

Significance
The bacterial genus Staphylococcus is a prominent member of the human skin microbiome, performing important and diverse functions such as tuning immunity, driving tissue repair, and preventing pathogen colonization. Each of these functions is carried out by a subset of staphylococcal strains, displaying differences in gene content and regulation. Delineating the genomic and functional diversity of Staphylococcus will enable researchers to unlock the potential of engineering skin communities to promote health. Here, we present a comprehensive multiomics analysis to characterize the inter- and intra-species diversity present in human skin-associated staphylococci. Our study conducts a detailed pan-genome comparison between prominent skin staphylococcal species giving valuable insight into gene sharing and providing an important resource.
Keywords: skin microbiota, staphylococci, 16S rRNA amplicon sequencing, pan-genomics, growth curve
Abstract
Human skin is stably colonized by a distinct microbiota that functions together with epidermal cells to maintain a protective physical barrier. Staphylococcus, a prominent genus of the skin microbiota, participates in colonization resistance, tissue repair, and host immune regulation in strain-specific manners. To unlock the potential of engineering skin microbial communities, we aim to characterize the diversity of this genus within the context of the skin environment. We reanalyzed an extant 16S rRNA amplicon dataset obtained from distinct body sites of healthy volunteers, providing a detailed biogeographic depiction of staphylococcal species that colonize our skin. S. epidermidis, S. capitis, and S. hominis were the most abundant staphylococcal species present in all volunteers and were detected at all body sites. Pan-genome analysis of isolates from these three species revealed that the genus-core was dominated by central metabolism genes. Species-restricted-core genes encoded known host colonization functions. The majority (~68%) of genes were detected only in a fraction of isolate genomes, underscoring the immense strain-specific gene diversity. Conspecific genomes grouped into phylogenetic clades, exhibiting body site preference. Each clade was enriched for distinct gene sets that are potentially involved in site tropism. Finally, we conducted gene expression studies of select isolates showing variable growth phenotypes in skin-like medium. In vitro expression revealed extensive intra- and inter-species gene expression variation, substantially expanding the functional diversification within each species. Our study provides an important resource for future ecological and translational studies to examine the role of shared and strain-specific staphylococcal genes within the skin environment.
The human epidermis and associated appendages including hair, nails, and glands (sebaceous, apocrine, and eccrine) form a physical and immunologic barrier between our body and the environment, protecting us against pathogens, injury, and unregulated water loss (1). The human skin microbiota, which stably colonize this epidermal interface, play an integral role in skin barrier function via microbe–microbe and host–microbe interactions (2–4). While early studies focused on the pathogenicity of skin microbes, more recent studies have explored the myriad ways in which microbial strains serve beneficial roles, including providing colonization resistance, wound healing, and tuning immune responses (5). There is growing recognition that microbial functions are often strain-specific, demonstrating a need to understand the full genetic diversity of the commensal microbiota. For example, strains of Staphylococcus epidermidis that produce the serine protease Esp were shown to limit Staphylococcus aureus colonization by selectively degrading proteins essential for biofilm formation (6). Many staphylococcal strains produce diverse antimicrobial peptides such as lugdunin and phenol-soluble modulins that restricted pathogen growth (7–10). In addition, colonizing S. epidermidis strains were shown to directly interact with the host by secreting enzymes that increased the level of skin lipids or ceramides, preventing excessive water loss (11). S. epidermidis strains shape and regulate skin immune function through coordinated action of resident immune cells in a highly dynamic fashion (12, 13). Intriguingly, new studies using genetically engineered S. epidermidis strains to express foreign antigens resulted in de novo immune responses, even demonstrating targeted activity against antigens expressed by melanoma cells (14). Together, these studies focused on S. epidermidis and related noncoagulase staphylococci (15) collectively demonstrate the diverse functions encoded by these species to maintain skin health and the future potential to engineer skin communities to restore health.
Despite their strain-dependent contribution to host health, current knowledge about healthy skin-associated staphylococcal genomic diversity is limited. Previous sequencing studies based on 16S rRNA gene amplicon (V1–V3) and shotgun metagenomics have revealed that Staphylococcus is one of the most prominent bacterial genera, along with Cutibacterium and Corynebacterium that persistently colonizes the human skin (2, 16). However, a detailed species-level picture of staphylococci and their distribution across different body sites is currently lacking. While previous studies characterized S. epidermidis pan-genomes (17–19), similar analyses in other skin-associated staphylococci have been limited. A pan-genome analysis captures the entire genetic diversity that is encoded within the sampled genomes for a given species by categorizing genes into core and accessory (20), and thus identifying lineage-specific versus strain-specific genes. In addition, studies in Escherichia coli demonstrated the role of differential gene expression in species diversification (21), highlighting the need to study gene expression to comprehend functional diversity.
In our current work, we seek to study skin-associated staphylococcal diversity from healthy volunteers using 16S rRNA amplicon sequencing, pan-genome, and gene expression analyses to gauge its functional potential. Based on our previous work which established that the V1–V3-based 16S rRNA analysis allows species-level discrimination of staphylococci (22), our current work aimed to characterize the distribution of staphylococcal species across skin sites of healthy volunteers. We show that the skin is colonized by multiple species but predominated by S. epidermidis, Staphylococcus hominis, and Staphylococcus capitis, for which human skin serves as the primary habitat (23). We focused on these three predominant species and sequenced nonclonal isolate genomes cultured from multiple skin sites, with an aim to understand species and genus-level pan-genome diversity. Most genes within the pan-genomes are variably shared, showing tremendous strain-level diversity as previously reported in skin S. epidermidis (24). Finally, we performed growth- and RNA-Seq analyses of isolates from all three species in conditions that mimic the skin environment. Gene expression analysis revealed extensive variation between isolates, highlighting the natural diversity in expression, which further contribute to the functional diversity in skin-associated staphylococci.
Results
Healthy Human Skin Harbors Multiple Staphylococcus Species.
To identify staphylococcal species that are resident members of the human skin microbiome, we reanalyzed an extant 16S rRNA amplicon dataset obtained from 14 body sites of 22 male and female healthy volunteers (between 18 and 40 y of age) that had previously been analyzed at the genus level (SI Appendix, Fig. S1 shows body sites sampled with abbreviations) (25). The selected body sites encompassed multiple sebaceous, moist, and dry skin surfaces from head to toe. In agreement with previously published results, Staphylococcus, Cutibacterium, and Corynebacterium were the most prominent genera represented in the microbiome of these skin sites (25) (SI Appendix, Fig. S2). The mean percent relative abundance of Staphylococcus varied widely across body sites from 4.3 ± 1.5% on back, to 64.1 ± 6.3% at the plantar heel. In general, Staphylococcus dominated the moist sites (37.3 ± 2.5%), where it was present at a significantly higher proportion than sebaceous sites (13.2 ± 1.4%; Wilcoxon rank-sum test, P value < 0.001) and dry sites (12.0 ± 2.1%; Wilcoxon rank-sum test, P value < 0.05). As this dataset interrogated the 5′ end of the 16S rRNA gene, sufficient high-confidence amplicon sequence variants (ASVs) exist to impute species-level relative abundance. Species present at ≥1% relative abundance at one or more body sites and detected in more than three individuals were considered skin-resident species for this study (26). Using these criteria for abundance and prevalence thresholds, a total of 17 staphylococcal species were identified as residents or indigenous to human skin (Fig. 1A). Each body site was colonized simultaneously by multiple skin-resident staphylococcal species, with the average ranging from 2.6 ± 0.3 species to 5.3 ± 0.4 species on back and toenails, respectively.
Fig. 1.
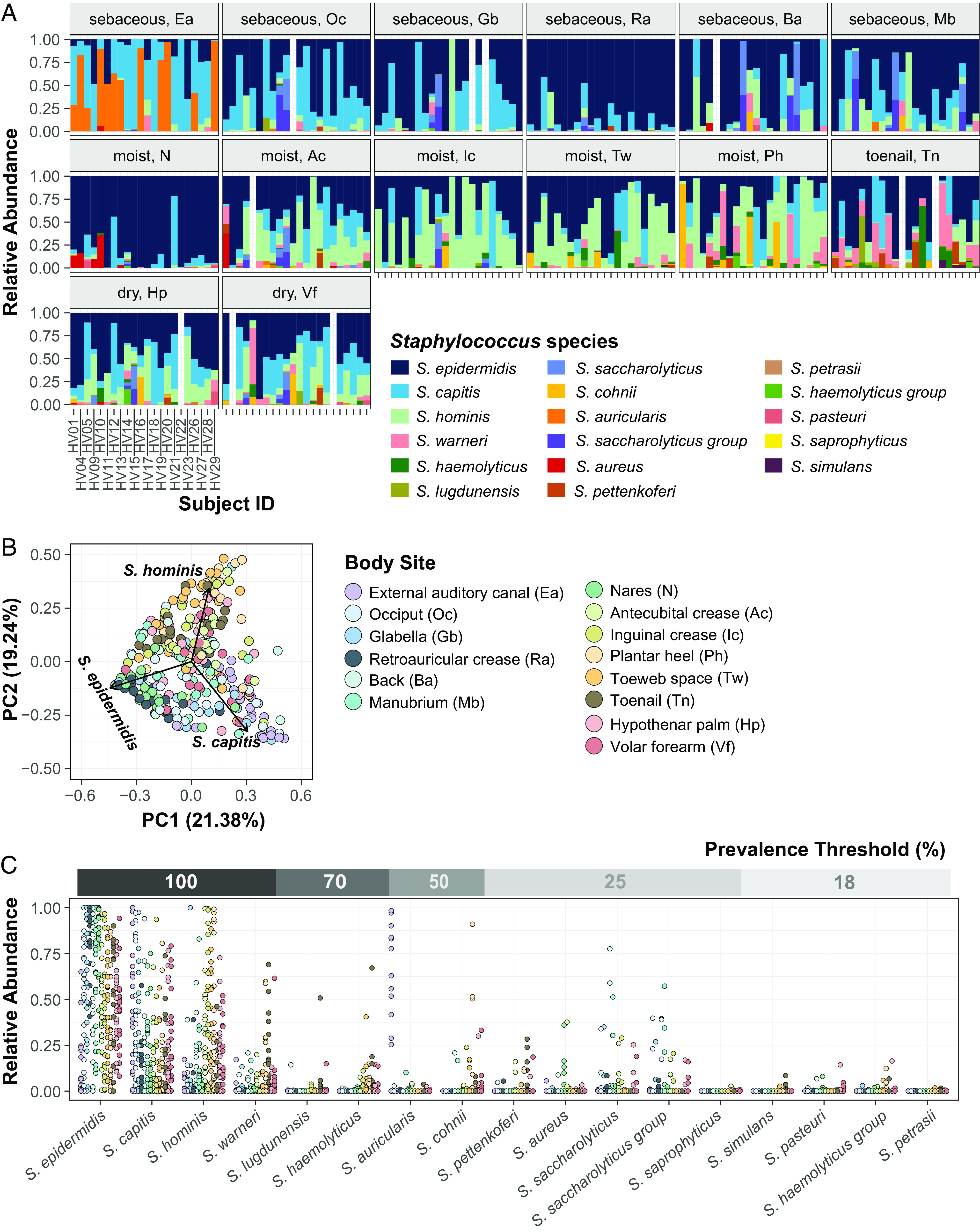
Diversity of staphylococcal species present on healthy human skin. (A) Barplots display the relative abundance of staphylococcal species at 14 body sites. Each color represents a distinct species as shown in the legend. Each bar represents one Subject/Healthy Volunteer (HV). Empty bars represent missing data. (B) Beta diversity analysis of staphylococcal communities present at different body sites (displayed as colored dots) using principal-coordinate analysis (PCoA) plot based on Bray–Curtis dissimilarity (PERMANOVA; R2 = 0.26804, P value < 0.0001). First two coordinates are shown accounting for 40.62% of the total variance. Individual staphylococcal ASVs driving the largest separation of body sites are shown as black arrows and are labeled by the corresponding species. Note that the body site legend is shared between figures B and C. (C) Relative abundance and prevalence of staphylococcal species detected in our dataset. Each dot represents proportion of a staphylococcal species relative to all other staphylococcal species in a sample. Dots are colored by body sites as shown in the shared legend. Percent prevalence threshold for a species to be considered part of the core staphylococcal community is shown in the upper gray bar (see text for details). Refer to SI Appendix, Fig. S1 for body site details.
Some body sites displayed distinct staphylococcal community composition, characterized by a higher proportion of select species and indicating site preference. The most intimate relationship observed was the external auditory canal (Ea) where ~55% healthy volunteers (12/22 Ea samples) were colonized by Staphylococcus auricularis at a mean relative abundance (MRA) of 66.4 ± 7.8%. S. auricularis was rarely detected in other samples from these healthy volunteers (15/156 samples, 2.3 ± 0.6 MRA, Wilcoxon rank-sum test, P value < 0.0001 for MRA of S. auricularis in positive Ea samples versus all other positive samples). Further, barring HV14 (toenail sample with 0.04% MRA), S. auricularis was completely absent in individuals that lacked Ea colonization.
S. epidermidis, S. capitis, and S. hominis species were present in most samples (S. epidermidis: ~95% samples at 52.3 ± 1.9 % MRA; S. capitis: ~75% samples at 26.3 ± 1.8 % MRA; S. hominis: ~70% samples at 23.7 ± 1.9% MRA). However, even for these prominent species, colonization across body sites was not uniform. S. epidermidis predominated the nares (N) and retroauricular crease (Ra) (MRA: N: 83.7 ± 4.3% and Ra: 86.0 ± 3.7% as compared to all other sites: 46.3 ± 1.9%; Wilcoxon rank-sum test, P value < 0.0001). S. capitis displayed a moderate site preference for the sebaceous head region (combined Ea/glabella/occiput: 45.4 ± 4.3% versus nonhead: 20.5 ± 1.7%; Wilcoxon rank-sum test, P value < 0.0001), whereas S. hominis preferred moist sites (combined antecubital crease/inguinal crease/plantar heel/toe web: 39.4 ± 3.4% versus nonmoist: 14.00 ± 1.6%; Wilcoxon rank-sum test, P value < 0.0001). To assess if site preferences shaped staphylococcal community composition at different body sites, we calculated the Bray–Curtis dissimilarity. Unsupervised ordination of the dissimilarity matrix using principal coordinate analysis (PCoA) revealed separation of body sites largely driven by the three most prominent staphylococcal species, as indicated by labeled arrows on the plot (PERMANOVA; R2 = 0.27, P value < 0.0001; Fig. 1B). Specifically, the sebaceous head region and the moist sites were separated by S. capitis and S. hominis, respectively, whereas the nares and retroauricular crease were grouped together by S. epidermidis colonization.
We next sought to define a core staphylococcal community by prevalence threshold, based on proportion of healthy volunteers being colonized by a species, irrespective of relative abundance. (Fig. 1C and SI Appendix, Table S1). Of the 17 skin-resident species, only S. epidermidis, S. capitis, S. hominis, and S. warneri were present on all volunteers, making them ubiquitous within our dataset. In addition to being most prevalent, S. epidermidis, S. capitis, and S. hominis also showed the highest MRA and could be detected at all skin sites in highly variable proportions (SI Appendix, Fig. S3). Decreasing the prevalence threshold below 100%, increased the number of species that can be considered core, with all species being included at a threshold of 18%; however, most of these low-prevalence species, including S. aureus, were present at low relative abundance and detected only at select body sites (Fig. 1C).
Collectively, our skin 16S rRNA amplicon sequencing analyses revealed diverse staphylococcal communities with body site–specific compositions. Given the predominance of S. epidermidis, S. capitis, and S. hominis on the skin, we focused on these three species for in-depth genomic and functional analyses.
Genome Sequencing Captures Intra-Species Diversity.
We sought to catalog the genetic makeup and diversity of human skin-associated S. epidermidis, S. capitis, and S. hominis by leveraging our extensive skin culture collection, which contains diverse bacterial isolates from multiple body sites of healthy volunteers. A total of 273 staphylococcal isolates were sequenced, and highly similar genomes were removed by de-replication (>99.9% ANI), to generate a final list of 22 S. capitis (9 volunteers, 10 body sites), 49 S. epidermidis (12 volunteers, 15 body sites), and 55 S. hominis (11 volunteers, 12 body sites) nonclonal complete genomes (N = 126 total) (SI Appendix, Fig. S1 and Dataset S1). Average nucleotide identity (ANI) values of dereplicated genomes revealed sharp species-specific boundaries as shown in SI Appendix, Fig. S4, with ANI values between 96.3 and 100.0% for conspecific genomes, and between 78.8 and 81.5% for nonconspecific pairs (27). Comparison of key genome statistics showed that S. hominis isolates had the smallest average genome size and the least number of predicted protein coding genes (Dataset S1 and SI Appendix, Table S2). We confirmed that the small genome size of S. hominis is not restricted to our dataset by analyzing publicly available genomes (SI Appendix, Fig. S5).
Species- and Genus-Level Pan-Genome Analysis Reveals Extensive Genetic Diversity.
Using a pan-genomics approach, we sought to identify orthologous genes conserved across all three species as well as those taxonomically restricted to one species or even a subset of strains. We initially calculated three independent species-level pan-genomes using Panaroo (28) to generate a nonredundant catalog of all protein coding sequences (CDS), along with their distribution. S. hominis has 5,118 total and 1,761 core genes: S. capitis has 3,203 total and 2,091 core genes; and S. epidermidis has 4,699 total and 1,902 core genes. Major pan-genome features of each species and sharing of genes among conspecific isolates are presented in SI Appendix, Table S3 and Dataset S2, respectively. Using these catalogs of conspecific genes, we calculated a genus-level pangenome using a reciprocal-best-blast approach to cluster cross-species orthologs at the protein level (≥40% identity and ≥50% coverage). This resulted in clustering of 78 to 93% of core genes from each species-level pan-genome to provide a merged genus pan-genome consisting of a total of 7,744 genes (Dataset S3). The genus pangenome comprised of 1,570 core genes (≥99% genomes), 90 soft-core genes (<99% to ≥95 % genomes), 1,116 shell genes (<95% to ≥15% genomes), and 4,968 cloud genes (<15% genomes) (Fig. 2A). The 7,744 genus pan-genome genes were divided into seven subsets according to their presence amongst one, two, or three species (Fig. 2B). A total of 2,085 genes were shared by isolates of all three species, out of which 1,660 genes were present in ≥95% of all the isolates and are termed as “genus-core genes” for the remainder of the manuscript. The six other subsets consisted of taxonomically restricted genes that were present in either 2/3 or 1/3 species. This included 359 genes shared between S. epidermidis and S. capitis isolates, 500 genes shared between S. epidermidis and S. hominis isolates, 146 genes shared between S. capitis and S. hominis isolates, 1,707 genes specific to S. epidermidis isolates, 2,353 genes specific to S. hominis, and 594 genes specific to S. capitis isolates. Interestingly, each of the taxonomically restricted subsets contained core genes shared by ≥95% of the subset genomes. These taxonomically restricted core genes are termed as “species-restricted-core genes” for the remainder of the manuscript, and their numbers are shown in brackets in Fig. 2B. The noncore genes that neither belonged to the genus-core nor the species-restricted-core are termed as accessory for the remainder of the manuscript. Distribution of genes within each genome, along with their sharing between genomes is demonstrated by a gene presence-absence matrix (Fig. 2C); sharing was quantified by Jaccard index measuring pair-wise gene sharing between all genomes (Fig. 2D). Since the genus-core of 1,660 genes accounted for ~70% of protein coding genes per genome (total: ~2,300 CDS/genome based on SI Appendix, Table S2), this indicated that majority of the genes encoded within a genome are conserved between all species.
Fig. 2.
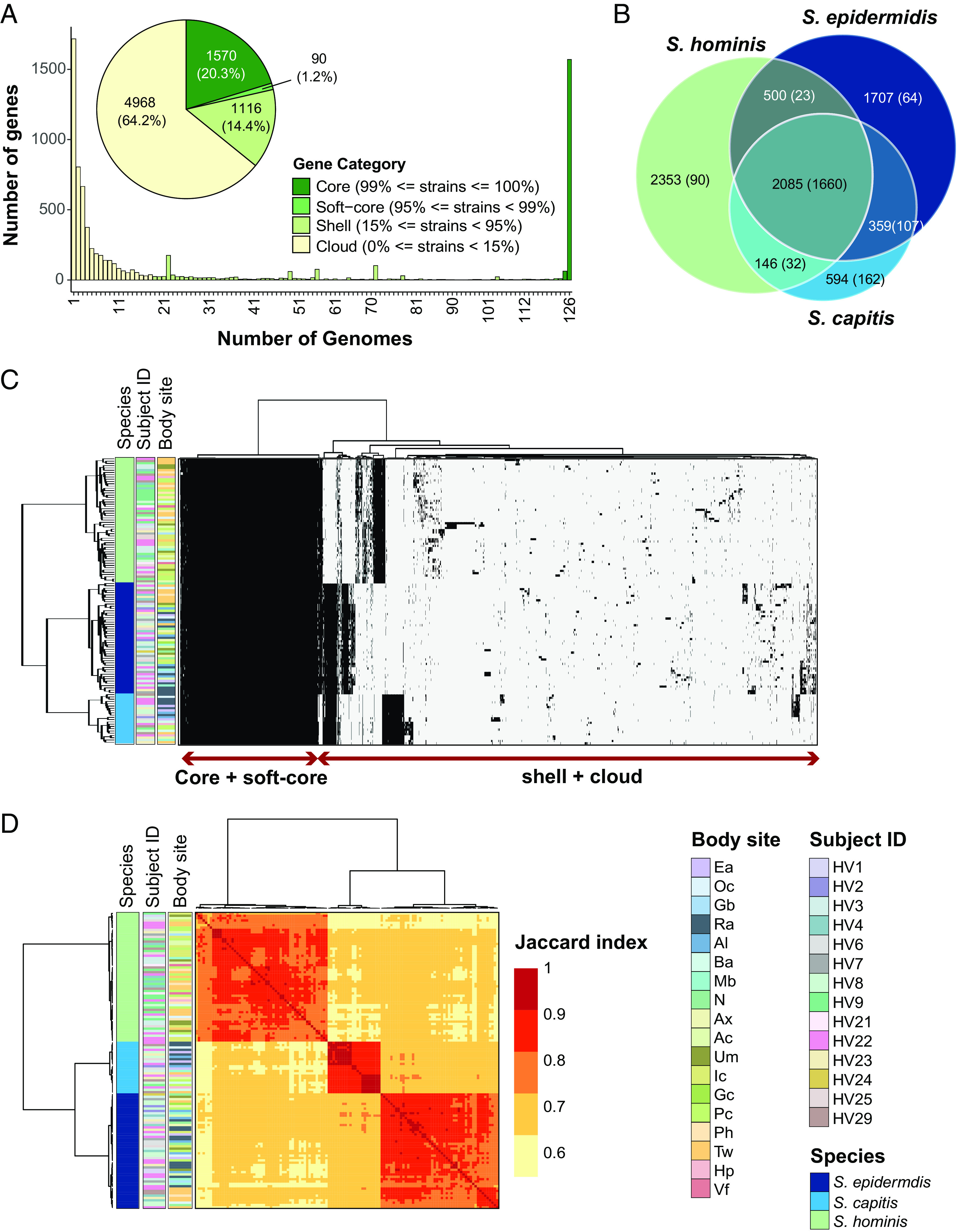
Genus-level pan-genome summary to quantify gene sharing between species. (A) Histogram of core, soft-core, shell, and cloud genes of the genus-pangenome. Pie chart shows the number of genes in each category as defined in the legend; percent of genes in each category are displayed in brackets. (B) Venn diagram displaying the distribution of 7,744 genus pan-genome genes into seven subsets based on their presence in one, two or three species. Number of total genes in each subset are shown outside the bracket and the core genes in each sub-set (genes detected in ≥95% genomes present in the sub-set) are shown inside brackets. (C) Clustering of all 126 genomes using Euclidean distance and Ward hierarchical clustering based on the presence/absence patterns of 7,744 genes detected in the pan-genome. Sidebars display species, volunteer, and body site of isolation for each genome. (D) Pairwise Jaccard index between genomes displaying the proportion of shared gene content. Note: Distance of 1.0 depicts complete gene overlap, with lower numbers representing lesser degree of gene sharing. Hierarchical clustering of genomes and the displayed sidebars are the same as that shown in figure C. Note that the legend is shared between figures C and D. Refer to SI Appendix, Fig. S1 for body site details.
Functional Analysis Shows Conserved Core Metabolic Functions between Species.
We next sought to annotate the genus pan-genome to dissect functional capabilities that are central to all three species versus species restricted. The percentage of genes that had an annotation or a predicted Pfam domain was ~63%, and this percentage progressively decreased from “core + soft-core” (93%) to shell (79%) to cloud (49%) genes (SI Appendix, Fig. S6A). A comparison of the top 20 most represented Pfam domains in each pan-genomic category revealed considerable differences between core + soft-core, shell, and cloud fractions (SI Appendix, Fig. S6B). Specifically, the core + soft-core gene’s domains were involved in transport and central metabolic enzymes, while the annotated cloud genes predominantly carried domains found in phages, transposable elements, and restriction modification systems. No Pfam domain was exclusive to or enriched in any one species. Overrepresentation of canonical metabolic pathways within the core was also supported by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (SI Appendix, Fig. S6C). This suggested that all species have similar central metabolic capabilities, which was confirmed by mapping genes onto KEGG modules to determine completeness of canonical pathways (SI Appendix, Fig. S7). Using a strict criterion of ≥75% module completeness in at least one genome, we found 62 modules, of which 46 were complete in all the genomes, irrespective of species, and were part of the genus-core genes. Three complete core metabolic modules were present in individual species as part of the species-restricted-core genes rather than the genus-core. These included M00026 (histidine biosynthesis) in S. epidermidis, M00045 (histidine degradation) in S. capitis, and M00127 (thiamine biosynthesis) in S. hominis genomes.
Along with these 46 metabolic pathways, the genus-core also carried several genes/operons with a predicted function in skin colonization including immune evasion (srtA, dltABCD, mprF, sepA, vraFG, vraX, graSRX, capABC, and oatA), acid response (ureABCEFGD and yut), osmotic tolerance (betA and opuCABCD), biofilm formation (atlE and pmtABCD), iron acquisition (yfmCDE and isdG), and niche occupancy (tagDXBGHA and gehC) (SI Appendix, Table S4).
Species-Restricted-Core Genes Carry Loci with Predicted Role in Skin Colonization.
We next analyzed all the species-restricted-core genes that were present as core in 2/3 or 1/3 species (Fig. 2B), as gene candidates under selection for conferring species-specific capabilities (Dataset S4). We focused on annotated genes, especially ones that play a role within the skin environment (either by themselves or in combination with genus-core genes, SI Appendix, Fig. S8). Some genes were present as paralogs of genus-core genes, with potentially divergent functionality. For example, the D- and L- methionine transport locus (metNIQ) was present as part of the genus-core, with an additional copy in all S. epidermidis genomes. Interestingly, some of the species-restricted-core genes were also present at a lower frequency in other species, indicating species-level differences in selection pressure to retain these genes. Of note is the biosynthetic pathway for the broad-spectrum metallophore, staphylopine, that was present in all S. epidermidis, but only a subset of S. capitis (N = 16/22) genomes (29). The pathway requires L-histidine, which can be synthesized by all S. epidermidis isolates (KEGG module M00026) or imported by S. capitis isolates (KEGG module M00045). Ni2+ is imported with high affinity by staphylopine, which then acts as a urease cofactor (genus-core genes) and plays an important role in survival within the acidic skin environment (30). Another well-known example is the icaABCDR locus involved in biofilm formation, that was core to S. capitis, and accessory to S. epidermidis (N = 17/49), where it is considered a virulence factor (19). Besides these examples, fitness determinants involved in resistance to reactive oxidants, host-generated polyamines and antimicrobial fatty acids, acid tolerance, biofilm formation, resistance to antibiotics, and osmoregulation were present as species-restricted-core genes as shown in SI Appendix, Fig. S8. Metabolic genes involved in fermentation pathways [glycerol, lactate, formate (31), and acetoin metabolism] that are important under glucose limiting and microaerobic skin conditions, and secreted proteases [metalloproteases: SepA (32), cysteine protease Ecp and serine protease Esp (33), and a sphingomyelinase [sph (11)] that play a role in microbe–microbe and host–microbe interactions were also detected. Well-characterized genes that were restricted to either S. capitis or S. hominis were rare. One example present in all S. hominis genomes is the patB gene, recently shown to be responsible for body odor-associated thioalcohol generation from the odorless precursor Cys-Gly-3M3SH secreted by apocrine glands (34). Although the biological role of thioalcohol generation is currently unknown, the inguinal crease is known to harbor a high density of apocrine glands, a preferred site for S. hominis colonization.
Apart from the genus-core and species-restricted-core genes, majority of the accessory genes (N = 5,401; ~68% of total genus pan-genome) were variably distributed in a subset of isolates or were singletons, likely conferring strain-specific functions. Most of these genes were uncharacterized with no functional annotation or encoded phage- and transposon-like functions.
Intraspecies Phylogenetic Clades Display Distinct Gene Sharing and Body Site Preferences.
Studies in other microbes have shown that the degree of gene sharing between two strains of the same species is primarily dictated by their evolutionary relatedness (35). To test this hypothesis in Staphylococcus, we carried out phylogenetic analysis of conspecific genomes using a maximum-likelihood tree based on nucleotide polymorphisms in core genes alignments. First, a genus-level phylogenetic tree was built using RAxML-NG and was based on a panaroo-generated alignment of select genus-core genes (N = 665). This genus-level tree was used to root individual species trees, whereby each species tree was rooted using the other two species as an outgroup (SI Appendix, Fig. S9). This outgroup-dependent rooting allowed us to define the presence of two divergent phylogenetic clades (along with potential sub-clades) in all three species with 100% bootstrap support (Fig. 3 A–C).
Fig. 3.
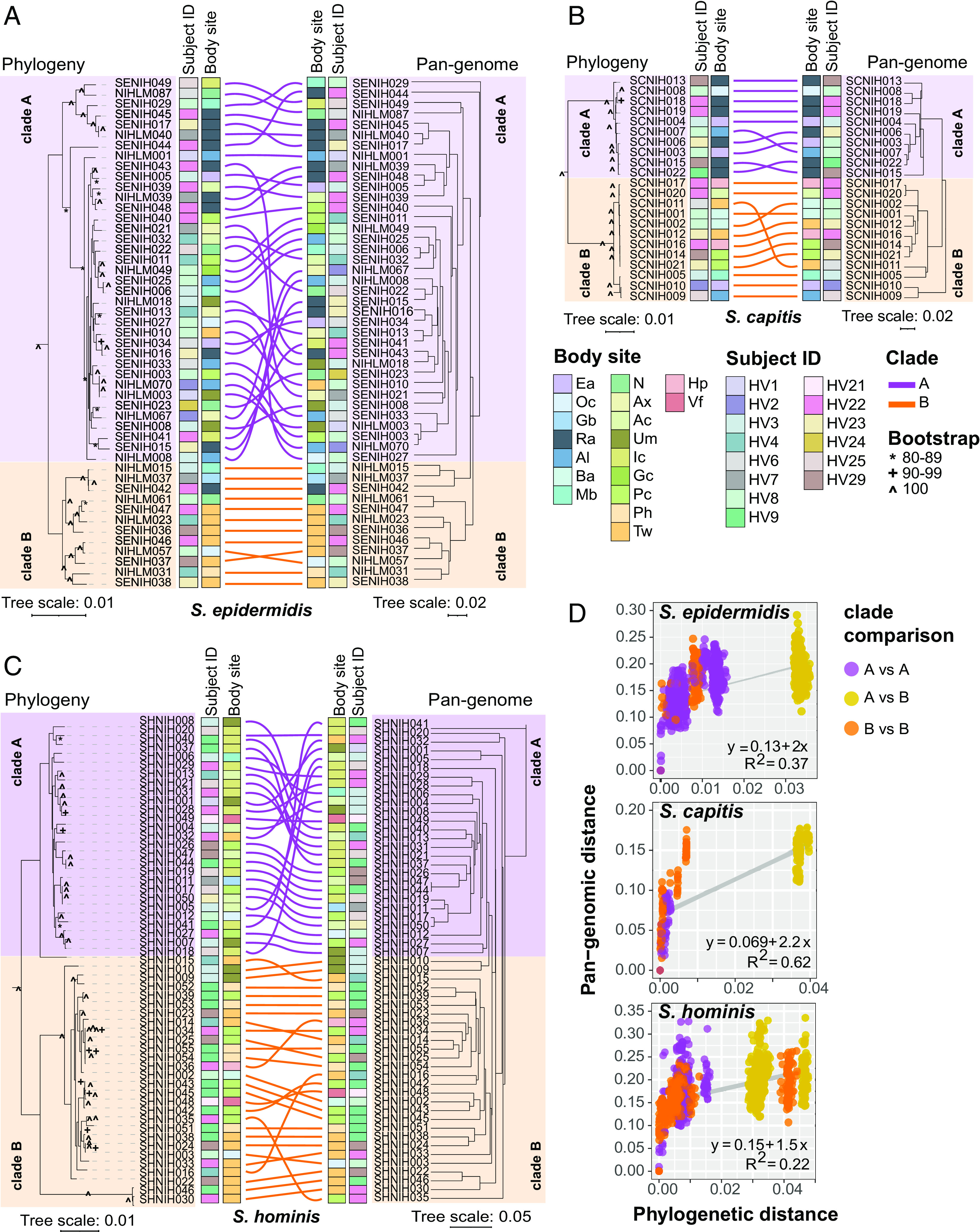
Relationship between phylogeny and pan-genome in conspecific staphylococcal genomes. (A–C) Tanglegrams depicting the relationship between the phylogenetic tree based on core genes sequence alignment (Left) and hierarchical clustering (binary distance; average linkage) of the pan-genome presence/absence matrix (Right). Clades A and B, as determined by rooting each species tree with an outgroup using a genus tree (SI Appendix, Fig. S9), are shown in purple and orange color, respectively. Colored lines are used to connect the same isolate on two trees of the tanglegram. Healthy volunteers and body sites of isolation for isolates are displayed as colored strips within the tanglegram. (D) Correlation between phylogenetic distance and pan-genome distance for all pair-wise genome comparisons within a species. Each dot represents one pair-wise comparison. Dot colors represent the clades to which the genomes being compared belong, with yellow color dots representing clade A versus clade B comparison. Refer to SI Appendix, Fig. S1 for body site details.
Grouping of isolates based on the pan-genome was tested by hierarchical clustering using the Jaccard distance (Fig. 3 A–C) and by PCA (SI Appendix, Fig. S10). This identified groups that broadly corresponded with the phylogenetic clades, and the overall pairwise pan-genomic distance within a clade was lower than between clades (Fig. 3D).
A pan-genome-wide-association study analysis was used to identify clade-specific gene enrichment in all three species (Dataset S5). For S. epidermidis and S. capitis, the results were concordant with previously published pan-genomes (18, 19, 36). Namely, formate dehydrogenase gene (fdhH) involved in formaldehyde detoxification was exclusively present in all S. epidermidis clade B isolates, whereas clade A, which covered isolates from bloodstream infections in earlier studies, was enriched for genes involved in host cell adhesion, antibiotic resistance, biofilm formation, and virulence genes. For S. capitis, the commensal S. capitis subspecies capitis overlap with clade A, and the more pathogenic S. capitis subspecies ureolyticus overlap with clade B. All clade A isolates carried the arginine catabolic mobile element, a histidine decarboxylase (hdcA) that synthesizes the neurotransmitter histamine, and Type I restriction-modification system (hsdRM). Clade B isolates were enriched for genes involved in the staphylopine biosynthesis (cntKLM), L-malate dehydrogenase (mqo), and hxlAB genes encoding formaldehyde detoxification system. For S. hominis, clade A encoded dhaKLM genes important for anaerobic utilization of glycerol, which is abundant on human skin, while clade B showed enrichment of lactate racemase (larABCDE), allowing the use of L- and D-lactate that is present at moist skin sites.
Intriguingly, clades showed significant enrichment by body sites (Fisher’s exact test, P value < 0.05), indicating site or habitat preferences (Fig. 3 A–C). S. epidermidis clade B was enriched for feet sites (7/12) compared to clade A (1/36), which did not show any habitat preference. All S. capitis clade A isolates were cultured from head sites (10/10), a region known to be the primary habitat of S. capitis colonization (37). Conversely, S. capitis clade B isolates were cultured from diverse skin sites, with only 2/12 coming from the head region. For S. hominis, each clade was enriched for a different body site, with clade A showing an over-representation of inguinal crease isolates (10/27 in A vs 0/28 in B) and clade B of feet region isolates (1/27 in A vs 15/28 in B).
Overall, our comparative pan-genome analysis between all three species revealed the presence of genus-core and species-restricted-core genes encoding central metabolic genes along with many skin-relevant functions for successful skin colonization. In addition, analysis of individual species pangenomes revealed the presence of phylogenetic clades, each enriched for a distinct set of genes, likely representing clade-driven niche specialization that allows clades to colonize specific body sites.
Growth Curves in Skin-Like Media Conditions Reveal Intraspecies Phenotypic Diversity.
In addition to pan-genomic differences, we were interested in identifying gene expression differences that may exist between staphylococcal isolates under similar skin-like growth conditions. We specifically wanted to test whether the shared genes showed similar expression patterns, with accessory genes driving the major gene expression differences between isolates.
We first compared the ability of our isolates to grow in conditions that mimic the skin environment, which can be broadly defined as acidic and nutrient-poor, with amino acids and lipids as the main energy sources (38). We tested an isolate collection (n = 11 to 16) from each species, which represented > 88% of the species pan-genome (without singletons). Kinetic growth analysis was carried out in two artificial skin media representing the acidic, low nutrient (Eccrine sweat; ES), and the sebaceous or oily (Eccrine sweat with lipids; ESL) conditions present on healthy human skin (SI Appendix, Table S5). Brain heart infusion supplemented with yeast extract (BHI-YE) was used as a control medium, which supported the robust growth of all isolates (Fig. 4 A–C). At the end of 24 h, we observed considerable intraspecies growth variation in nutrient-limited ES and ESL media, as quantified by area under the curve (AUC) (Fig. 4 D–F). Addition of lipids slightly improved the mean AUC for all species (1.3-fold, 1.5-fold, and 1.1-fold for S. epidermidis, S. capitis, and S. hominis, respectively), but was not statistically significant. We did not observe any clade-specific growth differences in S. epidermidis and S. hominis isolates. In contrast, clade B isolates of S. capitis grew consistently better than clade A isolates in all the media (P value < 0.001; t tests comparing mean AUC for clade A versus clade B isolates, independently in each medium), suggesting divergent growth requirements between the two phylogenetic clades.
Fig. 4.
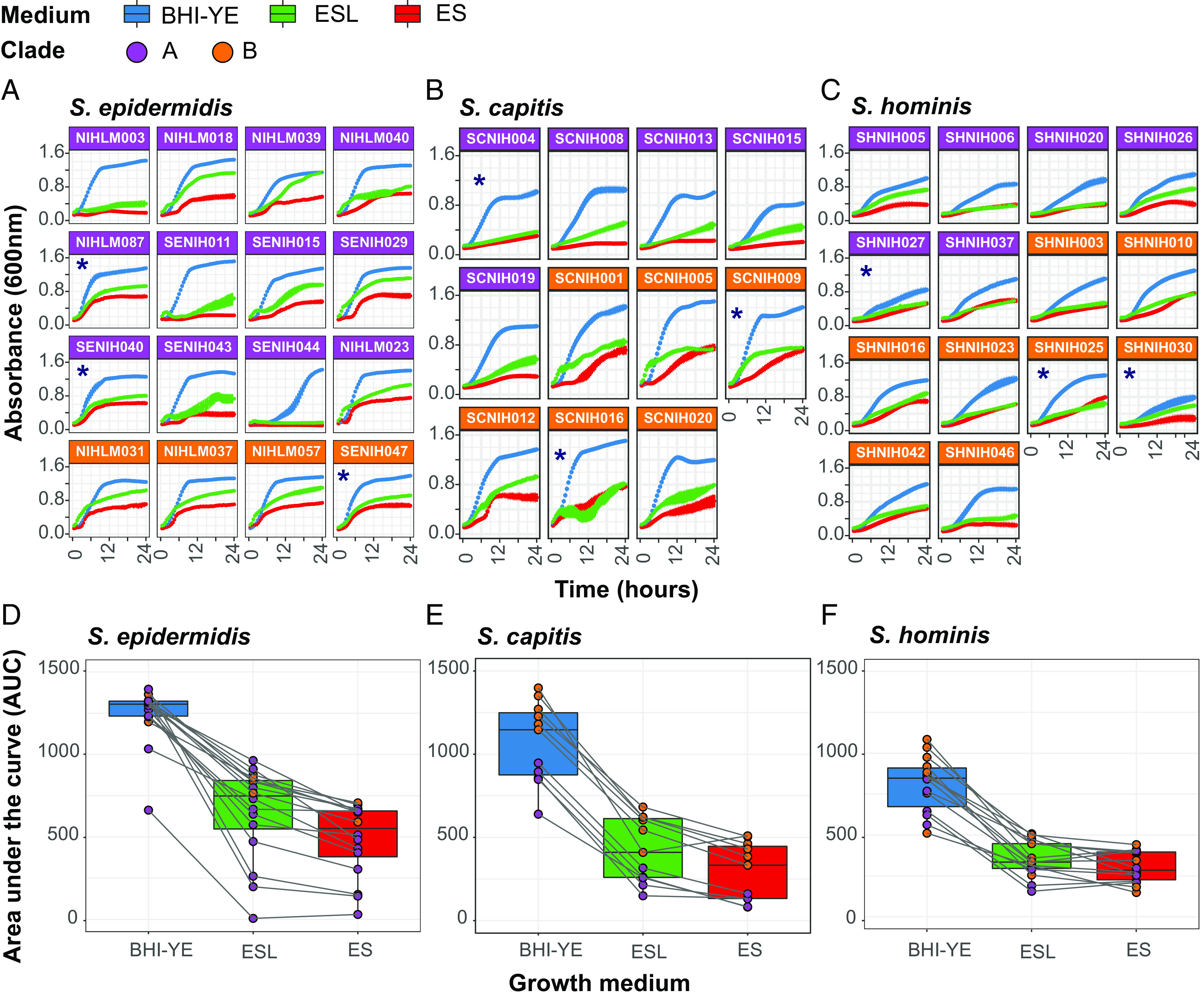
Kinetic growth analyses of select staphylococcal isolates in BHI-YE and artificial skin media, ES and ESL. (A–C) Growth curves of select S. epidermidis, S. capitis, and S. hominis isolates grown independently in three culture media, as measured by absorbance at 600 nm over 24 h. Each growth curve is an average of four independent biological replicates; error bars for each curve are shown. Isolates marked with an asterisk (*) were selected for RNA-Seq analyses. Purple and orange color of facets indicate the phylogenetic clade to which each isolate belongs. Growth curve color represents the growth medium. (D–F) The AUC was used to quantify the growth of each isolate shown in A–C. Boxplots depict combined AUC values for all isolates of a species in each growth medium, as depicted by the color of the boxplot. The center black line within each boxplot represents the median value, with edges showing the first and third quartiles. Individual AUC values are shown as dots colored by clade of the isolate. Note that the legend is shared between all figures.
RNA-Sequencing Analysis Shows Considerable Variation in Gene Expression between All Isolates.
Based on our growth curve analysis, we chose three isolates from each species for RNA-sequencing (RNA-Seq) (shown with an asterisk in Fig. 4 A–C), such that isolates belonged to different clades/subclades, and whenever possible, showed distinct growth phenotypes in our skin media. Each isolate was grown separately in triplicate in BHI-YE, ES, and ESL medium, and cells were harvested at mid-log phase for RNA-Seq.
To make preliminary comparisons between isolates in all the three media, we restricted our initial RNA-Seq analysis to 1,647 genus-core genes that had at least one read in all samples. This subsetting was done to prevent any clustering bias due to differences in gene presence/absence. A principal component analysis (PCA) (Fig. 5A) and hierarchical clustering (SI Appendix, Fig. S11) of variance stabilized read counts from the resulting dataset showed distinct clustering of BHI-YE samples from skin-like media. Further, samples from each medium grouped together by species, with S. hominis samples being more distant than the other two species. Given the overlapping profiles of ES and ESL samples for each species, only the ES samples were further analyzed.
Fig. 5.
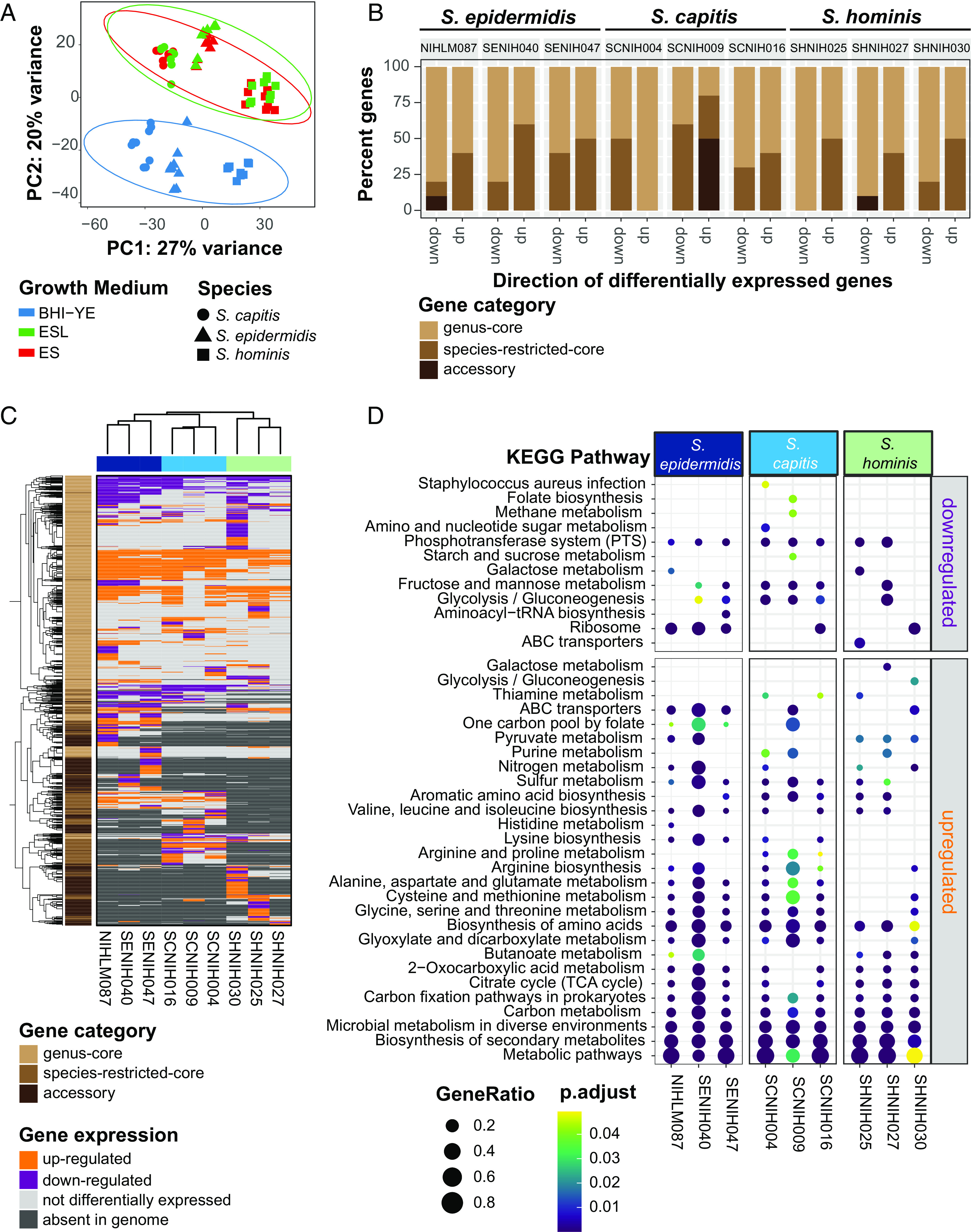
RNA-Seq analysis of nine distinct staphylococcal isolates in BHI-YE and in artificial skin media, ES, and ESL (N = 3 biological replicates). (A) Principal-component variance stabilizing transformation-normalized reads from all RNA-seq samples with 1,647 genus-core genes. Colors represent different growth media, and shapes represents staphylococcal species. (B) A stacked bar plot representing the proportion of genus-core, species-restricted-core, and accessory within the top 10 down- and up-regulated genes in each isolate in the ES medium relative to BHI-YE. (C) A discrete heatmap of log2-fold changes of all the genes that were differentially expressed (≥ or ≤ twofold change, adjusted P value < 0.05) in ES relative to BHI-YE. Columns are individual isolates and rows are DEGs. Genes up-regulated in the ES medium are shown in orange, down-regulated genes are in purple. Light gray cells represent genes that were encoded within an isolate genome, but not differentially expressed in that isolate. Dark gray cells denote genes that were absent in the isolate genome. The left sidebar depicts the category to which each gene belongs. Top and side dendrograms were generated by unsupervised clustering of the expression data (Euclidean distance; Ward). (D) KEGG pathway enrichment analysis of DEGs in ES medium relative to BHI-YE in each isolate. The X axis and Y axis show the isolate and pathway, respectively. Bubble size shows GeneRatio and color indicates adjusted P value.
To measure the contribution of species-specific-core and accessory genes, in addition to the genus-core genes, we next quantified the differential gene expression of each isolate individually. The number of differentially expressed genes (DEGs, ≥ or ≤ twofold change; adjusted P value < 0.05) in ES medium relative to BHI-YE, and their distribution between the genus-core, species-restricted-core and accessory gene categories for each isolate is shown in SI Appendix, Fig. S12 and Table S6 and Dataset S6. The majority of DEGs for each isolate belonged to the genus-core (median: 70%, range: 58 to 74%), followed by species-restricted-core (median: 18%, range: 13 to 29%) and then accessory (median: 13%, range: 3 to 17%), indicating that bulk of the DEGs belong to the conserved gene content. Interestingly, a closer look at the top 10 up-regulated and down-regulated genes in the ES medium relative to BHI-YE revealed that a disproportionately high percentage of up-regulated genes (median 50%) belonged to the species-restricted-core or accessory genes (Fig. 5B). Further, a comparison of gene expression patterns between all nine isolates in the ES medium relative to BHI-YE revealed extensive variation in all gene categories, even between conspecific isolates. A discrete heatmap displaying all 1,979 DEGs in the ES medium relative to BHI-YE (~26% genus pan-genome) across all isolates is shown in Fig. 5C. Within the DEGs,145, 216, and 102 genus-core genes and 58, 50, and 18 species-restricted-core genes have similar expression pattern in all three isolates of S. epidermidis, S. capitis, and S. hominis, respectively. Genes with similar gene expression patterns in all isolates included the upregulation of central metabolism genes involved in gluconeogenesis (pckA, gapB, ppdK, aldA, and S0553), tricarboxylic acid cycle (sdhA, sdhC, citB, citZ, and acnA), amino acid (metB, metC, yitJ, rocA, rocD, putA, ilvB, and yfmJ) and nucleotide (purE) metabolism, competence transcription factor (comK), and an alpha-glucosidase (yugT) in ES medium. This was accompanied by downregulation of genes involved in glycolysis (cggR, pgk, gapA, fruA, fruK, and ptsG) and zinc transport (adcA). Within the species-restricted-core genes, all S. capitis isolates showed upregulation of the lar operon, which is involved in utilization of L- and D-lactate that was present as the sole carbon source in the ES medium. Genes responding to the acidic conditions (pH 5.5) in the ES medium were up-regulated in all S. epidermidis isolates, including oppABCDF and ornithine biosynthesis: arcA, arcB, and argJBCD. In addition, genes involved in acetoin metabolism (lpdA and acoABC) that were core to S. epidermidis and S. hominis, but not S. capitis, were the most highly upregulated genes in all six isolates (median 254-fold change, range ~30 to 2,500), showing the importance of this pathway within the skin environment. Beyond these examples, however, most genes showed a wide variation in expression irrespective of the gene category. For example, mannitol metabolism pathway (mltABDF; >fourfold change), a fitness determinant under hyperosmotic conditions present on the skin (39), was encoded by all S. capitis, but only expressed by SCNIH004 in the ES medium. On the other hand, kdp operon (kdpABC; threefold to fourfold change), another S. capitis core locus that is induced by high NaCl concentration (40), was up-regulated in the ES medium in SCNIH009 and SCNIH016, but not in SCNIH004.
Finally, we sought to examine the broad functional classifications of genes that are significantly enriched in the ES medium relative to BHI-YE. KEGG enrichment analysis of DEGs identified overall enrichment in global metabolism pathways, including carbon and amino acid metabolism in all isolates (Fig. 5D). Despite the variation in DEGs, we saw a consistent enrichment in several amino acid metabolism pathways in S. epidermidis and S. capitis isolates, while butanoate metabolism genes, which encompasses acetoin metabolism, were enriched in all S. hominis and two S. epidermidis isolates.
Collectively, our data revealed a species-level signature to gene expression, accompanied with an extensive variability between isolates of the same species, which could be largely attributed to differential expression in the genus-core and species-restricted-core genes.
Discussion
The present study provides a detailed taxonomic and functional characterization of the most prevalent skin-associated staphylococci. Using the 16S rRNA gene as a taxonomic marker for species classification, our work revealed predominance of three staphylococcal species, viz. S. epidermidis, S. capitis, and S. hominis, for which humans serve as the primary host (23). In addition, many other clinically relevant species, including S. aureus, Staphylococcus lugdunensis, Staphylococcus pettenkoferi, Staphylococcus haemolyticus, Staphylococcus cohnii, and Staphylococcus saprophyticus were present in multiple volunteers at lower relative abundance. Further, S. aureus reached its highest relative abundance in the nares, where it was detected in 22% individuals, in agreement with known frequency of S. aureus nasal carriage (41).
Our amplicon data suggested that S. epidermidis, S. capitis, and S. hominis were generalists, colonizing multiple body sites at variable frequencies. However, a pan-genome analysis using isolates with detailed provenance tracking (both volunteer and body site of isolation) revealed that within each species isolates belonging to divergent phylogenetic clades exhibited body site preferences. Each clade was associated with distinct accessory genes, many of them encoding skin-relevant functions. Stable coexistence of genotypic clusters from the same species is thought to arise by adaptation to novel resources with nonuniform distribution within an environment (42). In-line with this hypothesis, human skin shows variations in temperature, pH, compositional differences in lipids, and in the expression of keratin and immune markers across different body sites (43), strongly suggesting the presence of spatially partitioned resources and antimicrobial factors, determining strain colonization. Future work directed towards identifying genes that are under selection in distinct staphylococcal clades should shed light on molecular mechanisms shaping body site preferences in these isolates.
Comparative pan-genome analysis between all three species showed that except for the essential and central metabolic genes, most of the genes were not uniformly shared across all species. One caveat of our pan-genome analysis is that some of the highly repetitive genes, including surface protein adhesins ebpS (elastin-binding) and srdH (unknown binding) (44), escaped ortholog clustering by reciprocal blast and were reported as distinct genes for each species. However, barring these few examples, most orthologs were successfully clustered together.
Functional analysis of the genus pan-genome showed that many of the species-restricted-core genes encoded skin-relevant functions, potentially allowing species to cocolonize by using distinct molecular pathways. While many of these genes have previously been studied as virulence determinants in pathogens such as S. aureus, these additional genomic data raise an intriguing possibility of these primarily being skin colonization factors that evolved in commensal staphylococci. For example, the arc gene cluster that is part of the arginine catabolic mobile element, and acts as a fitness determinant in MRSA, was part of the species-restricted-core genes in both S. epidermidis and S. capitis in our datasets. Recent studies speculate the role of arc genes in neutralizing the acidic skin condition, with S. epidermidis serving as the primary reservoir (45). Similar functional studies in other species-restricted-core genes should shed light on the selection pressures that retain and transfer these genes within the staphylococcal populations. In addition to these annotated genes, a large proportion (~19%) of the species-restricted-core genes were hypothetical, and future analysis of these genes could help identify novel functions unique to each species. For example, all S. capitis genomes carried a poorly characterized locus for gallidermin-type lantibiotic that might be crucial for competing with other skin resident microbes (46).
Finally, we used in vitro growth and RNA-Seq to characterize the inter- and intra-species phenotypic and transcriptional variation under similar growth conditions as a potential indicator of phylogenetic and ecological variation. Wide variation was observed in gene expression patterns between isolates, highlighting the importance of gene regulation in amplifying the genetically encoded differences between each isolate.
Overall, our current work, limited to a cohort of 22 healthy volunteers, presents a thorough analysis of the human skin-associated staphylococcal diversity that can serve as a reference for future studies of more geographically diverse human populations. Such studies would be essential to corroborate the universality of our findings for the larger human population.
Given the ease of culturing staphylococci, their survival outside the host environment, and our ability to genetically manipulate members of this genus (14), staphylococci hold great potential to provide strain-specific functions needed to maintain or restore a healthy skin barrier. For instance, the antibiotic-resistant pathogens such as methicillin-resistant S. aureus have reduced the efficacy of antibiotics and requires innovative solutions to combat this crisis. Traditional antagonism assays have shown that staphylococci are a rich source of antimicrobial peptides including lantibiotics and nonribosomally synthesized antibiotics such as lugdunin that have been shown to target S. aureus (7, 8, 47, 48). A systematic genome mining approach could enable the discovery of additional novel and unique functions to restrict colonization by deadly pathogens. Our current study provides an important resource for such ambitious future studies.
Materials and Methods
Healthy Volunteer Recruitment and Sampling.
Healthy volunteer recruitment, body site sampling, and 16S rRNA amplicon sequencing were performed as described previously (25). This natural history study was approved by the Institutional Review Board of the National Human Genome Research Institute (clinicaltrials.gov/NCT00605878) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (clinicaltrials.gov/NCT02471352), and all volunteers provided written informed consent prior to participation.
Microbiome Analyses.
16S rRNA amplicon (V1–V3) sequencing data were processed using the DADA2 pipeline version v1.2.0 (49) and downstream community analysis was carried out using phyloseq (50) in RStudio (R v4.2.0). Isolates for whole-genome sequencing (WGS) were obtained by plating samples on Tryptic Soy Blood Agar. WGS was carried out using Illumina MiSeq, PacBio Sequel II, or Oxford Nanopore Technology. Genomes were quality filtered according to completeness (≥98%) and contamination (≤5%) using CheckM (51) and dereplicated (>99.9% ANImf threshold) using dRep v3.2.2 (52). Genomes were annotated using prokka v1.14.6 (53), followed by functional analysis by eggNOG-mapper v2 (web version) and an in-house script to map completeness of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (54). Species-level pan-genome analysis was performed using panaroo v3.1.2 (28). A reciprocal-best-blast approach was used to cluster species-level pan-genome references at the protein level (≥40% identity and ≥50% coverage) to generate a merged genus pan-genome. Pan-genome analysis was conducted in RStudio. Maximum likelihood (ML) phylogenetic trees were constructed using RAxML-NG v1.1.0 (55), based on the core-gene alignment generated by panaroo. iTOL v6 (56) was used for tree display and annotation. Pan-genome-wide-association study analysis to identify clade-specific overrepresentation of genes was carried out using Scoary v1.6.16 (57). A greedy algorithm was used to select a subset of staphylococcal isolates for growth analysis.
Bacterial Isolate Growth and RNA-Seq Analysis.
Growth for kinetic analysis and RNA-Seq were carried out in rich BHI-YE and artificial skin media ES and ESL (Pickering Laboratories Catalog #s 1700-0023 and 1700-0556, respectively). Biological replicate data were pooled and AUC quantified using Growthcurver (58) in RStudio. For RNA-Seq, genomic DNA and Ribosomal RNA-depleted libraries were sequenced using Illumina NovaSeq 6000 platform. Rockhopper v 2.0.3 (59) was used to align reads from each sample to a reference genome. DESeq2 package (60) was used for raw read normalization and differential expression analysis. Detailed description of materials and methods is included in SI Appendix, Supplementary Text.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (DOCX)
Acknowledgments
We thank Lukian Robert and Qiong Chen for assisting with culturing, DNA extraction, and library preparation. The computational resources of the NIH High-Performance Computation Biowulf Cluster (https://hpc.nih.gov) were used for this study. This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Author contributions
P.J., S.C., H.H.K., and J.A.S. designed research; P.J., S.-Q.L.-L., C.D., and N.C.S.P. performed research; S.S.K. contributed new reagents/analytic tools; P.J. and S.C. analyzed data; H.H.K. and J.A.S. provided funding; N.C.S.P. group author with all authors listed in SI Appendix; H.H.K. collected samples; and P.J., S.C., and J.A.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Preprint server: https://www.biorxiv.org/content/10.1101/2023.06.22.546190v1.
Reviewers: M.H., University of St. Andrews; L.K., University of Wisconsin-Madison; and T.D.R., Emory University.
Contributor Information
Julia A. Segre, Email: jsegre@nhgri.nih.gov.
Collaborators: Beatrice B Barnabas, Sean Black, Gerard G Bouffard, Shelise Y Brooks, Juyun Crawford, Holly Marfani, Lyudmila Dekhtyar, Joel Han, Shi-Ling Ho, Richelle Legaspi, Quino L Maduro, Catherine A Masiello, Jennifer C McDowell, Casandra Montemayor, James C Mullikin, Morgan Park, Nancy L Riebow, Karen Schandler, Brian Schmidt, Christina Sison, Sirintorn Stantripop, James W Thomas, Pamela J Thomas, Meghana Vemulapalli, and Alice C Young
Data, Materials, and Software Availability
Genome data are deposited under the NCBI BioProjects PRJNA694925 and PRJNA986048 (61, 62). Some amplicon data were published previously (N = 145; PRJNA46333) (63) and the remainder are new to this study (N = 168; PRJNA46333). RNAseq data have been deposited under BioProject PRJNA694925. Code for this project along with the phyloseq object are available at https://github.com/skinmicrobiome/Joglekar_Staphylococcus_2023 (64).
Supporting Information
References
- 1.Proksch E., Brandner J. M., Jensen J. M., The skin: An indispensable barrier. Exp. Dermatol. 17, 1063–1072 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Oh J., et al. , Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh J., et al. , Temporal stability of the human skin microbiome. Cell 165, 854–866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris-Tryon T. A., Grice E. A., Microbiota and maintenance of skin barrier function. Science 376, 940–945 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Otto M., Staphylococci in the human microbiome: The role of host and interbacterial interactions. Curr. Opin. Microbiol. 53, 71–77 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Iwase T., et al. , Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Nakatsuji T., et al. , Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 9, eaah4680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zipperer A., et al. , Human commensals producing a novel antibiotic impair pathogen colonization. Nature 535, 511–516 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Cogen A. L., et al. , Staphylococcus epidermidis antimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS One 5, e8557 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill A. M., et al. , Identification of a human skin commensal bacterium that selectively kills cutibacterium acnes. J. Invest. Dermatol. 140, 1619–1628.e1612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y., et al. , Commensal Staphylococcus epidermidis contributes to skin barrier homeostasis by generating protective ceramides. Cell Host Microbe. 30, 301–313.e309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naik S., et al. , Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Q., et al. , Staphylococcus epidermidis contributes to healthy maturation of the nasal microbiome by stimulating antimicrobial peptide production. Cell Host Micr obe 27, 68–78.e65 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y. E., et al. , Engineered skin bacteria induce antitumor T cell responses against melanoma. Science 380, 203–210 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Becker K., Heilmann C., Peters G., Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27, 870–926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grice E. A., et al. , Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meric G., et al. , Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 7, 1313–1328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conlan S., et al. , Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol. 13, R64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espadinha D., et al. , Distinct phenotypic and genomic signatures underlie contrasting pathogenic potential of Staphylococcus epidermidis clonal lineages. Front. Microbiol. 10, 1971 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tettelin H., et al. , Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial "pan-genome". Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vital M., et al. , Gene expression analysis of E. coli strains provides insights into the role of gene regulation in diversification. ISME J. 9, 1130–1140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conlan S., Kong H. H., Segre J. A., Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS One 7, e47075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kloos W. E., Natural populations of the genus Staphylococcus. Annu. Rev. Microbiol. 34, 559–592 (1980). [DOI] [PubMed] [Google Scholar]
- 24.Zhou W., et al. , Host-specific evolutionary and transmission dynamics shape the functional diversification of Staphylococcus epidermidis in human skin. Cell 180, 454–470.e418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Findley K., et al. , Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price P. B., The bacteriology of normal skin; A new quantitative test applied to a study of the bacterial flora and the disinfectant action of mechanical cleansing. J. Infect. Dis. 63, 301–318 (1938). [Google Scholar]
- 27.Jain C., Rodriguez R. L., Phillippy A. M., Konstantinidis K. T., Aluru S., High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tonkin-Hill G., et al. , Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghssein G., et al. , Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 352, 1105–1109 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Zhou C., et al. , Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 15, e1007538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertrand B. P., et al. , Role of Staphylococcus aureus formate metabolism during prosthetic joint infection. Infect. Immun. 90, e0042822 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai Y., et al. , The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol. Microbiol. 63, 497–506 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Dubin G., et al. , Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol. Chem. 382, 1575–1582 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Rudden M., et al. , The molecular basis of thioalcohol production in human body odour. Sci. Rep. 10, 12500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konstantinidis K. T., et al. , Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc. Natl. Acad. Sci. U.S.A. 106, 15909–15914 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chong C. E., Bengtsson R. J., Horsburgh M. J., Comparative genomics of Staphylococcus capitis reveals species determinants. Front. Microbiol. 13, 1005949 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wesley K. H. S., Kloos E., Isolation and characterization of Staphylococci from human skin II. Descriptions of four new species: Staphylococcus warneri, Staphylococcus capitis, Staphylococcus hominis, and Staphylococcus simulans. Int. J. Syst. Bacteriol. 25, 50–61 (1975). [Google Scholar]
- 38.Byrd A. L., Belkaid Y., Segre J. A., The human skin microbiome. Nat Rev Microbiol 16, 143–155 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Nguyen T., et al. , Targeting mannitol metabolism as an alternative antimicrobial strategy based on the structure-function study of mannitol-1-phosphate dehydrogenase in Staphylococcus aureus. mBio 10, e02660-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price-Whelan A., et al. , Transcriptional profiling of Staphylococcus aureus during growth in 2 M NaCl leads to clarification of physiological roles for Kdp and Ktr K+ uptake systems. mBio 4, e00407-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Eiff C., Becker K., Machka K., Stammer H., Peters G., Nasal carriage as a source of Staphylococcus aureus bacteremia, Study Group. N Engl. J. Med. 344, 11–16 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Shapiro B. J., Polz M. F., Microbial speciation. Cold Spring Harb. Perspect. Biol. 7, a018143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merleev A. A., et al. , Biogeographic and disease-specific alterations in epidermal lipid composition and single-cell analysis of acral keratinocytes. JCI Insight 7, e159762 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otto M., Staphylococcus epidermidis–the "accidental" pathogen. Nat. Rev. Microbiol. 7, 555–567 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otto M., Coagulase-negative staphylococci as reservoirs of genes facilitating MRSA infection: Staphylococcal commensal species such as Staphylococcus epidermidis are being recognized as important sources of genes promoting MRSA colonization and virulence. Bioessays 35, 4–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dobson A., Cotter P. D., Ross R. P., Hill C., Bacteriocin production: A probiotic trait? Appl. Environ. Microbiol. 78, 1–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Janek D., Zipperer A., Kulik A., Krismer B., Peschel A., High frequency and diversity of antimicrobial activities produced by nasal Staphylococcus strains against bacterial competitors. PLoS Pathog. 12, e1005812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakatsuji T., et al. , Competition between skin antimicrobial peptides and commensal bacteria in type 2 inflammation enables survival of S. aureus. Cell Rep. 42, 112494 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callahan B. J., et al. , DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMurdie P. J., Holmes S., phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W., CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olm M. R., Brown C. T., Brooks B., Banfield J. F., dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seemann T., Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Saheb Kashaf S., et al. , Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Nat. Microbiol. 7, 169–179 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kozlov A. M., Darriba D., Flouri T., Morel B., Stamatakis A., RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Letunic I., Bork P., Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brynildsrud O., Bohlin J., Scheffer L., Eldholm V., Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17, 238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sprouffske K., Wagner A., Growthcurver: An R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17, 172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McClure R., et al. , Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41, e140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conlan S., Deming C., Segre J. A., NISC Comparative Sequencing Program, Uncovering new skin microbiome diversity through culturing and metagenomics. NCBI. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA694925. Deposited 26 January 2021.
- 62.Joglekar P., et al. , Coagulase negative staphylococci from human skin Genome sequencing and assembly. NCBI. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA986048. Deposited 21 June 2023.
- 63.Oh J., et al. , The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 23, 2103–2114 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joglekar P., Conlan S., Segre J. A., Joglekar_Staphylococcus_2023. Github. https://github.com/skinmicrobiome/Joglekar_Staphylococcus_2023. Deposited 16 June 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (DOCX)
Data Availability Statement
Genome data are deposited under the NCBI BioProjects PRJNA694925 and PRJNA986048 (61, 62). Some amplicon data were published previously (N = 145; PRJNA46333) (63) and the remainder are new to this study (N = 168; PRJNA46333). RNAseq data have been deposited under BioProject PRJNA694925. Code for this project along with the phyloseq object are available at https://github.com/skinmicrobiome/Joglekar_Staphylococcus_2023 (64).