

Abstract
Background:
Viral infections, especially those caused by rhinovirus, are the most common cause of asthma exacerbations. Previous studies have argued that impaired innate anti-viral immunity and, as a consequence, more severe infections contribute to these exacerbations.
Objective:
These studies explored the innate immune response in the upper airway of allergic rhinitis and asthmatic volunteers in comparison to healthy controls and interrogated how these differences corresponded to severity of infection.
Methods:
Allergic rhinitic, asthmatic, and healthy volunteers were inoculated with rhinovirus A16 and monitored for clinical symptoms. Tissue and nasal wash samples were evaluated for anti-viral signature and viral load.
Results:
Both allergic rhinitic and asthmatic subjects demonstrated more severe cold symptoms. Asthmatic subjects demonstrated worsened asthma control and increased bronchial hyperreactivity in the setting of higher exhaled breath FeNO and blood eosinophils. These studies confirmed reduced expression of interferons and virus-specific pattern recognition receptors in both atopic cohorts. However, despite this defect in innate immunity, allergic rhinitis/asthmatic volunteers demonstrated reduced rhinovirus concentrations in comparison to controls.
Conclusion:
These results confirm that the presence of an allergic inflammatory disorder of the airway is associated with reduced innate immune responsive to RV infection. Despite this, these allergic volunteers demonstrate reduced viral loads, arguing for the presence of a compensatory mechanism to clear the infection.
Keywords: rhinovirus, asthma, innate immunity, eosinophil, interferon, pathogen-associated molecular patterns
Introduction
Viral infections, especially those caused by rhinovirus (RV), are the most common cause of asthma exacerbations in children and can also cause exacerbations in adults.1–5 The immune mechanisms underlying this pathogenic response to RV largely remain an enigma. One proposed explanation recognizes an impaired innate anti-viral immune response in asthmatics. Healthy airway epithelial cells (EpCs) effectively recognize and respond to RV through various pattern recognition receptors (PRRs) including the Toll-like receptors (TLRs), specifically TLR2, TLR3, TLR7, and TLR8, the RIG-I-like receptors (RLR) retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), and also through engagement of the inflammasome.6–11 Inflammatory mediators released in response to these PRRs include the type I interferons (IFN) IFN-α/-ß, type III IFNs (IFN-λ), as well as numerous cytokines (including interleukin (IL)-1, IL-6, IL-18, and many others). Together these pathways orchestrate an anti-viral response within the EpC that is sufficient to eradicate the virus.12,13 An impairment of IFN production by EpCs has been proposed as a mechanism that would lead to enhanced RV replication, with the consequent heightened inflammation driving the asthma exacerbation. This impairment of IFN production by EpCs has been established in numerous studies.14–20 The current studies were designed to concomitantly explore the innate anti-viral immune response engaged by nasal EpCs and severity of subsequent viral infections. We focused on the nasal response as the nasopharynx is the primary sight of the RV infection and the practicality of having greater accessibility of tissue.21
Methods
Subjects.
To address the role of allergic (type 2high) inflammation in modulating the anti-viral immune response in the airway we enrolled healthy controls (n=11) and allergic subjects, including those with allergic rhinitis (AR; n=17) and with concomitant asthma (n=17; not all studies were done on all subjects.) AR and asthma subjects reported upper respiratory symptoms with exposure-relevant positive prick skin tests (sensitization to aeroallergens to which they were currently being exposed) and were studied when symptomatic. Use of oral or nasal antihistamines, intranasal corticosteroids (CCS), or leukotriene modifiers was not permitted. Healthy controls consisted of non-asthmatic, non-allergic subjects with negative prick skin tests to aeroallergens. Subject characteristics are summarized in Table 1. All subjects gave written informed consent under protocols approved by the University of Virginia Institutional Review Board (#19512 and #19517).
Table 1.
Subject Characteristics at Baseline
| Cohort | n | Age | Gender (%F) | Skin tests (# positive) | Absolute eosinophil count (cells/μL) | FEV1 (% predicted) | FEV1/FVC | FeNO (ppb) | Methacholine PC20 |
|---|---|---|---|---|---|---|---|---|---|
| Healthy Control | 11 | 22.8±0.4 | 54.5 | -- | 90±10 | 99.2±4.5 | .86±.01 | 12.9±2.0 | >25 |
| Allergic Rhinitis | 17 | 21.5±0.7 | 58.8 | 4.8±0.5 | 150±20 | 98.9±2.2 | .83±.01 | 24.1±3.3 | >25 |
| Asthma | 17 | 21.7±0.5 | 52.9 | 4.3±1.1 | 250±140 | 88.3±5.1 | .81±.03 | 53.8±18.0 | 1.89±0.7 |
Abbreviations: FeNO – fractional exhaled breath nitric oxide; FEV1 – forced expiratory volume, 1 second; FVC – forced vital capacity; PC – provocative concentration
Experimental infections with RV-A16.
Each subject was inoculated with 300 TCID50 of RV-A16 (IND# 15162). Nasal wash and nasal scraping samples were collected from each subject on days 0, 1, 2, 3, 4, and 7 post inoculation (dpi) as previously described.22,23 Nasal biopsies were collected on day 4 from the inferior and middle turbinates and the nasopharynx.
Clinical response to RV infection.
Upper respiratory tract symptom scores were evaluated as previously described.24 Briefly, this scoring system includes sneezing, nasal discharge, nasal obstruction, sore throat, headache, malaise, and chills with each scored on a severity scale of 0 to 3 (0 = symptoms not present; 1 = mild, but clearly present; 2 = moderate and uncomfortable; and 3 = severe, interfering with sleep or activity). Spirometry and fractional exhaled breath nitric oxide (FeNO) were measured at each visit. Methacholine reactivity and absolute eosinophil counts (AECs) were assessed on dpi 0 and 4.
Enzyme immunoassays.
EDN concentrations in nasal wash were quantified by enzyme immunoassay (EIA) according to manufacturer’s instructions (MBL, Nagoya, Japan). The lower limit of detection of the assay was 620 pg/ml.
Immunofluorescence staining.
Nasal biopsy samples were fixed in 4% paraformaldehyde, paraffin embedded and sectioned by the Histology Core Laboratory of the University of Virginia. For analyses, samples were deparaffinized in xylene and graded ethanol and then rehydrated in distilled water. Heat-induced antigen retrieval was performed by heating sections for 20 min in citrate buffer (Abcam; Cambridge, MA). Slides were blocked using 1% bovine serum albumin, 10% goat serum (Sigma, St. Louis, MO), 0.1% triton X-100 and 1 μg/ml Fc Block (BD Pharmingen; Sparks, MD) for 1.5 hrs, and then incubated with primary antibody for 16 hrs at 4°C (TLR3 (Novusbio, 40C1285.6, 1:100); RIG-I (Abcam, ab45428, 1:200); MDA5 (Abcam, ab79055, 1:200); human rhinovirus monoclonal antibody (QED Bioscience, 18758, 1:100); IFN-α (Proteintech, 18013–1-AP, 1:100); IFN-ß (Abcam, ab140211, 1:100); or appropriate isotype controls antibodies). Anti-major basic protein (MBP) (1:50) and anti-EDN (1:50) antibodies were gifts from Dr. Hirohito Kita. Sections were rinsed and then incubated with secondary allophycocyanin (APC) goat anti-rabbit/mouse IgG (1:200, Life Technologies) or Alexa Fluor 488 goat anti-rabbit IgG (1:200, Thermofisher) for 1 hour at room temperature. Nuclei were stained with 100 ng/ml DAPI (4’, 6-diamidino-2-phenylindole, Sigma) for 30 min at room temperature. Samples were washed and aqueous mounted with VectaMount AQ (Vector Laboratories; Burlington, CA).
Histological scoring.
The samples were analyzed using an EVOS FL Auto microscope (Life Technologies). EDN and MBP were quantified as positive area fraction (%) by ImageJ in multiple random high-power fields. IFN-α, IFN-ß, RIG-1, MDA5, TLR3 positive grade and RV positive cell numbers were scored in a blinded fashion based on multiple random high-power fields per slide. Grading was limited in the epithelial cells and scored as 0=no staining, 1=occasional positive staining/mild, 2=cluster of positive staining/moderate, 3=large area of positive staining/extensive; and 4=staining in all cells.
Quantitative real-time polymerase chain reaction (qPCR).
qPCR was performed on RNA extracted from nasal scrapes. Total RNA was extracted using TRI® reagent (Sigma, St. Louis, MO). Conversion of mRNA to cDNA was performed using a Taqman Reverse Transcription kit (Roche, Branchburg, NJ). Total RNA (200 ng) was added to each reaction along with oligo dT primers, 5.5 mM MgCl2, 2 mM dNTPs, RNase inhibitor, and reverse transcriptase. Reactions went through 10 min at 25° C, 30 min at 48° C and 5 min at 95° C in a Bio-Rad iCycler thermocycler (Bio-Rad). The PCR mix consisted of Sensimix (Bioline; Taunton, MA), cDNA and 400 nM of each primer. qPCR was performed for IFN-α, IFN-ß, IFN-λ, RIG-I, MDA5, and TLR3 using commercial primer pairs (Integrated DNA Technologies, Inc., Coralville, IA). Data were analyzed as the difference in CT (ΔCT) of each transcript from the CT ß-actin (ΔCT).
Proteomics.
EIAs performed on nasal lavage fluid for IFN-α, -ß, and -λ were all below the threshold of sensitivity of these assays, reflecting the extensive dilution. To address expression of other cytokines we utilized a commercial proximity extension assay (PEA)25 which included ability to assess cytokines associated with inflammasome activation (IL-1 and IL-18). The PEA is an affinity-based assay which semi-quantitatively characterizes protein abundance levels. For each measured protein, a pair of oligonucleotide-labeled antibody probes target the protein and if both probes are in close proximity, a PCR target sequence is formed by a proximity-dependent DNA polymerization event. The resulting sequence is then detected and quantified using standard real-time PCR.
RV viral titer determination.
RNA was extracted from nasal wash fluid using RNeasy RNA isolation kits (Qiagen, Crawley, United Kingdom) and cDNA was generated as previously described.26 The cDNA was amplified and detected via qPCR utilizing primers specific for conserved regions of RV and detected via qPCR (RV forward 5’- CCTCCGGCCCCTGAAT-3’; RV reverse 5’- AAACACGGACACCCAAAGTAGT-3’; Integrated DNA Technologies). The qPCR mix consisted of 2X SYBR green Supermix (BioRad), cDNA, and 400 nM of each primer. Nasal wash samples from the experimental RV-A16 challenge were evaluated in duplicate and the values compared to a standard curve to determine the copies of viral RNA/μL of nasal wash supernatant. The neat concentration of the RV-A16 pool used to generate the standard curve in this analysis contained 5×107 copies of viral RNA/μL.
Statistical analyses.
For comparisons between the cohorts and with baseline (dpi0) samples, data were compared using ANOVA or t-tests with Bonferroni correction for multiple comparisons. A p-value of <0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism 6 (La Jolla, CA).
Results
Clinical response to RV-A16 inoculation.
RV-A16 inoculation produced symptomatic infections in all volunteers. Allergic inflammation (AR and allergic asthma) was associated with both more severe and persistent symptoms (figure 1A; p<0.05 for days 2 and 3). Asthmatics demonstrated significant worsening of FEV1 (87% predicted at dpi0 to 76% at dpi4) and of FEV1/FVC ratio (.81 to .68; p<0.05; figure 1B). In addition, asthmatics had an approximately 2 log2 increase in bronchial hyperreactivity (PC20 1.89 to .40 mg/ml).
Figure 1:
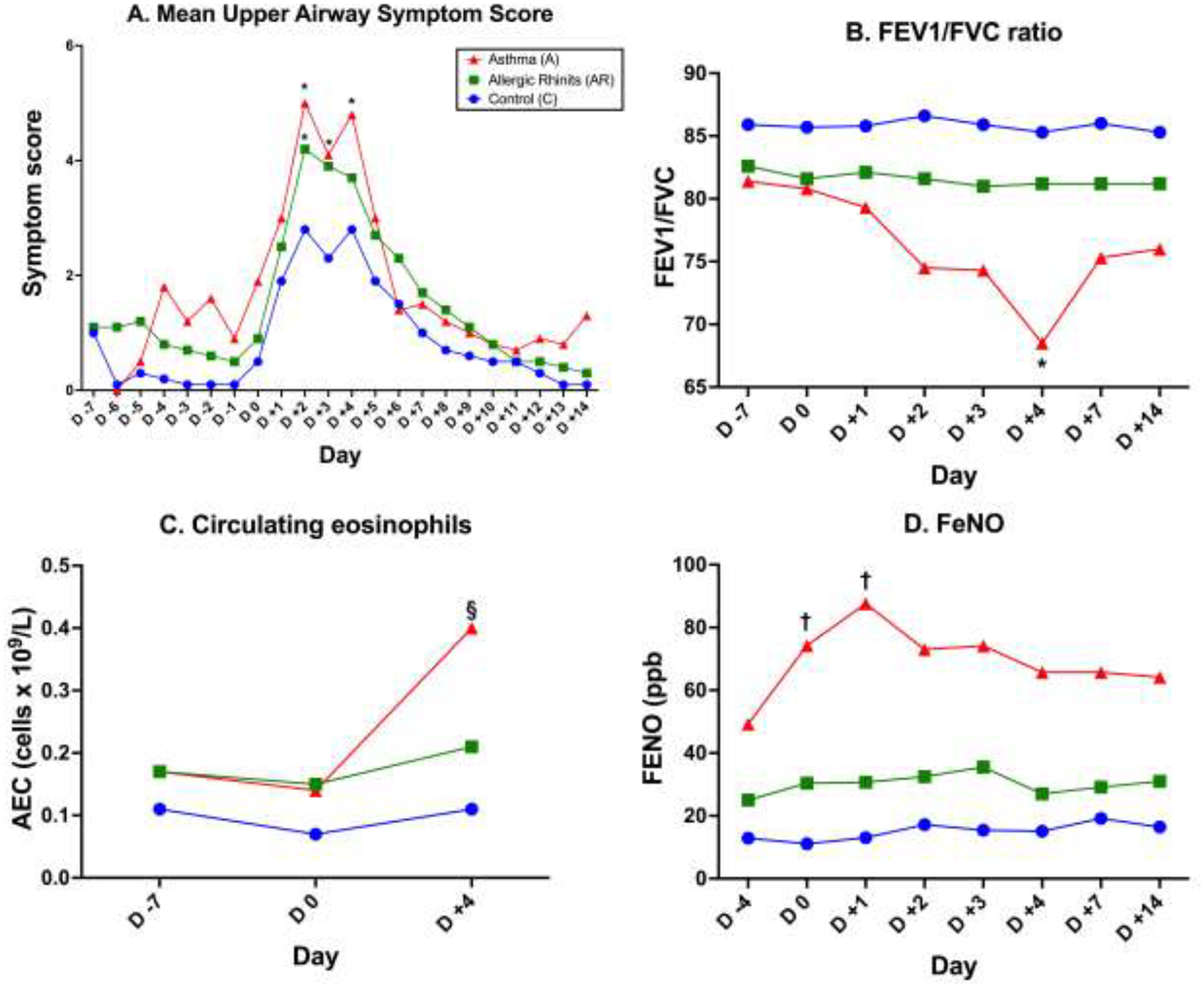
Clinical response to RV infection. 1A. Upper respiratory symptoms in control (C), AR, and asthmatics (A) post-viral inoculation. 1B. FEV1/FVC ratio. 1C. Circulating absolute eosinophil count. 1D. FeNO. *p<0.05 compared to control subjects; §p<0.01 compared to day 0; †p<0.05 compared to day 0.
Tissue eosinophilic inflammation post-infection.
We next investigated several parameters of eosinophilic inflammation as a correlate to the clinical impact observed in response to infection. Asthmatic (but not AR) subjects demonstrated a significant increase in AEC at day 4 (250 to 400/μL); figure 1C; p<0.01). Indirect evidence of increased type 2 inflammation in the lower airways of asthmatics was supported by significant increases in FeNO (figure 1D). Intact eosinophils were only rarely observed in nasal wash fluid or tissue samples. The presence of an eosinophilic inflammatory response was instead quantified in the day 4 biopsy samples by performing IF staining for EDN and MBP. Because of limitations in tissue sample availability, these tissue and proteomic analyses (below) were performed as a comparison between healthy controls and subjects with a type 2high upper airway inflammatory state (pooled samples from AR and allergic asthmatics). Figure 2A displays representative data from a healthy control and allergic subject. Negligible staining was observed with isotype control antibodies (on line supplement, eFigure 1). Blinded semiquantitative scoring for IF was performed as described. Little or no staining for either peptide was observed in the control specimens whereas elevated staining for both was observed in the allergic cohort (p<0.01 for EDN; figure 2B–C). Under the study protocol, we were not permitted to obtain baseline IF data regarding expression of EDN or MBP prior to inoculation, so it cannot be categorically argued that the IF data reflected de novo RV-induced expression of these peptides. However, induction of eosinophilic inflammation by RV was demonstrated in the allergic volunteers as demonstrated by the higher EDN expression in the allergic (AR and asthma) cohort (p<0.01) with significantly higher release of EDN into the nasal wash fluid observed at day 4 (p<0.05; figure 2D).
Figure 2:
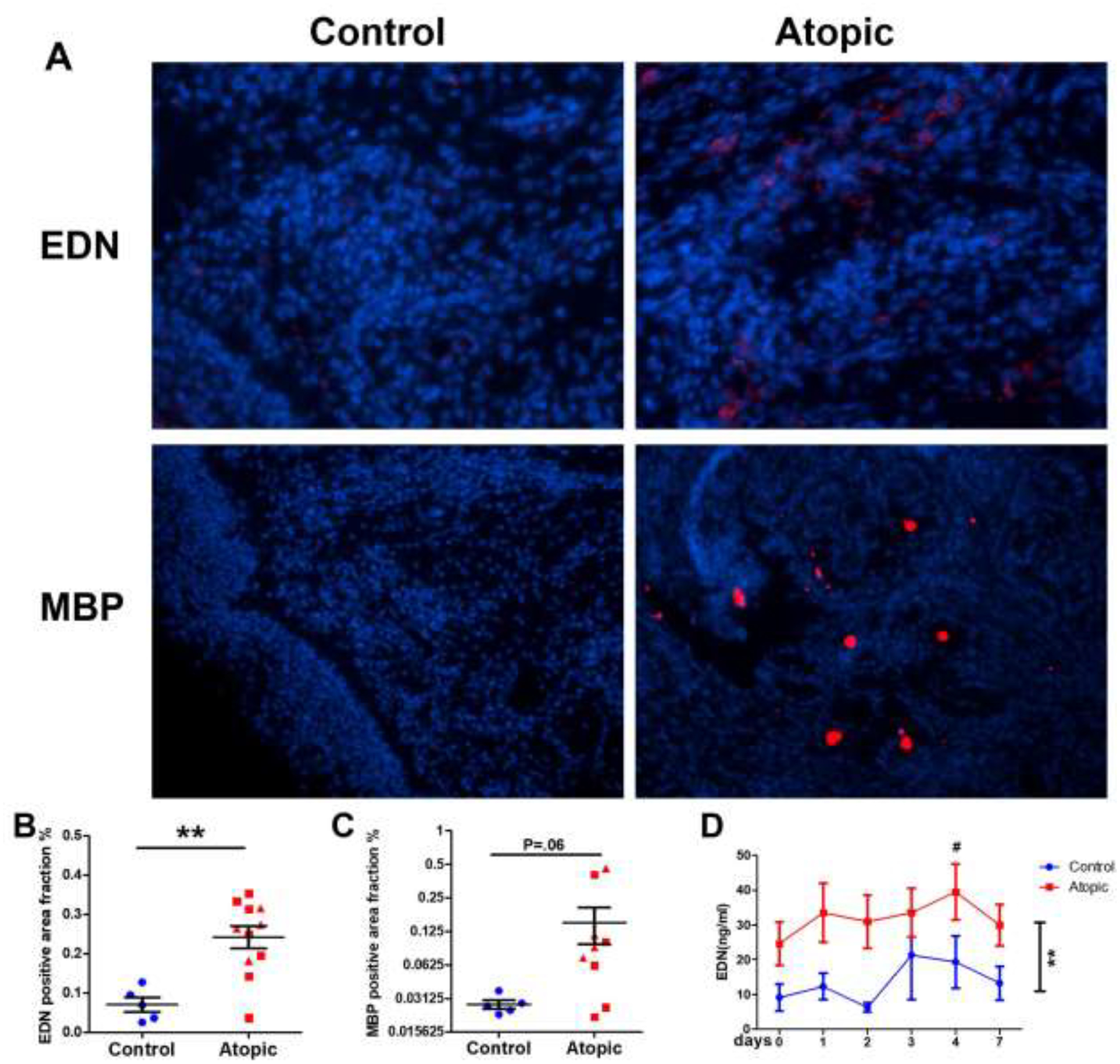
Immunofluorescence staining of eosinophil-derived EDN and MBP at dpi 4. 2A. Representative staining. 2B: Summary of expression of EDN. **p<0.01. 2C: Summary of expression of MBP. 2D: Nasal wash EDN concentrations as determined by EIA. **p<0.01 atopic compared to controls. #p<0.05 atopic subjects day 4 compared to day 0.
mRNA transcript expression of RV-detecting and responding molecules.
To explore the innate immune anti-viral signature in healthy controls and subjects with allergic inflammation of their upper airways, initially, we investigated the innate immune response of nasal epithelium at varying time points after inoculation. We examined innate molecules responsible for RV recognition (toll-like receptors for viral RNA and the RIG-I-like receptors RIG-I and MDA5) as well as expression of type I and III interferons (IFN-α, -ß, and -λ). RNA was extracted from nasal scrapings of infected individuals prior to inoculation (dpi 0) and post-infection on dpi 1–4 and 7 and gene expression determined by qPCR. Over the course of the infection expression of both type I (IFN-α/IFN-ß) and type III (IFN-λ) interferons was significantly higher in the control compared to the atopic subjects (p<0.01 for type I and <0.001 for type III IFNs). Expression of IFN-α and IFN-ß were similar at baseline whereas IFN-λ was significantly decreased in the allergic subjects (figure 3). After inoculation there was no change in expression of any of these IFN transcripts in the type 2high subjects. In contrast, statistically significant increases in transcript expression for IFN-α and IFN-ß were observed at dpi 2 and dpi 1, respectively in the healthy controls (p<0.05). Transcript expression for the TLRs and RLRs were similar in all 3 cohorts at baseline and no significant change in expression was observed with infection (data for TLR3, RIG-I and MDA5 are displayed in figure 3).
Figure 3.
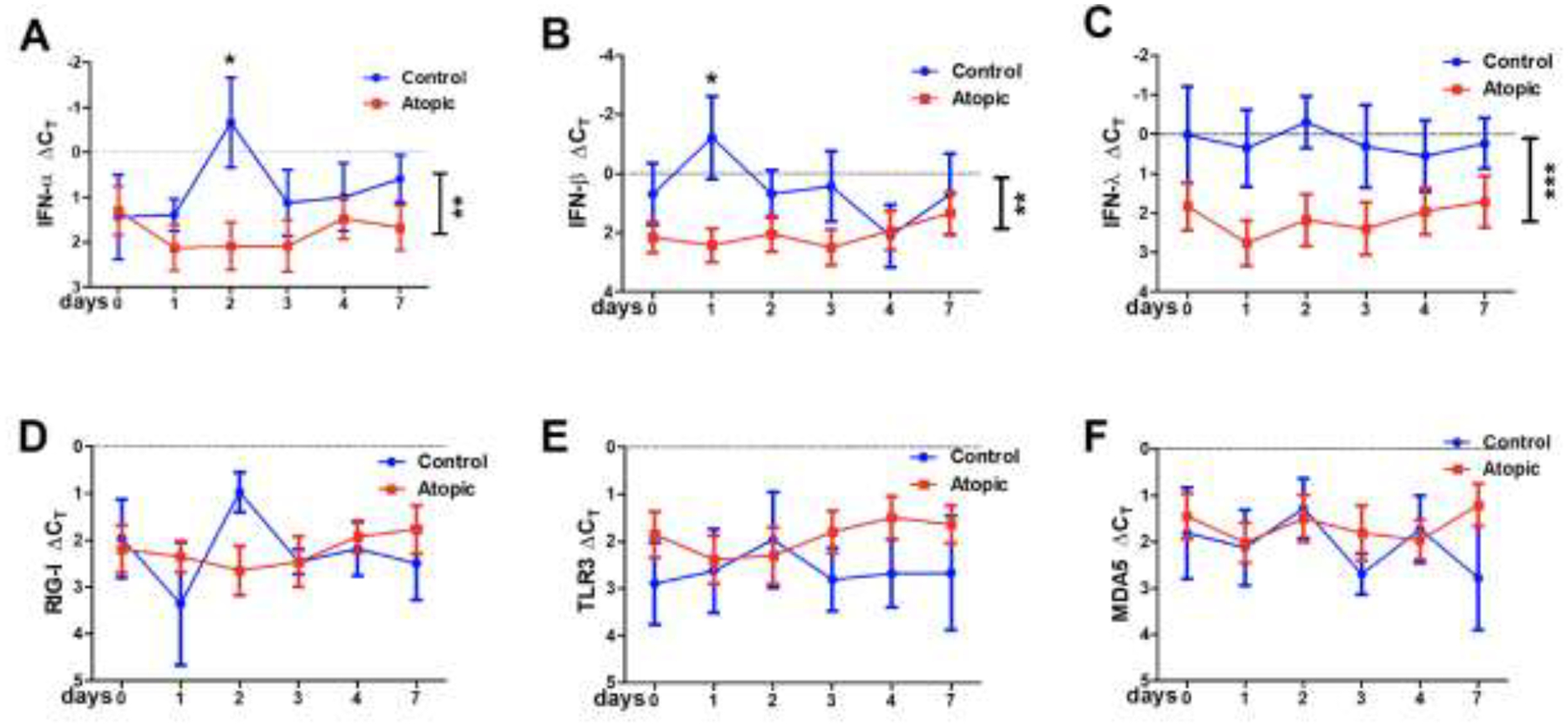
qPCR for RV sensing and responding molecules: 3A – IFN-α, 3B – IFN-ß, 3C – IFN-λ, 3D – RIG-I, 3E – TLR3, 3F – MDA5. Vertical bars on the right of each graph indicate significant difference between the two groups. and the * above, indicate the specific day these differences were significant. *p<0.05, **p<0.01, ***p<0.001.
Protein expression of RV-detecting and -responding molecules.
Enzyme immunoassays for IFN-α, -ß, and -λ were performed on nasal lavage fluid but were below the detection limit in all samples collected, presumably reflecting excessive sample dilution. We performed ultrasensitive proximity extension assays (PEA) to detect cytokines released into the nasal lavage fluid in response to inflammasome activation, specifically IL-1 and IL-18. While these proteins were both detectable via this methodology, no differences were observed between the control and allergic subjects and only a slight insignificant (1–2 log2 (~2–4-fold)) increase in IL-18 was observed in response to the infection (online supplement, eFigure 2).
Immunofluorescence.
Given the inadequacies of lavage fluid assays, protein expression was subsequently analyzed via immunofluorescence (IF) of nasal biopsies obtained at day 4. We performed IF staining to quantify expression of IFN-α, IFN-ß, TLR3, RIG-I, and MDA5. Representative IF data from a healthy control and allergic subject are displayed in figure 4A (with isotype control antibodies displayed in eFigure 1). Compared to controls, significantly reduced expression of IFN-α and IFN-ß (p<0.05) was observed in the type 2high (allergic) cohort (figure 4B) with smaller insignificant differences observed for TLR3, RIG-I, and MDA5 (p=0.09 for TLR3).
Figure 4:
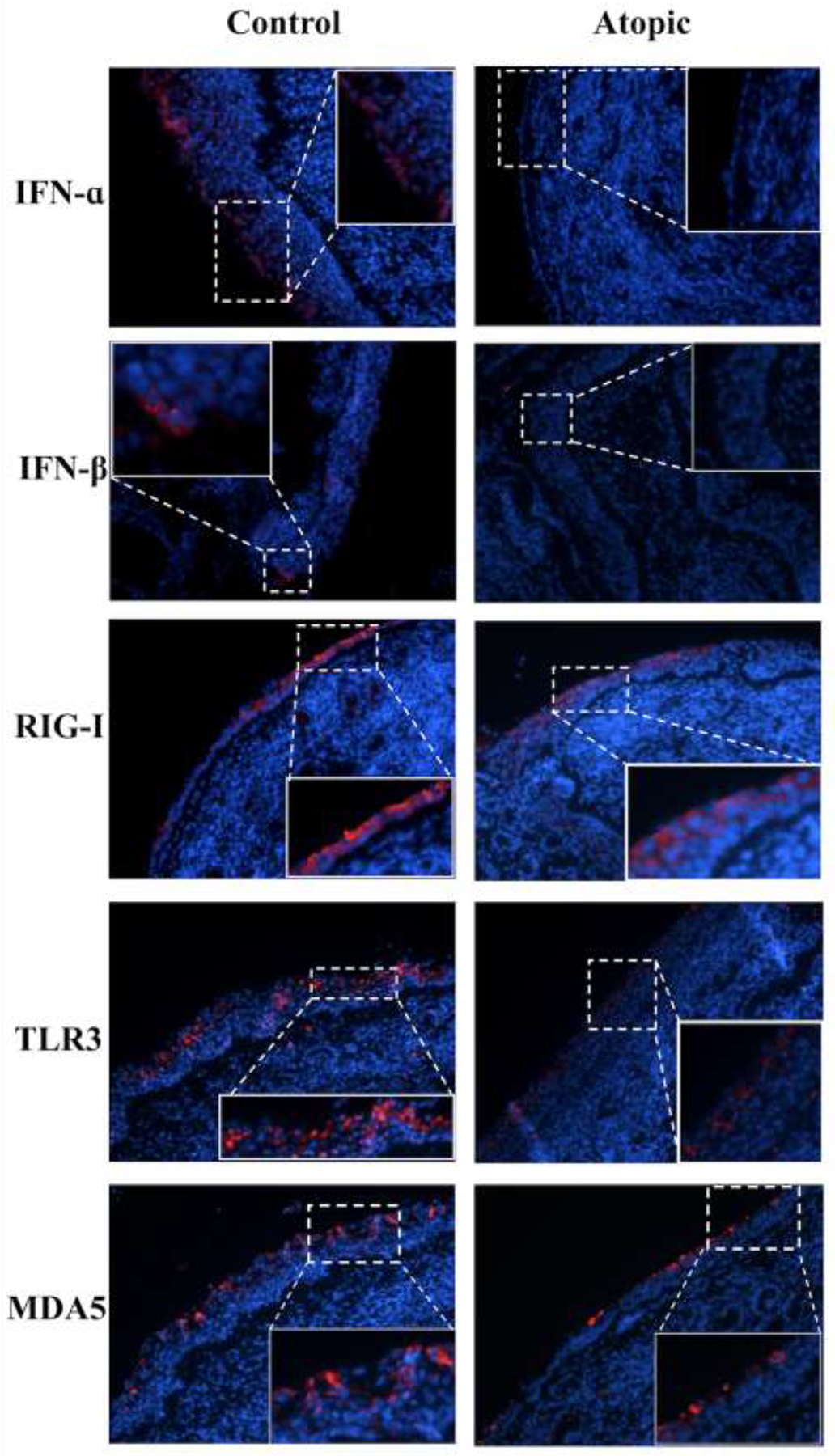
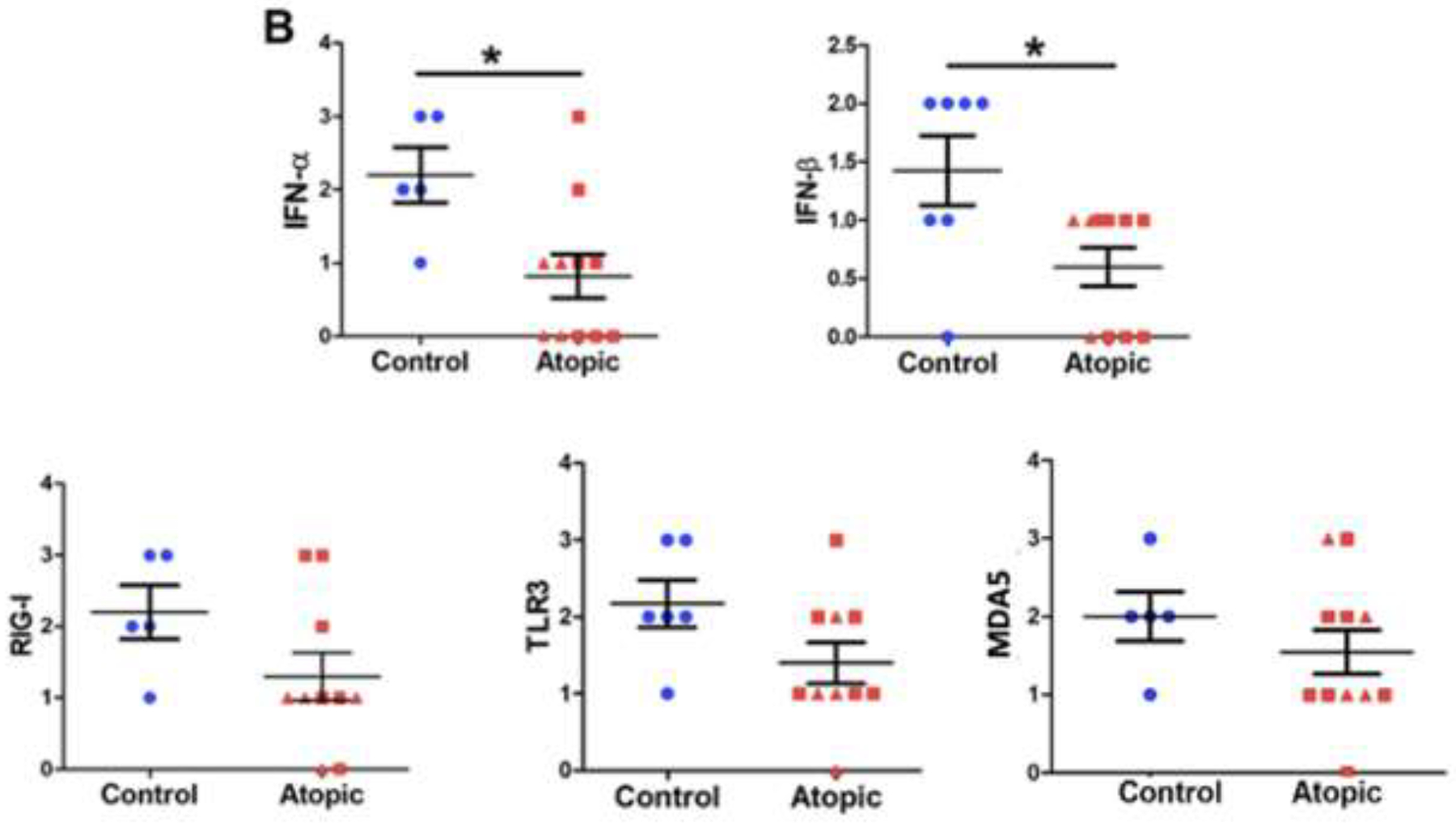
Immunofluorescent staining of RV-sensing and anti-viral responding molecules. 4A. Representative immunofluorescent staining. Images are shown at 200x with digital enlargement of key areas (dotted lines) to enhance visibility of details (inset, solid lines). 4B. Expression of RV-sensing and anti-viral responding molecules via IF. Statistical analyses involved unpaired t-tests. *p<0.05.
RV titer post inoculation.
Having confirmed previous studies demonstrating defects in components of the innate immune system in atopic subjects,14–20 we queried whether this would impact RV titer. RNA was extracted from nasal wash collected during the infection and viral titer determined via qPCR. In addition, we quantified RV-infected cells by IF in the nasal biopsy samples. In the day 4 biopsy samples we were no longer able to identify infected epithelial cells (presumably reflecting the rapid clearance of infected EpCs via apoptosis and engulfment).27,28 However, in contrast to epithelium, positive IF was observed in the submucosal tissue, especially in the healthy controls (figure 5A). Positive IF for RV was significantly reduced in the atopic subjects (p<0.05; figure 5B). This lower expression of RV in the atopic subjects despite their reduced expression of type I and type III IFNs was confirmed when we quantified virions in the nasal lavage fluid (p<0.01; figure 5C).
Figure 5:
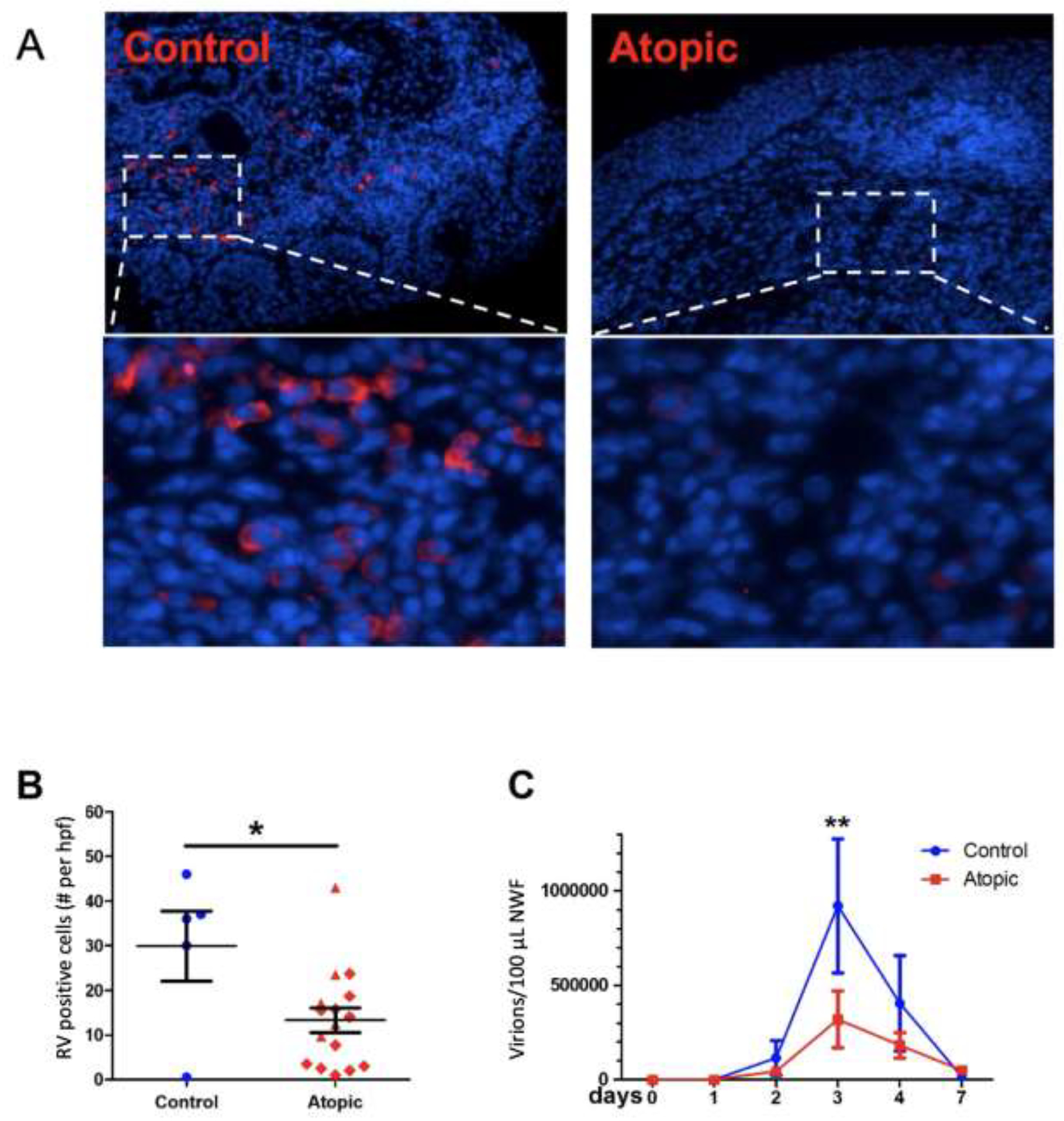
Rhinovirus expression post-inoculation. 5A. Representative RV immunofluorescence staining showing viral protein in submucosal (but not epithelial) tissue. 5B. Summary of IF of RV positive cells at dpi 4. *p<0.05. 5C. Viral load in nasal wash fluid. NWF – nasal wash fluid. **p<0.01.
Discussion
These studies further establish the experimental RV inoculation model as a robust methodology to explore the cellular and molecular mechanisms underlying the capability of this virus to induce asthma exacerbations. In contrast to previous studies dependent on a less potent RV (e.g., RV-A39),24,29 the current studies with RV-A16 demonstrated significant worsening of asthma as manifested as more severe respiratory symptoms, decreased lung function, and increases in FeNO and bronchial hyperreactivity (figure 1). In addition, in comparison to our more recent published studies using RV-A16,23 the current challenges were also more likely to have further induced worsening asthma as a result of the removal of volunteers from an aeroallergen-free environment and allowing volunteers to be exposed to relevant bystander allergens. This outcome is consistent with the concept that RV-induced exacerbations are mediated in large part by the tendency of the virus to potentiate an ongoing immune response to bystander allergens.3,23,29–33
In asthmatics, RV infection is associated with rapid expression of numerous cytokines/chemokines associated directly or indirectly with eosinophil activation and recruitment.34–37 It is well recognized that activation products of eosinophils damage airway epithelium, promote bronchospasm, and enhance bronchial hyperreactivity (reviewed in38). We therefore investigated the recruitment of eosinophils into the nasal airspace in a timeframe consistent with the clinical response to RV inoculation. Intact eosinophils were only rarely observed in nasal wash fluid or tissue samples. However, these studies demonstrated early expression of the eosinophil-derived peptide EDN in nasal lavage fluid (figure 2D) and biopsies obtained at day 4 post-inoculation confirm diffuse staining for both MBP and EDN in the AR and allergic asthmatic volunteers (figure 2B and 2C). It is intriguing to note that both this influx of eosinophilic mediators as well as the induction of blood eosinophilia (in asthmatics only; figure 1C), while within a timeframe consistent with the worsening of asthma symptoms, is well prior to the time required for an adaptive immune response to develop. Effector memory T cells will not be engaged until dpi 5–7 as suggested by our group’s previous publications.39,40 Mechanisms for epithelial recruitment of eosinophils include epithelial cell generation of innate lymphoid cell 2-activating cytokines and eosinophil-targeting chemokines.41,42 These data invite further investigations regarding the specific pathways initiated by RV-infected epithelium that drive the rapid induction of eosinophilic inflammation. This is of particular importance given the likely central relevance of these processes in driving the adverse clinical consequences.
Previous studies have provided evidence for decreased innate anti-viral immunity in the airways of asthmatics, suggesting an enhanced capacity for viral replication. This model argues that this would be associated with an increase in viral load after infection, thereby driving the asthma exacerbation. Despite this, our published studies quantifying RV in the nose showed that asthmatics displayed lower viral load in their nasal passages in comparison to either controls presenting to the ED with cold symptoms or after experimental viral inoculation.22,23 While RV does infect the lungs of asthmatics and infected bronchial epithelial cells can be identified,43 the primary target of RV infection is the nose.44 The reduction in innate immunity is thought to develop as a consequence of the type 2 inflammatory state present in asthma and – in the upper airway – also in allergic rhinitis.45,46 We therefore initially investigated whether the defect in innate anti-viral immunity extended to the nose, reflecting this “unified airway.” Our initial investigations confirmed a defect in the expression of type I and type III IFN immune responses in the upper airway of atopic volunteers (figures 3–4).14–20 However, despite this deficiency, we confirmed our earlier studies that the viral load observed in the nose of inoculated allergic volunteers was lower than in non-allergic healthy controls (figure 5).
These seemingly discordant findings signify that compensatory pathways are being engaged in the allergic subjects that must be responsible for eliminating the virus. One possibility is that it is the potentiated eosinophilic inflammation that provides this protection. Numerous studies demonstrate the capacity of eosinophils to eliminate respiratory RNA viruses including respiratory syncytial virus (RSV) and influenza. This reflects, at least in part, the potent RNase functionality of their cationic proteins,47–54 such as eosinophil-derived neurotoxin (EDN), that degrade the viral RNA.48,54 More recently, a favorable outcome to SARS-CoV2 infection was observed in asthmatics, but specifically only in those asthmatics with the highest eosinophil counts.55 An argument for the concept that eosinophils may target RV is supported by a study in which induction of eosinophilic inflammation led to less severe infections.56 And, more impressively, was the demonstration that depletion of eosinophils by administration of an anti-IL-5 biologic was associated with ~10-fold increased viral loads.57,58 The capacity of eosinophils to target rhinovirus has not been extensively explored. It should be noted, however, that in one conflicting study involving resting or fMLP-stimulated eosinophils, at least ex vivo, there was no reduction of viral load in an RV-infected epithelial cell line.59 And it is certainly plausible that a more robust response by, for example, NK cells or tissue memory cytotoxic lymphocytes, if present in the allergic volunteers, could provide alternative explanations for the decreased viral loads that were observed.
Together these findings suggest a specific pathogenic mechanism for RV-mediated asthma exacerbations (this model is displayed in figure 6). While induction of an eosinophil-mediated anti-viral response could compensate for reduced IFN-mediated anti-viral immunity, this compensation will occur at the cost of inducing the asthma exacerbation. This mechanism is supported by the paradox that whereas, eosinophil eradication via treatment with an anti-IL-5 monoclonal antibody, as noted, promoted viral replication, the concomitant eradication of the eosinophils prevented this from impacting symptoms.58 Indeed, the absence of worsened asthma despite much higher viral loads, points to the overarching significance of exacerbation of a bystander type 2 inflammatory state in driving RV-induced asthma exacerbations as opposed to any direct adverse impact from the RV infection itself. This concept is certainly consistent with the recognition that targeting type 2 inflammation with, in addition to anti-IL-5, either with anti-IL-4Rα or anti-IgE typically suffices to mitigate asthma exacerbations, many or most of which can be presumed to have been associated with natural RV infections.
Figure 6.
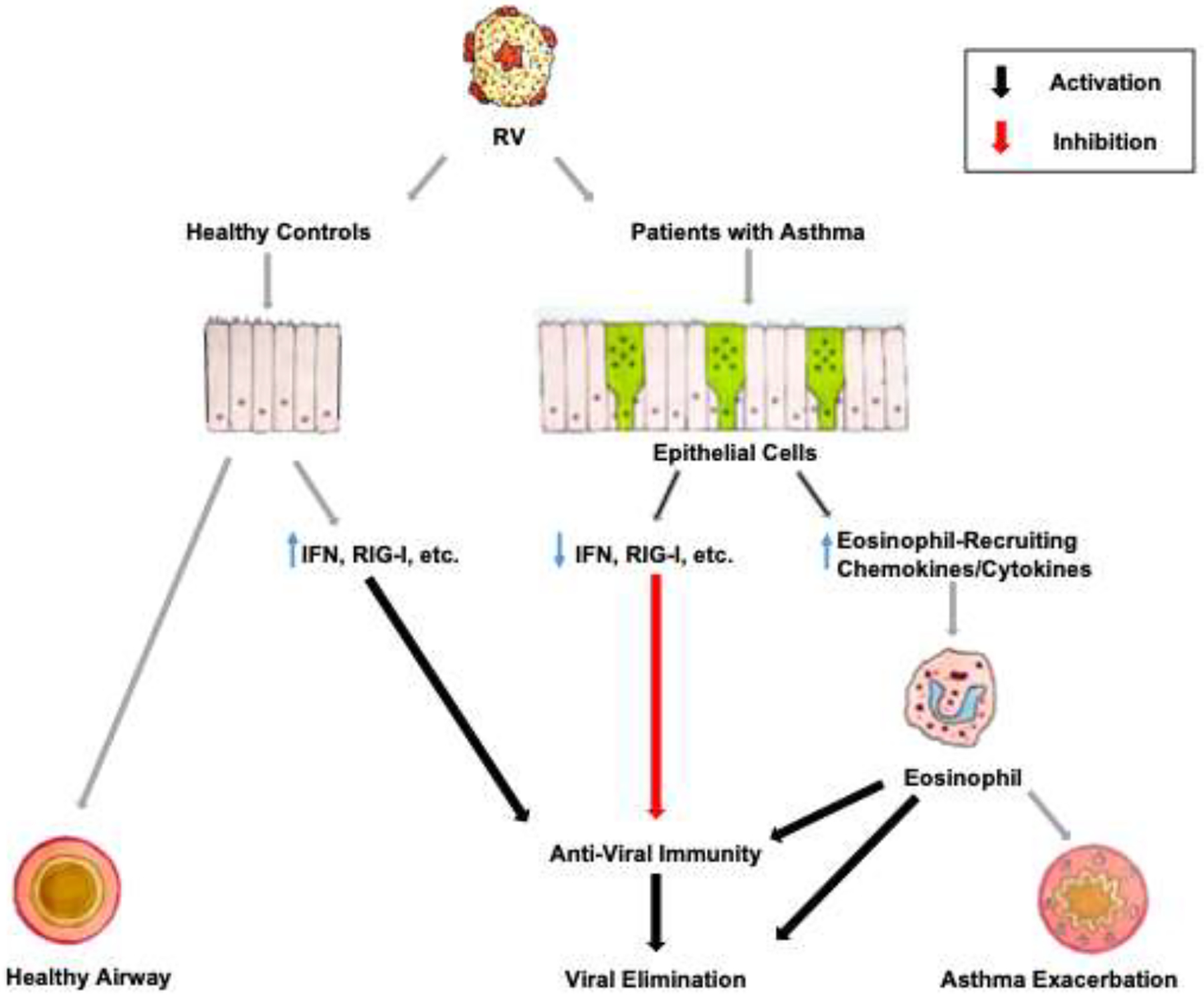
Model for the development of asthma exacerbations in response to RV infections. See text for details.
Supplementary Material
Acknowledgement
The authors gratefully acknowledge the gift of anti-major basic protein and anti-EDN antibodies from Dr. Hirohito Kita.
Funding Sources:
Supported by NIH UO1 AI123337, R21 AI151496, and UO1 AI100799 and by the National Natural Science Foundation of China (81700890, 82171106); Taishan Scholar Program of Shandong Province(tsqn202103166); Shandong Provincial Key Research and Development Program, China (2019GSF108227).
Abbreviations
- APC
allophycocyanin
- AR
allergic rhinitis
- CCS
corticosteroids
- DAPI
4’, 6-diamidino-2-phenylindole
- dpi
days post inoculation
- EDN
eosinophil-derived neurotoxin
- EpCs
epithelial cells
- IF
immunofluorescence
- IFN
interferon
- IL
interleukin
- MBP
major basic protein
- MDA5
melanoma differentiation-associated receptors
- PEA
primer extension assay
- PRR
pattern recognition receptors
- RIG-I
retinoic acid-inducible gene I
- RLR
RIG-I-like receptor
- qPCR
quantitative real-time polymerase chain reaction
- RSV
respiratory syncytial virus
- RV
rhinovirus
- TCID
tissue culture infective dose
- TLR
toll-like receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None of the authors report any conflict of interest relevant to the current submission.
Clinical Trial Registration: ClinicalTrials.gov: NCT02910401
References
- 1.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ 1993;307:982–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnston SL, Pattemore PK, Sanderson G, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995;310:1225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heymann PW, Carper HT, Murphy DD, et al. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol 2004;114:239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busse WW, Lemanske RF Jr., Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 2010;376:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol 2017;140:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slater L, Bartlett NW, Haas JJ, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog 2010;6:e1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Triantafilou K, Vakakis E, Richer EA, Evans GL, Villiers JP, Triantafilou M. Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2011;2:22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Q, Miller DJ, Bowman ER, et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog 2011;7:e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol 2013;49:923–34. [DOI] [PubMed] [Google Scholar]
- 10.Zaheer RS, Wiehler S, Hudy MH, et al. Human rhinovirus-induced ISG15 selectively modulates epithelial antiviral immunity. Mucosal Immunol 2014;7:1127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatchwell L, Collison A, Girkin J, et al. Toll-like receptor 7 governs interferon and inflammatory responses to rhinovirus and is suppressed by IL-5-induced lung eosinophilia. Thorax 2015;70:854–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie AI, Jackson DJ, Edwards MR, Johnston SL. Airway Epithelial Orchestration of Innate Immune Function in Response to Virus Infection. A Focus on Asthma. Ann Am Thorac Soc 2016;13 Suppl 1:S55–63. [DOI] [PubMed] [Google Scholar]
- 13.Warner SM, Wiehler S, Michi AN, Proud D. Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir Res 2019;20:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005;201:937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contoli M, Message SD, Laza-Stanca V, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med 2006;12:1023–6. [DOI] [PubMed] [Google Scholar]
- 16.Sykes A, Edwards MR, Macintyre J, et al. Rhinovirus 16-induced IFN-alpha and IFN-beta are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol 2012;129:1506–14 e6. [DOI] [PubMed] [Google Scholar]
- 17.Edwards MR, Regamey N, Vareille M, et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol 2013;6:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spann KM, Baturcam E, Schagen J, et al. Viral and host factors determine innate immune responses in airway epithelial cells from children with wheeze and atopy. Thorax 2014;69:918–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kicic A, Stevens PT, Sutanto EN, et al. Impaired airway epithelial cell responses from children with asthma to rhinoviral infection. Clin Exp Allergy 2016;46:1441–55. [DOI] [PubMed] [Google Scholar]
- 20.Simpson JL, Carroll M, Yang IA, et al. Reduced Antiviral Interferon Production in Poorly Controlled Asthma Is Associated With Neutrophilic Inflammation and High-Dose Inhaled Corticosteroids. Chest 2016;149:704–13. [DOI] [PubMed] [Google Scholar]
- 21.Winther B, Gwaltney JM Jr., Mygind N, Turner RB, Hendley JO. Sites of rhinovirus recovery after point inoculation of the upper airway. JAMA : the journal of the American Medical Association 1986;256:1763–7. [PubMed] [Google Scholar]
- 22.Kennedy JL, Shaker M, McMeen V, et al. Comparison of Viral Load in Individuals with and without Asthma during Infections with Rhinovirus. Am J Respir Crit Care Med 2014;189:532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heymann PW, Platts-Mills TAE, Woodfolk JA, et al. Understanding the asthmatic response to an experimental rhinovirus infection: Exploring the effects of blocking IgE. J Allergy Clin Immunol 2020;146:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zambrano JC, Carper HT, Rakes GP, et al. Experimental rhinovirus challenges in adults with mild asthma: response to infection in relation to IgE. J Allergy Clin Immunol 2003;111:1008–16. [DOI] [PubMed] [Google Scholar]
- 25.Enroth S, Berggrund M, Lycke M, et al. A two-step strategy for identification of plasma protein biomarkers for endometrial and ovarian cancer. Clin Proteomics 2018;15:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinke JW, Crouse CD, Bradley D, et al. Characterization of interleukin-4 stimulated nasal polyp fibroblasts. American Journal of Respiratory Cell and Molecular Biology 2004;30:212–9. [DOI] [PubMed] [Google Scholar]
- 27.Arruda E, Boyle TR, Winther B, Pevear DC, Gwaltney JM Jr., Hayden FG. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. The Journal of infectious diseases 1995;171:1329–33. [DOI] [PubMed] [Google Scholar]
- 28.Juncadella IJ, Kadl A, Sharma AK, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 2013;493:547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee WM, Lemanske RF Jr., Evans MD, et al. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med 2012;186:886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duff AL, Pomeranz ES, Gelber LE, et al. Risk factors for acute wheezing in infants and children: viruses, passive smoke, and IgE antibodies to inhalant allergens. Pediatrics 1993;92:535–40. [PubMed] [Google Scholar]
- 31.Rakes GP, Arruda E, Ingram JM, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med 1999;159:785–90. [DOI] [PubMed] [Google Scholar]
- 32.Busse WW, Morgan WJ, Gergen PJ, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. The New England journal of medicine 2011;364:1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soto-Quiros M, Avila L, Platts-Mills TA, et al. High titers of IgE antibody to dust mite allergen and risk for wheezing among asthmatic children infected with rhinovirus. The Journal of allergy and clinical immunology 2012;129:1499–505 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papadopoulos NG, Papi A, Meyer J, et al. Rhinovirus infection up-regulates eotaxin and eotaxin-2 expression in bronchial epithelial cells. Clin Exp Allergy 2001;31:1060–6. [DOI] [PubMed] [Google Scholar]
- 35.Perez GF, Pancham K, Huseni S, et al. Rhinovirus infection in young children is associated with elevated airway TSLP levels. Eur Respir J 2014;44:1075–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beale J, Jayaraman A, Jackson DJ, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med 2014;6:256ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson DJ, Makrinioti H, Rana BM, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med 2014;190:1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schatz M, Wasserman S, Patterson R. Eosinophils and immunologic lung disease. Med Clin North Am 1981;65:1055–71. [DOI] [PubMed] [Google Scholar]
- 39.Steinke JW, Liu L, Turner R, Braciale T, Borish L. Immune surveillance by rhinovirus-specific circulating CD4+ and CD8+ T lymphocytes. PLoS ONE 2013;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muehling LM, Mai DT, Kwok WW, Heymann PW, Pomes A, Woodfolk JA. Circulating Memory CD4+ T Cells Target Conserved Epitopes of Rhinovirus Capsid Proteins and Respond Rapidly to Experimental Infection in Humans. J Immunol 2016;197:3214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scanlon ST, McKenzie AN. Type 2 innate lymphoid cells: new players in asthma and allergy. Current opinion in immunology 2012;24:707–12. [DOI] [PubMed] [Google Scholar]
- 42.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nature immunology 2013;14:536–42. [DOI] [PubMed] [Google Scholar]
- 43.Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med 1997;155:1159–61. [DOI] [PubMed] [Google Scholar]
- 44.Arruda E, Hayden FG. Detection of human rhinovirus RNA in nasal washings by PCR. Mol Cell Probes 1993;7:373–9. [DOI] [PubMed] [Google Scholar]
- 45.Howell MD, Gallo RL, Boguniewicz M, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 2006;24:341–8. [DOI] [PubMed] [Google Scholar]
- 46.Contoli M, Ito K, Padovani A, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy 2015;70:910–20. [DOI] [PubMed] [Google Scholar]
- 47.Handzel ZT, Busse WW, Sedgwick JB, et al. Eosinophils bind rhinovirus and activate virus-specific T cells. J Immunol 1998;160:1279–84. [PubMed] [Google Scholar]
- 48.Rosenberg HF, Domachowske JB. Eosinophils, eosinophil ribonucleases, and their role in host defense against respiratory virus pathogens. J Leukoc Biol 2001;70:691–8. [PubMed] [Google Scholar]
- 49.Phipps S, Lam CE, Mahalingam S, et al. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood 2007;110:1578–86. [DOI] [PubMed] [Google Scholar]
- 50.Percopo CM, Dyer KD, Ochkur SI, et al. Activated mouse eosinophils protect against lethal respiratory virus infection. Blood 2014;123:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drake MG, Bivins-Smith ER, Proskocil BJ, et al. Human and Mouse Eosinophils Have Antiviral Activity against Parainfluenza Virus. Am J Respir Cell Mol Biol 2016;55:387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samarasinghe AE, Melo RC, Duan S, et al. Eosinophils Promote Antiviral Immunity in Mice Infected with Influenza A Virus. J Immunol 2017;198:3214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravanetti L, Dijkhuis A, Sabogal Pineros YS, et al. An early innate response underlies severe influenza-induced exacerbations of asthma in a novel steroid-insensitive and anti-IL-5-responsive mouse model. Allergy 2017;72:737–53. [DOI] [PubMed] [Google Scholar]
- 54.Domachowske JB, Dyer KD, Bonville CA, Rosenberg HF. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J Infect Dis 1998;177:1458–64. [DOI] [PubMed] [Google Scholar]
- 55.Eid R, Borish L. Eosinophils in antiviral immunity and (perhaps) a benefit of having asthma during the SARS-CoV2 pandemic. Ann Allergy Asthma Immunol 2021;127:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avila PC, Abisheganaden JA, Wong H, et al. Effects of allergic inflammation of the nasal mucosa on the severity of rhinovirus 16 cold. J Allergy Clin Immunol 2000;105:923–32. [DOI] [PubMed] [Google Scholar]
- 57.Sabogal Pineros YS, Bal SM, Dijkhuis A, et al. Eosinophils capture viruses, a capacity that is defective in asthma. Allergy 2019;74:1898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sabogal Pineros YS, Bal SM, van de Pol MA, et al. Anti-IL-5 in Mild Asthma Alters Rhinovirus-induced Macrophage, B-Cell, and Neutrophil Responses (MATERIAL). A Placebo-controlled, Double-Blind Study. Am J Respir Crit Care Med 2019;199:508–17. [DOI] [PubMed] [Google Scholar]
- 59.Mathur SK, Fichtinger PS, Kelly JT, Lee WM, Gern JE, Jarjour NN. Interaction between allergy and innate immunity: model for eosinophil regulation of epithelial cell interferon expression. Ann Allergy Asthma Immunol 2013;111:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.