

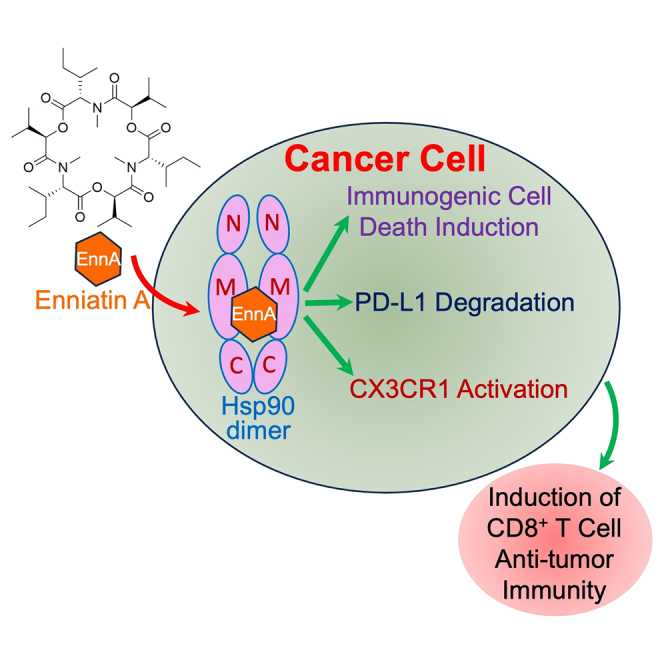
Summary
Low response rates and immune-related adverse events limit the remarkable impact of cancer immunotherapy. To improve clinical outcomes, preclinical studies have shown that combining immunotherapies with N-terminal Hsp90 inhibitors resulted in improved efficacy, even though induction of an extensive heat shock response (HSR) and less than optimal dosing of these inhibitors limited their clinical efficacy as monotherapies. We discovered that the natural product Enniatin A (EnnA) targets Hsp90 and destabilizes its client oncoproteins without inducing an HSR. EnnA triggers immunogenic cell death in triple-negative breast cancer (TNBC) syngeneic mouse models and exhibits superior antitumor activity compared to Hsp90 N-terminal inhibitors. EnnA reprograms the tumor microenvironment (TME) to promote CD8+ T cell-dependent antitumor immunity by reducing PD-L1 levels and activating the chemokine receptor CX3CR1 pathway. These findings provide strong evidence for transforming the immunosuppressive TME into a more tumor-hostile milieu by engaging Hsp90 with therapeutic agents involving novel mechanisms of action.
Subject areas: Microenvironment, Biological sciences, Molecular biology, Cancer
Graphical abstract
Highlights
-
•
Enniatin A targets the chaperone Hsp90 without inducing a heat shock response
-
•
Enniatin A induces immunogenic cell death in aggressive TNBC models
-
•
Enniatin A reduces immunosuppression by reducing PD-L1 protein and activating CX3CR1
-
•
Enniatin A promotes CD8+ T cell-mediated anti-tumor immunity against TNBC
Microenvironment; Biological sciences; Molecular biology; Cancer
Introduction
Triple-negative breast cancer (TNBC) is an aggressive disease that remains challenging with limited targeted therapies available. However, harnessing the power of the immune system to eliminate various aggressive cancers offers new hope to patients suffering from TNBC.1,2 Indeed, immune checkpoint inhibitors (ICIs) such as antibodies against programmed cell death 1 (PD-1), programmed cell death 1 ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) have shown striking clinical responses in various cancers, including advanced metastatic cases. Unfortunately, initial responses are often followed by relapse and disease progression in many patients.3,4,5 Thus, alternative treatments, including ICI combinations, have been developed. They have shown improved therapeutic efficacy compared to monotherapies in various human cancers and preclinical models such as E0771 and 4T1 syngeneic murine TNBC.6,7,8,9 Other strategies used involve combining immunotherapy with targeted chemotherapies. Indeed, several studies have demonstrated the potential therapeutic benefit of combining ICIs with N-terminal inhibitors of the chaperone Hsp90. For instance, the second-generation Hsp90 inhibitor ganetespib (STA-9090) enhances T-cell-mediated killing of patient-derived human melanoma cells by their autologous T cells in vitro and potentiates responses to anti-CTLA4 and anti-PD-1 therapy in vivo through upregulation of interferon target genes.10 Similar improvement in efficacy or restoration of sensitivity to immunotherapeutic agents has been reported in various cancers.11,12 These findings correlate with a recent report showing that Hsp90 N-terminal inhibitors of various chemical scaffolds including ganetespib, Luminespib (AUY-922), SNX-5422, PU-H71, and geldanamycin caused a significant decrease in the transcription and cancer cell-surface expression of immune checkpoint ligands PD-L1 and PD-L2.13 It is, however, well established that high doses of Hsp90 N-terminal inhibitors used in most preclinical and clinical studies induce an extensive heat shock response (HSR)14 driven by the transcription factor HSF1, which itself emerged as a promoter of various cancers including skin malignancies and hepatocarcinoma.15,16,17,18 Furthermore, activation of HSF1 drives the overexpression of Hsp70 and Hsp27, which are known to be anti-apoptotic factors.19,20,21,22,23 These pro-survival mechanisms are thought to lower the Hsp90 inhibitors’ antitumor efficacy. Yet, paradoxically, overexpression of Hsp70 and Hsp27 is used as a biomarker for Hsp90 inhibition in clinical trials. Overall, the clinical benefit of targeting Hsp90 to treat cancer remains largely unharnessed. Indeed, Hsp90 controls the stability and function of “client” proteins, many of which are oncoproteins and, thus, Hsp90 remains an attractive target for cancer treatment.24,25 Furthermore, accumulating evidence supports the emergence of a powerful concept that the biology of Hsp90 in cancer cells differs substantially from its normal function in non-transformed cells. Indeed, early studies have shown that biochemical complexes of Hsp90 in cancer cells contain higher levels of co-chaperones that confer a higher affinity state to certain Hsp90 N-terminal inhibitors.26 More recent and comprehensive studies have demonstrated the existence of a highly integrated chaperone network, referred to as an epichaperome, involving complex interconnections between Hsp90 and Hsp70 machines in cancer cells, promoting aggressive tumor states. Yet, this pathological molecular organization can confer sensitivity to Hsp90 inhibitors such as PU-71, which was shown to target the epichaperome and exhibit a more efficacious impact on aggressive tumors.27,28,29,30 Excitingly, the cytosolic Hsp90-selective inhibitor pimitespib (TAS-116) received the first approval in Japan for aggressive gastrointestinal stromal tumors that progressed after chemotherapy.31,32
Besides chemical optimization and dosing strategies of Hsp90 N-terminal inhibitors and their combination with other therapeutic agents, efforts to develop alternative small molecule inhibitors that lack detrimental side effects of N-terminal inhibitors have shown that targeting the C-terminal ATP-binding pocket does not induce extensive HSRs.25,33,34 Another strategy to inactivate the Hsp90 machine is to inhibit co-chaperones such as p23, the activator of Hsp90 ATPase (Aha1), the FK506-binding protein 52 (FKBP52), and the cell division cycle 37 (CDC37).35,36,37,38,39,40,41 However, to date, these inhibitors remain in early preclinical evaluation, and their clinical impact in treating cancer remains unknown.
To better harness the potential of the Hsp90 machine as a therapeutic target, we sought to identify small molecule inhibitors that cause cellular degradation of oncoprotein clients of Hsp90, without inducing an HSR. Here, we report our discovery that the natural product cyclohexadepsipeptide (Cyclo[N-methyl-L-isoleucyl-N-oxa-D-valyl-N-methyl-L-isoleucyl-N-oxa-D-valyl-N-methyl-L-isoleucyl-N-oxa-D-valyl]) Enniatin A (EnnA) is an Hsp90 inhibitor satisfying the above-specified criterion.
Previous studies have shown that EnnA has antifungal and antibiotic activity, as well as specific cytotoxicity against malignant cell types, including leukemia, human non-small cell lung cancer, and hepatoma.42 At the molecular level, EnnA inhibits several signaling pathways that are essential for cancer cell survival and has shown ionophoric properties.43 Interestingly, all of the pathways inhibited by EnnA involve Hsp90 client proteins such as the extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinases (PI3Ks)/Protein kinase B (AKT), and p53,44,45,46,47 which correlates with our finding that EnnA targets Hsp90 in vitro and in vivo. We show that compared to 17-N-allylamino-demethoxygeldanamycin (17-AAG, tanespimycin), the classical N-terminal Hsp90 inhibitor tested in Phase III clinical trials, EnnA exhibited superior efficacy without inducing an extensive HSR. Furthermore, EnnA triggered cancer cell immunogenic cell death (ICD) leading to reduced tumor growth through enhanced CD8+ T cell infiltration in the aggressive syngeneic E0771 and 4T1 murine breast cancer models. Following a lead from a bulk RNA-seq analysis of cancer cells, we established that the C-X3-C motif chemokine receptor 1 (CX3CR1) signaling pathway promotes EnnA’s antitumor activity in vivo. Furthermore, EnnA caused degradation of PD-L1 protein, preventing CD8+ T cell exhaustion and tumor immunosuppression. Our results suggest that EnnA is a previously unrecognized Hsp90 inhibitor with a unique antitumor mechanism of action that circumvents the detrimental effects of current Hsp90 inhibitors and activates the immune system to control tumor growth.
Results
Identification of EnnA as an inhibitor of the Hsp90 chaperone machine
Using a screening platform to identify potential inhibitors of the Hsp90 machine’s chaperoning activity,48 we screened a small library of 139 natural products mostly purified from extracts of medicinal plants and their symbiotic microorganisms (Table S1). The assay measures the ability of molecular chaperones to recover the hormone-binding activity of the progesterone receptor (PR) following heat inactivation. In this assay, we used either rabbit reticulocyte lysate (RRL) as a source of molecular chaperones, or purified Hsp90β (HSP90AB1), Hsp70 (HSPA1A), Hsp40 (DNAJB), Hsp70-Hsp90 organizing protein (HOP) (STIP1), and p23 (PTGES30) that have been shown to be essential for PR refolding.49 From this set of natural products, we identified several compounds with various chemical scaffolds showing strong inhibitory activity of Hsp90 machine chaperoning function. Figure S1A shows a typical dataset obtained with this assay. This report focuses on EnnA (Figure 1A), which was initially purified from natural sources, as described in STAR methods. Subsequently, EnnA was chemically synthesized as described in Data S1/Method S1 (supplemental information).
Figure 1.

EnnA inhibits PR refolding through Hsp90 inactivation via a different mechanism than that of 17-AAG
(A) Structure of EnnA.
(B) Dose-dependent effect of EnnA (in μM) on PR chaperoning using rabbit reticulocyte lysate (RL) as a source of chaperones. Green bar indicates 100% folding activity of RL without inhibitor. “PR22” represents the anti-PR antibody (PR22) without PR but with RL and EnnA (40 μM). “PR” represents only PR with PR22. Data are represented as mean ± S.E.M. and represent at least three independent studies.
(C) Coomassie blue staining of protein complex after PR reconstitution performed in B. “PR” is PR alone; “PR22” is PR22 + RL; “RL” is PR + RL, without inhibitor; “17AAG” is PR + RL with 1 μM 17-AAG; “EnnA” is PR + RL with 10 μM EnnA.
(D) EnnA inactivates the refolding of PR by the 5 purified chaperones (5P): Hsp90β, Hsp70, Hsp40, HOP, and p23; 17-AAG (10 μM) was used as control. Data are represented as mean ± S.E.M. and represent at least three independent studies.
(E and F) SPR analysis of EnnA binding to immobilized Hsp90β. E. Overlay of sensorgrams recorded at various EnnA concentrations. The association phase (kon) was monitored for 180s, and the dissociation phase (koff) was monitored for 300s. SPR experiments were repeated twice. Blank sensorgrams were subtracted. F. Plot of RU max values as a function of EnnA concentration. Kinetic parameters were estimated assuming a 1:1 binding model using Evaluation Software 4.1 (GE Healthcare).
(G) Proposed model of EnnA binding to the Hsp90 dimer. Hsp90 (gold/cyan) and p23/Sba1 (green) are shown as a ribbon diagram with EnnA (magenta) depicted as a stick model. Brackets mark boundaries of Hsp90 domains (NTD, MD, and CTD). EnnA binding was modeled in silico using the crystal structure of the closed-state Saccharomyces cerevisiae Hsp90-p23/Sba1 complex (PDB ID: 2CG9). Ligand docking was performed using AutoDock Vina [PMID: 19499576] with a docking box size of 68 × 78 × 66 Å3, which covers the Hsp90-MD. The highest-scoring solution is shown. Inset shows the predicted EnnA binding site with residues from both Hsp90 protomers contacting the bound ligand. The approximate localization of the 17-AAG and ATP binding sites on the cyan monomer is indicated with a dotted purple circle.
(H) Cellular thermal shift assay (CETSA) was performed with MCF7, Hs578T, or E0771 cells in the presence of 20 μM EnnA for 1 h. Cell lysates were separated by SDS-PAGE and analyzed by Western blotting.
(I) Effect of EnnA on the binding of p23, HOP, and FKBP52 to Hsp90β. H90.10 antibody was used to co-IP purified Hsp90β (0.5 μg) with 1 μg of p23, HOP, or FKBP52. Protein complexes were Western blotted using specific antibodies.
To validate EnnA’s specificity, we compared its PR reconstitution inhibitory activity to that of other cyclic peptides using RRL as a source of chaperones. As shown in Figures S1B and S1C, none of the cyclic peptides tested at the same molarity exhibited the potent activity shown by EnnA. This inhibition of PR hormone binding recovery with RRL is concentration–dependent with an IC50 of about 5 μM (Figure 1B). Protein complex analysis by SDS-PAGE followed by Coomassie blue staining showed that EnnA modifies the PR complexes in a manner distinct from that induced by the N-terminal Hsp90 inhibitor 17-AAG (Figure 1C). Remarkably, EnnA did not induce the accumulation of the intermediate chaperoning complexes rich with Hsp70 as observed with 17-AAG.
Characterization of EnnA interaction with Hsp90
To further characterize the molecular target of EnnA, we tested its inhibitory effect on the five purified human chaperones Hsp90β, Hsp70, Hsp40, HOP, and p23.39,49 As seen in Figure 1D, EnnA inhibited PR hormone binding recovery in a dose-dependent manner, with an IC50 of ∼7.5–10 μM. This suggests that EnnA’s main target is among the five chaperones used in the assay. To identify this target, we used Surface Plasmon Resonance (SPR) to evaluate the ability of EnnA to interact with the purified chaperones individually. To this end, proteins were covalently immobilized on a microchip.50 EnnA was tested in a range of 0.05–100 μM. Only Hsp90β showed dose-response sensorgrams (Figure 1E). From binding isotherms using one site assumption, and total and nonspecific binding equations we determined a KD of 14.5 ± 0.2 μM (Figure 1F). This is also confirmed by kinetic parameters (kon = 2.01 ∗103 1/ms and koff = 0.0322 1/s resulting in KD of 15.9 μM), which is in accordance with EnnA’s overall inhibitory efficacy in vitro (Figures 1B and 1D) and its cytotoxicity toward murine cancer cells (IC50 = 7.5 μM) (see below). For Hsp70, SPR signal variation occurred at high micromolar concentrations of EnnA without reaching saturation (Figure S2A). Under these conditions, kinetic parameters determined a KD of ∼246 μM, which is 16.5-fold lower affinity for EnnA compared to that for Hsp90β. SPR analysis did not indicate a dose-dependent response of EnnA binding to the other proteins (not shown). Thus, Hsp90 is the main target of EnnA among the five proteins tested.
To gain atomic insight into the Hsp90:EnnA interaction, we used in silico molecular docking. No crystal structure is available for the full-length human Hsp90β. Thus, docking was implemented using the crystal structure of the yeast Hsp90:p23 complex.51 As seen in Figure 1G, EnnA potentially binds to a new binding site composed of the interface between the two Hsp90 monomers in complex with the co-chaperone p23. This binding site is far from the 17-AAG binding site (Figure 1G) and involves amino acid residues belonging to the middle domain of both Hsp90 monomers, which makes EnnA a unique small molecule to potentially target the middle domain of the dimeric Hsp90. This finding is important because Hsp90 functions as a dimer, and the middle domain of Hsp90 is thought to be involved in its interaction with Hsp90 client proteins.52 The new binding site involves amino acid residues L331, E333, L427, I505, Y508, and Q596 in yeast protein numbering, which correspond to L332, E334, L429, I507, Y510, and Q598 in human Hsp90β. Strikingly, all of these amino acids are strictly conserved in human cytosolic Hsp90α (HSP90AA1) and β. They are also conserved in the Hsp90 orthologue in the endoplasmic reticulum glucose-regulated protein 94 (GRP94) (Figure S2B), suggesting that our binding predictions would be valid for the human protein and that EnnA might inhibit both Hsp90 cytosolic isoforms (i.e., α and β) (Figure S2B). Sequence analysis also suggests that GRP94 might bind to EnnA. They are, however, divergent in the Hsp90 mitochondrial tumor necrosis factor receptor-associated protein-1 (TRAP-1) suggesting that EnnA might not bind to TRAP-1.
To test whether Hsp90 is a cellular target of EnnA, we used the cellular thermal shift assay (CETSA), a well-established method to evaluate drug target engagement in cells.53 Human MCF7, Hs578T, and murine E0771 cells were treated with 20 μM EnnA for 1 h. Cell protein extracts were heated in a temperature gradient from 52o to 61°C to cause protein melting and denaturation. Cell extracts were then centrifuged to remove precipitated proteins. It is expected that if Hsp90 is a cellular target, EnnA binding will promote Hsp90 stability and resistance to temperature-induced melting compared to control. As seen in Figure 1H, while Hsp90β was almost completely precipitated at about 54°-56°C in control cell extracts, treatment with EnnA stabilized Hsp90β up to over 61°C, indicating that EnnA binds to Hsp90β in MCF7, Hs578T, and E0771 cell lines. This conclusion is substantiated by the finding that EnnA analogs, Enniatin B (AD96) and the cyclic peptide AD25, which did not inhibit the PR refolding (Figures S1A–S1C) did not stabilize Hsp90 in CETSA experiments using E0771 cells (Figure S2C).
Next, we tested the impact of EnnA on Hsp90β′s interaction with co-chaperones. Immunoprecipitation (IP) experiments using the Hsp90β monoclonal antibody H90.10 and purified proteins showed that, as with 17-AAG, EnnA disrupts Hsp90β binding to p23 (Figure 1I). However, unlike 17-AAG, EnnA does not affect HOP binding to purified Hsp90β. Furthermore, EnnA promotes in vitro FKBP52 (FKBP4) binding to Hsp90β, which was minimal under our experimental conditions (Figure 1I). These findings further strengthen the hypothesis that EnnA inhibits Hsp90β through a different mechanism than that of N-terminal inhibitors.
EnnA kills breast cancer cells without inducing a heat shock response
The above findings suggest that EnnA might interfere with Hsp90 function in cells. As shown in Figure 2A, EnnA caused cell death of various breast cancer cell lines including human the poorly aggressive luminal A molecular subtype member MCF7; HER2+ MDA-MB-453 cells, and TNBC Hs578T and MDA-MB-231, as well as TNBC murine E0771 and 4T1 cells with an IC50 ranging between 1.25 and 10 μM. EnnA cellular inactivation of Hsp90 was demonstrated by destabilization of Hsp90 client proteins such as kinases CDK4, Checkpoint kinase 1 (Chk1), AKT, human epidermal growth factor receptor 2 (HER2), and integrin-linked kinase (ILK) (Figure 2B). Importantly however, unlike 17-AAG, EnnA does not seem to induce an extensive HSR (i.e., upregulation of mRNA and protein levels of Hsp70 and Hsp27). Indeed, EnnA did not significantly change the mRNA levels of Hsp70 and Hsp27 in E0771 and Hs578T cells (Figure S3). A marked decrease of Hsp70 protein was seen in MCF7 but no substantial change was seen in the other cells (Figure 2B). However, the impact of EnnA on Hsp40 mRNA level is less consistent between the cell lines tested. EnnA treatment caused a slight decrease of Hsp40 mRNA in E0771, but a significant increase in Hs578T cells (Figure S3). This correlates with a decrease in Hsp40 protein level in E0771 but a slight increase in the Hs578T cell (Figure 2B). Consistently, however, EnnA induces much lower upregulation of Hsp40 compared to 17-AAG in all cell lines tested (Figure 2B). As expected, 17-AAG induced Hsp90 client protein degradation along with an upregulation of mRNA and protein levels of Hsp70, Hsp40, and Hsp27. Together, these data suggest that EnnA does not induce an extensive HSR and may have pharmacological advantages over N-terminal Hsp90 inhibitors such as 17-AAG.
Figure 2.

EnnA inhibits cancer cell lines proliferation in vitro
(A) Survival of human breast (MCF7, MDA-MB-453, Hs578T, MDA-MB-231) and murine (E0771 and 4T1) cancer cells upon treatment with EnnA for 48 h. Data are represented as mean ± S.E.M. and represent at least three independent studies.
(B) Western blot analyses for the indicated proteins using lysates from the cell types in A treated with DMSO, 0.5 μM 17-AAG, and EnnA (at EC50 concentrations determined in A).
EnnA displays robust antitumor activity that requires a functional adaptive immune system
To evaluate the antitumor activity of EnnA in vivo, we first used the E0771 syngeneic model implanted in the mammary glands of seven-week-old female C57BL/6 (B6) immunocompetent mice (Figure 3A). When tumors reached about 50 mm3, mice were treated with vehicle (indicated as Dimethyl sulfoxide, DMSO) or EnnA. A dose of 10 mg/kg was selected to deliver twice the IC50 determined in vitro every 48 h. Tumor growth and body weight of animals were monitored. As shown in Figure 3A, EnnA exhibited significant antitumor activity against E0771 tumors. Importantly, EnnA treatment does not cause any obvious toxic effects as indicated by the normal body weight of mice (Figure 3B), no evident signs of abnormal mouse behavior, and the absence of microscopic tissue damage or inflammatory lesions in livers, lungs, or kidneys (Figure S4).
Figure 3.

EnnA exhibits a powerful antitumor activity in vivo
(A–C) E0771 cells (1x105) were surgically implanted into the mammary fat pad of seven-week-old female B6 mice. Post-operative day 10 (POD10), mice were injected with either EnnA (10 mg/kg) or vehicle every 48 h. Tumor volumes (A) and body weight (B) were measured on the days indicated. C. Tumor lysates from six control (1–6) and six EnnA-treated mice (1–6) were used for Western blot analysis using specific antibodies to the indicated proteins.
(D) E0771 cells (5x104) were surgically implanted into the mammary fat pad of B6 mice. POD7, mice were injected with EnnA (10 mg/kg), 17-AAG (50 mg/kg), or vehicle every 48 h; tumor volumes were measured on days indicated.
(E) 4T1 cells (5x104) were surgically implanted into the mammary fat pad of BALBc mice. POD7, mice were injected with EnnA (10 mg/kg), 17-AAG (25 mg/kg), or vehicle every 48 h; tumor volumes were measured on days indicated.
(F) H&E staining of E0771 tumors from control and EnnA-treated mice from A. The scale bar represents 300 μm for 4x and 50 μm for 20x. G. Average of the number of immune-like cells found in two cm square necrotic areas from three controls and three EnnA-treated tumors from A.
(H) H&E staining of 4T1 tumors from control and EnnA-treated mice from E. (I) Average of the number of immune-like cells found in two cm square necrotic areas from three controls and three EnnA treated tumors from E. The scale bar represents 100 μm for 4x and 20 μm for 20x.
(J) Immunohistochemistry analysis of immune cell infiltration using anti-CD45 antibody staining of tumors from control and EnnA-treated mice from A. The scale bar represents 300 μm for 4x and 50 μm for 20x. (K) The corrected total cell fluorescence (CTCF) intensity was calculated, as indicated in Method, from 100 CD45+ cells from three different microscopic fields of control and EnnA-treated tumors.
(L) E0771 cells (5x104) were surgically implanted into the mammary fat pad of seven-week-old female nude mice. POD7, mice were injected with either EnnA (10 mg/kg) or vehicle every 48 h, and tumor volumes were measured on days indicated. Data are represented as the mean of at least two independent studies ±S.E.M. ∗∗p < 0.01 and ∗∗∗∗p < 0.0001, as determined by one-way unpaired tailed Student’s t test. n represents the number of mice per group.
In correlation with in vitro data, several Hsp90 client proteins (CDK4, ILK, phospho-AKT) were downregulated in most tumor lysates of EnnA-treated mice (Figure 3C), confirming that EnnA targets Hsp90 in vivo. Furthermore, we observed a decrease in Hsp40 protein expression in most EnnA-treated tumors. These data demonstrate that, in vivo, EnnA reduces tumor growth by inhibiting Hsp90 without inducing an excessive HSR.
To compare the antitumor activities of EnnA to that of 17-AAG in vivo, we used E0771 and 4T1 syngeneic mouse models. Clearly, EnnA was more efficacious than 17-AAG at reducing the tumor burden (Figures 3D and 3E). Histological analysis by H&E staining of tumors showed that EnnA-treated E0771 (Figure 3F) and 4T1 (Figure 3H) tumors were much more necrotic compared to DMSO controls (Figure S5A). We also observed that necrotic areas were filled with immune-like cells in EnnA-treated tumors but were largely void of cells in controls (Figures 3F–3I). Interestingly, 4T1 tumors from mice treated with 17-AAG were more necrotic than controls, but they were not as infiltrated as in the case of EnnA-treated animals (Figure S5B). Further characterization by immunofluorescence analysis of the infiltrates observed in EnnA-treated E0771 tumors confirmed that these are CD45+ immune cells (Figures 3J and 3K). These data suggest an important role of the immune system in the EnnA-induced antitumor activity against TNBC syngeneic models. This hypothesis was tested in vivo using the immunodeficient nude mouse model. Strikingly, the robust antitumor activity in the E0771 model seen in immunocompetent B6 mice (Figures 3A–3E) was greatly diminished (Figure 3L), suggesting that the adaptive immune system, which is deficient in nude mice, plays a key role in EnnA’s antitumor activity.
EnnA antitumor activity is mediated by CD8+ T cells
Immunofluorescence and flow cytometry analyses were performed to further characterize the tumor-infiltrating immune cells. As shown in Figures 4A and 4B, most of the CD45-positive E0771 tumor-infiltrating cells were also positive for CD8β. Similar results were found in 4T1 tumors, which became heavily infiltrated with CD8+ T cells in EnnA-treated animals and much less so in the 17-AAG-treated group (Figures 4C and 4D). The CD8β T cells infiltrating E0771 tumors heavily expressed granzyme B (Figures 4E and 4F), indicating that these cells are effector killer T cells. These immunohistochemistry (IHC) data were confirmed by flow cytometry analysis showing that E0771 EnnA-treated tumors contained at least three times as many CD45+CD8+ T cells compared to control tumors and these cells are expressing significantly higher levels of perforin (Figure 4G). Importantly, these cells showed fewer signs of exhaustion, as indicated by lower expression of PD-1 (Figure 4G). Furthermore, although EnnA induced a significant increase of CD45+CD4+ T cells in tumors, the compound seemed to reduce immune suppression by significantly lowering the number of CD4+FoxP3+ Tregs (Figure 4G) in treated tumors compared to controls.
Figure 4.

EnnA antitumor activity against E0771 tumors is mediated by CD8+ T cells
(A) Immunofluorescence analysis showing that EnnA induces recruitment of CD8β+ T cells to the E0771 tumor site and increases tumor infiltration compared to control. The scale bar represents 300 μm for 4x and 20 μm for 40x.
(B) Average of 180 cells from three different fields of control and EnnA-treated tumors (n = 3). Data are represented in the percentage of mean ± S.E.M.
(C) Immunofluorescence analysis of 4T1 tumors treated with EnnA (10 mg/kg/48 h) or 17-AAG (25 mg/kg/48 h) stained with anti-CD45 and anti-CD8α. The scale bar represents 50 μm.
(D) Average of 250 cells from three different fields of control and EnnA-treated tumors (n = 3). Data are represented in the percentage of mean ± S.E.M.
(E) Immunofluorescence analysis of E0771 tumors from control and EnnA-treated animals showing that CD8β+ T cells also express granzyme b. The scale bar represents 300 μm for 4x and 50 μm for 20x.
(F) Average of 135 cells from three different fields of control and EnnA-treated tumors (n = 3).
(G) Flow cytometric analysis of cells from tumors treated with EnnA or DMSO. Data are represented in the percentage of mean ± S.E.M.
(H) Depletion of CD8+ T cells from mice using a monoclonal antibody (InVivoMAb anti-mouse CD8α, clone 53–6.7) abrogates antitumor activity of EnnA. Data are represented in the percentage of mean ± S.E.M. n represents the number of mice per group.
(I) EnnA treatment increases the number of CD8+ T cells in spleens, which is abrogated by an anti-CD8 antibody. Data are represented as mean ± S.E.M. ∗p < 0.05; ∗∗p < 0.01 by one-way ANOVA or unpaired tailed Student’s t test and are a representation of at least two independent studies.
To validate whether the tumor-infiltrating CD8+ T cells play a key role in EnnA’s antitumor activity, we depleted these cells using anti-mouse CD8α antibody injections. As shown in Figure 4H, the antitumor effect of EnnA was abolished in mice depleted of CD8+ T cells. Efficient depletion of CD8+ T cells was validated by flow cytometry analysis of spleen cell suspensions. Indeed, EnnA treatment increased the percentages of spleen CD8+ T cells in treated animals as compared to the controls, but anti-CD8 antibody treatment reduced this population to similar levels as those observed in control mice (Figure 4I). Importantly, adoptive transfer of T cells from E0771 tumor-bearing EnnA-treated mice reduced the tumor growth in naive mice compared to T cells transferred from tumor-free mice (Figure S6). Taken together, these findings established that T lymphocyte populations, CD8+ T cells, in particular, play a key role in EnnA’s antitumor response.
EnnA induces features of immunogenic cancer cell death
To gain further insight into how EnnA treatment activates the immune system to fight tumors, we investigated whether EnnA induces ICD of cancer cells. ICD is a regulated process that renders dying cells more visible to the immune system. It involves changes in the cell surface composition and the release of soluble mediators that elicit antigen-specific adaptive immune responses. ICD is thought to be one of the prominent mechanisms through which effective conventional cancer therapies induce clinically relevant anti-cancer immunity. ICD is induced by certain chemotherapeutic drugs, oncolytic viruses, physicochemical therapies, photodynamic therapy, and radiotherapy.54 ICD induced by therapeutic agents is initiated by the emission of damage-associated molecular patterns (DAMPs)55 such as cell surface exposure of cytoplasmic chaperones (i.e., Hsp90 and Hsp70), endoplasmic reticulum (ER) chaperones (i.e., calreticulin), and the release of nuclear protein high-mobility group Box 1 (HMGB1) and ATP. Release of these markers is driven by cellular processes such as ER stress, autophagy, and necrotic plasma membrane permeabilization.56
According to the consensus guidelines for the detection of ICD,56 the gold-standard approach relies on vaccination assays to evaluate the ability of a specific therapeutic agent to induce ICD. Thus, we tested whether in vitro EnnA-treated E0771 tumor cells would immunize mice against a subsequent challenge with live E0771 cells. Mice received 4 s.c. injections of EnnA-treated tumor cells (10 days apart) and were challenged 2 days after the last injection with live, luciferase-expressing tumor cells (implanted in the opposite mammary gland). Strikingly, none of the mice that received the EnnA-treated tumor cells developed tumors after the challenge; however, all control mice did (Figure 5A).
Figure 5.

EnnA treatment triggers immunogenic cell death in vitro and in vivo
(A) EnnA-killed E0771 cells vaccinate mice against live cells. Three injections of E0771 cancer cells killed with EnnA (lower panel) are able to vaccinate mice against live cells (105) implanted in mammary fat pads of B6 mice (upper panel). Control mice receiving just culture media develop tumors, as expected.
(B) Electron microscopy (EM) analysis of E0771 cells treated with 5 μM EnnA for 24 h. Red arrows indicate autophagosomes. Blue arrows indicate vacuoles. The scale bar represents 1 μm.
(C) Western blot analysis of cell lysates from human Hs578T and MDA-MB-453 cells treated with indicated concentrations of EnnA. A specific antibody against total LC3B was used. β-actin was used as a loading control.
(D–G) Immunocytochemistry (IHC) analysis of E0771 cultured cells (D) and tumor samples (F), stained for autophagosomes (α-LC3BII) and the nucleus (DAPI). The scale bar represents 20 μm. E. 30 cells per field (n = 3) in control and EnnA-treated cells were analyzed for the presence of autophagic puncta. G. 70 cells per field (n = 3) in control and EnnA-treated tumors were analyzed for the presence of autophagic puncta. Data are represented in the percentage of mean ± S.E.M.
(H) Flow cytometry analysis of E0771 cells treated with 5 μM EnnA for 24 h. Cells were stained for surface expression of Hsp90β and calreticulin using specific antibodies. In each sample, a total of 10,000 cells were acquired. Gating strategies are shown in Figures S10–S12. Data are represented in the percentage of mean ± S.E.M.
(I) Flow cytometry analysis of E0771 tumor cell suspensions from control and EnnA-treated mice using indicated antibodies. In each sample, a total of 10,000 cells were acquired. Data are represented in the percentage of mean ± S.E.M. ∗∗p < 0.01; ∗∗∗∗p < 0.0001 by one-way ANOVA or unpaired tailed Student’s t test.
Autophagy is another key feature of ICD.57,58,59 We found that EnnA triggered autophagy in human breast cancer cells (Hsp578T and MDA-MB-453) as indicated by the accumulation of autophagic vacuoles seen by light microscopy (Figure S7A) and by electron microscopy (Figure 5B, blue arrowheads), which also identified autophagosomes (Figure 5B, red arrowheads). Accumulation of autophagosomes was further confirmed by Western blot analysis showing accumulation of the autophagosome marker LC3BII in EnnA treated cells (Figure 5C). EnnA induction of autophagy was further validated by immunofluorescence showing increased staining of LC3BII puncta in the murine cell line E0771 in vitro (Figures 5D and 5E) and in tumors (Figures 5F and 5G).
As mentioned above, molecular features of ICD include exposure of the cytoplasmic Hsp90 and the ER chaperone calreticulin (CRT) on the cell surface. Indeed, Hsp90 exposed on the cell surface is a DAMP signal linked to ICD.59 Hsp90 cell surface exposure facilitates the uptake of dying cells by dendritic cells (DCs), leading to tumor antigen cross-presentation and induction of tumor-specific cytotoxic T lymphocyte responses. We found that EnnA treatment caused Hsp90β to be exposed on the cell surface in a higher percentage of cancer cells in vitro (Figure 5H, upper panels) and in vivo (Figure 5I, upper panels) compared to controls. Correlating with the hypothesis that EnnA induces ICD, flow cytometry results showed that the EnnA treatment of E0771 cells enhanced the expression of CRT on the cell surface (Figure 5H, lower panels). Although not as drastic in vivo, tumor cells resected from EnnA-treated animals also showed a consistent increase in calreticulin surface expression (Figure 5I, lower panel). Using the highly aggressive murine and human TNBC cell lines, 4T1 and MDA-MB-231, respectively, we found that EnnA induces more pronounced cell surface exposure of Hsp90, calreticulin, and HMGB1 compared to 17-AAG (Figure S7B).
EnnA modulates cancer cell transcriptome to stimulate the immune system
To gain more insight into how EnnA triggers anti-cancer immunity, we analyzed the impact of EnnA treatment, in comparison with 17-AAG, on the transcriptome of the E0771 murine breast cancer cell line. Bulk RNA-seq analysis from cultured cells treated with IC25 doses for each compound was performed. Genes were then grouped based on the fold of up- or down-regulation [log2 fold change and an associated p value of 0.05 or less] (Figure 6A). This threshold criterion reduced the number of genes from 11024 to 251, which is about 2.3% of E0771 transcripts. The heatmap in Figure 6B shows that overall, the two Hsp90 inhibitors impact the E0771 transcriptome in very distinct ways. Surprisingly, this analysis revealed that just a few common genes are affected by both compounds. Using the above criteria, we determined that 241 and 251 genes were affected by 17-AAG or EnnA treatment, respectively (Figure 6C, upper panel). However, of these genes, only 34 (∼14%) were in common. Furthermore, their expression was not altered in the same way (Figure 6C, lower panel). These data confirmed that EnnA and 17-AAG inhibit the Hsp90 machine through different mechanisms.
Figure 6.

EnnA modulates E0771 cell transcriptome to activate the immune system
(A) Volcano plots of bulk RNA-seq data from E0771 cells treated with EnnA, 17-AAG or DMSO as control. Plots were generated based on EnnA versus control (upper, +/− 2-fold change and p < 0.065, as shown in dashed blue lines) and 17-AAG versus control (bottom, +/− 2-fold change and p < 0.05, as shown in dashed blue lines), after the removal of low-expressing genes.
(B) Heatmap generated based on the intersection of 4852 genes with the normalized FPKM counts from duplicates of control, EnnA-treated, and 17-AAG-treated cells, after the removal of low-expressing genes.
(C) Venn Diagram (upper panel) indicates 251 genes at or exceeding the cutoff of 2 log2 fold changes in response to EnnA versus control-treated cells and 241 genes at or exceeding the cutoff of 1.5 log2 fold changes in response to 17-AAG versus control-treated cells, with an intersection of 34 genes, which were used to plot a heatmap across control, EnnA, and 17-AAG groups (lower panel).
(D) Predicted biological impact: Two filtered fold-change tables, EnnA versus control (upper, +/− 3-fold change and p < 0.05) and 17-AAG versus control (bottom, +/− 2-fold change and p < 0.05), were imported into IPA software to predict biological effects (increases (orange) or decreases (blue)) on diseases and cell biology processes.
(E) List of top genes affected by EnnA treatment. Red: upregulated. Blue: downregulated.
(F) Plots of Heatmap and Enrichment Score by GSEA on immune responses involving KEGG pathways. Heatmap and Enrichment Scores were plotted using the 3,668 genes at a cutoff of 1.5-fold changes into the GSEA analysis with the duplicates of EnnA versus control to produce the immune response-associated KEGG pathways containing chemokine signaling pathways and cytokine-cytokine receptor interaction. Data are represented as a mean ± S.E.M. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by one-way ANOVA or unpaired tailed Student’s t test. Data represent at least two independent studies.
Focusing on the top 70 affected genes, Ingenuity Pathway Analysis (IPA) analysis showed a dramatic difference between the two compounds in the potential impact on disease and underlying signaling pathways (Figure 6D). For instance, while EnnA activates genes involved in the hematological system, 17-AAG seems to decrease their expression. Similarly, EnnA highly activates the inflammatory response and immune cell trafficking pathways, while 17-AAG decreases these functions. EnnA induces higher expression of genes involved in cellular movement compared to 17-AAG. In accordance with its mainly cytotoxic effect, 17-AAG upregulates the transcriptome of cell death and survival pathways.
Overall, these analyses indicate that EnnA might render tumor cells more visible to the immune system through activation of inflammatory responses and immune cell trafficking pathways. As seen in Figure 6E, EnnA significantly increased the level of IL23, which is a member of the inflammatory cytokine family. This has been validated by RTqPCR showing that EnnA upregulates other pro-inflammatory cytokines such as IL12b, TNFα, and IL1β (Figures S8A and S8B). In contrast, 17-AAG had a limited impact on these inflammatory cytokines (Figures S8A and S8C).
Interestingly, among the top genes highly upregulated by EnnA treatment (Figure 6E) are the CX3CR1 chemokine receptor, the CD74 MHC class II chaperone, and engulfment and cell motility protein 1 (ELMO1). CD74 plays a key role in MHC-I trafficking, regulates T cell and B cell development, DC motility, macrophage inflammation, and thymic selection.60 ELMO1 has been identified as a microbial sensor in epithelial and phagocytic cells that turns on inflammatory signals.61 Accordingly, a KEGG enrichment plot analysis showed a clear positive correlation of EnnA treatment with activation of the chemokine signaling and cytokine-cytokine receptor interaction pathways (Figure 6F), which is not the case when 17-AAG was used. Many of these genes are indeed downregulated (Figure S9A). Albeit to various extents, both EnnA and 17-AAG decreased mRNA expression of anti-inflammatory cytokines such as IL10 (Figure S8C), which validated the RNA-seq data and RTqPCR showing that EnnA dramatically reduced the level of IL19 (Figures 6E and S8A), a member of the IL10 family.
CX3CR1 is a positive mediator of EnnA antitumor activity in mice
The high upregulation of the chemokine receptor CX3CR1 expression (Figure 6E) was remarkable because a recent report has identified CX3CR1 as a marker for CD8+ T cells that withstand chemotherapy during cancer chemo-immunotherapy, suggesting that this chemokine receptor could be used as a marker for potential anti-PD-1 responsive patients.62 This observation correlates with a broader KEGG enrichment analysis showing a clear positive correlation between chemokine signaling and T cell receptor signaling pathways with EnnA treatment (Figures 6F and S9B). Strikingly, several chemokines are shown to be upregulated in E0771 cells treated with EnnA. These include CCL3, CXCL1, CCL4, CCL5, CXCL10, CCL25, and CSF2 (Figures 6F and S8). As shown below, further investigations indicated that the CX3CR1 pathway plays an important role in EnnA’s antitumor activity.
The upregulation of CX3CR1 upon treatment with EnnA was validated by RTqPCR using E0771 cells in vitro (Figure S6A). Flow cytometry analysis confirmed that CX3CR1 protein is upregulated at the surface of E0771 cells treated with EnnA compared to DMSO control (Figure 7A). Similar results were found in human Hs578T and MDA-MB-231 and murine 4T1 cell lines (Figure S5C). These findings were confirmed in E0771 tumor tissues as well. Indeed, using flow cytometry, we found that CD45-:CX3CR1+ cells are significantly upregulated in E0771 tumors treated with EnnA compared to tumors from the control group (Figure 7B, left panel). Interestingly, the number of infiltrating triple positive CD45+/CD8α+/CX3CR1+ cells isolated from EnnA-treated tumors was also significantly higher compared to cells isolated from the control group (Figure 7B, right panel). These findings prompted us to hypothesize that the CX3CR1 signaling pathway might be involved in EnnA’s antitumor activity. This was tested in vivo by combining EnnA with the CX3CR1 antagonist 5-[3-[4-(4-Chlorophenyl)-1-piperidinyl]propyl]pyrrolo[1,2-a]quinoxalin-4(5H)-one hydrochloride (JMS-17-2), an optimized compound that has been recently shown to reduce lung breast cancer metastasis.63 As shown in Figure 7C, when injected intraperitoneally at 10 mg/kg every 48 h, alternating with EnnA, JMS 17-2 significantly reduced the efficacy of EnnA. These data suggest that activation of the CX3CR1 signaling pathway is important for EnnA’s tumor-killing activity.
Figure 7.

CX3CR1 chemoattraction pathway and PD-L1 pathways are modulated by EnnA treatment
(A) Flow cytometry analysis of CX3CR1 surface expression on E0771 cells treated with 7.5 μM EnnA or DMSO control. Data are represented as a mean ± S.E.M.
(B) Flow cytometry analysis of CX3CR1 surface expression on cell suspensions from E0771 tumors treated with EnnA or control. Data are represented as a mean ± S.E.M.
(C) The CX3CR1 chemoattraction pathway is important for EnnA’s antitumor efficacy. 10 × 104 E0771 cells were injected into the mammary fat pad of seven-week-old female mice. EnnA and JMS-17-2 groups were injected intraperitoneally with 10 mg/kg of EnnA or JMS-17-2 every 48 h, and the control group (DMSO) received vehicle (mixture of 15% DMSO and 20% cremophor in PBS). The EnnA + JMS-17-2 group received the compounds at alternate 48-h time points. Tumor volume was measured as treatment progressed. n represents the number of mice per group. Data are represented as a mean ± S.E.M.
(D and E) Western blot analysis for PD-L1 expression in cell extracts from indicated cell lines treated with various concentrations of EnnA or 17-AAG. DMSO is used as control.
(F) Immunofluorescence analysis for PD-L1 expression in E0771 tumors from animals treated with DMSO control or EnnA. The scale bar represents 20 μm.
(G) The corrected total cell fluorescence (CTCF) intensity was calculated from 100 PD-L1+ cells from three different microscopic fields of control and EnnA-treated tumors.
(H) Western blot analysis of PD-L1 complexes isolated from E0771 cells by immunoprecipitation using anti-PD-L1 antibody. PD-L1 expression was induced with 10 ng/mL interferon-gamma for 24 h. Cells were treated with 10 μM EnnA or DMSO for an extra 5 h. Rat IgG was used as a control.
(I) Cells were treated as in H. After immunoprecipitation, part of the PD-L1 resin was stripped (with stripping) from endogenous interacting proteins using 250 mM KCl and then incubated with purified Hsp90. The other fraction was kept as is (No stripping) to visualize the endogenous Hsp90 binding to PD-L1. Mouse IgG was used as control.
(J) 10x104 E0771 cells were injected into 4th mammary fat pad of seven-week-old female B6 mice. Tumors were allowed to grow to about 50 mm3 before starting treatment. EnnA (10 mg/kg) and DMSO were injected every 48 h. Anti-PD-L1 (InVivoMAb anti-mouse PD-L1 (B7-H1) (BioXCell)) was injected intraperitoneally twice a week at 200 μg/mouse. n represents the number of mice per group.
(K) Model for EnnA’s antitumor activity. EnnA inhibits Hsp90, causing degradation of its oncogenic client proteins and autophagy. EnnA also causes exposure of calreticulin and Hsp90 on the surface of cancer cells, leading to cancer antigen cross-presentation to DCs and activation of CD8+ T cells and their mobilization through the CX3CR1 pathway to the tumor site. CD8 + T cell suppression by cancer cells is reduced by downregulation of PD-L1 expression. Data are represented as a mean ± S.E.M. ∗∗p < 0.01; ∗∗∗∗p < 0.0001 by one-way ANOVA or unpaired tailed Student’s t test.
EnnA treatment reduces PD-L1 levels in vitro and in vivo
To further strengthen the hypothesis that EnnA inhibition of Hsp90 reprograms the tumor microenvironment (TME) to promote CD8+ T cell antitumor immunity, we tested whether EnnA modulates key immunosuppressive mechanisms such as the PD-1/PD-L1 signaling pathway, which is a well-established potent suppressor of effector T cell function.64 This idea was prompt by a recent report showing that several Hsp90 inhibitors of various chemical scaffolds caused a decrease in both the mRNA and protein levels of PD-L1 in PHP-1 acute monocytic leukemia and MC-38 murine adenocarcinoma cell lines.13 Given the key importance of effector CD8+ T cells in EnnA-induced antitumor activity, we tested whether EnnA treatment affected PD-L1 expression. Indeed, Western blot and flow cytometry analyses showed that treating human and murine cell lines with EnnA significantly reduced the level of PD-L1 protein in several cell lines in vitro (Figures 7D and 7E) and in E0771 tumors in vivo (Figures 7F and 7G). However, contrary to the N-terminal inhibitors, EnnA does not reduce PD-L1 mRNA levels. Rather, RNA-seq and qPCR analyses showed a slight increase in PD-L1 mRNA level (Figure S8B), suggesting that EnnA acts by inducing degradation of PD-L1 protein, which is consistent with the hypothesis that PD-L1 might be a novel Hsp90 client protein. This hypothesis is supported by our immunoprecipitation results showing that Hsp90β is in protein complexes with PD-L1 in E0771 cell lysates (Figure 7H). Furthermore, treatment of cells with EnnA causes the dissociation of Hsp90β from PD-L1 complexes (Figure 7H), leading to PD-L1 cellular degradation (Figures 7D and 7E). To further test whether PD-L1 is a client of Hsp90, we tested its direct interaction with the chaperone in vitro (Figure 7I). To this end, PD-L1 was isolated from E0771 cell extracts using an anti-PD-L1 antibody. PD-L1 was stripped of its endogenous interacting proteins using high salt (250 mM KCl) and then incubated with purified Hsp90β. As shown in Figure 7I, PD-L1 binds specifically to purified Hsp90β compared to the background binding seen with IgGs. Non-stripped PD-L1 (left two columns) confirmed the results in Figure 7H, showing endogenous interaction of Hsp90 with PD-L1.
Seeking to improve EnnA’s antitumor activity, we tested whether the combination of EnnA with immunotherapeutic agents targeting the PD-L1 pathway, as previously reported for N-terminal inhibitors of Hsp90.13 Our results show that the combination of EnnA with anti-PD-L1 did not improve antitumor activity against the E0771 syngeneic mouse model (Figure 7J), suggesting that inactivation of this pathway by EnnA as indicated by the drastic reduction of PD-L1 protein (Figures 7D–7H) provides optimal benefit expected from inactivating this pathway.
Discussion
We present here compelling evidence that EnnA exhibits a powerful antitumor activity, at least in part, through inhibition of Hsp90 and activation of the immune system to recognize and destroy tumor cells. EnnA is a novel Hsp90 inhibitor with a distinct mechanism of action compared to inhibitors targeting the ATP binding site at the N-terminal of the chaperone. Indeed, EnnA does not induce an HSR and differently affects Hsp90 complexes with its client proteins and co-chaperones, compared to these other inhibitors. Although we have strong evidence that EnnA binds to Hsp90 in vitro and in cells, the exact molecular mechanism by which this binding occurs remains to be fully elucidated. Indeed, similarly to C-terminal inhibitors of Hsp90, EnnA does not induce an HSR. However, in silico docking studies suggest that EnnA binds to the middle domain of the Hsp90 dimer in a binding site formed by the two Hsp90 monomers. Interestingly, gambogic acid (GBA), another natural product with a different chemical scaffold, binds selectively to the middle domain of Hsp90β. However, only one Hsp90 monomer is sufficient for this interaction.65 Accordingly, GBA and EnnA exhibit distinct effects on cancer cells: GBA-induced cancer cell paraptosis.66 but EnnA triggered autophagy in various breast cancer models in vitro and in vivo (Figures 5A–5F and S7A). EnnA also induces apoptosis in vitro as indicated by surface expression of annexin V and 7AAD staining (Figure S7C). Clearly, EnnA kills cancer cells in a manner that exposes them to the immune system. Further studies have shown that in addition to autophagy, EnnA induces several other features of ICD including exposure of Hsp90, calreticulin, and HMGB1 on the surface of cancer cells. These futures are not induced by 17-AAG (Figure S7B). We thus propose that activation of the immune system through ICD induction contributes significantly to the superior antitumor efficacy of EnnA compared to 17-AAG. Indeed, the reprogramming of cancer cells and the TME caused by EnnA treatment promotes immune infiltration and reduced tumor immune suppression as reflected by increased mobilization of CD8+ T cells into the tumor site, decreased CD8+ T cell exhaustion, and a reduced CD4-FOXP3 regulatory T cell population in tumors.
The important role of Hsp90 in cancer immunology is becoming more appreciated, but its molecular underpinnings remain unclear. For instance, there is a consensus that exposure of Hsp90, and other intracellular chaperones, on the cell surface is an “eat me” signal for phagocytic cells. Using ovalbumin-derived antigens, it has been established that Hsp90 is essential for antigen cross-presentation by antigen-presenting cells (APCs). Cancer cells treated with ICD inducers such as EGFR antibody67 or bortezomib68 expose Hsp90 at the cell surface, which enhances DC-mediated phagocytosis and antigen presentation. Interestingly, however, the combination of bortezomib with geldanamycin (the parent compound of 17-AAG) enhanced tumor cell apoptosis but abrogated their immunogenicity,68 suggesting that N-terminal inhibitors of Hsp90 might interfere with its stimulation of the immune system.69 This correlates with an earlier report showing that treatment of cells with Hsp90 N-terminal inhibitors generates an ‘‘empty’’ MHC I, and inhibition of Hsp90 in antigen donor cells compromises their ability to cross-prime T cell responses.70 This, however, does not seem to be an issue for EnnA. It is worth noting that preclinical and clinical responses achieved with N-terminal Hsp90 inhibitors have been generated using doses that, in hindsight, have not been optimized to take into account the importance of the immune system in fighting tumors.14 Indeed, the acute high doses of Hsp90 N-terminal inhibitors needed to achieve tumor killing in immunodeficient mice have been shown to be immunosuppressive in immunocompetent mice and in clinical trials.69,71 Accordingly, a alternative treatment paradigm of a sustained low dose of N-terminal Hsp90 inhibitor was demonstrated to be more efficacious in combination with a single dose of nonspecific immune adjuvant in syngeneic immunocompetent mice.69 Thus, the importance of Hsp90 as a promotor of immunosuppression and how to manipulate this activity to stimulate the immune system needs more investigation.
Consistent with the idea that cytosolic Hsp90 promotes the cancer cell’s ability to escape an immune attack, recent studies have shown that targeting Hsp90 improved immunotherapy through induction of interferon target genes.10 Another study has found that Hsp90 N-terminal inhibitors of various scaffolds reduce PD-L1 and PD-L2 expression on the surface of cancer cells through inactivation of master transcriptional regulators and the Hsp90 client proteins the signal transducer and activator of transcription 3 (STAT-3) and c-Myc.13 However, EnnA-mediated reduction of PD-L1 level seems to be through causing PD-L1 protein dissociation from Hsp90 complexes and PD-L1 cellular degradation. Indeed, EnnA does not affect PD-L1 mRNA levels, suggesting that PD-L1 is a client protein of Hsp90. This is supported by binding of PD-L1 to Hsp90 in cells and in purified systems (Figures 7H and 7I). Therefore, the unique mechanism of action of EnnA allowed the discovery that PD-L1 is a novel Hsp90 client protein, which will have a significant impact on our understanding of how Hsp90 promotes an immunosuppressive microenvironment to facilitate tumor growth. A reduced level of PD-L1 on the surface of cancer cells is known to increase the number of activated CD8+ T cells within the TME. Thus, the loss of PD-L1 in cancer cells upon EnnA treatment is highly significant in promoting antitumor immunity.
Since EnnA induces degradation of oncoprotein clients of Hsp90, including PD-L1, and causes Hsp90 exposure on the cell surface, triggering CD8+ T cell-dependent antitumor activity, we propose that EnnA enhances antigen cross-presentation by DCs and mobilization of effector lymphocytes into the tumor bed (Figure 7K). This hypothesis is supported by our findings that EnnA induces significant activation of immune cell trafficking pathways and immune cell movement in vitro and in vivo. This seems to be promoted, at least in part, by an increase in CX3CR1 expression in cancer cells but also in CD8+ T cells. Indeed, inhibition of CX3CR1 using the antagonist JMS 17-2 significantly compromised EnnA’s efficacy against E0771 tumors (Figure 7C). In addition, RNA-seq and RTqPCR analyses showed that the two known ligands for CX3CR1, CX3CL1 and CCL26, are downregulated by EnnA treatment of cancer cells (Figures 6F and S8A). These data correlate with several lines of evidence, indicating the importance of this chemokine pathway in cancer: 1- High expressing CX3CR1 CD8+ T cells withstand chemotherapy and predict positive response to anti-PD-1 blockade therapy62; 2- High expression of CX3CL1 in tumors predicts poor prognosis in breast72 and hepatocellular73 carcinomas; 3- Absence of CX3CL1 prior to chemotherapy correlates with positive PD-L1 response blockades74; 4- downregulation of CX3CR1-CX3CL1 restricts cytotoxic T cells infiltration of solid tumors; and 5- High expression of CX3CL1 promotes monocytic myeloid-derived suppressor cell (M-MDSC) recruitment into the TME and increases PD-L1-independent immunosuppression.75 Therefore, EnnA holds great promise of combining the cytotoxic effect of Hsp90 inhibition with the stimulation of immune cell trafficking through modulation of the CX3CR1/CX3CL1 pathway to deliver a better antitumor efficacy in breast and other solid tumors. A better understanding of the molecular mechanism underlying EnnA’s induction of CX3CR1 and its cell membrane localization through the Hsp90 pathway would be of great interest to improve EnnA’s efficacy. Remarkably, a significant portion of CD8+ T cells remains in the tumor periphery (Figure 4A), suggesting the possibility of further enhancing EnnA’s efficacy using infiltration-promoting and immunotherapeutic agents.
Limitations of the study
Although this study describes the discovery of EnnA as a promising inhibitor of Hsp90, it remains that potential off-target effects of this natural product have not been fully addressed due to our inability to immobilize EnnA on a resin and perform pulldown experiments from cell lysates. Our initial efforts to functionalize EnnA for immobilization purposes resulted in a substantial loss of cytotoxic activity in vitro. Thus, developing analogs to optimize EnnA’s efficacy and further establish its Hsp90 specificity in cells and in vivo is needed. Furthermore, future pharmacodynamic and pharmacokinetic studies will address the potential toxicity of the compound and help optimize its therapeutic potential. These follow-up studies are important, especially because EnnA, like 17-AAG, does not discriminate between cancer and non-transformed immortalized cells in cell culture (Figure S10). Importantly, the lack of obvious in vivo toxicity at an efficacious dose as shown by the normal body weight of mice (Figure 3B), absence of signs of abnormal mouse behavior, and microscopic tissue damage or inflammatory lesions in vital organs (Figure S4) suggest that mechanisms of EnnA-induced cell death in normal versus cancer cells might be different. They also support the prediction that the benefits of EnnA treatment might outweigh its potential adverse effects as a potential therapeutic agent.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-Cdk4 (C-22) Polyclonal Antibody | Santa Cruz Biotechnology | Cat# sc-260; RRID:AB_631219 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB_626632 |
| Anti-Hsp70 (BB70 from Dr. David Toft, Mayo Clinic, Rochester, MN) | N/A | N/A |
| Anti-Hsp90β (H90.10 from Dr. David Toft, Mayo Clinic, Rochester, MN) | N/A | N/A |
| Anti-FKBP52 (His52C from Dr. David F. Smith, Mayo Clinic, Scottsdale, AZ) | N/A | N/A |
| Anti-P23 (JJ3 from David Toft) | N/A | N/A |
| Anti-HOP (F5 from David Smith) | N/A | N/A |
| Akt (pan) (C67E7) Rabbit mAb | Cell Signaling Technology | Cat# 4691; RRID:AB_915783 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| LC3B (D11) XP Rabbit mAb | Cell Signaling Technology | Cat# 3868; RRID:AB_2137707 |
| Autophagy Cleaved-LC3 Antibody (APG8a) | Abcepta | Cat# AP1805a; RRID:AB_2137587 |
| Rabbit Anti-HSP70 Polyclonal Antibody, Unconjugated | Bioss | Cat# bs-0126R; RRID:AB_10852592 |
| HSP27 antibody | Proteintech | Cat# 18284-1-AP; RRID:AB_2295540 |
| GAPDH (D-6) | Santa Cruz Biotechnology | Cat# sc-166545; RRID:AB_2107299 |
| ErbB-3 (C-17) (Anti-HER3) | Santa Cruz Biotechnology | Cat# sc-285, RRID:AB_2099723 |
| Calreticulin Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA3-900; RRID:AB_325990 |
| Human/Mouse CX3CR1 Affinity Purified Polyclonal Ab | R and D Systems | Cat# AF5825; RRID:AB_2292441 |
| Brilliant Violet 421(TM) anti-mouse CX3CR1 | BioLegend | Cat# 149023; RRID:AB_2565706 |
| Purified anti-mouse CD45 | BioLegend | Cat# 103102; RRID:AB_312967 |
| Brilliant Violet 605(TM) anti-mouse CD45 | BioLegend | Cat# 103139; RRID:AB_2562341 |
| ILK | BD Biosciences | Cat# 611803; RRID:AB_399283 |
| Purified anti-mouse CD8b.2 | BioLegend | Cat# 140402; RRID:AB_10645339 |
| Purified anti-mouse CD8a | BioLegend | Cat# 100801; RRID:AB_312762 |
| Alexa Fluor(R) 700 anti-mouse CD8a | BioLegend | Cat# 100729; RRID:AB_493702 |
| InVivoPlus anti-mouse PD-1 (CD279) | Bio X Cell | Cat# BE0146, RRID:AB_10949053 |
| APC/Cyanine7 anti-mouse CD279 (PD-1) | BioLegend | Cat# 135223, RRID:AB_2563522 |
| Purified anti-mouse FOXP3 | BioLegend | Cat# 126401, RRID:AB_1089120 |
| HMG-1 (HAP46.5) (HMGB-1) | Santa Cruz Biotechnology | Cat# sc-56698, RRID:AB_783817 |
| Chemicals, peptides, and recombinant proteins | ||
| 17-allylamino-demethoxygeldanamycin (17-AAG) | ChemieTek | Cat# #75747-14-7 |
| 5-[3-[4-(4-Chlorophenyl)-1-piperidinyl]propyl]pyrrolo[1,2-a]quinoxalin-4(5H)-one hydrochloride (JMS-17-2) | Tocris Bioscience | Cat# 6378/50 |
| Cyclo[N-methyl-L-isoleucyl-N-oxa-D-valyl-N-methyl-L-isoleucyl-N-oxa-D-valyl-N-methyl-L-isoleucyl-N-oxa-D-valyl] (Enniatin A) | Sigma | Cat #E9661 |
| Recombinant Mouse IFN-gamma Protein | R&D Systems | Cat #: 485-MI |
| Critical commercial assays | ||
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | Promega | Cat#: G3580 |
| MagniSort Mouse T cell Enrichment kit | Invitrogen | Cat#: 8804-6820 |
| RNA to cDNA EcoDry™ Premix (Double Primed) | Takara | Cat#: 639549 |
| PureLink RNA Mini Kit | ThermoFisher | Cat#: 12183018A |
| PureLink DNase Set | ThermoFisher | Cat#: 12185010 |
| TruSeq Stranded mRNA Library prep kit | Illumina | Cat#:20020594 |
| Experimental models: Cell lines | ||
| Hs 578T | ATCC | RRID:CVCL_0332 |
| EO771 | ATCC | Cat# CRL-3461, RRID:CVCL_GR23 |
| 4T1 | ATCC | CRL-2539, RRID:CVCL_0125) |
| MDA-MB-231 | ATCC | RRID:CVCL_0062 |
| MDA-MB-453 | ATCC | RRID:CVCL_0418 |
| MCF-7 | ATCC | RRID:CVCL_0031 |
| Experimental models: Organisms/strains | ||
| BALB/cJ | The Jackson Laboratory | Cat# JAX:000651, RRID:IMSR_JAX:000651 |
| C57BL/6J | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| NU/J | The Jackson Laboratory | Cat# JAX:002019, RRID:IMSR_JAX:002019 |
| Software and algorithms | ||
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| GraphPad Prism | http://www.graphpad.com/ | RRID:SCR_002798 |
| BIAevaluation Software | https://www.biacore.com/lifesciences/service/downloads/software_licenses/biaevaluation/ | RRID:SCR_015936 |
| FlowJo software | https://www.flowjo.com/solutions/flowjo | RRID:SCR_008520 |
| Oligonucleotides | ||
| mCx3cr1.F GAGTATGACGATTCTGCTGAGG | Eurofins | N/A |
| mCx3cr1.R CAGACCGAACGTGAAGACGAG | Eurofins | N/A |
| mCD74.F AGTGCGACGAGAACGGTAAC | Eurofins | N/A |
| mCD74.R CGTTGGGGAACACACACCA | Eurofins | N/A |
| mElmo1.F GAGAACAGCAGCCGAGAAGAT | Eurofins | N/A |
| mElmo1.R GTTGCAGGTCTCACTAGGCAG | Eurofins | N/A |
| mIl23a.F ATGCTGGATTGCAGAGCAGTA | Eurofins | N/A |
| mIl23a.R ACGGGGCACATTATTTTTAGTCT | Eurofins | N/A |
| mCxcr7.F AGCCTGGCAACTACTCTGACA | Eurofins | N/A |
| mCxcr7.R GAAGCACGTTCTTGTTAGGCA | Eurofins | N/A |
| mIL19. F CTCCTGGGCATGACGTTGATT | Eurofins | N/A |
| mIL19.R GCATGGCTCTCTTGATCTCGT | Eurofins | N/A |
| mCcl26.F TTCTTCGATTTGGGTCTCCTTG | Eurofins | N/A |
| mCcl26.R GTGCAGCTCTTGTCGGTGAA | Eurofins | N/A |
| mMtrf1l.F GGGACACGGAGTCATTGCTG | Eurofins | N/A |
| mMtrf1l.R TCTTCCGAAGGAACCAACAGT | Eurofins | N/A |
| mCCL4.F TTCCTGCTGTTTCTCTTACACCT | Eurofins | N/A |
| mCCL4.R CTGTCTGCCTCTTTTGGTCAG | Eurofins | N/A |
| mCxcl10.F CCAAGTGCTGCCGTCATTTTC | Eurofins | N/A |
| mCxcl10.R GGCTCGCAGGGATGATTTCAA | Eurofins | N/A |
| mCCL5.F GCTGCTTTGCCTACCTCTCC | Eurofins | N/A |
| mCCL5.R TCGAGTGACAAACACGACTGC | Eurofins | N/A |
| mCCL25.F TTACCAGCACAGGATCAAATGG | Eurofins | N/A |
| mCCL25.R CGGAAGTAGAATCTCACAGCAC | Eurofins | N/A |
| mCXCL1.F CTGGGATTCACCTCAAGAACATC | Eurofins | N/A |
| mCXCL1.R CAGGGTCAAGGCAAGCCTC | Eurofins | N/A |
| mTNF.R CCCTCACACTCAGATCATCTTCT | Eurofins | N/A |
| mTNF.R GCTACGACGTGGGCTACAG | Eurofins | N/A |
| mIL6.F TAGTCCTTCCTACCCCAATTTCC | Eurofins | N/A |
| mIL6.R TTGGTCCTTAGCCACTCCTTC | Eurofins | N/A |
| mIL12b.F TGGTTTGCCATCGTTTTGCTG | Eurofins | N/A |
| mIL12b.R ACAGGTGAGGTTCACTGTTTCT | Eurofins | N/A |
| mIL1B.F GCAACTGTTCCTGAACTCAACT | Eurofins | N/A |
| mIL1B.R ATCTTTTGGGGTCCGTCAACT | Eurofins | N/A |
| mIL4.F GGTCTCAACCCCCAGCTAGT | Eurofins | N/A |
| mIL4.R GCCGATGATCTCTCTCAAGTGAT | Eurofins | N/A |
| mIL10.F GCTCTTACTGACTGGCATGAG | Eurofins | N/A |
| mIL10.R CGCAGCTCTAGGAGCATGTG | Eurofins | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Ahmed Chadli (achadli@augusta.edu).
Materials availability
This study did not generate new unique reagents. Any other materials generated in this study are available from the lead contact Dr. Ahmed Chadli (achadli@augusta.edu).
Data and code availability
-
•
Data presented in this manuscript have been deposited in NCBI’s Gene Expression Omnibus under the GEO Series accession code GSE168875. All the other data are available within the article and the supplementary files.
-
•
No custom code or mathematical algorithm has been used in this study.
-
•
All raw data are available at: https://data.mendeley.com/drafts/g65wt6bf76.
Experimental model and subject details
Murine models
Six to eight weeks old wild-type female C57BL/6 (B6) (stock number 000664), BALB/c mice (stock number 000651), and BALB/C nude mice (stock number 002019) were purchased from The Jackson Laboratory (Bar Harbor, Maine). This study is focused on breast cancer in females but the authors expect EnnA to be efficient in male cancers as well. All animal experiments were performed according to ethical regulations and protocols approved by the Augusta University Institutional Animal Care and Use Committee. Mice are hosted in a facility that is fully accredited by Laboratory Animal Care International (AAALAC).
Cell lines
MDA-MB-231, MDA-MB-453, Hs578T, 4T1, and E0771 cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA) within the last 5 years. Large stocks were made and stored in liquid nitrogen. For the cells that are less frequently used, we are still using cells from the original stocks. The E0771 cell line is an ER−/PR−/HER2-, p53-mutated, basal-like murine breast cancer cell line, which qualifies it as a TNBC model.76 Murine 4T1 cells were originally isolated from a spontaneous mammary tumor in the BALB/c strain and show characteristics of the human TNBC subtype.77,78 When tested as a syngeneic model, the E0771 cell line is tumorigenic in C57BL/6 (B6) mice, and the 4T1 cell line is tumorigenic in BALB/c mice. All cell lines are regularly tested at least quarterly for mycoplasma using the Genetica Inc mycoplasma test and MycoAlert PLUS Mycoplasma Detection Kits.
Compounds
Our studies used 17-AAG (ChemieTek, Indianapolis, IN, #75747-14-7) and JMS-17-2 (Tocris Bioscience, Minneapolis, MN, Cat# 6378/50). Initial experiments with EnnA were performed using Enniatin purified from the fungal strains Fusarium tricinctum isolated from fresh, healthy roots of Aristolochia paucinervis growing in the Atlas Mountains of Morocco [S45]. We also have used EnnA purchased from Sigma (St. Louis, MO, Cat #E9661). Most of the studies, however, were done using EnnA synthesized and characterized in Dr. Brian Blagg’s laboratory (University of Notre Dame, IN) as fully described in Data S1/Method S1 (supplementarl information).
Method details
Progesterone receptor (PR) reconstitution assay
Purified PR was adsorbed onto PR22 antibody-protein A-Sepharose resin beads and was assembled into complexes. Rabbit reticulocyte lysate (RL) or purified chaperones were used to refold the PR. Briefly, approximately 0.05 μM PR was incubated with 1.4 μM Hsp70, 0.8 μM Hsp90β, 0.2 μM Hsp40 (Ydj), 0.08 μM HOP, and 2.6 μM p23 in reaction buffer (20 mM Tris/HCl, pH 7.5, 5 mM MgCl2, 2 mM DTT, 0.01% NP-40, 50 mM KCl, and 5 mM ATP). After incubation for 30 min at 30°C, 0.1 μM [3H]-progesterone (American Radiolabeled Chemicals, Inc #ART 0063, St. Louis, MO) was added. Samples were incubated on ice for 3h at 4°C. Complexes were then washed three times with 1 mL of reaction buffer and assessed for bound progesterone by liquid scintillation counting using a PerkinElmer Microbeta plate reader and for the composition of protein complexes by SDS-PAGE (10% gel) and Coomassie blue staining.
Co-immunoprecipitation of purified Hsp90β with co-chaperones
For these experiments, 2 μg purified Hsp90β was incubated with Hsp90 monoclonal antibody H90.10 adsorbed to protein A-Sepharose resin beads in 250 μL buffer (10 mM Tris/HCl, pH 7.5, 50 mM KCl, 0.03% NP40, 2.5% glycerol, and 5 mM MgCl2) for 30 min. Samples were shaken slowly at room temperature. Excess Hsp90 was washed away with 1 mL buffer twice; then 5 μg of p23, HOP, or FKBP52 was added to the resin, and the volume was adjusted to 200 μL. 5 μM 17A-AG or 40 μM EnnA in DMSO was then added, and the volume was adjusted to 200 μL. DMSO was used as a control. All samples received 5 mM ATP. Samples were further incubated for 30 min at 30°C, and the resin was resuspended every 5 min. Samples were washed three times with 1 mL of buffer and analyzed by SDS-PAGE (10% gel) and Western blotting using specific antibodies.
Immunoprecipitation from cell lysates
PD-L1 antibody (InVivoMAb, catalog number BE0101) was incubated with 30 μL slurry of protein A/protein G agarose beads (Pierce Recombinant Protein A-agarose, catalog number 20334 and Pierce Protein G agarose, catalog number 20399) for 2 h at room temperature. Following incubation, cell extracts (200 μg of protein) from E0771 cells were added to each sample and incubated overnight at 4°C with gentle rotation. The samples were then washed three times with 1X RIPA buffer along with protease inhibitors (Roche Applied Science, 11836170001), 10 mm NaF, 2 mm sodium pyrophosphate, 2 mm β-glycerophosphate, and 1 mm sodium orthovanadate, and 1mM phenylmethylsulfonyl fluoride (PMSF) and eluted with 2X sample buffer at 95°C for 5 min. To strip PD-L1 from the endogenous complexes, the immunoprecipitated PD-L1 was incubated with 300mM KCl for 30 min at 4°C. The resin was then washed 3 times and incubated with 20 μg purified Hsp90 in 250μL reaction volume for 30 min at 30°C. Protein complexes were eluted using SDS-PAGE sample buffer and analyzed by Western blot.
Cellular thermal shift assay (CETSA)
To analyze the engagement of EnnA and Hsp90 protein, a cellular thermal shift assay was performed. 1 × 107 Hs578T, E0771, or MCF7 cells were seeded in 10 cm culture dishes. After 24 h, cells were treated with DMSO (1%) or EnnA (20 μM) for 1 h. Following treatment, cells were trypsinized and collected with PBS containing protease inhibitors and transferred to PCR tubes. All samples were heated at 52°C - 60°C for 2 min using a Bio-Rad C1000 thermal cycler and lysed with 3–4 freeze−thaw cycles using liquid nitrogen. For each freeze-thaw cycle, the cells were exposed to liquid nitrogen for 1 min and placed in a water bath at 30°C for 1 min. Precipitated proteins and cell debris were then pelleted by centrifugation at 16,400 rpm for 15 min at 4°C and the cleared cell lysates were analyzed by Western blotting.
Protein expression and purification
All protein constructs were generously provided by Dr. David Toft (Mayo Clinic, MN). Hsp70 was prepared by the overexpression of human hsp70 in Sf9 cells. Cell extracts were run through the DEAE-Sepharose column. Fractions containing Hsp70 were further fractionated on a column of ATP-agarose. Pooled fractions of hsp70 were concentrated by precipitation with ammonium sulfate (75% saturation), and the precipitate was dissolved and dialyzed into Tris buffer (TB): 10 mM Tris-HCl, pH 7.5, 3 mM MgCl2, 50 mM KCl, and 2 mM dithiothreitol (DTT). Hsp70-containing fractions were applied to a 16/60 Superdex 200 sizing column equilibrated with TB. The hsp70 fractions eluting in the monomeric range were pooled, concentrated, and stored at −80°C.
Human hsp90β was overexpressed in SF9 cells and purified from cytosol extracts. Purification was performed by chromatography on columns of DEAE-Sepharose, heparin-agarose, and Mono Q. The purified hsp90 was stored at −80°C for later use.
Human Hop was expressed in bacteria using the pET-11 expression system. Bacterial lysates were fractionated by DEAE-Sepharose chromatography followed by hydroxyapatite column chromatography. Additional purification was achieved by fractionating the pool from hydroxyapatite on a Mono Q FPLC column that was eluted with a linear gradient of 0–0.5 M KCL. The fractions containing Hop were pooled, dialyzed into 10 mm Tris-HCl, 1 mm dithiothreitol, and 1 mm EDTA, pH 7.5, and stored at −80°C.
Ydj1 was expressed in a pJC25 bacterial expression system. Cell lysates were fractionated by DEAE-Sepharose column chromatography followed by hydroxyapatite column chromatography.
Human p23 was expressed in a pET-23 (+) bacterial expression system. The soluble fraction of bacterial lysate was fractionated by DEAE-Sepharose column chromatography followed by phenyl-Sepharose (hp1660) FPLC, dialyzed into 10 mm Tris-HCl, 1 mm dithiothreitol, and 1 mm EDTA, pH 7.5, and stored at −80°C.
Surface Plasmon Resonance (SPR)
Real-time binding assays were performed on a Biacore 3000 Surface Plasmon Resonance (SPR) instrument (GE Healthcare, Milan, Italy). Full-length recombinant proteins, Hsp90, Hsp70, HOP, and Hsp40 (what about p23??) were immobilized at similar immobilization levels (1503, 2410, 2172, 3100 RU, respectively) on a CM5 Biacore sensor chip, at ∼100μg/mL in 10 mM sodium acetate, pH 5.0, by using the EDC/NHS chemistry, with a flow rate of 2 μL/min and an injection time of 7 min. Binding assays were carried out by injecting 90 μL of the analyte, at 30 μL/min, with HBS (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, pH 7.4), and 1% (v/v) DMSO as running-buffer. The BIAevaluation analysis package (version 4.1, GE Healthcare), was used to subtract the signal of the reference channel, with a DMSO correction procedure.
RNA extraction and real-time RTqPCR
Total RNA was extracted using QIAzol lysis reagent (QIAGEN). RNA extracts were analyzed by a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). RNA quality was determined by the ratios of A260/A280 (close to 2) and A260/A230 (close to 2). RNA (1–5 μg) was used for generating cDNA using RNA to cDNA EcoDry Premix (Double Primed) (Clontech Laboratories, Inc.; now Takara Bio USA). cDNA was analyzed in triplicate using iQ TM SYBR Green Supermix (Bio-Rad) in a Bio-Rad CFX96 system. Eurofins Genomics (Louisville, KY) primers were used for the indicated genes. mRNA expression was normalized against that of beta-actin (internal control) (ΔCt = Ct (target gene) – Ct (Internal control gene)). The relative fold change was measured by the 2-ΔΔCt formula compared to control cells. Means and differences of the means with 95% confidence intervals were obtained using GraphPad Prism (GraphPad Software, Inc., San Diego, CA). The two-tailed Student’s t test was used for unpaired analysis comparing average expression between conditions. p values <0.05 were considered statistically significant.
Cell proliferation assay and drug treatment
Cell proliferation was monitored using The CellTiter 96 AQueous One Solution Cell Proliferation Assay reagent (Promega #G3580). Cells were grown to 40–60% confluency on 96-well tissue culture plates (Corning #3599, Glendale, AZ) followed by treatment with EnnA or DMSO control. For protein analysis by Western blot, cancer cell lines were grown for at least 48 h to be at 40–60% confluent in 6-well plates (Corning #3516) before drug treatment was started. Cells were then harvested at specified times, and cell lysates were made. For Western blotting, 15 μg of protein lysate was analyzed using the following specific antibodies.
Immunocytochemistry and fluorescence microscopy
The indicated cells were grown in 24-well plates (Corning #3337) on micro-cover glasses (Electron Microscopy Sciences) to about 50% confluency in MEM, 1X (Cellgro #10-010-CV) medium supplemented with 10% fetal bovine serum. Cells were treated with indicated concentrations of 17-AAG, EnnA, or DMSO control for 24 h. Cells were fixed with 0.1 M PIPES, pH 6.95; 1 mM EGTA, pH 8.0; 3 mM MgSO4; and 3% paraformaldehyde, then permeabilized with 0.1% Triton X-100, blocked with 10% fetal bovine serum with 5% glycerol, and stored at 4°C. The primary antibodies against LC3B, as well as mouse and rabbit secondary antibodies, were prepared in the blocking buffer.
Electron microscopy
Cell pellets were fixed in 4% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate (NaCac) buffer, pH 7.4, postfixed in 2% osmium tetroxide in NaCac, stained en bloc with 2% uranyl acetate, dehydrated with a graded ethanol series and embedded in Epon-Araldite resin. Thin sections were cut with a diamond knife on a Leica EM UC7 ultramicrotome (Leica Microsystems, Inc, Bannockburn, IL), collected on copper grids, and stained with uranyl acetate and lead citrate. Cells were observed in a JEM 1230 transmission electron microscope (JEOL USA Inc., Peabody, MA) at 110 kV and imaged with an UltraScan 4000 CCD camera & First Light Digital Camera Controller (Gatan Inc., Pleasanton, CA).
Quantification of immunofluorescence
ImageJ software was used to measure the area, mean fluorescence, and the integrated density of fluorescence for each cell. Mean fluorescence of areas void of cells was used to calculate the fluorescence background. The corrected total cell fluorescence (CTCF) was determined by using the following formula: CTCF = integrated density – the area of selected cell × mean fluorescence of background readings. Individual data points (n) were plotted in GraphPad Prism 9.4.1.
Flow cytometry
Twenty-five thousand E0771 cells were seeded in a 6-well plate and cultured in RPMI-1640 media containing 10% FBS and 1% antibiotic and antimycotic solution for 48 h. At about 60% confluency, cells were treated with 5μM EnnA dissolved in 1% DMSO for 24 h. After the treatment was completed, cells were collected using enzyme-free cell dissociation media and washed with PBS containing 2% FBS. The cell surface was stained with a specific primary antibody (1:200 dilution) for 30 min at 4°C. After washing, cells were stained with Alexa Fluor 488 (AF488) and Alexa Fluor 594 (AF594) secondary antibody specific for Hsp90β and calreticulin/Cx3Cr1, respectively, for 30 min at 4°C. Cells were washed twice with PBS containing 2% FBS and resuspended.
For tumor samples, about 1 cm3 of tissue was finely minced using a scalpel and was dissociated in RPMI 1640 media containing collagenase IV (1 mg/ml) and DNAse I (30 units/ml) for 30 min at 37°C with agitation. Tumor single-cell suspension was passed through a 70 μm cell strainer and washed twice with PBS containing 2% FBS. The cell surface was stained with the appropriate fluorescently conjugated antibodies for 30 min at 4°C. Following the washing steps, cells were treated with BD cyto-Fix/cyto-Perm buffer and then incubated with specific antibodies for intracellular staining. Cells were washed, resuspended, and acquired in a Thermo Fisher Attune NxT flow cytometer. Data were analyzed using FlowJo software. In each sample, a total of 10,000 cells were acquired and data are represented as a percentage of mean ± S.E.M. Unstained, and only secondary antibody-stained cells were used as a control for appropriate gating purposes. The gating strategy is shown in Figures S11–S13.
Animals and tumor cell implantation
A) C57BL/6 (B6) and BALB/cJ mice implantation: 5 × 104 E0771-luc or 4T1 cells were implanted in the mammary fat pad of seven-to eight-week-old C57BL/6 (B6) or BALB/c female mice, respectively. Ten days following implantation, mice were intraperitoneally injected for the time course of the experiment as follows: EnnA (10 mg/kg) or 17-AAG (50 mg/kg), or DMSO (control). Animal body weight and tumor volume were measured and recorded. At the end of the experiment, mice were necropsied, and tumor tissues and spleens were carefully dissected. Collected tissues were further processed for histopathological, flow cytometric, Western blotting, and RTqPCR analyses. B) Nude mouse implantation: 5 × 104 E0771-luc cells were implanted in the mammary fat pad of seven-to eight-week-old female nude mice. Ten days following implantation, mice were intraperitoneally injected for the time course of the experiment with EnnA (10 mg/kg) or DMSO (control). Animal body weight and tumor volume were measured and recorded. All mouse procedures were conducted in accordance with the University Committee on the Use and Care of Animals at Augusta University.
CD8+ T cell depletion
For testing the effect of in vivo CD8+ T cell depletion, InVivoMAb anti-mouse CD8α (cat# BE0004-1, Bio X Cell, NH, USA) was used. Briefly, 5 × 104 E0771-luc cells were implanted in the mammary fat pad of C57BL/6 (B6) mice. Ten days following implantation, mice were intraperitoneally injected for the time course of the experiment as follows: EnnA (10 mg/kg), EnnA (10 mg/kg) along with anti-mouse CD8α (250 μg/100 μL/mouse), DMSO, and DMSO along with anti-mouse CD8α (250 μg/100 μL/mouse). Anti-mouse CD8α-assigned groups were injected at day 25 and day 27 post-tumor implantation. Tumor volume was measured via a digital caliper and recorded.
Vaccination
E0771-luc cells were seeded in 10 cm dishes and were allowed to grow to 50–60% confluency. Cells were treated in vitro with EnnA (7.4 μM) for 48 h. Floating (detached) cells were solely collected and counted. Cells were spun down and resuspended in serum/antibiotic-free media. Each animal received two ventral subcutaneous injections of a total of 1 × 106 cells. Injections were prepared as follows: 500,000 cells/100 μL and a total of 2 injections/animal. Control mice received injections of media only. Each mouse received 3 separate injections: one every 10 days. Two days after the last injection, all mice were implanted in the mammary fat pad with 5 × 104 E0771-luc cells. Mice were monitored for tumor growth by imaging.
Adoptive cell transfer
5 × 104 E0771-luc cells were implanted in the mammary fat pad of C57BL/6 (B6) mice. Ten days following implantation, mice were treated with EnnA (10 mg/kg) or DMSO (control) for 14 days. For T cell processing, spleen, and lymph nodes were excised, and T cells were purified following the MagniSort Mouse T cell Enrichment kit (Cat# 8804-6820, Invitrogen, Thermo Fisher, USA) protocol. A new set of mice were injected with 2 × 106 T cells extracted from DMSO control mice (naive T cells) or EnnA-treated mice (Immune T cells). Mice were then implanted with 5 × 104 E0771-luc cells, as described above, and tumor volume was monitored via imaging. Measurement of luciferase activity was further used for statistical analysis.
Bioluminescence imaging
Isoflurane-anesthetized mice were intraperitoneally injected with 75 mg/kg D-luciferin substrate (Wako Pure Chemical Industries, Japan) and prepared in sterile PBS. Animals were imaged using an IVIS Lumina system (Caliper Life Sciences, Hopkinton, MA) 5 min after substrate administration.
mRNA sequencing and data analysis
E0771 murine cells were allowed to grow in 6-well tissue culture plates for 48 h supplemented with complete RPMI-1640 media (Corning, # 10-040-CV). Cells were treated with EnnA (7.4 μM) or 17-AAG (0.5 μM) for 24 h. Cells were collected by trypsinization, washed with ice-cold PBS, resuspended in QIAzol Lysis Reagent (Qiagen, #79306), and were immediately delivered to the Integrated Genomics core facility at Georgia Cancer Center, Augusta University.
RNA was isolated from mouse cells using PureLink RNA Mini Kit (ThermoFisher), and DNA was removed using the PureLink DNase Set (ThermoFisher). RNA purity and concentration are evaluated by spectrophotometry using the NanoDrop ND-1000 (ThermoFisher). RNA quality was assessed by the Tape Station (Agilent Technologies, Santa Clara, CA) and assured of an RNA Integrity Number (RIN) greater than 8. Total RNA samples were processed for cDNA library preparations using TruSeq Stranded mRNA Library prep kit (Illumina, San Diego, CA). Briefly, mRNA was captured using oligo-dT beads from 800 ng of total RNA. Following purification, the mRNA was fragmented into small segments, 300–350 bp in size, and converted to cDNA fragments. A single “A” base was added to these cDNA fragments and the adapter was ligated. The products were purified and enriched with PCR to create the final cDNA library. The prepared library was further examined by Tape Station and Qubit (ThermoFisher) to test for quality and quantity, respectively. The libraries were pooled and run on the NextSeq500 sequencing system using a 75-cycle paired-end protocol. BCL files generated by the NextSeq 500 were converted to FASTQ files for the downstream analysis. Reads passing quality control were aligned to the mouse reference genome of mm9, through the STAR (ultra-fast) aligner for obtaining alignment rates greater than 90%. The generated alignment files in a comparative setup were imported to the following Cufflinks and Cuffdiff tools for outputting differentially expressed genes with log2 fold changes and q values having replicates.
Pathway analysis was performed using Ingenuity Pathway Analysis, IPA (Qiagen), Gene Set Enrichment Analysis (GSEA), and Database for Annotation, Visualization, and Integrated Discovery (DAVID) v 6.7 to identify the relative Gene Ontology (GO) terms followed by discovering the enriched functional-related gene subgroups as shown in heatmaps by clustering.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism (9.1.0 (216). All data are represented as mean ± S.E.M. Statistical significance was determined using Student’s t test (two-tailed) or one-way ANOVA, as indicated in figure legends. p values less than 0.05 were considered significant. The number of animals (n) per group is indicated in figure legends. Animals were randomly assigned to groups. Mice that died and mice bearing tumors that were too large before the end of the experiment were excluded from the final analysis. Studies were not conducted blinded. p values of statistical significance are represented as: ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ∗∗, p < 0.01; and ∗, p < 0.05.
Acknowledgments
We thank Drs. Lisa Middleton and Rhea-Beth Markowitz for their thorough editing of the manuscript. We are grateful to Dr. Shelli R. McAlpin (University of New South Wales, Sydney, Australia) for providing the SM compounds. We thank the Georgia Cancer Center core facilities, especially the Integrated Genomics and Flow Cytometry shared resources, and the Augusta University Electron Microscopy and Histology cores, for their valuable expertise. This work was supported by NIH R01 grant GM102443-01 and R01CA249178, a Unite in The Fight Against Cancer Award, and Augusta University Startup Funds for A.C.; N.H.E was supported by a government scholarship fund from the Egyptian Cultural and Educational Bureau. V.C. was supported by fellowship award CA212467.
Author contributions
A.C. and B.B. conceived and designed experiments. A.C., N.H.E., V.M.C., A.E., V.K.K., M.A.S., T.L., S.L., K.K., Y.J., D.M., A.E.A., S.K.L., F.T.F.T., H.K., and A.D. performed the experiments. A.C., N.H.E., E.C., P.C.R., and D.M. analyzed and interpreted the data. A.C., N.H.E, A.D., and E.C. contributed to the preparation of the manuscript. C.E., A.D., B.B., P.C.R., and H.K. provided reagents.
Declaration of interests
A.C. and Augusta University have a patent (US 11,446,352 B2) related to this work. All other authors have no conflict of interest to declare for this research.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. We support inclusive, diverse, and equitable conduct of research.
Published: October 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108308.
Supplemental information
References
- 1.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins R.W., Barbie D.A., Flaherty K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer. 2018;118:9–16. doi: 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sade-Feldman M., Jiao Y.J., Chen J.H., Rooney M.S., Barzily-Rokni M., Eliane J.P., Bjorgaard S.L., Hammond M.R., Vitzthum H., Blackmon S.M., et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postow M.A., Chesney J., Pavlick A.C., Robert C., Grossmann K., McDermott D., Linette G.P., Meyer N., Giguere J.K., Agarwala S.S., et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P., et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolchok J.D., Kluger H., Callahan M.K., Postow M.A., Rizvi N.A., Lesokhin A.M., Segal N.H., Ariyan C.E., Gordon R.A., Reed K., et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crosby E.J., Wei J., Yang X.Y., Lei G., Wang T., Liu C.X., Agarwal P., Korman A.J., Morse M.A., Gouin K., et al. Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2017.1421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mbofung R.M., McKenzie J.A., Malu S., Zhang M., Peng W., Liu C., Kuiatse I., Tieu T., Williams L., Devi S., et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 2017;8:451. doi: 10.1038/s41467-017-00449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Ware M.B., Zaidi M.Y., Ruggieri A.N., Olson B.M., Komar H., Farren M.R., Nagaraju G.P., Zhang C., Chen Z., et al. Heat Shock Protein-90 Inhibition Alters Activation of Pancreatic Stellate Cells and Enhances the Efficacy of PD-1 Blockade in Pancreatic Cancer. Mol. Cancer Therapeut. 2021;20:150–160. doi: 10.1158/1535-7163.MCT-19-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song K.H., Oh S.J., Kim S., Cho H., Lee H.J., Song J.S., Chung J.Y., Cho E., Lee J., Jeon S., et al. HSP90A inhibition promotes anti-tumor immunity by reversing multi-modal resistance and stem-like property of immune-refractory tumors. Nat. Commun. 2020;11:562. doi: 10.1038/s41467-019-14259-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zavareh R.B., Spangenberg S.H., Woods A., Martínez-Peña F., Lairson L.L. HSP90 Inhibition Enhances Cancer Immunotherapy by Modulating the Surface Expression of Multiple Immune Checkpoint Proteins. Cell Chem. Biol. 2021;28:158–168.e5. doi: 10.1016/j.chembiol.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Neckers L., Blagg B., Haystead T., Trepel J.B., Whitesell L., Picard D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones. 2018;23:467–482. doi: 10.1007/s12192-018-0877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendillo M.L., Santagata S., Koeva M., Bell G.W., Hu R., Tamimi R.M., Fraenkel E., Ince T.A., Whitesell L., Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherz-Shouval R., Santagata S., Mendillo M.L., Sholl L.M., Ben-Aharon I., Beck A.H., Dias-Santagata D., Koeva M., Stemmer S.M., Whitesell L., Lindquist S. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin X., Moskophidis D., Mivechi N.F. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metabol. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai C., Whitesell L., Rogers A.B., Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saleh A., Srinivasula S.M., Balkir L., Robbins P.D., Alnemri E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 20.Beere H.M., Wolf B.B., Cain K., Mosser D.D., Mahboubi A., Kuwana T., Tailor P., Morimoto R.I., Cohen G.M., Green D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 21.Ravagnan L., Gurbuxani S., Susin S.A., Maisse C., Daugas E., Zamzami N., Mak T., Jäättelä M., Penninger J.M., Garrido C., Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 22.Garrido C., Brunet M., Didelot C., Zermati Y., Schmitt E., Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–2601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 23.Bruey J.M., Ducasse C., Bonniaud P., Ravagnan L., Susin S.A., Diaz-Latoud C., Gurbuxani S., Arrigo A.P., Kroemer G., Solary E., Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 24.Trepel J., Mollapour M., Giaccone G., Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez J., Carter T.R., Cohen M.S., Blagg B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets. 2020;20:253–270. doi: 10.2174/1568009619666191202101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamal A., Thao L., Sensintaffar J., Zhang L., Boehm M.F., Fritz L.C., Burrows F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors.[see comment] Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 27.Rodina A., Wang T., Yan P., Gomes E.D., Dunphy M.P.S., Pillarsetty N., Koren J., Gerecitano J.F., Taldone T., Zong H., et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature. 2016;538:397–401. doi: 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moulick K., Ahn J.H., Zong H., Rodina A., Cerchietti L., Gomes DaGama E.M., Caldas-Lopes E., Beebe K., Perna F., Hatzi K., et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pillarsetty N., Jhaveri K., Taldone T., Caldas-Lopes E., Punzalan B., Joshi S., Bolaender A., Uddin M.M., Rodina A., Yan P., et al. Paradigms for Precision Medicine in Epichaperome Cancer Therapy. Cancer Cell. 2019;36:559–573.e7. doi: 10.1016/j.ccell.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jhaveri K.L., Dos Anjos C.H., Taldone T., Wang R., Comen E., Fornier M., Bromberg J.F., Ma W., Patil S., Rodina A., et al. Measuring Tumor Epichaperome Expression Using [(124)I] PU-H71 Positron Emission Tomography as a Biomarker of Response for PU-H71 Plus Nab-Paclitaxel in HER2-Negative Metastatic Breast Cancer. JCO Precis. Oncol. 2020;4:1414–1424. doi: 10.1200/PO.20.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoy S.M. Pimitespib: First Approval. Drugs. 2022;82:1413–1418. doi: 10.1007/s40265-022-01764-6. [DOI] [PubMed] [Google Scholar]
- 32.Ohkubo S., Kodama Y., Muraoka H., Hitotsumachi H., Yoshimura C., Kitade M., Hashimoto A., Ito K., Gomori A., Takahashi K., et al. TAS-116, a highly selective inhibitor of heat shock protein 90alpha and beta, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Therapeut. 2015;14:14–22. doi: 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- 33.Garg G., Khandelwal A., Blagg B.S.J. Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects. Adv. Cancer Res. 2016;129:51–88. doi: 10.1016/bs.acr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhatia S., Diedrich D., Frieg B., Ahlert H., Stein S., Bopp B., Lang F., Zang T., Kröger T., Ernst T., et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood. 2018;132:307–320. doi: 10.1182/blood-2017-10-810986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L., Li L., Gu K., Xu X.L., Sun Y., You Q.D. Targeting Hsp90-Cdc37: a promising therapeutic strategy by inhibiting Hsp90 chaperone function. Curr. Drug Targets. 2017;18:1572–1585. doi: 10.2174/1389450117666160527125522. [DOI] [PubMed] [Google Scholar]
- 36.Smith J.R., Workman P. Targeting CDC37: an alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle. 2009;8:362–372. doi: 10.4161/cc.8.3.7531. [DOI] [PubMed] [Google Scholar]
- 37.Powers M.V., Clarke P.A., Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–262. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 38.Guy N.C., Garcia Y.A., Cox M.B. Therapeutic Targeting of the FKBP52 Co-Chaperone in Steroid Hormone Receptor-Regulated Physiology and Disease. Curr. Mol. Pharmacol. 2015;9:109–125. doi: 10.2174/1874467208666150519114115. [DOI] [PubMed] [Google Scholar]
- 39.Patwardhan C.A., Fauq A., Peterson L.B., Miller C., Blagg B.S.J., Chadli A. Gedunin Inactivates the Co-chaperone p23 Protein Causing Cancer Cell Death by Apoptosis. J. Biol. Chem. 2013;288:7313–7325. doi: 10.1074/jbc.M112.427328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chadli A., Felts S.J., Wang Q., Sullivan W.P., Botuyan M.V., Fauq A., Ramirez-Alvarado M., Mer G. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the Co-chaperone p23. J. Biol. Chem. 2010;285:4224–4231. doi: 10.1074/jbc.M109.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabai V.L., Yaglom J.A., Wang Y., Meng L., Shao H., Kim G., Colvin T., Gestwicki J., Sherman M.Y. Anticancer Effects of Targeting Hsp70 in Tumor Stromal Cells. Cancer Res. 2016;76:5926–5932. doi: 10.1158/0008-5472.CAN-16-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sy-Cordero A.A., Pearce C.J., Oberlies N.H. Revisiting the enniatins: a review of their isolation, biosynthesis, structure determination and biological activities. J. Antibiot. 2012;65:541–549. doi: 10.1038/ja.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tonshin A.A., Teplova V.V., Andersson M.A., Salkinoja-Salonen M.S. The Fusarium mycotoxins enniatins and beauvericin cause mitochondrial dysfunction by affecting the mitochondrial volume regulation, oxidative phosphorylation and ion homeostasis. Toxicology. 2010;276:49–57. doi: 10.1016/j.tox.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 44.Dornetshuber R., Heffeter P., Kamyar M.R., Peterbauer T., Berger W., Lemmens-Gruber R. Enniatin exerts p53-dependent cytostatic and p53-independent cytotoxic activities against human cancer cells. Chem. Res. Toxicol. 2007;20:465–473. doi: 10.1021/tx600259t. [DOI] [PubMed] [Google Scholar]
- 45.Wätjen W., Debbab A., Hohlfeld A., Chovolou Y., Kampkötter A., Edrada R.A., Ebel R., Hakiki A., Mosaddak M., Totzke F., et al. Enniatins A1, B and B1 from an endophytic strain of Fusarium tricinctum induce apoptotic cell death in H4IIE hepatoma cells accompanied by inhibition of ERK phosphorylation. Mol. Nutr. Food Res. 2009;53:431–440. doi: 10.1002/mnfr.200700428. [DOI] [PubMed] [Google Scholar]
- 46.Dornetshuber R., Heffeter P., Lemmens-Gruber R., Elbling L., Marko D., Micksche M., Berger W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009;53:1112–1122. doi: 10.1002/mnfr.200800571. [DOI] [PubMed] [Google Scholar]
- 47.Dornetshuber-Fleiss R., Heilos D., Mohr T., Richter L., Süssmuth R.D., Zlesak M., Novicky A., Heffeter P., Lemmens-Gruber R., Berger W. The naturally born fusariotoxin Enniatin B and Sorafenib exert synergistic activity against cervical cancer in vitro and in vivo. Biochem. Pharmacol. 2015;93:318–331. doi: 10.1016/j.bcp.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patwardhan C.A., Alfa E., Lu S., Chadli A. Progesterone Receptor Chaperone Complex-Based High-Throughput Screening Assay: Identification of Capsaicin as an Inhibitor of the Hsp90 Machine. J. Biomol. Screen. 2015;20:223–229. doi: 10.1177/1087057114549147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosano H., Stensgard B., Charlesworth M.C., McMahon N., Toft D. The assembly of progesterone receptor-hsp90 complexes using purified proteins. J. Biol. Chem. 1998;273:32973–32979. doi: 10.1074/jbc.273.49.32973. [DOI] [PubMed] [Google Scholar]
- 50.Terracciano S., Russo A., Chini M.G., Vaccaro M.C., Potenza M., Vassallo A., Riccio R., Bifulco G., Bruno I. Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Sci. Rep. 2018;8:1709. doi: 10.1038/s41598-017-14902-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ali M.M.U., Roe S.M., Vaughan C.K., Meyer P., Panaretou B., Piper P.W., Prodromou C., Pearl L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schopf F.H., Biebl M.M., Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 53.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 54.Ahmed A., Tait S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020;14:2994–3006. doi: 10.1002/1878-0261.12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casares N., Pequignot M.O., Tesniere A., Ghiringhelli F., Roux S., Chaput N., Schmitt E., Hamai A., Hervas-Stubbs S., Obeid M., et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kepp O., Senovilla L., Vitale I., Vacchelli E., Adjemian S., Agostinis P., Apetoh L., Aranda F., Barnaba V., Bloy N., et al. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3 doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michaud M., Martins I., Sukkurwala A.Q., Adjemian S., Ma Y., Pellegatti P., Shen S., Kepp O., Scoazec M., Mignot G., et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 58.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 59.Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 60.Su H., Na N., Zhang X., Zhao Y. The biological function and significance of CD74 in immune diseases. Inflamm. Res. 2017;66:209–216. doi: 10.1007/s00011-016-0995-1. [DOI] [PubMed] [Google Scholar]
- 61.Ravichandran K.S., Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 62.Yan Y., Cao S., Liu X., Harrington S.M., Bindeman W.E., Adjei A.A., Jang J.S., Jen J., Li Y., Chanana P., et al. CX3CR1 identifies PD-1 therapy-responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight. 2018;3 doi: 10.1172/jci.insight.97828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen F., Zhang Y., Jernigan D.L., Feng X., Yan J., Garcia F.U., Meucci O., Salvino J.M., Fatatis A. Novel Small-Molecule CX3CR1 Antagonist Impairs Metastatic Seeding and Colonization of Breast Cancer Cells. Mol. Cancer Res. 2016;14:518–527. doi: 10.1158/1541-7786.MCR-16-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yim K.H., Prince T.L., Qu S., Bai F., Jennings P.A., Onuchic J.N., Theodorakis E.A., Neckers L. Gambogic acid identifies an isoform-specific druggable pocket in the middle domain of Hsp90beta. Proc. Natl. Acad. Sci. USA. 2016;113:E4801–E4809. doi: 10.1073/pnas.1606655113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seo M.J., Lee D.M., Kim I.Y., Lee D., Choi M.K., Lee J.Y., Park S.S., Jeong S.Y., Choi E.K., Choi K.S. Gambogic acid triggers vacuolization-associated cell death in cancer cells via disruption of thiol proteostasis. Cell Death Dis. 2019;10:187. doi: 10.1038/s41419-019-1360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garrido G., Rabasa A., Sánchez B., López M.V., Blanco R., López A., Hernández D.R., Pérez R., Fernández L.E. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J. Immunol. 2011;187:4954–4966. doi: 10.4049/jimmunol.1003477. [DOI] [PubMed] [Google Scholar]
- 68.Spisek R., Charalambous A., Mazumder A., Vesole D.H., Jagannath S., Dhodapkar M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaeger A.M., Stopfer L., Lee S., Gaglia G., Sandel D., Santagata S., Lin N.U., Trepel J.B., White F., Jacks T., et al. Rebalancing Protein Homeostasis Enhances Tumor Antigen Presentation. Clin. Cancer Res. 2019;25:6392–6405. doi: 10.1158/1078-0432.CCR-19-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Callahan M.K., Garg M., Srivastava P.K. Heat-shock protein 90 associates with N-terminal extended peptides and is required for direct and indirect antigen presentation. Proc. Natl. Acad. Sci. USA. 2008;105:1662–1667. doi: 10.1073/pnas.0711365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Graner M.W. HSP90 and Immune Modulation in Cancer. Adv. Cancer Res. 2016;129:191–224. doi: 10.1016/bs.acr.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 72.Tsang J.Y.S., Ni Y.B., Chan S.K., Shao M.M., Kwok Y.K., Chan K.W., Tan P.H., Tse G.M. CX3CL1 expression is associated with poor outcome in breast cancer patients. Breast Cancer Res. Treat. 2013;140:495–504. doi: 10.1007/s10549-013-2653-4. [DOI] [PubMed] [Google Scholar]
- 73.Matsubara T., Ono T., Yamanoi A., Tachibana M., Nagasue N. Fractalkine-CX3CR1 axis regulates tumor cell cycle and deteriorates prognosis after radical resection for hepatocellular carcinoma. J. Surg. Oncol. 2007;95:241–249. doi: 10.1002/jso.20642. [DOI] [PubMed] [Google Scholar]
- 74.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okuma A., Hanyu A., Watanabe S., Hara E. p16(Ink4a) and p21(Cip1/Waf1) promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat. Commun. 2017;8:2050. doi: 10.1038/s41467-017-02281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y., Yang H.H., Hu Y., Watson P.H., Liu H., Geiger T.R., Anver M.R., Haines D.C., Martin P., Green J.E., et al. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget. 2017;8:30621–30643. doi: 10.18632/oncotarget.15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaur P., Nagaraja G.M., Zheng H., Gizachew D., Galukande M., Krishnan S., Asea A. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer. 2012;12:120. doi: 10.1186/1471-2407-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ouzounova M., Lee E., Piranlioglu R., El Andaloussi A., Kolhe R., Demirci M.F., Marasco D., Asm I., Chadli A., Hassan K.A., et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017;8 doi: 10.1038/ncomms14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data presented in this manuscript have been deposited in NCBI’s Gene Expression Omnibus under the GEO Series accession code GSE168875. All the other data are available within the article and the supplementary files.
-
•
No custom code or mathematical algorithm has been used in this study.
-
•
All raw data are available at: https://data.mendeley.com/drafts/g65wt6bf76.