

Abstract
This manuscript describes step-by-step procedures to establish and manage fresh and cryopreserved cultures of nerve-derived human Schwann cells (hSCs) at the desired scale. Adaptable protocols are provided to propagate hSC cultures through serial passaging and perform routine manipulations such as enzymatic dissociation, purification, cryogenic preservation, live-cell labeling, and gene delivery. Expanded hSCs cultures are metabolically active, proliferative, and phenotypically stable for at least three consecutive passages. Cell yields are expected to be variable as determined by the rate of growth of individual batches and the rounds of subculture. The purity, however, can be maintained high at >95% hSC regardless of passage. The cells obtained in this manner are suitable for various applications, including small drug screens, in vitro modeling of neurodevelopmental processes, and cell transplantation. One caveat of this protocol is that continued expansion of same-batch hSC populations is eventually restricted due to senescence-linked growth arrest.
Keywords: Ensheathing glia, Peripheral nerve, Cell culture, Mitogenic factors, Laminin, Serial passaging, Fibroblast contamination, Immunopanning, Magnetic-activated cell sorting, Cryopreservation, Scalability, Proliferation, Senescence
Background
Human Schwann cells (hSCs) from nerve tissues are fairly amenable cells for in vitro culture. Whereas preparing primary cells requires labor-intensive procedural steps, standard, general practices can be applied to manage the cultures successfully once the hSCs are established. hSCs are expandable under adherent conditions. Substantial scaling up of same-prep cultures can be achieved by supplementing the culture medium with specific mitogenic factors and plating the cells on matrix-coated dishes, preferably with laminin [reviewed in Monje (2020a and 2020b)].
The steady expansion of individual hSC populations is feasible up to the second or third passage because the rate of hSC proliferation diminishes thereafter in most preparations. However, a single harvest of adult nerve-derived fascicles may yield cultures able to expand at a >103 amplification rate and total yields may surpass 100 million cells within two or three rounds of subculture. Established hSC cultures maintain key characteristics of lineage-committed SCs, as evidenced by the expression of SC-specific markers such as NGFR, S100B, Nestin, and Sox10 (Peng et al., 2020). Aberrant growth or spontaneous hSC transformation has not been observed (Emery et al., 1999). Indeed, hSCs from expanded batches are deemed safe for transplantation partly due to their phenotypic stability and resistance to transform (Bastidas et al., 2017). In managing established hSC cultures, researchers should be mindful that the cells achieve an irreversible state of growth arrest or senescence by or around the fifth passage, or the equivalent of 20 population doublings, after their initial isolation from an adult nerve source (Levi et al., 1995; Levi, 1996).
This protocol describes our optimized approaches to propagate hSC cultures and perform various routine manipulations in vitro. A thorough description of materials, methods and procedures is presented here to obtain scalable hSC cultures from both fresh isolates and cryogenic stocks. The banking of hSCs is described in detail due to its multiple benefits for deferred experimentation, long-term storage, and transfer of cell cultures. Several purification and cell labeling methods are introduced along with recommendations on choosing certain methods for specific applications. The following sections describe step-by-step procedures to facilitate replication in other labs. Figure 1 presents a generic representation of suggested interventions at any given step of the culture workflow. This paper does not include comprehensive data sets. The microscopy image data in the figures and videos are provided for qualitative purposes only. Our publications contain more information about laboratory-scale experimentation using established hSC cultures (Monje et al., 2018; Peng et al., 2020). Investigators can use our papers to gather additional details on experimental design, use of positive and negative controls, and analysis of results from diverse assays. The hSC cultures prepared as described herein are reliable models for the development of cell-based platforms, including reconstituted co-culture systems, and studies of xenotransplantation. Lastly, these methodologies are intended for basic research, since they differ substantially from clinical protocols (Khan et al., 2021), and may be effective only when using hSC cultures prepared from normal nerve fascicles or ganglia. We have not used hSC cultures from unconventional sources.
Figure 1. Generation and management of established human Schwann cells (hSC) cultures: passaging, purification, transfer, and in vitro modifications.

The diagram summarizes the suggested steps described in each protocol. Primary, passage-zero (P0) hSC cultures (drop-plated dish, left panel) can be expanded at a 1:10 ratio (controlled passaging, upper panel) or an unfixed ratio (routine passaging) until enough cells are obtained or the cultures become senescent, as indicated. Purification, banking, and gene delivery (or labeling) can be attempted at any level of passage but preferably no later than passage-2 or -3, which is the optimal time for experimentation using cells that are both proliferative and pure. The indicated cell yields represent accumulated cell numbers in each passage as predicted by the expansion rate of a typical hSC culture containing 1 million cells at P0.
Materials and reagents
All materials, reagents, and solutions should be sterile and cell culture grade. If possible, consistently use materials and reagents of the same brand and maintain a record of lot numbers in case signs of cell toxicity are observed. The list below is not intended to be fully comprehensive or limit the use of products from certain manufacturers. The product information is provided for reference only. Follow the manufacturer’s recommendations and use best practices in cell culture to avoid microbial contamination and ensure the reproducibility of the results.
Supplies and consumables
Disposable serological pipettes (5, 10, and 25 mL), polystyrene, plugged, sterile, and individually wrapped (VWR, catalog numbers: 76201-710, 75816-100, and 75816-090)
Polystyrene Pasteur (transfer) pipettes, sterile and individually wrapped (VWR, Argos Technology, catalog number: 10122-560)
Petri dish, bacteriological grade, 10 cm, not tissue culture treated (Corning, catalog number: 351029)
Centrifuge tubes, 15 and 50 mL, polypropylene, conical-bottom (Corning, catalog numbers: 430791 and 430290)
Cell culture dishes (35, 60, and 100 mm), polystyrene, tissue culture treated (Corning, catalog numbers: 353001, 353002, and 353003)
Multi-well plates (6, 12, and 24-well), tissue culture treated, flat bottom (Corning, catalog numbers: 3506 and 3524)
Cell culture flasks (T25 and T75), canted neck with plug seal caps (Corning, catalog numbers: 430168 and 430720U)
Cryogenic vials, 2 mL, internal thread (Corning, catalog number: 430489)
Polycarbonate freezing container with a tube holder, Nalgene “Mr. Frosty” Freezing Container (VWR, catalog number: 55710-200)
Cell lifter, individually wrapped, polyethylene, 2 cm length blade, 18 cm long (VWR, catalog number: T-2443-4)
Ice pans (VWR, catalog number: 89233) containing wet ice and dry ice
Media, supplements, and other cell culture products
Distilled water, cell culture grade (Fisher Scientific, Gibco, catalog number: 15-230-147)
Hank’s balanced salt solution (HBSS), formulated without calcium or magnesium and containing phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14170-112)
Leibovitz’s L15 Medium (L15) (Thermo Fisher Scientific, Gibco, catalog number: 11415064)
Dulbecco’s Modified Eagle’s Medium (DMEM), with high glucose and phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 11965092). (Optional) Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) (Thermo Fisher Scientific, Gibco, catalog number: 11320033)
Gentamycin 1,000× provided as 50 mg/mL stock solution (Thermo Fisher Scientific, Gibco, catalog number: 15750-060). (Optional) Penicillin-Streptomycin, 10,000 U/mL stock solution (Thermo Fisher Scientific, Gibco, catalog number: 15140-148)
De-complemented fetal bovine serum (FBS) (HyClone, catalog number: SV 30014.03). Stored in aliquots at -80 °C; used in all applications requiring serum, including cryopreservation of cell stocks
GlutaMAX supplement (100×) consisting of 200 mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl (Thermo Fisher Scientific, Gibco, catalog number: 35050061)
Heregulin-β1, 177-244 amino acid peptide (Preprotech, catalog number: G-100-03); instructions on the preparation, storage, and use of heregulin-β1 stock solution (25 µM) can be found in our protocol Andersen and Monje (2018). The stock solution is best preserved when kept in aliquots at -80 °C and used only for media preparation, avoiding freezing/thawing cycles. Heregulin-β1 is the primary mitogenic factor for hSCs; it is referred to as “heregulin” in the text and figures
Forskolin powder (Sigma-Aldrich, catalog number: F68861); instructions on the preparation, storage, and use of forskolin stock solution (15 mM) can be found in our protocol Andersen and Monje (2018). Keep the forskolin stock solution in the -80 °C freezer for long-term preservation of the cAMP-inducing activity. Forskolin is a synergistic enhancer of heregulin-dependent hSC proliferation
0.5% Trypsin/EDTA (TE) solution (10×), without phenol red (Thermo Fisher Scientific, Gibco, catalog number: 15400054). (Optional) TrypLETM select enzyme, 1× (Thermo Fisher Scientific, Gibco, catalog number: 12563011)
Laminin stock, consisting of a sterile 1 mg/mL laminin solution from Engelbreth-Holm-Swarm murine sarcoma basement membrane (Sigma-Aldrich, catalog number: L2020), stored at -80 °C in aliquots for single use. Prepare laminin-coated dishes as described in Andersen and Monje (2018). Always use freshly coated laminin dishes for seeding hSCs. Nerve fibroblasts can be plated in regular cell culture–treated dishes without coating
Poly-L-lysine (PLL) powder (Sigma, catalog number: P-263). Prepare a PLL stock solution and use it for coating dishes as described in Andersen and Monje (2018). Store air-dried PLL-coated dishes at 4 °C for up to one month
Dulbecco’s phosphate-buffered saline (DPBS), pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14190)
Dimethyl sulfoxide (DMSO) (Invitrogen, catalog number: D12345). Used as a component of the freezing medium and solvent of lyophilized fluorophores
(Optional) RecoveryTM cell culture freezing medium, ready to use, stored in aliquots at -80 °C (Thermo Fisher Scientific, Gibco, catalog number: 12648010)
Isopropyl alcohol (Fisher Chemical, catalog number: A416P-4L)
Tris base, powder (Thermo Fisher Scientific, Invitrogen, catalog number: 15504020)
Hydrochloric acid (HCl) solution, 1 N, bioreagent suitable for cell culture (Sigma, catalog number: 7647-01-0); used to balance pH in buffers and cell culture medium
Antibodies, dyes, and assorted commercially available products
Anti-NGFR mouse IgG monoclonal antibody, produced from the HB-8737 hybridoma cell line (American Type Culture Collection, ATCC). See Ravelo et al. (2018) for technical details on our culture protocols for this and other hybridoma cell lines
Anti-O4 mouse IgM monoclonal antibody, produced from the O4 hybridoma cell line (Sommer and Schachner, 1981), kindly provided by Dr. Melitta Schachner. (Optional) Use purified O4 antibodies from a commercial source (Novus Biologicals, catalog number: NL637)
Anti-mouse immunoglobulins, goat polyclonal antibody (anti-IgG, IgA, and IgM), affinity purified, liquid, unconjugated (ICN/Cappel, catalog number: 55486)
CellTrackerTM Green CMFDA (5-chloromethylfluorescein diacetate) powder (Invitrogen, Molecular Probes, catalog number: C7025)
(Optional) Pluronic F-127 (Molecular Probes, catalog number: P-6866)
Hoeschst-34580 (Molecular probes, catalog number: H21486) prepared in water at 1 mg/mL
Basic NucleofectorTM kit for primary mammalian glial cells (Lonza, catalog number: VPI-1006)
GFP plasmids. The pmaxGFPTM expression vector (0.5 µg/µL in 10 mM Tris pH 8.0) can be used as provided in the Basic NucleofectorTM kit or expanded in house
Lentiviral particles, aliquoted and stored at -80 °C for up to six months. For a reference, we have used lentiviral expression vectors for the fluorescent reporters EGFP and mCherry (Monje et al., 2018) acquired as ultra-purified, ready-to-use, packaged lentiviruses from the Viral Vector Core Facility, The Miami Project to Cure Paralysis, Miami, FL. Follow the manufacturer’s recommendations on the safe storage and use of viral stocks, including the MOI determination for each virus lot. Multiple freeze/thaw cycles of the viral particles are not recommended
(Optional) Polybrene infection/transfection reagent, 10 mg/mL stock solution (Santa Cruz Biotechnology, catalog number: sc-134220)
(Optional) Eukaryotic antibiotics for the selection of virally-infected cells. Puromycin dihydrochloride (Santa Cruz Biotechnology, catalog number: sc-108071); Blasticidin S HCl solution (Santa Cruz Biotechnology, catalog number: sc-495389); Hygromycin B solution (Santa Cruz Biotechnology, catalog number: sc-29067)
Solutions
Low proliferation medium (LP) (see Recipes)
High proliferation medium (HP) (see Recipes)
TE dissociation solution (see Recipes)
Freezing medium (see Recipes)
Immunoglobulins solution (see Recipes)
Equipment
Biosafety cabinet, BL2 level (Thermo Fisher Scientific, model: 1300 Series A2)
CO2 cell incubator set up at 37 °C and 8%–9% CO2 (Thermo Fisher Scientific, model: Forma Steri-Cycle)
Benchtop centrifuge (Beckman Coulter, model: Allegra X-I2R)
Inverted phase contrast microscope with an attached digital camera (VWR, model: V5MP)
Cell counter for automated counting of cells in suspension (Bio-Rad, TC20). (Optional) Hemocytometer for manual cell counting
4 °C refrigerator and -80 °C laboratory freezer (Thermo Fisher Scientific, model: RLE Series)
Liquid nitrogen storage tank (Thermo Fisher Scientific, model: Locator JR Plus)
Single cuvette-based NucleofectorTM I device and certified cuvettes (formerly Amaxa Biosystems, now Lonza)
Inverted fluorescence microscope equipped with a UV, FITC, and TRITC filter sets (Olympus IX71) and attached digital camera
(Optional) Live-cell imaging system IncuCyte ZOOMTM (Essen BioScience, MI) for time-lapse microscopy of cultured cells plated in multi-well dishes
Software
(Optional) IncuCyte Zoom 2015A, Rev 1 for image capture and video assembly of time-lapse microscopy data
Image analysis software, ImageJ-FIJI (NIH), free-access software available at https://imagej.net/software/fiji/. Used to estimate the covered area (confluency) in selected phase contrast images using the area measuring tool
Procedure
Protocol 1: Trypsinization, plating, and propagation
Human Schwann cells isolated from nerve tissues are highly proliferative in the presence of added mitogens. At least two distinct mitogenic factors, namely heregulin (an ErbB agonist also known as neuregulin/NRG and glial growth factor/GGF) and forskolin (an agent that elevates intracellular cAMP), are needed for effective propagation (Bunge et al., 2017). For this reason, standard protocols for hSC culturing use a DMEM-based medium formulation containing heregulin (usually provided as a recombinant peptide at a nanomolar dose), forskolin (provided in the micromolar range), and serum (provided in the form of FBS) (Casella et al., 1996). This supplemented formulation (herein referred to as high proliferation or HP medium) supports 3–4 consecutive rounds of subculture of adult nerve-derived hSCs (Figure 1). The combination of heregulin and forskolin is optimal for hSCs. cAMP-elevating agents are known to both potently drive heregulin-dependent hSC proliferation and concomitantly reduce fibroblast growth (Rutkowski et al., 1992 and 1995).
The following sections explain how to propagate cultures from a confluent plate containing >90% hSCs, preferably from primary cells, as shown in the first paper of the series (Aparicio and Monje, 2023). A discrimination is made between routine (Protocol A) and controlled (Protocol B) serial passaging. Routine passaging at a non-constant ratio is practical for small-scale experimentation, but the lifespan of donor-relevant cultures may be hard to predict. Controlled passaging is suggested for preparing cryogenic stocks and comparing cultures from different donors (or lots), to ensure that cells have undergone roughly an equivalent number of cell divisions in culture by the time of experimentation. We recommend starting this protocol using cells at P0 to allow substantial propagation by passages-2 or -3. In all cases, researchers should pay attention to the growth attributes of each cell batch by performing daily observations and running appropriate tests. For reference, the growth characteristics of a representative, proliferative hSC culture are depicted in Figure 2. Videos 1 and 2 illustrate the distinctive dynamics of early-passage (non-senescent) and late-passage (senescent) hSC cultures, respectively.
Figure 2. Progression of a proliferative human Schwann cell (hSC) culture.

Images A–F and insets i–iii provide an example of the temporal course of changes in a typical, low-passage hSC culture plated in HP medium (Protocol 1A). Notice that the cells divide quickly and asynchronously as soon as they are plated (H). The development of a pattern of alignment (C–F), a defining characteristic of SC cultures in vitro, is time and density dependent under these conditions. Cell–cell alignment usually occurs at or around confluency. The time of arrival to confluency (C, F, G) depends on various factors intrinsic to the hSC populations and the extracellular environment. The phase contrast images were selected from automated video-imaging microscopy analysis using IncuCyte ZOOMTM (see Video 1). The covered area per time point (G, graph) was calculated from the respective mask images (D–F) generated with ImageJ software.
Video 2. Growth of a typical high-passage human Schwann cell (hSC) culture.

A single cell suspension of adult hSCs was obtained, plated, and imaged as described in Video 1. The cells adhere to the substrate and extend processes but most cells in this culture can be considered senescent. Senescent hSCs either fail to undergo cell division or do so at a very low rate, as revealed by the sporadic appearance of mitotic figures. Some senescent hSCs contain conspicuous vacuoles that are clearly observable at early time points and seem to be reduced over time. Whereas some individual cells develop an expanded cytoplasm and acquire a fibroblast-like morphology, others are bipolar and extend long processes resembling the phenotype of proliferative cells. Nevertheless, stable cell-to-cell alignment and pattern formation may not be achieved, as shown in this image series. Floating debris (high contrast puncta) are usually seen in senescent hSC cultures.
-
Routine passaging
Starting with a plate containing hSC cultures that have reached confluency (see Figure 2C–2F), remove the HP medium (see Recipe 2), and rinse the cultures with 10 mL of HBSS without calcium and magnesium.
Obtain a single cell suspension by controlled dissociation using TE dissociation solution (see Recipe 3). Use 5 mL of the 1× TE solution for a 10 cm tissue culture dish. Monitor the progression of the trypsinization by phase contrast microscopy to avoid overexposure of the cells to the action of the trypsin.
When the cells detach from the dish, add 10 mL of LP medium (see Recipe 1) directly onto the cultures to stop trypsinization.
Collect the cell suspension and transfer it to a 50 mL conical centrifuge tube. Rinse the dish with 5 mL of LP medium to collect the remaining cells. Confirm that no cells remain in the dish by phase contrast microscopy.
Centrifuge the cells at 200× g for 8–10 min at 4 °C and resuspend the cell pellet in HP medium for re-plating onto laminin-coated dishes.
Count the cells using an automated cell counting device or a hemocytometer and estimate viability according to the method of choice (e.g., Trypan blue or propidium iodide exclusion assays). The percentage of live cells should be high (>90%) after this and other routine operations. Obtaining a single-cell suspension is desirable to accurately estimate cell counts and allow an even distribution of the cells inside the culture dish.
Prepare laminin-coated dishes to be used fresh on the next day (applicable to all protocols). Briefly, to coat one 10 cm culture dish, thaw the laminin stock slowly at 4 °C and prepare a working solution containing 55 μg of laminin in 10 mL of DPBS. Incubate the plate ON at 4 °C on a flat surface up until use. Scale the volume of laminin solution according to the surface of the dish or well. It is recommended to use 0.7–1 μg of laminin per cm2 of coated surface. Sequential coating of dishes with PLL-laminin increases cell adhesion and recovery, e.g., after purification or transfection. Double coating is not needed for regular passaging if the goal is to simply expand the cultures.
Plate the cells in laminin dishes by homogeneously dispersing them at the ratio that best suits the experimental needs. A 1:3–1:5 plating ratio in HP medium is suitable for most applications. This density can render a confluent plate in <5 days (Figure 2), especially when using low-passage cells. Consider that batch variability can occur in this and other cellular responses.
Place the dishes inside a CO2 incubator immediately after seeding. hSCs normally attach within one hour after plating; use phase contrast microscopy to confirm that the cells have attached properly and extended processes after plating (Video 1) (see Note a).
Replace the medium two to three times per week with HP medium until the cultures reach confluency (see Notes b–c).
Repeat steps A1–A10 if further expansion is desired. Use the same conditions described above for trypsinization, plating, and growth regardless of the round of subculture. Monitor arrival to confluency as shown in Figure 2.
-
Controlled serial passaging
Starting with a confluent 10 cm plate of cells at P0, prepare a single-cell suspension, count the cells, and estimate their viability, as described in Protocol A. Next, plate 5 × 105 viable cells in a 10 cm dish in 10 mL of HP medium (see Note d).
Culture the cells in a CO2 incubator up until arrival to confluency. A confluent plate obtained in this manner can be considered an established culture at passage-1 (P1).
Proceed as described above to passage the cells but maintain the initial plating density at 5 × 105 cells per dish and per passage, which is the equivalent of a 1:10 expansion ratio in each round in proliferative cultures. If passaging is performed in this manner, each batch can be propagated effectively usually up to passage-4 (P4) (Figure 2).
Notes:
hSCs tend to spontaneously adhere to one another and form clumps of various sizes while in suspension. Cells within small clumps usually disperse evenly once the clumps attach to a laminin substrate, but big clumps may fail to attach. It is recommended to change the medium at this time only if dead cells, clumps, or excessive floating debris are observed.
It is recommended to allow cultures to reach confluency before sub-culturing them, so as to maximize cell yields in each passage. A confluent 10 cm plate usually contains 4 × 106–6 × 106 hSCs. The time needed to reach confluency can range from 4 to 10 days depending on the initial density, passage number, donor’s age, and other factors.
Two hallmarks of confluent cultures are the observation of an aligned configuration (explained above) and the disappearance or drastic reduction of mitotic figures (Figure 2). Confluent cultures must be used quickly or passaged to a new dish to prevent cell detachment.
Controlled passaging maximizes the efficiency of cell expansion per round of subculture. This protocol recommends using a plating density of 5 × 105 cells in each round (or a 1:10 expansion ratio) preferably starting with a confluent P0 culture. If passaging is performed as suggested, yields in the order of >108 cells can be obtained as soon as the second round of passage (Figure 1). The total cells obtained per batch are mainly linked to the cell yields from the initial harvest.
Protocol 2: Cryopreservation and transfer
This section describes the preparation of cryopreserved stocks of hSCs (Protocol 2A) and their re-plating after thawing (Protocol 2B). Cryogenic storage of hSCs is a standard practice that greatly facilitates in-house experimentation and the transfer of cell stocks to other laboratories. For this reason, this section describes simple protocols for transferring of cells in the form of live (adherent cultures in flasks) or banked stocks (frozen cells in cryovials) (Protocol 2C).
hSCs can be cryopreserved at any passage without detrimental effects on viability or adhesion post-recovery (our empirical observations). Cryopreserved hSCs have been used in various in vitro and in vivo approaches by our group (Monje et al., 2018) and others (Kohama et al., 2001; Bastidas et al., 2017). It is recommended to use healthy hSC cultures harvested in the logarithmic or exponential growth phase for optimal preservation and recovery. Another important recommendation is to prepare a master stock of hSCs at passages-1 or -2, as well as working stocks of the derivatives from these cells at passages-2, -3, and -4. Maintaining sufficient cryogenic stocks of cells collected at low passages can allow researchers to reinitiate the cultures at any time and use cells from the same batch and passage in independent experimental rounds (Monje et al., 2018).
Video 1. Growth of a typical low-passage human Schwann cell (hSC) culture.
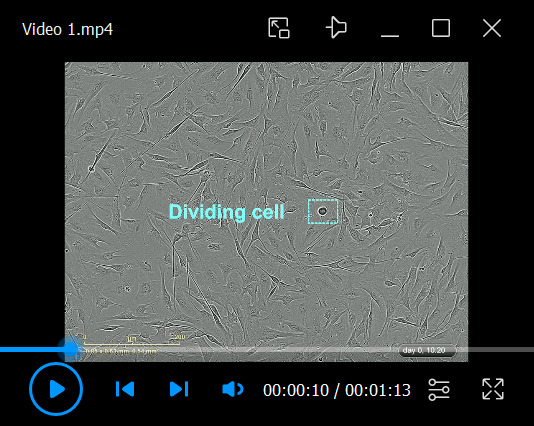
A single cell suspension of adult hSCs (proliferative) obtained by trypsin dissociation was plated on a laminin-coated dish and incubated in HP medium up until confluency, as described in Protocol 1A. Notice the fast adhesion of the cells, the changes in cell morphology (including process extension and alignment), the rapid cell migration, and the asynchronous appearance of mitotic figures. This culture can be considered confluent and ready to use by the third day of culture. The cells were imaged using IncuCyte ZOOMTM using 20× objective lenses. Individual images were taken every 20 min for a total time of 84 h from the onset of plating. Some relevant elements were highlighted in this and other videos.
-
Preparation and storage of cryogenic stocks
Once the hSC cultures are approximately 80% confluent, harvest the hSCs via trypsinization and collection by centrifugation (see Protocol 1, steps A1–A5).
Discard the supernatant and gently resuspend the cell pellet in ice-cold freezing medium (see Recipe 4) at a density of 1 × 106–2 × 106 cells/mL (see Note a).
Gently resuspend the cells by pipetting up and down using a 10 mL pipette (only once or twice) to obtain a homogeneous cell suspension.
Aliquot 1.5 mL of the cold cell suspension directly into properly pre-labeled cryogenic vials on ice. Work as fast as possible. This is a time- and temperature-sensitive step that can seriously affect the survival and recovery of the cells after thawing.
Immediately transfer the cryogenic vials to an isopropanol-filled polycarbonate freezing container to be placed in a -80 °C freezer.
Twenty-four hours later, transfer the cryogenic vials to a liquid nitrogen tank for long-term storage.
-
Retrieval of cryopreserved hSCs
Transfer the cryogenic vials directly from the liquid nitrogen tank to a safe container filled with dry ice for transportation to the biosafety cabinet.
Thaw the cells quickly by placing the vials in a 37 °C water (or bead) bath up until ~70% of the liquid volume has melted.
Transfer the cell suspension to a 50 mL conical centrifuge tube containing at least 15–20 mL of ice-cold LP medium.
Collect the cells by centrifugation at 200× g for 8 min at 4 °C. Remove the supernatant and resuspend the cell pellet in 10 mL of HP medium. Gently pipette the cells up and down no more than twice using a 10 mL pipette to obtain a homogeneous cell suspension.
Plate the cells in a 10 cm laminin-coated dish, incubate them in a CO2 incubator, and culture them as described in Protocol 1. Monitor the condition of the cells by phase contrast microscopy 1–3 h after plating. Most cells are expected to be attached to the substrate by this time. Lack of attachment may indicate poor viability or a problem with the substrate.
The following day, use a phase contrast microscope to confirm that the cells have attached properly and extended processes. Change the medium at this time only if floating dead cells or excessive debris are observed.
Monitor the progression of the culture daily. The increase in cell density should be obvious as soon as two days after plating (see Figure 2).
-
Packaging and shipping of cultured hSCs
To ship cryogenic stocks, retrieve the stocks from the liquid nitrogen tank and place them in a leak-proof plastic bag or conical tube (secondary container). Transfer the cells as soon as possible into a Styrofoam container filled with dry ice (see Note b).
To ship live cells, prepare a cell culture of hSCs in HP medium at 40%–60% confluency in a T25 or T75 culture flask double-coated with PLL and laminin for stronger adhesion. Use flasks with a plug-seal cap (not vented caps). On shipment day, fill the flasks with warm HP medium to full capacity. Tighten the cap and seal it with parafilm. Wrap the flasks with absorbent paper towels and place them inside a leak-proof bag or container. Position the flasks horizontally inside a small Styrofoam box surrounded by bubble wrap or soft paper, ensuring that the flask remains in place during transit. Ship the cells at room temperature (see Notes b–c).
Send the package using an express courier service for delivery between 24 and 72 h, preferably a next-day delivery.
Once the cells arrive at the destination, retrieve the cells as described in Protocol 2B for frozen stocks and Protocol 1A for live cell cultures.
Notes:
Ready-made cell freezing medium is available from various commercial sources. We have tested RecoveryTM with good results in the cryopreservation of rat SCs (Andersen and Monje, 2018). This medium is also suitable for hSCs and can be used in replacement of our home-made freezing media (Recipe 4).
Take all possible precautions to prevent leakages during transit by double-bagging and adding absorbent paper around the flasks. General recommendations for safe shipping of biohazards, such as using leak-proof packaging materials, must be followed, according to the requirements of postal service or customs authorities, as applicable. If possible, send a second cryogenic tube or flask as a backup.
Cells growing in flasks should be shipped at room temperature. Add thermal protection (insulation) to ensure safe transport. Do not add cold pads. hSCs proliferate even at room temperature in HP medium, so avoid shipping cultures that are close to reaching confluency.
Protocol 3: Purification methods
Various types of non-glial cells from the epi- and perineurial layers, the vasculature, and the endoneurial matrix can get introduced into the hSC cultures despite precautions taken during the dissection of the fascicles. Non-glial cells do not negatively influence hSC behavior in culture. Yet, contaminating cells may be undesirable experimentally or hamper data interpretation in some in vitro and in vivo studies. For instance, it has been argued that transplantation of hSCs containing a higher than acceptable number of fibroblasts leads to excessive collagen matrix deposition in the central nervous system (Brierley et al., 2001).
The hSC culture medium containing forskolin and heregulin selectively promotes hSC growth over that of fibroblasts and often causes a progressive enhancement in hSC purity (Levi et al., 1995). However, the selective pressure of media components is insufficient to prevent the spread of fibroblasts (Peng et al., 2020). Antibody-based technologies such as magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting are among the most efficient methods available for fibroblast removal (Morrissey et al., 1995b; Weiss et al., 2016). MACS is highly selective, scalable, and adaptable for direct purification of hSC cultures (Peng, Sant et al. 2020). However, there are cost-effective and simpler methods seemingly efficacious for fibroblast removal. Immunopanning, which utilizes a solid surface coated with a specific antibody (or protein ligand) immobilized onto the surface of a cell culture plate, is a traditional way to purify SCs from mice (Lutz, 2014) and humans (Fregien et al., 2005). The difference in cell size and adhesion properties between hSCs and fibroblasts, which leads to more expedited sedimentation and attachment of the latter cells, has been exploited to separate them from hSCs. This method is advantageous in clinical applications because it does not introduce reagents or chemicals that can pose a risk to patients (Khan et al., 2021). Controlled trypsinization and a shock of cold medium are also suitable for hSC enrichment (Haastert et al., 2007; Weiss et al., 2016). However, complement-mediated killing of Thy1+ positive fibroblasts, a widely used method to purify rat SCs (Brockes et al., 1979), is ineffective for hSC cultures because certain contaminating cells do not seem to express cell surface Thy1 (our empirical observations).
This section features two distinct panning protocols (i.e., cell culture plastic panning and immunopanning) and briefly introduces our nanobead-assisted MACS protocols, described previously in Ravelo et al. (2018), to help researchers balance options in dealing with fibroblast overgrowth. All protocols start with preparing hSCs in suspension (Protocol 1). Best results are obtained using established, myelin-free cell cultures (passage-1 or higher) that result in a clean, highly viable single-cell suspension whose numbers can be estimated properly. The methods described below are unsuitable for separating hSCs after nerve tissue dissociation because of interference with myelin and other tissue-derived debris. Before starting the purification procedures, make sure the cultures contain typical hSCs, as judged by cell surface expression of NGFR and/or O4 (Peng et al., 2020). For details on our suggested staining protocols, see the accompanying paper Monje (2023).
-
Fibroblast depletion by cell culture plastic panning
Rinse the hSC cultures with HBSS, obtain a single-cell suspension using a 1× TE solution, and collect them by centrifugation in LP medium, as described in Protocol 1. In this and all purification procedures, estimate the total number and viability of the cell suspensions before purification (Ravelo et al., 2018). The number of cells to be purified (step 2) should be based on the viable cell counts (see Note a).
Plate no more than 3 × 106 total cells suspended in a 7–10 mL of HP medium in an uncoated, cell culture-treated 10 cm dish and transfer the cells immediately to the CO2 incubator.
Incubate the cell suspensions at 37 °C for 15 min to allow large-diameter cells, mainly fibroblasts, to be deposited on the bottom of the dish. Do not leave the cells in the incubator for a longer time. Otherwise, hSCs will simultaneously attach to the plastic.
Remove the dish from the incubator and slowly aspirate the supernatant, which contains a cell suspension consisting mainly of hSCs. (Optional) Slowly add 5 mL of LP medium to one side of the culture dish and tilt the plate to collect the hSCs that are not attached, with the caveat that this procedure may also detach some of the fibroblasts.
Transfer the supernatant containing hSCs into a PLL-laminin-coated 10 cm dish for recovery in LP medium or use the cells directly in experimentation. Alternatively, transfer the cells into a 50 mL conical tube, collect them by centrifugation (200× g for 8 min) and plate them at the desired density in the medium of choice.
-
hSC enrichment by immunopanning
Immunopanning plates are prepared by pre-adsorbing secondary antibodies directly to the surface of a plastic Petri dish before the adsorption of primary antibodies (Protocol 3B1). A single-cell suspension is then plated onto the coated plates for antibody-mediated hSC binding (Protocol 3B2).
B1. Preparation of antibody-coated plates
Prepare 10 mL of anti-mouse immunoglobulins solution (unconjugated secondary antibody, see Recipe 5) for each dish to be coated.
Coat one or more 10 cm Petri dishes (non-cell culture treated plastic) with 10 mL of immunoglobulins solution each and incubate them ON in a 4 °C refrigerator. Place this dish on a flat surface ensuring that all areas are well-covered with liquid. Safeguard the dishes from contamination while stored at 4 °C by placing them inside a sterile container. Consider that the surface of untreated plates is hydrophobic compared with regular tissue culture plates, and movement of the dish will be needed to spread the liquid evenly.
The next day, remove the immunoglobulins solution by aspiration and rinse the dishes three times with cold L15 medium to remove unbound antibodies.
Immediately after, add 10 mL of the primary monoclonal antibody solution and incubate the plates for at least 2 h in the 4 °C refrigerator. As source of monoclonal antibodies, we use the culture supernatant (undiluted media) produced in house from hybridoma cell lines, HB-8737 (NGFR) and O4 (see Note b).
Remove the primary antibody solution by aspiration and rinse the dishes three times with cold L15 medium to remove unbound primary antibodies. These dishes can be stored ON at 4 °C on a flat surface. Do not remove the medium from the last wash up until the cells are ready for panning (Protocol B2).
-
B2. Cell purification
Gently resuspend a single cell preparation of hSCs in ice-cold L15 medium right before the panning experiment. For instance, prepare 3 × 106–5 × 106 cells in 10 mL of L15 medium. Maintain these cells on ice making sure no clumps are formed before panning.
Remove the L15 medium from the last wash (Protocol B1, step 5) and plate the cell suspensions on the antibody-coated dishes immediately after.
Place these dishes on a flat surface at 4 °C for 20 min without disturbing them. Use phase contrast microscopy to confirm that a proportion of the cells have adhered to the substrate by gently moving the plate from side to side. This movement of fluid also dislodges loosely bound cells. The time of incubation is determined empirically by visual observation. If attached cells are not observed after 20 min, incubate the dishes for an additional 10 min at 4 °C to allow more time. Do not prolong the incubation unnecessarily. Make sure the hSCs have settled on the plate but still maintain a rounded shape before proceeding with step 4.
Remove the media manually with a transfer pipette and gently wash the cells three times with L15 medium to rinse off non-attached cells (e.g., these cells will be mostly fibroblasts in HB-8737/NGFR immuno-panning experiments).
Add 10 mL of LP or HP medium and gently scrape the attached cells off the panning dish. Detach the cells gently from the 10 cm plate into the LP medium with a 2 cm blade cell lifter. Slightly tilt the dish and scrape the cells from the edges to the center to ensure all areas are covered. Confirm that the cells were detached by phase contrast microscopy. This is a time-sensitive step. Caution should be taken during the scraping procedure to prevent mechanical damage to the plasma membrane. Use a gentle dissociation reagent such as TrypLETM select to lift the cells if poor viability is observed while optimizing this step.
Transfer the cell suspension to a conical 15 mL tube for collection by centrifugation (200× g for 8 min) or plate them directly onto a PLL-laminin-coated dish for recovery, as explained in Protocol 3A, step 5.
-
hSC enrichment by magnetic-activated cell sorting
Prepare a single cell suspension of hSCs. Allocate at least 3 × 106 viable cells for each purification step.
Follow the instructions in our step-by-step MACS protocol for positive selection of NGFR+ and O4+ hSCs (Ravelo et al., 2018).
-
Analyze the resultant cell products by phase contrast and fluorescence microscopy to confirm the purity of the hSCs. Representative results are shown in Figure 3 and Videos 3, 4, and 5.
Video 3. Characteristics of unpurified human Schwann cell (hSC) cultures.
Download video file (81.7MB, mp4)
Video 4. Characteristics of magnetic-activated cell sorting (MACS)-purified human Schwann cell (hSC) cultures.
Download video file (87.4MB, mp4)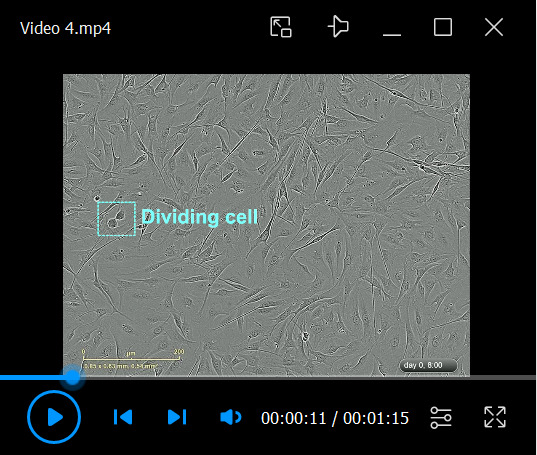 The MACS-purified hSCs (retained fraction) were derived from the mixed cultures shown in Video 3. The cells were plated immediately after purification. This video shows the fast adhesion, proliferation, and alignment of the hSCs, which can be considered evidence in support of the viability and biological activity of the purified cells.
The MACS-purified hSCs (retained fraction) were derived from the mixed cultures shown in Video 3. The cells were plated immediately after purification. This video shows the fast adhesion, proliferation, and alignment of the hSCs, which can be considered evidence in support of the viability and biological activity of the purified cells.Video 5. Characteristics of magnetic-activated cell sorting (MACS)-purified fibroblast cultures.
Download video file (78.7MB, mp4) The MACS-purified human fibroblasts (eluted fraction) were obtained from the cell cultures shown in Video 3. The cells were plated immediately after purification. Notice that the fibroblasts are highly proliferative, and their phenotype is clearly distinguishable from that of hSCs. Most fibroblasts exhibit a flat and expanded morphology with a conspicuous reticulated cytoplasm at confluency.
The MACS-purified human fibroblasts (eluted fraction) were obtained from the cell cultures shown in Video 3. The cells were plated immediately after purification. Notice that the fibroblasts are highly proliferative, and their phenotype is clearly distinguishable from that of hSCs. Most fibroblasts exhibit a flat and expanded morphology with a conspicuous reticulated cytoplasm at confluency.
Notes:
Perform all necessary controls during the optimization phase. For instance, it is useful to set out a sample of the original cell suspension and plate it in a multi-well dish to estimate the purity of the original populations in comparison to those obtained at the end of the purification procedure. One way to confirm the effectiveness of cell purification is to perform an immunostaining analysis using antibodies against hSC-specific markers before and after purification. An example of such analysis is provided in Figure 3. Our publication by Peng et al. (2020) provides additional experimental data on hSC purification methods, cell-based assays, and analysis of results.
The hybridoma ATCC #HB8737 was selected because it produces a monoclonal antibody against the extracellular domain of NGFR, a stable cell membrane marker for cultured hSCs (Peng et al., 2020). This antibody does not recognize rodent NGFR (Ross et al., 1984) and is suitable for live-cell labeling and immunopanning of hSCs. An alternative panning antibody is O4 (Bansal et al., 1989), which can be used for positive selection of stage-specific, O4-expressing hSCs (Ravelo et al., 2018).
Protocol 4: Cell labeling and gene delivery
SCs can be labeled directly with vital fluorophores or indirectly by introducing plasmids and viral vectors while retaining their normal phenotype (Mosahebi et al., 2000 and 2001; Hoyng et al., 2015). Like many other primary cells, hSC cultures are hard to transfect. In our experience, transient transfection of adherent nerve-derived hSCs with conventional lipid-based transfection reagents seldom overpasses 5% transfection efficiency regardless of the brand or formulation used (unpublished). However, we and others have found that Nucleofection technology is appropriate for introducing plasmid DNA into hSCs in suspension with efficiencies that can average 40%–60%, as determined by the levels of GFP expression from reporter plasmids (Haastert et al., 2007; Monje et al., 2008). Stable, long-term fluorescent labeling of cultured hSCs can be accomplished by lentiviral or retroviral infection (Monje et al., 2018). However, the initial transduction efficiency with retroviruses is generally low even when using low-passage cultures, as only hSCs in cell division can be infected (unpublished).
The issue of toxicity in relation to the efficiency of labeling or genetic modification is the most important challenge to overcome in any transfection or infection protocol. hSCs are particularly sensitive to changing environmental conditions. The additives needed for efficient transfection or transduction or cell selection (e.g., with antibiotics) can cause substantial cell loss. In certain cases, transient labeling with membrane-permeable fluorescent dyes is sufficient for the visualization of cells over a period of time, e.g., to monitor the morphology of SCs in vitro (Monje et al., 2009) or after transplantation in experimental animal models (Li et al., 2003).
The following sections describe our recommended methods for transient and long-lasting fluorescent labeling of hSC cultures for live-cell imaging and tracing. Our transfection methods are suitable to achieve overexpression of reporter genes and membrane receptors (Monje et al., 2008), and possibly other forms of genetic modification.
Figure 3. Purification of human Schwann cell (hSC) cultures.

The phase contrast images compare a representative adult nerve-derived hSC culture before (mixed culture, left panels) and after magnetic-activated cell sorting (MACS)-assisted cell separation (purified hSCs, middle panels), as per Protocol 3C. The fibroblast cultures are shown in parallel for comparison (purified fibroblasts, right panels). The phase contrast images were selected from IncuCyteTM Videos 3, 4, and 5, and the respective mask images (ImageJ, panels in magenta) were generated to denote the degree of confluency within the first- and third-days post-purification, as indicated. The hSCs were identified as NGFR+ cells (green) by immunofluorescence microscopy analysis (lower panels). NGFR- cells can be regarded generically as fibroblasts. Total cell nuclei were labeled with DAPI (blue), and proliferating nuclei were labeled with EdU (red). Notice that the post-MACS cell products (i.e., hSCs and fibroblasts) are both highly viable and proliferative cells.
-
Transient labeling with vital fluorophores
One practical way to fluorescently label hSCs with minimal cytotoxicity is with CellTrackerTM. This fluorescent dye is well-suited to detect dynamic changes in cell size and shape by fluorescence microscopy. CellTrackerTM freely passes through the plasma membrane and transforms into a cell membrane-impermeant fluorescent product that is maintained for several days in the hSCs’ cytoplasm. The labeling procedure involves the addition of the reagent in serum-free culture medium followed by washes to remove the soluble reagent. Cells are ready to use soon after the labeling treatment. The fluorescence intensity is strong even after fixation with aldehyde-based fixatives and can be combined with antibody-based staining or staining with nuclear dyes.
Prepare a 10 cm plate of cultured hSCs in HP medium, preferably at confluency (Figure 2).
Dissolve the lyophilized CellTrackerTM powder with high-quality DMSO to a concentration of 10 mM, as suggested by the manufacturer, immediately before use.
Rinse adherent hSCs with serum-free DMEM to remove traces of serum.
Dilute the freshly prepared CellTrackerTM stock solution to a working concentration of 6.5 μM in pre-warmed (37 °C) serum-free DMEM and directly add it to the cells. For 10 mL of medium, use 6.5 μL of 10 mM CellTrackerTM stock together with 30 μL of 20% (v/v) Pluronic F-127, a non-ionic detergent used as dispersing agent to enhance the labeling intensity. The addition of Pluronic is optional. Higher concentrations of Pluronic are toxic to hSCs.
Incubate the cells for 30 min in the CO2 incubator. Check the labeling efficiency by fluorescence microscopy before proceeding with the next step. Be strict with the labeling time so as not to compromise the health of the cells.
Rinse the cells with a sufficient volume of pre-warmed serum-free DMEM to remove the background fluorescence. Next, add 10 mL of HP medium and incubate the cells for at least 30 min at 37 °C (recovery) before experimentation.
(Optional) Include an additional incubation step with Hoeschst-34580 (1:1,000 dilution in LP medium) to concurrently stain the cell nuclei. Hoeschst-34580 is well-tolerated by hSC cultures. For reference, Figure 4A shows typical hSC cultures (live-cell imaging) after combined CellTrackerTM/Hoeschst-34580 staining.
(Optional) Re-stain the cells 48–72 h after by repeating steps A2–A6 if the fluorescence intensity declines.
-
Transient transfection using Nucleofection
Nucleofection is a technology that applies an electrical pulse to momentarily create small pores in the plasma and nuclear membranes to enable rapid delivery of nucleic acids into the nucleus. This technology has shown superior performance for transfecting various primary cells, including hSCs. The conditions outlined below were set up to transfect expanded, donor-derived hSCs in suspension using cuvettes and reagents provided by the manufacturer (Amaxa Biosystems, now Lonza). This low-throughput method, which uses high cell numbers (106–107 cells) per transfection reaction, is suitable for performing biochemistry (e.g., gene reporter assays and western blotting) and fluorescence microscopy studies using transfected hSCs (Monje et al., 2008). Variants on the original Nucleofection technology are currently available but have not been tested in our laboratory.
Prepare a PLL-laminin-coated 6-well plate containing 1.5 mL of HP medium and incubate it in the CO2 incubator for plating the hSCs immediately after transfection.
Starting with a confluent 10 cm plate of hSCs, harvest the cells by trypsinization, count the cells, and estimate their viability. Use a healthy hSC culture preferably collected at passage 1–3, as shown in Figure 2C–2F.
Prepare an aliquot of 5 × 106 viable cells in LP medium and collect them by low-speed centrifugation at room temperature to obtain a very loose cell pellet. This is a sensitive step. Centrifugation should not exceed 150× g (for up to 8 min) in a swinging bucket centrifuge.
Remove the supernatant completely, resuspend the cell pellet in Nucleofector Solution from the Basic NucleofectorTM kit for primary mammalian glial cells (100 µL per sample), and add 1 µg of pmaxGFPTM Vector (positive control) or 1–4 µg of plasmid DNA of choice (experimental) following the instructions provided by the manufacturer (see Note a).
Immediately transfer the cell/DNA suspension into a certified cuvette, remove air bubbles, and close the cuvette with the cap.
Select the appropriate Program (A-33 or O-17) for the NucleofectorTM I device or an equivalent program on other models. Insert the cuvette into the holder and apply the selected program. Program A-33 is preferred for better recovery of viable hSCs post-transfection. Program O-17 renders a higher transfection efficiency (>40% as determined by pmaxGFP expression) with the caveat of increased cell loss.
Promptly add 500 µL of pre-equilibrated HP medium directly to the cuvette and gently transfer the sample in a drop-by-drop manner directly into the wells of the 6-well plate. Work quickly, as the cells are very sensitive at this stage. Plate the product of one transfection reaction (initially 5 × 106 cells) into two wells of a 6-well plate, as substantial cell death is expected due to the electrical shock. Transfer the plate to the CO2 incubator for stabilization without delay.
Observe the plate 3–4 h post-transfection to confirm cell attachment and change the medium to remove floating (dead) cells and debris.
Assay the cells in the 6-well plate or re-plate them into a new multi-well dish after they have recovered for at least 24 h (see Note b). Substantial expression of pmaxGFP is expected in the positive control condition (Figure 4C).
-
Infection with lentiviral vectors
Lentiviral particles can be used to achieve stable overexpression of a transgene of interest in virtually any cell type, including non-proliferating, terminally differentiated cells. Once integrated into the DNA of the target cells, long-term constitutive expression of a gene product can be achieved. We have used lentiviruses to transduce rat and human SCs with vectors encoding fluorescent proteins for direct visualization of cells in isolation and in co-culture with neurons (Monje et al., 2018). Transducing hSC cultures at a low passage (e.g., P1) and expanding them for at least another round (e.g., 1:10 ratio) is feasible and recommended to create a working batch of transduced cells for experimentation and/or storage by cryopreservation.
Standard practices for viral transduction are applicable for the infection of hSCs. The first step is to empirically determine the suitable multiplicity of infection or MOI (i.e., the number of viral particles needed per cell to effectively achieve transduction) to optimize gene delivery in hSCs, as the MOI directly correlates with the number of integration events and the expression levels of the transgene. A MOI of 1 (one viral particle per cell) is commonly used, but a higher MOI (usually 5–10) is often needed to achieve >90% GFP expression in rat and human SCs (our empirical observations). In addition, it should be noted that high infection levels can lead to reduced hSC viability possibly linked to transgene overexpression.
We routinely perform preliminary experiments to assess the functional titer of viral stocks for each new virus type and lot (Protocol C1). We do so in replicate samples using at least two independent hSC cultures because the infection efficiency (and associated toxicity) can vary from batch to batch and donor to donor. Testing a range of viral concentrations in small wells (multi-well plates) can aid in determining the volume of viral stock needed for larger cultures. Once the infection conditions are optimized, the desired quantity of hSCs can be infected and the cells used as such or after being selected with antibiotics to enrich in the infected population (Protocol C2).
C1. Determination of the optimal virus dose
For the virus titration curve, seed the hSCs in a 24-well dish coated with PLL-Laminin and plate ~50,000 hSCs per well in HP medium to obtain a sub-confluent culture.
The next day, replace the medium with 300 µL of transduction medium (TM) containing increasing concentrations of the viral particles for EGFP, mCherry, or the virus of choice (see Note c).
Incubate the cells overnight at 37 °C in the CO2 incubator.
The following day, remove the viral particles by replacing the TM with 500 µL of HP medium per well for optimal growth and recovery of the cells.
Three days after infection, observe the expression of reporter proteins by fluorescence microscopy imaging.
Determine the optimal MOI to be used in subsequent experiments by calculating the percentage of cells expressing the gene reporter vs. the total number of cells by image analysis or other methods.
-
C2. Amplification of transduced hSCs
Plate hSCs at a density of 1 × 106–2 × 106 cells in a 10 cm plate double-coated with PLL and laminin in 10 mL of HP medium. (Optional) Scale the cell density and volume of medium up or down for other plate formats.
When the cells reach ~40%–50% confluency (usually within two days post-plating), transduce them by replacing the medium with 6 mL of TM at the desired MOI, as determined in Protocol C1.
Proceed as described above for managing virus-transduced cells.
Analyze the cells three days after infection to confirm expression of the gene reporters. By this time, >90% of the cells should display high levels of the reporter gene (Figure 4B). These cells are ready for use in experimentation as such, after additional expansion (step 5), or after antibiotic selection (step 6).
(Optional) Subculture the virally transduced cells as recommended in Protocol 1A or B to generate larger batches of cells for experimentation or cryogenic storage. Transgene expression is expected to be stable after 2–3 additional rounds of expansion.
(Optional) Enrich the infected populations at an early passage by treating the cells with an appropriate antibiotic as determined by the selection gene encoded in the viral vector. For reference, we have used 0.5 μg/mL puromycin, 100 μg/mL hygromycin, and 3 μg/mL blasticidin to select virally transduced hSCs established at passage-1 (unpublished). The antibiotic should be provided in HP medium for as long as needed to eliminate all non-transduced cells, per visual inspection under the phase contrast microscope.
Figure 4. Fluorescent cell labeling of cultured human Schwann cells (hSCs).

(A) Transient cell labeling with CellTrackerTM showing homogeneous staining of all viable cells. (B) Persistent cell labeling via infection with EGFP-encoding lentiviral vectors. (C–F). Transient transfection of GFP-encoding plasmid vectors via Nucleofection. In A, cells were treated for three days with combined heregulin (10 nM) and forskolin (2 µM) in the absence of serum, labeled with CellTrackerTM and Hoescht-34580, and imaged 1 h post-labeling. In B, cells were infected at passage-2 and subjected to another round of expansion in HP medium before imaging. Notice the nonhomogeneous levels of EGFP expression in individual cells. Lentiviral infection does not significantly alter the phenotype or the proliferation of hSCs in vitro even after several passages. In C–D, hSCs and rat SCs (control) were transfected via Nucleofection (program A-33, using pmaxGFP reporter plasmid), and visualization was performed four days after the procedure. Expression of pmaxGFP is maintained for at least one week. Yet, transfected hSCs can show signs of stress, as evidenced by the unusual stellate morphology of the cells (C–D).
Notes:
Strictly follow the instructions provided by the manufacturer for the preparation, storage, and use of reagents (including the quantity and quality of plasmid DNA) and the use of the equipment. During the optimization phase, perform pilot studies with at least two batches of cultured hSCs, as transfection efficiency varies from batch to batch. Introduce the necessary controls to estimate the transfection rate while observing cytotoxicity due to the procedure, the expressed gene product, or other variables.
Transfected cells can be harvested for analysis or assayed directly in the 6-well plates. To better control the cell density and obtain accurate replicas for experimentation with the genetically modified cells, re-plate the transfected hSCs into 96-, 12-, or 24-well assay plates. Perform cell-based assays preferably within three days post-transfection since the levels of transgene expression decline thereafter.
The TM consists of HP medium containing viral particles at a given MOI. Estimate the MOI based on the information provided by the manufacturer of viral particles (e.g., as it relates to the concentration of p24 capsid protein in the stock) and the number of cells plated in the test well. If there is no prior experience with hSCs or the virus, start by testing a broader range of MOIs (e.g., 0, 0.5, 1, 2, 5, 10, 15, 30). Transduction enhancers such as polybrene (8 μg/mL) may be added to the TM with caution because additives can be toxic to hSCs. Use duplicate or triplicate cultures for testing each MOI condition.
Recipes
-
Low proliferation (LP) medium
Reagent [Stock concentration] Final concentration Amount DMEM (or DMEM/F12) n/a 445 mL FBS [100%] 10% 50 mL GlutaMAX [100×] 1× 5 mL Gentamycin [1,000×] 1× 0.5 mL Total n/a 500 mL Note: Balance the pH of the LP medium to make it slightly acidic (pH = 6) using a cell culture grade HCl solution; then, sterilize it by filtration using a 0.22 µm filtration unit and store it at 4 °C for up to one month. Gentamycin is the preferred antibiotic but can be replaced by penicillin/streptomycin. Do not include antifungal reagents in this or any other culture media because of cytotoxicity to the hSCs.
-
High proliferation (HP) medium
Reagent [Stock concentration] Final concentration Amount Low proliferation medium n/a 500 mL Heregulin-β1 [25 µM] 10 nM 200 µL Forskolin [15 mM] 2 µM 69.25 µL Total 500 mL Note: Balance the pH, sterilize it by filtration, and store it as indicated for the LP medium. The HP medium induces optimal hSC proliferation when stored appropriately at 4 °C within a month of preparation. Do not freeze the HP medium.
-
TE dissociation solution
Reagent [Stock concentration] Final concentration Amount Trypsin/EDTA [10×] 1× 1 mL HBSS n/a 9 mL Total 10 mL Note: Dilute the 10× TE stock solution in ice-cold, sterile HBSS, and use it without delay to dissociate the cells. Do not freeze. Follow the instructions provided in our generic trypsinization protocol for storage and use of the TE stocks and working solutions (Andersen and Monje, 2018).
-
Freezing medium
Reagent [Stock concentration] Final concentration Amount Decomplemented FBS 90% (v/v) 90 mL DMSO [100%] 10% (v/v) 10 mL Total 100 mL Note: This medium is prepared fresh every time and maintained on ice up until the preparation of cryogenic hSC stocks. More information can be found in our generic cryopreservation protocol (Andersen and Monje, 2018).
-
Immunoglobulins solution
Reagent [Stock concentration] Final concentration Amount Tris base, pH = 9.5Immunoglobulins0.05 Mn/a99 mL1 mLTotal 100 mL Note: To prepare the 1 M Tris-buffer, dilute the Tris-base powder (121.14 g) in 800 mL of water, adjust the pH to 9.5 with 6 N or 1 N HCl (as needed), and add water to a final volume of 1,000 mL. Next, prepare 100 mL of a 0.05 M working solution from the 1 M Tris stock using sterile, cell culture grade water, and dilute the immunoglobulins at 1:100. Sterilize this solution by filtration and use it fresh to coat the immunopanning dishes.
General notes and troubleshooting
This section provides additional conceptual and technical information to understand the procedures, identify potentially problematic issues, and interpret the results. Implementing appropriate controls to verify the identity and function of the hSCs is highly recommended, as the cells that are passaged in vitro are expected to change (Figure 5, upper panel). Likewise, it is equally important to verify that all laboratory materials, including cell culture media, consist of endotoxin-free, sterile products optimal for hSC growth (Figure 5, lower panel).
Figure 5. Suggested quality control assessments in the propagation of human Schwann cells (hSCs).

The recommended routine controls for the cell cultures (above) and the materials (below) are highlighted in the diagram. Conducting purity checks for cellular (e.g., fibroblasts) and non-cellular (e.g., myelin) impurities is most critical during the establishment of the cultures (P0–P1). Late-passage hSCs cultures (P4–P5) should be tested for senescence as soon as the rate of proliferation begins to decline. Viability assays are valuable indicators of cell health after routine manipulations (e.g., enzymatic dissociation) and during the post-thaw recovery. Though rare, appropriate assays may be needed if growth becomes abnormal (e.g., cells lose contact inhibition or proliferate in the absence of heregulin) or propagation is extended over passage-5, as these observations may indicate transformation or contamination with proliferative cell lines. Researchers can refer to the accompanying paper (Monje, 2023) for detailed protocols on how to perform identity, purity, bioactivity, and authentication assays.
Expansion and subculture
Substrate. hSCs should be plated on freshly prepared laminin-coated dishes because they do not properly attach to non-coated dishes or dishes coated with simple substrates such as PLL. Laminin is unreplaceable for hSC adhesion and proliferation in vitro, which is an important difference when compared to rat SCs. Other groups have used alternative substrates such as fibronectin and collagen for the culturing of hSCs (Vleggeert-Lankamp et al., 2004). However, their effectiveness to sustain hSC expansion through passaging (this protocol) has not been determined experimentally.
Culture media. Effective expansion of hSCs in the absence of neurons strictly relies on the addition of media supplements. The mitogenic factors heregulin and forskolin are equally required for optimal propagation of hSCs. Serum factors do not promote extensive proliferation of isolated hSCs. Yet, a 10% concentration of FBS should be used consistently to maintain the hSCs viable and adherent for long periods of time. We have not identified additional factors with strong mitogenic activity for cultured hSCs that could replace or complement the abovementioned ones (Monje et al., 2018). Bovine pituitary extract, a source of heregulin-like activity, is used to supplement the culture medium of rat SCs (Brockes et al., 1979). However, pituitary extract is not mitogenic for hSCs (our unpublished observations). The source of cAMP can be variable. Forskolin is preferred because it is a non-cytotoxic and reversible inducer of cAMP that can be provided alone or together with cholera toxin for optimal cAMP elevation and proliferation in hSC cultures (Rutkowski et al., 1992; Levi et al., 1995; Morrissey et al. 1995b). It has been argued that cholera toxin should be avoided in clinical hSC preparations due to its irreversible effects on adenylyl cyclase activity [discussed in Bunge et al. (2017)].
Certain experimental procedures, such as kinase and proliferation assays, require manipulations to be performed in mitogen- and serum-free media. However, removing defined or undefined media components is not recommended for routine culture because it leads to substratum detachment and hSC apoptosis. We have found that stepwise mitogen and FBS deprivation over a 2–3-day period is effective in eliciting cell cycle withdrawal (quiescence) without significant cell loss (Monje et al., 2006). Overall, changing the concentration of serum and mitogenic factors is discouraged during the propagation, purification, and labeling of hSC cultures.
Good practices and routine controls. hSC cultures change daily (Videos 1–5). For this reason, the periodic inspection of the cultures by phase contrast microscopy is an unreplaceable practice to detect abnormalities such as cell clumping and detachment, fibroblast growth, and microbial contamination. The morphology of hSCs varies under standard growth conditions, and dynamic changes in cell size and shape are expected. Yet, the steady progression towards confluency should be clearly observable (Figure 2 and Video 1). Researchers should consider that a bipolar cell morphology and the formation of paralleled bundles of aligned cells may or may not occur in healthy hSC cultures, as this pattern of growth is often evident in confluent cultures only (Figure 2 and Video 1). Indeed, cell–cell alignment may be widespread across the dish or formed in local areas where the cell density is appropriate [for an example, see Monje (2020)]. In addition, variability should be expected in the expansion rate and other properties of hSC cultures due to cell intrinsic and extrinsic factors. Therefore, each batch- or donor-specific culture should be handled and investigated individually from the onset.
Always follow best cell culture practices and the recommendations from manufacturers in the preparation, storage, and use of culture media, supplements, buffers, and other reagents (Figure 5). Controlling the pH in buffers and media is critical to maintain hSC viability. hSCs are stable in acidic pH but are severely damaged if medium turns alkaline, especially while they are in suspension. Recalibrating the pH of buffers and media as frequently as needed and working fast while operations are conducted in the biosafety cabinet can avoid potentially harmful pH fluctuations. It is useful to set up the incubators at 8%–9% CO2 and adjust the pH of all solutions to be slightly acidic to prevent media alkalization during the handling of cells (see Recipes).
Limits to expandability. Passaging is the most defining factor affecting the expansion rate of established adult hSC populations because it eventually leads to hSC senescence. The age of the donor can be influential rather than decisive on the expandability of hSC stocks. Whereas cells from younger donors tend to proliferate faster (Boyer et al., 1994), advanced age does not preclude the derivation of proliferative hSC cultures (Levi, 1996). We have not discovered how to experimentally overcome hSC senescence in vitro but found that early passage hSCs can become senescent due to in vitro–induced stress rather than genetic influence (Monje et al., 2021). Senescent cultures may be discarded. A good practice is to establish and maintain enough cryogenic stocks (Protocol 2) to reinitiate the culture once signs of senescence are manifested.
Importantly, we have not observed spontaneous or induced (oncogene mediated) immortalization of cultured hSCs (unpublished). In fact, the creation of genetically modified hSC lines is restricted due to the hSCs’ resistance to immortalize in vitro by genetic ablation of tumor suppressors (Petrilli and Fernandez-Valle, 2018). There is precedent for hSC immortalization by ectopic co-expression of hTERT and SV40 large T-antigen (Lehmann et al., 2012). Yet, most available hSC lines are derived from peripheral nerve sheath tumors (Lee et al., 2004; Dilwali et al., 2014) rather than primary, normal hSCs. Perform an authentication analysis in case signs of aberrant growth (e.g., uncontrolled proliferation) become evident (Figure 5).
Cryogenic stocks. Cryopreservation is feasible and effective when using hSC cultures collected at any passage number, except for P0. P0 cultures are not yet established and usually contain abundant intracellular myelin and/or debris detrimental to the viability of the stocks (our empirical observations). A good practice is to create master stocks using purified hSCs from passages 1–2 along with working stocks using purified hSCs from passages 2–3 as per Protocol 1B, from each and all donors. Cultures established from cryopreserved hSCs are nearly identical to those obtained from fresh isolates as evidenced by their morphology, expression of SC-specific markers, and proliferation rates (Bastidas et al., 2017).
Standard safety practices for cell cryopreservation should be used to minimize the risk of microbial contamination, avoid temperature fluctuations, and maintain cell viability. Transfer the cells to the liquid nitrogen tank, preferably within 24–28 h, as the viability of hSCs declines if the stocks are stored at -80 °C for one week or longer. In addition, prevent unnecessary exposure of cells to the toxic effects of the DMSO during the freezing procedure and after thawing. It is good practice to dilute the freezing medium with LP medium (at 1:10 or a higher ratio) while thawing the cells to readily reduce the concentration of the cryo-protector before centrifugation and plating. Performing a stability study of the cryogenic stocks is recommended when cryopreservation methods are implemented for the first time.
Cell purification. Knowing the constitution of the cultures beforehand can help experimenters rationally choose an appropriate purification method. The panning protocols are relatively inexpensive and can be applied to reduce the content of contaminating cells in hSC cultures containing a low proportion (i.e., 30% or less) of fibroblasts. MACS is superior for the purification of heavily contaminated hSC cultures. Nevertheless, fibroblast elimination may only be achieved by performing repeated rounds of purification or combining physical (e.g., differential adhesion to plastic) and/or chemical (immunological) methods.
An important consideration is that cell culture plastic panning is a non-cell-type selective method. A proportion of hSCs is usually lost due to retention to the plastic and a proportion of fibroblasts is separated together with the hSC suspensions. If possible, collect the cells that adhere to the non-coated plates and analyze the expression of hSC- and fibroblast-selective markers to determine the efficiency of the separation. The immunopanning technique is easy to implement but may be suboptimal if the percentage of cells that exhibit the expression of the antigen used for panning is low in the cell population. Re-assess and optimize the panning conditions if a new batch of antibodies is used. During the optimization phase, it is important to run negative controls using non-coated plates (i.e., plates without pre-adsorbed antibodies) and plates coated with secondary antibodies only to determine non-specific cell attachment under the selected panning conditions.
Cell labeling and gene delivery
Vital fluorophores. hSC cultures can be stained with CellTrackerTM while adherent (monolayers) or in suspension immediately after enzymatic dissociation. This vital dye rapidly labels all cells except for dead cells with a homogeneous staining throughout the cytoplasm (Figure 4A). The more active the cells are, the higher the staining intensity will be. The hSCs typically stain with similar intensity but differences among treatments or among cell types, e.g., hSCs vs. fibroblasts, are evident. The fluorescent signal is maintained for at least 72 h inside the cells and decays thereafter. Importantly, the cultures can be re-stained without significantly affecting the viability, morphology, or migration of hSCs.
Transfection. The transfection efficiency can vary from prep to prep. Perform gene reporter controls to estimate the efficiency of transfection in each Nucleofection attempt. The pmaxGFP vector expresses GFP from the copepod Potellina sp. and is highly recommended because the fluorescence intensity is very strong and can be observed as soon as 5–6 h post-transfection in a proportion of the hSCs. The transfection efficiency would appear lower if other reporter constructs are used as transfection controls. The hSCs maintain high levels of pmaxGFP expression for at least 2–3 days with evenly distributed, mainly cytoplasmic signal localization. Although the fluorescence intensity declines with time, it is possible to observe pmaxGFP-expressing hSCs at 5–8 days post-transfection. Fading of fluorescence and granular expression of pmaxGFP can be revealed at later time points, possibly due to intracellular removal of excess pmaxGFP protein. The expression levels of other recombinant proteins are variable and likely determined by the characteristics of the protein (e.g., smaller proteins are usually expressed at higher levels), the type of vector used, and possibly environmental factors.
Lentiviral transduction. Follow best practices in handling viral particles to maintain their bioactivity and protect the operators. All operations involving lentiviruses should be performed in a BL2 biosafety cabinet. Infection of human cells with lentiviruses requires BL2+ or enhanced BL2 practices. Follow institutional guidelines to inactivate and dispose of unused viral agents, virally transduced cells, cell culture fluids, and other associated biohazardous products.
Ideally, the optimal MOI will be the minimum one that achieves 100% infection of the target hSCs. Experimentally, the most appropriate MOI may be limited by the levels of associated cytotoxicity. We have observed cytoplasm vacuolization and detachment in hSCs transduced at higher MOIs. Therefore, perform viability tests in the virus-treated conditions. We also recommend monitoring cultures for at least seven days post-infection to determine whether the transgene is toxic to the cells before seeding them for experimentation or preparing cryogenic stocks. If possible, perform the same titration experiment using more than one batch of hSCs to grasp donor-variability and select the best batches for future experiments. Additionally, the infection conditions should be re-tested in the following cases: (1) when a new stock of virus or hSC culture is used; (2) when the viral particles have been stored in the -80 °C freezer for >6 months; and (3) when the viral particles have been subjected to freezing-thawing.
Fluorescent proteins are useful gene reporters for direct image analysis of transduced cells. EGFP and mCherry are generally well tolerated by hSCs, but an excess of protein may be detrimental to cell physiology. For instance, the transduced cells may be more susceptible to die than wild-type cells upon changing environmental conditions (e.g., serum starvation) or manipulations, such as cryogenic storage. The expression of fluorescent proteins in cells infected at an appropriate MOI is not expected to change the proliferation, differentiation, and SC–axon interactions typical of non-infected SCs (Monje et al., 2018). Confirm the transgene expression by an appropriate method for those viral vectors that do not encode for fluorescent proteins.
Antibiotic selection can be used to enrich the number of transduced cells. For this, perform preliminary experiments (killing curve) to determine the minimum concentration of antibiotic that kills all non-infected cells. The optimal antibiotic concentration should be optimized in each cell preparation due to batch variability.
Acknowledgments
Natalia Andersen, Ketty Bacallao, and Kaiwen Peng provided expert technical assistance. Gabriela Aparicio assisted with data analysis and figure preparation, Lingxiao Deng with lab support, Valeria Nogueira with illustrations, Thomas Dolan with video editing, and Louise Pay with English editing. Patrick M. Wood and Mary Bunge contributed guidance and critical materials, including hybridoma cell lines and human Schwann cells, during our early investigations. The protocols described here were developed during a >15-year period supported by funding (to P.V.M.) from the National Institutes of Health NIH-NINDS (NS084326), the Craig H Neilsen Foundation, The Miami Project to Cure Paralysis and the Buoniconti Foundation from University of Miami, and the Indiana State Department of Health (grants 33997 and 43547). Grant NIH/NS009923 (to M.B., P.M.W., and P.V.M.) provided seminal funding for cell labeling and banking studies. P.V.M. is currently affiliated to and receives support from the Department of Neurosurgery from UK. The contents of this article are the responsibility of the author and do not necessarily represent the official views of the funding agencies. We are greatly indebted to the generosity of the anonymous individuals who donated their tissues for research.
Competing interests
P.V.M. is the founder of GliaBio LLC, a consulting company that focuses on glial cell research. The author declares that the research described here was conducted in the absence of commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical considerations
The experiments were performed with human Schwann cell cultures (anonymized, from non-pathological, post-mortem tissues) without regard to the gender or age of the donor. This research was deemed to constitute non-human subjects research by the Institutional Human Subjects Research Offices from University of Miami (years 2003–2018) and Indiana University (years 2019–2022). Procedures were further reviewed and approved by the Institutional Biosafety Committee of Indiana University. The original cell cultures used to optimize our expansion, purification, labeling, and cryopreservation methods were derived from nerve biospecimens made available by the Life Alliance Organ Recovery Agency (LAORA) of the University of Miami Miller School of Medicine to the laboratory of Patrick Wood (University of Miami). Requests for information and materials should be directed to P.V.M. Investigators are encouraged to provide feedback on the reported protocols.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1.Andersen N. D. and Monje P. V.(2018). Isolation, Culture, and Cryopreservation of Adult Rodent Schwann Cells Derived from Immediately Dissociated Teased Fibers. Methods Mol Biol 1739: 49-66. [DOI] [PubMed] [Google Scholar]
- 2.Aparicio G. I. and Monje P. V.(2023). Human Schwann cells in vitro I. Nerve Tissue Processing, Pre-degeneration, Isolation, and Culturing of Primary Cells. Bio Protoc 13(22): e4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bansal R., Warrington A. E., Gard A. L., Ranscht B. and Pfeiffer S. E.(1989). Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 24(4): 548-557. [DOI] [PubMed] [Google Scholar]
- 4.Bastidas J., Athauda G., G. De La Cruz, Chan W. M., Golshani R., Berrocal Y., Henao M., Lalwani A., Mannoji C., Assi M., et al.(2017). Human Schwann cells exhibit long-term cell survival, are not tumorigenic and promote repair when transplanted into the contused spinal cord. Glia 65(8): 1278-1301. [DOI] [PubMed] [Google Scholar]
- 5.Boyer P. J., Tuite G. F., Dauser R. C., Muraszko K. M., Tennekoon G. I. and Rutkowski J. L.(1994). Sources of human Schwann cells and the influence of donor age. Exp Neurol 130(1): 53-55. [DOI] [PubMed] [Google Scholar]
- 6.Brierley C. M., Crang A. J., Iwashita Y., Gilson J. M., Scolding N. J., Compston D. A. and Blakemore W. F.(2001). Remyelination of demyelinated CNS axons by transplanted human schwann cells: the deleterious effect of contaminating fibroblasts. Cell Transplant 10(3): 305-315. [PubMed] [Google Scholar]
- 7.Brockes J. P., Fields K. L. and Raff M. C.(1979). Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 165(1): 105-118. [DOI] [PubMed] [Google Scholar]
- 8.Bunge M. B., Monje P. V., Khan A. and Wood P. M.(2017). From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog Brain Res 231: 107-133. [DOI] [PubMed] [Google Scholar]
- 9.Casella G. T., Bunge R. P. and Wood P. M.(1996). Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia 17(4): 327-338. [DOI] [PubMed] [Google Scholar]
- 10.Haastert K., Mauritz C., Chaturvedi S. and Grothe C.(2007). Human and rat adult Schwann cell cultures: fast and efficient enrichment and highly effective non-viral transfection protocol. Nat Protoc 2(1): 99-104. [DOI] [PubMed] [Google Scholar]
- 11.Dilwali S., Patel P. B., Roberts D. S., Basinsky G. M., Harris G. J., Emerick K. S. and Stankovic K. M.(2014). Primary culture of human Schwann and schwannoma cells: improved and simplified protocol. Hear Res 315: 25-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emery E., Li X., Brunschwig J. P., Olson L. and Levi A. D.(1999). Assessment of the malignant potential of mitogen stimulated human Schwann cells. J Peripher Nerv Syst 4(2): 107-116. [PubMed] [Google Scholar]
- 13.Fregien N. L., White L. A., Bunge M. B. and Wood P. M.(2005). Forskolin increases neuregulin receptors in human Schwann cells without increasing receptor mRNA. Glia 49(1): 24-35. [DOI] [PubMed] [Google Scholar]
- 14.Hoyng S. A., De Winter F., Gnavi S., van Egmond L., Attwell C. L., Tannemaat M. R., Verhaagen J. and Malessy M. J.(2015). Gene delivery to rat and human Schwann cells and nerve segments: a comparison of AAV 1-9 and lentiviral vectors. Gene Ther 22(10): 767-780. [DOI] [PubMed] [Google Scholar]
- 15.Khan A., Diaz A., Brooks A. E., Burks S. S., Athauda G., Wood P., Lee Y. S., Silvera R., Donaldson M., Pressman Y., et al.(2021). Scalable culture techniques to generate large numbers of purified human Schwann cells for clinical trials in human spinal cord and peripheral nerve injuries. J Neurosurg Spine 36(1): 135-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohama I., Lankford K. L., Preiningerova J., White F. A., Vollmer T. L. and Kocsis J. D.(2001). Transplantation of cryopreserved adult human Schwann cells enhances axonal conduction in demyelinated spinal cord. J Neurosci 21(3): 944-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee P. R., Cohen J. E., Tendi E. A., Farrer R., Vries D. E. G. H., Becker K. G. and Fields R. D.(2004). Transcriptional profiling in an MPNST-derived cell line and normal human Schwann cells. Neuron Glia Biol 1(2): 135-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann H. C., Chen W., Mi R., Wang S., Liu Y., Rao M. and Hoke A.(2012). Human Schwann cells retain essential phenotype characteristics after immortalization. Stem Cells Dev 21(3): 423-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levi A. D.(1996). Characterization of the technique involved in isolating Schwann cells from adult human peripheral nerve. J Neurosci Methods 68(1): 21-26. [DOI] [PubMed] [Google Scholar]
- 20.Levi A. D., Bunge R. P., Lofgren J. A., Meima L., Hefti F., Nikolics K. and Sliwkowski M. X.(1995). The influence of heregulins on human Schwann cell proliferation. J Neurosci 15(2): 1329-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X., Dancausse H., Grijalva I., Oliveira M. and Levi A. D.(2003). Labeling Schwann cells with CFSE-an in vitro and in vivo study. J Neurosci Methods 125(1-2): 83-91. [DOI] [PubMed] [Google Scholar]
- 22.Lutz A. B.(2014). Purification of schwann cells from the neonatal and injured adult mouse peripheral nerve. Cold Spring Harb Protoc 2014(12): 1312-1319. [DOI] [PubMed] [Google Scholar]
- 23.Monje P. V.(2020). The properties of human Schwann cells: Lessons from in vitro culture and transplantation studies. Glia 68(4): 797-810. [DOI] [PubMed] [Google Scholar]
- 24.Monje P. V.(2020). Schwann Cell Cultures: Biology, Technology and Therapeutics. Cells 9(8): 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monje P. V., Andersen N., Sant D., Peng K., Wang G. and Xu X. M.(2021). Stability of lineage-specific attributes in senescent human peripheral glia. XV European Meeting on Glial Cells in Health and Disease(poster presentation).
- 26.Monje P. V., Athauda G. and Wood P. M.(2008). Protein kinase A-mediated gating of neuregulin-dependent ErbB2-ErbB3 activation underlies the synergistic action of cAMP on Schwann cell proliferation. J Biol Chem 283(49): 34087-34100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monje P. V., Bartlett Bunge M. and Wood P. M.(2006). Cyclic AMP synergistically enhances neuregulin-dependent ERK and Akt activation and cell cycle progression in Schwann cells. Glia 53(6): 649-659. [DOI] [PubMed] [Google Scholar]
- 28.Monje P. V., Deng L. and Xu X. M.(2021). Human Schwann Cell Transplantation for Spinal Cord Injury: Prospects and Challenges in Translational Medicine. Front Cell Neurosci 15: 690894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monje P. V., Rendon S., Athauda G., Bates M., Wood P. M. and Bunge M. B.(2009). Non-antagonistic relationship between mitogenic factors and cAMP in adult Schwann cell re-differentiation. Glia 57(9): 947-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monje P. V., Sant D. and Wang G.(2018). Phenotypic and Functional Characteristics of Human Schwann Cells as Revealed by Cell-Based Assays and RNA-SEQ. Mol Neurobiol 55(8): 6637-6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monje P. V.(2023). Human Schwann cells in vitro III. Analytical methods and a practical approach for quality control. Bio Protoc e4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrissey T. K., Kleitman N. and Bunge R. P.(1995). Human Schwann cells in vitro. II. Myelination of sensory axons following extensive purification and heregulin-induced expansion. J Neurobiol 28(2): 190-201. [DOI] [PubMed] [Google Scholar]
- 33.Morrissey T. K., Levi A. D., Nuijens A., Sliwkowski M. X. and Bunge R. P.(1995). Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc Natl Acad Sci U S A 92(5): 1431-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosahebi A., Woodward B., Green C., Martin R. and Terenghi G.(2000). Long-term effect of vital labelling on mixed Schwann cell cultures. Histochem J 32(6): 337-343. [DOI] [PubMed] [Google Scholar]
- 35.Mosahebi A., Woodward B., Wiberg M., Martin R. and Terenghi G.(2001). Retroviral labeling of Schwann cells: in vitro characterization and in vivo transplantation to improve peripheral nerve regeneration. Glia 34(1): 8-17. [DOI] [PubMed] [Google Scholar]
- 36.Peng K., Sant D., Andersen N., Silvera R., Camarena V., Pinero G., Graham R., Khan A., Xu X. M., Wang G. and Monje P. V.(2020). Magnetic separation of peripheral nerve-resident cells underscores key molecular features of human Schwann cells and fibroblasts: an immunochemical and transcriptomics approach. Sci Rep 10(1): 18433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravelo K. M., Andersen N. D. and Monje P. V.(2018). Magnetic-Activated Cell Sorting for the Fast and Efficient Separation of Human and Rodent Schwann Cells from Mixed Cell Populations. Methods Mol Biol 1739: 87-109. [DOI] [PubMed] [Google Scholar]
- 38.Ross A. H., Grob P., Bothwell M., Elder D. E., Ernst C. S., Marano N., Ghrist B. F., Slemp C. C., Herlyn M., Atkinson B., et al.(1984). Characterization of nerve growth factor receptor in neural crest tumors using monoclonal antibodies. Proc Natl Acad Sci U S A 81(21): 6681-6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rutkowski J. L., Kirk C. J., Lerner M. A. and Tennekoon G. I.(1995). Purification and expansion of human Schwann cells in vitro. Nat Med 1(1): 80-83. [DOI] [PubMed] [Google Scholar]
- 40.Rutkowski J. L., Tennekoon G. I. and McGillicuddy J. E.(1992). Selective culture of mitotically active human Schwann cells from adult sural nerves. Ann Neurol 31(6): 580-586. [DOI] [PubMed] [Google Scholar]
- 41.Sommer I. and Schachner M.(1981). Monoclonal antibodies(O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83(2): 311-327. [DOI] [PubMed] [Google Scholar]
- 42.Vleggeert-Lankamp C. L., Pego A. P., Lakke E. A., Deenen M., Marani E. and Thomeer R. T.(2004). Adhesion and proliferation of human Schwann cells on adhesive coatings. Biomaterials 25(14): 2741-2751. [DOI] [PubMed] [Google Scholar]
- 43.Weiss T., Taschner-Mandl S., Bileck A., Slany A., Kromp F., Rifatbegovic F., Frech C., Windhager R., Kitzinger H., Tzou C. H., et al.(2016). Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 64(12): 2133-2153. [DOI] [PubMed] [Google Scholar]