

Abstract
Adherens junctions (AJs) create spatially, chemically and mechanically discrete microdomains at cellular interfaces. Using a mechanogenetic platform that generates artificial AJs with controlled protein localization, clustering, and mechanical loading, we find that AJs also organize proteolytic hotspots for γ-secretase with a spatially-regulated substrate selectivity that is critical in the processing of Notch and other transmembrane proteins. Membrane microdomains outside of AJs exclusively organize Notch ligand-receptor engagement (LRE-µdomains) to initiate receptor activation. Conversely, membrane microdomains within AJs exclusively serve to coordinate regulated intramembrane proteolysis (RIP-µdomains). They do so by concentrating γ-secretase and primed receptors while excluding full-length Notch. AJs induce these functionally distinct microdomains by means of lipid-dependent γ-secretase recruitment and size-dependent protein segregation. By excluding full-length Notch from RIP-µdomains, AJs prevent inappropriate enzyme-substrate interactions and suppress spurious Notch activation. Ligand-induced ectodomain shedding eliminates size-dependent segregation, releasing Notch to translocate into AJs for processing by γ-secretase. This mechanism directs radial differentiative expansion of ventricular zone-neural progenitor cells in vivo and more broadly regulates the proteolysis of other large cell-surface receptors like amyloid precursor protein. These findings suggest an unprecedented role of AJs in creating size-selective spatial switches that choreograph γ-secretase processing of multiple transmembrane proteins regulating development, homeostasis, and disease.
Juxtacrine signaling occurs between cells that are in direct physical contact and orchestrates a wide range of cellular processes involved in development, physiology, and disease across multicellular organisms1,2. For example, Notch amplifies cellular differences during cell fate determination and pattern formation by a juxtacrine mechanism. Similarly, neural adhesion and immune receptors are critical in juxtacrine signaling events at neurological3–6 and immunological synapses7,8. Unlike diffusible ligands that interact with their receptors more uniformly across the cell surface, juxtacrine ligand-receptor pairs localize cell signaling activities to a signal-exchange interface. As two juxtaposed membranes are coupled chemically, spatially, and mechanically, the signal-exchange interface undergoes drastic reorganization that constrains the arrangement and activity of both protein and lipid components at the interface7–9. Cells can exploit these interfacial constraints to form specialized membrane compartments that regulate receptor activation, as exemplified by the kinetic segregation and liquid-liquid phase separation (LLPS) of immune receptor signaling10–16. However, many juxtacrine signaling interfaces beyond the immunological synapse can also create these constraints when coupled mechanically through the action of adhesion molecules. The structural and functional consequence of such constraints on signaling pathways outside those regulating immune cell function have not been explored extensively.
Here, we investigate specialized interfacial membrane compartments organized by adherens junctions (AJs) that create two physically and biochemically distinct microenvironments for the sequential molecular processing of Notch: one serves as a microdomain for ligand-receptor engagement (LRE-µdomain), and the other localizes the proteolytic activity of γ-secretase while effectively selecting only primed receptors for cleavage by size-dependent exclusion (RIP-µdomain). By employing mechanogenetics, spatial mutation, and CRISPR-Cas9 knockout approaches, we examine how this membrane microcompartmentalization choreographs the sequential activation of Notch both in vitro and in vivo. We further test whether these microdomains contribute to proteolytic processing of other large cell surface proteins such as amyloid precursor protein (APP) producing pathogenic amyloid beta (Aβ).
Notch activation steps occur in distinct membrane µdomains
Notch is a highly conserved mediator of contact-dependent cell-cell communication in metazoans17–19. Tight control of Notch activation is essential for many developmental processes20,21, while dysregulation of Notch activation can cause developmental, neurological, and immunological disorders and cancer19,22–25. Accordingly, to enable precise signal regulation, Notch activation occurs through multiple steps that are independently gated by sequential events including (i) ligand-receptor engagement, (ii) mechanical unfolding of the negative regulatory region (NRR) and proteolytic extracellular domain shedding (S2 cleavage), and (iii) regulated intramembrane proteolysis (S3 cleavage) finally releasing Notch intercellular domain (NICD) (Fig. 1a)17,19,26–30. Several macromolecular interactions are involved in this process, which include Notch ligands, metalloprotease (e.g., ADAM 10/17), and γ-secretase.
Fig. 1. Interfacial membrane compartmentalization organized by cadherin-based adherens junctions (AJs) creates two discrete microenvironments for the sequential molecular processing of Notch.
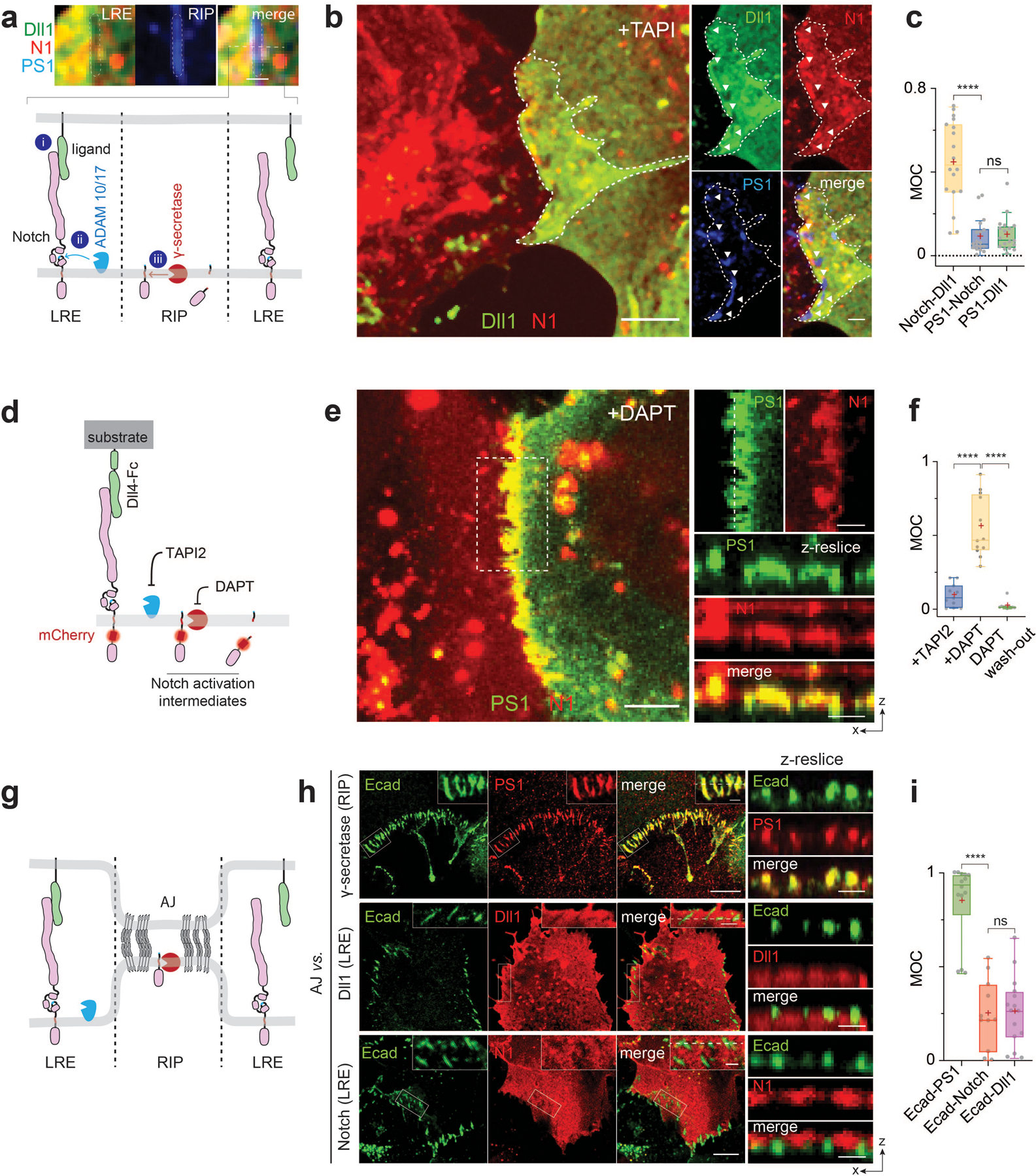
(a) A schematic showing membrane compartmentalization choreographing the sequential steps in cell-surface activation of Notch. LRE and RIP represent distinct membrane microdomains (µdomains) for Notch receptor-ligand engagement and regulated intramembrane proteolysis, respectively. (top) A representative image showing LRE and RIP µ-domains. Scale bar, 2 µm. (b) Representative confocal fluorescence images showing Dll1, Notch1 (N1), and presenilin1 (PS1) distributions at the interfacial membrane formed between cells co-expressing Notch1 and Dll1 in the presence of TAPI2. The area with a white dashed line indicates the cellular interface. (left) A maximum projection image of Dll1 and Notch1 constituting a LRE µdomain.[to authors: to save word space in your figure legend, you may combine the several descriptions at the end. I have provided an example of the scale bars; please verify if correct]. (right) Individual fluorescent channel and merged images for Dll1, N1, and PS1.[to authors: to save word space in your figure legend, you may combine the several descriptions at the end. I have provided an example of the scale bars; please verify if correct].. (c) Manders’ overlap coefficients (MOCs) of Notch with Dll1, PS1 with N1, and PS1 with Dll1, respectively. n = 18 cells examined for each condition, pooled over three independent experiments. (d) A schematic of spatial distribution of Notch intermediates during the cell-surface activation pathway. (e) Representative confocal images of N1 and PS1 within the RIP µdomains from the cells activated by culturing them on a Dll4-Fc immobilized substrate with DAPT. (left) A maximum projection image of enriched Notch-mCherry signal at RIP-µdomains. . (Top-right) Magnified individual fluorescence channel images of the boxed region.. (right) Z-resliced images showing the sections of the cellular interfaces. Scale bar, 4 µm. (f) MOCs of Notch1 with PS1 during sequential molecular processing of Notch1. n = (left to right) 11, 12, and 10 cells examined over two independent experiments. (g) A schematic showing AJ-mediated membrane compartmentalization that creates LRE- and RIP-microdomains. (h) Representative confocal fluorescence images of RIP- (PS1) and LRE- (Dll1 and N1) µdomains relative to AJs. (left) A maximum projection image. Inset shows a magnified image of the boxed area highlighting the membrane microdomains at cellular interfaces.. (right) z-resliced images. (i) MOCs of PS1, Dll1, and Notch1 localization with E-cadherin. n = (left to right) 15, 11, and 14 cells examined over two independent experiments. (c, f, i) For box and whisker plots, colored centre lines and (+) marks indicate median and mean, respectively. The boxes show the 25th to 75th percentiles, and the whiskers extend to the minima and the maxima. ****P < 0.0001, ns: non-significant; one-way ordinary ANOVA followed by Tukey’s multiple comparison. Scale bars are (a) 2 µm, (b, left) 5 µm, (b,right) 2 µm, (e, left) 10 µm, (e top right and right) 4 µm, (h) 2 µm.
To interrogate the spatial organization of these molecules, we formed Notch signaling interfaces by culturing cells co-expressing both SNAP-Notch and Halo-Dll1. We prevented Notch proteolysis and activation by treating cells with TAPI2 or shRNA cocktails against ADAM 10/17 (Extended Data Fig. 1b) to explicitly visualize full-length Notch. Enriched fluorescence signals for SNAP-Notch and Halo-Dll1 were seen at the cellular interface, indicating accumulation of the engaged ligand-receptor pairs at the interface (i.e., LRE-µdomain) (Fig. 1a,b; Extended Data Fig. 1a). Conversely, we also observed regions within the interfacial membrane depleted of both fluorescence signals, suggesting that certain interfacial microdomains exclude Notch ligands and receptors (Fig. 1a,b). Curiously, the punctuated exclusion patterns matched cell-surface γ-secretase distribution visualized with an anti-presenilin-1 (anti-PS1) antibody (Fig. 1a–c; Extended Data Fig. 1a)31,32. This result indicates that Notch ligands/receptors and γ-secretase were compartmentalized into two different membrane microdomains at the signaling interface, potentially preventing their direct interactions.
Considering the requirement of Notch proteolysis by γ-secretase for its activation, we reasoned that cell-surface molecular processing steps for Notch activation (ligand-receptor engagement followed by S2 and then S3 proteolysis) may occur within distinct membrane regions. To test this notion, we investigated how each cleavage step during activation alters the spatial distribution of Notch. To promote accumulation of specific Notch activation intermediates following ADAM 10/17 and γ-secretase cleavage, we induced Notch activation by culturing cells on a Dll4-coated substrate while selectively inhibiting protease activities with TAPI2 (S2 cleavage) or DAPT (S3 cleavage), respectively (Fig. 1d, Extended Data Fig. 1). With S2 inhibition (+TAPI2), we observed Notch exclusion from microdomains enriched with γ-secretase (Extended Data Fig. 1a,b). In contrast, with S2 activation (-TAPI2) and S3 inhibition (+DAPT), we observed strong enrichment of mCherry signal within the γ-secretase-containing microdomain (Fig. 1e). The change in spatial organization indicates that Notch with extracellular domain truncation (NEXT), the product of S2-cleavage, translocated to and was then concentrated within the microdomains. Finally, when γ-secretase activity was rescued by washing out DAPT, the mCherry signal previously enriched at the γ-secretase-containing microdomain disappeared (Extended Data Fig. 1c), presumably corresponding to release of NICD resulting from S3-proteolysis of accumulated Notch within microdomain (i.e., RIP-µdomain). Mander’s overlap coefficients of mCherry over PS1 also confirmed translocation of Notch from LRE- to RIP-µdomains, and then NICD release intracellularly (Fig. 1f). These results support the notion that these two membrane microdomains serve distinct and necessary functions in Notch activation. They also raise the possibility that movement of Notch between these domains serves as a spatial switch regulating the interaction between Notch intermediates and γ-secretase, thereby choreographing sequential steps in Notch proteolysis. According to this model, γ-secretase cannot process the full-length Notch before S2 cleavage because enzyme and substrate are concentrated in distinct regions of the cell surface. Following S2 cleavage, translocation of NEXT into the RIP-µdomain facilitates a productive Notch-γ-secretase interaction, S3 cleavage predominantly within AJs but also in endosomes resulting from RIP-µdomain internalization, and then downstream signaling (Extended Data Fig. 1e,f).
AJs organize Notch signaling molecules at cellular interface
To gain insight into molecular features responsible for organizing these distinct membrane microdomains we imaged γ-secretase distribution across cell membranes. Interestingly, we observed many microdomains with strong γ-secretase signals at cell-cell interface37, but detected negligible γ-secretase signals at cell membranes distal from cell-cell contact, suggesting that the RIP-µdomains were formed at cellular interface exclusively (Extended Data Fig. 2a). Since the cell-cell interface is established and maintained by AJs in many tissues33–36, we reasoned that AJs may organize LRE- and RIP-µdomains (Fig. 1g). To test this notion, we visualized these µdomains and AJs in cells recombinantly expressing Dll1, Notch, and E-cadherin. When compared with AJs, RIP-µdomains containing γ-secretase showed nearly identical spatial distribution with AJs (Fig. 1h,i and Extended Data Fig. 2b,c) 31,32, while LRE-µdomains containing Dll1 and Notch exhibited inverse distribution (Fig. 1h and Extended Fig. 2d–g). ADAM 10/17 exhibited no preferential distribution relative to AJs (Extended Fig. 2h–j). Notch exclusion from AJs was observed in multiple contexts, including endogenous vs. recombinant Notch (Extended Data Fig. 2k), different cadherins, cell types, and cell polarization (Extended Data Fig. 2l–n), supporting the generality of AJ-mediated microdomain formation. Moreover, we observed Notch translocation into AJs after S2-cleavage, consistent with Notch relocalization from LRE- to RIP-µdomains (Extended Data Fig. 3a–e). These observations suggest two mechanisms by which AJs might define the compartmentalized microdomains: first, AJs recruit γ-secretase to form RIP-µdomains; second, AJs segregate Notch ligands and receptors away from RIP-µdomains, limiting ligand-receptor engagement outside of AJs.
AJs recruit γ-secretase through ordered membrane phases
We then investigated how AJs form RIP-µdomains. Several reports have suggested possible engagement of AJs with spatially discrete lipid membrane phases35,37–39. Similarly, γ-secretase proteolytic activity is closely linked to detergent-resistant membranes40–47. Both of these membrane features preferentially associate with Flotillin-1 (Flot1)35,45–48. We therefore visualized Flot1 localization across the cell membrane. Strong Flot1 and γ-secretase signals were seen at AJs (Fig. 2a,b and Extended Data Fig. 4a,b). To validate the formation of discrete membrane microdomains at AJs, we mapped lipid order in membrane using di-4-ANEPPDHQ 49,50. AJs displayed significantly higher general polarization () values than non-junctional membrane regions (Fig. 2c,d), supporting the notion that both AJs and γ-secretase are associated with common and long-lived ordered phases, otherwise known to be short-lived and transient when alone35. Additionally, clustering of E-cadherin, as occurs at AJs, triggers F-actin polymerization at the cytoplasmic leaflet of the membrane30. Given the established interaction between F-actin and membrane constituents like phosphatidylserine that stabilize Flot1-containing lipid microdomains, we reasoned that AJ components may anchor phosphatidylserine leading to formation of the membrane microdomains35,51,52. To test this notion, we performed a coarse-grained molecular dynamic simulation of a lipid membrane comprising of 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DIPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), N-palmitoyl-O-phosphocholineserine (PPCS), 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (DPPS), and cholesterol (Chol, a key component of the discrete lipid membrane phase). We immobilized a portion of DPPS in the inner leaflet to reflect its interaction with AJ components (e.g., F-actin). DPPS immobilization resulted in a microdomain having increased transbilayer coupling (Fig. 2e) and decreased lipid diffusion, supporting microdomain stabilization (Fig. 2f,g) 51.
Fig. 2. AJs form RIP-µdomains by recruiting γ-secretase through ordered lipid assemblies.
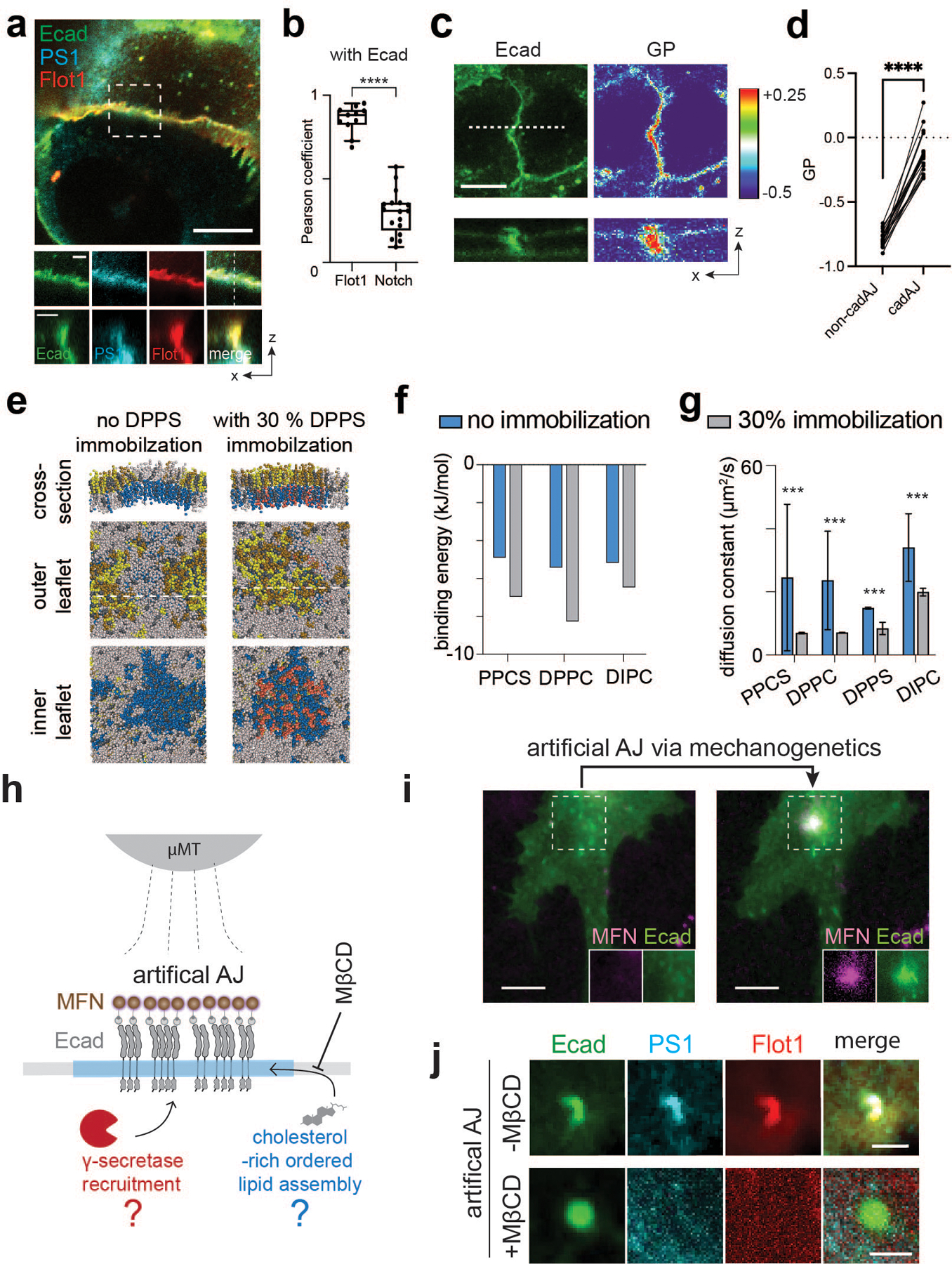
(a) Representative confocal fluorescence images showing the PS1 and Flot1 distribution relative to native AJs. (left) A maximum projection image of merged channels. Scale bar, 10 µm. (center) Magnified images of the boxed region. Scale bar, 2 µm. (right) z-resliced images showing the sections of the AJs. Scale bar, 2 µm. (b) MOCs of Flot1 and Notch colocalization with E-cadherin. For a box and whisker plot, colored centre lines and (+) marks indicate median and mean, respectively. The boxes show the 25th to 75th percentiles, and the whiskers extend to the minima and the maxima. n = (left to right) 11 and 17 cells examined over two independent experiments. (c) Di-4-ANEPPDHQ imaging of AJs. (top-left) a confocal fluorescence image of Ecad-GFP, and (top-right) the rainbow RGB -value image. (bottom) Z-resliced images showing a representative AJ (a white dashed line) of Ecad-GFP (left) and values (right). Scale bar, 4 µm. (d) A comparison of values from AJs and non-AJ regions. n = 11 cell-cell interfaces pooled from 3 independent experiments.(e) Snapshot images of coarse-grained MD simulation of a lipid bilayer comprising of DIPC (gray), DPPC (yellow), PPCS (green), DPPS (blue), immobilized DPPS (pink), and CHOL (grey). Left and right panels represent the simulation results without and with a partial (30%) DPPS immobilization, respectively. (f, g) Binding energy (f) and diffusion coefficients (g) of individual lipid components in a lipid bilayer with or without a partial (30%) DPPS immobilization in MD simulation shown in the panel (c). For diffusion coefficients (g), data are the mean ± s.d. of n = 3,000 particles analyzed over a MD simulation.(h) A schematic showing mechanogenetic interrogation of γ-secretase and ordered lipid microassemblies relative to the artificial AJs. Artificial AJs were formed by clustering Ecad-GFP labeled with magnetofluorescent nanoparticles (MFNs) by application of an external micromagnetic tweezer (µMT). MβCD was used for cholesterol depletion in the cell membrane before artificial AJ formation. (i) Epifluorescence images showing the formation of an artificial AJ by mechanogenetics. After stimulation by µMT, vivid MFN and E-cadherin signals at the magnetic focus were seen, indicating formation of AJs. Scale bar, 5 µm. Shown are representative from n = 4 independent experiments. (j) Confocal fluorescence images of E-cadherin, PS1, and Flot1 at the artificial AJ with and without MβCD treatment. After cholesterol depletion, no PS1 recruitment was seen at the AJ, suggesting that γ-secretase recruitment to the AJ requires lipid microdomain formation at the AJ. Scale bar, 2 µm. Shown are representative from n = 3 (-MβCD) and 5 (+MβCD) independent experiments. **** P<0.0001, ***P < 0.0001. For b,g: two-tailed unpaired Student’s t test; for d: two-tailed paired Student’s t test
To further confirm the role of discrete membrane phases in recruiting γ-secretase to AJs, we tested whether cholesterol-depletion disrupts γ-secretase localization within AJs. Because cholesterol depletion also destabilizes native AJs47, we instead generated artificial AJs by clustering E-cadherin via mechanogenetics33,58,59, while simultaneously depleting cholesterol with methyl-β-cyclodextrin (MβCD) (Fig. 2h,i). Similar to native AJs, vivid F-actin, Flot1, and PS1 signals were seen at the artificial AJ without MβCD treatment (Fig. 2j; Extended Data Fig. 4c–e), indicating that artificial AJs recapitulate the functional roles of native AJs including γ-secretase recruitment. In contrast, with MβCD, we observed no recruitments of F-actin, Flot1, or PS1 at the artificial AJ (Fig. 2j; Extended Data Fig. 4f,g). Further, artificial clusters of E-cadherin with intracellular domain truncation showed no recruitment of F-actin, Flot1, or PS1 (Extended Data Fig. 4h–j), suggesting that γ-secretase recruitment into AJs requires ordered lipid assemblies stabilized by molecular interactions and/or activities of the intracellular domain of E-cadherin. From these observations, we concluded that AJs form RIP-µdomains by recruiting and stabilizing γ-secretase through a common spatially discrete membrane assembly comprised of multiple lipid and protein (e.g., Flot1, actin) components. Nevertheless, these results do not preclude the possibility that more direct interactions between γ-secretase and other AJ-associated proteins also contribute to RIP-µdomain formation53.
AJs exclude Notch from RIP-µdomains due to their large size
We also visualized the spatial distribution of Notch signaling components relative to artificial AJs (Fig. 3a)30,54,55. Artificial AJs are free of membrane juxtaposition. Therefore, the influence of membrane juxtaposition on protein localization can be investigated by comparing receptor distributions in native AJs to artificial AJs. We imaged full-length Notch explicitly by treating cells with TAPI2. Surprisingly, we observed intense Notch localization at artificial AJs (Fig. 3b and Extended Data Fig. 5a–c), which contrasted starkly with the exclusion of Notch from native AJs (Fig. 1g–i). We reasoned that Notch could be excluded from native AJs because of the Notch extracellular domain (NECD, 136 nm) is much larger than the intermembrane cleft created by native AJs (20 nm)33,56. In contrast, artificial AJs lack a narrow intermembrane cleft, which permits access of Notch to the concentrated γ-secretase within the AJs57.
Fig. 3. Size-dependent protein segregation from AJs exclude Notch receptors from RIP-µdomains.
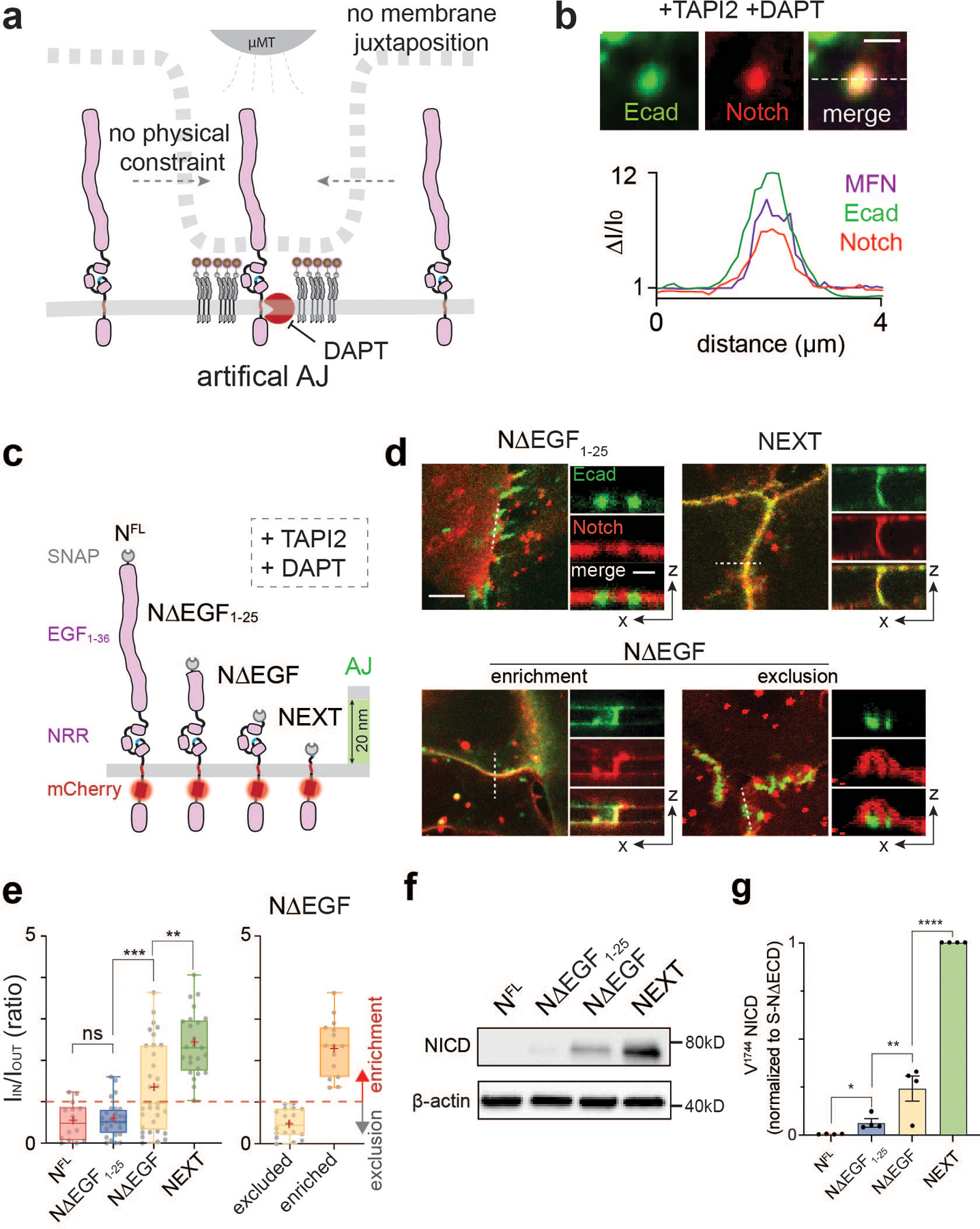
(a) A schematic showing mechanogenetic interrogation of Notch distribution relative to the artificial AJ. Cells treated with DAPT to inhibit γ-secretase activity. (b) Confocal fluorescence images showing E-cadherin and Notch distributions at the artificial AJs after µMT application. Line profiles of MFN, E-cadherin, and Notch signals along a white dashed line. ΔI/I0 represents a fold change relative to fluorescence intensity before stimulation. Scale bar, 2 µm. Shown are the representative images from n = 6 independent mechanogenetic experiments. (c) Schematics of Notch variants with different truncation lengths, in comparison with the AJ intermembrane cleft. All cells were treated with TAPI2 and DAPT to prevent any potential proteolysis. (d) Confocal fluorescence images showing spatial distribution of the Notch variants (red) relative to the AJs (green). (left) Maximum projections of confocal z-stacks. Scale bar, 5 µm. (right) Confocal z-resliced images along the white dashed lines in the maximum projection images. Scale bar, 2 µm. (e) Quantification of the enrichment factor (IIN/IOUT) of Notch variants relative to the AJs. A box-plot showing binary localization of NΔEGF which is defined as either excluded (yellow) or enriched (orange) is shown on the right. Colored lines and (+) marks indicate median and mean, respectively. The box limits indicate the 25th and 75th percentiles, and the whiskers extend to the minima and the maxima. n = (left to right) 14, 29, 33, 26, 18, and 15 cells examined over three independent experiments. ** P = 0.0002, *** P = 0.0006. (f,g) Western blot analyses of cleaved NICD levels in the cells stably expressing NFL, NΔEGF1–25, NΔEGF, and NEXT. β-actin levels represent the loading control. A representative image of immunoblotting (f), and quantification (g) of cleaved NICD levels. The average intensity of each NICD band relative to respective β-actin band was quantified and then normalized to that of NEXT. Data are the mean ± s.d. of n = 4 biological replicates. * P = 0.011, ** P = 0.041. (e,g) ****P < 0.0001, ns: non-significant; one-way ANOVA followed by Tukey’s multiple comparison test.
To test this size-dependent model for Notch exclusion from AJ-induced RIP-µdomains, we generated a series of U2OS cell lines stably expressing Notch variants with different truncation lengths: a partial deletion of the EGF repeats (NΔEGF1–25, approximate height: 48 nm), complete deletion of the EGF repeats but retention of NRR (NΔEGF, approximate height: 10 nm), and a complete removal of NECD (NEXT, approximate height: 4 nm) (Fig. 3c and Extended Data Fig. 5d,e). To quantify the spatial distribution of each Notch variant relative to the AJ, we measured the average mCherry fluorescence signal inside (IIN) and outside (IOUT) of the AJ and estimated an enrichment ratio (IIN/IOUT) where a value of 1 indicates homogenous distribution (Extended Data Fig. 5f,g). Consistent with predictions based on size-dependent protein segregation, NΔEGF1–25, which is taller than the height of the intermembrane AJ cleft, was excluded from AJs (IIN/IOUT = 0.57) (Fig. 3d,e). NEXT with an extracellular domain (ECD) smaller than the junctional height was enriched at AJs (IIN/IOUT = 2.39) (Fig. 3d,e). NΔEGF (intermediate height) exhibited a mixed binary localization pattern (exclusion or enrichment) relative to AJs (IIN/IOUT = 1.32) (Fig. 3d,e). These results suggest a role for the size-dependent protein segregation as a spatial switch that regulates the distribution of Notch intermediates relative to RIP-µdomain.
Notch localization into RIP-µdomains is sufficient for its activation regardless of S2 cleavage
We next interrogated the functional consequences concentrating or excluding a series of Notch variants having different extracellular domains into AJs (i.e., RIP-µdomains). We began by culturing cells in the presence of TAPI2 to decouple γ-secretase processing from S2 cleavage, and measured cleaved NICD levels by immunoblotting. Cells expressing NFL or NΔEGF1–25 resulted in no or minimal NICD, respectively (Fig. 3f,g, and Extended Data Fig. 5d), whereas cells expressing NΔEGF produced a significant amount of NICD (Fig. 3f,g). We validated these findings by measuring the dye-labeled extracellular SNAP signal and the intracellular mCherry-tag signals within AJs and within nuclei, respectively, after removing DAPT (Extended Data Fig. 5h–m). Importantly, we observed a dramatic decrease in the mCherry signal within AJs but not outside AJs, confirming that γ-secretase activity was localized within the RIP-µdomains (Extended Data Fig. 5j). Cells expressing NEXT exhibited the highest NICD production, about a four-fold increase compared with that of NΔEGF (Fig. 3f,g and Extended Data Fig. 5e). NICD production was proportional to the enrichment ratio (IIN/IOUT), suggesting the essential role of size-dependent protein segregation as a spatial switch to direct Notch activation. The substantial NICD production from the cells expressing NΔEGF indicates that, when localized together, γ-secretase can process Notch, bypassing S2 cleavage. Size-dependent but ligand-independent activation of Notch receptors with an intact S2 site was observed previously in Notch variants and synNotch constructs58–62, but the mechanism of this activation has been unclear. Our observations support the notion that colocalization of these Notch variants with γ-secretase is sufficient to trigger S3 proteolysis and signaling. According to this elaborated model, γ-secretase activity on full-length Notch is blocked by maintaining concentrations of Notch and its intermediates below the KM for γ-secretase due to their compartmentalization within LRE- and RIP-µdomains, respectively. Several hypotheses follow from this model which we evaluated experimentally.
First, the model suggests that enforced concentration of a Notch variant with an intact S2 site (e.g., NΔEGF) in AJs will enhance its processing by γ-secretase. To test this hypothesis, we employed a DNA-mediated crosslinking strategy to enhance NΔEGF enrichment of NΔEGF at the RIP-µdomain (i.e., AJ)63 (Fig. 4a, Extended Data Fig. 6a–c, See method). In the presence of TAPI2 and DAPT, we observed increased enrichment of NΔEGF at Ajs (IIN/IOUT = 1.89 ± 0.91) after DNA crosslinking (Fig. 4b). When DAPT was removed to rescue γ-secretase activity while maintaining TAPI2, we observed decreased mCherry signal at Ajs, indicating efficient S3 cleavage without S2 cleavage (Extended Data Fig. 6d–e). Consistently, in western blots, we observed increased NICD levels from the cells treated with DNA crosslinkers, compared with the untreated control (Fig. 4c). Considering that DNA crosslinking increases the ECD size of NΔEGF, the observed increase in NICD production cannot be explained by the prevailing steric repulsion model of γ-secretase-substrate selectivity62.
Fig. 4. Spatial mutations alter Notch activation, regardless of ligand presentation or S2 cleavage.
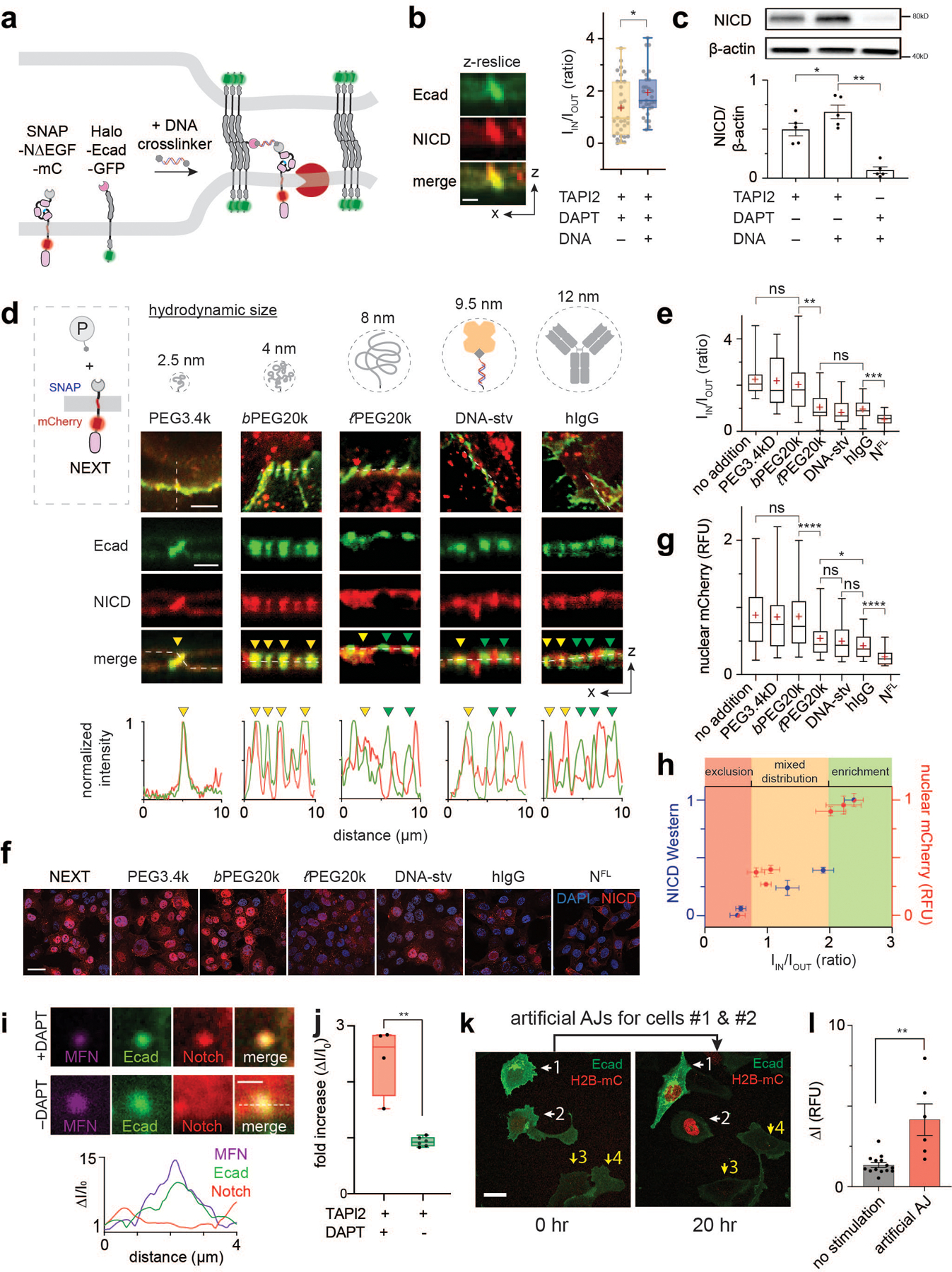
(a) The DNA-mediated crosslinking strategy to enhance NΔEGF localization at the AJ. (b) Confocal z-resliced images showing intense NΔEGF fluorescence (red) enriched at the AJ (green) after the DNA crosslinking. Quantification of enrichment (IIN/IOUT) without (n = 33 AJs pooled from 3 independent experiments) and with (n = 29 AJs pooled from 3 independent experiments) DNA crosslinker treatment (*P = 0.023; two-tailed Mann-Whitney-Wilcoxon test). (c) Western blot analyses of NICD in the NΔEGF cells treated with the DNA crosslinker. (top) A representative blot. (bottom) Quantification of cleaved NICD levels. Data are mean ± s.d. of n = 5 biological replicates (* P = 0.004, ** P = 0.005; ordinary one-way ANOVA followed by Tukey’s). (d) Spatial mutation of NEXT via chemical ligation of macromolecular pendants (denoted as P). Confocal images showing size-dependent spatial mutation of NEXT (red) at the AJs (green). The top row shows maximum projection images of the cells treated with the indicated pendants.The middle row shows confocal z-resliced images along the white dashed lines in the maximum projection images. Yellow and green arrowheads,AJs enrichment with and exclusion with [to authors: an edit for terseness]Notch, respectively. The bottom row, line profiles quantifying fluorescence signals from NEXT (red) and E-cadherin (green) along the white lines in the z-resliced images. (e)IIN/IOUT of NEXT with macromolecular pendants. n = 20 (no addition), 18 (PEG3.4k), 23 (b-PEG20k), 21 (l-PEG20k), 16 (DNA-stv), 32 (IgG), and 15 (NFL) cells examined over two independent experiments for (d, e) (** P = 0.002, *** P = 0.0016; ordinary one-way ANOVA). (f,g) Confocal fluorescence images (f) and quantification (g) of nuclear mCherry signal for the NEXT-expressing cells treated with macromolecular pendants.Cells expressing NFL were used as a negative control. n = 327 (no addition), 182 (PEG3.4k), 746 (b-PEG20k), 288 (l-PEG20k), 179 (DNA-stv), 412 (IgG), and 300 (NFL) cells examined over two independent experiments (* P = 0.013, **** P < 0.0001; ordinary one-way ANOVA). (h) NICD level of various Notch variants as a function of the enrichment factor. Red and blue dots are spatial mutations of NEXT and Notch variants with different truncation length, respectively. Data are median ± s.e.m. of n = 5 biological replicates (NICD Western) and same as (g) for nuclear mCherry. (i) Representative confocal fluorescence images of cells with an artificial AJ in the presence of TAPI2 and DAPT (upper), and TAPI2 only (lower). Line profiles of MFNs, E-cadherin, and Notch signal along the white dashed line. ΔI/I0: fold change relative to nonjunctional membrane signal. (j) Quantification of mCherry signal at artificial AJs after µMT application in the presence or absence of DAPT. n = 4 (with DAPT) and 6 (TAPI2 only) biologically independent samples (** P = 0.0003; two-tailed unpaired Student’s t test). (b, e, g, j) For box-and-whisker plots, the centre lines and (+) marks indicate median and mean, respectively. The box limits and whiskers indicate the interquartile and the full ranges, respectively. (k) Representative confocal fluorescence images of the reporter cells with artificial AJs. White and yellow arrows: cells with stimulation and the control cells, respectively. (l) Statistical analysis of stimulated cells (n = 6) vs. control cells (n = 14). Data are mean ± s.e.m.; ** P = 0.004; two-tailed unpaired Student’s t test. Scale bars are (b) 3 µm, (d, top) 5 µm, (d, middle) 3 µm, (f) 5 µm, (i) 2 µm, (k) 10 µm.
Second, the model suggests that for Notch variants bearing optimally positioned N-terminus for γ-secretase processing (e.g., NEXT), blocking them from accessing AJ will inhibit S3 processing. To test this hypothesis, we induced spatial mutation of NEXT by chemically conjugating it with BG-functionalized macromolecules of increasing hydrodynamic size: low molecular-weight polyethylene glycol (PEG3.4k, 2.5 nm), branched PEG20k (bPEG20k, 4.0 nm), linear PEG20k (PEG20k, 8.0 nm), DNA-streptavidin conjugates (DNA-stv, 9.5 nm), and human IgG (hIgG, 12 nm) (Fig. 4d; Extended Data Fig. 6h). Grafting of these macromolecular pendants onto NEXT increases the size of the Notch construct but does not modify the N-terminal amine for hydrogen bonding with nicastrin proposed in the existing model64. With DAPT, we observed a size-dependent distribution of NEXT at the AJ, where the larger pendants resulted in a greater decrease in mCherry signal at the AJ (Fig. 4d,e). We then examined the signaling consequences for each spatial mutation of NEXT by detecting nucleus mCherry signal resulting from NICD translocation after S3 cleavage. The PEG3.4k or bPEG20k addition did not significantly alter nuclear mCherry signal of NEXT, compared with cells with no pendant addition (Fig. 4f,g and Extended Data Fig. 6g). Conjugation of PEG20k, DNA-stv, and hIgG resulted in a substantial decrease in nuclear mCherry signal to 0.39, 0.37, and 0.27 fractional intensity, respectively. Similarly, immunoblots consistently showed a gradual decrease in the NICD production as a function of macromolecular graft size (Extended Data Fig. 6i). We summarized the NICD production for all Notch variants as a function of the Notch enrichment factor in Fig. 4h, visualizing the spatial dependence of S3 cleavage.
Third, the model predicts that permitting access of full-length Notch, which lacks a suitable N-terminal substrate for γ-secretase, into artificial AJs using mechanogenetics (Fig. 3b) is sufficient to activate the receptor, independent of S2 cleavage. We previously observed pronounced enrichment of full-length Notch at artificial AJs in presence of TAPI2 and DAPT. We repeated identical experiments but removed DAPT to allow γ-secretase activity. We observed no enrichment of mCherry signal at the artificial AJ, presumably due to S3 cleavage and NICD release (Fig. 4i,j and Extended Data Fig. 6j,k). To confirm that the loss of mCherry signal corresponded to bona fide signaling from Notch, we employed a UAS-Gal4 reporter system that detects Notch activation through expression of a nuclear-localized mCherry29,30,65,66. To a cell recombinantly expressing SNAP-NFL-Gal4 and Halo-Ecad-GFP, we again generated artificial AJs and measured the nuclear mCherry fluorescence every two hours. Note that no source of S2 cleavage (e.g., no ligand-immobilized substrate) was added. We observed strong nuclear mCherry signal from the cells with artificial AJs, but no signal from neighboring cells (Fig. 4k,l and Extended Data Fig. 6l). Together, these three experiments suggest that the new molecular interfaces produced by S2 cleavage are not necessary for S3 cleavage so long as γ-secretase is concentrated sufficiently with its substrate.
AJ-mediated RIP-µdomain is indispensable for Notch signaling
Given the significant role of AJs creating LRE- and RIP-µdomains, we next interrogated Notch activation in cells lacking AJs. We plated UAS-Gal4 reporter cells expressing SNAP-NFL-Gal4 on a Dll4-coated substrate in low- (to minimize physical contact between cells and hence AJ formation) or high- (to facilitate AJ formation) density, and then monitored Notch activation of the cells via the reporter signals. While cells with physical contacts with adjacent cells exhibited a robust increase in nuclear mCherry signal, those without cell-cell contact showed no increase in signal (Fig. 5a–c and Supplementary Video S1). Reestablishing AJs by plating cells on a substrate coated with Ecad-Fc and Dll4-Fc rescued Notch signaling in solitary cells (Fig. 5a,d and Supplementary Video S2). We further confirmed AJ-dependent Notch activation in cells cultured with varying densities across a Dll4-coated substrate (Fig. 5b). These results support a model wherein AJs are required for Notch processing at the cell surface and downstream signaling. Critically, E-cadherin seems to function by recruiting γ-secretase but independent of its role in mediating cell-cell contact. To further validate the role of AJs, we knocked out the gene encoding E- and N-cadherin (CDH1/2) in the reporter cell line using CRISPR-Cas9 (Extended Data Fig. 7a–d), then plated the cells at high density on Dll4-Fc coated plates. Strikingly, cadherin knockout (cad-KO) resulted in abrogation of Notch activation even in the presence of extensive cell-cell contacts (Fig. 5e,f and Extended Data Fig. 7e–g). Reintroduction of plasmids encoding E-cadherin or N-cadherin into Ecad-KO cells recovered Notch activation (Fig. 5e,f and Extended Data Fig. 7e–g). Single cell analysis of the nuclear fluorescence signal revealed a positive correlation with E-cadherin expression in the respective cells, confirming AJ-dependent Notch signaling (Fig. 5g).
Fig. 5. The AJ-mediated spatial switch regulates Notch signaling.
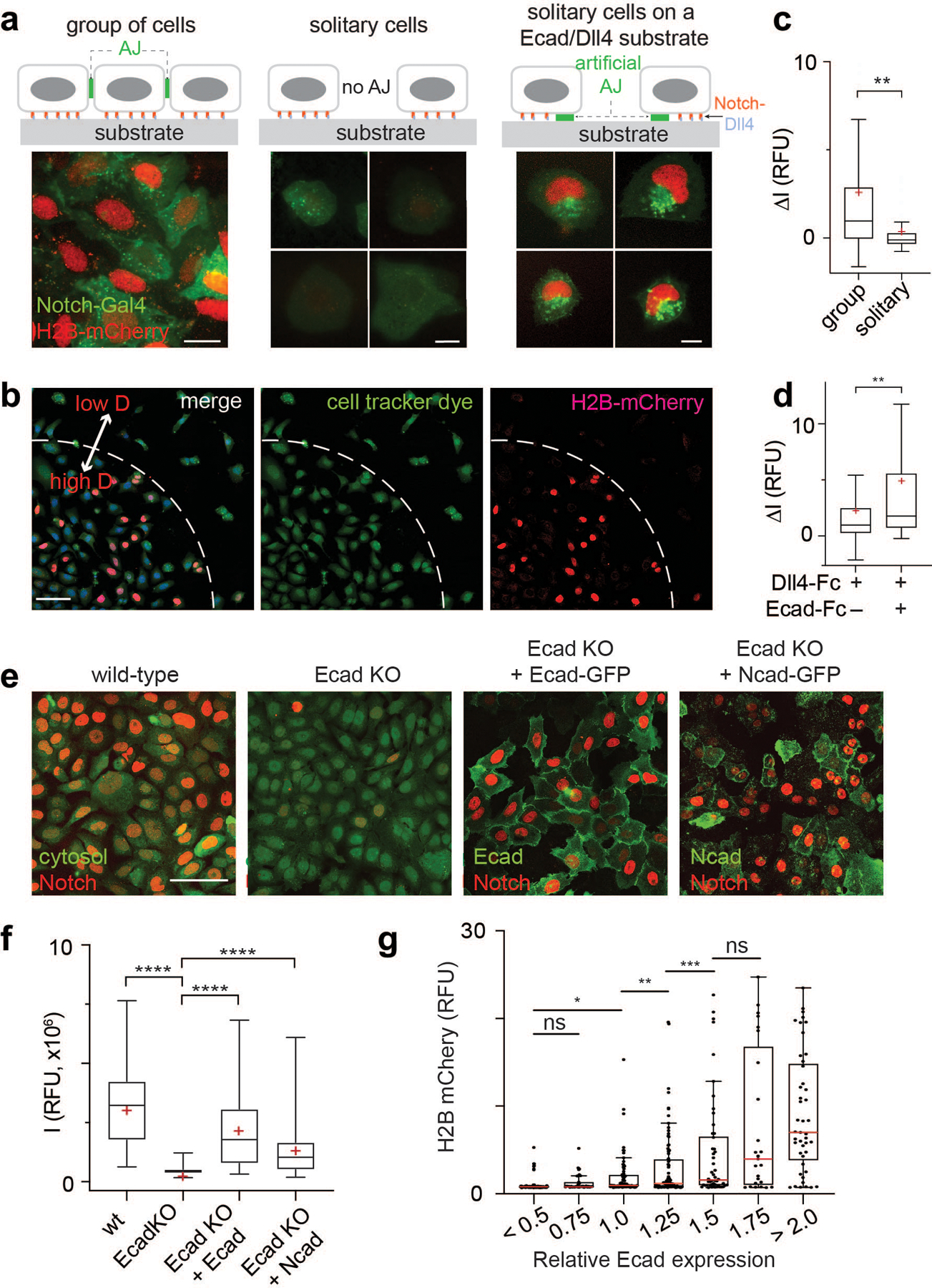
(a) Representative epi-fluorescence images showing Notch activation in U2OS SNAP-NFL-Gal4 reporter cell lines in different cellular environments: Group of cells on a Dll4-Fc coated substrate (left), solitary cells with no prior contact on a Dll4-Fc coated substrate (middle), and solitary cells plated on a Dll4-Fc and Ecad-Fc coated substrate (right). Scale bars, 20 µm. (b) Representative low magnification epi-fluorescence image showing both grouped cells and multiple solitary cells. Scale bar, 100 µm. (c) Quantification of Notch activation by measuring H2B-mCherry fluorescence changes in cells within a group (n = 152 cells from 3 independent experiments), solitary cells (n = 50 cells from 3 independent experiments). ** P = 0.0034 (unpaired two-tailed Student’s t test). (d) Quantification of Notch activation in solitary cells cultured on a Dll4-Fc coated substrate and those cultured on a Dll4-Fc and Ecad-Fc coated substrate (n = 27 cells for both conditions from 3 independent experiments). ** P = 0.005 (unpaired two-tailed Student’s t test). (e) Representative confocal images of H2B-mCherry fluorescence in U2OS SNAP-NFL-Gal4 reporter cells (wt), E-cadherin knockout cells (Ecad-KO), Ecad-KO cells with recombinant E-cadherin transfection (Ecad-KO + Ecad), and Ecad-KO cells with N-cadherin transfection (Ecad-KO + Ncad). Cytosol labeled with CMFDA dye was shown for wt and Ecad-KO cells. E-cadherin and N-cadherin were shown for Ecad-KO + Ecad and Ecad-KO + Ncad cells. Scale bar, 100 µm. (f) Quantification of Notch activation in the wt (n = 86), Ecad-KO (n = 100), Ecad-KO + Ecad (n = 52), and Ecad-KO + Ncad (n = 80) cells (all pooled from 2 independent experiments). **** P < 0.0001 (ordinary one-way ANOVA followed by Tukey’s). (c, d, and f) Boxes and whiskers indicate the interquartile and full ranges, respectively. Black lines and (+) marks indicate median and mean, respectively. (g) Comparison of Notch signal activation, readout by mean nuclear H2B-mCherry fluorescence, as a function of E-cadherin expression, readout by membrane GFP fluorescence signal. Each dot represents H2B-mCherry signal of a single cell, and cells are grouped into bins based on their levels of Ecad expression. (from left to right) n = 94, 35, 71, 87, 50, 25, and 45 cells examined across two independent experiments. * P = 0.019, ** P = 0.049, *** P = 0.036, ns, non-significant (ordinary one-way ANOVA followed by Tukey’s). In the box-whisker plot, the red lines indicate median. The boxes and whiskers indicate the 25th to 75th percentiles, and the 10th to 90th percentiles, respectively.
AJ-mediated RIP-µdomains regulate NPC differentiation in vivo
Notch signaling is essential for the maintenance of stemness, self-renewal, and differentiation of neural progenitor cells (NPCs)67,68. In the mammalian cerebral cortex, Notch signaling orchestrates developmental neurogenesis, where it modulates a balance between tangential proliferative and radial differentiative expansion of the apical ventricular-zone NPCs (VZ-NPCs) to establish a stratified neuronal organization69. Interestingly, radial expansion of VZ-NPCs accompanies its differentiation, suggesting that Notch signaling in VZ-NPCs may be coupled with cells’ spatial cues. Several reports also emphasize the critical role of apical-endfoot AJs in Notch signaling and in the decision-making process of VZ-NPC development70–72.
Given the essential role of the AJ-mediated microdomain formation for Notch signaling in cell line models, we reasoned that apical-endfoot AJs may also organize proteolytic hotspots for Notch activation. To test this hypothesis, we mapped the spatial distribution of Notch and γ-secretase relative to N-cadherin-based AJs in VZ-NPCs of the developing mouse brain (E13.5) (Fig. 6a–g). Notch and PS1 exhibited exclusion from and enrichment within AJs, respectively, confirming compartmentalization between LRE- and RIP-µdomains (Fig. 6c–g). We also captured the spatial distribution of the Notch activation intermediate by intracerebroventricular injection of DAPT into postnatal mice (P3). The immunostaining showed inclusion of Notch signal within AJs, presumably resulting from NEXT accumulation (Extended Data Fig. 8a) as observed in cell lines (Fig. 1d–f, Extended Data Fig. 3b,c). These results support the notion that AJs also drive compartmentalized microdomains and serve as a spatial switch regulating Notch signaling in vivo.
Fig. 6. The AJ-mediated spatial switch regulates neuronal progenitor cell differentiation in vivo.
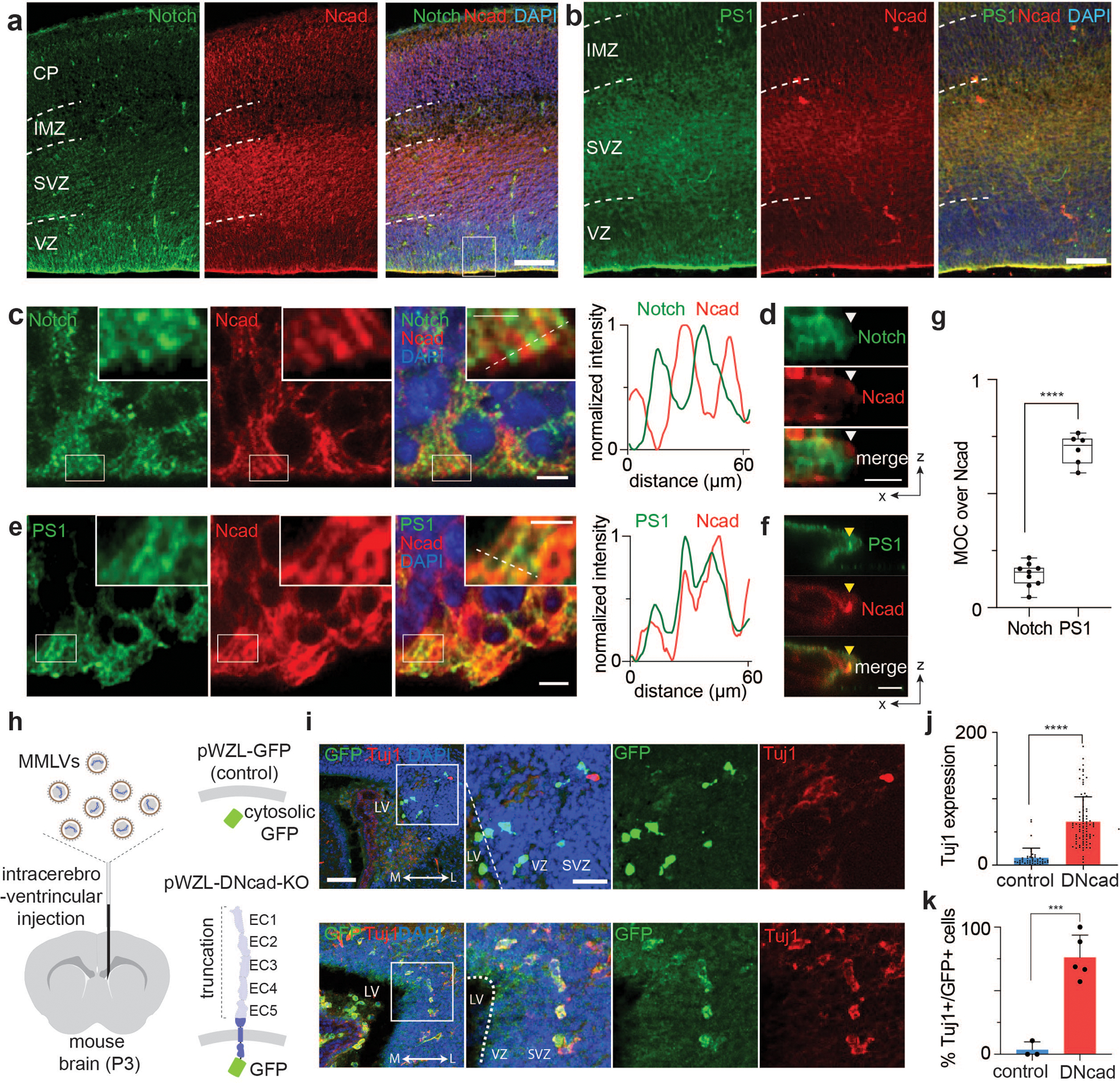
(a-f) Immunostaining of the subventricular zone (SVZ) in the lateral ventricle (LV) of the E13.5 mouse brain. Notch (a, c, d) and PS1 (b, e, f) distributions relative to AJs. Representative low (a,b) and high (c,e) magnification images. Scale bars, 100 µm and 5 µm, respectively. The boxed area in panels (c) and (e) is further magnified in the inset. Scale bar, 2.5 µm. Line profile analysis shown in panels (c) and (e). Representative confocal z-resliced image showing Notch exclusion (white arrowhead) and PS1 colocalization (yellow arrowhead) with the AJ (d,f). Scale bar, 3 µm. (g) Quantitative assessment of Notch and PS1 colocalization with N-cadherin in vivo. Each dot represents MOCs quantifying colocalized Notch (MOC = 0.14±0.05, n = 9 cells examined across 2 independent experiments) or PS1 (MOC = 0.69±0.07, n = 6 cells examined across 2 independent experiments) over selected AJs (**** P < 0.0001; two-tailed unpaired Student’s t test). In the box-whisker plot, the boxes show the 25th to 75th percentiles (with the black bar indicating the mean), and the whiskers extend to the maxima and minima. (h) Retroviral infection of a plasmid encoding control vector (EGFP) or dominant negative form of E-cadherin vector (DN-cad-EGFP) to developing P3 mice via intracerebroventricular injection. (i) Immunofluorescent staining images of brain slices of mice retrovirally infected with DN-cad-GFP with control plasmids, respectively. Cells differentiated into post-mitotic neurons can be identified as EGFP+/Tuj1+, while those which remained as NPCs with plasmid transfection are only EGFP+. Shown are representative from n = 3 (control) and 5 (DN-cad-GFP) independent animals. Scale bar, 50 µm. (j) Quantification of the expression of Tuj1 per single cells (n = 43 cells across 3 mice and n = 86 cells across 5 mice per control and DN-cad, respectively). Data are mean ± s.e.m. (**** P < 0.0001; two-tailed unpaired Student’s t test). (k) Quantification of the percentage of Tuj1-expressing post-mitotic neurons among all transfected EGFP+ cells were quantified. n = 3 (control plasmid) and 5 (DN-cad-GFP) biologically independent animals. Data are mean ± s.e.m. (*** P = 0.0005; two-tailed unpaired Student’s t test).
To understand the function of the AJs on VZ-NPC development, we disrupted AJs using dominant-negative cadherin expression, preventing RIP-µdomain formation. We retrovirally transduced VZ-NPCs in developing mice (P3) using a dominant-negative form of E-cadherin having an extracellular domain truncation (DN-cad)72 and a C-terminal GFP tag (Fig. 6h). While mice injected with control retroviruses showed negligible TuJ1-neuronal marker immunostaining, those with retroviruses encoding DN-cad exhibited robust TuJ1 expression, presumably through downregulation of Notch signaling (Fig. 6i–k and Extended Data Fig. 8b,c). These results support that AJ-mediated RIP-µdomains modulate NPC maintenance and differentiation via Notch signaling.
Size-dependent spatial dynamics and proteolysis of APPs
To test whether AJs serve as proteolytic hotspots with size-dependent substrate selectivity for other large cell surface proteins, we investigated the processing of amyloid precursor protein (APP). APP has a similar topology and proteolytic cleavage sequence to that of Notch. Like Notch, upon activation, APP is processed by two rounds of proteolysis: first α- or β-secretase and then γ-secretase releasing its extracellular and intracellular domains, respectively73–75. We generated U2OS cells co-expressing APP-GFP and SNAP-N-cadherin and monitored the cell surface spatial distribution of APP intermediates relative to N-cadherin-based AJs (NAJs) in the presence of protease inhibitors. Having an intermediate ECD size (80 kD), full-length APP showed binary localization (i.e., excluded or enriched) relative to AJs in the presence of inhibitors, similar to the Notch variant with EGF repeat truncation (i.e., NΔEGF) (Fig. 7a,b and Extended Data Fig. 9a,b). APP diffused into the NAJs after ECD shedding by α- or β-secretase, and then was processed by γ-secretase within it (Fig. 7a,b and Extended Data Fig. 9a,b).
Fig. 7. The AJ-mediated spatial switch regulates APP signaling.
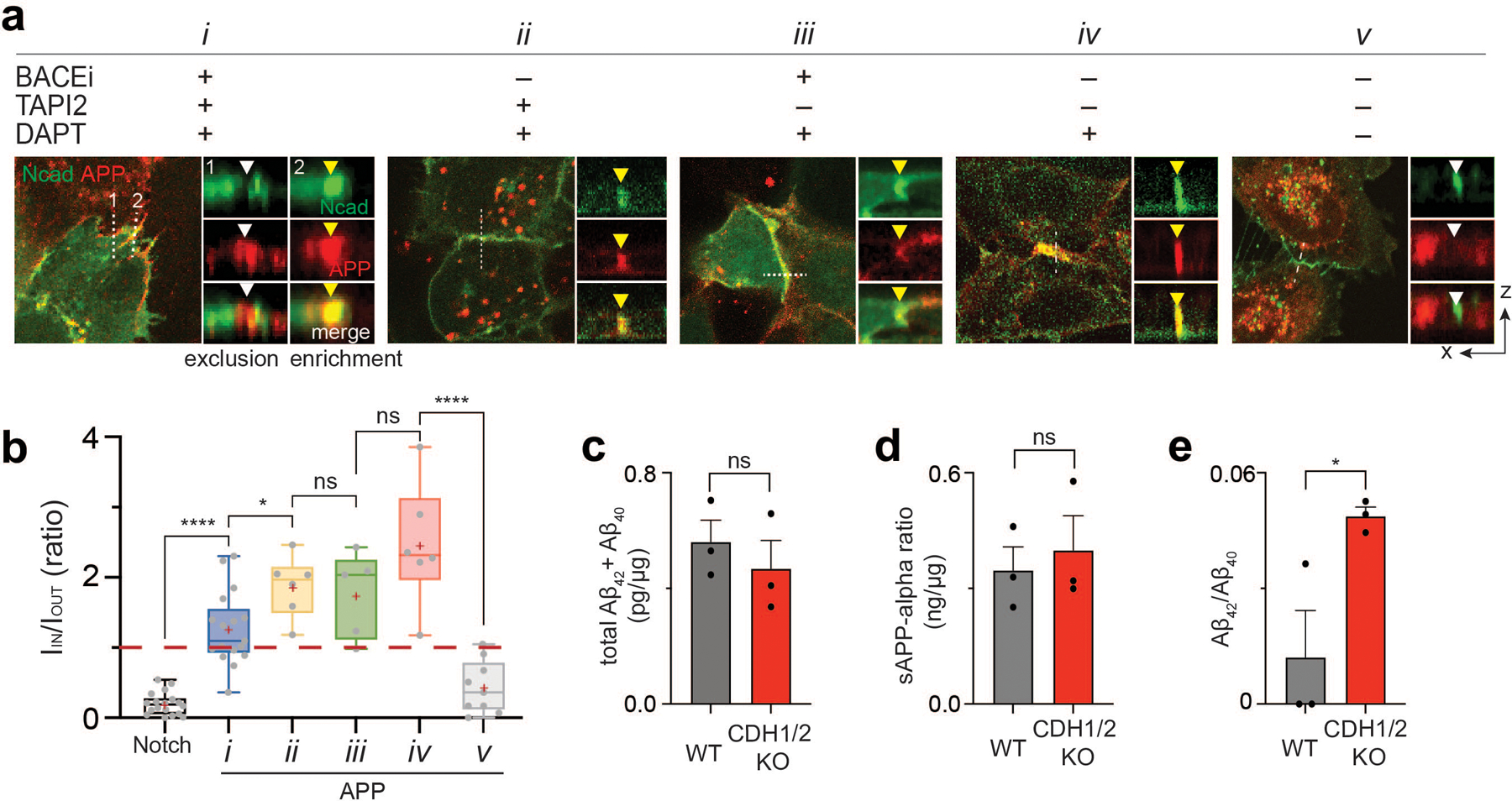
(a) Confocal fluorescence maximum projection (left) and z-resliced images (right) of U2OS cells co-expressing Ncad-mCherry (green) and APP-EGFP (red) in different combinations of α-, β-, and γ-secretase inhibitors. Scale bars, 10 µm (maximum projection) and 3 µm (inset). (b) Quantification of the enrichment factor (IIN/IOUT) of APP signal relative to the N-cadherin-based AJs. In the box-whisker plot, the boxes show the 25th to 75th percentiles, and the whiskers extend to the maxima and minima. Black lines and (+) marks indicate median and mean, respectively. n = (left to right) 18, 16, 6, 5, 6, 9 biologically independent cells examined across 2 independent experiments. *P=0.049; ****P < 0.0001; ns, not significant; ordinary one-way ANOVA followed by Tukey’s multiple comparison. (b-e). Total sum of Aβ42 and Aβ40 (c), soluble APPα (d), and Aβ42/Aβ40 ratio (e) measured by ELISA in wild-type cells or CDH1/2 KO cells. Data are mean ± s.e.m. of n = 3 biological replicates (*P = 0.041, ns, not significant; two-tailed unpaired Student’s t test).
APP proteolysis by γ-secretase produces more soluble p3 and Aβ40 predominately, along with less soluble and pathogenic Aβ42 and longer isoforms73,74,76. Previous reports showed that locally acidic microenvironment (e.g., pH 5.5) leads to a gain in the proportion of pathogenic Aβ species77. Additionally, N-cadherin expression in cells stabilizes an open conformation of PS1 that favors Aβ40 production over Aβ4278. Given our previous observation that loss of AJs leads to a decrease in cell-surface γ-secretase, we hypothesized that APP processing would be biased under these conditions towards Aβ42. We tested this hypothesis by measuring APP fragment production in U2OS cell lines recombinantly expressing APP but lacking both E- and N-cadherins (CDH1/2-KO cells) (Extended Data Fig. 7a–d). While no significant changes were observed in total Aβ(40+42) and soluble APPα (sAPPα) (Fig. 7c,d), CDH1/2-KO cells produced higher relative levels of Aβ42, compared to cells with endogenous cadherin expression53 (Fig. 7e).
Discussion
The Notch ligand-receptor interaction (a binding switch) is converted into intracellular signals only following multiple additional regulatory steps gated by mechanical, proteolytic, and spatial events. These include unfolding of NRR domain (a mechanical switch), S2- and S3- cleavage (proteolytic switches), and finally translocation of NICD from the cell membrane to the nucleus (a spatial switch)17,19,79. Our study reveals that Notch integrates an additional spatial switch by AJ-driven interfacial membrane compartmentalization to tightly choreograph the critical and irreversible proteolytic cleavage sequence prior to NICD release. Previously, it was thought that full-length Notch could interact with γ-secretase interaction at the cell surface80,81, where this proteolytic sequence was regulated by modification of the molecular interface between Notch and nicastrin after S2-cleavage62,64. Our model is not incompatible with a contribution of the nicastrin-Notch chemical interface on γ-secretase activity in that evidence suggests the S2 proteolysis lowers the KM of the enzyme for Notch. However, it strongly suggests that the AJ-driven membrane compartmentalization is the major regulator of Notch-γ-secretase interaction and signaling, functioning by increasing the concentration of the γ-secretase substrate to the point that it exceeds the KM and is efficiently processed by the enzyme.
The operating principle of this new spatial switch is closely related to another unique feature of Notch receptor: its unusually tall extracellular domain. The functional residues responsible for ligand binding are located near the N-terminus, which protrudes above the crowded cell surface, where they are poised to engage ligands on neighboring cells. However, it has also been shown that replacing the EGF-like domain repeats with a smaller ligand binding domain (e.g., synNotch) maintains receptor function82,83. Why then does Notch receptor bear such a massive ECD? Our study provides insight into this question, where the large ECD is crucial for its spatial segregation from γ-secretase thereby minimizing nonspecific ligand-independent activation. Low level NICD production was observed even for Notch variants with partial EGF truncation (NΔEGF1–25) and levels gradually increased upon successive truncations. NΔEGF1–25 has a comparable size to smaller Notch family homologs, including LIN-12/Notch and GLP-1/Notch, suggesting the relevance of a spatial switch across the Notch family and metazoans. Our model also explains previous observations where synNotch with a relatively small ECD exhibited significant ligand-independent activation (10–50% of ligand-induced activation)60.
We also show that size-dependent spatial segregation regulates APP cleavage and Aβ production. It has been previously shown that γ-secretase present in different subcellular compartments cleaves APP into diverse Aβ isoforms73,74,84. Our study shows that, after the ECD cleavage, AJ potentiates cell surface processing of APPs within the junction, yielding Aβ40 predominantly, while removal of AJ produces more Aβ4289. To establish the relevance of this observation to APP processing will require further investigation in a neuronal system, but our results suggest a potential role of the AJ-mediated APP-γ-secretase compartmentalization in Aβ pathology, possibly influenced by APOE-dependent intracellular and cerebral cholesterol levels85. More importantly, these finding suggest that AJ may represent proteolytic hotspots with size-dependent substrate selectivity across a more diverse range of cell-surface proteins.
Our study also suggests a critical role of the AJ-mediated membrane compartmentalization in VZ-NPC maintenance and differentiation during development. It has been previously proposed that apical-endfoot AJs promote Notch signaling in NPCs70–72, but the mechanism underlying precise Notch signal regulation was unclear. Our findings suggests that Notch signaling is maintained by creating RIP-µdomains within AJs, and disruption of the AJs downregulates Notch signaling and hence promotes NPC differentiation. This result also provides important insights on the molecular mechanism of how environmental spatial and physical changes of cells (i.e., VZ-NPC detachment and radial migration) direct cell signaling (i.e., Notch signaling) and differentiation, to facillitate spatiotemporally coordinated tissue development. Importantly, coupled delamination (due to AJ disruption) and differentiation (due to Notch inactivation) is not limited to this specific case, but seen in many other developmental processes, including intestinal stem cells86–88, supporting hair cells89, pituitary gland stem cells90, and epithelial-mesenchymal transition91. In these cases, loss of AJs could serve as a self-limiting mechanism for Notch signaling, enforcing proper tissue architecture during differentiation.
The size-dependent segregation of Notch from the RIP-µdomain has important analogies to the kinetic segregation model of immune cell activation, where the large CD45 phosphatase is excluded from T cell receptor (TCR) or FCγ receptor (FCγR) immunological synaptic clefts11,12,92,93. However, several distinct features of the Notch spatial switch exist. First, unlike the immunological synapse where TCR, FCγR and CD45 remain constant in size throughout activation, Notch undergoes a dramatic size change during activation, enabling its relocalization and sequential proteolysis. Second, the role of AJs in Notch signaling is not limited to creating a physical barrier, but also plays the critical role of recruiting and concentrating γ-secretase to provide proteolytic hotspots at the cell surface. Finally, the consequences of size-dependent segregation on signaling are reversed in comparison to the immunological synapse. While spatial segregation of CD45 enables sustained TCR/FCγR phosphorylation and downstream signaling, Notch segregation from AJs inhibits signal activation. Our result extends the relevance of size-dependent spatial segregation models beyond immune cells11,12,92, supporting the notion that size-dependent protein segregation can serve as a general mechanism for regulating a broad range of receptor signaling at the cell-cell interface. It is also important to note that our model may not be limited to the AJs, but may be extended to other cell-cell junctions that provides an environment for size-dependent protein segregation while effectively concentrating proteases.
Overall, AJ-mediated interfacial membrane compartmentalization sheds light on the mechanism underlying the sequential proteolysis of Notch and APPs. We anticipate further implications of our work in other areas of research such as providing new design principles for synthetic receptors like synNotch, as well as new therapeutic approaches that target Notch and APP signaling by spatial mutation in cancer and neurodegenerative diseases.
Methods
Plasmid construction
Plasmid constructs used in this study are listed in Supplementary Table 1. All constructs used in this paper were assembled using standard restriction enzyme-based cloning, in-fusion cloning, and/or Gibson isothermal assembly. The maps, sequences, and construction details of all plasmids are available upon request. All constructs were sequenced to confirm mutation. All primers were purchased from IDT (Integrated DNA Technology, USA). Complete details of all cloning procedures are available upon request.
Flag-human Notch1 (NFL)-Gal4 and pGF1-UAS-H2B-mCherry were gifts from S. Blacklow (Harvard University). Flag-human NFL-Gal4 was provided in a Tet-ON Flp-IN vector (pcDNA5). SNAP-NFL-mCherry and SNAP-NFL-Gal4 were constructed as previously reported30. All Notch1 variants with partial or full extracellular domain truncation were constructed by linearizing and amplifying SNAP-hN1-mCherry vector via inverse PCR while omitting the sequence corresponding the ECD truncation. Notch ectodomain sequences of amino acid 23–981, 23–1426, 23–1709 were deleted for SNAP-ΔEGF1–25-mCherry. SNAP-ΔEGF-mCherry. and SNAP-NEXT-mCherry, respectively. Note that similar Notch variants with partial ECD truncation were reported previously 82 where the structural integrity and function of Notch negative regulatory region (NRR) domain were preserved. Ecad-GFP was purchased from Addgene (plasmid # 28009; http://n2t.net/addgene:28009). SNAP-Ecad-GFP and Halo-Ecad-GFP were constructed first by linearizing and amplifying Ecad-EGFP vector via inverse PCR. SNAP- and Halo-tags were then inserted in frame with E-cadherin, downstream of the E-cadherin pro-peptide (amino acid #155) and upstream of the extracellular domain sequence, using Gibson assembly (NEB). pCMV6-Flotillin-1-Halo (Flot1-Halo) was constructed by replacing the myc-tag within the pCMV6-Flotillin1-myc (purchased from Origene (MR206823)) with the Halo-Tag sequence in frame with Flot1 using In-Fusion cloning (Clontech). Human amyloid precursor protein1-EGFP (APP-EGFP) was created by cloning human APP695 (a gift from C. Miller of King’s College London) into a pEGFP-N1 plasmid (Clontech). To facilitate membrane distribution mapping, we used APP constructs used in confocal imaging lack YENPTY motifs (Fig. 7a,b), and compared the result with full-length APP (Fig. 7a,b). APPΔYENPTY-EGFP was constructed by first linearizing the vector via inverse PCR, and deleting the sequence of final 15 amino acids upstream of C-terminus of APP (amino acid 681–695), which includes YENPTY motif (amino acid 682–687) using Gibson assembly.
Short hairpin RNA (shRNA) targeting ADAM10 was inserted into the lentiviral expression vector pLV-mTurquoise-MLC-IRES-neo (addgene plasmid #85145) using BamHI. The targeting sequences were gacatttcaacctacgaat for ADAM10 mRNA94. To form the shRNA, sequences were separated by a noncomplementary spacer (ttcaagaga) from their corresponding reverse complement sequence. Lentiviral particles for ADAM17 shRNA (sc-36604-V) were purchased from Santa Cruz Biotechnology.
Tissue culture
Human U2OS cells were cultured in McCoy’s 5A media supplemented with 10% heat-inactivated FBS (Life Technologies) and 1% Pen-Strep (Life Technologies) and passaged with 0.05% trypsin-EDTA. MDCK cells were grown in Eagle’s Minimum Essential Medium (UCSF cell culture facility) supplemented with 10% heat-inactivated FBS and 1% Pen-Strep. MDCK cells were lifted by treating for 10 minutes with PBS Ca2+ and Mg2+ free with 0.05% EDTA followed by trypsinization. HUVECs were purchased from ATCC (ATCC® CRL-1730™) and grown in Endothelial Cell Growth Medium-2 (EGM-2, Lonza CC-3162) for up to 4 passages (0.05% trypsin-EDTA) after thawing. Human immortalized keratinocyte cell line, HaCaT, were purchased from ATCC (ATCC® PCS-200–011) and grown in DMEM (Gibson) supplemented with 10% FBS and 1% Pen-Strep. All cell lines were maintained at 37°C in a humidified incubator with 5% CO2 and passaged every 2–3 days, depending on confluency, using 0.05% Trypsin-EDTA (UCSF cell culture facility). For polarized MDCK culture, cells were for use on a transwell filter (0.4 µm, collagen coated) at high density. All cells were authenticated based on their morphology, growth condition, and immunostaining with specific markers. The plain U2OS and Flp-In T-rex U2OS cell lines were previously authenticated in Seo et al. Cell (2016)30 and Kim et al. Nat Protocols (2017)91.
Transfection and cell line generation
All cell lines expressing recombinant proteins used in this study are listed in Supplementary Table 1. U2OS stable cell lines were constructed from parental U2OS T-rex cell lines (Flp-IN, Tet-ON engineered cell line, gift from S. Blacklow). Constructs were inserted into the engineered Flp-IN site by co-transfection with a plasmid containing the Flp-recombinase (pOG44) via electroporation with the Neon Transfection System (ThermoFisher) according to manufacturer’s protocol (Shock Conditions: 1230V, 10ms, 4 pulses, number of cells 5 × 106). The amount of total DNA used was 10 µg/well: 1 µg of DNA containing the desired construct and 9 µg pOG44. Cells transfected with desired plasmids were incubated in a selection medium containing 400 µg/mL hygromycin (Invitrogen) for at least 10 days. All cells with Notch truncation and reporter were further sorted for inducible expression of Notch variants via fluorescence-activated cell sorting (FACS) on a FacsAria2 (BD) by staining for the appropriate tag (SNAP or Halo) with fluorescently tagged antibody. For single-cell monoclonal population establishment, fluorescently-positive bulk-sorted populations were plated into 96 well plates at 0.2 cells/well by serial dilution and grown in selection medium. Each clonal cell population was tested and selected based on the levels of Notch reporter activity or Notch membrane expression. U2OS cells expressing recombinant proteins transiently were generated by transfecting plasmids encoding desired proteins using Neon-based electroporation. Cells were allowed to settle in a 6-well cell culture dish post electroporation for 6–8 hr. To remove dead cells, cells were lifted and re-plated on a fibronectin-coated glass bottom dish with 1 × 105 cells per well density. MDCK cells were plated at 70% density then transfected with NFL-mCherry utilizing Lipofectamine 3000 (ThermoFisher) or Neon electroporation according to the manufacturer’s protocol (Shock Conditions: 1,650V, 20ms, 1 pulse, number of cells 5 × 106). HUVEC cells were transfected via electroporation with SNAP-NFL via the BioRad Gene Pulser system (250 V, 20 ms square wave, 1×106 cells/mL Gene Pulser Electroporation Buffer, 5 µg/mL SNAP-NFL-mC). All cells transiently expressing recombinant proteins were incubated for 24–48 hr from the transfection, and then used for further analyses.
Live cell mechanogenetics experiment
Mechanogenetics experiments were performed as previously described in Kwak et al., 2019 with some modifications30,54,55. Monovalent magnetofluorescent nanoparticles (MFNs) were synthesized as previously described55.
Micro-magnetic tweezers (μMT) set up.
The μMT was set up and aligned on the inverted microscope with point-scanning confocal imaging capabilities (Nikon) as previously described30,55,95. The needle probe – NdFeB magnet assembly was attached to the z-translation stage (Sutter Instrument, MP-325) and its location was carefully aligned with the microscopic objective lens while observing the dummy substrate filled with DPBS. The µMT tip was positioned at the center of the objective oculus with bright-field illumination using the X-Y translation stage linked to PIMikroMove (Physik Instrumente) and µManager (UCSF). Using the z translation stage, the µMT was carefully lowered to set the height of the tip to 10 µm above the focal plane while recording the X-Y coordinates and the z-position of the needle probe.
Preparation of cells expressing recombinant Flotilin-1 for mechanogenetics experiments.
U2OS cells were co-transfected with SNAP-Ecad-GFP (5 µg) and Flot1-Halo (5 µg) plasmids using Neon electroporation. 24 hr later, cells were re-plated on a #1.5 glass-bottomed dish (MatTek, d = 10 mm) coated with collagen at a density of 1 × 105 cells per dish. To fluorescently label Flot1-Halo, cells were treated in a complete McCoy’s 5A medium containing 3.5 μM cell membrane permeable Halo-ligand 660 dye (Promega) for 30 minutes at 37°C. Cells were washed three times with DPBS, incubated with a phenol red-free complete medium, and then mechanogenetically stimulated (see below). For the cholesterol depletion experiment, we also treated cells 10 mM of methyl-β-cyclodextrine (MβCD) (Sigma-Aldrich) in serum-free McCoy’s 5A medium for 30 min at 37°C and washed with complete medium three times. To label SNAP-Ecad-GFP with MFNs, cells were first treated with 5 µM of an oligonucleotide bearing benzylguanine (BG-T60ACTG10) for 45 minutes at 37°C, washed two times with 10 ml of serum free medium, and then incubated with serum-free medium containing 10 nM monovalent MFNs bearing complementary sequence (T60CAGT10) and 0.5% alkali casein for 10 min at 37°C, 5% CO2. Cells were washed with 10 ml of complete medium two times, and then incubated with phenol red-free medium for mechanogenetic experiments on a confocal or wide-field epifluorescence microscope.
Preparation of cells expressing human Notch1 receptor for the mechanogenetic experiment.
Inducible U2OS cells stably integrated with SNAP-NFL-mCherry were transfected with Halo-Ecad-GFP (10 µg) using Neon electroporation. 24 h later, cells were re-plated on a collagen (or fibronectin)-coated glass-bottomed dish. To induce surface expression of SNAP-NFL-mCherry. cells were incubated with complete medium containing doxycycline (Sigma, 2 µg/mL) for 18 h. To inhibit γ-secretase activity, cells were treated with DAPT (5 µM) and further incubated for 6 h. Cells were treated with 5 µM of an oligonucleotide bearing chloroalkane (Cl-T60ACTG10) for 45 minutes at 37°C and labeled with MFNs via the procedure described above.
Mechanogenetic regulation of artificial E-cadherin junctions.
To induce MFN and hence cadherin clustering, the μMT was carefully directed towards a targeted subcellular location until the tip-to-membrane distance (d) reached 10 µm. As the tip approached the target membrane, the formation of an artificial E-cadherin junctions (AJs) was monitored every 5 minutes. After 30 min of mechanogenetic stimulation, the spatial distribution of MFNs and artificial AJs was monitored using time-lapse confocal fluorescence imaging. To investigate γ-secretase processing of full-length Notch, the spatial distribution of membrane mCherry (S-NFL-mC) or nuclear mCherry signals (S-NFL-Gal4) were monitored using time-lapse live cell confocal imaging. To observe localization of membrane microdomains, the spatial distribution of Flot1 fluorescence signal was monitored using live cell confocal imaging. Time-lapse live cell confocal imaging was performed using a 60x Plan-Apo oil objective (NA 1.4) on a Nikon A1 laser scanning confocal microscope equipped with an environmental chamber maintaining cells at 37°C, 5% CO2. Cells were immediately fixed with 4% paraformaldehyde (Life Technologies) in DPBS for 15 minutes and washed with DPBS 3 times for 5 minutes before immunostaining.
Fluorescence labeling and Immunostaining
Fluorescence labeling of cells expressing SNAP- or Halo-tag proteins.
Cells expressing SNAP- and/or Halo-tagged fusion proteins were labeled with BG- and/or chloroalkane functionalized fluorescence dyes, respectively. Dox-inducible cell lines grown on a collagen I-coated substrate were treated with doxycycline (2 µg/ml) 24 hr before labeling. Cells with transient receptor expression were labeled with dyes 48 hr post-transfection. Dye-labeling was performed by treating the cells with 5 μM fluorescence dye in serum containing media for 30 min. Cells were then washed 3 times with complete media. For live cell imaging, cells were incubated with phenol-red free complete media. For imaging of fixed cells, cells were washed with PBS, fixed with 4% PFA in PBS for 10 min, and then washed thoroughly with PBS.
Immunofluorescence staining.
Fixed cells were permeabilized with 0.5% tween-20 diluted in PBS for 15 minutes, then blocked by incubation with blocking buffer (5% normal goat serum and 1% BSA in 1xPBS) for 1 hr in room temperature. For immunostaining of surface ADAM10 and ADAM17 expression, the sample was directly blocked without a permeabilization step. Cells were incubated overnight at 4°C or 2 hr at 25°C with the primary antibodies: mouse monoclonal anti-ADAM10 (1:100; Santa Cruz Biotechnology), mouse monoclonal anti-ADAM17 (1:200, R&D Systems), rabbit polyclonal anti-PS1 antibody R222 (1:50)32, rabbit monoclonal anti-Nicastrin (1:200, Santa Cruz Biotechnology), mouse polyclonal anti-Paxillin (1:500; BD Bioscience), rabbit polyclonal anti-myc tag (1:200; abcam), mouse monoclonal anti VE-cadherin (1:400, BD Biosciences), and Alexa Fluor 488 Phalloidin (1:400 dilution of 200 units/mL stock ; life technologies). All antibodies were diluted in the 0.5x blocking buffer (2.5% normal goat serum and 0.5% BSA in 1xPBS). Following primary antibody incubation, cells were washed in PBS for 5 min four times and incubated with Alexa Fluor 405 goat anti-mouse (1:400, PS-1), Alexa Fluor 594 conjugated goat anti-mouse (1:400; Paxillin), Alexa Fluor 488-conjugated goat anti-mouse (1:400; VE-cadherin), Alexa Fluor 647-conjugated goat anti-mouse (1:500, ADAM10), and Alexa Fluor 647-conjugated goat anti-rabbit (1:500, nicastrin) as appropriate. Nucleus staining was performed using Hoechst 33342 (ThermoFisher) diluted in 1xPBS (1 µg/mL) for 15 min and then washed once. Cells were incubated at room temperature in the dark for 1 hr, washed with PBS for 5 min three times, and imaged by epifluorescence or confocal microscopy. Epifluorescence microscopy was performed with 60x Apo, 1.40 NA or 100x Apo, 1.49 NA oil objectives (Nikon) on a Nikon Ti Eclipse microscope equipped with OBIS solid-state lasers (488, 552, and 647 nm, Coherent Inc.), a 300W Xenon lamp (Sutter Instrument, Lambda LS), a motorized stage (ASI, MS-2000), and a temperature- and CO2-controlled stage top incubator (Okolab, Bold Line). Unless otherwise noted, confocal microscopy was performed using Plan-Apo 60x, 1.4 NA or Plan-Apo 100x, 1.4 NA oil objectives (Nikon) on a Nikon A1R laser scanning confocal microscope. Images were acquired using Galvano scanning mode and confocal zoom of 3–4x magnification.
Spatial distribution of Notch signaling molecules at cellular interface
We used three different cell culture model systems to investigate spatial distribution of Notch1 during its surface activation: Notch ligand- and receptor-expressing cells that form the signaling interface (#1; Fig. 1a–c), the signaling interface formed by the cells exclusively expressing either Notch ligand or receptor (#2; Extended Data Fig. 1a–b, d), and Notch-expressing cells cultured on a Dll4-coated substrate (#2; Fig. 1d–f).
The model system #1:
U2OS cells co-expressing SNAP-NFL-mCherry and Halo-Dll1 were plated on a collagen-coated #1.5 glass-bottomed Mattek dish at a density of 2×105 cells/mL. Cells were incubated with the appropriate inhibitors, either TAPI2 (100 μM), shRNA cocktails against ADAM10/17, and/or DAPT (5 μM). Different combinations of inhibitors were used to capture the respective intermediates. After 48 h, cells were labeled with SNAP-594 (NEB, 5 µM) and Janelia Fluor HaloTag 646 (Promega, 5 µM). Then cells were then fixed and immunostained for PS1 using the protocol described above. Briefly, following the permeabilization and blocking steps, cells were incubated overnight at 4 °C with rabbit polyclonal anti-PS1 and 1 h at 25 °C with AF405-conjugated goat anti-rabbit antibodies. Respective inhibitors were maintained during wash and fixation steps. Spatial distribution of respective Notch signaling molecules was then imaged with a confocal fluorescence microscope, as described above.
The model system #2:
We repeated identical experiments where cells exclusively expressing either SNAP-NFL-mCherry or Halo-Dll1 were co-cultured with the ratio of 1:1, to form trans ligand-receptor interactions exclusively. We observed amplified mCherry signals at some RIP µdomains upon rescuing ADAM10/17 activity in the presence of DAPT (Extended Data Fig. 1d; box (i) and (ii)), but we also observed decreased mCherry signals at other RIP µdomains (Extended Data Fig. 1d; box (iii)). We reasoned that because both full-length Notch and the Notch intermediate (i.e., S2-processed Notch1) bear mCherry tag, and spatial mapping of the activated Notch intermediate using mCherry fluorescence is challenging unless the fraction of the Notch intermediate is significantly higher than unprocessed full-length Notch. We reasoned that the relatively small cell-cell contact area (0.5–10 µm2) and low ligand density (< 300 molecules per µm2)96 limited receptor activation in this configuration, and hence the mCherry fluorescence signal from unprocessed Notch overwhelmed that from S2-prcessesed Notch in some areas of the cell.
The model system #3:
To maximize the ligand-receptor engagement and hence robust Notch activation, we used culture system #3 allowing high-density ligand loading (>3,000 molecules per µm2) and increased ligand-receptor contact area (100–500 µm2)7. To activate Notch, we plated cells expressing SNAP-NFL-mCherry on a substrate coated with Dll4 fused with a Fc fragment (Dll4-Fc). Briefly, a glass bottom dish (Lab-Tek II Chambered Coverglass, ThermoFisher, or 7-mm glass-bottomed dish, MatTek) was coated with fibronectin (Hamster, 5 µg/ml) and Dll4-Fc (2.5 µg/ml) for 1 hr at 37°C, and washed thoroughly with 10 ml of PBS. A negative control dish was also prepared by coating it with fibronectin only. U2OS cells co-expressing SNAP-NFL-mCherry and Ecad-GFP were plated and incubated with doxycycline (2 μg/mL), TAPI2 (100 μM)97, and/or DAPT (5 μM). Different combinations of inhibitors were used to capture the respective intermediates. After 48 hr, cells were labeled with SNAP-647 (NEB, 5 µM) and then fixed as detailed above. Inhibitor concentrations were maintained during wash and fixation steps. In this system, significantly more Notch becomes activated, and as a result, we observed strong enrichment of mCherry signals within the RIP microdomain as presented in Fig. 1d–f, indicating translocation of Notch after S2 cleavage.
To map spatial dynamics of Notch relative to AJs during activation (Extended Data Fig. 3a–e), we plated U2OS cells co-expressing SNAP-NFL-mCherry and Ecad-GFP on a Dll4-Fc coated substrate to trigger Notch activation as described above. Cells were also treated with TAPI2 (S2 cleavage inhibition) or DAPT (S3 cleavage inhibition) to capture Notch intermediates. For time-lapse imaging (Extended Data Fig. 3, d and e), we washed the cells with large volumes of PBS to remove DAPT, incubated cells with DAPT-free media, and imaged at each time points (0, 0.5, 1.5, 3, 6, and 12 hrs) , respectively. The spatial distribution of Notch intermediates and AJs was monitored using spinning disk confocal fluorescence microscopy (Zeiss Cell Observer Z1), equipped with Yokagawa spinning disk and Evolve 512 EMCCD Camera (Photometrics). Images were obtained with Plan-Apo 63x, 1.4 NA or Plan-Apo 100× 1.46 NA oil objectives (Zeiss) with solid-state lasers of 405, 488, 561 nm, and 647 nm. The microscope was controlled with Zeiss Zen software (Zeiss).
Spectral general polarization () imaging of the live-cell plasma membrane
Cells were prepared for di-4-ANEPPDHQ imaging as previously reported49. Briefly, U2OS cells expressing Ecad-GFP were plated on a fibronectin-coated glass bottom dish and incubated with di-4-ANEPPDHQ (Invitrogen, 2 µM) for 30 min at 37°C in a humidified 5% CO2 atmosphere. Cells were then washed 3 times with PBS. Spectral imaging was performed on a Nikon A1R laser scanning confocal microscope equippe with a 32-channel GaAsP detector array. Laser at 488 nm was selected for excitation and the detection range was set between 495 and 750 nm for di-4-ANEPPDHQ. Calculation and generation of images were performed as previously described using the provided ImageJ plug-in macro codes49. values for di-4-ANEPPDHQ imaging were calculated according to the following equation: and (the factor) =. For the imaging of cell membranes, is −0.85. is the value of di-4-ANEPPDHQ dye in pure DMSO measured with the same microscope setup. The plugin applies above calculation to produce a histogram of the map and a pseudo-colored map representing the value for each pixel of the image.
Single-cell cleavage kinetics of SNAP-NΔEGF-mC
A 6-channel µ-slide flow chamber (Ibidi, VI 0.4) was coated with fibronectin (2.5 µg/mL) for 1 hr at 37°C and washed with PBS four times. U2OS cells co-expressing SNAP-N∆EGF-mCherry and Ecad-GFP were plated on the µ-slide flow chamber by applying 60 µL of single cell suspension at a density of 3 × 105 cells/mL. After 3 hr, the channel was filled with a complete McCoy’s 5A medium containing doxycycline (2 µg/mL), TAPI2 (100 µM), and DAPT (5 µM). Cells were grown for 48 hr in normal growth medium to reach 70–80% confluency and form cadherin adherens junctions. Cells were labeled with BG-Alexa Fluor 647 (NEB) for 30 min to stain cell surface N∆EGF. Multiple cells with stable AJs were identified using large-area epi-fluorescence scanning (500 µm x 500 µm), and the spatial distribution of SNAP-NΔEGF-mCherry at AJs under TAPI2 and DAPT inhibition was imaged by confocal z-stack (step size = 0.2 µm, total range of z stacks = 10 µm) scanning from basal to apical membranes. Then, DAPT containing media was removed and replaced by flowing complete medium containing doxycycline and TAPI2 at a flow rate of 50 µl/min for 10 minutes using a syringe pump. Localization of extracellular (NECD) and intracellular (NICD) domain at the AJs before and during DAPT washout was monitored every 30 minutes in multiple color channels (NICD, mCherry; NECD, AF647; AJ, GFP) by time-lapse confocal z-stack microscopy for 12 hr. Time-lapse live cell confocal imaging was performed using a 60x Plan-Apo oil objective (NA 1.4) on a Nikon A1 laser scanning confocal microscope equipped with an environmental chamber maintaining cells at 37°C, 5% CO2.
Western Blot analysis
U2OS cells co-expressing Notch variants and Ecad-GFP (or Halo-Ecad-GFP) were incubated with culture media containing doxycycline (2 µg/mL) and TAPI2 (100 µM) in a 6-well plate at a density of 1×106 cells per well. After 24 hr, cells were washed with ice-cold DPBS twice and lysed in RIPA (Invitrogen) or 1% NP-40 (Invitrogen) supplemented with complete protease and phosphatase inhibitor cocktail (100x; Cell Signaling Technology) at 4°C while gently shaking for 30 minutes. Insoluble fractions were removed by centrifugation of the cell lysates at 13,000 r.p.m. for 10 minutes. Total protein concentrations in lysates were determined by a BCA assay (Bio-Rad). 20 µg of whole cell lysates were then mixed with 4x Laemmli sample buffer (Bio-Rad) with 10% β-mercaptoethanol (BME) and heated to 95°C for 5 minutes. For western blot analysis of DNA-crosslinked heterodimers, the cell lysates were mixed with 4x Laemmli sample buffer (Bio-Rad) without BME before boiling to denature. Samples were then loaded into a 4–15% Mini-Protein TGX precast gel (Bio-Rad) and were run at 70 V for 30 minutes and then 120 V for 45 minutes. Separated proteins were transferred to a PVDF membrane using Mini Trans-Blot Cell (100 V constant, 1 hr) or the Trans Turbo Blot system (Bio-Rad). Membranes were blocked for 1hr at room temperature in blocking solution (5% w/v nonfat dry milk in 1x TBST). The membranes were probed with anti-V1744 NICD antibody (1:1000; Cell Signaling Technology #4147), anti-SNAP (1:1000; NEB), anti-Notch1 (1:1000, Cell Signaling Technology #3447 or #4380), anti-mCherry (1:500, Abcam #167453), anti-E-cadherin (1:100, Santa Cruz Biotechnology, sc-8426), and anti-β-actin (1:5000; Cell Signaling Technology #4970) antibodies overnight at 4°C with gentle rocking. The membranes were washed in TBST three times for 5 minutes and incubated with an anti-rabbit (Cell Signaling Technology, # at 1:2000 for NICD, mCherry, SNAP detection and at 1:10000 for β-actin detection) or anti-mouse (Cell Signaling Technology, # at 1:2000 for E-cadherin detection) HRP conjugated antibody. The target proteins were visualized by chemiluminescence using an ECL detection kit and a ChemiDoc MP imaging system (Bio-Rad). Quantification of band intensities by densitometry was carried out using the Image Lab software (Bio-Rad). For quantification, the average intensity of NICD band was normalized to that of β-actin band in each sample, unless otherwise noted.
Spatial mutation of SNAP-NΔEGF-mCherry via DNA crosslinking
DNA-mediated crosslinking of SNAP-NΔEGF-mCherry with Halo-Ecad-GFP. DNA crosslinkers including benzylguanine (BG)- and chloroalkane (Cl) modified oligonucleotides were synthesized as previously described. To prepare 10x crosslinking DNA stock solution, complementary BG- and Cl-modified oligonucleotides were hybridized in situ. BG-T10(ACTG)5 and Cl-T10(CAGT)5 were mixed at equimolar concentration (20 µM) in PBS, incubated at 95°C on a dry heat block for 5 min, and slowly cooled down to room temperature for 2 hr. U2OS cells co-expressing SNAP-NΔEGF-mCherry and Halo-Ecad-GFP were cultured in a 6-well plate for western blot analysis at a density of 1 × 106 cells per mL or in a channel of an Ibidi µ-slide for confocal imaging analysis at a density of 3 × 105 cells per mL. Cells were grown to 70–80% confluency for typically 24 hr, followed by overnight incubation with complete medium containing doxycycline (2 µg/mL), TAPI2 (100 µM) and DAPT (5 µM). Cells were then serum starved with 2 ml of serum-free medium with doxycycline, DAPT, and TAPI2 for 6 hrs. Before adding DNA crosslinkers, cells were washed and placed in 450 µl of serum-free media. 50 µl of prewarmed 10x DNA crosslinker stock solution was added to each well and incubated at 37°C. Western blot analysis to validate receptor crosslinking were performed after 30-minute incubation of the DNA crosslinkers as detailed above.
Live cell confocal time-lapse imaging.
After overnight incubation with the DNA crosslinkers, imaging was performed on an inverted laser scanning confocal microscope (Nikon A1) equipped with an environmental chamber at 37°C and 5% CO2. Images were obtained with a Plan-Apochromat 60x, 1.4 NA oil objective (Nikon) with solid-state lasers of 405, 488, 561 nm, and 647 nm. Additionally, the microscope was equipped with Ti-E Perfect Focus System (Nikon). To examine the effect of DNA-mediated crosslinking on spatial distribution of SNAP-NΔEGF-mCherry at AJs, multiple AJs were imaged in entirety from basal to apical sides for Halo-Ecad-GFP and SNAP-NΔEGF-mCherry using a 488 nm and 561 nm laser respectively, for a 12 µm range at a z-step size of 0.25 µm. To monitor dissipation of SNAP-NΔEGF-mCherry at AJs upon removal of DAPT inhibition, fresh phenol red-free McCoy’s 5A medium containing doxycycline and TAPI2 was introduced into the channel using a syringe pump for 10 min, and confocal z-stack images of the previously selected AJs were acquired every 30 minutes for 6 hr. Images were acquired using NIS-element software (Nikon), and image post-processing and analyses were done using Fiji/ImageJ and custom-built scripts.
Spatial mutation of SNAP-NEXT-mCherry via molecular pendant addition
Synthesis of BG-modified polyethylene glycol (PEG).
Amine-functionalized PEGs with different molecular weights and structures were purchased from Creative PEGWorks (NH2-PEG3.4k), Sigma (NH2-bPEG20k), and NanoCS (NH2-PEG20k) and used without further purification. BG-functionalization of PEGs was performed by amine-NHS (N-hydroxysuccinimide; NEB) coupling reaction. Briefly, NH2-PEG (0.5 µmol), BG-GLH-NHS (2.4 mg, 5 µmol), and N,N-dimethylaminopyridine (0.73 mg, 6 µmol; Sigma) were dissolved in anhydrous dimethylsulfoxide (DMSO). The mixture allowed to react overnight with constant shaking. Crude products were recovered by evaporating DMSO using a Speed-Vac concentrator (Vacufuge, Eppendorf), reconstituted in 500 µl deionized water, and insoluble precipitates were removed by centrifugation at 14,000 r.p.m. for 10 minutes. The BG-modified PEG was then dissolved in 200 µl in deionized water and purified by reverse-phase high performance liquid chromatography with an Agilent Eclipse XDB C-18, 5 µm, 4.6 × 250 mm2 column using an elution gradient of 5–75% acetonitrile in 0.02% trifluoroacetic acid.
Synthesis of BG-modified DNA-streptavidin conjugates.
DNA oligonucleotides bearing biotin- and BG-functional groups were synthesized by reacting biotin-(ACTG)5-NH2 (IDT DNA) with BG-GLH-NHS as described above 63. Equimolar amounts of streptavidin (10 nmol) and BG-DNA-biotin (10 nmol) were dissolved in PBS (0.5 ml) for 2 hr, forming streptavidin-BG complex. The solution was concentrated to approximately 50 µl using an Amicon centrifugal filter (MWCO: 30k) and then diluted again with 0.45 ml of PBS. This concentration and reconstitution step was repeated three times to remove unconjugated DNA.
Synthesis of BG-modified human IgG.
hIgG (10 mg) and BG-GLA-NHS (0.82 mg) were dissolved in 850 µl of PBS and 150 µl of anhydrous DMSO, respectively. Two solutions were mixed and reacted for 2 hr at room temperature with gentle shaking. The solution was desalted with NAP-10 and then with NAP25 pre-equilibrated with PBS. The proteins were further concentrated until the final volume is 300–500 µl using Amicon centrifugal filter (MWCO: 30k). The IgG concentration was determined by measuring the absorbance at 280 nm.
Spatial mutation of SNAP-NEXT-mCherry using the BG-modified macromolecules.
U2OS cells co-expressing SNAP-NEXT-mCherry and Ecad-GFP were incubated in complete McCoy’s 5A medium containing doxycycline (2 µg/ml), TAPI2 (100 µM), DAPT (5 µM), and respective BG-modified macromolecules (10 µM). After 24 h, cells were fixed and imaged by confocal microscopy to determine the enrichment factor of SNAP-NEXT-mCherry at AJs. Images were taken with a 100x objective and 3x confocal zoom. 20 stage positions per each treatment were manually selected and their coordinates were stored in the computer. In each position, confocal z stacks of DAPI, Ecad-GFP and Notch-mCherry were acquired for a 12 µm range at a Z step-size of 0.25 µm to monitor the AJs in their entirety from basal to apical sides. To assess the levels of Notch activation, a set of identical experiment but without DAPT was performed. After 24 h, cells were fixed, stained with DAPI, and imaged by confocal microscopy to determine nuclear mCherry signal. Images were taken with a 60x objective and 1x confocal zoom. 5 stage positions per each condition were selected manually. For each position, a confocal large-area scan of DAPI, Ecad-GFP, and SNAP-NEXT-mCherry was acquired for a 1 mm x 1 mm area.
Plate-bound Dll4 Notch activation in high-density grouped versus solitary cells
To activate Notch, we plated SNAP-NFL-Gal4 reporter cells on a substrate coated with Dll4-Fc as detailed above. Two different cell seeding densities were used: We plated cells with a density of 1 × 103 cells per 10 mm glass-bottomed dish (MatTek, No. 1.5 glass), predominantly yielding solitary cells. We also plated cells with a density of 1 × 104 cells per dish, predominantly yielding high-density grouped cells.
Plate-bound E-cadherin Notch activation experiment.
Glass-bottomed dishes (MatTek, #1.5, D = 10 mm) were coated with recombinant human E-cadherin-Fc (50 µg/ml, R&D systems), recombinant human Dll4-Fc (2.5 µg/ml, Sino Biological), and fibronectin (5 µg/ml, Sino Biological) diluted in PBS for 1 hr at 37°C, and rinsed with 10 ml PBS with calcium and magnesium (UCSF cell culture facility). The U2OS SNAP-NFL-Gal4 reporter cells were transfected with Ecad-GFP (10 µg) via electroporation, incubated overnight, and re-plated onto a fibronectin, E-cadherin-Fc, and Dll4-Fc coated glass-bottomed dish at a density of 0.3 × 105 cells/ml, same as the solitary cell assay. Negative control experiment was performed with the cells plated on E-cadherin-Fc and fibronectin coated glass-bottomed dishes without Dll4-Fc coating.
Time-lapse epifluorescence imaging.
All cells were treated with 2 µg/ml doxycycline (sigma-aldrich) at the time of plating. 2 hr post-plating, cells were imaged using time-lapse microscopy. For a high-density cell seeding assay, several groups of cells having cell-cell contacts were manually identified and their coordinates were stored. For a solitary cell assay, a number of solitary cells without any prior cell-cell contact were manually identified and their coordinates were stored. While maintaining live cells on a microscope stage with a top stage incubator, time-lapse fluorescence images were acquired in GFP and mCherry channels. In each position, the microscope (Nikon) first found focuses using the Perfect Focus System (Nikon), took a DIC image, and two fluorescent images (GFP, mCherry). To image multiple solitary cells and grouped cells in one large image, cells were first plated at a high-density (2×105/ml) at the center, and after 15 min, cells were seeded at a low-density (2×103/ml) over the entire substrate area. After 24 h, cells were fixed and stained for membrane and nucleus. Epifluorescence images were obtained with an inverted microscope (Nikon, Ti Eclipse) equipped with 300W Xenon lamp (Sutter Instrument, Lambda LS), a motorized stage (ASI, MS-2000), and a temperature- and CO2-controlled stage top incubator (Okolab, Bold Line). Images were taken with 40x (CFI Plan fluor, N.A. 1.3, Nikon) objective lens. The microscopy setup was controlled using µ-manager software.
CRISPR editing to generate E-cadherin and N-cadherin knockout mutants
CRISPR/Cas9 was used to knock out E-cadherin and N-cadherin expression from U2OS SNAP-NFL-Gal4 reporter cells. The genes coding for the E-cadherin (CDH1) and N-cadherin (CDH2) protein from homo sapiens (gene ID: ENSG00000039068 and ENSG00000170558) were truncated by a CRISPR/Cas9 paired sgRNAs excision strategy98,99. For the fragment deletion of genomic DNA, we used a pair of gRNAs against the target locus of CDH1 (Exon 1 & 2 (940bp deletion) or 13 & 14 (~4712bp deletion) (Extended Data Fig. 5a) and CDH2 (Exon 1 & 2 (~29,255bp deletion) (Extended Data Fig. 5b) genes.
sgRNA design and expression vector cloning.
Cas9 and sgRNAs were expressed using the CMV promoter-driven Cas9–2A-mRFP-2A-Puro plasmid (hereafter, Cas9-puro vector) and the hU6 promoter-driven sgRNA plasmid (Toolgen), respectively. To design sgRNAs for fragmental deletion of target loci of genes, all candidate sgRNA target sites with a protospacer adjacent motif (PAM; 5’-NGG-3’) within the coding sequence of CDH1 and CDH2 were initially identified. For efficient deletion, selected sgRNAs for the candidate target sites were evaluated with DeepSpCas9 sgRNA prediction tool (http://deepcrispr.info/DeepSpCas9/)100. sgRNAs with high DeepSpCas9 score were selected and sgRNA oligonucleotides annealed and cloned into the vector as previously described (Ramakrishna et al., 2014a). Sequences of the vectors and sgRNAs listed here are available upon request.
Generation of single-cell derived knock-out clones.
For CDH1 knockout, SNAP-NFL-Gal4 reporter cells were transfected with plasmid mixtures containing Cas9-puro, U6-sgRNA encoding individual sgRNAs at a weight ratio of 1:2 using the Neon system. For CDH1/2 knockout, cells were transfected with plasmid mixtures containing Cas9-puro, U6-sgRNA targeting CDH1 loci, and U6-sgRNA targeting CDH2 loci at a weight ratio of 1:1:1 using the Neon system. One day after transfection, puromycin was added to the culture media at a final concentration of 2.5 µg ml−1. Three days after transfection, the pooled cells were analyzed for the indel efficiency of sgRNA pairs using T7E1 assay. To obtain single cell-derived clones containing the fragment deletion, we plated the cells after puromycin selection into 96-well plates at an average density of 0.25 cells/well. 14 days after plating, individual clones were isolated and analyzed using PCR and gel electrophoresis of genomic DNA to check the deletion and wild-type alleles. We next sequenced the genomic DNA of the clones containing targeted deletions to check if the two cleavage sites were joined by the generation of indels. Sequencing of genomic regions including the target sequence was performed as previously described102. Briefly, PCR amplicons that included the junction regions of the deleted lncRNA target sites were cloned into the T-Blunt vector (Promega) and sequenced using universal M13FP or RP primers.
T7E1 assay.
The T7E1 assay was performed as previously described103. Briefly, genomic DNA was isolated using the Wizard Genomic DNA purification Kit (Promega) according to the manufacturer’s instructions. The region including the target site was nested PCR-amplified using appropriate primers. The amplicons were denatured by heating and annealed to allow the formation of heteroduplex DNA, which was treated with 5 units of T7 endonuclease 1 (NEB) for 20 min at 37°C followed by analysis using 2% agarose gel electrophoresis. Mutation frequencies were calculated as previously described based on the band intensities using ImageJ software and the following equation103: mutation frequency (%) = 100 × (1 − (1 − fraction cleaved)1/2), where the fraction cleaved is the total relative density of the cleavage bands divided by the sum of the relative density of the cleavage bands and uncut bands.
RT-PCR.
Total RNA was extracted from wild-type SNAP-NFL-Gal4 reporter (WT) cells and knockout clonal cells using TRIzol (Ambion) or an RNeasy Kit (QIAGEN), after which complementary DNA (cDNA) synthesis was performed using a DiaStarTM RT Kit (SolGent Co., Ltd.). The synthesized cDNA was subjected to quantitative PCR (qPCR) in triplicate using an Applied Biosystems StepOnePlus Real Time PCR System with PowerSYBR Green PCR Master Mix (Applied Biosystems). Gene expression was normalized to that of the CDH1 gene in WT cells. Error bars represent the standard deviation (s.d.) of the mean of triplicate reactions. Primer sequences for qPCR are available upon request.
Notch activation assay.
WT cells, CRISPR CDH1 knock-out cells (CDH1−/−), CDH1−/− transfected with E-cadherin-GFP (CDH1−/− + E-cad), and CDH1−/− transfected with N-cadherin (CDH1−/− + N-cad) were plated on 8 well Nunc Lab-Tek II chambered coverglass pre-coated with recombinant human Dll4-Fc (2.5 µg/ml) and fibronectin (5.0 µg/ml) as previously described. All cells were plated at a density of 30,000 cells per well. After 24 hr incubation with doxycycline (2 µg/ml), cell cytoplasm and nucleus were stained with CellTracker CMFDA dye (Invitrogen) and Hoechst 33342 (ThermoFisher), respectively. The cells were then fixed with 4% PFA for 15 minutes at room temperature and proceeded to epifluorescence and confocal imaging.
Aβ40, Aβ42, and sAPPα ELISA
1×106 of wild-type or CDH1/CDH2 knockout cells were transfected with APP-mCherry (10 µg) and plated on 6-well tissue culture plate. After 48 hr incubation, conditioned media supplemented with 1x protease/phosphatase inhibitor cocktail (ThermoFisher) were centrifuged at 3,000xg 10 minutes at 4°C to remove cell debris and the supernatant were transferred to a new tube and stored at −80C. The conditioned media were analyzed for Aβ40, Aβ42, and sAPPα contents using Aβ (Invitrogen) and sAPPα (Kusa Biosci.) ELISA kits. All ELISAs were performed according to the manufacturer’s protocols. Briefly, 50 µl/well of samples, followed by 50 µl/well of detection antibodies, were applied to Aβ40, Aβ42, sAPPα coated 96-well plate, and incubated for 3 hr at room temp with shaking at 300 rpm. After washing step, 100 µl of HRP-IgG solution was applied and incubated for 30 min at room temp with shaking. After washing step, 100 µl of stabilized substrate solution was applied and incubated for 30 min at room temp with shaking. Finally, 100 µl of stop solution was applied. Absorbance at 450 nm was read and analyzed using a plate reader (Biotek Synergy 2). Wells were washed with wash buffer 4 times between each incubation step. Standard curves were generated using recombinant Aβ40, Aβ42, sAPPα provided by the manufacturer (Invitrogen).
In vivo experiments
Animals.
We used CD-1 embryonic day 13.5 (E13.5) and postnatal day three (P3) newly born mice for in vivo experiments. P3 pups (4 males and 4 females) were obtained by purchased of an untimed pregnant female mouse (E13–15) from Charles River Laboratories (Wilmington, MA) and waited for birth. Room temperature was maintained at 22°C ± 1°C with 30–70% humidity. All mice were housed under specific pathogen-free conditions under a 12h light-dark cycle, and all animal handling and use were in accordance with institutional guidelines approved by the University of California San Francisco Institutional Animal Care and Use Committee (IACUC, AN180609–02B).
Retrovirus injection.
To generate retrovirus, we used pWZL-GFP control vector (pWZL-Blast-GFP: addgene plasmid # 12269) and pWZL-dominant negative E-cadherin (pWZL-Blast-DN-E-cadherin addgene plasmid # 18800 and gift from Dr. Kenji Shimamura). GFP sequence was inserted in frame with dominant negative E-cadherin, downstream of C-terminus using In-Fusion cloning. For retrovirus production, we transfected retroviral vectors into Phoenix-Ampho cells using Calcium Phosphate transfection kit (Sigma, CAPHOS) with 50 µM chloroquine (Sigma, C6628), and collected supernatant from transfected cells after 48 h. Collected supernatant containing viral solutions was ultracentrifuged yielding concentrated solution of viral particles (25,000 rpm for 2 hours at 4°C). Approximately 107 transducing units per milliliter (TU/ml) viral solution was injected into the lateral ventricular of neonatal mouse pups (P3). After hypothermic anesthesia, viral solutions (5 µl) were slowly injected using IM-9B Narishige microinjector with 2 µl/min speed. After recovery on the warming pad, the pups were placed back to the cage. After additional 2 hours, mice were subject to intracardiac perfusion fixation using 4% paraformaldehyde in PBS.
DAPT injection.
10 µM DAPT was injected into neonatal mouse pups (P3) into a lateral ventricle (10 µl in each hemisphere). DMSO was injected into control mice. After 7 hours, mice were subject to fixation procedure using intracardiac perfusion of 4% paraformaldehyde in PBS.
Immunohistochemistry.
Mice were perfused with 4% paraformaldehyde in PBS (pH7.4) and the brains were subject to postfixation in the same fixative for 24 h. Brains were then cryoprotected in 30% sucrose in PBS, sectioned serially (20 μm) onto Superfrost plus glass slides (Fisher Scientific; Pittsburgh, PA). The brain slices were permeabilized and blocked with PBS solution containing 3% goat serum albumin and 0.3% Triton-X100, and then treated with anti-Ncadherin (1:200, Thermofisher), anti-Notch (1:200, Thermofisher), anti-PS1 antibody R22232 (1:50), and anti-beta III tubulin (1:500, Abcam) overnight at 4°C. The brain slices were washed three times with PBS and treated with secondary antibodies (1:1000, Thermofisher) for 30 min. Subsequently, the slices were washed with PBS, mounted and observed with a confocal microscope (Olympus, Fluoview 3000).
Image processing and analysis
Cadherin junction colocalization analysis.
Colocalization analysis was carried out in ImageJ, using thresholding to identify AJs and then applying the JACOP plugin to quantify colocalization using Pearson coefficient, Manders’ overlap coefficients, and cross-correlation analysis. All ImageJ macros and codes used for image post-processing and colocalization analysis have been deposited and are available at Gitihub (https://github.com/sukgi333/yonsei-notch-activation).
Confocal 3D z-stack image processing.
Custom python code was used for automatic segmentation and junction intensity ratio analysis for Notch activation and truncation studies. Code is available at (https://github.com/kmsouthard/JunctionAnalysis). In brief, resliced z-stacks of cell-cell interfaces were thresholded to identify the AJs and membrane Notch signal. To minimize the domination of high Notch intensity, we identified the membrane expressing Notch using a minimal threshold of membrane intensity just above background. An unbiased signal analysis window along each side the junction was selected, and the Notch membrane intensity was measured for each cell by averaging along the respective windows, while junctional intensity was measured within segments determined by cadherin junctional intensity. The ratio of junctional intensity was calculated as ratio = Ijunc /(Icell1+ Icell2) as deviations from the expected intensity at the junction is a function of the sum of each cell’s expression level.
Intracellular mCherry nuclear translocation analysis.
mCherry nuclear translocation analysis was carried out in ImageJ. GFP images were used for automated identification of cell edges and segmentation of single cells. DAPI images were used for automated identification of nucleus by implementing the Otsu thresholding method. Nuclear mCherry fluorescence data was extracted from nuclear segments by calculating the integrated fluorescence within the nucleus and subtracting a background scattering signal. In Fig. 4h, nuclear mCherry fluorescence intensities for NEXT cells treated with the macromolecular pendants were rescaled to make the intensity of NFL and NEXT to 0.002 and 1.0, respectively, which are identical to the normalized band intensities of NFL and NEXT measured by western blot.
Quantification of single-cell fluorescence.
Single-cell tracking and nuclear mCherry fluorescence signal analysis of UAS-Gal4 reporter cells were performed with ImageJ, as previously described 55,65,66.
Western blot quantification.
Quantification of band intensities by densitometry was carried out using the Image Lab software (Bio-Rad). Band intensities of NICD in each lane were normalized by band intensities of loading control, β-actin in the corresponding lane.
Estimation of protein heights
Protein heights including extended Notch height was estimated by measuring the structural size of each domain (i.e., EGF, NRR, SNAP) in Pymol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.) and then creating an additive estimate based on the number of domains in the full-length Notch construct and each Notch truncation.
Molecular dynamics simulations
All MD simulations were conducted using the GROMACS package104. Polarized MARTINI 2.2 parameters were used for the simulations105. The system was composed of 54 PPCS, 54 DPPC, 108 DPPS, 288 DIPC, 216 CHOL, and 10108 water molecules with a molar composition of lipid was CHOL:PPCS:DPPC:DIPC = 3.0 : 1.5 : 1.5 : 4.0 in upper layer and CHOL:DPPS:DIPC = 3.0 : 3.0 : 4.0 in lower layer. To create immobilized lipids, we increased the mass of the phosphorus atom within DPPS by a factor of 1,000, keeping everything else the same in the parameter file. The pressure was set at 1.0 bar with a semi-isotropic parrinello-rahman coupling with compressibility 4.5 × 10−5 bar−1and the temperature was set to 295 K using nose-hoover coupling. Each system was neutralized and brought to a concentration of 0.15 M with randomly placed sodium and chloride ions. We employed the LINCS algorithm to constrain to bond lengths106. A time step of 20 fs was used with an update of the neighbor list every 10 steps, which are typical values employed in MARTINI simulations. Each simulation was run afterwards for 12 µs, the last 3 µs of which was used for analysis. The MD simulations were analyzed using the in-built GROMACS tools. MDAnalysis libraries107,108 were used for calculating diffusion constant of lipid component and g_energy was used for calculating interlayer interaction.
Statistics and Reproducibility
Statistical analysis was performed in GraphPad Prism 8.0 (GraphPad) or Microsoft Excel. Figure legends indicate all statistical tests used in the figure. Unless otherwise noted in the figure legends, statistical differences were determined using Student’s t-test (two-tailed unpaired or paired t-test, depending on the experiment) when only two groups were compared or by ordinary one-way ANOVA followed by Tukey posthoc test when multiple groups were analyzed. The number of samples (‘n’) used for each experimental analysis is indicated in the figure legends. Sample sizes of sufficient power were chosen on the basis of general standards accepted by the field and previous published studies in the field to enable statistical analyses and ensure reproducibility (e.g. PMID: 27180907, 26051539, 29398116, 30628888). No statistical method was used to predetermine sample size. No data were excluded from the analyses. Data distribution was assumed to be normal, but this was not formally tested. Randomization was not necessary for this basic science study. All samples used in each set of experiments were equal, except the experimental condition being tested. All experiments were performed with appropriate control.
The number of independent experiments repeated for each representative result shown in Extended Data Figures is provided here: For extended data figure 1(a–c), n = 5 independent experiments; extended data figure 1(d), n = 3 independent experiments; extended data figure 1(e), n ≥ 7 biologically independent cells; extended data figure 2(a), n = 3 biologically independent samples; extended data fig 2(b), n = 18 cells over two independent experiments; extended data fig 2(c), n = 3 biological replicates; extended data fig 2(h, i), n = 5 biological replicates; extended data fig 2(k–n), n = 3 biological replicates; extended data fig 4(c), n = 6 independent experiments; extended data fig 4(e), n = 3 independent experiments; extended data fig 4(f), n = 5 independent experiments; extended data fig 4(h–j), n = 3 independent experiments; extended data fig 5(g), n = 31 cells examined across two independent experiments; extended data fig 5(l), n = 39 (+TAPI2, +DAPT) and n = 21 cells (+TAPI2, −DAPT) examined across three independent experiments; extended data fig 6(a, b); n = 2 independent samples; extended data fig 6(c), n = 33 (−DNA) and n = 29 (+DNA) cells over three independent experiments; extended data fig 6(h), n = 2 independently synthesized samples; extended data fig 6(i), n = 2 independent samples; extended data fig 6(j), n = 4 independent experiments; extended data fig 6(k), n = 6 independent experiments; extended data fig 7(f,g), n = 3 independent experiments; extended data fig 8(b), n = 3 (control) and n = 5 (DN Ecad) independent animals.
Data Availability
Previously published genomic sequence data that were re-analysed here are available from Ensembl for E-cadherin (CDH1) and N-cadherin (CDH2) protein from homo sapiens (gene ID: ENSG00000039068 and ENSG00000170558). Source data are provided with this study. All statistical source data have been provided as Source Data. All raw unprocessed gel images have been provided as Source Data. All raw images acquired using confocal, epifluorescence, and time-lapse microscopy, and additional data that support the findings of this study are available from the corresponding authors upon reasonable request. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code Availability
Custom python code used for automatic segmentation and junction intensity ratio analysis for Notch activation and truncation studies is available at (https://github.com/kmsouthard/JunctionAnalysis). Colocalization analysis was carried out in ImageJ and the JACOP plugin available at (https://imagej.nih.gov/ij/plugins/track/jacop.html). Custom ImageJ codes for other analyses, including quantification of Manders’ overlap coefficients, Pearson’s coefficients, and lipid polarization analyses, are available at (https://github.com/sukgi333/yonsei-notch-activation). Selected sgRNAs for the candidate target sites were evaluated with DeepSpCas9 sgRNA prediction tool (http://deepcrispr.info/DeepSpCas9/)100.
Extended Data
Extended Data Fig. 1. Interfacial membrane compartmentalization choreographs the sequential molecular processing of Notch.
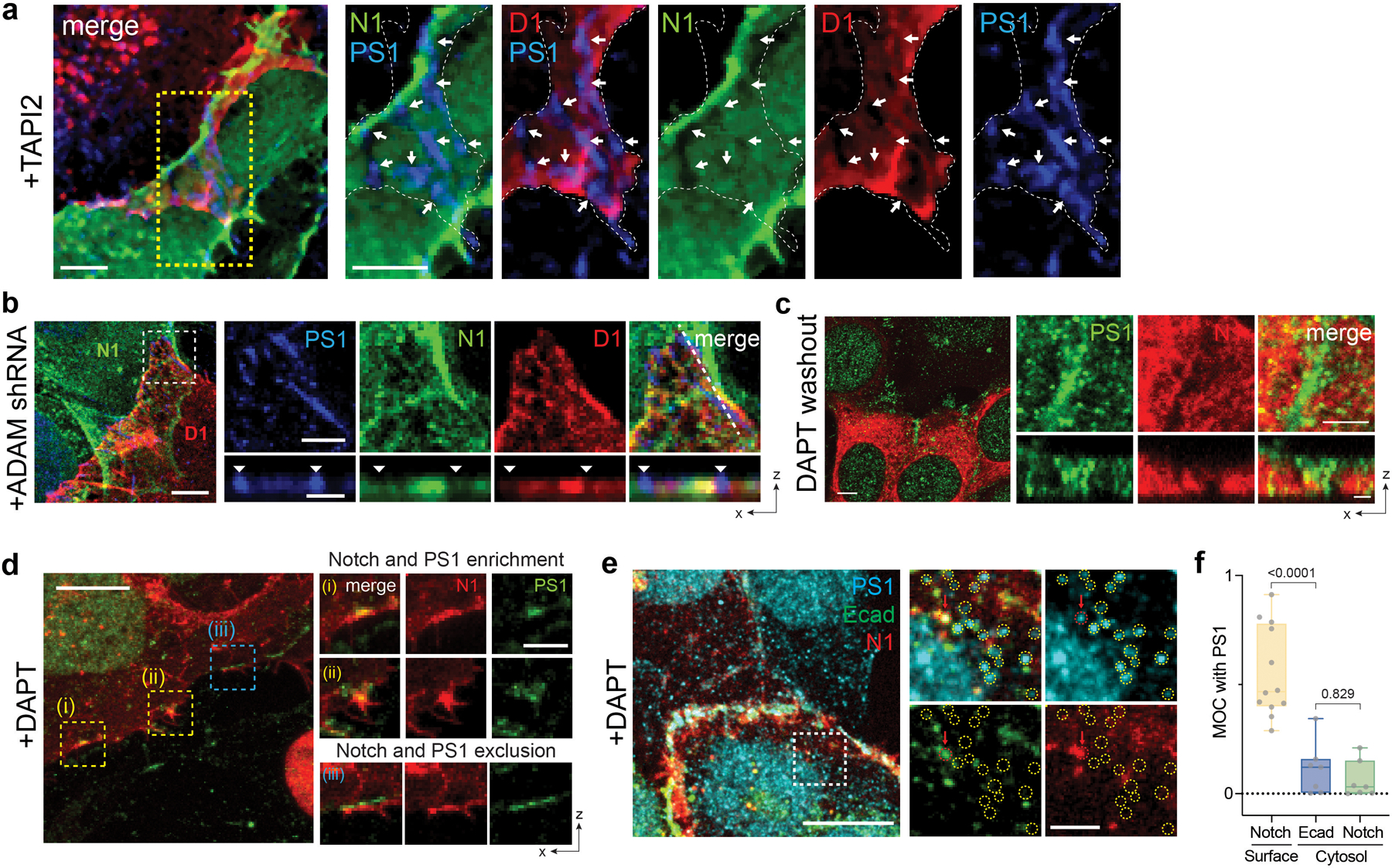
(a) Representative confocal images showing Dll1 (D1), Notch1 (N1), or presenilin1 (PS1) distributions at the interfacial junction between two cells exclusively expressing D1 or N1 in the presence of TAPI2. (left) A maximum intensity projection image of a LRE µdomains. Scale bar, 5 µm. (right) Magnified individual fluorescent channel and merged images of the yellow boxed region. The white dashed area and the white arrows indicate the cellular interface and the RIP µdomains, respectively. Scale bars, 3 µm. (b) Representative confocal images showing the interfacial junction between two cells exclusively expressing N1 and D1 in the presence of ADAM10/17 shRNA. The cell on top labeled ‘N1’ expresses N1 only, while the bottom cell labeled ‘D1’ expresses D1 only. (left) A maximum intensity projection image of a LRE µdomains. Scale bar, 5 µm. (top-right) Magnified individual fluorescence channel images of the boxed region. Scale bar, 3 µm. (bottom-right) Z-resliced images showing the sections of the cellular interfaces. Scale bar, 2 µm. (c) Representative confocal images showing N1, and PS1 distributions at the interfacial junction after washed out to remove TAPI2 and DAPT inhibition. With TAPI2 and siRNA treatment, Notch and γ-secretase compartmentalization was observed, while upon DAPT wash-out, Notch signals at the RIP µdomains disappeared. (left) A maximum intensity projection image of the cells showing enriched Notch mCherry signal at the RIP µdomains. Scale bar, 10 µm. (Top-right) Magnified individual fluorescence channel images of the boxed region. Scale bar, 2 µm. (right) Z-resliced images showing the sections of the cellular interfaces. Scale bar, 2 µm. (d) A representative confocal image showing Notch1 (N1) and presenilin1 (PS1) distribution at cellular interface. DAPT was added to inhibit S3 cleavage. (left) A maximum intensity projection image of a LRE µdomains. Scale bar, 10 µm. (right) Magnified individual fluorescent channel and merged images of the boxed region. Scale bar, 3 µm. (e) Representative confocal images showing intracellular distribution of PS1, E-cadherin (Ecad), and Notch1 (N1) in the presence of DAPT. (left) A maximum intensity projection image. Scale bar, 10 µm. (right) Magnified individual fluorescent channel and merged images of the specified intracellular region (a white dashed box). The yellow dashed circles represent intracellular puncta enriched with PS1 with no Ecad or N1 signals. The red dashed circle indicated with the red arrow represents an intracellular punctum showing all PS1, Ecad, and N1 fluorescence signals. Scale bar, 2 µm. (f) Manders’ overlap coefficients (MOCs) for quantitative assessment of PS1 with Notch1 at cell surface, PS1 with Ecad at cytosol, and PS1 with Notch1 at cytosol, respectively. Each dot represents the MOC of a selected cell surface or cytosol. In the box-whisker plot, the boxes show the 25th to 75th percentiles, and the whiskers extend to the maxima and the minima. Solid lines indicate median, respectively. n = 12 (Notch1 at surface), 7 (Ecad in cytosol), and 7 (Notch1 in cytosol) biologically independent cells across 2 independent experiments; ordinary one-way ANOVA with Tukey’s multiple comparison testing.
Extended Data Fig. 2. Adherens junctions reorganize Notch signaling biomolecules into two distinct membrane microdomains (µdomains).
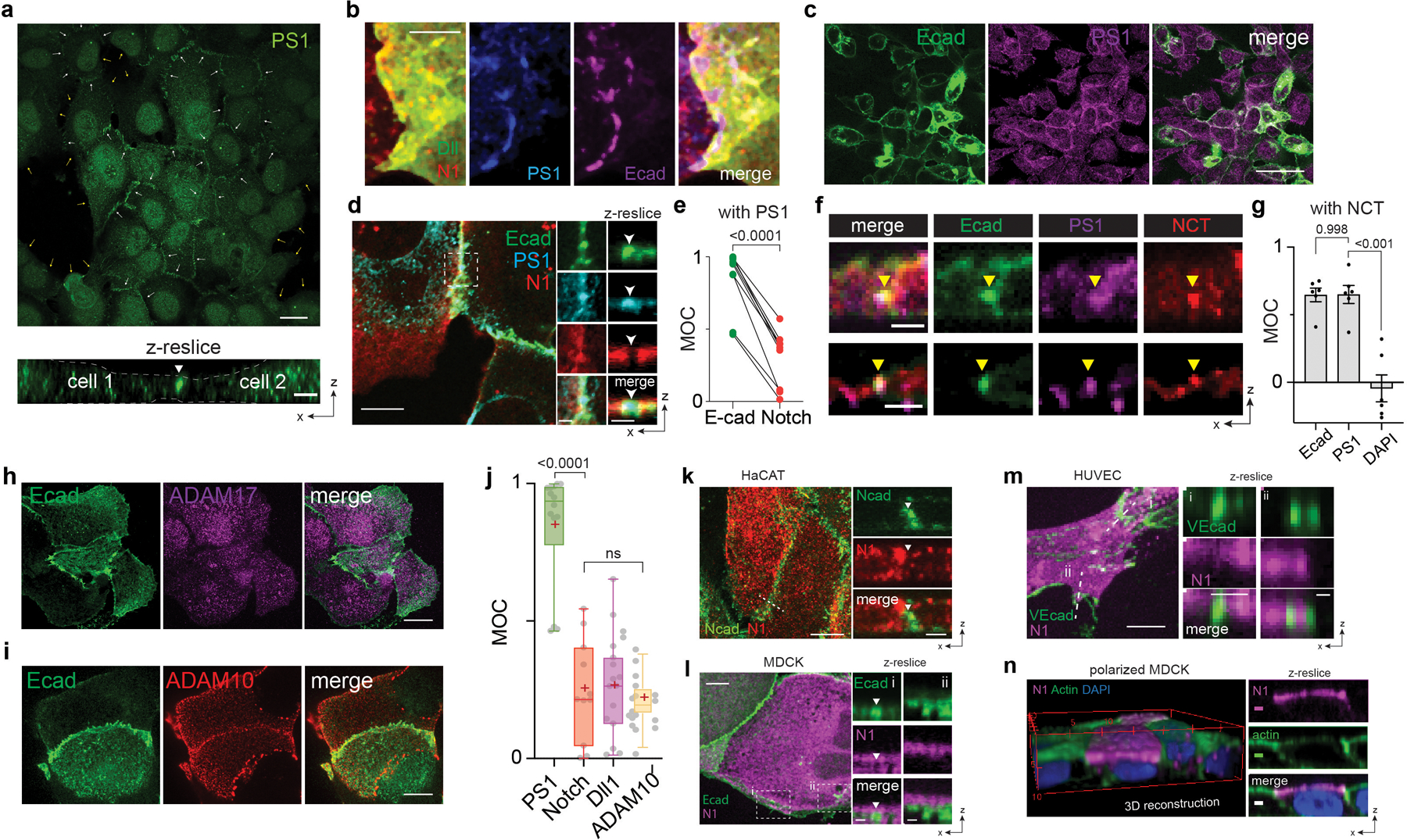
(a) Representative confocal immunofluorescence images showing distribution of presenilin1 (PS1) across entire cell membranes. White and yellow arrows indicate the cell-cell interfacial membranes and the cell membranes without contact, respectively. (top) A maximum intensity projection of the wide-field confocal z-stacks. Scale bar, 20 µm. (bottom) A confocal z-resliced image along the representative membranes of two contacting cells. A white arrow indicates the cell-cell interfacial membrane where strong PS1 signals were observed. Yellow dashed lines represent the cell membranes without cell-cell contacts. Scale bar, 3 µm. (b) Maximum intensity projection images showing distribution of PS1 and E-cadherin AJs (Ecad) relative to LRE-µdomain with Notch1-Dll1 pair enrichment at the interfacial membrane in the presence of TAPI2. PS1 and Dll1 were visualized by immunostaining, Notch and AJs by expression of their respective mCherry- or EGFP-fusion constructs. AJs showed nearly identical spatial distribution with the γ-secretase enriched RIP-µdomain, but exhibited inverse distribution with the LRE- µdomain. Scale bar, 5 µm. (c) Representative wide-field confocal immunofluorescence images showing PS1 (magenta) enrichment within AJs (green). Scale bar, 50 µm (low-magnification), 10 µm (zoom-in). (d) Confocal images of U2OS cells co-expressing Ecad-GFP (green) and SNAP-NFL-mCherry (red), and immunostained with PS1 antibody (cyan). Scale bars, 10 µm, 2 µm, and 2 µm for maximum intensity projection, zoomed-in, and z-resliced images, respectively. (e) Paired analysis of Manders’ overlap coefficients of E-cadherin and Notch signals over PS1 in multiple cells (n = 9 examined across 3 independent experiments). Two-tailed paired Student’s t test. (f) Confocal z-resliced images showing PS1 (magenta) and Nicastrin (NCT, red) distribution relative to cadAJs (green). Scale bar, 2 µm. (g) Analysis of Manders’ overlap coefficients (MOC) of E-cadherin, PS1, and DAPI over Nicastrin in multiple cells (n = 6 examined across 2 independent experiments). One-way ordinary ANOVA followed by Tukey’s multiple comparison testing. (h,i) Representative confocal immunofluorescence images showing (h) ADAM17 and (i) ADAM10 distribution relative to AJs. ADAM17 exhibited no preferential localization relative to AJs. Scale bar, 20 µm. (j) Box-whisker plots showing Manders’ overlap coefficients (MOCs) of PS1 (green), Notch (red), Dll1 (purple), and ADAM10 (yellow) relative to AJs. Each dot represents the MOC of a selected AJ. Boxes and whiskers denote the inner-quartile and full ranges. Colored lines and (+) marks indicate median and mean, respectively (n = 15 (Dll1), 11 (Notch1), 14 (PS1), and 19 (ADAM10) cells examined over two independent experiments; ns, not significant; ordinary one-way ANOVA with Tukey’s test). (k) Representative confocal fluorescence images of HaCaT cells immunostained with anti-Notch1 (red) and anti-N-cadherin (green). (l) Confocal images of MDCK cells expressing Ecad-GFP (green) and SNAP-NFL-mCherry. Notch receptors were labeled with BG-Alexafluor647 (magenta). (m) Confocal images of HUVECs expressing SNAP-NF-mCherry and immunostained with vascular endothelial cadherin (VE-cad) antibody. (l-m) Scale bar, 10 µm and 2 µm for maximum intensity projection and z-resliced images, respectively. (n) Polarized MDCK cells grown on a transwell filter. Notch, actin, and nucleus were immunostained with BG-AF647, phalloidin-488, and DAPI, respectively. Scale marked every 5 µm for 3D construction. Scale bar, 2 µm.
Extended Data Fig. 3. Spatial dynamics of Notch receptors relative to AJs during cell-surface activation.
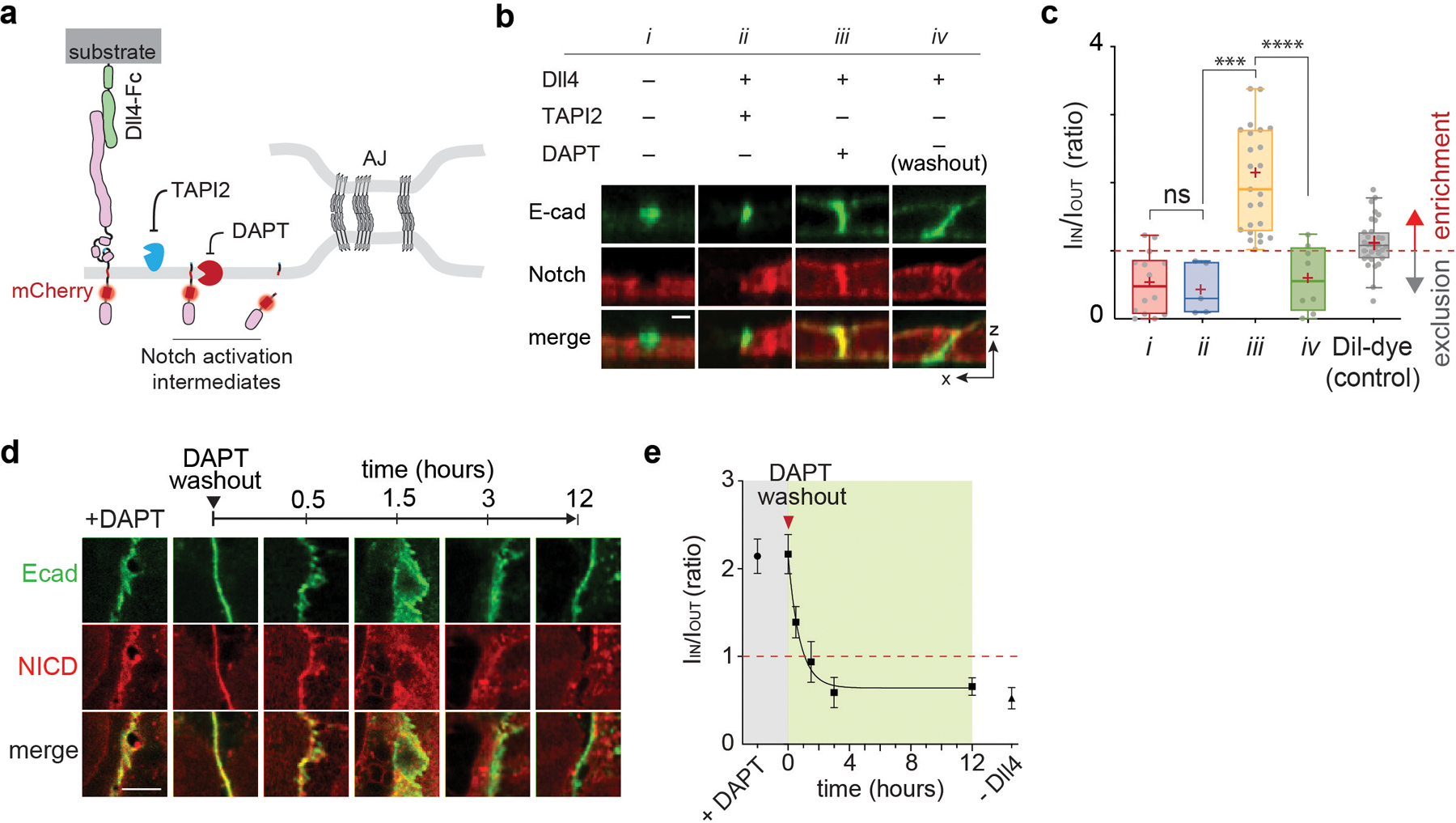
(a) A schematic to capture the spatial distribution of Notch intermediates during the cell-surface activation pathway. (b) Confocal z-resliced images showing Notch distribution (red) relative to AJ (green) from the cells without Dll4 activation (i), treated with Dll4 and TAPI2 (ii), treated with Dll4 and DAPT (iii), and washed out to remove DAPT inhibition (iv). Scale bar, 3 µm. (c) Quantification of Notch signal enrichment at the AJs during the activation. Notch enrichment (IIN/IOUT) is calculated as the ratio of average Notch fluorescence intensity within AJs (IIN) and outside AJ (IOUT). The enrichment factor of Dil is present as a control showing AJ-independent distribution. In the box-whisker plot, the boxes show the 25th to 75th percentiles, and the whiskers extend to the 10th and 90th percentiles, with individual data points above the whiskers shown for the lowest and highest 10% of each dataset. Solid lines and (+) marks indicate median and mean, respectively. n = (left to right) 13, 4, 25, 17 cells analyzed across three independent experiments. *** P = 0.0005, ****P < 0.0001, ns: non-significant, one-way ordinary ANOVA followed by Tukey’s multiple comparison testing. (d) Representative time-course confocal z-resliced images showing S2-cleaved Notch at AJs as a function of time after DAPT removal. The NICD signal (red) at the AJ gradually decreases, indicating NICD release. Images shown here are not from identical cells, but represent a general trend of NICD signal at AJs for each time point. Scale bar, 5 µm (e) Quantification IIN/IOUT ratio as a function of time after DAPT washout. Data are the mean ± s.d of n = 25 (+DAPT), 9 (0 hr), 10 (0.5 hr), 6 (1.5 hr), 8 (3 hr), 17 (12 hr), and 14 (-Dll4) biological replicates examined across 3 independent experiments.
Extended Data Fig. 4. Interrogation of the mechanism underlying γ-secretase recruitment into AJs.
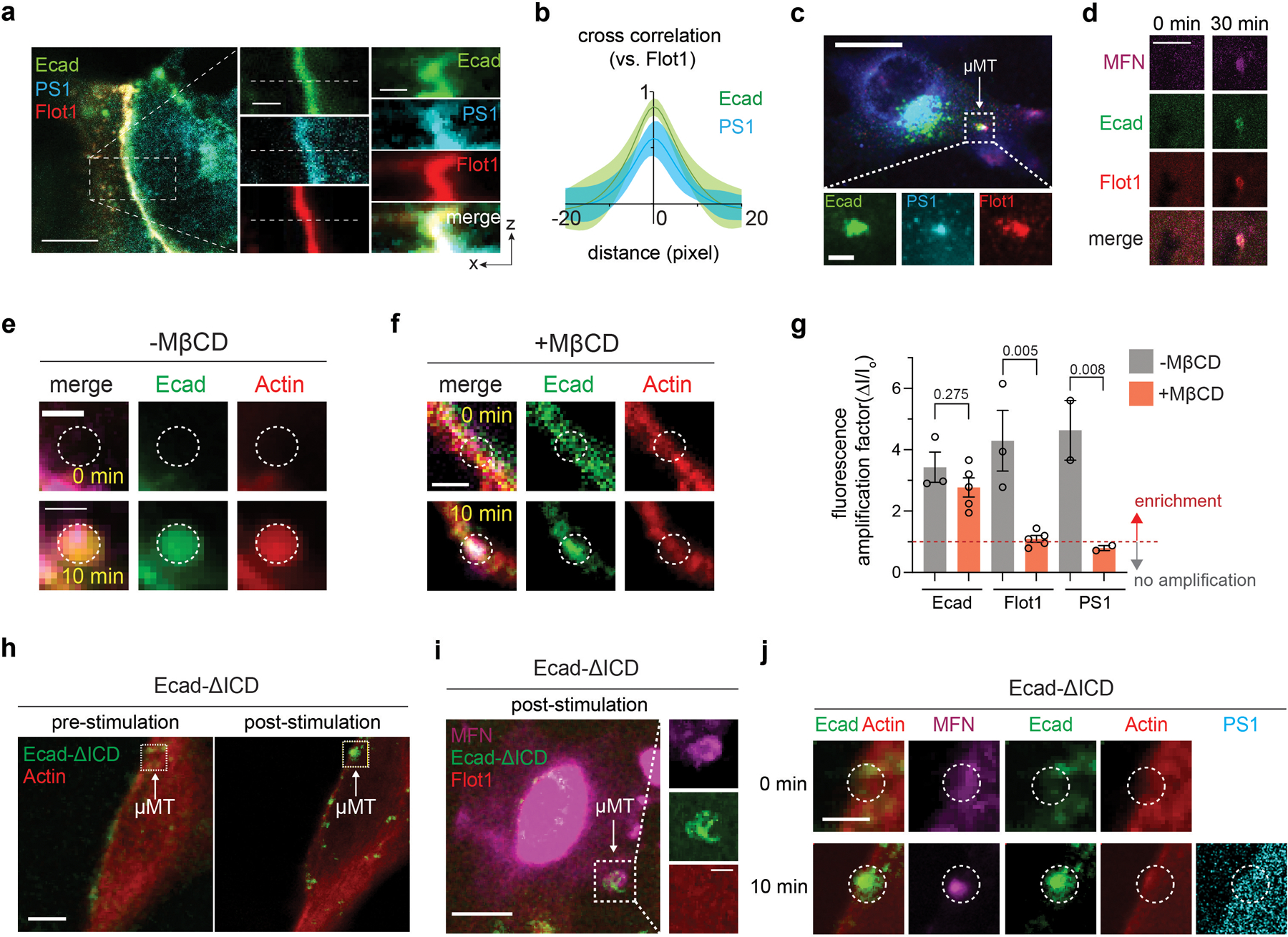
(a) Representative confocal fluorescence images showing the PS1 and Flot1 distribution relative to native cell-cell AJs. (left) A maximum intensity projection image of merged channels. Scale bar, 10 µm. (center) Magnified images showing greater details of the boxed region. Scale bar, 2 µm. (right) Z-resliced images showing the sections of the AJs. Scale bar, 2 µm. Line profiles of fluorescence signals from E-cadherin, PS1, and Flot1 along the white dashed lines in the z-resliced images. (b) Cross-correlation analysis of E-cadherin and PS1 over Flot1. Both Flot1 and PS1 fluorescence intensities exhibited strong positive correlation with the AJ. The solid curves and the shades indicate means and s.e.m, respectively. n = 7. (c) Confocal fluorescence images showing PS1 and Flot1 localization at artificial AJs by mechanogenetics. E-cadherin and Flot1 were labeled with fluorescent tags. Endogenous PS1 was immunostained after fixation. Scale bar, 20 µm. (d) Magnified confocal images showing PS1 and Flot1 localization before (0 min) and after (30 min) the formation of artificial AJs via mechanogenetics. Scale bar, 20 µm. (e) Magnified confocal images showing strong accumulation of Actin at the artificial AJ by mechanogenetics. Scale bar, 5 µm. (f) Representative confocal fluorescence images showing no enrichment of Actin signal at the artificial AJs during MβCD treatment. Scale bar, 5 µm. (g) Fluorescence amplification factors (I/Io) of Ecadherin, Flot1, and PS1 localization in response to the mechanogenetic formation of AJs quantified for multiple replicates. The effect of MβCD treatment on Flot1 and PS1 relocalization was assessed. Data are the mean ± s.e.m. of n = 3 (Ecad, -MβCD), 5 (Ecad, +MβCD), 3 (Flot1, -MβCD), 5 (Flot, +MβCD), 2 (PS1, -MβCD), and 2 (PS1, + MβCD) biologically independent mechanogenetic experiments. One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. (h) Representative fluorescence image showing the mechanogenetic formation of Ecad-ΔICD cluster. A white dashed box indicates the subcellular region where the µMT was applied. Scale bar, 10 µm. (i) Confocal image showing that the mechanogenetically induced Ecad-ΔICD clusters did not recruit Flot1. Scale bar, 20 µm. Zoom-in of the white frame-marked region of µMT application is shown on the right. Scale bar, 5 µm. (j) Spatial distribution of F-actin and PS1 in response to Ecad-ΔICD clustering. Neither F-actin nor PS1 were localized at the cluster region. Scale bar, 5 µm.
Extended Data Fig. 5. AJ-induced membrane juxtaposition drives Notch exclusion via size-dependent protein segregation.
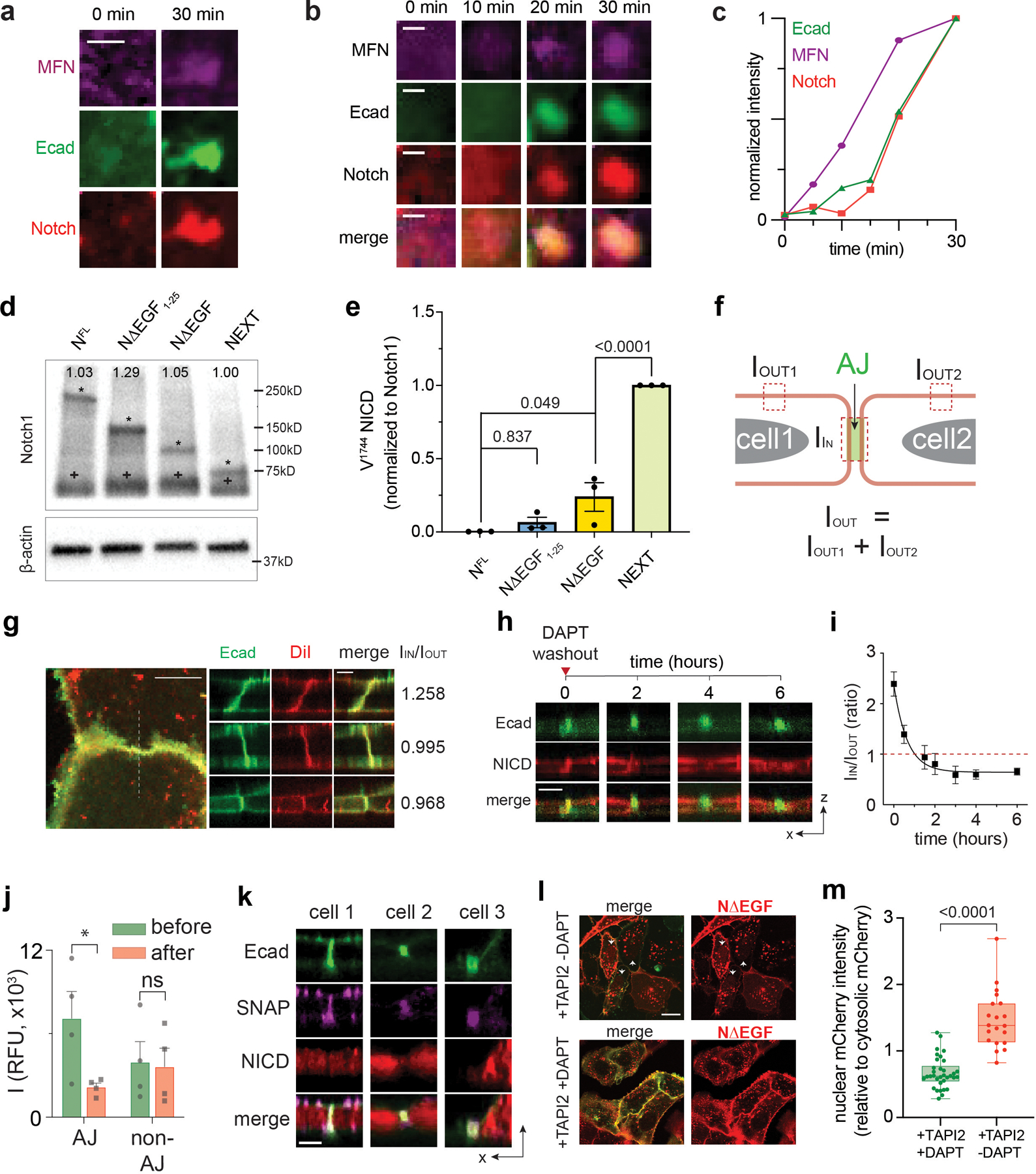
(a) Additional artificial AJs showing Notch recruitment. Scale bar, 2 µm. (b) Time-lapse epifluorescence images (were acquired before micromagnetic tweezer (µMT) stimulation and then at 10, 20, 30 minutes of the µMT application. Gradual MFN and E-cadherin clustering was clearly seen, followed by Notch accumulation at the AJ. Scale bar, 2 µm. (c) Kinetics of signal enrichments at the artificial AJ shown in the panel (b). This is a representative result from n = 3 artificial AJs from 3 independent experiments. (d) Representative western blot for total Notch ICD from the U2OS cells stably expressing Notch1 truncation variants. The blot was probed with anti-Notch1-ICD. The same lysates were used in (Fig. 3F). The asterisk (*, upper band) represent the intact Notch truncation variants. Expected molecular weight of NFL, NΔEGF1–25, NΔEGF, and NEXT are 250 kD, 150 kD, 110 kD, and 95 kD, respectively. The cross (+, lower) represents the reduced protein band of 70 kD. All variants contain the SDS/DTT-sensitive link that produces the protein band corresponding to the polypeptide of Notch ECD and transmembrane-intracellular domain (TMICD). The number shown in each lane indicates the quantified band intensity of the corresponding lane normalized to that of NEXT variant. The intensity is calculated by summing the intensities measured from two bands detected in each lane. (e) Western blot quantification of cleaved NICD levels over total Notch levels. Data are the mean ± s.e.m. of n = 3 experiments. One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. (f) Method to quantify Notch enrichment. Please see methods for more details. (g) Representative confocal images and enrichment factors (IIN/IOUT) of Dil membrane staining dye distribution relative to AJs. Scale bars, 10 µm (maximum intensity projection), 3 µm (z resliced images). (h) Time series of confocal z-resliced images showing the enrichment of NΔEGF (red) at the AJ (green) under DAPT treatment (t = 0), and the dissipation during DAPT washout (t ≥ 2). Scale bar, 3 µm. (i) Single-cell traces showing the time-course of the decline of NΔEGF enrichment factor at the AJs during DAPT washout (mean ± s.e.m.; n = 4 independent single-cell experiments). (j) Quantification of changes in NICD signal from these four cells at the AJs and non-AJ membrane, at t = 0 (green, before washout) and t = 6 hr (red, after DAPT washout). AJs and non-AJ membrane were detected based on thresholding and automatic segmentation using the custom-built script. Intracellular-mCherry signal significantly decreased at the AJs, but not at non-AJ membranes (*P = 0.035, ns: P = 0.075, Student’s t and Wilcoxon test, n = 4 cells examined across 2 independent experiments). (k) Confocal z-resliced images showing the distribution of extracellular SNAP (purple) and intracellular mCherry (red) tags of NΔEGF relative to AJs (green) after DAPT removal. Scale bar, 3 µm. (l, m) Nuclear location of NICD released from cell membrane that recombinantly expresses NΔEGF. (l) Confocal fluorescence images of U2OS cells expressing SNAP-NΔEGF-mCherry and Ecad-GFP. (Upper) Cells treated with TAPI2 only. White arrowheads indicate the cells with nuclear NICD-mCherry accumulation. (Lower) Cells treated with both TAPI2 and DAPT. Scale bar, 20 µm. (m) Quantification of the ratio of nucleus-to-cytosolic mCherry signals in cells with DAPT (n = 39 AJs) and those without DAPT (n = 21 AJs) from 3 independent experiments. A box and a whisker indicate the interquartile and the full range, respectively. Colored lines indicate median. Two tailed unpaired Student’s t-test.
Extended Data Fig. 6. Spatial mutations of Notch to study the effect of membrane compartmentalization on the signaling.
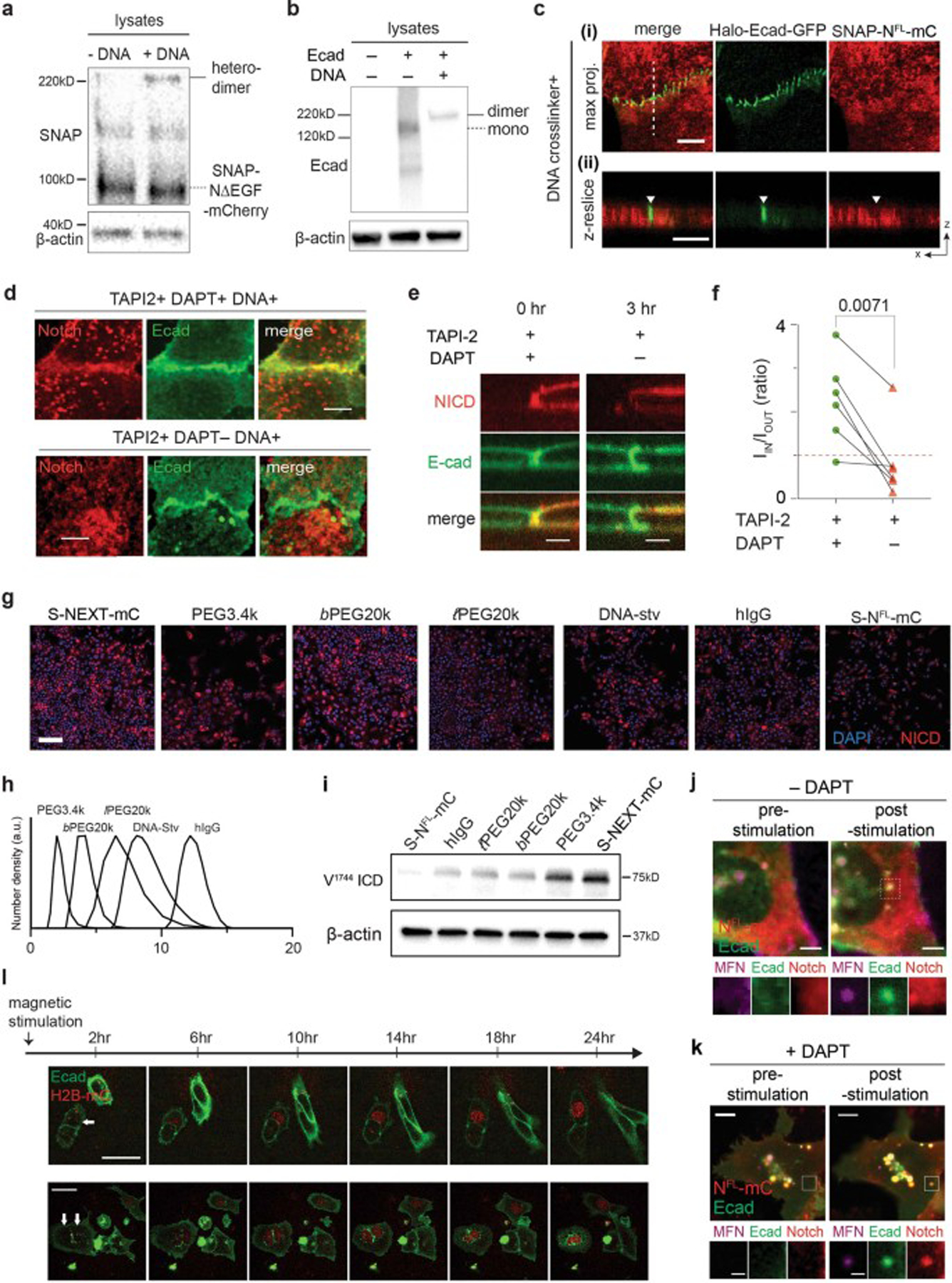
A representative western blot of lysate from cells expressing NΔEGF and Halo-Ecad-GFP after 2 hr incubation with or without DNA crosslinkers. The blot was labelled with anti-SNAP (a) and anti-Ecadherin (b) antibodies. The expected mass of NΔEGF, E-cadherin monomer, and the complex with the Notch construct and E-cadherin forming a heterodimer are 90 kD, 158 kD, and 230 kD, respectively. β-actin detection was used to assess protein loading. In both blots, predicted bands representing Notch-E-cadherin heterodimers (solid black lines) and SNAP-NΔEGF-mCherry or Halo-Ecad-GFP monomers (dashed black lines) are indicated. (c) A representative maximum intensity projection of XY images (i) and a XZ-resliced image (ii) showing exclusion of full-length Notch1 (SNAP-NFL-mCherry) from the AJs (green) after the DNA crosslinking. (d) Representative confocal maximum intensity projection images showing the distribution of NΔEGF relative to the AJs after crosslinking. Cells were treated with or without DAPT. Scale bar, 10 µm. (e) Single-cell confocal z-resliced images showing intracellular mCherry signal at the AJ under DNA and DAPT treatment (left) and after washing out DAPT (right). Removing DAPT elicited a significant reduction in mCherry signal intensity from the AJ. Scale bar, 5 µm. (f) Paired analysis of multiple cells expressing NΔEGF in enrichment factor (IIN/IOUT) after DAPT washout. Each dot represents IIN/IOUT value before and after DAPT washout from a single cell. Each line corresponds to the IIN/IOUT changes before and after DAPT washout in a same single cell (paired two-tailed Student’s t test; n = 6 cells examined across 2 independent experiments). (g) Larger area (1 ×1 mm2) confocal fluorescence images shown in Fig. 4f. Scale bar, 200 µm. (h) Dynamic light scattering spectra of BG-modified macromolecules used in the experiment to induce spatial mutation of NEXT in Fig. 4d–g. (i) Western blot analyses showing that spatial mutations of NEXT alter the level of Notch activation. Representative western blot from three independent experiments. The blot was probed with specific antibodies for cleaved NICD (Val1744) and β-actin. Each lane was loaded with the lysates from NEXT-expressing cells incubated with different BG-modified polymers or proteins for 20 h. The lysate from NFL was used as control. (j,k) Representative images of artificial AJs formed in live cells. Cells treated with both TAPI2 and DAPT (j) or with only TAPI2 but no DAPT (k). Magnified images were shown in lower panels. An intense mCherry signal was observed at the artificial AJ with TAPI2 and DAPT treatment, while no enrichment of Notch1 signal was seen from cells without DAPT. Scale bar, 5 µm (low-magnification), 2 µm (zoomed-in images). (l) Representative time-lapse images showing Notch signal activation in UAS-Gal4 reporter cells with artificial AJs (white arrows). Cells were cultured in the presence of TAPI2 and no source of S2 cleavage. Neighboring cells without magnetic stimulation were used as internal negative controls. Images were acquired using epifluorescence imaging every 2 hr for 24 hr. Scale bar, 50 µm.
Extended Data Fig. 7. Generation of U2OS SNAP-NFL-Gal4 fluorescence reporter cell lines lacking E-cadherin via CRISPR/Cas9.
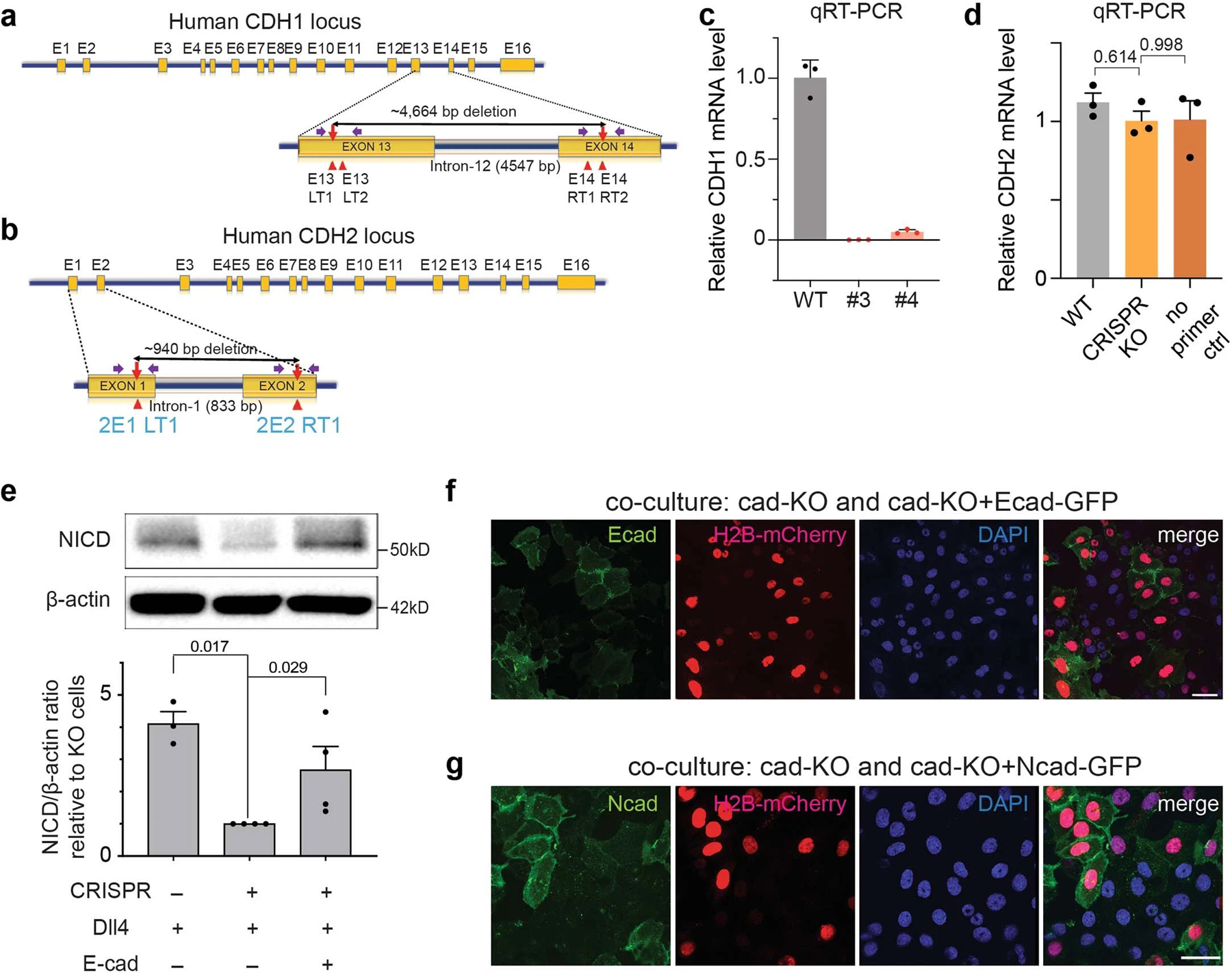
(a,b) Schematic representation of human CDH1 (a) and CDH2 (b) gene structure and targeted segmental deletion sites. The sixteen exons are shown in orange boxes (E1-E16). Red arrowheads indicate the sgRNA-binding sites (E13LT1, E13LT2, E14RT1 and E14RT2 for CDH1, 2E1LT1 and 2E2RT1 for CDH2). The targeted segmental deletions of 4.6 kb for CDH1 and 940 bp for CDH2 knockout are shown with a black line with red arrow tips, respectively. Purple arrows represent PCR primers used for the T7E1 assay and detection of alleles with targeted deletions, respectively. (c) CDH1 mRNA expression levels in CDH1 KO clone #3 and #4 were determined using qRT-PCR. CDH1 expression levels in the selected clones containing a segmental deletion were quantified relative to CDH1 mRNA levels of the wild-type U2OS SNAP-NFL-Gal4 cells. Clone #3 was used for subsequent experiments. Data are the mean ± s.d. of n = 3 biologically independent samples. (d) qRT-PCR analysis of CDH2 mRNA expression levels in U2OS SNAP-NFL-Gal4 reporter cells (WT), CDH1 KO clone #3 (Ecad-KO), and a negative control sample (no primer pair added). CDH2 mRNA levels in both WT and Ecad-KO cells were quantified relative to the negative control sample. Both WT and Ecad-KO cells showed negligible CDH2 mRNA levels, indicating that Ecad-KO cells have minimal mRNA expression of both CDH1 and CDH2. Data are the mean ± s.e.m. of n = 3 biologically independent samples; one-way ordinary ANOVA test. (e) Western blot analyses of cleaved NICD levels in the wild-type SNAP-NFL-Gal4 cells, CDH1 knock-out (Ecad-KO) cells, and Ecad-KO cells with recombinant E-cadherin transfection. (top) A representative image of immunoblotting. (bottom) Quantification of cleaved NICD levels. The average intensity of NICD bands relative to β-actin bands was quantified and then normalized to that of Ecad-KO cells. Data are the mean ± s.d. of n = 5 biological replicates; one-way ANOVA followed by Tukey’s test). (f,g) Representative epi-fluorescence images showing Notch activation in co-culture of Ecad-KO cells with Ecad-KO + Ecad cells (f) or with Ecad-KO + Ncad cells (g). Ecad-KO cells shows no GFP signal (green) while Ecad-KO + Ecad or Ecad-KO + Ncad cells show robust GFP signal indicative of reintroduction of E- or N-cadherin. Scale bar, 50 µm.
Extended Data Fig. 8. Additional immunofluorescence images showing that the AJ-mediated membrane compartmentalization modulates neural progenitor cell (NPC) differentiation.
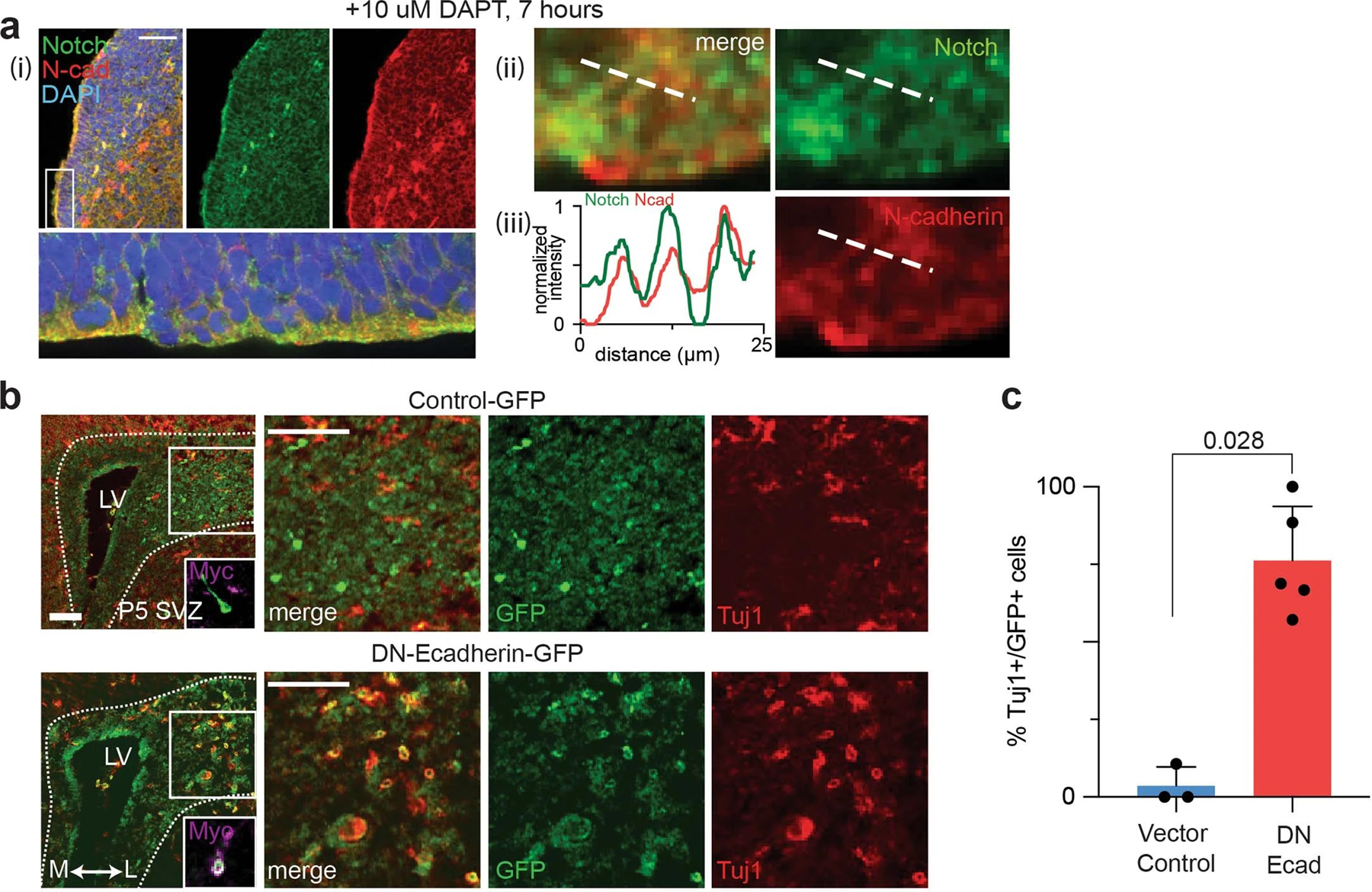
(a) Immunostaining of the subventricular zone (SVZ) in the lateral ventricle (LV) of the E13.5 DAPT-treated mouse brain. Notch was colocalized at NAJ, visualized by immunostaining with anti-N-cadherin and anti-Notch1 antibodies. (i) Representative lower magnification image. The indicated area (a white box) is magnified and rotated 90° clockwise in the lower panel. Scale bar, 100 µm. (ii) Magnified view of the region indicated with a white dashed box in the (i) lower panel. Scale bar, 2.5 µm. (iii) Line profiles of N-cadherin and Notch distributions. (b) Additional confocal images of coronal sections of developing mouse brain retrovirally infected with dominant negative form of E-cadherin vector (DN-Ecad-EGFP). Transduced cells differentiated into post-mitotic neurons can be identified as EGFP + /Tuj1 + , while those remained as NPCs with plasmid transfection are only EGFP + . (left) Low-magnification images. Insets show the magnified image of a representative single cell immunostained for myc-tag. (right) Magnified view of the boxed region. Scale bar, 50 µm. (c) Ratio of GFP/Tuj1-double positive cells to total Tuj1-positive post-mitotic neurons in these two conditions. Data are the mean ± s.e.m. of n = 3 (vector control) and 5 (DN-Ecad-EGFP) biologically independent animals. Two-tailed unpaired Student’s t test.
Extended Data Fig. 9. Amyloid precursor proteins (APPs) with intact YENPTY motif show size-dependent spatial segregation and membrane proteolysis, consistent with APP lacking the YENPTY motif.
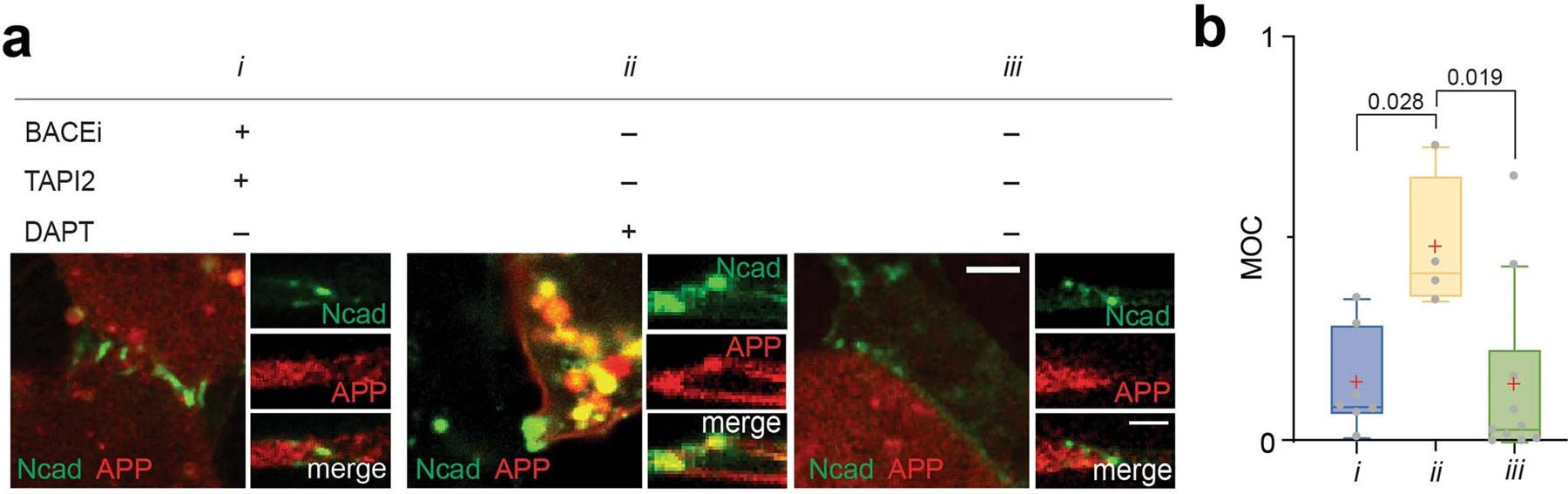
(a) Representative confocal maximum intensity projection (right) and z-resliced (left) images of U2OS cells co-expressing N-cadherin (green) and full-length APP (red). To capture the spatial distribution of the APP intermediates, cells were cultured with a combination of α-, β-, and γ-secretase inhibitors. Scale bar, 3 µm (max. projection) and 2 µm (z-resliced). (b) The spatial redistribution of APP relative to the NAJs was quantified using Manders’ overlap coefficient (MOC). Data are presented as boxes and whiskers, representing interquartile and min-to-max ranges, respectively; n = 7 (i), 4 (ii), and 10 (iii) NAJs examined over two independent experiments. each detected from a single cell. One-way ANOVA followed by Tukey’s multiple comparison test.
Supplementary Material
Acknowledgement
The authors thank Drs. S. Blacklow (Harvard U.), C. Miller (King’s College London), and K. Shimamura (Kumamoto U.) for the kind gifts of Notch, APP, and DN-cadherin plasmids, respectively. We also thank Drs. A. Balmain, M. Moasser, and E. Collison (UCSF) for sharing cell lines. Dr. Daniel Fletcher (UC Berkeley), Mr. Ari Joffe (UC Berkeley), and Dr. Duaa Al-Rawi (Stanford U.) provided insightful discussion. For reagents, technical support, and discussions we thank the Kim, Cheon, Gartner, and Jun laboratories, as well as the Nikon Imaging Center and Wynton at UCSF. M.K. was supported by a Life Science Research Foundation fellowship as the Shurl and Kay Curci Foundation fellow, and by Burroughs Wellcome Travel Fund. This work was supported by NRF- NRF-2021R1F1A1063378 (M.K.), NRF-2018R1A5A1025511 (D.S.) and NRF-2017R1A2B3004198 (H.K.), HI17C0676 from Korean Ministry of Health and Welfare (H.K.), 5R01NS047229 from National Institute on Aging (NIA) and the National Institute of Health (NIH) (A.G.), 5R01AG008200 from National Institute on Aging (NIA) and the National Institute of Health (NIH) (N.K.R.), IBS-R026-D1 from IBS (M.K., H.K., and J.C.), NRF-2019R1A2C1085712 (Y.H.K.), the UCSF Center for Cellular Construction (an NSF Science and Technology Center, no. DBI-1548297) (Z.J.G.), U01CA244109 from the National Cancer Institute (Z.J.G), 1R01GM112081, 1R01GM126542–01, and R35GM134948 from the National Institute of General Medical Science (NIGMS) and the NIH (Y.J.), 1R21AG072232–01 from the National Institute on Aging (NIA) and the NIH (M.L.K. and Y.J.), R00CA226366 from the National Cancer Institute (M.L.K) and NIH, and the UCSF Program for Breakthrough Biomedical Research (PBBR) funded in part by the Sandler Foundation (M.L.K and Y.J.). Z.J.G. is a Chan Zuckerberg BioHub Investigator.
Footnotes
Competing Interests
Z.J.G. is an equity holder in Scribe Biosciences and Provenance Bio and is an advisor for Serotiny.
References
- 1.Manz BN & Groves JT Spatial organization and signal transduction at intercellular junctions. Nature Reviews Molecular Cell Biology 2010 11:5 11, 342–352 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen CS, Tan J & Tien J Mechanotransduction at Cell-Matrix and Cell-Cell Contacts. Annu Rev Biomed Eng 6, 275–302 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Südhof TC Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 171, 745–769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Südhof TC & Malenka RC Understanding Synapses: Past, Present, and Future. Neuron 60, 469–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng M et al. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 174, 1172–1187.e16 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Sanes JR & Zipursky SL Synaptic Specificity, Recognition Molecules, and Assembly of Neural Circuits. Cell 181, 536–556 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Narui Y & Salaita K Membrane Tethered Delta Activates Notch and Reveals a Role for Spatio-Mechanical Regulation of the Signaling Pathway. Biophys J 105, 2655–2665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid EM et al. Size-dependent protein segregation at membrane interfaces. Nature Physics 2016 12:7 12, 704–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salaita K et al. Restriction of receptor movement alters cellular response: Physical force sensing by EphA2. Science (1979) 327, 1380–1385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustin M & Shaw A Costimulation: building an immunological synapse. Science 283, 649–650 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Groves JT & Kuriyan J Molecular mechanisms in signal transduction at the membrane. Nature Structural & Molecular Biology 2010 17:6 17, 659–665 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Merwe PA & Dushek O Mechanisms for T cell receptor triggering. Nature Reviews Immunology 2011 11:1 11, 47–55 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Belardi B, Son S, Felce JH, Dustin ML & Fletcher DA Cell–cell interfaces as specialized compartments directing cell function. Nature Reviews Molecular Cell Biology 2020 21:12 21, 750–764 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Case LB, Zhang X, Ditlev JA & Rosen MK Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science (1979) 363, 1093–1097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James JR & Vale RD Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature 2012 487:7405 487, 64–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case LB, Ditlev JA & Rosen MK Regulation of Transmembrane Signaling by Phase Separation. Annu Rev Biophys 48, 465–494 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovall RA, Gebelein B, Sprinzak D & Kopan R The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Developmental Cell vol. 41 228–241 Preprint at 10.1016/j.devcel.2017.04.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopan R & Ilagan MXG The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell vol. 137 216–233 Preprint at 10.1016/j.cell.2009.03.045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray SJ Notch signalling in context. Nature Reviews Molecular Cell Biology vol. 17 722–735 Preprint at 10.1038/nrm.2016.94 (2016). [DOI] [PubMed] [Google Scholar]
- 20.van Es JH et al. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005 435:7044 435, 959–963 (2005). [DOI] [PubMed] [Google Scholar]
- 21.S Artavanis-Tsakonas MRRL Notch signaling: cell fate control and signal integration in development. Science (1979) 284, 770–776 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Boukhatmi H, Martins T, Pillidge Z, Kamenova T & Bray S Notch Mediates Inter-tissue Communication to Promote Tumorigenesis. Current Biology 30, 1809–1820.e4 (2020). [DOI] [PubMed] [Google Scholar]
- 23.South AP, Cho RJ & Aster JC The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol 23, 458–464 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolas M et al. Notch1 functions as a tumor suppressor in mouse skin. Nature Genetics 2003 33:3 33, 416–421 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Lai EC Notch signaling: control of cell communication and cell fate. Development 131, 965–973 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Haines N & Irvine KD Glycosylation regulates Notch signalling. Nature Reviews Molecular Cell Biology 2003 4:10 4, 786–797 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Stanley P & Okajima T Roles of Glycosylation in Notch Signaling. Curr Top Dev Biol 92, 131–164 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Wang X & Ha T Defining Single Molecular Forces Required to Activate Integrin and Notch Signaling. Science (1979) 340, 991–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon WR et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev Cell 33, 729–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo D et al. A Mechanogenetic Toolkit for Interrogating Cell Signaling in Space and Time. Cell 165, 1507–1518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marambaud P et al. A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21, 1948–1956 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgakopoulos A et al. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell 4, 893–902 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Brasch J, Harrison OJ, Honig B & Shapiro L Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol 22, 299–310 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honig B & Shapiro L Adhesion Protein Structure, Molecular Affinities, and Principles of Cell-Cell Recognition. Cell 181, 520–535 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yap AS, Gomez GA & Parton RG Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev Cell 35, 12–20 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Falo-Sanjuan J & Bray SJ Membrane architecture and adherens junctions contribute to strong Notch pathway activation. bioRxiv 2021.05.26.445755 (2021) doi: 10.1101/2021.05.26.445755. [DOI] [PMC free article] [PubMed]
- 37.Guillaume E et al. Flotillin microdomains stabilize cadherins at cell–cell junctions. J Cell Sci 126, 5293–5304 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Taulet N et al. N-cadherin/p120 Catenin Association at Cell-Cell Contacts Occurs in Cholesterol-rich Membrane Domains and Is Required for RhoA Activation and Myogenesis *. Journal of Biological Chemistry 284, 23137–23145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Causeret M, Taulet N, Comunale F, Favard C & Gauthier-Rouvière C N-Cadherin Association with Lipid Rafts Regulates Its Dynamic Assembly at Cell-Cell Junctions in C2C12 Myoblasts. Mol Biol Cell 16, 2168–2180 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szaruga M et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 170, 443–456.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Lee S-J et al. A detergent-insoluble membrance compartment contains Aβ in vivo. Nature Medicine 1998 4:6 4, 730–734 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Vetrivel KS et al. Spatial Segregation of γ-Secretase and Substrates in DistinctMembraneDomains *. Journal of Biological Chemistry 280, 25892–25900 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chow VW, Mattson MP, Wong PC & Gleichmann M An Overview of APP Processing Enzymes and Products. NeuroMolecular Medicine 2009 12:1 12, 1–12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ehehalt R, Keller P, Haass C, Thiele C & Simons K Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. Journal of Cell Biology 160, 113–123 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wahrle S et al. Cholesterol-Dependent γ-Secretase Activity in Buoyant Cholesterol-Rich Membrane Microdomains. Neurobiol Dis 9, 11–23 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Vetrivel KS et al. Association of γ-Secretase with Lipid Rafts in Post-Golgi and Endosome Membranes *. Journal of Biological Chemistry 279, 44945–44954 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urano Y et al. Association of active γ-secretase complex with lipid rafts. J Lipid Res 46, 904–912 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Galbiati F, Razani B & Lisanti MP Emerging Themes in Lipid Rafts and Caveolae. Cell 106, 403–411 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Owen DM, Rentero C, Magenau A, Abu-Siniyeh A & Gaus K Quantitative imaging of membrane lipid order in cells and organisms. Nat Protoc 7, 24–35 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Komura naoko et al. raft-based interactions of gangliosides with a gpi-anchored receptor. Nat Chem Biol 12, (2016). [DOI] [PubMed] [Google Scholar]
- 51.Raghupathy R et al. Transbilayer Lipid Interactions Mediate Nanoclustering of Lipid-Anchored Proteins. Cell 161, 581–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kusumi A et al. Paradigm Shift of the Plasma Membrane Concept from the Two-Dimensional Continuum Fluid to the Partitioned Fluid: High-Speed Single-Molecule Tracking of Membrane Molecules. Annu Rev Biophys Biomol Struct 34, 351–378 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Kouchi Z et al. p120 Catenin Recruits Cadherins to γ-Secretase and Inhibits Production of Aβ Peptide. Journal of Biological Chemistry 284, 1954–1961 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J, Jeong H, Southard KM, Jun Y & Cheon J Magnetic Nanotweezers for Interrogating Biological Processes in Space and Time. Acc Chem Res 51, 839–849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwak M et al. Small, Clickable, and Monovalent Magnetofluorescent Nanoparticles Enable Mechanogenetic Regulation of Receptors in a Crowded Live-Cell Microenvironment. Nano Lett 19, 3761–3769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrison OJ et al. The Extracellular Architecture of Adherens Junctions Revealed by Crystal Structures of Type I Cadherins. Structure 19, 244–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polacheck WJ et al. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017 552:7684 552, 258–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Struhl G & Adachi A Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol Cell 6, 625–636 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Palmer WH & Deng W-M Ligand-Independent Mechanisms of Notch Activity. Trends Cell Biol 25, 697–707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Z, Yu Z, Cai Y, Du R & Cai L Engineering of an enhanced synthetic Notch receptor by reducing ligand-independent activation. Communications Biology 2020 3:1 3, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mumm JS et al. A Ligand-Induced Extracellular Cleavage Regulates γ-Secretase-like Proteolytic Activation of Notch1. Mol Cell 5, 197–206 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Bolduc DM, Montagna DR, Gu Y, Selkoe DJ & Wolfe MS Nicastrin functions to sterically hinder γ-secretase–substrate interactions driven by substrate transmembrane domain. Proceedings of the National Academy of Sciences 113, E509–E518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang SI et al. Phosphorylated EGFR Dimers Are Not Sufficient to Activate Ras. Cell Rep 22, 2593–2600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shah S et al. Nicastrin Functions as a γ-Secretase-Substrate Receptor. Cell 122, 435–447 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Sprinzak D et al. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature 2010 465:7294 465, 86–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nandagopal N et al. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 172, 869–880.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ables JL, Breunig JJ, Eisch AJ & Rakic P Not(ch) just development: Notch signalling in the adult brain. Nature Reviews Neuroscience 2011 12:5 12, 269–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.A Louvi SA-T Notch signalling in vertebrate neural development. Nature Rev. Neurosci 7, 93–102 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Franco SJ & Müller U Shaping Our Minds: Stem and Progenitor Cell Diversity in the Mammalian Neocortex. Neuron 77, 19–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das RM & Storey KG Apical Abscission Alters Cell Polarity and Dismantles the Primary Cilium During Neurogenesis. Science (1979) 343, 200–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujita I et al. Endfoot regeneration restricts radial glial state and prevents translocation into the outer subventricular zone in early mammalian brain development. Nature Cell Biology 2019 22:1 22, 26–37 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Hatakeyama J et al. Cadherin-based adhesions in the apical endfoot are required for active Notch signaling to control neurogenesis in vertebrates. Development (Cambridge) 141, 1671–1682 (2014). [DOI] [PubMed] [Google Scholar]
- 73.de Strooper B & Karran E The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Brien RJ & Wong PC Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu Rev Neurosci 34, 185–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Brien RJ & Wong PC Amyloid Precursor Protein Processing and Alzheimer’s Disease 10.1146/annurev-neuro-061010-113613 34, 185–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quintero-Monzon O et al. Dissociation between the Processivity and Total Activity of γ-Secretase: Implications for the Mechanism of Alzheimer’s Disease-Causing Presenilin Mutations. Biochemistry 50, 9023–9035 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uemura K et al. N-cadherin-based adhesion enhances Aβ release and decreases Aβ42/40 ratio. J Neurochem 108, 350–360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bray SJ Notch signalling: A simple pathway becomes complex. Nature Reviews Molecular Cell Biology vol. 7 678–689 Preprint at 10.1038/nrm2009 (2006). [DOI] [PubMed] [Google Scholar]
- 80.J, R. W. et al. Evidence for a physical interaction between presenilin and Notch. Proceedings of the National Academy of Sciences 96, 3263–3268 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ray WJ et al. Cell surface presenilin-1 participates in the γ-secretase-like proteolysis of Notch. Journal of Biological Chemistry 274, 36801–36807 (1999). [DOI] [PubMed] [Google Scholar]
- 82.Morsut L et al. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roybal KT et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang G et al. Structural basis of Notch recognition by human γ-secretase. Nature 2018 565:7738 565, 192–197 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Hao W et al. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proceedings of the National Academy of Sciences 118, e2102191118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maeda K, Takemura M, Umemori M & Adachi-Yamada T E-cadherin prolongs the moment for interaction between intestinal stem cell and its progenitor cell to ensure Notch signaling in adult Drosophila midgut. Genes to Cells 13, 1219–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 87.Soh BS et al. N-cadherin prevents the premature differentiation of anterior heart field progenitors in the pharyngeal mesodermal microenvironment. Cell Research 2014 24:12 24, 1420–1432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kwon C et al. A regulatory pathway involving Notch1/β-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol 11, 951–957 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Collado MS et al. The Postnatal Accumulation of Junctional E-Cadherin Is Inversely Correlated with the Capacity for Supporting Cells to Convert Directly into Sensory Hair Cells in Mammalian Balance Organs. Journal of Neuroscience 31, 11855–11866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshida K, Murata M, Yamaguchi T, Matsuzaki K & Okazaki K Reversible Human TGF-β Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. Journal of Clinical Medicine 2016, Vol. 5, Page 7 5, 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arefin B, Parvin F, Bahrampour S, Stadler CB & Thor S Drosophila Neuroblast Selection Is Gated by Notch, Snail, SoxB, and EMT Gene Interplay. Cell Rep 29, 3636–3651.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 92.Dustin ML & Groves JT Receptor Signaling Clusters in the Immune Synapse. Annu Rev Biophys 41, 543–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bakalar MH et al. Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 174, 131–142.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 94.Pruessmeyer J et al. A Disintegrin and Metalloproteinase 17 (ADAM17) Mediates Inflammation-induced Shedding of Syndecan-1 and −4 by Lung Epithelial Cells*. Journal of Biological Chemistry 285, 555–564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim J et al. Single-cell mechanogenetics using monovalent magnetoplasmonic nanoparticles. Nature Protocols 2017 12:9 12, 1871–1889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dudok B et al. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nature Neuroscience 2014 18:1 18, 75–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moss ML & Rasmussen FH Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem 366, 144–148 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Lee HJ et al. En bloc and segmental deletions of human XIST reveal X chromosome inactivation-involving RNA elements. Nucleic Acids Res 47, 3875–3887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pulido-Quetglas C et al. Scalable Design of Paired CRISPR Guide RNAs for Genomic Deletion. PLoS Comput Biol 13, e1005341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim HK et al. SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Sci Adv 5, eaax9249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramakrishna S et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 24, 1020–1027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gopalappa R, Suresh B, Ramakrishna S & Kim H (Henry). Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res 46, e71–e71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guschin DY et al. A Rapid and General Assay for Monitoring Endogenous Gene Modification. Methods in Molecular Biology 649, 247–256 (2010). [DOI] [PubMed] [Google Scholar]
- 104.van der Spoel D et al. GROMACS: Fast, flexible, and free. J Comput Chem 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- 105.de Jong DH et al. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J Chem Theory Comput 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 106.Hess B, Bekker H, Berendsen HJC & Fraaije JGEM LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18, 1463–1472 (1997). [Google Scholar]
- 107.Michaud-Agrawal N, Denning EJ, Woolf TB & Beckstein O MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J Comput Chem 32, 2319–2327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chavent M et al. Methodologies for the analysis of instantaneous lipid diffusion in md simulations of large membrane systems. Faraday Discuss 169, 455–475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Previously published genomic sequence data that were re-analysed here are available from Ensembl for E-cadherin (CDH1) and N-cadherin (CDH2) protein from homo sapiens (gene ID: ENSG00000039068 and ENSG00000170558). Source data are provided with this study. All statistical source data have been provided as Source Data. All raw unprocessed gel images have been provided as Source Data. All raw images acquired using confocal, epifluorescence, and time-lapse microscopy, and additional data that support the findings of this study are available from the corresponding authors upon reasonable request. All other data supporting the findings of this study are available from the corresponding author on reasonable request.