

Abstract
Hedgehog (Hh) signaling controls growth and patterning during embryonic development and homeostasis in adult tissues. Hh binding to the receptor Patched (Ptc) elicits intracellular signaling by relieving Ptc-mediated inhibition of the transmembrane protein Smoothened (Smo). We uncovered a role for the lipid phosphatidic acid (PA) in the regulation of the Hh pathway in Drosophila melanogaster. Deleting the Ptc C-terminal tail or mutating the predicted PA binding sites within it prevented Ptc from inhibiting Smo in wing discs and in cultured cells. The C-terminal tail of Ptc directly interacted with PA in vitro, an association that was reduced by Hh, and increased the amount of PA at the plasma membrane in cultured cells. Smo also interacted with PA in vitro through a binding pocket located in the transmembrane region, and mutating residues in this pocket reduced Smo activity in vivo and in cells. By genetically manipulating PA amounts in vivo or treating cultured cells with PA, we demonstrated that PA promoted Smo activation. Our findings suggest that Ptc may sequester PA in the absence of Hh and release it in the presence of Hh, thereby increasing the amount of PA that is locally available to promote Smo activation.
Introduction
Initially discovered in Drosophila melanogaster as a segment polarity gene, Hedgehog (Hh) is not only involved in embryo patterning but is critical in post-developmental tissue homeostasis (1, 2). Aberrant signaling through the highly conserved Hh pathway is implicated in various human diseases, including several types of cancers (3–7). Among the signaling components, Smoothened (Smo), an atypical G protein–coupled receptor (GPCR) family member, acts as a key regulator of the pathway for both insects and vertebrates. Smo is an attractive therapeutic target (8, 9) because abnormal activation of Smo results in basal cell carcinoma (BCC) and medulloblastoma. Given the critical roles of Smo in Hh signaling, the amount of this protein on the cell surface is precisely controlled to fine-tune Hh signal transduction in both vertebrates and invertebrates (1, 10–13).
In Drosophila, the Hh reception system at the plasma membrane includes the receptor complex consisting of Patched (Ptc) and Interference hedgehog (Ihog) and the signal transducer Smo (14, 15). In the absence of Hh, Ptc inhibits Smo, which is subjected to inactivation by ubiquitylation and subsequent degradation (16, 17). The downstream Zn-finger transcription factor Cubitus interruptus (Ci) becomes truncated and functions as a transcriptional repressor (18). In the presence of Hh, binding of Hh to Ptc-Ihog relieves the inhibition of Smo by Ptc and Smo becomes phosphorylated by multiple kinases (19, 20), resulting in a conformational change and the activation of Smo on the cell surface (21, 22). Full-length Ci is activated to induce the expression of Hh target genes, including ptc, decapentaplegic (dpp), and engrailed (en) (2, 18). This dual activity – the formation of transcriptional repressors in the absence of Hh and formation of transcriptional activators in the presence of Hh – is conserved in vertebrates, in which transcription factors is the Gli family function analogously to Ci (2, 18).
In the Drosophila wing disc, posterior (P) compartment cells produce and secrete Hh proteins that act upon neighboring anterior (A) compartment cells located adjacent to the A/P boundary to induce the expression of target genes, including ptc (23–26), resulting in the high abundance of Ptc along the A/P boundary in addition to the basal amounts of Ptc in the A-compartment cells. Ci is present only in A-compartment cells, whereas Smo is present throughout the wing disc, but its abundance is lower in the A-compartment than in the P-compartment. In the absence of Hh, as in the A-compartment, Smo is subjected to ubiquitination-mediated degradation (16, 17). In response to Hh stimulation, as in the P-compartment cells and in the A-compartment cells near the A/P boundary, Smo accumulates due to phosphorylation (19, 27), a conformational change (22), and interaction with the Costal2-Fused protein complex (28–31).
The mechanisms by which Ptc inhibits Smo in the absence of Hh and how Hh stimulation alleviates this inhibition are not well-defined. Studies suggest that Ptc likely inhibits Smo through an indirect mechanism that does not require Ptc-Smo contact (32, 33). Inhibition occurs even when Smo is present in 50-fold molar excess of Ptc, and substochiometric amounts of Ptc can repress Smo activation (33, 34), indicating that the inhibition process is catalytic. Ptc may inhibit the production of positive regulator(s) or promote the synthesis of inhibitory molecules (33).
Lipids have been implicated in Smo regulation. Cholesterol directly interacts with (35) and activates Smo by binding to its extracellular cysteine-rich domain (CRD) (36–38). However, cholesterol was found to be sufficient to activate Smo in an in vitro system even in the absence of the CRD domain (39), implying that the CRD is not likely required for Ptc-Smo communication. In vertebrates, Hh signaling occurs in the primary cilium, and increasing the amount of cholesterol in the cilium results in Smo activation (40). Studies report that Ptc has cholesterol transporter activity (41, 42) and that cholesterol can influence Smo through a cholesterol site that is independent from the CRD (39). In Drosophila, Hh promotes the production of phosphatidyl-inositol 4-phosphate (PI(4)P) (43, 44), which directly interacts with Smo through the arginine motifs in its C-terminal tail (C-tail), thereby promoting Smo phosphorylation and activation (43). Hh also promotes the production of oxysterols (45) that activate Smo in vertebrate cilium (46). However, the amount of oxysterol produced in response to Hh stimulation is substantially lower than the EC50 for Hh pathway activation (47). Furthermore, oxysterols do not interact with Drosophila Smo (48). Currently, no single unifying mechanism exists for Ptc-mediated Smo inhibition and activation of Smo by Hh, indicating that other molecule(s) are likely involved in Ptc-Smo communication.
Here, we found that the C-tail of Ptc was required for Ptc to inhibit Smo and identified phosphatidic acid (PA) binding sites in C-tail of Ptc. Mutations in these PA-binding sites attenuated Ptc inhibitory activity on Smo. In addition, we found that PA promoted the activation of Smo and identified PA binding sites in Smo. With studies fly imaginal discs, culture cells, and in vitro lipid-protein binding assays, our findings imply a mechanism by which Ptc inhibits Smo in the absence of Hh stimulation by regulating the pool of PA that is available at the cell membrane, restricting it in the absence of Hh and increasing it in the presence of Hh.
Results
The C-terminal tail of Ptc is required for it to limit Smo abundance
Initially identified in Drosophila as the product of an anterior-posterior (A-P) patterning gene (49, 50), Ptc is a twelve transmembrane domain protein containing a sterol-sensing domain (SSD), two large extracellular loops that bind Hh, and a C-terminal tail (C-tail) (51–53)(Fig. 1A). SSD is required for Hh signaling (54), and mutations that affect this domain increase the amounts of Smo, but these mutations do not compromise the binding and internalization of Hh (33, 55). Deletion of second extracullular loop (loop2) abolishes Ptc interaction with Hh but preserves the inhibitory activity toward Smo (34, 56). The C-tail is involved in Ptc localization and self-association (57), ubiquitination-mediated degradation (58–60), and phosphorylation (61). To further characterize the roles of these domains in regulating Smo, we generated mutants of Ptc lacking each domain individually and assessed their ability to regulate Smo in the Drosophila wing disc.
Fig. 1. The C-tail is required for Ptc to inhibit Smo.
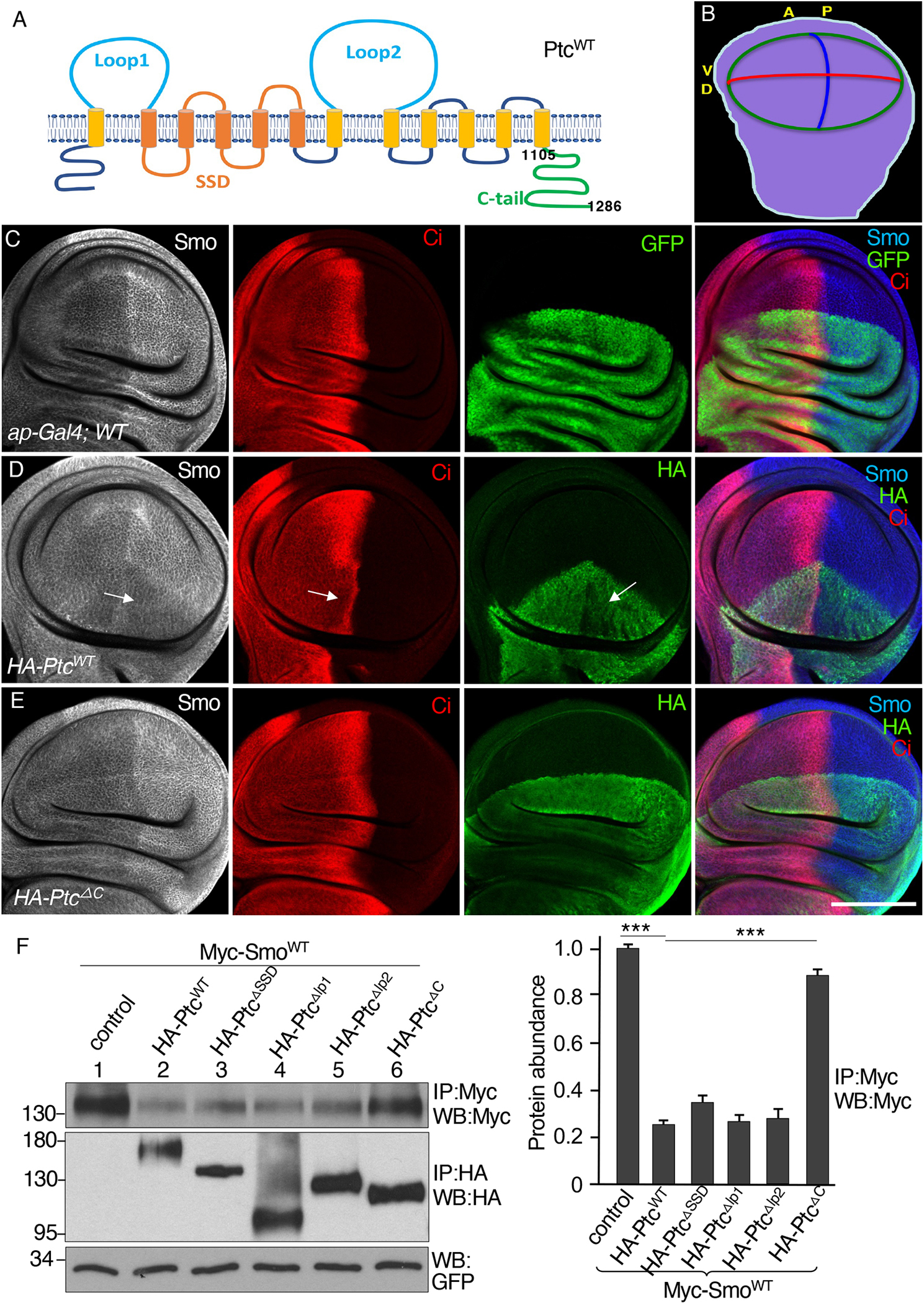
(A) Ptc domain organization, with numbers indicating the amino acids positions of the C-tail. SSD, sterol-sensing domain. (B) A schematic drawing of a third instar larval wing disc, with anterior (A), posterior (P), dorsal (D), and ventral (V) compartments indicated. The blue line indicates the A/P boundary, the red line indicates the D/V boundary, and the green circle indicates the wing pouch region. (C) A wild-type (WT) wing disc expressing GFP under control of the dorsal compartment–specific ap-Gal4 driver and stained for Smo and Ci. N=3 wing discs. (D and E) Wing discs expressing HA-PtcWT (D) or HA-PtcΔC (E) under control of the ap-Gal4 driver stained for Smo, Ci and HA. Arrows in (D) indicate the region of the HA-PtcWT expression domain in the anterior compartment with decreased Smo, Ci, and HA-PtcWT. N=5 (D) or 4 (E) wing discs. (F) S2 cells were transfected with Myc-SmoWT in combination with the indicated HA-tagged Ptc variants. Identical volumes of cell lysates were immunoprecipitated (IP) and blotted (WB) with antibodies specific for Myc or HA. GFP served as transfection and lysate control. The density of each Myc-SmoWT band was quantified using ImageJ software. N=3 independent experiments. *** indicates a p < 0.001 (Student’s t test). Scale bar, 50 μm.
We used the wing disc model (Fig. 1B) with the dorsal compartment–specific ap-Gal4 driver to overexpress HA-tagged wild-type Ptc (PtcWT) or the mutant forms under the control of a UAS promoter and monitored Smo and Ci abundance in the dorsal compartment cells, with Smo and Ci abundance in the ventral compartment cells as controls (Fig. 1C). With this model, we found that the expression of PtcWT in the dorsal compartment blocked the accumulation of Smo and restricted the amount of Ci within the dorsal region where PtcWT was overexpressed (Fig. 1D). The amount of PtcWT in P-compartment cells was lower than that in the A-compartment cells, indicating that Hh stimulation in P-compartment cells promoted PtcWT degradation (Fig. 1D, green signal). The dorsal compartment was decreased in size (Fig. 1D) because Ptc reduces the expression of dpp as well as the genes encoding the cell cycle regulators CycD and E, leading to decreased cell growth (62). Overexpression of the mutant lacking the C-tail (PtcΔC) had no effect on Smo accumulation or the distribution of Ci (Fig. 1E), suggesting that this mutant lacked inhibitory activity toward Smo. It has been reported that Ptc1130X, a truncation at amino acid 1130 that preserves the first 25 amino acids of the C-tail, can sequester Hh but fails to inhibit Hh target gene expression (63). In our overexpression assay using the ap-Gal4 driver, we found that Ptc1130X expression failed to inhibit Smo accumulation and that Ptc1130X rather increased Smo accumulation and Hh target gene expression (Fig. S1C and D, compared to S1A and S1B, respectively). Consistently, a previous study showed that Ptc1130X has dominant negative activity, relieves inhibitory activity toward Smo, and is unable to rescue ptc mutant embryos (57). These data are consistent with the previous finding that Ptc lacking the C-tail retains the capability to bind Hh but cannot repress the Hh pathway (63).
As an orthogonal approach, we evaluated the ability of Ptc mutants to influence Smo abundance when both proteins were co-expressed in cultured Drosophila S2 cells. We co-transfected constructs encoding epitope-tagged forms of both proteins and used immunoprecipitation to examine their abundance because the amounts of endogenous Smo and Ptc are low and because we do not have a good antibody for direct Western blotting to examine their abundance. Using GFP as a control for transfection and the amount of cell lysate, we immunoprecipitated either Myc-tagged wild-type Smo (Myc-SmoWT) using an antibody against the Myc tag or the HA-tagged wild-type Ptc (HA-PtcWT) molecule using an antibody against the HA tag. We evaluated the relative amount of Smo in the immunoprecipitates as an indicator of Ptc inhibitory activity. Consistent with the findings in the wing discs, co-expression of PtcWT reduced the amount of Smo compared with the amount immunoprecipitated from control cells that did not express HA-PtcWT, and co-expression of PtcΔC had no effect on the amount of Smo immunoprecipitated from the cells (Fig. 1F). Thus, deletion of the C-tail abolished the inhibitory activity of Ptc. We further found that co-expression of Smo with Ptc mutants lacking the SSD domain (PtcΔSSD), loop 1 (PtcΔlp1), or loop 2 (PtcΔlp2) resulted in reduced amounts of Smo compared with control cells (Fig. 1F), suggesting that these regions of Ptc were dispensable for the inhibitory effect of Ptc on Smo.
Putative PA binding motifs in Ptc contribute to Smo inhibition
Because Ptc does not directly interact with Smo and a lipid transporter function of Ptc may mediate the inhibitory effects of Ptc on Smo (41, 42), we examined whether removing lipids from the medium affected Hh signaling in S2 cells. To detect Hh signaling activity, we used a ptc-luciferase (ptc-luc) reporter in S2 cells coexpressing Ci to enable Hh-stimulated transcriptional responses when HhN, an active form of Hh (29), was also coexpressed. HhN-mediated induction of the reporter was reduced in serum-free medium (fig. S2A). However, in immunoprecipitation experiments, PtcWT consistently decreased the amount of Smo in cells cultured with normal medium, serum-free medium, or medium containing lipid-depleted serum (fig. S2B), suggesting that an endogenous molecule was involved in the destabilizing effect of Ptc on Smo.
We hypothesized that Ptc may remove an endogenous molecule to reduce Smo abundance. Through sequence analysis, we found that the Drosophila Ptc C-tail contains four arginine/lysine motifs very similar to the consensus PA-binding motif (R/K/H)-(R/K/H) or (R/K/H)-x(1–3)-(R/K/H), where x is any amino acid (64)(Fig. 2A), and the C-tails of mouse and human Ptc contain 5 consensus PA-binding motifs (fig. S3A). PA is one of the simplest membrane phospholipids with unique biophysical properties of a small headgroup, negative charge, and a phosphomonoester group. Upon interaction with arginine or lysine, the charge of PA increases, which stabilizes protein-lipid interactions. To determine whether these motifs in the Ptc C-tail indeed played roles in regulating Ptc activity, we generated mutations within full-length Ptc: PtcM1 with mutations in the first PA binding motif, PtcM2 with mutations in the second, PtcM3 with mutations in the third, PtcM4 with mutations in the fourth, PtcM24 with mutations in the second and fourth, and PtcM124 with mutations in the first, second, and fourth (Fig. 2A). We generated transgenic lines with these HA-tagged Ptc variants and performed rescue experiments to examine their in vivo activity to rescue a ptcS2 mutant using mosaic analysis with a repressible cell marker (MARCM) (65). The expression of exogenous HA-Ptc or its variants with mutated PA binding motifs was driven by tub-Gal4 to express Ptc in amounts similar to that of the endogenous Ptc protein. As expected, ptcS2 mutant cells exhibited increased amounts of Smo (Fig. 2B). The expression of HA-PtcWT, but not HA-PtcΔC, rescued the ptcS2 mutation by inhibiting Smo accumulation (Fig. 2, C and D), indicating that the C-tail of Ptc was essential for Ptc to inhibit Smo in vivo. We further examined the capacity of different mutant forms of Ptc to rescue the increase in Smo in ptcS2 mutant cells. We found that, compared to the inhibitory activity Ptc with a single PA binding motif mutated (Fig. 2E), mutating two or three of the PA binding motifs substantially reduced its ability to inhibit Smo accumulation (Fig. 2, F and G). HA-PtcM2, HA-PtcM3, and HA-PtcM4, each of which has a single site mutated, behaved like HA-PtcM1. Furthermore, using the overexpression approach, we found that mutating two or three PA binding motifs in Ptc dramatically limited the ability of overexpressed HA-Ptc to inhibit Smo in the dorsal compartment of wing discs (Fig. 2H), compared to the activity of single site mutations (Fig. 2, I to L). Together, our findings suggest that PA binding motifs are required for Ptc inhibitory activity.
Fig. 2. Mutations in the putative PA binding motifs of Ptc redcue Ptc-mediated Smo inhibition.
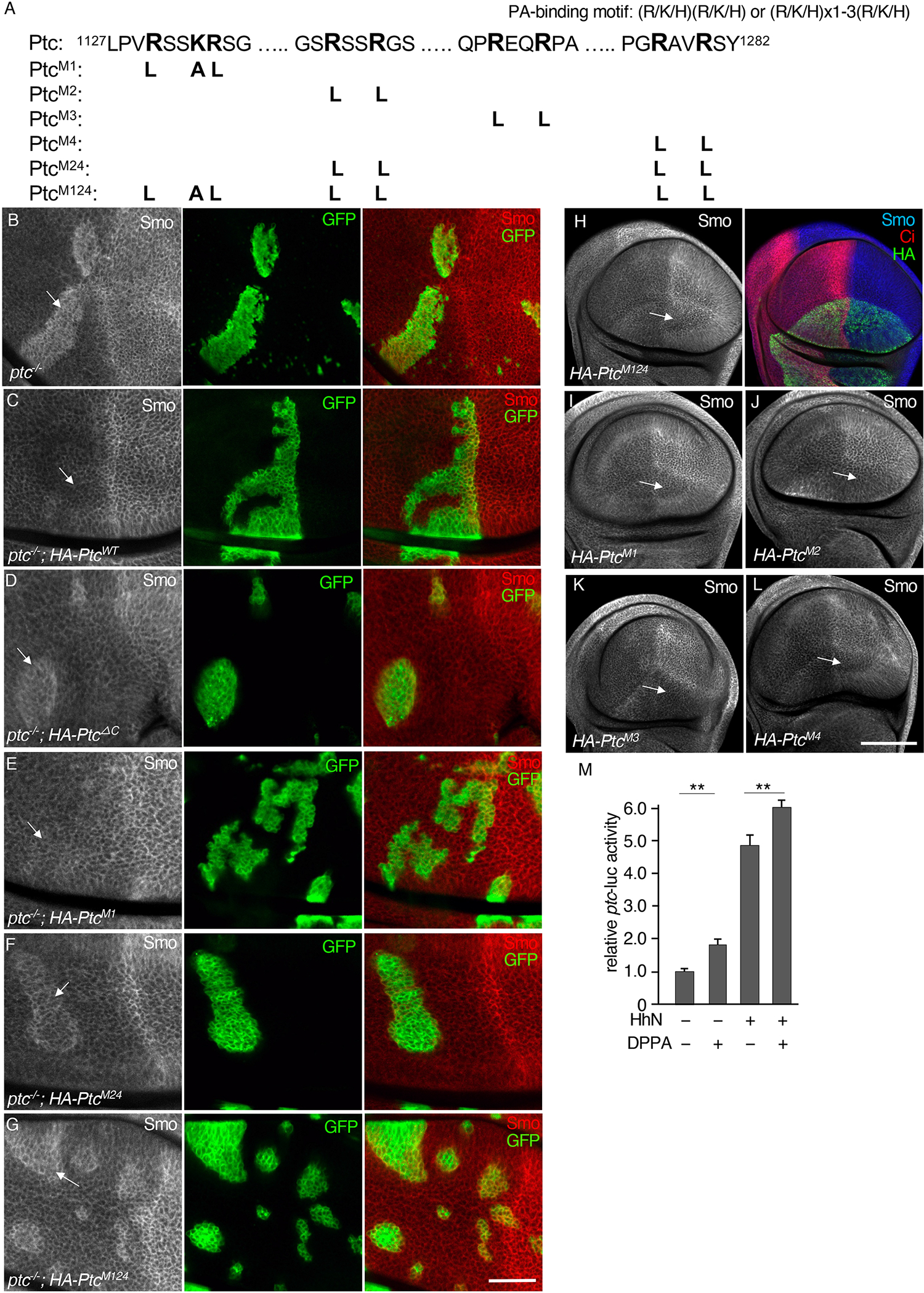
(A) A schematic drawing of the predicted PA binding motifs in PtcC and the mutants generated in this study. (B) A wing disc containing clones of GFP-expressing ptc−/− cells was immunostained for Smo and GFP. Arrow indicates the accumulation of Smo in a ptc−/− clone. N=5 wing discs. (C to G) Rescue of Smo accumulation in ptc−/− cells by Ptc PA binding motif mutants. Wing discs containing ptc−/− clones expressing HA-tagged PtcWT (C), HA-PtcΔC (D), HA-PtcM1 (E), HA-PtcM24 (F), or HA-PtcM124 (G) under control of the tub-Gal4 driver were generated using the MARCM technique and immunostained for Smo and GFP. Arrows indicate the positions of the clones. N=4 wing discs for each genotype. (H) A wing disc expressing HA-PtcM124 in the dorsal compartment using the ap-Gal4 driver was stained for Smo, Ci, and HA. Arrow indicates the domain of HA-Ptc expression in the posterior compartment. N=5 wing discs. (I to L) Wing discs expressing HA-PtcM1 (I), HA-PtcM2 (J), HA-PtcM3 (K), or HA-PtcM4 (L) under control of the ap-Gal4 driver were stained for Smo. Arrows indicate areas of reduced Smo accumulation in the domain of HA-Ptc expression in the posterior compartment. N=5 wing discs for each genotype. (M) Quantification of ptc-luc reporter activity in S2 cells expressing Ci constructs and treated with DPPA or co-transfection of HhN. N=3 independent experiments. ** indicates a p < 0.01 (Student’s t test). Scale bars, 25 μm (B to G) and 50 μm (H to L).
To determine whether PA plays a role in Hh signaling, we used the ptc-luc reporter assay in S2 cells co-expressing Ci. We found that the treatment with DPPA, a saturated form of PA, significantly increased ptc-luc activity (Fig. 2M). HhN co-expression expectedly increased ptc-luc reporter activity (Fig. 2M), which was further increased by treating the HhN-expressing cells with DPPA (Fig. 2M). These data suggest that PA plays a positive role in Hh signal transduction.
To further examine the effects of different forms of Ptc for their inhibitory activity, we turned to cultured S2 cells to determine how forms of Ptc with mutated PA-binding motifs affected the amounts of Smo. Using the immunoprecipitation assay in S2 cells co-expressing Myc-SmoWT with the mutated Ptc proteins, we found that PtcWT or Ptc carrying a single-site mutation (PtcM1, PtcM2, PtcM3, or PtcM4) decreased the amounts of Smo as well as or nearly as well as did PtcWT, whereas Ptc bearing mutations in two or three PA binding sites (PtcM24, PtcM124) had relatively little effect on the amount of immunoprecipitated Smo (Fig. 3A). Because PtcM124 was the most compromised in inhibiting Smo, we used PtcM124 for the subsequent experiments.
Fig, 3. Mutations in the Ptc PA binding motifs reduce Smo inhibition and Ptc binding to PA.
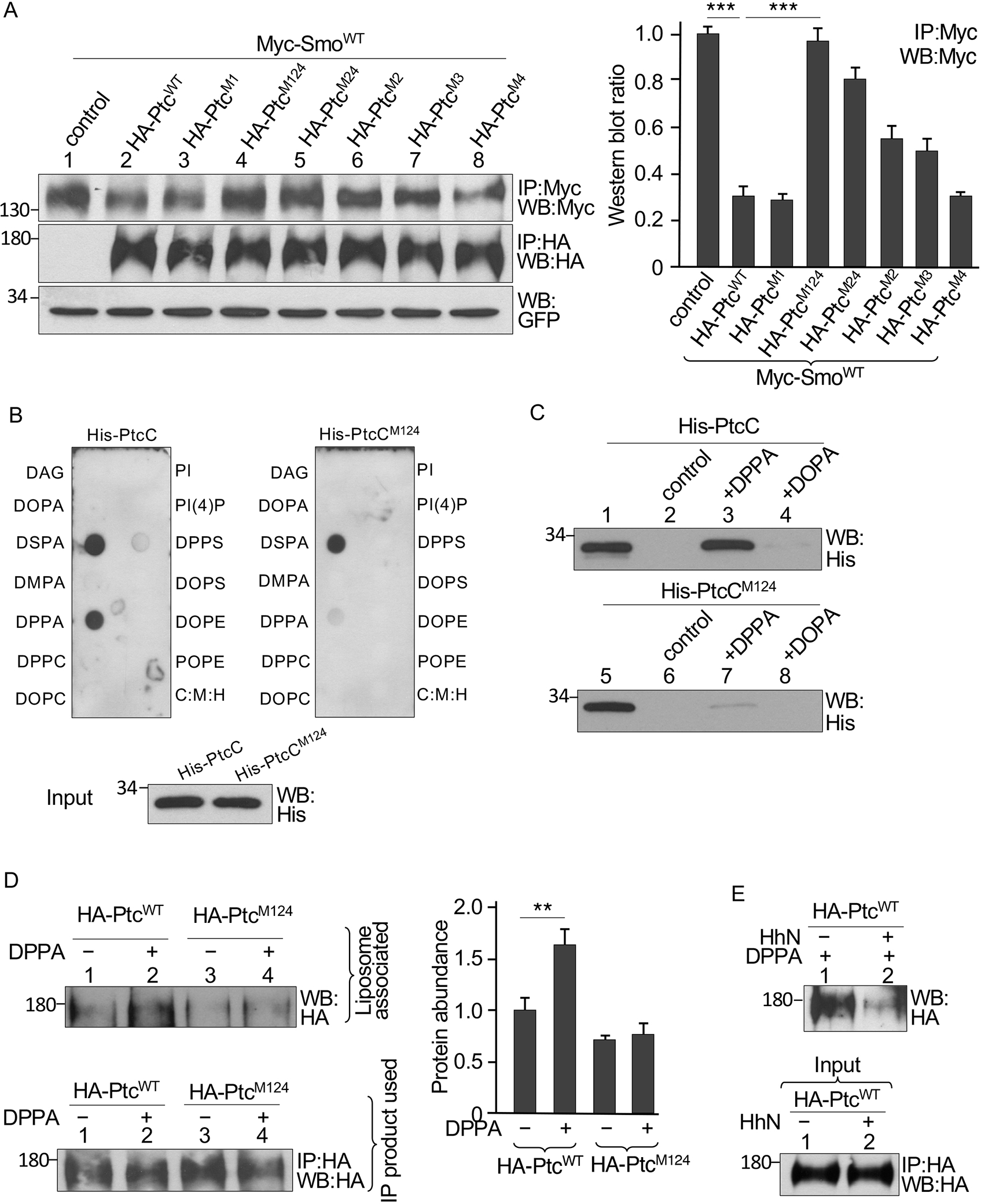
(A) S2 cells were transfected with Myc-SmoWT in combination with the indicated HA-tagged Ptc constructs. Identical volumes of lysate were immunoprecipitated (IP) and blotted (WB) with antibodies specific for Myc or HA. GFP served as transfection and lysate control. The density of each Myc-SmoWT band was quantified. N=3 independent experiments. *** indicates a p < 0.001 (Student’s t test). (B) PtcC interaction with lipids in a solid phase lipid binding assay. Membranes dotted with the indicated lipids were incubated with bacterially expressed and purified His-tagged PtcC or PtcCM124 and immunoblotted for His. C:M:H is a vehicle control. Blotting of a gel loaded with the same amount of His-PtcC and His-PtcCM124 applied to the lipid-dotted membranes (Input) is shown. N=3 independent experiments. (C) Bacterially expressed and purified His-PtcC or His-PtcCM124 was incubated with liposomes containing DOPA or DPPA and proteins that pelleted with liposomes were evaluated by Western blot. Control liposomes contained DOPC and POPE. Blots are representative of N=3 independent experiments. (D) Full-length HA-PtcWT or HA-PtcM124 was expressed in S2 cells, immunoprecipitated from lysates with the HA antibody, and incubated with liposomes with or without DPPA. Protein bound to the liposomes was detected by Western blot. The amount of Ptc in an equal amount of immunoprecipitation product is shown. N=3 independent experiments. The amounts of Ptc proteins bound to liposomes were quantified. ** indicates a p < 0.01 (Student’s t test). (E) Lysates of S2 cells transfected with HA-PtcWT with or without the HhN construct were immunoprecipitated with an antibody specific for HA and incubated with liposomes containing DPPA. Protein that bound to the liposomes was detected by Western blot. The amount of Ptc in an equal amount of immunoprecipitation product is shown (Input). N=3 independent experiments. Statistical analysis is shown in the Supplementary Materials (fig. S3E).
To confirm that mutation of the three PA binding motifs in the Drosophila Ptc C-tail did not interfere with other Ptc properties, we compared protein stability, Hh responsiveness, and self-association for HA-PtcWT and PtcM124 expressed in S2 cells. We found that the stability of PtcM124 was increased compared to PtcWT (fig. S3B), indicating that the mutant form did not fail to inhibit Smo due to being inherently instable. Co-expression of HhN decreased the amounts of PtcWT and PtcM124, indicating that, like PtcWT, the mutant responded to Hh stimulation (fig. S3C). We tested each of the PA binding motif mutants, including PtcM124, for homo-oligomerization by co-expressing each with Myc-tagged wild-type Ptc (Myc-PtcWT). All of the mutants co-immunoprecipitated with Myc-PtcWT, indicating that mutation of these motifs did not interfere with Ptc self-association (fig. S3D). Thus, the functional differences that we observed for PtcM124 were not related to unexpected defects in the protein.
PA interacts with the Ptc C-tail at three specific sites
We tested whether PA interacted with Ptc at these potential binding sites using a solid-phase lipid-binding assay to test the interaction of purified His-tagged Ptc C-tail (His-PtcC) with 13 lipids: DAG (diacylglycerol); DOPA (dioleoylphosphatidic acid), DPPA (1,2-palmitoyl-phosphatidic acid), DSPA (distearoylphosphatidic acid), DMPA (dimyristoylphosphatidic acid), DPPC (1,2-palmitoyl-phosphatidylcholine), DOPC (dioleoylphosphatidylcholine), DPPS (1,2-palmitoyl-phosphatidylserine), POPS (palmitoyloleoylphosphatidylserine]), DOPE (dioleoylphosphatidylserine), POPE (palmitoyloleoylphosphatidylethanolamine), PI (phosphatidylinositol), and PI(4)P (phosphatidylinositol 4-phosphate). His-PtcC interacted only with DSPA and DPPA, both of which are saturated forms of PA (Fig. 3B). In contrast, His-PtcCM124 barely interacted with DPPA (Fig. 3B).
Binding to artificial liposomes, which are a lipid bilayer surrounding an inner aqueous compartment, is often used to assess the specificity and affinity of phospholipid-protein interactions (66). To further evaluate the PtcC interaction with PA, we performed liposome-binding assays with purified recombinant His-PtcC and His-PtcCM124 and liposomes containing DPPA or DOPA. His-PtcC strongly interacted with liposomes containing DPPA but weakly interacted with liposomes containing DOPA (Fig. 3C), whereas His-PtcM124 had a very weak interaction only with liposomes containing DPPA (Fig. 3C). These data suggested that the C-tail directly interacts with PA.
We also examined the interaction using full-length forms of Ptc using HA-PtcWT and HA-PtcM124 immunoprecipitated from transfected S2 cells and mixed with liposomes. Similar to the isolated C-tail, HA-PtcWT strongly interacted with DPPA-containing liposomes, whereas HA-PtcM124 interacted weakly if at all with the liposomes (Fig. 3D). To examine whether Hh stimulation affected the interaction between Ptc and PA in the context of liposomes, we immunoprecipitated HA-PtcWT from S2 cells with or without co-expression of HhN. Hh decreased the interaction of Ptc with DPPA-containing liposomes (Fig. 3E and fig. S3E). These data indicated that Ptc interacts with PA and that this interaction is negatively regulated by Hh.
Membrane-tethered PA binding domains enhance the abundance of both PA and Smo
The expression of PtcC alone did not have any effect on the distribution of Ci and Smo in the wing disc (fig. S4A). However, expression of a membrane-tethered form of PtcC — generated by fusion of a myristoylation signal at the N-terminus of PtcC (myr-PtcCWT) — increased Smo and Ci in the wing disc (Fig. 4A). In contrast, the expression of myr-PtcCM124 with three PA binding motifs mutated did not increase either Smo or Ci (Fig. 4B). This was an unexpected finding, because it indicates that the C-tail stimulates, rather than inhibits, Smo and does so in a PA-dependent manner. We hypothesized that the recruitment of PA to the membrane by myr-PtcCWT might affect membrane organization (64). Armadillo (Arm), the Drosophila homolog of β-catenin, is evenly distributed on the plasma membrane of wing disc cells. We examined the cell membrane morphology and the distribution of Arm on the membrane in cells of wing discs and found that the expression of myr-PtcCWT consistently increased the amounts of Ci but did not change the morphology of the membrane or Arm distribution (Fig. 4C). This finding suggests that the effects of myr-PtcCWT on Smo abundance were not due to changes cell membrane organization. It possible that PtcWT undergoes constitutive endocytosis that could carry away PA from the plasma membrane, whereas membrane-tethered myr-PtcCWT could bring PA to the plasma, enabling it to accumulate.
Fig. 4. Membrane-tethered PA binding domains of Ptc enrich PA at the cell surface.
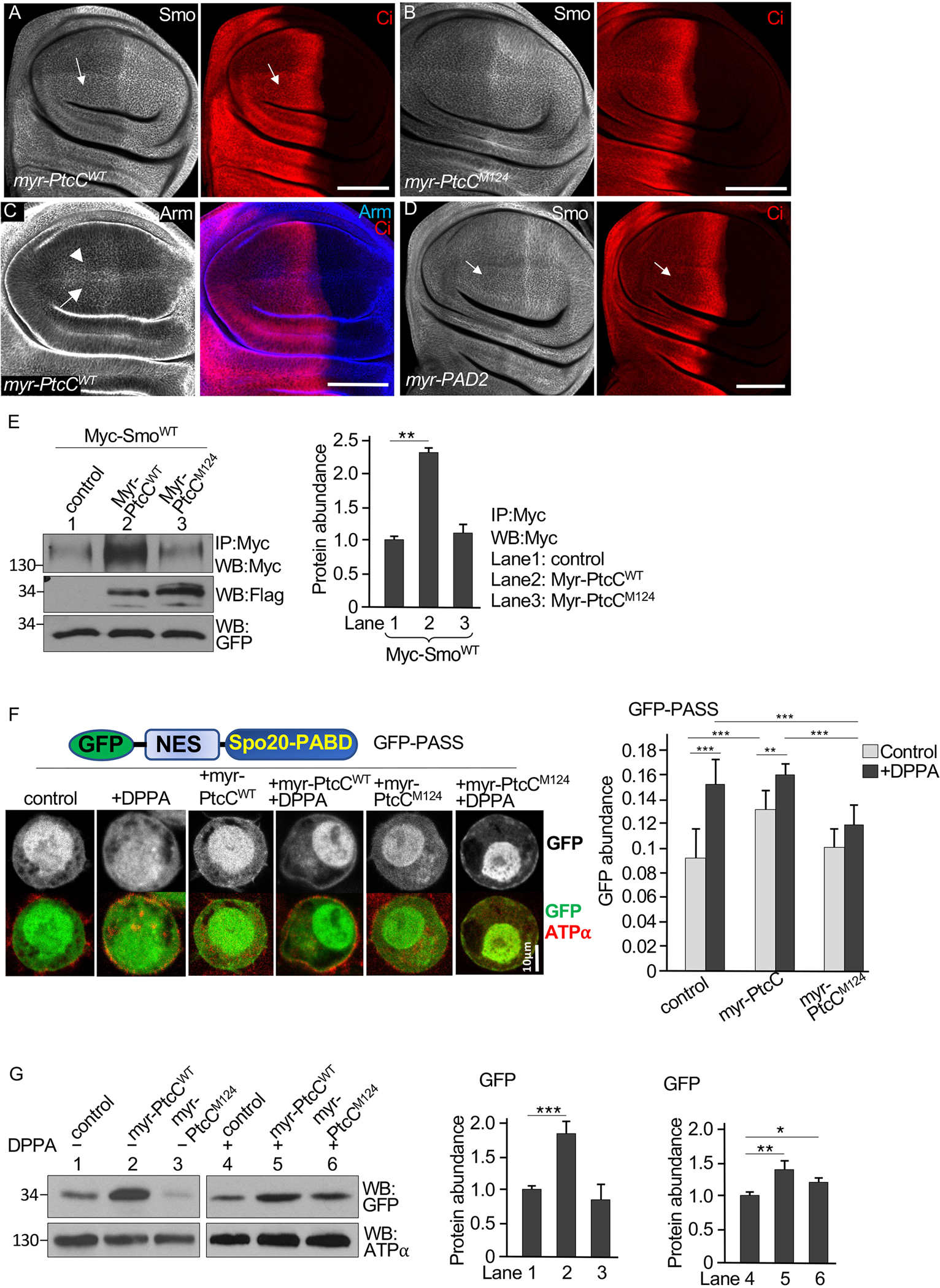
(A and B) Wing discs expressing a membrane-tethered form of the WT Ptc C-tail (myr-PtcCWT, A) or the M124 mutant form (myr-PtcCM124, B) in the dorsal compartment using the ap-Gal4 driver were stained for Smo and Ci. Arrows indicate increased Smo accumulation in the domain of myr-PtcCWT expression in the anterior portion of the dorsal compartment. The flag tag in myr-PtcC was not detectable by immunostaining. N=5 wing discs per genotype. (C) A wing disc expressing myr-PtcCWT under control of the ap-Gal4 driver stained for Arm and Ci. Arrow and arrowhead indicate Arm expression in the dorsal and ventral compartment, respectively. N=3 wing discs. (D) A wing disc expressing myr-PAD2 under control of the ap-Gal4 driver stained for Smo and Ci. Arrows indicate the accumulation of Smo and Ci in the myr-PAD2 expression domain. N=5 wing discs. (E) S2 cells were transfected with Myc-SmoWT, either myr-PtcCWT or myr-PtcCM124, and a control plasmid, UAST-GFP. Lysates were immunoprecipitated (IP) and blotted (WB) with an antibody specific for Myc. Identical volumes of lysates were evaluated on separate blots with an antibody specific for Flag to detect myr-PtcCWT or with an antibody specific for GFP. N=3 independent experiments. The amounts of Myc-SmoWT were quantified. ** indicates a p < 0.01 (Student’s t test). (F) S2 cells were transfected with the GFP-PASS construct (schematic drawing) in combination with myr-PtcCWT or myr-PtcCM124 and treated with DPPA or not. Cells were fixed and stained for the plasma membrane marker ATP⍺. Representative images in individual S2 cells are shown. N=23–25 cells per group from 4 independent experiments. The ratio of cell surface GFP to total GFP (membrane GFP/total GFP) was calculated using the ImageJ software. ** indicates an adjusted p < 0.01, *** indicates an adjusted p < 0.001 (Student’s t test with Bonferroni correction for multiple comparisons adjustment). (G) Membrane fractions from lysates of the cells in (F) were immunoblotted (WB) for GFP and GFP abundance was quantified. ATP⍺ is a loading control. N=3 independent experiments. *** indicates an adjusted p < 0.001; ** indicates a p < 0.01; * indicates a p < 0.05 (Student’s t test with Bonferroni correction). Scale bars, 50 μm (A to D) and 10 μm (F).
To further test the hypothesis that the effect of myr-PtcCWT on Smo and Ci depends on interactions with PA, we examined a membrane-tethered PA binding motif from the Saccharomyces cerevisiae protein Opi, which specifically binds PA (67, 68). Like the expression of myr-PtcCWT, expression of a membrane-tethered form of two copies of the Opi PA binding domain (myr-PAD2) increased Smo and Ci in the wing disc (Figs. 4D and S4B) and increased the amounts of Smo in S2 cells (fig. S4C), although the effects were not as strong as those from myr-PtcCWT. The weak effect may reflect differences in PA affinity between myr-PtcCWT and myr-PAD2, or the requirement for a minimum length or sequence context for PA binding in myr-PAD2. To examine whether the specific mode of membrane anchoring affected the regulatory interaction of PtcC with Smo, we generated a chimeric protein that contained the transmembrane and extracellular domain of Sevenless (Sev) fused to PtcC, resulting in Sev-PtcC, and found that the expression of Sev-PtcC increased the amounts of Smo in S2 cells (Fig. S4D), a similar effect to that caused by the expression of myr-PtcCWT.
To address whether endogenous Ptc played a role in the observed regulation of Smo by myr-PtcCWT, we examined the activity of myr-PtcCWT in the absence of endogenous Ptc protein in wing discs with clonal populations of cells lacking Ptc using the null allele ptcIIw. We were unable to use ap-Gal4 to drive the expression of myr-PtcCWT in the dorsal compartment because the driver is located on the same chromosome as ptc. We therefore used the wing-specific ms1096-Gal4 driver, which has stronger expression in the dorsal compartment than in the ventral compartment (31), to drive the expression of myr-PtcCWT. As expected, Smo accumulated in clones of cells lacking ptc in the A-compartment (fig. S4E). The expression of myr-PAD2 by ms1096-Gal4 increased Smo accumulation in dorsal compartment ptc mutant clones, compared to Smo accumulation in ventral compartment clones with low myr-PAD2 expression (fig. S4F). Similarly, myr-PtcCWT expression increased Smo accumulation in dorsal compartment ptc mutant cells (fig. S4G). We also examined the effect of myr-PtcCM124 expression, and we did not observe further increase in Smo in ptc mutant clones (fig. S4H). Consistently, using the immunoprecipitation assay, we found an increase in Smo in cells co-expressing myr-PtcCWT but not in those expressing myr-PtcCM124 (Fig. 4E). Taken together, our findings suggested that the effects of myr-PAD2 and myr-PtcCWT expression were independent of endogenous Ptc protein and that the increase of PA abundance near the plasma membrane mediated by membrane-anchored PtcC increased Smo accumulation.
To further examine the effect of the Ptc C-tail in regulating PA more precisely, we used the PA biosensor PASS (phosphatidic acid biosensor with superior sensitivity). PASS contains the PA binding domain from yeast Spo20 (Spo20-PABD), which has increased selectivity for PA compared to other phospholipids (69). We incorporated GFP and a nuclear export sequence (NES) with PASS (referred to as GFP-PASS) and expressed GFP-PASS in Drosophila S2 cells. To detect changes in the distribution of GFP-PASS, we compared the GFP-PASS signal from the region of the cell membrane defined by staining for the α subunit of Na+/K+-ATPase (ATPα), a marker for the plasma membrane. We found that exposing the cells to DPPA increased the plasma membrane GFP signal (Fig. 4F). Coexpression of myr-PtcCWT with GFP-PASS significantly increased the plasma membrane GFP signal in the absence of DPPA and further increased it upon the addition of DPPA (Fig. 4F). In contrast, co-expression of myr-PtcCM124 did not increase plasma membrane GFP signals in the absence of DPPA and significantly reduced the plasma membrane GFP signal in the presence of added DPPA compared to control cells expressing GFP-PASS alone or cells coexpressing GFP-PASS and myr-PtcCWT (Fig. 4F). As an orthogonal method, we quantified the amount of GFP in the membrane fractions of cell lysates by Western blotting. The coexpression of myr-PtcCWT with GFP-PASS in S2 cells appeared to increased the amount of GFP-PASS detected in the plasma membrane fraction from cells that were untreated and in those exposed to DPPA (Fig. 4G). In contrast, the amount of GFP-PASS in the plasma membrane fraction was similar to the amount in the controls for cells co-expressing myr-PtcCM124 (Fig. 4G). Although the difference in the GFP signal between control cells and cells expressing myr-PtcCWT in the presence of DPPA was not statistically significant in the immunostaining assay (Fig. 4F), it was significant in the Western blot assay (Fig. G), which was likely a result of differences in sensitivity between the methods. Based on these results with the S2 cells, we hypothesized that the C-tail of Ptc regulates Smo activity by changing the pool of PA at the plasma membrane.
PA influences Smo activity and Hh signaling
A major pathway for PA metabolism at the plasma membrane involves the hydrolysis of phosphatidylcholine (PC) by phospholipase D (Pld) to produce PA and conversion of PA to diacylglycerol by the phosphatase Lipin (Fig. 5A). We examined the functions of Pld and Drosophila Lipin (dLipin) in regulating Smo in wing discs that are wild type for Ptc and found no effect on Smo or Ci upon overexpression of either Pld or dLipin and knockdown of Pld or dLipin by RNAi (fig. S5, A–D). In wing discs bearing clones of cells homozygous for the null allele ptcIIw, we found a mild increase in Smo accumulation in the mutant clones (Fig. 5B), and the expression of Pld with the ms1096-Gal4 driver increased Smo accumulation in the ptc mutant cells (Fig. 5C). In contrast, the expression of dLipin decreased Smo accumulation caused by ptc mutation (Fig. 5D, compare Smo staining in dorsal and ventral clones). These results indicated that PA contributed to the increase in Smo abundance that occurred in the absence of Ptc. Neither Pld nor dLipin has been identified as a modulator of Hh signaling through conventional genetic screens, and their overexpression or RNAi alone had no effect on Smo accumulation in the wing disc (figs. S5, A–D). Changing the amounts of Pld or Lipin in a background with endogenous enzymes may not result in changes in Smo, which is likely caused by the existence of exogenous PA from nutrients, or the presence of endogenous Ptc protein, or both.
Fig. 5. Perturbations in PA metabolism affect Smo activity.
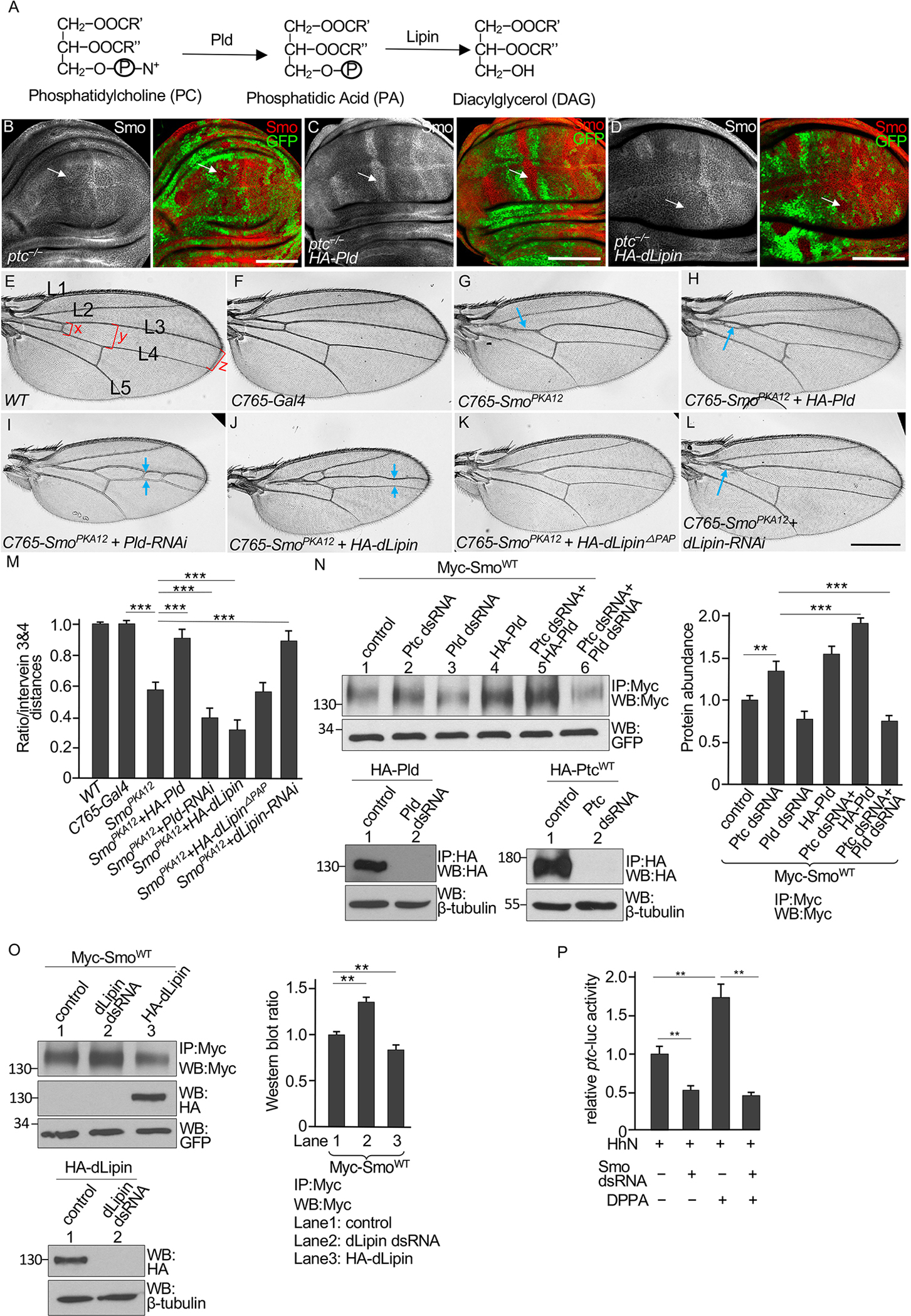
(A) The PA metabolic pathway, showing biosynthesis by phospholipase D (Pld) from PC and hydrolysis by Lipin to DAG. (B) A wing disc bearing clones of ptcIIw −/− mutant cells, marked by the loss of GFP, was stained for Smo and GFP. The arrow indicates Smo accumulation in a mutant clone in the anterior compartment. N=5 wing discs. (C and D) Wing discs bearing GFP− ptcIIw −/− mutant clones and expressing HA-Pld or HA-dLipin under control of the ms1096-Gal4 driver were stained for Smo and GFP. Arrows indicate Smo accumulation in mutant clones in the anterior portion of the dorsal compartment. N=7 (HA-Pld) or 6 (HA-dLipin) wing discs. (E) A wild-type adult wing showing longitudinal veins 1–5 (L1–L5). Red brackets indicate the intervein distances between veins 3 and 4 measured at the anterior crossvein (x), adjacent to the posterior crossvein (y), and near the wing margin (z), which were used for statistical analysis. (F) Wing from a fly expressing only the C765-Gal4 driver. N = 25 wings. (G to L) Wings from flies expressing SmoPKA12 driven by the C765-Gal4 driver alone (G) or in combination with HA-Pld (H), Pld-RNAi (I), HA-dLipin (J), HA-dLipinΔPAP (K), or dLipin-RNAi (L). Blue arrows indicate vein fusions. N= 21 (G), 18 (H), 20 (I), 18 (J), 25 (K), and 19 (L) wings. (M) Statistical analysis of the intervein distances, presented as the average intervein distance (x+y+z/3) relative to WT. N = 5 adult wings for each genotype. *** indicates an adjusted p < 0.001, (Student’s t test with Bonferroni correction for multiple comparisons adjustment). (N) S2 cells were transfected with Myc-SmoWT alone (control) or in combination with Ptc dsRNA, Pld dsRNA, or HA-Pld. Lysates were immunoprecipitated (IP) with an antibody specific for Myc and immunoblotted (WB) for Myc and GFP. GFP serves as transfection and lysate control. The amounts of Myc-SmoWT were quantified. RNAi efficiency was confirmed by Western blotting of HA immunoprecipitates from cells expressing HA-Pld or HA-Ptc. β-tubulin is a loading control. N=3 independent experiments. ** indicates an adjusted p < 0.01; *** indicates an adjusted p < 0.001 (Student’s t test with Bonferroni correction for multiple comparisons adjustment). (O) S2 cells were transfected with Myc-SmoWT and HA-dLipin or dLipin dsRNA, followed by Myc immunoprecipitation and Western blotting for Myc and HA. RNAi efficiency was confirmed by Western blotting for HA in cells coexpressing HA-dLipin and dLipin dsRNA. N=3 independent experiments. Statistical analysis of the western blot is shown in the right panel. ** indicates an adjusted p < 0.01 (Student’s t test with Bonferroni correction for multiple comparisons adjustment). (P) Quantification of ptc-luc reporter activity in S2 cells expressing Ci and HhN, in combination with Smo dsRNA as indicated and treated with DPPA or not. N=3 independent experiments. ** indicates an adjusted p < 0.01 (Student’s t test with Bonferroni correction for multiple comparisons adjustment). Scale bars, 50 μm (B to D) and 500 μm (E to L).
To further examine the effects of Pld and dLipin in regulating Smo, we turned to a more sensitive assay using a sensitized genetic background in which a partial dominant negative Smo is expressed. Our previous study indicated that mutating two protein kinase A (PKA) phosphorylation sites in Smo (SmoPKA12) reduced Smo signaling activity and turned Smo into a weak dominant negative form (21). Compared to wings from wild-type adults or adults carrying the C765-Gal4 driver, which is expressed throughout the larval wing disc (Fig. 5, E and F), wings expressing SmoPKA12 in the C765-Gal4 domain showed in a reproducible phenotype with partial fusion of veins 3 and 4 (Fig. 5G), a phenotype indicating a partial loss of Hh signaling activity (70). Coexpression of HA-Pld with SmoPKA12 by the C765-Gal4 driver weakened the phenotype compared to expressing SmoPKA12 alone (Fig. 5H). Knocking down Pld expression by RNAi in SmoPKA12-expressing wings by the C765-Gal4 driver resulted in smaller wings with further fusion of veins 3 and 4 (Fig. 5I), a phenotype resembling that caused by a more severe loss of Hh signaling (23). We examined Hh target gene expression in wing discs from third instar larvae and found that the accumulation of Ptc adjacent to the A/P boundary that results from Hh-dependent stimulation of ptc expression (fig. S5, E and F) was decreased by C765-driven expression of SmoPKA12 (fig. S5G), which was partially restored by expression of HA-Pld (fig. S5H), whereas Pld RNAi further blocked this Ptc accumulation (fig. S5I). Similarly, the expression of SmoPKA12 and HA-dLipin, but not the expression of a catalytically dead form of dLipin (HA-dLipinΔPAP) (71), enhanced vein fusions compared to wings expressing only SmoPKA12 fusion of vein 3 and vein 4 in wings also expressing SmoPKA12 (Fig. 5, J and K). In contrast, RNAi-mediated knockdown of dLipin weakened the dominant negative effect caused by SmoPKA12 (Fig. 5L). Statistical analysis of the intervein distances suggested significant changes caused by the activity of Pld or dLipin (Fig. 5M). We also found that knockdown of Pld attenuated the effect of myr-PtcCWT in the wing disc (fig. S5, J to K), suggesting that the effect of myr-PtcCWT in regulating Smo accumulation was indeed caused by accumulating PA.
We also examined the effect of manipulating PA biosynthesis and metabolism in cultured Drosophila S2 cells. Knocking down Ptc by RNAi or overexpressing HA-Pld increased the amount of Myc-SmoWT that immunoprecipitated from the cells, and the combination of Ptc knockdown and HA-Pld overexpression promoted a greater increase in Myc-SmoWT (Fig. 5N). Knockdown of Pld alone or in combination with Ptc knockdown reduced Myc-SmoWT abundance (Fig. 5N). HA-tagging and immunoprecipitation of HA-Pld were used because an antibody for Pld was unavailable. These data suggested that Smo stability was more sensitive to Pld activity in the absence of Ptc or when Ptc was limited. Knockdown of dLipin increased the amount of Myc-SmoWT that immunoprecipitated from S2 cells, whereas overexpression of dLipin decreased it (Fig. 5O). These data suggested that Smo in S2 cells was more sensitive to PA metabolism than was the Smo in wing discs, likely due to differences between the amounts of endogenous Ptc or in the accumulation or depletion of PA by manipulating Pld and dLipin activity.
To determine whether PA activates Smo to stimulate Ci transcriptional targets, we used the ptc-luc reporter assay in S2 cells also expressing Ci. As expected, knocking down Smo by RNAi significantly decreased ptc-luc reporter activity stimulated by the coexpression of HhN (Fig. 5P). Exposing the cells to DPPA significantly increased reporter activity but not if Smo was knocked down (Fig. 5P). These data indicated that Smo is required for PA to activate Hh pathway.
To explore whether Hh may regulate Pld and/or dLipin, we examined the transcription and protein stability for Pld and dLipin in S2 cells. Knocking down Ptc by RNAi did not change the amounts of the Pld or dLipin proteins (fig. S5M and S5N). We also examined the distribution of GFP-PASS in wing discs and found no difference in the GFP signal between the A- and P-compartment (fig. S5O). These data suggest that Hh likely does not not regulate PA metabolism by Pld and dLipin.
Smo interacts with PA through a pocket in the transmembrane domains
We hypothesized that the Ptc C-tail altered PA distribution or accessibility to affect Smo stability and activation state. One possible mechanism is that the Ptc C-tail sequesters PA in the absence of Hh and that Hh ligand binding to Ptc promotes the release and transfer of PA from Ptc to Smo. We predicted that this mechanism would involve PA binding motif(s) in Smo. One possible mechanism for PA to regulate Smo directly is through interaction with the sterol binding pocket, which was originally identified within the seven transmembrane domains of vertebrate Smo and determined to be functionally important for Gli activation through mutation studies (72). Using the new insights from AlphaFold (73, 74), we examined the structure of this form of Smo mutant and compared it to the AlphaFold model of Drosophila Smo (Figs. 6A and 6B). The model of Drosophila Smo is predicted with high confidence and structurally aligns closely with the known structure of mouse Smo, with an R.M.S.D. = 1.2 Å. The structure of vertebrate Smo has been solved (72, 75, 76). We next examined the sterol binding pocket in the seven-transmembrane core, which has been proposed to be directly coupled to Smo activation (72) (Fig. 6C). Examination of the sterol binding pocket allowed us to design mutations to block binding. Mutation to larger residues that maintain the same overall biophysical character and were not predicted to significantly perturb protein stability were selected to occlude the sterol binding site (Figs. 6D and S6A). To test the hypothesis that the sterol binding pocket is responsible for the stabilizing effect of DPPA and binds PA in Drosophila Smo, we generated a mutant form of Smo with M354F, L427F, L431F, and A489Q (SmoMLLA), containing bulkier side chains matching the mutations tested for vertebrate Smo (Fig. 6E). The mutant protein was produced in both cultured S2 cells and Drosophila tissues, with changes in the activity of Smo (see below), indicating that it was properly folded.
Fig. 6. Identification of a PA binding pocket in Smo.
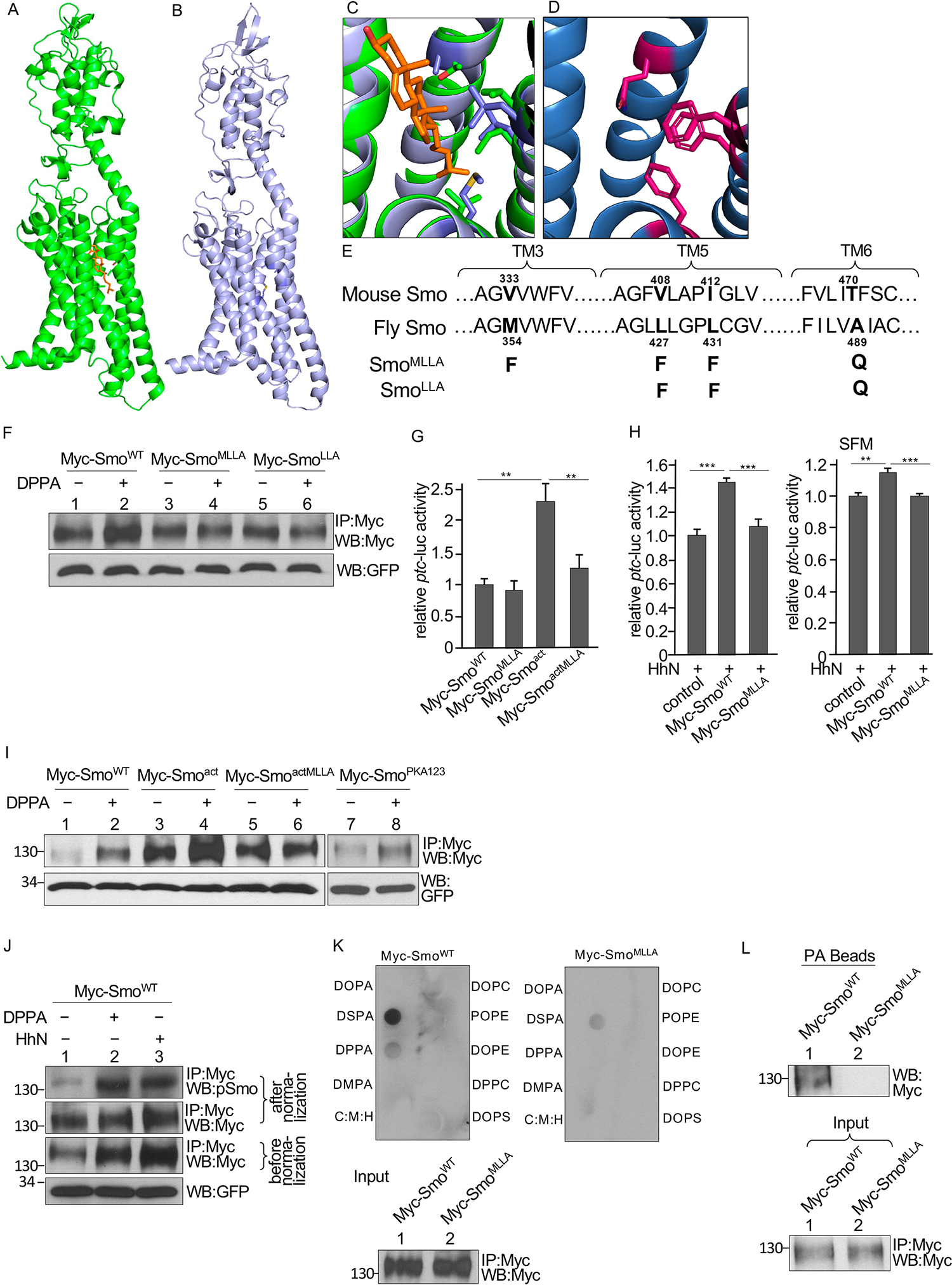
(A) Structure of mouse Smo with the cholesterol molecule in orange. (B) Alphafold model of Drosophila Smo. (C) The sterol binding site of mouse Smo (green) with the Drosophila model (blue) superimposed. (D) Alphafold model of the sterol binding site in the Drosophila SmoMLLA mutant. (E) Residues in Smo TM3, TM5, and TM6 domains that contribute to a pocket located near the sterol binding site and predicted to bind PA. Sequences of Drosophila and mouse Smo are shown. Mutations tested in fly Smo are indicated. (F) Myc immunoprecipitates (IP) from S2 cells transfected with either WT Smo or the indicated mutant forms of Smo and treated with DPPA were blotted (WB) for Myc and GFP. GFP served as transfection and lysate control. N=3 independent experiments. Statistical analysis of Smo abundance is shown in the Supplementary Materials (fig. S6B). (G) Quantification of ptc-luc reporter activity in S2 cells were cotransfected with the reporter, Ci, and the indicated Smo constructs. N=3 independent experiments. ** indicates a p < 0.01, versus control (Student’s t test). (H) Quantification of ptc-luc reporter activity in S2 cells cotransfected with the reporter, Ci, HhN, and the indicated Smo constructs and cultured in either regular medium or serum-free medium (SFM). N=3 independent experiments. *** indicates a p < 0.001, versus control; ** indicates a p < 0.01, versus control (Student’s t test). (I) Myc immunoprecipitates (IP) from S2 cells transfected with the indicated Smo constructs and treated with DPPA were blotted for Myc and GFP. N=3 independent experiments. Statistical analysis of Smo abundance is shown in the Supplementary Materials (fig. S6C). (J) Myc immunoprecipitates from S2 cells transfected with Myc-SmoWT and either cotransfected with HhN or treated with DPPA were blotted for Myc and phosphorylated Smo (pSmo). Samples were normalized by adjusting loading to quantify Smo phosphorylation fig. S6D). N=3 independent experiments. (K) Smo interaction with lipids in a solid phase lipid binding assay. Membranes dotted with the indicated lipids were incubated with Myc-SmoWT or Myc-SmoMLLA immunoprecipitated from S2 cells and immunoblotted for Myc. C:M:H is a vehicle control. The amounts of Myc-SmoWT and Myc-SmoMLLA added to each membrane (Input) are shown in the lower blot. N=3 independent experiments. (L) Myc immunoprecipitates of lysates from S2 cells expressing Myc-SmoWT or Myc-SmoMLLA were incubated with PA beads followed by Western blotting for Myc antibody to detect the bound Smo. The amounts of each immunoprecipitated protein applied to the beads (Input) is shown in the lower blot. N=3 independent experiments.
Using the immunoprecipitation assay in S2 cells, we found that the addition of DPPA to the S2 cells expressing Myc-SmoWT increased the amount of immunoprecipitated Smo (Fig. 6F and fig. S6B), indicating a stabilizing effect on Smo. When expressed in S2 cells, the amount of Myc-SmoMLLA that immunoprecipitated was similar in the presence or absence of DPPA (Fig. 6F and fig. S6B), and mutation of just the last three of these residues [L427F, L431F, and A489Q (SmoLLA)] also blocked the DPPA-induced increase in Myc-SmoLLA (Fig. 6F and fig. S6B). To match the vertebrate mutant (72), we used SmoMLLA for subsequent experiments.
To examine the activity of SmoMLLA, we performed the luciferase assay using coexpression of the ptc-luc reporter and Ci in S2 cells and stimulated Hh signaling by also expressing NHh. In the absence of NHh, the activity of Myc-SmoMLLA and Myc-SmoWT Smo was similar (Fig. 6G). We also tested a constitutively active mutant of Smo (Smoact), which has three phosphorylation clusters in its C-tail mutated to aspartate to mimic phosphorylation (21), and a form with the MLLA mutations in the background of this constitutively active form of Smo (SmoactMLLA). Whereas Myc-Smoact had increased activity compared to Myc-SmoWT, the activity of SmoactMLLA was significantly decreased (Fig. 6G). Furthermore, SmoMLLA failed to respond to NHh when the cells were grown in standard medium with lipids or in serum-free medium with low amounts lipids (Fig. 6H). These results suggested that the sterol binding pocket of Smo was necessary for Smo activation and downstream signaling.
We evaluated the effect of the MLLA mutation on the stabilizing effect of DPPA on constitutively active Smo expressed in S2 cells. The amount of Myc-Smoact immunoprecipitated was higher than that of Myc-SmoWT, indicating that the activated mutant not only had higher signaling activity but also had greater stability that was further promoted by PA (Fig. 6I and fig. S6C). A previous report showed that Myc-Smoact is further stabilized and activated by Hh stimulation (21). Our findings suggest that PA-mediated enhancement of Smo activation may account for the additional regulation of Smo beyond phosphorylation. Whereas immunoprecipitation of both Myc-SmoWT and Myc-Smoact was increased by the presence of DPPA, the amount of SmoactMLLA immunoprecipitated was similar in the presence or absence of DPPA (Fig. 6I and fig. S6C), suggesting that the effect of PA on Smo stability can be separated from the effect of phosphorylation. In support of this hypothesis, immunoprecipitation of the phosphorylation-deficient form of Smo (SmoPKA123) was also increased by DPPA treatment (Fig. 6I and fig. S6C). DPPA increased the amount of phosphorylated Myc-SmoWT immunoprecipitated from S2 cells similarly to the effect that HhN coexpression increased Smo phosphorylation (Fig. 6J and fig. S6D) (31). Taken together, these data suggested that the effect of PA on Smo did not require phosphorylation and that PA increased Smo stability and phosphorylation.
To determine whether Smo directly interacts with PA, we performed a solid-phase lipid-binding assay using Smo immunoprecipitated from S2 cells. We found that Myc-SmoWT strongly interacted with DSPA and weakly interacted with DPPA (Fig. 6K). However, Myc-SmoMLLA did not interact with DPPA and had a weak interaction with DSPA compared to SmoWT (Fig. 6K). Smo interaction with PA was further confirmed with a pulldown assay using PA-coated beads with Myc-SmoWT or Myc-SmoMLLA immunoprecipitated from S2 cells. Whereas Myc-SmoWT bound to the PA beads, Myc-SmoMLLA did not (Fig. 6L). Together, these findings suggested that, instead of interacting with an arginine/lysine PA-binding motif, PA interacted with Smo through a pocket within the transmembrane domains.
To explore whether there is another motif in Smo to bind PA, we examined other arginine/lysine motifs in Smo. There are four arginine/lysine clusters in the Smo C-tail, which regulates Smo activity (22); however, similar to Myc-SmoWT responding to DPPA in S2 cells (Fig. 6F), expression of a mutant in the four arginine clusters (SmoRA1234) in S2 cells did not change Smo responsiveness to PA stimulation (fig. S6E). Mutation of lysines and arginines to alanine in two other motifs, one containing Lys564 and Arg565 (SmoKRA), and the other containing Lys580, Lys583, and Lys585 (Smo3KA), also did not impair the increase of Smo in response to DPPA treatment (fig. S6F and S6G) and did not change Smo activity in ptc-luc assays. Our findings suggested that these residues were not involved in PA regulation of Smo and did not function as PA binding sites.
Mutation of the PA binding pocket reduces Smo activity in vivo
In previous studies, we established a platform to precisely examine changes in Smo activity in Drosophila wing discs in vivo using a ptc-LacZ reporter gene as the readout (31). Using this system, we generated transgenic lines of Smo variants using the attP recombinase site at the 75B1 locus to ensure equivalent expression of the Smo proteins, without positional effect of the transgenes. Compared to wild-type expressing the ptc-lacZ reporter (Fig. 7A), Myc-SmoWT transgenic flies had an increase in the number of cells positive for ptc-lacZ expression (Fig. 7B), whereas Myc-SmoMLLA transgenic flies did not have an expanded zone of ptc-lacZ expression in wing discs (Fig. 7C), suggesting that the responsiveness of SmoMLLA to Hh stimulation was decreased. Consistent with our previous observations (21, 31), the expression of Smoact induced a greatly expanded zone of ptc-lacZ along with the Hh transcriptional target En in cells away from the A/P border (Fig. 7D), indicating that Smoact was constitutively active. The MLLA mutation in the context of Smoact (SmoactMLLA) substantially decreased the intensity of the ptc-lacZ and En signals (Fig. 7E), suggesting that loss of PA binding impaired the constitutive activity of the phosphorylation-mimicking Smo mutant.
Fig. 7. Mutation of the PA binding pocket decreases Smo activity.
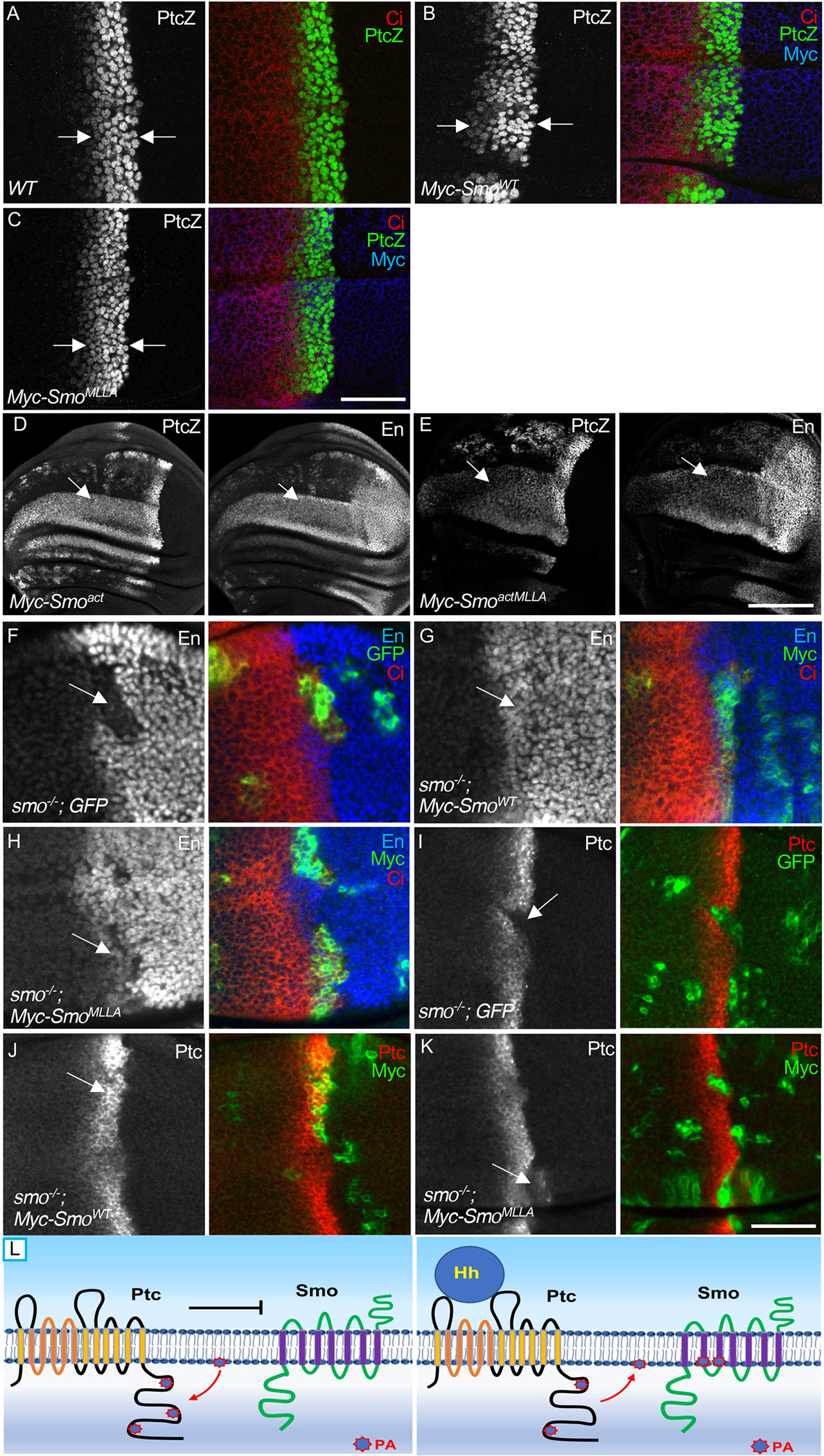
(A) A WT wing disc was stained to show expression of the ptc-lacZ reporter (PtcZ) and Ci. Arrows indicate the strip of cells in the A compartment near the A/P boundary in which endogenous Hh induces ptc-lacZ expression. N=3 wing discs. (B and C) Expression of PtcZ, Ci and Myc in wing disc expressing Myc-SmoWT (B) or Myc-SmoMLLA (C) under control of the ms1096-Gal4 driver. Arrows indicate the width of the Ptz+ zone for comparison. N=5 wing discs per genotype. (D) Wing discs expressing either Myc-Smoact (D) or Myc-SmoactMLLA (E) were stained for PtcZ and En. Arrows in indicate areas of expansion of ptc-lacZ and En expression anterior to the A/P compartment boundary. N=3 wing discs for each genotype. (F) A wing disc bearing GFP+ smo−/− clones was immunostained for En, Ci, and GFP. Arrow indicates the loss of En expression in smo mutant cells. N=5 wing discs. (G and H) Wing discs carrying smo−/− clones expressing Myc-SmoWT (G) or Myc-SmoMLLA (H) were stained for En, Ci, and Myc. Positive Myc staining labels the mutant cells. N=5 wing discs for each genotype. (I) A wing disc beaing GFP+ smo−/− clones was immunostained for Ptc and GFP. Arrow indicates the loss of Ptc expression in smo−/− cells. N=5 wing discs. (J and K) Wing discs carrying smo−/− clones expressing Myc-SmoWT or Myc-SmoMLLA were stained for Ptc and Myc. N=5 wing discs for each genotype. Scale bars, 25 μm (A, C, F to K), 50 μm (D and E). (L) Our findings are consistent with a model in which Ptc binds PA and sequesters it from Smo in the absence of Hh, thus reducing Smo activation. Hh binding may cause Ptc to release the PA, thus increasing its availability to Smo and promoting Smo activation. This model for Ptc-dependent regulation of Smo activation by PA is supported by the observation that myr-PtcCWT brings PA close to the membrane and promotes Smo activation.
To further examine the activity of a mutant form Smo, we performed a rescue assay with the smo3 null mutant. We found that En expression was blocked in anterior cells lacking smo (Fig. 7F), which was fully rescued by Myc-SmoWT expression driven by C765-Gal4 (Fig. 7G). The strategy of using the C765-Gal4 in combination with the MARCM technique is a very sensitive approach previously reported to examine the activity of exogenously expressed Smo (77). With this platform, we found that Myc-SmoMLLA partially rescued the smo null mutant, indicating a decrease in the activity of the Smo protein (Fig. 7H). Similarly, whereas Ptc expression was blocked in smo mutant clones (Fig. 7I), Myc-SmoWT fully rescued (Figs. 7J) and Myc-SmoMLLA partially rescued (Figs. 7K) Ptc expression in smo mutant cells. These data indicate that mutating the PA binding sites in Smo decreased its activity in vivo.
We also analyzed wing discs of flies expressing Myc-Smo3KA and found that the domain of ptc-lacZ expression was similar to that caused by Myc-SmoWT expression (fig. S7A), indicating that these other arginine motifs are unlikely to be involved in Smo regulation by PA. Taken together, our data suggested that PA is a positive regulator for Smo through direct interaction with the MLLA pocket and that Hh promotes the release of PA from Ptc or the transfer of PA from Ptc to Smo to promote Smo stabilization and activation.
Discussion
Hh signal transduction is highly conserved at the plasma membrane, where Ptc inhibits Smo in the absence of Hh stimulation; however, the molecular mechanisms through which Ptc inhibits Smo are unclear. This study uncovers a mechanism for Ptc to bind PA and affect the accessibility of PA to Smo (Fig. 7L). We propose that, in the absence of Hh stimulation, Ptc attracts PA and reduces the accessibility of PA to Smo. The binding of Hh to Ptc causes conformational change in Ptc, which may release PA and allow PA to activate Smo (Fig. 7L). A membrane-tethered form of PtcC (myr-PtcCWT) or a PA binding domain from yeast increases the activity of Smo by enriching PA at the membrane (Fig. 7L). It is notable that Smo showed increased sensitivity to PA in the absence of Ptc, because Pld or dLipin affected Smo activity in cells lacking Ptc but not in cells with Ptc.
How does Hh regulate PA in order to activate Smo? Hh does not seemingly affect the production or degradation of PA. Several lines of evidence support this idea. First, RNAi of Ptc did not change the amounts of dLipin and Pld in cultured cells (figs. S5, I and J), suggesting that Hh signaling may not affect the production of these enzymes. Second, GFP-PASS signals were evenly present in both the A- and P-compartment cells (fig. S5K), suggesting that Hh in P-compartment cells does not change the amounts of PA. Although Hh signaling does not appear to regulate PA biosynthesis or catabolism, binding of Hh to Ptc may cause changes in Ptc-PA association. Indeed, we found that Hh stimulation decreased the interaction between Ptc and PA (Fig. 3E). Structural studies have indicated that, upon binding of Hh, Ptc protein undergoes twisting in the transmembrane domains (52, 53), which may cause conformational change(s) in the C-tail, perhaps releasing PA from the Ptc C-tail. To put PA regulation of Smo in the context of Hh signaling, it is possible that Hh binding causes the Ptc C-tail to release PA, which becomes enriched in the inner leaflet of the membrane, allowing PA to interact with Smo and ultimately activate Smo through the MLLA pocket in the transmembrane domain. It is also possible that Ptc controls the influx or efflux of PA or changes the distribution of PA in the two leaflets of the membrane.
Studies have suggested that the Ptc C-tail mediates endocytosis (57, 58, 60); however, independent studies found that a Ptc mutant failing to endocytose could still inhibit Hh signaling (78) and that Ptc1130X, lacking the tail after amino acid 1130, still sequesters Hh but fails to inhibit Hh target gene expression (63). These studies suggest that Ptc endocytosis mediated Hh sequestration is neither necessary (78) nor sufficient (63) for Hh pathway inhibition. The fact that Ptc1130X removes all of the PA binding sites in the C-tail suggests a critical role of the binding sites in regulating Ptc inhibitory activity.
Upon Hh stimulation, Smo undergoes a conformational change in its C-tail, which involves the four arginine motifs counteracted by phosphorylation (19, 22). Hh stimulation also induces structural changes in the extracellular CRD of Smo (48, 79, 80); however, the CRD is unlikely to be required for Ptc-Smo communication (39). Hh stimulation may also induce structural changes in transmembrane domains of Smo. In this study, we found that, instead of interacting with a consensus binding motif, PA interacted with Smo through a pocket within the transmembrane domains. This may be a more sensitive mechanism for PA to regulate Smo, in consideration of Ptc and Smo structural changes in their transmembrane domains upon Hh stimulation.
Many studies have indicated that cholesterol directly interacts with and activates Smo through different binding sites (35–38). It has also been suggested by studies in mammalian cells that Ptc inhibits Smo by transporting cholesterol. Our study suggests that a Ptc-PA-Smo axis provides an additional mechanism for Ptc to inhibit Smo, and this mechanism may act in parallel with the cholesterol transport mechanism. Further studies are needed address the relationship between cholesterol and PA in terms of their capacities to activate Smo in Drosophila.
Materials and Methods
Drosophila Lines and Mosaic Analysis
All Drosophila strains and plasmids are listed in the Key resources table (table S1). Transgenic lines were generated using the 75B1 attP locus to ensure proteins were expressed at similar amount without positional effects. These include transgenic lines of Myc-SmoWT, Myc-Smoact (a mutant form of Smo in which three clusters of PKA and CK1 sites were mutated to Asp to mimic phosphorylation and thus the activation of Smo), and HA-PtcWT described previously (21, 31, 43), transgenic lines of HA-PtcΔC , HA-PtcM124 , HA-PtcM1, HA-PtcM2, HA-PtcM4, HA-PtcM24; Myc-SmoMLLA and Myc-SmoactMLLA; HA-Pld; HA-dLipin; myr-PAD2, myr-PtcCWT, and myr-PtcCM124, and transgenic lines of GFP-PASS. Null allele of ptcIIw was a gift from Dr. Joan Hooper. ptcS2 mutant stock was from BSC (#6332). Ptc1130X was from BSC (#44612). Genotypes for examining the activity of a specific transgene (myr-PtcCWT, myr-PtcCM124, myr-PAD2, HA-Pld, or HA-dLipin) in ptc clones: yw hsp-flp/+ or Y; ptcIIw FRT42/hs-GFP FRT42; transgene/+. The wing dorsal-compartment-specific ap-Gal4 and the wing-specific ms1096-Gal4 have been described previously (81). For rescue experiments using the MARCM: yw hsp-flp/+; ptcS2 FRT42D/tub-Gal80 UAS-GFP FRT42D; transgene/tub-Gal4. Genotypes for examining the activity of a Smo transgene (Myc-SmoWT or Myc-SmoMLLA) in smo clones: yw hsp-flp/+ or Y; smo3 tub-Gal80FRT40/FRT40; transgene/+ or tub-Gal4-UAS-GFP (control). Pld RNAi (VDRC v106137) and dLipin RNAi (BSC #63614 and #77170).
Constructs and mutants
UAST-2HA-PtcWT has been described (43). HA-PtcΔlp1 and HA-PtcΔlp2 were generated on the background of UAST-2HA-PtcWT by deleting amino acids 98–427 and 738–939, respectively. HA-PtcΔC was generated by PCR to delete residues 1105 –1286 of 2HA-PtcWT. Ptc mutants were generated by PCR-based mutagenesis with the following site mutations: PtcM1, R1130L, K1133A, R1134L; PtcM2, R1145L, R1148L; PtcM3, R1195L, R1198L; PtcM4, R1277L, R1280L. UAST-myr-PtcCWT was constructed by inserting the Ptc C-tail (amino acids 1105 –1286) into UAST-flag-myr vector (82). UAST-Sev-PtcC was constructed by fusing Sevenless transmembrane and extracellular domains (82) with the Ptc C-tail. pET-30a-His-PtcC was generated by subcloning the Ptc C-tail amino acids 1105 –1286 into pET-30a vector.
HA-Pld was generated by fusion of Pld sequence (clone GH07346) into the UAST-2HA vector. HA-dLipin was generated by fusion of dLipin sequence (clone GH19076) into UAST-2HA vector.
SmoMLLA was generated by PCR-based mutagenesis and contains M354F, L427F, L431F and A489Q mutations. SmoKRA and Smo3KA were generated by the same approach and contain K564A + K565A and K580A + K583A + K585A, respectively.
GFP-PASS construct was made by subcloning NES-PAD sequence (gift from Dr. Guangwei Du) into UAST-GFP vector. The UAST-GFP construct was generated by subcloning the GFP coding sequence to the UAST vector. Of note, all of the constructs are designed in the background of attB-UAST vector and are designed to be used for both S2 cells and transgenic fly lines. Primers used for generating the constructs in this study are shown in Table S2.
Solid phase lipid-binding assay, PA bead pull-down assay, and liposome assay
His-PtcC fusion proteins expressed in bacteria were purified by His GraviTrap (GE Healthcare), then eluted with 5 volumes of elution buffer (50 mM Tris HCl, 300 mM NaCl, 500 mM Imidazole, pH 6.8) at 4°C. HA-Ptc and mutants were expressed in S2 cells and then immunoprecipitated with anti-HA antibody and protein A ultralink resin, elution was performed twice in 5 volumes of HA peptide elution buffer (50mM HEPES pH7.9, 100mM NaCl, 1.5 mM MgCl2, 0.05%Triton X-100, 300 μg/ml HA peptide). Myc-tagged Smo proteins expressed in S2 cells were immunoprecipitated with anti-Myc antibody combined with beads of protein A ultralink resin, followed by two sequential elution with Myc peptide (Abmart, 100 μg/ml Myc peptide, 50 mM Tris HCl,150 mM NaCl, pH 7.4). The eluted purified proteins were concentrated by the Centrifugal filter units (Millipore), followed by dialyzed overnight at 4°C in 25 mM Tris HCl pH7.4, 125 mM KCl, 10% glycerol, 1mM DTT.
Solid phase lipid-binding assay was carried out according to the method Adachi et al. (83). Briefly, all lipids were first solved in chloroform: methanol: H2O (1:1:0.1) at 50 μM, 2 μl of each lipid was spotted onto PVDF membrane, membranes were dried at room temperature and then blocked in PBST (PBS + 1% Tween 20) with 3% fatty acid free BSA for 1 h. 1 μg/ml His-PtcC was then incubated with membrane in PBS overnight at 4°C. Membrane was then washed with PBST (PBS + 0.05% Tween 20) 3 times, followed by Western blotting with the anti-His (ThermoFisher), anti-HA (Santa Cruz) or anti-Myc (Santa Cruz) antibodies. Lipids used in this assay were DOPA ( Avanti Polar Lipids,1,2-dioleoyl-sn-glycero-3-phosphate (sodium salt)), DSPA (Cayman Chemical,1,2-Distearoyl-sn-glycero-3-phosphate (sodium salt)), DPPA (Avanti Polar Lipids,1,2-dipalmitoyl-sn-glycero-3-phosphate (sodium salt)), DMPA (Echelon Biosciences, 1,2-Dimyristoyl-sn-glycero-3-phosphate, sodium salt), DOPC (Cayman Chemical,1,2-Dioleoyl-sn-glycero-3-Phosphocholine), POPE (Cayman Chemical,1-Palmitoyl-3-oleoyl-sn-glycero-2-Phosphoethanolamine), DOPE (Cayman Chemical,1,2-Dioleoyl-sn-glycero-3-Phosphoethanolamine), DPPC (Cayman Chemical,1,2-Dipalmitoyl-sn-glycero-3-Phosphatidylcholine), DOPS (Cayman Chemical,1,2-Dioctadecenoyl-sn-glycero-3-Phosphoserine), DAG (Echelon Biosciences, Dipalmitoyl-sn-glycerol), DPPS (Cayman Chemical,1,2-Dipalmitoyl-sn-glycero-3-phospho-L-serine), PI (Cayman Chemical, PtdIns-(1,2-dioctanoyl) (sodium salt)), PI4P (Cayman Chemical, PtdIns-(4)-P1 (1,2-dioctanoyl) (ammonium salt)).
PA bead pull-down experiments were performed according to Echelon Biosciences Lipid Bead-Protein Pull-down Protocol. Briefly, 20 μg protein was incubated with 50 μl PA beads (Echelon cat #P-B0PA) for 4 h at 4°C, then gently washed twice with 10× wash/binding buffer (10mM HEPES, pH 7.4, 150mM NaCl, 0.25% Igepal). Laemmli sample buffer (50 μl 2× stock solution) was added to the PA beads and the samples were heated to 95°C for 5 mins. Samples were then loaded into SDS-PAGE for Western blot analysis.
Liposomes were prepared according to the methods described by Putta et al. (84). Each liposome contains 400 nmol lipids, control liposomes contain 55 mol% DOPC and 45 mol% POPE; liposomes with DPPA contain 45 mol% DOPC, 45 mol% POPE and 10mol% DPPA. DOPC and POPE were stocked in chloroform at 10mg/ml, DPPA was stocked in chloroform: methanol 1:1 at 1 mg/ml. Lipids were mixed in a glass tube then dried under nitrogen. After that, lipids were suspended in 200 μl freshly prepared extrusion buffer (250 mM Raffinose pentahydrate, 25 mM Tris–HCl pH 7.5, and 1 mM DTT) with occasional vertexing and brief sonication, and then left at room temperature for 40 min to hydrate. Lipids were then diluted with 3 volumes of freshly prepared 1× binding buffer (150 mM KCl, 25 mM Tris–HCl pH7.5, 1 mM DTT, and 0.5 mM EDTA), then centrifuged at 21000g for 45 min to collect liposomes. His-PtcC (0.5 μM) or Myc-Smo (1 μM) was incubated with liposomes at room temperature for 40 min in 100 μl binding buffer. Liposomes were collected by centrifuged at 16000g for 30 min, then washed once with 300 μl binding buffer and transferred into a new tube, centrifuged at 16000g for 30 min to pellet the liposomes. Liposomes were resuspended in 30 μl of 1 × Laemmli sample buffer, heated to 95 °C for 5 min before running on SDS-PAGE.
Cell culture, immunoprecipitation, Western blot, and luciferase reporter assay
S2 cells were cultured in Schneider’s Drosophila medium (VWR) and transfections were carried out using Effectene transfection reagent (Qiagen). Forty-eight hours post-transfection, cells were treated with lysis buffer followed by centrifugation at 15000g for 10 min. For each sample, 6 × 106 cells were harvested and lysed in 450 μL lysate buffer (50 mM Tris–HCl pH8.0, 100 mM NaCl, 1% NP40, 10% glycerol, 1.5 mM EDTA pH8.0, protease inhibitor tablet. Adding 10 mM NaF and 1 mM Na3VO4 when examining phosphorylation). Samples were divided: 50 μL was saved for Western blots (4 μL per lane) and 400 μL was used for immunoprecipitation assays (30 μL per immunoprecipitation and 5 μL of immunoprecipitated sample per lane). UAST-GFP was a separate construct co-transfected with the other constructs in each experiment. For immunoprecipitation, the cell lysate was incubated with the proper primary antibody for 2 h in the presence of beads of protein A ultralink resin (Thermo). Protein samples were resolved by SDS-PAGE and transferred onto PVDF membranes (Millipore) for Western blot, which was performed using the indicated antibodies and the enhanced chemiluminescence (ECL) protocol. For all the experiments with cultured S2 cells, N=3 independent experiments, and statistical analysis of the density from western blot bands was performed using the ImageJ software followed by Student’s t test analysis.
For ptc-luc reporter assays, S2 cells were cultured in 6-well plates and transfected with 50 ng tub-Ci (31) and 150 ng ptc-luc reporter constructs. 48 h post-transfection, cells were lysed for luciferase activity analysis using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). Renilla was used to normalize the luciferase activity. The measurements of Dual-Luciferase were performed using a GLOMAX Multi Detection System (Promega). Presented data are representative of three assays, with standard deviation (SD) bars indicated from four replicates. For statistical analysis, Student’s t test was used for cell culture studies involving two independent groups.
For treatment of S2 cells with different forms of PA, cells were changed to serum-free medium for 16 h before treatment. PA was first dissolved in chloroform/methanol 1:1 at 1 mM. Aliquots of PA were dried under nitrogen and dried pellets were stored at 4°C. PA was then dissolved in PBS at 1 mM and heated to 70°C to ensure no residual solvent remained. PA in PBS was added to cells at a final concentration of 50 μM (unless otherwise indicated) for 4 h.
Treating S2 cells with dsRNA has been described (31, 85). dsRNA was synthesized against Ptc (nucleotides 1–781), Pld (nucleotides 1091–1610), and Lpin (nucleotides 450–950) and RNAi efficiency for each individual RNAi was monitored by Western blot with anti-HA antibody to detect the expression levels of cotransfected Ptc/Pld/Lpin.
For Real-Time PCR, total RNA was extracted using Trizol reagent (Invitrogen). cDNA was synthesized using SuperScript III First Strand Synthesis kit (Invitrogen) from 1.0 μg total RNA according to the manufacturer’s instructions. Quantitative Real-Time PCR reactions were carried out using SYBR Green PCR master mix reagents (Thermo) on the ABI QStudio Real-Time PCR System (Applied Biosystems). Thermal cycling was conducted at 95°C for 30sec, followed by 40 cycles of amplification at 95°C for 5sec, 55°C for 30sec and 72°C for 15sec. The following primers were used to amplify: L32: 5’-GCTAAGCTGTCGCACAAATG-3’ and 5’- GTTCGATCCGTAACCGATGT −3’; Ptc: 5’-ATGCTGTGCTTCAATGTGCT-3’ and 5’-CGACGTGGAGAGCATGAACA-3’.
PA membrane accumulation analysis with GFP-PASS
GFP-PASS construct was made by subcloning NES-PAD sequence (gift from Dr. Guangwei Du) into attB-UAST-GFP vector at enzyme site BglII and Asp718. S2 cells were transfected with GFP-PASS construct with or without the co-transfection of myr-PtcCWT or myr-PtcCM124, followed by treatment with or without PA (final concentration 50 μM for 4 h). For immunostaining, cells were washed with PBS, fixed with 4% formaldehyde for 20 min, stained with antibody that recognizes the alpha subunit of the Na-K-ATPase (ATPα), and mounted on slides in 80% glycerol. Fluorescence signals were acquired with the 100× objective on a Nikon A1+-Ti2 confocal microscope. GFP was excited at 488nm wavelength, and the emission was collected through at 514 nm. Plasma membrane region was defined by the staining of ATPα. ImageJ software was used to calculate the density within the ATPα defined membrane vs total GFP signal in the cell. The intensity changes of membrane GFP/ total GFP were analyzed using the Image J software. Each experiment was based on 20 individual cells. For Western blot to detect membrane-associated GFP-PASS, each sample was collected from one 100-mm dish of S2 cells cultured in 10% FBS for 3 days. Plasma membrane was separated according to manufacturer’s instruction (Abcam, Plasma Membrane Protein Extraction kit). Loading amount was measured based on the membrane ATPα detected by the anti-ATPα antibody. GFP-PASS signal was detected by the mouse anti-GFP antibody.
Immunostaining of wing imaginal disc and confocal microscopy
Wing discs from third instar larvae were dissected in PBS and fixed with 4% formaldehyde in PBS for 20 min. After permeabilization with 1% PBST, discs were incubated with primary antibodies for 3 h, appropriate secondary antibodies for 1 h, and washed three times with PBST after each incubation. Discs were then subjected for secondary antibody labeling using the affinity-purified secondary antibodies from Jackson ImmunoResearch. Fluorescence signals were acquired on a Nikon A1+-Ti2 confocal microscope and images processed with NIS-Elements software. About 20 imaginal discs were screened and 3–6 disc images were taken for each experiment/genotype.
Structural analysis of Smo and Smo mutants
Structural coordinates were obtained from the Protein Data Bank (PDB=6O3C) and AlphaFold database (alphafold.ebi.ac.uk P91682) (73, 74). The effect of mutations was predicted using mCSM-membrane (86). Structural models were analyzed, and figures prepared using PyMOL (The PyMOL Molecular Graphics System, Version 2.4.1 Schrödinger, LLC.).
Quantification and statistical analysis
All experiments were repeated multiple times to confirm reproducibility. Images shown are representative of at least three independent repeats. All quantifications are presented as the mean ± standard error of mean (SEM). Statistical tests for each bar graph in figures were determined by unpaired two-tailed t test with n.s. or ns, no significant; * p< 0.05; ** p < 0.01; *** p<0.001.
Supplementary Material
Acknowledgements:
We are grateful to Dr. Guangwei Du for the GFP-PASS construct; Dr. Michael Lehmann for the dLipin and dLipinΔPAP flies; Dr. Jin Jiang for the MARCM flies and MARCM combined with C765-Gal4 flies; Bloomington Drosophila Stock Center (BSC) and Vienna Drosophila Resource Center (VDRC) for fly stocks; and the Developmental Studies Hybridoma Bank (DSHB) for the anti-Ci and anti-β-tubulin antibodies. We also thank Dr. Feng Yan for help with generating the Sev-PtcC construct during manuscript revision.
Funding:
This study was supported by the National Institutes of Health grants (MIRA R35GM131807) to J.J. This study was also supported by the Shared Resource Facilities of the University of Kentucky Markey Cancer Center (P30CA177558), and the Imaging Core and Pilot funds from the COBRE (P20GM121327).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Briscoe J, Therond PP, The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14, 416–429 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Jiang J, Hui CC, Hedgehog signaling in development and cancer. Dev Cell 15, 801–812 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villavicencio EH, Walterhouse DO, Iannaccone PM, The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet 67, 1047–1054 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taipale J, Beachy PA, The Hedgehog and Wnt signalling pathways in cancer. Nature 411, 349–354 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Pasca di Magliano M, Hebrok M, Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 3, 903–911 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Pak E, Segal RA, Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev Cell 38, 333–344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang J, Hedgehog signaling mechanism and role in cancer. Semin Cancer Biol, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guha M, Hedgehog inhibitor gets landmark skin cancer approval, but questions remain for wider potential. Nature reviews. Drug discovery 11, 257–258 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Sekulic A, Von Hoff D, Hedgehog Pathway Inhibition. Cell 164, 831 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Torroja C, Gorfinkiel N, Guerrero I, Mechanisms of Hedgehog gradient formation and interpretation. J Neurobiol 64, 334–356 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Qi X, Li X, Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends Biochem Sci 45, 397–410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong JH, Siebold C, Rohatgi R, Biochemical mechanisms of vertebrate hedgehog signaling. Development 146, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VanHook AM, Focus issue: fine-tuning Hedgehog signaling in development and disease. Sci Signal 4, eg10 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Lum L, Beachy PA, The Hedgehog response network: sensors, switches, and routers. Science 304, 1755–1759 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Zheng X, Mann RK, Sever N, Beachy PA, Genetic and biochemical definition of the Hedgehog receptor. Genes Dev 24, 57–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Chen Y, Shi Q, Yue T, Wang B, Jiang J, Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol 10, e1001239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia R, Jia H, Fan J, Liu Y, Jia J, USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol 10, e1001238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hooper JE, Scott MP, Communicating with Hedgehogs. Nat Rev Mol Cell Biol 6, 306–317 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Jiang J, Decoding the phosphorylation code in Hedgehog signal transduction. Cell research 23, 186–200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Liu Z, Jia J, Mechanisms of Smoothened Regulation in Hedgehog Signaling. Cells 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia J, Tong C, Wang B, Luo L, Jiang J, Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Tong C, Jiang J, Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450, 252–258 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Basler K, Struhl G, Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature 368, 208–214 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Tabata T, Kornberg TB, Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76, 89–102 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Strigini M, Cohen SM, A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development 124, 4697–4705 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Vervoort M, Crozatier M, Valle D, Vincent A, The COE transcription factor Collier is a mediator of short-range Hedgehog-induced patterning of the Drosophila wing. Curr Biol 9, 632–639 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Jiang K, Jia J, Smoothened regulation in response to Hedgehog stimulation. Front Biol (Beijing) 10, 475–486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia J, Tong C, Jiang J, Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev 17, 2709–2720 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, Weis-Garcia F, Gong R, Wang B, Beachy PA, Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell 12, 1261–1274 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Ogden SK, Ascano M Jr., Stegman MA, Suber LM, Hooper JE, Robbins DJ, Identification of a functional interaction between the transmembrane protein Smoothened and the kinesin-related protein Costal2. Curr Biol 13, 1998–2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan J, Liu Y, Jia J, Hh-induced Smoothened conformational switch is mediated by differential phosphorylation at its C-terminal tail in a dose- and position-dependent manner. Dev Biol 366, 172–184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denef N, Neubuser D, Perez L, Cohen SM, Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102, 521–531 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Taipale J, Cooper MK, Maiti T, Beachy PA, Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Casali A, Struhl G, Reading the Hedgehog morphogen gradient by measuring the ratio of bound to unbound Patched protein. Nature 431, 76–80 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Byrne EF, Luchetti G, Rohatgi R, Siebold C, Multiple ligand binding sites regulate the Hedgehog signal transducer Smoothened in vertebrates. Curr Opin Cell Biol 51, 81–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao X, Tang JJ, Peng C, Wang Y, Fu L, Qiu ZP, Xiong Y, Yang LF, Cui HW, He XL, Yin L, Qi W, Wong CC, Zhao Y, Li BL, Qiu WW, Song BL, Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol Cell 66, 154–162 e110 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Byrne EF, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, Sansom MS, Newstead S, Rohatgi R, Siebold C, Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, Salic A, Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 166, 1176–1187 e1114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myers BR, Neahring L, Zhang Y, Roberts KJ, Beachy PA, Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proceedings Of The National Academy Of Sciences Of The United States 114, E11141–E11150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinnebrew M, Iverson EJ, Patel BB, Pusapati GV, Kong JH, Johnson KA, Luchetti G, Eckert KM, McDonald JG, Covey DF, Siebold C, Radhakrishnan A, Rohatgi R, Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bidet M, Joubert O, Lacombe B, Ciantar M, Nehme R, Mollat P, Bretillon L, Faure H, Bittman R, Ruat M, Mus-Veteau I, The hedgehog receptor patched is involved in cholesterol transport. PLoS One 6, e23834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Bulkley DP, Xin Y, Roberts KJ, Asarnow DE, Sharma A, Myers BR, Cho W, Cheng Y, Beachy PA, Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 175, 1352–1364 e1314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang K, Liu Y, Fan J, Zhang J, Li XA, Evers BM, Zhu H, Jia J, PI(4)P Promotes Phosphorylation and Conformational Change of Smoothened through Interaction with Its C-terminal Tail. PLoS Biol 14, e1002375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U, Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell 19, 54–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Findakly S, Daggubati V, Garcia G, LaStella SA, Choudhury A, Tran C, Li A, Tong P, Garcia JQ, Puri N, Reiter JF, Xu L, Raleigh DR, Sterol and oxysterol synthases near the ciliary base activate the Hedgehog pathway. Journal Of Cell Biology. 220, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raleigh DR, Sever N, Choksi PK, Sigg MA, Hines KM, Thompson BM, Elnatan D, Jaishankar P, Bisignano P, Garcia-Gonzalo FR, Krup AL, Eberl M, Byrne EFX, Siebold C, Wong SY, Renslo AR, Grabe M, McDonald JG, Xu L, Beachy PA, Reiter JF, Cilia-Associated Oxysterols Activate Smoothened. Mol Cell 72, 316–327 e315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, Bazan JF, Beachy PA, Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell 26, 346–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, Rohatgi R, Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2, e01340 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakano Y, Guerrero I, Hidalgo A, Taylor A, Whittle JR, Ingham PW, A protein with several possible membrane-spanning domains encoded by the Drosophila segment polarity gene patched. Nature 341, 508–513 (1989). [DOI] [PubMed] [Google Scholar]
- 50.Hooper JE, Scott MP, The Drosophila patched gene encodes a putative membrane protein required for segmental patterning. Cell 59, 751–765 (1989). [DOI] [PubMed] [Google Scholar]
- 51.Qi X, Schmiege P, Coutavas E, Wang J, Li X, Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 560, 128–132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qi X, Schmiege P, Coutavas E, Li X, Two Patched molecules engage distinct sites on Hedgehog yielding a signaling-competent complex. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong X, Qian H, Cao P, Zhao X, Zhou Q, Lei J, Yan N, Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 361, (2018). [DOI] [PubMed] [Google Scholar]
- 54.Martin V, Carrillo G, Torroja C, Guerrero I, The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr Biol 11, 601–607 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Strutt H, Thomas C, Nakano Y, Stark D, Neave B, Taylor AM, Ingham PW, Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr Biol 11, 608–613 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Briscoe J, Chen Y, Jessell TM, Struhl G, A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol Cell 7, 1279–1291 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Lu X, Liu S, Kornberg TB, The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev 20, 2539–2551 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang S, Zhang Z, Zhang C, Lv X, Zheng X, Chen Z, Sun L, Wang H, Zhu Y, Zhang J, Yang S, Lu Y, Sun Q, Tao Y, Liu F, Zhao Y, Chen D, Activation of Smurf E3 ligase promoted by smoothened regulates hedgehog signaling through targeting patched turnover. PLoS Biol 11, e1001721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S, Li S, Wang B, Jiang J, Hedgehog reciprocally controls trafficking of Smo and Ptc through the Smurf family of E3 ubiquitin ligases. Sci Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brigui A, Hofmann L, Arguelles C, Sanial M, Holmgren RA, Plessis A, Control of the dynamics and homeostasis of the Drosophila Hedgehog receptor Patched by two C2-WW-HECT-E3 Ubiquitin ligases. Open Biol 5, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan J, Gao Y, Lu Y, Wu W, Yuan S, Wu H, Chen D, Zhao Y, PKAc-directed interaction and phosphorylation of Ptc is required for Hh signaling inhibition in Drosophila. Cell Discov 5, 44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duman-Scheel M, Weng L, Xin S, Du W, Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417, 299–304 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Johnson RL, Milenkovic L, Scott MP, In vivo functions of the patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Mol Cell 6, 467–478 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Zhukovsky MA, Filograna A, Luini A, Corda D, Valente C, Phosphatidic acid in membrane rearrangements. FEBS Lett 593, 2428–2451 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Wu JS, Luo L, A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nat Protoc 1, 2583–2589 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Julkowska MM, Rankenberg JM, Testerink C, Liposome-binding assays to assess specificity and affinity of phospholipid-protein interactions. Methods Mol Biol 1009, 261–271 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Loewen CJ, Gaspar ML, Jesch SA, Delon C, Ktistakis NT, Henry SA, Levine TP, Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science 304, 1644–1647 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Kassas N, Tanguy E, Thahouly T, Fouillen L, Heintz D, Chasserot-Golaz S, Bader MF, Grant NJ, Vitale N, Comparative Characterization of Phosphatidic Acid Sensors and Their Localization during Frustrated Phagocytosis. Journal Of Biological Chemistry. 292, 4266–4279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang F, Wang Z, Lu M, Yonekubo Y, Liang X, Zhang Y, Wu P, Zhou Y, Grinstein S, Hancock JF, Du G, Temporal production of the signaling lipid phosphatidic acid by phospholipase D2 determines the output of extracellular signal-regulated kinase signaling in cancer cells. Molecular And Cellular Biology. 34, 84–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jia H, Liu Y, Xia R, Tong C, Yue T, Jiang J, Jia J, Casein kinase 2 promotes Hedgehog signaling by regulating both smoothened and Cubitus interruptus. Journal Of Biological Chemistry. 285, 37218–37226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmitt S, Ugrankar R, Greene SE, Prajapati M, Lehmann M, Drosophila Lipin interacts with insulin and TOR signaling pathways in the control of growth and lipid metabolism. J Cell Sci 128, 4395–4406 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Deshpande I, Liang J, Hedeen D, Roberts KJ, Zhang Y, Ha B, Latorraca NR, Faust B, Dror RO, Beachy PA, Myers BR, Manglik A, Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Zidek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D, Velankar S, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qi X, Liu H, Thompson B, McDonald J, Zhang C, Li X, Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, Rohatgi R, Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li S, Ma G, Wang B, Jiang J, Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci Signal 7, ra62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Torroja C, Gorfinkiel N, Guerrero I, Patched controls the Hedgehog gradient by endocytosis in a dynamin-dependent manner, but this internalization does not play a major role in signal transduction. Development 131, 2395–2408 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, Sansom MSP, Newstead S, Rohatgi R, Siebold C, Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rana R, Carroll CE, Lee HJ, Bao J, Marada S, Grace CR, Guibao CD, Ogden SK, Zheng JJ, Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nature communications 4, 2965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang K, Liu Y, Zhang J, Jia J, An intracellular activation of Smoothened that is independent of Hedgehog stimulation in Drosophila. J Cell Sci 131, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jia J, Tong C, Jiang J, Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev 17, 2709–2720 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, Sesaki H, Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol Cell 63, 1034–1043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Putta P, Rankenberg J, Korver RA, van Wijk R, Munnik T, Testerink C, Kooijman EE, Phosphatidic acid binding proteins display differential binding as a function of membrane curvature stress and chemical properties. Biochim Biophys Acta 1858, 2709–2716 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Jia H, Liu Y, Yan W, Jia J, PP4 and PP2A regulate Hedgehog signaling by controlling Smo and Ci phosphorylation. Development 136, 307–316 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pires DEV, Rodrigues CHM, Ascher DB, mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res 48, W147–W153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.