

Abstract
Background:
Both Major Depressive Disorder (MDD) and Primary Insomnia (PI) have been linked to deficiencies in cortical γ-aminobutyric acid (GABA) and glutamate (Glu) thus suggesting a shared neurobiological link between these two conditions. The extent to which comorbid insomnia contributes to GABAergic or glutamatergic deficiencies in MDD remains unclear.
Methods:
We used single-voxel proton magnetic resonance spectroscopy (1H MRS) at 4 Tesla to examine GABA+ and Glu relative to creatine (Cr) in the dorsal anterior cingulate cortex (dACC) and in the parieto-occipital cortex (POC) of 51 non-medicated adults with MDD, 24 adults with Primary Insomnia (PI), and 25 age- and sex-matched good sleeper controls (HC). Measures of depression severity and subjective and objective sleep quality were compared with 1H MRS metabolite measures.
Results:
MDD subjects exhibited a 15% decrease in Glu/Cr in the dACC compared to HC. Within the MDD group, there was a trend inverse correlation between dACC Glu/Cr and anhedonia ratings. We observed no significant association between measures of sleep quality with dACC Glu/Cr in those with MDD.
Limitations:
The protocol and data interpretation would have been enhanced by the recruitment of MDD subjects with a broader range of affect severity and a more comprehensive assessment of clinical features.
Conclusions:
These findings support the role of cortical glutamatergic mechanisms in the pathophysiology of MDD. Insomnia severity did not further contribute to the relative deficiency of glutamatergic measures in MDD.
Keywords: GABA, Glutamate, Magnetic Resonance Spectroscopy, Depression, Insomnia, Sleep
INTRODUCTION
Recent advances in neuroimaging techniques, such as the increasing use of proton magnetic resonance spectroscopy (1H MRS), have made it possible to examine in vivo an array of major CNS neurochemicals including gamma-amino-butyric acid (GABA), glutamate (Glu), and a composite measure (Glx) of glutamate (Glu) and glutamine (Gln). Many of these 1H MRS studies of GABA and Glu have suggested that both neurotransmitters are involved in the neurobiology of PI and MDD. There is both indirect and direct evidence consistent with this conclusion.
With respect to indirect lines of evidence, GABA and Glu neurons discharge maximally in association with discrete sleep-wake states (Jones, 2017). From a treatment perspective, broad support for GABA’s role in sleep regulation derives from the observation that benzodiazepine receptor agonists, which are efficacious in the treatment of insomnia, increase activity at GABA neurons (Gottesmann, 2002). In an analogous manner, the role of Glu in the neurobiology of MDD is advanced by the therapeutic benefit of ketamine, an NMDA Glu receptor antagonist whose mechanism of action remains obscure (Lener et al., 2017; Zanos and Gould, 2018).
Direct evidence of GABA’s role in the neurobiology of PI is suggested by previous work in our laboratory. Using 1H MRS, we demonstrated global brain reductions in GABA (Winkelman et al., 2008) and locally specific reductions in the occipital (OC) and dorsal anterior cingulate cortices (dACC) in subjects with PI (Plante et al., 2012). Moreover, within those with PI, reductions in GABA were inversely correlated with the severity of insomnia as indexed by polysomnographic wake after sleep onset (WASO). In a more recent study, exploratory data suggested that GABA levels in the dACC were associated with habitual sleep duration (Spiegelhalder et al., 2016). In addition to GABAergic abnormalities in PI, sleep impairment has also been associated with abnormalities in glutamatergic systems. Relative to healthy controls, 1H MRS measures of Glu were reduced in the occipital cortex of patients with insomnia notable for short sleep durations (Miller et al., 2017).
In addition to studies of the neurobiology of PI, a range of reports over the past two decades directly documented abnormalities in 1H MRS-detected GABA and Glu in MDD. A recent meta-analysis of 1H MRS studies in MDD found that MDD is associated with reductions in GABA, especially in occipital brain regions, and that these reductions are state-dependent; depressed individuals show significantly lower levels of GABA compared to those with remitted MDD (Schur et al., 2016). With respect to glutamatergic abnormalities, reviews and meta-analyses are largely in agreement that Glu and/or Glx is decreased in MDD (Yuksel and Ongur, 2010; Luykx et al., 2012; Moriguchi et al., 2019), particularly in anterior brain regions such as the prefrontal cortex (Michael et al., 2003; Hasler et al., 2007) and ACC (Auer et al., 2000; Zhang et al., 2013).
Broadly stated, these studies of 1H MRS-detected abnormalities in both GABAergic and glutamatergic brain chemistry suggest some overlap in the underlying neurobiology of PI and MDD. This suggestion is reinforced by the interrelationship of PI and MDD on the clinical level. Sleep disturbance is a diagnostic feature of MDD with insomnia occurring in nearly 75% of patients with depression (Nutt et al., 2008). Furthermore, the relationship between PI and MDD appears to be bidirectional. For example, pre-morbid insomnia increases the risk of subsequently developing MDD (Chang et al., 1997; Riemann and Voderholzer, 2003). Furthermore, residual insomnia following resolution of a major depressive episode increases the risk of depressive relapse (Dombrovski et al., 2007; Cho et al., 2008).
These clinical observations coupled with overlapping GABA and Glu abnormalities in both PI and MDD suggest that the GABAergic and glutamatergic abnormalities observed in MDD might better reflect the co-morbid sleep disturbance in this disorder rather than core mood or affective symptoms. The objective of the present study was to compare regional 1H MRS-derived measures of GABA and Glu in three groups: healthy, good sleeping controls (HCs), subjects with PI, and subjects with MDD. We hypothesized that GABA and Glu levels would be reduced in both patient groups relative to controls. Furthermore, we hypothesized that increasing levels of sleep disturbance in MDD would correspond with decreased 1H MRS-measured cortical GABA and Glu levels, independent of the degree of mood disturbance. The primary outcome measures included 1H MRS-derived measures of GABA+ and Glu relative to creatine (Cr), in the parieto-occipital cortex (POC) and dorsal anterior cingulate cortex (dACC). Secondary outcome measures included questionnaire assessments of sleep quality and depressive affect as well as measures of sleep quality derived from subjects’ diaries and overnight polysomnography.
METHODS
Participants
One hundred adult (ages 18–70 years) subjects were recruited from the greater Boston, MA area from February 2013 to September 2016, primarily using online and poster advertisements as well as hospital-based recruitment tools available through the Depression Clinical Research Program at Massachusetts General Hospital. Efforts were made to match subjects by age and sex across MDD, PI and HC groups. All subjects were evaluated with an unstructured clinical interview for history of sleep and medical disorders. Lifetime history of Axis I and II psychiatric disorders was evaluated using the Structured Clinical Interview for DSM-IV (SCID-I/SCID-II) (First et al., 2002; First et al., 1997). Subjects were also evaluated with the 17-item Hamilton Rating Scale for Depression (HAM-D) (Hamilton, 1960), the Insomnia Severity Index (ISI) (Morin, 1993), Beck Depression Inventory (BDI-IA) (Beck and Steer, 1993), Pittsburgh Sleep Quality Index (PSQI) (Buysse et al., 1989), Dysfunctional Beliefs and Attitudes about Sleep (DBAS-16) (Morin et al., 2007), Perceived Stress Scale (PSS) (Levenstein et al., 1993), and the Fatigue Severity Scale (FSS) (Krupp et al., 1989). A measure of Anhedonia was derived by summing items 4, 12, 15 and 21 from the BDI-IA.
Major Depressive Disorder subjects met DSM-IV criteria for MDD (296) and required a HAM-D score ≥ 11 (not including insomnia items). Primary Insomnia subjects met DSM-IV criteria for PI (307.42). Inclusion criteria for PI also required an ISI ≥14, a HAM-D score ≤8 (excluding 3 insomnia items), and a BDI-IA ≤10. Age and sex-matched healthy control subjects (HC) without sleep complaints or history of psychiatric conditions were also recruited. Inclusion criteria for HC subjects specified an ISI ≤ 4, a HAM-D ≤ 8, and a BDI-IA ≤ 10.
All subjects were required to be free of CNS active agents for two weeks (or 5 half-lives) prior to the first study visit or willing to taper off CNS active agents prior to beginning the study. Use of CNS active agents was prohibited for the duration of the study. Moderate consumption of alcohol (no more than 2 standard alcoholic drinks per day for a period > 1 month in the preceding year) with no history of alcohol abuse was permitted. Subjects were excluded if caffeinated beverage/pill consumption was estimated to contain more than 300 mg of caffeine daily (approximately 3 standard cups of coffee). Subjects were also excluded for current smoking of more than 10 cigarettes per day. A total of 5 smokers were included in the study: 1 of 25 controls, 4 of 51 in the MDD group, and 0 in the PI group. Of these 5 smokers, only 2 smoked at least one cigarette per day; both were in the MDD group (2 of 51 Ss). Proscriptions against CNS agents were assessed using urine toxic screens at baseline and end of study which tested for benzodiazepines, opioids, THC, and amphetamines. Use of other CNS medications was assessed through interview at each of the study visits and through daily sleep diary reports of tobacco, caffeine, alcohol, or medication consumption. Exclusion criteria for all subjects included clinical evidence of any moderate to severe sleep disorder other than insomnia (e.g. obstructive sleep apnea, restless legs syndrome, narcolepsy, etc.) within the preceding year; apnea + hypopnea index (AHI) > 25 during the first night of polysomnographically-recorded home sleep testing; lifetime history of Axis I DSM-IV schizophrenia, psychotic disorders, or panic disorder, or significant Personality Disorder (PD); lifetime history of Axis I DSM-IV alcohol or illicit substance dependence; history of significant medical or neurologic conditions that may exacerbate or cause depressive symptoms or insomnia (including menopause); and women who were pregnant, lactating, or planning to become pregnant during the study. Baseline laboratories included a urine pregnancy test (for female subjects).
The study was approved by the Institutional Review Board of Partner’s Healthcare, the parent organization of Massachusetts General Hospital and McLean Hospital, and carried out in accordance with the Declaration of Helsinki. All subjects provided written informed consent and were compensated for their participation in this study.
Actigraphy and Sleep Diaries
Following initial evaluation, subjects wore a wrist actigraph (Actiwatch AW-64, Minimitter, Bend OR) and completed sleep-wake diaries for two weeks. Wrist actigraphy provided a continuous record of sleep patterns in the home environment while sleep diaries reported sleep-related parameters such as bed- and rise-time, estimated Sleep Onset Latency (SOL), Wake After Sleep Onset (WASO), Total Sleep Time (TST), and Sleep Efficiency (SE%), as well as caffeine/alcohol/medication consumption and a visual analog scale (VAS) of subjective sleep quality. HC subjects were excluded if they reported ≤7or ≥10 hours of sleep or SOL or WASO >30 minutes on ≥ 5/14 nights of sleep diaries. To be included in the PI group, subjects had to report ≤ 7 hours of sleep and SOL or WASO>45 minutes or SOL+WASO>60 minutes on at least 7/14 nights of sleep diaries. Actigraphy corroborated sleep diaries but were not used to determine subject inclusion/exclusion.
Polysomnography
Subjects completed two consecutive nights of home polysomnography (PSG) using an ambulatory Somte PSG device (Compumedics, Charlotte, NC). The first night was an acclimation night to assess for comorbid sleep disordered breathing and periodic limb movements of sleep. On the second night of home sleep recording, subjects were equipped with an abbreviated recording montage, to assess sleep architecture. All sleep recordings were scored using the American Academy of Sleep Medicine sleep scoring criteria (Iber et al., 2007) by the same experienced, registered PSG technologist, blind to subject diagnostic group.
Magnetic Resonance Imaging
MR imaging was performed as soon as logistically possible after completion of actigraphy/diaries and the second home PSG (average = 2 days; range = 0–21 days). Subjects were re-scanned if spectra were not obtained due to movement artifact or technical failure. Consistent with prior studies in MDD (Bhagwagar et al., 2008), female subjects were scanned during the follicular phase of their menstruation cycle to control for confounding hormonal effects on GABA.
For those MDD subjects who underwent a 2-week washout of CNS active agents prior to initial evaluation, the completion of actigraphy/diaries and 2 home PSGs extended the washout interval prior to MR imaging to a minimum of 4 weeks. Subjects continued sleep diaries and actigraphy until the day of their scans to confirm that typical sleep-wake patterns continued prior to MRS. The sleep diary data shown in Table 1 constitute the within-subject average of the final 7 days of diary documentation.
Table 1.
Demographics, Rating Scales, and Sleep-Wake Measures in Healthy Controls, Primary Insomnia, and Major Depression.
| Healthy Controls (Mean (SD)) (n = 25) | Primary Insomnia (Mean (SD)) (n = 24) | Mayor Depressive Disorder (Mean (SD)) (n = 51) | |
|---|---|---|---|
|
| |||
| Demographics | |||
| Age, y | 33.9 (14.6) | 39.5 (14.2) | 33.2 (14.4) |
| Sex (F:M) | 17:8 | 17:7 | 33:18 |
| BMI, kg/m | 22.7 (3.2) | 24.4 (4) | 23.6 (4.1) |
| Rating Scales | |||
| Insomnia Severity Index | 1.32 (1.57) | 17.58 (2.80) | 14.86 (5.86) |
| HAM-D (without sleep items) | 0.44 (0.71) | 1.92 (1.74) | 16.49 (4.07) |
| Beck Depression Inventory1 | 2.21 (0.98) | 5.13 (2.80) | 23.55 (7.68) |
| Anhedonia | 0.16 (0.37) | 0.54 (0.88) | 4.37 (1.91) |
| Pittsburgh Sleep Quality Index | 1.56 (1.33) | 10.58 (2.10) | 9.67 (3.56) |
| DBAS 162 | 2.73 (1.26) | 4.70 (1.22) | 5.14 (1.47) |
| Perceived Stress Scale3 | 18.5 (2.47) | 18.61 (3.46) | 22.43 (4.07) |
| Fatigue Severity Scale4 | 2.12(0.85) | 3.48 (1.31) | 4.57 (1.19) |
| Sleep-Wake Diaries | |||
| Sleep Onset Latency, min | 11.62 (6.27) | 38.37(21.23) | 37.98 (30.81) |
| Wake After Sleep Onset, min | 9.18(7.41) | 68.29 (39.40) | 31.16(30.75) |
| Total Sleep Time, hours | 7.80 (0.54) | 5.55 (0.88) | 6.52 (1.21) |
| Sleep Efficiency. % | 93.44 (2.68) | 69.65 (9.27) | 80.87(11.65) |
| VAS SIecp Quality | 73.57(11.62) | 38.70 (10.03) | 47.98 (15.68) |
| Polysomnography | |||
| Apoca/Hypopnea Index | 0.94 (1.43) | 3.23 (9.49) | 2.68 (5.51) |
| LPS, min | 14.22 (19.21) | 26.23 (29.22) | 41.23(38.89) |
| SOI min* | 11.20(19.48) | 18.38 (22.41) | 3S.37 (32.49) |
| Wake After Sleep Onset, min | 44.16 (36.62) | 87.50 (88.55) | 61.48 (57.30) |
| Total Sleep Time, hours5 | 7.59 (0.72) | 6.09 (1.72) | 6.79 (1.18) |
| Sleep Efficiency, % | 89.47 (7.26) | 78.17 (17.72) | 81.18(10.18) |
All MRS data were collected at McLean Hospital in Belmont, MA. We utilized a whole body 4-Tesla MR scanner (Agilent Technologies; Santa Clara, CA) using a 16-rung, volumetric birdcage design RF head coil (XLR Imaging, London, Canada) operating at 170.3MHz for proton imaging and spectroscopy. The axial and sagittal high-resolution, T1-weighted anatomical images were utilized to systematically place single voxels in the bilateral dorsal anterior-cingulate cortex (dACC) (35 × 25 × 20 mm) and the bilateral parieto-occipital cortex (POC) (30 × 25 × 25 mm) (See Figure 1). Voxel placement was visually checked by an experienced user (JEJ).
Figure 1.
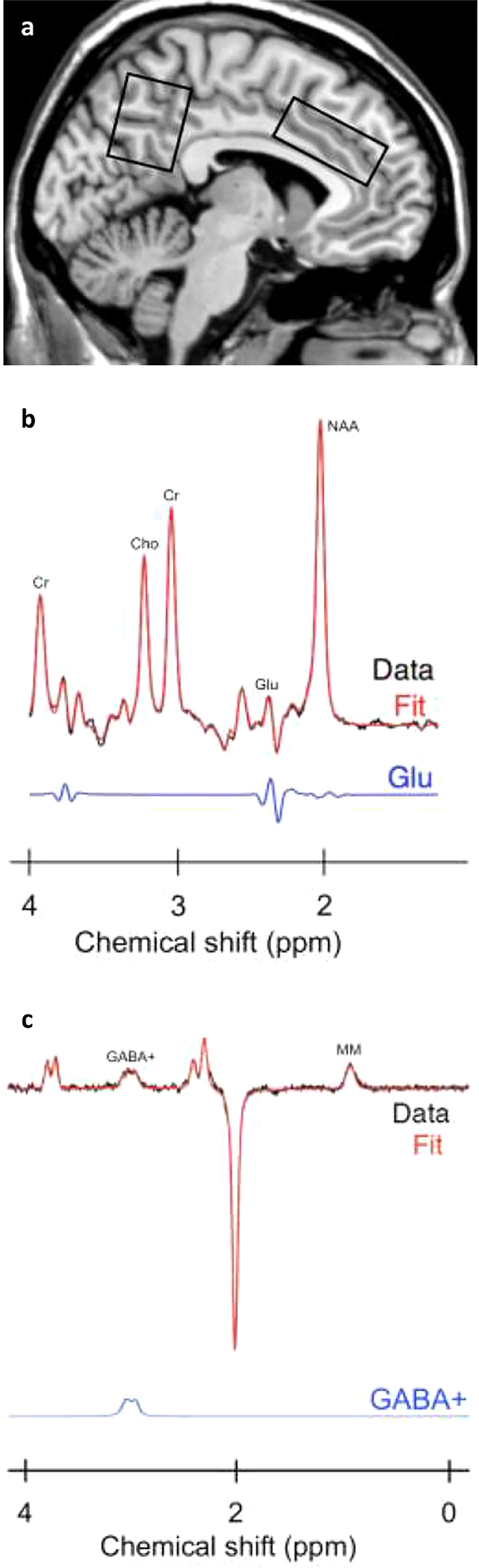
(a) Mid-sagittal slice showing anatomical placement of MRS voxels in the parieto-occipital cortex and anterior cingulate cortex from a single subject. “Off” spectrum (b) and difference-edited spectrum (c) from a study subject. The two spectra are displayed with no filtering. Metabolites shown include: total creatine (Cr). choline (Cho). glutamate (Glu), N-acetylaspartate (NAA), γ-ami-nobutyric acid plus co-edited macromolecule resonance (GABA +), and co-edited macromolecule resonance at −0.9 ppm (MM).
Proton spectroscopy employed a GABA-optimized MEGAPRESS sequence (Mescher et al., 1998) for measures of GABA using difference-editing as well as measures of glutamate using the 68ms sub-spectrum. The transmitter frequency was set onto the creatine resonance at 3.00 ppm to minimize chemical-shift displacement artifact for each spectral acquisition. The MEGAPRESS sequence used the following acquisition parameters: TR = 2s, TE = 68ms, spectral bandwidth = 2 kHz, readout duration = 512 ms, NEX = 384, total scan duration = 13min.
Subsequent data processing and analyses were conducted on a LINUX workstation using in-house software as well as commercial fitting software. To quantify difference-edited GABA and glutamate with the MEGAPRESS data, the difference-edited spectra were fitted with LCModel (Provencher et al., 1993; Provencher et al., 2001). All phase and frequency-corrected ‘ON’ and ‘OFF’ spectra were averaged separately to produce a single 68 ms ‘ON’ and ‘OFF’ spectrum, which were subsequently subtracted to produce the final, optimized, difference-edited GABA spectrum. The appropriate LCModel templates were used to fit the 68ms ‘OFF’ spectrum for the measurement of glutamate and the difference-edited spectrum for GABA measurement. The difference-edited GABA resonance area at 3.00ppm, as well as the fitted 68 ms glutamate resonance were normalized to the LCModel fitted 68 ms ‘OFF’ spectrum creatine resonance area and left as simple ratios. As the edited GABA signal includes contributions from GABA as well as macromolecules, we refer to the signal obtained as GABA+.
The high resolution T1-weighted axial images were segmented into gray matter (GM), white matter (WM), and cerebrospinal fluid (CSF) compartments employing FSL 4.1 (FMRIB Software Library; Analysis Group, FMRIB; Oxford, UK). Subsequently, segmented images were input to an automated voxel co-registration and partial-volume analysis program to calculate volumetric contributions of GM, WM and CSF.
Statistical Analysis
The primary outcome variables were the GABA+/Cr and Glu/Cr metabolite concentrations in the two regions of interest (dACC, POC). HC, PI, and MDD group differences were tested with one-way ANOVA. Post-hoc contrasts were assessed by the Tukey-Kramer (T-K) test. Voxels exhibiting a Cramér-Rao index >20% or data values falling 3 standard deviations outside of the group mean were excluded from analyses. Between group differences in our secondary outcome variables i.e., psychometric scores and PSG/sleep-wake diary variables, were also compared using one-way ANOVA with post-hoc contrasts assessed by T-K tests. Finally, the relationship of our secondary outcome variables to MRS metabolite levels were explored using Pearson’s correlations. Statistical analyses were performed using R (Version 3.6.0), Excel (Version 2013, Microsoft Corp, Redmond, WA), SPSS (Version 2016, IBM Corp, Armonk, N.Y.).
RESULTS
Table 1 reports demographic, psychometric, and sleep-wake measures for HC, PI, and MDD groups. The MDD group (n=51) included 33 women. The mean age of the MDD group was 33.2 ± 14.4 (range = 18–70) years. Comorbid diagnoses included anxiety disorders (n=23), dysthymia (n=6), eating disorders (n=5), personality disorders (n=9), and ADHD (n=1). The PI group (n=24) included 17 women, with a mean age of 39.5±14.2 (range = 21–69) years. The age and sex-matched HC group (n=25, 68% female) had a mean age of 33.9 ± 14.6 (range = 19–68) years.
ANOVA documented significant (p<0.001) between-group differences for all rating scale variables shown in Table 1. Post-hoc tests revealed that subjects with MDD, compared to HCs, reported significantly greater levels of depression on the HAM-D and BDI, greater levels of Anhedonia as derived from the BDI, and increased stress, as measured by the PSS (p<0.001). Compared to HCs, subjects with MDD also reported significantly worse sleep quality as measured by the ISI and PSQI, greater dysfunctional beliefs and attitudes about sleep as measured by the DBAS-16, and increased fatigue as measured by the FSS (p<0.001). Relative to HCs, subjects with PI reported significantly worse sleep quality measured by the ISI and PSQI, greater dysfunctional beliefs and attitudes about sleep reflected by the DBAS-16, and increased fatigue as measured by the FSS (p<.001). Finally, relative to those with PI, subjects with MDD reported greater levels of depression measured by the HAM-D, BDI, and Anhedonia subscale, as well as higher levels of stress (PSS) (p<.001). Notably, MDD and PI subjects did not differ on ratings of sleep quality by ISI or PSQI.
ANOVA of all measures derived from the sleep-wake diaries also documented significant between group differences (p<0.001). Post-hoc tests revealed that both MDD and PI subjects, compared to HCs, reported significantly greater sleep disturbance, i.e., longer SOL, less TST, poorer SE% and reduced sleep quality (p<0.001); MDD subjects also reported more WASO than HCs (p<0.05). Relative to those with MDD, subjects with PI reported even greater sleep disturbance, i.e., more WASO, less TST, and poorer SE% (p<0.001). In large part, objective sleep quality, measured by PSG and subjected to ANOVA, corroborated many of the diary entries. Compared to HCs, MDD exhibited increased SOL (p<0.01), decreased TST (p<0.05), and poorer SE% (p<0.05) whereas subjects with PI exhibited significantly more WASO (p<0.05), less TST (p<0.001), and poorer SE% (p<0.01). However, latency to the first epoch of sleep was significantly longer for those with MDD relative to those with PI (p<0.05).
Voxels excluded from analysis due to the Cramér-Rao index exceeding 20% included 18 for GABA+/Cr in the dACC (4 HC subjects, 10 MDD subjects, and 4 PI subjects), 2 for Glu/Cr in the POC (2 PI subjects), and 3 for Glu/Cr in the dACC (1 HC subject, 2 MDD subjects). Additional data excluded from analysis due to falling 3 standard deviations outside of the mean included 1 for GABA+/Cr in the dACC (1 HC subject), 1 for Glu/Cr in the dACC (1 MDD subject), and 1 for Glu/Cr in the POC (1 MDD subject). We observed no significant between-group differences for creatine, or grey or white matter (as percent of grey matter plus white matter) in either POC or dACC.
Our primary outcome measures are shown in Table 2 and Figure 2a–c. A significant between-group difference was observed for Glu/Cr in the dACC (F=4.97, df=95, p <.009). Post-hoc Tukey-Kramer contrasts determined that dACC Glu/Cr was significantly lower (~15%) in MDD subjects than in HCs (q=4.41, p<.01). No significant between group differences were observed for GABA+/Cr in the dACC nor for GABA+/Cr or Glu/Cr in the POC. However, there was a trend in the POC suggesting a modest reduction in Glu/Cr in those with PI relative to healthy controls and those with MDD.
Table 2.
1H MRS Measures in Healthy Controls, Primary Insomnia, and Major Depression.
| REGION OF INTEREST | 1H MRS Measuresa | Healthy Controls (Mean, SD) | Primary Insomnia (Mean, SD) | Major Depressive Disorder (Mean, SD) | F | P-value (two tailed) | df | Tukey Kramer Post-Hoc Contrasts |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Anterior Cingulate Cortex | GABA + /Cr | 0.19 (0.03) | 0.19 (0.06) | 0.18 (0.04) | 0.75 | 0.474 | 80 | |
| Glu/Cr | 1.00 (0.20) | 0.93 (0.19) | 0.86 (0.18) | 4.97 | 0.009 | 95 | MDD<HC <.01 | |
| Creatine | 161.48 (42.25) | 170.93 (42.34) | 165.97 (37.72) | 0.34 | 0.71 | 99 | ||
| Parieto-Occipital Cortex | GABA + /Cr | 0.21 (0.03) | 0.20 (0.05) | 0.22 (0.04) | 1.21 | 0.302 | 98 | |
| Gtu/Cr | 0.83 (0.10) | 0.79 (0.13) | 0.84 (0.08) | 2.60 | 0.080 | 95 | ||
| Creatine | 219.2 (40.78) | 214.83 (45.27) | 217.44 (34.02) | 0.08 | 0.93 | 98 | ||
Values reportd only include subjects with Cramér-Rao < 20%.
Figure 2.
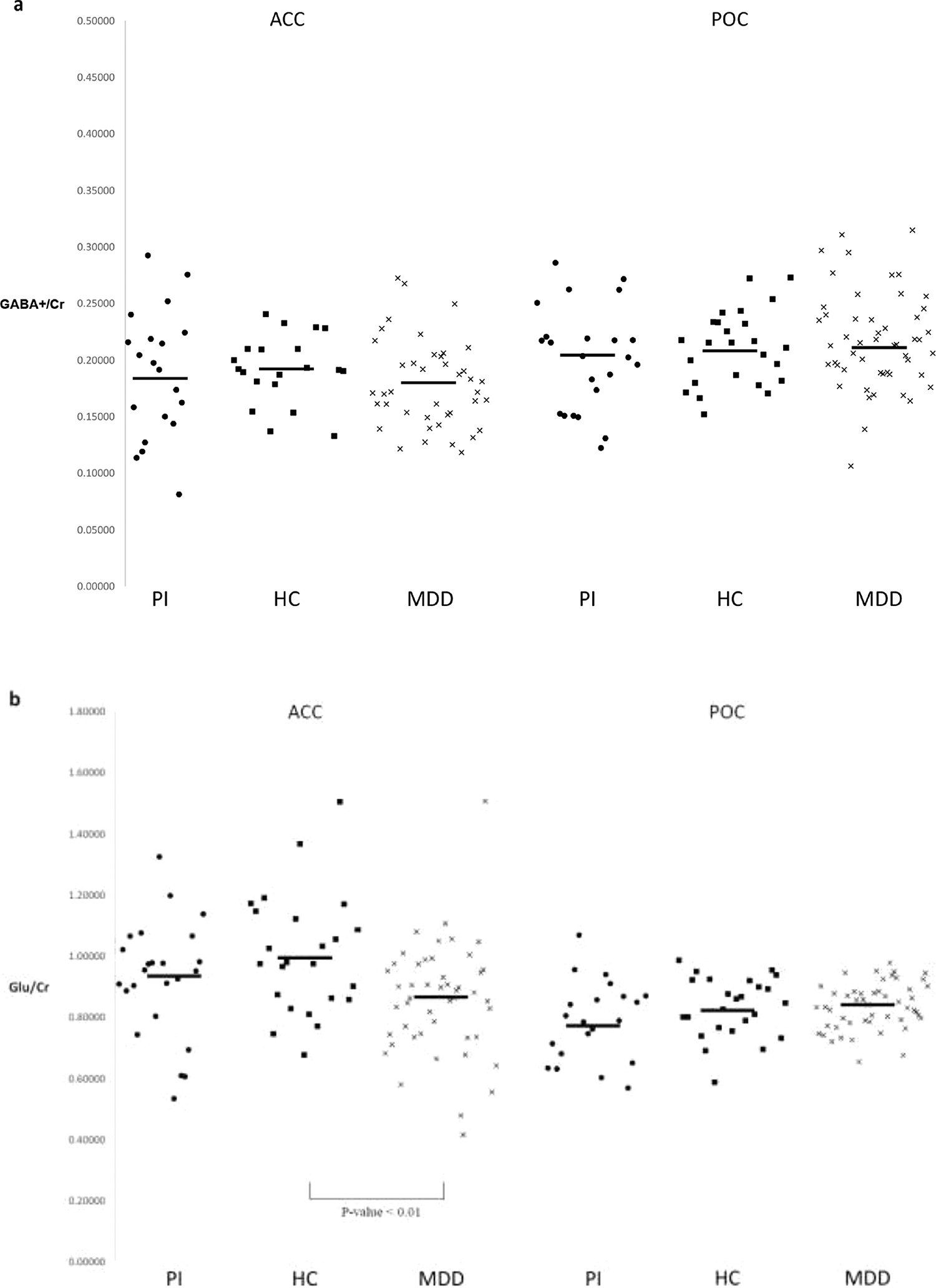
Individual subject GABA+/Cr (a) and Glu/Cr (b) by brain region of interest. Horizontal bars denote group means.
To further explore the significant decrement in dACC Glu/Cr levels in MDD relative to controls, we examined the relationship between the rating scale measures and dACC Glu/Cr levels within the MDD sample. In the dACC, Glu/Cr was not significantly associated with age, sex, ISI, PSQI, HAM-D, BDI, or PSS. There was, however, a trend negative correlation between dACC Glu/Cr and Anhedonia (r=−0.24, p=0.10). Also, of note, no sleep diary or PSG measure was significantly correlated with MDD dACC Glu/Cr. Within the PI sample, we partially confirmed our previous finding (Winkelman et al., 2008) of an inverse relationship between GABA/Cr levels and PSG measures of WASO; there was a strong trend towards an inverse relationship between dACC GABA+/Cr and WASO (r= −0.43 p=0.06). However, we failed to demonstrate a significant inverse relationship between POC GABA+/Cr and WASO (r=0.05, p=0.82). Finally, we observed a significant, but positive correlation (r=0.47, p < 0.01) between POC GABA+/Cr and WASO in the MDD sample.
DISCUSSION
In this study, we demonstrated glutamatergic abnormalities in the dACC of subjects with diagnosed MDD. Relative to controls, Glu/Cr was significantly reduced by about 15 percent; this decrement did not occur in those with PI. Broadly stated, our observations concur with most (Yuksel and Ongur, 2010; Moriguchi et al., 2019), but not all (Sanacora et al., 2004; Godlewska et al., 2017) 1H MRS investigations of Glu and/or Glx concentrations in those with MDD. Commonly noted are reports of Glx deficits in the dACC and several neighboring loci in the prefrontal cortex in unmedicated MDD subjects (Pfleiderer et al., 2003; Hasler et al., 2007; Chen et al., 2014). Glu and Glx deficits have also been observed in the ACC in symptomatic but medicated patients with MDD (Auer et al., 2000; Horn et al., 2010). This report of elevated levels of glutamine in the putamen of those with MDD, and no decrement in either the OC or dACC (Godlewska et al., 2017), argue for a broadened inquiry into potential regional glutaminergic abnormalities.
Our demonstration of Glu/Cr deficits in the dACC represents only a partial confirmation of the hypotheses we articulated in the introduction. A secondary hypothesis suggested that Glu/Cr decrements in MDD would be related to the extent of sleep disturbance in that group. As shown in Table 1, subjects with MDD were clearly characterized by poor sleep quality, a diagnostic feature of MDD. Despite this demonstration of disordered sleep in those with MDD and, contrary to our secondary hypothesis, dACC Glu/Cr was not significantly correlated with any subjective or objective measure of sleep disturbance.
Although sleep quality was not a significant covariate of Glu/Cr decrements in the dACC, the extent of depressive symptoms remains a potential alternative covariate contributing to the Glu and Glx values in those with MDD. A recent meta-analysis including over 1000 MDD patients determined that Glx deficits in patients relative to controls could not be explained by symptom severity (Moriguchi et al., 2019). This conclusion was based on several studies. Glu and Glx levels in the ACC did not correlate with HAMD ratings (Auer et al., 2000). Also, in MDD patients, Glx deficits in prefrontal areas did not correlate with MADRS (Montgomery-Asberg Rating Scale) scores (Hasler et al., 2007). Likewise, Glx deficits in the ACC of MDD patients did not correlate with HAMD ratings (Chen et al., 2014). Our data provides mixed support for these conclusions as we observed no association between Glu/Cr levels with neither the HAMD nor the BDI rating scales in our MDD sample. In contrast, the role of the dACC in mood per se (Stan et al., 2014) is modestly supported by our findings. Not only were Glu/Cr levels in the dACC significantly reduced in our MDD subjects relative to controls, but Glu/Cr levels, as a trend, were inversely correlated with levels of anhedonia, a defining feature of MDD. This latter finding is consistent with the observation that anhedonia in MDD is associated with decreased Glu in the pregenual ACC (Walter et al., 2009).
We observed no differences in GABA+/Cr levels in either the POC or the dACC between MDD relative to HCs. These findings are consistent with other reports documenting no GABA differences in the OC (Godlewska et al., 2017) or in the dACC (Walter et al., 2009; Brennan et al., 2017) in MDD relative to controls. These reports stand in contrast to other MRS studies documenting reduced GABA levels in OC (Sanacora et al., 1999; Sanacora et al., 2004; Price et al., 2009), prefrontal loci (Hasler et al., 2007), and dACC (Gabbay et al., 2012) in actively symptomatic, unmedicated MDD subjects.
Among these studies, discrepant GABA findings might be attributed to differences in MRS data acquisition and processing or to heterogeneity in the MDD samples. Within our study and the eight studies cited above, acquisition and processing of MRS data varied in several respects including magnet strength, dimension and placement of voxels in the ROIs (OC, dACC, and prefrontal cortex), as well as referencing of GABA or GABA+ levels either to creatine or to unsuppressed voxel tissue water. Broadly speaking, none of these acquisition and/or processing factors consistently differentiated positive from negative GABA findings. A more compelling explanation of disparate findings might be found in the heterogeneity of the MDD samples, in particular, illness severity and symptom profile. Cogent evidence for the impact of illness severity and symptom profile on GABA is suggested by studies of GABA levels in the OC. In our protocol, MDD subjects were largely recruited through community advertisements, were characterized by mild to moderate depression severity, and few had a history of treatment resistance. In Sanacora’s seminal study (Sanacora et al., 1999), GABA levels in the OC were reduced by 52% in the MDD sample compared to controls. The depressives in that sample were markedly ill and were largely recruited from an inpatient psychiatric unit. In a subsequent study from the same group, GABA levels in the OC were reduced by an average of only 15% in the MDD sample relative to controls (Sanacora et al, 2004). That sample, like ours, was recruited through advertisements and community referrals. The combined studies suggested that GABA deficits were particularly prevalent in those with more severe depression.
The role of GABA in sleep regulation is supported by a range of evidence. Through direct study, GABAergic neurons, notably in the ventrolateral preoptic nucleus, have been shown to suppress CNS arousal (Saper et al., 2005). Indirect confirmation of the involvement of GABAergic mechanisms in the pathophysiology of insomnia derives from the efficacy of benzodiazepine and nonbenzodiazepine hypnotics in the treatment of insomnia. These observations are consistent with our previous MRS studies of disordered sleep in PI. These studies revealed a significant inverse relationship between sleep disturbance, notably the amount of polysomnographically-recorded WASO, and GABA/Cr levels both globally (Winkelman et al., 2008) as well as in both the OC and dACC (Plante et al., 2012). In contrast to these previous findings, our current study did not demonstrate a POC or dACC GABA+/Cr deficit in our PI subjects or in our MDD subjects although both groups demonstrated significant comorbid sleep disturbance. We observed a trend inverse association between dACC GABA+/Cr and WASO in the PI group. However, within our MDD sample, we did not observe an inverse correlation between dACC or POC GABA+/Cr and either subjectively or objectively recorded sleep disturbance. We cannot readily explain our finding of a positive association between PSG WASO measures of sleep disturbance and GABA+/Cr levels in the POC.
Across psychiatric conditions, a cohesive picture of the relationship of disordered sleep to GABA mechanisms has failed to emerge. In a study contrasting subjects with PI to good sleeper controls, sleep disturbance in the insomniacs, quantified by PSG WASO, was inversely correlated with OC GABA despite the observation that GABA levels were higher in the insomniacs than in the controls (Morgan et al., 2012). In a more recent study comparing patients with PI to good sleeper controls, GABA and Glx were measured in dACC and dorsolateral prefrontal cortex (DLPFC). GABA levels did not differentiate insomniacs from controls; however, GABA levels in dACC were positively associated with sleep duration suggesting that GABA deficits in the dACC might herald objective sleep disturbances (Spiegelhalder et al., 2016). Two studies of patients with PTSD, a diagnosis characterized by heightened levels of disordered sleep, have also corroborated an association between GABA deficits and poor sleep quality. In a comparison of those with PTSD to trauma-exposed controls without PTSD, GABA levels in the parieto-occipital cortex were significantly reduced in those with PTSD, and, in this patient group, these reductions were associated with poor sleep quality as measured by the Insomnia Severity Index (Meyerhoff et al., 2014). Finally, PTSD subjects have demonstrated not only a lower dACC GABA/H2O level relative to controls, but also an inverse correlation between these GABA deficits and subjective measures of sleep disturbance (Sheth et al., 2019).
In contrast to the well-studied role of GABA in sleep regulation, our understanding of the role of Glu and Gln in sleep regulation is less well advanced, which is puzzling given that Gln is a major substrate for the synthesis of both Glu and GABA and for the maintenance of their neurotransmitter pools. However, in a recent MRS study of insomnia patients with objectively verified short sleep duration, in contrast to insomnia patients with normal sleep duration and to good sleeper controls, Glu levels in the left OC were significantly reduced in the short sleep duration insomniacs relative to controls and relative to the insomnia group with normal sleep duration. In addition, Glu concentration in the left OC was significantly correlated with total sleep duration and negatively correlated with WASO (Miller et al., 2017). These findings are consistent with our hypothesis (although not supported in our current data) that GABAergic and potentially glutamatergic mechanisms might mediate disordered sleep in MDD.
Our study design reflected several important strengths. We employed 1H MRS to assess in vivo potential GABAergic and glutamatergic abnormalities in both POC and dACC while utilizing a 4 Tesla MRS scanner with greater spectral signal dispersion compared to lower field strengths. Relative to other 1H MRS studies of GABA and Glu abnormalities in depression, our protocol involved one of the largest MDD sample sizes to date (51 depressed). They were age and gender-matched to 25 healthy controls, and 24 enrollees with PI. All subjects with MDD were free from psychoactive medications for a minimum of 4 weeks at the point of MRS scanning, and most for more than a year (44 of 51 participants). Sleep quality was extensively assessed utilizing both subjective and objective measures: ISI and PSQI questionnaires, a minimum of 2 weeks of sleep diaries, 2 weeks of actigraphy monitoring, and 2 overnight polysomnograms which included an assessment of comorbid disordered breathing and periodic limb movements.
In contrast to the strengths of our study design, we also acknowledge the following limitation. Adherence to exclusion criteria proscribing CNS active agents was assessed by urine toxic screens at baseline and end of study, by interview at each study visit, and through daily sleep diary self-report. Use of CNS agents was not confirmed through any other means. The protocol and attendant analytic capabilities would have been enhanced by a more comprehensive assessment of the clinical features of the MDD sample as well as by the recruitment of MDD subjects with a broader range of depression severity.
Summary
This study documented a 15% reduction in Glu/Cr levels in the dACC in MDD compared to healthy controls. In those with PI, there was a trend reduction in Glu/Cr levels in the POC. We did not observe GABA+/Cr decrements in either the POC or the dACC in either patient group compared to controls or to each other.
Although glutamatergic abnormalities may lack diagnostic validity for MDD, they may track specific symptoms or comorbidities both within and across this and other psychiatric disorders. In our protocol, Glu/Cr decrements in the dACC suggested greater ratings of anhedonia, a core feature of MDD. Because of our previous findings linking GABA/Cr deficits in the OC and dACC to disordered sleep in PI, we hypothesized that GABAergic and glutamatergic deficits in unipolar depression might reflect comorbid sleep disturbance rather than the affective component of depressive illness. The results of our current protocol did not confirm this hypothesis. However, coupling our previous findings to subsequent reports of an inverse relationship between occipital GABA levels and PSG measures of WASO (Morgan et al., 2012), as well as self-rated sleep quality (Meyerhoff et al., 2014), suggests that our hypothesis warrants further investigation.
Highlights:
1H MRS was used to examine GABA+ and Glu relative to creatine (Cr) in the dorsal anterior cingulate cortex (dACC) and parieto-occipital cortex (POC) in those with Major Depressive Disorder (MDD), Primary Insomnia (PI), and Healthy Controls (HC).
Relative to HCs, subjects with MDD exhibited decreased Glu/Cr in the dACC.
In those with MDD, Glu/Cr levels in the dACC were inversely correlated with anhedonia ratings.
In those with MDD, sleep quality was not associated with Glu/Cr in the dACC.
Relative to HCs, subjects with PI exhibited no significant differences in GABA+/Cr or Glu/Cr.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F, 2000. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol. Psychiatry 47, 305–313. 10.1016/S0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- Beck A, Steer RA, 1993. Manual for the Beck Depression Inventory. Psychological Corporation, San Antonio, TX. [Google Scholar]
- Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Boorman E, P, J.C P,MM., 2008. Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int. J. Neuropsychopharmacol. 11, 255–260. 10.1017/s1461145707007924. [DOI] [PubMed] [Google Scholar]
- Brennan BP, Admon R, Perriello C, LaFlamme EM, Athey AJ, Pizzagalli DA, Hudson JI, Pope HG Jr., Jensen JE, 2017. Acute change in anterior cingulate cortex GABA, but not glutamine/glutamate, mediates antidepressant response to citalopram. Psychiatry Res. Neuroimaging 269, 9–16. 10.1016/j.pscychresns.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buysse DJ, Reynolds CF 3rd, Monk TH, Berman SR, Kupfer DJ, 1989. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 28, 193–213. 10.1016/0165-1781(89)90047-4. [DOI] [PubMed] [Google Scholar]
- Chang PP, Ford DE, Mead LA, Cooper-Patrick L, Klag MJ, 1997. Insomnia in young men and subsequent depression. The Johns Hopkins Precursors Study. Am. J. Epidemiol. 146, 105–114. 10.1093/oxfordjournals.aje.a009241. [DOI] [PubMed] [Google Scholar]
- Chen LP, Dai HY, Dai ZZ, Xu CT, Wu RH, 2014. Anterior cingulate cortex and cerebellar hemisphere neurometabolite changes in depression treatment: A 1H magnetic resonance spectroscopy study. Psychiatry Clin. Neurosci. 68, 357–364. 10.1111/pcn.12138. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Lavretsky H, Olmstead R, Levin MJ, Oxman MN, Irwin MR, 2008. Sleep disturbance and depression recurrence in community-dwelling older adults: a prospective study. Am. J. Psychiatry 165, 1543–1550. 10.1176/appi.ajp.2008.07121882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrovski AY, Mulsant BH, Houck PR, Mazumdar S, Lenze EJ, Andreescu C, Cyranowski JM, Reynolds CF 3rd, 2007. Residual symptoms and recurrence during maintenance treatment of late-life depression. J. Affect. Disord. 103, 77–82. 10.1016/j.jad.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First M, Gibbon M, Spitzer RL, Williams JBW, Benjamin LS, 1997. Structured Clinical Interview for DSM-IV Axis II Personality Disorders, (SCID-II). American Psychiatric Press, Inc., Washington, D.C. [Google Scholar]
- First M, Spitzer RL, Gibbon M, and Williams JBW, 2002. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition with Psychotic Screen (SCID-I/P W/ PSY SCREEN). Biometrics Research, New York State Psychiatric Institute, New York. [Google Scholar]
- Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, Alonso CM, Shungu DC, 2012. Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch. Gen. Psychiatry 69, 139–149. 10.1001/archgenpsychiatry.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewska BR, Clare S, Cowen PJ, Emir UE, 2017. Ultra-high-field magnetic resonance spectroscopy in psychiatry. Front. Psychiatry 8, 123. 10.3389/fpsyt.2017.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesmann C, 2002. GABA mechanisms and sleep. Neuroscience 111, 231–239. 10.1016/S0306-4522(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Hamilton M, 1960. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 23, 56–62. 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC, 2007. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 64, 193–200. 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Horn DI, Yu C, Steiner J, Buchmann J, Kaufmann J, Osoba A, Eckert U, Zierhut KC, Schiltz K, He H, Biswal B, Bogerts B, Walter M, 2010. Glutamatergic and resting-state functional connectivity correlates of severity in major depression - the role of pregenual anterior cingulate cortex and anterior insula. Front. Syst. Neurosci 4. 10.3389/fnsys.2010.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iber C, Ancoli-Israel S, Chesson AL Jr, Quan S, 2007. The new sleep scoring manual - The evidence behind the rules, J. Clin. Sleep Med. 3, 107–107. [Google Scholar]
- Jones BE, 2017. Principal cell types of sleep-wake regulatory circuits. Curr. Opin. Neurobiol. 44, 101–109. 10.1016/j.conb.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD, 1989. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch. Neurol. 46, 1121–1123. 10.1001/archneur.1989.00520460115022. [DOI] [PubMed] [Google Scholar]
- Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, Zarate CA Jr., 2017. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol. Psychiatry 81, 886–897. 10.1016/j.biopsych.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenstein S, Prantera C, Varvo V, Scribano ML, Berto E, Luzi C, Andreoli A, 1993. Development of the Perceived Stress Questionnaire: a new tool for psychosomatic research. J. Psychosom. Res. 37, 19–32. 10.1016/0022-3999(93)90120-5. [DOI] [PubMed] [Google Scholar]
- Luykx JJ, Laban KG, van den Heuvel MP, Boks MP, Mandl RC, Kahn RS, Bakker SC, 2012. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of (1)H-MRS findings. Neurosci. Biobehav. Rev. 36, 198–205. 10.1016/j.neubiorev.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Mescher M, Merkle H, Kirsch J, Garwood M, Gruetter R, 1998. Simultaneous in vivo spectral editing and water suppression. NMR Biomed. 11, 266–272. . [DOI] [PubMed] [Google Scholar]
- Meyerhoff DJ, Mon A, Metzler T, Neylan TC, 2014. Cortical gamma-aminobutyric acid and glutamate in posttraumatic stress disorder and their relationships to self-reported sleep quality. Sleep 37, 893–900. 10.5665/sleep.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael N, Erfurth A, Ohrmann P, Arolt V, Heindel W, Pfleiderer B, 2003. Metabolic changes within the left dorsolateral prefrontal cortex occurring with electroconvulsive therapy in patients with treatment resistant unipolar depression. Psychol. Med. 33, 1277–1284. 10.1017/S0033291703007931. [DOI] [PubMed] [Google Scholar]
- Miller CB, Rae CD, Green MA, Yee BJ, Gordon CJ, D’Rozario AL, Kyle SD, Espie CA, Grunstein RR, Bartlett DJ, 2017. An objective short sleep insomnia disorder subtype is associated with reduced brain metabolite concentrations in vivo: a preliminary magnetic resonance spectroscopy assessment. Sleep 40. 10.1093/sleep/zsx148. [DOI] [PubMed] [Google Scholar]
- Morgan PT, Pace-Schott EF, Mason GF, Forselius E, Fasula M, Valentine GW, Sanacora G, 2012. Cortical GABA levels in primary insomnia. Sleep 35, 807–814. 10.5665/sleep.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, Plitman E, Sano Y, Tarumi R, ElSalhy M, Katayama N, Ogyu K, Miyazaki T, Kishimoto T, Graff-Guerrero A, Meyer JH, Blumberger DM, Daskalakis ZJ, Mimura M, Nakajima S, 2019. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 24, 952–964. 10.1038/s41380-018-0252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin C, 1993. Insomnia: Psychological Assessment and Management. Guilford Press, New York. [Google Scholar]
- Morin CM, Vallieres A, Ivers H, 2007. Dysfunctional beliefs and attitudes about sleep (DBAS): validation of a brief version (DBAS-16). Sleep 30, 1547–1554. 10.1093/sleep/30.11.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt D, Wilson S, Paterson L, 2008. Sleep disorders as core symptoms of depression. Dialogues Clin. Neurosci. 10, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleiderer B, Michael N, Erfurth A, Ohrmann P, Hohmann U, Wolgast M, Fiebich M, Arolt V, Heindel W, 2003. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. 122, 185–192. 10.1016/S0925-4927(03)00003-9. [DOI] [PubMed] [Google Scholar]
- Plante DT, Jensen JE, Schoerning L, Winkelman JW, 2012. Reduced gamma-aminobutyric acid in occipital and anterior cingulate cortices in primary insomnia: a link to major depressive disorder? Neuropsychopharmacology 37, 1548–1557. 10.1038/npp.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, Murrough JW, Charney DS, Mathew SJ, 2009. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biol. Psychiatry 65, 792–800. 10.1016/j.biopsych.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher SW, 1993. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med. 30, 672–679. 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Provencher SW, 2001. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 14, 260–264. 10.1002/nbm.698. [DOI] [PubMed] [Google Scholar]
- Riemann D, Voderholzer U, 2003. Primary insomnia: a risk factor to develop depression? J. Affect. Disord. 76, 255–259. 10.1016/S0165-0327(02)00072-1. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF, 2004. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch. Gen. Psychiatry 61, 705–713. 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH, 1999. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 56, 1043–1047. 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J, 2005. Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263. 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schur RR, Draisma LW, Wijnen JP, Boks MP, Koevoets MG, Joels M, Klomp DW, Kahn RS, Vinkers CH, 2016. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of (1) H-MRS studies. Hum. Brain Mapp. 37, 3337–3352. 10.1002/hbm.23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth C, Prescot AP, Legarreta M, Renshaw PF, McGlade E, Yurgelun-Todd D, 2019. Reduced gamma-amino butyric acid (GABA) and glutamine in the anterior cingulate cortex (ACC) of veterans exposed to trauma. J. Affect. Disord. 248, 166–174. 10.1016/j.jad.2019.01.037. [DOI] [PubMed] [Google Scholar]
- Spiegelhalder K, Regen W, Nissen C, Feige B, Baglioni C, Riemann D, Hennig J, Lange T, 2016. Magnetic resonance spectroscopy in patients with insomnia: a repeated measurement study. PLoS One 11, e0156771. 10.1371/journal.pone.0156771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan AD, Schirda CV, Bertocci MA, Bebko GM, Kronhaus DM, Aslam HA, LaBarbara EJ, Tanase C, Lockovich JC, Pollock MH, Stiffler RS, Phillips ML, 2014. Glutamate and GABA contributions to medial prefrontal cortical activity to emotion: implications for mood disorders. Psychiatry Res. 223, 253–260. 10.1016/j.pscychresns.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U, Schnepf B, Boeker H, Boesiger P, Northoff G, 2009. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch. Gen. Psychiatry 66, 478–486. 10.1001/archgenpsychiatry.2009.39. [DOI] [PubMed] [Google Scholar]
- Winkelman JW, Buxton OM, Jensen JE, Benson KL, O’Connor SP, Wang W, Renshaw PF, 2008. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-MRS). Sleep 31, 1499–1506. 10.1093/sleep/31.11.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuksel C, Ongur D, 2010. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol. Psychiatry 68, 785–794. 10.1016/j.biopsych.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Gould TD, 2018. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 23, 801–811. 10.1038/mp.2017.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Narr KL, Woods RP, Phillips OR, Alger JR, Espinoza RT, 2013. Glutamate normalization with ECT treatment response in major depression. Mol. Psychiatry 18, 268–270. 10.1038/mp.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]