

Abstract
The construction of 1,2‐position‐connected azulene oligomers was achieved. In the crystal packing structure of the terazulene, two molecules of (R a)‐ and (S a)‐configurations formed a pair. Variable temperature NMR measurements and theoretical calculations of the quaterazulene suggest that the helical and syn‐type structure with terminal azulene overlap is more stable. Two kinds of fused terazulenes (1,2′′‐closed and 1,8′′‐closed) were also synthesized by intramolecular Pd‐catalyzed C−H/C−Br arylation of the terazulene moieties. X‐ray structure analysis of 1,2′′‐closed terazulene revealed a planar structure, while an analysis of 1,8′′‐closed terazulene performed on a C60 co‐crystal revealed a curved structure forming a 1 : 1 complex covering the co‐crystal. Nucleus‐independent chemical shift (NICS) calculations carried out for the central seven‐membered ring of 1,8′′‐closed terazulene showed a positive value, suggesting anti‐aromatic properties.
Keywords: antiaromaticity, azulenes, helical structure, oligomers, polycyclic hydrocarbons
Twisted or helical shape 1,2‐position‐connected terazulene and quaterazulene were synthesized. Variable temperature NMR measurements and theoretical calculations of the quaterazulene suggest that the syn‐type structure with terminal azulene overlap is more stable. Planar or bent shape fused terazulenes were also synthesized. Nucleus‐independent chemical shift (NICS) calculations carried out for the central seven‐membered ring of the bent shape fused terazulene showed a positive value, suggesting anti‐aromatic properties.

Introduction
Organic semiconductor materials have recently attracted the attention of the scientific community as next‐generation electronic materials to replace inorganic materials owing to their superior light weight properties and flexibility. [1] Numerous studies have been conducted on such systems linked with acenes, having relatively small HOMO‐LUMO gaps, for application in fields like organic electronics and photonics. [2] For example, studies on naphthalene, [3] the simplest of the acenes, highlight the potential of its helical chiroptical properties [3b] and charge transfer layers in organic electroluminescent (EL) devices. [3d]
Azulene [4] is a structural isomer of naphthalene, and is known to have a relatively high HOMO and low LUMO, [4f] and it exhibits a bright blue color due to its low symmetry and the contribution of its specific polarized structure. Despite there being fewer studies on azulene‐linked systems than on naphthalene, some reports on the former have attracted significant attention. Theoretical calculations predict that azulene oligomers bonded at the 1,3‐ and 2,6‐positions would show conductivity via the polaron mechanism.[ 4j , 4k , 4l ] Recent papers report that species in which azulene is connected at the 2,6‐position of a trimer exhibit n‐type or ambipolar semiconductor properties. [4h] Polycyclic hydrocarbons incorporating an azulene skeleton have also recently been the subject of studies because of their antiaromaticity and/or biradical character. [5] To date, we have synthesized a dimer in which an azulene with an aryl group at the 2‐position is connected at the 1‐position [6] and elucidated its crystal structures and rotational behavior and electronic properties of the azulene‐linked site, along with its complexation behavior with metals.[ 4b , 6 ] On the other hand, reports on 1,2‐connected systems have mainly focused on the synthesis of 1,2′‐biazulene, and their electronic properties and geometric structures are yet to be defined. [7]
In light of this, in this paper, a trimer 1 with an azulene connected at the 1‐ and 2‐positions was synthesized, the same procedure was extended to the tetramer 2, and azulene fused polycyclic hydrocarbon compounds 3 and 4 were synthesized by means of ring closing reactions of 1. Their geometric structures and electronic properties were then elucidated.
Results and Discussion
Homologation of the azulene moiety was carried out by utilizing the Suzuki–Miyaura cross‐coupling reaction of 1‐position brominated azulene with 2‐azulenylboronate 5 [8] (Scheme 1). After reacting 1 equiv. of N‐bromosuccinimide (NBS) with 1,2′‐biazulene, the resulting compound was then coupled with 5 using a palladium catalyst, producing terazulene 1 in 61 % yield. Subsequently, bromination with 1 equiv. of NBS was performed again on 1, followed by a reaction with 5, resulting in the formation of quaterazulene 2 (8 %). Interestingly, this reaction also produced the ring‐closed trimer 3 (36 %). The fused terazulene 3 was thought to be generated by the progression of intramolecular C−H/C−Br arylation favored by the Pd catalyst of 1. [9] A more detailed study of the Pd‐catalyzed arylation of 1 showed that the product yield depended on the phosphine ligand used (Table S1). The 1,2′′‐position ring‐closed 3 was obtained in 69 % yield by using 2‐dicyclohexylphosphino‐2′,6′‐dimethoxybiphenyl (SPhos) as a phosphine ligand for Pd. On the other hand, by using PPh3 or 1,1′‐bis(diphenylphosphino)ferrocene (DPPF), the 1,8′′‐position ring‐closed 4 was generated in 12 % and 18 % yields, respectively.
Scheme 1.

Synthesis of 1–4. Reagents and conditions: (i) 1) NBS, CH2Cl2, −78 °C, 3 h, 2) 2‐(azulen‐2‐yl)‐4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolane (5)*, [Pd(OAc)2], SPhos, K3PO4, DME/H2O (10 : 1), reflux; (ii) 1) NBS, CH2Cl2, −78 °C, 3 h, 2) [Pd(dba)2], ligand, Cs2CO3, toluene, reflux.
All NMR signals of 1–4 were assigned using HSQC, HMBC, 1H‐1H COSY, and NOESY experiments (Figures S1–S6, Supporting Information). A comparison of the 1H NMR spectra of azulene, 1,2′‐biazulene, 1, and 2 is shown in Figure 1. The proton signals corresponding to H2, H8, and H3′ in 1,2′‐biazulene (protons neighboring the azulene‐linked positions) tended to shift to a lower magnetic field after dimerization. These protons were de‐shielded by the linked azulene unit. The signals of H8′ in 1 and H3′′ and H8′′ in 2 also showed downfield shifts. On the other hand, the signals of H2 in 1 and H2 and H6–H8 in 2 show a strong upfield shift compared to those observed before the azulene units were subjected to homologation. The signals of H3′′′–H5′′′ in 2 also appeared in a lower magnetic field compared to those of H3–H5 in azulene. Furthermore, these protons were derived from terminal azulene moieties and should be shielded by overlapping azulene units. Variable‐temperature (VT)‐1H NMR measurements of 2 also showed upfield shifts, particularly for H2, H6–H8, and H3′′′ (Figure S7), suggesting that these protons predominantly existed within the shielded region at lower temperatures.
Figure 1.
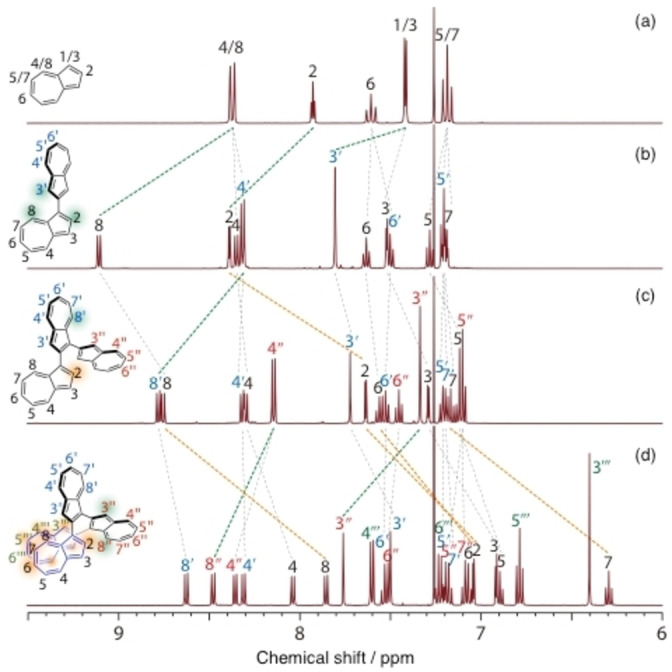
Comparison of 1H NMR spectra of (a) azulene, (b) 1,2′‐biazulene, (c) 1, and (d) 2.
Theoretical calculations suggest that the syn‐type conformer of 2 [(R a,R a)−/(S a,S a)−2] with terminal azulene overlap is 2.8 kcal mol−1 more stable than the anti‐forms [(R a,R a)−/(S a,S a)−2] (Figures 2a and S8). Thus, the Boltzmann distribution of 2 corresponds to a 99 % syn‐type conformer at 300 K. The azulene terminals of the optimized structure are stacked at the closest C⋅⋅⋅C distance (3.08 Å), which is similar to the most stable azulene dimer arrangement of #6 in a stacked parallel type structure studied by Grimme et al. [10] It is considered that there is an attractive interaction between the terminal azulene moieties. Conversely, among the four sets of conformer forms in quaternaphthalene 6 (anti1‐: [(R a,R a,R a)−/(S a,S a,S a)−], anti2‐: [(R a,R a,S a)−/(S a,S a,R a)−], syn1‐: [(R a,S a,S a)−/(S a,R a,R a)−], and syn2‐: [(R a,S a,R a)−/(S a,R a,S a)−]), it was predicted by theoretical calculations that the syn2‐form was the most stable (Figures 2b and S9). The overlapping area of terminal groups of the syn2‐form was smaller and the distance between proximal carbon atoms (3.22 Å) was greater compared to those of syn‐2 (3.08 Å). Sparr et al. reported an exceptionally high rotational barrier of ΔG ≠ 453 K=36.8 kcal mol−1 for tri‐ortho‐substituted oligo‐1,2‐naphthylenes and concluded that the rotation of their aryl‐aryl bonds was sufficiently restricted to form discrete stereoisomers. [3c] Therefore, the conformers of quaternaphthalene 6 are also considered to exist as discrete stereoisomers, which is in contrast to those of quaterazulene 2. The 1H NMR spectra of the ring‐closed terazulenes 3 and 4 (Figures S4–S6) showed that the signals corresponding to the protons neighboring the azulene‐linked positions (H3 and H8 in 3 and H2, H7, H3′′, and H8′′ in 4) tended to shift to a lower magnetic field compared to those of 1 (Figure S10).
Figure 2.

Relative energies (E/kcal mol−1) for the conceivable conformers of (a) quaterazulene 2 and (b) quaternaphthalene 6 calculated at M06‐2X/6‐31G* level.
Compounds 1, 3, and 4 were successfully characterized using single crystal X‐ray structure analysis (Table S2). The dihedral angles formed by the connected azulene planes of 1 were approximately 40° and 46° (Figure 3), and the carbon‐carbon bonds around the bonded carbon were slightly elongated (in blue). The area around the two azulene‐connecting axes in the molecule presented the same axial chirality. In the packing structure, two molecules of (R a)‐ and (S a)‐configurations formed a pair along the direction of the a‐axis (Figure S20). The closest C⋅⋅⋅H distance between the molecules was measured to be 2.72 Å, suggesting a C−H/π interaction. This pair also formed a two‐dimensional sheet in the directions of the b and c axes, with a network thought to originate from the C−H/π interaction. Concentration dependence of the 1H NMR signal chemical shift of 1 also suggests stepwise self‐association (Figure S13).
Figure 3.
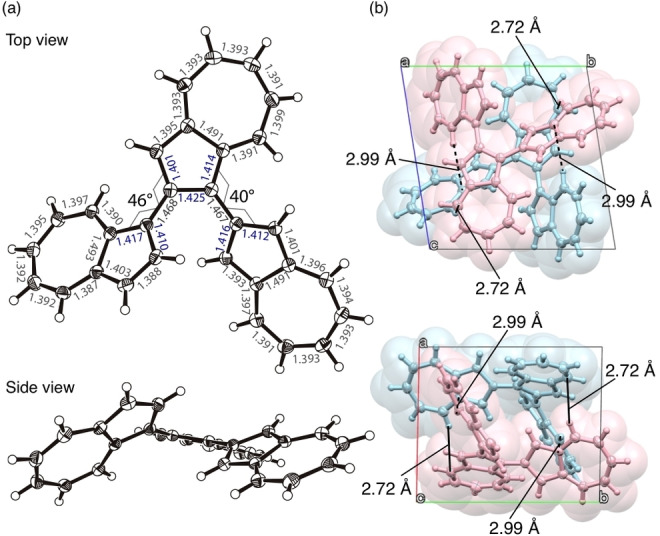
(a) ORTEP drawings of top and side view of 1 (thermal ellipsoids shown at 50 % probability) with bond distances (Å). (b) Crystal packing of 1.
The fused terazulene 3 features an almost planar structure, which is in contrast with the naphthalene based structural isomer, benzo[c]naphtho[2,1‐p]‐chrysene (Figure S14). [11] In Figure 4, the bond lengths (in green) are shown to be smaller than the average C−C distance (1.41 Å) in the regions where a bond alternation was observed. The bond lengths of the central six‐membered ring showed relatively close values although relatively long. These bond lengths suggest that 3 behaves as if three heptafulvenes and a weakly π‐bonded benzene had formed a condensed ring. The packing structure of 3 shows a dihedral angle (near‐neighbor interplanar angle) with a molecular plane intersection of 79° (Figure S15), and the shortest lattice axis length of 11.0 Å. These results suggest a π‐stacked (γ)‐type structure close to the herringbone type. [12] Furthermore, the γ‐/herringbone hybrid type layer of this b‐c plane was laminated in the a‐axis direction.
Figure 4.
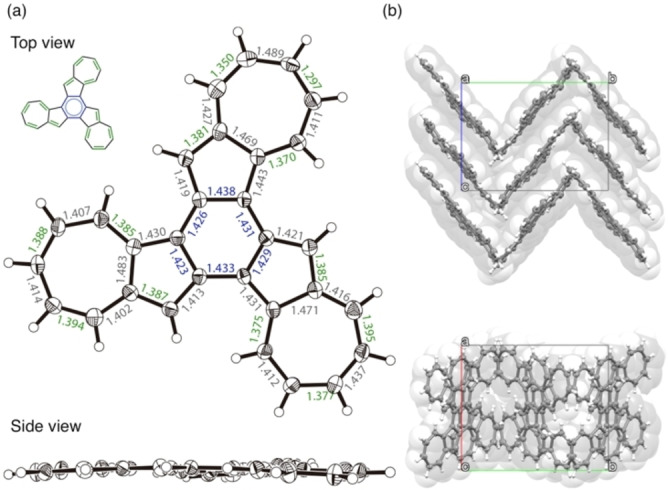
(a) ORTEP drawings of top and side view of 3 (thermal ellipsoids shown at 50 % probability) with bond distances (Å). (b) Crystal packing of 3.
Compound 3 is characterized by a slightly lower solubility in solvents such as benzonitrile, chlorobenzene, chloroform, tetrahydrofuran, and toluene, while featuring a very high crystallinity. On the other hand, 4 showed a high solubility in those solvents, but a low crystallinity. Therefore, X‐ray structural analysis of 4 was performed on a C60 co‐crystal, which showed that the curved compound 4 formed a 1 : 1 complex covering the C60 co‐crystal (Figure 5). Subsequently, the co‐crystal was analyzed with five types of disorder for C60, each with an occupancy of 20 % (Figure S16a), and was thought to be rotating at high speed even at a temperature of 120 K. [13] This suggested the possibility of multiple π−π interactions existing mainly between 4 and fullerene. Crystal packing shows that 4 and C60 have a segregated‐stack structure (Figure S16b), with the C60 co‐crystal arranged along the a‐axis, resulting in the shortest intermolecular carbon‐carbon distance of 2.95 Å. This value is smaller than the one measured in co‐crystals with porphyrin and various cyclic compounds, [14] and is comparable to that of the C60 single crystal. [15]
Figure 5.

(a) ORTEP drawings of top and side view of 4 ⋅ C60 (thermal ellipsoids shown at 50 % probability) with atomic distances (Å). Chlorobenzene and disorder component of C60 are omitted for clarity. (b) Crystal packing of 4 ⋅ C60 ⋅ PhCl.
To evaluate the aromaticities of 1, 3, and 4, the harmonic oscillator model of aromaticity (HOMA) value was calculated (Figure S17).[ 14j , 16 ] The results suggested that performing ring closure from 1 to 3 reduced the HOMA value of the five‐membered ring and reduced aromaticity. Meanwhile, when ring closure was performed from 1 to 4, the newly formed seven‐membered ring featured smaller bond alternation than the five‐membered one.
Nucleus‐independent chemical shift (NICS) [17] calculations (Figure S18) showed that the aromaticity of the five‐membered ring was shown to decrease with ring closure from 1 to 3, with a six‐membered ring formation in the center. Interestingly, the central seven‐membered ring of 4 showed a positive value [NICS(0): 8.42, NICS(+1): 3.17, NICS(−1): 3.89], suggesting anti‐aromatic properties, albeit not very strong. A seven‐membered ring surrounded by one naphthalene and two five‐membered rings derived from azulenes has been previously reported to also present a small but nonzero paratropic ring current [NICS(0): 7.22, NICS(+1)/NICS(−1): 2.43],[ 5f , 18 ] and the system under investigation featured a slightly larger value. The paratropic ring current in 4 can be explained by the polarization of the two seven‐membered rings condensed in the heptalene skeleton, along with the central seven‐membered ring contributing to the negative 8π antiaromatic resonance structure (Figure S19).
Ultraviolet‐visible absorption spectra were recorded in THF (Figure S20a). When azulene was connected at the 1,2‐position, a relatively strong absorption was observed near 450 nm, which is not observed for azulene alone. This result showed relatively good agreement with the spectral shape predicted by TD‐DFT calculations (Figures S20b and S21). The onsets of the absorptions of 1, 3, and 4 were all approximately 850 nm, while that of 4 extended to as far as 950 nm.
The redox properties of 1–4 were examined together with azulene and 1,2′‐biazulene via square wave voltammetry (SWV) measurements, which were carried out using ferrocene as an internal standard at the same concentration (Table 1, Figure S22). As the number of azulene linkages increased, especially from dimer to tetramer, the first oxidation potential and the first reduction potential of successive homologs remained essentially constant. These behaviors, also for 3 and 4, were relatively consistent with the HOMO‐LUMO levels obtained from theoretical calculations (Figure S23), indicating the smaller HOMO‐LUMO gaps compared to those of the ring‐closed compounds of 1,2‐position connected ternaphthalene, benzo[c]naphtho[2,1‐p]chrysene, and benzene‐based truxene. Truxene with C 3h symmetry is expected to find applications in various fields, [19] such as photoluminescence and organic electronics.[ 19a , 19b ] Benzo[c]naphtho[2,1‐p]chrysene, exhibit a C 3 symmetry and have attracted attention as potential precursors for the synthesis of bowl‐shaped fullerene substructures. [11] In addition, based on the peak areas, all the peaks are believed to be derived from one‐electron redox reactions, suggesting the presence of electronic interactions between the units. Compound 1 was found to be susceptible to oxidation and reduction via ring closure, and 4 had the smallest redox potential difference. This was consistent with the absorption spectra trends.
Table 1.
Redox Potentials of 1–4.[a]
|
Compd |
ox E 1 |
red E 1 |
red E 2 |
red E 3 |
|---|---|---|---|---|
|
azulene |
0.56 |
−2.13 |
– |
– |
|
1 |
0.32 |
−1.95 |
−2.25 |
−2.34 |
|
2 |
0.29 |
−1.97 |
−2.09 |
−2.37 |
|
3 |
0.08 |
−1.83 |
−2.28 |
−2.40 |
|
4 |
0.07 |
−1.77 |
−2.21 |
−2.30 |
[a] All potentials in V vs. Cp2Fe0/+, measured in PhCN, n Bu4NPF6 as supporting electrolyte.
Reacting azulene with an acid is known to protonate an electron‐rich five‐membered ring to produce a relatively stable tropylium cation (Figure 6b). [20] The absorption spectrum of 3 was thus measured while adding 0–15 equiv. of methanesulfonic acid (Figure 6a), resulting in a decrease in the absorption near 397 nm and 457 nm, and the appearance of a new absorption peak between 500–800 nm. In this system, protonation was thought to occur at the 3‐position of the five‐membered ring with a large HOMO coefficient, and there was relatively good agreement with the spectral shape predicted by TD‐DFT calculations (Figure 6c). The azulene titration plot shows that absorption continued to increase with the addition of acid, with an inflection point observed close to 3–5 equiv. of acid for 3 (Figure 6a, inset), after which the change in absorption tended to saturate. This demonstrated that 3 is more reactive to acids than azulene.
Figure 6.

UV‐vis spectra of (a) 3 and (b) azulene in the presence of methanesulfonic acid (0–15 eq.) in CH2Cl2. [substance]0: 1.6×10−5 m. Inset: titration plots of 3 and azulene with methanesulfonic acid. (c) Simulated absorption spectra of 3 and 3 ⋅ H+.
Conclusion
In this study, 1,2‐position connected terazulene and quaterazulene, and ring‐closed compounds of terazulene were successfully synthesized, and their structures and electronic properties were investigated. Quaterazulene 2 was revealed to exist as a helical structure, and the syn‐type structure with terminal azulene overlap was observed to be more stable. A method to determine the optical resolution of 2 is currently underway in our laboratory. Compound 3 was characterized by a smaller HOMO‐LUMO gap compared to benzo[c]naphtho[2,1‐p]chrysene and truxene, while exhibiting the same threefold rotation axis; therefore, its application in device materials is anticipated. Interestingly, compound 4 showed high solubility due to its curved structure and a smaller HOMO‐LUMO gap compared to 3. Such systems could provide a novel design concept for functional electronic and optical devices or catalysts, which can find application in technologies such as supramolecular architectures, hole transport materials, and chiral recognition agents.
Experimental Section
General: 1H and 13C NMR spectra were recorded on Bruker AVANCE‐III‐400 (400 MHz for 1H, 100 MHz for 13C) or Bruker AVANCE‐III‐600 (600 MHz for 1H, 150 MHz for 13C). Spectra are reported (in δ) referenced to internal Me4Si. Mass spectra were recorded on Thermo Scientific, Exactive Plus Orbitrap Mass Spectrometer for electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI), or ITQ 700 GC/MS for electronic impact ionization (EI). IR spectra were recorded on JASCO FT/IR‐610 Spectrometer. Melting points were determined with Yanaco melting point apparatus. Absorption spectra were recorded on Shimadzu UV‐3600 spectrometer. Elemental analyses were performed on Perkin Elmer PE 2400‐II CHNS/O analyzer and Yanaco MT‐6 analyzer. Cyclic voltammetry (CV) measurements were performed on BAS Electrochemical Analyzer (Model 630E). Column chromatography was carried out using Kanto chemical silica gel 60 N, 60–210 μm meshes. Preparative gel permeation chromatography was carried out using HPLC LC‐918 system (Japan Analytical Industry, Co. Ltd) equipped with polystyrene gel column JAIGEL‐1H.
Data of 1 and 3 were collected using a Bruker APEX II CCD diffractometer with Mo Kα radiation (λ=0.71073 Å). Data of 4 ⋅ C60 were collected using a Rigaku HyPix‐600 diffractometer with Cu Kα radiation (λ=1.54187 Å). Single crystals were mounted on MiTeGen Dual‐Thickness MicroMounts using a trace of mineral oil. Frames were collected, reflections were indexed and processed, and the files were scaled and corrected for absorption using Bruker APEX3 program for 1 and 3 and Rigaku CrysAlisPro program for 4 ⋅ C60. The space groups were assigned, and the structures were solved by direct methods using XPREP within the SHELXTL suite of programs and refined by full‐matrix least‐squares against F 2 with all reflections using SHELXL‐2014 (1 and 2) or SHELXL‐2018 (4 ⋅ C60) with the graphical interface SHELXLE.
Deposition Numbers 1993531 (for 1), 1993535 (for 3), and 1993536 (for 4 ⋅ C60) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
1,2′ : 1′,2′′‐terazulene (1). To a dichloromethane (10 mL) solution of 1,2′‐biazulene (100 mg, 0.39 mmol) was slowly dropped a dichloromethane (30 mL) solution of N‐bromosuccinimide (70 mg, 0.39 mmol) at −78 °C under argon. After completion of the dropwise addition, the mixture was stirred for 3 h. Thereafter, a saturated aqueous solution of sodium hydrogen carbonate (20 mL) was added, and the dichloromethane layer was washed with brine. After the dichloromethane solution was dried over sodium sulfate, 80 mL of hexane was added, and the mixture was filtered through aluminium oxide. The solvent of the filtrate was replaced with a mixed solvent of 1,2‐dimethoxyethane/water (10 : 1, 44 mL) using a rotary evaporator. To this solution were added 2‐(azulen‐2‐yl)‐4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolane (5) (100 mg, 0.39 mmol), palladium(II) acetate (2 mg, 0.01 mmol), 2‐dicyclohexylphosphino‐2′,6′‐dimethoxybiphenyl (SPhos, 8 mg, 0.02 mmol) and tripotassium phosphate (170 mg, 0.79 mmol). The mixture was refluxed for 5 h under argon. Then, the reaction mixture was filtered through celite, extracted with hexane, and washed with brine. The hexane layer was dried over sodium sulfate, and the solution was concentrated using a rotary evaporator. The obtained crude product was purified by silica gel column chromatography using a mixed solvent of hexane/dichloromethane (4 : 1) as an eluent to obtain a green solid of 1. (91 mg, 0.24 mmol 61 %); green solid; m.p. 104–108 °C; 1H NMR (600 MHz, CDCl3): δ 8.74 (d, J=10.2 Hz, 1H), 8.72 (d, J=9.6 Hz, 1H), 8.28 (d, J=9.6 Hz, 1H), 8.26 (d, J=9.0 Hz, 1H), 8.11 (d, J=9.0 Hz, 2H), 7.69 (s, 1H), 7.60 (d, J=3.6 Hz, 1H), 7.53 (t, J=10.2 Hz, 1H), 7.49 (t, J=10.2 Hz, 1H), 7.42 (t, J=9.0 Hz, 1H), 7.30 (s, 2H), 7.26 (d, J=4.2 Hz, 1H), 7.18 (t, J=9.0 Hz, 1H), 7.16 (t, J=9.0 Hz, 1H), 7.13 (t, J=9.6 Hz, 1H), 7.10 (t, J=9.6 Hz, 1H), 7.08 (t, J=9.6 Hz, 2H); 13C NMR (150 MHz, CDCl3): δ 147.0, 146.3, 142.7, 141.8, 140.6, 140.2, 138.8, 138.2, 137.0, 136.9, 136.9, 136.4, 135.4, 135.4, 134.9, 134.8, 127.2, 125.7, 124.98, 124.54, 124.1, 123.9, 123.2, 119.6, 119.4, 118.3; IR (KBr): νmax 1567, 1509, 1380, 897, 781, 732 cm−1; MS (ESI) m/z: 381.16 [M+H]+; UV‐vis (THF) λmax (ϵ): 234 (41500), 262 (51500), 289 (56000), 316 (49700), 432 (29600), 580 (1170) nm; Anal. Calcd for C30H20: C, 94.70; H, 5.30. Found: C, 94.80; H,5.49.
1,2′ : 1′,2′′ : 1′′,2′′′‐quaterazulene (2) and azuleno[1,2‐e]dicyclohepta[a , h ]‐as‐indacene (3). To a dichloromethane (15 mL) solution of 1 (175 mg, 0.46 mmol) was slowly dropped a dichloromethane (45 mL) solution of N‐bromosuccinimide (80 mg, 0.46 mmol) at −78 °C under argon. After completion of the dropwise addition, the mixture was stirred for 3 h. Thereafter, a saturated aqueous solution of sodium hydrogen carbonate (30 mL) was added, and the dichloromethane layer was washed with brine. After the dichloromethane solution was dried over sodium sulfate, 80 mL of hexane was added, and the mixture was filtered through aluminium oxide. The solvent of the filtrate was replaced with a mixed solvent of 1,2‐dimethoxyethane/water (10 : 1, 11 mL) using a rotary evaporator. To this solution were added 2‐(azulen‐2‐yl)‐4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolane (5) (115 mg, 0.45 mmol), palladium(II) acetate (3 mg, 0.01 mmol), 2‐dicyclohexylphosphino‐2′,6′‐dimethoxybiphenyl (SPhos, 9 mg, 0.023 mmol) and tripotassium phosphate (190 mg, 0.9 mmol). The mixture was refluxed for 15 h under argon. Then, the reaction mixture was filtered through celite, extracted with hexane, and washed with brine. The hexane layer was dried over sodium sulfate, and the solution was concentrated using a rotary evaporator. The obtained crude product was purified by preparative gel permeation chromatography with chloroform to obtain a green solid of 2 (18 mg, 0.036 mmol 8 %) and a black solid of 3 (63 mg, 0.16 mmol, 36 %).
2: brown solid; m.p. 178–181 °C; 1H NMR (400 MHz, CDCl3): δ 8.62 (d, J=9.7 Hz, 1H), 8.46 (d, J=9.8 Hz, 1H), 8.36‐8.29 (m, 2H), 8.03 (d, J=9.4 Hz, 1H), 7.84 (d, J=9.8 Hz, 1H), 7.75 (s, 1H), 7.57 (d, J=9.2 Hz, 2H), 7.50‐7.48 (m, 3H), 7.24–7.15 (m, 4H), 7.10–7.03 (m, 3H), 7.02–6.89 (m, 2H), 6.75 (t, J=9.8 Hz, 2H), 6.39 (s, 2H), 6.29 (t, J=9.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 147.5, 147.0, 146.6, 142.1, 141.7, 141.6, 139.8, 138.8, 138.4, 137.8, 137.4, 136.8, 136.1, 135.7, 135.6, 135.2, 135.1, 134.7, 134.4, 134.3, 133.8, 127.8, 126.9, 126.0, 124.33, 124.29, 124.0, 123.9, 123.1, 122.7, 122.3, 120.2, 118.5, 118.2, 117.5; IR (KBr): νmax 1722, 1568, 1512, 1511, 1460, 1384, 1261, 1096, 1028, 799, 726 cm−1; MS (EI) m/z: 507.26 [M+H]+; UV‐vis (THF) λmax (ϵ): 258 (53600), 290 (57300), 430 (30300), 580 (1230) nm; Anal. Calcd for C40H26: C, 94.83; H, 5.17. Found: C, 94.71; H, 5.09.
3: black solid; m.p. 245–250 °C; 1H NMR (600 MHz, CDCl3): δ 9.37 (d, J=9.5 Hz, 3H), 8.55 (d, J=9.5 Hz, 3H), 8.49 (s, 3H), 7.67 (t, 3H, J=9.5 Hz), 7.61 (t, J=9.5 Hz, 3H), 7.41 (t, J=9.5 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 143.9, 142.3, 139.1, 134.6, 134.0, 131.3, 125.8, 125.1, 120.0, 114.7; IR (KBr): νmax 1574, 1531, 1414, 804, 757, 710, 687 cm−1; MS (ESI) m/z: 379.16 [M+H]+; UV‐vis (THF) λmax (ϵ): 257 (24400), 304 (26000), 397 (46500), 456 (61700), 743 (250, sh) nm; Anal. Calcd for C30H18: C, 95.21; H, 4.79. Found: C, 95.12; H, 4.70.
azuleno[1,2‐e]dicyclohepta[a , h ]‐as‐indacene (3) and diazuleno[1,2‐a : 1′,2′‐c]‐cyclopenta[ef]heptalene (4). To a dichloromethane (15 mL) solution of 1 (175 mg, 0.46 mmol) was slowly dropped a dichloromethane (45 mL) solution of N‐bromosuccinimide (80 mg, 0.46 mmol) at −78 °C under argon. After completion of the dropwise addition, the mixture was stirred for 3 h. Thereafter, a saturated aqueous solution of sodium hydrogen carbonate (30 mL) was added, and the dichloromethane layer was washed with brine. After the dichloromethane solution was dried over sodium sulfate, 80 mL of hexane was added, and the mixture was filtered through aluminium oxide. The solvent of the filtrate was replaced with toluene (60 mL) using a rotary evaporator. To this solution were added tris(dibenzylideneacetone)dipalladium(0) (18 mg, 0.02 mmol), ligand (0.024 mmol) and caesium carbonate (290 mg, 0.9 mmol). The mixture was refluxed for 20 h under argon. Then, the reaction mixture was filtered through celite and washed with brine. The hexane layer was dried over sodium sulfate, and the solution was concentrated using a rotary evaporator. The obtained crude product was purified by preparative gel permeation chromatography with chloroform to obtain black solids of 3 (16–69 %) and 4 (12–18 %); black solid; m.p. 134–138 °C; 1H NMR (600 MHz, CDCl3): δ 8.84 (d, J=9.6 Hz, 1H), 8.74 (d, J=9.6 Hz, 1H), 8.22 (d, J=4.2 Hz, 1H), 8.01 (d, J=9.6 Hz, 1H), 7.89 (d, J=9.6 Hz, 1H), 7.87 (d, J=9.6 Hz, 1H), 7.74 (s, 1H), 7.61 (d, J=10.8 Hz, 1H), 7.36 (t, J=9.6 Hz, 1H), 7.31 (s, 1H), 7.24‐7.16 (m, 3H), 7.10 (t, J=9.6 Hz, 1H), 7.04 (t, J=9.6 Hz, 1H), 6.98‐6.94 (m. 2H), 6.66 (t, J=9.6 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 150.0, 149.9, 147.8, 145.4, 143.8, 143.0, 141.64, 141.60, 141.2, 138.1, 137.0, 136.6, 135.4, 134.5, 134.2, 133.0, 132.9, 131.5, 127.8, 127.6, 126.9, 126.2, 126.1, 125.9, 125.8, 123.0, 120.8, 119.1, 117.9, 117.5, 114.7; IR (KBr): νmax 1562, 1508, 1407, 1026, 800, 713 cm−1; MS (ESI) m/z: 379.16 [M+H]+.; UV‐vis (THF) λmax (ϵ): 240 (24900), 350 (16000), 461 (30200), 870 (80, sh) nm; Anal. Calcd for C30H18: C, 95.21; H, 4.79. Found: C, 95.25; H, 4.66.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant JP15K05435 in the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Tsuchiya T., Higashibeppu M., Mazaki Y., ChemistryOpen 2023, 12, e202100298.
Contributor Information
Prof. Dr. Takahiro Tsuchiya, Email: ttsuchi@kitasato-u.ac.jp.
Prof. Dr. Yasuhiro Mazaki, Email: mazaki@kitasato-u.ac.jp.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Kousseff C. J., Halaksa R., Parr Z. S., Nielsen C. B., Chem. Rev. 2022, 122, 4397–4419; [DOI] [PubMed] [Google Scholar]
- 1b. Burmeister D., Trunk M. G., Bojdys M. J., Chem. Soc. Rev. 2021, 50, 11559–11579; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Freudenberg J., Jansch D., Hinkel F., Bunz U. H. F., Chem. Rev. 2018, 118, 5598–5689. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Yang X., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2021, 60, 7941–7946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Hayashi H., Kato Y., Matsumoto A., Shikita S., Aizawa N., Suzuki M., Aratani N., Yasuda T., Yamada H., Chem. Eur. J. 2019, 25, 15565–15571; [DOI] [PubMed] [Google Scholar]
- 2c. Kumaki D., Umeda T., Suzuki T., Tokito S., Org. Electron. 2008, 9, 921–924; [Google Scholar]
- 2d. Raghunath P., Reddy M. A., Gouri C., Bhanuprakash K., Rao V. J., J. Phys. Chem. A 2006, 110, 1152–1162; [DOI] [PubMed] [Google Scholar]
- 2e. Ito K., Suzuki T., Sakamoto Y., Kubota D., Inoue Y., Sato F., Tokito S., Angew. Chem. Int. Ed. 2003, 42, 1159–1162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1191–1194. [Google Scholar]
- 3.
- 3a. Castrogiovanni A., Herr P., Larsen C. B., Guo X., Sparr C., Wenger O. S., Chem. Eur. J. 2019, 25, 16748–16754; [DOI] [PubMed] [Google Scholar]
- 3b. Yamamoto T., Ishibashi A., Koyanagi M., Ihara H., Eichenauer N., Suginome M., Bull. Chem. Soc. Jpn. 2017, 90, 604–606; [Google Scholar]
- 3c. Lotter D., Neuburger M., Rickhaus M., Häussinger D., Sparr C., Angew. Chem. Int. Ed. 2016, 55, 2920–2923; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2973–2976; [Google Scholar]
- 3d. Schwabegger G., Dingemans T., Resel R., Sitter H., Simbrunner C., Appl. Phys. A 2014, 115, 731–735; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Tsubaki K., Miura M., Morikawa H., Tanaka H., Kawabata T., Furuta T., Tanaka K., Fuji K., J. Am. Chem. Soc. 2003, 125, 16200–16201. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Xin H., Hou B., Gao X., Acc. Chem. Res. 2021, 54, 1737–1753; [DOI] [PubMed] [Google Scholar]
- 4b. Tsuchiya T., Umemura R., Kaminaga M., Kushida S., Ohkubo K., Noro S. I., Mazaki Y., ChemPlusChem 2019, 84, 655–664; [DOI] [PubMed] [Google Scholar]
- 4c. Ito H., Itami K., Chem. Commun. 2019, 55, 9606–9609; [DOI] [PubMed] [Google Scholar]
- 4d. Shoji T., Ito S., Chem. Eur. J. 2017, 23, 16696–16709; [DOI] [PubMed] [Google Scholar]
- 4e. Koide T., Takesue M., Murafuji T., Satomi K., Suzuki Y., Kawamata J., Terai K., Suzuki M., Yamada H., Shiota Y., ChemPlusChem 2017, 82, 1010–1014; [DOI] [PubMed] [Google Scholar]
- 4f. Xin H., Gao X., ChemPlusChem 2017, 82, 945–956; [DOI] [PubMed] [Google Scholar]
- 4g. Tobe Y., Chem. Rec. 2015, 15, 86–96; [DOI] [PubMed] [Google Scholar]
- 4h. Yamaguchi Y., Ogawa K., Nakayama K.-i., Ohba Y., Katagiri H., J. Am. Chem. Soc. 2013, 135, 19095–19098; [DOI] [PubMed] [Google Scholar]
- 4i. Amir E., Amir R. J., Campos L. M., Hawker C. J., J. Am. Chem. Soc. 2011, 133, 10046–10049; [DOI] [PubMed] [Google Scholar]
- 4j. Nöll G., Lambert C., Lynch M., Porsch M., Daub J., J. Phys. Chem. C 2008, 112, 2156–2164; [Google Scholar]
- 4k. Dias J. R., J. Phys. Org. Chem. 2007, 20, 395–409; [Google Scholar]
- 4l. Wang F., Lai Y.-H., Kocherginsky N., Kosteski Y. Y., Org. Lett. 2003, 5, 995–998; [DOI] [PubMed] [Google Scholar]
- 4m. Schmitt S., Baumgarten M., Simon J., Hafner K., Angew. Chem. Int. Ed. 1998, 37, 1077–1081; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1129–1133. [Google Scholar]
- 5.
- 5a. Liu J., Mishra S., Pignedoli C. A., Passerone D., Urgel J. I., Fabrizio A., Lohr T. G., Ma J., Komber H., Baumgarten M., J. Am. Chem. Soc. 2019, 141, 12011–12020; [DOI] [PubMed] [Google Scholar]
- 5b. Konishi A., Horii K., Shiomi D., Sato K., Takui T., Yasuda M., J. Am. Chem. Soc. 2019, 141, 10165–10170; [DOI] [PubMed] [Google Scholar]
- 5c. Skidin D., Eisenhut F., Richter M., Nikipar S., Krüger J., Ryndyk D. A., Berger R., Cuniberti G., Feng X., Moresco F., Chem. Commun. 2019, 55, 4731–4734; [DOI] [PubMed] [Google Scholar]
- 5d. Mishra S., Lohr T. G., Pignedoli C. A., Liu J., Berger R., Urgel J. I., Müllen K., Feng X., Ruffieux P., Fasel R., ACS Nano 2018, 12, 11917–11927; [DOI] [PubMed] [Google Scholar]
- 5e. Hieulle J., Carbonell-Sanromà E., Vilas-Varela M., Garcia-Lekue A., Guitián E., Pena D., Pascual J. I., Nano Lett. 2018, 18, 418–423; [DOI] [PubMed] [Google Scholar]
- 5f. Uehara K., Mei P., Murayama T., Tani F., Hayashi H., Suzuki M., Aratani N., Yamada H., Eur. J. Org. Chem. 2018, 2018, 4508–4511; [Google Scholar]
- 5g. Murai M., Iba S., Ota H., Takai K., Org. Lett. 2017, 19, 5585–5588. [DOI] [PubMed] [Google Scholar]
- 6. Tsuchiya T., Katsuoka Y., Yoza K., Sato H., Mazaki Y., ChemPlusChem 2019, 84, 1659–1667. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Shoji T., Miyashita K., Araki T., Tanaka M., Maruyama A., Sekiguchi R., Ito S., Okujima T., Synthesis 2016, 48, 2438–2448; [Google Scholar]
- 7b. Dyker G., Borowski S., Heiermann J., Körning J., Opwis K., Henkel G., Köckerling M., J. Organomet. Chem. 2000, 606, 108–111; [Google Scholar]
- 7c. Morita T., Takase K., Bull. Chem. Soc. Jpn. 1982, 55, 1144–1152. [Google Scholar]
- 8. Kurotobi K., Miyauchi M., Takakura K., Murafuji T., Sugihara Y., Eur. J. Org. Chem. 2003, 2003, 3663–3665. [Google Scholar]
- 9.
- 9a. Kim J., Hong S. H., ACS Catal. 2017, 7, 3336–3343; [Google Scholar]
- 9b. Segawa Y., Maekawa T., Itami K., Angew. Chem. Int. Ed. 2015, 54, 66–81; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 68–83. [Google Scholar]
- 10. Piacenza M., Grimme S., J. Am. Chem. Soc. 2005, 127, 14841–14848. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Pérez D., Guitián E., Chem. Soc. Rev. 2004, 33, 274–283; [DOI] [PubMed] [Google Scholar]
- 11b. Pena D., Pérez D., Guitián E., Castedo L., Org. Lett. 1999, 1, 1555–1557; [Google Scholar]
- 11c. Hagen S., Bratcher M. S., Erickson M. S., Zimmermann G., Scott L. T., Angew. Chem. Int. Ed. 1997, 36, 406–408; [Google Scholar]; Angew. Chem. 1997, 109, 407–409; [Google Scholar]
- 11d. Hagen S., Scott L. T., J. Org. Chem. 1996, 61, 7198–7199; [DOI] [PubMed] [Google Scholar]
- 11e. Laarhoven W., van Broekhoven J., Tetrahedron Lett. 1970, 11, 73–76. [Google Scholar]
- 12.
- 12a. Tiekink E. R. T., Zukerman-Schpector J., The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering, John Wiley & Sons; 2012; [Google Scholar]
- 12b. Valiyev F., Hu W.-S., Chen H.-Y., Kuo M.-Y., Chao I., Tao Y.-T., Chem. Mater. 2007, 19, 3018–3026; [Google Scholar]
- 12c. Desiraju G. R., Gavezzotti A., Acta Crystallogr. Sect. B 1989, 45, 473–482; [Google Scholar]
- 12d. Fawcett E., Trotter J., Proc. Roy. Soc. A 1966, 289, 366–376. [Google Scholar]
- 13. Matsuno T., Nakai Y., Sato S., Maniwa Y., Isobe H., Nat. Commun. 2018, 9, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Ke X.-S., Kim T., Brewster J. T., Lynch V. M., Kim D., Sessler J. L., J. Am. Chem. Soc. 2017, 139, 4627–4630; [DOI] [PubMed] [Google Scholar]
- 14b. Yokoi H., Hiraoka Y., Hiroto S., Sakamaki D., Seki S., Shinokubo H., Nat. Commun. 2015, 6, 8215; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Yamamura M., Saito T., Nabeshima T., J. Am. Chem. Soc. 2014, 136, 14299–14306; [DOI] [PubMed] [Google Scholar]
- 14d. Xia J., Bacon J. W., Jasti R., Chem. Sci. 2012, 3, 3018–3021; [Google Scholar]
- 14e. King B. T., Olmstead M. M., Baldridge K. K., Kumar B., Balch A. L., Gharamaleki J. A., Chem. Commun. 2012, 48, 9882–9884; [DOI] [PubMed] [Google Scholar]
- 14f. Iwamoto T., Watanabe Y., Sadahiro T., Haino T., Yamago S., Angew. Chem. Int. Ed. 2011, 50, 8342–8344; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8492–8494; [Google Scholar]
- 14g. Song J., Aratani N., Shinokubo H., Osuka A., J. Am. Chem. Soc. 2010, 132, 16356–16357; [DOI] [PubMed] [Google Scholar]
- 14h. Nobukuni H., Shimazaki Y., Tani F., Naruta Y., Angew. Chem. Int. Ed. 2007, 46, 8975–8978; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9133–9136; [Google Scholar]
- 14i. Olmstead M. M., Costa D. A., Maitra K., Noll B. C., Phillips S. L., Van Calcar P. M., Balch A. L., J. Am. Chem. Soc. 1999, 121, 7090–7097; [Google Scholar]
- 14j. Kruszewski J., Krygowski T., Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar]
- 15. David W. I. F., Ibberson R. M., Matthewman J. C., Prassides K., Dennis T. J. S., Hare J. P., Kroto H. W., Taylor R., Walton D. R. M., Nature 1991, 353, 147–149. [Google Scholar]
- 16. Krygowski T. M., Szatylowicz H., Stasyuk O. A., Dominikowska J., Palusiak M., Chem. Rev. 2014, 114, 6383–6422. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. AbuSalim D. I., Lash T. D., J. Phys. Chem. A 2018, 123, 230–246; [DOI] [PubMed] [Google Scholar]
- 17b. Fischer L. J., Dutton A. S., Winter A. H., Chem. Sci. 2017, 8, 4231–4241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Gershoni-Poranne R., Stanger A., Chem. Soc. Rev. 2015, 44, 6597–6615; [DOI] [PubMed] [Google Scholar]
- 17d. Chen Z., Wannere C. S., Corminboeuf C., Puchta R., v R Schleyer P., Chem. Rev. 2005, 105, 3842–3888; [DOI] [PubMed] [Google Scholar]
- 17e. v R Schleyer P., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J., J. Am. Chem. Soc. 1996, 118, 6317–6318. [DOI] [PubMed] [Google Scholar]
- 18. Uehara K., Mei P., Murayama T., Tani F., Hayashi H., Suzuki M., Aratani N., Yamada H., Eur. J. Org. Chem. 2019, 2019, 860–861. [Google Scholar]
- 19.
- 19a. Huang C., Fu W., Li C.-Z., Zhang Z., Qiu W., Shi M., Heremans P., Jen A. K.-Y., Chen H., J. Am. Chem. Soc. 2016, 138, 2528–2531; [DOI] [PubMed] [Google Scholar]
- 19b. Goubard F., Dumur F., RSC Adv. 2015, 5, 3521–3551; [Google Scholar]
- 19c. Otero G., Biddau G., Sánchez-Sánchez C., Caillard R., López M. F., Rogero C., Palomares F. J., Cabello N., Basanta M. A., Ortega J., Nature 2008, 454, 865–868. [DOI] [PubMed] [Google Scholar]
- 20. Mikheev Y. A., Guseva L., Ershov Y. A., Russ. J. Phys. Chem. A 2013, 87, 1645–1653. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.