

A joint computational and experimental effort unravels a peptide scaffold for self-assembled nanomaterials.
Abstract
Peptide self-assembly inspired by natural superhelical coiled coils has been actively pursued but remains challenging due to limited helicity of short peptides. Side chain stapling can strengthen short helices but is unexplored in design of self-assembled helical nanofibers as it is unknown how staples could be adapted to coiled coil architecture. Here, we demonstrate the feasibility of this design for pentapeptides using a computational method capable of predicting helicity and fiber-forming tendency of stapled peptides containing noncoded amino acids. Experiments showed that the best candidates, which carried an aromatically substituted staple and phenylalanine analogs, displayed exceptional helicity and assembled into nanofibers via specific head-to-tail hydrogen bonding and packing between staple and noncoded side chains. The fibers exhibited sheet-of-helix structures resembling the recently found collapsed coiled coils whose formation was sensitive to side chain flexibility. This study expands the chemical space of coiled coil assemblies and provides guidance for their design.
INTRODUCTION
Bioinspired nanoarchitecture has recently emerged as a versatile tool to bridge fundamental biology and nanobiotechnology disciplines. Among these tools, peptides, as the fundamental component of proteins, have surfaced as a versatile candidate for the construction of nanomaterials. Notably, the use of short peptides as building blocks has raised tremendous attention due to their unique physical, chemical, and biocompatible properties, which can be applied to diverse functional applications in biomedicine, optical engineering, and electronic nanotechnology (1–4).
It is well documented that peptides exhibit enormous variation in both sequences and secondary structures. Peptides with various secondary structures could allow for different types of molecular packing, giving rise to a wide range of organized supramolecular structures (5). In particular, ultrashort peptides are an important type of building block. Because of their diversity, flexibility, and ease of synthesis, these peptide building blocks are especially good candidates for de novo design of self-assembled materials that can be fabricated in a controllable and modular manner (6, 7).
Most of the self-assembling short peptides have been demonstrated to form supramolecular assemblies rich in β sheet structures (8). However, α-helical structures, the most prominent secondary structures of proteins, are less exploited for fabrication of materials assembled by short peptides. Thus far, the design of helical peptide–assembled nanostructures has been largely inspired by one type of naturally occurring α-helix-rich structure, coiled coils, which are composed of multiple helical peptide segments associated laterally into bundles (9, 10). The sequence-structure relationship for coiled coil structures has been extensively studied (11, 12). A special pattern of heptad repeats in amino acid sequences was identified as a key feature of helical peptide segments capable of forming coiled coils. The amino acids at specific positions of the heptad repeats participate in the lateral interfaces between the helical segments, and the impact of amino acid substitutions at these positions on the ability of peptide segments to form coiled coils is highly predictable. This sequence-structure relationship has provided invaluable guidance for the design of coiled coil–based nanostructures, including superhelical nanofibers in particular (11).
Naturally occurring coiled coil supramolecular structures are often assembled by large peptide segments containing up to hundreds of amino acids. There have been continuous efforts seeking short peptide segments that could be used as building blocks that would permit large-scale synthesis and controlled chemical modification for improved functionality. Pioneering studies have demonstrated that, with properly designed peptide termini, peptide segments as short as three to four heptad repeats can assemble into long superhelical fibers via end-to-end stacking (13, 14). However, the weak intrinsic helical stability of shorter peptides containing only naturally occurring amino acids is one of the main obstacles to the formation of self-assembled helical structures.
There have been numerous efforts in designing self-assembling ultrashort peptides using helix-promoting noncoded amino acids such as β–amino acids and α-aminoisobutyric acid (15, 16). For example, it has recently been shown that α-aminoisobutyric acids could stabilize a single helical heptad that can self-assemble into dimeric fibrous structures (17, 18). This helical heptad has served as a template for the design of other types of assemblies (19). Similarly, a pentapeptide containing α-aminoisobutyric acids was found to assume a kinked short helical structure that self-assembled into superhelical column structures (20). In another study, a pentapeptide was observed to fold into an α helix upon dimerization through the formation of an intermolecular disulfide bond (21). The helical dimers were further assembled into nanosheet structures.
Despite these advances, the molecular design of self-assembling ultrashort helical peptides remains challenging. In many cases, there is a lack of detailed structural models for supramolecular structures assembled by the short peptides. In addition, it is often unclear how the assembled structures would change in response to mutation or chemical modification. In theory, the rational design of these ultrashort peptides could be enhanced by the design rules of long α-helical peptides envisioned from coiled coil structures. Unlike long peptide segments, however, the helical propensity of ultrashort peptides is less tolerant to mutations or chemical modification. A further complication is that short peptides with noncoded amino acid substitutions, due to direct modification to their backbones, can often deviate from the canonical helical structures seen in coiled coils (20). As a result, with few exceptions (18, 21), the empirical rules derived for naturally occurring coiled coils are rarely applicable to the design of self-assembling ultrashort peptides.
Side chain stapling is a common strategy for enhancing the helical propensity of peptides. Recently, we have shown that a thioether-containing staple can promote the folding of peptides as short as five amino acids into canonical α-helical structures (22). Another special characteristic of this staple is its additional substitution site, which enriches the chemical space. When decorated with aromatic groups on their staples, the peptides are capable of assembling into large-scale assemblies (23). Despite these initial applications, the rational design of this stapled peptide remains hindered by multiple factors. Apart from the sensitivity of their helical propensity to chemical modification (22), there remains an insufficient understanding of how stapled peptides self-assemble. It is especially unclear whether and how a pentapeptide with a bulky staple would fit into the coiled coil framework derived from heptad repeats of naturally occurring amino acids, which thus impedes the application of this stapled peptide system in the molecular design of peptide-based nanofibers.
In this study, we demonstrate the feasibility of these designs with a combined computational and experimental approach. By adapting the structural rules for coiled coils to our stapled peptide system, we constructed a library of stapled pentapeptides and screened them with multiscale computational methods. In particular, the use of a hybrid-resolution model (24, 25) allowed us to assess the ability of a given peptide to assemble into nanofibers without the need to perform countless experiments. This method also allowed us to predict the assembly behavior of peptides, which is normally experimentally intractable for aggregation-prone systems. This detailed information assisted us in understanding the structural details of peptide assemblies, rationalizing possible modifications to peptides, and interpreting their outcomes. With this approach, we have designed an ultrashort stapled peptide that can self-assemble into nanofibers. The molecular architecture of these assembled fibers has been characterized computationally and experimentally. We also investigated the importance of specific hydrophobic and hydrogen bonding interactions and peptide helicity for fiber formation.
RESULTS
Molecular design of self-assembling stapled helical pentapeptides
Our peptide scaffold consists of an N-terminally acetylated, C-terminally amidated pentapeptide (Fig. 1A). A thioether-containing staple is then introduced to bridge the first and the fifth amino acid residues, aimed at reducing the entropy cost of helix folding. The variable parts of this scaffold include three side chains (R2-R4 shown in Fig. 1A) and a substituent group (R1) attached to the staple. Our goal was thus to search for the substitutions at R1-R4, which would confer the peptide with the capacity to self-assemble into fibrous helical structures. In theory, one could manage this by exhausting all possible combinations of substitutions of these groups. This daunting task was simplified by applying the empirical rules for coiled coil fibers to guide our design.
Fig. 1. Design of a stapled peptide as a building block of helical assemblies.
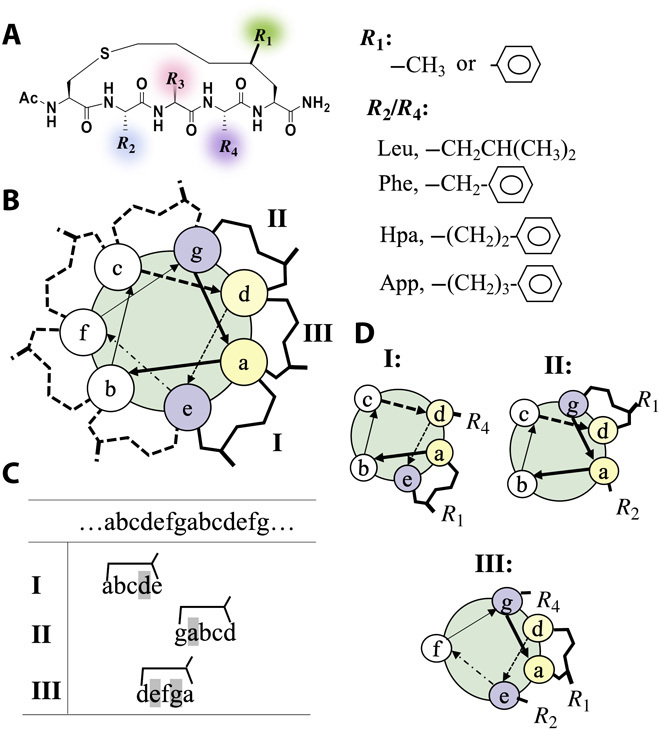
(A) Chemical structures of the stapled peptide motif investigated in this study with its four modifiable groups highlighted in colors. (B) Helical wheel view of the stapled peptide. Seven faces of an ideal helix, which are respectively occupied by heptad sequence, are denoted as a to g. The bridges surrounding the wheel denote possible positions of the staple depending on how the pentapeptide sequence is mapped onto this ideal helical wheel. Those dashed bridges indicate the placement of the staple on the hydrophilic faces of the helix, which are thus discarded. The three potentially workable ways of mapping pentapeptide onto the helix and their corresponding helical wheel representations are shown in (C) and (D), respectively, with possible chemical modifications to the peptides summarized in (A).
Coiled coils are formed through lateral associations between helical peptides. How these helical peptides interact with each other is primarily determined by the nature of amino acids on their lateral faces. Because a single helical turn comprises 3.6 residues, a helix can be thought to display seven distinct lateral faces (denoted as a-b-c-d-e-f-g; Fig. 1B), each of which harbors amino acids with a seven-residue spacing in sequence. This is why coiled coil–forming peptides usually contain heptad repeats in their sequence. The sequence pattern of the heptad repeat encodes the ability for self-assembly ability of these peptides and thereby forms the basis for the empirical rules of design for self-assembled coiled coils (11, 12). For instance, positions a and d in the heptad repeat must be occupied by hydrophobic amino acids to form the hydrophobic core of the coiled coils (11, 12). In some cases, hydrophobic residues can also be placed at positions e and g to enable additional hydrophobic interactions (26), while hydrophilic residues or small helix-promoting residues such as alanine, rather than bulky hydrophobic residues, are preferred at positions b, c, and f (12).
Our pentapeptide motif differs from conventional coiled coil peptides in that it has only five amino acid residues. Hence, of the seven lateral faces of helix, only five are occupied, and two of them are taken by the bulky hydrophobic staple. To adapt the empirical rules discussed above to this type of peptide motif, we needed to consider various ways of arranging the five amino acids on the seven lateral faces of helix while keeping nonhydrophobic lateral faces clear of the staple. Figure 1 (B and C) illustrates three possible arrangements of side chains (termed modes I to III). In mode I, for instance, residues 1 to 5 are placed on the a-b-c-d-e faces of the helix such that a and e are occupied by the staple and d is occupied by the side chain (R4) of residue 4. Thus, for this mode, hydrophobic substitutions at R1 and R4 were the most likely to increase the chance that the peptide would assemble into fibrous structures (Fig. 1D).
A library of pentapeptides was then created by varying R1, R2, and R4 for the three modes (Fig. 1A). Several possible hydrophobic substitutions for these groups were included. Both methyl and phenyl (Ph) substitutions at R1 were previously found to be crucial for helix formation of the pentapeptide, whereas other bulkier substituents greatly weakened the ability of the peptide to form a helix (22). As R2 and R4 could occupy the a or d face of the helix, leucine (Leu) and phenylalanine (Phe) substitutions were considered for both of these groups. Leu is among the most frequently observed hydrophobic amino acids at the hydrophobic faces of conventional coiled coil peptides (11, 12), while Phe is often incorporated into ultrashort helical peptides to promote self-assembly via π-π stacking of Ph rings (17, 19–21). In total, there were 20 stapled peptide candidates that were investigated in the present study.
Computational screening for self-assembling stapled peptides
To further narrow down which candidates to test experimentally, we conducted molecular dynamics (MD) simulations to assess the ability of each pentapeptide in the library to self-assemble into fibrous helical structures. Ideally, this assessment would be carried out by directly simulating self-assembly of a large number of peptide molecules using all-atom models that capture full atomic details of systems. This simulation, however, is impractical because of long time scale and enormously large configurational space associated with peptide assembly that is inaccessible to atomic simulations. Nonetheless, several studies have shown that a key prerequisite for an ultrashort helical assembling peptide is its high helical propensity (17, 19, 20). The helical propensity of ultrashort peptides is highly sensitive to mutation (17, 22), whereas that of long coiled coil–forming peptides is not. It was thus important to rule out any mutation that would hinder the helical stability of the peptide. Hence, we adopted a two-stage screening strategy to overcome the aforementioned computational challenge. We first examined the helical content of the peptide candidates through helix folding simulations. As helical content is a property pertaining to single peptide molecule, its simulation is manageable with all-atom models. In the present study, the all-atom chemistry at Harvard macromolecular mechanics (CHARMM) force field (version 27) was used. Replica exchange MD technique was used to further accelerate sampling convergence (27). The candidates predicted to have high helical contents entered the second screening stage in which a computationally less expensive model was used to explore their self-assembly behaviors.
Table 1 summarizes the calculated helicities for all of the pentapeptide candidates in our library. A select few of pentapeptides were also synthesized and characterized to validate the helicity calculation (fig. S1, A to C). An excellent correlation (r2 = 0.95) between the simulated and measured helicities was observed (fig. S1E), indicating the robustness of our helicity calculation for each of the pentapeptides. It is notable that despite the variation in helicity with substitutions, the folded helical structures remained unchanged, as all were similar to the canonical helical structure seen in the crystal of a pentapeptide with R1 = Me and R4 = Ile (fig. S1, F and G).
Table 1. Summary of the helicity and morphologies.
The simulated and experimental helicity of pentapeptides examined in the present study and the morphologies of assemblies by these peptides according to simulations.
| ID | R 1 | R 2 | R 4 |
Computed
helicity |
Computed
relative helicity |
Experiment
relative helicity |
S shape | S θ |
| 1 | -Me | Ala | Leu | 0.71 ± 0.02* | 0.77 | 0.78 | 0.49 | 0.29 |
| 2 | -Ph | Ala | Leu | 0.54 ± 0.09 | 0.59 | 0.59 | 0.39 | 0.35 |
| 3 | -Me | Leu | Ala | 0.39 ± 0.08 | 0.42 | –† | – | – |
| 4 | -Ph | Leu | Ala | 0.41 ± 0.09 | 0.45 | – | 0.48 | 0.39 |
| 5 | -Me | Ala | Phe | 0.27 ± 0.09 | 0.29 | – | – | – |
| 6 | -Ph | Ala | Phe | 0.19 ± 0.05 | 0.21 | 0.27 | 0.38 | 0.38 |
| 7 | -Me | Ala | Hpa | 0.68 ± 0.10 | 0.74 | – | 0.44 | 0.36 |
| 8 | -Ph | Ala | Hpa | 0.58 ± 0.08 | 0.63 | 0.65 | 0.53 | 0.32 |
| 9 | -Me | Ala | App | 0.93 ± 0.03 | 1.01 | – | 0.62 | 0.35 |
| 10 | -Ph | Ala | App | 0.92 ± 0.06 | 1.00 | 1.00 | 0.97‡ | 0.06 |
| 11 | -Me | Phe | Ala | 0.22 ± 0.07 | 0.24 | – | – | – |
| 12 | -Ph | Phe | Ala | 0.26 ± 0.04 | 0.28 | 0.35 | – | – |
| 13 | -Me | Hpa | Ala | 0.34 ± 0.10 | 0.37 | – | – | – |
| 14 | -Ph | Hpa | Ala | 0.30 ± 0.10 | 0.33 | – | – | – |
| 15 | -Me | App | Ala | 0.34 ± 0.09 | 0.37 | – | 0.59 | 0.39 |
| 16 | -Ph | App | Ala | 0.42 ± 0.05 | 0.46 | – | – | – |
| 17 | -Me | Leu | Leu | 0.70 ± 0.06 | 0.76 | – | 0.37 | 0.32 |
| 18 | -Ph | Leu | Leu | 0.66 ± 0.02 | 0.72 | – | 0.34 | 0.42 |
| 19 | -Me | App | App | 0.94 ± 0.08 | 1.02 | – | 0.25 | 0.36 |
| 20 | -Ph | App | App | 0.82 ± 0.10 | 0.89 | – | 0.25 | 0.36 |
| Derivatives of peptide 10 | ||||||||
| 21 | Ala2Lys (A2K) | 0.86 ± 0.08 | 0.93 | 0.96 | 0.94 | 0.06 | ||
| 22 | Ala3Lys (A3K) | 0.95 ± 0.04 | 1.03 | 1.01 | 0.98 | 0.096 | ||
| 23 | Ala2Glu (A2E) | 0.94 ± 0.05 | 1.02 | 0.88 | 0.98 | 0.08 | ||
| 24 | Ala3Glu (A3E) | 0.95 ± 0.08 | 1.03 | 0.92 | 0.91 | 0.08 | ||
*Uncertainty was determined as absolute difference in predicted helicities between REMD runs starting with full helical and nonhelical structures.
†Not determined.
‡The results for peptides that can assemble into fiber segments are highlighted in boldface.
A few trends regarding the effect of substitutions on helicity emerged from the simulation results. For instance, compared with Leu substitutions (in peptides 1 to 4), Phe substitutions (in peptides 5, 6, 11, and 12) disfavored the helix formation, which is in agreement with the previously reported general trend of helical propensity for these two amino acid types (28). The substitution effects depended on positions. The Leu substitution at R4 (1 and 2) resulted in a greater increase in helical contents than the same substitution at R2 (3 and 4) (54 to 71% versus ~40%). This can be attributed to the fact that R4 is closer than R2 to the hydrophobic staple when the peptide folds into a helical conformation. A Leu side chain at R4 likely interacted favorably with the staple to stabilize the helical structure. In contrast, a Phe substitution at R4 (5 and 6) abolished the helicity of the peptide (reduced from 54 to 71% to 19 to 27%). In this case, the Ph ring at R4 seemed too bulky for the staple, causing steric clash instead (fig. S1H). The sensitivity of peptide helicity to the specific side chain–staple interactions suggests that there could be room for further improvement of the helical propensity of our peptide motif via optimization of the substitution at R4.
Given the important role of Ph groups in self-assembly of short peptides, we also examined whether other Ph-containing amino acids such as homophenylalanine (Hpa) and 2-amino-5-phenylpentanoic acid (App) could serve as an alternative to Phe (Fig. 1A). Compared to Phe, these amino acids have longer hydrocarbon linkers. We postulated that, with a longer linker, the Ph ring could avoid steric hindrance with the backbone and the staple and, perhaps, even be positioned properly to allow for favorable interaction with the staple. Thus, we simulated the helix folding of peptides with Hpa or App at R4 (7 to 10) or R2 (13 to 16). As expected, these peptides exhibited much higher helical content than their counterparts with Phe substitutions (5, 6, 11, and 12). In particular, one peptide (10) with an App substitution at R4 permitted hydrophobic contacts between its App side chain and the staple, thereby displaying the highest helical content of all the peptides examined.
Of all the peptides investigated, only those with high helical contents (>50%) were selected for further testing. We examined whether these helix-prone peptides could assemble into fibrous structures through long-time MD simulations. The direct observation of assembly of peptides into ordered supramolecular structures through simulations was accomplished with a highly efficient hybrid-resolution model called protein model with atomic details in coarse-grained environment (PACE) (24, 25). This model was originally designed to study protein folding and aggregation (24), and extended later to self-assembly of dipeptides (25). As will be demonstrated in Materials and Methods, it can also be used to faithfully model the formation of de novo designed coiled coil bundles.
All of the assembly simulations started with 20 randomly dispersed copies of peptides at ~16 mM and lasted for 3 to 5 μs. In the simulations, the peptides were kept in helical conformations to avoid sampling both folding and assembly at the same time. This strategy is reasonable given that the peptides were observed to assume predominantly helical structures in solution. The formation of ordered fibrous assemblies was monitored across two order parameters, namely, Sshape and Sθ. Sshape describes the overall shape of the aggregate with a value of unity indicating linear morphology (see Materials and Methods) (29). Sθ describes the average relative orientation of individual helical axes with zero indicating parallel or antiparallel alignment. The assembly process typically took place in two stages (fig. S2, A and B). The dispersed peptides first agglomerated within a few hundred nanoseconds. The aggregates that were formed at this stage had globular shapes, with Sshape fluctuating between 0.2 and 0.6, and were lacking in ordered alignments of individual peptides (Sθ ~ 0.3 to 0.4). From among all the peptides investigated, only one (peptide 10) with an App side chain at R4 and Ph at R1 underwent reorganization and further self-assembled into stable helical fiber segments on a microsecond time scale (Sshape > 0.9; Sθ < 0.1) (fig. S2, A and B).
To rule out the possibility that nonhelical structures could also have been observed in the assembly of peptide 10 but did not arise because of the helical constrains, we conducted a simulation of two dispersed peptide chains without any constraints. This system is simple enough for converged sampling of conformational change and association of the peptides. As shown in fig. S2C, unbound peptides could sample both helical and nonhelical structures but, when associated, assumed predominantly helical structures. This result suggests that our use of fixed helical conformations in the assembly simulations should be reasonable.
Structures of short fiber segments formed by designed stapled peptide 10
To confirm the observed assembly behavior of peptide 10, we also conducted multiple assembly simulations, each containing 20 to 40 peptide copies (table S1). In most of these simulations, a fiber segment was observed. To rule out that these observed fibers were metastable only due to the limited simulation time, we conducted some simulations in which the system was repeatedly heated and annealed to avoid kinetical traps (30) and the same fiber segment was observed in these simulations (fig. S2D). Last, the stability of the fiber structures obtained with the hybrid-resolution model was tested using more accurate atomistic simulations (see Materials and Methods). During a 500-ns restraint-free simulation of the fiber segment, the root mean square deviation of the system was kept around 3.8 Å (fig. S2, E to G), indicating that the observed fiber structure here was robust.
We next analyzed the basic structural features of the observed fiber segments. These segments were formed through the lateral associations between multiple helical columns (Fig. 2, A and B). Within each helical column, peptide chains stacked up via head-to-tail hydrogen bonds (HBs) in the direction of the fiber axis. In the fibers, these helical columns ran antiparallel to each other. The number of helical columns in each fiber varied from four to six (table S1). The binding interfaces between helical columns were composed of the App side chains at R4 and the staples, in accordance with our design. Several specific interactions between these groups were observed (Fig. 2C and fig. S2H): The Ph rings at R4 from laterally associated peptides interacted with one another to form an interdigitated ladder (box 1 in Fig. 2C). These direct contacts were observed less often between the Ph rings positioned at R1. Instead, these rings interacted with the staples and the App side chains of neighboring peptides (boxes 2 and 3).
Fig. 2. Molecular architecture of fiber segment of the designed stapled pentapeptide observed in simulations.
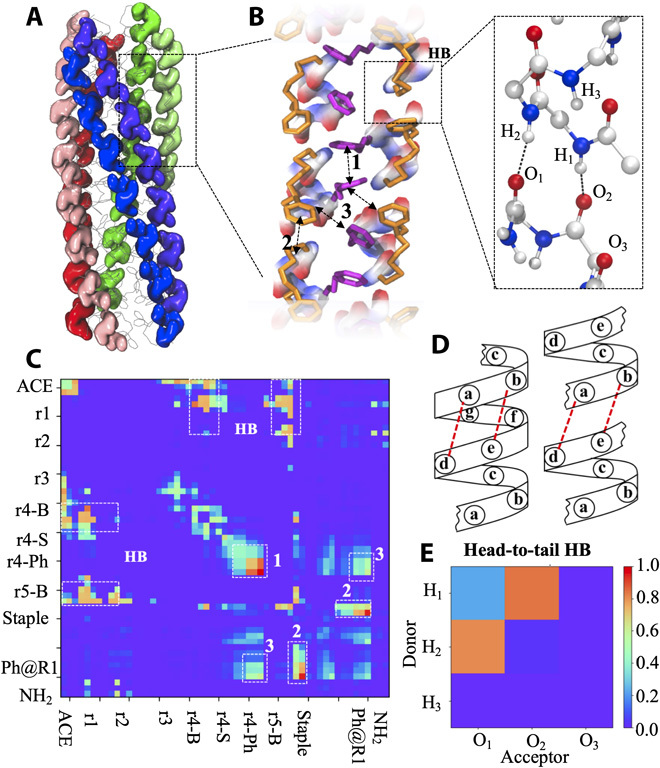
(A) Representative structure of fiber segment. Shown as ellipsoids are peptide backbones. Peptides in the same helical columns are shown in the same color, and different helical columns are shown in different colors. (B) Details of local packing between peptides. O, N, C, and H atoms are shown in red, blue, gray, and white, respectively. Shown as orange and purple sticks are the staples and the App side chains at R4, respectively. Dashed double-headed arrows indicate the interchain side chain packing. Inset is a close-up view of head-to-tail interactions between peptides with the H1─O2 and H2─O1 HBs shown as dashed lines. (C) Map of atomic contact between peptides. In the axes of the plot, “r” denotes residue number, and “B” and “S” denote backbone and side chain, respectively. “Ph@R1” denotes the Ph at R1. “ACE” denotes the acetylated N-terminus. Two atoms of different peptides were thought to form contact if their distance is shorter than 4.5 Å. Dashed boxes highlighted the interpeptide contacts illustrated in (B). (D) Comparison of a long helix of heptad repeats (left) and a helical column of the pentapeptides (right). The head-to-tail HBs seen in (B) and their counterparts in the long helix are shown as dashed red lines. (E) Map of head-to-tail HBs. An HB is thought to occur if the donor-acceptor distance is <3.5 Å and the donor-hydrogen-acceptor angle is >120°. The chance of a particular HB type is calculated as nHB/(npep − ncol), where nHB is the number of HBs of this type and npep and ncol are the numbers of peptides and helical columns, respectively, seen in the aggregates. The results were averaged over the last microseconds of simulations. Shown on the right is the scale bar of probability.
Note that in order for our design to work, all of the App side chains and the staples needed to occupy the same lateral faces of the helical columns. This was made possible by two specific head-to-tail HBs between neighboring peptides that arose between the first/second carbonyl oxygen atom at the C terminus of one peptide and the second/first amide hydrogen atom at the N terminus of another (namely, O1─H2 and O2─H1 shown in Fig. 2, B and D). Because an α helix has three unpaired carbonyl oxygen atoms at its C terminus and three unpaired amide hydrogens at its N terminus, these specific HBs do not necessarily arise. During the course of self-assembly of peptide 10, these two types of HBs seldomly formed in the initial agglomerate stage but became dominant HB types once the fibrous structures arose (yellow and dark blue lines in fig. S2A). Of the peptides investigated here, only peptide 10 showed a high tendency to form these HBs (Fig. 2E and figs. S2I and S3, A to L). Therefore, apart from specific side chain interactions, specific interactions between the peptide backbones are also essential for the self-assembly of our ultrashort peptides into fibrous structures.
Simulation of fiber growth by the designed stapled peptide
The fibrous assemblies investigated above contained a relatively small number of peptides (~20), likely corresponding to seeds of fibers formed at an early stage. To gain insight into how these early assemblies could evolve into more mature fiber structures and the molecular details of these structures, we simulated the growth of these fibers. Specifically, a fiber segment with 20 peptide copies was taken from one of the self-assembly simulations above to serve as the initial structure. We then performed a series of sequential simulations, each of which started with the fiber structure obtained from the last simulation and 10 additional free monomers, and ended after the monomers had been incorporated into the growing fiber. In this way, the concentration of monomers in simulations was kept below ~2 mg/ml throughout, mimicking low peptide concentrations normally adopted in self-assembly experiments (23). Following this procedure, two independent sets of growth simulations were conducted.
As shown in Fig. 3, the free peptides were either added to the tips of the fiber to elongate existing helical columns or to the side of the fiber to create new helical columns (Fig. 3A). However, as the fiber became larger in size, its longitudinal growth outpaced its lateral growth. As a result, the aspect ratio of the fiber increased (from 2.0 to 4.0) as the fiber grew (Fig. 3B).
Fig. 3. Growth of fiber segment into ribbon-like fiber observed in seeded simulations.
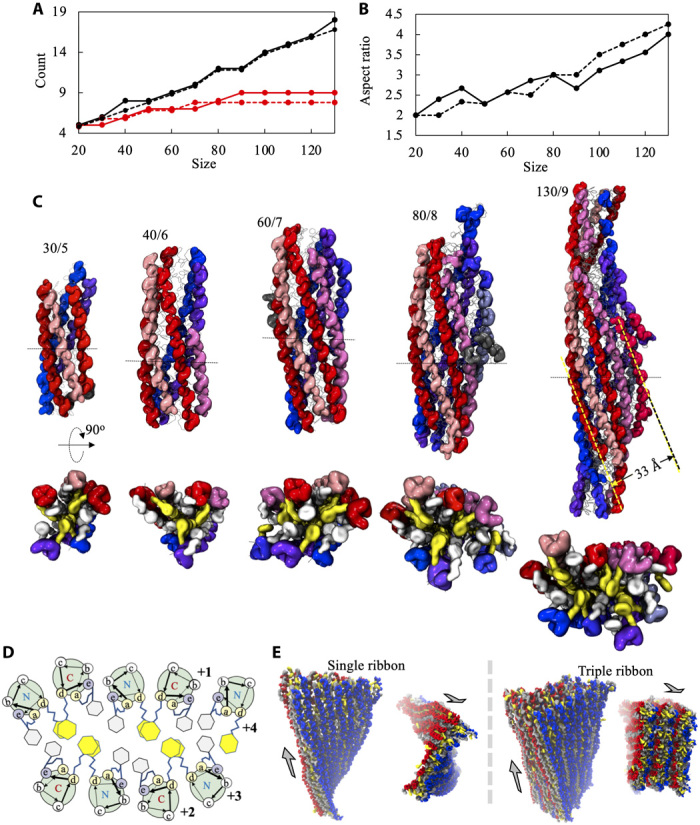
(A and B) Shape of fiber assembly as a function of fiber size. Shown in (A) are the number of helical columns (red dots) and that of peptides in the longest column (black dots). Shown in (B) is the aspect ratio of the assembly. The results of two independent simulations are shown as solid and dashed lines, respectively. (C) Representative structures of fiber assemblies in different sizes and their cross sections. Shown as ellipsoids are peptide backbones with those in the same helical columns being in the same color and those in different helical columns being in different colors. The staples and the App side chains are shown as white and yellow ellipsoids, respectively. The numbers shown above each structure denote the count of peptides and helical columns, respectively. (D) Helical wheel representation of ribbon-like fiber observed in the simulations. The numbers in boldface indicate the order in which the columns emerged at the lateral edge of the fiber during the growth simulations. (E) Structures of single-ribbon (left) and triple-ribbon (right) models of fibers obtained at t = 100 ns of simulations. Gray arrows indicate the directions of fiber axes.
Our simulations also showed that the fiber changed shape while growing laterally, evolving from a cylinder shape into a ribbon-like shape with a left-handed twist (Fig. 3C). The ribbon-like fiber appeared to be composed of two sheets of helical columns packing together with their hydrophobic faces, reminiscent of the recently reported collapsed coiled coil structures (Fig. 3D) (10). Within each sheet, the helical columns were aligned antiparallel to each other with a spacing of approximately 8 Å. Notably, despite the different overall shapes, the local interactions between specific groups observed in the small fiber segments were largely retained in the ribbon-like fiber as well (fig. S4A versus Fig. 2C).
As new helical columns continued to emerge at the lateral edges of the ribbon-like fiber, it was possible for the fibers to grow much wider. It remained impractical to probe more realistic structures of the fibers directly through assembly simulations due to the large system size and long simulation time required. Instead, we constructed, according to the results of the fiber growth simulations (see Materials and Methods), two sheet-of-helix models of large fibers, one representing a single-ribbon fiber (containing 360 peptides) and the other (containing 1080 peptides) representing a three-ribbon bundle. Both models retained the details of molecular packing seen in the growth simulations. Similar to previous computational studies of ribbon-like assembly twisting (31), the ribbons were constructed so as to be flat and straight to examine the spontaneous twisting of the ribbons. Our simulations revealed that these fibers were all stable (Fig. 3E). They were observed to twist in a left-handed manner, consistent with our growth simulations. Nonetheless, this twist was found to be much less profound than for the fiber made of packed ribbons (Fig. 3E). These results suggest that the fibers may tend to be straight when growing thicker and wider.
Experimental examination of the designed stapled peptide assembly structure
To validate the predicted assembly ability of peptide 10, we synthesized the peptide and characterized its self-assembly behavior. The circular dichroism (CD) spectrum of a freshly prepared solution of peptide 10 displayed a positive maximum at 195 nm and negative minima at 206 and 215 nm, indicating the formation of helical conformations by short peptides, as previously suggested (17, 32). The helical content of the peptide solution, derived using CD (Fig. 4A), was much higher than that of any other previously reported homologous peptides (22).
Fig. 4. Experimental characterization of molecular structures of fibrous assemblies of the designed stapled peptide.
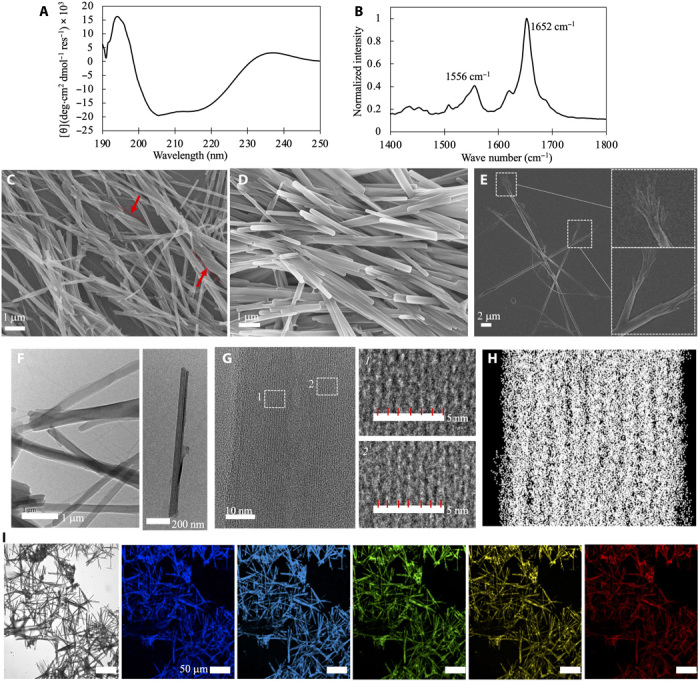
(A) CD spectrum of freshly prepared solution (2 mg/ml) of peptide 10. (B) Fourier transform infrared spectroscopy (FTIR) spectrum of assemblies of peptide 10 prepared at 2 mg/ml with sonification. SEM images of assemblies of peptide 10 prepared at 2 mg/ml with sonification (C) and through annealing (D). Red arrows in (C) highlighted fibers with a left-handed twist. (E) SEM images of annealed fibers prepared at 0.5 mg/ml. Insets show the branching ends of the fibers. (F) High-resolution TEM images of annealed fibers prepared at 0.5 mg/ml. (G) Close-up view of the fiber shown in the right panel of (F). The regions highlighted by dashed white boxes are amplified on the right. Short red lines illustrate the spacing between discrete strands aligned in the direction of fiber axis. (H) Projection of the triple-ribbon structure obtained from simulations onto the plane of sheet of helix. Each white dot represents a heavy atom. (I) Optical microscopy of the fibrous assemblies. From left to right, light microscopy and fluorescence microscopy using standard excitation wavelengths at 405, 440, 488, 514, and 561 nm.
Linear filaments were observed after the solution (2 mg/ml) of peptide 10 was treated with ultrasonication. Scanning electron microscopy (SEM) showed that these filaments were approximately tens of micrometers in length and around 0.25 to 1 μm in diameter, showing a high aspect ratio (Fig. 4C). Notably, these filaments were predominantly straight, but filaments with a left-handed twist were also observed on occasion (highlighted by red arrows). This observation is in line with our computational finding that the fibers of peptide 10 tend to twist but that this tendency is diminished when the fibers grow thicker. The secondary structures of peptides within the assemblies were probed by infrared spectroscopy. The observation of a strong absorption peak at 1652 cm−1 in the amide I region and a second absorption peak at 1556 cm−1 in the amide II region (Fig. 4B) indicated that the peptide predominantly adopted α-helical conformations in the fibrous assembly (33).
Filaments with a similar morphology were also observed when the solution was initially heated and then slowly cooled to the room temperature (Fig. 4D), suggesting that the formation of fibrous assemblies could be thermodynamically favorable. This experimental finding corroborates our computational observation that the peptides can assemble into fibers during both unbiased and annealing simulations. Compared to those obtained after sonification, the surface structures of the annealed filaments appeared more uniform, between 200 and 400 nm in diameter. A similar ordering of supramolecular structures of self-assembled coiled coil fibers upon annealing was also reported previously (34). The annealed filaments tended to cluster, but at a lower concentration (0.5 mg/ml), they were more scattered. This allowed us to examine the structural details of individual filaments (Fig. 4E). Here, the ends of the filaments displayed a broomstick-like appearance, branching into multiple finer threads. These filaments likely resulted from a lateral association between thinner threads whose diameter was in tens of nanometers. Still, these thinner threads were rather wide and likely contained structures composed of multiple ribbons as seen in the simulations. Such a ripening of filaments is common for self-assembled coiled coil fibers and has been reported previously (13, 17, 34).
To further gain insight into the molecular packing of the observed fibers, we characterized their internal structures with high-resolution transmission electron microscopy (TEM). Figure 4F shows TEM images of fibers of peptide 10 prepared with the annealing method at 0.5 mg/ml. These fibers exhibited morphologies very similar to those imaged with SEM (Fig. 4, C to E). A close-up view of the TEM fiber images revealed many discrete strands running in the direction of the fiber axis (Fig. 4G). The spacing between the strands was around 8 to 9 Å. This structural feature may be in agreement with our sheet-of-helix model. To confirm this, we projected all of the heavy atoms in the fiber model containing triple ribbons onto a plane parallel to the helical sheet (shown in Fig. 4H). Each white dot in this figure represents a heavy atom. This projection mimicked the electron microscope image of a fiber and was found to exhibit lanes with a similar spacing to those observed in TEM. It is thus likely that each helical column in our fiber model corresponds to a single lane observed in TEM imaging (Fig. 4H versus Fig. 4G). Nevertheless, because of the resolution limit of our TEM measurements, we were unable to distinguish between parallel and antiparallel strand arrangements. Together, our experimental characterization suggested that peptide 10 can assemble into fibers in the manner we predicted computationally.
Our previous studies showed that our stapled peptide scaffold, when containing an aromatic group, could assemble into aggregates with fluorescence, likely due to intermolecular π-π interactions (23). Given its one more aromatic ring and its extensive π-π stacking seen in the assembled states, the fibrous assemblies of peptide 10 may also be fluorescent. To test this possibility, we performed fluorescence microscopy on the fibers of peptide 10. The intrinsic fluorescence of the structures was observed at a broad range of excitation wavelengths, suggesting that peptide 10 could be potentially useful for optical and imaging applications.
Roles of helicity and side chain interactions in the formation of fibrous assemblies
According to our assembly simulations, from all of the peptide variants examined, only peptide 10 was capable of forming fibers. To validate whether the fiber-forming ability is unique to this peptide, and to gain insights into key factors determining this ability, we synthesized several close variants of peptide 10.
We first replaced the App at R4 with Leu (peptide 2), Phe (peptide 6), or Hpa (peptide 8) to examine the importance of App to the self-assembly of peptide 10. The morphologies of assemblies of these variants appeared different from one another, but all were disordered, lacking any one-dimensional structures (figs. S5, A to C). A straightforward explanation for the loss of the assembly ability in this peptide is a reduction in helicity caused by the mutations. CD measurement showed that the helical contents of these variants were considerably lower than that of peptide 10, in line with our calculations (Table 1 and fig. S1A). To further confirm the importance of helicity on fiber-forming ability of peptides and validate the exclusion of candidates with low helical content during our computational screening, we also synthesized another peptide (peptide 12) that was predicted to be among those with the lowest helicity. Again, no fibrous structures were observed for this peptide, which was in accord with our expectation (fig. S5D). Therefore, the ability of peptide 10 to form fibers could stem from its extremely high helicity resulting from the stabilization of its helical conformation by App-staple interactions.
Note that in our assembly simulations, none of the three variants could assemble into fibers although they were already restrained in helical confirmations. This suggests that peptide helicity may not be the only factor determining the self-assembly of the peptides. Instead, the correct packing of side chains could also be of importance. In particular, compared to App, the hydrocarbon linkers in Hpa and Phe are shorter by only one or two bridging methylene groups. A contact analysis of assembled aggregates of these peptides revealed that the shorter the hydrocarbon linker, the less likely the Ph rings at R4 participate in interchain aromatic interactions (Fig. 2C and fig. S4, A to D). It is possible that the Ph rings could be sterically hindered and thereby unbale to orient themselves properly for intermolecular packing when tethered too close to the backbone with a short linker, especially when the peptides are in helical states. Together, these results suggested that both high helicity and rather specific interactions involving Ph groups at R4 are required to allow peptide 10 to assemble into fibers.
Formation of thin fibers by stapled peptides with charged substitution
In order for peptide 10 to be of practical use, it is crucial that its fiber-forming ability is able to tolerate certain chemical modifications. In light of its special assembly ability from among the large number of peptides examined, it is important to examine whether there could exist any other fiber-forming derivatives of this peptide.
On the basis of the requirements for peptide helicity and specific interpeptide packing, we proposed four mutants of peptide 10, namely, A2E, A2K, A3E, and A3K, which replaced Ala at R2 or R3 with either Glu or Lys (Table 1, peptides 21 to 24). In these mutants, Glu and Lys are known to exhibit the highest helical propensity of all polar amino acids, close to that of Ala (35). Thus, it is unlikely that the high helical content of peptide 10 would be affected by these mutations. In addition, our fiber model (Fig. 3D) suggests that these mutations should be positioned on the exposed b and c faces of the helical cylinder. They should not therefore perturb the formation of the hydrophobic cores in the ribbon-like fiber, although the packing between the ribbons may be affected.
We thus investigated the self-assembly behavior of these four peptide variants both computationally and experimentally. In accordance with our expectation, both the replica exchange MD simulations and CD measurement showed that these peptides displayed similar helical contents to peptide 10 (Table 1 and fig. S1C). Self-assembly simulations showed that these variants also spontaneously formed linear segments with similar molecular packing and head-to-tail HBs as those found for peptide 10 (Table 1 and figs. S3, M to P, and S4, E to H), although the average chance of finding these specific head-to-tail HBs was decreased as compared to the case of peptide 10. In particular, these HBs were less likely to occur for the peptides in the terminal regions of the linear segments formed by these variants (fig. S3Q), perhaps due to unfavorable repulsion between like-charged side chains aligned on the same helical faces. Nonetheless, they were well conserved in the cores of the linear segments (fig. S3Q).
In SEM (Fig. 5), samples of the variants prepared at 0.5 to 2.0 mg/ml under the annealing condition also gave rise to filaments, although these assemblies were less abundant than those formed by peptide 10 under similar conditions presumably, perhaps due to the increase in solubility of the peptides caused by the inclusion of charged residues or, as discussed above, repulsive forces between like-charged side chains that may weaken the driving force for self-assembly. The filaments formed by the variants were also thinner and more curved than those of peptide 10, comprising only a few threads around 20 to 40 nm in diameter, often exhibiting a left-handed twist. These structural features of the variant assemblies were consistent with the molecular architecture observed in fibers formed from peptide 10 (Fig. 3, C to E). The thinner appearance of these filaments may be attributed to the presence of side chains with like charges on their surface, which may have impeded further lateral stacking. Collectively, the findings above suggest that certain polar substitutions at R2 or R3 could be compatible with retaining the fiber-forming ability of peptide 10.
Fig. 5. Fibrous assemblies formed by designed derivatives of peptide 10.

SEM images of annealed fibrous assemblies formed by (A) A2K, (B) A3K, (C) A2E, and (D) A3E mutants of peptide 10. In each case, the right panel is a close-up view of the highlighted region in the left panel.
DISCUSSION
The molecular design of short peptides as principle building blocks for self-assembled materials represents an important direction for nanotechnology (6). In particular, α-helical peptides have proved to be versatile building blocks in various applications (21). The design of α-helical assembling peptides has been inspired by naturally occurring coiled coil structures but remains challenging for ultrashort peptides, which are more amenable to chemical synthesis and modification but lack stability in helical conformations. Although such a design has been managed for a few short peptides using noncoded helix-promoting amino acids, these peptides usually contain the minimum length of seven amino acids to furnish at least one heptad characteristic of coiled coil–forming sequences (17).
In this study, we have demonstrated de novo design of coiled coil nanofibers from peptides with only five amino acids. Our design is based on a peptide scaffold with a thioether-containing side chain staple that is capable of restraining a single helical turn in canonical helical conformations (22). The main challenge for this design is to identify chemical modifications that can enhance both helical propensity and assembly ability of the peptide scaffold that contains a side chain staple and amino acids, likely noncoded. These moieties have been unexplored previously for the design of self-assembled coiled coil structures. This challenge was overcome by adapting the empirical structural rules for natural coiled coils and using a multiscale computational method to assess the helical propensity of peptides and their ability to self-assemble and to predict assembled structures. This method greatly reduced the efforts required to search for viable candidates. The best pentapeptide predicted by computation, which contained an App substitution in the fourth residue and a Ph substitution in the staple, was shown experimentally to assemble into nanofibers. Several additional derivatives with charged substitutions were further rationally designed and validated.
The combination of self-assembly simulations and various spectroscopic characterizations also offered molecular insights into the self-assembly behavior of this peptide and its organization into fibers. The overall molecular architecture of the fibers formed by our stapled peptides was similar to those formed by other short peptides (14, 18, 34). The peptides stacked on top of one another in a head-to-tail manner, forming helical columns that further assembled laterally.
Unlike previously reported peptides that formed helical bundles or barrels (14, 34), the newly designed pentapeptides, based on our self-assembly simulation, assembled into ribbon-like fibers comprising two sheets of helical columns, stacked along their hydrophobic faces. Within each sheet, the helical columns ran antiparallel to each other in the direction of the fiber axis with an approximately 8 Å lateral spacing. This structural feature was also suggested by our TEM measurements. Although the ribbon-like fibers observed here are atypical, they appear to be consistent with the collapsed coiled coil bundles reported in recent crystallographic studies (10). According to these studies, the designed heptad repeats with a and d occupied by bulky hydrophobic amino acids, as well as e and g occupied by a small hydrophobic amino acid, namely, Ala, can form two sheets of three to four helices each, especially when Phe was introduced at a or d. In our case, a Ph group is also present on the d position of the pentapeptide. Nevertheless, our simulation of fiber growth suggested that the fiber can keep growing laterally, as new helical columns continued to emerge on the fiber’s lateral edges. This may be attributed to the large App side chain at d and the bulky staple across a and e that, together, created exposed hydrophobic patches on the lateral edges of the fiber. The lateral growth may also help to explain why the actual fibers seen in SEM or TEM were much wider than individual helical columns or bundles.
The consensus on the longitudinal growth of coiled coil peptide fibers is that short helical peptides must have uncapped end termini to promote favorable head-to-tail attraction because their capped counterparts often fail to do so (14, 17, 18, 34). In one de novo designed crystal of coiled coils, capped peptides stacked on the top of each other via HB interactions between their end termini (36). The assembling pentapeptide reported here also provides another example that uncapped termini are not always necessary for peptides to assemble into coiled coil fibers. Our assembly simulations further revealed the molecular details of these HB interactions between capped termini. We found that two specific HBs were formed between the first/second amide groups of one peptide and the second-to-last/last carbonyl groups of another. These HBs allow the pentapeptides to stack on the top of each other in such a way that the resulting column of helices mimics a long helix of regular heptad repeats. This geometric argument suggests that similar head-to-tail HBs may be observed in helical columns formed by other terminally capped peptides, so long as their length differs from our pentapeptide by multiples of seven residues. In this regard, it is intriguing that the same head-to-tail HBs can be observed in the aforementioned designed crystal of coiled coils (36): In this case, the assembling peptide contains 26 amino acids, exactly five more residues than three complete heptad repeats. Together, our study suggests that there may be a relationship between peptide length and modes of head-to-tail stacking in coiled coil fibers assembled from end-capped peptides.
Of particular interest is the sensitivity of fiber-forming ability in the stapled peptide to the details of chemical substitutions. When the App residue at d was replaced with other App analogs that had hydrocarbon linkers shorter by only one (Hpa) or two bridging methylene groups (Phe), the peptide’s fiber-forming ability was abolished. Thus, even the small difference of a single methylene group can have a great impact on the assembly ability of the peptide. Our simulations and experiments suggest that this substitution effect can be attributed to the peptide helicity. Phe has the shortest linker, and its Ph group caused steric hindrance to the helical backbone. This hindrance was avoided with the Hpa substituent, whose linker is slightly longer. Nevertheless, only App has a linker long enough to allow for a favorable interaction between its Ph and the staple, which further stabilizes the helical conformation. Apart from the better helicity, our simulations further suggested that the long linker in the App substituent may also permit better intermolecular aromatic stacking of the Ph groups. In contrast, the formation of aromatic packing between Phe/Hpa side chains would be incompatible to the helical conformation of backbone, consistent with previous reports (18). Together, the current findings above add a crucial layer of details to our understanding of the substitution effects on the self-assembly of ultrashort helical peptides.
In summary, we have extended the design of coiled coil nanofibers to chemically stapled short peptides with noncoded amino acid side chains. Given the growing number of stapled peptide motifs that exhibit a diverse range of physical, chemical, and biological activities in general, this work opens an avenue to an unexplored chemical space for the design of assembling helical peptides. In these designs, one needs to systematically determine how chemical modifications would affect the helicity and assembly propensity of stapled peptides. The combined computational and experimental approach presented here shows promise toward making this goal attainable.
MATERIALS AND METHODS
Computational models
Helicity of peptides was examined through simulations of helix folding with the CHARMM27 atomistic force field (37). The parameters for Hpa, App, and the thioether-containing staple were assigned using analogous atom types defined in the force field. In each system, a single peptide was solvated in a dodecahedron box that had a 12-Å paddling distance and was filled with water molecules modeled with three-site transferable intermolecular potential (TIP3P).
For self-assembly systems, peptides were modeled with a hybrid-resolution model called PACE (24). In PACE, proteins and peptides are represented at a united-atom level, i.e., hydrogen atoms except for those in amide groups are combined with the heavy atoms that they are attached to. This representation provides an accurate description of packing of side chains and directionality of HB interactions. The water solvent is modeled with the MARTINI coarse-grained (CG) water model that represents four water molecules with a single CG site (38). The details of potential energy terms of PACE and their parameterization have been discussed in our previous works (24, 25). The PACE has been successfully applied to the study of folding of small proteins (with a size of 20 to 80 amino acids) (24) and aggregation of peptides (39). Recently, the PACE was further optimized to reproduce molecular packing of short peptides in assemblies (25). The improved PACE, whose ability of modeling protein folding remains unaffected, has been used to simulate self-assembly of diphenylalanine peptides into nanotubes, yielding insights into the relationship between peptide conformation and molecular packing. Therefore, this version of PACE was used in the present study. Again, the PACE parameters for Hpa, App, and the staple were obtained by analogy. The files containing the CHARMM and PACE parameters generated for the staple, Hpa, and App can be found via Internet (https://github.com/Hel-assembly/procedure). Other files containing peptide topologies, their initial coordinates, and simulation configurations that are required for reproducing computational results presented here are also provided.
Self-assembly simulations started with 20 to 40 copies of dispersed peptides randomly placed and orientated in a 113 Å by 113 Å by 113 Å box. The resulting concentration varied between 16 and 32 mg/ml. Simulations of fiber growth were conducted in a 170 Å by 170 Å by 170 Å box with a fiber placed at the center of the box and another 10 randomly dispersed peptides with at least 15-Å clearance off the fiber. In these self-assembly simulations, peptides were kept in their helical states. Following our previous work (39), we restrained backbone dihedrals (φ, ψ) at their corresponding helical regions using harmonic potential expressed as
| (1) |
where φi and ψi are the backbone dihedrals of the ith residue and kφ,i/kψ,i and φi,0/ψi,0 are the corresponding force constants and reference dihedral values of harmonic forces applied on these dihedrals, respectively. These parameters were optimized to reproduce the dihedral distributions obtained from atomistic simulations of the same peptide in helical conformations. The resulting parameters are shown in table S2.
To further validate the applicability of the protocol described above for the study of self-assembly of helical peptides, we also performed simulations of self-assembly of a de novo designed peptide termed CC-pII that is known to self-assemble into a three-stranded coiled coil (34). The simulation started with three dispersed CC-pII peptides restrained in helical states and ended up with a stable trimer helical bundle whose structure deviated from the crystal structure (Protein Data Bank ID: 4DZL) by only ~2.0 Å (fig. S6).
Last, to construct a fiber model on a more realistic scale, we took a basic unit of four peptides from one of the sheet structures in the assembled fiber containing 130 peptides (Fig. 3C). This unit consisted of two laterally associated helical segments, each containing two peptides stacked through head-to-tail HBs. All the peptides were aligned in the direction of z axis and placed in the xz plane. Then, we replicated it in the z direction with a 19.9-Å spacing and in the x direction with a 16.8-Å spacing to generate one sheet of helical columns. This sheet was then replicated, rotated about the z axis by 180°, translated in y by 14.5 Å and in x by 8.0 Å to create a double-layered ribbon. The triple-ribbon fiber was generated by replicating two additional ribbons in y with a 26.1-Å spacing. The fibers were modeled with PACE and solvated in a 140 Å by 140 Å by 200 Å box. The harmonic distance restraints with a force constant of 2.0 kJ mol−1 Å−2 were applied to maintain all the H1─O2 and H2─O1 HBs during simulations to avoid disruption of the model caused by steric clash introduced initially in the model construction.
Simulation setup and analysis
All simulations were performed with the GROMACS package (version 4.5.4) (40). For atomistic simulations, nonbonded interactions were truncated at 12 Å. Long-range electrostatics was evaluated with the particle mesh Ewald method. For PACE simulations, van der Waals interactions were smoothly switched off by applying a shift function from 9 to 12 Å. Electrostatic interactions were switched off by applying the shift function from 0 to 12 Å. The time step was set to 2 fs for atomic simulations with linear constraint solver (LINCS) and 5 fs for PACE simulations. The Nosé-Hoover method and the Parrinello-Rahman method were used for simulations at constant temperature and pressure, respectively.
REMD simulations (27) with the atomistic model were conducted to examine peptide helicity. In each REMD run, 24 parallel replicas were performed at temperatures between Tmin = 310 K and Tmax = 450 K with the ith replica being conducted at Ti = Tmin(Tmax/Tmin)(i − 1)/(24 − 1). Exchanges between replicas were attempted every 2 ps with an average acceptance ratio of ~30%. For each REMD run, the trajectory obtained at Tmin = 310 K was used for analysis. The helical content was estimated as the frequency of finding in the trajectory peptide backbone deviate by less than 2.0 Å from that of the crystal structure of our pentapeptide scaffold (22). Peptide helicity obtained with converged REMD simulations should not depend on initial structures used. To examine the sampling convergence, we performed, for each peptide, two independent REMD runs with one starting with nonhelical conformations selected at random from a 5-ns simulation at 700 K and the other starting from full helical structures built upon the crystal helical structure. We initially performed 4.8-μs simulations (200 ns per replica) for both REMD runs. Then, we iteratively extended both runs by 1.2 μs (50 ns per replica) each until the helicities obtained from the last 100 ns of the two REMD runs differed from each other by less than 10% (fig. S1D). The final helicity of the peptide was calculated as the average over the two runs, and the difference in helicity between the two runs was used to estimate calculation error (Table 1).
All the self-assembly simulations were performed at 310 K and 1.0 atm. The length of each simulation and the last snapshot were shown in table S1 and fig. S3. The Sshape score was used to monitor the assembly morphologies. This score is expressed as (29)
| (2) |
where λ1, λ2, and λ3 are three eigenvalues of the rational inertia tensor of the system. Here, the rotational inertia tensor was estimated using all Ph groups. A score of Sshape = 1 indicates the formation of fibrous structures, and a score of Sshape = 0 indicates a spherical shape of the assemblies. In addition, we used Sθ to examine the orientation of peptides in the assemblies. Sθ was given by
| (3) |
where N is the number of peptides and and are the unit vectors in the direction of the principle axes of peptide i and peptide j, respectively. A score of Sθ = 0 indicates that all helices are aligned in the same direction (either in a parallel manner or in an antiparallel manner).
Solid-phase peptide synthesis
All solvents and reagents were purchased from commercial suppliers including GL Biochem (Shanghai) Ltd., Shanghai Hanhong Chemical Co., J&K Co. Ltd., Shenzhen Tenglong Logistics Co., or Energy Chemical Co. and were used without further purification unless otherwise stated. All peptides were synthesized by manual Fmoc-based [9-fluorenyl methyloxycarbonyl (Fmoc)] solid-phase synthesis (fig. S7A). Peptide synthesis was performed manually on Rink amide 4-methylbenzhydrylamine hydrochloride (MBHA) resin (loading capacity, 0.54a mmol/g) (GL Biochem Ltd.) by standard Fmoc-based solid-phase peptide synthesis. In general, Rink amide amino methylpolystyrene (AM) resin was preswelled with equal volume dichlorometane (DCM) and N-methyl pyrrolidone (NMP) for 30 min. Fmoc deprotection was performed with morpholine (50% in NMP) for 30 min × 2. Then, the resin was washed with NMP (five times), DCM (five times), and NMP (five times). According to the initial loading of the resin, 6.0-equivalent (eq) Fmoc-protected amino acids and 5.9-eq 2-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyl uranium hexafluorophosphate were dissolved in NMP, followed by 12.0-eq diisopropylethylamine (DIPEA). The mixture was preactivated for 1 min and added to the resin for 1 to 2 hours, and then the resin was washed with NMP (five times), DCM (five times), and NMP (five times). Upon completion of peptide synthesis, peptides were N-terminally acetylated with a solution of acetic anhydride and DIPEA in NMP (1:1:8 in volume) for 1 hour.
Intramolecular thiol-ene reactions were used for constructing cyclic peptides. Thiol-ene reaction is conducted at ultraviolet light (365 nm) with 1-eq 1:1 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone/4-methoxyacetophenone catalysis in dimethylformamide for 2 hours. Peptides were cleaved from the resin with a mixture of 94:2.5:2.5:1 trifluoroacetic acid (TFA)/deionized water (H2O)/1,2-ethanedithiol/triisopropylsilane for 2 hours and concentrated under a stream of nitrogen. The crude peptides were then precipitated with 1:1 hexane/diethyl ether at −20°C, isolated by centrifugation then dissolved in water/acetonitrile, purified by semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC), analyzed by liquid chromatography–mass spectrometry (LC-MS), and lyophilized to get the final products.
Peptides were analyzed and purified by RP-HPLC (SHIMAZU Prominence LC-20AT) using a C18 analytic column (Agilent ZORBAX SB-Aq; 4.6 mm by 250 mm, 5 μm; flow rate, 1.0 ml/min) and a C18 semipreparative column (Agilent Eclipse XDB-C18; 9.4 mm by 250 mm, 5 μm; flow rate, 5 ml/min). H2O (containing 0.1% TFA) and pure acetonitrile (containing 0.1% TFA) were used as solvents in linear gradient mixtures. LC-MS spectra were carried out on SHIMAZU LC-MS 8030 (electrospray ionization). The MS results for the peptides examined in the present studies are summarized in fig. S8.
Synthesis of noncoded amino acids
The key to the aforementioned synthetic route is the construction of a peptide containing at its fifth position in sequence a noncoded amino acid, namely, Fmoc-S5(R′)-OH in a R configuration. R′ corresponds to R1 group of our pentapeptide scaffold. The synthesis of this amino acid was described with details previously (22, 23) and is briefly illustrated in fig. S7B. In addition, other noncoded amino acids including Fmoc-Hpa-OH [Chemical Abstract Service (CAS) Registry Number: 132684-59-4] and Fmoc-App-OH (CAS: 959578-11-1) were purchased from GL Biochem (Shanghai) Ltd. and J&K Co. Ltd. They were used in peptide synthesis without further purification unless otherwise stated.
Preparation of peptide assemblies
Lyophilized pentapeptide powder was first thawed with less than 0.5% dimethyl sulfoxide, and deionized water was added to target concentration. Self-assembly took place while the concentrated peptide solutions were either sonicated for 30 min or heated in a metal bath to 80°C for 30 min and then naturally cooled to room temperature.
CD spectroscopy
CD spectra were recorded by Chirascan Plus Circular Dichroism Spectrometer (Applied Photophysics) at 25°. Peptides were dissolved in deionized water at an appropriate concentration, respectively. Parameters used in the experiment are as followed: wavelengths from 250 to 190 nm were measured with a resolution of 0.5 nm at a scan speed of 0.5 nm/s. Each sample was scanned twice, and the averaged spectrum was smoothed using Pro-Data Viewer by Applied Photophysics with a smooth window of 10. CD data were presented as mean residual ellipticity [θ] in degree square centimeters per decimole. The α-helical content of each peptide was calculated using ellipticity at 215 nm as was performed previously (32). The final helical content was presented as a relative ratio to that of peptide 10.
Fourier transform infrared spectroscopy
Qualitative analysis of the samples was performed by analyzing the infrared absorption spectrum of each sample to learn the interaction inside the samples. KBr pellet was prepared before measurement. Bruker Vertex 70 FT-IR spectrometer was used for Fourier transform infrared spectroscopy (FTIR) analysis with wave number range from 4000 to 400 cm−1.
Scanning electron microscopy
Samples of peptide assemblies were placed on silicon slides, were left to dry at room temperature, and then were viewed using an SEM (ZEISS Supra 55, Oxford X-Max 20; 20 kV).
High-resolution TEM
The samples (10 μl) of peptide assemblies were placed on Formvar/carbon mesh 400 copper grids (Electron Microscopy Sciences) for 20 s and then removed using a piece of nitrocellulose paper. The Cu grids were left to dry at room temperature. TEM images were recorded by high-resolution TEM (JEOL, JEM-3200FS; 200 kV).
Fluorescence microscopy
After peptide 10 was assembled, samples about 100 μl were dropped on a coverslip and got dried at room temperature. The coverslip with the samples was fastened to a slide glass and was imaged at different excitation wavelengths using Nikon A1R.
Acknowledgments
We thank R. Wang from the School of Advanced Materials at Peking University for assistance in conducting the SEM test. The computational work is supported by high-performance computing platform of Peking University. Funding: This work was supported by the National Science Foundation of China (grant nos. 21977011 and 21673013 to W.H., 21778009 to Z.L., and 21671137 to Q.Z.), National Key Research and Development Program (grant no. 2018YFA0902504 to Z.L.), Natural Science Foundation of Guangdong Province (grant no. 2019A1515011958 to Q.Z.), the Shenzhen Science Technology and Innovation Commission (grant nos. KQTD2015032709315529 and JCYJ20170818085409785 to W.H. and JCYJ20170817172023838 to Z.L.), Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institute (grant no. 2019SHIBS0004 to Z.L.), and China Postdoctoral Science Foundation (grant no. 2020 M672770 to Y.J.). Author contributions: Y.J., F.Y., C.W., J.L., and Q.Z. conducted experiments and analyzed experimental data. W.Z. conceived the computational molecular design, conducted the computation, and analyzed computational data. X.C. optimized model parameters. W.H. and Z.L. designed the research. W.H., Z.L., W.Z., Y.J., and F.Y. drafted and revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabd0492/DC1
REFERENCES AND NOTES
- 1.Hill R. B., Raleigh D. P., Lombardi A., DeGrado W. F., De novo design of helical bundles as models for understanding protein folding and function. Acc. Chem. Res. 33, 745–754 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mravic M., Thomaston J. L., Tucker M., Solomon P. E., Liu L., DeGrado W. F., Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 363, 1418–1423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty P., Tang Y., Yamamoto T., Yao Y. F., Guterman T., Zilberzwige-Tal S., Adadi N., Ji W., Dvir T., Ramamoorthy A., Wei G. H., Gazit E., Unusual two-step assembly of a minimalistic dipeptide-based functional hypergelator. Adv. Mater. 32, 1906043 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Basavalingappa V., Guterman T., Tang Y. M., Nir S., Lei J. T., Chakraborty P., Schnaider L., Reches M., Wei G. H., Gazit E., Expanding the functional scope of the fmoc-diphenylalanine hydrogelator by introducing a rigidifying and chemically active urea backbone modification. Adv. Sci. 6, 1900218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamley I. W., Peptide nanotubes. Angew. Chem. Int. Ed. 53, 6866–6881 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Gazit E., Self-assembled peptide nanostructures: The design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 36, 1263–1269 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Frederix P. W. J. M., Scott G. G., Abul-Haija Y. M., Kalafatovic D., Pappas C. G., Javid N., Hunt N. T., Ulijn R. V., Tuttle T., Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 7, 30–37 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kwon S., Kim B. J., Lim H.-K., Kang K., Yoo S. H., Gong J., Yoon E., Lee J., Choi I. S., Kim H., Lee H.-S., Magnetotactic molecular architectures from self-assembly of β-peptide foldamers. Nat. Commun. 6, 8747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joh N. H., Wang T., Bhate M. P., Acharya R., Wu Y., Grabe M., Hong M., Grigoryan G., DeGrado W. F., De novo design of a transmembrane Zn2+−transporting four-helix bundle. Science 346, 1520–1524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhys G. G., Wood C. W., Lang E. J. M., Mulholland A. J., Brady R. L., Thomson A. R., Woolfson D. N., Maintaining and breaking symmetry in homomeric coiled-coil assemblies. Nat. Commun. 9, 4132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apostolovic B., Danial M., Klok H. A., Coiled coils: Attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 39, 3541–3575 (2010). [DOI] [PubMed] [Google Scholar]
- 12.D. N. Woolfson, in Advances in Protein Chemistry (Elsevier, 2005), vol. 70, pp. 79–112. [DOI] [PubMed] [Google Scholar]
- 13.Sharp T. H., Bruning M., Mantell J., Sessions R. B., Thomson A. R., Zaccai N. R., Brady R. L., Verkade P., Woolfson D. N., Cryo-transmission electron microscopy structure of a gigadalton peptide fiber of de novo design. Proc. Natl. Acad. Sci. U.S.A. 109, 13266–13271 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong H., Paramonov S. E., Hartgerink J. D., Self-assembly of α-helical coiled coil nanofibers. J. Am. Chem. Soc. 130, 13691–13695 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Bera S., Gazit E., Self-assembly of functional nanostructures by short helical peptide building blocks. Protein Pept. Lett. 26, 88–97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mándity I. M., Monsignori A., Fülöp L., Forró E., Fülöp F., Exploiting aromatic interactions for β-peptide foldamer helix stabilization: A significant design element. Chem. A Eur. J. 20, 4591–4597 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Mondal S., Adler-Abramovich L., Lampel A., Bram Y., Lipstman S., Gazit E., Formation of functional super-helical assemblies by constrained single heptad repeat. Nat. Commun. 6, 8615–8622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mondal S., Basavalingappa V., Jacoby G., Shimon L. J. W., Beck R., Gazit E., Functional coiled-coil-like assembly by knob-into-hole packing of single heptad repeat. ACS Nano 13, 12630–12637 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Mondal S., Varenik M., Bloch D. N., Atsmon-Raz Y., Jacoby G., Adler-Abramovich L., Shimon L. J. W., Beck R., Miller Y., Regev O., Gazit E., A minimal length rigid helical peptide motif allows rational design of modular surfactants. Nat. Commun. 8, 14018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maity S., Jana P., Maity S. K., Haldar D., Mesoporous vesicles from supramolecular helical peptide as drug carrier. Soft Matter 7, 10174–10181 (2011). [Google Scholar]
- 21.Lee J., Choe I. R., Kim N.-K., Kim W.-J., Jang H.-S., Lee Y.-S., Nam K. T., Water-floating giant nanosheets from helical peptide pentamers. ACS Nano 10, 8263–8270 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Hu K., Geng H., Zhang Q., Liu Q., Xie M., Sun C., Li W., Lin H., Jiang F., Wang T., Wu Y.-D., Li Z., An in-tether chiral center modulates the helicity, cell permeability, and target binding affinity of a peptide. Angew. Chem. Int. Ed. 55, 8013–8017 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Hu K., Jiang Y. X., Xiong W., Li H., Zhang P. Y., Yin F., Zhang Q. L., Geng H., Jiang F., Li Z., Wang X. W., Li Z. G., Tuning peptide self-assembly by an in-tether chiral center. Sci. Adv. 4, eaar5907 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han W., Schulten K., Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: Improved backbone hydration and interactions between charged side chains. J. Chem. Theory Comput. 8, 4413–4424 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong Q., Jiang Y., Cai X., Yang F., Li Z., Han W., Conformation dependence of diphenylalanine self-assembly structures and dynamics: Insights from hybrid-resolution simulations. ACS Nano 13, 4455–4468 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Liu J., Zheng Q., Deng Y., Cheng C.-S., Kallenbach N. R., Lu M., A seven-helix coiled coil. Proc. Natl. Acad. Sci. U.S.A. 103, 15457–15462 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugita Y., Okamoto Y., Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151 (1999). [Google Scholar]
- 28.Chakrabartty A., Kortemme T., Baldwin R. L., Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 3, 843–852 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tribello G. A., Giberti F., Sosso G. C., Salvalaglio M., Parrinello M., Analyzing and driving cluster formation in atomistic simulations. J. Chem. Theory Comput. 13, 1317–1327 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Frederix P. W. J. M., Patmanidis I., Marrink S. J., Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 47, 3470–3489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai C. T., Rosi N. L., Schatz G. C., All-atom molecular dynamics simulations of peptide amphiphile assemblies that spontaneously form twisted and helical ribbon structures. J. Phys. Chem. Lett. 8, 2170–2174 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Shepherd N. E., Hoang H. N., Abbenante G., Fairlie D. P., Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 127, 2974–2983 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Nevskaya N. A., Chirgadze Y. N., Infrared spectra and resonance interactions of amide-I and II vibrations of α-helix. Biopolymers 15, 637–648 (1976). [DOI] [PubMed] [Google Scholar]
- 34.Burgess N. C., Sharp T. H., Thomas F., Wood C. W., Thomson A. R., Zaccai N. R., Brady R. L., Serpell L. C., Woolfson D. N., Modular design of self-assembling peptide-based nanotubes. J. Am. Chem. Soc. 137, 10554–10562 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Pace C. N., Scholtz J. M., A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanci C. J., MacDermaid C. M., Kang S.-g., Acharya R., North B., Yang X., Qiu X. J., DeGrado W. F., Saven J. G., Computational design of a protein crystal. Proc. Natl. Acad. Sci. U.S.A. 109, 7304–7309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackerell A. D. Jr., Feig M., Brooks C. L. III, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., De Vries A. H., The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Jiang X. H., Cao Y., Han W., In silico study of recognition between Aβ40 and Aβ40 fibril surfaces: An N-terminal helical recognition motif and its implications for inhibitor design. ACS Chem. Nerosci. 9, 935–944 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Hess B., Kutzner C., Van Der Spoel D., Lindahl E., GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabd0492/DC1