

Abstract
The dramatic increase in intracranial pressure after subarachnoid hemorrhage leads to a decrease in cerebral perfusion pressure and a reduction in cerebral blood flow. Mitochondria are directly affected by direct factors such as ischemia, hypoxia, excitotoxicity, and toxicity of free hemoglobin and its degradation products, which trigger mitochondrial dysfunction. Dysfunctional mitochondria release large amounts of reactive oxygen species, inflammatory mediators, and apoptotic proteins that activate apoptotic pathways, further damaging cells. In response to this array of damage, cells have adopted multiple mitochondrial quality control mechanisms through evolution, including mitochondrial protein quality control, mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer, to maintain mitochondrial homeostasis under pathological conditions. Specific interventions targeting mitochondrial quality control mechanisms have emerged as promising therapeutic strategies for subarachnoid hemorrhage. This review provides an overview of recent research advances in mitochondrial pathophysiological processes after subarachnoid hemorrhage, particularly mitochondrial quality control mechanisms. It also presents potential therapeutic strategies to target mitochondrial quality control in subarachnoid hemorrhage.
Keywords: mitochondrial biogenesis, mitochondrial dynamics, mitochondrial dysfunction, mitochondrial fission and fusion, mitochondrial quality control, mitophagy, subarachnoid hemorrhage
Introduction
Subarachnoid hemorrhage (SAH) is an acute hemorrhagic stroke; about 80% of SAH cases are caused by ruptured intracranial aneurysms, while the remaining mainly come from vascular malformations and vasculitis (Andersen et al., 2019). According to the latest epidemiological data, the global incidence of SAH is 7.9 per 100,000 person-years (Etminan et al., 2019), accounting for 3.1% of all strokes, and ranks first among stroke patients under 18 years of age, making it a critical health threat to the population (Wang et al., 2022b).
Current treatments (i.e., craniotomy or endovascular treatments) target the structural abnormalities of the brain vessels that cause SAH to prevent rebleeding; however, there is no effective treatment for brain injury caused by hemorrhage. Early brain injury (EBI) followed by SAH is mediated by the effects of acute cerebral ischemia during bleeding, along with the effects of hemoglobin in subarachnoid. Increased intracranial pressure, intracerebral hemorrhage-induced brain tissue destruction, brain shift, and herniation are secondary effects that all contribute to the pathology (Rass and Helbok, 2019; Fragata et al., 2020; Xu et al., 2022). Many patients survive after these events but, in approximately one-third of cases, the state of patients deteriorates days later from delayed cerebral ischemia (DCI) (Rass and Helbok, 2021). According to current theories, vasospasm, arteriolar constriction, thrombosis, and EBI-induced processes all contribute to DCI (Macdonald, 2014).
As the brain is the most energy-intensive organ, neurons strongly depend on mitochondria to provide the energy for maintaining their normal function (Dienel, 2019; Du et al., 2021). After SAH, direct damage from ischemia, hypoxia, and the toxicity of hemoglobin (Hb) and its degradation products lead to the collapse of the mitochondrial membrane potential (ΔΨm). The subsequent excessive production of reactive oxygen species (ROS), the release of apoptotic proteins, and the activation of mitochondria-associated inflammation further damage the cells (Forbes and Thorburn, 2018; Pinti et al., 2019). Mitochondrial dysfunction is thought to play an important mediating role in these pathophysiological processes. In response to this series of assaults, eukaryotic cells have adopted multiple mitochondrial quality control (MtQC) mechanisms throughout evolution, including protein quality control, mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer to maintain mitochondrial homeostasis under stress conditions (Pickles et al., 2018). Dysfunction of these regulatory mechanisms can damage mitochondria, leading to cell death or neurological dysfunction (Picca et al., 2018; Tang et al., 2021; Eldeeb et al., 2022).
Motivated by the critical role of mitochondrial integrity and the MtQC system in SAH, we summarize here the latest research on the pathophysiological mechanisms of SAH involving mitochondria, particularly MtQC. Additionally, we evaluate potential therapeutic strategies for SAH targeting mitochondria.
Search Strategy
We gathered studies on SAH, mitochondrial dysfunction, and MtQC published from January 2001 to December 2022 from the PubMed database using the following search terms: “mitochondrial quality control” AND “SAH;” “mitochondrial dynamics” AND “SAH;” “mitophagy” AND “SAH;” “mitochondrial biogenesis” AND “SAH;” “intercellular mitochondrial transfer” AND “SAH,”. Based on the search results, we expanded the search by adding key molecules and other terms as search terms. Finally, we selected relevant articles based on their title and abstract and ended up with 132 included papers.
Direct Factors Causing Mitochondrial Dysfunction
After an SAH event, blood enters the subarachnoid space, dramatically increasing intracranial pressure and decreasing cerebral perfusion pressure and cerebral blood flow. Next, vasospasm exacerbates global cerebral ischemia and hypoxia; besides, the blood components and their degradation products are neurotoxic (Osgood, 2021). In this process, many factors directly or indirectly damage mitochondria; here, we focus on the factors directly damaging mitochondria (Figure 1).
Figure 1.
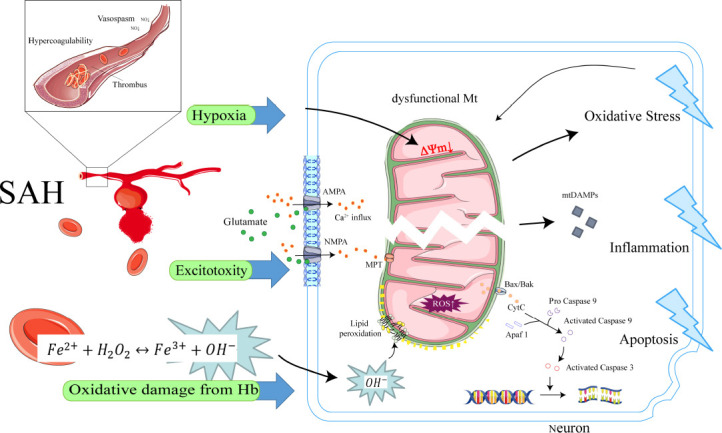
Initiation and amplification of mitochondrial dysfunction in SAH.
After SAH, mitochondria are directly affected by ischemia and hypoxia, excitotoxicity, and toxicity of hemoglobin and its degradation products, which trigger mitochondrial dysfunction and lead to the collapse of ΔΨm. Subsequently, mitochondrial dysfunction leads to the massive production of mtROS, release of mtDAMP, and activation of the mitochondrial apoptotic pathway. Simultaneous excitotoxicity increases the Ca2+ inward flow, triggering calcium-dependent MPT, which further increases ROS production and apoptotic protein release. This process ultimately leads to oxidative stress, inflammation, and apoptosis, amplifying cellular damage. Created using used Adobe Illustrator 2021, with material from Servier Medical Art. AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; Apaf1: apoptotic protease-activating factor 1; Bax/Bak: BCL2-associated X/BCL2 antagonist/killer 1; CytC: cytochrome c; Hb: hemoglobin; MPT: mitochondrial permeability transition; mtDAMP: mitochondrial damage-associated molecular patterns; mtROS: mitochondrial reactive oxygen species; NMPA: N-methyl-D-aspartate; ROS: reactive oxygen species; SAH: subarachnoid hemorrhage; ΔΨm: mitochondrial membrane potential.
Hypoxia
In the initial stage after SAH, small artery spasms and microperfusion to the brain are significantly reduced (Lenz et al., 2021). Uhl et al. (2003) found that single or bead-like multisegmental “microvasospasm” occurred in small arteries during aneurysm clamping procedures, with a decrease in the diameter of affected small arteries of up to 75.1% (mean 30.1%). Subsequent microcirculation disturbance can induce thrombophilia, intracapillary platelet aggregation, and leukocyte embolism formation, exacerbating brain tissue ischemia (Clarke et al., 2020). Animal studies have reported the prevalence of microthrombi in the cerebral vasculature after SAH, which is usually observed in areas of small arterial stenosis. In addition, a decrease in nitric oxide (an endogenous vasodilator substance) exacerbates this process (Tewari et al., 2021). Hypoxia due to microcirculation disturbance aggravates the pathophysiological changes after SAH. For the vast majority of patients with SAH, cranial imaging reveals no ischemic foci early in the course of their disease. Even for the poorly reported SAH with graded severity, only about 21% of patients will suffer from cerebral infarction (Taran et al., 2020). However, a metabolomics study found that 2-hydroxyglutaric acid, a biomarker of tissue hypoxia, can be used to predict long-term outcomes in patients with SAH (Lu et al., 2018). Brain microdialysis and multimodal neuromonitoring studies have shown that more than 60% of patients with SAH suffer from brain tissue hypoxia (partial pressure of brain tissue oxygen < 20 mmHg) within 24 hours of onset (Helbok et al., 2015).
Within minutes, mitochondrial dysfunction induced by neuronal hypoxia and glucose deficiency leads to a reduction of adenosine triphosphate (ATP) production and excessive production of ROS. Neurons have a larger energy consumption than other brain cells, but their energy reserves are limited. ATP depletion is one of the main initiators of ischemic cascade reactions, such as ΔΨm collapse, cellular potassium efflux, membrane depolarization, and sodium, chloride, and water inward flow (Lee et al., 2000; Hofmeijer and van Putten, 2012; Dharmasaroja, 2016).
Excitotoxicity
Excitotoxicity plays an important role in both EBI and DCI after SAH. In EBI, aneurysm rupture increases intracranial pressure, and the subsequent whole-brain hypoperfusion causes energy depletion, metabolism failure, and ion homeostasis disturbance, leading to the depolarization of the plasma membrane, which causes the excessive and uncontrolled release of neurotransmitters such as glutamate (Kanamaru and Suzuki, 2019). Experimental evidence suggests that neurons release glutamate when the cerebral blood flow is below 20 mL/100 g brain/min (Mindt et al., 2020). In DCI, cerebral ischemia also triggers excessive glutamate release. The massive release of glutamate overactivates α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, N-methyl-D-aspartate, and kainate receptors on neuron membranes, together with other cellular components of the neurovascular unit (Choi, 2020).
The overactivation of glutamate receptors mediates intracellular Ca2+ overload through two mechanisms: ionotropic glutamate receptors overactivation increases Ca2+ inward flow from the extracellular space, while metabotropic glutamate receptors overactivation increases intracellular Ca2+ release from the endoplasmic reticulum (ER) (Zhang et al., 2018b). Subsequently, excitotoxicity or cortical spreading depolarization causes mitochondrial dysfunction, impaired energy metabolism, and ionic imbalance; this mismatch of metabolic supply and demand results in relative cerebral ischemia (van Lieshout et al., 2018). Continued cortical spreading depolarizations are accompanied by a diffuse inhibition of cortical electrical activity and an increase in glutamate release, the latter leading to excitotoxicity (Suzuki et al., 2021). This process results in a positive feedback loop (Ehlert et al., 2020).
Oxidative damage from hemoglobin
Hb and its metabolites trigger a toxic cascade in EBI and DCI after SAH (Akeret et al., 2021). Hb is the protein transporting oxygen. It consists of four chains, two alpha chains and two beta chains, each with a heme containing one iron atom. Oxyhemoglobin and its metabolites are thought to be the main source of ROS during SAH (Zeineddine et al., 2022). After SAH erythrocytes enter the subarachnoid space and hemolysis occurs, erythrocytes release tetrameric Hb, which gradually degrades into toxic compounds (Stokum et al., 2021). The degradation product of Hb, heme further releases ferrous iron (Fe2+). Fe2+ can react with hydrogen peroxide in a Fenton reaction to produce more toxic hydroxyl radicals that may attack mitochondria. The massive generation of free radicals damages phospholipid bilayers such as the mitochondrial membrane, leading to phospholipid hydrogen peroxide production and, eventually, ferroptosis (Otasevic et al., 2021). In conclusion, Hb-derived ROS disrupt the structure and function of mitochondria, causing further propagation of intracellular oxidative damage.
Mitochondrial Dysfunction Amplifies Damage after Subarachnoid Hemorrhage
A series of initial damage factors after SAH contribute to the onset of mitochondrial dysfunction. Dysfunctional mitochondria produce large amounts of ROS, release inflammatory mediators, and release apoptotic proteins that activate apoptotic pathways, causing further cellular damage (Figure 1).
Mitochondria and oxidative stress
As previously mentioned, after SAH, cellular ischemia and hypoxia lead to mitochondrial respiratory dysfunction (Zhang et al., 2022b). Mitochondria damaged by Hb-associated ROS cause further intracellular ROS-induced damage. The disruption of mitochondrial respiration may well be the main cause of intracellular ROS production. A potentially deleterious effect of ROS is the promotion of Ca2+-dependent mitochondrial permeability transition (MPT) in mitochondrial membranes. MPT facilitates the passing of Ca2+ through the membrane. The uptake of Ca2+ by the MPT pores alters the morphological and functional activity of mitochondria, amplifying ROS production (Bonora et al., 2022). The sudden and massive production of ROS far exceeds the scavenging capacity of the antioxidant defense system, leading to oxidative stress in the cells.
Massive amounts of intracellular ROS extensively oxidate cellular lipids, proteins, and DNA. Besides, excess ROS can attack mitochondria, disrupting their important functions, such as the maintenance of ΔΨm and oxidative phosphorylation (Ehlert et al., 2020).
Mitochondria-related inflammation
Mitochondria do play an important role in the control of inflammation after SAH. Together, the two mitochondrial membranes—the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM)—provide a double barrier separating mitochondrial damage-associated molecular patterns (DAMPs) from their cognate pattern recognition receptors (Marchi et al., 2023). Under pathological situations, such as cell injury or stress, mitochondrial components, such as ATP, mitochondrial DNA (mtDNA), formyl peptides, and cardiolipin, can be released into the cytoplasmic or extracellular environment, where they act as DAMPs and trigger downstream inflammation (Meyer et al., 2018).
A clinical study found that cerebrospinal fluid (CSF) mtDNA levels on admission were associated with the outcome of aneurysmal SAH (Wang et al., 2013). Also, serum mtDNA may directly or indirectly influence complications and clinical outcomes after SAH, such as pneumonia, hydrocephalus, and epilepsy (Chaudhry et al., 2019). Another study found that plasma-free mtDNA and Toll-like receptor-9/mitogen-activated protein kinase expression were simultaneously increased in SAH rats, suggesting that mtDNA induces systemic and local inflammatory responses in the brain (Liu et al., 2017).
Mitochondria-dependent apoptosis
After SAH, dysfunctional mitochondria produce large amounts of ROS, causing oxidative stress, which can trigger mitochondria-dependent apoptosis through the release of pro-apoptotic proteins, such as cytochrome c (Cyt C) or apoptosis-inducing factors (Gao et al., 2022c).
The mitochondria-dependent apoptosis pathway is an important aspect of the intrinsic apoptotic pathway. The increased permeability of the mitochondrial membrane results in an elevation of Ca2+ flow, triggering calcium-dependent MPT through various factors, allowing apoptotic proteins, such as P53, to flow into the cytoplasm, releasing Cyt C, and activating caspase-9 and -3, leading to cell death (Su et al., 2019; Bock and Tait, 2020). Singh et al. (2019) have proved that when Bax and Bak are activated and accumulate at the OMM, they oligomerize and mediate OMM permeation, resulting in the release of pro-apoptotic factors, such as Cyt C. Cyt C can interact with apoptotic protease activator-1 to form apoptotic bodies and activate caspase-9. Caspase-9 can cleave and activate downstream effector caspases, such as caspase-3 and -7, which cleave various protein substrates, eventually causing cell death (Tian et al., 2023). After SAH, patients have high cleaved caspase-3 levels in the hippocampus and cerebral cortex (He et al., 2018). An earlier study reported the presence of Cyt C in the subarachnoid hemolysis product, which was associated with subsequent neuronal cell death (Matz et al., 2001).
Mitochondrial dysfunction commonly leads to neuronal apoptosis in patients with SAH, and it is considered an important therapeutic target for EBI after SAH (Zhang et al., 2022b). Many animal experiments have shown that, after SAH-associated whole-brain ischemia, apoptosis occurs in most areas of the brain, including the hippocampus and basal cortex (Fan et al., 2021). Puerarin can attenuate neurological dysfunction via the Bcl-2/Bax/cleaved caspase-3 pathway in SAH mice (Zhang et al., 2019c). SS31 treatment increased mitochondrial Cyt C levels and increased Bax levels, suggesting that regulating the mitochondrial apoptosis pathway can alleviate EBI after SAH (Shen et al., 2020).
Mitochondrial Quality Control and Subarachnoid Hemorrhage
MtQC systems
Neuronal survival critically depends on mitochondrial integrity and function. A hierarchical system of cellular monitoring mechanisms protects mitochondria from stress and damage. MtQC mechanism regulates protein and organelle levels (Roca-Portoles and Tait, 2021). MtQC relies on mitochondrial protein hydrolases and chaperone proteins to maintain mitochondrial protein homeostasis. These proteins prevent the accumulation of toxic folding intermediates or aggregates, which are detrimental to the cell, by facilitating correct protein folding. Regarding organelle levels, MtQC involves mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer. Maintaining mitochondrial morphology and function and clearing damaged mitochondria are important processes to prevent mitochondrial dysfunction and cell death. Therefore, the MtQC system plays a crucial role in maintaining intracellular homeostasis and function (Pickles et al., 2018). Here we will focus on MtQC at the organelle level (Figure 2).
Figure 2.
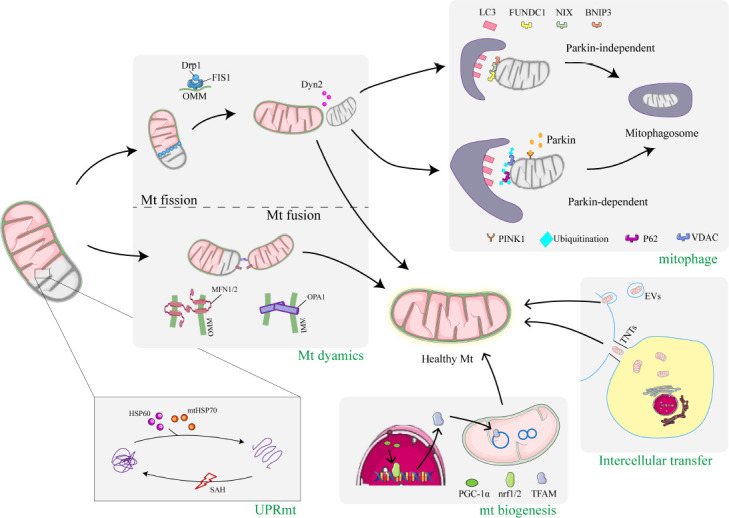
Mitochondrial quality control in SAH.
This schematic represents the quality control mechanisms of mitochondria after SAH, including UPRmt, mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer. First, at the protein level, the MtQC process called UPRmt involves HSP60 and mtHSP70 entering the mitochondria and restoring the normal conformation of misfolded proteins. Second, at the organelle level, dysfunctional mitochondria can undergo mitochondrial outer and inner membrane fusion via MFN1, MFN2, or OPA1. Mitochondrial fusion prevents the accumulation of damaged contents in individual mitochondria through the exchange of mtDNA, proteins, and metabolites between healthy and damaged mitochondria. Dysfunctional and damaged mitochondria can also be separated by fission. Drp1 translocates from the cytoplasm to the outer mitochondrial membrane and mediates fission through Dyn2 to form two unequal mitochondria. The damaged mitochondria produced by fission are then degraded by mitophagy via the PINK1/Parkin-dependent or PINK1/Parkin-independent pathway. Various stimuli lead to the activation of PGC-1α, which then binds to Nrf1/2 to promote the expression of several mitochondrial proteins, including TFAM, ultimately leading to increased mitochondrial biogenesis. Mitochondria are transported between cells by actin-based TNT or EVs released from healthy neurons (or donor cells) that are then internalized into injured neurons. Mitochondrial transfer rescues cells containing damaged mitochondria by translocating healthy mitochondria from neighboring cells. Created using used Adobe Illustrator 2021, with material from Servier Medical Art. BNIP3: Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3; Drp1: dynamin-related protein 1; Dyn2: dynamin 2; EVs: extracellular vesicles; FIS1: fission 1; FUNDC1: FUN14 domain containing 1; HSP60: heat shock protein 60; IMM: inner mitochondrial membrane; LC3: light chain 3; MFN1/2: mitofusin 1/2; mtHSP70: mitochondrial heat shock protein 70; NIX: Nip3-like protein X; Nrf1/2: nuclear factor E2-related factor1/2; OMM: outer mitochondrial membrane; OPA1: optic atrophy 1; P62: sequestosome-1; PGC-1α: AMPK-proliferator-activated receptor cofactor 1; PINK1: phosphatase-and-tensin-homolog-induced putative kinase 1; SAH: subarachnoid hemorrhage; TFAM: mitochondrial transcription factor A; TNTs: tunneling nanotubes; VDAC: voltage-dependent anion channel.
Mitochondrial unfolded protein response
The mitochondrial unfolded protein response (UPRmt) is an important protein homeostasis regulatory system, and a protein-level MtQC mechanism. UPRmt can occur in mitochondria at an early stage of oxidative stress and is a cytoprotective mechanism (Shpilka and Haynes, 2018). In eukaryotes, polypeptides are synthesized by ribosomes and need to be folded correctly to properly perform their respective physiological functions. When cells are stimulated by external factors such as oxidative stress, the polypeptides are misfolded and further degraded, and the undegraded polypeptides form cytotoxic aggregates that can contribute to the unfolded protein response. The unfolded protein response can occur in the ER and mitochondria. Mitochondria are not only the energy factories of cells, but also the main organelle where oxidative stress occurs; besides, misfolded proteins accumulate in mitochondria when oxidative stress occurs. However, UPRmt is an important regulator for mitochondria to maintain the homeostasis of their internal protein network, which plays an important role in the refolding and clearance of misfolded proteins (Shpilka and Haynes, 2018).
When oxidative stress occurs, mitochondrial protein homeostasis is disrupted, and many unfolded or misfolded proteins accumulate in the mitochondrial matrix. The excessive accumulation of unfolded proteins lead to the formation of mitochondrial protein aggregates, which can help maintain mitochondrial homeostasis by promoting the degradation of misfolded proteins (Lin et al., 2018). The caseinolytic mitochondrial matrix peptidase proteolytic subunit proteolytic complex recognizes and binds misfolded or unassembled proteins to degrade them into polypeptides. Subsequently, these peptides are translocated to the cytoplasm. Meanwhile, the expression of various mitochondrial molecular chaperones, such as heat shock protein 60 (HSP60) and mitochondrial heat shock protein 70 (mtHSP70), is upregulated; these proteins then enter the mitochondria to help restore the normal conformation of the misfolded proteins (Kumar et al., 2022; Song et al., 2022). Therefore, promoting the degradation and refolding of mitochondrial protein aggregates may be a viable strategy to inhibit the onset of EBI after unfolded protein response.
A recent study by Ma et al. (2022) showed that the levels of HSP60 and mtHSP70 in the overall brain tissue did not change significantly at 24 hours after SAH. Meanwhile, their levels in brain mitochondria started to increase at 6 hours. This result illustrates the activation of UPRmt, which facilitates the transport of HSP60 into the mitochondria due to the onset of oxidative stress after SAH. Besides, they found that GrpE-like 1 (GrpEL1) expression was unchanged and the levels of GrpEL1-mtHSP70 complex were decreased after SAH. Overexpressing GrpEL1 promoted the binding of GrpEL1 and mtHSP70, forming the GrpEL1-mtHSP70 complex and promoting neuronal mitochondrial homeostasis. Thus, increasing GrpEL1 to promote binding of GrpEL1 and mtHSP70 might be an important clinical intervention target for EBI after SAH (Ma et al., 2022).
Mitochondrial dynamics
A large amount of evidence suggests that mitochondria are not static, discrete organelles. Instead, mitochondria form a highly dynamic network of organelles that can remodel themselves in order to maintain mitochondrial mass (Lai et al., 2020; Chen et al., 2022; Wu et al., 2022). Mitochondrial dynamics, referring to mitochondrial fusion and fission, are important adaptive responses to metabolic or environmental stresses and key defense mechanisms that protect the health of mitochondrial populations, especially when the damage exceeds the repair capacity of the protein-level quality control mechanisms. Mitochondrial fusion promotes the exchange of material between mitochondria. The reversible fusion of damaged and healthy mitochondria restores damaged mitochondrial function by diluting accumulated mutant mtDNA and oxidized proteins. Meanwhile, mitochondrial fission is required to separate mitochondria that are about to enter daughter cells during cell proliferation and to isolate the damaged fraction of dysfunctional mitochondria for mitophagy. Mitochondrial fission is more prone to promoting apoptosis in cells under severe stress than mitochondrial fusion is (Chan, 2020).
Mitochondrial fusion and fission are controlled by members of the conserved GTPase family. The central protein mediating mitochondrial fission is dynamin-related protein 1 (Drp1), a guanosine triphosphate (GTP) hydrolase (Rosdah et al., 2020) predominantly distributed in the cytoplasm. It responds to pathological stimuli through post-translational modifications (mainly phosphorylation, dephosphorylation, ubiquitination, and sumoylation) (Tsushima et al., 2018; Ma et al., 2020; Gao et al., 2022b). Generally, the phosphorylation of Ser616 (S616) accelerates the recruitment of Drp1 to the mitochondrial membrane, whereas the phosphorylation of S637 inhibits it (Chen et al., 2020a; Miao et al., 2022). Then, Drp1 is recruited to the OMM with the help of its receptors mitochondrial fission protein 1, mitochondrial fission factor, and mitochondrial dynamics protein of 49 and/or 51 kDa (Wu et al., 2021a). Subsequently, Drp1 oligomerizes and forms a ring around the mitochondria using the energy generated by GTP hydrolysis to contract the organelle (Kalia et al., 2018). Finally, the OMM is cleaved after the recruitment of dynamin 2 (Kamerkar et al., 2018). In addition, under stress conditions, such as oxidative stress and hypoxia, Drp1 is upregulated and translocated, causing an imbalance in mitochondrial division and fusion that damages the mitochondrial network and concomitantly induces apoptosis (Wu et al., 2011).
Mitochondrial fission is the initial event of SAH, and Drp1-mediated oxidative mitochondrial damage is involved in the pathological process of acute neurological injury due to SAH. The dysregulation of mitochondrial dynamics after SAH shifts the balance of mitochondrial division and fusion toward division, leading to neuronal injury (Wu et al., 2017). Wu et al. (2017) observed an increase in Drp1 levels in mice after SAH, whereas the experiments of Fan et al. (2017) demonstrated that Drp1 levels were significantly increased in mitochondria and decreased in the cytoplasm, suggesting that SAH triggered Drp1 translocation to the mitochondria. Wu et al. (2017) also found that the Drp1 inhibitor mitochondrial division inhibitor 1 (Mdivi-1) significantly improved neurological dysfunction, reduced cerebral edema and blood-brain barrier permeability, and attenuated apoptosis. Besides, Mdivi-1 reduced Drp1 levels and S616 phosphorylation. Moreover, it inhibited excessive mitochondrial fission and restored the ultrastructure of mitochondria (Wu et al., 2017). In addition, Mdivi-1 inhibited the nuclear translocation of nuclear factor kappa B, reducing the expression of tumor necrosis factor-α, interleukin-6, and interleukin-1β, decreasing the levels of matrix metalloproteinase 9, and preventing the degradation of tight junction proteins (occludin, claudin-5, and zona occludens 1). Finally, it decreased cleaved caspase-3 and Bax levels and increased Bcl-2 levels. Thus, the neuroprotective effect of Mdivi-1 may be mediated by the suppression of inflammation-associated blood-brain barrier disruption (Fan et al., 2017).
Drp1 regulates mitochondrial fission after SAH through multiple endogenous factors. Among them, the adenosine monophosphate-activated protein kinase (AMPK) pathway might play an essential role in the regulation of Drp1, and several studies have focused on this pathway. HSP22 downregulates Drp1 expression and its activation of mitochondrial apoptosis in an AMPK-proliferator-activated receptor cofactor 1 (PGC-1α)-dependent manner (Fan et al., 2021). In another study, the activation of the melanocortin 1 receptor with BMS-470539 significantly attenuated EBI after SAH by inhibiting oxidative stress, apoptosis, and mitochondrial fission via the AMPK/Sirtuin 1/PGC-1α signaling pathway (Xu et al., 2021). Additionally, metformin, a widely used drug, also modulates Drp1. Metformin restored the homeostasis between mitochondrial fusion and fission by downregulating Drp1 in an AMPK-dependent manner and attenuated EBI after SAH in rats; it also promoted mitochondrial autophagy (Zhang et al., 2022a). In addition, Drp1 is affected by various post-transcriptional modifications. After SAH, excess nitric oxide is converted to S-nitrosoglutathione and stored in cells, increasing the S-nitrosylation of neuronal intracellular proteins and causing nitrosative stress. S-nitrosoglutathione reductase promotes S-nitrosoglutathione degradation and attenuates Drp1 S-nitrosylation, reducing mitochondrial division and neuronal apoptosis, thus exerting a neuroprotective effect (Wang et al., 2022a). Studies by our group have demonstrated that Drp1 and Drp1-mediated mitochondrial fission also play an important role in the activation of microglia after SAH. The activation of P2X purinoceptor 7 leads to the dephosphorylation of Drp1 at S637, increasing mitochondrial fission and reducing mitochondrial function, which may cause the reduction in microglia phagocytosis. The drug-induced inhibition of P2X purinoceptor 7 activation in mice rescued mitochondrial function and improved phagocytosis after SAH (Tao et al., 2022).
Various drugs have been used to target mitochondrial fission. Rapamycin targeted inhibition of the mechanistic target of rapamycin alleviated excessive mitochondrial fission by downregulating Drp1, restoring mitochondrial function, thus reducing apoptosis and having a neuroprotective effect after SAH (Li et al., 2020). Docosahexaenoic acid ameliorates mitochondrial dysfunction in vitro and in vivo and prevents oxidative stress-based apoptosis after SAH by upregulating the mitochondrial fusion-associated protein optic atrophy 1 (OPA1), downregulating Drp1, and lowering its phosphorylation at S616 (Zhang et al., 2018a). The main bioactive component of tea catechins, (-)-epigallocatechin-3-gallate, alleviates SAH-induced mitochondrial kinetic damage by downregulating the expression of Drp1 and fission protein 1 and upregulating OPA1, mitofusin 1 (MFN1), and mitofusin 2 (MFN2) (Chen et al., 2018). T0901317 activates liver X receptors and attenuates neuronal apoptosis via the liver X receptors/interferon regulatory factor-1/P53 upregulated apoptosis regulator/Drp1 pathway in SAH rats (Dai et al., 2021).
Mitochondrial fusion is regulated by GTPase superfamily members and involves two aspects: the fusion of mitochondrial membranes and the remixing of mitochondrial contents. Mitochondrial fusion includes OMM fusion—mediated by MFN1 and MFN2—and IMM fusion—mediated by OPA1. After fusion initiation, MFNs in adjacent mitochondrial OMMs interact to tether the organelles. GTP hydrolysis causes conformational changes in MFNs, facilitating the docking of OMMs and increasing the contact surface area. Subsequent MFN oligomerization ensures OMM fusion. After OMM fusion, the interaction between OPA1 and cardiolipin tethers the IMM for fusion (Gao and Hu, 2021). OPA1 has five different protein isoforms: two long isoforms (L-OPA1) that mediate intramembrane fusions and three short fusion inactive isoforms (S-OPA1). This pattern is due to the cleavage at the S1 and S2 sites of OPA1, releasing S-OPA1 into the membrane gap (Guillery et al., 2008). Matrix-AAA protease isoenzymes and OMA1, two kinds of mitochondrial metallopeptidase, are major mitochondrial factors that sense and respond to cellular stress (Guo et al., 2020). When ΔΨm is intact, the L-OPA1 heterodimer induces IMM fusion. OMA1 responds to stress by cleaving L-OPA1 into S-OPA1 heterodimers, which lose fusion activity, leading to the collapse of the mitochondrial network into a fragmented population of organelles (Gilkerson et al., 2021).
Several studies have investigated mitochondrial fusion in SAH. An animal study showed that MFN1 and MFN2 levels in mouse brain tissue decreased significantly 24 hours after SAH, and that the Sirtuin3 agonist honokiol increased the expression of the mitochondrial fusion proteins MFN1 and MFN2, thereby maintaining mitochondrial morphological integrity and promoting neuron survival. Furthermore, the selective inhibition of AMPK eliminated these protective effects, suggesting that they are AMPK-dependent (Wu et al., 2020). Another animal experiment showed that SAH rats had significantly lower L-OPA1 expression levels than control rats 24 hours after SAH, and these levels continued to decrease over 72 hours. SAH rats also had slightly lower S-OPA1 expression levels, but this result was not significant.
In addition, mitoquinone treatment after SAH in rats improved mitochondrial morphology and protected the blood-brain barrier via the nuclear factor E2-related factor 2 (Nrf2)/prohibitin 2/OPA1 pathway (Zhang et al., 2019b).
Mitophagy
Mitophagy is the selective degradation of excess and defective mitochondria through autophagy. Under physiological conditions, mitophagy eliminates unwanted mitochondria during development and adjusts the number of mitochondria in response to changes in metabolic demand (Killackey et al., 2020). Under stress conditions, mitophagy, as a key mechanism of the MtQC, identifies and labels severely damaged mitochondria for rapid elimination (Pickles et al., 2018).
Mitophagy requires efficient recognition and engulfment of target mitochondria in the mitophagosome. In mammalian cells, two signaling pathways are associated with mitophagy regulatory mechanisms: the phosphatase-and-tensin-homolog-induced putative kinase 1 (PINK1)/parkin-dependent pathway (Barazzuol et al., 2020) and PINK1/parkin-independent pathway (Di Rita et al., 2018).
Various stress conditions, such as calcium overload and ΔΨm collapse, can activate the Parkin-dependent mitotic pathway (Rimessi et al., 2013; Georgakopoulos et al., 2017). Active PINK1 initiates PINK1/parkin-dependent mitophagy by phosphorylating ubiquitin at the Ser 65 position and recruiting cytoplasmic parkin to the impaired mitochondria (Yang et al., 2018). Parkin is an E3 ubiquitin ligase that ubiquitinates OMM substrates and linker proteins, such as sequestosome-1 (p62), 52 kDa nuclear dot protein, and optineurin (Kane et al., 2014). Meanwhile, the light chain 3 (LC3) precursor molecule is cleaved by autophagy-associated protein (Atg) into LC3-I and further processed into LC3-II. The ubiquitinated adaptor protein connects to the microtubule-associated protein LC3-II and binds to the ER membrane to form a mitophagosome (Wesch et al., 2020). Thus, mitochondria are enclosed by the ER isolation membrane and fuse with lysosomes during degradation (Towers et al., 2021).
The mitochondrial receptor Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3), Nip3-like protein X (NIX), and FUN14-domain-containing protein 1 are the most significant factors in parkin-independent mitophagy. Hypoxic stress conditions elevate Bnip3 and NIX levels, promoting mitophagy (Wu et al., 2021b). BNIP3 and NIX are located in the OMM and interact with Atg8 and LC3 through its N-terminal region, thereby promoting autophagy. FUN14-domain-containing protein 1, another OMM protein, contains a typical LC3 interaction region that binds to Atg8 or LC3II to induce mitophagy.
Increasing evidence suggests that mitophagy participates in the pathogenesis of SAH. After SAH, mitophagy levels rise and levels of marker proteins associated with the PINK1/Parkin-dependent and PINK1/Parkin-independent pathways are elevated (e.g., PINK1, Parkin, LC3-II/LC3-I, Atg5, and NIX) (Cao et al., 2017; Zhang et al., 2020). For the most part, mitophagy is considered an adaptive or defensive mechanism. However, inappropriate or uncontrolled mitophagy can also be harmful since it destroys healthy mitochondria, which are necessary for energy metabolism. Thus, whether mitophagy is beneficial or detrimental in SAH remains a matter of debate.
Current evidence leans toward a beneficial effect of mitophagy after SAH, especially in the EBI phase. Several studies reported that enhancing mitochondrial autophagy after SAH exerted neuroprotective effects. The endogenous Nrf2 antioxidant system appears to be a major regulatory target of mitophagy. Zhang et al. (2019a) used mitoquinone to activate mitophagy via the Kelch-like epichlorohydrin-associated protein 1/Nrf2/prohibitin 2 pathway after SAH to inhibit oxidative stress-related neuronal death. Liu et al. (2022a) increased LC3II, PINK1, Parkin, and Nrf1 levels via the cannabinoid receptor 1/Nrf1/PINK1 pathway after SAH by treating rats with arachidonyl-2-chloroethylamide, which promoted the formation of mitophagosomes, maintained the mitochondrial morphology, and had anti-oxidative stress and anti-apoptotic effects (Liu et al., 2022a). Melatonin increased Nrf2 expression and induced mitophagy, protecting against brain injury after SAH (Sun et al., 2018). Another study found that melatonin mediated the upregulation of mitophagy-related proteins (Parkin and PINK-1, LC3-II/LC3-I, and Atg5). Additionally, melatonin alleviated morphological changes in mitochondria with decreased ROS production (Cao et al., 2017). The AMPK pathway is also involved in the regulation of mitophagy. Metformin promoted mitophagy in an AMPK-dependent manner and attenuated EBI. Meanwhile, the authors observed by transmission electron microscopy that metformin treatment improved the morphology of mitochondria after SAH and promoted the formation of mitophagosomes (Zhang et al., 2022a). In addition, other drugs (such as triiodothyronine) enhanced mitochondrial autophagy through the PINK1/Parkin pathway, increased ΔΨm, improved mitochondrial morphology, and protected neurons after SAH (Chang et al., 2022). Rapamycin also activated voltage-dependent anion channels, induced mitophagy, and reduced apoptosis and necrosis in neurons after SAH (Li et al., 2014).
However, two studies on CSF samples from patients showed that increased mitophagy levels seemed to be associated with a poor outcome. Youn et al. (2022) found that patients with DCI had significantly higher Bnip3L and PINK1 CSF mRNA levels than patients without DCI. In another study from the same group, patients with DCI had significantly elevated mitophagy flux, accumulation of autophagic vesicles, and p62 and LC3-II expression levels during DCI. Additionally, they had significantly higher Bnip3 and PINK1 mRNA expression levels than patients without DCI, suggesting that increased mitophagy-related mitochondrial dysfunction plays an important role in the pathogenesis of DCI (Youn et al., 2021). Nevertheless, the role of mitophagy in SAH is not fully understood, and further studies are needed to determine whether mitophagy facilitates recovery after SAH.
Mitochondrial biogenesis
Mitochondrial biogenesis refers to the generation of new mitochondria and mtDNA replication through the proliferation of existing mitochondria (Cardanho-Ramos and Morais, 2021). Newly generated mitochondria replace dysfunctional or damaged mitochondria that have been selectively degraded by mitophagy (Ding et al., 2021). Mitochondrial biogenesis increases the cellular energy supply, improving clinical outcomes in SAH (Fan et al., 2021; Tu et al., 2021; Zhou et al., 2021).
The PGC-1α/Nrf1/2/mitochondrial transcription factor A (TFAM) pathway is a major regulator of mitochondrial biogenesis. Peroxisome PGC-1α also plays a key role in this process. It activates Nrf1 and Nrf2, inducing the expression of related mitochondrial genes, including TFAM, thus increasing mitochondrial respiration and mtDNA replication/transcription (Cardanho-Ramos and Morais, 2021). Several signaling cascades activate mitochondrial biogenesis through the PGC-1α/Nrf1/2/TFAM pathway. Among them, the AMP/ATP ratio and Ca2+ levels are the main initiating factors. High AMP levels activate AMPK, which phosphorylates PGC-1α. Phosphorylated PGC-1α regulates the expression of its own genes, as well as genes related to mitochondrial biogenesis (Sun et al., 2022). The NAD+/NADH ratio is another important signal for mitochondrial biogenesis; when it is elevated, Sirtuin 1 deacetylates PGC-1α, resulting in its activation (Zhao et al., 2020).
Recent studies have demonstrated that PGC-1α levels start to increase significantly 6 hours after SAH in rats, reach a peak at 24 hours, and then gradually decrease until 72 hours. Besides, elevated PGC-1α levels were observed in the temporal cortex of rats after SAH, along with an increase in mitochondrial biogenesis (Zhou et al., 2021). Several studies have attempted to mitigate SAH damage by promoting mitochondrial biogenesis. HSP22 rescues mitochondrial function in an AMPK-PGC-1α-dependent manner, exerts neuroprotection, regulates the TFAM/Nrf1 pathway in a positive feedback manner to promote mitochondrial biogenesis, and attenuates oxidative stress and brain injury (Fan et al., 2021). Meanwhile, resveratrol upregulated the expression of PGC-1α, Nrf1, and TFAM and promoted the nuclear translocation of PGC-1α, suggesting that resveratrol promotes mitochondrial biogenesis after experimental SAH in mice by activating the PGC-1α signaling pathway (Zhou et al., 2021). Finally, irisin improved mitochondrial biogenesis via mitochondrial uncoupling protein-2 and had a neuroprotective effect against experimental SAH in mice (Tu et al., 2021).
Intercellular mitochondrial transfer
For a long time, mitochondria were thought to remain inside the cell during their whole lifetime. However, recent studies have found that damaged neurons could send mitochondria as distress signals to neighboring glial and endothelial cells, prompting them to respond accordingly (Hayakawa et al., 2016; Gao et al., 2022a). After an acute injury in the central nervous system, astrocytes and microglia may exchange mitochondria with neighboring neurons to protect them (Hayakawa et al., 2016). Structures such as tunneling nanotubes (TNTs) and extracellular vesicles can perform the intercellular exchange of membrane vesicles and organelles, and transport mitochondria between cells (Nasoni et al., 2021; Peruzzotti-Jametti et al., 2021). F-actin makes up the structure of TNTs and mediates the transport of mitochondria in TNTs (Liu et al., 2022b). Although little is known about the mechanism, the transfer of mitochondria out and in between cells appears to be a regulated process. Indeed, calcium-dependent CD38 signaling can upregulate mitochondrial release from astrocytes. Besides, blocking integrin and tyrosine protein kinase/spleen tyrosine kinase signaling can reduce mitochondrial entry into neurons (Hayakawa et al., 2018a). A recent study showed that exogenous mitochondrial transplantation increases cell viability and decreases ROS levels and apoptosis in an oxygen-dependent manner. Mitochondria tracking showed that some of the exogenous mitochondria fused with those of the host cell, while others were incorporated into lysosomes. Transcriptome analysis of mitochondrial transplanted neural cells showed that differences in the expressed genes were closely associated with lipid metabolism compared to controls. Mitochondrial transfer can be facilitated by drug treatment. For example, melatonin can promote mitochondrial transfer between damaged HT22 cells by increasing the connectivity of TNTs after ischemia-reperfusion injury (Nasoni et al., 2021).
The detection of extracellular mitochondria in CSF indicates a possible role of intercellular mitochondrial transfer in SAH. Chou et al. (2017) isolated respiratory mitochondria in CSF samples from SAH patients. They found that JC1 levels were reduced after SAH. In addition, a higher ΔΨm in the CSF was associated with a good outcome 3 months after SAH (Chou et al., 2017). Meanwhile, Youn et al. (2020) suggested that the ΔΨm of extracellular mitochondria in the CSF was a biomarker of DCI.
Transplantation of healthy exogenous mitochondria into damaged cell is an emerging treatment for mitochondrial dysfunction (Park et al., 2021). Exogenous transplantation to replace damaged mitochondria through the mitochondrial transfer mechanism can rescue mitochondrial dysfunction in damaged cells. For example, the microinjection of healthy mitochondria at the site of injury in an ischemic stroke model has been reported (Chen et al., 2020b). Recent studies have shown that mitochondria transfer occurs from astrocytes to neuronal cells after ischemia. This suggests that mitochondrial transfer from astrocytes to damaged neurons is a recovery mechanism and provides a theoretical basis for therapeutic interventions for stroke patients. In that study, the CD38 signaling pathway mediated the release of functional mitochondria from activated astrocytes to damaged neurons, thereby restoring ATP levels, neuronal survival, plasticity, and behavioral function (Hayakawa et al., 2016). However, this endogenous mitochondrial transfer from astrocytes to damaged neurons is transient and insufficient to induce robust and stable neuroprotection. In fact, without exogenous therapeutic intervention, mitochondrial transfer from astrocytes cannot impede the secondary cell death triggered by a stroke. Therefore, a stem cell-based approach to mitochondrial transfer may be an effective strategy to support endogenous healthy mitochondria transfer.
Mitochondrial transplantation therapy is currently available in two ways. First, stem cells can be used to induce the repair of damaged cells. For example, the mitochondrial transfer from endothelial progenitor cells to ischemic brain endothelial cells increases mtDNA copy and ATP levels (Hayakawa et al., 2018b). After ischemic injury, mesenchymal stem cells maintain aerobic respiration of damaged endothelial cells through mitochondrial transfer and inhibit endothelial cell apoptosis (Liu et al., 2014). Second, healthy exogenous mitochondria can be transplanted into the damaged tissue. Studies showed that transplantation of exogenous mitochondria into cerebral ischemic rats restored motor function, reduced the size of cerebral infarcts, and decreased neuronal cell death (Huang et al., 2016). Another study showed that transplantation of muscle-derived autologous mitochondria via the lateral ventricle reduced cellular oxidative stress and apoptosis, attenuated reactive astrocyte proliferation, promoted neurogenesis, reduced cerebral infarct volume, and reversed neurological dysfunction after ischemic stroke in rats (Zhang et al., 2019d). In SAH, the therapeutic role of mitochondrial transplantation has not been studied. Nevertheless, this is a promising research area for developing treatments for SAH injury.
Future Perspectives and Conclusion
Mitochondrial dysfunction is a central event in the pathophysiology of SAH. Early in the disease, mitochondria are directly hit by ischemia and hypoxia, excitotoxicity, the toxicity of Hb and its degradation products, and other direct factors that cause mitochondrial dysfunction. Subsequently, mitochondrial dysfunction triggers oxidative stress, inflammation, and apoptosis, amplifying cellular damage. Meanwhile, the MtQC system, a series of relatively balanced events (namely mitochondrial fission and fusion, mitophagy and mitochondrial biogenesis, mitochondrial transfer, and mitochondrial protein quality control), maintains mitochondrial integrity and neuronal homeostasis in multiple ways.
Despite the advances in our understanding of the mechanisms of MtQC after SAH and the resulting treatment strategies, several questions remain. First, the mitochondrial pathophysiology during injury and repair after SAH remains incompletely understood. For example, in the resting state, mitochondrial fission and fusion each have their benefits and drawbacks. It is now generally accepted that excessive mitochondrial fission impairs the function of the mitochondrial network after SAH. However, whether moderate mitochondrial fission exerts beneficial effects by excluding damaged mitochondrial components and preserving the function of most mitochondria is a critical question. Besides, this balance remains to be identified and controlled. Similarly, proper mitochondrial autophagy appears to have a neuroprotective effect by removing damaged mitochondria, whereas excessive mitochondrial autophagy disrupts energy production and mitochondria-related signaling pathways. Therefore, the balance between mitochondrial dynamics and mitochondrial autophagy is more critical than the absolute level of each process. In addition, the brain is a heterogeneous organ that includes neurons, various types of glial cells, and circulating immune cells involved in brain injury and repair. However, existing studies have focused on neuronal cells, and the mitochondrial physiology and pathophysiology of other cell types remain unclear. In particular, mitochondrial dysfunction in microglia after SAH and the role of endothelial and glial cells in the development of SAH require further investigation. New evidence from other organs suggests that differences in MtQC mechanisms in different types of cells lead to different or even opposite effects on tissue injury and repair. For instance, PINK1 deficiency in lung epithelial cells leads to mitochondrial depolarization, pro-fibrotic factor expression, and pulmonary fibrosis, suggesting a mitogenic protective function of the PINK1/parkin pathway in idiopathic pulmonary fibrosis (Bueno et al., 2015). In contrast, parkin-mediated mitosis in alveolar macrophages appears to help these cells resist apoptosis and promote pulmonary fibrosis (Larson-Casey et al., 2016). Therefore, it is necessary to determine the impact of an altered MtQC capacity of different cell types in SAH.
Second, research on the many kinds of MtQC mechanisms remains in its initial stage. Although emerging evidence links mitochondrial transfer and UPRmt to disease mechanisms following SAH, their exact role in these pathological conditions and the molecular mechanisms involved are still largely unknown. The present study on MtQC highlights the following points: The MtQC mechanism operates through an integrated network of hierarchical pathways. Changes in any of the MtQC mechanisms may affect other mechanisms, and thus the whole system. For instance, mitochondrial fission is essential for mitophagy and is also closely linked to mitochondrial biogenesis (Kleele et al., 2021). Furthermore, although much progress has been made in this field in terms of basic research, translating mitochondria-targeting drugs into clinical applications for SAH remains a great challenge. Indeed, their potential adverse effects and mechanisms are unclear, and the mitochondrial physiology and pathophysiology of their multiple targets are still unclear.
Therefore, future studies should concentrate on determining the role and regulation of MtQC mechanisms in SAH in various brain cell types to facilitate the discovery of drugs for the prevention and treatment of EBI and DCI and improve the outcome of SAH.
Limitations remain in our review design. First, the mechanisms by which SAH leads to mitochondrial dysfunction are intricate, and we described only representative mechanisms. Second, due to space, we did not describe each MtQC mechanism in detail, such as UPRmt and intercellular mitochondrial transfer. Finally, we did not describe the interconnections and interactions between MtQC mechanisms.
Funding Statement
Funding: This work was supported by the National Natural Science Foundation of China, Nos. 82130037 (to CH), 81971122 (to CH), 82171323 (to WL), and the Natural Science Foundation of Jiangsu Province of China, No. BK20201113 (to WL).
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement: Data availability is not applicable to this article as no new data were created or analyzed in this study.
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
References
- 1.Akeret K, Buzzi RM, Schaer CA, Thomson BR, Vallelian F, Wang S, Willms J, Sebök M, Held U, Deuel JW, Humar R, Regli L, Keller E, Hugelshofer M, Schaer DJ. Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury. J Cereb Blood Flow Metab. 2021;41:3000–3015. doi: 10.1177/0271678X211020629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen CR, Presseau J, Saigle V, Etminan N, Vergouwen MDI, English SW. Core outcomes for subarachnoid haemorrhage. Lancet Neurol. 2019;18:1075–1076. doi: 10.1016/S1474-4422(19)30412-0. [DOI] [PubMed] [Google Scholar]
- 3.Barazzuol L, Giamogante F, Brini M, Calì T. PINK1/Parkin mediated mitophagy, Ca(2+) signalling and ER-mitochondria contacts in Parkinson's disease. Int J Mol Sci. 2020;21:1772. doi: 10.3390/ijms21051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 5.Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. 2022;23:266–285. doi: 10.1038/s41580-021-00433-y. [DOI] [PubMed] [Google Scholar]
- 6.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, Ying G, Gu C, Wang L, Chen G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep. 2017;7:2417. doi: 10.1038/s41598-017-02679-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardanho-Ramos C, Morais VA. Mitochondrial biogenesis in neurons: how and where. Int J Mol Sci. 2021;22:13059. doi: 10.3390/ijms222313059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–259. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 10.Chang H, Lin C, Li Z, Shen Y, Zhang G, Mao L, Ma C, Liu N, Lu H. T3 alleviates neuroinflammation and reduces early brain injury after subarachnoid haemorrhage by promoting mitophagy via PINK 1-parkin pathway. Exp Neurol. 2022;357:114175. doi: 10.1016/j.expneurol.2022.114175. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhry SR, Frede S, Seifert G, Kinfe TM, Niemelä M, Lamprecht A, Muhammad S. Temporal profile of serum mitochondrial DNA (mtDNA) in patients with aneurysmal subarachnoid hemorrhage (aSAH) Mitochondrion. 2019;47:218–226. doi: 10.1016/j.mito.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Chen XY, Wang QL, Yang SJ, Zhou H, Ding LS, Qing LS, Luo P. Astragaloside IV derivative (LS-102) alleviated myocardial ischemia reperfusion injury by inhibiting Drp1(Ser616) phosphorylation-mediated mitochondrial fission. Front Pharmacol. 2020a;11:1083. doi: 10.3389/fphar.2020.01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Huang J, Hu Y, Khoshnam SE, Sarkaki A. Mitochondrial transfer as a therapeutic strategy against ischemic stroke. Transl Stroke Res. 2020b;11:1214–1228. doi: 10.1007/s12975-020-00828-7. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Chen J, Sun X, Shi X, Wang L, Huang L, Zhou W. Evaluation of the neuroprotective effect of EGCG: a potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018;9:6349–6359. doi: 10.1039/c8fo01497c. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Chai E, Mou Y, Roda RH, Blackstone C, Li XJ. Inhibiting mitochondrial fission rescues degeneration in hereditary spastic paraplegia neurons. Brain. 2022;145:4016–4031. doi: 10.1093/brain/awab488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi DW. Excitotoxicity: still hammering the ischemic brain in 2020. Front Neurosci. 2020;14:579953. doi: 10.3389/fnins.2020.579953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, Lo EH, Hayakawa K. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke. 2017;48:2231–2237. doi: 10.1161/STROKEAHA.117.017758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke JV, Suggs JM, Diwan D, Lee JV, Lipsey K, Vellimana AK, Zipfel GJ. Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: a review and synthesis. J Cereb Blood Flow Metab. 2020;40:1565–1575. doi: 10.1177/0271678X20921974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai J, Xu S, Okada T, Liu Y, Zuo G, Tang J, Zhang JH, Shi H. T090131, an agonist of liver X receptors, attenuates neuronal apoptosis in early brain injury after subarachnoid hemorrhage in rats via liver X receptors/interferon regulatory factor/P53 upregulated modulator of apoptosis/dynamin-1-like protein pathway. Oxid Med Cell Longev. 2021;2021:8849131. doi: 10.1155/2021/8849131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dharmasaroja PA. Fluid intake related to brain edema in acute middle cerebral artery infarction. Transl Stroke Res. 2016;7:49–53. doi: 10.1007/s12975-015-0439-1. [DOI] [PubMed] [Google Scholar]
- 21.Di Rita A, Peschiaroli A, P DA, Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M, Melino G, Papaleo E, Dengjel J, El Alaoui S, Campanella M, Dötsch V, Rogov VV, Strappazzon F, Cecconi F. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun. 2018;9:3755. doi: 10.1038/s41467-018-05722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019;99:949–1045. doi: 10.1152/physrev.00062.2017. [DOI] [PubMed] [Google Scholar]
- 23.Ding Q, Qi Y, Tsang SY. Mitochondrial biogenesis, mitochondrial dynamics, and mitophagy in the maturation of cardiomyocytes. Cells. 2021;10:2463. doi: 10.3390/cells10092463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du YH, Sun Y, Yang RY, Wang LY, Cai M. Mechanisms of neuroinflammation in mild cognitive impairment. Zhongguo Zuzhi Gongcheng Yanjiu. 2021;25:4743–4749. [Google Scholar]
- 25.Ehlert A, Starekova J, Manthei G, Ehlert-Gamm A, Flack J, Gessert M, Gerss J, Hesselmann V. Nitric oxide-based treatment of poor-grade patients after severe aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2020;32:742–754. doi: 10.1007/s12028-019-00809-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA. Mitochondrial quality control in health and in Parkinson's disease. Physiol Rev. 2022;102:1721–1755. doi: 10.1152/physrev.00041.2021. [DOI] [PubMed] [Google Scholar]
- 27.Etminan N, Chang HS, Hackenberg K, de Rooij NK, Vergouwen MDI, Rinkel GJE, Algra A. Worldwide incidence of aneurysmal subarachnoid hemorrhage according to region, time period, blood pressure, and smoking prevalence in the population: a systematic review and meta-analysis. JAMA Neurol. 2019;76:588–597. doi: 10.1001/jamaneurol.2019.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan H, Ding R, Liu W, Zhang X, Li R, Wei B, Su S, Jin F, Wei C, He X, Li X, Duan C. Heat shock protein 22 modulates NRF1/TFAM-dependent mitochondrial biogenesis and DRP1-sparked mitochondrial apoptosis through AMPK-PGC1αsignaling pathway to alleviate the early brain injury of subarachnoid hemorrhage in rats. Redox Biol. 2021;40:101856. doi: 10.1016/j.redox.2021.101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan LF, He PY, Peng YC, Du QH, Ma YJ, Jin JX, Xu HZ, Li JR, Wang ZJ, Cao SL, Li T, Yan F, Gu C, Wang L, Chen G. Mdivi-1 ameliorates early brain injury after subarachnoid hemorrhage via the suppression of inflammation-related blood-brain barrier disruption and endoplasmic reticulum stress-based apoptosis. Free Radic Biol Med. 2017;112:336–349. doi: 10.1016/j.freeradbiomed.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nature reviews Nephrology. 2018;14:291–312. doi: 10.1038/nrneph.2018.9. [DOI] [PubMed] [Google Scholar]
- 31.Fragata I, Bustamante A, Penalba A, Ferreira P, Nunes AP, Canhão P, Montaner J. TNF-R1 correlates with cerebral perfusion and acute ischemia following subarachnoid hemorrhage. Neurocrit Care. 2020;33:679–687. doi: 10.1007/s12028-020-01082-3. [DOI] [PubMed] [Google Scholar]
- 32.Gao L, Liu F, Hou PP, Manaenko A, Xiao ZP, Wang F, Xu TL, Hu Q. Neurons release injured mitochondria as “Help-Me“signaling after ischemic stroke. Front Aging Neurosci. 2022a;14:785761. doi: 10.3389/fnagi.2022.785761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Q, Tian R, Han H, Slone J, Wang C, Ke X, Zhang T, Li X, He Y, Liao P, Wang F, Chen Y, Fu S, Zhang K, Zeng F, Yang Y, Li Z, Tan J, Li J, Lu Y, et al. PINK1-mediated Drp1(S616) phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct Target Ther. 2022b;7:103. doi: 10.1038/s41392-022-00933-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao S, Hu J. Mitochondrial fusion: the machineries in and out. Trends Cell Biol. 2021;31:62–74. doi: 10.1016/j.tcb.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Gao X, Gao YY, Yan HY, Liu GJ, Zhou Y, Tao T, Yue TT, Pang C, Chen XX, Gao S, Wu LY, Hang CH, Li W. PDK4 decrease neuronal apoptosis via inhibiting ROS-ASK1/P38 pathway in early brain injury after subarachnoid hemorrhage. Antioxid Redox Signal. 2022c;36:505–524. doi: 10.1089/ars.2021.0083. [DOI] [PubMed] [Google Scholar]
- 36.Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol. 2017;13:136–146. doi: 10.1038/nchembio.2287. [DOI] [PubMed] [Google Scholar]
- 37.Gilkerson R, De La Torre P, St Vallier S. Mitochondrial OMA1 and OPA1 as gatekeepers of organellar structure/function and cellular stress response. Front Cell Dev Biol. 2021;9:626117. doi: 10.3389/fcell.2021.626117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombès A, Belenguer P, Arnoult D, Rojo M. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell. 2008;100:315–325. doi: 10.1042/BC20070110. [DOI] [PubMed] [Google Scholar]
- 39.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579:427–432. doi: 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 2018a;75:119–122. doi: 10.1001/jamaneurol.2017.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. doi: 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayakawa K, Chan SJ, Mandeville ET, Park JH, Bruzzese M, Montaner J, Arai K, Rosell A, Lo EH. Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells. 2018b;36:1404–1410. doi: 10.1002/stem.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He X, Sun J, Huang X. Expression of caspase-3, Bax and Bcl-2 in hippocampus of rats with diabetes and subarachnoid hemorrhage. Exp Ther Med. 2018;15:873–877. doi: 10.3892/etm.2017.5438. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Helbok R, Schiefecker AJ, Beer R, Dietmann A, Antunes AP, Sohm F, Fischer M, Hackl WO, Rhomberg P, Lackner P, Pfausler B, Thomé C, Humpel C, Schmutzhard E. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Crit Care. 2015;19:75. doi: 10.1186/s13054-015-0809-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmeijer J, van Putten MJ. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke. 2012;43:607–615. doi: 10.1161/STROKEAHA.111.632943. [DOI] [PubMed] [Google Scholar]
- 46.Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, Chang JC, Pan HC, Lin SZ, Liu CS, Su HL. Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 2016;25:913–927. doi: 10.3727/096368915X689785. [DOI] [PubMed] [Google Scholar]
- 47.Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. 2018;558:401–405. doi: 10.1038/s41586-018-0211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun. 2018;9:5239. doi: 10.1038/s41467-018-07543-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanamaru H, Suzuki H. Potential therapeutic molecular targets for blood-brain barrier disruption after subarachnoid hemorrhage. Neural Regen Res. 2019;14:1138–1143. doi: 10.4103/1673-5374.251190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Killackey SA, Philpott DJ, Girardin SE. Mitophagy pathways in health and disease. J Cell Biol. 2020;219:e202004029. doi: 10.1083/jcb.202004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, Ruberto FP, Nemir M, Wai T, Pedrazzini T, Manley S. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. 2021;593:435–439. doi: 10.1038/s41586-021-03510-6. [DOI] [PubMed] [Google Scholar]
- 53.Kumar R, Chaudhary AK, Woytash J, Inigo JR, Gokhale AA, Bshara W, Attwood K, Wang J, Spernyak JA, Rath E, Yadav N, Haller D, Goodrich DW, Tang DG, Chandra D. A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60. J Clin Invest. 2022;132:e149906. doi: 10.1172/JCI149906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M, Liu J, Du H, Chen R, Pan X, Liu N, Chen H. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020;34:101503. doi: 10.1016/j.redox.2020.101503. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016;44:582–596. doi: 10.1016/j.immuni.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lenz IJ, Plesnila N, Terpolilli NA. Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice. J Cereb Blood Flow Metab. 2021;41:1669–1681. doi: 10.1177/0271678X20973787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Lu J, Mi Y, Shi Z, Chen C, Riley J, Zhou C. Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 2014;1573:74–83. doi: 10.1016/j.brainres.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Wu P, Dai J, Zhang T, Bihl J, Wang C, Liu Y, Shi H. Inhibition of mTOR alleviates early brain injury after subarachnoid hemorrhage via relieving excessive mitochondrial fission. Cell Mol Neurobiol. 2020;40:629–642. doi: 10.1007/s10571-019-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin S, Xing H, Zang T, Ruan X, Wo L, He M. Sirtuins in mitochondrial stress: indispensable helpers behind the scenes. Ageing Res Rev. 2018;44:22–32. doi: 10.1016/j.arr.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 61.Liu B, Tian Y, Li Y, Wu P, Zhang Y, Zheng J, Shi H. ACEA attenuates oxidative stress by promoting mitophagy via CB1R/Nrf1/PINK1 pathway after subarachnoid hemorrhage in rats. Oxid Med Cell Longev. 2022a;2022:1024279. doi: 10.1155/2022/1024279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu JP, Xia Y, Li J, You C. Plasma free mtDNA and expression of TLR-9/MAPK in brain tissues of rats with subarachnoid hemorrhage. Sichuan Da Xue Xue Bao Yi Xue Ban. 2017;48:225–229. [PubMed] [Google Scholar]
- 63.Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014;92:10–18. doi: 10.1016/j.mvr.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Liu Z, Sun Y, Qi Z, Cao L, Ding S. Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci. 2022b;12:66. doi: 10.1186/s13578-022-00805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu AY, Damisah EC, Winkler EA, Grant RA, Eid T, Bulsara KR. Cerebrospinal fluid untargeted metabolomic profiling of aneurysmal subarachnoid hemorrhage: an exploratory study. Br J Neurosurg. 2018;32:637–641. doi: 10.1080/02688697.2018.1519107. [DOI] [PubMed] [Google Scholar]
- 66.Ma C, Gao B, Wang Z, You W, Yu Z, Shen H, Li X, Li H, Zhang X, Wang Z, Chen G. GrpEL1 regulates mitochondrial unfolded protein response after experimental subarachnoid hemorrhage in vivo and in vitro. Brain Res Bull. 2022;181:97–108. doi: 10.1016/j.brainresbull.2022.01.014. [DOI] [PubMed] [Google Scholar]
- 67.Ma R, Ma L, Weng W, Wang Y, Liu H, Guo R, Gao Y, Tu J, Xu TL, Cheng J, Zhu MX, Zhou A, Li Y. DUSP6 SUMOylation protects cells from oxidative damage via direct regulation of Drp1 dephosphorylation. Sci Adv. 2020;6:eaaz0361. doi: 10.1126/sciadv.aaz0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10:44–58. doi: 10.1038/nrneurol.2013.246. [DOI] [PubMed] [Google Scholar]
- 69.Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23:159–173. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matz PG, Fujimura M, Lewen A, Morita-Fujimura Y, Chan PH. Increased cytochrome c-mediated DNA fragmentation and cell death in manganese-superoxide dismutase-deficient mice after exposure to subarachnoid hemolysate. Stroke. 2001;32:506–515. doi: 10.1161/01.str.32.2.506. [DOI] [PubMed] [Google Scholar]
- 71.Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, Pottecher J, Sibilia J, Geny B. Mitochondria: an organelle of bacterial origin controlling inflammation. Front Immunol. 2018;9:536. doi: 10.3389/fimmu.2018.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miao Z, Tian W, Ye Y, Gu W, Bao Z, Xu L, Sun G, Li C, Tu Y, Chao H, Lam SM, Liu N, Ji J. Hsp90 induces Acsl4-dependent glioma ferroptosis via dephosphorylating Ser637 at Drp1. Cell Death Dis. 2022;13:548. doi: 10.1038/s41419-022-04997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mindt S, Tokhi U, Hedtke M, Groß HJ, Hänggi D. Mass spectrometry-based method for quantification of nimodipine and glutamate in cerebrospinal fluid. Pilot study with patients after aneurysmal subarachnoid haemorrhage. J Clin Pharm Ther. 2020;45:81–87. doi: 10.1111/jcpt.13028. [DOI] [PubMed] [Google Scholar]
- 74.Nasoni MG, Carloni S, Canonico B, Burattini S, Cesarini E, Papa S, Pagliarini M, Ambrogini P, Balduini W, Luchetti F. Melatonin reshapes the mitochondrial network and promotes intercellular mitochondrial transfer via tunneling nanotubes after ischemic-like injury in hippocampal HT22 cells. J Pineal Res. 2021;71:e12747. doi: 10.1111/jpi.12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Osgood ML. Aneurysmal subarachnoid hemorrhage: review of the pathophysiology and management strategies. Curr Neurol Neurosci Rep. 2021;21:50. doi: 10.1007/s11910-021-01136-9. [DOI] [PubMed] [Google Scholar]
- 76.Otasevic V, Vucetic M, Grigorov I, Martinovic V, Stancic A. Ferroptosis in different pathological contexts seen through the eyes of mitochondria. Oxid Med Cell Longev. 2021;2021:5537330. doi: 10.1155/2021/5537330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park A, Oh M, Lee SJ, Oh KJ, Lee EW, Lee SC, Bae KH, Han BS, Kim WK. Mitochondrial transplantation as a novel therapeutic strategy for mitochondrial diseases. Int J Mol Sci. 2021;22:4793. doi: 10.3390/ijms22094793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peruzzotti-Jametti L, Bernstock JD, Willis CM, Manferrari G, Rogall R, Fernandez-Vizarra E, Williamson JC, Braga A, van den Bosch A, Leonardi T, Krzak G, Kittel Á, Benincá C, Vicario N, Tan S, Bastos C, Bicci I, Iraci N, Smith JA, Peacock B, et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021;19:e3001166. doi: 10.1371/journal.pbio.3001166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, Leeuwenburgh C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15:543–554. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab. 2019;316:E268–E285. doi: 10.1152/ajpendo.00314.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rass V, Helbok R. Early brain injury after poor-grade subarachnoid hemorrhage. Curr Neurol Neurosci Rep. 2019;19:78. doi: 10.1007/s11910-019-0990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rass V, Helbok R. How to diagnose delayed cerebral ischaemia and symptomatic vasospasm and prevent cerebral infarction in patients with subarachnoid haemorrhage. Curr Opin Crit Care. 2021;27:103–114. doi: 10.1097/MCC.0000000000000798. [DOI] [PubMed] [Google Scholar]
- 84.Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, Pinton P. Perturbed mitochondrial Ca2+signals as causes or consequences of mitophagy induction. Autophagy. 2013;9:1677–1686. doi: 10.4161/auto.24795. [DOI] [PubMed] [Google Scholar]
- 85.Roca-Portoles A, Tait SWG. Mitochondrial quality control: from molecule to organelle. Cell Mol Life Sci. 2021;78:3853–3866. doi: 10.1007/s00018-021-03775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosdah AA, Smiles WJ, Oakhill JS, Scott JW, Langendorf CG, Delbridge LMD, Holien JK, Lim SY. New perspectives on the role of Drp1 isoforms in regulating mitochondrial pathophysiology. Pharmacol Ther. 2020;213:107594. doi: 10.1016/j.pharmthera.2020.107594. [DOI] [PubMed] [Google Scholar]
- 87.Shen R, Zhou J, Li G, Chen W, Zhong W, Chen Z. SS31 attenuates oxidative stress and neuronal apoptosis in early brain injury following subarachnoid hemorrhage possibly by the mitochondrial pathway. Neurosci Lett. 2020;717:134654. doi: 10.1016/j.neulet.2019.134654. [DOI] [PubMed] [Google Scholar]
- 88.Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2018;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- 89.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193. doi: 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song YF, Zheng H, Luo Z, Hogstrand C, Bai ZY, Wei XL. Dietary choline alleviates high-fat diet-induced hepatic lipid dysregulation via UPRmt modulated by SIRT3-mediated mtHSP70 deacetylation. Int J Mol Sci. 2022;23:4204. doi: 10.3390/ijms23084204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stokum JA, Cannarsa GJ, Wessell AP, Shea P, Wenger N, Simard JM. When the blood hits your brain: the neurotoxicity of extravasated blood. Int J Mol Sci. 2021;22:5132. doi: 10.3390/ijms22105132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F, Peng ZY. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. doi: 10.1155/2019/5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun B, Yang S, Li S, Hang C. Melatonin upregulates nuclear factor erythroid-2 related factor 2 (Nrf2) and mediates mitophagy to protect against early brain injury after subarachnoid hemorrhage. Med Sci Monit. 2018;24:6422–6430. doi: 10.12659/MSM.909221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun J, Song FH, Wu JY, Zhang LQ, Li DY, Gao SJ, Liu DQ, Zhou YQ, Mei W. Sestrin2 overexpression attenuates osteoarthritis pain via induction of AMPK/PGC-1α-mediated mitochondrial biogenesis and suppression of neuroinflammation. Brain Behav Immun. 2022;102:53–70. doi: 10.1016/j.bbi.2022.02.015. [DOI] [PubMed] [Google Scholar]
- 95.Suzuki H, Kanamaru H, Kawakita F, Asada R, Fujimoto M, Shiba M. Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol Histopathol. 2021;36:143–158. doi: 10.14670/HH-18-253. [DOI] [PubMed] [Google Scholar]
- 96.Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nature reviews Nephrology. 2021;17:299–318. doi: 10.1038/s41581-020-00369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tao T, Chen X, Zhou Y, Zheng Q, Gao S, Wang J, Ding P, Li X, Peng Z, Lu Y, Gao Y, Zhuang Z, Hang CH, Li W. Continued P2X7 activation leads to mitochondrial fission and compromising microglial phagocytosis after subarachnoid haemorrhage. J Neurochem. 2022;163:419–437. doi: 10.1111/jnc.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taran S, Trivedi V, Singh JM, English SW, McCredie VA. The use of standardized management protocols for critically Ill patients with non-traumatic subarachnoid hemorrhage: a systematic review. Neurocrit Care. 2020;32:858–874. doi: 10.1007/s12028-019-00867-5. [DOI] [PubMed] [Google Scholar]
- 99.Tewari D, Sah AN, Bawari S, Nabavi SF, Dehpour AR, Shirooie S, Braidy N, Fiebich BL, Vacca RA, Nabavi SM. Role of nitric oxide in neurodegeneration: function, regulation and inhibition. Curr Neuropharmacol. 2021;19:114–126. doi: 10.2174/1570159X18666200429001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tian Q, Liu S, Han SM, Zhang W, Qin XY, Chen JH, Liu CL, Guo YJ, Li MC. The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage. Neural Regen Res. 2023;18:244–252. doi: 10.4103/1673-5374.346542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Towers CG, Wodetzki DK, Thorburn J, Smith KR, Caino MC, Thorburn A. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev Cell. 2021;56:2029–2042.e5. doi: 10.1016/j.devcel.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res. 2018;122:58–73. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tu T, Yin S, Pang J, Zhang X, Zhang L, Zhang Y, Xie Y, Guo K, Chen L, Peng J, Jiang Y. Irisin contributes to neuroprotection by promoting mitochondrial biogenesis after experimental subarachnoid hemorrhage. Front Aging Neurosci. 2021;13:640215. doi: 10.3389/fnagi.2021.640215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Uhl E, Lehmberg J, Steiger HJ, Messmer K. Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery. 2003;52:1307–1315. doi: 10.1227/01.neu.0000065154.04824.9e. discussion 1315-1317. [DOI] [PubMed] [Google Scholar]
- 105.van Lieshout JH, Dibué-Adjei M, Cornelius JF, Slotty PJ, Schneider T, Restin T, Boogaarts HD, Steiger HJ, Petridis AK, Kamp MA. An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg Rev. 2018;41:917–930. doi: 10.1007/s10143-017-0827-y. [DOI] [PubMed] [Google Scholar]
- 106.Wang HC, Yang TM, Lin WC, Lin YJ, Tsai NW, Liou CW, Kwan AL, Lu CH. The value of serial plasma and cerebrospinal fluid nuclear and mitochondrial deoxyribonucleic acid levels in aneurysmal subarachnoid hemorrhage. J Neurosurg. 2013;118:13–19. doi: 10.3171/2012.8.JNS112093. [DOI] [PubMed] [Google Scholar]
- 107.Wang L, Wang Z, You W, Yu Z, Li X, Shen H, Li H, Sun Q, Li W, Chen G. Enhancing S-nitrosoglutathione reductase decreases S-nitrosylation of Drp1 and reduces neuronal apoptosis in experimental subarachnoid hemorrhage both in vivo and in vitro. Brain Res Bull. 2022a;183:184–200. doi: 10.1016/j.brainresbull.2022.03.010. [DOI] [PubMed] [Google Scholar]
- 108.Wang YJ, Li ZX, Gu HQ, Zhai Y, Zhou Q, Jiang Y, Zhao XQ, Wang YL, Yang X, Wang CJ, Meng X, Li H, Liu LP, Jing J, Wu J, Xu AD, Dong Q, Wang D, Wang WZ, Ma XD, et al. China Stroke Statistics: an update on the 2019 report from the National Center for Healthcare Quality Management in Neurological Diseases, China National Clinical Research Center for Neurological Diseases, the Chinese Stroke Association, National Center for Chronic and Non-communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention and Institute for Global Neuroscience and Stroke Collaborations. Stroke Vasc Neurol. 2022b;7:415–450. doi: 10.1136/svn-2021-001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wesch N, Kirkin V, Rogov VV. Atg8-family proteins-structural features and molecular interactions in autophagy and beyond. Cells. 2020;9:2008. doi: 10.3390/cells9092008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu M, Gu X, Ma Z. Mitochondrial quality control in cerebral ischemia-reperfusion injury. Mol Neurobiol. 2021a;58:5253–5271. doi: 10.1007/s12035-021-02494-8. [DOI] [PubMed] [Google Scholar]
- 111.Wu P, Li Y, Zhu S, Wang C, Dai J, Zhang G, Zheng B, Xu S, Wang L, Zhang T, Zhou P, Zhang JH, Shi H. Mdivi-1 alleviates early brain injury after experimental subarachnoid hemorrhage in rats, possibly via inhibition of Drp1-activated mitochondrial fission and oxidative stress. Neurochem Res. 2017;42:1449–1458. doi: 10.1007/s11064-017-2201-4. [DOI] [PubMed] [Google Scholar]
- 112.Wu Q, Liu J, Mao Z, Tian L, Wang N, Wang G, Wang Y, Seto S. Ligustilide attenuates ischemic stroke injury by promoting Drp1-mediated mitochondrial fission via activation of AMPK. Phytomedicine. 2022;95:153884. doi: 10.1016/j.phymed.2021.153884. [DOI] [PubMed] [Google Scholar]
- 113.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011;278:941–954. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 114.Wu X, Luo J, Liu H, Cui W, Feng D, Qu Y. SIRT3 protects against early brain injury following subarachnoid hemorrhage via promoting mitochondrial fusion in an AMPK dependent manner. Chin Neurosurg J. 2020;6:1. doi: 10.1186/s41016-019-0182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W, Yan W, Cao M, Zheng W, Jiang L, Wu J, Han F, Qin Z, Fang L, Hu W, Chen Z, Zhang X. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy. 2021b;17:1934–1946. doi: 10.1080/15548627.2020.1802089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu J, Li Q, Xu CY, Mao S, Jin JJ, Gu W, Shi Y, Zou CF, Ye L. Obstructive sleep apnea aggravates neuroinflammation and pyroptosis in early brain injury following subarachnoid hemorrhage via ASC/HIF-1αpathway. Neural Regen Res. 2022;17:2537–2543. doi: 10.4103/1673-5374.339000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu W, Yan J, Ocak U, Lenahan C, Shao A, Tang J, Zhang J, Zhang JH. Melanocortin 1 receptor attenuates early brain injury following subarachnoid hemorrhage by controlling mitochondrial metabolism via AMPK/SIRT1/PGC-1αpathway in rats. Theranostics. 2021;11:522–539. doi: 10.7150/thno.49426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018;16:263–275. doi: 10.1016/j.redox.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Youn DH, Kim BJ, Kim Y, Jeon JP. Extracellular mitochondrial dysfunction in cerebrospinal fluid of patients with delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2020;33:422–428. doi: 10.1007/s12028-019-00895-1. [DOI] [PubMed] [Google Scholar]
- 120.Youn DH, Kim BJ, Hong EP, Jeon JP. Bioinformatics analysis of autophagy and mitophagy markers associated with delayed cerebral ischemia following subarachnoid hemorrhage. J Korean Neurosurg Soc. 2022;65:236–244. doi: 10.3340/jkns.2021.0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Youn DH, Kim Y, Kim BJ, Jeong MS, Lee J, Rhim JK, Kim HC, Jeon JP. Mitochondrial dysfunction associated with autophagy and mitophagy in cerebrospinal fluid cells of patients with delayed cerebral ischemia following subarachnoid hemorrhage. Sci Rep. 2021;11:16512. doi: 10.1038/s41598-021-96092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zeineddine HA, Honarpisheh P, McBride D, Pandit PKT, Dienel A, Hong SH, Grotta J, Blackburn S. Targeting hemoglobin to reduce delayed cerebral ischemia after subarachnoid hemorrhage. Transl Stroke Res. 2022;13:725–735. doi: 10.1007/s12975-022-00995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang J, Yuan G, Liang T, Pan P, Li X, Li H, Shen H, Wang Z, Chen G. Nix plays a neuroprotective role in early brain injury after experimental subarachnoid hemorrhage in rats. Front Neurosci. 2020;14:245. doi: 10.3389/fnins.2020.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang T, Wu P, Budbazar E, Zhu Q, Sun C, Mo J, Peng J, Gospodarev V, Tang J, Shi H, Zhang JH. Mitophagy reduces oxidative stress via Keap1 (kelch-like epichlorohydrin-associated protein 1)/Nrf2 (nuclear factor-E2-related factor 2)/PHB2 (prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke. 2019a;50:978–988. doi: 10.1161/STROKEAHA.118.021590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang T, Wu P, Zhang JH, Li Y, Xu S, Wang C, Wang L, Zhang G, Dai J, Zhu S, Liu Y, Liu B, Reis C, Shi H. Docosahexaenoic acid alleviates oxidative stress-based apoptosis via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell Mol Neurobiol. 2018a;38:1413–1423. doi: 10.1007/s10571-018-0608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang T, Xu S, Wu P, Zhou K, Wu L, Xie Z, Xu W, Luo X, Li P, Ocak U, Ocak PE, Travis ZD, Tang J, Shi H, Zhang JH. Mitoquinone attenuates blood-brain barrier disruption through Nrf2/PHB2/OPA1 pathway after subarachnoid hemorrhage in rats. Exp Neurol. 2019b;317:1–9. doi: 10.1016/j.expneurol.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Y, Yang X, Ge X, Zhang F. Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid hemorrhage mice. Biomed Pharmacother. 2019c;109:726–733. doi: 10.1016/j.biopha.2018.10.161. [DOI] [PubMed] [Google Scholar]
- 128.Zhang Y, Zhang T, Li Y, Guo Y, Liu B, Tian Y, Wu P, Shi H. Metformin attenuates early brain injury after subarachnoid hemorrhage in rats via AMPK-dependent mitophagy. Exp Neurol. 2022a;353:114055. doi: 10.1016/j.expneurol.2022.114055. [DOI] [PubMed] [Google Scholar]
- 129.Zhang Z, Ma Z, Yan C, Pu K, Wu M, Bai J, Li Y, Wang Q. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav Brain Res. 2019d;356:322–331. doi: 10.1016/j.bbr.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Z, Zhang A, Liu Y, Hu X, Fang Y, Wang X, Luo Y, Lenahan C, Chen S. NeW MECHANISMS AND TARGETS OF SUBARACHNOID HEMORRHAGE: A FOCUS ON MITOCHondria. Curr Neuropharmacol. 2022b;20:1278–1296. doi: 10.2174/1570159X19666211101103646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang Z, Liu J, Fan C, Mao L, Xie R, Wang S, Yang M, Yuan H, Yang X, Sun J, Wang J, Kong J, Huang S, Sun B. The GluN1/GluN2B NMDA receptor and metabotropic glutamate receptor 1 negative allosteric modulator has enhanced neuroprotection in a rat subarachnoid hemorrhage model. Exp Neurol. 2018b;301:13–25. doi: 10.1016/j.expneurol.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 132.Zhao Q, Tian Z, Zhou G, Niu Q, Chen J, Li P, Dong L, Xia T, Zhang S, Wang A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics. 2020;10:4822–4838. doi: 10.7150/thno.42387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhou J, Yang Z, Shen R, Zhong W, Zheng H, Chen Z, Tang J, Zhu J. Resveratrol improves mitochondrial biogenesis function and activates PGC-1αpathway in a preclinical model of early brain injury following Subarachnoid Hemorrhage. Front Mol Biosci. 2021;8:620683. doi: 10.3389/fmolb.2021.620683. [DOI] [PMC free article] [PubMed] [Google Scholar]