

Significance
A single residue in Lmod3 was identified to be essential for the opposite actin filament regulatory functions of tropomodulin (actin filament shortening) and leiomodin (actin filament elongation). This Lmod3 residue is particularly significant since it was previously identified as a mutation [G326R] in patients with nemaline myopathy (NM). As such, this investigation reveals a potential mechanism underlying the development of NM.
Keywords: actin, thin filament, sarcomere, skeletal muscle, nemaline myopathy
Abstract
Actin is a highly expressed protein in eukaryotic cells and is essential for numerous cellular processes. In particular, efficient striated muscle contraction is dependent upon the precise regulation of actin-based thin filament structure and function. Alterations in the lengths of actin-thin filaments can lead to the development of myopathies. Leiomodins and tropomodulins are members of an actin-binding protein family that fine-tune thin filament lengths, and their dysfunction is implicated in muscle diseases. An Lmod3 mutation [G326R] was previously identified in patients with nemaline myopathy (NM), a severe skeletal muscle disorder; this residue is conserved among Lmod and Tmod isoforms and resides within their homologous leucine-rich repeat (LRR) domain. We mutated this glycine to arginine in Lmod and Tmod to determine the physiological function of this residue and domain. This G-to-R substitution disrupts Lmod and Tmod’s LRR domain structure, altering their binding interface with actin and destroying their abilities to regulate thin filament lengths. Additionally, this mutation renders Lmod3 nonfunctional in vivo. We found that one single amino acid is essential for folding of Lmod and Tmod LRR domains, and thus is essential for the opposing actin-regulatory functions of Lmod (filament elongation) and Tmod (filament shortening), revealing a mechanism underlying the development of NM.
Actin is one of the most highly expressed proteins in eukaryotic cells. The assembly and disassembly of actin filaments is crucial for the regulation of several cellular processes, including cell motility, maintenance of cell shape, and muscle contraction. Actin-binding proteins play an important role in regulating these processes and, if perturbed, can result in a wide range of diseases. Actin dynamics are thus highly regulated, especially in striated muscle where actin-based thin filaments are finely tuned to enable muscle contraction.
Striated muscle cells are composed of repeating contractile units known as sarcomeres. Contraction occurs when myosin (thick) filaments and actin (thin) filaments overlap and interact with one another. The lengths of thin and thick filaments are critical for efficient contractile activity (for reviews, see refs. 1 and 2). The contractile apparatus is dependent upon the precise assembly of actin in order to maintain proper thin filament lengths, the alterations of which are linked to the development of various skeletal and cardiac myopathies in mice and humans (3–8).
Leiomodins (Lmods) and tropomodulins (Tmods) are members of the Tmod family of actin-binding proteins and are expressed in both skeletal and cardiac muscle tissue (for reviews, see refs. 9 and 10). Lmod and Tmod play an important role in regulating the lengths of thin filaments by modulating actin polymerization at their pointed ends (near the center of the sarcomere). Lmod and Tmod are related in their domain organization (Fig. 1), with both containing actin-binding sites one and two (ABS1 and ABS2). Additionally, Tmod and Lmod contain tropomyosin-binding site one (TpmBS1); Tmod also possesses a second tropomyosin-binding site (TpmBS2). Unlike Tmod, Lmod has a C-terminal extension that contains a poly-proline-rich region along with a third actin-binding site (ABS3) represented by the Wiskott–Aldrich homology 2 domain (WH2).
Fig. 1.

Schematic of Lmod and Tmod domain organization and the LRR domain homology. Lmods and Tmods possess similar yet distinct domains. Lmod and Tmod both contain ABS1; ABS2 along with TpmBS1. ABS2, or the LRR domain, contains 5 LRRs and the C-terminal α-helix. TpmBS2 is unique to Tmods, while Lmods possess a C-terminal extension containing a poly-proline-rich region, as well as a ABS3 represented by the WH2 (for review, see ref. 11). Sequence alignment of human Lmod3 (hLmod3), mouse Lmod (mLmod) isoforms 2 and 3, and mouse Tmod1 (mTmod1) LRRs is illustrated below the domain schematic. Multiple sequence alignment results were viewed using JalView 2 (12). The glycine residue mutated to arginine is marked by a red box, corresponding to G326 (hLmod3), G337 (mLmod3), G291 (mLmod2), and G268 (mTmod1). Dark purple highlights residues that are identical in all four sequences, lighter purple highlights residues that are identical in at least two of the four sequences, and white highlights residues that are not identical.
Despite their high sequence and domain homology, Lmod and Tmod have opposite actin regulatory functions. Lmods are thought to promote actin polymerization and elongate thin filaments, while Tmods shorten thin filaments by effectively capping the pointed end and inhibiting actin polymerization (13–15). It has therefore been proposed that Lmod and Tmod are antagonists and compete with one another for binding at the pointed ends in order to maintain correct thin filament lengths (16).
Based on previous studies, it is clear that both Lmod and Tmod are necessary for proper striated muscle development and function. Knockout (KO) of the predominantly expressed cardiac isoforms, Tmod1 or Lmod2, results in severe cardiac defects in mice. Lmod2-KO mice die at about 3 wk of age due to cardiac defects (5) and cardiac-specific KO of Lmod2 in adult mice results in a rapid loss of contractile force and cardiac failure (17). KO of Tmod1 is lethal by embryonic day 10 due to cardiac abnormalities (18). Additionally, KO of the major skeletal muscle isoform, Lmod3, in mice results in nemaline myopathy (NM), which is the most common nondystrophic congenital myopathy in humans and is characterized by severe skeletal muscle weakness (19, 20).
Analogous to what is observed in mouse models, mutations in the LMOD genes have been linked to the development of disease in human patients (7, 21–23). Several mutations in Lmod3, most of which are nonsense or frameshifts leading to truncation, have been identified in patients with NM (24–28). Of particular interest, however, is a missense variant in Lmod3 that results in severe NM. This missense mutation [G326R], which was found to be compound heterozygous with truncating mutations in two patients (24), resides in Lmod3’s leucine-rich repeat (LRR) domain. The LRR domain is homologous within Lmod and Tmod proteins and houses their ABS2. Interestingly, the G326 residue is also conserved in Tmod1 and Lmod2 as G268 and G291, respectively. This residue’s high conservation within a homologous domain suggests noteworthy functional importance for the Tmod protein family. Therefore, we decided to introduce this G-to-R mutation in the striated muscle isoforms of Tmod and Lmod in order to study how this residue and the LRR domain it is harbored in play a role in their respective functions.
Utilizing molecular dynamics simulations (MDSs), circular dichroism (CD), actin polymerization assays as well as cellular and in vivo experiments, we examined how a clinically relevant mutation in a conserved residue can affect Lmod and Tmod’s regulation of actin filament lengths. We found that not only does a single, disease-causing [G326R] mutation perturb Lmod and Tmod’s homologous LRR domain structure but also their interaction with actin and ability to regulate thin filament lengths in striated muscle. Specifically, the G-to-R mutation disrupts Lmod’s actin filament-elongation and Tmod’s actin filament-shortening abilities in myocytes. Additionally, we created a patient model of disease by expressing G-to-R mutant Lmod3 in the skeletal muscle of Lmod3-KO mice via adeno-associated virus (AAV). Using this approach, we found that the G-to-R mutation renders Lmod3 nonfunctional in vivo. Conversely, expression of WT Lmod3 improved numerous NM phenotypes observed in Lmod3-KO mice, including overall body and muscle weights, grip strength, motor coordination, myosin isoform composition, sarcomeric disorganization, Z-line widening, and thin filament lengths predominantly in fast-twitch skeletal muscle fibers. Our results uncover a mechanism for how patients harboring mutations in Lmod3 develop severe NM.
Results
The G-to-R Mutation Affects Tmod’s Actin Filament Capping Ability and Lmod’s Ability to Nucleate Actin Polymerization.
We used pyrene-actin polymerization assays to examine whether the NM–linked (G-to-R) mutation in the LRR domain affects Tmod’s capping ability and Lmod’s ability to nucleate actin polymerization. For this purpose, we introduced the G-to-R mutation into the corresponding positions in mLmod3, Lmod2, and Tmod1 proteins [see alignment of mouse Tmod and Lmod mutations with the human [G326R] NM-causing mutation: Fig. 1]. The mouse equivalent amino acid position of the human mutation for Lmod3 is G337, G291 for Lmod2, and G268 for Tmod1.
Tmod1 has been previously shown to reduce actin polymerization in vitro by acting as a cap at the pointed ends of filaments (13). Therefore, to assess the impact of this mutation on Tmod1’s capping ability, we prepolymerized actin filaments capped at the barbed end with gelsolin and monitored pointed end polymerization by utilizing pyrene-actin polymerization assays. As expected, Tmod1 reduced actin polymerization rate, as indicated by a decrease in fluorescence intensity of pyrene-labeled actin over time, compared to actin polymerization rate at the pointed ends of barbed end-capped filaments in the absence of Tmod1 (control) (Fig. 2A). Notably, the capping ability of Tmod1 harboring the [G268R] mutation was considerably impaired in comparison to Tmod1 (~fivefold weaker, SI Appendix, Table S1), demonstrating that the G-to-R mutation interferes with Tmod1’s ability to bind actin at the pointed end (Fig. 2 A and D).
Fig. 2.

The G-to-R mutation disrupts Tmod1’s ability to cap the pointed end of actin filaments and Lmod2’s and Lmod3’s ability to nucleate actin polymerization. Representative curves of pyrene-actin fluorescence (A.U.: arbitrary units) over time (min) in the presence of (A) 400 nM Tmod1 or Tmod1 [G268R], (B) 50 nM Lmod2 or Lmod2 [G291R], and (C) 200 nM Lmod3 or Lmod3 [G337R]. Blue: WT protein, red: G-to-R mutant protein, black: actin only control (actin polymerization at the pointed ends of filaments capped at their barbed ends in the absence of Tmod1 in panel A and spontaneous actin polymerization in the absence of Lmod in panels B and C). Concentration-dependent actin polymerization rates in the presence of (D) Tmod or Tmod1 [G268R], (E) Lmod2 or Lmod2 [G291R], and (F) Lmod3 or Lmod3 [G337R]. Actin polymerization rates relative to the control were calculated as the first derivatives at time zero after exponential fit. n = 3 to 4, Mean ± SD, *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t test.
Conversely to Tmods, Lmods are potent nucleators of actin polymerization in vitro (15). We therefore used pyrene-actin polymerization assays to examine the effect of the G-to-R mutation on Lmod’s ability to nucleate actin polymerization. We confirmed that Lmod2 and Lmod3 increased actin polymerization rates compared to spontaneous actin polymerization (control), as indicated by an increase in fluorescence intensity (Fig. 2 B and C). Strikingly, both Lmod2 [G291R] and Lmod3 [G337R] demonstrated dramatically lower activity in nucleating actin polymerization compared to their WT counterparts; neither were able to reach the maximum activity of WT proteins at any concentrations tested (Fig. 2 E and F and SI Appendix, Table S1). The mutation appeared to weaken the function of each Lmod isoform to a similar degree (~60-fold weaker than their WT counterparts). From this, we conclude that the G-to-R mutation impairs the LRR domain of both Lmod2 and Lmod3 and disrupts their ability to nucleate actin polymerization by significantly reducing their affinities for binding actin.
Lmod’s LRR Domain Is More Flexible than Tmod’s LRR Domain.
Crystal structures of the LRR domains of Tmod1 or Lmod2 alone or in complex with actin have been solved [Protein Data Bank (PDB) ID codes: 4PKI and 5WFN, respectively]. We used these crystal structures as templates to examine the effect of the G-to-R mutation on the structure of both the LRR domains in complex with actin, as well as the LRR domain alone in silico. [G268R] and [G291R] substitutions were made in Tmod1 and Lmod2, respectively, using the molecular modeling program, UCSF Chimera. LRR domains of WT Tmod1 or Lmod2 as well as their mutated forms, for a total of four different LRR domains, were subjected to an energy minimization followed by a 400-ns MDS run. Graphs showing rmsd of atomic positions for all LRR domains are shown in SI Appendix, Fig. S1. Surprisingly, while the introduced mutations did not result in any significant difference between simulated structures, deviation of the simulated Lmod2’s LRR domains from the original crystal structure reached ~3.5 Å after 100 ns. For Tmod1’s LRR domains, the deviation from the crystal structure was ~2.5 Å reached after 50 ns. These results indicate that Lmod2’s LRR domain is likely more flexible than Tmod1’s LRR domain, a feature that may be useful when Lmod binds to F-actin.
The G-to-R Mutation Destabilizes Lmod2’s LRRs Asparagine Ladder More than Tmod1’s.
An important feature that adds stability to LRRs is the asparagine ladder (29). In the LRR domains of Tmod1 and Lmod2, the ladder is represented by four hydrogen bonds (H-bonds) across the α-β loops of the structure (30, 31) (SI Appendix, Figs. S2A and S3A). For a more detailed perspective on the stability/flexibility of each LRR domain, distances between atoms participating in these H-bonds were examined. Within the Tmod1 LRRs, distances between all four pairs of hydrogen bonding atoms remain below the bond cutoff, with few exceptions, over the simulation’s duration (SI Appendix, Fig. S2 B–E). The results with the G-to-R simulation are similar, except for H-bond 3, which is destabilized. On the other hand, the H-bonds in the Lmod2 LRRs are less stable compared to the Tmod1 LRRs and are destabilized even further by the G-to-R mutation (SI Appendix, Fig. S3 B–E). In the Lmod2 LRRs, the hydrogen bonding atoms in bonds 1, 3, and 4 remain below the cutoff (with some exceptions) over the simulation’s duration, while H-bond 2 is not stable. The G-to-R mutation destabilizes both H-bonds 3 and 4, leaving H-bond 1 the only stable bond in the Lmod2 mutant LRR asparagine ladder. These results demonstrate in greater detail the flexibility of the Lmod2 LRR domain compared to the Tmod1 LRR domain and explain the differential effect of the G-to-R mutation.
The G-to-R Mutation Interferes with Binding Interactions between the LRR Domains and Actin.
Based on PDB structures 4PKI and 5WFN, the interactions of the Tmod1 and Lmod2 LRR domains with actin are characterized by a few key salt bridges and hydrogen bonds. To determine how well suited the Tmod1 and Lmod2 G-to-R mutant LRR domains are for actin binding, each final simulated structure was docked onto actin by alignment with its crystal structure at the beginning of the simulation and the interactions were examined. After the simulation, 9 of the existing 16 side chains of interacting residues in the Tmod1-LRR [G268R] no longer remained in position to bind with actin (Fig. 3 A and B and SI Appendix, Table S2). For Lmod2-LRR [G291R], 12 of the existing 15 interactions did not remain in position to bind with actin. In both structures, side chains of some residues were positioned to make new interactions with actin such that 5 contacts were added between Tmod1-LRR [G268R] and actin, and 7 contacts were added between Lmod2-LRR [G291R] and actin (Fig. 3 C and D and SI Appendix, Table S2). While the G-to-R mutation has a greater effect on the Lmod2 LRR domain than the Tmod1 LRR domain, it appears that the flexibility of the Lmod2 LRR domain allows it to recover more contacts after the perturbation. With these results, we identified specific binding residues that are predicted to lose contact between the LRR domain and actin when the G-to-R mutation is introduced in Tmod1 and Lmod2 (SI Appendix, Table S3); this reflects a weakened binding affinity of Tmod1 and Lmod2 to actin in agreement with the results from our actin polymerization assays (Fig. 2).
Fig. 3.

Representation of the loss of contacts between actin and Tmod1 or Lmod2 LRR domains with the G-to-R mutation. Distances between atoms of salt bridges formed with actin Glu125 and Asp363 and either (A) Tmod1-LRR Arg343 or (B) Tmod1-LRR [G268R] Arg343. Distances between atoms of salt bridges or hydrogen bonds formed with actin Glu84 and Thr126 and either (C) Lmod2-LRR Arg330 or (D) Lmod2-LRR [G291R] Arg330. LRR ribbon is in cyan, and actin ribbon is in green. Oxygen atoms are red; nitrogen atoms are blue; and hydrogen atoms are white. Bond distances are represented by dashed lines, and the bonds predicted to lose contact with actin with the G-to-R mutation are marked by red dashed lines.
The G-to-R Mutation Reduces the Stability of Lmod3, Lmod2, and Tmod1 LRR Domains.
We purified the Lmod3, Lmod2, and Tmod1 LRR domains with and without the G-to-R mutation to study its effect on the structure and stability of the LRR domain by CD. Lmod3, Lmod2, and Tmod1 LRR domains display mostly α-helical spectra at 20 °C, consistent with previous spectra of the Tmod1 LRR domain (32). Notably, the Lmod2 LRR domain has a higher content of disordered regions than the Tmod1 LRR domain as its CD spectrum exhibits a lower minimum at ~210 nm than its minimum at 222 nm (SI Appendix, Fig. S4A). The CD spectrum of Tmod1’s LRR domain, on the other hand, exhibits a higher minimum at ~210 nm than its minimum at 222 nm (SI Appendix, Fig. S4A). Introduction of the mutation into Tmod1 and Lmod2 LRR domains does not affect their ability to fold at 20 °C, as their CD spectra are nearly identical to their original structures (SI Appendix, Fig. S4A). However, when the temperature is increased to 37 °C, the domains with the G-to-R mutation lose their integrity and form visible aggregates unlike their WT counterparts, which largely stay intact (SI Appendix, Fig. S4B). Introduction of the mutation into the Lmod3 LRR domain destabilizes it drastically, causing aggregation at both 20 °C and 37 °C. Because of this, we were unable to obtain CD spectra of the aggregated mutant LRR domains. Taken together, the G-to-R mutation dramatically destabilizes the structure of Tmod1, Lmod2, and Lmod3 LRR domains.
It is known that the LRR domains aggregate during thermal unfolding at temperatures above 60 °C and this prevents assessing the stability of the LRR domains quantitatively from melting curves (33, 34). Therefore, we instead performed chemical denaturation of the proteins by titration with urea in order to compare the stabilities of the WT and mutated LRR domains (Fig. 4A). We observed that all the WT LRR domains have similar stability, with midpoints of transition from folded to unfolded at ~4 M urea. Introduction of the G-to-R mutation into Lmod2 and Tmod1 LRR domains shifts the midpoints of transition from folded to unfolded down to ~2 and ~1 M urea, respectively, indicating a significant loss in stability. It is important to note that we could not obtain an accurate chemical denaturation curve for the mutated Lmod3’s LRR domain because of its tendency to aggregate in the absence of urea. This observation suggests that the impact of the G-to-R mutation is potentially more detrimental to the LRR domain of Lmod3 compared to that of Tmod1 and Lmod2.
Fig. 4.

The G-to-R mutation destabilizes Tmod and Lmod LRR domains. (A) Urea-dependent chemical denaturation of Tmod and Lmod LRR domains. Mean residue ellipticities [θ] at 222 nm were measured at 20 °C at a range of urea concentrations in 10 mM sodium phosphate, pH 7.6. WT LRR domains demonstrated 50% unfolding at ~4 M urea while 50% of Lmod2-LRR [G291R] and Tmod1-LRR [G268R] were unfolded at ~2 and ~1 M urea, respectively. Note that chemical denaturation data could not be obtained for Lmod3-LRR [G337R] due to its heavy aggregation in the absence of urea. (B) Representative sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels for 20 μg LRR domains after incubation with 0.4 μg Staphylococcus aureus V8 protease at 20 °C over time (C) Timed-course proteolysis of LRR domains was quantified from the band density of remaining fragments relative to the band density of WT LRRs without protease (time = 0). O/N: overnight incubation; dashed boxes indicate the areas of the gel that were analyzed. Blue indicates WT LRR domains, and red indicates G-to-R mutant LRR domains. Mean ± SD, n = 4, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, Student’s t test.
In order to gain more insight about the structural instability of the mutant LRR domains, we also performed limited proteolysis experiments. We incubated the LRR domains of WT or mutant Tmod and Lmods with V8 protease (Endoproteinase Glu-C), a serine protease that preferentially cleaves peptide bonds at the C terminus of aspartic or glutamic acid residues (35). The amount of remaining uncleaved protein over time was assessed by SDS-PAGE. The mutant LRR domains were digested by V8 protease remarkably sooner, with significantly less protein present compared to the WT counterparts by 60 min (Fig. 4 B and C). This suggests that the cleavage sites on mutant LRR domains were more accessible to V8 protease due to their disordered structure compared to WT LRR domains. Note that the proteolysis time course for the mutant LRR domain of Lmod3 is different from its WT LRR domain; no lower fragment was formed at the first minute. This can be explained by the abovementioned aggregation caused by the mutation. These results are in agreement with our observations from CD experiments that demonstrated that the G-to-R mutation makes the LRR domain more disordered and dramatically perturbs its structural stability.
The G-to-R Mutation Disrupts Both Tmod and Lmod’s Ability to Effectively Shorten or Elongate Thin Filament Lengths, Respectively.
In order to assess the effect of the G-to-R mutation on Tmod and Lmod’s abilities to alter actin-thin filament lengths, we transfected either green fluorescent protein (GFP)-Tmod1 or GFP-Tmod1 [G268R]; GFP-Lmod2 or GFP-Lmod2 [G291R]; and GFP-Lmod3 or GFP-Lmod3 [G337R] into isolated neonatal rat cardiomyocytes. Myocytes transduced with GFP alone were used as a control. We imaged filamentous actin (F-actin) by staining with fluorescent phalloidin and examined overall sarcomere assembly by staining for α-actinin (Fig. 5A). Note, the G-to-R mutation did not appear to alter Tmod and Lmod’s thin filament pointed end assembly. Strikingly, when we measured actin-thin filament lengths, we observed that the mutated proteins were unable to either shorten or elongate thin filament lengths like their WT counterparts. Specifically, GFP-Tmod1 [G268R] was unable to restrict actin-thin filament lengths (0.72 ± 0.01 μm) like GFP-Tmod1 (0.69 ± 0.01 μm) and was not significantly different from the expression of GFP alone (0.73 ± 0.01 μm) (Fig. 5B). GFP-Lmod2 expression, conversely, elongates thin filament lengths (0.81 ± 0.01 μm) when compared to GFP alone (0.76 ± 0.01 μm); note that GFP-Lmod2 [G291R] was unable to elongate thin filament lengths (0.75 ± 0.01 μm) like GFP-Lmod2 (Fig. 5C). Likewise, GFP-Lmod3 [G337R] (0.75 ± 0.01 μm) was unable to significantly elongate thin filament lengths like GFP-Lmod3 (0.78 ± 0.01 μm) and was not significantly different from GFP alone (0.76 ± 0.01 μm) (Fig. 5D). These data reveal that 1) a single amino acid substitution is responsible for the loss of respective elongation and actin capping activities of Lmod and Tmod, and 2) the homologous LRR domain is essential for enabling Tmod and Lmod to properly regulate actin-thin filament architecture in myocytes.
Fig. 5.

The G-to-R mutation disrupts Tmod and Lmod’s respective thin filament length-regulatory functions but does not alter their thin filament pointed end assembly in cardiomyocytes. (A) Immunostaining was performed using anti-GFP (green) antibodies, anti-α-actinin (red) antibodies, and phalloidin to probe for F-actin. Teal line marks a gap in the F-actin staining across the M-line (center of sarcomere), Z: Z-disc; M: M-line. (Scale bar, 2 μm.) Actin thin filament (TF) length measurements that were taken from rat neonatal cardiomyocytes expressing (B) GFP control (0.73 ± 0.01 μm) GFP-Tmod1 (0.69 ± 0.01 μm), GFP-Tmod1 [G268R] (0.72 ± 0.01 μm), (C) GFP control (0.76 ± 0.01 μm), GFP-Lmod2 (0.81 ± 0.01 μm), GFP-Lmod2 [G291R] (0.75 ± 0.01 μm), or (D) GFP control (0.76 ± 0.01 μm), GFP-Lmod3 (0.78 ± 0.01 μm), GFP-Lmod3 [G337R] (0.75 ± 0.01 μm). Mean ± SEM, n = 10 to 20 cells per culture, 3 cultures, **P < 0.01, ***P < 0.001, and ****P < 0.0001, one-way ANOVA.
Lmod3 [G337R] Does Not Rescue Skeletal Muscle Disorders in Lmod3-KO Mice.
In order to investigate the extent to which the NM–linked (G-to-R) mutation impairs Lmod3 function and ascertain the behavior of mutant protein in vivo, we introduced Lmod3 [G337R] as well as wild-type (WT) Lmod3 for comparison via AAV into Lmod3-KO mice (SI Appendix, Fig. S5 A, B, D, and E). Lmod3-KO mice have been previously characterized; they present with reductions in body weights, muscle weights, and grip strength as well as develop severe NM with sarcomeric disorganization, formation of rod bodies, and myosin fiber type switch in their skeletal muscle (19, 20). Remarkably, introduction of WT Lmod3, but not mutant Lmod3, into Lmod3-KO mice improved all parameters tested including body and muscle weights, grip strength, and motor coordination (Fig. 6 and SI Appendix, Fig. S6). Body weight along with all individual muscle weights were significantly improved and either completely or nearly restored to normal levels in Lmod3-KO mice injected with GFP-Lmod3-AAV (Fig. 6 A–C and SI Appendix, Fig. S6 A–E). Conversely, Lmod3-KO mice injected with GFP-Lmod3 [G337R]-AAV did not demonstrate significant improvement in any of the measured parameters compared to Lmod3-KO mice injected with GFP-AAV alone (control). These observations were consistent with grip strength and rotarod test measurements in which Lmod3-KOs injected with GFP-Lmod3-AAV appeared to have improved function, unlike mice injected with the GFP-Lmod3 [G337R]-AAV (Fig. 6 D–F and SI Appendix, Fig. S6F).
Fig. 6.
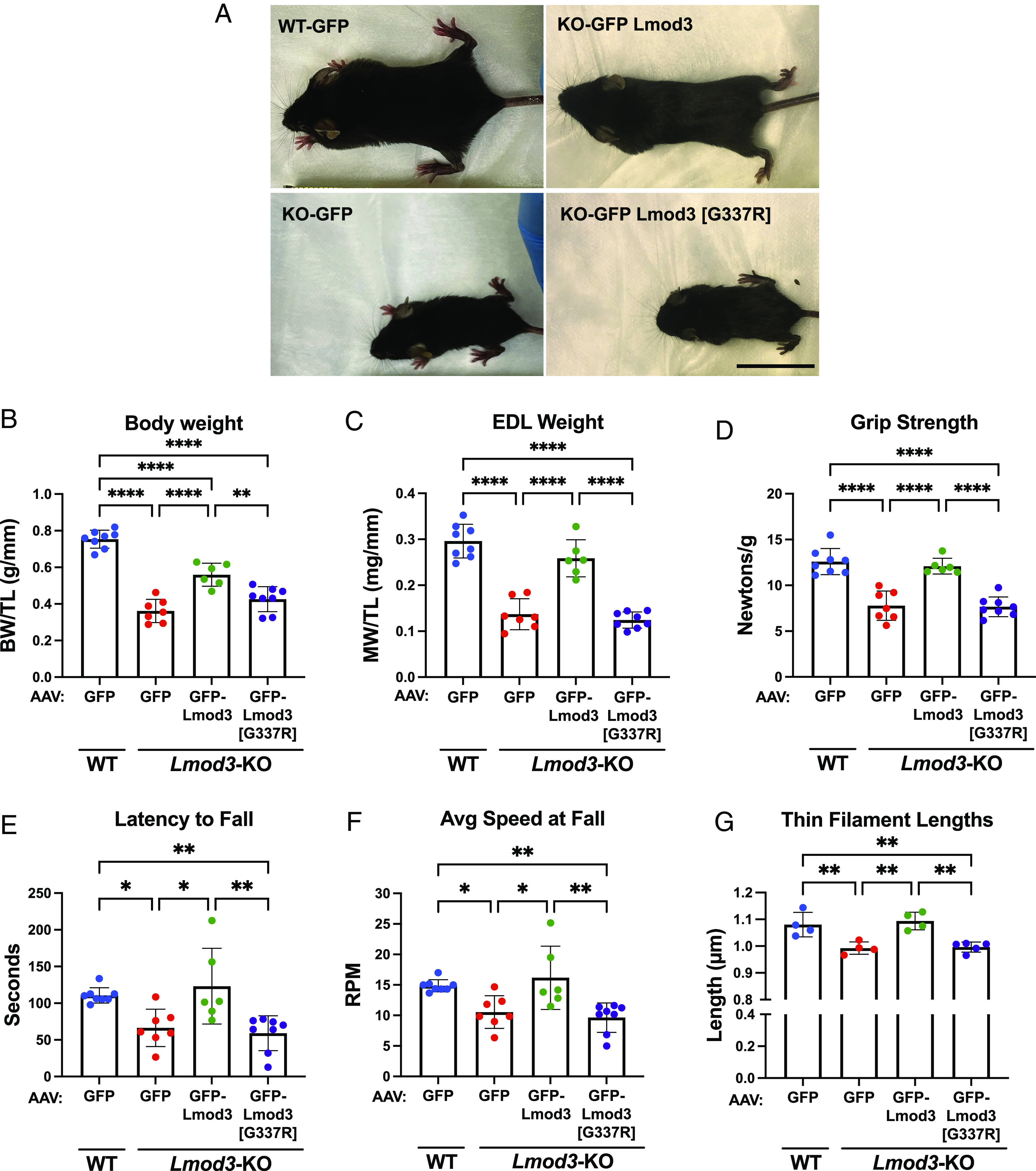
Expression of GFP-Lmod3, but not GFP-Lmod3 [G337R], improves body and muscle weights, grip strength, motor coordination, and skeletal actin-thin filament lengths of Lmod3-KO mice. (A) Representative images of injected postnatal day (PD) 21 WT or Lmod3-KO mice, (B) Body weight (BW) (WT+GFP-AAV, 0.75 ± 0.05 g/mm; KO+GFP-AAV, 0.36 ± 0.06 g/mm; KO+GFP-Lmod3-AAV, 0.56 ± 0.06 g/mm; KO+GFP-Lmod3 [G337R]-AAV, 0.42 ± 0.07 g/mm), and (C) extensor digitorum longus (EDL) muscle weight (WT+GFP-AAV, 0.30 ± 0.04 mg/mm; KO+GFP-AAV, 0.14 ± 0.03 mg/mm; KO+GFP-Lmod3-AAV, 0.26 ± 0.04 mg/mm; KO+GFP-Lmod3 [G337R]-AAV, 0.12 ± 0.02 mg/mm) normalized to tibia length (TL), (D) grip strength from all limbs (WT+GFP-AAV, 12.59 ± 1.43 N/g; KO+GFP-AAV, 7.8 ± 1.6 N/g; KO+GFP-Lmod3-AAV, 12.1 ± 0.9 N/g; KO+GFP-Lmod3 [G337R]-AAV, 7.7 ± 1.1 N/g), (E) latency to fall from rotarod test (WT+GFP-AAV, 110.6 ± 10.5 s; KO+GFP-AAV, 66.5 ± 25.5s; KO+GFP-Lmod3-AAV, 123.2 ± 51.6s; KO+GFP-Lmod3 [G337R]-AAV, 59.0 ± 23.8s), (F) average speed at fall from rotarod test (WT+GFP-AAV, 14.9 ± 1.0 RPM; KO+GFP-AAV, 10.6 ± 2.7 RPM; KO+GFP-Lmod3-AAV, 16.2 ± 5.2 RPM; KO+GFP-Lmod3 [G337R]-AAV, 9.6 ± 2.4 RPM), and (G) thin filament lengths taken from EDL muscle (WT+GFP-AAV, 1.08 ± 0.05 μm; KO+GFP-AAV, 0.99 ± 0.02 μm; KO+GFP-Lmod3-AAV, 1.09 ± 0.03 μm; KO+GFP-Lmod3 [G337R]-AAV, 1.00 ± 0.02 μm). All measurements were taken from PD21 WT or Lmod3-KO mice injected with either GFP, GFP-Lmod3, or GFP-Lmod3 [G337R] AAV. (B–F): n = 6 to 8 mice per group, (G) n = 4 to 5 mice per group. Mean ± SD, *P < 0.05, **P < 0.01, and ****P < 0.0001, one-way ANOVA. N/g: Newtons/gram, s: seconds, RPM: rotations per minute. (Scale bar, 2.5 cm.)
Additional transmission electron microscopy (TEM) analysis revealed that introduction of GFP-Lmod3, but not GFP-Lmod3 [G337R] by AAV, prevents the development of structural disorganization and hallmarks of NM in EDL (extensor digitorum longus) muscle of Lmod3-KO mice (Fig. 7A). On the other hand, the ultrastructure of the SOL muscle was largely unaffected in Lmod3-KO mice, and expression of either Lmod3 construct did not affect sarcomere assembly in this muscle type (SI Appendix, Fig. S7A). Furthermore, immunofluorescence analysis of α-actinin organization in the EDL muscle of Lmod3-KO mice demonstrated Z-line streaming (nonuniform widening), another hallmark of NM (Fig. 7 A and B). Peak profiles obtained from line scans (Fig. 7C) revealed α-actinin striations in Lmod3-KO mice to be ~50% wider compared to their WT counterparts, and this alteration was completely prevented by introducing WT Lmod3, but not mutant Lmod3, into the Lmod3-KO EDL muscles (Fig. 7D). Additionally, SDS-PAGE revealed a change in the myosin heavy chain (MHC) composition in the EDL muscles of Lmod3-KO, with a switch from type IIB into types IIA/IIX compared to WT mice (Fig. 7 E and F). This switch was also completely prevented by introduction of GFP-Lmod3, but not GFP-Lmod3 [G337R] via AAV injection. The same analysis revealed no significant change in MHC distribution in SOL muscles of WT and Lmod3-KO mice, and introducing WT Lmod3, or Lmod3 [G337R] had no further effect on the fiber type composition (~60% type IIA/IIX, ~40% type I) in this muscle type (SI Appendix, Fig. S7 B and C). We also found that thin filament lengths in the EDL muscle were significantly decreased in Lmod3-KO mice (0.99 ± 0.02 μm) compared to WT mice (1.08 ± 0.05 μm) and restored to normal levels in GFP-Lmod3-AAV injected mice (1.09 ± 0.03 μm) (Fig. 6G). Thin filament lengths in the EDL muscle in GFP-Lmod3 [G337R]-AAV injected mice (1.00 ± 0.02 μm) were not statistically different from the KO mice injected with GFP-AAV (0.99 ± 0.02 μm). This suggests that the G-to-R mutation destroys Lmod3’s ability to regulate thin filament lengths in the EDL muscle.
Fig. 7.

Expression of GFP-Lmod3, but not GFP-Lmod3 [G337R], prevents sarcomeric disorganization and myosin isoform switch in the EDL muscle of Lmod3-KO mice. (A) Representative images of fixed EDL muscle sections taken by TEM (scale bar, 1 µm.) (B) Representative immunofluorescence images (scale bar, 5 µm) and (C) line profile for α-actinin assembly at the Z-lines of sarcomeres from EDL muscle. (D) Expression of GFP-Lmod3, but not GFP-Lmod3 [G337R], prevented Z-line disorganization and widening in EDL muscles of the Lmod3-KO mice, determined from α-actinin half peak width measurements relative to WT+GFP. Mean ± SD, n = 5 to 9 mice per group, ****P < 0.0001, one-way ANOVA. (E) 8% poly-acrylamide SDS-gel showing MHC distribution (Top: type IIA/IIX, middle: type IIB, Bottom: type I) after separation by electrophoresis in EDL muscle from WT or Lmod3-KO mice expressing either GFP, GFP-Lmod3, or GFP-Lmod3 [G337R]. (F) Quantification of MHC distribution in EDL muscle is shown as a percent of total myosin. Mean ± SD, n = 4 to 6 mice per group, ****P < 0.0001, two-way ANOVA, ns: not significant.
In summary, no improvement in attributes associated with NM were observed in the Lmod3-KO mice when Lmod3 [G337R] was expressed, indicating the destructive nature of the mutation in vivo and revealing a potential actin thin filament length-dependent mechanism for disease progression in NM patients.
Discussion
Actin-thin filament length regulation is essential for a plethora of different intracellular processes. In particular, striated muscle function is dependent upon the fine-tuning of thin filaments in order to enable the propagation of muscle contraction. Dysregulated thin filament lengths are implicated in the development of both human skeletal and cardiac myopathies. Tmods and Lmods are part of the actin-binding Tmod protein family. They have been shown to modulate actin-thin filament lengths from their pointed ends, with Tmods shortening lengths and Lmods elongating lengths. However, it is not yet completely understood what contributes to the opposite thin filament length-regulating functions of different members of the Tmod protein family.
It has been previously reported that several mutations identified in the LMOD3 gene result in shorter thin filament lengths and the development of NM in humans. Among the mutations identified is a compound heterozygous missense variant [G326R] that lies within the LRR domain, a homologous actin-binding domain of Tmod and Lmod (24). Of note, this is the only missense variant in Lmod3 recorded to date that results in severe NM. Interestingly, this glycine residue is conserved in the LRR domains of all Tmod family members. In this study, we introduced this NM–linked glycine to arginine (G-to-R) mutation specifically in the Tmod and Lmod isoforms expressed in striated muscle in order to investigate a potential mechanism for disease development. We found that the single G-to-R mutation disrupted the LRR domain structure and inhibited both the actin filament elongating function of Lmod and the actin filament shortening function of Tmod.
The LRR domain of Tmod and Lmod consists of alternating α-helices and β-sheets. The mutated G-to-R residue investigated in this study resides in the α-helix of the fourth LRR. MDS and CD experiments in this study allowed us to identify that the LRR domain of Lmod is more flexible compared to the LRR domain of Tmod. This identification allows us to predict that this feature allows for the different actin filament length regulating functions of the Lmods and Tmods. Specifically, the increased flexibility of Lmod may enable it to be more extensible at the pointed end in order to recruit and bind additional actin monomers. This prediction is in agreement with Lmod’s role as a “leaky-cap” which allows for filament elongation at the pointed end (36) and ability to bind to the sides of actin filaments (37). Conversely, Tmod1’s ability to cap actin is likely enabled by less flexibility of its LRR domain and thus leading to greater rigidity in structure at the pointed end. Therefore, we propose that the differences in LRR domain flexibility are an important contribution to the divergent functions of Tmod and Lmod. While we were only able to perform MDS with Tmod1 and Lmod2 since the crystal structure for Lmod3 has not yet been solved, we would predict similar results with Lmod3 given its high sequence homology and functional similarity with Lmod2.
Our MDS, CD, and limited proteolysis results revealed that the G-to-R mutation destabilizes the LRRs structure. It is important to note that the glycine residue affected by this mutation does not make a direct contact with actin (SI Appendix, Fig. S8). Based on this, the G-to-R mutation is predicted to have an allosteric effect in weakening the LRR’s binding interface with actin. In support of this hypothesis, we identified specific residues on the actin-binding interface (SI Appendix, Table S3) that are predicted to no longer interact with actin after the G-to-R mutation was introduced. Furthermore, our actin polymerization assay results showed that Tmod1’s actin capping and Lmod2 and Lmod3’s abilities to nucleate actin polymerization were significantly inhibited upon introduction of the G-to-R mutation. Consequently, in addition to disrupting the structure of the LRR domain, this mutation also weakens the affinities of Tmod and Lmod for binding to actin. Interestingly, this result is in contrast to our previous work that showed that mutating other single residues within the LRR domain of Tmod (V232D or L313D) preserved its domain structure but weakened its interacting sites/ability to cap the pointed ends of actin filaments (38); this demonstrates that weakening of the actin-binding ability without an alteration in the total domain structure is possible.
In order to further validate our findings on a cellular level, we also examined how the G-to-R mutation affects the subcellular localization and functions of Tmod and Lmod. A model has previously been proposed that Tmod1 and Lmod2 compete with one another for binding at the pointed end in order to adjust actin-thin filament lengths accordingly in cardiac muscle (16). While the G-to-R mutation did not alter Tmod and Lmod’s localization at or near the pointed ends, we found that both Tmod and Lmod’s ability to effectively shorten or elongate thin filament lengths, respectively, was abolished in cells. Binding to Tpm was previously established to be the major determinant for the pointed end assembly of both Tmod1 (39) and Lmod2 (36). Since the G-to-R mutant proteins harbor the Tpm-binding site(s), it is not surprising that they were able to localize similarly to the WTs. However, the single amino acid change in the LRR domain amazingly rendered Tmod1, Lmod2, and Lmod3 nonfunctional as length regulators since thin filament lengths of the cells expressing the mutated isoforms were not statistically significantly different from that of the control cells. Taken together, our data allow us to conclude that the homologous LRR domain is crucial for the respective thin filament length regulatory functions of Tmod and Lmod both biochemically and in the context of myocytes.
To examine the extent to which the G-to-R mutation could affect Lmod3’s physiological in vivo function, we implemented the Lmod3-KO mouse model which had been previously shown to present with skeletal muscle defects representative of NM (19). Strikingly, we demonstrated here that introduction of GFP-Lmod3, but not GFP-Lmod3 [G337R], in the skeletal muscle of Lmod3-KO mice rescues deficits in body weight, muscle weights, grip strength, motor coordination, sarcomeric organization, MHC composition, and thin filament lengths in skeletal muscle. In fact, mice expressing GFP-Lmod3 [G337R] were overall identical to the Lmod3-KO mice expressing GFP alone. This suggests that the putative nature of the G-to-R mutation results in the expression of a nonfunctional Lmod3 variant in vivo. Interestingly, the extent to which Lmod3-KO mice were rescued by introducing Lmod3 varied. For instance, GFP-Lmod3 expression was able to fully improve the deficits in grip strength and motor coordination in the Lmod3-KO mice, while it partially restored decreased skeletal muscle weights; the exception was the EDL muscle weight which was fully rescued. This partial rescue in muscle weights could be due to the delayed expression of GFP-Lmod3 [which is introduced via AAV injection at postnatal day (PD) 4], and thus, tissue remodeling may have already begun to occur in certain skeletal muscle types. Skeletal muscle consists of fast-twitch and slow-twitch myofiber types, which are differentiated in part by the process in which they produce energy for muscle movement (for review, see ref. 40). Skeletal muscle fiber types have the potential to be preferentially affected under diseased conditions. Therefore, we compared the MHC composition and actin-thin filament lengths in examples of fast-twitch (EDL) and slow-twitch (SOL) skeletal muscles from Lmod3-KO mice. Here, we report that Lmod3-KO mice have altered myosin isoform expression (switched from type IIB to type I and IIA/IIX) and significantly shorter actin-thin filament lengths in the fast-twitch EDL muscle. Strikingly, the expression of GFP-Lmod3 was able to rescue myosin isoform expression and thin filament lengths to normal levels, while GFP-Lmod3 [G337R] expression did not. As little as 12% of endogenous Lmod3 levels in the Lmod3-KO mice was sufficient to significantly restore the myosin composition and actin-thin filament lengths in the EDL muscle (Fig. 6G and SI Appendix, Fig. S5B). This observation shows a direct link between the function of Lmod3’s LRR domain and regulation of myofibrillogenesis and thin filament lengths in vivo. Therefore, we propose that perturbation of length regulation as a direct result of a deleterious mutation in the LRR domain in Lmod3 leads to alterations of sarcomeric components and disease development in skeletal muscle. While we observed a significant change in sarcomere alignment, thin filament lengths, and myosin composition in the EDL muscle of Lmod3-KO mice, these parameters were unchanged in the SOL muscle of these mice (SI Appendix, Figs. S5F and S7). Furthermore, introducing GFP-Lmod3 to the SOL muscle of Lmod3-KO mice did not affect sarcomeric organization, thin filament lengths, or myosin composition compared to WT mice. Thus, we suggest that Lmod3 affects thin filament lengths and sarcomeric assembly predominantly in fast-twitch myofiber types (EDL), but not in the slow-twitch myofiber types (SOL).
In addition to Lmod3, Lmod2 is also expressed in the EDL and SOL muscles, and it has been shown to contribute to thin filament length regulation in slow-twitch myofibers (41). In light of these findings and our observation here that the absence of Lmod3 has no effect on thin filament lengths in the SOL muscle, Lmod2 is likely to play a more key role in the regulation of slow-twitch myofiber types compared to Lmod3. Based on this, it is clear that the importance of the actin-regulatory role of Lmod3 varies depending on skeletal muscle myofiber type. Moreover, we also observed an upregulation of Lmod2 protein expression in the EDL muscle of Lmod3-KO mice (SI Appendix, Fig. S5C). However, this increase in Lmod2 expression was evidently not able to compensate for the lack of functional Lmod3, specifically in the EDL muscle. This indicates that Lmod isoforms expressed in skeletal muscle have unique individual roles in the different myofiber types. We previously showed that Lmod2 is important for actin-thin filament organization and maintenance in both juvenile (5) and adult hearts (17). It is evident from our study that expression of Lmod3 in mice between PDs 4 to 21 is critical for growth and function of skeletal muscle. Therefore, we propose a similar role for Lmod3; that it is an essential component of actin assembly during myofibrillogenesis in skeletal muscle, in addition to maintaining the organization and lengths of mature thin filaments.
In summary, studying the NM–linked (G-to-R) mutation has enabled us to further decipher the mechanisms of thin filament length regulation in the Tmod family of proteins. We have demonstrated how exceptionally damaging the G-to-R mutation is to the structures and respective actin-regulatory functions of Tmod and Lmod and reveal that the homologous LRR domain is crucial for maintaining these functions. Additionally, by expressing exogenous GFP-Lmod3 and GFP-Lmod3 [G337R] in Lmod3-KO mice, we show how detrimental the G-to-R mutation is to Lmod3’s function in vivo, mirroring the potential disease condition in muscles of human patients harboring this mutation. More variants continue to be identified in the Tmod family of proteins (particularly Lmod3) in patients with skeletal and cardiac myopathies [(42) see references therein]. This further accentuates that understanding the Tmod protein family’s molecular and cellular functions is essential for elucidating the mechanisms underlying muscle disease development.
Materials and Methods
Immunofluorescence Microscopy.
Cryosections and cells were permeabilized with 0.2% Triton X-100/phosphate-buffered saline (PBS) for 20 min at room temperature and then blocked for 1 h at room temperature in 2% bovine serum albumin plus 1% normal donkey serum/PBS. Samples were then incubated with mouse monoclonal anti-α-actinin (1:200) (EA-53; Sigma) primary antibody diluted in blocking solution for 1.5 h at room temperature and then washed with TBST for 3 × 5 min. Secondary antibodies including Alexa Fluor™ 405–conjugated goat anti-mouse IgG (1:200) (Thermo Fisher Scientific) and Texas Red™-X Phalloidin (1:50) (Thermo Fisher Scientific) were diluted in 1x Tris-Buffered Saline, 0.1% Tween®-20 Detergent (TBST) and incubated for 1.5 h at room temperature. Following incubation, cryosections and cells were then washed with TBST for 3 × 5 min and mounted onto slides with Aqua Poly/Mount (Polysciences). Images of EDL and SOL tissue sections were taken at room temperature using a Deltavision RT system (Applied Precision) with a 100× NA 1.3 objective and a CCD camera (CoolSNAP HQ; Photometrics). Images were deconvolved using SoftWoRx software and then processed using ImageJ (v1.52, NIH). Images of cultured cardiomyocytes were taken at room temperature using a Nikon Eclipse Ti microscope with a 100 × NA 1.5 objective and a digital complementary metal oxide semiconductor camera (ORCA-flash4.0; Hamamatsu Photonics, Shizuoka Prefecture, Japan). Images were deconvolved using NIS offline deconvolution software (Nikon Corporation, Tokyo, Japan) and were processed using Photoshop CC (Adobe, San Jose, CA). Thin filament lengths and sarcomere lengths were measured using the DDecon plugin for ImageJ (v1.52, NIH) (43, 44). Sarcomere lengths were not statistically significantly different between the groups. Line scan measurements of α-actinin striations were taken using ImageJ (v1.52, NIH). The peak half-width values were averaged from ~10 sarcomeres per image and 10 images per mouse to obtain a single average value per mouse. The half-width values were normalized relative to the WT+GFP control, which was set to 1.
Fluorescence Measurements.
Actin polymerization was measured by the change in pyrene actin fluorescence using an Agilent Cary Eclipse fluorescence spectrometer (Santa Clara, CA). In order to measure inhibition rates of actin polymerization at the pointed ends by WT and mutant mTmod1 (mouse Tmod1) isoforms, 10 μM G-actin was polymerized into F-actin in the presence of 0.1 μM gelsolin in F-buffer (25 mM Imidazole, pH 7.0, 100 mM KCl, 2 mM MgCl2, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 2 mM Tris–HCl, 0.2 mM CaCl2, 0.2 mM adenosine triphosphate, and 0.5 mM dithiothreitol for 2 h at room temperature to prepare actin filaments capped at their barbed ends with gelsolin. Filaments were incubated for 30 min at room temperature in the presence of 10 μM phalloidin then kept overnight at 4 °C. Phalloidin-stabilized F-actin seeds were diluted to 1 μM with 1 μM G-actin (10% pyrenyl-actin) in F-buffer in the presence of varying concentrations of either WT or mutant mTmod1, and actin polymerization was observed by the increase in fluorescence.
Nucleation of actin polymerization by Lmod was measured from 1.5 μM G-actin (10% pyrenyl-actin) in the absence or presence of differing concentrations of mLmod2 or mLmod3 (WT or mutant) in F-buffer. Exponential growth curves to maximum were fitted to the polymerization data using SigmaPlot 12.0, and initial rates relative to control (spontaneous actin polymerization) were calculated as the first derivatives at time zero. The Kd values (the concentration required for 50% maximal activity of WT) were calculated from semi-log linear plots of relative rates vs. concentration.
CD.
CD measurements were performed using an Aviv model 420 spectropolarimeter (Lakewood, NJ). The spectra of ~0.2 mg/mL LRR proteins in 10 mM sodium phosphate, pH 7.6, were recorded in a 1-mm cuvette at 20 °C or at 37 °C. For normalization, data were recalculated to mean residue ellipticity. For unfolding experiments, the ellipticity of ~0.2 mg/mL LRR domains in 10 mM sodium phosphate, pH 7.6, containing urea from 0 M up to 6 M, in 0.5 M steps, was recorded in a 1-mm cuvette at 20°C. Separate samples were prepared for each urea concentration, and the ellipticity at 222 nm value of each sample was recorded.
Grip Strength and Rotarod.
Grip strength relative to body weight and rotarod measurements were performed in order to assess muscle strength and motor coordination. For grip strength, the Chatillon Force Measurement DFEII (Columbus Instruments) system was used. The system is composed of a steel pull bar assembly with a surface area of 100 mm × 60 mm, which is attached to a precision force gauge. We measured grip strength from the forelimbs (front limbs) as well as the forelimbs and hindlimbs (all limbs) together. The test was performed 7 times for both the forelimbs and all limbs with resting in between. The highest and lowest values were removed, and then, the remaining forces were averaged. Motor coordination was also tested utilizing the Rota Rod Rotamex 5 (Columbus Instruments). Mice were placed on a rotarod spindle; once the system software was started, the spindles gradually increased in acceleration by increments of 10 rpm. An optical sensor that is mounted over the rotarod assembly is utilized to determine when a mouse has fallen from the spindle, upon which the system logged the information for current speed (rpm) and total time running (s). The mice were given 15 min to rest in between runs, and a total of 3 runs were completed. Both grip strength and rotarod measurements were taken prior to mouse collection at PD20-22. Due to the sensitivity of these mice and age of tissue collection, no time was given for the mice to equilibrate to the experiment.
Further information regarding Plasmid Construction and Site-Directed Mutagenesis, Protein Expression and Purification, MDSs, Limited Proteolysis, AAV Generation, Isolation and Transfection of Neonatal Cardiomyocytes and Mouse Skeletal Muscle Tissue, Immunoblotting, Electron Microscopy, MHC Gels, Animal Work, and Statistical Analysis can be found in SI Appendix, SI Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Dr. Miensheng Chu for AAV generation along with Drs. Stefanie Novak and Christopher Pappas for their scientific feedback and helpful input. We would also like to thank Dr. Eric Olson, University of Texas Southwestern Medical Center, for providing us with the Lmod3-KO mice. We thank Dr. Joshua Strom for aiding with the grip strength and rotarod data collection, Yaeren Hernandez, MS, for providing the skeletal muscle dissection protocol, and Dr. Paula Tonino for help with TEM. This work was funded by the NIH grants F31AR076209 and T32 HL007249-40 (L.E.S.), R01HL12307801 (C.C.G.), R01GM120137 (A.S.K.), T32 GM0008336 (G.E.S.) and AHA grants GR00007474 (G.E.S.),19POST34450023 (M.C.), 835980 (T.M.L.), as well as the Czarina M. & Humberto S. Lopez Endowed Chair for Excellence in Cardiovascular Research (C.C.G.) and Finley and Florence Brown Endowed University of Arizona Sarver Heart Research Grant (L.E.S.). Portions of this work were developed from the doctoral dissertation of L.E.S.
Author contributions
A.S.K. and C.C.G. funding acquisition and conceptualization; L.E.S., M.C., G.E.S., R.M.M., and T.M.L. experiments were performed; and L.E.S., M.C., G.E.S., T.M.L., A.S.K., and C.C.G. writing and editing of manuscript.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. P.W.G. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Henderson C. A., Gomez C. G., Novak S. M., Mi-Mi L., Gregorio C. C., Overview of the muscle cytoskeleton. Compr. Physiol. 7, 891–944 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prill K., Dawson J. F., Assembly and maintenance of sarcomere thin filaments and associated diseases. Int. J. Mol. Sci. 21, 542 (2020), 10.3390/ijms21020542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang M. L., et al. , Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 173, 905–916 (2006), 10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ottenheijm C. A. C., et al. , Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum. Mol. Genet. 18, 2359–2369 (2009), 10.1093/hmg/ddp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pappas C. T., et al. , Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc. Natl. Acad. Sci. U.S.A. 112, 13573–13578 (2015), 10.1073/pnas.1508273112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter J. M. D., et al. , Mutation-specific effects on thin filament length in thin filament myopathy. Ann. Neurol. 79, 959–969 (2016), 10.1002/ana.24654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahrens-Nicklas R. C., et al. , Disruption of cardiac thin filament assembly arising from a mutation in LMOD2: A novel mechanism of neonatal dilated cardiomyopathy. Sci. Adv. 5, eaax2066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mi-Mi L., et al. , In vivo elongation of thin filaments results in heart failure. PLoS One 15, e0226138 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colpan M., Moroz N. A., Kostyukova A. S., Tropomodulins and tropomyosins: Working as a team. J. Muscle Res. Cell Motil. 34, 247–260 (2013), 10.1007/s10974-013-9349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fowler V. M., Dominguez R., Tropomodulins and leiomodins: Actin pointed end caps and nucleators in muscles. Biophys. J. 112, 1742–1760 (2017), 10.1016/j.bpj.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tolkatchev D., Gregorio C. C., Kostyukova A. S., The role of leiomodin in actin dynamics: A new road or a secret gate. FEBS J. 289, 6119–6131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J., Jalview Version 2–A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber A., Pennise C. R., Babcock G. G., Fowler V. M., Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol. 127, 1627–1635 (1994), 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregorio C. C., Weber A., Bondad M., Pennise C. R., Fowler V. M., Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature 377, 83–86 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Chereau D., et al. , Leiomodin is an actin filament nucleator in muscle cells. Science 320, 239–243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukada T., et al. , Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J. Cell Sci. 123, 3136–3145 (2010), 10.1242/jcs.071837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pappas C. T., Farman G. P., Mayfield R. M., Konhilas J. P., Gregorio C. C., Cardiac-specific knockout of Lmod2 results in a severe reduction in myofilament force production and rapid cardiac failure. J. Mol. Cell Cardiol. 122, 88–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz-Six K. L., et al. , Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. J. Cell Biol. 163, 1033–1044 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cenik B. K., et al. , Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Invest. 125, 1569–1578 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian L., et al. , Leiomodin-3-deficient mice display nemaline myopathy with fast-myofiber atrophy. DMM Dis. Model. Mech. 8, 635–641 (2015), 10.1242/dmm.019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halim D., et al. , Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. U.S.A. 114, E2739–E2747 (2017). 10.1073/pnas.1620507114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenway S. C., et al. , Early death of 2 siblings related to mutations in LMOD2, a recently discovered cause of neonatal dilated cardiomyopathy. CJC Open. 3, 1300–1302 (2021), 10.1016/j.cjco.2021.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuen M., et al. , Neonatal-lethal dilated cardiomyopathy due to a homozygous LMOD2 donor splice-site variant. Eur. J. Hum. Genet. 30, 450–457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuen M., et al. , Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Invest. 124, 4693–4708 (2014), 10.1172/JCI75199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michael E., et al. , Long-term follow-up and characteristic pathological findings in severe nemaline myopathy due to LMOD3 mutations. Neuromuscul. Disord. 29, 108–113 (2019), 10.1016/j.nmd.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Berkenstadt M., et al. , LMOD3-associated nemaline myopathy: Prenatal ultrasonographic, pathologic, and molecular findings. J. Ultrasound. Med. 37, 1827–1833 (2018), 10.1002/jum.14520. [DOI] [PubMed] [Google Scholar]
- 27.Abbott M., et al. , Neonatal fractures as a presenting feature of LMOD3-associated congenital myopathy. Am. J. Med. Genet. Part A. 173, 2789–2794 (2017), 10.1002/ajmg.a.38383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schatz U. A., et al. , Evidence of mild founder LMOD3 mutations causing nemaline myopathy 10 in Germany and Austria. Neurology 91, e1690–e1694 (2018), 10.1212/WNL.0000000000006428. [DOI] [PubMed] [Google Scholar]
- 29.Pettersen E. F., et al. , UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004), 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 30.Lee T. S., et al. , GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J. Chem. Inf. Model. 58, 2043–2050 (2018), 10.1021/acs.jcim.8b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier J. A., et al. , ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015), 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kostyukova A., Maeda K., Yamauchi E., Krieger I., Maéda Y., Domain structure of tropomodulin: Distinct properties of the N-terminal and C-terminal halves. Eur. J. Biochem. 267, 6470–6475 (2000), 10.1046/j.1432-1327.2000.01738.x. [DOI] [PubMed] [Google Scholar]
- 33.Kostyukova A. S., Tiktopulo E. I., Maéda Y., Folding properties of functional domains of tropomodulin. Biophys. J. 81, 345–351 (2001), 10.1016/S0006-3495(01)75704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillaud L., et al. , Role of tropomodulin’s leucine rich repeat domain in the formation of neurite-like processes. Biochemistry 53, 2689–2700 (2014), 10.1021/bi401431k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breddam K., Meldal M., Substrate preferences of glutamic-acid-specific endopeptidases. Eur. J. Biochem. 206, 103–107 (1992). [DOI] [PubMed] [Google Scholar]
- 36.Tolkatchev D., et al. , Leiomodin creates a leaky cap at the pointed end of actin-thin filaments. PLoS Biol. 18, e3000848 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skwarek-Maruszewska A., et al. , Different localizations and cellular behaviors of leiomodin and tropomodulin in mature cardiomyocyte sarcomeres. Mol. Biol. Cell. 21, 3352–3361 (2010), 10.1091/mbc.E10-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsukada T., et al. , Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. J. Biol. Chem. 286, 2194–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moroz N. A., et al. , Alteration of tropomyosin-binding properties of tropomodulin-1 affects its capping ability and localization in skeletal myocytes. J. Biol. Chem. 288, 4899–4907 (2013), 10.1074/jbc.M112.434522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talbot J., Maves L., Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 5, 518–534 (2016). 10.1002/wdev.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiss B., et al. , Nebulin and Lmod2 are critical for specifying thin-filament length in skeletal muscle. Sci. Adv. 6, eabc1992 (2020), 10.1126/sciadv.abc1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y., et al. , Compound heterozygosity for novel truncating variants in the LMOD3 gene as the cause of polyhydramnios in two successive fetuses. Front. Genet. 10, 835 (2019), 10.3389/fgene.2019.00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Littlefield R., Fowler V. M., Measurement of thin filament lengths by distributed deconvolution analysis of fluorescence images. Biophys. J. 82, 2548–2564 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gokhin D. S., Fowler V. M., Software-based measurement of thin filament lengths: An open-source GUI for Distributed Deconvolution analysis of fluorescence images. J. Microsc. 265, 11–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information.