

SUMMARY
CRISPR-Cas9 genome editing has enabled advanced T cell therapies, but occasional loss of the targeted chromosome remains a safety concern. To investigate whether Cas9-induced chromosome loss is a universal phenomenon and evaluate its clinical significance, we conducted a systematic analysis in primary human T cells. Arrayed and pooled CRISPR screens revealed that chromosome loss was generalizable across the genome and resulted in partial and entire loss of the targeted chromosome, including in preclinical chimeric antigen receptor T cells. T cells with chromosome loss persisted for weeks in culture, implying the potential to interfere with clinical use. A modified cell manufacturing process, employed in our first-in-human clinical trial of Cas9-engineered T cells (NCT03399448), reduced chromosome loss while largely preserving genome editing efficacy. Expression of p53 correlated with protection from chromosome loss observed in this protocol, suggesting both a mechanism and strategy for T cell engineering that mitigates this genotoxicity in the clinic.
Graphical Abstract

In brief
CRISPR-Cas9 genome editing induces persistent chromosome loss in human primary T cells and engineered CAR T cells for clinical use, a phenomenon that can be mitigated by modifying T cell preparation protocols prior to clinical application.
INTRODUCTION
The precision of CRISPR-Cas9 genome editing is paramount to ensure clinical safety and avoid unintended and permanent genotoxicities. Promiscuous off-target genome editing at unintended sites has been extensively studied1–3 and mitigated4,5 in vitro and in vivo. However, unintended chromosomal abnormalities following on-target genome editing, such as chromosome loss, have not been systematically investigated or prevented. Thus, these potential concerns for genome editing continue to persist, including in the clinic, at an unknown frequency.
T cells have been extensively engineered using Cas9 to develop potent immunotherapies for cancer6–9 and autoimmune diseases.10,11 In a previous study, low-level chromosome 14 aneuploidy was detected in primary human T cells after Cas9-mediated genome editing of the T cell receptor alpha constant (TRAC) gene using one clinically relevant guide RNA (gRNA).12 However, the extent to which chromosome loss occurs at other target sites, the determinants of this phenomenon, the behavior of T cells with Cas9-induced chromosome loss, and the clinical relevance of these findings remains unknown. Along with understanding this phenomenon, strategies to reduce or eliminate chromosome loss as an outcome of genome editing would improve the safety of engineered T cell therapies for patients.
Here, we analyzed chromosome loss following Cas9-induced genome editing at hundreds of sites across every somatic chromosome in the human genome to determine the frequency, determinants, and consequences of this phenomenon. We found that Cas9-induced chromosome loss was a generalizable phenomenon, occurring at targeted sites across the genome although specific to the chromosome targeted by Cas9. T cells with Cas9-induced chromosome loss had a fitness and proliferative disadvantage, yet could persist over multiple weeks of ex vivo culture. Surprisingly, chromosome loss also occurred during preclinical production of chimeric antigen receptor (CAR) T cells but was minimal or undetectable in Cas9-edited patient T cells from a first-in-human phase 1 clinical trial. Further experimentation demonstrated that a modified T cell editing protocol employed in our clinical trial increased levels of the DNA damage response protein p53 while decreasing chromosome loss, suggesting a possible mechanism for Cas9-induced chromosome loss and an unexpected strategy to avoid this unintended outcome in patients.
RESULTS
scRNA-seq reveals chromosome loss in Cas9-edited T cells
The TRAC locus, which encodes the T cell receptor (TCR) responsible for CD4 and CD8 T cell reactivity to peptide-major histocompatibility complex (MHC) complexes, is of immense interest for genome editing applications in human T cells. For engineered adoptive T cell therapies, where T cells are armed with a transgenic receptor for targeted immunotherapy, disrupting endogenous TCR expression limits graft-versus-host toxicity associated with mispairing of transgenic and endogenous TCRs.13 Additionally, abrogating the TCR is an important step toward developing allogeneic ‘‘off-the-shelf’’ T cell therapies that could reduce cell-manufacturing costs and expand patient accessibility.14
To quantify chromosome loss after genome editing of TRAC, primary human T cells were electroporated with S. pyogenes Cas9 ribonucleoprotein (RNP), including 1 of 11 different gRNAs tiled across the first exon of TRAC or a non-targeting gRNA (Figure 1A). Reduced TCR expression occurred in 60%–99% of all cells as measured by flow cytometry (Figure S1A), and editing at the TRAC locus was observed in 62%–97% of all genomic DNA sequencing reads (Figure S1B), depending on the gRNA. 4 days after Cas9 RNP introduction, T cells were subjected to multiplexed single-cell RNA sequencing (scRNA-seq) to detect reduced transcript levels resulting from chromosome loss caused by Cas9-mediated double-strand DNA breaks (DSBs) (Figure S1C).15 Transcriptome-wide analysis using existing computational methods revealed a reduction in gene dosage, specifically on chromosome 14, in cells with a TRAC-targeting gRNA compared with cells with a non-targeting gRNA (Figure 1B).16 We developed a novel algorithm that analyzes changes in gene dosage to predict the genome-wide chromosomal breakpoint location, finding the highest frequency to be near the known genomic location of our TRAC-targeting gRNAs on chromosome 14 (Figure 1C). Additionally, we wondered whether Cas9-induced chromosome loss was an arm-level or entire-chromosome-level event. After developing a computational pipeline to map changes in gene dosage along a given chromosome, we observed cells with lower gene dosages originating at the Cas9 target site (partial chromosome loss), as well as cells with lower gene dosage across the entire chromosome (whole chromosome loss) (Figure 1D). Overall, ~5%–20% of T cells exhibited partial or whole loss of chromosome 14 depending on the TRAC-targeting gRNA (Figures 1E and S1D–S1F).
Figure 1. CRISPR-Cas9 genome editing of TRAC results in whole and partial chromosome loss.

(A) Cas9 gRNA target sites tiled across the first exon of TRAC on chromosome 14.
(B) Gene dosage from transcriptome-wide scRNA-seq of T cells treated with Cas9 and a non-targeting gRNA (top heatmap) or TRAC-targeting gRNA (bottom heatmap). Each individual row corresponds to a single cell and each column corresponds to a specific gene and its genomic position, grouped by chromosome (outlined in black). Red represents increase in gene dosage, while blue represents decrease in gene dosage. Rows outlined in black represent cells treated with different TRAC-targeting gRNAs. Blue arrows highlight chromosome 14, where TRAC is located.
(C) Distribution of computationally predicted chromosome 14 breakpoints in cells predicted to have a chromosomal loss event. The distribution is an aggregate of 11 different TRAC-targeting gRNAs (all within ~300 bp) in cells with partial chromosome loss.
(D) Representative single cell chromosome 14 gene dosage plots illustrating a cell with no chromosome loss (left), whole chromosome loss (middle), or partial chromosome loss (right). Gene dosage was normalized to non-targeting samples. Gray shaded area (gene dosage of 0.95–1.05) represents normal gene dosage (2 copies). Blue shaded area (gene dosage of <0.95) represents reduction in gene dosage (1 copy). Dotted lines represent the centromere, black lines represent the Cas9 target site, and the red line represents the computationally predicted breakpoint.
(E) Quantification of whole and partial chromosome 14 loss across all gRNAs from scRNA-seq. NT indicates non-targeting gRNA.
(F) Schematic of ddPCR assay to measure partial chromosome loss. The yellow lightning bolt represents the Cas9 target site. The detection of both HEX and FAM probes indicates no DSB or repaired DSB (top illustration). The detection of the HEX probe but not the FAM probe indicates a non-ligated DSB that represents partial chromosome loss (bottom illustration). Primers and probes were positioned ~30–120 bp from the Cas9 cut site (see Figure S1G).
(G) Quantification of partial chromosome loss at the Cas9 target site across all gRNAs from ddPCR (n = 3, n = 2 biological donors). Numbers next to each point represent the TRAC-targeting gRNA. NT indicates non-targeting gRNA and represents samples from four different ddPCR amplicons. Error bars represent the standard deviation from the mean. Dashed line represents linear regression line of best fit and shaded region represent 95% confidence intervals (slope = 1.082; R2 = 0.9853).
(H) Relationship between genome editing efficacy and partial chromosome loss for two TRAC-targeting gRNAs. 5-fold dilutions of Cas9 RNP were electroporated into T cells (n = 3). Genome editing efficacy was measured at the genomic level, via next-generation sequencing (NGS), or at the protein expression level, via flow cytometry, while chromosome loss was measured via ddPCR. Error bars represent the standard deviation from the mean.
See also Figures S1 and S7.
DNA-based ddPCR is an orthogonal method to detect partial chromosome loss
As an orthogonal approach to detect chromosome loss, we used droplet digital PCR (ddPCR) to directly quantify genomic DNA copy number, eliminating potential interference from transcriptional or epigenetic factors that may affect the scRNA-seq results. Two primer/probe sets were designed to amplify nearby regions of the target gene, one as a control (hexachlorofluorescein-labeled [HEX]) and the other spanning the Cas9 gRNA target site (6-fluorescein-labeled [FAM]) so that a DSB without religation would inhibit amplicon generation (Figures 1F and S1G). Detection of the control amplicon without detection of the Cas9 target amplicon was considered partial chromosome loss. 3 days after Cas9 electroporation, genomic DNA harvested from TRAC-targeted T cells resulted on average in ~4%–22% partial chromosome loss; these losses were highly reproducible across biological T cell donors (Figure 1G). Importantly, primers and probes in the amplicon spanning the intended Cas9 target site were positioned to avoid binding-site disruption by small insertions and deletions (indels), the most common outcome after Cas9 genome editing (Figure S1G).
Both Cas9-induced gene disruption and chromosome loss are dependent on DSB generation. To investigate the relationship between these two outcomes, we electroporated dilutions of Cas9 RNP into primary human T cells. For two representative gRNAs, we observed the expected dose-dependent decrease in gene disruption and partial chromosome loss (Figure 1H). Both indel formation, at the genetic level, and TCR disruption, at the protein expression level, showed mirrored trends with partial chromosome loss across all dilutions. Based on the observation that Cas9 preferentially remains bound to the DNA end opposite of the protospacer adjacent motif (PAM) after cleavage,17,18 we also wondered whether orientation of Cas9 relative to the centromere influenced chromosome loss (Figure S1H). We found no significant difference between chromosome loss and whether Cas9 was expected to be bound to the centromeric or distal end of the chromosome (Figure S1I). Furthermore, the rates of chromosome 14 loss measured by scRNA-seq or ddPCR did not correlate with the genomic position targeted by different gRNAs (Figure S1J).
Target-specific chromosome loss is a general phenomenon following genome editing
To determine the generality of chromosome loss after genome editing, we conducted a comprehensive CRISPR screen using a library of 384 gRNAs targeting 3–7 genes on every somatic chromosome (92 genes in total) with 4 unique gRNA sequences targeting each gene (Figures 2A, S2A, and S2B). The edited cells were profiled using CROP-seq, an assay that enabled the simultaneous identification of a lentivirus-delivered gRNA and the profiling of the transcriptome expression for each individual cell using scRNA-seq.19 We chose to utilize CROP-seq in our experiment because it minimizes template switching from lentiviral transduction observed with other methods20 and because it has been successfully deployed in primary human T cells.21 Targets relevant to T cell genome engineering were prioritized, such as TRAC,6,22,23 TRBC,6 PDCD1,6 B2M,24 IL2RA,23 CXCR4,25 and CIITA,26 as well as other targets of interest for clinical genome editing such as BCL11A27,28 and HBB29,30 for the treatment of sickle cell disease, TTR to treat transthyretin amyloidosis,31 HTT to treat Huntington’s disease,32 and SER-PINA1 to treat alpha-1-antitrypsin deficiency.33
Figure 2. Genome-scale CRISPR-Cas9 screen reveals target-specific chromosome loss.
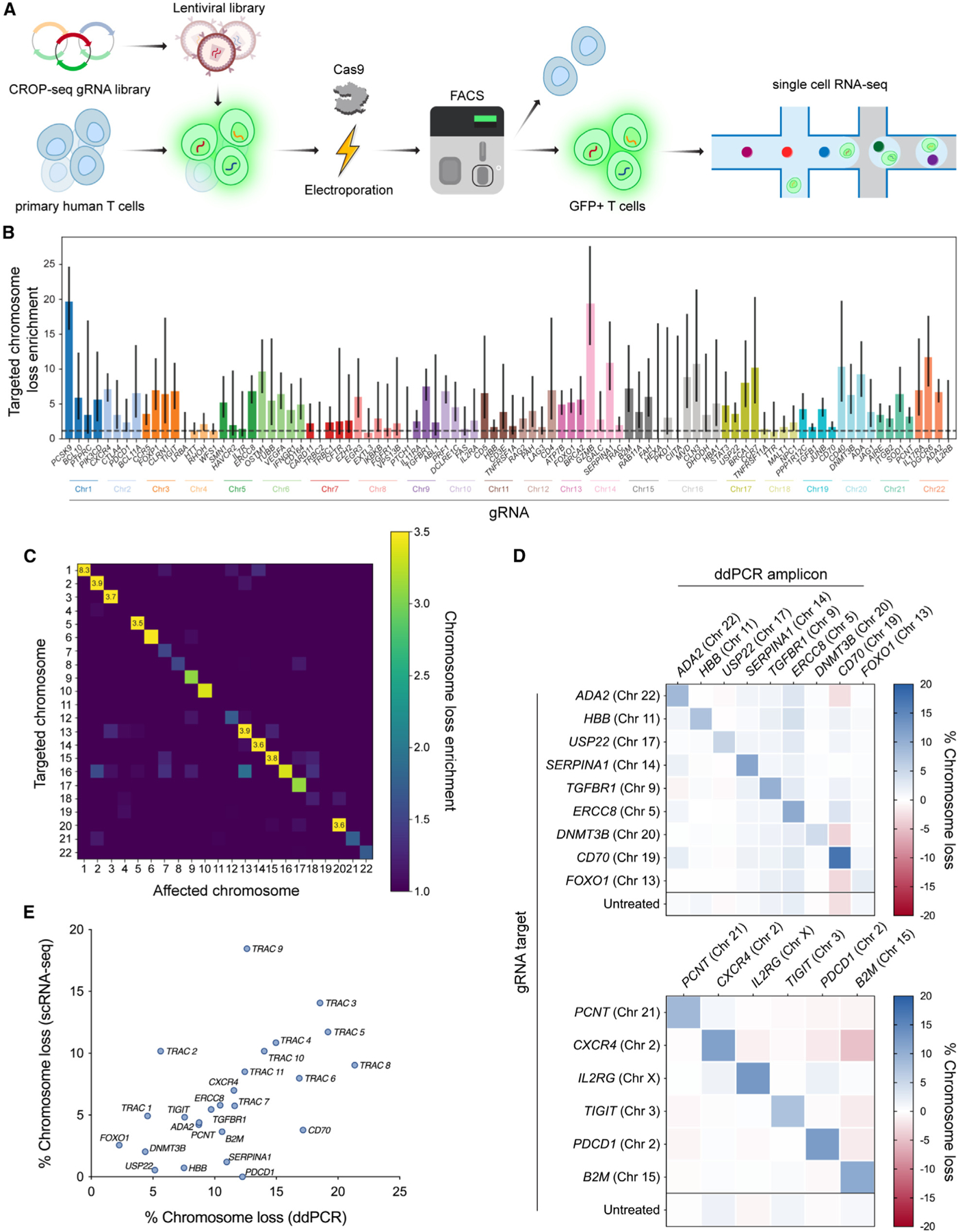
(A) Workflow for a CRISPR-Cas9 screen to estimate chromosome loss in T cells. Primary human T cells were transduced with a CROP-seq lentiviral library expressing one of 384 gRNAs. Cells were then electroporated with Cas9 protein, before GFP+ cells (co-expressed on the CROP-seq gRNA vector) were enriched via fluorescence-activated cell sorting. Enriched cells were subject to scRNA-seq and downstream analysis.
(B) Quantification of targeted chromosome loss enrichment for each target gene. Each of the 92 bars represents the combination of four unique gRNAs targeting the same gene. Chromosome loss enrichment was calculated relative to the baseline loss per chromosome in cells containing a gRNA targeting a different chromosome. Error bars represent 95% confidence intervals.
(C) Chromosomal loss enrichment at each somatic chromosome across all gRNAs. Rows represent the chromosome targeted by the Cas9 gRNA. Columns represent the chromosome analyzed for chromosome loss. Heatmap values are based on multiple gRNAs targeting multiple genes on a specified chromosome (all target genes appear in B).
(D) Partial chromosome loss measured by ddPCR at 15 different Cas9 target sites across the genome. Row titles indicate the identity of the gRNA used. Column titles indicate the site in the genome that was analyzed via ddPCR. Heatmap values represent the mean of replicates (n = 3, except n = 2 for B2M target column).
(E) Correlation between chromosome loss from 25 gRNAs as measured by scRNA-seq and ddPCR. Spearman’s correlation = 0.59; **p = 0.0017 (two-tailed).
See also Figures S2, S3, and S7.
Using our previously established computational pipeline on the CROP-seq dataset, we determined the breakpoints and gene dosage for 92 different genes targeted by Cas9 (Figures S2C–S2F). For numerous genes targeted in our screen, we observed significant enrichment for loss of the targeted chromosome in cells with a corresponding gRNA compared with cells with a gRNA targeting a different chromosome (Figure 2B). Chromosome loss above background levels occurred with 55% (201/364) of all gRNAs, for 89% (82/92) of all genes targeted, and in 100% (22/22) of all chromosomes targeted. Across all gRNAs, 3.25% of targeted cells had detectable whole or partial chromosome loss (Figure S3A). For cells with a non-targeting gRNA, no detectable chromosome loss was identified on any somatic chromosome. Enrichment for chromosome loss was much higher in chromosomes targeted by Cas9 compared with non-targeted chromosomes (Figure 2C), suggesting that this phenomenon is an outcome of target-specific cleavage during Cas9-mediated genome editing.
We validated this genome-scale chromosome loss by selecting 15 gRNAs from the library and individually electroporating them as Cas9 RNPs into T cells; ddPCR was used to measure partial chromosome loss at various sites in the genome and showed greater levels at the targeted site compared to non-targeted sites for nearly all gRNAs (Figure 2D). Additionally, rates of chromosome loss were highly correlated (Spearman’s correlation = 0.59) with those estimated by scRNA-seq (Figure 2E).
Further analyses of the CROP-seq screen to investigate the contribution of the Cas9 gRNA sequence revealed no influence by gRNA binding orientation (Figure S3B), nucleotide sequence (Figures S3C and S3D), or GC content on chromosome loss (Figure S3E). The computationally predicted dominant end-joining repair pathway, namely nonhomologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ), for each gRNA target also did not show a strong correlation with chromosome loss (Figures S3F and S3G). However, we did observe a moderate correlation between the distance of each targeted gene from the centromere and the rate of chromosome loss induced (Figure S3H), with gRNAs targeting closer to the centromere showing higher levels of chromosome loss.
Chromosome loss accompanies transcriptional signatures of DNA damage response, apoptosis, and quiescence
To assess the functional effects of Cas9-induced chromosome loss, we performed differential gene expression analysis between Cas9-edited T cells with or without chromosome loss. We identified genes that were differentially expressed across groups of cells with different chromosomes lost (Figures 3A and S4A). CD70, for example, was significantly upregulated in every group of cells with chromosome loss regardless of which chromosome was lost, and MDM2 was significantly upregulated in every group of cells with chromosome loss except those that lost chromosomes 13 or 18. Meanwhile, PHGDH was downregulated in every group of cells with chromosome loss except those that lost chromosomes 6, 10, 18, or 21. The numerous genes that were differentially expressed across cells with various chromosomes lost suggests that these findings are not a result of expression changes from lowered dosage of the target gene, but are a general influence of Cas9-induced chromosome loss.
Figure 3. Genetic and epigenetic factors influence Cas9-induced chromosome loss.
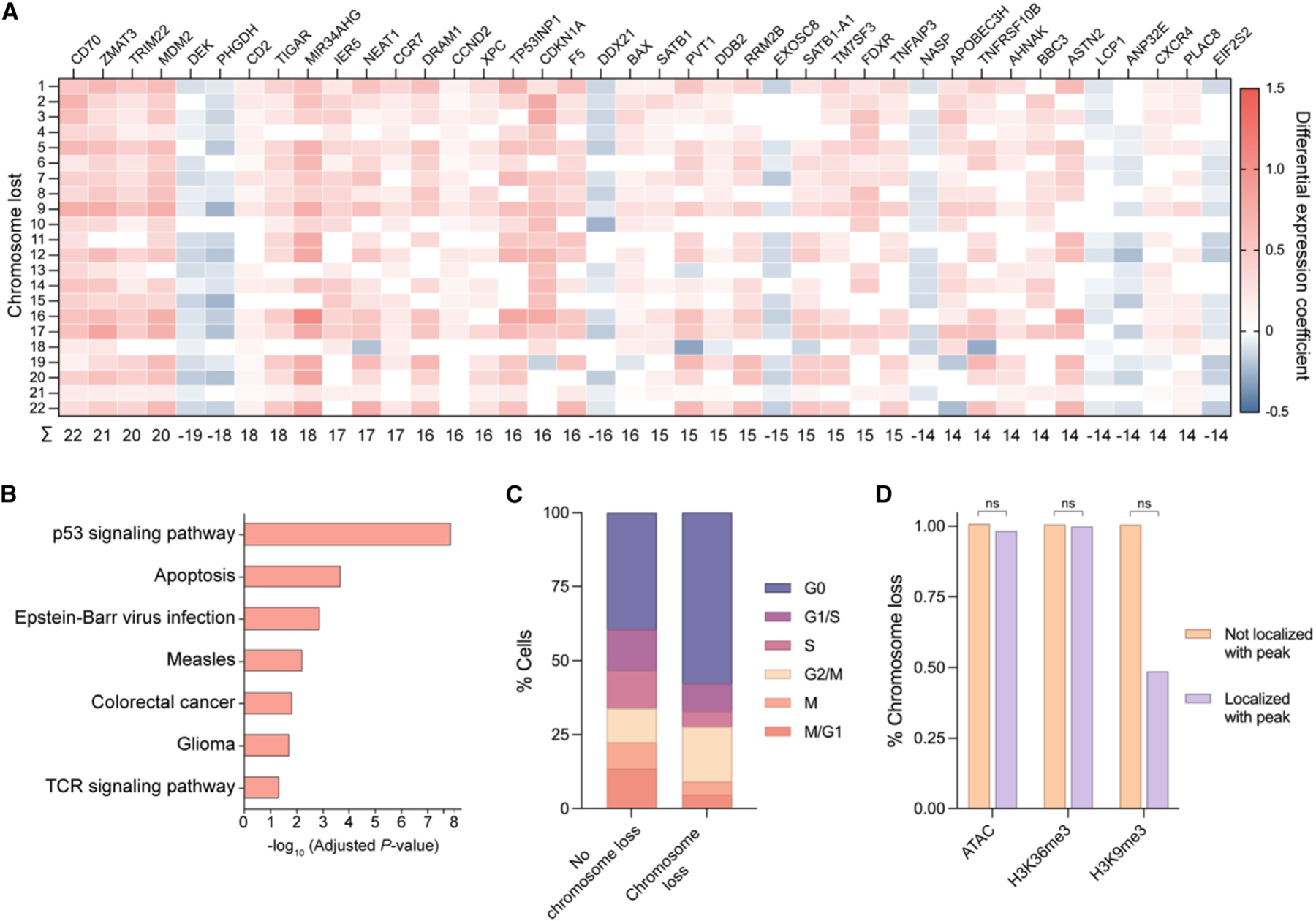
(A) Heatmap of differentially expressed genes in cells with chromosome loss compared with cells without chromosome loss. Cells with chromosome loss were divided into 22 groups depending on which somatic chromosome was lost (rows), and differentially expressed genes were individually investigated (columns). Upregulated genes are shown in red while downregulated genes are shown in blue. Genes were given a score of 1 (upregulated), —1 (downregulated), or 0 (no difference) for each chromosome loss group. Summed gene scores across all chromosome loss groups are shown below; genes with a score >|13| are displayed.
(B) Gene ontology analysis based on differential gene expression. The most significantly upregulated modules are displayed.
(C) Cell cycle analysis based on expression profiles. The percentage of cells in each cell cycle phase were quantified for cells with no chromosome loss or cells with chromosome loss.
(D) Influence of epigenetic marks on chromosome loss. The gRNA sequence for cells with or without chromosome loss was analyzed for localization within ±75 bp of an epigenetic marker peak. p values were calculated using a two-sided Fisher’s exact test and are from left to right (ATAC) 0.365496, (H3K36me3) 0.789824, and (H3K9me3) 0.305706. ns = not significant.
See also Figures S4 and S7.
Previous studies have demonstrated that a single Cas9-induced DSB can lead to p53 upregulation.34 Consistent with this finding, gene ontology analysis revealed the p53 DNA damage response and general apoptosis pathways were the most significantly overexpressed gene modules in cells with Cas9-induced chromosome loss (Figure 3B). We also observed an increase in cell cycle markers associated with the G0 phase and a decrease in those associated with the S phase for T cells with chromosome loss compared with those without (Figures 3C, S4B, and S4C), suggesting p53-induced cell cycle arrest. The results of both the differential gene expression analysis and cell cycle analyses indicate reduced fitness in T cells with Cas9-induced chromosome loss.
We further investigated the relationship between epigenetic markers and chromatin accessibility with Cas9-induced chromosome loss in primary human T cells; however, no significant correlation was found between these epigenetic factors and chromosome loss (Figure 3D).
T cells with chromosome loss persist ex vivo but with reduced fitness and proliferation
To determine whether T cells with Cas9-induced chromosome loss persist over time, we used ddPCR to measure the extent of partial chromosome loss at various time points during ex vivo culture. We chose time points over 2–3 weeks, which is a similar length of ex vivo culture compared with the manufacturing protocols of clinical trials with Cas9-edited T cells.6,7 As expected, T cells treated with Cas9 RNP targeting TRAC showed the highest levels of DSBs 1 day after electroporation (data not shown), when Cas9 RNP is still present within cells and DNA repair is ongoing.35,36 By day 3 post-treatment, DSBs plateaued until day 14 with most conditions showing a slight downward trend (Figure 4A). Since Cas9 RNP-mediated cleavage and DNA repair go to completion within 24 h,37 we posited that DSBs measured at day 3 and beyond have been resolved without religation of the cleaved genomic ends, which we considered partial chromosome loss.
Figure 4. Cas9-induced chromosome loss persists for weeks but results in reduced fitness and proliferation.

(A) ddPCR measurements of partial chromosome loss at the Cas9 TRAC target site over 14 days. Error bars represent the standard deviation from the mean (n = 3). Day 3 results were additional used as the donor 2 (female) results shown in Figure 1G.
(B) ddPCR measurements of partial chromosome loss for 15 different gRNAs targeted to sites across the genome over 14 days. Error bars represent the standard deviation from the mean (n = 3, except n = 2 for B2M). Day 3 results were additionally used for the diagonal values in the heatmaps of Figure 2D.
(C) Measurement of chromosome loss across T cells of varying proliferative capacity. T cells were stained with CellTrace Violet (CTV) and cultured for 5 days before sorting the top and bottom quartile (top panel). ddPCR was used to measure partial chromosome loss in lowly proliferative (CTV high) and highly proliferative (CTV low) populations (bottom panel). NT = non-targeting gRNA. Non-targeted samples evaluated for chromosome loss at the gRNA 2 or gRNA 8 amplicon were combined into a single column (n = 3 for each of the two different ddPCR amplicons). p values are from Welch’s unpaired t tests and from left to right are 0.002970, 0.002970, and 0.275572.
See also Figure S4.
We evaluated the temporal dynamics of chromosome loss over a longer period by repeating the experiment over the course of 21 days using 4 of the 11 TRAC-targeting gRNAs. Again, levels of partial chromosome loss showed a slight reduction over the 3 weeks, with detectable chromosome loss at the last time point remaining above that of non-targeting controls (Figure S4D).
To test the possibility that targeting a specific gene or chromosome may affect chromosome loss or T cell viability, we repeated the Cas9 electroporation and genomic DNA ddPCR with 15 gRNAs targeting other genes throughout the genome. Culturing the genome edited T cells for 14 days and measuring partial chromosome loss at various time points throughout, we once again observed a gradual reduction in chromosome loss over time (Figure 4B); 20% of gRNAs showed a >50% decrease in chromosome loss, 67% of gRNAs showed a ≤50%–25% decrease in chromosome loss, and 13% of gRNAs showed a <25% decrease in chromosome loss between days 3 and 14. These findings show that chromosome loss in T cells is observable as far out as 2–3 weeks, across multiple targeted genes and chromosomes. However, the gradual decrease in chromosome loss over time suggests a fitness disadvantage for T cells with this genomic aberration.
We additionally tested whether chromosome loss was associated with a proliferative disadvantage in T cells. TRAC-edited T cells were stained with a cell proliferation dye and cultured for 5 days. Cells that underwent the highest and lowest amounts of proliferation, based on dye intensity, were sorted and partial chromosome loss was measured between the two groups. Chromosome loss in the highest proliferating quartile was identifiable but statistically lower than chromosome loss in the lowest proliferating quartile (Figure 4C), which suggests that Cas9-induced chromosome loss confers a proliferative disadvantage.
Gene insertion via HDR results in chromosome loss
Thus far, we have shown that targeted chromosome loss can occur when using Cas9 to disrupt a desired gene, which predominantly occurs through end-joining DNA repair pathways. Tremendous effort has also been invested toward using Cas9 genome editing to correct a mutation or insert a gene by homology-directed repair (HDR). Since end-joining repair and HDR are divergent DNA repair pathways that involve different proteins, undergo different amounts of resection of the DSB ends, and occur in different stages of the cell cycle,38 we wanted to determine whether chromosome loss after Cas9-mediated genome editing also occurs during HDR. To explore this, we used Cas9 RNPs with a gRNA targeting CD5 and various oligonucleotide HDR templates to integrate a short hemagglutinin (HA) tag inframe with CD5.39 Successful generation of CD5+/HA+ cells via HDR peaked as high as ~40% 3 or 5 days after electroporation (Figure S5A). We performed ddPCR to quantify partial chromosome loss rates at both time points and observed similar levels of chromosome loss in CD5-targeted cells with an HDR template compared with CD5-targeted cells without an HDR template (Figure S5B). Additionally, using CD5 and other T cell surface markers, we attempted to use fluorescence-activated cell sorting (FACS) to enrich for cells without chromosome loss; however, we observed no reduction in chromosome loss, likely because loss of one allele may not be sufficient for detection via protein expression (Figures S5C and S5D).
Preclinical CAR T cell generation results in chromosome loss
While CAR T cells and transgenic TCR T cells are currently manufactured using a retrovirus or lentivirus to semirandomly integrate the retargeting transgene, researchers have also used Cas9 and HDR to precisely insert the transgene within the TRAC locus.22,39,40 This approach, which utilizes the native TRAC promoter to control expression of the CAR or retargeted TCR, offers advantages including uniform expression, minimal tonic signaling, and simultaneous disruption of the endogenous TCR. To explore whether chromosome loss occurs when generating preclinical CAR T cells via HDR, we electroporated primary human T cells with Cas9 complexed with one of two TRAC-targeting gRNAs or a non-targeting gRNA. Just after electroporation, recombinant adeno-associated virus 6 (AAV6) encoding a 1928z CAR as an HDR template was added to the T cells (Figures 5A and 5B).22,41 Both the reduction of TCR expression and the gain of CAR expression were observed in two independent electroporations; cells from one electroporation were subjected to scRNA-seq at day 4 post-manufacturing, while cells from the other were subjected to scRNA-seq at day 7 post-manufacturing. Overall rates of CAR integration via HDR were ~34%–69% (Figure S5E). In all conditions that received a TRAC-targeting gRNA, regardless of day or whether an HDR template was present, we observed an enrichment in chromosome 14 loss compared with conditions with a non-targeting gRNA (Figures 5C, S5F, and S5G). When chromosome 14 loss enrichment was normalized to TCR disruption—since day 4 and day 7 CAR T cells were engineered independently—we observed a slight decrease in chromosome 14 loss enrichment over time (Figure 5D). Together, these data suggest that chromosome loss is a general phenomenon that occurs in Cas9-edited T cells, regardless of the DNA repair pathway involved. Our findings also indicate that cells with chromosome loss are present among preclinical, Cas9-edited CAR T cells, highlighting the importance of understanding and mitigating this potential genotoxicity in the context of engineered T cell therapies.
Figure 5. Preclinical CAR T cell production via homology-directed repair results in chromosome loss.

(A) Strategy to generate CAR T cells via HDR with Cas9. AAV6 encoding a 1928ζ CAR between left and right homology arms (LHA and RHA, respectively) serves as a template for HDR after Cas9 cleavage (yellow lightning bolt) of TRAC.
(B) Three potential genomic outcomes after Cas9 HDR: indels that disrupt TCR expression (top), insertion of the CAR transgene that simultaneously disrupts TCR expression (middle), and chromosome loss that disrupts TCR expression (bottom).
(C) Quantification of chromosome 14 loss enrichment across two TRAC-targeting gRNAs with or without an AAV HDR template from scRNA-seq (n = 2 biological donors). Two separate batches of CAR T cells were manufactured, before being subjected to scRNA-seq 4 or 7 days after generation. Chromosome 14 loss enrichment was calculated relative to T cells treated with Cas9 and a non-targeting gRNA.
(D) Chromosome 14 loss enrichment over time, normalized to Cas9 editing efficacy (n = 2 biological donors). Editing efficacy was determined by the percentage of TCR negative cells as measured via flow cytometry (see Figure S5E).
See also Figures S5 and S7.
Investigation of chromosome loss in Cas9-edited T cells from clinical trial patients
Our studies thus far have focused on ex vivo culturing of T cells; it is not yet known how these findings translate in vivo. We conducted a first-in-human phase 1 clinical trial (clinicaltrials.gov, NCT03399448) where Cas9-genome-edited T cells were administered to patients with advanced, refractory cancer.6 Autologous T cells from three cancer patients were collected and electroporated with three different Cas9 RNPs, simultaneously targeting TRAC, TRBC, and PDCD1. These edited T cells were then transduced with a lentivirus encoding an HLA-A2*0201-restricted TCR specific to a peptide from the NY-ESO1 and LAGE-1 cancer antigens, resulting in engineered T cells (NYCE). NYCE cells were infused back into the patients and were found to be well tolerated.
To investigate whether clinical manufacturing of a Cas9-edited adoptive T cell therapy results in levels of chromosome loss similar to those observed in our laboratory studies, we analyzed scRNA-seq data from NYCE cells of two patients at various time points throughout the clinical trial. Cells were collected from patient UPN35 prior to infusion and at day 28 post-infusion, while cells from patient UPN39 were collected prior to infusion as well as at days 10 and 113 post-infusion (Figure 6A). Similar to our laboratory experiments, we inferred gene dosage at each of the target chromosomes (TRAC, Chr14; TRBC, Chr7; PDCD1, Chr2) and looked for partial and whole chromosome loss. Surprisingly, we observed extremely low levels of chromosome loss at the targeted chromosomes (<1%, data not shown), even before infusion, which were not enriched compared with background levels at non-targeted chromosomes (Figure 6B).
Figure 6. Clinical CRISPR-Cas9 genome editing protocol in patient T cells mitigates chromosome loss.

(A) Strategy to investigate chromosome loss in two clinical trial patients with CRISPR-edited T cells. Two patients with refractory cancer had T cells isolated, electroporated with TRAC, TRBC, and PDCD1-targeting Cas9 RNPs, and transduced with a lentivirus encoding an NY-ESO-1 TCR (NYCE cells). Cells were subjected to scRNA-seq prior to infusion (day 0) as well as at different time points postinfusion (days 10, 28, and/or 113).
(B) Chromosome loss enrichment on chromosome 14 (TRAC), chromosome 7 (TRBC), or chromosome 2 (PDCD1) at different time points for both patients. Enrichment was calculated relative to non-targeted chromosomes (all chromosomes but 2, 7, and 14). Day 0 represents NYCE cells prior to infusion, while other later time points represent NYCE cells that were collected after circulation in vivo.
(C) Diagram of two different protocols for Cas9 genome editing of primary human T cells. The activated T cell editing protocol (top) consisted of activating/stimulating cells prior to Cas9 electroporation and was used throughout this study along with most clinical trials. The non-activated T cell editing protocol (bottom) consisted of electroporating cells with Cas9 prior to activating/stimulating and is representative of our unique clinical trial.
(D) Relative partial chromosome loss with 11 different TRAC-targeting gRNAs using the activated T cell editing or non-activated T cell editing protocol in primary human T cells. Partial chromosome loss was normalized to the indel efficacy (see Figure S6B) (n = 3 biological donors).
(E) Fold TP53 mRNA expression during the activated T cell editing or non-activated T cell editing protocols for Cas9 genome editing of primary human T cells (n = 5 biological donors). Data points are the mean of two technical replicates. x-axis letters correspond to time points in (C). Cas9 DSBs represents the timepoints in the two protocols where Cas9 was electroporated into T cells to generate DSBs.
See also Figures S6 and S7.
Order of operations during Cas9 genome editing impacts chromosome loss
We wondered whether the discrepancy between the high rates of chromosome loss in our laboratory studies versus the low rates in our clinical trial could be attributed to the engineered T cell manufacturing protocol. In our laboratory studies, along with nearly all clinical trials of genome edited T cells, activation and stimulation of the T cells occurred prior to introducing Cas9 RNP or other genome editors to generate DSBs (hereafter referred to as the activated T cell editing protocol), to maximize editing efficacy.8,9,42 However, in our clinical trial we introduced Cas9 RNP and generated DSBs prior to activating and stimulating the T cells (hereafter referred to as the non-activated T cell editing protocol) (Figure 6C).6 We therefore performed Cas9 genome editing of TRAC in primary human T cells with the widely utilized activated T cell editing protocol as well as our unique non-activated T cell editing protocol, in parallel. Across all 11 TRAC-targeting gRNAs, we observed lower levels of partial chromosome loss using our non-activated T cell editing protocol compared with our activated T cell editing protocol (Figure S6A). However, genome editing with the non-activated T cell editing protocol on average resulted in ~13% lower rates of indels compared with the activated T cell editing protocol (Figure S6B). To control for this difference, we normalized the rate of chromosome loss to the rate of indels generated per gRNA and still observed a reduction in chromosome loss with our non-activated T cell editing protocol as compared with our activated T cell editing protocol with 10 out of 11 gRNAs (Figure 6D). During the non-activated T cell editing protocol, we observed a small decrease in T cell viability after activation/stimulation, which could arise from cells with chromosome loss experiencing apoptosis at a cell cycle checkpoint (Figure S6C).43
Previous studies have shown that p53, a key protein in cell cycle regulation and apoptosis, also regulates T cell activation. Downregulation of p53 is critical for murine T cell proliferation.44
Additionally, p53-mediated apoptosis has been shown as a mechanism for selecting against aneuploid cells.45 Therefore, we tested whether differences in manufacturing protocol influenced p53 levels, and how that related to the chromosome loss we observed. Expression of TP53, which encodes p53, was measured via RT-qPCR at different time points throughout both T cell genome editing protocols. Similar to what was observed in murine T cells, expression of p53 was lowered in both protocols after human T cell activation/stimulation (Figure 6E). The mean TP53 expression across five biological donors was >10 times higher immediately prior to Cas9-induced DSBs in our non-activated T cell editing protocol compared with our activated T cell editing protocol (Figure 6E). Thus, TP53 expression during Cas9-induced DSBs inversely correlated with rates of chromosome loss in T cells between our two protocols. For our non-activated T cell editing protocol, the higher expression of this key DNA damage response factor during Cas9-induced DSBs could select against cells with chromosome loss and explain the dramatically lower rates we observed. Implementation of this modified protocol for Cas9 genome editing in T cells represents a simple adjustment that could substantially mitigate chromosome loss in future research and clinical studies.
DISCUSSION
In this study, we comprehensively investigated the frequency and consequences of Cas9-induced chromosome loss in primary human T cells, taking a genome-scale approach to understand what influences this phenomenon and investigating both preclinical and clinical T cell therapies. Targeting Cas9 across the TRAC locus, we estimated chromosome loss in ~5%–20% of cells depending on the gRNA. We discovered that Cas9-induced chromosome loss was a generalizable phenomenon; chromosome loss was observed across the genome in an average of 3.25% of T cells that were targeted by Cas9. These T cells showed detectable levels of chromosome loss over 2–3 weeks of ex vivo culture, though they displayed a fitness and proliferative disadvantage. These disadvantages could cause cells without chromosome loss to outgrow those with chromosome loss, explaining the gradual reduction in this chromosomal aberration measured over the multiweek culture. Importantly, we still detected chromosome loss in nearly all conditions at our final time point, and this 2–3 week timeframe is similar to current clinical adoptive T cell therapy manufacturing protocols.6,7 This suggests that T cells with Cas9-induced chromosome loss could persist throughout ex vivo manufacturing and end up in the final product administered to patients. In addition, continued efforts aim to further shorten the engineered T cell manufacturing process, which has been shown to improve T cell activity and persistence but could result in higher levels of chromosome loss.46
To date, no study has investigated Cas9-induced chromosome loss in a clinical setting. In order to determine clinical significance, we generated CAR T cells using Cas9-mediated HDR, an approach being used in a growing number of clinical trials,8,9 and found a significant enrichment in chromosome loss compared with non-targeted cells. We also investigated Cas9-edited T cells of two patients enrolled in a first-in-human phase 1 clinical trial. We previously reported detectable levels of Cas9-induced translocations, another unintended genomic aberration, in these patient T cells, although levels reduced to the limit-of-detection after in vivo circulation.6 Surprisingly, when we investigated the patients’ T cells for chromosome loss, we saw no enrichment above background levels, marking two of the few cases where we did not find Cas9-induced chromosome loss.
Comparing the results from our laboratory experiments (where substantial chromosome loss was detected) and our clinical trial (where we did not observe chromosome loss above background levels), there were multiple technical differences in the parameters used for chromosome loss estimation. We tried to account for these differences by downsampling the CROP-seq screen dataset so that its parameters were similar to those of the clinical trial dataset, which was sparser (Figure S6D). Even upon down-sampling, our estimations of chromosome loss in the CROP-seq screen were comparable to the original complete dataset (Figure S6E). This supports the conclusion that biological rather than technical reasons explain the dramatic difference in chromosome loss estimation.
We considered and eliminated multiple factors that might correlate with or potentially explain Cas9-induced chromosome loss including Cas9 binding orientation, gRNA sequence, chromatin accessibility, and targeted gene or chromosome. Instead, we found that introducing Cas9-mediated DSBs prior to T cell activation/stimulation (the non-activated T cell editing protocol), a protocol used in our clinical trial but not in our laboratory experiments or most other clinical trials, influenced this phenomenon by significantly diminishing chromosome loss. It is possible that high levels of transcription in activated T cells during our activated T cell editing protocol may predispose cells to chromosome loss due to genome instability caused by active transcription.47 This effect could also be explained by levels of the DNA damage response protein p53 at the time of DSB generation, since we found TP53 expression and chromosome loss were inversely correlated. Consistent with this hypothesis, a report in immortalized fibroblasts showed that knockout of p53 increased Cas9-induced chromosomal truncations.48 Although most clinical trials with T cells rely on activation/stimulation prior to genome editing (activated T cell editing protocol), using the manufacturing protocol described here in which cells are activated after delivery of Cas9 (non-activated T cell editing protocol) could become standard practice to minimize chromosome loss in the manufactured product. While current trials using HDR to integrate a transgene of interest utilize the activated T cell editing protocol,8,9 and HDR has been shown to be minimally efficient in non-activated T cells, NHEJ-based methods such as homology-independent targeted insertion (HITI) could be further investigated to integrate transgenes into non-activated T cells.8,49 Our non-activated T cell editing protocol does not require additional equipment, modification of Cas9 or its gRNA, or additional cost, meaning that it can be easily and immediately integrated into clinical practice. In addition, our finding that major genotoxicities in T cells can be influenced by a small change in manufacturing protocol emphasizes the need for replicating and evaluating clinical protocols during preclinical laboratory experiments.
Recently, several other Cas9-mediated chromosomal abnormalities such as translocations,6 large deletions,50,51 loss of heterozygosity,52 and chromothripsis53 have been reported. Of these, only methods and technologies for limiting translocations have been demonstrated, including serial rather than simultaneous multiplexed genome editing,54 use of Cas12 nucleases,55 fusion of Cas9 to an exonuclease to limit repeat cleavage,56 or utilizing base editors that do not generate DSBs.57 Along with our modified non-activated T cell editing protocol, additional technologies could be developed to similarly mitigate chromosome loss.
CRISPR-based technologies that do not generate a DSB, such as base editors or epigenome editors, would likely avoid high levels of chromosome loss.58,59 However, base editing can only modify one or a few nucleotides and epigenetic editing lacks permanence; neither of these technologies are ideal for permanent gene disruption or gene insertion. The use of CRISPR-Cas9 genome editing that creates DSBs is still highly advantageous and will continue to expand in clinical use. Therefore, mitigating genomic aberrations from DSBs, such as chromosome loss, will have substantial value to avoid potential genotoxicity in patients. Our comprehensive study suggests that, although chromosome loss is a universal consequence of site-specific Cas9 genome editing, protocol adjustments and further exploration of underlying mechanisms can minimize its occurrence and impact.
Limitations of the study
Our data represent a systematic analysis of Cas9-induced chromosome loss. However, we do not differentiate between loss of one or both chromosomes, which could be investigated with different techniques. Additionally, from our scRNA-seq data we could differentiate between large deletions (<70% of genes reduced in gene dosage) and chromosome loss (≥70% of genes reduced in gene dosage) (see STAR Methods for additional detail). However, ddPCR does not provide chromosome-wide data and therefore cannot differentiate between large deletions and chromosome loss (false positives) and can only measure partial, and not whole chromosome loss (false negatives) (Figure S1G). Based on our comparisons in Figure 2E, chromosome loss as measured by ddPCR is a slight overestimation compared with scRNA-seq. However, based on the correlation between the two assays, it is likely that chromosome loss estimated by scRNA-seq contributes to the majority of genomic outcomes measured by ddPCR.
To determine the generalizability of Cas9-induced chromosome loss, we performed a CROP-seq CRISPR screen targeting several genes on each somatic chromosome. The emergence of higher throughput scRNA-seq may allow this study to be expanded to a genome-wide screen in the future. Additionally, where possible we selected highly active and specific gRNAs from previously reported studies to include in our CROP-seq library. However, since we cannot reliably measure genome editing efficacy in our pooled format, it is possible that gRNAs with low chromosome loss detected simply had low cleavage activity.
Finally, sequencing quality and computational gene calling varied between experiments (Figure S7A). Our estimates of chromosome loss were heavily dependent on the computational definitions we applied. It should be noted that changing the required percentage or minimum number of genes would influence estimations of chromosome loss. Based on our definitions, the ability to estimate chromosome loss was dependent on the number of genes reported by inferCNV. Analyzing several parameters that could influence the number of genes reported by inferCNV, we found that the median number of genes per cell obtained from scRNA-seq was the best indicator (Figure S7B). One scenario with a limited number of genes was shorter chromosomes (mostly chromosomes 18–22). Accordingly, we observed more background noise in these chromosomes. Other scenarios with a limited number of genes were the CAR T cell and clinical trial scRNA-seq datasets. Since this could influence our chromosome loss measurements, we primarily displayed relative chromosome loss enrichment, which was normalized to a non-targeting gRNA or untreated sample, rather than absolute chromosome loss.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jennifer A. Doudna (doudna@berkeley.edu).
Materials availability
This study did not generate new reagents.
Data and code availability
scRNA-seq data original to this study is available at the Gene Expression Omnibus (GEO) under the accession numbers GSE241838, GSE241839, and GSE241882. Clinical scRNA-seq data (NCT03399448) was accessed and is available through the NIH database of Genotypes and Phenotypes (dbGaP) under the accession number PHS001707.v2.
All original code used in this study has been deposited online and is publicly available on GitHub at www.github.com/yelabucsf/Cas9_chromosome_loss_scRNA-seq.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
Human Embryonic Kidney 293T (HEK293T) cells were obtained from the UC Berkeley Cell Culture Facility and cultured in Dulbecco’s Modified Eagle Medium (Gibco) with 10% FBS and 1% penicillin/streptomycin (Gibco). HEK293T cells were grown at 37 °C and 5% CO2. HEK293T cells were originally derived from a female donor.
Primary adult peripheral blood mononuclear cells
Primary adult peripheral blood mononuclear cells (PBMCs) were obtained as cryopreserved vials from Allcells Inc. CD3+ T cells were isolated from PBMCs using EasySep Human T Cell Isolation Kits (StemCell Technologies) according to the manufacturer’s instructions. Isolated CD3+ T cells were cultured in X-VIVO 15 medium (Lonza) with 5% fetal bovine serum (FBS) (VWR), 50 μM 2-mercaptoethanol, and 10 mM N-acetyl L-cysteine (Sigma-Aldrich). One day post-isolation, CD3+ T cells were activated and stimulated with a 1:1 ratio of anti-human CD3/CD28 magnetic Dynabeads (Thermo Fisher) to cells, as well as 5 ng/mL IL-7 (PeproTech), 5 ng/mL IL-15 (R&D Systems), and 300 U/mL IL-2 (PeproTech) for 3 days. After the initial activation and stimulation, magnetic beads were removed and T cells were cultured in medium with 300 U/mL IL-2. Medium was replaced every other day and T cells were maintained at a density of ~0.5–1×106 cells/mL.
For CAR T cell experiments, primary adult PBMCs were obtained as Leukopaks (StemCell Technologies) from deidentified healthy donors and cryopreserved in RPMI medium supplemented with 20% human serum and 10% DMSO. T cells were isolated as described before and cultured in X-VIVO 15 medium supplemented with 5% human serum, 5 ng/mL IL-7 (Miltenyi Biotec), and 5 ng/mL IL-15 (Miltenyi Biotec). Immediately after isolation, T cells were stimulated for 2 days with anti-human CD3/CD28 magnetic Dynabeads (Thermo Fisher) using a 1:1 bead-to-cell ratio.
All PBMCs were grown at 37 °C and 5% CO2. Various PBMCs from both male and female donors were used throughout this paper.
METHOD DETAILS
Cas9 ribonucleoprotein electroporation
100 pmol Alt-R crRNA and 100 pmol Alt-R tracrRNA (IDT) were diluted in IDT Duplex Buffer, incubated at 90° C for 5 min, and then slow cooled to room temperature (Tables S1, S7, and S8). 50 pmol S. pyogenes Cas9 V3 (IDT) was diluted in RNP buffer (20 mM HEPES, 150 mM NaCl, 10% Glycerol, 1 mM MgCl2, pH 7.5). Cas9 and duplexed gRNA (1:2 molar ratio) were incubated at 37° C for 15 min. Primary human T cells were washed once with PBS (−/−) before 250,000 cells were resuspended in P3 Buffer (Lonza). 50 pmol Cas9 RNP was added to the cells before electroporation in a Lonza 4D-Nucleofector with pulse code EH-115. X-VIVO 15 medium with 300 U/mL IL-2 was added to the electroporated cells before a 30 min recovery at 37° C. Electroporated T cells were plated at a density of ~0.5–1×106 cells/mL in 96-well U-bottom plates.
TCR flow cytometry
T cells were resuspended in Cell Staining Buffer (BioLegend) with Ghost Dye Red 780 (1:1,000, TonboBio) and anti-human TCR α/β Brilliant Violet 421 (1:100, BioLegend). Cells were stained for 30 min at 4° C in the dark. After staining, cells were washed with Cell Staining Buffer and analyzed on a Thermo Fisher Attune NXT Flow Cytometer with an autosampler. Over 20,000–100,000 cells were routinely collected and analyzed with FlowJo.
Next-generation sequencing of TRAC genome editing
Genomic DNA from T cells was extracted with QuickExtract DNA Extraction Solution (Lucigen) by incubating resuspended cells for 10 minutes at room temperature before heating lysates at 65° C for 20 minutes then 95° C for 20 min. The region of TRAC containing the Cas9 target site was amplified from genomic DNA with Q5 High-Fidelity DNA polymerase (NEB) to add universal adaptors (Table S2). Amplicons were cleaned with SPRIselect beads (Beckman Coulter) before a second round of PCR was performed to add unique i5 and i7 Illumina indices to each sample. Subsequent amplicons were cleaned again, and libraries were sequenced on an Illumina iSeq 100 (2×150 bp). FASTQ files were trimmed, merged, and analyzed for indels with CRISPResso2 (crispresso2.pinellolab.org).60 For non-targeting conditions, sequencing from cells receiving Cas9 and a non-targeting gRNA was used as input to check for indels at a given region.
Single-cell RNA sequencing with MULTI-seq barcoding
Four days post-electroporation, T cells were labeled via MULTI-seq as previously described.15 Briefly, a lipid-modified oligonucleotide (LMO) was combined with a unique oligonucleotide barcode (Tables S3 and S6) at a 1:1 molar ratio in PBS (−/−). 500,000 cells or fewer were washed twice with PBS (−/−) and then resuspended in PBS (−/−). The LMO/barcode solution was mixed with each cell suspension and incubated on ice for 5 minutes before addition of a co-anchor LMO and incubation on ice for an additional 5 minutes. Cold 1% BSA in PBS (−/−) was added to sequester free LMOs before washing cells twice with cold 1% BSA in PBS (−/−). Uniquely labeled cells were pooled in equal numbers in 1% BSA in PBS (−/−) to a final concentration of ~1,600 cells/mL. 10x Genomics Chromium Next GEM Single Cell 3′ Gene Expression Kits (v3.1) were utilized according to the manufacturer’s instructions, with the following modifications. Lanes of a standard Chromium chip were ‘‘super loaded’’ with ~50,000 cells to yield a target cell recovery of ~25,000 cells. During the cDNA amplification, 1 μL of a 2.5 mM MULTI-seq primer (see McGinnis et al.) was added. Supernatant from the cDNA bead cleanup was saved because it contained the MULTI-seq barcode amplicon. Supernatants were further cleaned by addition of SPRIselect beads and isopropanol with a conventional magnetic bead cleanup protocol. 3.5 ng of each cleaned amplicon was used in a PCR reaction to add sequencing indices; the reactions included KAPA HiFi HotStart ReadyMix (Roche), a unique i5 primer, and a unique RPI i7 primer (Table S3). The PCR reactions were cleaned with SPRIselect beads before final library QC. Gene expression and MULTI-seq barcode libraries were pooled 6:1 (molar ratio) and sequenced on an Illumina NovaSeq 6000 S1 Flow Cell.
Single-cell RNA sequencing analysis
Cell Ranger (v7.0) was used to process Chromium single cell data. cellranger count was performed with the parameters –r1-length=28 and –r2-length=90. For the first TRAC-targeting experiment the –force-cells parameter was set to 15,000 cells. To demultiplex the different pools of cells using the MULTI-seq barcode, cellranger multi was performed. The results from the different pools were aggregated using cellranger aggr. The results from cellranger were parsed with scanpy and converted to a h5ad file format. The demultiplexed results were added as metadata to the h5ad file.
For the CROP-seq screen, we counted the number of reads within each cell aligning to each of the gRNAs used in the screen. To determine which cells were targeted by a single, unique gRNA, we tested whether the gRNA with the highest number of reads had significantly more reads than the second highest. Specifically, let and be the number of reads for the first and second most common gRNAs in a cell, respectively. To test whether is significantly greater than , we calculated a P-value based on a binomial distribution with parameters and (i.e. ). If the probability of was smaller than 0.05, the cell was determined to be transduced by a single gRNA.
Quantification of chromosome loss from scRNA-seq
To assess the dosage of each gene in each cell, inferCNV of the Trinity CTAT project (https://github.com/broadinstitute/infercnv) was executed in R (version 4.1) with default parameters over the h5ad dataset created by cellranger (see previous section) for every scRNA-seq dataset.16 Each cell with a gRNA was labeled as a ‘‘treatment’’ and each cell with a non-targeted gRNA was labeled as a ‘‘control.’’ To successfully run inferCNV for the CROP-seq screen, inferCNV was performed in multiple batches of 30,000 cells.
The output of inferCNV was the estimated dosage for each gene; according to the software’s specifications, values below 0.95 were considered loss of at least one copy of the gene. inferCNV values for each gene were binarized as <0.95 or ≥ 0.95. Each chromosome in each cell was then searched for the interval between two genes that maximizes the difference between the average binarized inferCNV values on either side of the interval. This interval was the candidate breakage point for a particular chromosome in a cell.
We used the inferCNV values for all genes on a given chromosome within each cell (with respect to each of the 22 somatic chromosomes) to estimate the loss status of that chromosome. Specifically, we estimated whether there was 1) no chromosome loss, 2) whole chromosome loss, or 3) partial chromosome loss. If at least 70% of a minimum 150 genes to either the left or right of the candidate breakage point were below the 0.95 threshold, but less than 70% of the genes on the other side were below the threshold, the cell was labeled as partial chromosome loss for that chromosome. Otherwise, if at least 70% of all the genes throughout the entire chromosome were below the threshold, the cell was labeled as having whole chromosome loss for that chromosome. If neither were true, the cell was labeled as having no chromosome loss for that chromosome.
For the clinical trial scRNA-seq analysis, cells were genotyped as having an edit or no edit in TRAC, TRBC, or PDCD1, as previously described.6 Cells genotyped as having an edit in a particular gene were analyzed identically to as described above to evaluate potential chromosome loss as the corresponding chromosome.
Downsampling of the CROP-seq screen dataset was performed to assess the dependency of chromosome loss enrichment on the number of genes in the inferCNV output. To do this, 4,000 or 1,000 genes contained within the 9,639 gene output of the full CROP-seq inferCNV output were randomly sampled. Our chromosome loss calling pipeline, as described above, was then performed on these downsampled datasets.
Droplet digital quantitative PCR
Genomic DNA was collected from T cells at different times post-electroporation with QuickExtract DNA Extraction Solution, identical to as described earlier. The ddPCR setup was similar to what has been previously described.37 For multiplexed ddPCR, two ~200 bp amplicons for each target gene were designed (Tables S4, S7, and S8). Amplicon 1 was located proximal to the centromere and utilized a hexachlorofluorescein-labeled (HEX) oligonucleotide probe (PrimeTime qPCR probes, Zen double quencher, IDT). Amplicon 2 was located ~200–500 bp away from amplicon 1, was distal relative to the centromere, and utilized a 6-fluorescein-labeled (FAM) oligonucleotide probe (PrimeTime qPCR probes, Zen double quencher, IDT). Amplicon 1 served as a control, which should be un-affected by Cas9 genome editing or chromosome loss and would signal whether the gene of interest was in a given droplet. Amplicon 2 spanned the Cas9 target site, with the probe located ~30–60 bp away from the cleavage site. If the target site was not successfully repaired after Cas9 cleavage, amplicon 2 would not be able to be amplified and the FAM probe would be unable to dissociate from its quencher. ddPCR reactions were assembled with ddPCR Supermix for Probes (No dUTP, Bio-Rad), 900 nM of each primer, 250 nM of each probe, and 10–30 ng of genomic DNA. Droplets were formed using a Bio-Rad QX200 Droplet Generator following the manufacturer’s instructions before thermal cycling. The following day, ddPCR droplets were analyzed on a Bio-Rad QX200 Droplet Reader. Data were analyzed with the QX Manager Software (Bio-Rad), and thresholds were set manually based on wells with untreated samples. The percentage of alleles with chromosome loss was calculated based on droplets that had the target amplicon 1 (HEX+) but were unable to produce the neighboring amplicon 2 (FAM-). The equation utilized is as follows: .
Genome-scale CROP-seq CRISPR screen design
The CROP-seq library was designed to contain multiple gRNAs that target multiple genes on every chromosome. When possible, validated gRNA sequences from previous publications were utilized (Table S5). The gRNA library was ordered as an oPool oligo pool (IDT) and Golden Gate cloned into a custom CROP-seq vector that co-expressed GFP. To analyze the library, primers were used to amplify the gRNA spacer from either the plasmid library or genomic DNA library before sequencing on an Illumina iSeq 100. MAGeCK (https://sourceforge.net/p/mageck/wiki/) was used to quantify the representation of each gRNA in the library.61
CROP-seq CRISPR screen lentiviral production
For lentivirus production, HEK293Ts were transfected at 70–90% confluency with 10 μg CROP-seq gRNA plasmid, 10 μg Gag-pol expression plasmid (psPax2, gift from Didier Trono, Addgene plasmid #12260), and 1 μg pCMV-VSV-G plasmid (gift from Bob Weinberg, Addgene plasmid #121669) using polyethylenimine (PEI, Polysciences Inc.) at a 3:1 PEI:plasmid ratio. Approximately 6–8 hours after transfection, the medium was aspirated from cells and replaced with Opti-Mem (Gibco). Supernatant containing lentivirus was collected 48 hours after transfection, the medium was replaced, and medium was collected once more after an additional 48 hours. Viral supernatants were filtered through a 0.45 μm PES membrane bottle top filter (Thermo Fisher) and then concentrated with Lenti-X Concentrator (Takara) according to the manufacturer’s instructions. Purified and concentrated lentivirus was used immediately or stored at −80° C. Lentivirus was titered by counting the number of initially transduced cells, adding serial dilutions of lentivirus to primary human T cells, and measuring the percentage of GFP+ cells after 3 days (only in conditions with <30% GFP+ cells to ensure a majority were single transduction events).
CROP-seq CRISPR screen
For the CROP-seq screen, primary human T cells were isolated and stimulated as stated previously. 24 hours after stimulation, lentivirus was added to the cells at a multiplicity of infection (MOI) of ~0.3. MOI was confirmed via flow cytometry 2 days later, at day 3 post-stimulation. Dynabeads were then removed from T cells and Cas9 was electroporated as stated previously. For the full CROP-seq library experiment, 4 days post-electroporation, T cells were subject to fluorescence-activated cell sorting (FACS) on a Sony SH800S cell sorter to enrich for GFP+ cells. Genomic DNA was harvested from a small number of cells, as previously described, to assess the library representation. The rest of the live/GFP+ cells were arbitrarily divided into six pools and subject to MULTI-seq barcoding and 10x Genomics scRNA-seq, as previously described. The CROP-seq gRNA was enriched from the resulting cDNA similar to what has been previously described (Table S6).20 Briefly, 25 ng of cDNA was added to eight separate KAPA HiFi HotStart ReadyMix PCR reactions and amplified for 10 cycles with an annealing temperature of 65° C to enrich for the gRNA. Individual PCR reactions were pooled together and cleaned with SPRIselect beads. 8 μL of cleaned PCR1 product was added to a second KAPA HiFi HotStart ReadyMix PCR reaction and amplified for 10 cycles with an annealing temperature of 65° C to add Illumina sequencing adaptors. Gene expression, MULTI-seq barcode, and CROP-seq enrichment libraries were sequenced on an Illumina NovaSeq 6000. Multiple iterations of library sequencing were concatenated to achieve the desired sequencing depth.
Strand and MMEJ analyses
Each gRNA was mapped using the GRCh38 genome assembly. A two-sided Fisher’s Exact Test was performed to determine whether gRNAs binding distal or proximal to the centromere, relative to the gRNA spacer sequence, affected chromosome loss. MMEJ analyses were performed using inDelphi (indelphi.giffordlab.mit.edu), which computationally predicts Cas9-induced indels and groups them into ‘‘likely from NHEJ’’ or ‘‘likely from MMEJ’’, allowing for an estimate of MMEJ strength based on this ratio.62 The cell type was set to K562s and the MMEJ strength was measured for each unique gRNA sequence.
Differential gene expression analysis
To identify genes differentially expressed between cells with and without chromosome loss, we used the memento algorithm with default parameters (capture rate = 0.07).65 We tested each gene for differential expression with respect to each of the 22 somatic chromosomes separately, and only reported genes that were consistently over- or underexpressed across most chromosomes. We further ensured that the differences we observed were specific to the tested gene and were not the result of overall lower or greater levels of gene expression in cells with chromosome loss. To do this we accounted for the total expression of transcripts sharing the same chromosome as the tested gene by including the overall count of all transcripts on that chromosome as a covariate. This covariate was defined with respect to the chromosome containing the gene tested for differential expression and not with respect for the chromosome determining the two compared groups (cells with or without loss of that chromosome). Since memento supports only discrete covariates, we discretized the total transcript count into 10 decile bins.
We corrected the results of memento for multiple testing using FDR over the combined set of all tested genes over all tested chromosomes. We considered a gene to be statistically significant with respect to a chromosome (i.e. the gene to be over- or underexpressed in cells losing that chromosome) if its corrected P-value was below 0.05. Accordingly, we assigned the significance status of that gene-chromosome combination to be 1, 0, or −1 if it was significantly overexpressed, not significant, or under expressed, respectively. We then assigned each gene a total score between −22 and 22 by summing the significance status of that gene with respect to each of the 22 somatic chromosomes. 613 genes obtained a total score ≥5 and were considered the top overexpressed genes, while 590 obtained a total score ≤ −5 and were considered the top underexpressed genes.
We identified pathways enriched among the 613 top overexpressed genes by searching through the pathway terms defined in the KEGG database using the GSEApy Python package.63,64
Cell cycle analysis
Cell cycle states were defined using data and methods as previously described.67 In short, a set of marker genes was defined for each of the cell cycle states (M, M/G1, G1/S, S, G2/M; 603 marker genes in total). Only marker genes correlated with the average normalized expression across the other marker genes in the dataset were retained. Each cell was then given a score for each of the five cell cycle states equal to the average normalized expression of the marker genes of that state, which was then normalized across cells to obtain z-values. If all five z-values were negative, the cell was labeled non-cycling (G0). Otherwise, the cell was labeled according to the cell cycle state with the greatest z-value.
Epigenetic analyses
Datasets (ENCFF233TXT, ENCFF055FYI, and ENCFF129GAM) corresponding to activated T cells from a male donor (43 years old) were selected from the ENCODE Portal (www.enccodeproject.org).66,68 The presence of open chromatin from ATAC-seq data and the location of epigenetic marks (H3K9me3 and H3K36me3) from ChIP-seq data were determined within a 75 bp window around the GRCh38 coordinates of each gRNA and a P-value <10−5, according to best practices.69
T cell proliferation tracking
After isolation and stimulation, primary human T cells were electroporated with Cas9 RNPs identical to what was described earlier. Immediately after electroporation recovery, cells were pelleted and resuspended in 5 μM CellTrace Violet (Invitrogen). Cells were incubated in CellTrace Violet for 20 min at 37° C, prior to diluting in 4x volume of complete X-VIVO 15 medium to absorb unbound dye and incubating again for 5 min at 37° C. Cells were pelleted and resuspended in complete X-VIVO 15 with 300 U/mL IL-2. T cells were passaged every other day to refresh medium and maintain a density of ~0.5–1×106 cells/mL. Cells were sorted on a BD FACSAria II to obtain the approximate bottom and top quartile of cells according to CellTrace Violet signal.
Cas9-mediated CD5 homology-directed repair
HA tag insertion was achieved with either a single-stranded DNA HDR template (ssDNA HDRT), a double-stranded DNA HDR template (dsDNA HDRT), or a single-stranded DNA HDR template with Cas9 target sequences (ssCTS HDRT) (Table S9).39 Equimolar HDRT oligonucleotides were diluted in IDT Duplex Buffer, heated to 95° C for 5 minutes, then allowed to slow cool to room temperature. 100 pmol of HDRT was added to Cas9 RNP electroporations of primary human T cells, identical to as described above. Cells were analyzed on a Thermo Fisher Attune NXT Flow Cytometer with an autosampler, identical to as described above, except with the antibodies anti-human CD5 (UCHT2)-PE (1:100, Invitrogen) and anti-HA (6E2)-AF647 (1:100, Cell Signaling Technology). Over 20,000–100,000 cells were routinely collected and analyzed with FlowJo.
Fluorescence-activated cell sorting of CD5, CD81, and CD3E
Primary human T cells were electroporated with Cas9 RNPs targeting CD5 identical to what was described before. Seven days post-electroporation, cells were stained with anti-human TCR α/β Brilliant Violet 421 (BioLegend), anti-human CD5 (UCHT2)-PE (Invitrogen), anti-human CD81 (5A6)-FITC (BioLegend), and anti-human CD3E (SK7)-APC (Invitrogen), all at a 1:100 dilution. Cells were sorted on a BD FACSAria II to isolate different populations.
CAR adeno-associated virus production
An AAV transgene plasmid encoding the inverted terminal repeats, a 1928ζ CAR, a truncated human EGFR (EGFRt) tag, and TRAC homology arms for HDR was used as previously described (Table S10).22 The AAV plasmid was packaged into AAV6 by transfection of HEK293T cells together with pHelper and pAAV Rep-Cap plasmids using PEI. The AAVs were purified using iodixanol gradient ultracentrifugation. The titration of the AAV was determined by quantitative PCR on DNaseI (NEB) treated and proteinase K (Qiagen) digested AAV samples, using primers against the left homology arm. The quantitative PCR was performed with SsoFast EvaGreen Supermix (Bio-Rad) on a StepOnePlus Real-Time PCR System (Applied Biosystems).
CAR T cell production
gRNAs targeting exon 1 of the TRAC locus (TRAC gRNA 12), the intron preceding the TRAC locus (TRAC gRNA 13), or a non-targeting control gRNA were purchased from Synthego and resuspended in TE buffer (Table S11). Cas9 RNP was generated by incubating 60 pmol of Cas9 protein with 120 pmol sgRNA. T cells were counted, resuspended in P3 buffer at 2×106 per 20 mL, mixed with 3 mL of RNPs and added to a 96-well nucleofection plate. Cells were electroporated using a Lonza 4D-Nucleofector 96-well unit with the EH-115 protocol and immediately recovered by adding pre-warmed X-VIVO 15 medium without human serum. Recombinant AAV6 encoding the HDR template was added to the culture 30 to 60 min after electroporation at an MOI of 105, and incubated with the cells overnight. The day after the electroporation and transduction, edited cells were resuspended in medium and expanded using standard culture conditions, keeping a density of 106 cells/mL. TCR disruption and CAR HDR efficiency was evaluated by flow cytometry by staining the TCR with anti-TCRα/β (BW242/412)-PE (1:50, Miltenyi) and the CAR with goat anti-mouse IgG (H+L) AlexaFluor 647 Fab (1:100, Jackson ImmunoResearch).
CAR T cell scRNA-seq
CAR T cells were harvested at two time points after independent electroporations (day 4 and day 7 post-electroporation). TotalSeq-A0251-1 anti-human Hashtag reagents (BioLegend) were used to label different cell conditions. For the experiment, 500,000 cells from each condition were labeled with the hash antibodies in Cell Staining Buffer at 4° C for 30 min. After labeling, cells were washed three times with Cell Staining Buffer at 4° C and then resuspended in PBS (−/−) containing 0.04% BSA. Labeled cells were pooled and 50,000 cells were ‘‘super loaded’’ into four lanes (two lanes for day 4 samples and the other two lanes for day 7 samples) of a 10x Chromium Single-Cell G Chip. A 10x Genomics Chromium Next GEM Single Cell 3’ Gene Expression Kit (v3.1) was utilized according to the manufacturer’s instructions, and the subsequent library was sequenced on an Illumina NovaSeq 6000 S4 Flow Cell.
Activated T cell versus non-activated T cell manufacturing
For the activated T cell editing protocol, T cells were activated and stimulated identical to what was described earlier. After electroporation of Cas9 RNP, T cells were cultured in X-VIVO 15 medium with 5 ng/mL IL-7, 5 ng/mL IL-15, and 300 U/mL IL-2.
For the non-activated T cell editing protocol, we followed a protocol similar to what was used in our phase 1 clinical trial.6 After T cell isolation, cells were cultured in X-VIVO 15 medium with 5 ng/mL IL-7 and 5 ng/mL IL-15 for two days. Non-activated T cells were electroporated with 50 pmol Cas9 RNP using a Lonza 4D-Nucleofector with pulse code EH-115. After electroporation, cells were incubated in X-VIVO 15 medium with 5 ng/mL IL-7 and 5 ng/mL IL-15 for a 30 min recovery at 37° C. Electroporated T cells were plated at a density of ~0.5–1×106 cells/mL in 96-well U-bottom plates. Two days after electroporation, cells were counted and activated/stimulated with a 1:1 ratio of anti-human CD3/CD28 magnetic Dynabeads to cells, as well as 5 ng/mL IL-7, 5 ng/mL IL-15, and 300 U/mL IL-2 for an additional 3 days.
T cells were stained to measure viability using 7-Aminoactinomycin D (Thermo Fisher) according to the manufacturer’s instructions. Stained cells were analyzed on a Thermo Fisher Attune NXT Flow Cytometer with an autosampler. Over 20,000–50,000 cells were routinely collected and analyzed with FlowJo.
T cell RT-qPCR
During the activated or non-activated T cell editing protocols, 500,000 cells were periodically pelleted and resuspended in TRIzol (Invitrogen). RNA was isolated via phenol-chloroform extraction, precipitated by addition of isopropanol, washed with 75% ethanol, and resuspended in nuclease-free water. Isolated RNA was treated with TURBO DNase (Invitrogen) and SUPERase-In RNase inhibitor (Thermo Fisher) for 30 min at 37° C before addition of DNase Inactivation Reagent according to the manufacturer’s instructions. DNA-free RNA underwent cDNA synthesis using SuperScript III Reverse Transcriptase (Invitrogen) and Random Primers (Promega) according to the manufacturer’s instructions. qPCRs were performed with the resulting cDNA using iTaq Universal SYBR Green Supermix (Bio-Rad) on a Bio-Rad CFX96 Real-Time PCR Detection System (Table S12). TP53 expression levels were normalized to the expression levels of the housekeeping gene GAPDH, and to time point A (where the activated and non-activated T cell editing protocols start identically) using the ΔΔCt method.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed by Python or Graphpad Prism 9.4.1. Information on replicate numbers, biological donors, error bars, P values, and statistical tests used are listed in the figure legends.
ADDITIONAL RESOURCES
All information regarding the clinical trial analyzed in this study can be found at clinicaltrials.gov, under trial NCT03399448.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| anti-human TCR α/β Brilliant Violet 421 | BioLegend | Cat#306722; RRID: AB_2562805 |
| anti-human CD5 (UCHT2)-PE | Invitrogen | Cat#12-0059-42 |
| anti-human CD81 (5A6)-FITC | BioLegend | Cat#349504; RRID: AB_10642825 |
| anti-human CD3E (SK7)-APC | Invitrogen | Cat#17-0036-42 |
| anti-TCRα/β (BW242/412)-PE | Miltenyi | Cat#130-113-531; RRID: AB_2733903 |
| goat anti-mouse IgG (H+L) AlexaFluor 647 Fab | Jackson ImmunoResearch | Cat#115-607-003: RRID: AB_2338931 |
| Anti-HA (6E2)-AF647 | Cell Signaling Technology | Cat#3444S |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Dulbecco’s Modified Eagle Medium | Gibco | Cat#11965092 |
| Fetal Bovine Serum | VWR | Cat#1500-500GH |
| Penicillin/streptomycin | Gibco | Cat#15140122 |
| X-VIVO 15 Serum-free Hematopoietic Cell Medium | Lonza | Cat#04-418Q |
| 2-Mercaptoethanol | Gibco | 21985-023 |
| N-Acetyl-L-cysteine | Sigma-Aldrich | Cat#A7250 |
| DNase I Solution | STEMCELL Technologies | Cat#7900 |
| EasySep Buffer | STEMCELL Technologies | Cat#20144 |
| Dynabeads Human T-Activator CD3/CD28 for T cell Expansion and Activation | Gibco | Cat#11131D |
| Recombinant Human IL-2 | PeproTech | Cat#200-02 |
| Recombinant Human IL-7 | PeproTech | Cat#200-07 |
| Recombinant Human IL-7 | Miltenyi Biotec | Cat#130-095-367 |
| Recombinant Human IL-15 | R&D Systems | Cat#247-ILB-005/CF |
| Recombinant Human IL-15 | Miltenyi Biotec | Cat#130-095-760 |
| Alt-R S. pyogenes Cas9 Nuclease V3 | IDT | Cat#1081059 |
| Cell staining buffer | BioLegend | Cat#420201 |
| Ghost Dye Violet 780 | Tonbo Bioscience | Fisher Scientific Cat#50-105-2988 |
| QuickExtract DNA Extraction Solution | Lucigen | Cat#QE09050 |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat#M0491L |
| SPRIselect | Beckman Coulter | Cat#B23318 |
| KAPA HiFi Hotstart ReadyMix | Roche | Cat#KK2601 |
| ddPCR Supermix for Probes (No dUTP) | Bio-Rad | Cat#1863023 |
| Polyethylenimine | Polysciences Inc. | Cat#24314 |
| Opti-MEM Reduced Serum Medium | Gibco | Cat#31985070 |
| Lenti-X Concentrator | Takara | Cat#631231 |
| CellTrace Violet Cell Proliferation Kit | Invitrogen | Cat#C34571 |
| EvaGreen Supermix | Bio-Rad | Cat#186-4033 |
| 7-Aminoactinomycin D | Invitrogen | Cat#A1310 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| TURBO DNase | Invitrogen | Cat#AM2238 |
| SUPERase-In RNase inhibitor | Invitrogen | Cat#AM2694 |
| SuperScript III Reverse Transcriptase | Invitrogen | Cat#18080044 |
| Random primers | Promega | Cat#C1181 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#1725124 |
|
| ||
| Critical commercial assays | ||
|
| ||
| EasySep Human T Cell Isolation Kit | STEMCELL Technologies | Cat#17951 |
| P3 Primary Cell 96-well Nucleofector Kit | Lonza | Cat#V4SP-3096 |
| iSeq 100 i1 Reagent v2 | Illumina | Cat#20031371 |
| NovaSeq 600 S1 v1.5 | Illumina | Cat#20028317 |
| NovaSeq 600 S4 v1.5 | Illumina | Cat#20028312 |
| Chromium Next GEM Single Cell 3’ GEM Kit v3.1 | 10x Genomics | Cat#1000123 |
| Chromium Next GEM Single Cell 3’ Library Kit v3.1 | 10x Genomics | Cat#1000157 |
| Chromium Next GEM Single Cell 3’ Gel Bead Kit v3.1 | 10x Genomics | Cat#1000122 |
| TotalSeq-A0251-1 anti-human Hashtag Reagents | BioLegend | Cat#394601 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and processed single cell RNA sequencing data | This paper | GEO: GSE241838, GSE241839, and GSE241882 |
| Clinical single cell RNA sequencing data | Stadtmauer et al.6 | dbGaP: PHS001707.v2 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human embryonic kidney 293T cells | UC Berkeley Cell Culture Facility | N/A |
| Primary adult peripheral blood mononuclear cells | Allcells Inc. | Fisher Scientific Cat#50-156-841 or 50-156-574. |
| Primary adult peripheral blood mononuclear cells | StemCell Technologies | Cat#70500 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Cas9 gRNAs | IDT/Synthego | Tables S1, S7, S8, and S11 |
| MULTI-seq barcodes | IDT | Table S3 and S6 |
| CROP-seq library | IDT | Table S5 |
| ddPCR primers/probes | IDT | Tables S4, S7, and S8 |
| RT-qPCR primers | IDT | Table S12 |
| NGS primers | IDT | Tables S2 and S7 |
|
| ||
| Recombinant DNA | ||
|
| ||
| CROP-seq-opti-EGFP (modified from CROP-seq-opti) | This paper, modified from Addgene | Addgene#106280 |
| psPax2 (Gag-pol) | Addgene | Addgene#12260 |
| pCMV-VSV-G | Addgene | Addgene #8454 |
| pAAV-TRAC-1928z | Eyquem et al.22 | N/A |
| pHelper | Eyquem et al.22 | N/A |
| pAAV Rep-Cap | Eyquem et al.22 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo v10.5.3 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| CRISPResso2 | Clement et al.60 | https://github.com/pinellolab/CRISPResso2 |
| 10x Cell Ranger | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Scanpy v1.9.3 | Virshup et al. 2023 | https://github.com/scverse/scanpy |
| inferCNV | inferCNV of the Trinity CTAT Project | https://github.com/broadinstitute/infercnv |
| MAGeCK | Li et al.61 | https://sourceforge.net/p/mageck/wiki |
| inDelphi | Shen et al.62 | https://indelphi.giffordlab.mit.edu/about |
| KEGG | Kanehisa and Goto63 | https://www.genome.jp/kegg/ |
| GSEApy v1.0.3 | Fang et al.64 | https://github.com/zqfang/GSEApy |
| Prism v9.4.1 | GraphPad | https://www.graphpad.com/ |
| Memento | Kim et al.65 | https://doi.org/10.1101/2022.11.09.515836 |
| R v4.1 | The Comprehensive R Archive Network | https://www.R-project.org/ |
| Python v3.9.12 | Python Software Foundation | https://www.python.org/ |
| ENCODE Portal: Encyclopedia of DNA Elements | Sloan et al.66 | www.enccodeproject.org |
| Scripts for chromosome loss estimation (gene-expression matrix generation, sgRNA assignments, demultiplexing, inferCNV analysis, breakpoint estimation) | This paper | www.github.com/yelabucsf/Cas9_chromosome_loss_scRNA-seq |
Highlights.
Cas9 genome editing in T cells results in unintended but targeted chromosome loss
Chromosome loss from T cell genome editing is generalizable across target sites
Cas9-induced chromosome loss persists for weeks in cultured T cells
A modified protocol mitigates chromosome loss in T cells used for a clinical trial
ACKNOWLEDGMENTS
The authors thank Jennifer Hamilton, Matthew Kan, Elizabeth Abby Stahl, and David Colognori for thoughtful discussion throughout this project. We thank Christopher McGinnis and Zev Gartner for providing the MULTI-seq reagents; Yoon Gi Justin Choi (QB3 Functional Genomics Laboratory) for assistance with the scRNA-seq; Scarleth Chalen Ulloa for assistance with the FACSAria; Netra Krishnappa (IGI Center for Translational Genomics), Carrianne Miller (QB3 Genomics Sequencing Laboratory), and Eric Chow (UCSF Center for Advanced Technology) for assistance with NGS; MinCheol Kim for assistance with the differential gene expression analysis; Francois Aguet for assistance with the cell cycle analysis; and Brian Shy for assistance with the CD5 HDR assay. We thank the patients, their families, and the clinical study team at the Abramson Cancer Center Clinical Research Unit for their involvement in the clinical trial. We thank all members of the Satpathy, Cate, Eyquem, Fraietta, June, Chang, Ye, and Doudna laboratories for their input and advice. C.A.T. is supported by a National Institutes of Health (NIH) Ruth L. Kirschstein National Research Service Award F31 Pre-Doctoral Fellowship (National Heart Lung and Blood Institute, F31HL156468–01) and the Siebel Scholarship (Siebel Foundation). N.B. is supported by a Cancer Research Institute Irvington Fellowship (Cancer Research Institute, CRI4499). B.Y. is supported by the Parker Institute for Cancer Immunotherapy Parker Bridge Fellowship and the V Foundation. L.H. is supported by the U/SELECT program (Utrecht University). This project was supported by the California Institute for Regenerative Medicine (DISC2–13212, A.T.S.), the Parker Institute for Cancer Immunotherapy (A.T.S., J.E., C.H.J., H.Y.C., and C.J.Y.), the NIH National Institute of General Medical Sciences (R01-GM065050, J.H.D.C.), the NIH National Cancer Institute (R35CA209919, H.Y.C.), the NIH Centers for Excellence in Genomic Science (RM1HG007735, H.Y.C.; RM1HG009490, J.A.D.), the Howard Hughes Medical Institute (H.Y.C. and J.A.D.), the NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR071522, C.J.Y.), the NIH National Institute of Allergy and Infectious Diseases (R01AI136972, C.J.Y.), the Chan Zuckerberg Initiative and Chan Zuckerberg Biohub (C.J.Y.), and the NIH Somatic Cell Genome Editing Program of the Common Fund (U01AI142817–02, J.A.D.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.08.041.
DECLARATION OF INTERESTS
C.A.T., J.A.D., and the Regents of the University of California have patents pending or issued related to the use of CRISPR genome editing technologies. R.B. is an employee of BioMarin Pharmaceutical Inc., J.L. is an employee of Altos Labs, and K.R.P. is a co-founder and employee of Cartography Biosciences. A.T.S. is a co-founder of Immunai and Cartography Biosciences. A.T.S. has received research support from Arsenal Biosciences, Allogene Therapeutics, and 10x Genomics. J.H.D.C. is a co-founder of Initial Therapeutics. J.E. is a co-founder of Mnemo Therapeutics, a scientific advisory board member of Cytovia Therapeutics, and a consultant for Casdin Capital, Resolution Therapeutics, IndeeLabs, and Treefrog Therapeutics. J.E. has received research support from Cytovia Therapeutics, Mnemo Therapeutics, and Takeda Pharmaceutical Company. J.A.F. has received research support from Tmunity. C.H.J. and the University of Pennsylvania have patents pending or issued related to the use of gene modification in T cells for adoptive T cell therapy. C.H.J. is a co-founder of Tmunity. H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, and Orbital Therapeutics, and an advisor to 10x Genomics, Arsenal Biosciences, Chroma Medicine, Spring Discovery, and Vida Ventures. C.J.Y. is a co-founder of Survey Genomics, and a scientific advisory board member of Related Sciences and Immunai. C.J.Y. is a consultant for Maze Therapeutics, TReX Bio, ImYoo, and Santa Ana Bio. C.J.Y. has received research support from the Chan Zuckerberg Initiative, Chan Zuckerberg Biohub, Genentech, BioLegend, ScaleBio, and Illumina. J.A.D. is a co-founder of Editas Medicine, Intellia Therapeutics, Caribou Biosciences, Mammoth Biosciences, and Scribe Therapeutics, and a scientific advisory board member of Intellia Therapeutics, Caribou Biosciences, Mammoth Biosciences, Scribe Therapeutics, Vertex Pharmaceuticals, Felix Biosciences, The Column Group, Inari, and Isomorphic Labs. J.A.D. is the Chief Science Advisor at Sixth Street and a Director at Johnson & Johnson, Tempus, and Altos Labs. J.A.D. has sponsored research projects through Apple Tree Partners, Genentech, and Roche.
REFERENCES
- 1.Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, and Joung JK (2017). CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 14, 607–614. 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wienert B, Wyman SK, Richardson CD, Yeh CD, Akcakaya P, Porritt MJ, Morlock M, Vu JT, Kazane KR, Watry HL, et al. (2019). Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289. 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae S, Park J, and Kim J-S (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, and Joung JK (2016). High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017). Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550, 407–410. 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365. 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y, et al. (2020). Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med 26, 732–740. 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, Zhang L, Wei G, Tian Y, Zhao K, et al. (2022). Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature 609, 369–374. 10.1038/s41586-022-05140-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foy SP, Jacoby K, Bota DA, Hunter T, Pan Z, Stawiski E, Ma Y, Lu W, Peng S, Wang CL, et al. (2023). Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615, 687–696. 10.1038/s41586-022-05531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, Völkl S, Simon D, Kleyer A, Munoz L, et al. (2022). Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med 28, 2124–2132. 10.1038/s41591-022-02017-5. [DOI] [PubMed] [Google Scholar]
- 11.Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, Böltz S, Manger B, Mackensen A, and Schett G (2021). CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med 385, 567–569. 10.1056/NEJMc2107725. [DOI] [PubMed] [Google Scholar]
- 12.Nahmad AD, Reuveni E, Goldschmidt E, Tenne T, Liberman M, Horovitz-Fried M, Khosravi R, Kobo H, Reinstein E, Madi A, et al. (2022). Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol 40, 1807–1813. 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser ADM, Pouw N, Debets R, Kieback E, et al. (2010). Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med 16, 565–570. 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 14.Depil S, Duchateau P, Grupp SA, Mufti G, and Poirot L (2020). ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov 19, 185–199. 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 15.McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z, et al. (2019). MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626. 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata M, Nishimasu H, Kodera N, Hirano S, Ando T, Uchihashi T, and Nureki O (2017). Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun 8, 1430. 10.1038/s41467-017-01466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill AJ, McFaline-Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, and Trapnell C (2018). On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274. 10.1038/nmeth.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15. 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, and Sadelain M (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117. 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton JR, Tsuchida CA, Nguyen DN, Shy BR, McGarrigle ER, Sandoval Espinoza CRS, Carr D, Blaeschke F, Marson A, and Doudna JA (2021). Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep 35, 109207. 10.1016/j.celrep.2021.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou P, Chen S, Wang S, Yu X, Chen Y, Jiang M, Zhuang K, Ho W, Hou W, Huang J, et al. (2015). Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep 5, 15577. 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, et al. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol 37, 252–258. 10.1038/s41587-019-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, et al. (2019). Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med 25, 776–783. 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and b-thalassemia. N. Engl. J. Med 384, 252–260. 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 29.Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, et al. (2016). CRISPR/Cas9 b-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389. 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo S-J, Mitros T, Muñoz DP, Boffelli D, et al. (2016). Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med 8, 360ra134. 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med 385, 493–502. 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 32.Dabrowska M, Juzwa W, Krzyzosiak WJ, and Olejniczak M (2018). Precise excision of the CAG tract from the huntingtin gene by Cas9 nickases. Front. Neurosci 12, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odate S, Strapps W, and Lescarbeau RM (2023). Compositions and methods for treating alpha-1 antitrypsin deficiency https://patents.google.com/patent/WO2018119182A1/en.
- 34.Schiroli G, Conti A, Ferrari S, della Volpe L, Jacob A, Albano L, Beretta S, Calabria A, Vavassori V, Gasparini P, et al. (2019). Precise Gene Editing Preserves hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 24, 551–565.e8. 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S, Kim D, Cho SW, Kim J, and Kim J-S (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019. 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 7, 1765–1771. 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose JC, Stephany JJ, Valente WJ, Trevillian BM, Dang HV, Bielas JH, Maly DJ, and Fowler DM (2017). Rapidly inducible Cas9 and DSB-ddPCR to probe editing kinetics. Nat. Methods 14, 891–896. 10.1038/nmeth.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeh CD, Richardson CD, and Corn JE (2019). Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol 21, 1468– 1478. 10.1038/s41556-019-0425-z. [DOI] [PubMed] [Google Scholar]
- 39.Shy BR, Vykunta VS, Ha A, Talbot A, Roth TL, Nguyen DN, Pfeifer WG, Chen YY, Blaeschke F, Shifrut E, et al. (2022). High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol 41, 521–531. 10.1038/s41587-022-01418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen DN, Roth TL, Li PJ, Chen PA, Apathy R, Mamedov MR, Vo LT, Tobin VR, Goodman D, Shifrut E, et al. (2020). Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol 38, 44–49. 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med 5, 177ra38. 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, et al. (2017). Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med 9, eaaj2013. 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 43.Barnum KJ, and O’Connell MJ (2014). Cell cycle regulation by checkpoints. Methods Mol. Biol 1170, 29–40. 10.1007/978-1-4939-0888-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe M, Moon KD, Vacchio MS, Hathcock KS, and Hodes RJ (2014). Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 40, 681–691. 10.1016/j.immuni.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, and Zhang P (2010). The ATM–p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA 107, 14188–14193. 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svoboda J, Gerson JN, Landsburg DJ, Chong EA, Barta SK, Dwivedy Nasta S, Ruella M, Hexner EO, Marshall A, Leskowitz R, et al. (2022). Interleukin-18 secreting autologous anti-CD19 CAR T-cells (huCART19-IL18) in patients with non-Hodgkin lymphomas relapsed or refractory to prior CAR T-cell therapy. Blood 140, 4612–4614. 10.1182/blood-2022-162393. [DOI] [Google Scholar]
- 47.Gómez-González B, and Aguilera A (2019). Transcription-mediated replication hindrance: a major driver of genome instability. Genes Dev 33, 1008–1026. 10.1101/gad.324517.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, Teichmann M, Rousseau E, Lamrissi-Garcia I, Guyonnet-Duperat V, et al. (2019). CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun 10, 1136. 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149. 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, Beard MR, Hughes J, Pomerantz RT, and Thomas PQ (2018). Large deletions induced by Cas9 cleavage. Nature 560, E8–E9. 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 51.Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol 36, 765–771. 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alanis-Lobato G, Zohren J, McCarthy A, Fogarty NME, Kubikova N, Hardman E, Greco M, Wells D, Turner JMA, and Niakan KK (2021). Frequent loss of heterozygosity in CRISPR-Cas9–edited early human embryos. Proc. Natl. Acad. Sci. USA 118, e2004832117. 10.1073/pnas.2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, Zhang CZ, Weiss MJ, and Pellman D (2021). Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet 53, 895–905. 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bothmer A, Gareau KW, Abdulkerim HS, Buquicchio F, Cohen L, Viswanathan R, Zuris JA, Marco E, Fernandez CA, Myer VE, et al. (2020). Detection and modulation of DNA translocations during multi-gene genome editing in T cells. CRISPR J 3, 177–187. 10.1089/crispr.2019.0074. [DOI] [PubMed] [Google Scholar]
- 55.Xin C, Yin J, Yuan S, Ou L, Liu M, Zhang W, and Hu J (2022). Comprehensive assessment of miniature CRISPR-Cas12f nucleases for gene disruption. Nat. Commun 13, 5623. 10.1038/s41467-022-33346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yin J, Lu R, Xin C, Wang Y, Ling X, Li D, Zhang W, Liu M, Xie W, Kong L, et al. (2022). Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing. Nat. Commun 13, 1204. 10.1038/s41467-022-28900-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webber BR, Lonetree CL, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, Lahr WS, Draper GM, Slipek NJ, Smeester BA, et al. (2019). Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun 10, 5222. 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porto EM, Komor AC, Slaymaker IM, and Yeo GW (2020). Base editing: advances and therapeutic opportunities. Nat. Rev. Drug Discov 19, 839–859. 10.1038/s41573-020-0084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura M, Gao Y, Dominguez AA, and Qi LS (2021). CRISPR technologies for precise epigenome editing. Nat. Cell Biol 23, 11–22. 10.1038/s41556-020-00620-7. [DOI] [PubMed] [Google Scholar]
- 60.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, et al. (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanehisa M, and Goto S (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fang Z, Liu X, and Peltz G (2023). GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757. 10.1093/bioinformatics/btac757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim MC, Gate R, Lee DS, Lu A, Gordon E, Shifrut E, Marson A, Ntranos V, and Ye CJ (2022). Memento: generalized differential expression analysis of single-cell RNA-seq with method of moments estimation and efficient resampling 10.1101/2022.11.09.515836. [DOI]
- 66.Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. (2016). ENCODE data at the ENCODE portal. Nucleic Acids Res 44, D726–D732. 10.1093/nar/gkv1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq data original to this study is available at the Gene Expression Omnibus (GEO) under the accession numbers GSE241838, GSE241839, and GSE241882. Clinical scRNA-seq data (NCT03399448) was accessed and is available through the NIH database of Genotypes and Phenotypes (dbGaP) under the accession number PHS001707.v2.
All original code used in this study has been deposited online and is publicly available on GitHub at www.github.com/yelabucsf/Cas9_chromosome_loss_scRNA-seq.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.