

Abstract
High-fat diet (HFD) consumption leads to obesity and a chronic state of low-grade inflammation, named metainflammation. Notably, metainflammation contributes to neuroinflammation due to the increased levels of circulating free fatty acids and cytokines. It indicates a strict interplay between peripheral and central counterparts in the pathogenic mechanisms of obesity-related mood disorders. In this context, the impairment of internal hypothalamic circuitry runs in tandem with the alteration of other brain areas associated with emotional processing (i.e., hippocampus and amygdala). Palmitoylethanolamide (PEA), an endogenous lipid mediator belonging to the N-acylethanolamines family, has been extensively studied for its pleiotropic effects both at central and peripheral level.
Our study aimed to elucidate PEA capability in limiting obesity-induced anxiety-like behavior and neuroinflammation-related features in an experimental model of HFD-fed obese mice.
PEA treatment promoted an improvement in anxiety-like behavior of obese mice and the systemic inflammation, reducing serum pro-inflammatory mediators (i.e., TNF-α, IL-1β, MCP-1, LPS). In the amygdala, PEA increased dopamine turnover, as well as GABA levels. PEA also counteracted the overactivation of HPA axis, reducing the expression of hypothalamic corticotropin-releasing hormone and its type 1 receptor. Moreover, PEA attenuated the immunoreactivity of Iba-1 and GFAP and reduced pro-inflammatory pathways and cytokine production in both the hypothalamus and hippocampus. This finding, together with the reduced transcription of mast cell markers (chymase 1 and tryptase β2) in the hippocampus, indicated the weakening of immune cell activation underlying the neuroprotective effect of PEA. Obesity-driven neuroinflammation was also associated with the disruption of blood–brain barrier (BBB) in the hippocampus. PEA limited the albumin extravasation and restored tight junction transcription modified by HFD. To gain mechanistic insight, we designed an in vitro model of metabolic injury using human neuroblastoma SH-SY5Y cells insulted by a mix of glucosamine and glucose. Here, PEA directly counteracted inflammation and mitochondrial dysfunction in a PPAR-α-dependent manner since the pharmacological blockade of the receptor reverted its effects.
Our results strengthen the therapeutic potential of PEA in obesity-related neuropsychiatric comorbidities, controlling neuroinflammation, BBB disruption, and neurotransmitter imbalance involved in behavioral dysfunctions.
Keywords: High-fat diet, Metabolic impairment, Inflammation, Mood disorders, Astrogliosis, Microgliosis, Mastocytosis, Blood–brain barrier permeability, N-acylethanolamines, Peroxisome proliferator-activated receptor-α
1. Introduction
Fat overnutrition leads to metabolic dysfunction at the peripheral level due to multiple and parallel mechanisms involving different compartments and converging to a state of low-grade inflammation, known as metainflammation (Li et al., 2018). Furthermore, high-fat diet (HFD) intake leads to the loss of gut barrier integrity, causing the transfer of macromolecules, such as microbial- or pathogen-associated molecular patterns (i.e., lipopolysaccharide, LPS) into the systemic circulation.
The inflammatory response to overnutrition at the central level, above all in the hypothalamus, develops more rapidly than peripheral tissues, damaging the neurocircuits related to fasting and feeding before overt obesity (Waise et al., 2015). Hypothalamic inflammation is sustained by the peripheral inflammatory and immune signals, activating microglial cells and astrocytes (Valdearcos et al., 2017). In this scenario, mast cells act as early players of neuroinflammation through the production and the release of neurotoxic factors that impair blood–brain barrier (BBB) integrity, already undermined by systemic inflammation (Skaper et al., 2017; Wang et al., 2020). Following long-term HFD feeding, inflammation may affect extra-hypothalamic brain areas involved in mood regulation and anxiety (i.e., prefrontal cortex, amygdala or AMY, hippocampus) (Guillemot-Legris and Muccioli, 2017). The bidirectional mechanisms linking obesity and anxiety shared pathophysiological processes converging in metabolic dysfunction and neuroinflammation. Obesity and insulin resistance, secondary to the excessive consumption of HFD, are events often associated with neuropsychiatric illnesses (Baker et al., 2017; Milaneschi et al., 2019; Tsan et al., 2021).
During the last years, many studies have focused on both metabolic and neuroprotective effects of palmitoylethanolamide (PEA), an endogenous lipid mediator whose activity is mainly mediated by peroxisome proliferator-activated receptor (PPAR)-α (Locci and Pinna, 2019; Mattace Raso et al., 2014a). PEA improves metabolic alterations in several murine models of obesity: it restores hypothalamic leptin signaling, modulates orexigenic and anorexigenic neurocircuitries (Mattace Raso et al., 2014b), and counteracts the metabolic dysfunction caused by lipid overload in an AMP-activated protein kinase-dependent manner (Annunziata et al., 2020). Recently, we have highlighted that PEA limits the depressive- and anhedonic-like behavior in obese mice, stimulating synaptic plasticity and neurogenesis (Lama et al., 2021).
Here, we investigated the effects of PEA on HFD-induced anxiety-like behavior in mice in relation to its capability in limiting immune cell activation and neuroinflammation in the hypothalamus and hippocampus. We also performed in vitro experiments on human SH-SY5Y cells insulted by a metabolic challenge to provide evidence for the involvement of PPAR-α. Here, we determined PEA modulation of inflammation and mitochondrial bioenergetics.
2. Materials and methods
2.1. Animals and treatments
Male C57Bl/6J mice (Charles River, Wilmington, MA, USA) at six weeks of age housed in stainless steel cages in a room kept at 22 ± 1 °C with a 12:12 h lights-dark cycle. They were randomly divided into three groups (11 animals for each group) as follows: control group (STD) receiving chow diet (Mucedola Srl, Milan, Italy) and vehicle; HFD (Research Diets Inc, NJ, USA) group receiving vehicle; HFD group treated with PEA (HFD + PEA, 30 mg/kg daily per os). The treatments started 12 weeks after HFD feeding and lasted seven weeks along with HFD (Fig. 1). STD diet had 17% fat without sucrose, while HFD had 45% energy derived from fat and 7% sucrose. We used PEA in the ultra-micronized formulation provided by Epitech Group Research Labs (Padua, Italy), dissolved in carboxymethyl cellulose (1.5%). Preliminary experiments showed no significant modification of body weight, fat mass, or energy intake in PEA-treated STD animals compared to the STD group (Figure S1). Furthermore, central variables depending on obesity (i.e., anxiety-like behavior and neuroinflammation) were not induced in STD mice. Therefore, the STD diet-fed group treated with PEA was not included. Animals were sacrificed by an i.p. injection of a mixture of ketamine/xylazine followed by cervical dislocation. All animal experiments were conducted under license number 982/2017-PR released by the Italian Ministry of Health. All procedures involving the animals were carried out following the Institutional Guidelines and complied with the Italian D.L. no.116 of January 27, 1992 of Ministero della Salute and associated guidelines in the European Communities Council (86/609/ECC and 2010/63/UE).
Fig. 1. The experimental protocol.

Mice of the experimental HFD groups received HFD for 12 weeks, while the STD group was fed with a standard chow diet. After 12 weeks on diet, the STD group received the vehicle, while HFD groups received either vehicle or ultra-micronized PEA (30 mg/kg daily p.o.) for seven weeks along with HFD.
2.2. Open field test
The open field test is one of the most used to obtain general information about locomotor activity and emotionality of animals (Seibenhener and Wooten, 2015). Indeed, the experimenter can evaluate anxiety-like behavior by evaluating mice’s propensity to avoid open areas perceived as dangerous, namely thigmotaxis. Briefly, mice were placed in an open-field arena and allowed to explore for 30 min. After each trial, the arena floor was cleaned with 70% ethanol to delete odor cues for the next subject. The movements of mice were recorded by the infrared video camera and analyzed by Any-maze-tracking software (Stoelting Co., Wood Dale, IL, U.S.A.). The evaluated parameters from the open field test were: 1) the percentage of covered distance in the central area [(distance travelled in the centre/total travelled distance) × 100]; 2) the number of entries in the central area; 3) total travelled distance (in m).
2.3. Neurochemical quantification (monoamine analyses)
Dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-HT, and 5 hydroxy-indol acetic acid (5-HIAA) levels were measured in the AMY of all mice by HPLC coupled with an electrochemical detector (Ultimate ECD, Dionex Scientific, Milan, Italy) as already reported (Lama et al., 2021). The separation was performed by a LC18 reverse phase column (Kinetex, 150 mm × 3.0 mm, ODS 5 μm; Phenomenex, Castel Maggiore- Bologna, Italy). The detection was accomplished by a thin-layer amperometric cell (Dionex, Thermo-Scientific, Milan, Italy) with a 5-mm diameter glassy carbon electrode at a working potential of 400 mV vs. Pd. The mobile phase consisted of an aqueous buffer containing 75 mM NaH2PO4, 1.7 mM octane sulfonic acid, 0.3 mM EDTA, acetonitrile 10%, buffered at pH 3.0. The flow rate was maintained by an isocratic pump (Shimadzu LC-10 AD, Kyoto, Japan) at 0.7 ml/min. Data were acquired and integrated using Chromeleon software (version 6.80, Dionex, Thermo Scientific, San Donato Milanese, Italy). Glutamic acid (GLU) and γ-aminobutyric acid (GABA) were quantified by using HPLC coupled with fluorimetric detection (emission length, 4.60 nm; excitation length, 3.40 nm). Chromatographic detection was accomplished by ODS-3 column (150 × 4.6 mm, 3 μm; INERTSIL) after derivatization with ophthalaldehyde/mercaptopropionic acid. The mobile phase gradient was 50 mM sodium acetate buffer, pH 6.95, with methanol increasing linearly from 2 to 30% (v/v) over 40 min. The flow rate was maintained by a pump (JASCO, Tokyo, Japan) at 0.5 ml/min. Results were analyzed by Borwin software (version 1.50; Jasco), and substrate concentration was expressed as μM.
2.4. Serum parameters analysis
Serum samples, collected at the end of the experimental period, were assayed for LPS, monocyte chemoattractant protein (MCP)-1, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, corticotropin-releasing hormone (CRH), and adrenocorticotropic hormone (ACTH).
LPS was measured using the Limulus amebocyte lysate (LAL QCL-1000; Lonza Group Ltd., Basel, Switzerland) technique. IL-1β (Thermo Fisher Scientific, Rockford, IL), TNF-α, and MCP-1 (Biovendor R&D, Brno, Czech Republic) were measured using commercially available ELISA kits. Corticosterone (Cat#ab108821) and ACTH (Cat#ab263880) were quantified using commercially available ELISA kits (Abcam, Cambridge, UK) according to the manufacturer’s instructions. All samples and standards were analyzed in duplicate to avoid intra-assay variations.
2.5. Peroxidase immunohistochemistry and morphometric analysis
Mice from each experimental group were anesthetized with the intraperitoneal ketamine/xylazine solution and transcardially perfused with a saline solution (0,9% NaCl in dH2O) containing heparin (10 mg/l) (Cat#2106; Sigma-Aldrich, Milan, Italy) followed by fixative (4% paraformaldehyde, 15% picric acid, 0,1% glutaraldehyde in PBS). Brains were carefully removed from the skull, post-fixed in the same fixative solution for 24 h at 4 °C and washed in PBS. Free-floating 40-μm-thick coronal brain sections were cut with a Leica VT1200S vibratome (Leica Microsystems, Vienna, Austria) and kept in PBS (pH 7.4) at 4 °C until use in immunohistochemical experiments.
Free-floating brain sections were reacted with 1% NaOH and 1% H2O2 (20 min), 0.3% glycine (10 min), and 0.03% sodium dodecyl sulphate (10 min). After rinsing in PBS, they were blocked with 3% normal serum (in 0,2% Triton X-100; 60 min) and incubated with mouse monoclonal IgG anti-Iba1 (Wako, Shanghai, China, # 019–19741; dilution at 1:1500) or mouse monoclonal IgG anti-glial fibrillary acidic protein (GFAP) (Merck, St. Louis, Missouri, USA; #G3893; dilution at 1:1000) in PBS, overnight at 4°C. The next day, after a thorough rinse in PBS, sections were incubated in 1:200 v/v biotinylated secondary antibody solution (in PBS; 30 min), rinsed in PBS, and incubated in avidin–biotin peroxidase complex (ABC Elite PK6100, Vector Laboratories, Burlingame, CA, USA), washed several times in PBS, and finally incubated in 3,3′ diaminobenzidine tetrahydrochloride (0.05% in 0.05 M Tris with 0.03% H2O2; 5 min). After immunohistochemical staining, sections were mounted on slides, air-dried, dehydrated in ethanol, cleared with xylene, and covered with Eukitt. Staining was never detected when the primary antibody was omitted.
Morphometric analyses were performed on five mice for each experimental group. From each mouse, three non-adjacent and corresponding brain coronal sections containing the tuberal hypothalamus and the hippocampus (from bregma – 1.46 mm to bregma – 1.94 mm according to Paxinos and Franklin, 2019) were immunostained for Iba1 and GFAP. Adjacent sections were used to identify the exact location of the arcuate nucleus of the hypothalamus, the dentate gyrus (DG), and the stratum radiatum of the CA1 of the hippocampus by Nissl staining. The number of Iba1-positive microglial cells was manually counted at 10x magnification. In contrast, GFAP immunostaining was scored in 40x photographs by assessing the percentage of GFAP staining in astrocyte somata and processes with the ImageJ software using threshold values to standardize intensity (NIH, Bethesda, MD, USA). GFAP positive processes of astrocytes are often entangled with each other and for this reason a more affordable method of morphometric evaluation is the assessment of the area of GFAP specific staining, as widely reported (Berkseth et al., 2014; Douglass et al., 2017). Measures were taken in each hemisphere’s left, and right values were added up for each slice and averaged in each mouse. Measurements were performed by an experimenter who was blinded to the identity of the treatment group.
2.6. Immunofluorescence, confocal microscopy, and morphometric analysis
For double fluorescent immunohistochemistry, free-floating brain sections were processed as above up to incubation with the primary antibody. They were then incubated overnight in a mixture of two primary antibodies: goat polyclonal IgG anti-albumin (Novus Biologicals, Centennial, CO, USA, Cat # NB600–41532; dilution at 1:2000 v/v) and mouse monoclonal IgG anti-NeuN (Millipore, Burlington, MA, USA, Cat # MAB377; dilution at 1:100 v/v). The next day, sections were washed twice with PBS and incubated in a cocktail of fluorophore-linked secondary antibodies at a dilution of 1:400 in PBS for one hour at room temperature. The secondary antibodies were Alexa Fluor® 488 donkey anti-goat IgG and Alexa Fluor® 555 donkey anti-mouse IgG (both from Invitrogen, Carlsbad, CA, USA). Subsequently, sections were washed twice with PBS, counterstained with TO-PRO3-iodide, mounted on standard glass slides, air-dried, and coverslipped using Vectashield mounting medium (Vector Laboratories). Sections were viewed under a motorized Leica DM6000 microscope at different magnifications. Fluorescence was detected with a Leica TCS-SL spectral confocal microscope equipped with an Argon and He/Ne mixed gas laser. Fluorophores were excited with the 488 nm, 543 nm, and 649 nm lines and imaged separately. Images (1024 × 1024 pixels) were obtained sequentially from two channels using a confocal pinhole of 1.1200 and stored as TIFF files. The brightness and contrast of the final images were adjusted using Photoshop 6 (Adobe Systems, Mountain View, CA, USA).
Morphometric analysis was performed on corresponding hippocampus-containing brain coronal sections processed in parallel. One section for each mouse and five mice for each experimental group was analyzed. The number of albumin-immunoreactive neuronal profiles are shown from both left and right DG and CA1 hippocampal subfield.
2.7. Cell culture and treatments
SH-SY5Y neuroblastoma cells (ATCC® number CCL-2266) were cultured in DMEM-F12 medium with 10% FBS, 100 IU/ml penicillin, and 100 mg/ml streptomycin at 37 °C in a humidified atmosphere with 5% CO2. After 6-hour starvation in 2% FBS medium, cells were incubated for 24 h with a mix of 55 mM glucose (CARLO ERBA Reagents, Milan, Italy) and 2 mM glucosamine (Gluc2, Merck Life Science, Milan, Italy) to mimic the metabolic impairment induced by HFD in mice (Mi et al., 2017). One hour before Gluc2 challenge, cells were pretreated with PEA (10 μM) in the presence or not of PPAR-α antagonist, GW6471 (2 μM, Tocris Bioscience, Bristol, UK), added 30 min before PEA. After treatments, cells were collected, and mRNA extracts were obtained for additional analysis. The results were obtained from three different experiments.
2.8. Seahorse assay
SH-SY5Y cells underwent XF Cell Mito Stress Test kit using Seahorse Bioscience XFe24 Analyzer (Agilent Technologies, Santa Clara, CA, USA) to reveal critical parameters of cellular metabolism, as previously described (Pirozzi et al., 2020). Briefly, SH-SY5Y cells were seeded in mini plates at 80,000 cells/well in growth medium overnight, stimulated with Gluc2, in the presence or not of PEA and/or GW6471. Before starting XF assay, the medium was replaced with Seahorse XF DMEM assay medium (pH 7.4), and the cells were equilibrated at 37 °C in a CO2-free incubator for 1 h. Mito Stress Assay evaluates the oxygen consumption rate (OCR, pmol/min) under baseline or stressed conditions. The proton leak and ATP-linked respiration were determined after the injection of 1 μM oligomycin (as an inhibitor of the F0/F1 ATPase). Afterward, the mitochondrial electron transport chain was maximally stimulated by adding the uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 0.5 μM). The extra-mitochondrial respiration was determined after injecting mixed rotenone (1 μM)/antimycin A (0.5 μM), complexes I and III inhibitors. All parameters were calculated through Seahorse Wave Desktop Software (Agilent Technologies, Santa Clara, CA, USA). OCR was normalized to the protein content (μg/ml) of each well for all measurements by Bradford assay. The results were obtained from three different experiments.
2.9. Quantification of gene expression using Real-Time PCR
Total RNA, isolated from the mouse hypothalamus and hippocampus, and SH-SY5Y cells, was extracted using TRIzol Reagent (Bio-Rad Laboratories, Hercules, CA, USA) and assayed as previously described (Lama et al., 2019; Lama et al., 2021). Each sample contained 500 ng cDNA in 2X QuantiTect SYBR Green PCR Master Mix and primers pairs to amplify mouse chymase 1 (Cma1), tryptase β2 (Tpsb2), NF-KB (NfKb1), IL-1β (Il1b), CRH (Crh), CRH receptor 1 (Crhr1), TNF-α (Tnf), myeloid differentiation primary response gene 88 (Myd88), toll-like receptor (TLR)2 (Tlr2), tight junction protein 1 (Tjp1), occludin (Ocln), and claudin 5 (Cldn5) (Qiagen, Hilden, Germany), human NLR family pyrin domain containing 3 (NLRP3), TLR4 (Qiagen, Hilden, Germany), and TNF-α (5′- GGC AGT CAG ATC ATC TTC TCG AA –3′; 5′ – TGA AGA GGA CCT GGG AGT AGA TG – 3′) (IDT Technologies, Coralville, IA, USA) in a final volume of 50 μl. The relative amount of each studied mRNA was normalized to actin as a housekeeping gene, and data were analyzed according to the 2−ΔΔCT method.
2.10. Western blotting
Hippocampal lysates were subjected to SDS-PAGE. The blot was performed by transferring proteins from a slab gel to a nitrocellulose membrane at 240 mA for 60 min at room temperature. The filter was then blocked with 1X PBS (pH 7.4) and 5% non-fat dried milk for 60 min at room temperature and probed with rabbit polyclonal antibody against anti-TLR4 (1:1000; Cat#Sc-3002; Santa Cruz Biotechnology, Dallas, TX, USA). Western blot for anti-β-actin (1:5000; Cat#A5441; Sigma-Aldrich, Milan, Italy) was performed to ensure equal sample loading. The detection of the filter was performed by ChemiDoc Imaging System (Bio-Rad Laboratories, Hercules, CA, USA).
2.11. Data and statistical analysis
Data are presented as mean ± SEM. All experiments were analyzed using analysis of variance (ANOVA) for multiple comparisons followed by Bonferroni’s post hoc test, using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA). Normality was tested using the Shapiro-Wilk test. Bonferroni’s post hoc test was run when F was significant. Differences among groups were considered significant at values of p < 0.05.
3. Results
3.1. PEA improves anxiety-like behavior induced by HFD and modulates DA turnover and GABA level in AMY.
During open field test, obese mice showed a significant increase of thigmotaxis and latency to enter the central zone of the open field, as demonstrated by the reduced total distance travelled in the centre (Fig. 2A, F (2, 30) = 0.2204; STD: 8.523 ± 0.668; HFD: 4.071 ± 0.529; p < 0.0001) and the number of entries into this area (Fig. 2B, F (2, 30) = 0.4252; STD: 78.36 ± 6.742; HFD: 41.36 ± 5.501; p = 0.0002). This tendency was counteracted by PEA treatment, that stimulated mice to explore the whole arena, increasing both the total distance (HFD + PEA: 6.936 ± 0.565; p = 0.2008 vs STD; p = 0.0053 vs HFD) and the number of entries in the centre (HFD + PEA: 62.18 ± 4.647; p = 0.1608 vs STD; p = 0.0446 vs HFD). In open field test, no significant change in mice movement was shown between PEA-treated and untreated obese mice in total distance travelled (Fig. 2C, F (2, 30) = 1.490; STD: 53.71 ± 3.193; HFD: 37.85 ± 2.781, p = 0.0008 vs STD; HFD + PEA: 46.67 ± 2.029; p = 0.2278 vs STD; p = 0.0859 vs HFD). Moreover, all mice were subjected to rotarod test, revealing no difference among groups in motor coordination (data not shown).
Fig. 2. PEA limits the anxiety-like behavior of obese mice.
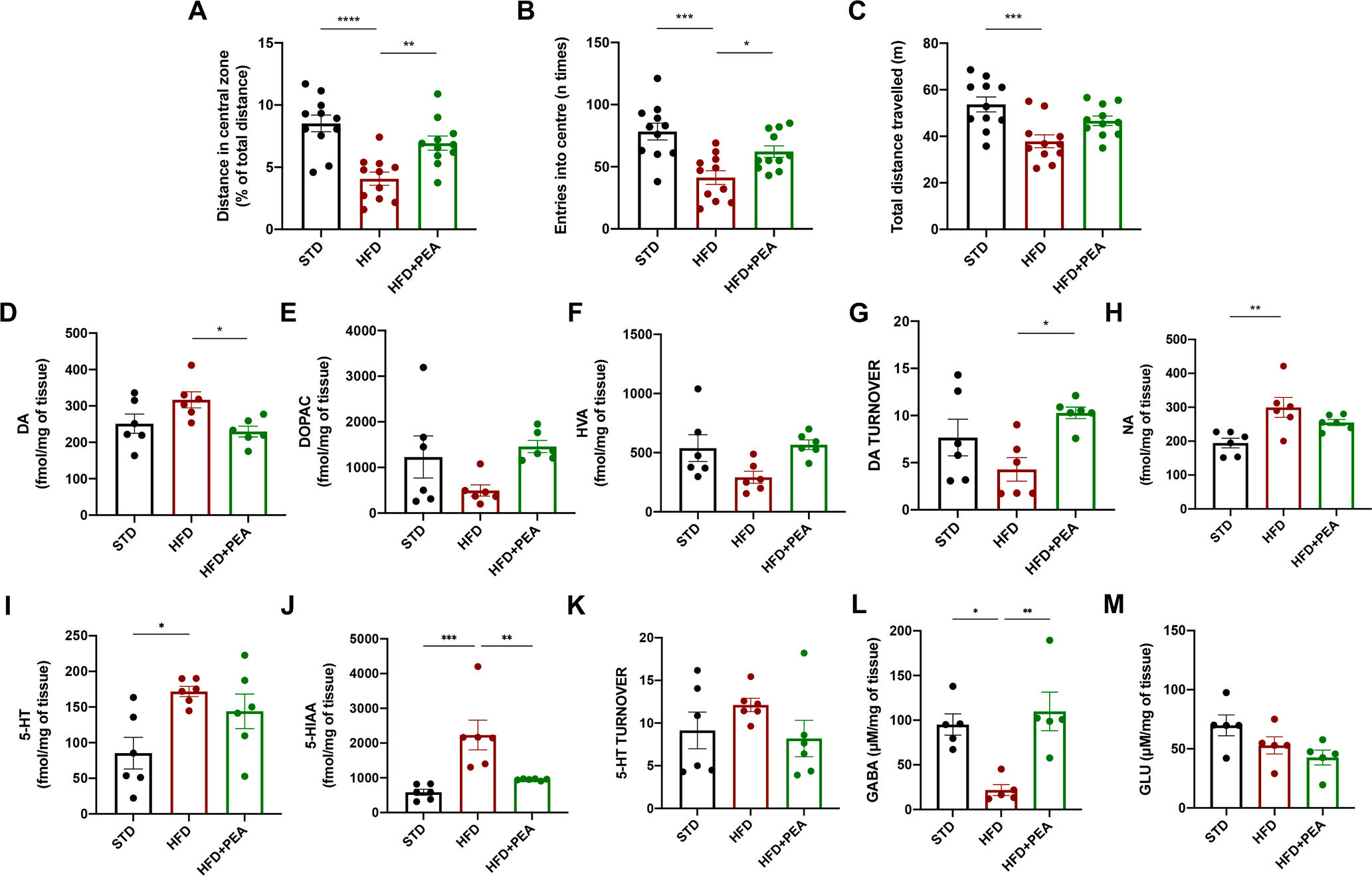
(A) Total distance in the centre, (B) the number of entries into the centre, and (C) the total distance travelled by mice in the open field test (n = 11 per group). The levels of (D) DA, (E) DOPAC, (F) HVA, and (G) DA turnover, (H) NA, (I) 5-HT, (J) 5-HIAA, and (K) 5-HT turnover (n = 6 per group) in AMY of all mice. The levels of (L) GABA and (M) GLU in AMY of all experimental groups (n = 5 per group). All data are shown as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Considering the pivotal role of AMY in the response to stressors, we evaluated the levels of many neurotransmitters in this area obtained from all animal groups. To this purpose, we evaluated the levels of DA, and its metabolites (DOPAC and HVA), noradrenaline (NA), and 5-HT, together with its metabolite 5-HIAA. After 7 weeks of PEA treatment, mice showed a reduction of DA levels (Fig. 2D, F (2, 15) = 0.7377; STD: 251.2 ± 26.42; HFD: 316.9 ± 22.00, p = 0.1442 vs STD; HFD + PEA: 229.6 ± 14.68, p > 0.9999 vs STD; p = 0.0357 vs HFD). Although the levels of DOPAC (Fig. 2E, F (2, 15) = 4.711; STD: 1230 ± 462; HFD: 494.7 ± 124.1, p = 0.2696 vs STD; HFD + PEA: 1458 ± 132.6; p > 0.9999 vs STD p = 0.0936 vs HFD) and HVA (Fig. 2F, F (2, 15) = 1.190; STD: 538.6 ± 113.3; HFD: 291.9 ± 50.65, p = 0.1061 vs STD; HFD + PEA: 567.2 ± 40,89; p > 0.9999 vs STD p = 0.0627 vs HFD) did not significantly differ among groups, PEA increased the turnover of DA (Fig. 2G, F (2, 15) = 2.790, STD: 7.664 ± 1.944; HFD: 4.293 ± 1.249, p = 0.3136 vs STD; HFD + PEA: 10.28 ± 0.608; p = 0.6004 vs STD p = 0.0234 vs HFD). Furthermore, PEA slightly decreased the levels of NA (Fig. 2H, F (2, 15) = 1.268; STD: 194.4 ± 14.13; HFD: 299.5 ± 29.34, p = 0.0051 vs STD; HFD + PEA: 254.9 ± 8.969; p = 0.1337 vs STD p = 0.3809 vs HFD) and 5-HT (Fig. 2I, F (2, 15) = 2.107; STD: 85.29 ± 22.20; HFD: 171.9 ± 7.218, p = 0.0197 vs STD; HFD + PEA: 144.0 ± 24.21; p = 0.1482 vs STD p = 0.9785 vs HFD), strongly altered by HFD. Although the levels of 5-HIAA resulted high in obese mice and reduced by PEA (Fig. 2J, F (2, 15) = 2.636; STD: 584.7 ± 87.15; HFD: 2233 ± 426.1, p = 0.0010 vs STD; HFD + PEA: 950.7 ± 10.45; p = 0.9576 vs STD p = 0.0077 vs HFD), 5-HT turnover did not differ among group (Fig. 2K, F (2, 15) = 2.157; STD: 9.148 ± 2.152; HFD: 12.12 ± 0.783, p = 0.7883 vs STD; HFD + PEA: 8.186 ± 2.135; p > 0.9999 vs STD p = 0.4334 vs HFD). In addition, aminoacidic neurotransmitter levels, such as GLU and GABA, were also evaluated. HFD induced a significant decrease in GABA content in the AMY of obese mice, while PEA administration restored GABA levels (Fig. 2L, F (2, 12) = 0.6647; STD: 95.14 ± 11.95; HFD: 21.85 ± 6.107, p = 0.0107 vs STD; HFD + PEA: 109.8 ± 21.58; p = 0.7643 vs STD p = 0.0031 vs HFD). Conversely, GLU levels did not differ among all groups (Fig. 2M, F (2, 12) = 0.0490; STD: 69.86 ± 8.765; HFD: 52.93 ± 7.287, p = 0.2861 vs STD; HFD + PEA: 42.63 ± 6.299; p = 0.0603 vs STD p = 0.6096 vs HFD).
3.2. PEA counteracts the systemic inflammation in obese mice.
Because of the HFD-induced gut leakiness (Araujo et al., 2017), we also evaluated the anti-inflammatory and immunomodulatory activity of PEA in serum samples of all experimental groups. In particular, we showed that PEA limited the endotoxemia induced by HFD, reducing the serum levels of LPS (Fig. 3A, F (2, 15) = 20.49; STD: 0.7652 ± 0.033; HFD: 1.645 ± 0.096, p < 0.0001 vs STD; HFD + PEA: 1.169 ± 0.022; p = 0.0008 vs STD; p = 0.0001 vs HFD) and the chemoattractant MCP-1 (Fig. 3B, F (2, 15) = 1.301; STD: 27.67 ± 0.882; HFD: 79 ± 1.732, p < 0.0001 vs STD; HFD + PEA: 47.67 ± 2.525; p < 0.0001 vs STD; p < 0.0001 vs HFD). Furthermore, PEA confirmed its remarkable anti-inflammatory properties, as demonstrated by the reduction of serum TNF-α (Fig. 3C, F (2, 15) = 0.9185; STD: 0.9233 ± 0.049; HFD: 3.867 ± 0.095, p < 0.0001 vs STD; HFD + PEA: 1.473 ± 0.126; p = 0.0030 vs STD; p < 0.0001 vs HFD) and IL-1β (Fig. 3D, F (2, 15) = 3.783; STD: 1.820 ± 0.032; HFD: 6.335 ± 0.218, p < 0.0001 vs STD; HFD + PEA: 3.681 ± 0.070; p < 0.0001 vs STD; p < 0.0001 vs HFD).
Fig. 3. PEA improves systemic parameters altered by HFD.
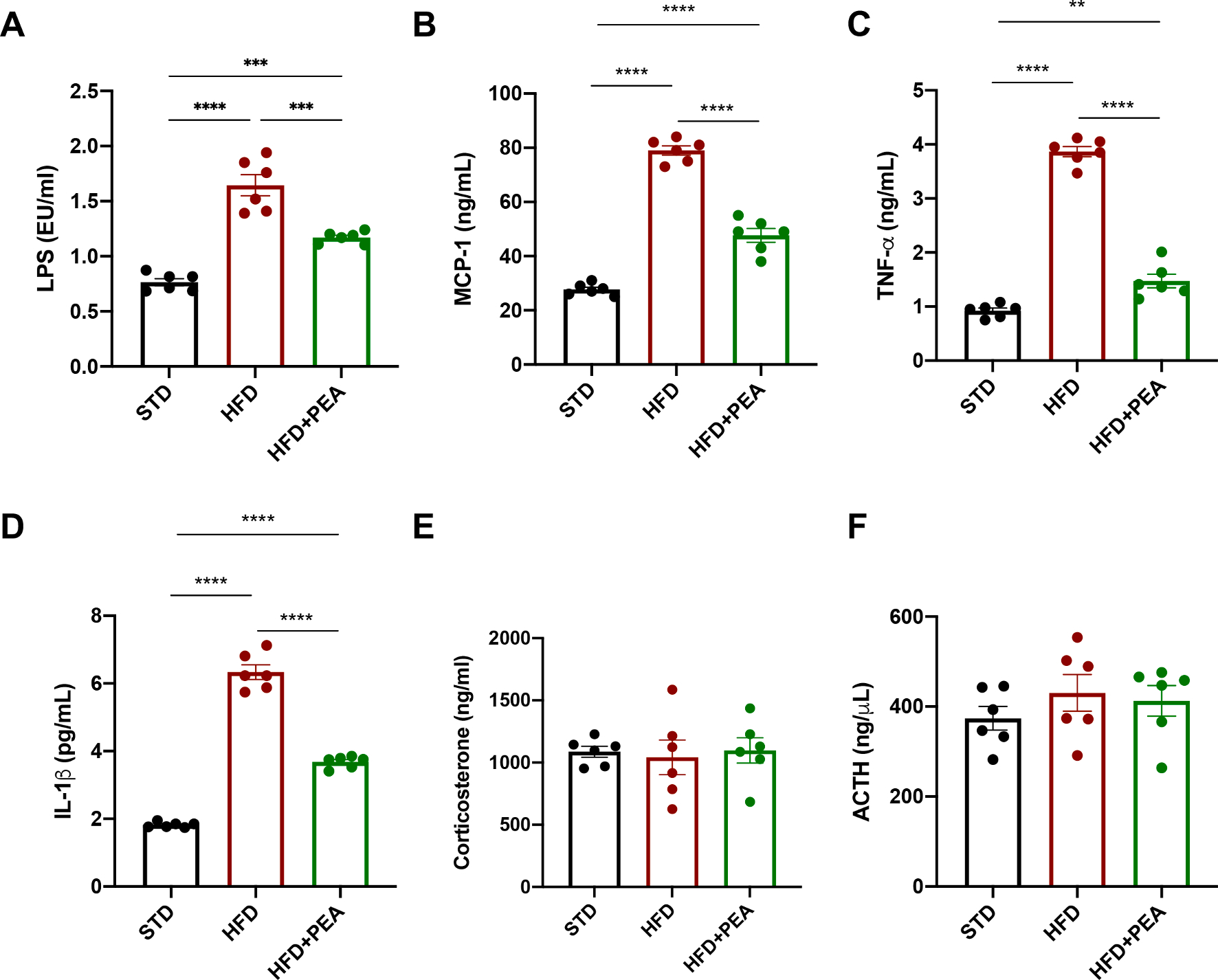
Serum levels of (A) LPS, (B) MCP-1, (C) TNF-α, (D) IL-1β, (E) corticosterone and (F) ACTH (n = 6 per group). All data are shown as mean ± S.E.M. **p < 0.01, ***p < 0.001, and ****p < 0.0001.
The serum levels of corticosterone (Fig. 3E, F (2, 15) = 2.343; STD: 1087 ± 43.55; HFD: 1042 ± 139.8, p > 0.9999 vs STD; HFD + PEA: 1098 ± 101.3; p > 0.9999 vs STD; p > 0.9999 vs HFD) and ACTH (Fig. 3F, F (2, 15) = 0.7741; STD: 374 ± 26.44; HFD: 430.4 ± 40.71, p = 0.7836 vs STD; HFD + PEA: 412.8 ± 33.89; p > 0.9999 vs STD; p > 0.9999 vs HFD) did not differ among the groups.
3.3. PEA attenuates HFD-induced hypothalamic injury of obese mice.
After 19 weeks of HFD intake, obese mice showed an increased transcription of NF-KB (Fig. 4A, F (2, 12) = 0.695; STD: 1.000 ± 0.060; HFD: 1.910 ± 0.174, p = 0,0022 vs STD) and IL-1β (Fig. 4B, F (2, 12) = 0.9217; STD: 1.000 ± 0.091; HFD: 3.548 ± 0,315, p < 0,0001 vs STD) in hypothalamus, while PEA treatment limited these pro-inflammatory mediators (for NF-KB: HFD + PEA: 1.311 ± 0.166; p = 0.4510 vs STD; p = 0.0357 vs HFD; for IL-1β: HFD + PEA: 1.957 ± 0.308; p = 0.0689 vs STD; p = 0.0029 vs HFD). Then, we also evaluated mRNA expression of CRH and its receptor CRHR1, whose central alteration suggests a dysregulation of hypothalamic–pituitaryadrenal (HPA) axis. PEA limited the increased transcription of CRH (Fig. 4C, F (2, 12) = 1.085; STD: 1.000 ± 0.104; HFD: 1.947 ± 0.286, p = 0.0242 vs STD; HFD + PEA: 1.036 ± 0.202; p > 0.9999 vs STD; p = 0.0303 vs HFD) and CRHR1 (Fig. 4D, F (2, 12) = 1.415; STD: 1.000 ± 0.106; HFD: 1.800 ± 0.234, p = 0.0110 vs STD; HFD + PEA: 0.822 ± 0.089; p > 0.9999 vs STD; p = 0.0026 vs HFD) in obese mice. By peroxidase immunohistochemistry, we evaluated the number of Iba1-positive microglial cells (Fig. 4E) and the immunoreactive area (%) of GFAP-positive astrocytes (Fig. 4F) in the arcuate nucleus, taken as indices of HFD-induced hypothalamic inflammation. As expected (Buckman et al., 2013; Valdearcos et al., 2014), HFD produced a significant microgliosis and astrogliosis in the hypothalamic arcuate nucleus, that were counteracted by PEA treatment (Fig. 4E, F (2, 12) = 0.2209; STD: 119.6 ± 4.534; HFD: 162.6 ± 3.669, p < 0.0001 vs STD; HFD + PEA: 136.2 ± 5.286; p = 0.0719 vs STD; p = 0.0044 vs HFD; Fig. 4F, F (2, 12) = 0.0323; STD: 19.43 ± 1.280; HFD: 24.71 ± 1.257, p = 0.0325 vs STD; HFD + PEA: 18.18 ± 1.185; p > 0.9999 vs STD; p = 0.0087 vs HFD).
Fig. 4. PEA counteracts hypothalamic injury in HFD-induced obese mice.
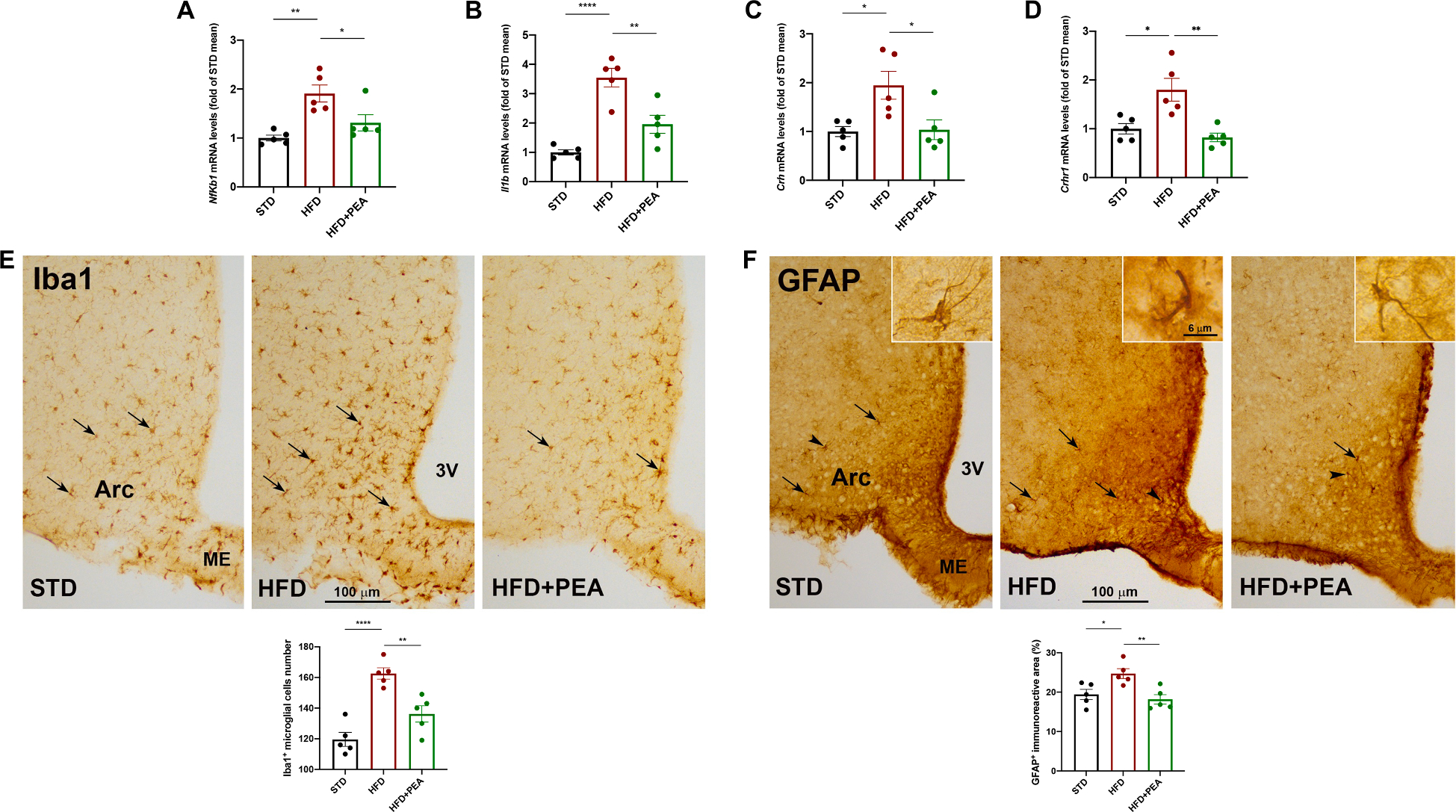
The mRNA transcription of (A) Nfkb1, (B) Il1b, (C) Crh, and (D) Crhr1 in the hypothalamus (n = 5 per group). Immunohistochemical evaluations and morphometric analyses of (E) the number of IBA1-positive microglial cells and (F) GFAP immunoreactive area in the hypothalamic arcuate nucleus (Arc) (n = 5 per group). 3 V, third ventricle, ME, median eminence. All data are shown as mean ± S.E.M. *p < 0.05, **p < 0.01, and ****p < 0.0001.
3.4. PEA limits hippocampal inflammation, astrogliosis, and microgliosis of obese mice.
Since the alteration of bidirectional network between hypothalamus and hippocampus represents the pathogenetic basis for neuropsychiatric disorders related to obesity (Miller and Spencer, 2014), we also evaluated inflammation, astrogliosis and microgliosis in hippocampi of all animal groups. PEA reduced mRNA expression of TNF-α (Fig. 5A, F (2, 12) = 6.133; STD: 1.000 ± 0.110; HFD: 4.307 ± 0.858, p = 0.0017 vs STD; HFD + PEA: 0.603 ± 0.118; p > 0.9999 vs STD; p = 0.0007 vs HFD) and IL-1β (Fig. 5B, F (2, 12) = 4.923; STD: 1.000 ± 0.970; HFD: 4.173 ± 0.921, p = 0.0043 vs STD; HFD + PEA: 1.505 ± 0.177; p > 0.9999 vs STD; p = 0.0140 vs HFD), altered by HFD. Then, we analyzed the distribution of Iba1-positive cells and the percentage of GFAP specific staining in the DG and in the stratum radiatum of the CA1 hippocampus (Fig. 5C and F). HFD caused a marked microgliosis, as shown by the significant increase of the number of Iba1-positive cells in both DG and stratum radiatum. Interestingly, PEA attenuated this detrimental condition in a significant manner in the stratum radiatum, and only partially in the DG (Fig. 5D, F (2, 12) = 0.1838; STD: 97.60 ± 7.109; HFD: 123.2 ± 4.283, p = 0.0280 vs STD; HFD + PEA: 113.9 ± 5.828; p = 0.2161 vs STD; p = 0.8547 vs HFD; Fig. 5G, F (2, 12) = 0.0087; STD: 76.47 ± 2.284; HFD: 90.27 ± 1.971, p = 0.0017 vs STD; HFD + PEA: 79.67 ± 2.035; 0.9082 vs STD; p = 0.0116 vs HFD). Accordingly, HFD strongly induced an increase of GFAP-positive immunoreactive area (%) in DG, that was reverted by PEA (Fig. 5E, F (2, 12) = 2.342; STD: 7.980 ± 0.101; HFD: 10.64 ± 0.473, p = 0.0015 vs STD; HFD + PEA: 8.030 ± 0.496; p > 0.9999 vs STD; p = 0.0018 vs HFD). HFD involved a tendency to an increase of GFAP-positive immunoreactive area (%) in stratum radiatum, that was partially reduced by PEA (Fig. 5H, F (2, 12) = 1.035; STD: 6.608 ± 0.477; HFD: 7.150 ± 0.592, p > 0,9999 vs STD; HFD + PEA: 5.684 ± 0.269; p = 0.5582 vs STD; p = 0.1380 vs HFD).
Fig. 5. PEA limits neuroinflammation, astrogliosis and microgliosis in the hippocampus of HFD mice.
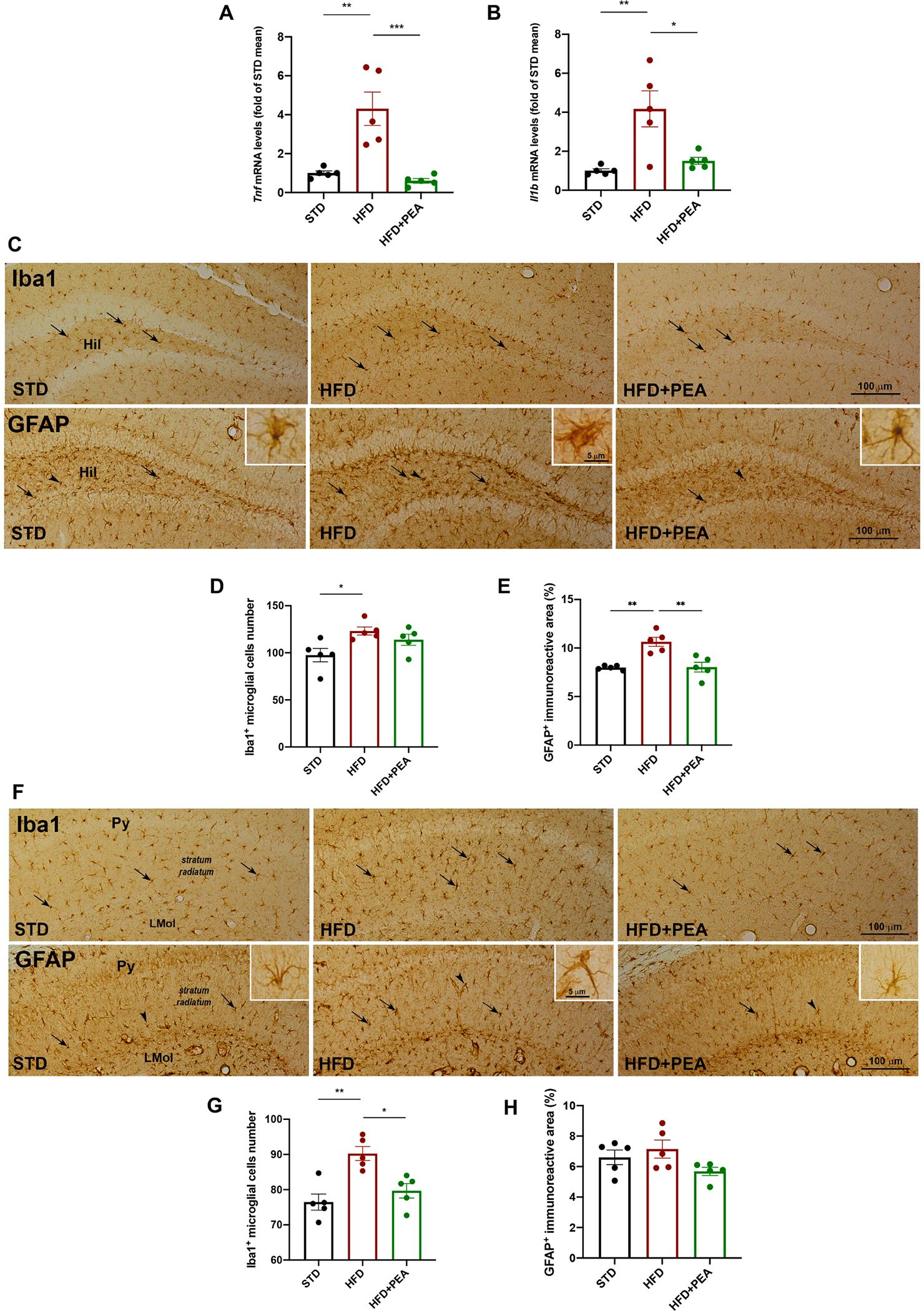
The mRNA expression of (A) Tnf and (B) Il1b in the hippocampus of all experimental groups (n = 5 per group). The immunohistochemical analyses of Iba1-positive microglial cells and GFAP-positive astrocytes in (C) the DG and (F) stratum radiatum (n = 5 per group). The count of the number of positive cells for (D, G) Iba1 and (E, H) of GFAP immunoreactive in DG and stratum radiatum, respectively (n = 5 per group). Hil, hilus of the DG; Py, pyramidal cell layer; LMol, lacunosum molecular layer. All data are shown as mean ± S.E.M. *p < 0.05, **p < 0.01, and ***p < 0.001.
3.5. PEA effects on neuroinflammation, mast cell proteases, and BBB permeability in the hippocampus of obese mice.
The peripheral endotoxemia caused by HFD intake also reflected in hippocampus, where the lipid overload caused an increase of protein TLR4 (Fig. 6A, F (2, 13) = 0.5691; STD: 0.596 ± 0.112; HFD: 1.173 ± 0.067, p = 0.0019 vs STD; HFD + PEA: 0.698 ± 0.082; p > 0.9999 vs STD; p = 0.0060 vs HFD), its downstream gene Myd88 (Fig. 6B, F (2, 12) = 2.811; STD: 1.000 ± 0.045; HFD: 1.552 ± 0.133, p = 0.0013 vs STD; HFD + PEA: 0.851 ± 0.018; p = 0.6627 vs STD; p = 0.0002 vs HFD), and the transcription of TLR2 (Fig. 6C, F (2, 12) = 0.1345; STD: 1.000 ± 0.009; HFD: 1.151 ± 0.012, p < 0.0001 vs STD; HFD + PEA: 0.812 ± 0.014; p < 0.0001 vs STD; p < 0.0001 vs HFD), all immune factors reduced by PEA treatment. Furthermore, PEA reduced mRNA expression of two specific proteases of murine mast cells, Cma1 (Fig. 6D, F (2, 12) = 2.925; STD: 1.000 ± 0.232; HFD: 2.037 ± 0.391, p = 0.0498 vs STD; HFD + PEA: 0.408 ± 0.042; p = 0.4148 vs STD; p = 0.0027 vs HFD) and Tpsb2 (Fig. 6E, F (2, 12) = 1.641; STD: 1.000 ± 0.252; HFD: 2.031 ± 0.245, p = 0.0127 vs STD; HFD + PEA: 0.748 ± 0.073; p > 0.9999 vs STD; p = 0.0027 vs HFD), increased by HFD. The detrimental process of neuroinflammation triggered by HFD caused the loss of BBB integrity, as shown by the reduced transcription of Tjp1 (Fig. 6F, F (2, 12) = 0.2657; STD: 1.000 ± 0.048; HFD: 0.733 ± 0.037, p = 0.0042 vs STD; HFD + PEA: 0.963 ± 0.051; p > 0.9999 vs STD; p = 0.0116 vs HFD), Ocln (Fig. 6G, F (2, 12) = 1.091; STD: 1.000 ± 0.059; HFD: 0.724 ± 0,020, p = 0.0042 vs STD; HFD + PEA: 1.030 ± 0.053; p > 0.9999 vs STD; p = 0.0019 vs HFD), and Cldn5 (Fig. 6H, F (2, 12) = 0.9122; STD: 1.000 ± 0.029; HFD: 0.731 ± 0.060, p = 0.0037 vs STD; HFD + PEA: 0.964 ± 0.042; p > 0.9999 vs STD; p = 0.0103 vs HFD), whose levels were improved by PEA. To provide further evidence of PEA-mediated effect on BBB integrity, we resorted to immunoreactivity against endogenous albumin which extravasates following disruption of the BBB (Saunders et al., 2015; Venema et al., 2020) and is taken up by neuronal and glial cells (Braganza et al., 2012; van Vliet et al., 2007). Results showed that HFD mice exhibited an increase in the number of scattered albumin-positive neurons in both DG (Fig. 6I, F (2, 12) = 0.1667; STD: 4.200 ± 0.3742; HFD: 7.000 ± 0.7746, p = 0.0261 vs STD; HFD + PEA: 2.600 ± 0.6782; p = 0.2967 vs STD; p = 0.0011 vs HFD) and CA1 hippocampal subfield compared to STD mice (Fig. 6J, F (2, 12) = 2.211; STD: 4.200 ± 0.800; HFD: 6.600 ± 0.245, p = 0.0242 vs STD; HFD + PEA: 3.400 ± 0.400; p = 0.9346 vs STD; p = 0.0035 vs HFD). PEA treatment reduced all these parameters.
Fig. 6. The impact of PEA on immune activation, mast cell degranulation, and BBB integrity in the hippocampus of HFD mice.
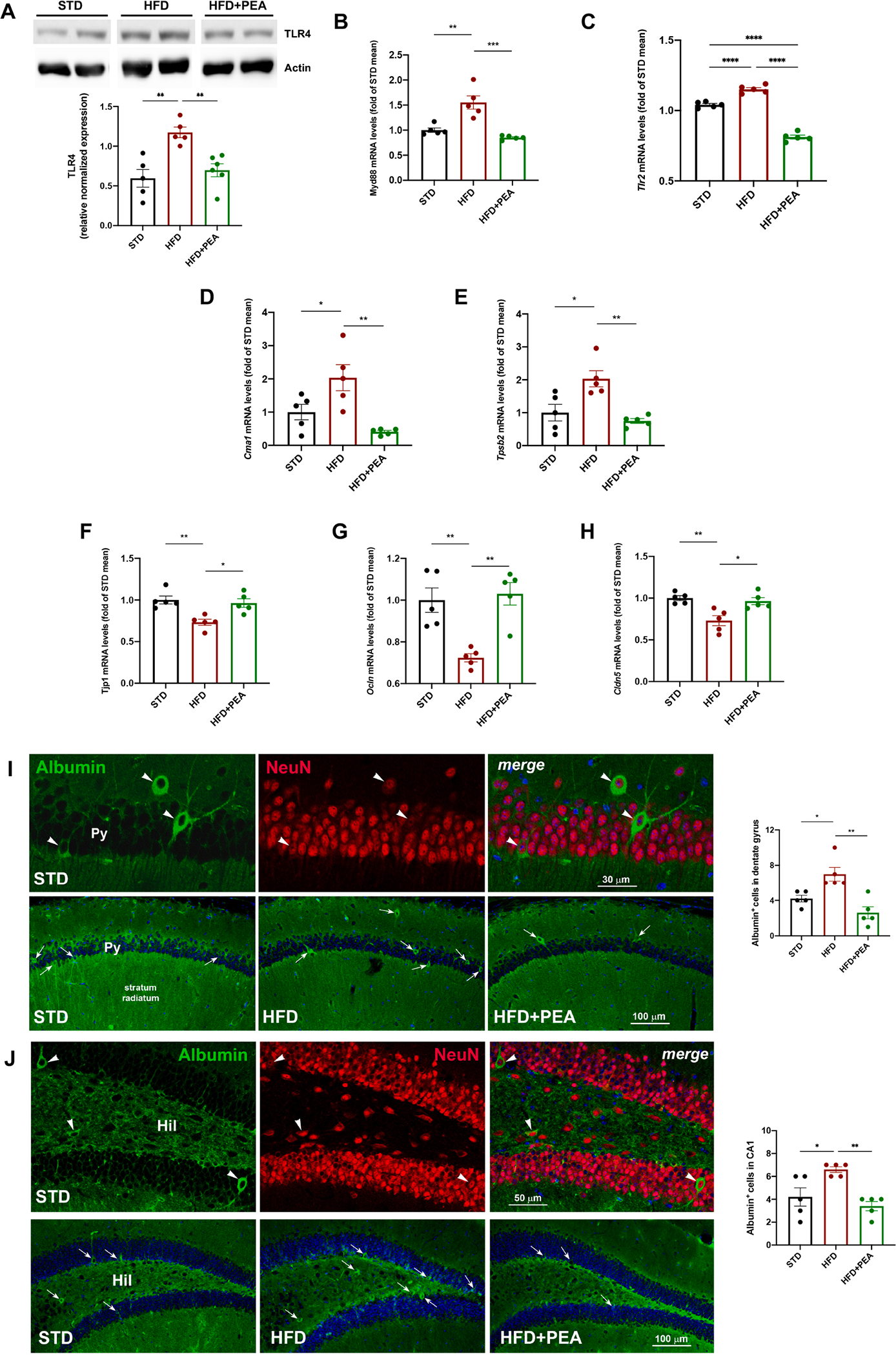
Western blot for (A) TLR4, and Real-Time PCR analysis of (B) Myd88, and (C) Tlr2, (D) Cma1, (E) Tpsb2 (F) Tjp1, (G) Ocln, and (H) Cldn5 in the hippocampus of all experimental groups (at least n = 5 per group for western blot; n = 5 per group for Real-Time PCR). Morphometric analysis of albumin-immunoreactive cells in hippocampal (I) DG and (J) CA1 subfield. As shown in the upper panels of I and J, most albumin-positive cells (green, arrowheads) are also positive for the neuronal marker NeuN (red, arrowheads). The lower panels of I and J represent images depicting albumin staining in the DG and, respectively, CA1 subfield for each experimental condition. Hil, hilus; Py, pyramidal cell layer. In the merged images, cell nuclei are stained with TO-PRO3. The semiquantitative evaluation of albumin-positive neuronal-like profiles (n = 5 per group) is also shown. All data are shown as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.6. The direct effect of PEA on neuroinflammation and mitochondrial dysfunction in Gluc2-insulted SH-SY5Y cells: PPAR-α involvement
An in vitro neuronal model of insulin resistance and metabolic inflammation was used for mechanistic studies (Mi et al., 2017). Human SH-SY5Y cells were challenged with a mix of glucose and glucosamine (Gluc2) to study the direct effect of PEA on neuroinflammation and mitochondrial dysfunction. Moreover, to evaluate the involvement of PPAR-α in PEA activity, the PPAR-α antagonist GW6471 was used (Fig. 7A). Cell treatment with PEA limited Gluc2-induced inflammation, reducing the transcription of TNF-α (Fig. 7B, F (3, 8) = 1.735; CON: 1.000 ± 0.004; Gluc2: 2.817 ± 0.186p < 0.0001 vs CON; Gluc2 + PEA: 1.414 ± 0.078, p < 0.0001 vs Gluc2; Gluc2 + GW + PEA: 2.262 ± 0.056, p = 0.0027 vs Gluc2 + PEA), TLR4 (Fig. 7C, F (3, 8) = 0.5453; CON: 1.000 ± 0.016; Gluc2: 2.320 ± 0.186, p = 0.0043 vs CON; Gluc2 + PEA: 0.580 ± 0.044, p = 0.0007 vs Gluc2; Gluc2 + GW + PEA: 2.337 ± 0.294, p = 0.0006 vs Gluc2 + PEA), and NLRP3 (Fig. 7D, F (3, 8) = 1.345; CON: 1.000 ± 0.003; Gluc2: 2.365 ± 0.185, p = 0.0002 vs CON; Gluc2 + PEA: 1.501 ± 0.058, p = 0.0056 vs Gluc2; Gluc2 + GW + PEA: 2.104 ± 0.141, p = 0.045 vs Gluc2 + PEA). The anti-inflammatory activities of PEA were blunted by GW6471. In another set of experiments, we evaluated PEA effect on mitochondrial dysfunction of insulted SH-SY5Y cells, in presence or not of GW6471, using Mito Stress assay by Seahorse analyzer (Fig. 7E). PEA treatment recovered mitochondrial energy metabolism altered by Gluc2 stimulation, improving basal and maximal respiration, promoting ATP production, and increasing proton leak (Fig. 7F). The co-incubation with GW6471 caused the loss of PEA effects, confirming the key role of PPAR-α (Basal respiration: F (3, 8) = 0.5116; CON: 87.43 ± 6.177; Gluc2: 48.03 ± 6.924, p = 0.0012 vs CON; Gluc2 + PEA: 81.41 ± 3.223, p = 0.0047 vs Gluc2; Gluc2 + GW + PEA: 50.35 ± 3.944, p = 0.081 vs Gluc2 + PEA; ATP production: F (3, 8) = 0.5016; CON: 76.28 ± 5.694; Gluc2: 39.81 ± 6.119, p = 0.001 vs CON; Gluc2 + PEA: 69.03 ± 2.816, p = 0.0059 vs Gluc2; Gluc2 + GW + PEA: 40.65 ± 3.665, p = 0.0074 vs Gluc2 + PEA; proton leak: F (3, 8) = 0.2326; CON: 11.14 ± 0.685; Gluc2: 8.213 ± 0.846, p = 0.0664 vs CON; Gluc2 + PEA: 12.38 ± 0.465, p = 0.0065 vs Gluc2; Gluc2 + GW + PEA: 9.7 ± 0.709, p = 0.1066 vs Gluc2 + PEA; maximal respiration: F (3, 8) = 0.6235; CON: 116.8 ± 9.752; Gluc2: 48.20 ± 7.207, p < 0.0001 vs CON; Gluc2 + PEA: 95.91 ± 4.976, p = 0.0021 vs Gluc2; Gluc2 + GW + PEA: 75.84 ± 3.853, p = 0.359 vs Gluc2 + PEA).
Fig. 7. The role of PPAR-α in PEA effects on inflammation and mitochondrial function in glucose þ glucosamine (Gluc2)-insulted SH-SY5Y cells.
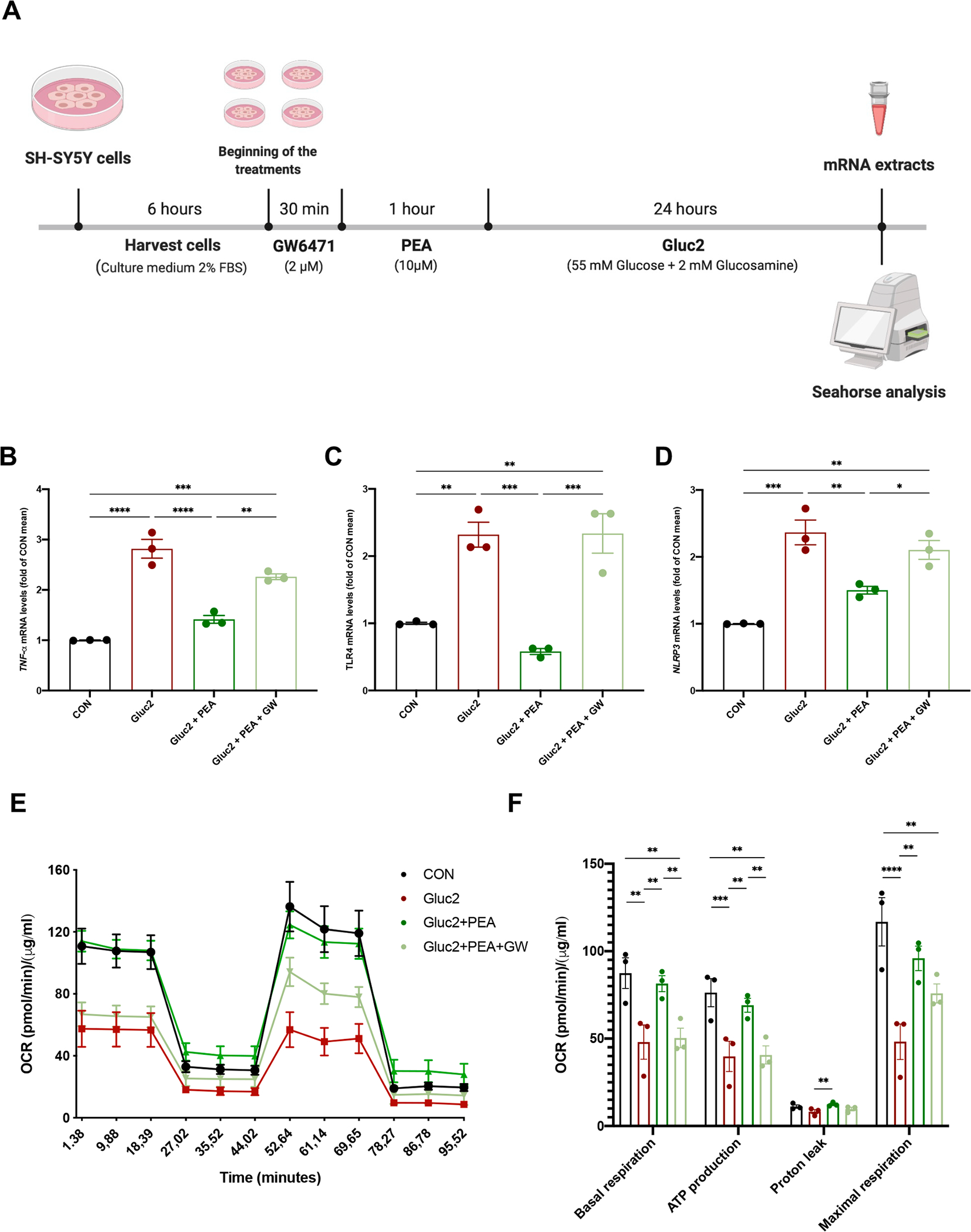
(A) The experimental protocol on human SH-SY5Y cells. The mRNA extracts of all treated cells for (B) TNF-α, (C) TLR4, and (D) NLRP3. (E) SH-SY5Y cells were undergone to Cell Mito Stress Test in Seahorse analyzer, evaluating the basal oxygen consumption rate (OCR), after the addition of oligomycin, the uncoupler FCCP, rotenone, and antimycin A. (F) Basal respiration, ATP production, proton leak, and maximal respiration were determined. OCR was normalized with the protein amount by Bradford assay. Data are the mean ± SEM of three different experiments with three replicates. All data are shown as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, and **** p < 0.0001.
4. Discussion
Obesity is often associated with a high prevalence of mood disorders besides being a risk factor for other comorbidities, such as cardiovascular diseases, type 2 diabetes, and cancer (Farzi et al., 2019). Animal models of HFD feeding have already pinpointed how the inflammatory process associated with obesity may be involved in peripheral and brain disorders. Our recent data demonstrated the capability of PEA in limiting weight gain and hepatic metabolic dysfunction and inflammation, restoring glucose and lipid homeostasis in obese mice (Annunziata et al., 2020). PEA also showed beneficial effects on depressive-like phenotype and cognitive deficit in obese mice, related to virtuous pathways, which led to synaptic strength and neurogenesis (Lama et al., 2021).
Here, we show that HFD animals exhibited altered serum parameters, characterized by elevated levels of cytokines (TNF-α, IL-1β), chemokines (MCP-1), and endotoxemia, defining metainflammation. Interestingly, PEA treatment counteracted the increase of circulating inflammatory factors, limiting the “knock-on effect” on peripheral or central tissues and related dysfunctions.
Fat overnutrition initially induces hypothalamic inflammation and dysfunction in response to excessive fatty acid flux (Cai and Khor, 2019; Lee et al., 2020). The hypothalamus also generates adaptative feedback signals to inflammation in other brain structures. Long-term HFD feeding induces hippocampal neuroinflammation (Wang et al., 2018) due to BBB leakage specifically in the hippocampus, rather than other brain structures, i.e., prefrontal cortex or cerebellum (Guillemot-Legris and Muccioli, 2017). In our study, PEA treatment not only limited inflammation in both hypothalamus (NF-κB and IL-1β) and hippocampus (TNF-α and IL-1β) but also improved hippocampal BBB integrity restoring tight junction transcription and reducing endogenous albumin extravasation and neuronal uptake. The bolstering effect of PEA on BBB integrity was associated with a reduction of central immune cells, evidenced in both hypothalamus and hippocampus. Very recent findings pinpoint the protective effect of PEA in BBB dysfunction-related diseases. After cerebral ischaemic/reperfusion, PEA regulated paracellular permeability through the prevention of cytoskeletal alterations (Kong et al., 2021). Moreover, Li et al. (2021) showed that the inhibition of N-acylethanolamine acid amidase (NAAA), a specific deactivating PEA enzyme, significantly limited BBB disruption and alleviated the traumatic brain injury in mice, proposing a new therapeutic approach focused on NAAA/PEA/PPAR-α pathway.
Microglia and astrocytes play a pivotal role in obesity-induced neuroinflammation (Le Thuc et al., 2017; Yu et al., 2019). PEA showed its anti-inflammatory effects by the decreased GFAP and Iba1 positive cells in both areas compared to HFD-fed mice. Interestingly, glial cells are generally located close to mast cells in inflamed brain areas. Unlike resident microglia, mast cells reside in the brain parenchyma and can cross BBB when impaired (Skaper, 2017). Since the perivascular location and the capability to release a broad range of pre-formed detrimental factors, mast cells play a role in the early phase of neuroinflammation, preceding glial activation (Wang et al., 2020). In our experimental conditions, besides glial marker decrease, PEA also reduced the transcription of murine specific chymase 1 and tryptase β2 in the hippocampus, as a likely limited mast cell activation. These data suggest PEA capability to blunt the unwholesome interaction between mast cells and glia, whose activation strongly contributes to BBB leakage and neuroinflammation (Hendriksen et al., 2017).
Consistent with reducing hippocampal inflammation, PEA treatment down-regulated TLR2 and TLR4 with its downstream coactivator MyD88. In obesity-induced neuroinflammation TLR4 was found overexpressed in hypothalamus and hippocampus (Guillemot-Legris and Muccioli, 2017), where TLR4 pathway can be activated by saturated fatty acids similarly to LPS (Rocha et al., 2016). The activation of TLR4/MyD88 pathway triggers the release of cytokines and chemokines which induces innate immune response and the following neuroinflammation (Leitner et al., 2019). Therefore, the reduced expression of TLR4 by PEA is consistent with the shutdown of the inflammatory cascade. Notably, TLR4 and TLR2 activation on mast cells increases cytokine release, favouring microglial pro-inflammatory response, which in turn amplifies mast cell-induced TLR overexpression (Pietrzak et al., 2011). As our data demonstrated, the blunted synthesis of pro-inflammatory cytokines by PEA can stop this vicious cycle among brain immune cells, limiting the progression of enduring neuroinflammation. On the other hand, PEA showed a neuroprotective effect on the hippocampus by improving neurogenesis and synaptic plasticity (Cristiano et al., 2018; Lama et al., 2021).
Lines of evidence showed that obesity is associated with HPA axis dysregulation (Dionysopoulou et al., 2021), which contributes to the pathogenesis of anxiety (Tafet and Nemeroff, 2020). Neuroinflammation affects hypothalamic inter-connections with brain regions regulating mood and anxiety and increases CRH release (Silverman et al., 2005). Consistently, our data demonstrated an increased transcription of CRH and its receptor CRHR1 in the hypothalamus of obese mice, normalized by PEA treatment. Nevertheless, in our experimental conditions, no change in serum ACTH and corticosterone levels was shown among groups, according to previous data on HFD-fed mice (Lizarbe et al., 2018; Pierre et al., 2019). These findings suggest an impairment of the HPA system, mostly implicating the hypothalamus.
Several preclinical studies have already demonstrated the anxiolytic effect of PEA, such as chronic corticosterone exposure (Crupi et al., 2013) or the socially isolated mouse exposed to fear conditioning (Locci and Pinna, 2019) without any metabolic dysfunction. Here, we demonstrate the counteractive effect of PEA on anxiety-like behavior induced by HFD in mice. In open field test, PEA-treated mice were more prone to explore “dangerous” areas and likely exposed to potential threats than untreated obese mice.
Since the amygdalar complex is considered an important site in managing the stress response and anxiety-related behaviors (Roozendaal et al., 2009), we evaluated neurochemical monoamine and neurotransmitter patterns in this brain area. The behavioral improvement induced by PEA was paralleled by a non-significant trend of reduction of noradrenaline and 5-HT. Conversely, PEA-induced decrease in DA levels, without significant change in DOPAC and HVA, leads to an increase in DA turnover, thus suggesting the involvement of different pathways other than catabolic enzymes. Evidence on the mechanisms underlying mood disorders has shown an impairment of the excitatory-inhibitory balance involving glutamatergic and GABAergic neurotransmission in the amygdala. Notably, GLU levels were unchanged under overnutrition, while the reduction of GABA levels was reverted by PEA treatment, indicating a restoration of GABAergic inhibitory control. This finding is consistent with PEA capability in inducing an increase in allopregnanolone levels (Locci and Pinna, 2019; Sasso et al., 2012), leading to a reinforcement of GABAergic tone through the positive modulation of GABA(A) receptor by the hormone.
Our data show a relevant effect of PEA on the metainflammation in obese mice, reducing systemic detrimental factors which impair the BBB and trigger neuroinflammation. However, we cannot exclude a direct central effect of PEA since it can cross the BBB after peripheral administration (Petrosino and Di Marzo, 2017), even more in the ultra-micronized formulation with improved oral bioavailability and efficacy (Petrosino et al., 2018). As previously shown by pharmacokinetics studies, 20 min after oral administration, PEA is mainly accumulated in the rat hypothalamus (Artamonov et al., 2005). Similarly, Beggiato et al. (2020) showed more recently that orally administered ultramicronized PEA is adsorbed and distributed in the mouse hypothalamus, measuring a significant peak of plasmatic and tissue concentrations between 1.0- and 1.5-hour post-administration and a drop to baseline values after 4 h. Consistently, we found similar concentrations of PEA 4 h-post administration in the hypothalamus of treated and untreated HFD mice, and a similar pattern of concentrations was shown in the hippocampus. However, while no relevant difference was shown in hippocampal levels among groups, a significant increase of PEA levels was revealed in the hypothalamus from PEA-treated HFD animals than STD mice (Figure S2).
The possible direct central effect is supported by in vitro experiments on neuronal cells. We have demonstrated PEA capability in reducing the expression of different inflammatory markers (i.e., TNF-α, TLR4, and NLRP3) associated with the metabolic challenge of glucose and glucosamine. In addition, PEA significantly restored mitochondrial bioenergetics in terms of basal and maximal respiration, ATP-linked respiration, and proton leak. Indeed, mitochondrial dysfunction has been closely associated with stress-related central disorders, leading to altered brain metabolism, synaptic function, and plasticity (Daniels et al., 2020; Reddy and Beal, 2008). We have recently demonstrated that HFD consumption led to mitochondrial dysfunction in the brain synaptosomal fraction. This alteration paralleled with the reduction of brain-derived neurotrophic factor (Cavaliere et al., 2019), a key brain signaling molecule stimulated by PEA treatment in different animal models (Lama et al., 2021; Cristiano et al., 2018). Notably, in in vitro experiments, we showed the involvement of PPAR-α activation in the PEA effects since the blockade of the receptor by the selective antagonist GW6471 blunted PEA anti-inflammatory activity and reversed the improvement of mitochondrial bioenergetics. Other findings have demonstrated the direct PPAR-α-mediated effects of PEA at central level. Locci and Pinna (2019) revealed that PEA induced corticolimbic allopregnanolone synthesis in an animal model of post-traumatic stress disorder, evidencing the obligatory role of PPAR-α in PEA effects. These findings were consistent with our previous data on the de novo synthesis of neurosteroids by PEA in astrocytes and the modulation of pentobarbital-evoked hypnotic effect in mice (Mattace Raso et al., 2011; Sasso et al., 2010). Although our study showed for the first time PEA effect on HFD-induced anxiety-like behavior, there are some limitations. It would be interesting to discriminate between central and peripheral mechanisms, that are strictly interconnected in this model, and to address the different impact of their contribution in the neuroprotective and anti-inflammatory effect of PEA. However, the pharmacological control of inflammation, immunity, and neurotransmitter imbalance by PEA strengthens its therapeutic potential for neuropsychiatric comorbidities of obesity, also considering its pleiotropic properties, safety profile, and converging homeostatic mechanisms affecting central targets.
Supplementary Material
Acknowledgments
This article is dedicated to the memory of dr. Francesco Della Valle, who passed away while this paper was being peer-reviewed. Those who have known him, miss his devotion to science and life. We thank Mr. Giovanni Esposito, Mr. Angelo Russo, and Dr. Antonio Baiano for animal care and technical assistance. Giuseppina Mattace Raso and Rosaria Meli were recipients of the “Programme of International Exchange (2017) between the University of Naples Federico II and Foreign Universities or Research centres for short-term mobility” to collaborate with Sabrina Diano (Yale University, New Haven). Adriano Lama received a 1-year outgoing international fellowship issued by the Italian Society of Pharmacology (2017) to join Sabrina Diano’s lab. Italian Ministry of Economic Development (MISE) partially financed the study (Project n. F/200052/01/X45).
Funding
This work was partially supported by a grant assigned to Prof. Rosaria Meli from Epitech Group S.p.A. Dr. Adriano Lama acknowledges that he has benefited from a fellowship sponsored by Epitech Group S.p. A. Prof. Sabrina Diano was supported by the NIH DK120321 and DK097566. Dr. Stefania Petrosino is an employee of Epitech Group SpA and is co-inventor on patent on Adelmidrol, which is unrelated to the present study.
Abbreviations:
- 5-HIAA
5-hydroxy-indol acetic acid
- ACTH
adrenocorticotropic hormone
- AMY
amygdala
- BBB
blood-brain barrier
- Cldn5
claudin 5
- CRH
corticotropin-releasing hormone
- DA
dopamine; DG, dentate gyrus
- DOPAC
3,4-dihydroxyphenylacetic acid
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- GABA
γ-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- GLU
glutamic acid
- HFD
high-fat diet
- HPA
hypothalamic-pituitaryadrenal
- HVA
homovanillic acid
- IL
interleukin
- LPS
lipopolysaccharide
- MCP
monocyte chemoattractant protein
- MyD
myeloid differentiation primary response gene
- NA
noradrenaline
- NLRP3
NLR family pyrin domain containing 3
- Ocln
occludin
- OCR
oxygen consumption rate
- PEA
palmitoylethanolamide
- PPAR
peroxisome proliferator-activated receptor
- STD
control group; Tjp1, tight junction protein 1
- TLR
toll-like receptor
- TNF
tumor necrosis factor
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2022.02.008.
References
- Annunziata C, Lama A, Pirozzi C, Cavaliere G, Trinchese G, Di Guida F, Nitrato Izzo A, Cimmino F, Paciello O, De Biase D, Murru E, Banni S, Calignano A, Mollica MP, Mattace Raso G, Meli R, 2020. Palmitoylethanolamide counteracts hepatic metabolic inflexibility modulating mitochondrial function and efficiency in diet-induced obese mice. FASEB J. 34 (1), 350–364. 10.1096/fj.201901510RR. [DOI] [PubMed] [Google Scholar]
- Araujo JR, Tomas J, Brenner C, Sansonetti PJ, 2017. Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie 141, 97–106. 10.1016/j.biochi.2017.05.019. [DOI] [PubMed] [Google Scholar]
- Artamonov M, Zhukov O, Shuba I, Storozhuk L, Khmel T, Klimashevsky V, Mikosha A, Gula N, 2005. Incorporation of labelled N-acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under x-ray irradiation. Ukr. Biokhim. Zh. 1999 (77), 51–62. [PubMed] [Google Scholar]
- Baker KD, Loughman A, Spencer SJ, Reichelt AC, 2017. The impact of obesity and hypercaloric diet consumption on anxiety and emotional behavior across the lifespan. Neurosci. Biobehav. Rev. 83, 173–182. 10.1016/j.neubiorev.2017.10.014. [DOI] [PubMed] [Google Scholar]
- Beggiato Sarah, Tomasini Maria Cristina, Cassano Tommaso, Ferraro Luca, 2020. Chronic oral palmitoylethanolamide administration rescues cognitive deficit and reduces neuroinflammation, oxidative stress, and glutamate levels in a transgenic murine model of Alzheimer’s Disease. Journal of Clinical Medicine 9(2), 428. 10.3390/jcm9020428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkseth KE, Guyenet SJ, Melhorn SJ, Lee D, Thaler JP, Schur EA, Schwartz MW, 2014. Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: a combined immunohistochemical and magnetic resonance imaging study. Endocrinology 155, 2858–2867. 10.1210/en.2014-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braganza O, Bedner P, Huttmann K, von Staden E, Friedman A, Seifert G, Steinhauser C, 2012. Albumin is taken up by hippocampal NG2 cells and astrocytes and decreases gap junction coupling. Epilepsia 53, 1898–1906. 10.1111/j.1528-1167.2012.03665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman LB, Thompson MM, Moreno HN, Ellacott KLJ, 2013. Regional astrogliosis in the mouse hypothalamus in response to obesity. J. Comp. Neurol. 521 (6), 1322–1333. 10.1002/cne.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Khor S, 2019. “Hypothalamic Microinflammation” paradigm in aging and metabolic diseases. Cell Metab. 30 (1), 19–35. 10.1016/j.cmet.2019.05.021. [DOI] [PubMed] [Google Scholar]
- Cavaliere G, Trinchese G, Penna E, Cimmino F, Pirozzi C, Lama A, Annunziata C, Catapano A, Mattace Raso G, Meli R, Monda M, Messina G, Zammit C, Crispino M, Mollica MP, 2019. High-fat diet induces neuroinflammation and mitochondrial impairment in mice cerebral cortex and synaptic fraction. Front. Cell. Neurosci. 13, 509. 10.3389/fncel.2019.00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristiano C, Pirozzi C, Coretti L, Cavaliere G, Lama A, Russo R, Lembo F, Mollica MP, Meli R, Calignano A, Mattace Raso G, 2018. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: contribution of central and peripheral mechanisms. Brain Behav. Immun. 74, 166–175. 10.1016/j.bbi.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Crupi R, Paterniti I, Ahmad A, Campolo M, Esposito E, Cuzzocrea S, 2013. Effects of palmitoylethanolamide and luteolin in an animal model of anxiety/depression. CNS Neurol. Disord. Drug Targets 12, 989–1001. 10.2174/18715273113129990084. [DOI] [PubMed] [Google Scholar]
- Daniels TE, Olsen EM, Tyrka AR, 2020. Stress and Psychiatric Disorders: The Role of Mitochondria. Annu. Rev. Clin. Psychol. 16 (1), 165–186. 10.1146/annurev-clinpsy-082719-104030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionysopoulou S, Charmandari E, Bargiota A, Vlahos NF, Mastorakos G, Valsamakis G, 2021. The role of hypothalamic inflammation in diet-induced obesity and its association with cognitive and mood disorders. Nutrients 13 (2), 498. 10.3390/nu13020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP, 2017. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 6, 366–373. 10.1016/j.molmet.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzi A, Hassan AM, Zenz G, Holzer P, 2019. Diabesity and mood disorders: Multiple links through the microbiota-gut-brain axis. Mol. Aspects Med. 66, 80–93. 10.1016/j.mam.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Guillemot-Legris O, Muccioli GG, 2017. Obesity-induced neuroinflammation: beyond the hypothalamus. Trends Neurosci. 40 (4), 237–253. 10.1016/j.tins.2017.02.005. [DOI] [PubMed] [Google Scholar]
- Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA, 2017. Mast cells in neuroinflammation and brain disorders. Neurosci. Biobehav. Rev. 79, 119–133. 10.1016/j.neubiorev.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Kong D, Xie B, Li Y, Xu Y, 2021. PEA prevented early BBB disruption after cerebral ischaemic/reperfusion (I/R) injury through regulation of ROCK/MLC signaling. Biochem. Biophys. Res. Commun. 566, 164–169. 10.1016/j.bbrc.2021.06.019. [DOI] [PubMed] [Google Scholar]
- Lama A, Annunziata C, Coretti L, Pirozzi C, Di Guida F, Nitrato Izzo A, Cristiano C, Mollica MP, Chiariotti L, Pelagalli A, Lembo F, Meli R, Mattace Raso G, 2019. N-(1-carbamoyl-2-phenylethyl) butyramide reduces antibiotic-induced intestinal injury, innate immune activation and modulates microbiota composition. Sci. Rep. 9, 4832. 10.1038/s41598-019-41295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama A, Pirozzi C, Annunziata C, Morgese MG, Senzacqua M, Severi I, Calignano A, Trabace L, Giordano A, Meli R, Mattace Raso G, 2021. Palmitoylethanolamide counteracts brain fog improving depressive-like behaviour in obese mice: possible role of synaptic plasticity and neurogenesis. Br. J. Pharmacol. 178 (4), 845–859. 10.1111/bph.15071. [DOI] [PubMed] [Google Scholar]
- Le Thuc O, Stobbe K, Cansell C, Nahon JL, Blondeau N, Rovere C, 2017. Hypothalamic inflammation and energy balance disruptions: spotlight on chemokines. Front. Endocrinol. (Lausanne) 8, 197. 10.3389/fendo.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Suk K, Yu R, Kim MS, 2020. Cellular contributors to hypothalamic inflammation in obesity. Mol. Cells 43, 431–437. 10.14348/molcells.2020.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner GR, Wenzel TJ, Marshall N, Gates EJ, Klegeris A, 2019. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 23 (10), 865–882. 10.1080/14728222.2019.1676416. [DOI] [PubMed] [Google Scholar]
- Li C, Xu MM, Wang K, Adler AJ, Vella AT, Zhou B, 2018. Macrophage polarization and meta-inflammation. Transl. Res. 191, 29–44. 10.1016/j.trsl.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou P, Hu T, Ren J, Xu Y, Qiu Y, Lu C, Li Y, 2021. NAAA inhibitor F96 attenuates BBB disruption and secondary injury after traumatic brain injury (TBI). Eur. J. Pharmacol. 912, 174561. 10.1016/j.ejphar.2021.174561. [DOI] [PubMed] [Google Scholar]
- Lizarbe B, Soares AF, Larsson S, Duarte JMN, 2018. Neurochemical modifications in the hippocampus, cortex and hypothalamus of mice exposed to long-term high-fat diet. Front. Neurosci. 12, 985. 10.3389/fnins.2018.00985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locci A, Pinna G, 2019. Stimulation of peroxisome proliferator-activated receptor-alpha by N-palmitoylethanolamine engages allopregnanolone biosynthesis to modulate emotional behavior. Biol. Psychiatry 85, 1036–1045. 10.1016/j.biopsych.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Mattace Raso G, Esposito E, Vitiello S, Iacono A, Santoro A, D’Agostino G, Sasso O, Russo R, Piazza PV, Calignano A, Meli R, 2011. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: involvement of peroxisome-proliferator activated receptor-alpha. J. Neuroendocrinol. 23, 591–600. 10.1111/j.1365-2826.2011.02152.x. [DOI] [PubMed] [Google Scholar]
- Mattace Raso G, Russo R, Calignano A, Meli R, 2014a. Palmitoylethanolamide in CNS health and disease. Pharmacol. Res. 86, 32–41. 10.1016/j.phrs.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Mattace Raso G, Santoro A, Russo R, Simeoli R, Paciello O, Di Carlo C, Diano S, Calignano A, Meli R, 2014b. Palmitoylethanolamide prevents metabolic alterations and restores leptin sensitivity in ovariectomized rats. Endocrinology 155, 1291–1301. 10.1210/en.2013-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Y, Qi G, Fan R, Qiao Q, Sun Y, Gao Y, Liu X, 2017. EGCG ameliorates high- fat- and high-fructose-induced cognitive defects by regulating the IRS/AKT and ERK/CREB/BDNF signaling pathways in the CNS. FASEB J. 31 (11), 4998–5011. 10.1096/fj.201700400RR. [DOI] [PubMed] [Google Scholar]
- Milaneschi Y, Simmons WK, van Rossum EFC, Penninx BW, 2019. Depression and obesity: evidence of shared biological mechanisms. Mol. Psychiatry 24 (1), 18–33. 10.1038/s41380-018-0017-5. [DOI] [PubMed] [Google Scholar]
- Miller AA, Spencer SJ, 2014. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav. Immun. 42, 10–21. 10.1016/j.bbi.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Cordaro M, Verde R, Schiano Moriello A, Marcolongo G, Schievano C, Siracusa R, Piscitelli F, Peritore AF, Crupi R, Impellizzeri D, Esposito E, Cuzzocrea S, Di Marzo V, 2018. Oral ultramicronized palmitoylethanolamide: plasma and tissue levels and spinal anti-hyperalgesic effect. Front. Pharmacol. 9, 249. 10.3389/fphar.2018.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V, 2017. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 174 (11), 1349–1365. 10.1111/bph.13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre A, Regin Y, Van Schuerbeek A, Fritz EM, Muylle K, Beckers T, Smolders IJ, Singewald N, De Bundel D, 2019. Effects of disrupted ghrelin receptor function on fear processing, anxiety and saccharin preference in mice. Psychoneuroendocrinology 110, 104430. 10.1016/j.psyneuen.2019.104430. [DOI] [PubMed] [Google Scholar]
- Pietrzak A, Wierzbicki M, Wiktorska M, Brzezińska-Błaszczyk E, 2011. Surface TLR2 and TLR4 expression on mature rat mast cells can be affected by some bacterial components and proinflammatory cytokines. Mediators Inflamm. 2011, 1–11. 10.1155/2011/427473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirozzi C, Lama A, Annunziata C, Cavaliere G, De Caro C, Citraro R, Russo E, Tallarico M, Iannone M, Ferrante MC, Mollica MP, Mattace Raso G, De Sarro G, Calignano A, Meli R, 2020. Butyrate prevents valproate-induced liver injury: In vitro and in vivo evidence. FASEB J. 34 (1), 676–690. 10.1096/fj.201900927RR. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF, 2008. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14 (2), 45–53. 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH, 2016. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 244, 211–215. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S, 2009. Stress, memory and the amygdala. Nat. Rev. Neurosci. 10 (6), 423–433. 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Sasso O, La Rana G, Vitiello S, Russo R, D’Agostino G, Iacono A, Russo E, Citraro R, Cuzzocrea S, Piazza PV, De Sarro G, Meli R, Calignano A, 2010. Palmitoylethanolamide modulates pentobarbital-evoked hypnotic effect in mice: involvement of allopregnanolone biosynthesis. Eur. Neuropsychopharmacol. 20 (3), 195–206. 10.1016/j.euroneuro.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Sasso O, Russo R, Vitiello S, Raso GM, D’Agostino G, Iacono A, La Rana G, Vallee M, Cuzzocrea S, Piazza PV, Meli R, Calignano A, 2012. Implication of allopregnanolone in the antinociceptive effect of N-palmitoylethanolamide in acute or persistent pain. Pain 153, 33–41. 10.1016/j.pain.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Mollgard K, Habgood MD, 2015. Markers for blood-brain barrier integrity: how appropriate is Evans blue in the twenty-first century and what are the alternatives? Front. Neurosci. 9, 385. 10.3389/fnins.2015.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Wooten MC, 2015. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. e52434 10.3791/52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman MN, Pearce BD, Biron CA, Miller AH, 2005. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 18 (1), 41–78. 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, 2017. Impact of inflammation on the blood-neural barrier and blood-nerve interface: from review to therapeutic preview. Int. Rev. Neurobiol. 137, 29–45. 10.1016/bs.irn.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Zusso M, Giusti P, 2017. Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist 23 (5), 478–498. 10.1177/1073858416687249. [DOI] [PubMed] [Google Scholar]
- Tafet GE, Nemeroff CB, 2020. Pharmacological treatment of anxiety disorders: the role of the HPA Axis. Front. Psychiatry 11, 443. 10.3389/fpsyt.2020.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsan L, Decarie-Spain L, Noble EE, Kanoski SE, 2021. Western diet consumption during development: setting the stage for neurocognitive dysfunction. Front. Neurosci. 15, 632312 10.3389/fnins.2021.632312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos M, Robblee M, Benjamin D, Nomura D, Xu A, Koliwad S, 2014. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 9 (6), 2124–2138. 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP, Koliwad SK, 2017. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 26 (1), 185–197.e3. 10.1016/j.cmet.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araujo S, Redeker S, van Schaik R, Aronica E, Gorter JA, 2007. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130 (2), 521–534. 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Venema W, Severi I, Perugini J, Di Mercurio E, Mainardi M, Maffei M, Cinti S, Giordano A, 2020. Ciliary neurotrophic factor acts on distinctive hypothalamic arcuate neurons and promotes leptin entry into and action on the mouse hypothalamus. Front. Cell. Neurosci. 14, 140. 10.3389/fncel.2020.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waise TMZ, Toshinai K, Naznin F, NamKoong C, Md Moin AS, Sakoda H, Nakazato M, 2015. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 464 (4), 1157–1162. 10.1016/j.bbrc.2015.07.097. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sha H, Zhou L, Chen Y, Zhou Q, Dong H, Qian Y, 2020. The mast cell is an early activator of lipopolysaccharide-induced neuroinflammation and blood-brain barrier dysfunction in the hippocampus. Mediators Inflamm. 2020, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yuan J, Yu Z, Lin L.i., Jiang Y, Cao Z, Zhuang P, Whalen MJ, Song B.o., Wang X-J, Li X, Lo EH, Xu Y, Wang X, 2018. FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol. Neurobiol. 55 (6), 4702–4717. 10.1007/s12035-017-0663-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Huang H, Dong S, Sha H, Wei W, Liu C, 2019. High mobility group box-1 mediates hippocampal inflammation and contributes to cognitive deficits in high-fat high-fructose diet-induced obese rats. Brain Behav. Immun. 82, 167–177. 10.1016/j.bbi.2019.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.