

Significance
Transmembrane Cav2.2 voltage-gated calcium channels play a central role in pain through providing the Ca2+ for sustained neuronal firing and neurotransmitter release. Therapeutics targeting Cav2.2 include the blocking peptide toxin Ziconotide and the gabapentinoids [e.g., Gabapentin (GBP)], which target an auxiliary subunit of Cav2.2; however, both drugs are encumbered with numerous side effects. Collapsin response mediator protein 2 (CRMP2) is a key cytosolic regulator of Cav2.2. CBD3063, a peptidomimetic that we devised, selectively curbs Cav2.2 activity, paralleling GBP in alleviating neuropathic and inflammatory pain across sexes. In contrast to GBP, CBD3063 upheld the protective/adaptive role of pain, without negatively affecting sedative, depressive, or cognitive behaviors. In short, CBD3063 may be a superior successor in pain management.
Keywords: Cav2.2, peptidomimetic, electrophysiology, chronic pain, analgesia
Abstract
Transmembrane Cav2.2 (N-type) voltage-gated calcium channels are genetically and pharmacologically validated, clinically relevant pain targets. Clinical block of Cav2.2 (e.g., with Prialt/Ziconotide) or indirect modulation [e.g., with gabapentinoids such as Gabapentin (GBP)] mitigates chronic pain but is encumbered by side effects and abuse liability. The cytosolic auxiliary subunit collapsin response mediator protein 2 (CRMP2) targets Cav2.2 to the sensory neuron membrane and regulates their function via an intrinsically disordered motif. A CRMP2-derived peptide (CBD3) uncouples the Cav2.2–CRMP2 interaction to inhibit calcium influx, transmitter release, and pain. We developed and applied a molecular dynamics approach to identify the A1R2 dipeptide in CBD3 as the anchoring Cav2.2 motif and designed pharmacophore models to screen 27 million compounds on the open-access server ZincPharmer. Of 200 curated hits, 77 compounds were assessed using depolarization‐evoked calcium influx in rat dorsal root ganglion neurons. Nine small molecules were tested electrophysiologically, while one (CBD3063) was also evaluated biochemically and behaviorally. CBD3063 uncoupled Cav2.2 from CRMP2, reduced membrane Cav2.2 expression and Ca2+ currents, decreased neurotransmission, reduced fiber photometry-based calcium responses in response to mechanical stimulation, and reversed neuropathic and inflammatory pain across sexes in two different species without changes in sensory, sedative, depressive, and cognitive behaviors. CBD3063 is a selective, first-in-class, CRMP2-based peptidomimetic small molecule, which allosterically regulates Cav2.2 to achieve analgesia and pain relief without negative side effect profiles. In summary, CBD3063 could potentially be a more effective alternative to GBP for pain relief.
Cav2.2 (N-type) voltage-gated calcium channels are expressed in key sites for nociceptive transmission including the dorsal root ganglia (DRG) and spinal cord dorsal horn (DH). Within the DH, Cav2.2 channels are localized in the central terminals of primary afferent fibers where they mediate the release of excitatory neurotransmitters (1). Cav2.2 deficient mice show reduced responses to intraplantar formalin and increased acute thermal nociceptive thresholds (2–4). In animal models of neuropathic pain, the expression and function of Cav2.2 channels increase concurrently with the development of mechanical and thermal allodynia (5, 6). Genetic ablation (4) or pharmacological blockade of Cav2.2 reduces behavioral signs of neuropathic pain (7, 8), demonstrating the relevance of this channel for therapeutic intervention.
Three drugs targeting Cav2.2 are commercially available for managing neuropathic pain. Ziconotide (Prialt®)—a synthetic version of the cone snail toxin ω-conotoxin MVIIA—is a Cav2.2 blocker and was the first nonopioid intrathecal (i.t.) analgesic approved by the US Food and Drug Administration for treatment of intractable chronic pain (9). Its use is hampered by its invasive route of administration, narrow therapeutic window, and a panoply of side effects (10). Gabapentin (GBP) (Neurontin®) and Pregabalin (Lyrica®)—ligands of the α2δ-1 auxiliary subunit—alleviate chronic pain by disrupting the Cav2.2–α2δ-1 interaction to prevent channel trafficking to the plasma membrane (11, 12). Both gabapentinoids have low efficacy and present with serious side effects (13). Gabapentinoids misuse led to increased overdose-related deaths from 2019 to 2020 (14). Nevertheless, gabapentinoids remain first-line neuropathic pain treatments. Developing effective pain management with minimal side effects is crucial.
We previously identified the microtubule-binding collapsin response mediator protein 2 (CRMP2) as a regulator of Cav2.2 trafficking and function (15–17). CRMP2 overexpression leads to enhanced Cav2.2 currents and surface expression (16, 17) and enhanced neurotransmitter release (16, 17) in hippocampal (16) and DRG neurons (17). We identified an intrinsically disordered 15-amino-acid peptide (designated CBD3, for calcium channel binding domain 3) from CRMP2 that interferes with the Cav2.2–CRMP2 interaction and decreases calcium influx, transmitter release, and acute, inflammatory, and neuropathic pain (18). Our analysis of CBD3 revealed an antinociceptive core in the first six amino acids with two residues (Ala1 and Arg4) accounting for most of the binding affinity (19).
Due to incomplete structural information on the Cav2.2 intracellular domains important for interaction with CRMP2 to guide rational design of small molecule inhibitors, we developed a pipeline that leverages molecular dynamics of the core CBD3 peptide and identified its most stable conformational motif as the A1R2 dipeptide. Based on well-established biophysical principles (20), we took this motif to be the anchor of the interaction responsible for molecular recognition of Cav2.2 and used the A1R2 ensemble as a scaffold to design pharmacophore models to search for compounds to disrupt the CRMP2–Cav2.2 interaction. Screening 27 million compounds led to identification of a first-in-class small molecule peptidomimetic [(3R)‐3‐acetamido‐N‐[3‐(pyridin‐2‐ylamino)propyl]piperidine‐1‐carboxamide; hereafter designated CBD3063] that disrupted the Cav2.2–CRMP2 interaction, antagonized Cav2.2 function, and blocked experimental neuropathic and inflammatory pain.
Results
Stability Analysis of CBD3 and Identification of a Small Molecule Peptidomimetic.
Underlying molecular recognition is the ability of a ligand to present a suitable structural motif for at least a few nanoseconds to efficiently engage its receptor (20). A far-western assay of CRMP2 peptides revealed that the highest binding to Cav2.2 was attained by peptide ARSRLAELRGVPRGL (CBD3) (18). Subsequent work identified the first six amino acids ARSRLA as the core peptide with two residues (Ala1 and Arg4) critical for binding affinity (19). To predict the recognition motif in CBD3, we performed three independent molecular dynamics simulations (MDS) of the core peptide, scanned the trajectories for the most stable dipeptide conformation, and assessed whether the side chains are blocked by intrapeptide contacts or exposed to solvent and hence free to interact. The A1R2 dipeptide formed the most stable solvent-exposed motif, while the rest of the peptide was mostly cluttered (Fig. 1 A and B and SI Appendix, Fig. S1). This peptide, identified from the conformation having the largest 1 Å radius cluster, encompassed about 44% of our total runs. Since previously reported cell-based experiments used the cell-penetrating tat sequence conjugated to CBD3, we ran MDS for tat-ARSRLA, obtaining the same structural motif (SI Appendix, Fig. S1). The robustness of the A1R2 motif led us to use it as the basis for design of a pharmacophore model (Fig. 1C) which we used in the open-access server ZincPharmer (http://zincpharmer.csb.pitt.edu/) to search more than 27 million commercially available compounds from the ZINC database (21). Based on availability and manual curation, we selected 77 peptidomimetics for experimental validation (Fig. 1D and SI Appendix, Fig. S2).
Fig. 1.
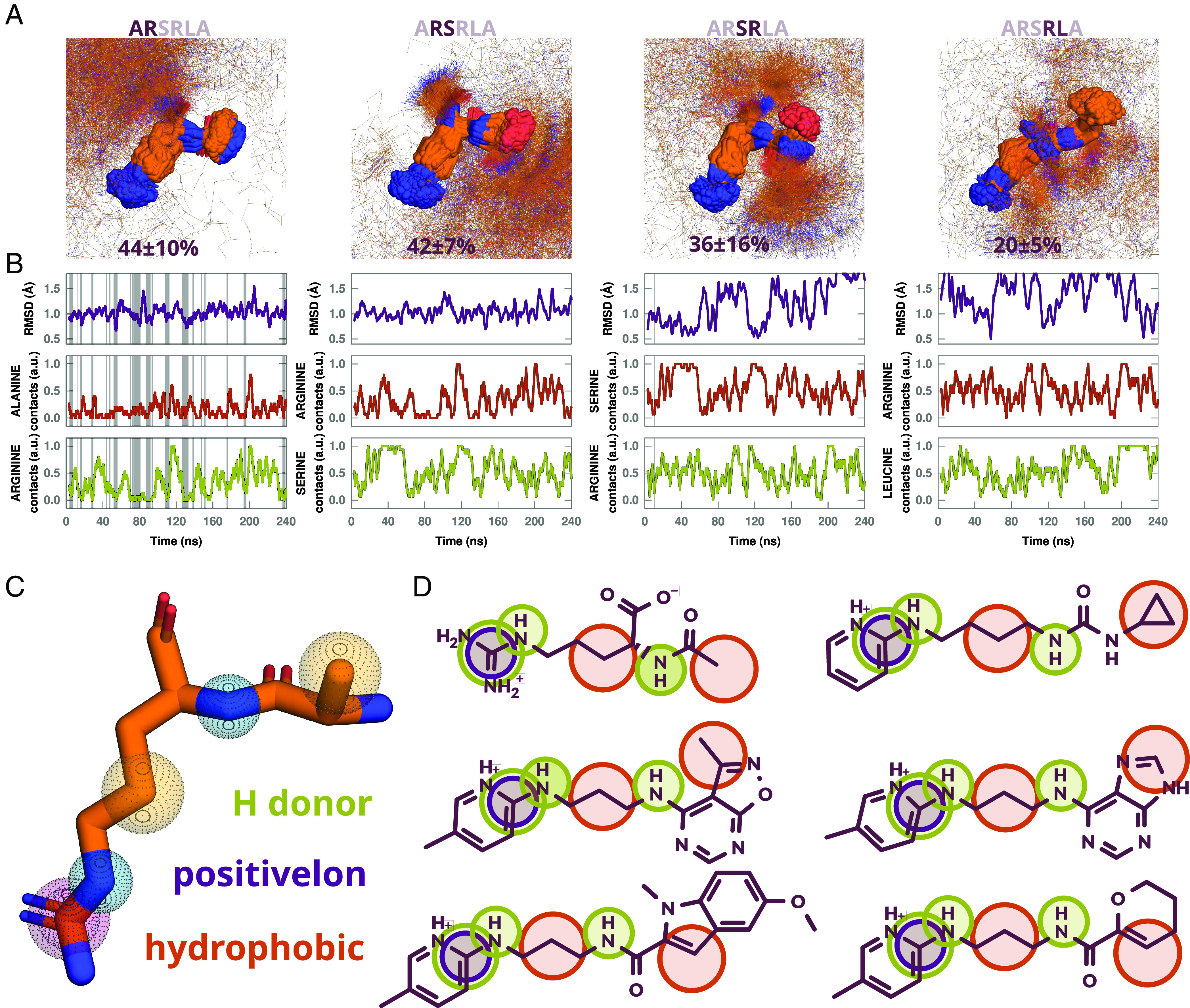
Stability analysis of CBD3 dipeptides, pharmacophore model on average A1R2 dipeptide cluster center, and sample of matched hits. (A) Largest cluster of MD snapshots with rmsd less than 1 Å for dipeptides A1R2, R2S3, S3R4, and R4L5, with average percentage of cluster size in 3 independent simulations. (B) rmsd relative to cluster center and times the amino acid side chains are free of contacts (3.8 Å for hydrogen bond donors and 4 Å for hydrophobic side chains) within 3-ns windows in a representative MD trajectory. Gray regions highlight times for which RMSD of snapshots are less than 1 Å from cluster center and side chains are 80% or more free of contacts within 3 ns windows. (C) Pharmacophores radii used: guanidine group, 0.78 Å for positive ion and 1 Å for hydrogen bond donor; 0.78 Å for other hydrogen bond donors, and 1.0 Å for hydrophobic atoms. (D) Sample of compounds that matched all pharmacophores.
We reported that targeting the Cav2.2–CRMP2 interaction with tat-CBD3 (18) or a membrane-tethered myristoylated variant (myr-tat-CBD3) (22) peptides reduces depolarization-induced Ca2+-influx in sensory neurons. However, acute application (5 to 30 min) of myr-tat-CBD3 peptide had no effect on Ca2+ influx due to the time required for peptide uptake and/or Cav2.2 trafficking alterations. In contrast, chronic application (~24 h) of myr-tat-CBD3 peptide reduced Ca2+ currents by ~40% (22). Hence, we incubated test compounds overnight at 20 µM and used Fura 2-AM-based ratiometric Ca2+-imaging to screen for altered Ca2+-influx in rat DRG neurons (DRGs). To activate high-voltage-activated Ca2+ channels, we challenged DRGs of all sizes with 90 mM KCl (23). We identified nine compounds that inhibited Ca2+ influx by more than 50% relative to control (SI Appendix, Fig. S3). All nine compounds (SI Appendix, Fig. S4) can be assigned to three chemotypes. Specifically, CBD3018, 3026, 3062, 3065, and 3074 are guanidines; CBD3033, 3038, and 3039 are 2-aminopyridylpropylcarboxamides; and CBD3062 and 3063 are 2-aminopyridylpropylureas.
Next, we tested the top nine compounds in small-to-medium rat DRG neurons using patch-clamp recordings. DRGs were exposed overnight to 20 µM of compounds or vehicle (0.1% dimethylsulfoxide (DMSO)). Ba2+ mediated the inward current, denoted as Ca2+ currents. From a holding potential of −90 mV, 200-ms depolarization steps from −70 to +60 mV in 10 mV increments elicited a family of Ca2+ currents (SI Appendix, Fig. S3B). Comparing peak current density (pA/pF), CBD3063, 3,065, and 3,074 notably decreased Ca2+ currents vs. vehicle-treated cells (SI Appendix, Fig. S3C). Among these three compounds, CBD3063 and CBD3065 were unique (SI Appendix, Table S1) and chosen for further study. However, CBD3065 lacked analgesic effects due to differences in physicochemical properties (SI Appendix, Table S1) and was not tested further. CBD3063 (20 µM) decreased Ca2+ current density from 0 to 30 mV (SI Appendix, Fig. S5 A and B). At peak density, CBD3063 inhibited currents by ~46.5% compared to control. Analysis of voltage dependence and steady-state inactivation showed no significant differences between groups (SI Appendix, Fig. S5D and Table S2). These data show that CBD3063 inhibits Ca2+ currents in small-to-medium, likely nociceptive, sensory neurons.
CBD3063 Disrupts Cav2.2–CRMP2 Binding and Impairs Membrane Trafficking of Cav2.2 Channels without Affecting CRMP2 Posttranslational Modifications.
We previously showed that tat-CBD3 (18) and myr-tat-CBD3 (22) peptides hinder the Cav2.2–CRMP2 interactions. Therefore, we investigated whether CBD3063 could achieve similar interference (1, Fig. 2A) in CAD cells, a mouse neuronal line expressing both CRMP2 and Cav2.2. Immunoprecipitation assays showed that CBD3063 (20 µM) reduced Cav2.2 protein association with CRMP2 by ~35%, compared to control (Fig. 2 B and C). CBD3063 also decreased the closeness of Cav2.2–CRMP2 in neurons by the proximity ligation assay (PLA) (Fig. 2 D and E). Thus, CBD3063 disrupts association of CRMP2 with Cav2.2.
Fig. 2.
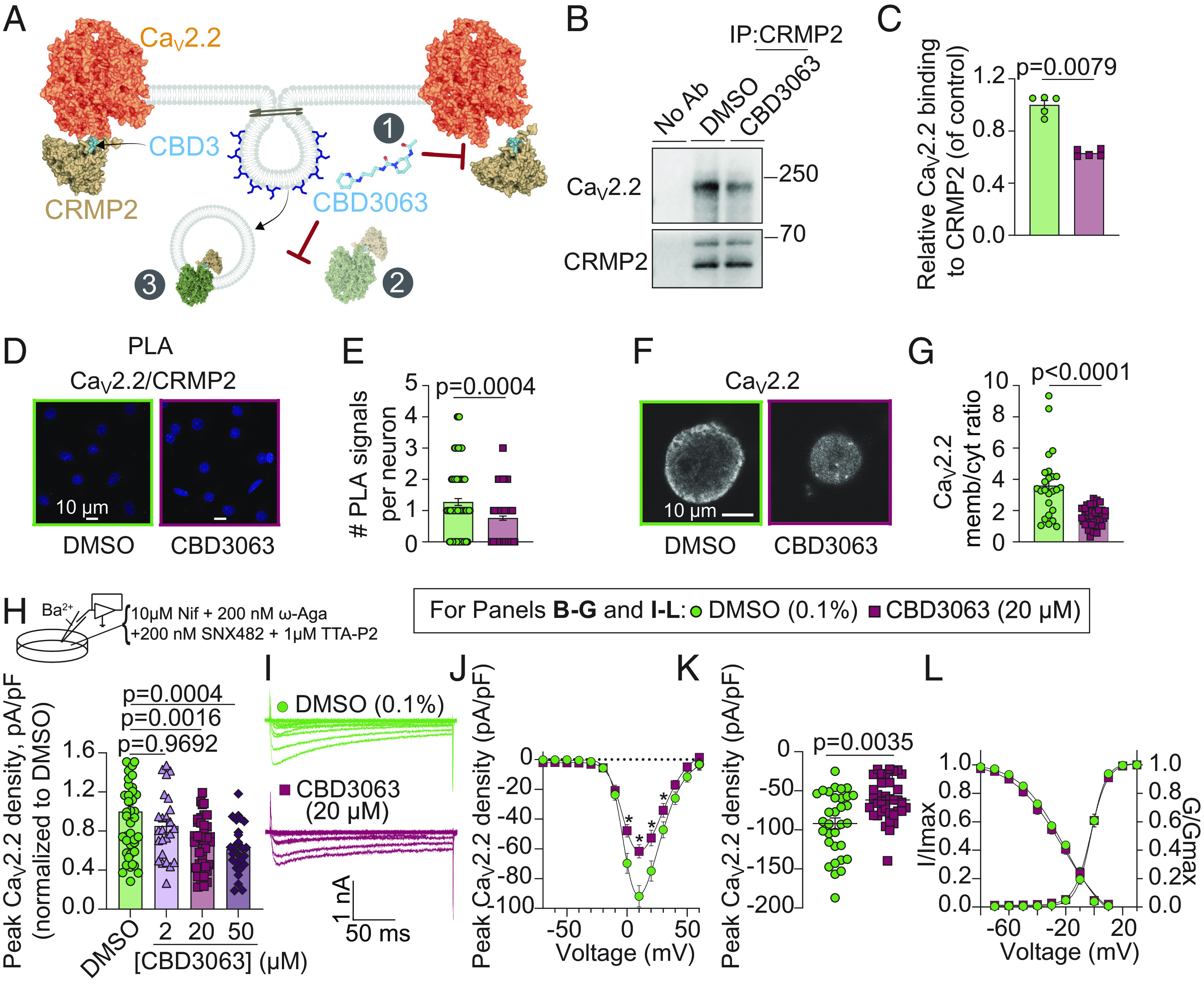
CBD3063 suppresses Cav2.2–CRMP2 interaction, surface trafficking of Cav2.2, and N-type (Cav2.2) calcium currents. (A) Schematic representation of CBD3063 modulation of CaV2.2–CRMP2 interaction. 1, CBD3063 disrupts CaV2.2–CRMP2 binding. 2, CBD3063 prevents CRMP2-mediated surface trafficking of CaV2.2 channels to the plasma membrane via CaV2.2 endocytosis (3). Representative immunoblots (B) and summary (C) of CRMP2 immunoprecipitation (IP) to detect Cav2.2 from CAD cells treated overnight with CBD3063 (20 µM) (n = 5 independent assays). P value as indicated; Mann-Whitney test. (D) Representative images of rat DRGs incubated overnight with 0.1% DMSO (control) or 20 µM CBD3063 following the PLA. PLA immunofluorescence labeled sites of interaction between CRMP2 and Cav2.2 (red puncta). In addition, nuclei are labeled with DAPI. (Scale bar: 10 µm.) (E) PLA analysis reveals fewer CaV2.2–CRMP2 interactions in CBD3063-treated DRGs than in controls (0.1% DMSO) (n = 102 to 141 cells). (F) Representative images of CaV2.2-labeled DRGs. (G) Overnight treatment with 20 µM CBD3063 affects CaV2.2 membrane/cytosol ratio in DRGs (n = 25 to 38 cells). (H) Schematic highlights bath solution for CaV2.2 current isolation. Graph summarizes peak N-type ICa2+ density in DRGs treated with CBD3063 or DMSO (n = 25 to 46 cells from five rats). (I) Example N-type calcium current traces from small-to-medium sized DRGs treated overnight with 20 µM CBD3063. Currents activated by a 200-ms pulse between −70 and +60 mV. (J) Double Boltzmann curve fits for current density–voltage. Asterisk (*) indicates P < 0.05. (K) Summary graph of peak N-type calcium current densities. (L) Boltzmann fits for voltage-dependent activation and inactivation. Half-maximal activation potential (V1/2) and slope values (k) detailed in SI Appendix, Table S2. Data derived from 32 to 36 cells across six rats. Error bars show mean ± SEM. See Dataset S1 for full statistics.
We previously showed that CRMP2 facilitates and that cell-penetrant CBD3 peptides decrease trafficking of Cav2.2 to the plasma membrane (16–18, 22). We next asked whether CBD3063 alters Cav2.2 subcellular localization (2 and 3, Fig. 2A). DRGs were incubated with vehicle (0.1% DMSO) or CBD3063 (20 µM) overnight prior to immunofluorescent microscopy. In DRGs treated with vehicle, Cav2.2 fluorescence appeared as an annulus at the plasma membrane (Fig. 2F). Cav2.2 localization at the membrane was significantly decreased in cells incubated with CBD3063 (Fig. 2G), showing that CBD3063 reduces cell-surface trafficking of Cav2.2 to the plasma membrane of sensory neurons.
We examined whether CBD3063 affects the phosphorylation and small ubiquitin-like modifier (SUMO)ylation of CRMP2, both of which were reported to influence its cellular functions(24). Immunoblot tests on CAD cell lysates treated with CBD3063 showed no change in CRMP2’s phosphorylation (SI Appendix, Fig. S6 A–F) or SUMOylation (SI Appendix, Fig. S6 G and H) levels compared to DMSO-treated cells. Thus, CBD3063 does not affect these posttranslational modifications of CRMP2.
CBD3063 Selectively Decreases Cav2.2 (N-type) Ca2+ Currents in Sensory Neurons.
CBD3 primarily blocks N-type currents (18), so we investigated whether CBD3063 acts similarly in patch-clamp recordings using a calcium channel blocker cocktail on DRGs treated with varying CBD3063 concentrations or a 0.1% DMSO control. CBD3063, at 20 and 50 µM, reduced peak N-type channel current density compared to control (Fig. 2H). Given minimal difference between 20 and 50 µM (Fig. 2 I and J), we chose 20 µM for further studies. Given minimal difference between 20 and 50 µM, we chose 20 µM for further studies. At peak current density (+10 mV; Fig. 2K), CBD3063 decreased N-type currents by ~33% in female and ~34% in male DRGs (SI Appendix, Fig. S7 A and B), vs. control. Activation and inactivation curves showed no noticeable variations (Fig. 2L and SI Appendix, Fig. S7C and Table S2). Acute CBD3063 exposure did not impact N-type currents (SI Appendix, Fig. S8), indicating no direct channel binding.
Current density–voltage curves and peak current densities were unaltered for Cav1 (L-type; SI Appendix, Fig. S9 A and B), Cav2.1 (P/Q-type; SI Appendix, Fig. S9 C and D), Cav2.3 (R-type; SI Appendix, Fig. S9 E and F), and Cav3 (T-type; SI Appendix, Fig. S9 G and H) when CBD3063 was incubated overnight. CBD3063 did not affect the voltage dependence of activation or inactivation of these channels (SI Appendix, Table S2).
We previously reported that CRMP2 regulates voltage-gated Na+ channels (25–27). However, CDB3063 did not affect Na+ current density or gating properties of Na+ channels in DRGs (SI Appendix, Fig. S10 A–C and Table S2). We also found that CBD3063 did not affect total potassium currents (SI Appendix, Fig. S10 D and E), fast inactivating (IKA, SI Appendix, Fig. S10 F and G) and slowly inactivating (IKS, SI Appendix, Fig. S10 H and I) potassium currents, or hyperpolarization-activated cyclic, nucleotide-gated channels (SI Appendix, Fig. S10 J and K). Therefore, CBD3063 specifically inhibits Cav2.2, leaving other DRG-expressed voltage-gated ion channels untouched.
Disrupting Cav2.2–CRMP2 Interaction Decreases Sensory Neuron Excitability and Spinal Cord Neurotransmission.
Modulating Cav2.2 channel activity influences DRG neuron excitability (6, 22, 28). We investigated whether CBD3063, targeting Cav2.2, impacts sensory neuron excitability. While CBD3063 did not alter resting membrane potential, it increased the rheobase (minimum current to elicit an AP) and decreased the number of evoked APs over varied current injections (SI Appendix, Fig. S11). This indicates that CBD3063 inhibits Cav2.2 channels, reducing sensory neuron excitability.
Cav2.2 regulates neurotransmitter release in sensory neurons connecting to second-order neurons in the DH. We explored whether CBD3063 could disrupt this communication (Fig. 3A). Treatment of mouse spinal cord slices for ~30 min with CBD3063 (20 µM) decreased the frequency and amplitude of sEPSCs (spontaneous excitatory postsynaptic currentsf) compared to controls (Fig. 3 B–D). Cumulative probability plots revealed an increase in interevent intervals and a decrease in amplitude during CBD3063 treatment (Fig. 3 E and F). This suggests that targeting the Cav2.2–CRMP2 interaction in the spinal cord can inhibit neuronal activity.
Fig. 3.

CBD3063 decreases spinal cord excitability and excitatory neurotransmitter release. (A) Hypothesis: In the DH of the spinal cord, CBD3063 interrupts the Cav2.2–CRMP2 interaction, leading to reduced pronociceptive transmitter release. (B) Representative traces of sEPSC recordings from substantia gelatinosa (SG) neurons in control conditions (Vehicle-ACSF) and after perfusing CBD3063 (20 µM) for 30 min. (C) Bar graph with scatter plot showing sEPSC frequency reduction after CBD3063 perfusion. (D) Bar graph with scatter plot showing that sEPSC amplitude is also decreased after perfusing spinal cord slices with CBD3063. P values as indicated; paired t test; n = 9 cells. (E) Cumulative distribution of sEPSC interevent intervals recorded from SG neurons. (F) Cumulative distribution of sEPSC amplitude intervals recorded from SG neurons. P values as indicated; Kolmogorov–Smirnov test; n = 9 cells. (G) KCl depolarization-evoked immunoreactive calcitonin gene–related peptide (CGRP) release was measured from spinal cords isolated from naive female rats following preincubation and incubation with 0.1% DMSO (control) or CBD3063 (20 μM) for 30 min. Graph showing iCGRP levels observed in bath solution normalized to the weight of each spinal cord tissue. P values as indicated; two-way ANOVA; n = 5 rats. (H) The bar graph shows that the peak evoked CGRP release of the treatment fraction was decreased by CBD3063 (20 µM) treatment compared with control (0.1% DMSO). P values as indicated; unpaired t test; n = 5 rats. For all data, error bars indicate mean ± SEM. Refer to Dataset S1 for full statistics.
Presynaptic Cav2.2 channels influence neurotransmitter release in sensory neurons (29). We tested whether CBD3063 could reduce the release of the excitatory pronociceptive neurotransmitter calcitonin gene–related peptide (CGRP) (30) using an ex vivo assay with KCl-induced depolarization. CBD3063 (20 µM, 30 min pretreatment) decreased iCGRP release by ~63% (Fig. 3 G and H). This aligns with prior results using tat-CBD3 (18), suggesting that disrupting Cav2.2–CRMP2 interaction lessens spinal neurotransmitter release.
Intraperitoneal (i.p.) Administration of CBD3063 Is Antinociceptive in Mouse Models of Neuropathic Pain.
The primary neuropathic pain analgesics, gabapentinoids like GBP and Pregabalin, clinically alleviate pain by presumably disrupting the interaction between Cav2.2 and α2δ-1 subunits. (11, 12). Therefore, we evaluated the potential antinociceptive properties of CBD3063 in mice against the first-line neuropathic pain analgesic, GBP (30 mg/kg, i.p.) (31), using the spared nerve injury (SNI) model of neuropathic pain (Fig. 4A). SNI reliably produced robust mechanical allodynia in male and female mice at the 21-d post-surgical timepoint (Fig. 4B). Intraperitoneal CBD3063 administration dose-dependently mitigated SNI-induced mechanical allodynia (Fig. 4 B and C) with an ED50 of 1.02 mg/kg (Fig. 4D). Similarly, CBD3063 exhibited a dose-dependent reversal of SNI-induced cold allodynia (Fig. 4 E and F) with an ED50 of 1.50 mg/kg (Fig. 4G). CBD3063 (10 mg/kg) effectively paralleled the antinociceptive strength of GBP (30 mg/kg) in alleviating the behavioral signs of neuropathic pain (Fig. 4).
Fig. 4.
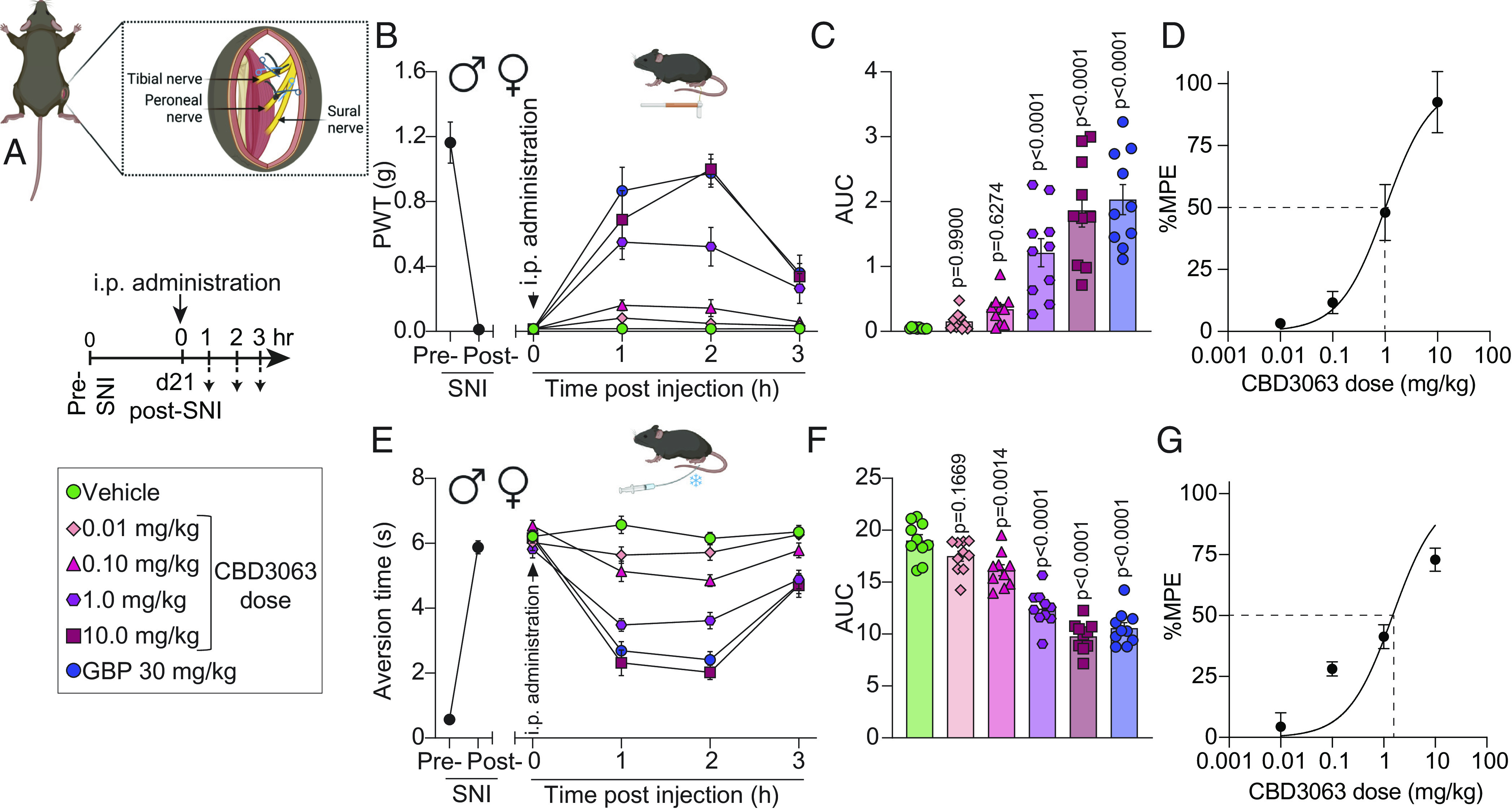
CBD3063 reverses mechanical and cold allodynia in two models of neuropathic pain. (A) SNI model schematic. Timeline and treatment conditions indicated. (B) Baseline (BL) paw withdrawal threshold (PWT) measurements were conducted before (pre-SNI) and after (post-SNI) nerve injury. Dose–response curves of single i.p. injections of vehicle, CBD3063, or GBP were measured from 0 to 3 h post injection on day 21 post-SNI. n = 10 mice. Results were compared using two-way ANOVA, factors; time*treatment, and Tukey post hoc test. (C) Quantification of area under the curve (AUC) in panel B from 0 to 3 h. CBD3063 (1 to 10 mg/kg) reversed SNI-induced mechanical allodynia at a similar fashion as GBP. P values as indicated; one-way ANOVA with the Dunnett post hoc test. n = 10 mice. (D) Dose–response curve analysis for the antinociceptive activity induced by CBD3063. MPE (%) = Antinociception as a percentage of the maximum possible effect. (E) BL measurements of aversion time to acetone stimulation were conducted before (pre-SNI) and after (post-SNI) nerve injury. Aversion time responses were measured from 0 to 3 h post injection. n = 10 mice. Results were compared using two-way ANOVA, time*treatment, and Tukey post hoc test. (F) Quantification of AUC in panel E between 0 and 3 h. CBD3063 (1 to 10 mg/kg) and GBP exhibited comparable reversal of SNI-induced cold allodynia. P values as indicated; one-way ANOVA followed by the Dunnett post hoc test. n = 10 mice. (G) Dose–response curve analysis for cold aversion induced by CBD3063 as a percentage of the maximum possible effect. For all panels, error bars indicate mean ± SEM, groups composed of 50% males and 50% females. See Dataset S1 for full statistics.
Next, we investigated whether CBD3063’s antinociceptive effects were observable along the central pain processing pathway. In rodents, the majority of nociceptive stimuli are transmitted via the spino-parabrachio-amygdaloid pathway, making the parabrachial nucleus (PBN) a crucial brainstem hub responsible for transmitting nociceptive signals to higher-order brain regions, where they are integrated with cognitive and emotional input (32). We utilized in vivo fiber photometry to monitor calcium dynamics of glutamatergic neurons in the PBN during mechanical stimulation before and after induction of neuropathic pain (33) as well as after administration of either CBD3063 (10 mg/kg, i.p.) or GBP (30 mg/kg, i.p.) (Fig. 5 A and B). Glutamatergic PBN neurons showed heightened calcium responses to mechanical stimuli after SNI (Fig. 5 C–E), mirroring the increase in SNI-induced pain-like behaviors (Fig. 4B). Remarkably, both CBD3063 and GBP reduced the SNI-evoked increases in calcium transients in response to mechanical stimulation (Fig. 5 C–E). These results parallel our in vivo behavioral testing results (Fig. 4 B–G) and further indicate the potential of CBD3063 as a pain-relieving agent.
Fig. 5.

CBD3063 reduces SNI-evoked increases in parabrachial glutamatergic neuron activity. (A) Timeline describing the order of in vivo fiber photometry experiments. (B) Representative GCamp6s viral expression and fiber optic track in the PBN. (C–E) Summary of quantified AUC in response to (C) 0.07 g filament, (D) 1.0 g filament, and (E) pinprick stimuli at BL, post SNI, and following i.p. administration of CBD3063 (10 mg/kg) or GBP (30 mg/kg); Friedman tests followed by Dunn’s multiple comparisons (0.07 g, P = 0.0064, 1.0 g, P = 0.0064, pin prick, P = 0.0022. Dunn’s P values as indicated, N = 8 mice). (F–I) Traces showing average normalized fluorescence calcium response to 0.07 g filament, (J–M) 1.0 g filament, and (N–Q) pin prick; black trace represents the mean of all animals’ responses, gray cloud represents SEM, vertical line at x = 0 represents stimulus application. Groups composed of 50% males and 50% females. See Dataset S1 for full statistics.
We next tested CBD3063 in a model of chemotherapy-induced peripheral neuropathy. Paclitaxel (PAC) (8 mg/kg, i.p. for a total of 4 doses) but not vehicle produced robust mechanical and cold allodynia in male and female mice (Fig. 6 A–D). CBD3063 (9 mg/kg, i.p.) significantly reversed PAC-induced mechanical (Fig. 6 A and B) and cold allodynia (Fig. 6 C and D). Importantly, the antinociceptive dose of CBD3063 did not alter locomotor activity in naive (SI Appendix, Fig. S12) or chemotherapy induced peripheral neuropathy (CIPN) mice (SI Appendix, Fig. S13). Overall, these results show that targeting the Cav2.2–CRMP2 interaction provides significant pain relief in preclinical models of neuropathic pain.
Fig. 6.

CBD3063 reverses PAC-induced mechanical and cold hypersensitivity. (A) PWT measurements were performed at BL before 4 doses of i.p. injection of PAC (8 mg/kg) or vehicle, and again 21 d after the first PAC-injection (time 0). Mice were then injected i.p. with CBD3063 (9 mg/kg) or vehicle (10% DMSO in saline) and tested for mechanical sensitivity at 1, 3, 6, and 24 h. CBD3063 reversed PAC-induced mechanical allodynia. P value as indicated; two-way ANOVA, factors; time*treatment, and Tukey post hoc test (*P < 0.05 vs. corresponding PAC/vehicle group). (B) Quantification of AUC in panel A between 0 and 24 h after i.p. injection. P value as indicated; one-way ANOVA followed by the Tukey post hoc test. (C) Pre-PAC BL measurements of aversion time were assessed before 4 i.p. injections of PAC (8 mg/kg), and again on day 21 after the first PAC-injection (time 0). Mice were then injected i.p. with CBD3063 (9 mg/kg) or vehicle and tested for cold sensitivity to an acetone drop stimulus as above. CBD3063 reversed PAC-induced cold allodynia. Results were compared using two-way ANOVA, factors; time*treatment, and Tukey post hoc test (*P < 0.05 vs. correspondent PAC/vehicle group). (D) Quantification of AUC in panel C between 0 and 24 h after i.p. injection. P value as indicated; one-way ANOVA followed by the Tukey post hoc test. N = 8 mice per condition. For all panels, error bars indicate mean ± SEM, and groups were composed of 50% males and 50% females. See Dataset S1 for full statistics.
CBD3063 Is Antinociceptive in Inflammatory and Neuropathic Pain Via Multiple Routes of Administration.
We next explored CBD3063 administration across multiple models of pain, species, and routes of administration. First, we tested intranasal administration of CBD3063 in male and female rats in a model of trigeminal neuropathic pain. Chronic constriction injury of the infraorbital nerve (CION) reliably produced facial mechanical hypersensitivity in both male and female rats (Fig. 7 A–D). Notably, administration of CBD3063 (200 µg/20 µL, intranasal) but not saline reduced CION-induced mechanical hypersensitivity as assessed by both von Frey filaments (Fig. 7 A and B) and pinprick responsiveness (Fig. 7 C and D). Therefore, intranasally delivered CBD3063 blunts trigeminal neuropathic pain in rats. Next, we tested single intraplantar injection of CBD3063 in a mouse model of persistent inflammatory pain. Administration of Complete Freund’s adjuvant (CFA, 5 µg) into the hind paw of male and female mice (Fig. 7E) produced robust inflammation and mechanical hypersensitivity 2 d post-injection (Fig. 7F). CBD3063 (25 µg/5 µL, intraplantar) but not vehicle (10% DMSO in saline) administration robustly attenuated CFA-induced mechanical hypersensitivity for up to 4 h post-administration (Fig. 7 F and G). Last, we tested repeated i.t. administration of CBD3063 in the SNI model of neuropathic pain in male rats. CBD3063 (0.3 μg/kg, i.t.) was administered on days 3, 5, 7, 10, and 14 post nerve injury before von Frey testing 2 h postinjection (Fig. 7H). CBD3063 produced a reproducible reversal of SNI-induced mechanical hypersensitivity at an equivalent magnitude at all time points (Fig. 7H). These results demonstrate that CBD3063 does not produce tolerance (loss of antinociception) in a model of chronic neuropathic pain.
Fig. 7.
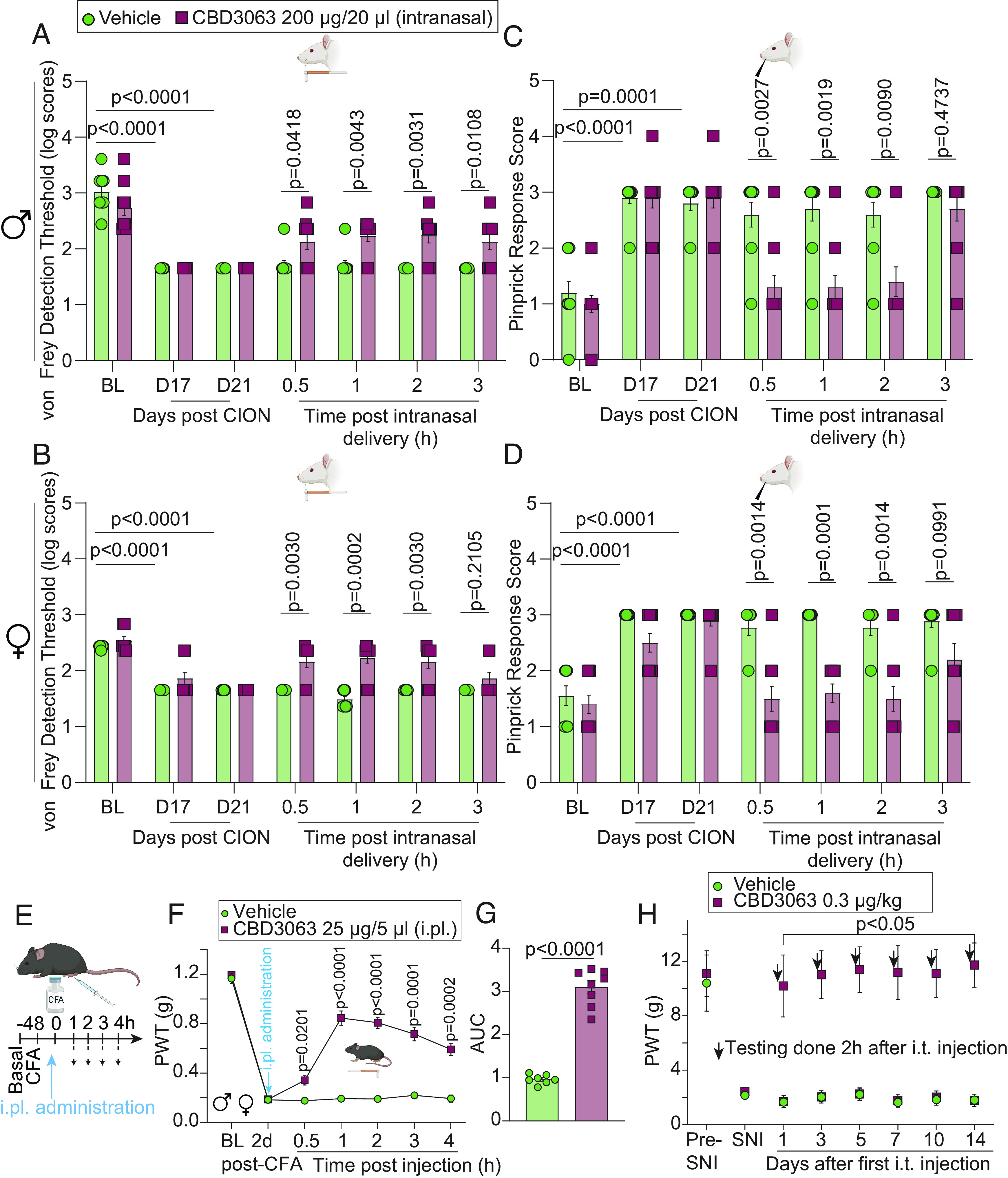
CBD3063 effectively alleviates pain-like behavior through multiple routes of administration. Male (A) and female (B) rats developed significant hypersensitivity to von Frey monofilaments at days 17 and 21 post CION compared to BL. Intranasal CBD3063 resulted in a significant increase in the von Frey detection threshold in the ipsilateral side of male and female rats compared to vehicle (10% DMSO in saline) at 0.5 to 3 h post CBD3063 administration in males and from 0.5 to 2 h pot-CBD3063 in females. Male (C) and female (D) rats developed significant hypersensitivity as indicated by increased pinprick response scores at days 17 and 21 post CION compared to BL. Intranasal CBD3063 resulted in a significant decrease in pinprick response scores in the ipsilateral side compared to vehicle-treated rats at 30 min, 1, and 2 h post CBD3063 administration in males and up to 3 h in females. P value as indicated; Multiple Mann-Whitney tests. n = 10 rats. (E) Male and female mice developed significant hypersensitivity to von Frey filaments 2 d after intraplantar injection of CFA (5 µg) into the left hindpaw. (F) Two days after CFA injection, CDB3063 or vehicle (10% DMSO in saline) was administrated intraplantarly (25 µg/5 µL; i.pl.) in the inflamed paw and mechanical sensitivity was recorded for the following 4 h. Administration of CBD3063 resulted in a significant increase in the PWT compared to vehicle. n = 7 to 8 mice per group (~50% females). Results were compared using two-way ANOVA, factors; time*treatment, and Tukey post hoc test. P values as indicated. (G) Quantification of AUC in panel F between 2 d post-CFA and 4 h after i.pl. injection of CBD3063. P values as indicated; unpaired t test; n = 7 to 8 mice (~50% females). (H) Repeated i.t. injection of CBD3063 (0.3 µg/kg) caused a significant increase in PWTs in SNI-injured male rats compared to vehicle treatment (10% DMSO in saline) at 1, 3, 5, 7, 10, and 14 d after the first injection. n = 5 male rats per group. Results were compared using two-way ANOVA, factors; time*treatment, and Bonferroni post hoc test. P values as indicated. For all panels, error bars indicate mean ± SEM. See Dataset S1 for full statistics.
CBD3063 Preserves the Protective Function of Pain in Uninjured Mice.
We next profiled the antinociceptive CBD3063 dose (10 mg/kg, i.p., Fig. 4) in comparison to GBP (30 mg/kg, i.p.) for responsiveness to mechanical and thermal sensory stimuli in naive (uninjured) male and female mice. First, neither CBD3063 nor GBP induced alterations in mechanical sensitivity assessed by von Frey filaments (Fig. 8 A and B). Second, GBP but not CBD3063 increased tail withdrawal latency to a 52 °C nociceptive stimulus (Fig. 8C). Third, neither CBD3063 nor GBP induced alterations in cool sensitivity assessed by acetone droplet application (Fig. 8 D and E). Fourth, GBP, but not CBD3063, increased latency of mice to withdrawal on a 52 °C hot-plate test (Fig. 8F). Importantly, these findings demonstrate that CBD3063, in contrast to GBP, does not affect somatosensation nor compromise the protective role of pain in naive mice.
Fig. 8.
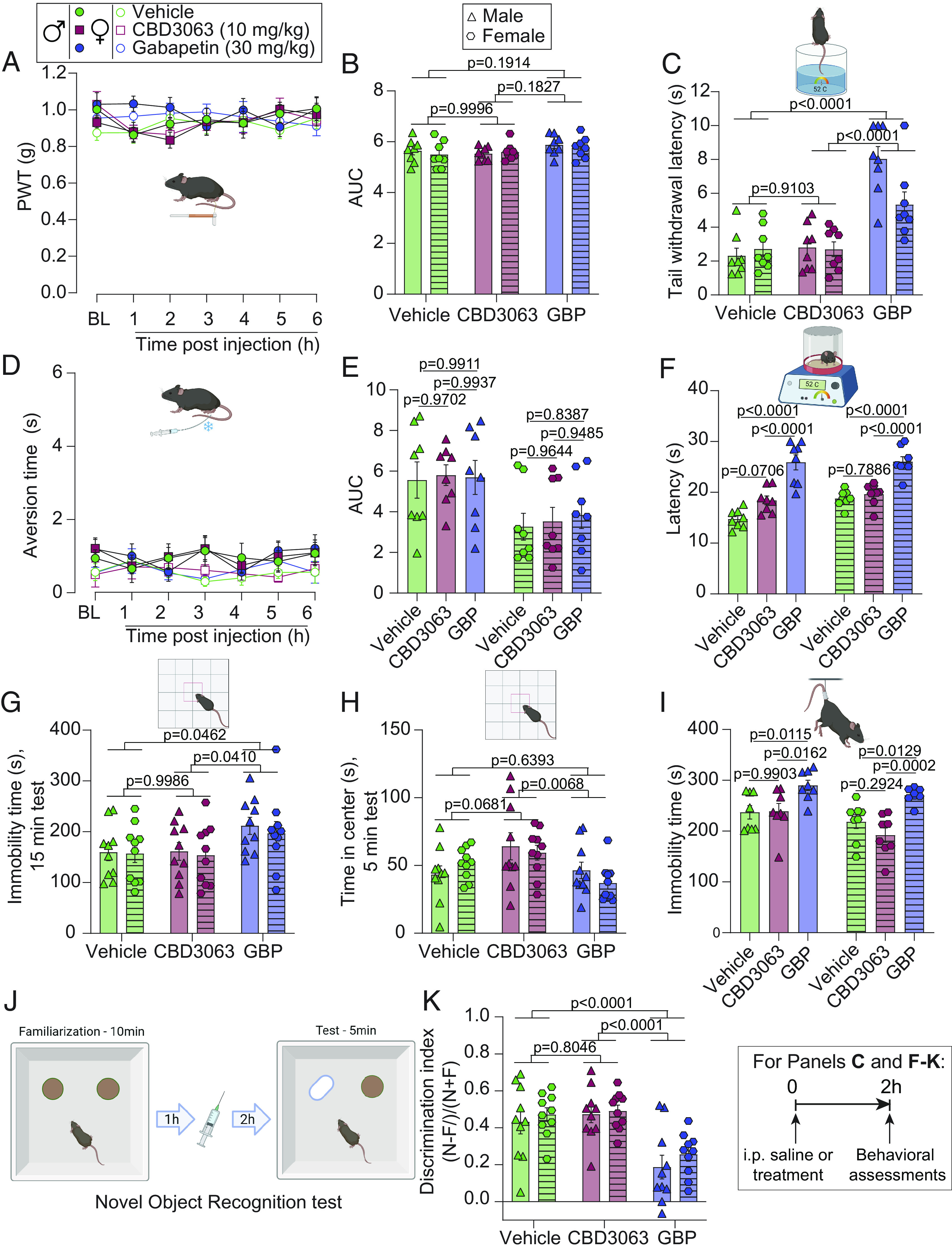
Intraperitoneal CBD3063 preserves adaptive pain without eliciting neurological side effects. (A) Time course of PWT after vehicle (10% DMSO in saline), CBD3063, or GBP in naive mice. BL measurements were taken before i.p. injection. (B) Quantification of AUC in panel A between BL and 6 h after i.p. injection. Neither CBD3063 nor GBP modified PWTs when compared to vehicle-treated mice. (C) Tail withdrawal latency measurements of mice. GBP, but not CBD3063, increased tail withdrawal latency compared to vehicle- and CBD3063-treated mice. (D) Time course of aversion time in response to acetone stimulation after vehicle, CBD3063, or GBP administration in mice. BL measurements were taken before injection. (E) Quantification of AUC in panel D between BL and 6 h after i.p. injection. Neither CBD3063 nor GBP modified aversion time when compared to vehicle-treated mice. (F) Withdrawal latency measurements revealed that GBP, but not CBD3063, increased latency compared to vehicle- or CBD3063-treated mice. (G) GBP produced sedative-like behaviors when compared with vehicle or CBD3063, assessed by duration of immobility in the Open Field during a 15-min test. (H) CBD3063 showed anxiolytic-like effects when compared with GBP and vehicle, indicated by time spent in the center of the Open Field during a 5-min test. (I) Immobility time evaluation in the Tail Suspension Test showing that GBP increased immobility time compared to vehicle and CBD3063-injected mice. CBD3063 did not affect immobility time compared to vehicle. (J) Cartoon depicting assessment of cognitive-like behaviors using the Novel Object Recognition (NOR) assay. Naive mice were first familiarized with two similar objects, followed by i.p. injection 1 h later (vehicle, CBD3063 or GBP), and 2 h after treatment tested for discrimination of a novel (N) vs. familiar (F) object. Discrimination-index = (N-F)/(N+F). (K) GBP showed a significant decrease in cognitive-like performance, suggested by decreased exploration of the novel object. CBD3063 did not compromise cognitive-like behavior. Injections were given intraperitoneally and 2 h before the test for panels B, C, E–I, and K. N = 10 mice per group for panels G, H, and K. N = 8 mice per group for panels A–F and I. Results from panels A and D were compared using two-way ANOVA, factors; time*treatment, and Tukey post hoc test. Quantification of AUC in panel B and E between 0 and 6 h after i.p. injection was analyzed by one-way ANOVA followed by the Tukey post hoc test. P values as indicated. Results from panels C, F, G–I, and K were compared using one-way ANOVA followed by the Tukey post hoc test. P values as indicated. For all panels, error bars indicate mean ± SEM. Vehicle = 10% DMSO in saline, CBD3063 (10 mg/kg) and Gabapentin = GBP (30 mg/kg). See Dataset S1 for full statistics.
Intraperitoneal Administration of CBD3063 Does Not Induce Sedative, Affective, or Cognitive Compromises in Male and Female Naïve Mice.
At pain-treatment doses, GBP commonly leads to sedation and cognitive impairment (34). To assess whether CBD3063 might have similar sedative effects, we compared CBD3063 (10 mg/kg, 10-fold higher than the reported ED50 in Fig. 4D) with GBP [30 mg/kg, approximately 2.6-fold higher than the published ED50 of 11.6 mg/kg for mice (35)] in naive mice, 2 h after administration. The Open Field Test revealed increased immobility in GBP-treated mice, indicative of sedation, as seen clinically (36). In contrast, CBD3063 did not alter immobility compared to the vehicle (Fig. 8G and SI Appendix, Fig. S12A).
Both clinical and preclinical evidence suggest that GBP can be anxiolytic (36). In addition to assessing sedative effects, our results from the OFT showed significant effects of treatment on the time spent in the center of the arena (first 5 min; Fig. 8H, full 15-min test; SI Appendix, Fig. S12B), a proxy for reduced anxiety-like behavior. Importantly, comparisons between treatment groups showed that CBD3063 resulted in significantly more exploration of the center zone than GBP, suggesting significant anxiolytic-like effects of CBD3063 compared to GBP and trending in comparison with vehicle (P = 0.068). These findings were seen in the initial 5 min of testing, and at 15 min (SI Appendix, Fig. S12B). We did not detect any significant differences in anxiety-like behavior effects between GBP and vehicle treatment.
Next, we examined whether CBD3063 affected behavioral despair or “depressive-like” behaviors. Our findings show that CBD3063 did not increase immobility time in the tail suspension test (Fig. 8I), suggesting that it does not induce depressive-like behaviors. GBP-treated mice exhibited a higher level of depressive-like behavior as suggested by increased immobility (Fig. 8I), but this may reflect the sedative effects from this drug, as seen in the OFT.
Finally, to assess the potential effects of CBD3063 on cognitive-like outcomes, we conducted the Novel Object Recognition test (Fig. 8J). Naive mice were first exposed to two similar objects and 1 h later injected with vehicle, CBD3063 (10 mg/kg), or GBP (30 mg/kg). Two hours after drug treatment, they were again exposed to the arena with one familiar and one new object. A discrimination index was calculated based on the time exploring the novel vs. familiar object. Consistent with previous studies (34), we detected a significant decrease in recognition of the novel object following treatment with GBP (Fig. 8K), reflecting impaired cognitive-like performance. Treatment with CBD3063 not disrupt recognition of the novel object, compared to vehicle. Potential sedative effects did not confound these results, as immobility was found to be a nonsignificant covariate (P = 0.991, ANCOVA) (Dataset S1). These results provide evidence that CBD3063 has no discernible impact on cognitive functions.
Discussion
Cav2.2 channels are almost exclusively expressed in neuronal tissue (37) and are abundant at presynaptic nerve terminals where they trigger the release of neurotransmitters via physically interacting with synaptic release machinery (38). For this reason, Cav2.2 channels are critical determinants of increased neuronal excitability and neurotransmission that accompany chronic neuropathic pain (5, 6). In 2018, Nieto-Rostro et al. reported a mouse expressing Cav2.2 channels with an extracellularly accessible hemagglutinin epitope tag engineered into their pore-forming Cav2.2 α1 subunit permitting identification of endogenous Cav2.2 channels in the plasma membrane of sensory neurons for the first time (39). These mice revealed disease-associated changes in Cav2.2 subcellular distribution in pain that confirmed these channels as attractive therapeutic targets. Indeed, this therapeutic potential has been demonstrated in Cav2.2 deficient mice which have reduced responses to mechanical stimuli, radiant heat, and chemical-induced inflammatory pain (2, 3).
Despite compelling evidence, clinical development of N-type Ca2+ channel blockers has proven challenging. Modulators of the Cav2.2–α2δ interaction (i.e., GBP and Pregabalin) are recommended as first-line treatment for neuropathic pain (40), but they only partially alleviate chronic pain, are implicated in overdose deaths (14), and cause a litany of side effects (29). A Cav2.2-selective, state-dependent inhibitor—N-triazole oxindole—was reported by Merck but cardiovascular and motor impairment hampered its further development (41). Another study reported a sulfonamide-derived, state-dependent inhibitor of Cav2.2, but this compound was limited by structural liabilities of this class of compounds (42). Targeting Cav2.2 interaction partners can reverse pain symptoms. The β3 auxiliary subunits boost Cav2.2 activation, membrane localization, and neurotransmitter release (43, 44). β3 protein expression rises in neuropathic pain (45), and β3-null mice show reduced pain from decreased Cav2.2 currents (46). Consistent with these findings, we reported that a small molecule targeting the Cav2.2–β interaction reduces currents through Cav2.2 channels, inhibits spinal neurotransmission, and alleviates neuropathic pain (15, 47, 48). Collectively, these studies converge on the idea that targeting auxiliary subunits of Cav2.2 channels is beneficial for pain management. Along these lines, accumulating evidence points to CRMP2 as an auxiliary subunit of Cav2.2 channels (49).
We demonstrated that disrupting the interaction between Cav2.2 and CRMP2 with a short peptide from CRMP2 (CBD3) is efficacious in reversing pain (18, 19, 22). Disrupting Cav2.2–CRMP2 binding with tat-CBD3 or myr-tat-CBD3 peptides has neuroprotective effects (50) without affecting memory, motor functions, anxiety/depression, or producing addictive behaviors (18, 22). Tat-CBD3 (51) and R9-CBD3 (50) also disrupt the CRMP2–NMDAR interaction to attenuate NMDAR-mediated currents, have neuroprotective effects against glutamate-induced Ca2+ dysregulation (51), and protect against neurotoxicity caused the toxic fragment of amyloid-β (Aβ)25-35 (50). These studies demonstrate that interfering with Cav2.2–CRMP2 binding is safe, beneficial, and does not produce unwanted side effects.
Despite CBD3’s documented analgesic success without side effects (18, 19, 22), lack of structural insight has so far hampered the development of small molecules capable of overcoming the shortcomings of peptide-based therapeutics such as i) a short half-life due to in vivo instability (52) from peptidases and excretion and ii) decreased bioavailability from digestive enzymes. To address these issues, we used a biophysical approach based on pharmacophore modeling to generate small molecule peptidomimetics that enhance CBD3 peptide’s bioactivity. Our MDS-based pipeline identifies key peptide residues (anchor residues) for pharmacophore modeling (20). We recently used this approach to discover a small molecule neuroprotectant that interferes with Kv2.1 binding to syntaxin1a (53). Here, using this pipeline, we identified the anchor motif that mediates CBD3 and Cav2.2 complex formation and used this motif for a pharmacophore screen to identify first-in-class small molecule peptidomimetics to disrupt the Cav2.2–CRMP2 interaction.
Pharmacokinetic (PK) analysis (conducted by Global DMPK Services, WuXi AppTec) demonstrated that CBD3063 (i.p., 10 mg/kg) reached a peak plasma concentration within 0.25 h (15 min; Tmax) with a mean maximum plasma concentration of 6327 ng/ml (~19 µM; Cmax) and a half-life of 3.1 h in mice. Consistent with this PK profile, CBD3063 was antinociceptive following i.p., intranasal, i.t., and intraplantar administration in male and female mice and rats. Importantly, CBD3063 could be repeatedly administered to effectively alleviate nerve injury–induced mechanical allodynia, in stark contrast to opioids which fail to alleviate chronic pain due to downregulation of receptors in neuropathic pain states and development of tolerance (54). CBD3063’s rapid effects may arise from its involvement in Cav2.2 endocytosis and plasma membrane trafficking which occur within ~15 min as reported for myr-tat-CBD3 (22). We demonstrate that CBD3063 parallels GBP in reversing pain-like behaviors and is as effective as GBP at reducing SNI-induced increases in net glutamatergic PBN neuron activity. Notably, CBD3063 did not induce sedation or impair affective and cognitive behaviors but exhibited anxiolytic properties when compared to GBP.
Previously we found that a single amino acid point mutant of CBD3 (tat-CBD3A6K) decreased R- and T-type Ca2+ currents in DRGs (55) and that tat-CBD3 peptide also decreased T-type currents (55) in sensory neurons. Importantly, in the present study, CBD3063 did not exert an effect on any other voltage-gated ion channels expressed in DRGs, which argues for selectivity for Cav2.2. We have shown that Cdk5-mediated CRMP2 phosphorylation at residue S522 increases its binding to Cav2.2, which leads to an increase in calcium influx (56). Here, we found no effect of CBD3063 on pS522 CRMP2. These data correlate with our previous findings that interrupting Cav2.2–CRMP2 interaction with myr-tat-CBD3 does not affect CRMP2 phosphorylation (22). We also investigated the regulation of sodium channels by CBD3063 since we previously described that phosphorylation and SUMOylation of CRMP2 regulates Nav1.7 channel trafficking and activity (25–27). Here, we found that CBD3063 does not directly affect sodium channel currents, nor does it exert an indirect influence through effects on CRMP2 phosphorylation or SUMOylation. These findings collectively suggest that CBD3063 neither inhibits other pain-relevant ion channels nor dysregulates CRMP2 posttranslational modifications.
A limitation of our study is CBD3063’s decrease of DRG neuron excitability. Although past studies show that inhibiting Cav2.2 channels can reduce DRG excitability (6, 22, 28), explaining this is complex due to the channels’ peak at depolarized potentials. We exposed DRGs to nerve growth factor, potentially boosting Cav2.2 expression (57), which might also reduce hyperexcitability. Calcium ions’ role in gene expression suggests that CBD3063 might influence other proteins affecting neuronal excitability (58). Additionally, CBD3063 might impact calcium-activated potassium channels like BK and SK channels. Delving deeper into these mechanisms requires further research.
Although CBD3063 exhibits promising drug-likeness (SI Appendix, Table S1), it does have marginal predicted blood–brain barrier penetration. Nevertheless, we noted CBD3063-induced anxiolytic properties comparable to GBP, suggesting efficacious CNS penetration. CBD3063 was also effective in reducing trigeminal neuropathic pain-like mechanical allodynia and acute nociception via the intranasal delivery route which can efficiently transport drugs to the brain through olfactory and trigeminal pathways, useful for targeted treatment of orofacial disorders with fewer systemic side effects. In upcoming research, enhancements aimed at optimizing the central nervous system exposure and the absorption, distribution, metabolism, and excretion characteristics of CBD3063 will be undertaken. This optimization process will involve reducing the topological polar surface area. This may be achieved through various strategies, including the modification or removal of acetamide and urea bonds, augmenting hydrophobic properties, or strategically introducing fluorine atoms to reduce the capacity of N-H donors. Further, compound potency/target affinity will be improved by exploring constraints (cyclization, ring size) to minimize loss of conformational entropy upon binding, and we will also explore changes of the piperidine and acyl groups (pink and blue areas, respectively; SI Appendix, Fig. S14) to improve its properties and efficacy.
In summary, our study revealed that CBD3063 disrupted the Cav2.2–CRMP2 interaction, reduced membrane Cav2.2 expression and Ca2+ currents, and decreased neurotransmission. Notably, CBD3063, via four different routes of administration, reversed nociceptive behaviors in four distinct pain models across two different species and both sexes. Importantly, this occurred without altering sensory, sedative, affective, or cognitive behaviors. Additionally, CBD3063 induced anxiolytic-like effects. These compelling findings not only validate our approach but also offer a strong foundation for potential application to other therapeutic targets.
Materials and Methods
Detailed descriptions of experiments and associated references are available in SI Appendix, SI Materials and Methods. This study aimed to create selective blockers of CaV2.2 channels for treating chronic pain. Leveraging the known regulation of CaV2.2 by the CRMP2 protein, we designed a molecule to mimic an antinociceptive CRMP2-derived peptide that detaches the CaV2.2–CRMP2 bond. Using a molecular dynamics method, we identified the CaV2.2 recognition motif, and then designed models to screen 27 million compounds on ZincPharmer. We validated the interaction in cell cultures, studied its effects on CaV2.2 trafficking, and examined the functional outcomes in DRG neurons and spinal cord slices using electrophysiological methods. Furthermore, we investigated the effects of our disruption strategy in naive animals as well as in three neuropathic pain and an inflammatory model to assay the off-target and on-target actions of this approach. Four routes of administration were used, including i.p., i.t., intraplantar, and intranasal. All electrophysiology, biochemistry, in vivo fiber photomometry, and behavior experiments were performed according to established protocols (18, 19). All procedures involving electrophysiology, biochemistry, and behavior adhered to established protocols. Animal protocols received approval from the College of Medicine’s Institutional Animal Care and Use Committees of the involved Universities, aligning with the NIH’s Guide for Care and Use of Laboratory Animals. We determined sample sizes from our past experiments. Researchers were blind to treatments, and animals were allocated to groups at random.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
Supported by NIH awards (NINDS NS098772 and NS120663, NIDA DA042852 to R.K.); A.M. by NINDS NS119263; J.W. by NIGMS GM115384 and NINDS NS121776; G.L. by the Rita Allen Foundation and NINDS NS121259; T.K. by NIGMS P30GM145497 to Ian Meng; and M.I.D. by NCI R01CA219637. All data are available in the main text, figures, SI Appendix.
Author contributions
K.G., U.S., T.S.N., H.N.A., A.C.-R., S.H., E.J.R.P., E.Z., P.S.A., M.P., M.K., H.H., G.L., T.K., J.W., M.I.D., O.A.K., C.J.C., and R.K. designed research; K.G., U.S., T.S.N., H.N.A., A.C.-R., S.H., E.J.R.P., Y.Z., P.D., S.L.-L., E.Z., U.K., R.S., E.K., B.M., D.G., W.Z., K.E.I., A.D., A.C., D.R., Y.L., X.L., Handoko, A.M., H.H., G.L., T.K., O.A.K., C.J.C., and R.K. performed research; Handoko, P.S.A., and R.K. contributed new reagents/analytic tools; K.G., T.S.N., H.N.A., A.C.-R., S.H., E.J.R.P., Y.Z., P.D., S.L.-L., E.Z., U.K., R.S., E.K., B.M., D.G., W.Z., K.E.I., A.D., A.C., S.P.-M., Y.L., X.L., Handoko, M.P., A.M., H.H., G.L., T.K., M.I.D., O.A.K., and R.K. analyzed data; U.S. performed computational screening; J.W. supervised student in his lab; C.J.C. performed in silico screening; R.K. provided funding and oversight of all work; and K.G., T.S.N., H.N.A., S.H., S.P.-M., M.K., T.K., J.W., M.I.D., O.A.K., C.J.C., and R.K. wrote the paper.
Competing interests
M.K. and R.K. are the co-founders of Regulonix LLC, a company developing non-opioids drugs for chronic pain.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Carlos J. Camacho, Email: ccamacho@pitt.edu.
Rajesh Khanna, Email: rk4272@nyu.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Heinke B., Balzer E., Sandkühler J., Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons Eur. J. Neurosci. 19, 103–111 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Kim C., et al. , Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol. Cell Neurosci. 18, 235–245 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Hatakeyama S., et al. , Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca(2+) channels. Neuroreport 12, 2423–2427 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Saegusa H., et al. , Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. Embo. J. 20, 2349–2356 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cizkova D., et al. , Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 147, 456–463 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Yang J., et al. , Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain. Behav. Immun. 71, 52–65 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Chaplan S. R., Pogrel J. W., Yaksh T. L., Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther. 269, 1117–1123 (1994). [PubMed] [Google Scholar]
- 8.Scott D. A., Wright C. E., Angus J. A., Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur. J. Pharmacol. 451, 279–286 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Doggrell S. A., Intrathecal ziconotide for refractory pain. Expert. Opin. Investig. Drugs 13, 875–877 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Nair A. S., Poornachand A., Kodisharapu P. K., Ziconotide: Indications, adverse effects, and limitations in managing refractory chronic pain. Ind. J. Palliat. Care 24, 118–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrich J., et al. , Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc. Natl. Acad. Sci. U.S.A. 105, 3628–3633 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer C. S., et al. , The anti-allodynic alpha(2)delta ligand pregabalin inhibits the trafficking of the calcium channel alpha(2)delta-1 subunit to presynaptic terminals in vivo. Biochem. Soc. Trans. 38, 525–528 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Evoy K. E., Peckham A. M., Covvey J. R., Tidgewell K. J., Gabapentinoid pharmacology in the context of emerging misuse liability. J. Clin. Pharmacol. 61 (suppl. 2), S89–s99 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Kuehn B. M., Gabapentin increasingly implicated in overdose deaths. Jama 327, 2387 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Khanna R., et al. , Targeting the CaValpha-CaVbeta interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain 160, 1644–1661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brittain J. M., et al. , An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi X. X., et al. , Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 122, 4351–4362 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brittain J. M., et al. , Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nat. Med. 17, 822–829 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moutal A., et al. , Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. Br. J. Pharmacol. 175, 2244–2260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajamani D., Thiel S., Vajda S., Camacho C. J., Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 11287–11292 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sterling T., Irwin J. J., ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model 55, 2324–2337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.François-Moutal L., et al. , A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156, 1247–1264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez K., et al. , Stereospecific effects of benzimidazolonepiperidine compounds on T-Type Ca(2+) channels and pain. ACS Chem. Neurosci. 13, 2035–2047 (2022), 10.1021/acschemneuro.2c00256. [DOI] [PubMed] [Google Scholar]
- 24.Moutal A., et al. , Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Mol. Neurobiol. 56, 6736–6755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dustrude E. T., Wilson S. M., Ju W., Xiao Y., Khanna R., CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dustrude E. T., et al. , Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc. Natl. Acad. Sci. U.S.A. 113, E8443–E8452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dustrude E. T., et al. , A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin) 11, 316–328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pitake S., Middleton L. J., Abdus-Saboor I., Mishra S. K., Inflammation induced sensory nerve growth and pain hypersensitivity requires the N-type calcium channel Cav2.2. Front. Neurosci. 13, 1009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zamponi G. W., Striessnig J., Koschak A., Dolphin A. C., The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S. E., Kim J. H., Involvement of substance P and calcitonin gene-related peptide in development and maintenance of neuropathic pain from spinal nerve injury model of rat. Neurosci. Res. 58, 245–249 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Nelson T. S., et al. , Spinal neuropeptide Y Y1 receptor-expressing neurons are a pharmacotherapeutic target for the alleviation of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 119, e2204515119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauriau C., Bernard J. F., Pain pathways and parabrachial circuits in the rat. Exp. Physiol. 87, 251–258 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Sun L., et al. , Parabrachial nucleus circuit governs neuropathic pain-like behavior. Nat. Commun. 11, 5974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suto T., Eisenach J. C., Hayashida K., Peripheral nerve injury and gabapentin, but not their combination, impair attentional behavior via direct effects on noradrenergic signaling in the brain. Pain 155, 1935–1942 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miranda H. F., et al. , Antinociceptive synergism of gabapentin and nortriptyline in mice with partial sciatic nerve ligation. Pharmacology 95, 59–64 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Hong J. S. W., et al. , Gabapentin and pregabalin in bipolar disorder, anxiety states, and insomnia: Systematic review, meta-analysis, and rationale. Mol. Psychiatry 27, 1339–1349 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nowycky M. C., Fox A. P., Tsien R. W., Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443 (1985). [DOI] [PubMed] [Google Scholar]
- 38.Evans A. R., Nicol G. D., Vasko M. R., Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 712, 265–273 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Nieto-Rostro M., Ramgoolam K., Pratt W. S., Kulik A., Dolphin A. C., Ablation of alpha2delta-1 inhibits cell-surface trafficking of endogenous N-type calcium channels in the pain pathway in vivo. Proc. Natl. Acad. Sci. U.S.A. 115, E12043–E12052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Attal N., et al. , EFNS guidelines on pharmacological treatment of neuropathic pain. Eur. J. Neurol. 13, 1153–1169 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Abbadie C., et al. , Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J. Pharmacol. Exp. Ther. 334, 545–555 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Shao P. P., et al. , Aminopiperidine sulfonamide Cav2.2 channel inhibitors for the treatment of chronic pain. J. Med. Chem. 55, 9847–9855 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Scott V. E., et al. , Beta subunit heterogeneity in N-type Ca2+ channels. J. Biol. Chem. 271, 3207–3212 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Richards M. W., Butcher A. J., Dolphin A. C., Ca2+ channel beta-subunits: Structural insights AID our understanding Trends Pharmacol. Sci. 25, 626–632 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Li L., et al. , Up-regulation of Cavbeta3 subunit in primary sensory neurons increases voltage-activated Ca2+ channel activity and nociceptive input in neuropathic pain. J. Biol. Chem. 287, 6002–6013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami M., et al. , Pain perception in mice lacking the beta3 subunit of voltage-activated calcium channels. J. Biol. Chem. 277, 40342–40351 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Khanna R., et al. , Targeting the CaVα-CaVβ interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain 160, 1644–1661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ran D., et al. , Comparison of quinazoline and benzoylpyrazoline chemotypes targeting the CaVα-β interaction as antagonists of the N-type CaV2.2 channel. Channels (Austin) 15, 128–135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Striessnig J., Getting a handle on Ca(V)2.2 (N-type) voltage-gated Ca(2+) channels Proc. Natl. Acad. Sci. U.S.A. 115, 12848–12850 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ji Y., et al. , CRMP2-derived peptide ST2-104 (R9-CBD3) protects SH-SY5Y neuroblastoma cells against Abeta25-35-induced neurotoxicity by inhibiting the pCRMP2/NMDAR2B signaling pathway. Chem. Biol. Interact. 305, 28–39 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Brustovetsky T., Pellman J. J., Yang X. F., Khanna R., Brustovetsky N., Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity J. Biol. Chem. 289, 7470–7482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruckdorfer T., Marder O., Albericio F., From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future. Curr. Pharm. Biotechnol. 5, 29–43 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Yeh C. Y., et al. , Defining the Kv2.1-syntaxin molecular interaction identifies a first-in-class small molecule neuroprotectant. Proc. Natl. Acad. Sci. U.S.A. 116, 15696–15705 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan M. M., Christie M. J., Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 164, 1322–1334 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piekarz A. D., et al. , CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol. Pain 8, 54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brittain J. M., Wang Y., Eruvwetere O., Khanna R., Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with Cav2.2. FEBS Lett. 586, 3813–3818 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Baldelli P., Forni P. E., Carbone E., BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons Eur. J. Neurosci. 12, 4017–4032 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Fields R. D., Effects of ion channel activity on development of dorsal root ganglion neurons. J. Neurobiol. 37, 158–170 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information.