19,20 To counteract this, concerted endeavors are being directed toward the development of LAG3-specific agonists such as IMP761, poised to orchestrate upstream activation of T lymphocytes while concurrently assuaging autoimmune maladies.21 Comprising a subset of immune-negative regulatory entities, LAG3+T cells have garnered scrutiny owing to their secretion of TGF-β3 and additional immune-negative regulatory factors. Research spearheaded by Okamura et al. has unveiled that CD4+CD25−LAG3+ regulatory T cells evoke substantial TGF-β3 production through a mechanism reliant upon zinc finger transcription factor early growth response 2 (EGR2) and Fas-mediated modalities. Instances of these LAG3+Tregs being administered to murine models afflicted with lupus nephritis have evinced noteworthy attenuation in renal disease progression, a phenomenon notably countered by neutralizing antibodies targeting TGF-β3, thereby underscoring the therapeutic efficacy of LAG3+T cells vis-à-vis the ailment.22 Notably, the study by Gertel et al. found that CD4+LAG3+T cells decreased in patients with psoriatic arthritis.23 the LAG3+Tregs cell population was reduced in the context of rheumatoid arthritis.24 This posits that LAG3+Tregs could potentially deter the progression of autoimmune pathogenesis.
In the specific context of renal transplantation murine models, LAG3 deficiency has been implicated in fibrotic sequelae, albeit without impeding the instigation of tolerance induction toward spontaneously accepted allografts, hence emblematic of its plausible therapeutic role in regard to transplantation outcomes.25 A more nuanced interrogation is imperative to plumb the depths of immune dysregulation that foments the transition from inflammatory processes within tissues to the mantle of fibrotic transformation, an intricate orchestration observable within the terrain of rheumatic pathologies.
Considering these considerations, the current investigation undertook a multifaceted approach that amalgamated bioinformatics analyses with experiment methodologies. Our primary objective entailed a pioneering endeavor to discern the intricate nexus shared between CD4+LAG3+T cells and the milieu of SSc-ILD. Concomitant with this, we embarked upon a comprehensive exploration of the influences exerted by CD4+LAG3+T cells upon lung fibroblasts and macrophages within an in vitro milieu. This systematic inquiry led to an expansive elucidation of their manifold impacts upon the intricacies of the pulmonary fibrotic process. The comprehensive panorama thus delineated is substantiated by corroborative evidence stemming from in vivo experimentation conducted on animal models. This collective scholarly exertion culminates in the provision of novel empirical insights, thus furnishing a fresh vantage point and potential target within the broader landscape of immune regulatory processes intertwined with organ fibrogenesis.
Results
We collected data of 23 patients with SSc and 36 patients with SSc-ILD who visited Guangdong Provincial People's Hospital from 2021 to 2022 (Table 1). The patients’ lung tissues were examined by morphological staining and immunofluorescence (Figures S1A and S1B) and we analyzed the patients’ laboratory tests. The data amassed herein demonstrate that the affirmative occurrence of anti-Scl-70 antibodies among SSc patients reached 34.78%, whereas within the subset of SSc-ILD patients, this rate escalated to 58.33%. However, the statistical significance of this observation was not attained as denoted by the p value. The presence of anti-Scl-70 antibodies, a well-established hallmark of SSc, has been explored in relation to its potential linkage to ILD.26 Among the cohort of 23 SSc patients, 8 individuals (34.8%) exhibited poikiloderma, whereas a distinct contrast manifested within the SSc-ILD stratum where only 4 patients (11.1%) evidenced such dermatological manifestations. This accord between our observations and extant literature corroborates the etiological relevance of poikiloderma within the purview of SSc’s immunopathological processes and its consequential vascular compromise. Pertinent to the biochemical milieu, it was ascertained that 8.7% of SSc patients exhibited heightened ESR, while within the cohort of 36 SSc-ILD patients, 25 individuals (69.4%) demonstrated elevated ESR levels. A meticulous scrutiny of the data, as assessed by the χ2 test, renders evident a statistically significant distinction (χ2 = 5.563, p = 0.018) (see STAR Methods for additional details). ESR, functioning as a conventional non-specific biomarker, bears utility in gauging the activity of the disease as well as the extent of inflammatory engagement. It is conjectured that the persistence of inflammatory responses precipitates sustained morphological changes within the pulmonary tissue architecture. The cascade of lung inflammation serves as a stimulus for lung cells to unleash extracellular matrix constituents, typified by collagen, a phenomenon concomitant with fibrotic transformations. In concordance with the proinflammatory milieu, IL-6 occupies a prominent role, both as an instigator of inflammation and an orchestrator of alveolar wall fibrosis. Remarkably, the current study has unveiled a substantial correlation between elevated IL-6 levels and SSc-ILD patients. Precisely, 39.1% of SSc patients exhibited elevated IL-6 concentrations, while a notable 69.4% of SSc-ILD patients displayed a comparable elevation. This discrepancy is strikingly significant in terms of its statistical validation. Collectively, our observations suggest that augmented ESR, a constellation of clinical signs indicative of ILD in SSc patients, as well as the presence of positive anti-Scl-70 antibodies might collectively serve as predictive indicators for the presence of ILD among this patient cohort. A comprehensive summation of these findings is encapsulated within Table 2 for reference.
Table 1.
Basic information of SSc patients with and without ILD
| Element | Diagnosis |
χ2 | p value | ||
|---|---|---|---|---|---|
| SSc | SSc-ILD | ||||
| Gender | Female | 18 (78.3%) | 26 (72.2%) | 0.270 | 0.603 |
| Male | 5 (21.7%) | 10 (27.8%) | |||
| Age | <65 | 18 (78.3%) | 24 (66.7%) | 0.920 | 0.338 |
| ≥65 | 5 (21.7%) | 12 (33.3%) | |||
Data are represented as forms of count and percentage. Statistical significance was analysis by chi-square test for comparison between groups.
Table 2.
Comparison of laboratory findings in SSc patients with and without ILD
| Group | SSc | SSc-ILD | χ2 | p value |
|---|---|---|---|---|
| Dysphagia | 4 (17.4%) | 5 (13.9%) | 0.133 | 0.715 |
| Skin Sclerosis | 22 (95.7%) | 29 (80.6%) | 2.729 | 0.099 |
| Raynaud phenomenon | 19 (82.6%) | 28 (77.8%) | 0.202 | 0.653 |
| Arthritis | 8 (34.8%) | 17 (47.2%) | 0.889 | 0.346 |
| Poikiloderma | 8 (34.8%) | 4 (11.1%) | 4.853 | 0.028 |
| Cutaneous Ulcer | 6 (26.1%) | 3 (8.3%) | 3.422 | 0.064 |
| Myalgia | 3 (13.0%) | 1 (2.8%) | 2.34 | 0.126 |
| Hydropericardium | 4 (17.4%) | 3 (8.3%) | 1.101 | 0.294 |
| ANA positive | 21 (91.3%) | 35 (97.2%) | 1.018 | 0.313 |
| RF positive | 1 (4.3%) | 3 (8.3%) | 0.353 | 0.553 |
| Anti cent B antibody positive | 4 (17.4%) | 6 (16.7%) | 0.005 | 0.942 |
| Anti Scl-70 antibody positive | 7 (34.78%) | 19 (58.33%) | 2.842 | 0.092 |
| Urinary protein | 4 (17.4%) | 8 (22.2%) | 1.727 | 0.422 |
| AST | 2 (8.7%) | 8 (22.2%) | 1.824 | 0.177 |
| CK | 3 (13.0%) | 4 (11.1%) | 0.05 | 0.823 |
| LDH | 4 (17.4%) | 15 (41.7%) | 3.788 | 0.052 |
| CRP | 3 (13.0%) | 11 (30.6%) | 2.378 | 0.123 |
| ESR | 2 (8.7%) | 13 (36.1%) | 5.563 | 0.018 |
| IL-6 | 9 (39.1%) | 25(69.4%) | 5.281 | 0.022 |
Data are represented as forms of count and percentage. Statistical significance was analysis by chi-square test for comparison between groups.
Bioinformatics analysis of LAG3 and EGR2 expression in SSc-ILD disease
EGR2 serves as a pivotal transcription factor intricately engaged in the orchestration of T cell differentiation and activation processes. The literature delineates that deviations in EGR2 functionality are intimately linked to the etiology and progression of autoimmune disorders. LAG3, functioning as a counteractive regulatory molecule, contributes substantively to the regulation of immune retorts exhibited by T cells and immunocytes at large. Diminished LAG3 expression imparts an impetus for heightened autoimmune responses. Considering these insights, it is plausible to posit that EGR2 and LAG3 could potentially occupy pivotal roles within the pathogenic landscape of SSc-ILD. In this inquiry, we delved into the analysis of the Gene Expression Omnibus database: dataset GSE76808.27 The ensuing outcomes manifest as a potent indicator, reflecting a pronounced decrease in the expression levels of EGR2 and LAG3 within the peripheral blood milieu of SSc-ILD patients (Figure 1A). Further dissection of the data reveals a robust negative correlation between LAG3 expression and the prevalence of M2-type macrophages, which aroused our great research interest (Figures 1B–1D) (see STAR Methods). To fortify the integrity of our analytical findings, we sought to corroborate our deductions through an assessment of mRNA expression profiles. The evaluation of LAG3 and EGR2 mRNA expression within peripheral blood mononuclear cells (PBMCs) sourced from both SSc and SSc-ILD cohorts ensued. Remarkably, our empirical findings were demonstrative of alignment with our initial analysis. The expression levels of LAG3 and EGR2 demonstrated a significant reduction in the peripheral blood of SSc-ILD patients, with a further discernible decrement noted among those afflicted with concurrent ILD (Figures 1E and 1F).
Figure 1.

Bioinformatics analysis combined with PCR was used to verify the expression of EGR2 and LAG3 in patients with SSc-ILD
(A)The expressions of EGR2 and LAG3 in lung tissue of SSc-ILD patients were decreased. Data are represented as the form of boxplots. Statistical significance was analyzed by unpaired two-tail t-test, ∗∗p < 0.01.
(B–D) There was no significant difference between LAG3 and M0 and M1 macrophages, but LAG3 was significantly correlated with M2 macrophages. Data are represented as the form of correlation scatterplots. Statistical significance was analysis by chi-square test for comparison between groups.
(E and F) The expression of EGR2 and LAG3 was decreased in peripheral blood of patients with SSc-ILD. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05.
TGF-β3 and CD4+LAG3+T cells are downregulated and mesenchymal markers are upregulated in SSc-ILD patients
To investigate the role of CD4+LAG3+T cells, we first examined the expression of CD25 in CD4+LAG3+T cells from peripheral blood of healthy individuals. The results showed that the majority of CD4+LAG3+T cells were CD25-(5.1%), while CD25+ accounted for only 0.62%. (Figure S2A). In addition, CD25, also known as the IL-2 receptor, was not significantly correlated with TGF-β3 and LAG3 expression (Figures S2B and S2C). Okamura et al.’s study showed that >90% of LAG3 cells belong to CD4+CD25−,28 similarly, Rika Kato’s study also found that usually CD4+LAG3+T cells do not express CD25,29 our results are like previous findings. Considering these results and the activity of sorted cells, our subsequent experimental protocol selected two targets, CD4 and LAG3, while abandoning CD25.
The investigation revealed a notable discrepancy in the population of CD4+LAG3+ T cells present within the peripheral blood of individuals afflicted by rheumatoid arthritis, as compared to their healthy counterparts.24 Insight into the realm of in vivo experimentation evinced that the introduction of CD4+LAG3+T cells into animal models yielded tangible suppressive effects on the progression of maladies including rheumatoid arthritis, chronic enteritis, and chronic graft-versus-host disease.21,30 This body of evidence tentatively suggests that CD4+LAG3+ T cells may command a pivotal role in exerting negative regulatory influences upon the sustained abnormal inflammatory responses characteristic of the bodily milieu. Within this context, our present inquiry aimed to ascertain the distribution of CD4+LAG3+ T cells within the peripheral blood of individuals burdened by SSc-ILD. Our empirical assessment yielded intriguing results, wherein the proportions of CD4+LAG3+ T cells were ascertained to be 3.64% in normal control, 2.61% in SSc patients, and notably dwindling further to 1.57% among those concurrently afflicted by ILD (Figure 2A). We then assessed lung infiltrates for CD4, LAG3, and TGF-β3 expression. We observed a substantial increase in the fluorescence expression of CD4 and LAG3, alongside a notable reduction in the fluorescence intensity of TGF-β3 within the SSc-ILD group when compared to the control group. This phenomenon is likely associated with the initial onset of pulmonary infection or inflammation in patients, obvious infiltration of CD4+T cells, which subsequently triggers an activation of the immune response, leading to an elevated expression of LAG3 (Figures 2B and 2C). However, it remains unclear whether CD4+LAG3+T cells function by secreting TGF-β3, warranting our further investigation in this regard.
Figure 2.

The proportion of CD4+LAG3+T cells in peripheral blood and its correlation with laboratory indexes
Expression of CD4, LAG3, and TGF-β3 in clinical samples.
(A) Compared with control group, the proportion of CD4+LAG3+T cells was decreased in SSc patients, and further decreased in complex with ILD. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05.
(B and C) Compared with control group, CD4 and LAG3 were up-regulated and TGFβ3 was down-regulated in lung tissues of SSc-ILD patients.
(D) CD4+LAG3+T cell proportion was not significantly correlated with Scl-70 and inflammatory markers CRP and ESR but was significantly negatively correlated with IL-6.
(E) Compared with control group, the TGF-β3 content in peripheral blood was decreased in SSc patients, and further decreased in complex with ILD. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 5), ∗p < 0.05.
To enrich our understanding, we undertook an exploration of the correlation between the proportions of CD4+LAG3+T cells and pertinent inflammatory markers in patients, alongside the presence of Scl-70 antibodies (see STAR Methods). Evidently, a statistically significant negative correlation emerged between the proportions of CD4+LAG3+ T cells and IL-6 levels (Figure 2D). It is germane to note that extant research underscores the close nexus between IL-6 levels and the progression of SSc-ILD,31 Additionally, another investigation posits the independent prognostic potential of IL-6 in predicting carbon monoxide diffusing capacity.32 Nevertheless, it remains ambiguous whether LAG3+T cells possess the capacity to inhibit IL-6 secretion22, thereby warranting further elucidation.22 Complementing these insights, our enzyme-linked immunosorbent assay (ELISA) analyses disclosed a noteworthy reduction in TGF-β3 content within the peripheral blood of SSc patients, a diminishment further pronounced among patients concurrently harboring ILD (Figure 2E).
Regulation of CD4+LAG3+T cells on the function and phenotypic conversion of human lung fibroblasts
To explicate the involvement of CD4+LAG3+T cells in the context of SSc-ILD, a meticulous stratagem encompassing magnetic bead sorting was undertaken, yielding a proportion of CD4+LAG3+T cells increased from 2.89% to 32.89% following this procedure (Figure 3A) (see STAR Methods). To find out whether the ability to secrete TGF-β3 was related to LAG3, we examined the supernatants of CD4+LAG3+T and CD4+LAG3−T cells. ELISA results showed that CD4+LAG3+T cells could secrete higher levels of TGF-β3 compared with control group. However, the content of TGF-β3 was not detected in CD4+LAG3−T cells supernatants (Figure 3B). In view of this situation, there was no difference between control group and CD4+LAG3−cells, so we did not consider CD4+LAG3−T cells as the experimental intervention group in our subsequent experiments. Collagen I and collagen III are components of the extracellular matrix and markers of activated myofibroblasts. PCR results showed that CD4+LAG3+T cells supernatants can inhibit collagen secretion of TGF-β1-induced lung fibroblasts (Figure 3C). These cells secreted supernatants were addition to TGF-β1 stimulated pHLFs, and a significant reduction in the number of proliferating cells was observed (Figure 3D). Similarly, the number of migrating and invading cells was also reduced (Figure 3E). Considering that TGF-β1 activation of the Smad2/3 pathway is a classic mechanism for mediating organ fibrosis, we use TGF-β1 treatment to establish a cell model, western blot results showed that CD4+LAG3+T cells supernatants inhibited Smad3 phosphorylation induced by TGF-β1 and inhibited the expression of fibronectin and α-SMA. This indicates that CD4+LAG3+T cells supernatants can inhibit the activation of lung fibroblasts mediated by TGF-β1 through the Smad3 pathway (Figure 3F). To understand the reason for the changes in lung fibroblast function, we analyzed the changes in F-actin using phalloidin labeling, which is one of the main components of the cell skeleton and plays an important role in cell movement. Immunofluorescence observation showed that after TGF-β1 stimulation, the fluorescence intensity of F-actin increased significantly, and the cell microfilaments were significantly visible. However, after treatment with CD4+LAG3+T cells supernatants, the cell microfilament structure was disrupted, indicating that the effect of CD4+LAG3+T cells supernatants on cell function is exerted by affecting the cell skeleton (Figure 3G). Surprisingly, these changes in collagen secretion, cell function, pathway proteins and cytoskeleton were all significantly attenuated by the addition of anti-TGF-β3 antibody, indicating that CD4+LAG3+T cells regulate the function and phenotype transition of lung fibroblasts through the secretion of TGF-β3.
Figure 3.

CD4+LAG3+T cells can inhibit TGF-β1-induced activation and mesenchymal transition of lung fibroblasts
(A) Flow cytometry shows that the percentage of CD4+LAG3+T cells isolated by magnetic beads increased from 2.83% to 32.89%.
(B) ELISA results show that the control group and CD4+LAG3−T cells did not significantly secrete TGF-β3, while CD4+LAG3+T cells could secrete high levels of TGF-β3. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗∗∗∗p < 0.0001.
(C–G) The supernatant of CD4+LAG3+T cells can inhibit collagen secretion, proliferation, migration, invasion, mesenchymal transition, and F-actin expression of pHLFs stimulated by TGF-β1, while antagonistic TGF-β3 can weaken these effects. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 5), ∗p < 0.05, ∗∗p < 0.01.
The effect of CD4+LAG3+T cells on the transition of macrophages into myofibroblasts
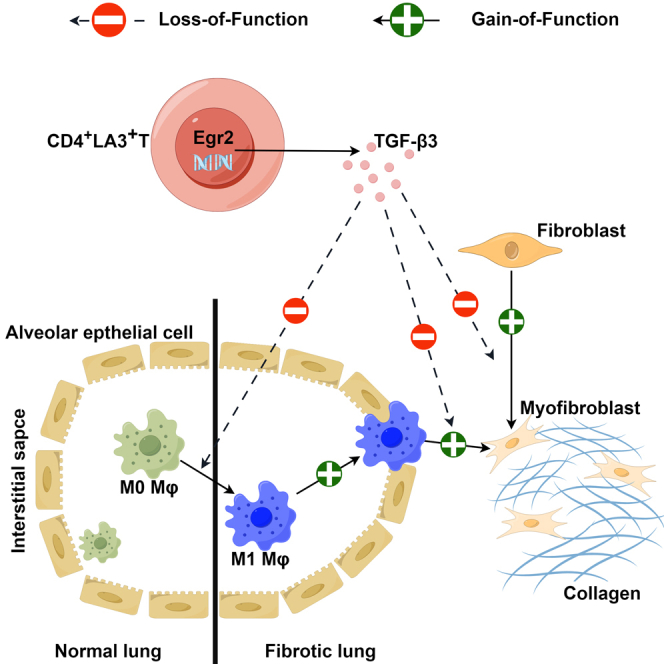
Macrophage’s role in organ fibrosis through polarization toward the proinflammatory M1 Phenotype.33 The substantial scholarly focus on macrophages’ pivotal role in organ fibrosis highlights their essential contribution. These versatile immune effectors undergo distinct polarization, adopting the proinflammatory M1 phenotype. Concurrently, CD4+LAG3+T cell subsets, recognized as regulatory T cells, have emerged as significant participants. In our study above, we found that these subsets secrete considerable TGF-β3, enabling intricate modulation of lung fibroblasts; however, it is not known whether they regulate macrophage polarization dynamics. This regulatory interplay significantly impacts the pulmonary fibrotic landscape. In experimental studies, murine bone marrow-derived macrophages were exposed to CD4+LAG3+T cell-derived supernatant from the murine spleen (see STAR Methods for additional details). These cells underwent specific polarization cues, driven toward M1 and M2 phenotypes through stimuli like LPS, IFN-γ, and IL-4. The results incontrovertibly showcased CD4+LAG3+T cells’ potential to hinder M0-to-M1 macrophage transition, while enhancing the shift of M0 macrophages to the M2 polarization state (Figure 4A). Functional assays demonstrated decreased M1 macrophage cell migration (Figure 4B). M2 macrophages are characterized by interleukin-10 (IL-10) and arginase-1 (Arg1). IL-10 activates the STAT3 pathway to facilitate M2 macrophage formation and activation, while Arg1 supports their immunosuppressive and reparative functions via metabolic pathways. Notably, IL-10 and Arg1 exhibit synergistic effects on M2 macrophage dynamics. In exploring broader impacts, the effect of CD4+LAG3+T cell supernatant on macrophage cytokine secretion was investigated. Results revealed that CD4+LAG3+T cells supernatants enhance the secretion of IL-10 and Arg1 by M2 macrophages (Figure 4C). Based on the changes in cell proportion, migration ability and the enhancement of characteristic secretion factors, we added one group, the anti-TGF-β3 group, in the above experiments, and found that these changes in the cells were weakened. Accordingly, we suggest that CD4+LAG3+Tcells can significantly regulate the polarization of macrophages by secretion TGF-β3.
Figure 4.

CD4+LAG3+T cells regulate macrophage polarization and mesenchymal transition
(A) Flow cytometry results show that adding CD4+LAG3+T cells supernatant to macrophages inhibited macrophages polarization to M1 and promoted macrophages polarization to M2.
(B) Transwell results show thatCD4+LAG3+T cells inhibited TGF-β1-induced M1 macrophage migration, while after antagonism TGF-β3 the effects were weakened. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05, ∗∗p < 0.01.
(C) ELISA results show that CD4+LAG3+T cells promote TGF-β1-induced M2 macrophage secretion of IL-10 and Arg1, while after antagonism TGF-β3 the effects was weakened. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05, ∗∗∗p < 0.001.
(D and E) PCR results show that CD4+LAG3+T cells inhibited TGF-β1 induced collagen Ⅰ and Ⅲ secretion, and inhibited the expression of interstitial markers fibronectin, vimentin, and α-SMA mRNA expression, while the effect was weakened after antagonist TGF-β3. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(F) Western blot results show that CD4+LAG3+T cells inhibited TGF-β1 induced mesenchymal transition, while the effect was weakened after antagonist TGF-β3. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ∗p < 0.05, ∗∗p < 0.01.
(G) Western blot results show that CD4+LAG3+T cells regulated macrophages through non-contact secretion of TGF-β3. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 3), ns = not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Recent findings have illuminated TGF-β1’s role in driving macrophages-to-myofibroblast transition (MMT), highlighting its pivotal function in fibrotic processes.34 We found that CD4+LAG3+T cell supernatant has been recognized for its regulatory potential in this context. Quantitative PCh analyses exhibited reduced Colla1、Col3a1、fibronectin、vimentin, and α-SMA mhNA levels (Figures 4D and 4E). Exposure to CD4+LAG3+T cell supernatant prompted a down-regulation of fibrotic markers protein levels, including fibronectin, vimentin, and α-SMA (Figure 4F). However, this effect was mitigated when anti-TGF-β3 were introduced concurrently, suggesting a nuanced regulatory interplay. In order to ascertain whether the TGF-β3 ability to contact the dependence, our results show that when the CD4+LAG3+T in the upper chamber or the lower chamber intervention on TGF-β1 induced macrophage, both got the same result, in chamber on the secretion of TGF-β3 can penetrate the membrane of the small chamber and cells in contact with the chamber. In the chamber to add recombinant TGF-β3 and add LAG3+CD4+T cells obtained similar results, antagonism TGF-β3 effect is abated, proving that CD4+LAG3+T cells secrete TGF-β3 for non-contact dependencies (Figure 4G). Intriguingly, the impact of recombinant TGF-β3 closely mirrored that of CD4+LAG3+T cells, underscoring their similar regulatory potency. These findings underscore the role of CD4+LAG3+T cells, known for their elevated TGF-β3 expression in governing crucial macrophage processes such as macrophage polarization and macrophage-to-myofibroblast transition.
Effect and significance of CD4+LAG3+T cells on bleomycin-induced human pulmonary fibrosis in mice
In vitro results demonstrate CD4+LAG3+T cells’ positive impact on lung fibroblasts and macrophages. However, their potential to ameliorate experimental pulmonary fibrosis in vivo remains unclear. Here, we induced pulmonary fibrosis in mice using bleomycin (see STAR Methods for additional details). The flow chart of animal experiments is shown in Figure 5A. Histological staining revealed disrupted alveolar structures, significantly thickened lung interstitial, and heightened collagen deposition in bleomycin-exposed mice, compared with pure bleomycin model group, through the tail vein injection of LAG3 +CD4+T cells or recombinant TGF-β3 can be observed that the improvement of lung tissue pathology, including alveolar structure identifiable, lung interval thinning, decrease collagen deposition; however, antagonistic TGF-β3 aggravates the disease (Figure 5B). Hydroxyproline, an amino acid specific to collagen employed to assess tissue collagen metabolism, like histological staining results, exhibited a substantial elevation in Bleomycin, decreased in CD4+LAG3+T and TGF-β3 groups and increased again after administration of TGF-β3 antibody (Figure 5C). Following two weeks of bleomycin administration, marked macrophage infiltration was evident within the lungs of the experimental group, CD4+LAG3+T cells or recombinant TGF-β3 treated infiltrating macrophages decreased significantly and antagonistic TGF-β3 treated macrophages increased again (Figure 5E). Given the spleen’s recognized significance as a pivotal lymphoid organ, a combination of ELISA and flow cytometry methodologies were employed to assess TGF-β3 levels (Figure 5D) as well as the proportions of CD4+LAG3+T cells within both the peripheral blood and spleen of both control and experimental murine subjects. The outcomes unequivocally unveiled a discernible reduction in the percentages of CD4+LAG3+T cells within the spleen (5.36%) and peripheral blood (2.86%) of experimental mice, in stark contrast to their control group counterparts (spleen: 8.69%, peripheral blood: 4.13%) (Figure 5F). To track whether the adoptively transferred CD4+LAG3+T cells migrated to the lungs, immunofluorescence assay showed significant fluorescence staining in mice infused with CD4+LAG3+T cells (Figure 5G). In Figure 4, we have substantiated the influence of CD4+LAG3+T cells on MMT. To garner further in vivo evidence, we simultaneously employed markers CD68 and α-SMA, with CD68 serving as a pivotal indicator of macrophages. Immunofluorescence analysis unveiled a substantial elevation in the fluorescence intensity of CD68 and α-SMA in lungs of bleomycin-induced pulmonary fibrosis mice. However, subsequent treatment with CD4+LAG3+T cells or recombinant TGF-β3 prominently mitigated this fluorescence intensity. Conversely, the therapeutic efficacy was diminished upon antagonism of TGF-β3 (Figure 5H). Similarly, there was a notable upsurge in the fluorescence intensity of α-SMA and fibronectin within the lungs of the bleomycin-treated mice in comparison to lung tissues from the normal control group (Figure 5I). Both the infusion of CD4+LAG3+T cells and the administration of recombinant TGF-β3 exhibited akin therapeutic outcomes. These outcomes were discernible through the amelioration of lung tissue architecture, evident alveolar cavities, enhanced lung histopathology, decreased hydroxyproline content, and abated macrophage infiltration. It is worth noting that the favorable influence exerted by CD4+LAG3+T cells on pulmonary fibrosis encountered partial attenuation in the presence of concurrent TGF-β3 antibody treatment. Our findings underscore the inhibitory capacity of CD4+LAG3+T cells in curtailing collagen accumulation and mitigating lung macrophage infiltration. This regulatory effect is chiefly mediated through the secretion of TGF-β3, thereby contributing to the amelioration of pathological processes within pulmonary tissue.
Figure 5.

CD4+LAG3+T cells improved bleomycin-induced pulmonary fibrosis in mice
(A) Experiment protocol for pulmonary fibrosis model.
(B, C, and E) Morphological staining and ELISA results show that compared with Bleomycin group, tail vein injection of CD4+LAG3+T cells or TGF-β3 improved mouse lung pathological morphology (200X) and reduced hydroxyproline content, while antagonistic TGF-β3 had the opposite effect. Data are represented as mean ± SD. Statistical significance was analyzed by one-way ANOVA (n = 5), ∗p < 0.05, ∗∗p < 0.01.
(D and F) ELISA and flow cytometry results showed respectively, compared with the control group, Bleomycin group of peripheral blood TGF - beta 3 content decreased significantly, CD4+LAG3+T cells in peripheral blood and spleen ratio decreased significantly. Data are represented as mean ± SD. Statistical significance was analyzed by unpaired two-tail t-test (n = 5), ∗p < 0.05.
(G) Immunofluorescence results showed that compared with Bleomycin group, the expression of CD4+LAG3+T cells in lung tissue were significantly increased after tail vein injection.
(H) Immunofluorescence results showed that the expression of α-SMA and CD68 in Bleomycin group was significantly higher than that in the control group, and the expression of MMT markers was downregulated after the treatment of CD4+LAG3+T cells or TGF-β3, and up-regulated after the antagonism of TGF-β3.
(I) Immunofluorescence results showed that the expression of α-SMA and fibronectin in Bleomycin group was significantly higher than that in the control group, and the expression of mesenchymal markers was downregulated after the treatment of CD4+LAG3+T cells or TGF-β3, and upregulated after the antagonism of TGF-β3.
Discussion
Pulmonary fibrosis represents a prevailing sequela inherent to numerous advanced pulmonary afflictions. Within this context, connective tissue disease-associated interstitial lung disease (CTD-ILD) emerges as a noteworthy manifestation of pulmonary fibrosis, notably with SSc-ILD standing out as the predominant rheumatologic subtype.35 The prognosis for SSc-ILD remains unfavorable, exerting a profound impact on the overall quality of life experienced by afflicted individuals.36 Although the multifarious origins of pulmonary fibrosis give rise to its intricate etiological landscape, the ensuing pathogenic progression uniformly bespeaks initial diffuse alveolitis. Initial impairment of the capillary barrier precipitates the recruitment of inflammatory cellular constituents, comprising granulocytes, lymphocytes, and macrophages. This influx is accompanied by the discharge of inflammatory cytokines, orchestrating the metamorphosis of pulmonary fibroblasts and macrophages within the interstitial milieu into contractile myofibroblasts. The culmination of this process is characterized by the extensive accretion of collagen and extracellular matrix components within the pulmonary parenchymal confines, culminating inexorably in the pathogenesis of pulmonary fibrosis.35 Consequently, inflammation stands as the incipient nexus catalyzing the trajectory of pulmonary fibrotic progression.
In a meticulously executed endeavor, we methodically amassed blood specimens and clinical data emanating from a cohort of SSc-ILD patients who were under hospitalization within our clinical purview spanning the interval between 2021 and 2022. Our scrutiny of this dataset unveiled a distinctive profile, whereby individuals afflicted with SSc-ILD evinced a heightened rate of erythrocyte sedimentation, concomitant with elevated interleukin-6 concentrations when contrasted against their SSc counterparts. This collective evidence underscores the proposition that inflammation may wield a pivotal agency in precipitating the trajectory toward pulmonary fibrogenesis. Within the immunological landscape, the CD4+LAG3+T cell cohort assumes a role of regulatory distinction, its emergence and differentiation within the peripheral immune milieu being endowed with a cardinal mantle of immune orchestration. Insight into this niche was gleaned through the pioneering work of Okamura et al., who delineated that while constituting a mere 2–8% fraction of the total CD4+T cell constituency within murine splenic architecture, the LAG3+T cell subset emerges as an indispensable architect of immune modulation, thereby underscoring its vital regulatory role.37 Subsequent investigations have elucidated a notable decline in the CD4+CD25−LAG3+T cell population among patients afflicted with rheumatoid arthritis, particularly evident within subsets characterized by elevated clinical disease activity index scores. Notably, intervention employing abatacept over a span of six months yielded a discernible surge in the frequency of these specific cells, concomitant with an augmentation in EGR2 expression discerned within the CD4+ cell milieu.24 Similarly, a decrease in CD4+LAG3+T cells was also observed in active psoriatic arthritis.23 Correspondingly, our observations underscore congruent trends, whereby the relative abundance of CD4+LAG3+T cells within the peripheral blood milieu of SSc-ILD patients is markedly diminished, paralleled by a concurrent reduction in TGF-β3 levels. In the context of pulmonary fibrogenesis, the role of immune dysregulation emerges as a pivotal instigator, a notion accentuated by the backdrop of diffuse connective tissue ailments that inherently harbor autoimmune components. These autoimmune reactions encompass the recruitment and activation of assorted inflammatory cell lineages such as T cells, B cells, and macrophages, culminating in the release of an array of inflammatory mediators and auto-antibodies. This collective cascade perpetuates a state of enduring systemic inflammation, consequently impinging upon the body’s faculty for effective immune regulation. Given this intricate immunological scenario, the decrement in CD4+LAG3+T cell proportions—essentially entrusted with the orchestration of negative immune regulation—affords insights into the potential mechanisms underpinning immune dysregulation in afflicted individuals.
With the intention of delving deeper into the functional underpinnings of CD4+LAG3+T cells within the context of SSc-ILD, we meticulously procured CD4+LAG3+T cell subsets from the peripheral blood pool of patients. As anticipated, our investigation unveiled a discernible attenuation in the abundance of CD4+LAG3+T cells among individuals diagnosed with SSc, with a pronounced exacerbation of this decrement noted within the subset concomitantly afflicted with ILD. Moreover, a statistically significant inverse correlation materialized, linking the levels of CD4+LAG3+T cells with those of IL-6—a multifunctional cytokine renowned for its capacity to incite a panoply of cellular responses encompassing both inflammatory cells and fibroblasts. This orchestrated cascade precipitates alveolar wall inflammation alongside the instigation of pulmonary interstitial fibrosis, thereby substantiating the mechanistic alliance between CD4+LAG3+T cell attenuation and the dysregulated cytokine milieu operative within this clinical milieu.38,39 Nonetheless, an evident research gap persists concerning the intricate interplay between CD4+LAG3+T cells and IL-6, necessitating further exhaustive inquiry to elucidate this intricate relationship. Notwithstanding this lacuna, it remains unequivocal that CD4+LAG3+T cells wield the capacity to elicit substantial secretion of the protective cytokine TGF-β3. Our ELISA outcomes consistently manifest heightened TGF-β3 levels among individuals characterized as normal. To date, a solitary study has demonstrated the potential of TGF-β3 to abrogate IL-6 expression stimulation in vitro within human cleft lip and palate fibroblasts.40 Our findings serve to propose that the inhibitory action of TGF-β3 upon IL-6 might conceivably manifest as a ubiquitous phenomenon traversing the broader physiological landscape—an intriguing proposition yet to be fully unveiled in the realm of human biology. Given the validated clinical utility of IL-6 inhibitors within the context of SSc-ILD, the ramifications of our investigations carry therapeutic implications germane to the management of this clinical entity. Consequently, we are emboldened to surmise that CD4+LAG3+T cells could potentially orchestrate the suppression of inflammatory cascades underpinning the trajectory of pulmonary fibrosis via their pronounced exudation of TGF-β3, thus warranting further exploration of this putative regulatory mechanism. The findings derived from our investigation serve to posit the potential ubiquity of the inhibitory influence exerted by TGF-β3 upon IL-6 within the broader physiological milieu, thus presenting a novel observation within the realm of human biology. Notably, the clinically established efficacy of IL-6 inhibitors in addressing the pathophysiology of SSc-ILD augments the relevance of our discoveries by engendering therapeutic implications for the management of this pathological state. Consequently, our observations provide a cogent basis for conjecture, wherein we posit that the pronounced exudation of TGF-β3 by CD4+LAG3+T cells potentially endows these cells with the capability to curtail the trajectory of inflammatory progression characteristic of pulmonary fibrosis.
The TGF/Smad3 signaling cascade constitutes an archetypal pro-fibrotic pathway that assumes salience within the ambit of organ fibrosis, manifesting prominently across diverse anatomical domains such as the cardiac, pulmonary, and hepatic tissues.41 Within the purview of the present study, we embarked upon an inquiry to discern the influence wielded by CD4+LAG3+T cells upon lung fibroblasts. Evidentially, our investigations unearth a discernible propensity of these specific cells to effectuate a pronounced repression of TGF-β1-triggered mesenchymal transition within lung fibroblasts. This transformative shift is epitomized by the attenuation in the expression of p-Smad3, fibronectin, and α-SMA protein moieties, culminating in the amelioration of both cellular function and architectural integrity, coalescing to mitigate the exudation of collagen. This salutary impact can be principally attributed to the bioactive involvement of TGF-β3. In the trajectory of lung fibrosis, macrophage infiltration within the pulmonary milieu emerges as a pivotal driver of the inflammatory dynamics that undergird the transition toward fibrotic processes—often observed to transpire during the nascent stages of the disease progression. Within the milieu of organ fibrosis at large, a marked propensity is witnessed for the conversion of M0 macrophages into the pro-inflammatory M1 phenotype, thereby affording a potent mechanism underpinning the excessive inflammatory milieu characteristic of this pathological continuum.
A corpus of antecedent scholarly works has unveiled that within neoplastic microenvironments, LAG3+PD1+CD8+T cells are closely linked to heightened M2 macrophage expressions within the tumor milieu.42 In congruence with these paradigms, our current investigation similarly uncovers a parallel scenario, wherein CD4+LAG3+T cells partake in the constriction of M0-to-M1 transition while concurrently fostering the transition to M2, thereby potentiating an augmented gamut of immune-regulatory modalities. Although this result is contrary to the results of
Figures 1B–1D in this paper, it may be due to two reasons. One is because the sample size of the dataset GSE76808 is too small, including only 4 SSc-ILD patients and 4 control patients. The second is to analyze the overall expression level of LAG3 in lung tissue, while the present study is to study the effects of LAG3+ and CD4+ T cells on macrophages, which are not exactly the same. This orchestrated response serves to potentiate a comprehensive mitigation of immune-inflammatory drivers operative within the pulmonary confines. Within the purview of renal fibrosis, the process of MMT has emerged as a salient facet intrinsic to the chronic inflammatory milieu, attaining heightened significance within the landscape of fibrogenic progression.34,43 In consonance with these delineations, our study extends its purview to assess the impact on macrophage mesenchymal transition, wherein we ascertain a pivotal function attributed to CD4+LAG3+T cells in conferring a substantial inhibition upon the expression of mesenchymal transition marker proteins incited by TGF-β1. Furthermore, the transcriptional levels of Collagens I and III are correspondingly quelled, concomitant with the restraint of cellular migratory attributes. Notably, these effects demonstrate susceptibility to reversal following the introduction of TGF-β3-neutralizing antibodies. This observation underscores the bioactive role of CD4+LAG3+T cells in exuding TGF-β3 to effectuate their inhibitory impact upon macrophage differentiation and activation dynamics.
To validate the in vivo efficacy of CD4+LAG3+T cells, we observed a discernible macrophage infiltration within lung tissues of the model group after a two-week regimen of intratracheal bleomycin administration. In contrast, the infusion of CD4+LAG3+T cells observed significantly infiltrate the lung, resulting in a reduction of inflammation and ameliorated pathological alterations within the lung tissue. In alignment with the preceding in vitro findings, we also obtained evidence that CD4+LAG3+T cells inhibited MMT in vivo, and this beneficial effect was abrogated upon antagonizing TGF-β3, thereby implicating the pivotal role of TGF-β3 secretion as the principal mediator of the inhibitory effect exerted by CD4+LAG3+T cells on pulmonary fibrosis. In synthesis, our empirical evidence substantiates the favorable influence of CD4+LAG3+T cells both in vitro and in the in vivo context. To the best of our knowledge, this constitutes the inaugural investigation into the regulatory role of CD4+LAG3+T cells within the domain of organ-specific pulmonary fibrosis. Evidently, CD4+LAG3+T cells may emerge as salient negative regulators of organ fibrosis, thereby harboring the potential to serve as discerning biomarkers germane to the spectrum of SSc-ILD.
Limitations of the study
First, the small number of cases we collected from patients with SSc-ILD may not provide a representative disease population profile. Furthermore, this study focused only on patients with SSc-ILD, and the results may not apply to other types of pulmonary fibrosis. In addition, this study mainly investigated the role of TGF-β3 secreted by CD4+LAG3+T cells in cell mesenchymal transition, cell function, and collagen secretion under TGF-β1 stimulation. Other potential mechanisms and factors that promote the development of pulmonary fibrosis may not have been thoroughly explored. Finally, this study did not investigate the potential adverse effects of CD4+LAG3+T cells as a therapeutic approach for treating pulmonary fibrosis. Therefore, the safety and efficacy of this method require further research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-Human CD4 | BD Biosciences | Cat# 550628, RRID: AB_393789 |
| PE anti-Human LAG3 | Biolegend | Cat# 369306, RRID: AB_2629592 |
| FITC anti-Human CD4 | Biolegend | Cat# 317408, RRID: AB_571951 |
| PE anti-Mouse CD206 | Biolegend | Cat# 141705, RRID: AB_10896421 |
| PerCP/Cy5.5 anti F4/80 | Biolegend | Cat# 123127, RRID: AB_893496 |
| FITC anti-Mouse CD11c | Biolegend | Cat# 117305, RRID: AB_313774 |
| PE anti-Mouse LAG3 | Biolegend | Cat# 125208, RRID: AB_2133343 |
| PE-Cy7 anti-Mouse Cd11b | Biolegend | Cat# 101205, RRID: AB_312788 |
| TGF-β3 antibody | R and D Systems | Cat# AB-244-NA, RRID: AB_354312 |
| TGF-β3 antibody | Affinity Biosciences | Cat# AF0261, RRID: AB_2833435 |
| Anti-Fibronectin antibody | Abcam | Cat# ab45688, RRID: AB_732380 |
| Anti-α-SMA antibody | Abcam | Cat# ab7817, RRID: AB_262054 |
| Smad3 Rabbit mAb | Cell Signaling Technology | Cat# 9523, RRID: AB_2193182 |
| Phospho-Smad3 Rabbit mAb | Cell Signaling Technology | Cat# 9520, RRID: AB_2193207 |
| Anti-Vimentin | Cell Signaling Technology | Cat# 12826, RRID: AB_2798037 |
| Anti-GAPDH | Abcam | Cat# ab181602, RRID: AB_2630358 |
| Anti-FITC MultiSort Kit | Miltenyi Biotec | Cat# 130-058-701, RRID: AB_244372 |
| Anti-PE MicroBeads | Miltenyi Biotec | Cat# 130-048-801, RRID: AB_244373 |
| Gout anti-Mouse IgG 488 | Abcam | Cat# ab150113, RRID: AB_2576208 |
| Gout anti-Rabbit IgG 594 | Abcam | Cat# ab150080, RRID: AB_2650602 |
| CD4 antibody | Proteintech | Cat# 67786-1-Ig, RRID: AB_2918550 |
| LAG-3 antibody | Proteintech | Cat# 16616-1-AP, RRID: AB_2133350 |
| CD68 antibody | Proteintech | Cat# 25747-1-AP, RRID: AB_2721140 |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | ACMEC | Cat# AC0065 |
| Fluoroshield | Sigma-Aldrich | Cat# F6182 |
| Bleomycin | Sigma-Aldrich | Cat# 9041-93-4 |
| M-CSF | PEPROTECH | Cat# 315-02 |
| LPS | Sigma | Cat# L6529 |
| IFN-γ | PEPROTECH | Cat# 315-05 |
| IL-4 | PEPROTECH | Cat# 214-14 |
| TGF-β1 | PEPROTECH | Cat# 100-21 |
| TGF-β3 | PEPROTECH | Cat# 100-36E |
| Biological samples | ||
| Human peripheral blood serum | The study cohort was inpatients from the Department of Rheumatology and immunology of Guangdong Provincial People’s Hospital | N/A |
| Critical commercial assays | ||
| Lymphoprep | STEMCELL TECHNOLOGIES | Cat# 07851 |
| Total RNA Kit | Omega Bio-tek | Cat# R6834-01 |
| Evo M-MLV Reverse Transcriptase | Accurate Biology | Cat# AG11706 |
| SYBR Green Pro Taq HS qPCR | Accurate Biology | Cat# AG11701 |
| TGF-β3 ELISA Kit | Joyee Biotechnics | N/A |
| EdU | RIBOBIO | Cat# R11053.9 |
| Cell Culture Insert | CORNING | Cat# 353097 |
| Hydroxyproline Content Kit | Geruisi Bio | Cat# G1205W |
| BCA protein assay | Beyotime Biotechnology | Cat# P0009 |
| Fitc-labeled phalloidin | Solarbio | Cat# CA1620 |
| Mouse IL-10 ELISA Kit | MEIMIAN | Cat# MM-0176M2 |
| Mouse Arg-1 ELISA Kit | MEIMIAN | Cat# MM-44685M2 |
| Fetal Bovine Serum | ZETA LIFE | Cat# Z7010FBS-500 |
| Experimental models: Cell lines | ||
| pHLFs | This paper | N/A |
| BMDM | This paper | N/A |
| Experimental models: Organism/strains | ||
| Mouse:C57BL/6J | GemPharmatech | RRID: IMSR_GPT: N000013 |
| Deposited data | ||
| Sequencing Data | National Center for Biotechnology Information | Number: GSE76808 |
| Oligonucleotides | ||
| Hu-EGR2-Forward: CTTCCCCAGCTTCAACCACA | This paper | N/A |
| Hu-EGR2-Reverse: GTAGAGGTCTCCTGCACAGC | This paper | N/A |
| Hu-LAG3-Foorward: GAACAGCAGCTCAATGCCAC | This paper | N/A |
| Hu-LAG3-Reverse: AAACTCCTCTGGGATGGGGT | This paper | N/A |
| Hu-GAPDH-Forward: TTCGTCATGGGTGTGAACCA | This paper | N/A |
| Hu-GAPDH-Reverse: GTCTTCTGGGTGGCAGTGAT | This paper | N/A |
| Hu-Colla1-Forward: CGGAGCAGACGGGAGTTT | This paper | N/A |
| Hu-Colla1-Reverse: TACGCAGGTGATTGGTGGG | This paper | N/A |
| Hu-Col3a1-Forward: GCTCCTACTCGCCCTCCTAA | This paper | N/A |
| Hu-Col3la1-Reverse: GTCCTCCTACTGCTACTCCAGAC | This paper | N/A |
| Hu-18S rRNA-Forward: Hu-GACTCAACACGGGAAACCTC | This paper | N/A |
| Hu-18S rRNA-Reverse: AGACAAATCGCTCCACCAAC | This paper | N/A |
| Mo-Fibronectin-Forward: ACTCCTTGCTGGTGTCATGG | This paper | N/A |
| Mo-Fibronectin-Reverse: GTGTACTCGGTTCCTGGCTC | This paper | N/A |
| Mo-Vimentin-Forward: TGCTTCAAGACTCGGTGGAC | This paper | N/A |
| Mo-Vimentin-Reverse: AAGCGCACCTTGTCGATGTA | This paper | N/A |
| Mo-α-SMA-Forward: CCACCATGTACCCAGGCATT | This paper | N/A |
| Mo-α-SMA-Reverse: GAAGGTAGACAGCGAAGCCA | This paper | N/A |
| Mo-GAPDH-Forward: ATGTGTCCGTCGTGGATCTG | This paper | N/A |
| Mo-GAPDH-Reverse: AAGTCGCAGGAGACAACCTG | This paper | N/A |
| Mo-Col1a1-Forward: TTCTCCTGGCAAAGACGGAC | This paper | N/A |
| Mo-Colla1-Reverse: CGGCCACCATCTTGAGACTT | This paper | N/A |
| Mo-Col3a1-Forward: AGTCAGGAAGACCTGGACGA | This paper | N/A |
| Mo-Col3a1-Reverse: TTGCGTCCATCAAAGCCTCT | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 8.0.2.263 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | NIH | RRID: SRC_003070 |
| CytoExpert 2.4 | Beckman Coulter | https://resources.mybeckman.cn/ |
| R version 4.3.0 or higher | R Development Core Team, 2011 | https://www.r-project.org/ |
| SPSS 25.0 | IBM | http://www.spss.com.cn |
| ZEN 2 | Carl Zeiss Stiftung | www.zeiss.com/microscopy |
| Adobe Illustrator | Adobe | https://www.adobe.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiao Zhang (13922255387@163.com).
Materials availability
-
•
Cell generated in this study are available from the lead contact upon request.
Data and code availability
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report any original code.
Experimental model and study participant details
Human subjects
Among the patients hospitalized in our hospital from 2021 to 2022, 23 patients with SSc were selected according to the diagnostic criteria of the American College of Rheumatology in 1980 and confirmed by the rheumatology and immunology Department of Guangdong Provincial People’s Hospital. The diagnosis of ILD in 36 patients with SSc-ILD was confirmed by clinical dry cough, shortness of breath, cyanosis, fine wet rales in the lungs, interstitial lesions and fibrosis, reticular and nodular shadows formed by interstitial fibrosis proliferation, or pulmonary CT scan, and moderate or severe ventilation and diffusion dysfunction in pulmonary function test. Peripheral blood from patients was centrifuged to obtain PBMCs and serum. The recruitment of this study was approved by the Medical Research Ethics Committee of Guangdong General Hospital (Guangdong Academy of Medical Sciences) [Authorization number: No. GDREC20160679H(R1)] and written informed consent documents were obtained and signed by all subjects.
Extraction and culture of pHLFs
Lung tissues of 8 healthy controls were obtained from patients suspected of lung cancer in the Department of Thoracic Surgery, Guangdong Provincial People’s Hospital, and more than 5cm away from the lesion site. The lung tissues of 3 patients with SSc-ILD were also obtained from patients who underwent lung biopsy in the Thoracic Surgery Department of Guangdong Provincial People’s Hospital and were later diagnosed as SSc-ILD by rheumatologists. All use of biological samples was approved by the Research Ethics Committee of Guangdong General Hospital (Guangdong Academy of Medical Sciences) [License No. GDREC20160679H(R1)].
The tissue was cut into 1 mm³ piece, evenly spread in culture dishes, and placed upside down in a 37°C and 5% CO2 incubator for 4 hours so that the lung tissue could adhere to the bottom of the dish, and then high-glucose DMEM (Dulbecco’s Modified Eagle Medium) culture medium containing 20% fetal bovine serum and 1% penicillin-streptomycin mixture was added. About 3–4 days, when the cells crawled out of the tissue, they are replaced with fresh medium. When the crawling cells become a colony, the tissue block is removed, and the fresh culture medium was replaced every other day until the cell density reached 90% for passage. Cell in passages 4 to 8 were used for subsequent experiments. It is worth to mention that not every tissue block is able to climb out of the cells, and tissue blocks that are not completely dried and floating can be re-manipulated to attach to the bottom of the dish as described above.
Mouse BMDM were extracted and cultured
Collect the femurs and tibias of mice under sterile conditions, remove the muscle tissue, and cut the bone at both ends. Rinse with 1 mL of 1640 medium using a syringe. Centrifuge the cell suspension at 300g for 10 minutes, collect the centrifugal precipitate, and induce monocytes to adhere to the wall under the condition of 10% fetal bovine serum, 1640 medium, and 5% CO2 by adding 20 ng/mL M-CSF. Add 1 mL of fresh medium to each well of a 6-well plate every other day. After the cells adhere to the wall for 6 days, induce M1 polarization by adding 100 ng/mL LPS and 20 ng/mL IFN-γ for 24 hours or induce M2 polarization by adding 20 ng/mL IL-4 for 24 hours.
Method details
PBMC were extracted
The patient’s peripheral blood was diluted with an equal volume of normal saline or Phosphate Buffer Saline (PBS), thoroughly mixed, and the 1:1 diluted blood was slowly added to a 15 mL centrifuge tube containing 3–4 mL Lymphoprep using a bus tube. At this time, the liquid can be seen to have obvious stratification, do not add quickly, in case of breaking through the liquid level boundary. The centrifuge tube was placed in the centrifuge at room temperature, 1500rpm, acceleration 6, deceleration 0, and centrifuged for 25min, then the white film layer in the middle of the liquid was carefully collected with Pasteur pipette, and then re-suspended with 5 times the volume of normal saline or PBS, 1500rpm, centrifuge for 5min, repeat washing twice. The supernatant is sucked dry and precipitated as PBMCs, which can be followed by magnetic bead sorting, flow cytometry analysis or total RNA extraction.
qPCR
Total cellular RNA was extracted using a Total RNA Kit (Omega Bio-tek) according to the manufacture’s instructions, and reverse transcription system were prepared using a reverse transcriptase kit (Accurate Biology) according to the manufacturer’s instructions. A two-step PCR reaction system was employed with the following parameters: one cycle at 95°C for 30 seconds, followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. The ΔΔCT method were used as reference to calculate relative expression levels between samples. Primer pairs used in this paper are shown in key resources table. GAPDH, 18shNA and β-Actin were reference genes. 3 repeat holes were set in the sample, and if the CT value was greater than 24, it was excluded.
Flow cytometry
CD4+LAG3+T cells were detected
Add 100μL of cell suspension into each tube of the flow tube. Then, add 5μL of, PerCP Anti-CD4, and PE Anti-LAG3 for surface staining. Incubate at 4°C for 30 minutes and wash with PBS solution. Centrifuge at 1500rpm for 6 minutes and discard the supernatant. Repeat this washing step twice. Finally, add 100μL of PBS solution and test the sample using the CytoExpert machine.
Macrophages were detected
24h after the addition of polarization inducer, the medium was discarded, washed 3 times with PBS, digested with 0.25% trypsin for 1min, and the digestion was terminated by adding the whole culture. The macrophages were blown down by pipette, the suspension was collected, centrifuged at 1000rpm for 6min, and resuspended in PBS for 3 times. Macrophages were labeled with 5μL Anti-Mouse F4/80 PerCP-Cy 5.5 and Anti-Mouse CD11b PE-Cy7, and M1 macrophages were labeled with 5μL Anti-Mouse CD11c FITC, respectively. M2 macrophages were labeled with 5μL of Anti-Mouse CD206 PE and incubated with flow antibodies as described previously, followed by on-machine examination.
Immunofluorescence
The lung tissue paraffin sections were heated at 60°C for 4 hours, rehydrated using gradient alcohol, subjected to antigen retrieval with citric acid, cleaned with PBS, permeabilized with Triton X-100, blocked with BSA, primary antibodies were incubated at a concentration of 1:500 for 24 hours at 4°C, and subsequently incubated with a secondary antibody at room temperature for 1 hour. The sections were counterstained with DAPI and imaged using a confocal microscope.
ELISA
Using Mouse IL-10 ELISA Kit and Mouse Arg-1 ELISA Kit [Jiangsu Meimian industrial Co., Ltd] and TGF-β3 ELISA Kit. Samples were removed from the −80° C freezer, 100μL of samples and standards were added to the 96-well plate, incubated at 37° C for 90min, the washing solution was prepared, 350μL was added to each well of the incubated well plate, left for 30s, blotted dry with absorbent paper, and the washing was repeated three times. Freshly prepared 50μL of biotinylated antibody working solution was added to each well, incubated at 37° C for 60min, and the above washing steps were repeated three times. Add 50μL enzyme binding solution to each well, incubate at 37°C for 30min, wash three times repeatedly, add 90μL chromo-developing substrate to each well, incubate in the dark for 15min, add 100μL termination solution to each well, put into the microplate reader for detection, record OD value, and calculate the concentration of the tested sample according to the standard curve.
Magnetic bead sorting and T cell activation
PBMCs were counted and incubated with FcR blocking agent, followed by the addition of FIITC-Anti-CD4 and PE-Anti-LAG3. The mixture was then incubated in the dark at 4°C for 10 minutes, followed by the addition of buffer and centrifugation at 300g for 10 minutes. After discarding the supernatant, 8μg Anti-FITC MultiSort microbeads were added to every 107 cells and incubated at 4°C for 15 minutes, followed by repeated centrifugation. The solution in the column was CD4+T cells by rapidly pushing the piston. To obtain CD4+LAG3+T cells, the magnetic beads were dissociated, and the above magnetic sorting process was repeated according to the manufacturer’s instructions. Cells were intervened for 12h in well plates precoated with CD3 mAb (5mg/mL) and CD28 mAb (20mg/mL) to allow cell activation.
CD4+LAG3+T cells supernatant were addition to pHLFs and BMDM
The logarithmic growth phase pHLFs were uniformly seeded into a 24-well plate. When the cell density reached 60%, the cells were grouped as follows, Control group: DMSO was added; CD4+LAG3+T group: stimulated with 5 × 105 T cells supernatant for 24 hours; TGF-β1 group: treated with 4ng/mL TGF-β1 for 24h; CD4+LAG3+T+TGF-β1 group: stimulated with 5 × 105 T cells supernatant and 4ng/mL TGF-β1 for 24h; CD4+LAG3+T+Anti-TGF-β3 group: stimulate with 5 × 105 T cells supernatant and 2μg/mL TGF-β3 for 24 hours.
On the day prior to inducing polarization of macrophages towards M1 and M2, the 24-well plates were divided into the following 5 groups. Control group: M-CSF was added before inducing cell polarization without LPS, IFN-γ and IL-4; Induction group: M1 macrophages were induced by adding LPS and IFN-γ, while M2 macrophages were induced by adding IL-4; CD4+LAG3+T group: 5 × 105 T cells supernatant were added one day before inducing polarization; CD4+LAG3+T+Anti-TGF-β3 group: 5 × 105 T cells supernatant and 2μg/mL Anti-TGF-β3 were added one day before inducing polarization; TGF-β3 group: 40ng/mL TGF-β3 was added one day before inducing polarization.
EdU
The pHLFs cells (4×103∼10×105 cells per well) were seeded into 96-well plates and then treated with TGF-β3 (5ng/mL) for 24h. After that, 100μL of 50 μmol/L EdU solution was added to each well and incubated for 4h. The cells were then fixed and stained according to the kit instructions. Finally, the cells were photographed and counted under a fluorescence microscope.
Migration and Transwell
The cell density of pHLFs was adjusted to 2×105/mL, and 200μL of cell suspension was added to the upper chamber of the Transwell, and 3×104 cells were added to the upper chamber for macrophages. Then, 600μL of the medium was added to the lower chamber, and the cells were incubated for 24 hours. After incubation, the cells on the upper surface of the membrane were removed using a cotton swab. The membrane was then fixed with methanol, stained with crystal violet, and observed under a microscope. Five fields were randomly selected, and photos were taken to count the number of migrated cells.
Western blot
Total protein was extracted from tissues and cells, and its concentration was determined using BCA method. Protein denaturation was carried out, followed by electrophoresis and membrane transfer. primary antibody (concentration 1:1000) was incubated overnight, and the secondary antibody was incubated at room temperature for 1 hour. Finally, a photo analysis was performed. Total tissue and cell proteins were extracted, protease inhibitors were added, and the concentration was determined by BCA assay.
Protein denaturation was first performed at 100° C for 10min, followed by the addition of 10 to 15ug of protein for electrophoresis and membrane transfer. Anti-fibronectin, anti-α-SMA, anti-vimentin, anti-GAPDH, anti-p-Smad3 and anti-Smad3 primary antibodies were incubated at a concentration of 1:1000 for at least 12 hours, washed three times in Tris solution containing Tween20, and then incubated with secondary antibodies for 1 hour at room temperature. Repeat washing and the protein was developed using ECL luminescent solution (TIANYA BIOTEC), detected on an imager (LAS 500), and the antibodies used are listed in Table.
Phalloidin-stained
The grouping and intervention of lung fibroblasts are as described above. When the density reached 50%, the culture-medium was removed, the cells were washing twice at 37°C with PBS, fixed at room temperature for 10 minutes with 4% methanol solution, the cells were covered with FITC-labeled Phalloidin working solution, incubated at room temperature for 30 minutes without light, washing 3 times with PBS for 5 minutes each time, and the nucleus were stained with DAPI solution for 1 minute. PBS was washing 3 times, 5 minutes each time. Sealing with an Fluoroshield reagent, the cell climbing tablets were observed under confocal microscope, and the images were preserved and analyzed.
Contact-independent assay
The differentiated macrophages were divided into 6 groups: the control group was treated with DMSO, the experimental group and the 4 treatment groups were treated with TGF-β1 for 48 hours, and then treated. All four treatment groups used chambers; one group was supplemented with activated CD4+LAG3+ cells. In one group, the supernatant of CD4+LAG3+ cells were added under the chamber (1000 rpm/min and centrifuged for 5min to remove cell sediment); In one group, recombinant TGF-β3 was added into the upper compartment. One group was co-added with recombinant TGF-β3 factor and Anti-TGF-β3 in the upper compartment. The cells were treated for 24h, after which the whole protein of macrophages was extracted for western blot analysis.
Mouse pulmonary fibrosis and ethics
Thirty 8-week-old male C57BL/6J mice were housed in the Laboratory Animal Research Center of South China University of Technology (License number: SYXK (Guangdong) 2022-0178) according to the requirements of "Laboratory animal Requirements of environment and housing facilities" (GB14925-2010). First, the mice were anesthetized by intraperitoneal injection of pentobarbital sodium saline solution [body weight(g)∗10–100(μL)]. After anesthesia took effect, the limbs of the mice were fixed, the incisors were suspended on a thin rope, and the mouth of the mice was placed in an upward position. The left hand was used to carefully hold the mouse tongue with forceps to fully expose the epiglottic. A mouse model of pulmonary fibrosis was established by instillation of 100μL bleomycin aqueous solution (3 mg/kg body weight) into the trachea with a 1 mL syringe held in the right hand. The mice were randomly divided into 5 groups, 6 mice in each group. Control group: received an equivalent volume of normal saline; bleomycin group: intratracheally injected with 3 mg/kg bleomycin; CD4+LAG3+T group: received 1×106 CD4+LAG3+T cells via tail vein injection on day 1 and day 14 before bleomycin injection; CD4+LAG3+T+Ati-TGF-β3 group: received 1×106 CD4+LAG3+T cells and 2 μg/kg Anti-TGF-β3 antibody via tail vein injection; TGF-β3 group that received 100 μg/kg TGF-β3 via tail vein injection on day 1 and day 14 before bleomycin injection. On day 14 after the injection, the mice were sacrificed for lung tissue macrophage immunohistochemical staining, and on day 28, blood and spleen were collected for further experiments on lung tissue. This study was approved by the Research Ethics Committee of Guangdong General Hospital (Guangdong Academy of Medical Sciences) and the Experimental Animal Ethics Committee of South China University of Technology with authorization numbers No. GDREC2016079A and AEC2019044.
Detection of hydroxyproline content
Mouse lung tissue samples were obtained after blood removal by perfusion with normal saline (including heparin sodium). 200mg of right lung tissue samples were taken and homogenized at 4°C according to the proportion of 100μL SOD sample preparation solution added to every 10mg tissue. Centrifuge at 4°C about 12000g for 5 minutes, take the supernatant as the sample to be tested and proceed according to the kit instructions.
Morphological staining of lung tissue
H&E staining: Mouse lung tissue samples were embedded in paraffin, continuously and uniformly cut into 4μm thick sections, deparaffinized to water, stained with hematoxylin and eosin, dehydrated and sealed, observed under a microscope, and pictures were collected for analysis.
Masson staining: The tissue samples were baked at 60°C for 2 hours, then dewaxed using xylene and rehydrated with a gradient of alcohol. After soaking in hematoxylin for 20 minutes, the samples were soaked in Lichun red dye, followed by soaking in a 2% acetic acid aqueous solution and then a 1% phosphomolybdate aqueous solution for differentiation. The samples were then soaked in an aniline blue solution, followed by soaking in a 2% acetic acid solution. After dehydration with ethanol and clearing with xylene, the samples were sealed.
Immunohistochemical staining
Mouse lung tissues were fixed, embedded, sectioned, deparaffinized, rehydrated and incubated with primary antibodies followed by horseradish peroxidase-labeled secondary antibodies. Color was developed with DAB, observed under a microscope and photographed.
Quantification and statistical analysis
Statistical analysis
Statistical analysis and visualization were carried out using GraphPad Prism 8 software. SPSS 25.0 software was used to analyze the correlation of clinical data of patients with Spearman coefficient. Quantitative data was illustrated in forms of count and percentage, and was compared through chi-square test between groups. For laboratory experiments, Data are presented as Mean ± SD. Comparisons between two groups were performed using the t-test, while comparisons between multiple groups were conducted using the ANOVA test. p values<0.05 were considered statistically significant.
Acknowledgments
This research was supported by the Natural Science Foundation of Guangdong Province (grant numbers 2019A1515010927 & 2019A1515010047), National Natural Science Foundation of China (grant numbers 81771734 & 82271822), and the Science and Technology Program of Guangzhou (grant number 202102021181). We also thank Figdraw platform, the graph abstract of this paper is drawn by Figdraw (www.home-for-researchers.com).
Author contributions
L.Q., H.L., and F.Z. conceived the experiments and drafted and revised the manuscript. J.W. and H.C. performed the statistical analysis. T.X. and G.Z. conducted the formal analysis. X.Z. revised the study. All authors made substantial, direct, and intellectual contributions to the work and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108225.
Supplemental information
References
- 1.Zhao M., Wang L., Wang M., Zhou S., Lu Y., Cui H., Racanelli A.C., Zhang L., Ye T., Ding B., et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target. Ther. 2022;7:206. doi: 10.1038/s41392-022-01070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karamichos D., Hutcheon A.E.K., Zieske J.D. Reversal of fibrosis by TGF-β3 in a 3D in vitro model. Exp. Eye Res. 2014;124:31–36. doi: 10.1016/j.exer.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathai S.C., Danoff S.K. Management of interstitial lung disease associated with connective tissue disease. BMJ. 2016;352:h6819. doi: 10.1136/bmj.h6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spagnolo P., Distler O., Ryerson C.J., Tzouvelekis A., Lee J.S., Bonella F., Bouros D., Hoffmann-Vold A.M., Crestani B., Matteson E.L. Mechanisms of progressive fibrosis in connective tissue disease (CTD)-associated interstitial lung diseases (ILDs) Ann. Rheum. Dis. 2021;80:143–150. doi: 10.1136/annrheumdis-2020-217230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King T.E., Jr., Bradford W.Z., Castro-Bernardini S., Fagan E.A., Glaspole I., Glassberg M.K., Gorina E., Hopkins P.M., Kardatzke D., Lancaster L., et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L., du Bois R.M., Raghu G., Azuma A., Brown K.K., Costabel U., Cottin V., Flaherty K.R., Hansell D.M., Inoue Y., et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 7.Border W.A., Noble N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994;331:1286–1292. doi: 10.1056/nejm199411103311907. [DOI] [PubMed] [Google Scholar]
- 8.Lodyga M., Hinz B. TGF-β1 - A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020;101:123–139. doi: 10.1016/j.semcdb.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Batlle E., Massagué J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity. 2019;50:924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H., Wang B., Zhang A., Hassounah F., Seow Y., Wood M., Ma F., Klein J.D., Price S.R., Wang X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019;27:571–583. doi: 10.1016/j.ymthe.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J., Zhou R., Liang Y., Fu X., Wang D., Wang C. Blockade of lncRNA-ASLNCS5088-enriched exosome generation in M2 macrophages by GW4869 dampens the effect of M2 macrophages on orchestrating fibroblast activation. FASEB J. 2019;33:12200–12212. doi: 10.1096/fj.201901610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ask K., Bonniaud P., Maass K., Eickelberg O., Margetts P.J., Warburton D., Groffen J., Gauldie J., Kolb M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int. J. Biochem. Cell Biol. 2008;40:484–495. doi: 10.1016/j.biocel.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L., Xiong S., Guo R., Yang Z., Wang Q., Xiao F., Wang H., Pan X., Zhu M. Transforming growth factor β3 attenuates the development of radiation-induced pulmonary fibrosis in mice by decreasing fibrocyte recruitment and regulating IFN-γ/IL-4 balance. Immunol. Lett. 2014;162:27–33. doi: 10.1016/j.imlet.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y., Peng Y., Gao D., Feng C., Yuan X., Li H., Wang Y., Yang L., Huang S., Fu X. Mesenchymal stem cells suppress fibroblast proliferation and reduce skin fibrosis through a TGF-β3-dependent activation. Int. J. Low. Extrem. Wounds. 2015;14:50–62. doi: 10.1177/1534734614568373. [DOI] [PubMed] [Google Scholar]
- 15.Miyashita-Ishiwata M., El Sabeh M., Reschke L.D., Afrin S., Borahay M.A. Hypoxia induces proliferation via NOX4-Mediated oxidative stress and TGF-β3 signaling in uterine leiomyoma cells. Free Radic. Res. 2022;56:163–172. doi: 10.1080/10715762.2022.2061967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao S., Liu M., Shen D., Zhang W., Wang T., Bai Y. TGF-β/Smads Signaling Affects Radiation Response and Prolongs Survival by Regulating DNA Repair Genes in Malignant Glioma. DNA Cell Biol. 2018;37:909–916. doi: 10.1089/dna.2018.4310. [DOI] [PubMed] [Google Scholar]
- 17.Shi A.P., Tang X.Y., Xiong Y.L., Zheng K.F., Liu Y.J., Shi X.G., Lv Y., Jiang T., Ma N., Zhao J.B. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.785091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews L.P., Marciscano A.E., Drake C.G., Vignali D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017;276:80–96. doi: 10.1111/imr.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogishi M., Yang R., Aytekin C., Langlais D., Bourgey M., Khan T., Ali F.A., Rahman M., Delmonte O.M., Chrabieh M., et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat. Med. 2021;27:1646–1654. doi: 10.1038/s41591-021-01388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu S., Liu X., Li T., Li Z., Hu F. LAG3 (CD223) and autoimmunity: Emerging evidence. J. Autoimmun. 2020;112 doi: 10.1016/j.jaut.2020.102504. [DOI] [PubMed] [Google Scholar]
- 21.Chen S.Y., Hsu W.T., Chen Y.L., Chien C.H., Chiang B.L. Lymphocyte-activation gene 3(+) (LAG3(+)) forkhead box protein 3(-) (FOXP3(-)) regulatory T cells induced by B cells alleviates joint inflammation in collagen-induced arthritis. J. Autoimmun. 2016;68:75–85. doi: 10.1016/j.jaut.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Okamura T., Sumitomo S., Morita K., Iwasaki Y., Inoue M., Nakachi S., Komai T., Shoda H., Miyazaki J.I., Fujio K., Yamamoto K. TGF-β3-expressing CD4+CD25(-)LAG3+ regulatory T cells control humoral immune responses. Nat. Commun. 2015;6:6329. doi: 10.1038/ncomms7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gertel S., Polachek A., Furer V., Levartovsky D., Elkayam O. CD4(+) LAG-3(+) T cells are decreased in active psoriatic arthritis patients and their restoration in vitro is mediated by TNF inhibitors. Clin. Exp. Immunol. 2021;206:173–183. doi: 10.1111/cei.13646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakachi S., Sumitomo S., Tsuchida Y., Tsuchiya H., Kono M., Kato R., Sakurai K., Hanata N., Nagafuchi Y., Tateishi S., et al. Interleukin-10-producing LAG3(+) regulatory T cells are associated with disease activity and abatacept treatment in rheumatoid arthritis. Arthritis Res. Ther. 2017;19:97. doi: 10.1186/s13075-017-1309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraaijeveld R., de Graav G.N., Dieterich M., Litjens N.H.R., Hesselink D.A., Baan C.C. Co-inhibitory profile and cytotoxicity of CD57(+) PD-1(-) T cells in end-stage renal disease patients. Clin. Exp. Immunol. 2018;191:363–372. doi: 10.1111/cei.13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khanna D., Tashkin D.P., Denton C.P., Renzoni E.A., Desai S.R., Varga J. Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2020;201:650–660. doi: 10.1164/rccm.201903-0563CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christmann R.B., Sampaio-Barros P., Stifano G., Borges C.L., de Carvalho C.R., Kairalla R., Parra E.R., Spira A., Simms R., Capellozzi V.L., Lafyatis R. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014;66:714–725. doi: 10.1002/art.38288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamura T., Fujio K., Shibuya M., Sumitomo S., Shoda H., Sakaguchi S., Yamamoto K. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl. Acad. Sci. USA. 2009;106:13974–13979. doi: 10.1073/pnas.0906872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato R., Sumitomo S., Tsuchida Y., Tsuchiya H., Nakachi S., Sakurai K., Hanata N., Nagafuchi Y., Kubo K., Tateishi S., et al. CD4(+)CD25(+)LAG3(+) T Cells With a Feature of Th17 Cells Associated With Systemic Lupus Erythematosus Disease Activity. Front. Immunol. 2019;10:1619. doi: 10.3389/fimmu.2019.01619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sega E.I., Leveson-Gower D.B., Florek M., Schneidawind D., Luong R.H., Negrin R.S. Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PLoS One. 2014;9 doi: 10.1371/journal.pone.0086551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Lauretis A., Sestini P., Pantelidis P., Hoyles R., Hansell D.M., Goh N.S.L., Zappala C.J., Visca D., Maher T.M., Denton C.P., et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J. Rheumatol. 2013;40:435–446. doi: 10.3899/jrheum.120725. [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa M. Biomarkers in systemic sclerosis: Their potential to predict clinical courses. J. Dermatol. 2016;43:29–38. doi: 10.1111/1346-8138.13156. [DOI] [PubMed] [Google Scholar]
- 33.Lu H., Wu L., Liu L., Ruan Q., Zhang X., Hong W., Wu S., Jin G., Bai Y. Quercetin ameliorates kidney injury and fibrosis by modulating M1/M2 macrophage polarization. Biochem. Pharmacol. 2018;154:203–212. doi: 10.1016/j.bcp.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Tang P.M.K., Zhang Y.Y., Xiao J., Tang P.C.T., Chung J.Y.F., Li J., Xue V.W., Huang X.R., Chong C.C.N., Ng C.F., et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc. Natl. Acad. Sci. USA. 2020;117:20741–20752. doi: 10.1073/pnas.1917663117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wells A.U., Denton C.P. Interstitial lung disease in connective tissue disease--mechanisms and management. Nat. Rev. Rheumatol. 2014;10:728–739. doi: 10.1038/nrrheum.2014.149. [DOI] [PubMed] [Google Scholar]
- 36.Fischer A., du Bois R. Interstitial lung disease in connective tissue disorders. Lancet. 2012;380:689–698. doi: 10.1016/s0140-6736(12)61079-4. [DOI] [PubMed] [Google Scholar]
- 37.Okamura T., Fujio K., Sumitomo S., Yamamoto K. Roles of LAG3 and EGR2 in regulatory T cells. Ann. Rheum. Dis. 2012;71:i96–i100. doi: 10.1136/annrheumdis-2011-200588. [DOI] [PubMed] [Google Scholar]
- 38.Jones S.A., Jenkins B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018;18:773–789. doi: 10.1038/s41577-018-0066-7. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka T., Narazaki M., Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baroni T., Carinci P., Bellucci C., Lilli C., Becchetti E., Carinci F., Stabellini G., Pezzetti F., Caramelli E., Tognon M., Bodo M. Cross-talk between interleukin-6 and transforming growth factor-beta3 regulates extracellular matrix production by human fibroblasts from subjects with non-syndromic cleft lip and palate. J. Periodontol. 2003;74:1447–1453. doi: 10.1902/jop.2003.74.10.1447. [DOI] [PubMed] [Google Scholar]
- 41.Liu M., Zeng X., Wang J., Fu Z., Wang J., Liu M., Ren D., Yu B., Zheng L., Hu X., et al. Immunomodulation by mesenchymal stem cells in treating human autoimmune disease-associated lung fibrosis. Stem Cell Res. Ther. 2016;7:63. doi: 10.1186/s13287-016-0319-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gusak A., Fedorova L., Lepik K., Volkov N., Popova M., Moiseev I., Mikhailova N., Baykov V., Kulagin A. Immunosuppressive Microenvironment and Efficacy of PD-1 Inhibitors in Relapsed/Refractory Classic Hodgkin Lymphoma: Checkpoint Molecules Landscape and Macrophage Populations. Cancers. 2021;13 doi: 10.3390/cancers13225676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang P.M.K., Nikolic-Paterson D.J., Lan H.Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019;15:144–158. doi: 10.1038/s41581-019-0110-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report any original code.