

Abstract
Within the last several years, molecular techniques have uncovered numerous 16S rRNA gene (rDNA) sequences which represent a unique and globally distributed lineage of the kingdom Crenarchaeota that is phylogenetically distinct from currently characterized crenarchaeotal species. rDNA sequences of members of this novel crenarchaeotal group have been recovered from low- to moderate-temperature environments (−1.5 to 32°C), in contrast to the high-temperature environments (temperature, >80°C) required for growth of the currently recognized crenarchaeotal species. We determined the diversity and abundance of the nonthermophilic members of the Crenarchaeota in soil samples taken from cultivated and uncultivated fields located at the Kellogg Biological Station’s Long-Term Ecological Research site (Hickory Corners, Mich.). Clones were generated from 16S rDNA that was amplified by using broad-specificity archaeal PCR primers. Twelve crenarchaeotal sequences were identified, and the phylogenetic relationships between these sequences and previously described crenarchaeotal 16S rDNA sequences were determined. Phylogenetic analyses included nonthermophilic crenarchaeotal sequences found in public databases and revealed that the nonthermophilic Crenarchaeota group is composed of at least four distinct phylogenetic clusters. A 16S rRNA-targeted oligonucleotide probe specific for all known nonthermophilic crenarchaeotal sequences was designed and used to determine their abundance in soil samples. The nonthermophilic Crenarchaeota accounted for as much as 1.42% ± 0.42% of the 16S rRNA in the soils analyzed.
The kingdom Crenarchaeota is one of the two kingdoms that comprise the archaeal domain. The members of the Crenarchaeota that have been isolated to date are extreme thermophiles that have optimal growth temperatures of more than 80°C. With certain exceptions, these extreme thermophiles are obligate anaerobes with sulfur-dependent metabolisms. Within the last several years, however, increasing numbers of crenarchaeotal 16S rRNA gene (rDNA) sequences have been recovered from low- to moderate-temperature environments. These sequences represent a unique lineage of the Crenarchaeota and have been obtained from environments that include the Pacific and Atlantic oceans (10–13, 22, 28, 34), freshwater sediments of North American lakes (16, 19, 31), the gut of a sea cucumber (24), the tissues of a sponge (28), agricultural soils from North America and Japan (5, 37), and forest soils from Europe and South America (7, 17). This collection of more than 100 16S rDNA sequences represents a diverse and globally distributed group of organisms that belong to the kingdom Crenarchaeota but are phylogenetically distinct from the thermophilic Crenarchaeota.
No member of the nonthermophilic Crenarchaeota group has been isolated and cultivated; therefore, the physiological characteristics of these organisms and their roles in ecosystems are unknown. It is presumed that these members of the Crenarchaeota are nonthermophilic based on the environments in which they have been found (temperatures, −1.5 to 32°C), their phylogenetic distance from the thermophilic members of the Crenarchaeota, and the low G+C contents of their 16S rDNA (51 to 58%) compared to the G+C contents of the thermophilic organisms (60 to 69%) (10, 16, 28). In addition, the abundance of nonthermophilic Crenarchaeota in the marine water column and in the oxic region of freshwater sediments suggests that certain members of the nonthermophilic Crenarchaeota are tolerant to oxygen (11, 19). The abundance of nonthermophilic crenarchaeotal rRNA found in picoplankton from cold ocean waters suggests that these organisms are ecologically relevant members of marine microbial communities (11, 19, 22). Members of the nonthermophilic Crenarchaeota have recently been identified in soils, but the abundance and significance of these organisms in soil microbial communities have not been assessed (5, 7, 17, 37).
In this paper we describe recovery, phylogenetic analysis, and quantification of crenarchaeotal 16S rRNA sequences in soil samples. Soil samples were taken from plots that historically had been cultivated with intensive agricultural practices or from nearby native plots that had never been cultivated. Total-community DNA was extracted from the soil, and 16S rDNA was amplified, cloned, and characterized by restriction fragment length polymorphism (RFLP) and sequence analyses. An oligonucleotide probe specific for all of the nonthermophilic Crenarchaeota was designed and tested. Total RNA was extracted from the soils, and the relative abundance of crenarchaeotal rRNA was determined by quantitative hybridization.
MATERIALS AND METHODS
Strains used.
The microorganisms used in this study were Arthrobacter globiformis ATCC 8010, Bacillus subtilis ATCC 6051, Cytophaga johnsonae ATCC 17061, Haloferax volcanii ATCC 29605, Methanobrevibacter sp. strain RFM-3 (18), Nitrosomonas europaea ATCC 25978, Pseudomonas aeruginosa ATCC 10145, and Serratia marcesens ATCC 13880. Most of the strains were cultivated by using the conditions recommended by the American Type Culture Collection (14); the only exception was Methanobrevibacter sp. strain RFM-3, which was grown as described by Leadbetter and Breznak (18).
Soil sampling.
Soil samples were obtained in May 1997 from the Michigan State University W. K. Kellogg Biological Station (KBS) Long-Term Ecological Research site located in Hickory Corners, Mich. Soil samples were obtained from both native and cultivated fields (descriptions of native [treatment 8] and cultivated [treatment 1] plots may be accessed at http://www.kbs.msu.edu). The native fields have never been farmed and are generally covered with vegetation consisting of a variety of perennial herbs and grasses. The cultivated fields have been farmed for more than 50 years and since 1988 have been under a regimen characterized by high levels of fertilization, herbicide addition, annual tillage, and a wheat-corn-soybean crop rotation. At the time of sampling, soybeans had been sown in the cultivated fields but had not germinated. Soil cores (depth, 10 cm; diameter, 2.5 cm) were taken from three replicate plots for each of the two treatments.
Soil cores were homogenized by using a 4-mm sieve, immediately frozen in liquid nitrogen, and stored at −80°C. Portions of samples were saved at 4°C in order to determine moisture contents, microscopically visible cell numbers, and numbers of CFU per gram of soil. The moisture content of a sample was determined by baking 10 g of soil at 80°C for more than 48 h and determining the decrease in mass due to desiccation. The total number of cells per gram of soil was determined by using the fluorescent stain 5-([4,6-dichlorotriazin-2-yl[amino)-fluorescein (DTAF) (Sigma) as previously described (6). The number of CFU per gram of soil was determined by diluting and dispersing cells in a buffered salt solution (0.85% sodium chloride, 50 mM sodium phosphate; pH 8) and plating the solution onto R2A agar medium (Difco). The plates were incubated at 30°C, and the colonies were counted after 48 h.
Nucleic acid extraction and analysis.
Sufficient quantities of DNA suitable for use in PCR amplification experiments were readily obtained from 1 g of soil by using the method of Purdy et al. (29); however, this method did not provide sufficient amounts of nucleic acids for filter hybridization experiments. Therefore, a modified method was used to obtain total nucleic acids from soils, as described below. Ten grams of soil was suspended in 20 ml of homogenization buffer (4 M guanidium isothiocyanate, 200 mM sodium phosphate [pH 8], 25 mM sodium citrate, 0.5% N-lauryl sarcosine) (26) and then combined with 20 g of 0.1-mm-diameter zirconia-silica beads (Biospec Products). To lyse the soil microorganisms, samples were disrupted in a bead beater (Biospec Products) for two 1-min cycles on ice. The particulate matter fraction was removed by centrifugation at 5,000 × g for 10 min. The supernatant fraction was collected, and the pellet was washed with 20 ml of homogenization buffer. The supernatants were pooled, combined with 0.1 volume of 5 M sodium chloride and 0.5 volume of 50% polyethylene glycol 8000, and incubated for 2 h at 4°C to precipitate the nucleic acids. The nucleic acids were recovered by centrifugation at 15,000 × g for 30 min. The pellet was washed with 70% ethanol and resuspended in 2 ml of 120 mM sodium phosphate buffer (pH 7.2). Then the nucleic acids were extracted with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1) (pH 4.7). Hydroxyapatite spin columns were used to remove humic acids by the method of Purdy et al. (29), with the following modifications: (i) 3-ml syringe barrels were used to provide a 2-ml hydroxyapatite bed volume; (ii) the columns were prewashed three times with 1 ml of 120 mM sodium phosphate (pH 7.2); (iii) 2-ml aqueous samples were added to hydroxyapatite columns; (iv) the loaded columns were then washed again as described above; and (v) the nucleic acids were eluted with 1 ml of 300 mM potassium phosphate (pH 7.2). The nucleic acids were desalted and precipitated (29) and then resuspended in 200 μl of RNase-free water. RNA was extracted from cultures by using a conventional bead beating protocol (33).
Nucleic acids were analyzed with a Lambda 3B spectrophotometer (Perkin-Elmer). The absorption of light by samples was determined at wavelengths between 220 and 320 nm, and point measurements were taken at 230, 260, and 280 nm. Absorption of light by humic acids occurs throughout the UV spectrum but can be most conveniently measured at 230 nm; therefore, absorption at 260 nm (A260)/A230 values provided an indication of humic acid contamination in nucleic acid samples (41). The total RNA concentrations of samples were estimated by using an orcinol reaction to determine ribose concentrations (9).
PCR amplification and cloning of Crenarchaeota 16S rDNA.
DNA purified from soil was used as a template for PCR. The archaea-specific primers used in the PCR included primer 89Fb (5′-ACGGCTCAGTAACRC-3′), modified from the primer described by Hershberger et al. (16), and primer Arc915R (5′-GTGCTCCCCCGCCAATTCCT-3′) (32). This primer pair was designed to amplify DNA from the nonthermophilic Crenarchaeota but may also amplify DNA from some members of the thermophilic Crenarchaeota and the Euryarchaeota. PCR were performed by using 100-μl mixtures containing 200 ng of template DNA, each primer at a concentration of 30 pM, each deoxynucleoside triphosphate at a concentration of 50 μM (Boehringer Mannheim), 0.05% Nonidet P-40, 0.05% bovine serum albumin, 2.5 U of Taq polymerase (Gibco), and 10 μl of PCR buffer (supplied with enzyme). The reactions were performed with a Gene Amp model 9600 thermocycler (Perkin-Elmer). Each PCR amplification included a 4-min hold at 94°C, followed by 30 cycles consisting of 1.5 min at 94°C, 1.5 min at 48°C, and 2 min at 72°C. After amplification, an additional extension step consisting of 15 min at 72°C was performed. Positive controls (mixtures containing 200 ng of SCA1145 plasmid as the template [5]) and negative controls (mixtures containing no template) were included. Amplified products from environmental samples were directly cloned by using a TOPO-TA cloning kit (Invitrogen).
As a preliminary screening step to eliminate redundancy, the amplified 16S rDNA of 25 clones from each soil sample were digested with a pair of restriction enzymes to determine RFLP patterns. PCR amplification of clones was performed as described above except that an inoculating loopful of colony material from each clone was used as the template. The amplified 16S rDNA fragments were digested with HinP1I and MspI (New England Biolabs) and resolved on a 2.5% NuSieve gel (FMC BioProducts). Six clones from each soil sample, representing unique restriction patterns, were selected for sequencing. The clones were sequenced with a model 373A DNA sequencer (Applied Biosystems Inc.) by using dideoxy dye terminator chemistry. The primers used for sequencing were primers 89Fb, Arc915R, and Cren745R (see below). The clones were screened for the presence of chimeras with the CHIMERA_CHECK algorithm (www.cme.msu.edu/RDP) (21).
Phylogenetic analyses.
Phylogenetic analyses were performed by using the programs ARB (www.mikro.biologie.tu-muenchen.de) (35), PAUP (36), and MacClade (20). Previously published crenarchaeotal clone sequences were obtained from public databases and were inserted along with our cloned sequences into the ARB environment. The sequences were initially aligned by using the ARB automatic aligner and then were verified and corrected manually. Regions of ambiguous alignment were identified and excluded from subsequent phylogenetic analyses. Phylogenetic trees were generated by performing neighbor-joining (30), parsimony (33), and maximum-likelihood analyses (27). Phylogenetic trees were assembled by using MacClade and were rearranged manually to generate the most parsimonious trees. In addition, transversion distance analyses were performed in ARB by considering only transversion events during construction of trees by the neighbor-joining method. During tree construction the sequences belonging to the Crenarchaeota and outgroup sequences were varied.
Oligonucleotide probe design and characterization.
The oligonucleotide probe Cren745 was designed with the ARB program (35) to target 16S rRNA from members of the nonthermophilic Crenarchaeota. The Oligonucleotide Probe Database (www.cme.msu.edu/OPD) (1) designation for Cren745 is S-*-Cren-0745-a-A-19. The dissociation temperature of Cren745 was determined empirically by membrane hybridization as previously described (32) by using rRNA transcribed in vitro from clone SCA1145 (5). To transcribe the SCA1145 clone rRNA, the pGEM-11ZF (Promega) backbone was cut by using EcoRI and HindIII (Boehringer Mannheim), and the rRNA was transcribed by using SP6 RNA polymerase, as indicated by the Riboprobe system (Invitrogen). The specificity of Cren745 was determined empirically by hybridizing the probe to 100, 50, and 25 ng of either the transcribed SCA1145 target RNA or various nontarget RNA (see above) by using the hybridization conditions described below.
Quantitative filter hybridization.
Quantitative filter hybridizations were performed as previously described, with certain exceptions (10). Nucleic acids from soil samples and cultures were denatured with 0.5% glutaraldehyde–50 mM Na2HPO4 and were serially diluted to provide a range of sample concentrations for blotting. Nucleic acids were blotted onto nylon membranes with a dot blot device and were immobilized by using UV cross-linking (Stratalinker; Stratagene). The membranes were prehybridized and hybridized by using 32P-labeled oligonucleotide probes as previously described (10). Replicate filters were prepared and used for hybridization with either Univ1390 (2), Arc915 (31), or Cren745. All hybridizations were carried out for more than 12 h at 45°C; the filters were washed for 30 min at 45°C and then for an additional 30 min at 45°C for Univ1390, at 56°C for Arc915, or at 60°C for Cren745 (10). Specifically bound probe was quantified by using a radioanalytic imaging system (AMBIS, Inc.).
To calculate the relative abundance of nonthermophilic Crenarchaeota in samples, the slopes of the probe binding curves were determined for serial dilutions of controls and environmental samples. The abundance was then calculated by determining the ratio of probe Cren745 binding to probe Univ1390 binding; controls were used to account for nonspecific binding and differences in probe-specific activities, as previously described (10, 15). To calculate the amount of Crenarchaeota 16S rRNA (in nanograms) per gram of soil, the relative abundance determined for samples was normalized to the estimated total amount of 16S rRNA present in soil samples.
Nucleotide sequence accession numbers.
The nucleotide sequences of KBS 16S rDNA clones have been deposited in the GenBank database under accession no. AFO58719 through AFO58730.
RESULTS
Soil extraction protocol.
Table 1 lists some of the characteristics of the soils and extracted nucleic acids analyzed in this study. Soil samples from the native field possessed a notably higher moisture content than soil samples from the cultivated field. This is not surprising as the native field had a dense vegetation cover capable of retaining moisture, while the cultivated field was devoid of vegetation at the time of sampling. The native field and cultivated field samples supported similar numbers of microorganisms, as determined by microscopic counts and by CFU counts on R2A agar media. For samples from both sites the proportion of microscopically visible cells growing on plates was quite low (∼0.33%), as demonstrated previously for soil samples (2). Despite the similarities in population sizes, the total RNA yields from native field samples were considerably higher than the total RNA yields from cultivated fields. The nucleic acids isolated were relatively free of proteins and humic acids, as demonstrated by high A260/A280 and A260/A230 values. A possible source of bias when nucleic acids are extracted from soil is the potential for differential cell lysis, which could lead to misrepresentation of nucleic acid concentrations from certain populations. To assess the extent of this problem, the lysis efficiency of the extraction procedure was measured. The bead beating protocol which we used disrupted 97.3% ± 0.8% of the cells present, as determined by microscopic counting of DTAF-stained cells before and after homogenization. The efficiency of RNA extraction from soil was estimated to be 19% ± 5.3%, as determined by spiking soil samples with known quantities of RNA and comparing actual RNA yields to expected yields.
TABLE 1.
Comparison of soils and nucleic acids extracted from native and cultivated plotsa
| Sample | H2O content (%) | CFU (106)/g (dry wt) of soil | Cells (109)/g (dry wt) of soilb | RNA concn (μg/g)c | A260/A230d | A260/A280d |
|---|---|---|---|---|---|---|
| Native | 9.2 ± 0.2 | 6.70 ± 0.90 | 2.37 ± 0.38 | 3.00 ± 0.62 | 1.71 ± 0.04 | 1.95 ± 0.07 |
| Cultivated | 5.9 ± 0.7 | 7.77 ± 2.27 | 2.02 ± 0.23 | 0.96 ± 0.06 | 1.89 ± 0.05 | 1.52 ± 0.11 |
The values are the means and standard deviations of multiple values obtained from three replicate plots.
Number of DTAF-stained cells per gram (dry weight) of soil, as determined by microscopy.
RNA concentration per gram (dry weight) of soil, as estimated by the orcinol reaction (9).
Nucleic acid sample absorption ratios.
Analysis of 16S rDNA clones.
Analysis of 35 16S rDNA clones resulted in identification of seven unique RFLP patterns. A total of 12 clones were sequenced; these clones included representatives exhibiting all of the RFLP patterns obtained from each sampling site. The phylogenic positions of clones from the KBS soils were determined relative to the positions of all previously described crenarchaeotal clones (Fig. 1). The environments in which the clones were found, the accession numbers, and references are presented in Table 2. Generation of the phylogenetic tree in Fig. 1 was complicated by the fact that many of the crenarchaeotal clones have been sequenced only partially and many of the sequences do not overlap. To overcome this difficulty, maximum-likelihood analysis was used to construct a tree that included 67 clones for which sequence data between Escherichia coli 16S rDNA positions 1 and 915 were available. Overlapping partial sequences were then added to this backbone tree by using the parsimony method and considering only regions in which there were sequence data for the clones. The validity of the tree was tested by generating alternative trees by the distance and parsimony analysis methods with various subsets of sequences that shared regions of sequence data. Parsimony and maximum-likelihood analyses were used to generate bootstrap values for the phylogenetic clusters containing nonthermophilic Crenarchaeota by using representative sequences from each group (Fig. 2).
FIG. 1.

Phylogenetic tree showing the relationships of nonthermophilic Crenarchaeota 16S rDNA sequences. The environments from which clones were recovered and the studies in which the clones were examined are listed in Table 2. The symbols indicate the specificities of crenarchaeotal probe Cren499R (8) (⧫), probes Cren667 (11) and GI-554 (22) (■), and probe Cren745 (this study) (•). The sequences determined in this study are indicated by boldface type. C. symbiosum, Cenarchaeum symbiosum.
TABLE 2.
Information concerning the crenarchaeotal clones shown in Fig. 1
| Cluster | Designation prefix(es) | Environment where found | Accession no. | Reference |
|---|---|---|---|---|
| Uncertain | pSL | Hot spring, Yellowstone National Park, Wyoming | U46338-U46371 | 3 |
| pJP | Hot spring, Yellowstone National Park, Wyoming | L25300-L25309, L25852 | 4 | |
| Freshwater | pLemB | Freshwater sediment, Lake Lemon, Indiana | —a | 16 |
| pGrfB | Freshwater sediment, Lake Griffy, Indiana | — | 16 | |
| pLAW | Freshwater sediment, Lawrence Lake, Michigan | U77568-U77575 | 31 | |
| Marine | SB95 | Marine picoplankton, Santa Barbara Channel, California | U78195-U78206 | 22 |
| SBAR | Marine picoplankton, Santa Barbara Channel, California | M88057, M88058 | 10 | |
| NH | Marine picoplankton, Pacific Ocean, San Diego, California | Z11569-Z11573 | 12 | |
| PVA | Marine picoplankton, Pacific Ocean, Hawaii | U46679, U46680 | 25 | |
| Fosmid 4B7 | Marine picoplankton, Pacific Ocean, Oregon | U39635 | 34 | |
| ANT12 | Marine picoplankton, Arthur Harbor, Antarctica | U11043 | 11 | |
| WHARQ | Marine picoplankton, Atlantic Ocean, Woods Hole, Massachusetts | M88079 | 10 | |
| C. symbiosum | Marine sponge tissue, Pacific Ocean | U51469 | 28 | |
| JM | Sea cucumber midgut, Atlantic Ocean | L24195-L24201 | 24 | |
| LMA | Freshwater sediment, Lake Michigan, Wisconsin | U87515-U87520 | 19 | |
| Terestrial | KBS | Agricultural soil, Michigan | AFO58719-AFO58730 | This study |
| PAD16, FIE16 | Agricultural soil, Japan | D26206, D26266 | 37 | |
| SCA | Agricultural soil, Wisconsin | U62811-U62820 | 5 | |
| pM17, pP17 | Forest soil, eastern Amazon, Brazil | U68605, U68654 | 7 | |
| pLemA | Freshwater sediment, Lake Lemon, Indiana | — | 16 | |
| pGrfA | Freshwater sediment, Lake Griffy, Indiana | — | 16 | |
| FFSB | FFSB | Forest soil, northern Finland | X96688-X96696 | 17 |
FIG. 2.

Phylogenetic tree generated by using maximum-likelihood analysis for 740 nucleotide positions between E. coli 16S rDNA positions 1 and 915. The bootstrap value to the left of each backslash was generated by using maximum-likelihood analysis, and the value to the right was generated by using parsimony analysis. Bootstrap values that were less than 50% are indicated by two asterisks. Scale bar = 10% difference between nucleotide sequences. S. shibatae, Sulfolobus shibatae; P. occultum, Pyrodictum occultum.
All trees supported the monophyletic grouping of clones within the FFSB, marine, and terrestrial clusters, as well as the relative branching order of these groups, as indicated by the bootstrap values associated with the groups (Fig. 2). The phylogenetic positions of the freshwater cluster and the group composed of clones pSL1, pSL69, pSL123, pJP44, and pJP89 were somewhat variable, as reflected by the poor bootstrap values associated with the positions of these organisms (Fig. 2). Although the members of these two groups typically exhibited close affiliations with one another, their ancestries alternated between affiliation with the thermophilic Crenarchaeota and affiliation with the nonthermophilic Crenarchaeota, depending on the method used to generate the tree. Crenarchaeotal clones pGrfB286, pSL4, pSL17, pSL22, pSL55, pSL78, and pSL79 are not shown in Fig. 1 as their phylogenetic positions were found to be highly variable, alternating between positions close to the freshwater cluster, positions within the thermophilic Crenarchaeota, and positions ancestral to the Crenarchaeota (3).
Cren745 design and characterization.
Several 16S rRNA-targeted probes that are specific for certain crenarchaeotal taxa have been described (Fig. 1). None of these probes, however, is complementary to crenarchaeotal sequences that have been found in the soil. The probe which we designed, Cren745, recognized more than 95% of the 16S rRNA sequences of members of the nonthermophilic Crenarchaeota, including sequences found in the soil (Fig. 1). The melting profile of Cren745 hybridized to target RNA was empirically determined in order to determine the hybridization conditions required for stringency. The temperature at which one-half of the bound probe was removed was found to be 61°C. Outside the nonthermophilic crenarchaeotal lineage, Cren745 is not complementary to any known rRNA sequence. The negative controls used for hybridization with Cren745 were chosen to represent phylogenetically diverse microorganisms but included organisms (H. volcanii, Methanobrevibacter sp. strain RFM-3) that represented the most similar nontarget rRNA sequences known (Fig. 3). Hybridization experiments in which Cren745 was tested with target and nontarget nucleic acids demonstrated that the probe provided the desired specificity when it was used as described above (Fig. 3).
FIG. 3.

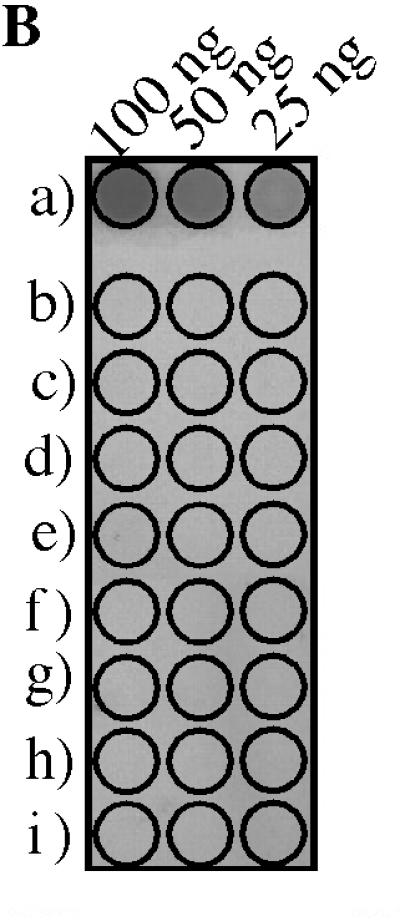
Cren745 oligonucleotide probe sequence aligned with its target sequence from nonthermophilic crenarchaeotal 16S rRNA and nontarget sequences used as negative controls. Bases not shared with the target sequence are indicated, while bases shared with the target sequence are indicated by dots. To demonstrate specificity, Cren745 was hybridized to 100, 50, and 25 ng of total RNA from each of the controls. M. RFM-3, Methanobrevibacter sp. strain RFM-3.
Quantification of moderate crenarchaeotal 16S rRNA in soil samples.
Probe Cren745 was used to determine the contribution of nonthermophilic crenarchaeotal 16S rRNA to total community rRNA. The relative abundance of nonthermophilic crenarchaeotal rRNA was lower in the native soils (0.37% ± 0.13%) than in the cultivated soils (1.42% ± 0.59%) (Fig. 4A), although the difference was not found to be significant by an unpaired t test. In cultivated soils, Archaea 16S rRNA comprised 1.5% ± 0.59% of the total 16S rRNA, as determined with the domain level archaeal probe Arc915 (Fig. 4A). It should be noted, however, that the amounts of nonthermophilic crenarchaeotal 16S rRNA per gram (dry weight) of soil were practically the same in native fields (12.1 ± 10.2 ng/g) and cultivated fields (15.3 ± 11.7 ng/g) (Fig. 4B). This finding reflected the relationships between the relative abundance of 16S rRNA and the total amount of rRNA in the soil samples.
FIG. 4.

(A) Relative abundance of nonthermophilic crenarchaeotal 16S rRNA in soil samples from native (Nat Cren) or cultivated (Cul Cren) fields, as well as relative abundance of archaeal 16S rRNA in the cultivated field samples (Cul Arc). (B) Amounts of crenarchaeotal 16S rRNA per gram (dry weight) of soil, as estimated by normalizing the abundance of 16S rRNA to the total amount of community 16S rRNA recovered from the soils. The error bars indicate sample standard errors; the sample sizes were 9 for native field samples and 8 for cultivated field samples.
DISCUSSION
Phylogenetic analysis of crenarchaeotal 16S rDNA clones recovered from low- to moderate-temperature environments revealed that these clones belong to at least four distinct groups which appear to have a common ancestry. We described this group of environmental clones as the nonthermophilic Crenarchaeota to distinguish them from other members of the Crenarchaeota. The majority of the sequences used in this phylogenetic analysis have been described previously (Table 2). For the sake of consistency, the names of groups which contained sequences from independent studies were chosen based on the environments from which the majority of the clones were obtained. The terrestrial cluster contains sequences that were primarily recovered from soil samples, although it should be noted that some sequences found in freshwater sediments also fell in the terrestrial cluster (pGrfA, pLemA). Most of the sequences in the marine cluster were found in marine systems; the only exceptions were four sequences recovered from freshwater sediments (LMA137, LMA226, LMA229, and LMA238). The FFSB cluster is limited to the sequences identified in a single study of boreal soil from Finland (17), and the freshwater cluster contains sequences found exclusively in freshwater sediments.
On the basis of a phylogenetic analysis that included the pLEM and pGrf environmental 16S rDNA clones, Hersberger et al. (16) proposed that the ability to grow at low to moderate temperatures arose independently at least three times in the Crenarchaeota. The results of analyses that included the additional rDNA sequences currently available are consistent with a monophyletic grouping of the nonthermophilic Crenarchaeota provided that clones pSL12 and pSL77, which were recovered from a hot spring, represent allochtonous organisms that were washed into the hot spring from a moderate-temperature environment. A finding which supports this hypothesis is the observation that the G+C contents of pSL12 and pSL77 16S rDNA (57 and 58%, respectively) are similar to the G+C contents of the rDNAs of nonthermophilic Crenarchaeota (51 to 58%) and fall outside the range of the G+C contents of the rDNAs of thermophilic Crenarchaeota (60 to 69%). Another complication in resolving the evolution of low- to moderate-temperature growth in the Crenarchaeota is the uncertain placement of the sequences of the freshwater cluster and clone pGrfB286. The relative positions of these sequences are dependent on the method and sequences used to generate phylogenetic trees. The phylogeny presented here indicates that members of the nonthermophilic Crenarchaeota are distinct from members of the thermophilic Crenarchaeota, but the data are not sufficient to conclude whether the ability to grow in low- to moderate-temperature environments has evolved once or multiple times in the crenarchaeotal lineage.
In soil samples taken from fields with distinct treatment histories, amplification, cloning, and RFLP screening of 16S rDNA resulted in identification of 12 unique crenarchaeotal 16S rDNA sequences. Phylogenetic analysis of the KBS sequences revealed that they were associated with the sequences in the terrestrial cluster (Fig. 1). The KBS sequences do not appear to have a common ancestor in the terrestrial cluster but are distributed throughout this group. In addition, there appears to be no relationship between the history of treatment of a soil and the phylogenetic position of the sequences from that soil. The clones from the native field are as likely to be related to clones from the cultivated field as they are to other native field clones, and the opposite is true as well.
Using oligonucleotide probes specific for 16S rRNA, we determined the relative abundance of nonthermophilic crenarchaeotal 16S rRNAs in the native and cultivated soil samples. The concentration of rRNA in a cell generally increases with growth rate, and so the abundance of rRNA in an environmental sample is a function of both the growth rate and the population size of the organism under consideration (39). The contribution of the nonthermophilic crenarchaeotal 16S rRNA to the total community 16S rRNA was lower in native samples (0.37% ± 0.13%) than in cultivated samples (1.42% ± 0.59%) (Fig. 4A). This observation could be explained either by lower amounts of crenarchaeotal rRNA or by larger contributions of rRNA from bacterial populations in native samples. When the percentage of nonthermophilic crenarchaeotal 16S rRNA was normalized to the total amount of rRNA per gram (dry weight) of soil, the actual sizes of the nonthermophilic crenarchaeotal 16S rRNA pools were similar in the native and cultivated samples (Fig. 4B). The differences in abundance were therefore due to increased contribution of 16S rRNA from organisms other than Crenarchaeota.
In samples from cultivated fields, archaeal 16S rRNA was found to account for 1.5% ± 0.59% of the total community rRNA. A previous study in which fluorescent in situ hybridization was used showed that Archaea account for 0.21% ± 0.65% of the microscopically detectable cells in a forest soil (40). These two values, determined by independent methods, confirm that the Archaea represent a measurable component of soil microbial communities. The archaeal 16S rRNA abundance determined for cultivated fields was nearly equivalent to the abundance of nonthermophilic Crenarchaeota in the same fields (Fig. 4A). The similarity in the abundance values for the Archaea and nonthermophilic Crenarchaeota in cultivated fields suggests that these Crenarchaeota represent a majority of the archaea in the cultivated field soil samples.
Molecular approaches have demonstrated that nonthermophilic Crenarchaeota are found in diverse environments and are globally distributed; however, the physiological characteristics and ecological significance of these organisms remain unknown. The phylogeny presented in this paper suggests that the nonthermophilic Crenarchaeota may have a common ancestor and that there are several distinguishable groups within this lineage. We describe the use of a new probe that is specific for all of the currently identified members of the nonthermophilic Crenarchaeota and the presence and abundance of this group in soil samples from the KBS in Hickory Corners, Mich. There were no detectable differences in the diversity or abundance of Crenarchaeota in the fields sampled despite considerable differences in the disturbance history and plant community diversity associated with these plots. Further investigations are needed to characterize the distribution and abundance of the globally distributed nonthermophilic Crenarchaeota, to understand the ecological significance of these organisms, and to help design strategies for their enrichment and isolation.
ACKNOWLEDGMENTS
We are grateful to Robert M. Goodman and Scott B. Bintrim for providing crenarchaeotal 16S rDNA clones, to Jared R. Leadbetter for providing cultures of Methanobrevibacter sp. strain RFM-3, to Bernard M. Schroeter for sequencing, and to Bradley S. Stevenson, Joel A. Klappenbach, and John W. Urbance for their helpful comments.
This research was sponsored by grant DE-FG02-96ER62210 from the Department of Energy as part of the NABIR program and by grant DEB 9120006 from the NSF Center for Microbial Ecology.
ADDENDUM
A recent study identified four nonthermophilic Crenarchaeota 16S rDNA sequences obtained from suspended particulate matter in the North Sea and one sequence obtained from the digestive tract of a flounder. The phylogeny of these five 16S rDNA sequences indicates that they fall in the marine cluster of the nonthermophilic Crenarchaeota (38). In addition, McInerney et al. (23) identified 10 Crenarchaeota sequences not included in our analyses that also fall in the marine cluster. A phylogenetic analysis of the Crenarchaeota marine cluster (marine archaea group I) by McInerney et al. (23) indicated that there is great phylogenetic distance between these sequences and the thermophilic Crenarchaeota and supported the hypothesis that the nonthermophilic Crenarchaeota are distinct from the thermophilic Crenarchaeota.
REFERENCES
- 1.Alm E W, Oerther D B, Larsen N, Stahl D A, Raskin L. The oligonucleotide probe database. Appl Environ Microbiol. 1996;62:3557–3559. doi: 10.1128/aem.62.10.3557-3559.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann R I, Ludwig W, Schleifer K-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barns S M, Delwiche C F, Palmer J D, Pace N R. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc Natl Acad Sci USA. 1996;93:9188–9193. doi: 10.1073/pnas.93.17.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barns S M, Fundyga R E, Jeffries M W, Pace N R. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc Natl Acad Sci USA. 1994;91:1609–1613. doi: 10.1073/pnas.91.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bintrim S B, Donohue T J, Handelsman J, Roberts G P, Goodman R M. Molecular phylogeny of archaea from soil. Proc Natl Acad Sci USA. 1997;94:277–282. doi: 10.1073/pnas.94.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloem J, de Ruiter P C, Koopman G J, Lebbink G, Brussard L. Microbial numbers and activity in dried and rewetted arable soil under integrated and conventional management. Soil Biol Biochem. 1992;24:655–665. [Google Scholar]
- 7.Borneman J, Triplett E W. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl Environ Microbiol. 1997;63:2647–2653. doi: 10.1128/aem.63.7.2647-2653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burggraf S, Mayer T, Amann R, Schadhauser S, Woese C R, Stetter K O. Identifying members of the domain Archaea with rRNA-targeted oligonucleotide probes. Appl Environ Microbiol. 1994;60:3112–3119. doi: 10.1128/aem.60.9.3112-3119.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniels L, Hanson R S, Phillips J A. Chemical analysis. In: Gerhardt P, Murray R G E, Wood W A, Krieg N R, editors. Methods for general and molecular bacteriology. Washington, D.C: American Society for Microbiology; 1994. p. 536. [Google Scholar]
- 10.DeLong E F. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLong E F, Wu K Y, Prezelin B B, Jovine R V M. High abundance of Archaea in Antarctic marine picoplankton. Nature. 1994;371:695–697. doi: 10.1038/371695a0. [DOI] [PubMed] [Google Scholar]
- 12.Fuhrman J A, McCallum K, Davis A A. Novel major archaebacterial group from marine plankton. Nature. 1992;356:148–149. doi: 10.1038/356148a0. [DOI] [PubMed] [Google Scholar]
- 13.Fuhrman J A, McCallum K, Davis A A. Phylogenetic diversity of subsurface marine microbial communities from the Atlantic and Pacific oceans. Appl Environ Microbiol. 1993;59:1294–1302. doi: 10.1128/aem.59.5.1294-1302.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gherna R, Pienta P, Cote R, editors. Catalogue of bacteria and phages. 18th ed. Rockville, Md: American Type Culture Collection; 1992. [Google Scholar]
- 15.Giovannoni S J, Britsschgi T B, Moyer C L, Field K G. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- 16.Hershberger K L, Barns S M, Reysenbach A-L, Dawson S C, Pace N R. Wide diversity of Crenarchaeota. Nature. 1996;384:420. doi: 10.1038/384420a0. [DOI] [PubMed] [Google Scholar]
- 17.Jurgens G, Lindstrom K, Saano A. Novel group within the kingdom Crenarchaeota from boreal forest soil. Appl Environ Microbiol. 1997;63:803–805. doi: 10.1128/aem.63.2.803-805.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leadbetter J R, Breznak J A. Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl Environ Microbiol. 1996;62:3620–3631. doi: 10.1128/aem.62.10.3620-3631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacGregor B J, Moser D P, Alm E W, Nealson K H, Stahl D A. Crenarchaeota in Lake Michigan sediment. Appl Environ Microbiol. 1997;63:1178–1181. doi: 10.1128/aem.63.3.1178-1181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maddison W P, Maddison D R. MacClade: analysis of phylogeny and character evolution, 3.0. Sunderland, Mass: Sinauer Associates, Inc.; 1992. [Google Scholar]
- 21.Maidak B L, Olsen G J, Larsen N, Overbeek R, McCaughey M J, Woese C R. The Ribosomal Database Project. Nucleic Acids Res. 1997;25:109–111. doi: 10.1093/nar/25.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massana R, Murray A E, Preston C M, DeLong E F. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara channel. Appl Environ Microbiol. 1997;63:50–56. doi: 10.1128/aem.63.1.50-56.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McInerney J O, Mullarkey M, Wernecke M E, Powell R. Phylogenetic analysis of group I marine archaeal rRNA sequences emphasizes the hidden diversity within the primary group Archaea. Proc R Soc Lond B Biol Sci. 1997;264:1663–1669. [Google Scholar]
- 24.McInerney J O, Wilkinson M, Patching J W, Embley T M, Powell R. Recovery and phylogenetic analysis of novel archaeal rRNA sequences from a deep-sea deposit feeder. Appl Environ Microbiol. 1995;61:1646–1648. doi: 10.1128/aem.61.4.1646-1648.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moyer C L, Tiedje J M, Dobbs F C, Karl D M. Diversity of deep-sea hydrothermal vent Archaea at Loihi seamount, Hawaii. Deep Sea Res. 1998;45:303–317. [Google Scholar]
- 26.Ogram A, Sun W, Brockman F J, Fredrickson J K. Isolation and characterization of RNA from low-biomass deep-subsurface sediments. Appl Environ Microbiol. 1995;61:763–768. doi: 10.1128/aem.61.2.763-768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen G J, Matsuda H, Hagstrom R, Overbeek R. FasDNAml: a tool for construction of phylogenetic trees of DNA sequences using maximum liklihood. Comput Appl Biosci. 1994;10:41–48. doi: 10.1093/bioinformatics/10.1.41. [DOI] [PubMed] [Google Scholar]
- 28.Preston C M, Wu K Y, Molinski T F, DeLong E F. A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc Natl Acad Sci USA. 1996;93:6241–6246. doi: 10.1073/pnas.93.13.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purdy K J, Embley T M, Takii S, Nedwell D B. Rapid extraction of DNA and rRNA from sediments by a novel hydroxyapatite spin-column method. Appl Environ Microbiol. 1996;62:3905–3907. doi: 10.1128/aem.62.10.3905-3907.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 31.Schleper C, Holben W, Klenk H-P. Recovery of crenarchaeotal ribosomal DNA sequences from freshwater-lake sediments. Appl Environ Microbiol. 1997;63:321–323. doi: 10.1128/aem.63.1.321-323.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stahl D A, Amann R. Development and application of nucleic acid probes in bacterial systematics. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. Chichester, England: John Wiley & Sons Ltd.; 1991. pp. 205–248. [Google Scholar]
- 33.Stahl D A, Flesher B, Mansfield H R, Montgomery L. Use of phylogenetically based probes for studies of ruminal microbial ecology. Appl Environ Microbiol. 1988;54:1079–1084. doi: 10.1128/aem.54.5.1079-1084.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein J L, Marsh T L, Wu K Y, Shizuya H, DeLong E F. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol. 1996;178:591–599. doi: 10.1128/jb.178.3.591-599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strunk O, Ludwig W. ARB: a software environment for sequence data, 2.1.1. Munich, Germany: Department of Microbiology, Technical University of Munich; 1996. [Google Scholar]
- 36.Swofford D L. PAUP, 3.0. Champaign: Illinois Natural History Survey; 1991. [Google Scholar]
- 37.Ueda T, Suga Y, Matsuguchi T. Molecular phylogenetic analysis of a soil microbial community in a soybean field. Eur J Soil Sci. 1995;46:415–421. [Google Scholar]
- 38.van der Maarel M J E C, Artz R R E, Haanstra R, Forney L J. Association of marine Archaea with the digestive tracts of two marine fish species. Appl Environ Microbiol. 1998;64:2894–2898. doi: 10.1128/aem.64.8.2894-2898.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward D M, Bateson M M, Weller R, Ruff-Roberts A L. Ribosomal RNA analysis of microorganisms as they occur in nature. Adv Microb Ecol. 1992;12:219–268. [Google Scholar]
- 40.Zarda B, Hahn D, Chatzinotas A, Schonhuber W, Neef A, Amann R I, Zeyer J. Analysis of bacterial community structure in bulk soil by in situ hybridization. Arch Microbiol. 1997;168:185–192. [Google Scholar]
- 41.Zhou J, Bruns M A, Tiedje J M. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62:316–322. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]