

Abstract
Several evolved properties of adeno-associated virus (AAV), such as broad tropism and immunogenicity in humans, are barriers to AAV-based gene therapy. Most efforts to re-engineer these properties have focused on variable regions near AAV’s 3-fold protrusions and capsid protein termini. To comprehensively survey AAV capsids for engineerable hotspots, we determined multiple AAV fitness phenotypes upon insertion of six structured protein domains into the entire AAV-DJ capsid protein VP1. This is the largest and most comprehensive AAV domain insertion dataset to date. Our data revealed a surprising robustness of AAV capsids to accommodate large domain insertions. Insertion permissibility depended strongly on insertion position, domain type, and measured fitness phenotype, which clustered into contiguous structural units that we could link to distinct roles in AAV assembly, stability, and infectivity. We also identified engineerable hotspots of AAV that facilitate the covalent attachment of binding scaffolds, which may represent an alternative approach to re-direct AAV tropism.
Keywords: adeno-associated virus, capsid engineering, domain insertional profiling, capsid assembly, HUH tags, gene therapy
Graphical abstract
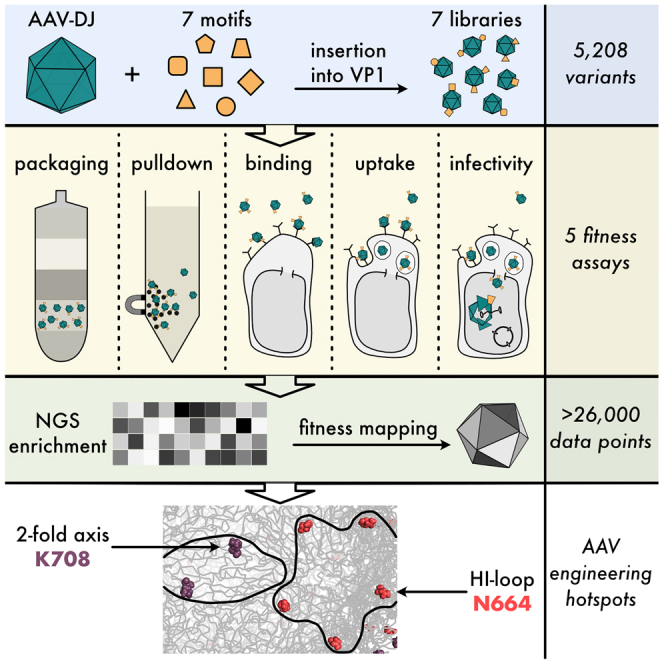
Schmidt and colleagues comprehensively mapped AAV-DJ capsid protein VP1 tolerance to insertion of several motifs with retargeting abilities. Assessment of multiple fitness phenotypes—including packaging, binding, and infectivity fitness—led to the discovery of engineerable hotspots near the capsid’s 2-fold axis and in the HI loop.
Introduction
Recombinant adeno-associated virus (rAAV) has proved to be safe and able to drive long-term expression in dividing and non-dividing human cells. Several AAV-based therapeutics have been approved by the US Food and Drug Administration (FDA) and numerous clinical trials using AAV for the treatment of genetic diseases are underway.1,2,3,4 Despite the exceptional clinical potential of naturally evolved AAV serotypes, they could be substantially improved with respect to production yield, DNA packaging capacity, immunogenicity, cell type specificity, and infectivity.5
Addressing AAV’s drawbacks through engineering is facilitated by its relatively simple structural and genetic organization. The ∼4.7-kb single-stranded DNA (ssDNA) genome comprises two genes, rep and cap, flanked by inverted terminal repeats (ITRs). One open reading frame (ORF) of the capsid gene cap encodes for three viral proteins VP1 (737 amino acids [aa], 87 kDa), VP2 (600 aa, 72 kDa), and VP3 (535–503 aa, 62 kDa). It is expressed from the p40-promoter (C terminus of the rep gene) and translated from overlapping ORFs in a way that VP2 is lacking the N terminus of VP1 and VP3 is missing the N-terminal part of VP1/VP2.6 Other cap ORFs express the assembly-activating protein (AAP)7 and membrane-associated accessory protein (MAAP8). For recombinant production of AAV, the rep and cap genes between the ITRs are replaced by the transgene of interest. The capsid assembly takes place in the nucleus and entails a stochastic oligomerization process forming an empty capsid in the nucleus first,9,10,11 followed by packaging of the ssDNA payload.9,10,11 The capsid shell is composed of 60 VP monomers at an average ratio of 1:1:10 VP1, VP2, and VP3, respectively.12,13,14,15 This ratio is highly divergent and assembly is stochastic, such that every capsid has a unique structural assembly.11 The icosahedral capsid features a cylindrical pore at the 5-fold interface, depressions surrounding the 5-fold pore continuing through the 2-fold axis, as well as protrusions at the 3-fold axis.16 Although the overall topology of AAV capsids is conserved across serotypes, Govindasamy et al. determined variable regions (VRs; VR1–9) mapping to surface loops of the capsid.17 These VRs are commonly involved in virus binding to (co-)receptors on the surface of a target cell, such as heparan sulfate or AAVR,18,19 and the subsequent process of endocytosis.20,21,22 The unique N-terminal portion of VP1, also known as VP1u, is located inside the capsid and is indispensable for infection. Upon infection, acidification during endosomal trafficking causes unfolding of the VP1u domain so that it can be externalized through the pore.23,24,25,26,27 A conserved phospholipase A2 domain (PLA228) and nuclear localization signals (NLSs29), which are part of VP1u, can then facilitate endosomal escape and nuclear entry.22 Upon interaction of the AAV with the nuclear pore complex, the virions enter the nucleus, where they are forwarded to the nucleus and the genetic payload is released.22,27
A variety of capsid engineering approaches have been applied in the past to improve the natural infection efficiency of AAVs ranging from shuffling of natural AAV serotypes, recovery of ancestral serotypes, and peptide display. These methods resulted in significantly advanced capsid variants, such as AAV-DJ,30 Anc80,31 AAV-PHP.eB,32 or AAV2.7m8.33 Moreover, there have been successes with incorporating larger, structured protein domains. The first was fusing the green fluorescent protein (GFP) N terminally to VP2, which was useful for visualizing intracellular trafficking of AAV particles.34 In the same manner, Gaussia luciferase35 and an ankyrin repeat protein (DARPin36) were successfully incorporated into the capsid. Other studies incorporated domains for a cell type-specific targeting into VR4 of VP1 or VP2, including nanobodies,37 HUH tags,38 or DARPins.39 While the aforementioned studies focus on the termini and VR4 or VR8 for domain insertions or peptide insertions, respectively, few studies attempted to comprehensively survey permissive capsid regions for peptide and domain insertions. Among them, Judd et al. constructed a random insertion library of mCherry into the VP3-encoding section of VP1 of AAV2, identifying only a single clone, in VR4, that tolerated insertion.40 Another example is the study from Koerber et al.,41 in which a transposon-based saturation mutagenesis approach was used to insert a hexahistidine peptide in AAV2 and AAV8, again pointing to VR8 as the prime peptide insertion spot. Taken together, there is still a paucity of large-scale domain insertional datasets that comprehensively assess (1) the effect on distinct biologically relevant AAV properties, i.e., assembly, binding, and infection, as well as (2) the effect of inserting domains with different physicochemical properties. In the absence of these data, the boundaries of AAV capsid plasticity with respect to accepting domain insertions while maintaining fitness (i.e., assembly, packaging, cell entry, etc.) are yet to be fully understood.
Our goal in this study was to survey as many proxies (AAV fitness phenotypes) for these distinct virion functions as possible. We posit that a comprehensive multiparametric AAV fitness dataset will facilitate the optimization of engineering AAV along multiple axes.8,42,43 Furthermore, based on our previous studies in other protein models,44,45 we hypothesized that systematic domain insertion (i.e., perturbation scanning) across different measured phenotypes may identify capsid determinants for assembly, stability, and dynamics, thereby revealing the topological organization of virus functions.
To achieve this goal, we here combined saturated programmable insertion engineering (SPINE46) with sequencing-based fitness assays to comprehensively determine multiple AAV fitness phenotypes upon insertion of the FLAG peptide tag as well as several large, structured protein domains into VP1 of AAV-DJ. Our data revealed a surprising robustness of AAV viral capsids to accommodate large protein domain insertions. We also found strong positional, domain-type, and fitness phenotype dependence of insertion permissibility, which we can map to contiguous structural units of the AAV capsid and link to distinct roles in AAV assembly, stability, and infectivity. We also identified additional engineerable hotspots of AAV accepting insertion of small protein tags that facilitate the covalent attachment of antibodies. These hotspots may enable alternative approaches to re-direct AAV tropism.
Results
Domain insertional profiling in AAV-DJ VP1
We turned to AAV-DJ30 as the testbed for inserting a peptide tag (FLAG) and six different protein domains (Figure S1) in between every two residues of VP1. Our rationale for focusing on VP1, as opposed to the more abundant VP3 or the non-essential VP2, was as follows: as the least abundant VP isoform, there are, on average, between one and five copies of VP1 incorporated per capsid.12,13,14,15 Note that this is an average copy number based on bulk measurements12,13,14,15; because assembly is a stochastic process, there are many particles with copy numbers at the extreme tails (i.e., 0 copies or >10 copies).11 We reasoned that keeping a low number of VPs carrying inserted domains, which are potentially very disruptive, is more likely to result in assembled and functional virions. This is akin to applying a low or intermediate amount of selection pressure in directed protein evolution experiments, which can reveal more facetted fitness phenotypes.47 Furthermore, we had observed in a prior study that inserting large domains into VR4 (part of the VP common region) completely abolished AAV production unless it was limited to VP1 (or VP2) only.38 Most importantly, focusing on VP1 allows us to interrogate the effect of domain insertion on AAV cell entry; VP1 is required for AAV infectivity as it contains the PLA2 domain that mediates endosomal escape.26,28,48
To generate separate expression constructs for the VP1 domain insertion library and cap expressing only VP2 and VP3, we duplicated the AAV-DJ cap gene and introduced mutations (M1K; T138A/M203K/M211L/M235L) to suppress expression of VP1 or VP2/VP3, respectively (Figure 1A; and Table S1). We also replaced the VP1 heparin binding domain (HBD: residues 587–590) with a hemagglutinin (HA) tag—originally intended but not used—for western blot tracking. The VP1-only cap gene was then subjected to SPINE.46 In brief, the VP1 gene was divided into 14 fragments and insertion variants carrying a temporary genetic handle at each residue position within each fragment were micro-array synthesized as oligo libraries. The genetic handle was subsequently replaced by the FLAG peptide tag or one of the six domains. This cloning approach resulted in a library of VP1 variants with a peptide tag or protein domain, flanked by short linkers, inserted in between every two residues. We chose the FLAG peptide tag (DYKDDDDK) as one type of insertion because of its size similar to prior AAV peptide insertion screens.49,50,51 With an eye toward redirecting AAV tropism, we focused on protein domains that themselves have retargeting abilities (nanobody binding to GFP52) or that enable covalent linkage of retargeting moieties (SNAP tag to link O6-benzylguanine derivatives;53 SpyCatcher to link SpyTag fusions54), and three different HUH tags (WDV, DCV, mMobA) to covalently link ssDNA-conjugated molecules in a sequence specific manner.55,56,57,58
Figure 1.

Design and analysis of AAV domain insertion libraries
(A) Schematic of library design. (B) Plasmids used for the triple transfection in AAV production. (C) Schematic of AAV capsid assembly. (D) Purification of AAV via gradient purification. (E–H) Schematic of workflows analyzing the fitness of AAV domain insertion libraries (i.e., pull-down, binding, uptake, and infectivity assays). (I) Quantification of packaging titer via qPCR. Data are means ± SEM. One-way ANOVA test p value 0.19, not significant (ns). (J) Representative western blot image of AAV domain insertion libraries stained with A1 antibody (detecting VP1 subunits). Predicted sizes for VP1 and its domain fusions are VP1, 81.9 kDa; VP1-nanobdy, 96.0 kDa; VP1-SNAP, 102.4 kDa; VP1-SpyCatcher, 92.7 kDa; VP1-mMobA, 104.0 kDa; VP1-WDV, 98.7 kDa; VP1-FLAG, 83.8 kDa; VP1-DCV, 95.5 kDa. (K) Western blot quantification of VP1, VP2, and VP3 subunits. Data are means (n = 3). Two-way ANOVA test p value 0.641, not significant (ns). (L) Tm of AAV libraries obtained by DSF assay. Data are means (n = 2).
Since insertions can affect any of the steps in AAV packaging and infection, we attempted to independently assay different insertion variant fitness phenotypes. The relatively large insertion variant library size (744 AAV positions (including HA tag) × 7 motifs = 5,208 variants for each assay) necessitated a high-throughput format. We therefore devised assays in which the different fitness phenotypes of an insertion variant were assessed by next-generation (NextGen) sequencing (NGS) variant populations before and after a fitness test.59 A requirement for our approach was a stringent linkage between genotype (the insertion variant) and the measured phenotype (determined by properties of the capsid into which this VP1 variant is assembled). We achieved this by flanking the VP1 variant library with ITRs such that the gene encoding a specific VP1 variant is likely to become packaged into the capsid that incorporated this variant following assembly (payload sizes for the different libraries are shown in Figure S1C). As a measure to reduce cross-packaging (a mismatch between packaged VP1 variant gene and VP1 variant protein that is incorporated into the capsid), we transfected producer cells with a significantly reduced plasmid copy number per cell. This has been demonstrated to reduce cross-packaging.60
We then used these VP1 variant input libraries, stratified by inserted motif, for helper-free virus production followed by gradient purification (Figures 1B–1D). Using NGS of packaged genomes from full capsids, we assayed AAV packaging fitness. Note, that the term “packaging” convolutes two processes, assembly of preformed capsid in the nucleus, which we cannot measure directly, and DNA packaging (requiring a preformed capsid) mediated by Rep52/40.61 Other AAV properties, such as its propensity to aggregate and reducing yield from producer cells, are captured by packaging fitness as well. We calculated the packaging fitness by counting the frequency of a given VP1 variant (i) after packaging (s) relative to the frequency of that variant in the input library (u), normalized to wild-type AAV (wt):
We measured absolute wild-type fitness by spiking in an AAV-DJ ITR-flanked VP1-only cap gene containing 10 synonymous mutations into the VP1-library mix (Table S1). Wild-type AAV-DJ and AAV-DJ with silent mutations showed no difference in production titers (Figure S2). Using a similar approach to count VP1 variants before and after selection, we established assays that determine pull-down fitness (using affinity purification material), cell binding, cell uptake, and infectivity fitness (Figures 1E–1H). Infectivity assays were based on transduction of HEK293FT cells with miRFP670nano62 expressed from the AAV payload (Figure 1A). This enabled flow sorting of infectious variants (enriched in miRFP670nanohigh cells) (Figure S3).
VP1 insertion library quality and completeness
Overall, coverage for all phenotype assays and inserted motifs was excellent (median dataset completeness is 98.5%; see Table S2 for sequencing statistics). Biological replicates were highly correlated for plasmid library, packaging, and pull-down assays (Pearson correlation coefficient: 0.67–0.99; Figure S4A). The infection assay was very noisy for some domains (Pearson correlation coefficient 0.38–0.88), likely related to small number of cells collected for the miRFP670nanohigh cell pool (Figure S3). For all tested motifs, the majority of missing insertion positions (no data in either replicate; for example, positions 428–445) were missing in all phenotypes and the input library, suggesting that the dropout rate was related to library construction (Figures S4B and S4C). Since replicate 2 had a higher read depth and completeness overall, we used this dataset for further analysis. Median read depth across tested motifs and phenotype assays was 635 reads per position (Figure S4D; Table S2).
Domain insertions do no alter bulk properties
After helper-free production and iodixanol gradient purification, we measured virus titers by qPCR. While titers appeared somewhat lower for insertion libraries, this difference was not significant (Figure 1I; one-way ANOVA p value 0.19). Western blot with the A1 antibody (which recognizes a VP1-unique epitope) confirmed that VP1 was incorporated into some virions for all libraries (Figure 1J). However, we observed VP1 bands at various smaller sizes, suggesting that the libraries contained some unstable VP1 domain fusions that are susceptible to degradation. We next used the B1 antibody densitometry to determine bulk VP1/VP2/VP3 ratios (Figures 1K and S5). We found that wild-type AAV-DJ virion contains on average three VP1 copies per 60-mer capsid, in line with prior studies.11,15 There was no significant difference in VP composition for the different insertion libraries (two-way ANOVA p value 0.641), nor VP1 content (one-way ANOVA p value 0.206). To further characterize our libraries on the bulk level, we measured capsid melting temperatures using differential scanning fluorimetry. While SNAP and FLAG libraries had slightly elevated melting points (Figures 1L and S6), all libraries were overall remarkably similar and within a 1°C range of AAV-DJ, suggesting that domain insertions in VP1 did not significantly affect bulk capsid stability of the insertion library. We next used negative-stain electron microscopy to compare full/empty capsid ratios of wild-type AAV-DJ, SNAP, and nanobody libraries and found that full particles comprised between 80% and 90% of all samples (Figure S7).
Note that the western blot, capsid melting assays, and electron microcopy data merely determine average properties of our libraries; with these bulk measurements, we cannot determine VP ratio at the individual capsid level or measure how individual VP1 variants and their copies affect capsid stability or full/empty ratios. Previous studies have shown that the actual single-capsid stoichiometry varies tremendously, even without any modification, and up to 60% of capsids lack either VP1 or VP2 altogether.11 Taken together, our data suggest that on average (i.e., considering the entire insertion library) bulk properties are not altered by domain insertions. This suggests either that assembly of AAV capsid with VP1 variants is a rare event (such that bulk properties are mostly determined by VP2/VP3-only capsids) or that many VP1 variants are compatible with capsid assembly, suggesting a certain degree of conformational plasticity. To resolve this question, we analyzed our libraries for their insertional fitness on the single-capsid level.
High-resolution AAV fitness profiles across different phenotypes
By measuring enrichment or depletion of individual VP1 variants in different selection assays, we found that different insertion types at different sites do have divergent impacts on AAV fitness. These data are summarized in Figure 2, showing a heatmap of insertion fitness of all seven motifs in all 744 VP1 positions, segregated by measured fitness phenotypes (packaging, pull-down, binding, uptake, and infectivity). Fitness values are mapped from magenta over white to green, corresponding to lower to higher than wild-type AAV-DJ fitness (white). Poisson errors were generally low (Figure S8) and only slightly elevated at the junctions between the 14 different fragments used in the SPINE-mediated assembly of the AAV insertion library. Median error with respect to dynamic range of each fitness assay was (0.102/2) log units = 5.1%, which is typical for these types of high-throughput protein fitness assays.63 While overall there was a strong dependence on insertion position and type of inserted domain for all assays, uptake and infection fitness were decreased for nearly every position. One notable exception was the increased fitness of the WDV domain between positions 480 and 530. We currently do not know the underlying mechanism, but domain-idiosyncratic fitness effects support the idea that AAV capsid structure and function are sensitive to the structural context of a perturbation.
Figure 2.

Fitness of AAV domain insertion libraries
(A) Heatmap representing the fitness of each motif insertion in each VP1 position for AAV packaging, pull-down, binding, uptake, and infectivity assays. Green indicates higher and magenta lower fitness than AAV-DJ (white). Yellow denotes positions without data. VP1 secondary structure elements and VR1-9 are indicated on top. (B) Fitness distributions of all AAV libraries compared to AAV-DJ (fitness = 0) ± standard error (red dashed lines).
Effects of domain insertions in VP1u
Focusing on packaging fitness, we saw a distinct increase in fitness when motifs (in particular, nanobody, SNAP tag, or DCV HUH tag) were inserted into the PLA2 domain of VP1u (Figure 3). For the same positions, a pull-down with the respective affinity materials (e.g., GFP-agarose beads) showed significant enrichment when the nanobody, mMobA, DCV, and SNAP tag were inserted into VP1u (residues 1-160; Figures 4 and S9). Both observations can only be true if these VP1 variants are incorporated in the capsid such that the inserted motif is accessible on the capsid exterior. While speculative, a likely explanation is that the inserted motif forces VP1u to remain external, thus removing a steric barrier to genome packaging, which would result in increased recovery of this variants after both fitness assay selections. FLAG tag insertions into VP1u, which, because of their small size, presumably did not interfere with internalization, did not increase pull-down fitness. Neither did WDV insertions, which is consistent with their deleterious impact on packaging fitness. Overall binding fitness was neutral (peaked around wild-type fitness; Figures 2 and S10), which was expected as most VP subunits (VP2 and VP3) contain the wild-type determinants of proteoglycan and AAVR binding.21,64 However, we observed a notable drop for binding fitness for VP1u insertions of motifs that package well (e.g., nanobody; Pearson correlation coefficient −0.641), possibly due to steric hindrance of virus binding when it carries large external motifs. The only motifs that were not impaired for binding upon VP1u insertions are the FLAG tag, which can be explained by its small size, as well as SpyCatcher and WDV, which are motifs that our pull-down data suggest were not compatible with virus packaging when inserted into VP1u.
Figure 3.

Packaging fitness of AAV domain insertion libraries mapped to the capsid structure
(A) Top left corner: AAV-DJ capsid structure view from the inside (left) and outside (right) radially color cued. The 2-, 3-, and 5-fold axes are indicated. VP1u domain was modeled using RoseTTAFold98 and manually positioned. All other structures show packaging fitness heatmaps of the indicated domain insertions. Green indicates higher and magenta lower fitness than AAV-DJ (RCSB PDB: 7KFR). (B) Zoom of the outside structures from (A). The 2-, 3-, and 5-fold axes are outlined.
Figure 4.

Pull-down fitness of AAV domain insertion libraries mapped to the capsid structure
(A) Top left corner: AAV-DJ capsid structure view from the inside (left) and outside (right) radially color cued. The 2-, 3-, and 5-fold axes are indicated. VP1u domain was modeled using RoseTTAFold98 and manually positioned. All other structures show pull-down fitness heatmaps of the indicated domain insertions. Green indicates higher and magenta lower fitness than AAV-DJ (white) (RCSB PDB: 7KFR). (B) Zoom of the outside structures from (A). The 2-, 3-, and 5-fold axes are outlined.
Consistent with the role and required timing of VP1u in virus trafficking upon endocytosis, uptake fitness (our assay measures presence of viral genome in any compartment inside the cell65) was impaired for all motifs inserted into the N terminus of VP1 (Figures 2 and S11). It has previously been demonstrated that premature exposition of VP1u decreases infectivity,66 meaning that variants with (as we speculate) pre-externalized VP1u escape less efficiently from the endosome and are degraded in the lysosomal compartment.22 We observed the same for infectivity fitness, with the notable exceptions of FLAG and WDV.
Effects of domain insertions near AAV’s 3-fold axis
Insertion into protrusion near the 3-fold axis (residues 420–620; including VR4–8) generally impaired virus packaging (Figures 2 and 3), which is consistent with the highly interdigitated structure of this region and its role in early assembly of VP trimers that are then forwarded to the nucleus as capsid building blocks.9,10,27 VP1 with insertions in this region may interfere with efficient trimer assembly (through a kinetic mechanism or by promoting off-pathway products), which would lower the overall capsid assembly efficiency. Nevertheless, several lines of evidence suggested that there appears to be some plasticity in trimer assembly to accommodate VP1 insertion variants so that these trimers can be incorporated into assembling capsid. For one, the overall hit to packaging fitness depended on the specific inserted motif. As expected, we saw that FLAG peptide insertions were relatively benign, but so were insertions of two HUH tags (DCV and WDV) and SpyCatcher (Figures 2 and 3). There was no clear correlation with motif size, hinting at more complex determinants for insertion fitness in this region. Second, despite impaired packaging fitness, pull-down fitness was greater than wild type for all motifs, suggesting that they can become incorporated into purified AAV capsids, albeit at a lower overall efficiency (Figures 2 and 4).
Similar to what we observed for insertions into the N terminus of VP1, binding fitness was not impaired for insertions in the 3-fold protrusion (Figures 2 and S10). In fact, the three HUH tags showed increased binding, which could be due to their generally higher cationic surface charge (Figure S1B) aiding interaction with negatively charged components of the extracellular matrix, such as proteoglycans. Consistent with higher surface binding, we found higher uptake fitness than wild-type AAV-DJ in the case of DCV and WDV when inserted into VR5 or VR8 (Figures 2 and S11). We note that binding and uptake fitness measured in this high-throughput assay for insertion of mMobA into VR4 match our results from our previous engineering of this region.38 Despite higher binding and uptake, none of the insertions in this region could achieve wild-type infection efficiency (Figure 2), suggesting that domain insertion affected later steps of virus trafficking to the nucleus.
Effects of domain insertions near AAV’s 2-fold valleys
The neighborhood near the 2-fold symmetry center, which includes VR9, emerged as another region with distinct motif-specific phenotypes. This is consistent with earlier studies showing that dynamics of the 2-fold regions are essential for genome packaging67 and AAV infectivity.68 Here, we observed both strongly deleterious fitness (e.g., SNAP tag) and strongly beneficial fitness (SpyCatcher and WDV). In all cases, pull-down fitness was positive (Figure 4), suggesting some degree of VP1 variant incorporation in this region. Interestingly, binding, uptake, and infectivity were impaired for most motifs.
Unbiased clustering of insertion fitness reveals contiguous functional units in AAV capsids
Taking our fitness measurements across all motifs, all insertion positions, and all measured phenotypes in aggregate, we noticed patterns in fitness variance that appear correlated in contiguous regions of the AAV capsid. For example, packaging fitness varied predominantly in the PLA2 domain of VP1u and the 2-fold symmetry axis (Figure 5A). Focusing on residues unique to the 2-fold interface, we found that packaging fitness distributions of interface and non-interface residues were not significantly different when DCV or FLAG peptide were inserted (Figure 5C). However, fitness was significantly improved for mMobA, SpyCatcher, or WDV insertions into interface residues. SNAP tag insertions were strongly deleterious. The variance at the 3-fold symmetry axis was markedly different. As described above, all motifs except WDV lowered packaging fitness, which resulted in lower overall fitness variance at this interface (Figure 5D). Remarkably, uptake fitness variance was greatest in the protrusion around the 3-fold axis and still considerably high along the 2-fold axis (Figure 5B). For all measured phenotypes, variance was relatively low around the 5-fold symmetry axis (Figure S12).
Figure 5.

Variance of packaging and uptake fitness
(A and B) Variance of packaging fitness (A) and uptake fitness (B) from all domain insertion libraries mapped to the AAV capsid structure. The capsid inside (top) and the capsid outside (bottom) are shown (RCSB PDB: 7KFR). (C and D) Empirical cumulative density insertional fitness of residues within (petrol green) and outside (red) the 2-fold axis (C) and 3-fold axis (D). Significance of distribution differences was tested using a two-sided, two-sample Kolmogorov-Smirnov test. Significance level and p values are shown. (E) Venn diagram showing which of the 335 AAV interface residues are unique and shared among interfaces.
If we think of different inserted motifs as different degrees of perturbation (e.g., weak for FLAG peptide insertion, strong for large nanobody insertion), then the positional and domain-type dependence of insertion permissibility we observed in our data suggests that we were measuring spatially resolved information of how different aspects of AAV fitness responds to these different degrees of perturbation. To link insertion permissibility phenotypes to mechanistic structure/function relationships, we used an unbiased clustering approach (Uniform Manifold Approximation and Projection [UMAP]).69 This resulted in five robust clusters that map to regions of the capsid with distinct roles in AAV biology (Figure 6A). Importantly, these clusters mapped to structurally contiguous (not interspersed) regions of the AAV capsid (Figures 6B and 6C). Cluster 1 contains VP1u in addition to residues lining the bases of the 3-fold and 5-fold axes. Cluster 2 forms an extended network that comprises the HI loop and connects to the 3-fold axis protrusions. Cluster 3 represents protrusion at the 3-fold axis and residues on the external turns of the DE loop that line the pore at the 5-fold symmetry axis. Cluster 4 maps to the depression near the 2-fold symmetry axis and buried regions, which are part of the 3-fold axis. Cluster 5 predominantly maps to residues that line the capsid interior and the 5-fold pore, or that interdigitate HI loop, external residues of the 2-fold valley, and 3-fold protrusions (Figure 6B).
Figure 6.

Unbiased clustering of insertion fitness
(A) UMAP cluster analysis of the AAV domain insertional profiling data resulting in five distinct clusters. (B) Cluster map to distinct capsid regions: (1) N terminus of VP1 and bases of the 3-fold and 5-fold interface in red; (2) the HI loop, the pore and the inner connecting residue layer in yellow; (3) protrusions of the 3-fold interface in green; (4) 2-fold axis in blue; and (5) the surrounding of the 5-fold pore in light lilac. (C) Mean domain insertion fitness of packaging, pull-down, binding, and uptake shown by residues aligned to the UMAP clusters in (A). (D) Distribution of insertion fitness of packaging, pull-down, and uptake for each cluster.
To understand the underlying mechanisms that drive clustering, we segregated fitness phenotype distributions by cluster identity (Figure 6D). Considering packaging fitness, we found that insertion into two clusters (1 and 5; containing VP1u and the network of residues that connect HI loop to the base of the 3-fold protrusion) were associated with improved packaging fitness, while clusters 3 and 4, which represent the interdigitated external region 3-fold and 2-fold axes, were associated with poor packaging fitness. Insertions into capsid-lining regions (cluster 2) were neutral with respect to packaging. With different measured phenotypes, these association patterns change: considering pull-down fitness, we found that clusters representing buried residues or those lining the capsid interior (i.e., clusters 1, 2, and 5) were associated with poor fitness compared to those in externally accessible regions (cluster 3 and 4). For uptake, cluster 1 (which contains VP1u) had the worst fitness and cluster 3 (the 3-fold protrusion) was closest to wild-type fitness.
Disulfide crosslinking to probe conformational flexibility
Conformational dynamics between different capsid regions plays a central role in AAV structure and function, including packaging and infectivity.9,24,26,67 Thus, clusters may emerge from our data because they represent units of correlated conformational plasticity that are also correlated in the way they respond to a perturbation (i.e., domain insertion). We therefore would expect that changing inter-cluster dynamics affect packaging and/or infectivity more than intra-cluster changes. One way to test this idea is by replacing two proximal residues by cysteines, such that a cystine disulfide link is potentially formed once AAV is exposed to an oxidizing environment (i.e., after release from producer cells). If the two mutated residues that undergo a cystine disulfide link are part of the same cluster of correlated conformational plasticity, and assuming that the individual cysteine substitutions are benign, we expect a less significant effect on packaging and/or infectivity compared to when the two residues belong to different clusters. Our choices of residue pairs are summarized in Figure S13A. Unlike domain insertions, which were only done in VP1, cysteine substitutions were introduced into all VPs. All single and double mutants produced near to wild-type titers (Figure S13A; one-way ANOVA not significant [NS]; Dunnett’s test with wild-type AAV-DJ as control, NS).
Some single cysteine mutants had dominant deleterious effects on infectivity (e.g., W608C, H292C). For double mutants in the “within cluster” set that had wild-type single-mutant infectivity, infectivity was comparable to wild-type (e.g., H625C/Y426C; Figure S13B), suggesting the minimal disruption of conformational dynamics. For pairs that belong to different clusters, all but one (H643C/Y350C) showed effects on infectivity that differed from what was predicted based on the individual single mutants (Figure S13B; see the Note S2 for a description of individual variants). While disulfide bond formation is also influenced by the protein structure and surrounding amino acids and we are not testing an exhaustive set of potential crosslinks, our results support the idea that at least the variation of infectivity fitness that contributes to cluster identity has a basis in different conformational dynamics.
Engineerable hotspots near the 2-fold axis and in the HI loop
We previously used the HUH tag mMobA in AAV-DJ VR4 to covalently link targeting scaffolds to the AAV capsid, which redirected AAV tropism in vitro.38 Here, we interrogated the entire AAV capsid for suitable HUH tag insertion hotspots. Comparing fitness maps for all three HUH tags, there were many differences among tags, which is likely related to their different biophysical properties (Figure S1). We noticed that WDV was remarkably different from all other inserted domains in several regards. For one, packaging fitness was improved over wild-type when this domain was inserted into HI loops or along the 2-fold axis (Figure 3). Binding fitness was generally strong, but insertions into the 2-fold axis were deleterious (Figure S10); this was the only insertion type for which we saw a deleterious phenotype for this assay. For uptake fitness, we observed a strong segregation in fitness between 3-fold protrusion and 2-fold and 5-fold axes (Figure S11). Given that most previous studies have investigated VRs in the 3-fold protrusion for capsid engineering, we turned our attention to insertion sites near the 2-fold and 5-fold axes that had near wild-type packaging fitness in the NGS-based assay. We produced 10 VP1 WDV insertion variants individually as crude cell lysates and measured titers, which all were comparable to wild-type (Figure 7A, one-way ANOVA NS). Testing each crudely enriched variant for the ability to infect HEK293FT cells, we found that several WDV variants inserted into surface exposed sites retained infection potency (Figure 7B). Among those were insertions into the 3-fold protrusions (S268), DE loop of the 5-fold pore (T331), HI loop (N664), and two sites along the 2-fold axis (Y702, K708). All sites had positive fitness in the NGS-based pull-down assay, suggesting the WDV-VP1 does become incorporated into AAV capsid (Figures 2 and 4). For two variants at particularly non-intuitive regions, namely N664 in the HI loop and K708 at the 2-fold axis (Figure 7C), we produced iodixanol-gradient-purified virus, which both trended to produce at higher titer compared to wild-type (Figure 7D). All variants incorporated VP1 as confirmed by western blot (Figure S14), albeit at levels that varied for N664 but not K708 (one-way ANOVA for VP1 content, p value 0.00993; Dunnett’s test for pairwise comparison with wild-type AAV-DJ as control, N664 p value 0.0074; K708 p value 0.4755). As we have previously shown,38 HUH tags mediate the attachment of ssDNA antibodies to AAV, which in turn increased infectivity in cells that express, on the cell surface, the antigen recognized by the antibody. Using surface-expressed GFP (GFP-GPI) as a test case, we tested infectivity of the two purified WDV variants and wild-type AAV-DJ with and without conjugation to an ssDNA-linked anti-GFP antibody. Note that expression of GFP-GPI alone reduced cell health, likely related to endoplasmic reticulum (ER) stress. Although infectivity of WDV variants was unaffected by conjugation to anti-GFP just like for wild-type AAV-DJ (expected as it was a mock conjugation since it does not contain an HUH tag), we saw a boost to infectivity for both N664-WDV and K708-WDV upon co-expression of surface-expressed GFP (Figure 7E), suggesting that anti-GFP became conjugated to WDV and then enhanced infectivity by directing AAV toward surface-expressed GFP as a binding receptor (two-way mixed measures ANOVA p value 0.0063).
Figure 7.

Fitness of WDV insertion variants
(A) Quantification of packaging titer of AAV-DJ and 10 different WDV insertion variants via qPCR. Data are means ± SEM. (B) Infection fitness of the WDV insertion variants in (A) quantified by measuring the percentage of tdTomato-positive cells 48 h post transduction at the MOIs of 1 × 102, 1 × 103, 5 × 103 and 4 × 104 vg/cell. Data are mean ± SEM. (C) Zoom to the 2-fold and 5-fold axes (outlined) of the capsid surface. Positions of N664 and K708 are shown as red spheres. (D) Quantification of packaging titers of AAV-DJ and WDV insertion variants N664 and K708. Data are mean ± SEM. (E) Infection fitness of the WDV insertion variants N644 and K708 quantified by measuring the percentage of tdTomato-positive cells 48 h post transduction at an MOI of 1 × 104 vg/cell. If indicated, cells were co-expressing GFP-GPI and/or an ssDNA-anti-GFP antibody was added. Data shown as boxplots. Lower and upper hinges of boxes indicate 25th and 75th percentile, respectively. Mean is indicated by a horizontal bar in each box. Whiskers extend 1.5 × IQR. Two-way mixed measures ANOVA was used to test the significance of variance between means (AAV-DJ, N664, K708) for conditions with and without GFP present. Presence of the monoclonal antibody (mAb) was a significant source of variation when GFP-GPI was expressed (p value 0.0063) but not without (p value 0.514). Significance levels and p values for pairwise comparisons using a Bonferroni correction are shown.
Discussion
Viral vectors are an essential component for gene delivery in therapies treating inherited disorders and cancer. AAV is widely used in both approved therapies and in ongoing clinical trials because of its good safety in humans and ability to drive long-term expression in both dividing and non-dividing cells. Unfortunately, several evolved properties of AAV are mismatched to clinical needs (e.g., broad tropism, limited payload capacity, existing human serum-immunity) or they pose biomanufacturing challenges (e.g., scale-up of helper virus-free production, yield of full virions to maximize potency).
Motivated by these challenges, there have been extensive efforts to re-engineer AAV properties in the past, including directed-evolution approaches, such as repeated mutagenesis, capsid shuffling,70 viral display of short peptides,49,50,51 and adding larger, structured targeting scaffolds, such as antibodies, nanobodies, DARPins, or affibodies.36,37,38,39,71 Most of these studies have focused on regions in the 3-fold protrusion, commonly VR8, VR4, or VP termini. Recently, deep mutagenesis of the entire capsid protein, combined with machine learning,8,42,72 demonstrated that learned sequence and function relationships can aid the prediction of sequence variation to improve desired AAV traits. For example, Ogden et al. performed a comprehensive single-amino-acid substitution, insertion, and deletion screen.8 They identified a higher tolerance in the VRs and particularly in the 3-fold protrusions. While comprehensive, this study focused on single-amino-acid changes. Based on prior work in other protein systems,44,45 we hypothesized that the domain insertion permissibility differs substantially considering the larger size, structure/folding, and polarity of entire domains.
We thus combined the concept of deep mutagenesis with scaffold insertion to systematically measure the fitness of AAV containing VP1 with seven different motifs inserted in between every two residues. This comprehensive analysis quantitatively links where different structured motifs can be inserted into VP1 to retain compatibility with AAV virion packaging, cell binding, and uptake. Note that fitness measurements are derived from AAV capsids with a variable copy number of VP1 located at random faces, which could affect phenotype penetrance; e.g., a higher number of VP1 proteins might lower the packaging fitness due to steric hinderance of genome packaging but could increase the cell type-specific infection potency due to improved receptor binding mediated by the inserted domain or large number of PLA2 domains. The fitness profiles show that there is a strong dependence on insertion position and type of inserted motif, with several regions showing diverging fitness for different measured phenotypes (e.g., VP1u for packaging vs. cell uptake; Figure 2). This highlights that the outcome of sequence variation (encompassing single-residue changes to large-scale insertions) can have multi-facetted impact on AAV properties, and it calls for integration of assays across several clinically relevant AAV attributes to safeguard against inadvertent optimization for undesired traits. In recent years, we have gained broad access to precision variant library engineering,46,73,74 NextGen sequencing (enabling counting number of sequence variants in a highly diverse population before and after applying a test for fitness59), unified analytical frameworks to interpret these large dataset,75 and machine learning approaches for deciphering sequence/function relationships.76 Taken together, generating and interrogating large AAV variant libraries has become feasible. Our focus can now shift toward what these datasets tell us about AAV biology and how they can guide and accelerate viral vector engineering.
For example, while fitness of many insertion sites is consistent with known AAV structure and function (e.g., the importance of trimer assembly along 3-fold axis in capsid assembly9), the high packaging fitness when motifs are inserted into the PLA2 domain of VP1u was surprising. Several studies have elucidated VP1u and VP2 dynamics as part of events after virus uptake by the cell, in which the externalization of VP1u PLA2 is a required step for endosomal escape.28,48 VP1u internalization during maturation of AAV virions is less well understood but appears to be coordinated with genome packaging.67 The existence of such mechanism would reconcile recently proposed models of stochastic assembly11 (which should result in both internalized and externalized VP1u) and the well-known requirement for heating or pH lowering to induce VP1u externalization before it can be detected by VP1-specific antibodies.24,26,77,78 It is plausible that domain insertions interfere with this process, leaving VP1u externalized, which would leave more room inside the AAV capsid for genome packaging by Rep52/40 due to lack of steric hindrance (one VP1 occupies 85 × 103 Å3 or one-thirty-fifth of the available space inside the capsid), but at the loss of infectivity. Interestingly, VP1u and VP2 of the related parvovirus B19V have acquired a receptor-binding domain insertion just upstream of PLA2 and are always external,79,80,81,82 supporting the idea that addition of extra domains into VP1u leaves it externalized.
Given that precise timing of VP1u externalization in the correct endosomal compartment is required for maximum infectivity (premature externalization reduced infectivity66), this may point to an evolutionary mechanism that balances virion packaging efficiency with infectivity. If the full/empty ratio is fundamentally constrained by a packaging/infectivity balance, this would have implications for biomanufacturing of AAV in which one of the major ongoing efforts is to find ways to enrich full particles. One prediction of our hypothesis is that full particles may contain fewer VP1, on average, compared to empty particles, and this is negatively affecting infection potency. The observation that overexpression of VP1 inhibits rAAV packaging is consistent with this idea.83 There may exist a Goldilocks regimen, just the right copy number of VP1, that maximizes both genome content and endosomal escape. Further research is required to fully test this hypothesis, including measuring VP stoichiometry and genome content at the single-capsid level.
Unbiased clustering of insertion fitness across several phenotypes also revealed a topological organization of AAV into regions that can be linked to correlates in AAV capsid assembly, genome packaging, and infectivity. Given that many of these roles have a basis in distinct regimes of capsid stability and dynamics, we hypothesize that inserting different domains (which represent different degrees of perturbation) probes conformational plasticity at or near the insertion site. Put simply, it probes whether the insertion site is conformationally rigid (allowing no insertions) or conformationally flexible (allowing some or all insertions). Clusters emerge because conformational plasticity of different capsid regions impinges differently with different measured phenotypes (e.g., capsid flexing required for efficient genome packaging67 vs. flexing during externalization of VP1u/PLA2 after cell uptake, which only happens in full, but not empty, capsids24).
Similar ideas of spatially contiguous protein regions linked to specific functions have been proposed in the past, including protein “sectors” mapped through measuring amino acid co-evolution,84,85 regional conformational flexibility mapped by circular permutation profiling86 and domain insertion,44,45,73 or revealing the functional architecture of an enzyme from high-throughput enzyme variant kinetics.87 At their core, all these approaches use mutations to perturb sequence/function relationships. Similarly, by perturbing VP1 through domain insertion and measuring how it responds (in terms of packaging, infectivity, etc.), we learn how AAV structure intersects with AAV function. Going forward, domain insertional profiling in the background of different genetic backgrounds (i.e., serotypes) may further separate general principles of AAV packaging, function, and serotype-specific properties (e.g., stability, immune evasion, tropism). As we have done here, these should focus on alterations of VP1 or VP2, which have a significantly lower copy number in the assembled capsid and are more likely to be benign compared to VP3 or all VP modifications.
Our systematic domain insertion approach also revealed additional opportunities for viral engineering. Following the intuition that engineering non-conserved, surface-exposed, and tropism-determining loops is the likeliest path to change AAV properties, much of AAV engineering so far has focused on N termini of capsid proteins or variable loops of the 3-fold protrusions.36,38,40,71,88,89,90,91,92 However, comprehensive studies we and others conducted45,86 suggest that there is significantly more to explore. We found that permissibility to domain insertion is not correlated with conservation, surface exposure, or other static, structural features. Instead, dynamic features, such as regional flexibility, are predictive as to where a domain insertion is tolerated. In this study, we identified two regions near the 2-fold and 5-fold axes that can tolerate the insertion of HUH tags (15 kDa), which in turn enable the covalent linkage of antibodies (150 kDa). In a proof of principle, we found that insertions have little impact on production levels and provide a modest boost to infecting cells that express the antibody’s cognate antigen. Further research and engineering are required to fully leverage the potential of these engineerable hotspots. They represent an exciting opportunity to sidestep the constraint of directed evolution of targeting the region near the 3-fold axis. As this region is important for receptor binding, it is also the most antigenic region. In fact, the binding sites for several proteoglycans, AAVR, and neutralizing antibodies overlap.18,19,21,93 Approaches that shuffle the sequence of this region must apply selection pressure to selectively remove antibody binding while retaining the mode of cell binding and uptake. By providing an alternative site to which a targeting scaffold can be linked, it may be possible to address this challenge more effectively.
Materials and methods
Cloning and library generation
All plasmids and libraries used in this study are listed in Table S1 and were generated either by classical restriction enzyme cloning or Golden Gate Assembly.94 Restriction enzymes were obtained from NEB; standard oligos, labeled oligos, as well as gBlocks from IDT; and oligo pools for library cloning from Agilent Technologies. For amplification of DNA sequences for cloning and NGS, the PrimeSTAR Max DNA polymerase (Takara Bio) and for colony PCRs the OneTaq Quick-Load Master Mix Polymerase was used (NEB). PCR products were analyzed on 1% TAE agarose gels, cut out and purified using the Zymoclean Gel DNA Extraction Kit (Zymo Research) by following the manufacturer’s instructions. Post cloning, plasmids were transformed into NEB Stable Competent Escherichia coli cells and libraries into MegaX DH10B T1R Electrocomp Cells (Thermo Fisher), before plated on LB plates containing either carbenicillin (100 μg/mL) alone or a combination with chloramphenicol (25 μg/mL) depending on the selection marker(s) on the plasmids and libraries. To assess coverage of libraries, a small amount from the transformed cells was taken, and serial dilutions were prepared and plated on LB plates with the respective selection marker(s). The next day, colonies were counted to estimate coverage and colony PCRs were run to verify library diversity. Plasmids were isolated using the Zyppy Plasmid Miniprep Kit, ZymoPURE II Midiprep Kit, or the ZymoPURE II Maxiprep Kit (all Zymo Research).
For the generation of AAV-DJ insertion libraries, an altered cap-DJ gene sequence was used with mutated start sites for VP2 (T138A) and VP3 (M203K, M211L, and M235L), a T176A mutation eliminating the BsmBI cutting site, and a replacement of the HBD domain (R587-R590) by an HA tag (AYPYDVPDYAA). The libraries were created using SPINE.46 In brief, the cap DJ-VP1 sequence was split up into 14 fragments, and oligos containing a genetic handle behind every amino acid position were designed. Every oligo contained barcodes for amplification, matching BsmBI restriction sites to assemble the 14 fragments, and BsaI restriction sites to swap out the handle. The genetic handle was first replaced by a chloramphenicol expression cassette flanked by BsmBI cutting sites to insert a selection marker for library presence. Next, the library was transferred into an AAV plasmid backbone encoding an elongation factor-1 alpha short (EFS)-driven miRFP670nano sequence terminated by an SV40-polyA, a p40 promoter with BsmBI sites for the library insertion and flanked by ITRs. Last, the chloramphenicol was replaced by different domains (nanobody, SpyCatcher, SNAP, mMobA, WDV, or DCV) or a FLAG tag. While the domains had 5-aa SGGGG-domain-GGGGS linkers, the FLAG tag was flanked by short SG-FLAG-GS linkers only. The DJ-VP1 silent mutation plasmid, which was used as a reference, was designed by introducing 10 silent mutations into the VP1-only DNA sequence of AAV-DJ. To this end, codons for either arginine, serine, or leucine were altered by two nucleotides each at positions that were 200–300 bp apart from each other. The silent mutation VP1 coding sequence was ordered as a gBlock and cloned into the same backbone as the plasmid libraries, i.e., miRFP670nano expression cassette and a p40 promoter to drive VP1 expression and flanked by ITRs. Cysteine point mutations were introduced by site-directed mutagenesis of the wild-type rep2-capDJ plasmid. WDV insertion variants were generated by inserting the domain into the above-mentioned p40-driven and altered AAV-DJ-VP1-only sequence by Golden Gate Cloning.
Tissue culture
HEK293FT cells (Invitrogen) and 293AAV cells (Cell Biolabs) were maintained in DMEM (Gibco) containing 4.5 g/L D-glucose, L-glutamine, 110 mg/L sodium pyruvate, and supplemented with 10% fetal bovine serum (FBS) (Gibco) and 100 U per mL penicillin/100 μg per mL streptomycin (Gibco). Cells were kept in a humidified cell culture incubator at 5% CO2 and 37°C and passaged every 2–3 days when reaching 70%–90% confluency. For experiments with HEK293FT cells, plates were pre-coated with growth factor-reduced basement membrane matrix Matrigel (Corning) prior to seeding. If applicable, HEK293FT cells were transfected with a plasmid encoding GFP-GPI using Turbofect (Invitrogen) while seeding and according to the manufacturer’s protocol. The amounts of DNA used for transfection are further specified in the sections of the different assays. For AAV productions, 293AAV cells were used only until reaching passage 10.
AAV crude lysate production
293AAV cells were seeded into six-well plates at a density of 500,000 cells per well. The next day, cells were transfected with 2.5 μg of DNA using PEI and an equimolar ratio of the plasmids necessary for the respective AAV production. Three days post transfection, cells were harvested by flushing off the cells by pipetting. Cells were washed with PBS once and then subjected to five freeze and thaw cycles by alternating between liquid nitrogen and a 37°C water bath. Cell debris was pelleted by centrifugation at 18,000 rpm at 4°C for 10 min and the supernatant containing the AAV particles was stored at −20°C until use.
Purified AAV production
Large-scale productions of AAV were either done by the University of Minnesota Viral Vector and Cloning Core using a sucrose gradient or in lab following published iodixanol gradient density protocols.95,96 In brief, 4 million 293AAV cells were seeded into 15-cm dishes and transfected using PEI and 47 μg of total DNA per dish 48 h post seeding. For the AAV-DJ library control, an equimolar triple transfection was used composed of an Adeno-helper plasmid, a plasmid encoding the rep2 and capDJ genes, and a transgene plasmid encoding an EFS promoter-driven miRFP670nano sequence flanked by ITRs. For AAV-DJ insertion library productions, a plasmid ratio of 1:0.1:0.1 of an adeno-helper plasmid, a plasmid encoding rep2 and only VP1 of cap-DJ, as well as the respective AAV-DJ insertion library in which the AAV-DJ silent mutation variant was spiked in was transfected. To top up to 47 μg of total DNA, a pUC19 stuffer plasmid was added. At 72 h post transfection, cells were detached with a cell lifter and cells pelleted by centrifugation at 400 × g for 15 min. The cell pellet was washed once with PBS and resuspended in a buffer containing 2 mM MgCl2, 0.15 M NaCl, and 50 mM Tris-HCl at pH 8.5. Cells were cracked open using five freeze and thaw cycles. Free genomic and plasmid DNA was digested with a Benzonase Nuclease (Sigma-Aldrich). Cell debris was removed by centrifugation and, subsequently, the lysate transferred into ultracentrifugation tubes (Beckman Coulter). The iodixanol discontinuous gradient (15%, 25%, 40%, and 60% iodixanol concentration) was layered underneath the cell lysate. Density gradient centrifugation was done at 50,000 rpm for 2 h at 4°C using a 70.1 Ti rotor (Beckman Coulter). Post centrifugation, the 40% iodixanol phase containing the AAV was isolated, aliquoted, and stored at −80°C until use. For pull-down, binding, and uptake, as well as the differential scanning fluorimetry (DSF) assays, AAV samples were dialyzed to PBS supplemented with 5% glycerol using 10-kDa Amicon Ultra-15 Centrifugal Filter Units (Millipore, Sigma). Certain assays required preparation of purified virus: for the pull-down assay, we observed an increased stickiness to the beads in iodixanol preps compared to preps in PBS. For the binding and internalization assay, we decided to use PBS as buffer, because we think that the iodixanol slows down the settlement of AAV particles and consequently the attachment of the particles to the cells. For the binding and internalization assays, the settle time matters as the AAV particles are only incubated for 1 h at 4°C before unbound particles are washed away. For the DSF, a fluorescent dye is used and the iodixanol quenches the fluorescent signal.
qPCR
To determine the titer of crude lysate AAV samples, 1–5 μL of the crude lysate were mixed with PBS supplemented with 2 mM MgCl2 to a final volume of 50 μL. Then, 0.1 μL of ultrapure Benzonase Nuclease (Sigma-Aldrich) was added. Samples were incubated for 30 min at 37°C to digest non-encapsidated DNA. Next, 5 μL of a 10× Proteinase K buffer (100 nM Tris-HCl, pH 8.0, 10 mM EDTA, and 10% SDS) and 1 μL of Proteinase K (20 mg/mL; Zymo Research) were added to stop the DNA digest and start the protein digest to free the ssDNA from the AAV particles. Samples were incubated for 20 min at 50°C, followed by a heat inactivation of the enzymes for 5 min at 95°C. The DNA was purified using the DNA Clean & Concentrator-5 kit (Zymo Research) according to the manufacturer’s instructions for ssDNA purification. For purified AAV samples, the Benzonase digest step was skipped and only the Proteinase K and DNA purification steps were performed. All samples were diluted 1:1,000 in H2O prior to qPCR. The viral genome (vg) quantification was done on a QuantStudio5 Real-Time PCR System (Applied Biosystems) using the PowerUp SYBR Green Master Mix (Applied Biosystems) and following the manufacturer’s instructions. To calculate the viral titer (vg/mL), a plasmid standard with a known concentration of plasmid copies was used. Primer sets binding either the cytomegalovirus (CMV) enhancer or the p40 promoter of the AAV genomes, as well as in the plasmid standard, were selected (Table S3).
Pull-down assay of libraries
Between 1 × 109 and 1 × 1010 vg were used as input material to bind to different magnetic beads for pull-down assays: SNAP-Capture Magnetic Beads (NEB) for SNAP tag insertion; Pierce Anti-DYKDDDDK Magnetic Agarose (Thermo Scientific) for FLAG tag insertion; and Streptavidin Magnetic Beads (NEB) for nanobody, SpyCatcher, and HUH tag insertions. The 80-μL bead slurry for SNAP pull-downs and 50-μL bead slurry for FLAG pull-downs were washed three times with 300 μL of wash buffer I (0.15 M NaCl, 20 mM Tris-HCl pH 7.5, 1 mM EDTA). Then, AAV libraries were mixed with wash buffer I and the beads to a final volume of 300 μL before incubated on a slow shaker for 30 min at room temperature. Beads were washed again three times with 300 μL of wash buffer I for SNAP beads and with PBS (pH 7.4, Gibco) for FLAG beads to remove unbound AAV particles and finally resuspended in 50 μL of PBS. For pull-down assays with streptavidin beads, 100 μL of bead slurry was washed three times with 300 μL of wash buffer II (0.15 M NaCl, 20 mM Tris-HCl pH 7.5, 1 mM EDTA). For nanobody and SpyCatcher binding, beads were pre-incubated with either 320 pmol of biotinylated superfolder-GFP or 1,000 pmol of biotinylated SpyTag, respectively, for 30 min on a slow shaker at room temperature in a total volume of 300 μL in wash buffer II. Afterward, unbound superfolder-GFP and SpyTag were removed by washing the beads three times with 300 μL of wash buffer II. For HUH tag pull-downs, AAV libraries were first reacted with 1 nmol of biotinylated ssDNA oligos (sequences are given in Table S3) in PBS supplemented with 0.05% v/v salmon sperm DNA (Invitrogen), 1 mM MgCl2, and 1 mM MnCl2 for 15 min at 37°C. Next, beads were incubated with AAV libraries for 30 min on a slow shaker at room temperature, before unbound AAV particles were removed by washing three times with 300 μL wash buffer II. Beads with bound AAV particles were resuspended in 50 μL of PBS. Viral genomes of samples after the pull-down were purified using the Quick-DNA Microprep Plus Kit (Zymo research) and by following the manufacturer’s protocol.
Binding and uptake assays of libraries
The binding and uptake assays were performed as previously described65 but using HEK293FT cells. In brief, 375,000 HEK293FT cells were seeded into six-well plates using 2 mL of culturing medium per well. The next day, cells were incubated for 30 min at 4°C. Afterward, the media was aspirated and 200 μL of cold DMEM containing AAV particles at an MOI of 1e4 vg/cell added. The cells were further incubated for 1 h at 4°C. Next, cells were washed three times with ice-cold PBS to eliminate unbound AAV particles. For the binding assay, 150 μL of ice-cold PBS was added, the cells detached with a cell scraper, and the cell suspension transferred to a microcentrifuge tube. For the uptake assay, 1 mL of pre-warmed culturing medium was added immediately after the PBS wash and the cells incubated for 2 h in a cell culture incubator to allow for uptake of the AAV particles. Then, the cells were detached by trypsinization and collected in a microcentrifuge tube before being washed three times with 200 μL of PBS. The DNA from binding and uptake samples was purified using the Quick-DNA Microprep Plus Kit (Zymo research) and by following the manufacturer’s protocol.
Infectivity assay of libraries
Here, 150,000 HEK293FT cells were seeded into 12-well plates using 1 mL of culturing medium per well. The next day, cells were transduced with purified AAV (in PBS supplemented with 5% glycerol) at an MOI of 2 × 105 vg/cell. To this end, the medium was aspirated and cells were washed with 500 μL of PBS once. Purified AAVs were mixed with DMEM without supplements to a final volume of 500 μL and added onto the cells. After 2 h of incubation in the cell culture incubator, 1.5 mL of culturing medium (with supplements) was added. At 24 h post transduction, the temperature was reduced to 33°C to promote protein expression rather than cell growth.97 At 72 h post transduction, cells were prepared for cell sorting as follows. Medium was aspirated, cells were washed with 500 μL of PBS, and 500 μL of Accutase solution (Sigma-Aldrich) was added and incubated at room temperature until all cells detached. The cell suspension was transferred to a microcentrifuge tube and centrifuged for 3 min at 400 × g to pellet cells. Cells were washed two times with 500 μL of PBS, before being resuspended in 650 μL of cell sorting buffer (PBS supplemented with 5 mM EDTA and 2.5% FBS) and passed through a 35-μm cell strainer to avoid cell clumps. Cell sorting was performed by the University of Minnesota Flow Cytometry Resource (UFCR) on a FACS Aria II instrument (BD Biosciences) with an 85-μm nozzle by sorting miRFP670nano positive (excitation 640-nm laser, emission 670/30-nm bandpass filter). Post sorting, the DNA was extracted from the cells using the Quick-DNA Microprep Plus Kit (Zymo research) and by following the manufacturer’s protocol.
NGS preparation
Purified DNA samples (library plasmid DNA, ssDNA from purified AAV, and DNA extracted after pull-down, binding, uptake, and infectivity assays) were amplified using primers binding 50 base pairs up- and downstream of the VP1 coding sequence (Table S3). For amplification, the PrimeSTAR Max DNA Polymerase (Takara Bio) was used according to the manufacturer’s recommendation with a 25-μL reaction volume, an annealing temperature of 62°C, and an elongation time of 15 s. At least five reactions were pooled for each DNA sample, whereas the cycle number was kept at a minimum to obtain >50 ng per sample. PCR products were purified using the DNA Clean & Concentrator-5 kit (Zymo Research) and by following the manufacturer’s instructions for dsDNA purification. The DNA was eluted in a 10 mM Tris buffer with pH 8.0. Prior to sequencing, the DNA was quantified using the Qubit 1× dsDNA HS assay kit and a Qubit 4 Fluorometer (both Invitrogen), and the DNA of the insertion libraries from the same assay were pooled in an equimolar ratio. Then, >50 ng of each DNA pool was submitted to the University of Minnesota Genomics Center, where Nextera XT libraries were created, and samples sequenced using a NovaSeq SPrime 150 paired-end run.
Sequencing data analysis and enrichment calculation
Forward and reverse reads were aligned individually using a DIP-seq pipeline,73 slightly modified for SPINE compatibility and for updated python packages. The code for handling data from domain insertion library sequencing is available at: https://github.com/SavageLab/dipseq. This pipeline results in.csv spreadsheets (available as processed data, along scripts to reproduce manuscript figures, which are available at https://github/com/Schmidt-lab/AAV_Insertion_Profiling), indicating insertion position, direction, and whether it was in frame. Fitness was calculated from the frequency of a given VP1 variant (i) after packaging (s) relative to the frequency of that variant in the input library (u), normalized to wild-type AAV (wt):
Fitness standard error for each variant was calculated assuming a Poisson distribution.
Mapping of fitness data onto the AAV capsid structure
Fitness data were mapped onto the structure of AAV-DJ (PDB: 7KFR) using Pymol. VP1u, which is not resolved in the cryoelectron microscopy (cryo-EM) structure of AAV-DJ, was modeled using RoseTTAFold.98 The highest-scored predicted VP1u structure was manually positioned in Pymol adjusting peptide-backbone bond dihedral angles to avoid clashes with the experimentally determined capsid structure.
Western blot
For VP protein analysis 3 × 109 to 1 × 1010 vg of purified AAV were mixed with 12.5 μL of 4× Laemmli Sample Buffer (Bio-Rad, supplemented with 10% 2-mercaptoethanol) and topped up to a final volume of 50 μL with PBS. Samples were denatured for 10 min at 95°C and afterward chilled on ice. Protein samples, and 5 μL of the Precision Plus Protein Dual Color Standard (Bio-Rad), were separated by molecular weight on a 7.5% precast polyacrylamide gel (Bio-Rad) in Tris/Glycine/SDS Electrophoresis Buffer (Bio-Rad) for 85 min at 120 V. Next, proteins were transferred to a nitrocellulose membrane (pore size 0.45 μm; Thermo Scientific) in an ice-cold blotting buffer (25 mM Tris base, 96 mM glycine, 20% methanol) for 80 min at 110 V. The membrane was washed once in TBS-T (20 mM Tris base, 137 mM NaCl, pH 7.6, 0.05% Tween 20) and incubated in 5% skim milk solution in TBS-T for 2 h at room temperature on a slow shaker to block non-specific binding. A primary antibody detecting either all three VP proteins (anti-AAV VP1/VP2/VP3 mouse monoclonal, B1, supernatant, Progen) or only VP1 (anti-AAV VP1 mouse monoclonal, A, lyophilized, purified, Progen) was diluted 1:250 in 5% skim milk solution in TBS-T and incubated overnight at 4°C. The next day, the membrane was washed four times for 5 min in TBS-T on a shaker before the secondary antibody was added (anti-mouse immunoglobulin [Ig] G-peroxidase antibody produced in goat, Sigma-Aldrich). The secondary antibody was diluted 1:50,000 in 5% skim milk solution in TBS-T and incubated for 2 h at room temperature on a slow shaker. Afterward, the membrane was washed again four times for 5 min in TBS-T at room temperature to remove unbound antibodies before the SuperSignal West Dura Extended Duration Substrate kit solution (Thermo Scientific) was applied and incubated for 2 min at room temperature. The chemiluminescence signal was detected with an Amersham Imager 600 (GE Healthcare) using exposure times between 1 s and 10 min, depending on the signal intensities. Quantification of VP expression was done using ImageJ.
DSF
First, 5,000× SYPRO Orange dye (Invitrogen) was diluted 1:100 in PBS with 5% glycerol. Then, each sample was prepared by mixing 50× SYPRO Orange dye 1:10 with >5 × 109 vg of purified AAV samples in PBS with 5% glycerol to a final volume of 25 or 50 μL. Samples were mixed by pipetting up and down and into a 0.1-mL MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems). The plate was sealed with an optical adhesive cover (Applied Biosystems) and spun down for 2 min at 1,000 × g. A melt-curve experiment was run on a QuantStudio5 Real-Time PCR instrument (Applied Biosystems) using the x1-m4 filter set (excitation filter, 470/15 nm; emission filter, 623/14nm) and the following settings: 30°C for 2 min, temperature increase from 30°C to 99°C in 0.5°C and 30-s increments, and a final incubation step of 2 min at 99°C. Lysozyme at a concentration of 0.1 mg/mL with a determined melting temperature of 70°C was used as a reference control within each run. Post-processing of the melt-curve data was done in MATLAB R2021a (The Mathworks). A smoothing spline (smoothing parameter p = 0.9) was fitted to the data before calculating the numerical gradient .
Transmission electron microscopy
To quantify the empty to full capsid ratio, negative staining and transmission electron microscopy was performed by the Characterization Facility, University of Minnesota. At least 300 AAV particles were manually counted per sample.
Infectivity assay of cysteine mutants and WDV variants
Here, 75,000 HEK293FT cells were seeded per well of a 24-well plate using 0.5 mL of culturing medium. If applicable, cells were transfected with 100 ng of GFP-GPI plasmid while seeding. The next day, medium was exchanged and cells transduced with crude lysates or purified AAV at the indicated MOIs. At 48 h post transduction, cells were prepared for flow cytometry as follows. Medium was aspirated, cells were washed with 500 μL of PBS, 250 μL Accutase solution (Sigma-Aldrich) was added, and they were incubated at room temperature until all cells detached. Cell suspension was transferred into a microcentrifuge tube and centrifuged for 3 min at 400 × g to pellet cells. Cells were washed two times with 300 μL of PBS before being resuspended in 600 μL of flow cytometry buffer (PBS supplemented with 5 mM EDTA and 2.5% FBS) and passed through a 35-μm cell strainer to avoid cell clumps. Flow cytometry was performed either on a LSRFortessa X-20 or a FACSymphony A3 Cell Analyzer (both BD Biosciences) equipped with 561- and 488-nm lasers to detect tdTomato- and GFP-positive cells, respectively. A minimum of 10,000 single cell events were recorded per sample. Data analysis was performed using the FlowJo 10.8.0 software (BD Biosciences).
Data and code availability
Library staging and accessory plasmids (including sequence information) are available upon request. Sequencing data generated in this study have been deposited in the Sequence Raw Archive (https://www.ncbi.nlm.nih.gov/sra) under accession code PRJNA950466; refer to for corresponding metadata and read statistics. The code for handling data from domain insertion library sequencing is available at https://github.com/SavageLab/dipseq. The SPINE code is available at https://github.com/schmidt-lab/SPINE. The version used for this study is archived at https://zenodo.org/badge/latestdoi/223953195. Processed read count data, along with R scripts to reproduce manuscript figures, are available at https://github.com/Schmidt-lab/AAV_Insertion_Profiling. A Shiny app to display 3D views of AAV insertion fitness maps in a web browser is available at https://github.com/Schmidt-lab/shinyAAViewerR.
Acknowledgments
The project described was supported by a Career Development Award from the American Society of Gene & Cell Therapy (M.D.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Society of Gene & Cell Therapy. This work was supported by the National Institutes of Health (R01GM141152 to D.S.) and (P30DA048742 to D.S.). Some AAV productions used in this study were generated by the University of Minnesota Viral Vector and Cloning Core (Minneapolis, MN). We thank the University of Minnesota Genomics Center for providing NGS service and technical support. This work was also supported by the resources and staff at the University of Minnesota University Imaging Centers (UIC). The UIC RRID is SCR_020997. Transmission electron microscopy was carried out in the Characterization Facility, University of Minnesota, which receives partial support from the NSF through the MRSEC (award number DMR-2011401) and the NNCI (award number ECCS-2025124) programs.
Author contributions
M.D.H., A.C.Z., and D.S. designed the study with input from W.R.G. and G.A. M.D.H., A.C.Z., D.N., and D.S. conducted the experiments with assistance from Y.H. M.D.H. and D.S. analyzed the data and authored the manuscript. All authors have given approval to the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.101143.
Contributor Information
Mareike D. Hoffmann, Email: hoff1570@umn.edu.
Daniel Schmidt, Email: schmida@umn.edu.
Supplemental information
References
- 1.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoy S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs. 2019;79:1255–1262. doi: 10.1007/s40265-019-01162-5. [DOI] [PubMed] [Google Scholar]
- 3.Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012;20:1831–1832. doi: 10.1038/mt.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 6.Jay F.T., Laughlin C.A., Carter B.J. Eukaryotic translational control: adeno-associated virus protein synthesis is affected by a mutation in the adenovirus DNA-binding protein. Proc. Natl. Acad. Sci. USA. 1981;78:2927–2931. doi: 10.1073/pnas.78.5.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonntag F., Schmidt K., Kleinschmidt J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA. 2010;107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogden P.J., Kelsic E.D., Sinai S., Church G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366:1139–1143. doi: 10.1126/science.aaw2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateu M.G. Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 2013;531:65–79. doi: 10.1016/j.abb.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 10.Medrano M., Fuertes M.Á., Valbuena A., Carrillo P.J.P., Rodríguez-Huete A., Mateu M.G. Imaging and Quantitation of a Succession of Transient Intermediates Reveal the Reversible Self-Assembly Pathway of a Simple Icosahedral Virus Capsid. J. Am. Chem. Soc. 2016;138:15385–15396. doi: 10.1021/jacs.6b07663. [DOI] [PubMed] [Google Scholar]
- 11.Wörner T.P., Bennett A., Habka S., Snijder J., Friese O., Powers T., Agbandje-McKenna M., Heck A.J.R. Adeno-associated virus capsid assembly is divergent and stochastic. Nat. Commun. 2021;12:1642. doi: 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buller R.M., Rose J.A. Characterization of adenovirus-associated virus-induced polypeptides in KB cells. J. Virol. 1978;25:331–338. doi: 10.1128/jvi.25.1.331-338.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson F.B., Ozer H.L., Hoggan M.D. Structural proteins of adenovirus-associated virus type 3. J. Virol. 1971;8:860–863. doi: 10.1128/jvi.8.6.860-863.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose J.A., Maizel J.V., Inman J.K., Shatkin A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971;8:766–770. doi: 10.1128/jvi.8.5.766-770.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snijder J., van de Waterbeemd M., Damoc E., Denisov E., Grinfeld D., Bennett A., Agbandje-McKenna M., Makarov A., Heck A.J.R. Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. J. Am. Chem. Soc. 2014;136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Q., Bu W., Bhatia S., Hare J., Somasundaram T., Azzi A., Chapman M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Govindasamy L., Padron E., McKenna R., Muzyczka N., Kaludov N., Chiorini J.A., Agbandje-McKenna M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 2006;80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerch T.F., O’Donnell J.K., Meyer N.L., Xie Q., Taylor K.A., Stagg S.M., Chapman M.S. Structure of AAV-DJ, a retargeted gene therapy vector: cryo-electron microscopy at 4.5 Å resolution. Structure. 2012;20:1310–1320. doi: 10.1016/j.str.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer N.L., Chapman M.S. Adeno-associated virus (AAV) cell entry: structural insights. Trends Microbiol. 2022;30:432–451. doi: 10.1016/j.tim.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillay S., Meyer N.L., Puschnik A.S., Davulcu O., Diep J., Ishikawa Y., Jae L.T., Wosen J.E., Nagamine C.M., Chapman M.S., Carette J.E. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer N.L., Hu G., Davulcu O., Xie Q., Noble A.J., Yoshioka C., Gingerich D.S., Trzynka A., David L., Stagg S.M., Chapman M.S. Structure of the gene therapy vector, adeno-associated virus with its cell receptor. Elife. 2019;8 doi: 10.7554/eLife.44707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riyad J.M., Weber T. Intracellular trafficking of adeno-associated virus (AAV) vectors: challenges and future directions. Gene Ther. 2021;28:683–696. doi: 10.1038/s41434-021-00243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bleker S., Sonntag F., Kleinschmidt J.A. Mutational analysis of narrow pores at the fivefold symmetry axes of adeno-associated virus type 2 capsids reveals a dual role in genome packaging and activation of phospholipase A2 activity. J. Virol. 2005;79:2528–2540. doi: 10.1128/JVI.79.4.2528-2540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kronenberg S., Böttcher B., von der Lieth C.W., Bleker S., Kleinschmidt J.A. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J. Virol. 2005;79:5296–5303. doi: 10.1128/JVI.79.9.5296-5303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nam H.J., Gurda B.L., McKenna R., Potter M., Byrne B., Salganik M., Muzyczka N., Agbandje-McKenna M. Structural studies of adeno-associated virus serotype 8 capsid transitions associated with endosomal trafficking. J. Virol. 2011;85:11791–11799. doi: 10.1128/JVI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkatakrishnan B., Yarbrough J., Domsic J., Bennett A., Bothner B., Kozyreva O.G., Samulski R.J., Muzyczka N., McKenna R., Agbandje-McKenna M. Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 2013;87:4974–4984. doi: 10.1128/JVI.02524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotmore S.F., Tattersall P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014;1:517–537. doi: 10.1146/annurev-virology-031413-085444. [DOI] [PubMed] [Google Scholar]
- 28.Zádori Z., Szelei J., Lacoste M.C., Li Y., Gariépy S., Raymond P., Allaire M., Nabi I.R., Tijssen P. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell. 2001;1:291–302. doi: 10.1016/s1534-5807(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 29.Grieger J.C., Snowdy S., Samulski R.J. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J. Virol. 2006;80:5199–5210. doi: 10.1128/JVI.02723-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zinn E., Pacouret S., Khaychuk V., Turunen H.T., Carvalho L.S., Andres-Mateos E., Shah S., Shelke R., Maurer A.C., Plovie E., et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015;12:1056–1068. doi: 10.1016/j.celrep.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalkara D., Byrne L.C., Klimczak R.R., Visel M., Yin L., Merigan W.H., Flannery J.G., Schaffer D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013;5 doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 34.Warrington K.H., Gorbatyuk O.S., Harrison J.K., Opie S.R., Zolotukhin S., Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004;78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asokan A., Johnson J.S., Li C., Samulski R.J. Bioluminescent virion shells: new tools for quantitation of AAV vector dynamics in cells and live animals. Gene Ther. 2008;15:1618–1622. doi: 10.1038/gt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Münch R.C., Janicki H., Völker I., Rasbach A., Hallek M., Büning H., Buchholz C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013;21:109–118. doi: 10.1038/mt.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eichhoff A.M., Börner K., Albrecht B., Schäfer W., Baum N., Haag F., Körbelin J., Trepel M., Braren I., Grimm D., et al. Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors. Mol. Ther. Methods Clin. Dev. 2019;15:211–220. doi: 10.1016/j.omtm.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zdechlik A.C., He Y., Aird E.J., Gordon W.R., Schmidt D. Programmable Assembly of Adeno-Associated Virus-Antibody Composites for Receptor-Mediated Gene Delivery. Bioconjugate Chem. 2020;31:1093–1106. doi: 10.1021/acs.bioconjchem.9b00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michels A., Frank A.M., Günther D.M., Mataei M., Börner K., Grimm D., Hartmann J., Buchholz C.J. Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8. Mol. Ther. Methods Clin. Dev. 2021;23:334–347. doi: 10.1016/j.omtm.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judd J., Wei F., Nguyen P.Q., Tartaglia L.J., Agbandje-McKenna M., Silberg J.J., Suh J. Random Insertion of mCherry Into VP3 Domain of Adeno-associated Virus Yields Fluorescent Capsids With no Loss of Infectivity. Mol. Ther. Nucleic Acids. 2012;1:e54. doi: 10.1038/mtna.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koerber J.T., Jang J.H., Yu J.H., Kane R.S., Schaffer D.V. Engineering adeno-associated virus for one-step purification via immobilized metal affinity chromatography. Hum. Gene Ther. 2007;18:367–378. doi: 10.1089/hum.2006.139. [DOI] [PubMed] [Google Scholar]
- 42.Bryant D.H., Bashir A., Sinai S., Jain N.K., Ogden P.J., Riley P.F., Church G.M., Colwell L.J., Kelsic E.D. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 2021;39:691–696. doi: 10.1038/s41587-020-00793-4. [DOI] [PubMed] [Google Scholar]
- 43.Ravindra Kumar S., Miles T.F., Chen X., Brown D., Dobreva T., Huang Q., Ding X., Luo Y., Einarsson P.H., Greenbaum A., et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods. 2020;17:541–550. doi: 10.1038/s41592-020-0799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coyote-Maestas W., He Y., Myers C.L., Schmidt D. Domain insertion permissibility-guided engineering of allostery in ion channels. Nat. Commun. 2019;10:290. doi: 10.1038/s41467-018-08171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coyote-Maestas W., Nedrud D., Suma A., He Y., Matreyek K.A., Fowler D.M., Carnevale V., Myers C.L., Schmidt D. Probing ion channel functional architecture and domain recombination compatibility by massively parallel domain insertion profiling. Nat. Commun. 2021;12:7114. doi: 10.1038/s41467-021-27342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coyote-Maestas W., Nedrud D., Okorafor S., He Y., Schmidt D. Targeted insertional mutagenesis libraries for deep domain insertion profiling. Nucleic Acids Res. 2020;48:1010. doi: 10.1093/nar/gkz1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bershtein S., Segal M., Bekerman R., Tokuriki N., Tawfik D.S. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 48.Girod A., Wobus C.E., Zádori Z., Ried M., Leike K., Tijssen P., Kleinschmidt J.A., Hallek M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 2002;83:973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 49.Perabo L., Büning H., Kofler D.M., Ried M.U., Girod A., Wendtner C.M., Enssle J., Hallek M. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol. Ther. 2003;8:151–157. doi: 10.1016/s1525-0016(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 50.Varadi K., Michelfelder S., Korff T., Hecker M., Trepel M., Katus H.A., Kleinschmidt J.A., Müller O.J. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Ther. 2012;19:800–809. doi: 10.1038/gt.2011.143. [DOI] [PubMed] [Google Scholar]
- 51.Waterkamp D.A., Müller O.J., Ying Y., Trepel M., Kleinschmidt J.A. Isolation of targeted AAV2 vectors from novel virus display libraries. J. Gene Med. 2006;8:1307–1319. doi: 10.1002/jgm.967. [DOI] [PubMed] [Google Scholar]
- 52.Rothbauer U., Zolghadr K., Tillib S., Nowak D., Schermelleh L., Gahl A., Backmann N., Conrath K., Muyldermans S., Cardoso M.C., Leonhardt H. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods. 2006;3:887–889. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- 53.Juillerat A., Gronemeyer T., Keppler A., Gendreizig S., Pick H., Vogel H., Johnsson K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 54.Zakeri B., Fierer J.O., Celik E., Chittock E.C., Schwarz-Linek U., Moy V.T., Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Everett B.A., Litzau L.A., Tompkins K., Shi K., Nelson A., Aihara H., Evans Iii R.L., Gordon W.R. Crystal structure of the Wheat dwarf virus Rep domain. Acta Crystallogr. F Struct. Biol. Commun. 2019;75:744–749. doi: 10.1107/S2053230X19015796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lovendahl K.N., Hayward A.N., Gordon W.R. Sequence-Directed Covalent Protein-DNA Linkages in a Single Step Using HUH-Tags. J. Am. Chem. Soc. 2017;139:7030–7035. doi: 10.1021/jacs.7b02572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smiley A.T., Tompkins K.J., Pawlak M.R., Krueger A.J., Evans R.L., Shi K., Aihara H., Gordon W.R. Watson-Crick Base-Pairing Requirements for ssDNA Recognition and Processing in Replication-Initiating HUH Endonucleases. mBio. 2023;14 doi: 10.1128/mbio.02587-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tompkins K.J., Houtti M., Litzau L.A., Aird E.J., Everett B.A., Nelson A.T., Pornschloegl L., Limón-Swanson L.K., Evans R.L., Evans K., et al. Molecular underpinnings of ssDNA specificity by Rep HUH-endonucleases and implications for HUH-tag multiplexing and engineering. Nucleic Acids Res. 2021;49:1046–1064. doi: 10.1093/nar/gkaa1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmit P.F., Pacouret S., Zinn E., Telford E., Nicolaou F., Broucque F., Andres-Mateos E., Xiao R., Penaud-Budloo M., Bouzelha M., et al. Cross-Packaging and Capsid Mosaic Formation in Multiplexed AAV Libraries. Mol. Ther. Methods Clin. Dev. 2020;17:107–121. doi: 10.1016/j.omtm.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.King J.A., Dubielzig R., Grimm D., Kleinschmidt J.A. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 2001;20:3282–3291. doi: 10.1093/emboj/20.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oliinyk O.S., Shemetov A.A., Pletnev S., Shcherbakova D.M., Verkhusha V.V. Smallest near-infrared fluorescent protein evolved from cyanobacteriochrome as versatile tag for spectral multiplexing. Nat. Commun. 2019;10:279. doi: 10.1038/s41467-018-08050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmiedel J.M., Lehner B. Determining protein structures using deep mutagenesis. Nat. Genet. 2019;51:1177–1186. doi: 10.1038/s41588-019-0431-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stagg S.M., Yoshioka C., Davulcu O., Chapman M.S. Cryo-electron Microscopy of Adeno-associated Virus. Chem. Rev. 2022;122:14018–14054. doi: 10.1021/acs.chemrev.1c00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berry G.E., Tse L.V. Virus Binding and Internalization Assay for Adeno-associated Virus. Bio. Protoc. 2017;7 doi: 10.21769/BioProtoc.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonntag F., Bleker S., Leuchs B., Fischer R., Kleinschmidt J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006;80:11040–11054. doi: 10.1128/JVI.01056-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerlach B., Kleinschmidt J.A., Böttcher B. Conformational changes in adeno-associated virus type 1 induced by genome packaging. J. Mol. Biol. 2011;409:427–438. doi: 10.1016/j.jmb.2011.03.062. [DOI] [PubMed] [Google Scholar]
- 68.Horowitz E.D., Finn M.G., Asokan A. Tyrosine cross-linking reveals interfacial dynamics in adeno-associated viral capsids during infection. ACS Chem. Biol. 2012;7:1059–1066. doi: 10.1021/cb3000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McInnes L., Healy J., Melville J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- 70.Wu P., Xiao W., Conlon T., Hughes J., Agbandje-McKenna M., Ferkol T., Flotte T., Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hagen S., Baumann T., Wagner H.J., Morath V., Kaufmann B., Fischer A., Bergmann S., Schindler P., Arndt K.M., Müller K.M. Modular adeno-associated virus (rAAV) vectors used for cellular virus-directed enzyme prodrug therapy. Sci. Rep. 2014;4:3759. doi: 10.1038/srep03759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Becker J., Fakhiri J., Grimm D. Fantastic AAV Gene Therapy Vectors and How to Find Them-Random Diversification, Rational Design and Machine Learning. Pathogens. 2022;11:756. doi: 10.3390/pathogens11070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nadler D.C., Morgan S.A., Flamholz A., Kortright K.E., Savage D.F. Rapid construction of metabolite biosensors using domain-insertion profiling. Nat. Commun. 2016;7 doi: 10.1038/ncomms12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plesa C., Sidore A.M., Lubock N.B., Zhang D., Kosuri S. Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science. 2018;359:343–347. doi: 10.1126/science.aao5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rubin A.F., Gelman H., Lucas N., Bajjalieh S.M., Papenfuss A.T., Speed T.P., Fowler D.M. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 2017;18:150. doi: 10.1186/s13059-017-1272-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang K.K., Wu Z., Arnold F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods. 2019;16:687–694. doi: 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- 77.DiPrimio N., Asokan A., Govindasamy L., Agbandje-McKenna M., Samulski R.J. Surface loop dynamics in adeno-associated virus capsid assembly. J. Virol. 2008;82:5178–5189. doi: 10.1128/JVI.02721-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wobus C.E., Hügle-Dörr B., Girod A., Petersen G., Hallek M., Kleinschmidt J.A. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cotmore S.F., McKie V.C., Anderson L.J., Astell C.R., Tattersall P. Identification of the major structural and nonstructural proteins encoded by human parvovirus B19 and mapping of their genes by procaryotic expression of isolated genomic fragments. J. Virol. 1986;60:548–557. doi: 10.1128/jvi.60.2.548-557.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaufmann B., Simpson A.A., Rossmann M.G. The structure of human parvovirus B19. Proc. Natl. Acad. Sci. USA. 2004;101:11628–11633. doi: 10.1073/pnas.0402992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawase M., Momoeda M., Young N.S., Kajigaya S. Most of the VP1 unique region of B19 parvovirus is on the capsid surface. Virology. 1995;211:359–366. doi: 10.1006/viro.1995.1418. [DOI] [PubMed] [Google Scholar]
- 82.Ros C., Gerber M., Kempf C. Conformational changes in the VP1-unique region of native human parvovirus B19 lead to exposure of internal sequences that play a role in virus neutralization and infectivity. J. Virol. 2006;80:12017–12024. doi: 10.1128/JVI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Q., Wu Z., Zhang J., Firrman J., Wei H., Zhuang Z., Liu L., Miao L., Hu Y., Li D., et al. A Robust System for Production of Superabundant VP1 Recombinant AAV Vectors. Mol. Ther. Methods Clin. Dev. 2017;7:146–156. doi: 10.1016/j.omtm.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McLaughlin R.N., Poelwijk F.J., Raman A., Gosal W.S., Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rivoire O., Reynolds K.A., Ranganathan R. Evolution-Based Functional Decomposition of Proteins. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Atkinson J.T., Jones A.M., Zhou Q., Silberg J.J. Circular permutation profiling by deep sequencing libraries created using transposon mutagenesis. Nucleic Acids Res. 2018;46:e76. doi: 10.1093/nar/gky255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Markin C.J., Mokhtari D.A., Sunden F., Appel M.J., Akiva E., Longwell S.A., Sabatti C., Herschlag D., Fordyce P.M. Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Science. 2021;373 doi: 10.1126/science.abf8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Girod A., Ried M., Wobus C., Lahm H., Leike K., Kleinschmidt J., Deléage G., Hallek M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999;5:1438. doi: 10.1038/71021. [DOI] [PubMed] [Google Scholar]
- 90.Michelfelder S., Lee M.K., deLima-Hahn E., Wilmes T., Kaul F., Müller O., Kleinschmidt J.A., Trepel M. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp. Hematol. 2007;35:1766–1776. doi: 10.1016/j.exphem.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 91.Müller O.J., Kaul F., Weitzman M.D., Pasqualini R., Arap W., Kleinschmidt J.A., Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 92.Münch R.C., Muth A., Muik A., Friedel T., Schmatz J., Dreier B., Trkola A., Plückthun A., Büning H., Buchholz C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015;6:6246. doi: 10.1038/ncomms7246. [DOI] [PubMed] [Google Scholar]
- 93.Large E.E., Chapman M.S. Adeno-associated virus receptor complexes and implications for adeno-associated virus immune neutralization. Front. Microbiol. 2023;14 doi: 10.3389/fmicb.2023.1116896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Engler C., Kandzia R., Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3 doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 96.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 97.Lin C.Y., Huang Z., Wen W., Wu A., Wang C., Niu L. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PLoS One. 2015;10 doi: 10.1371/journal.pone.0123562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D., et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Library staging and accessory plasmids (including sequence information) are available upon request. Sequencing data generated in this study have been deposited in the Sequence Raw Archive (https://www.ncbi.nlm.nih.gov/sra) under accession code PRJNA950466; refer to for corresponding metadata and read statistics. The code for handling data from domain insertion library sequencing is available at https://github.com/SavageLab/dipseq. The SPINE code is available at https://github.com/schmidt-lab/SPINE. The version used for this study is archived at https://zenodo.org/badge/latestdoi/223953195. Processed read count data, along with R scripts to reproduce manuscript figures, are available at https://github.com/Schmidt-lab/AAV_Insertion_Profiling. A Shiny app to display 3D views of AAV insertion fitness maps in a web browser is available at https://github.com/Schmidt-lab/shinyAAViewerR.